
ibuprofénu vo forme suspenzie.
Pre vydávanie bez lekárskeho predpisu je maximálna denná dávka obmedzená na 1,2 g ibuprofénu. Ak u detí (od 6 rokov do 12 rokov) je tento liek potrebné podávať viac ako 3 dni alebo sa príznaky
ochorenia zhoršujú, je potrebné poradiť sa s lekárom.
U dospelých sa nemá tento liek užívať viac ako 3 dni v prípade horúčky a nie viac ako 4 dni pri liečbe bolesti. Ak príznaky pretrvávajú alebo sa zhoršujú, pacientovi sa odporúča, aby sa obrátil na lekára. Má sa použiť najnižšia účinná dávka počas najkratšieho obdobia, ktoré je potrebné na zmiernenie symptómov (pozri časť 4.4).
Spôsob podávani a
Filmom obalené tablety sa prehĺtajú celé, zapíjajú sa dostatočným množstvom tekutiny.
Ak sa objavia gastrointestinálne ťažkosti, odporúča sa podať tablety s malým množstvom potravy či zapiť ich mliekom.
4.3 Kontraindikácie
- Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1, alebo precitlivenosť na kyselinu acetylsalicylovú a iné nesteroidové antiflogistiká prejavujúca sa ako astma, urtikária a iné alergické reakcie.
- Gastrointestinálne krvácanie alebo perforácia súvisiaca s liečbou NSAID v anamnéze, aktívny alebo rekurentný peptický vred/krvácanie v anamnéze (dva alebo viac prípadov potvrdených
vredov alebo krvácanie).
- Závažné srdcové zlyhanie (trieda IV NYHA), poruchy hemokoagulácie a hemopoézy.
- Tretí trimester gravidity.
Ibalgin 400 nie je určený deťom do 12 rokov vzhľadom na veľkosť jednotlivej dávky.
Ibalgin 200 sa nepodáva deťom mladším ako 6 rokov. Pre deti mladšie ako 6 rokov je určený liek
Ibalgin Baby vo forme suspenzie.
4.4 Osobitné upozornenia a opatrenia pri používaní
Ibalgin sa nesmie užívať súbežne s inými NSAID vrátane selektívnych inhibítorov cyklooxygenázy-2. Nežiaduce účinky sa môžu minimalizovať užitím najnižšej účinnej dávky počas najkratšieho obdobia, ktoré je potrebné na kontrolu symptómov (pozri GI a kardiovaskulárne riziko nižšie).
Sta rší paci ent i
Starší pacienti majú zvýšenú frekvenciu nežiaducich účinkov spôsobených NSAID, zvlášť gastrointestinálneho krvácania a perforácie, ktoré môžu byť aj smrteľné (pozri časť 4.8).
Gastrointestinálne krvácanie, ulcerácia a perforácia: GIT krvácanie, ulcerácia alebo perforácia, ktoré môžu byť smrteľné, sú zaznamenané u všetkých NSAID a môžu vzniknúť kedykoľvek počas liečby, s predchádzajúcimi varovnými príznakmi, alebo bez nich, alebo bez predchádzajúcej anamnézy závažných gastrointestinálnych stavov. Riziko vzniku gastrointestinálneho krvácania, ulcerácie či
perforácie sa zvyšuje so stúpajúcou dávkou NSAID, u pacientov s anamnézou vredovej choroby, predovšetkým, pokiaľ bola vredová choroba komplikovaná krvácaním alebo perforáciou (pozri časť
4.3) a u starších pacientov. U týchto pacientov treba začať liečbu s čo najnižšou možnou dávkou.
U týchto pacientov a tiež u pacientov vyžadujúcich súčasnú liečbu nízkymi dávkami kyseliny acetylsalicylovej alebo iných látok zvyšujúcich gastrointestinálne riziko sa má zvážiť podanie protektívnej liečby (napr. misoprostolom alebo inhibítormi protónovej pumpy) (pozri časť 4.5). Pacienti s anamnézou GIT toxicity, predovšetkým starší pacienti, majú byť poučení, aby hlásili akékoľvek nezvyčajné abdominálne príznaky (predovšetkým gastrointestinálne krvácanie) a to zvlášť v začiatočných štádiách liečby.
Zvýšená opatrnosť je nevyhnutná u pacientov užívajúcich súčasne lieky zvyšujúce riziko vzniku ulcerácie alebo krvácania, ako sú perorálne kortikosteroidy, antikoagulanciá ako warfarín, selektívne
inhibítory spätného vychytávanie sérotonínu alebo doštičkové inhibítory ako je kyselina
acetylsalicylová (pozri časť 4.5).
Pokiaľ sa u pacienta liečeného Ibalginom objaví gastrointestinálne krvácanie alebo ulcerácia, musí sa liek vysadiť.
NSAID sa majú podávať opatrne u pacientov s anamnézou gastrointestinálneho ochorenia (ulcerózna kolitída, Crohnova choroba), pretože by mohlo dôjsť k exacerbácii týchto chorôb (pozri časť 4.8). Pred začatím liečby u pacientov s hypertenziou a/alebo zlyhávaním srdca je potrebná opatrnosť
(konzultácia s lekárom alebo lekárnikom), pretože v súvislosti s liečbou NSAID boli hlásené retencia tekutín, hypertenzia a edémy.
Zvýšená opatrnosť je potrebná pri srdcovej, renálnej a hepatálnej insuficiencii, u astmatikov, pri systémovom lupus erythematosus a iných ochoreniach spojivového tkaniva (riziko aseptickej meningitídy).
U rizikových pacientov, t.j. u pacientov s obmedzením funkcie srdca a obličiek, liečených diuretikami či pri dehydratácii akejkoľvek etiológie sa odporúča kontrola renálnych funkcií. Ak sa objavia poruchy vízusu, nezreteľné videnie, skotómy alebo poruchy farbocitu, treba liečbu prerušiť. Pri dlhodobej terapii je vhodná kontrola krvného obrazu a rutinné sledovanie pečeňových funkcií.
Pri zhoršení pečeňových funkcií v súvislosti s podávaním ibuprofénu je vhodné terapiu vysadiť, potom sa zvyčajne stav normalizuje. U pacientov užívajúcich kumarínové antikoagulanciá je vhodná častejšia kontrola hemokoagulačných parametrov. Takisto je vhodná občasná kontrola glykémie.
K ar di ovas kul ár ne a c er ebr ovas kul ár n e úči nky
Klinické štúdie naznačili, že používanie ibuprofénu, najmä vo vysokých dávkach (2 400 mg/deň) môže byť spojené s malým zvýšeným rizikom arteriálnych trombotických udalostí (napríklad infarkt myokardu alebo mozgová príhoda). Epidemiologické štúdie celkovo nenaznačujú, že by nízke dávky ibuprofénu (napr. 1 200 mg/deň) boli spojené so zvýšeným rizikom arteriálnych trombotických udalostí.
Pacienti s nekontrolovanou hypertenziou, kongestívnym zlyhaním srdca (triedy II – III NYHA),
diagnostikovaným ischemickým ochorením srdca, ochorením periférnych artérií
a cerebrovaskulárnym ochorením majú byť liečení ibuprofénom len po dôkladnom zvážení a nemajú sa používať vysoké dávky (2 400 mg/deň).
Pred začatím dlhodobej liečby pacientov s rizikovými faktormi pre vznik kardiovaskulárnych udalostí
(napr. hypertenzia, hyperlipidémia, diabetes mellitus, fajčenie) je potrebné dôsledné zváženie, najmä
v prípade, že sú potrebné vysoké dávky ibuprofénu (2 400 mg/deň).
Zá važné ko žné r ea kci e
V súvislosti s liečbou NSAID boli veľmi zriedkavo hlásené závažné kožné reakcie, niektoré z nich smrteľné, vrátane exfoliatívnej dermatitídy, Stevensovho-Johnsonovho syndrómu a toxickej
epidermálnej nekrolýzy (pozri časť 4.8.). U pacientov, u ktorých pravdepodobne existuje najvyššie
riziko týchto reakcií na začiatku liečby, sa reakcia vyskytuje vo väčšine prípadov v prvom mesiaci liečby. V súvislosti s liekmi obsahujúcimi ibuprofén bola hlásená akútna generalizovaná
exantematózna pustulóza (AGEP). Ibalgin je potrebné vysadiť pri prvom výskyte prejavov a
príznakov závažných kožných reakcií, ako je kožná vyrážka, lézie na slizniciach alebo akýkoľvek iný
prejav precitlivenosti.
Ma skovani e s ympt ómo v exi stuj úci ch i nf ekcií
Ibalgin môže maskovať symptómy infekcie, čo môže viesť k oneskorenému začatiu vhodnej liečby, a tým aj k zhoršeniu výsledku infekcie. Táto skutočnosť sa pozorovala v prípade bakteriálnej pneumónie získanej v komunite a bakteriálnych komplikácií súvisiacich s ovčími kiahňami. Ak sa Ibalgin podáva na zníženie horúčky alebo zmiernenie bolesti súvisiacej s infekciou, odporúča sa sledovanie infekcie.
V podmienkach mimo nemocnice je potrebné, aby sa pacient obrátil na lekára, pokiaľ symptómy pretrvávajú alebo sa zhoršujú.
Počas ovčích kiahní sa odporúča vyhnúť sa užívaniu lieku Ibalgin. Počas liečby nie je vhodné pitie alkoholických nápojov a fajčenie.
Pedi at ri cká popul áci a
Existuje riziko poškodenia obličiek u dehydratovaných detí a dospievajúcich.
Tento liek obsahuje menej ako 1 mmol (23 mg) sodíka v jednej tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Pri súbežnom podávaní ibuprofénu (najmä vo vysokých dávkach) s antikoagulanciami, ako je warfarín dochádza k predĺženiu protrombínového času a zvýšenému riziku krvácania. Fenobarbital zrýchľuje metabolizáciu ibuprofénu; ibuprofén zvyšuje plazmatické hladiny lítia, digoxínu a fenytoínu, zvyšuje toxicitu metotrexátu a baklofénu. Súbežné podanie kortikoidov alebo ďalších nesteroidných antireumatík zvyšuje riziko krvácania do GIT a riziko vzniku vredovej choroby, antiagregačné lieky
a SSRI (inhibítory spätného vychytávania sérotonínu) zvyšujú riziko gastrointestinálneho krvácania. Ibuprofén znižuje urikozúrický účinok probenecidu a sulfínpyrazónu.
Môže sa znížiť účinok diuretík a antihypertoník a mifepristónu. Súbežné podanie káliumšetriacich diuretík môže viesť k hyperkaliémii. Pri súčasnom podávaní chinolónových antibiotík a nesteroidných antireumatík sa môže zvýšiť riziko vzniku kŕčov. Zásahom ibuprofénu do syntézy prostaglandínov
v obličkách sa môže zvýšiť toxicita cyklosporínu.
Kys eli na acet ylsali cyl ová
Súbežné podávanie ibuprofénu a kyseliny acetylsalicylovej sa neodporúča z dôvodu možných zvýšených nežiaducich udalostí.
Experimentálne údaje naznačujú, že ibuprofén môže pri súčasnom dávkovaní kompetitívne inhibovať účinok nízkej dávky kyseliny acetylsalicylovej na agregáciu trombocytov. Hoci existujú nejasnosti
s ohľadom na extrapoláciu týchto údajov na klinickú situáciu, nedá sa vylúčiť možnosť, že pravidelné,
dlhodobé používanie ibuprofénu môže znížiť kardioprotektívny účinok nízkej dávky kyseliny acetylsalicylovej. V prípade príležitostného užívania ibuprofénu nie je klinicky relevantný účinok pravdepodobný (pozri časť 5.1).
4.6 Fertilita, gravidita a laktácia
Gravidita
Inhibícia syntézy prostaglandínov môže nepriaznivo ovplyvniť graviditu a/alebo vývoj embrya alebo plodu. Údaje z epidemiologických štúdií poukazujú na zvýšené riziko potratu, malformácií srdca
a gastroschízy po užívaní inhibítorov syntézy prostaglandínov na začiatku gravidity. Absolútne riziko
kardiovaskulárnych malformácií bolo zvýšené z menej než 1 % na približne 1,5 %. Predpokladá sa, že riziko sa zvyšuje s dávkou a dĺžkou liečby. U zvierat ukázalo podávanie inhibítorov syntézy prostaglandínov zvýšenie pre- a postimplantačných strát a embryo-fetálnu letalitu. Naviac u zvierat,
ktoré dostávali v priebehu organogenézy inhibítory syntézy prostaglandínov bola popísaná zvýšená incidencia rôznych malformácií, vrátane kardiovaskulárnych.
Pokiaľ to nie je jednoznačne nevyhnutné, ibuprofén sa nemá podávať počas prvého a druhého
trimestra gravidity. Pokiaľ ibuprofén užíva žena, ktorá sa snaží otehotnieť alebo v priebehu prvého
a druhého trimestra gravidity, má užívať nízke dávky a liečba má byť čo najkratšia.
Počas tretieho trimestra gravidity môžu všetky inhibítory syntézy prostaglandínov vystaviť
plod:
- kardiopulmonálnej toxicite (s predčasným uzáverom ductus arteriosus a pulmonárnej
hypertenzie);
- renálnej dysfunkcii, ktorá môže progredovať do zlyhania obličiek s oligohydramniónom;
matku a plod na konci gravidity:
- možnému predĺženiu času krvácania; antiagregačnému
- inhibícii kontrakcií maternice vedúcich k oneskorenému alebo predĺženému pôrodu.
Preto je ibuprofén kontraindikovaný počas tretieho trimestra gravidity.
Doj če ni e
V nedostatočnom počte doteraz dostupných štúdií sa preukázalo, že ibuprofén prestupuje do materského mlieka len vo veľmi nízkych koncentráciách. Neodporúča sa, aby ibuprofén užívali dojčiace matky.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ibalgin 200 a Ibalgin 400 nemá žiadny alebo má zanedbateľný vplyv na schopnosti viesť vozidlá
a obsluhovať stroje.
4.8 Nežiaduce účinky
Ibuprofén môže spôsobiť nasledujúce nežiaduce účinky (rozdelené do skupín podľa terminológie MedDRA s uvedením frekvencie výskytu: veľmi časté (³ 1/10); časté (³ 1/100 až < 1/10); menej časté (³ 1/1 000 až < 1/100); zriedkavé (³ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (nie je možné určiť z dostupných údajov).
T
riedy orgánových
systémov podľa databázy
MeDRA
Poruchy krvi
a lymfatického systému
Poruchy imunitného systému
Frekvencia
výskytu Nežiaduci účinok
veľmi zriedkavé poruchy krvotvorby (neutropénia, agranulocytóza, aplastická alebo hemolytická anémia, trombocytopénia)
zriedkavé hypersenzitívna reakcia (horúčka, vyrážka,
hepatotoxicita)
Poruchy metabolizmu
a výživy
veľmi zriedkavé retencia sodíka a tekutín
Psychické poruchy veľmi zriedkavé nespavosť, depresia, emočná labilita
Poruchy nervového
systému
menej časté závraty, bolesti hlavy
zriedkavé aseptická meningitída (zvlášť u pacientov so systémovým lupus erythematosus a u niektorých kolagenóz)
Poruchy oka zriedkavé poruchy vízu, poruchy percepcie farieb, toxická
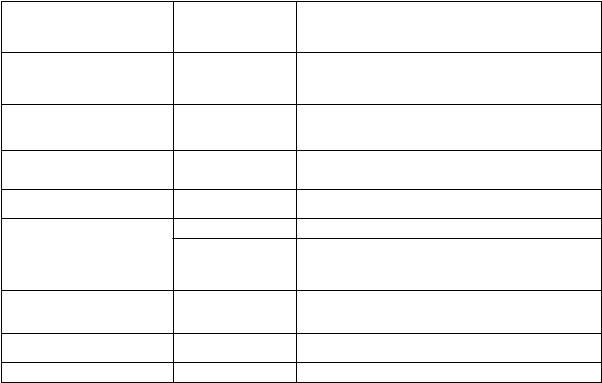
amblyopia
Poruchy ucha a labyrintu neznáme porucha sluchu
zriedkavé zlyhávanie srdca
Poruchy srdca a srdcovej
činnosti
|
veľmi zriedkavé
|
palpitácie
|
Poruchy ciev
|
veľmi zriedkavé
|
hypertenzia
|
Poruchy dýchacej sústavy,
hrudníka a mediastína
|
zriedkavé
|
bronchospazmus (hlavne u astmatikov)
|
Poruchy
gastrointestinálneho traktu
|
veľmi časté
|
nauzea, vracanie, pálenie záhy, hnačka,
obstipácia, nadúvanie
|
|
časté
|
bolesti v epigastriu
|
|
zriedkavé
|
gastritída, žalúdočný vred, duodenálny vred,
krvácanie z GIT (meléna, hemateméza),
perforácia gastrointestinálneho traktu
|
|
veľmi zriedkavé
|
ulcerózna stomatitída, exacerbácia Crohnovej
choroby, exacerbácia ulceróznej kolitídy
|
Poruchy pečene a žlčových
ciest
|
zriedkavé
|
poruchy pečeňových funkcií (zvyčajne
reverzibilné)
|
Poruchy kože
a podkožného tkaniva
|
veľmi zriedkavé
|
bulózna reakcia vrátane Stevensovho-
Johnsonovho syndrómu a toxickej epidermálnej nekrolýzy
|
|
neznáme
|
lieková reakcia s eozinofíliou a systémovými
príznakmi (syndróm DRESS)
|
|
neznáme
|
fotosenzitívna reakcia kože
Akútna generalizovaná exentematózna pustulóza
(AGEP)
|
Poruchy obličiek
a močových ciest
|
veľmi zriedkavé
|
cystitída, hematúria, poruchy obličkových funkcií
vrátane intersticiálnej nefritídy alebo nefrotického syndrómu
|
Celkové poruchy a reakcie
v mieste podania
|
zriedkavé
|
edémy
|
Laboratórne a funkčné
vyšetrenia
|
veľmi zriedkavé
|
pokles krvného tlaku
|
Medzi nežiaducimi účinkami sú gastrointestinálne nežiaduce účinky najčastejšie. Môže sa vyskytnúť
žalúdočný vred, perforácia alebo GI krvácanie, ktoré môžu byť fatálne.'
V súvislosti s terapiou NSAID boli hlásené edémy, hypertenzia a zlyhávanie srdca.
Klinické štúdie naznačili, že používanie ibuprofénu, najmä vo vysokých dávkach (2 400 mg/deň) môže byť spojené s malým zvýšením rizika arteriálnych trombotických udalostí (napríklad infarkt myokardu alebo mozgová príhoda) (pozri časť 4.4).
Hl ás eni e podozr ení na nežiaduc e r ea kcie Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hl ásenia uvede né v
Prí l ohe V .4.9 PredávkovanieIbuprofén v dávke do 100 mg/kg telesnej hmotnosti je netoxický, v dávke nad 400 mg/kg telesnej hmotnosti môže spôsobiť závažnú intoxikáciu: môžu vzniknúť poruchy CNS – bolesti hlavy, závraty, nystagmus, kŕče, ktoré sa môžu vystupňovať až do bezvedomia. Ďalej sa môžu objaviť bolesti brucha, nevoľnosť, vracanie. V závažných prípadoch môže dôjsť k hypotenzii, acidóze, zastaveniu dychu
a cyanóze.
Pri závažnej otrave sa môže vyskytnúť metabolická acidóza.
Terapia akútneho predávkovania: čo najskôr uskutočniť výplach žalúdka s podaním aktívneho uhlia a preháňadlá či vyvolať dáviaci reflex. Terapia je podporná a symptomatická – kontrola a úprava bilancie tekutín a elektrolytov, udržiavanie respiračných a kardiovaskulárnych funkcií, pri kŕčoch je možné podať diazepam, pri hypotenzii plazmaexpandéry, prípadne dopamín či norepinefrín. Forsírovaná diuréza a hemodialýza sa ukázali neúčinné, o hemoperfúzii nie sú údaje.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmaceutická skupina: antiflogistiká a antireumatiká, deriváty kyseliny propiónovej, ATC kód:
M01AE01
Ibuprofén, derivát kyseliny propiónovej, je nesteroidové antireumatikum s dobrým analgetickým, protizápalovým a antipyretickým účinkom. V nižších dávkach pôsobí analgeticky, vo vyšších protizápalovo. Protizápalový účinok je daný inhibíciou cyklooxygenázy s nasledovnou inhibíciou biosyntézy prostaglandínov. Zápal sa zmierňuje znížením uvoľňovania mediátorov zápalu
z granulocytov, bazofilov a žírnych buniek. Ibuprofén znižuje citlivosť ciev voči bradykinínu
a histamínu, ovplyvňuje produkciu lymfokínov v T-lymfocytoch a potláča vazodilatáciu. Tlmí aj
agregáciu krvných doštičiek. Vyznačuje sa dobrou gastrointestinálnou znášanlivosťou. Analgetický účinok nastupuje po polhodine, maximálny antipyretický účinok sa dosiahne po
2 – 4 hodinách. Antipyretický účinok trvá 4 – 8 aj viac hodín, analgetický 4 – 6 hodín.
Experimentálne údaje naznačujú, že ibuprofén môže pri súčasnom dávkovaní kompetitívne inhibovať účinok nízkej dávky kyseliny acetylsalicylovej na agregáciu trombocytov.
Niektoré farmakodynamické štúdie preukázali, že pri jednej dávke ibuprofénu 400 mg užitého
v priebehu 8 hodín pred alebo v priebehu 30 minút po užití dávky kyseliny acetylsalicylovej
s okamžitým uvoľňovaním (81 mg) došlo k zníženému účinku kyseliny acetylsalicylovej na tvorbu tromboxánu alebo agregáciu trombocytov.
Hoci existujú nejasnosti s ohľadom na extrapoláciu týchto údajov na klinickú situáciu, nedá sa vylúčiť možnosť, že pravidelné, dlhodobé používanie ibuprofénu môže znížiť kardioprotektívny účinok nízkej
dávky kyseliny acetylsalicylovej. V prípade príležitostného používania ibuprofénu sa žiaden klinicky
relevantný účinok nepovažuje za pravdepodobný (pozri časť 4.5).
5.2 Farmakokinetické vlastnosti
Po perorálnom podaní sa rýchlo a dobre vstrebáva, pri podaní nalačno dosahuje vrchol plazmatickej koncentrácie už po 45 minútach, pri podaní s jedlom cca po 1 – 3 hodinách. Po rektálnej aplikácii sa ibuprofén vstrebáva pomalšie, maximálna koncentrácia v sére sa dosiahne 2 hodiny po aplikácii. Ibuprofén sa viaže na plazmatické proteíny, ale väzba je reverzibilná. Pomerne rýchlo sa metabolizuje v pečeni a vylučuje močom, najmä vo forme metabolitov a ich konjugátov, menšia časť sa vylučuje žlčou do stolice. Biologický polčas je asi 2 hodiny. Ak sa zníži vylučovanie, môže dôjsť ku kumulácii lieku v organizme. Exkrécia ibuprofénu sa ukončí 24 hodín po podaní poslednej dávky. Biologická dostupnosť sa minimálne alteruje prítomnosťou stravy. Ibuprofén prestupuje cez placentárnu bariéru, vylučuje sa do materského mlieka v množstve menšom ako 1 mg/ml.
5.3 Predklinické údaje o bezpečnosti
Akút na toxicita
LD50 u myši p.o. 800 mg/kg telesnej hmotnosti a 320 mg/kg intraperitoneálne.
LD50 u potkana p.o. 1 600 mg/kg telesnej hmotnosti a 1 300 mg/kg subkutánne. U všetkých uhynutých
zvierat (hlodavcov) bola manifestne vyjadrená depresia CNS a nájdené ulcerogénne zmeny
gastrointestinálneho traktu.
Ibuprofén sa ďalej podával psom v dávke 125 mg/kg telesnej hmotnosti a vyššie, toxické účinky sa
prejavili eróziami žalúdka a albuminúriou. V dávke 20 a 50 mg/kg sa nepreukázali žiadne toxické zmeny. Výsledky ukazujú, že ibuprofén v letálnych dávkach spôsobil postihnutie CNS u hlodavcov, zatiaľ čo ulcerogénny účinok sa pozoroval v oboch skupinách zvierat (aj nehlodavcov). Ulcerogénne účinky sú výsledkom systémového aj lokálneho pôsobenia ibuprofénu - gastrointestinálne lézie sa pozorovali po parenterálnej aj perorálnej aplikácii.
Chr onická t oxi cit a
Desiatim potkanom sa podával ibuprofén v dávke 180 mg/kg (5 zvierat) a 60 mg/kg telesnej hmotnosti počas 26 a 13 týždňov. 1 samček uhynul v dôsledku intestinálnej lézie. Na konci terapie sa u samcov
aj u samíc zistila anémia. Ibuprofén takisto alteroval pomer hmotnosť orgánu/celková telesná
hmotnosť - pri pečeni, obličkách, gonádach a pri druhotných pohlavných orgánoch. Histologicky sa nezistili signifikantné zmeny s výnimkou 1 samca a 3 samíc, kde sa našli intestinálne ulcerácie. Zväčšenie pečene a obličiek pravdepodobne súvisí s metabolizmom a exkréciou ibuprofénu.
Po podaní ibuprofénu psom v dávke 16 mg/kg telesnej hmotnosti na deň počas 30-tich dní sa nenašli klinické príznaky toxicity, pri pitve sa však zistili erózie a ulcerácie žalúdka a zápaly čriev. Podobné
lézie sa našli v dávke 8 mg/kg/deň, ale nie v dávke 4 mg/kg/deň.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
mikrokryštalická celulóza kukuričný škrob
sodná soľ kroskarmelózy
stearát horečnatý hydratovaný oxid kremičitý hypromelóza
makrogol mastenec
oxid titaničitý E171
erytrozín E127
simetikónová emulzia SE4
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25 ºC v pôvodnom vnútornom obale, vnútorný obal uchovávajte v škatuľke.
Uchovávajte mimo dohľadu a dosahu detí.
6.5 Druh obalu a obsah balenia
Druh obalu:
Ibalgin 200 (10, 12, 24 a 30 filmom obalených tabliet): blister (PVC/Al), písomná informácia pre používateľa, papierová škatuľka.
Ibalgin 400 (10, 12, 24, 30, 36, 48, 96 a 100 filmom obalených tabliet): blister (PVC/Al), písomná informácia pre používateľa, papierová škatuľka.
Ibalgin 400 (100 filmom obalených tabliet): PE fľaša so závitovým PE uzáverom, písomná informácia pre používateľa, papierová škatuľka.
V eľkosti bal eni a:
Ibalgin 200: 10, 12, 24 a 30 filmom obalených tabliet
Ibalgin 400: 10, 12, 24, 30, 36, 48, 96 a 100 filmom obalených tabliet
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Opella Healthcare Slovakia s.r.o. Einsteinova 24, 851 01
Bratislava, Slovenská republika
8. REGISTRAČNÉ ČÍSLA
Ibalgin 200: 29/0140/89-CS Ibalgin 400: 29/0154/88-CS
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Ibalgin 200:
Dátum prvej registrácie: 5. apríla 1989
Dátum posledného predĺženia registrácie: 24. mája 2004
Ibalgin 400:
Dátum prvej registrácie: 24. augusta 1988
Dátum posledného predĺženia registrácie: 24. mája 2004
10. DÁTUM REVÍZIE TEXTU
12/2022