
iánom sledovania až do 3 rokov bola vyššia incidencia lymfómu pozorovaná u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu. Lymfóm
sa diagnostikoval u 11 osôb (1 v skupinách liečených s 50 mg golimumabu a 10 v skupinách liečených so 100 mg golimumabu) s incidenciou (95 % IS) na 100 pacientskych rokov následného sledovania
0,03 (0,00; 0,15) udalostí pre golimumab 50 mg a 0,13 (0,06; 0,24) udalostí pre golimumab 100 mg
a 0,00 (0,00; 0,57) udalostí pre placebo. Väčšina lymfómov sa vyskytla v štúdii GO-AFTER, v ktorej boli zaradení pacienti s predošlou expozíciou anti-TNF liečivám, ktorí mali dlhšie trvajúce
a refraktérnejšie ochorenie (pozri časť 4.4).
Malignity iné ako lymfóm
V kontrolovaných obdobiach pivotných skúšaní a počas približne 4 rokov následného sledovania bola podobná incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) medzi golimumabom a kontrolnými skupinami. Počas približne 4 rokov následného sledovania bola incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) podobná všeobecnej populácii.
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov bol nemelanómový karcinóm kože diagnostikovaný u 5 osôb v skupine dostávajúcej placebo,
10 s golimumabom 50 mg a 31 s golimumabom 100 mg s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,36 (0,26; 0,49) pre kombinovaný golimumab a 0,87 (0,28; 2,04) pre
placebo.
V kontrolovanom a nekontrolovanom období pivotných skúšaní s mediánom sledovania až do 3 rokov boli malignity okrem melanómového, nemelanómového karcinómu kože a lymfómu diagnostikované
u 5 osôb v skupine dostávajúcej placebo, 21 s golimumabom 50 mg a 34 s golimumabom 100 mg s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,48 (0,36; 0,62) pre
kombinovaný golimumab a 0,87 (0,28; 2,04) pre placebo (pozri časť 4.4).
Prípady hlásené v klinických štúdiách s astmou
V prieskumnej klinickej štúdii bol pacientom so závažnou perzistujúcou astmou podávaný golimumab v záťažovej dávke (150 % určenej terapeutickej dávky) subkutánne v týždni 0 a následne v dávkach
200 mg golimumabu, 100 mg golimumabu alebo 50 mg golimumabu každé 4 týždne subkutánne až do
52. týždňa. Hlásilo sa osem malignít v kombinovanej skupine liečenej golimumabom (n = 230)
a žiadna v skupine dostávajúcej placebo (n = 79). Lymfóm sa hlásil u 1 pacienta, nemelanómový karcinóm kože u 2 pacientov a iné malignity u 5 pacientov. U žiadneho typu malignity nevzniklo
špecifické zoskupovanie.
Počas placebom kontrolovanej časti štúdie bola incidencia (95 % IS) všetkých malignít na
100 pacientskych rokov v následnom sledovaní 3,19 (1,38; 6,28) v skupine s golimumabom. V tejto štúdii bola incidencia (95% IS) na 100 pacientskych rokov v následnom sledovaní u osôb liečených
golimumabom 0,40 (0,01; 2,20) pre lymfóm, 0,79 (0,10; 2,86) pre nemelanómové karcinómy kože
a 1,99 (0,64; 4,63) pre iné malignity. U osôb s placebom bola incidencia (95 % IS) na
100 pacientskych rokov v následnom sledovaní týchto malignít 0,00 (0,00; 2,94). Význam tohto nálezu nie je známy.
Neurologické udalosti
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov sa pozorovala väčšia incidencia demyelinizácie u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu (pozri časť 4.4).
Zvýšenia hepatálnych enzýmov
V kontrolovanom období pivotných skúšaní RA a PsA sa vyskytlo mierne zvýšenie ALT (> 1
a < 3 x horná hranica normálu (ULN)) v štúdiách s RA a PsA v podobnej miere u pacientov s golimumabom a u kontrolných pacientov (22,1 % k 27,4 % pacientov). V štúdiách s AS
a nr-axiálnou SpA malo mierne zvýšenie ALT viac pacientov liečených golimumabom (26,9 %) než
pacienti v kontrolnej skupine (10,6 %). V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní RA a PsA s mediánom sledovania približne 5 rokov bola v štúdiách s RA a PsA incidencia
mierneho zvýšenia ALT podobná u pacientov liečených golimumabom a u kontrolných pacientov.
V kontrolovanom období pivotných skúšaní zameraných na nasadenie golimumabu pri UC sa vyskytli mierne zvýšenia ALT (> 1 a < 3 x ULN) v podobnej miere u pacientov liečených golimumabom
(8,0 %) a u pacientov v kontrolnej skupine (6,9 %). V kontrolovaných a nekontrolovaných obdobiach
pivotných skúšaní UC s mediánom sledovania približne 2 roky bol podiel pacientov s miernymi zvýšeniami ALT 24,7 % u pacientov dostávajúcich golimumab počas udržiavacej časti štúdie UC.
Zvýšenie ALT ≥ 5 x ULN bolo v kontrolovanom období pivotných skúšaní RA a AS menej časté a pozorovalo sa častejšie u pacientov liečených golimumabom (0,4 % a 0,9 %) než u kontrolných pacientov (0,0 %). Táto tendencia sa nepozorovala v skupine s PsA. V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní RA, PsA a AS s mediánom sledovania 5 rokov bola incidencia zvýšenia ALT ≥ 5 x ULN podobná u pacientov liečených golimumabom aj u kontrolných
pacientov. Vo všeobecnosti boli tieto zvýšenia asymptomatické a abnormality poklesli alebo vymizli,
či už pri pokračovaní liečby alebo po ukončení podávania golimumabu alebo po modifikácii súbežne podávaných liekov. V kontrolovaných a nekontrolovaných obdobiach štúdie nr-axiálnej SpA (až do
1 roka) sa nehlásili žiadne prípady. V kontrolovaných obdobiach pivotných skúšaní zameraných na
nasadenie golimumabu pri UC sa vyskytli zvýšenia ALT ≥ 5 x ULN v podobnej miere u pacientov liečených golimumabom (0,3 %) a u kontrolných pacientov (1,0 %). V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní UC s mediánom sledovania približne 2 roky bol
podiel pacientov so zvýšeniami ALT ≥ 5 x ULN 0,8 % u pacientov dostávajúcich golimumab počas udržiavacej časti štúdie UC.
Počas pivotných skúšaní RA, PsA, AS a nr-axiálnej SpA sa u jedného pacienta v skúšaní RA liečeného golimumabom s existujúcimi hepatálnymi abnormalitami a zamenenými liekmi vyvinula fatálna neinfekčná hepatitída so žltačkou. Úloha golimumabu ako prispievajúceho alebo zhoršujúceho činiteľa nemôže byť vylúčená.
Reakcie v mieste podaniaV kontrolovaných obdobiach pivotných skúšaní malo 5,4 % pacientov liečených golimumabom reakciu v mieste podania v porovnaní s 2,0 % kontrolných pacientov. Prítomnosť protilátok proti
golimumabu môže zvýšiť riziko reakcií v mieste podania. Väčšina reakcií v mieste podania bola
mierna alebo stredne závažná a najčastejšie sa manifestovala ako erytém v mieste podania. Reakcie v mieste podania všeobecne nemusia nutne viesť k ukončeniu liečby.
V kontrolovaných klinických skúšaniach fázy IIb a/alebo III s RA, PsA, AS, nr-axiálnou SpA, závažnou perzistujúcou astmou a v skúšaniach fázy II/III s UC sa u žiadneho pacienta liečeného golimumabom neobjavili anafylaktické reakcie.
Autoimunitné protilátkyV kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní boli počas 1 roka následného sledovania nanovo ANA-pozitívne 3,5 % pacientov liečených golimumabom a 2,3 % kontrolných pacientov (s titrom 1:160 alebo vyšším). Frekvencia protilátok anti-dsDNA za 1 rok následného sledovania bola 1,1 % u pacientov, ktorí boli na začiatku anti-dsDNA negatívni.
Pediatrická populáciaPolyartikulárna juvenilná idiopatická artritída (pJIA)Bezpečnosť golimumabu sa skúmala v štúdii fázy III so 173 pacientmi s pJIA vo veku 2 až 17 rokov. Priemerná doba sledovania bola približne 2 roky. V tejto štúdii boli typ a frekvencia hlásených nežiaducich udalostí vo všeobecnosti podobné tým pozorovaným v štúdiách u dospelých s RA.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinickej štúdii boli jednorazovo intravenózne podané dávky až do 10 mg/kg bez toxicity limitujúcej dávku. V prípade predávkovania sa odporúča sledovanie pacienta na akékoľvek prejavy a príznaky nežiaducich účinkov a okamžite sa má začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNF-α), ATC kód: L04AB06
Mechanizmus účinkuGolimumab je ľudská monoklonálna protilátka, ktorá vytvára stabilné komplexy s vysokou afinitou so solubilnými aj transmembránovými bioaktívnymi formami ľudského TNF-α, čím zabraňuje naviazanie TNF-α na jeho receptory.
Farmako
d
ynamické
účinky
Naviazaním golimumabu na ľudský TNF sa preukázala neutralizácia expresie adhezívnych molekúl na povrchu bunky indukovaná TNF-α – E-selektínu, adhezívnej molekuly cievnych buniek (VCAM)-1
a intercelulárnej adhezívnej molekuly (ICAM)-1 ľudských endotelových buniek.
In vitro golimumab
tiež inhiboval TNF indukovanú sekréciu interleukínu (IL)-6 a IL-8 a faktoru stimulujúceho kolónie granulocytov a makrofágov (GM-CSF) ľudských endotelových buniek.
V porovnaní so skupinami s placebom sa pozorovalo relatívne zlepšenie hladín C-reaktívneho proteínu (CRP) a liečba Simponi viedla v porovnaní s kontrolnou liečbou k významnému zníženiu sérových hladín IL-6, ICAM-1, matrixovej metaloproteinázy (MMP)-3 a vaskulárneho endotelového rastového faktoru (VEGF) oproti východiskovým hodnotám. Došlo aj ku zníženiu hladín TNF-a u pacientov
s RA a AS a hladiny IL-8 sa znížili u pacientov s PsA. Tieto zmeny sa pozorovali pri prvom hodnotení
(4. týždeň) po úvodnom podaní Simponi a všeobecne pretrvávali až do 24. týždňa.
Klinická účinnosťPolyartikulárna juvenilná idiopatická artritídaBezpečnosť a účinnosť Simponi sa hodnotili v randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii s ukončením liečby skúšaným liekom (GO-KIDS) u 173 detí (vo veku 2 až
17 rokov) s aktívnou pJIA s najmenej 5 aktívnymi kĺbmi a neadekvátnou odpoveďou na MTX. Do štúdie boli zaradené deti s polyartikulárnym priebehom JIA (polyartritída s negatívnym alebo pozitívnym reumatoidným faktorom, rozšírená oligoartritída, juvenilná psoriatická artritída alebo
systémová JIA bez systémových príznakov v súčasnosti). Východiskový medián počtu aktívnych kĺbov bol 12 a medián CRP bol 0,17 mg/dl.
Časť 1 štúdie pozostávala zo 16-týždňovej otvorenej fázy, v ktorej 173 zaradených detí dostávalo každé 4 týždne Simponi v dávke 30 mg/m2 (maximálne 50 mg) subkutánne a MTX. 154 detí, ktoré dosiahli odpoveď Ped ACR (American College of Rheumatology) 30 v 16. týždni vstúpilo do časti
2 štúdie, randomizovanej fázy s ukončením liečby skúšaným liekom a dostávalo Simponi v dávke
30 mg/m2 (maximálne 50 mg) + MTX alebo placebo + MTX každé 4 týždne. Po vzplanutí ochorenia dostávali deti Simponi v dávke 30 mg/m2 (maximálne 50 mg) + MTX. V 48. týždni deti vstúpili do dlhodobého predĺženia štúdie.
U detí v tejto štúdii sa preukázali odpovede Ped ACR 30, 50, 70, a 90 od 4. týždňa.
V 16. týždni malo 87 % detí odpoveď Ped ACR 30, 79 % odpoveď Ped ACR 50, 66 % odpoveď Ped ACR 70 a 36 % odpoveď Ped ACR 90. V 16. týždni malo 34 % detí neaktívne ochorenie definované ako prítomnosť všetkého z nasledujúcich: žiadny kĺb s aktívnou artritídou; bez horúčky, vyrážky, serozitídy, splenomegálie, hepatomegálie alebo generalizovanej lymfadenopatie pravdepodobne spojenej s JIA; bez aktívnej uveitídy; normálne hodnoty FW (< 20 mm/hodina) alebo CRP
(< 1,0 mg/dl); celkové hodnotenie aktivity ochorenia lekárom (≤ 5 mm na VAS); dĺžka trvania rannej stuhnutosti < 15 minút.
V 16. týždni všetky zložky Ped ACR preukázali klinicky relevantné zlepšenie oproti východiskovým hodnotám (pozri tabuľku 3).
Tabuľka 3: Zlepšenia oproti východiskovým hodnotám zložiek Ped ACR v 16. týždniaMedián zlepšenia v percentách
Zlepšenia oproti východiskovým hodnotám zložiek Ped ACR v 16. týždniaMedián zlepšenia v percentách Simponi 30 mg/m2 nb = 173
Celkové hodnotenie ochorenia lekármi (VASc
0−10 cm)
Celkové hodnotenie celkovej pohody pacientom/rodičom (VAS 0−10 cm)
88 %
67 %

Počet aktívnych kĺbov 92 % Počet kĺbov s obmedzeným rozsahom pohybu 80 % Telesné funkcie podľa CHAQd 50 % FW (mm/h)e 33 % a východisková hodnota = týždeň 0
b „n“ odráža zaradených pacientov
c VAS: vizuálna analógová stupnica
d CHAQ (Child Health Assessment Questionaire): dotazník hodnotiaci zdravie dieťaťa
e FW (mm/h): rýchlosť sedimentácie erytrocytov (milimetre za hodinu)
Primárny koncový ukazovateľ, podiel detí, ktoré dosiahli odpoveď Ped ACR 30 v 16. týždni
a u ktorých nedošlo k vzplanutiu ochorenia medzi 16. a 48. týždňom, sa nedosiahol. U väčšiny detí nedošlo k vzplanutiu ochorenia medzi 16. a 48. týždňom (59 % v skupine Simponi + MTX a 53 % v skupine placebo + MTX; hodnota p = 0,41).
Analýzy primárneho koncového ukazovateľa vo vopred definovaných podskupinách podľa východiskových hladín CRP (≥ 1 mg/dl oproti < 1 mg/dl) u osôb s východiskovou hladinou CRP
≥ 1 mg/dl preukázali vyššie miery vzplanutia ochorenia v skupine dostávajúcej placebo + MTX oproti
skupine so Simponi + MTX (87 % oproti 40 % p = 0,0068).
V 48. týždni dosiahlo odpoveď Ped ACR 30 53 % detí v skupine Simponi + MTX a 55 % detí
v skupine placebo + MTX a neaktívne ochorenie 40 % detí v skupine Simponi + MTX a 28 % detí v skupine placebo + MTX.
Reumatoidná artritída u dospelýchÚčinnosť Simponi sa preukázala v troch multicentrických randomizovaných dvojito zaslepených, placebom kontrolovaných štúdiách s vyše 1 500 pacientmi vo veku ≥ 18 rokov so stredne závažnou
a závažnou aktívnou RA, diagnostikovanou podľa kritérií American College of Rheumatology (ACR)
najmenej 3 mesiace pred skríningom. Pacienti mali najmenej 4 opuchnuté a 4 bolestivé kĺby. Simponi alebo placebo sa podávali subkutánne každé 4 týždne.
V GO-FORWARD sa hodnotilo 444 pacientov, ktorí mali aktívnu RA napriek stabilnej dávke MTX aspoň 15 mg/týždeň a ktorí sa predtým neliečili žiadnym anti-TNF liečivom. Pacienti boli randomizovaní a dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. Po 24. týždni boli pacienti, ktorí dostávali placebo + MTX, prevedení na Simponi 50 mg + MTX. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia.
V GO-AFTER sa hodnotilo 445 pacientov, ktorí sa už predtým liečili jedným alebo viacerými anti- TNF liečivami adalimumabom, etanerceptom alebo infliximabom. Pacienti boli randomizovaní
a dostávali placebo, Simponi 50 mg alebo Simponi 100 mg. Počas tejto štúdie mohli pacienti
pokračovať v už prebiehajúcej liečbe DMARD s MTX, sulfasalazínom (SSZ) a/alebo hydroxychlorochínom (HCQ). Stanovené dôvody na ukončenie predchádzajúcej anti-TNF liečby boli nedostatočná účinnosť (58 %), intolerancia (13 %) a/alebo iné dôvody než bezpečnosť alebo účinnosť (29 %, zväčša finančné dôvody).
V GO-BEFORE sa hodnotilo 637 pacientov s aktívnou RA, ktorí sa predtým neliečili MTX ani anti- TNF liečivom. Pacienti boli randomizovaní tak, aby dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia, v ktorom pacienti dostávajúci placebo + MTX s aspoň 1 citlivým alebo opuchnutým kĺbom, boli prestavení na Simponi 50 mg + MTX.
Primárnymi spoločnými koncovými ukazovateľmi v GO-FORWARD boli stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni a zlepšenie v 24. týždni v dotazníku hodnotiacom zdravie (Health Assessment Questionnaire, HAQ) oproti východiskovej hodnote.
V GO-AFTER bolo primárnym koncovým ukazovateľom stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni. V GO-BEFORE boli primárnymi spoločnými koncovými
ukazovateľmi stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 50 v 24. týždni a zmena
skóre podľa Sharpa modifikovanom van der Heijdeovou (vdH-S) v 52. týždni oproti východiskovej hodnote. Okrem primárnych koncových ukazovateľov sa vykonali ďalšie hodnotenia vplyvu liečby Simponi na prejavy a príznaky artritídy, rádiografickú odpoveď, telesnú funkciu a kvalitu života spojenú so zdravím.
Celkovo sa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg so súbežným MTX až do 104. týždňa v GO-FORWARD
a GO-BEFORE a až do 24. týždňa v GO-AFTER. Podľa dizajnu každej z RA štúdií mohli byť pacienti
v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznaky
Kľúčové výsledky ACR pre dávku Simponi 50 mg v 14., 24. a 52. týždni GO-FORWARD,
GO-AFTER a GO-BEFORE sú zobrazené v tabuľke 4 a sú popísané nižšie. Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po úvodnej aplikácii Simponi.
V skupine 89 osôb randomizovaných na Simponi 50 mg + MTX v GO-FORWARD bolo stále na tejto liečbe 48 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 40 pacientov, odpoveď ACR 50
u 33 pacientov a odpoveď ACR 70 u 24 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
V GO-AFTER bolo percento pacientov, ktorí dosiahli odpoveď ACR 20, vyššie u pacientov dostávajúcich Simponi než u pacientov dostávajúcich placebo, bez ohľadu na hlásený dôvod ukončenia jednej alebo viacerých predchádzajúcich terapií anti-TNF.
Tabuľka 4:
Kľúčové výsledky účinnosti z kontrolovaných častí GO-FORWARD, GO-AFTER
a GO-BEFORE
GO-FORWARD Aktívna RA napriek MTX
Simponi
GO-AFTER Aktívna RA, predtým liečená jedným alebo viacerými anti-TNF liečivami
GO-BEFORE Aktívna RA, neliečená MTX
Simponi
Placebo
+ MTX
50 mg
+
MTX Placebo
Simponi
50 mg
Placebo
+ MTX
50 mg
+ MTX
na 133 89 150 147 160 159
Respondenti, % pacientov
ACR 20
14. týždeň 33 % 55 %* 18 % 35 %* NA NA
24. týždeň 28 % 60 %* 16 % 31 %

p = 0,002
49 % 62 %
52. týždeň NA NA NA NA 52 % 60 %
ACR 50
14. týždeň 10 % 35 %* 7 % 15 %
p = 0,021
NA NA
24. týždeň 14 % 37 %* 4 % 16 %* 29 % 40 %
52. týždeň NA NA NA NA 36 % 42 %
ACR 70
14. týždeň 4 % 14 %
p = 0,008
2 % 10 %
p = 0,005
NA NA
24. týždeň 5 % 20 %* 2 % 9 % p = 0,009 16 % 24 %
52. týždeň NA NA NA NA 22 % 28 %

a n vyjadruje randomizovaných pacientov; aktuálny počet pacientov hodnotiteľných pre každý koncový ukazovateľ sa môže líšiť podľa časového bodu.
* p ≤ 0,001
NA: Neaplikovateľné
V GO-BEFORE nebola primárna analýza v 24. týždni u pacientov so stredne závažnou až závažnou
reumatoidnou artritídou (kombinované skupiny Simponi 50 a 100 mg + MTX vs. samotný MTX pre ACR50) štatisticky signifikantná (p = 0,053). V 52. týždni bolo v celkovej populácii celkovo vyššie percento pacientov, ktorí dosiahli odpoveď ACR, v skupine so Simponi 50 mg + MTX, ale
v porovnaní so samotným MTX nebolo signifikantne odlišné (pozri tabuľku 4). Ďalšie analýzy sa vykonali v podskupinách reprezentujúcich indikovanú populáciu pacientov so závažnou aktívnou
a progresívnou RA. V indikovanej populácii bol preukázaný celkovo väčší účinok Simponi 50 mg + MTX oproti samotnému MTX v porovnaní s celkovou populáciou.
V GO-FORWARD a GO-AFTER sa v každom dopredu určenom časovom bode pozorovali klinicky významné a štatisticky signifikantné odpovede na stupnici aktivity ochorenia (Disease Activity Scale, DAS) 28, v 14. týždni a v 24. týždni (p ≤ 0,001). V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 od
104. týždňa až do 256. týždňa podobné.
V GO-BEFORE bola meraná výrazná klinická odpoveď, definovaná ako udržanie odpovede ACR 70 nepretržite počas 6-mesačného obdobia. V 52. týždni dosiahlo výraznú klinickú odpoveď 15 % pacientov v skupine so Simponi 50 mg + MTX v porovnaní so 7 % pacientov v skupine s placebom + MTX (p = 0,018). V skupine 159 osôb randomizovaných na Simponi 50 mg + MTX bolo stále na tejto liečbe 96 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 85 pacientov, odpoveď ACR 50
u 66 pacientov a odpoveď ACR 70 u 53 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
Rádiografická odpoveď:V GO-BEFORE bola na stanovenie stupňa štrukturálneho poškodenia použitá zmena vo vdH-S skóre oproti východiskovej hodnote, čo je kombinované skóre štrukturálneho poškodenia, ktoré rádiograficky stanovuje počet a veľkosť erózií kĺbov a stupeň zúženia kĺbovej štrbiny (joint space narrowing, JSN) na rukách/zápästiach a nohách. Kľúčové výsledky pre dávku Simponi 50 mg v 52. týždni sú uvedené v tabuľke 5.
Počet pacientov bez nových erózií alebo so zmenou oproti východiskovej hodnote v celkovom vdH-S skóre ≤ 0 bol signifikantne vyšší v skupine liečenej Simponi ako v kontrolnej skupine (p = 0,003). Rádiografické účinky pozorované v 52. týždni boli udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli rádiografické účinky od 104. týždňa až do 256. týždňa podobné.
Tabuľka 5: Priemerné (štandardná odchýlka) rádiografické zmeny oproti východiskovej hodnote v celkovom vdH-S skóre v 52. týždni v celkovej populácii v GO-BEFOREPlacebo + MTX Simponi 50 mg + MTX
Priemerné (štandardná odchýlka) rádiografické zmeny oproti východiskovej hodnote v celkovom vdH-S skóre v 52. týždni v celkovej populácii v GO-BEFOREPlacebo + MTX Simponi 50 mg + MTXn
a 160 159Celkové skóreVýchodisková hodnota 19,7 (35,4) 18,7 (32,4) Zmena oproti
východiskovej hodnote 1,4 (4,6) 0,7 (5,2)*
Skóre erózieVýchodisková hodnota 11,3 (18,6) 10,8 (17,4) Zmena oproti
východiskovej hodnote 0,7 (2,8) 0,5 (2,1)
Skóre JSN

Východisková hodnota 8,4 (17,8) 7,9 (16,1) Zmena oproti
východiskovej hodnote 0,6 (2,3) 0,2 (2,0)**
a n vyjadruje randomizovaných pacientov
* p = 0,015
** p = 0,044
Telesná funkcia a kvalita života spojená so zdravímTelesná funkcia a neschopnosť sa posudzovali ako samostatný koncový ukazovateľ v GO-FORWARD a GO-AFTER používajúc index neschopnosti HAQ DI. V týchto štúdiách preukázal Simponi klinicky významné a štatisticky signifikantné zlepšenie v HAQ DI od základného stavu oproti kontrole v 24. týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, sa udržalo zlepšenie v HAQ DI až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii
a liečených Simponi bolo zlepšenie v HAQ DI od 104. týždňa až do 256. týždňa podobné.
V GO-FORWARD sa preukázali klinicky významné a štatisticky signifikantné zlepšenia kvality života spojenej so zdravím, merané pomocou skóre telesných komponentov SF-36 u pacientov liečených Simponi oproti placebu v 24. týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, bolo udržané zlepšenie telesných komponentov SF-36 až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi bolo zlepšenie telesných komponentov SF-36 od 104. týždňa až do 256. týždňa podobné. V GO-FORWARD
a GO-AFTER sa pozorovali štatisticky signifikantné zlepšenia únavy, merané škálou únavy-funkčné posúdenie liečby chronickej choroby (FACIT-F).
Psoriatická artritída u dospelýchBezpečnosť a účinnosť Simponi bola hodnotená v multicentrickej randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-REVEAL) so 405 dospelými pacientmi s aktívnou PsA (≥ 3
opuchnuté kĺby a ≥ 3 bolestivé kĺby) napriek liečbe nesteroidovými antiflogistikami (NSAID) alebo
DMARD. Pacienti v tejto štúdii mali diagnózu PsA najmenej 6 mesiacov a mali aspoň miernu psoriatickú chorobu. Boli zaradení pacienti s každým podtypom psoriatickej artritídy, vrátane polyartikulárnej artritídy bez reumatoidných uzlíkov (43 %), asymetrickej periférnej artritídy (30 %), distálnej artritídy interfalangeálnych kĺbov (DIP) (15 %), spondylitídy s periférnou artritídou (11 %)
a arthritis mutilans (1 %). Predchádzajúca liečba anti-TNF liečivom nebola prípustná. Simponi alebo placebo sa podávali subkutánne každé 4 týždne. Pacientom bolo náhodne pridelené placebo, Simponi
50 mg alebo Simponi 100 mg. Po 24. týždni boli pacienti, ktorí dostávali placebo, prevedení na
Simponi 50 mg. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia. Približne štyridsaťosem percent pacientov pokračovalo na stabilnej dávke metotrexátu (≤ 25 mg/týždeň). Spoločnými primárnymi koncovými ukazovateľmi boli percento pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni a zmena v celkovom vdH-S skóre modifikovanom pre PsA v 24. týždni oproti východiskovej hodnote.
Celkovo sa až do 104. týždňa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg. Podľa dizajnu štúdie mohli byť pacienti
v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznakyKľúčové výsledky pre dávku 50 mg v 14. a 24. týždni sú zobrazené v tabuľke 6 a opísané nižšie.
Tabuľka 6:
Kľúčové výsledky účinnosti z GO-REVEAL

Placebo
Simponi
50 mg*
na 113 146
Respondenti, % pacientov
ACR 20
14. týždeň 9 % 51 %
AC
R 50
AC
R 70
PASI
b
75
c
24. týždeň 12 % 52 %
14. týždeň 2 % 30 %
24. týždeň 4 % 32 %
14. týždeň 1 % 12 %
24. týždeň 1 % 19 %
14. týždeň 3 % 40 %
24. týždeň 1 % 56 %
* p < 0,05 pre všetky porovnávania;
a n vyjadruje randomizovaných pacientov; aktuálny počet hodnotiteľných pacientov sa môže pre každý výsledný ukazovateľ líšiť podľa časového bodu.
b Oblasť postihnutá psoriázou a stupeň závažnosti
c Založené na podskupine pacientov so vstupným postihnutím BSA ≥ 3 %, 79 pacientov
(69,9 %) zo skupiny s placebom a 109 pacientov (74,3 %) zo skupiny so Simponi 50 mg.
Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po prvom podaní Simponi. Podobné odpovede
ACR 20 sa pozorovali v 14. týždni u pacientov s polyartikulárnou artritídou bez reumatoidných uzlíkov a pri asymetrickej periférnej artritíde, podtyp PsA. Počet pacientov s inými podtypmi PsA bol
príliš malý na zmysluplné vyhodnotenie. Odpovede pozorované v skupinách liečených Simponi boli
podobné u pacientov užívajúcich a neužívajúcich súčasne metotrexát. V skupine 146 pacientov randomizovaných na Simponi 50 mg bolo stále na tejto liečbe 70 pacientov v 104. týždni. Z týchto
70 pacientov bola odpoveď ACR 20 u 64 pacientov, odpoveď ACR 50 u 46 pacientov a odpoveď
ACR 70 u 31 pacientov. V skupine pacientov zostávajúcich v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede ACR 20/50/70.
Štatisticky významné odpovede v DAS 28 sa pozorovali aj v 14. a 24. týždni (p < 0,05).
V skupine pacientov liečených Simponi sa v 24. týždni pozorovali zlepšenia parametrov periférnej aktivity, charakteristických pre psoriatickú artritídu (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída). Liečba Simponi viedla k výraznému zlepšeniu telesných funkcií hodnotenej podľa HAQ DI, ako aj k signifikantnému zlepšeniu kvality života spojenej so zdravím ako bolo namerané pomocou sumárneho skóre telesných a mentálnych údajov podľa SF-36. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 a HAQ DI udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 a HAQ DI od 104. týždňa až do
256. týždňa podobné.
Rádiografická odpoveď:
Štrukturálne poškodenie oboch rúk a nôh sa rádiograficky hodnotilo zmenou vo východiskovej hodnote oproti vdH-S skóre modifikovanom pre PsA pridaním distálnych interfalangeálnych (DIP)
kĺbov rúk.
Liečba Simponi 50 mg znížila v 24. týždni rýchlosť progresie poškodenia periférnych kĺbov
v porovnaní s podávaním placeba podľa zmeny v celkovom modifikovanom vdH-S skóre oproti východiskovej hodnote (priemer ± SD skóre bolo 0,27 ± 1,3 v skupine s placebom v porovnaní
s -0,16 ± 1,3 v skupine so Simponi; p = 0,011). Zo 146 pacientov, ktorí boli randomizovaní na
Simponi 50 mg, boli RTG údaje dostupné u 126 pacientov v 52. týždni, z ktorých u 77 % sa nepreukázala žiadna progresia v porovnaní s východiskovou hodnotou. V 104. týždni boli RTG údaje
dostupné u 114 pacientov a u 77 % sa nepreukázala žiadna progresia oproti východiskovej hodnote.
V skupine pacientov zostávajúcich v štúdii a liečených Simponi v období od 104. týždňa až do 256. týždňa podobný pomer pacientov nepreukázal žiadnu progresiu oproti východiskovej hodnote.
Imunogenita
Počas štúdií fázy III s RA, PsA a AS boli až do 52. týždňa použitím metódy enzýmovej imunoanalýzy
(EIA) zistené protilátky proti golimumabu u 5 % (105/2 062) pacientov liečených golimumabom a kde
sa testovali, boli takmer všetky protilátky neutralizujúce in vitro. Podobný pomer sa pozoroval vo všetkých reumatologických indikáciách. Súbežná liečba s MTX mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez MTX (približne 3 % [41/1 235] oproti 8 % [64/827] v uvedenom poradí).
Pri nr-axiálnej SpA sa protilátky proti golimumabu zistili použitím metódy EIA do 52. týždňa u 7 % (14/193) pacientov liečených golimumabom.
V štúdiách fázy II a III s UC boli až do 54. týždňa použitím metódy EIA zistené protilátky proti golimumabu u 3 % (26/946) pacientov liečených golimumabom. Šesťdesiatosem percent (21/31) pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky. Súbežná liečba
s imunomodulátormi (azatioprin, 6-merkaptopurín a MTX ) mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez
imunomodulátorov (1 % (4/308) oproti 3 % (22/638) v uvedenom poradí). Z pacientov, ktorí pokračovali v predĺžení štúdie a mali hodnotiteľné vzorky v 228. týždni, sa preukázali protilátky proti
golimumabu u 4 % (23/604) pacientov liečených golimumabom. Osemdesiatdva percent (18/22)
pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky.
Na stanovenie protilátok proti golimumabu sa v štúdii pJIA použila metóda drug-tolerant EIA. Pri použití metódy drug-tolerant EIA v porovnaní s metódou EIA sa očakávalo zistenie väčšieho výskytu protilátok proti golimumabu z dôvodu vyššej citlivosti a lepšej tolerancie lieku. V štúdii fázy III pJIA sa počas 48. týždňa použitím metódy drug-tolerant EIA zistili protilátky proti golimumabu u 40 % (69/172) detí liečených golimumabom, z ktorých väčšina mala titer nižší ako 1:1 000. Vplyv na sérové koncentrácie golimumabu sa pozoroval pri titroch > 1:100, zatiaľ čo vplyv na účinnosť sa nepozoroval do titrov > 1:1 000, aj keď počet detí s titrom > 1:1 000 bol nízky (N = 8). U detí, u ktorých sa preukázali protilátky proti golimumabu malo 39 % (25/65) neutralizujúce protilátky. Keďže boli prítomné nízke titre protilátok, vyšší výskyt protilátok pri použití metódy drug-tolerant EIA nemal zjavný vplyv na hladiny liečiva, účinnosť a bezpečnosť a preto nepredstavuje žiadny nový bezpečnostný signál.
Prítomnosť protilátok proti golimumabu môže zvýšiť riziko reakcií v mieste injekcie (pozri časť 4.4). Malý počet pacientov pozitívnych na protilátky proti golimumabu limituje schopnosť vyvodiť definitívne závery, týkajúce sa vzťahu medzi protilátkami proti golimumabu a klinickou účinnosťou alebo bezpečnostné opatrenia.
Pretože analýzy imunogenity sú špecifické pre daný liek a daný test, porovnanie frekvencie výskytu protilátok s frekvenciami pri ostatných liekoch nie je vhodné.
5.2. Farmakokinetické vlastnosti
Absorpcia
Po jednorazovej subkutánnej aplikácii golimumabu zdravým osobám alebo pacientom s RA bol priemerný čas na dosiahnutie maximálnych koncentrácií v sére (Tmax) od 2 do 6 dní. Subkutánna injekcia 50 mg golimumabu zdravým osobám priniesla priemernú hodnotu ± smerodajná odchýlka maximálnych koncentrácií (Cmax) v sére 3,1 ± 1,4 μg/ml.
Po jednorazovej subkutánnej injekcii 100 mg bola absorpcia golimumabu v hornej časti ramena, na bruchu a na stehne podobná, s priemernou absolútnou biologickou dostupnosťou 51 %. Keďže golimumab preukázal približnú dávkovo úmernú FK po subkutánnom podaní, dá sa očakávať, že absolútna biologická dostupnosť po dávke 50 mg alebo 200 mg golimumabu bude podobná.
Distribúcia
Po jednorazovom i.v. podaní bol priemerný distribučný objem 115 ± 19 ml/kg.
Eliminácia
Systémový klírens golimumabu bol odhadnutý na 6,9 ± 2,0 ml/deň/kg. Hodnota terminálneho polčasu u zdravých osôb bola odhadnutá na približne 12 ± 3 dni a podobné hodnoty sa pozorovali u pacientov s RA, PsA, AS alebo UC.
U pacientov s RA, PsA alebo AS, ktorí dostávali 50 mg golimumabu každé 4 týždne, sa dosiahol rovnovážny stav koncentrácií v sére v 12. týždni. Pri súbežnom podávaní MTX priniesla liečba
s 50 mg golimumabu subkutánne každé 4 týždne priemerné (± smerodajná odchýlka) minimálne
sérové koncentrácie v rovnovážnom stave približne 0,6 ± 0,4 μg/ml u pacientov s RA s aktívnou RA napriek liečbe MTX a približne 0,5 ± 0,4 μg/ml u pacientov s aktívnou PsA a približne 0,8 ± 0,4 μg/ml u pacientov s AS. Priemerné minimálne sérové koncentrácie golimumabu v rovnovážnom stave
u pacientov s nr-axiálnou SpA boli podobné koncentráciám, ktoré sa pozorovali u pacientov s AS po subkutánnom podaní 50 mg golimumabu každé 4 týždne.
Pacienti s RA, PsA alebo AS, ktorí nedostávali súbežne MTX, mali približne o 30 % nižšie hodnoty minimálnych koncentrácií golimumabu v rovnovážnom stave než tí, ktorí dostávali golimumab
s MTX. U obmedzeného počtu pacientov s RA liečených subkutánnym golimumabom počas 6- mesačného obdobia súbežné používanie MTX znížilo zdanlivý klírens golimumabu o približne 36 %.
Populačná farmakokinetická analýza však naznačila, že súbežné používanie NSAID, perorálnych kortikosteroidov alebo sulfasalazínu nemalo vplyv na zdanlivý klírens golimumabu.
U pacientov s UC sa po indukčných dávkach 200 mg golimumabu v 0. týždni a 100 mg golimumabu v 2. týždni a udržiavacích dávkach 50 mg alebo 100 mg golimumabu subkutánne následne každé
4 týždne dosiahli sérové koncentrácie golimumabu v rovnovážnom stave približne 14 týždňov po začiatku liečby. Liečba 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne počas
udržiavania viedla k priemernej minimálnej sérovej koncentrácii v rovnovážnom stave približne
0,9 ± 0,5 mg/ml a 1,8 ± 1,1 mg/ml.
Súbežné užívanie imunomodulátorov u pacientov s UC liečených 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne nemalo významný vplyv na minimálne hladiny golimumabu
v rovnovážnom stave.
Pacienti, u ktorých sa vyvinuli protilátky proti golimumabu, mali v rovnovážnom stave všeobecne nízke minimálne sérové koncentrácie golimumabu (pozri časť 5.1).
LinearitaGolimumab vykazoval približne dávkovo úmernú farmakokinetiku u pacientov s RA v rozsahu dávok
0,1 mg až 10,0 mg/kg po jednorazovej intravenóznej dávke. Po jednorazovej subkutánnej dávke
u zdravých osôb sa v rozmedzí dávok 50 mg až 400 mg pozorovala farmakokinetika, ktorá bola tiež približne proporcionálna k dávke.
Vplyv telesnej hmotnosti na farmakokinetikuZistil sa trend k vyšším hodnotám zdanlivého klírensu golimumabu s narastajúcou telesnou hmotnosťou (pozri časť 4.2).
 Pediatrická populácia
Pediatrická populáciaFarmakokinetika golimumabu sa stanovila u 173 detí s pJIA s vekovým rozpätím od 2 do 17 rokov.
V štúdii pJIA mali deti, ktoré dostávali subkutánne golimumab v dávke 30 mg/m2 (maximálne 50 mg) každé 4 týždne, hodnoty mediánu minimálnych koncentrácií golimumabu v rovnovážnom stave, ktoré boli podobné v rôznych vekových skupinách a tiež podobné alebo trochu vyššie ako tie, pozorované
u dospelých pacientov s RA, ktorí dostávali 50 mg golimumabu každé 4 týždne.
Farmakokinetické/farmakodynamické modelovanie populácie a simulácia u detí s pJIA potvrdili vzťah medzi expozíciami golimumabu v sére a klinickou účinnosťou a podporili dávkovací režim golimumabu 30 mg/m2 každé 4 týždne u detí s pJIA.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
S golimumabom sa nevykonali žiadne štúdie mutagenity, štúdie fertility na zvieratách ani dlhodobé štúdie karcinogenity.
V štúdiách fertility a všeobecných reprodukčných funkcií myší, pri použití analogickej protilátky selektívne inhibujúcej funkčnú aktivitu myšieho TNFα, sa znížilo množstvo gravidných myší. Nie je známe, či bol tento nález spôsobený vplyvom na samce a/alebo samice. V štúdii vývinovej toxicity vykonanej na myšiach pri aplikácii rovnakej analógnej protilátky u myší a pri použití golimumabu
u opíc druhu Cynomolgus neboli zaznamenané žiadne dôkazy maternálnej toxicity, embryotoxicity alebo teratogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sorbitol (E420) Histidín
Monohydrát histidíniumchloridu
Polysorbát 80
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
24 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Naplnené pero uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
Simponi sa môže uchovávať pri teplotách do maximálne 25 °C počas jedného obdobia až do 30 dní, nesmie však presiahnuť pôvodný dátum exspirácie vytlačený na škatuli. Nový dátum exspirácie sa
musí napísať na škatuľu (až do 30 dní od dátumu vybratia z chladničky).
Ak sa Simponi začne uchovávať pri izbovej teplote, nesmie sa vrátiť späť do chladničky. Ak sa
Simponi nepoužije počas 30 dní uchovávania pri izbovej teplote, musí sa zlikvidovať.
6.5 Druh obalu a obsah balenia
Simponi 45 mg/0,45 ml injekčný roztok
0,45 ml roztoku naplneného v injekčnej striekačke (sklo typu 1) s napevno nasadenou ihlou (nerezová oceľ) a krytom ihly (kaučuk obsahujúci latex) v naplnenom pere. Každé naplnené pero môže dodať
0,1 ml až 0,45 ml v čiastkach po 0,05 ml.
Veľkosť balenia po 1 naplnenom pere.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Simponi sa dodáva ako jednorazové naplnené pero nazývané VarioJect. Každé balenie obsahuje návod na použitie, ktorý úplne popisuje spôsob používania pera. Po vybratí naplneného pera z chladničky sa má pero nechať dosiahnuť izbovú teplotu počas 30 minút pred podaním Simponi. Perom sa nesmie triasť.
Roztok je číry až mierne opaleskujúci, bezfarebný až svetložltý a môže obsahovať malé množstvo priehľadných alebo bielych čiastočiek proteínov. Tento vzhľad nie je ničím nezvyčajným pre roztoky s obsahom proteínov. Simponi sa nesmie použiť pri zmene farby roztoku, zakalení alebo ak obsahuje viditeľné cudzie častice.
Podrobné inštrukcie o spôsobe prípravy a podaní Simponi v naplnenom pere sú súčasťou balenia. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/09/546/009 1 naplnené pero
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 1. októbra 2009
Dátum posledného predĺženia registrácie: 19. júna 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Simponi 50 mg injekčný roztok naplnený v injekčnom pere
Simponi 50 mg injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Simponi 50 mg injekčný roztok naplnený v injekčnom pere
Jedno 0,5 ml naplnené pero obsahuje 50 mg golimumabu*.
Simponi 50 mg injekčný roztok naplnený v injekčnej striekačke
Jedna 0,5 ml naplnená injekčná striekačka obsahuje 50 mg golimumabu*.
* Ľudská monoklonálna protilátka IgG1κ získaná z bunkovej línie hybridómu myši technológiou rekombinantnej DNA.
Pomocná látka so známym účinkom
Každé naplnené pero obsahuje 20,5 mg sorbitolu v 50 mg dávke.
Každá naplnená injekčná striekačka obsahuje 20,5 mg sorbitolu v 50 mg dávke.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok naplnený v injekčnom pere (injekcia), SmartJect Injekčný roztok naplnený v injekčnej striekačke (injekcia) Roztok je číry až mierne opaleskujúci, bezfarebný až svetložltý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidná artritída (RA)
Simponi je v kombinácii s metotrexátom (MTX) indikovaný na:
· liečbu stredne závažnej až závažnej aktívnej reumatoidnej artritídy dospelým, u ktorých sa nedosiahla dostatočná odpoveď na liečbu ochorenie modifikujúcimi antireumatikami (disease-modifying anti-rheumatic drug, DMARD) vrátane MTX.
· liečbu závažnej aktívnej a progresívnej reumatoidnej artritídy dospelým ešte neliečeným MTX.
Preukázalo sa, že Simponi v kombinácii s MTX znižuje rýchlosť progresie poškodenia kĺbov hodnotenú RTG a zlepšuje telesnú funkciu.
Juvenilná idiopatická artritída
Polyartikulárna juvenilná idiopatická artritída (pJIA)
Simponi je v kombinácii s MTX indikovaný na liečbu polyartikulárnej juvenilnej idiopatickej artritídy deťom vo veku 2 roky a starším, ktoré mali nedostatočnú odpoveď na predchádzajúcu liečbu MTX.
Psoriatická artritída (PsA)
Simponi, samostatne alebo v kombinácii s MTX, je indikovaný na liečbu aktívnej a progresívnej psoriatickej artritídy dospelým pacientom, u ktorých sa nedosiahla dostatočná odpoveď na liečbu
DMARD. Preukázalo sa, že Simponi znižuje rýchlosť progresie poškodenia periférnych kĺbov
hodnotenú RTG u pacientov s polyartikulárnymi symetrickými podtypmi ochorenia (pozri časť 5.1)
a zlepšuje telesnú funkciu.
Axiálna spondyloartritída
Ankylozujúca spondylitída (AS)
Simponi je indikovaný na liečbu závažnej, aktívnej ankylozujúcej spondylitídy dospelým, ktorí dostatočne neodpovedali na konvenčnú liečbu.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axiálna SpA)
Simponi je indikovaný na liečbu závažnej, aktívnej axiálnej spondyloartritídy bez rádiografického dôkazu dospelým s objektívnymi prejavmi zápalu na základe indikácie zvýšenou hladinou C-
reaktívneho proteínu (CRP) a/alebo dôkazom pri zobrazení magnetickou rezonanciou (MR),
s nedostatočnou odpoveďou na nesteroidové protizápalové liečivá (NSAID) alebo tým, ktorí tieto liečivá netolerujú.
Ulcerózna kolitída (UC)
Simponi je indikovaný na liečbu stredne závažnej až závažnej aktívnej ulceróznej kolitídy dospelým pacientom s nedostatočnou odpoveďou na konvenčnú liečbu vrátane kortikosteroidov a 6-
merkaptopurínu (6-MP) alebo azatioprinu (AZA), alebo pacientom, ktorí takéto liečby netolerujú alebo sú u nich zdravotne kontraindikované.
4.2 Dávkovanie a spôsob podávania
Liečbu majú iniciovať a dohliadať na ňu iba lekári špecialisti skúsení v diagnostike a liečbe reumatoidnej artritídy, polyartikulárnej juvenilnej idiopatickej artritídy, psoriatickej artritídy, ankylozujúcej spondylitídy, axiálnej spondyloartritídy bez rádiografického dôkazu alebo ulceróznej kolitídy. Pacienti, ktorí sa liečia Simponi, majú dostať kartu s pripomienkami pre pacienta.
Dávkovanie
Reumatoidná artritída
Simponi 50 mg sa podáva raz mesačne, vždy v rovnaký deň v mesiaci. Simponi sa má podávať súbežne s MTX.
Psoriatická artritída, ankylozujúca spondylitída alebo axiálna spondyloartritída bez rádiografického dôkazu
Simponi 50 mg sa podáva raz mesačne, vždy v rovnaký deň v mesiaci.
Pre všetky z vyššie uvedených indikácií dostupné údaje naznačujú, že sa klinická odpoveď dosiahne zvyčajne v priebehu 12. až 14. týždňa liečby (po 3-4 dávkach). U pacientov, u ktorých sa počas tohto obdobia nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Pacienti s telesnou hmotnosťou vyššou ako 100 kg
Pre všetky z vyššie uvedených indikácií sa u pacientov s RA, PsA, AS alebo nr-axiálnou SpA
s telesnou hmotnosťou vyššou ako 100 kg, ktorí po 3 alebo 4 dávkach nedosiahli primeranú klinickú odpoveď, môže zvážiť zvýšenie dávky na 100 mg golimumabu raz mesačne, pričom treba vziať do
úvahy zvýšené riziko niektorých závažných nežiaducich reakcií pri dávke 100 mg v porovnaní
s dávkou 50 mg (pozri časť 4.8). U pacientov, u ktorých sa po podaní 3 až 4 ďalších dávok po 100 mg nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Ulcerózna kolitída
Pacienti s telesnou hmotnosťou nižšou ako 80 kg
Simponi sa podáva ako 200 mg úvodná dávka, po ktorej nasleduje 100 mg v 2. týždni. Pacienti, ktorí majú dostatočnú odpoveď, majú dostať 50 mg v 6. týždni a následne každé 4 týždne. U pacientov,
ktorí nemajú dostatočnú odpoveď, môže byť prospešné pokračovanie so 100 mg v 6. týždni a následne každé 4 týždne (pozri časť 5.1).
Pacienti s telesnou hmotnosťou vyššou ako 80 kg alebo rovnou 80 kg
Simponi sa podáva ako 200 mg úvodná dávka, po ktorej nasleduje 100 mg v 2. týždni, potom 100 mg každé 4 týždne (pozri časť 5.1).
Počas udržiavacej liečby sa dávka kortikosteroidov môže znižovať v súlade s usmerneniami pre klinickú prax.
Dostupné údaje naznačujú, že sa klinická odpoveď zvyčajne dosiahne v priebehu 12. – 14. týždňa liečby (po 4 dávkach). U pacientov, u ktorých sa počas tohto obdobia nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Vynechanie dávky
V prípade, že si pacient zabudne podať Simponi v plánovanom termíne, vynechaná dávka sa má podať hneď, ako si pacient spomenie. Pacienti majú byť poučení o tom, aby si nepodali dvojnásobnú dávku
ako náhradu za vynechanú dávku.
Ďalšia dávka sa má podať podľa nasledujúcich pokynov:
· ak sa podanie oneskorí o menej ako 2 týždne, pacient si má podať vynechanú dávku a pokračovať podľa pôvodnej schémy.
· ak sa podanie oneskorí o viac ako 2 týždne, pacient si má podať vynechanú dávku a má sa stanoviť nová schéma, počínajúc dňom podania tejto injekcie.
Osobitné skupiny pacientov
Staršie osoby (≥ 65 rokov)
U starších osôb sa úprava dávky nevyžaduje.
Porucha funkcie obličiek a pečene
Simponi sa neskúmal v týchto skupinách pacientov. Odporúčanú dávku nie je možné stanoviť.
Pediatrická populácia
Bezpečnosť a účinnosť Simponi u pacientov mladších ako 18 rokov pri indikáciách iných ako pJIA
neboli stanovené.
Polyartikulárna juvenilná idiopatická artritída
Simponi 50 mg sa podáva raz mesačne, vždy v rovnaký deň v mesiaci, deťom s telesnou hmotnosťou najmenej 40 kg. Pre deti s polyartikulárnou juvenilnou idiopatickou artritídou s telesnou hmotnosťou nižšou ako 40 kg je k dispozícii 45 mg/0,45 ml naplnené pero.
Dostupné údaje naznačujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12. až 14. týždňa liečby (po 3 – 4 dávkach). U detí, u ktorých sa počas tohto obdobia nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Spôsob podávania
Simponi je na subkutánne použitie. Po dôkladnom oboznámení sa s technikou subkutánnej aplikácie si ho môžu pacienti podať sami, ak to ich lekár uzná za vhodné, s následným lekárskym dohľadom podľa
potreby. Pacienti majú byť poučení o podaní celej dávky Simponi podľa podrobných inštrukcií
o použití, ktoré sú súčasťou písomnej informácie pre používateľa. Ak sa vyžaduje podanie viacerých injekcií, injekcie sa majú podať do rôznych miest na tele.
Návod na použitie, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza (TBC) alebo iné závažné infekcie ako je sepsa a oportúnne infekcie (pozri časť
4.4).
Stredne závažné alebo závažné zlyhávanie srdca (NYHA trieda III/IV) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Na zlepšenie sledovateľnosti biologických liekov sa má jasne zaznamenať.názov a číslo šarže podaného lieku.
Infekcie
Pacienti musia byť pred liečbou, počas liečby a po liečbe golimumabom dôkladne sledovaní, či sa u nich neobjaví infekcia vrátane tuberkulózy. Pretože eliminácia golimumabu môže trvať až
5 mesiacov, sledovanie má pokračovať počas celého obdobia. Ak sa u pacienta rozvinie závažná
infekcia alebo sepsa, ďalšia liečba golimumabom sa nesmie podať (pozri časť 4.3).
Golimumab sa nemá podávať pacientom s klinicky závažnou aktívnou infekciou. Opatrnosť je potrebná, keď sa zvažuje použitie golimumabu u pacientov s chronickou infekciou alebo s anamnézou opakovaných infekcií. Pacientov treba poučiť a podľa možnosti sa vyhýbať potenciálnym rizikovým faktorom infekcií.
Pacienti užívajúci TNF-blokátory sú náchylnejší na závažné infekcie.
U pacientov, ktorým bol podaný golimumab, sa hlásili bakteriálne infekcie (vrátane sepsy
a pneumónie), mykobakteriálne infekcie (vrátane TBC), invazívne mykotické a oportúnne infekcie, vrátane smrteľných prípadov. Niektoré z týchto závažných infekcií sa vyskytli u pacientov so
súbežnou imunosupresívnou liečbou, ktorá ich, okrem vlastného ochorenia, môže urobiť
náchylnejšími na vznik infekcií. Pacienti, u ktorých sa vyvinula nová infekcia počas liečby golimumabom, sa majú starostlivo monitorovať a podstúpiť kompletné diagnostické vyšetrenie. Podávanie golimumabu sa má prerušiť, ak sa u pacienta vyvinie nová ťažká infekcia alebo sepsa a má sa začať vhodná antimikrobiálna alebo antimykotická liečba, pokým nie je infekcia pod kontrolou.
U pacientov, ktorí žili alebo cestovali do oblastí, kde sú endemické invazívne mykotické infekcie, ako je histoplazmóza, kokcidioidomykóza alebo blastomykóza, sa majú pred začatím liečby golimumabom starostlivo zvážiť prínosy a riziká liečby golimumabom. U rizikových pacientov liečených golimumabom, u ktorých sa vyvinie závažné systémové ochorenie, sa má zvážiť podozrenie na invazívnu mykotickú infekciu. Ak je to možné, diagnostika a podanie empirickej antimykotickej
liečby sa má u týchto pacientov vykonať po konzultácii s lekárom so skúsenosťami v starostlivosti o pacientov s invazívnymi mykotickými infekciami.
Tuberkulóza
U pacientov používajúcich golimumab sa hlásili prípady tuberkulózy. Je potrebné zdôrazniť, že vo väčšine týchto hlásení bola tuberkulóza extrapulmonálna, prebiehajúca buď ako lokálne alebo ako
diseminované ochorenie.
Pred začatím liečby golimumabom sa musia všetci pacienti vyhodnotiť na aktívnu i neaktívnu („latentnú“) tuberkulózu. Toto vyhodnotenie má zahŕňať detailnú anamnézu s osobnou anamnézou tuberkulózy alebo možného predchádzajúceho kontaktu s tuberkulózou a predchádzajúcu a/alebo súčasnú imunosupresívnu liečbu. U všetkých pacientov sa majú urobiť vhodné skríningové vyšetrenia, t.j. tuberkulínový kožný test alebo krvné vyšetrenie a RTG hrudníka (možno aplikovať miestne odporúčania). Odporúča sa, aby sa vykonanie týchto testov zaznamenalo do karty s pripomienkami pre pacienta. Upozorňujeme predpisujúcich lekárov na riziko falošne negatívnych výsledkov tuberkulínových kožných testov, najmä u pacientov, ktorí sú ťažko chorí alebo imunokompromitovaní.
Ak sa diagnostikuje aktívna tuberkulóza, liečba golimumabom sa nesmie začať (pozri časť 4.3). Ak je podozrenie na latentnú tuberkulózu, treba to konzultovať s lekárom s odbornosťou v liečbe
tuberkulózy. Vo všetkých situáciách popísaných nižšie sa má veľmi starostlivo zvážiť vyváženie pomeru prínosu/rizika liečby golimumabom.
Ak sa diagnostikuje neaktívna („latentná“) tuberkulóza, musí sa, predtým ako sa iniciuje liečba golimumabom, začať antituberkulózna liečba latentnej tuberkulózy, a to v súlade s miestnymi odporúčaniami.
U pacientov, ktorí majú niekoľko rizikových faktorov alebo významné rizikové faktory pre vznik tuberkulózy a majú negatívny test na latentnú tuberkulózu, sa musí pred nasadením golimumabu zvážiť antituberkulózna liečba. Použitie antituberkulóznej liečby sa má tiež zvážiť pred nasadením golimumabu u pacientov s latentnou alebo aktívnou tuberkulózou v anamnéze, u ktorých nie je možné potvrdiť adekvátny priebeh liečby.
U pacientov liečených golimumabom počas a po liečbe latentnej tuberkulózy sa vyskytli prípady aktívnej tuberkulózy. Pacienti liečení golimumabom sa majú starostlivo sledovať na prejavy
a príznaky aktívnej tuberkulózy, vrátane pacientov s negatívnym testom na latentnú tuberkulózu, pacientov, ktorí sú na latentnú tuberkulózu liečení, alebo pacientov, ktorí boli predtým liečení na
tuberkulózu.
Všetci pacienti majú byť informovaní o tom, že musia vyhľadať lekársku pomoc, ak sa u nich počas liečby alebo po liečbe golimumabom objavia prejavy/príznaky, ktoré poukazujú na tuberkulózu (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, zvýšená teplota).
Reaktivácia vírusovej hepatitídy B
U pacientov liečených TNF-antagonistom vrátane golimumabu, ktorí sú chronickými nositeľmi tohto vírusu (t.j. pozitívny povrchový antigén), sa objavila reaktivácia hepatitídy B. Niektoré prípady mali
fatálne následky.
Pred začatím liečby golimumabom majú byť pacienti vyšetrení na infekciu HBV. Pacientom s pozitívnym výsledkom vyšetrenia na infekciu HBV sa odporúča konzultácia s lekárom
s odbornosťou na liečbu hepatitídy B.
Nositelia HBV, u ktorých je potrebná liečba golimumabom, sa majú starostlivo sledovať na prejavy
a príznaky aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby. Nie sú dostupné dostatočné údaje o liečbe pacientov, ktorí sú nositeľmi HBV a dostávajú antivírusovú liečbu
v kombinácii s liečbou TNF-antagonistom, aby sa zabránilo reaktivácii HBV. U pacientov, u ktorých
došlo k reaktivácii HBV, je potrebné golimumab ukončiť a má sa začať účinná antivírusová liečba s príslušnou podpornou liečbou.
Malignity a lymfoproliferatívne poruchy
Potenciálna úloha liečby blokujúcej TNF v rozvoji malignít nie je známa. Podľa súčasných vedomostí nemožno u pacientov liečených TNF-antagonistom vylúčiť riziko vzniku lymfómov, leukémie alebo
iných malignít. Keď sa zvažuje liečba blokujúca TNF u pacientov s malignitou v anamnéze alebo pri zvažovaní pokračovania liečby u pacientov, u ktorých sa vyvinula malignita, je potrebná opatrnosť.
Malignity u detí a dospievajúcich
Malignity, niektoré fatálne, sa hlásili u detí, dospievajúcich a mladých dospelých (do veku 22 rokov)
liečených liečivami blokujúcimi TNF (začatie liečby vo veku ≤ 18 rokov) v období po uvedení lieku na trh. Približne polovica týchto prípadov boli lymfómy. Ďalšie prípady predstavovali rôzne druhy iných malignít a zahŕňali zriedkavé malignity zvyčajne súvisiace s imunosupresiou. Riziko rozvoja malignít u detí a dospievajúcich liečených TNF-blokátormi nie je možné vylúčiť.
Lymfóm a leukémia
V kontrolovaných častiach klinických skúšaní všetkých liečiv blokujúcich TNF vrátane golimumabu, sa pozorovalo viac prípadov lymfómu u pacientov, ktorí dostávali anti-TNF liečbu, v porovnaní
s pacientmi v kontrolnej skupine. V priebehu fázy IIb a fázy III klinických skúšaní Simponi pri RA,
PsA a AS, bola incidencia lymfómu u pacientov liečených golimumabom vyššia, ako sa očakáva
v bežnej populácii. U pacientov liečených golimumabom sa hlásili prípady leukémie. U pacientov
s reumatoidnou artritídou s dlhotrvajúcim, vysoko aktívnym zápalovým ochorením je zvýšené základné riziko vzniku lymfómov a leukémie, čo komplikuje odhad rizika.
U pacientov liečených inými liečivami blokujúcimi TNF sa po ich uvedení na trh hlásili zriedkavé prípady hepatosplenického lymfómu T-buniek (HSTCL) (pozri časť 4.8). Tento zriedkavý typ lymfómu T-buniek má veľmi agresívny priebeh ochorenia a je zvyčajne smrteľný. Väčšina prípadov sa objavila u dospievajúcich a mladých dospelých mužov, z ktorých takmer všetci užívali súbežnú liečbu azatioprinom (AZA) alebo 6-merkaptopurínom (6-MP) na zápalové ochorenie čreva. Možné riziko pri kombinácii AZA alebo 6-MP a golimumabu sa má starostlivo zvážiť. Riziko vzniku hepatosplenického lymfómu T-buniek u pacientov liečených blokátormi TNF nie je možné vylúčiť.
Malignity iné ako lymfóm
V kontrolovaných častiach klinických skúšaní Simponi fázy IIb a fázy III s RA, PsA, AS a UC, bola incidencia nelymfómových malignít (okrem nemelanómového karcinómu kože) podobná po
golimumabe a v kontrolných skupinách.
Dysplázia/karcinóm hrubého čreva
Nie je známe, či má liečba golimumabom vplyv na riziko vzniku dysplázie alebo rakoviny hrubého čreva. Všetci pacienti s ulceróznou kolitídou, u ktorých je zvýšené riziko dysplázie alebo rakoviny
hrubého čreva (napríklad pacienti s dlhodobo pretrvávajúcou ulceróznou kolitídou alebo primárnou
sklerotizujúcou cholangitídou) alebo s dyspláziou alebo karcinómom hrubého čreva v anamnéze, majú byť pred liečbou a počas celého priebehu ich ochorenia v pravidelných intervaloch vyšetrovaní na prítomnosť dysplázie. Toto vyšetrenie má zahŕňať kolonoskopiu a biopsie podľa miestnych odporúčaní. U pacientov s novo diagnostikovanou dyspláziou, ktorí sa liečia golimumabom, sa musia pozorne prehodnotiť riziká a prínosy pre konkrétneho pacienta a má sa zvážiť, či sa má v liečbe pokračovať.
V prieskumnom klinickom skúšaní, ktoré hodnotilo používanie golimumabu u pacientov so závažnou perzistujúcou astmou, bolo hlásených viac malignít u pacientov liečených golimumabom v porovnaní s pacientmi v kontrolnej skupine (pozri časť 4.8). Význam tohto nálezu nie je známy.
V prieskumnom klinickom skúšaní, ktoré hodnotilo používanie iného anti-TNF liečiva, infliximabu, u pacientov s mierne závažnou až závažnou chronickou obštrukčnou chorobou pľúc (CHOCHP), sa hlásilo viac malignít u pacientov liečených infliximabom, prevažne v pľúcach alebo na hlave a krku, v porovnaní s pacientmi v kontrolnej skupine. Všetci pacienti mali v anamnéze intenzívne fajčenie. Z tohto dôvodu je potrebná opatrnosť pri používaní akéhokoľvek TNF-antagonistu u pacientov
s CHOCHP, tak ako aj u pacientov so zvýšeným rizikom vývoja malignity z dôvodu intenzívneho fajčenia.
Rakovina kože
U pacientov liečených blokátormi TNF vrátane golimumabu sa hlásil výskyt melanómu a karcinómu z Merkelových buniek (pozri časť 4.8). Odporúča sa vykonávať pravidelné vyšetrenie kože, najmä
u pacientov s rizikovými faktormi pre vznik rakoviny kože.
Kongestívne zlyhávanie srdca (congestive heart failure, CHF)
Pri TNF-blokátoroch vrátane golimumabu boli hlásené prípady zhoršenia kongestívneho zlyhávania srdca (CHF) a nový vznik CHF. Niektoré prípady mali fatálne následky. V klinickom skúšaní s iným TNF-antagonistom sa pozorovalo zhoršenie kongestívneho zlyhávania srdca a zvýšenie mortality
z dôvodu CHF. Golimumab sa u pacientov s CHF neskúmal. Golimumab sa má použiť s opatrnosťou u pacientov s miernym zlyhávaním srdca (NYHA trieda I/II). Pacienti, u ktorých sa rozvinú nové
alebo sa zhoršia príznaky zlyhávania srdca, sa majú dôkladne sledovať a podávanie golimumabu sa musí ukončiť (pozri časť 4.3).
Neurologické udalosti
Použitie liečiv blokujúcich TNF vrátane golimumabu bolo spojené s prípadmi nového vzniku alebo exacerbácie klinických príznakov a/alebo rádiografickým dôkazom demyelinizačných porúch centrálneho nervového systému vrátane roztrúsenej sklerózy a periférnych demyelinizačných porúch.
U pacientov s existujúcimi alebo nedávno diagnostikovanými demyelinizačnými poruchami sa majú pred začatím liečby golimumabom starostlivo zvážiť prínosy a riziká anti-TNF liečby. Ak sa tieto poruchy vyvinú, má sa zvážiť ukončenie liečby golimumabom (pozri časť 4.8).
Chirurgickývýkon
Skúsenosti o bezpečnosti liečby golimumabom u pacientov, ktorí sa podrobili chirurgickému výkonu vrátane artroplastiky, sú obmedzené. Ak sa plánuje chirurgický výkon, má sa zohľadniť dlhý biologický polčas. Pacient, ktorý počas liečby golimumabom potrebuje chirurgický výkon, sa má pozorne sledovať z dôvodu infekcií a majú sa vykonať náležité opatrenia.
Imunosupresia
Existuje možnosť, že liečivá blokujúce TNF vrátane golimumabu ovplyvňujú obranyschopnosť organizmu voči infekciám a malignitám, keďže TNF sprostredkúva zápalový proces a moduluje
bunkové imunitné odpovede.
Autoimunitné procesy
Relatívny deficit TNFα spôsobený anti-TNF liečbou môže viesť k rozvoju autoimunitného procesu. Ak sa u pacienta po liečbe golimumabom vyvinú príznaky pripomínajúce lupusu podobný syndróm a ak má pacient pozitívne protilátky proti dvojvláknovej DNA, liečba golimumabom sa má ukončiť (pozri časť 4.8).
Hematologické reakcie
U pacientov dostávajúcich TNF-blokátory vrátane golimumabu boli hlásené pancytopénia, leukopénia, neutropénia, agranulocytóza, aplastická anémia a trombocytopénia. Všetkým pacientom sa má
odporučiť okamžite vyhľadať lekársku starostlivosť, ak sa rozvinú prejavy a príznaky naznačujúce
dyskrázie krvi (napr. pretrvávajúca horúčka, tvorba modrín, krvácanie, bledosť). U pacientov
s potvrdenými signifikantnými hematologickými abnormalitami sa má zvážiť ukončenie liečby golimumabom.
Súbežné podávanie TNF-antagonistov a anakinry
V klinických štúdiách so súbežným používaním anakinry a iného liečiva blokujúceho TNF, etanerceptu, sa pozorovali závažné infekcie a neutropénia, bez prídavného klinického prínosu. Kvôli charakteru nežiaducich udalostí, pozorovaných pri tejto kombinovanej liečbe, podobné toxicity môžu tiež vyústiť z kombinácie s anakinrou a inými liečivami blokujúcimi TNF. Kombinácia golimumabu
a anakinry sa neodporúča.
Súbežné podávanie TNF-antagonistov a abataceptu
V klinických štúdiách sa súbežné podávanie TNF-antagonistov a abataceptu spájalo so zvýšeným rizikom infekcií, vrátane závažných infekcií, v porovnaní s TNF-antagonistami samotnými, bez zvýšeného klinického prínosu. Kombinácia golimumabu a abataceptu sa neodporúča.
Súbežné podávanie s inými biologickými liečivami
Existujú nedostatočné informácie týkajúce sa súbežného používania golimumabu s inými biologickými liečivami používanými na liečbu rovnakých ochorení ako golimumab. Súbežné používanie golimumabu s týmito biologickými liečivami sa neodporúča vzhľadom na možnosť zvýšeného rizika vzniku infekcie a iných potenciálnych farmakologických interakcií.
Zámena jednotlivých biologických DMARDs
Pri prechode z jedného biologického liečiva na iné sa má postupovať s opatrnosťou a pacienti majú byť naďalej monitorovaní, pretože prekrývanie biologického účinku môže ďalej zvyšovať riziko
vzniku nežiaducich udalostí vrátane infekcie.
Očkovania/infekčné látky na terapeutické účely
Pacienti liečení golimumabom môžu byť súčasne očkovaní, s výnimkou živých vakcín (pozri časti 4.5
a 4.6). U pacientov, ktorí dostávajú anti-TNF liečbu, sú dostupné obmedzené údaje týkajúce sa odpovede na očkovanie živými vakcínami alebo sekundárneho prenosu infekcie živými vakcínami. Použitie živých vakcín môže viesť k vzniku klinických infekcií vrátane diseminovaných infekcií.
Iné použitia infekčných látok na terapeutické účely, ako sú napr. živé atenuované baktérie (napr. BCG na instiláciu do močového mechúra na liečbu rakoviny) môžu viesť ku vzniku klinických infekcií vrátane diseminovaných infekcií. Odporúča sa, aby sa infekčné látky na terapeutické účely nepodávali súbežne s golimumabom.
Alergické reakcie
Po uvedení lieku na trh boli po podaní golimumabu hlásené závažné systémové hypersenzitívne reakcie (vrátane anafylaktickej reakcie). Niektoré z týchto reakcií sa vyskytli po prvom podaní
golimumabu. Ak sa objaví anafylaktická reakcia alebo iné závažné alergické reakcie, podávanie
golimumabu sa má okamžite ukončiť a má sa iniciovať príslušná liečba.
Precitlivenosť na latex
Ochranný kryt ihly na naplnenom pere alebo naplnenej injekčnej striekačke je vyrobený zo suchého prírodného kaučuku, ktorý obsahuje latex a môže vyvolať alergické reakcie u osôb precitlivených na latex.
Osobitné skupiny pacientov
Staršie osoby (≥ 65 rokov)
Vo fáze III štúdií s RA, PsA, AS a UC nebol pozorovaný výrazný rozdiel v nežiaducich udalostiach (adverse event, AE), v závažných nežiaducich udalostiach (serious adverse event, SAE) a v závažných infekciách u pacientov vo veku 65 rokov alebo starších, ktorí dostávali golimumab v porovnaní
s mladšími pacientmi. Je však potrebná opatrnosť v liečbe starších osôb a zvláštna pozornosť sa musí venovať vzhľadom k výskytu infekcií. V štúdii nr-axiálnej SpA neboli žiadni pacienti vo veku
45 rokov a starší.
Porucha funkcie obličiek a pečene
Špecifické štúdie golimumabu u pacientov s poruchou funkcie obličiek alebo pečene sa nevykonali. Golimumab sa má používať s opatrnosťou u osôb s poruchou funkcie pečene (pozri časť 4.2).
Deti a dospievajúci
Očkovania
Odporúča sa, aby detskí a dospievajúci pacienti pred začatím liečby golimumabom, ak je to možné, absolvovali všetky očkovania v súlade so súčasnými odporúčaniami pre imunizáciu (pozri
Očkovania/infekčné látky na terapeutické účely vyššie).
Pomocné látky
Simponi obsahuje sorbitol (E420). U pacientov s dedičnou neznášanlivosťou fruktózy sa musí vziať do úvahy aditívny účinok súbežne podávaných liekov obsahujúcich sorbitol (alebo fruktózu) a príjem
sorbitolu (alebo fruktózy) v strave (pozri časť 2).
Možnosť chýb pri podávaní lieku
Simponi je registrovaný v silách 50 mg a 100 mg na subkutánne podanie. Je dôležité, aby sa použila správna sila na podanie správnej dávky, ako je to indikované v dávkovaní (pozri časť 4.2). Má sa dbať
na to, aby sa poskytla správna sila, aby sa zaistilo, že u pacientov nedôjde k poddávkovaniu alebo predávkovaniu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Súbežné používanie s inými biologickými liečivami
Kombinácia golimumabu s inými biologickými liečivami používanými na liečbu rovnakých ochorení ako golimumab vrátane anakinry a abataceptu sa neodporúča (pozri časť 4.4).
Živé vakcíny/infekčné látky na terapeutické účely
Živé vakcíny sa nemajú podávať súbežne s golimumabom (pozri časti 4.4 a 4.6).
Infekčné látky na terapeutické účely sa nemajú podávať súbežne s golimumabom (pozri časť 4.4). Metotrexát
I napriek tomu, že súbežné použitie MTX vyvoláva u pacientov s RA, PsA a AS vyššie minimálne koncentrácie golimumabu v rovnovážnom stave, údaje nenaznačujú, že je potrebná úprava dávky či už
golimumabu alebo MTX (pozri časť 5.2).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať vhodnú antikoncepciu na zabránenie gravidity a pokračovať v jej používaní najmenej 6 mesiacov po poslednej liečbe golimumabom.
Gravidita
Nie sú dostatočné údaje o použití golimumabu u gravidných žien. Z dôvodu jeho inhibície TNF môže golimumab podávaný počas gravidity ovplyvniť normálne imunitné odpovede novorodenca. Štúdie na
zvieratách nepreukázali priame alebo nepriame škodlivé účinky s ohľadom na graviditu,
embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3). Použitie golimumabu
u gravidných žien sa neodporúča; golimumab sa môže podať gravidnej žene len ak je to jednoznačne nevyhnutné.
Golimumab prechádza placentou. Po liečbe monoklonálnou protilátkou blokujúcou TNF počas gravidity sa počas 6 mesiacov zistili protilátky v sére dojčaťa narodeného liečenej matke. V dôsledku toho môžu mať tieto dojčatá zvýšené riziko infekcie. Podanie živých vakcín dojčatám vystaveným golimumabu in utero sa neodporúča po dobu 6 mesiacov po matkinej poslednej injekcii golimumabu počas gravidity (pozri časti 4.4 a 4.5).
Dojčenie
Nie je známe, či sa golimumab vylučuje do ľudského mlieka alebo či sa po užití systémovo absorbuje. Ukázalo sa, že golimumab prestupuje do materského mlieka opíc, a pretože ľudské imunoglobulíny
prestupujú do materského mlieka, ženy nesmú dojčiť v priebehu liečby a najmenej 6 mesiacov po
liečbe golimumabom.
Fertilita
S golimumabom sa nevykonali žiadne štúdie fertility na zvieratách. Štúdia fertility na myšiach pri použití analogickej protilátky selektívne inhibujúcej funkčnú aktivitu myšieho TNFα nepreukázala žiadne významné účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Simponi má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní Simponi sa však môže objaviť závrat (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V kontrolovanom období pivotných skúšaní RA, PsA, AS, nr-axiálnej SpA a UC bola najčastejšou nežiaducou reakciou (adverse reaction, AR) infekcia horných dýchacích ciest, ktorá sa hlásila
u 12,6 % pacientov liečených golimumabom v porovnaní s 11,0 % pacientov v kontrolnej skupine.
Najzávažnejšie AR, ktoré boli hlásené pri golimumabe, zahŕňajú závažné infekcie (vrátane sepsy, pneumónie, tuberkulózy, invazívnych mykotických a oportúnnych infekcií), demyelinizačné poruchy, reaktiváciu HBV, CHF, autoimunitné procesy (lupusu podobný syndróm), hematologické reakcie, závažnú systémovú hypersenzitivitu (vrátane anafylaktickej reakcie), vaskulitídu, lymfóm a leukémiu (pozri časť 4.4).
Tabuľkový zoznam nežiaducich reakcií
AR pozorované v klinických štúdiách a celosvetovo hlásené po uvedení golimumabu na trh sú uvedené v tabuľke 1. V rámci stanovených tried orgánových systémov sú AR vymenované pod
hlavičkami frekvencií výskytu s použitím nasledovnej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100
až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (nedá sa odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
Tabuľka 1
Tabuľkový zoznam AR
Veľmi časté: Infekcie horných dýchacích ciest (nazofaryngitída, faryngitída, laryngitída a rinitída)
Časté: Bakteriálne infekcie (ako je flegmóna), infekcie dolných
dýchacích ciest (ako je pneumónia), vírusové infekcie (ako je chrípka a herpes), bronchitída, sinusitída, povrchové mykotické infekcie, absces
Menej časté: Sepsa vrátane septického šoku, pyelonefritída
Zriedkavé: Tuberkulóza, oportúnne infekcie (ako sú invazívne mykotické infekcie [histoplazmóza, kokcidioidomykóza, pneumocystóza],
bakteriálna, atypická mykobakteriálna a protozoálna infekcia),
reaktivácia hepatitídy B, bakteriálna artritída, infekčná burzitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov
Menej časté: Neoplazmy (ako sú karcinóm kože, skvamocelulárny karcinóm a melanocytový névus)
Zriedkavé: Lymfóm, leukémia, melanóm, karcinóm z Merkelových buniek
Neznáme: Hepatosplenický lymfóm T-buniek* Poruchy krvi a lymfatického systému
Časté: Leukopénia (vrátane neutropénie), anémia
Menej časté: Trombocytopénia, pancytopénia
Zriedkavé: Aplastická anémia, agranulocytóza
Poruchy imunitného systému

Časté: Alergické reakcie (bronchospazmus, hypersenzitivita, urtikária), pozitívne autoprotilátky
Zriedkavé: Závažné systémové hypersenzitívne reakcie (vrátane
anafylaktickej reakcie), vaskulitída (systémová), sarkoidóza
Poruchy endokrinného systému
Menej časté: Poruchy štítnej žľazy (ako sú hypotyreoidizmus, hypertyreoidizmus a struma)
Poruchy metabolizmu a výživy
Menej časté: Zvýšenie glukózy v krvi, zvýšenie lipidov
Psychické poruchy
Časté: Depresia, insomnia
Poruchy nervového systému
Časté: Závrat, bolesť hlavy, parestézia
Menej časté: Poruchy rovnováhy
Zriedkavé: Demyelinizačné poruchy (centrálne a periférne), poruchy chuti
Poruchy oka
Menej časté: Poruchy zraku (ako je zahmlené videnie a zníženie zrakovej ostrosti), konjunktivitída, alergia oka (ako je svrbenie
a podráždenie)
Poruchy srdca a srdcovej činnosti
Menej časté: Arytmia, ischemické poruchy koronárnych artérií
Zriedkavé: Kongestívne zlyhávanie srdca (nové alebo zhoršujúce sa)
Poruchy ciev
Časté: Hypertenzia
Menej časté: Trombóza (ako je hĺbková venózna alebo aorty), sčervenenie
Zriedkavé: Raynaudov fenomén
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté: Astma a pridružené príznaky (ako sú sipot a bronchiálna hyperaktivita)
Menej časté: Intersticiálna choroba pľúc
Poruchy gastrointestinálneho traktu
Časté: Dyspepsia, gastrointestinálna a abdominálna bolesť, nauzea, gastrointestinálne zápalové poruchy (ako sú gastritída a kolitída), stomatitída
Menej časté: Zápcha, gastroezofágová refluxová choroba
Poruchy pečene a žlčových ciest
Časté: Zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza
Menej časté: Cholelitiáza, poruchy pečene
Poruchy kože a podkožného tkaniva
Časté: Svrbenie, vyrážka, alopécia, dermatitída
Menej časté: Bulózne kožné reakcie, psoriáza (nová alebo zhoršená psoriáza, palmárna/plantárna a pustulárna), urtikária
Zriedkavé: Lichenoidné reakcie, odlupovanie kože, vaskulitída (kutánna)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Zriedkavé: Lupusu podobný syndróm
Poruchy obličiek a močových ciest
Zriedkavé: Poruchy močového mechúra, poruchy obličiek
Poruchy reprodukčného systému a prsníkov
Menej časté: Poruchy prsníkov, menštruačné poruchy
Celkové poruchy a reakcie v mieste podania
Časté: Pyrexia, asténia, reakcie v mieste podania injekcie (ako sú erytém, urtikária, zdurenie, bolesť, modrina, svrbenie, podráždenie
a parestézia v mieste vpichu), diskomfort na hrudníku
Zriedkavé: Zhoršené hojenie
Úrazy, otravy a komplikácie liečebného postupu
Časté: Zlomeniny kostí
* pozorované u iných liečiv blokujúcich TNF
V celej tejto časti je pre všetky spôsoby použitia golimumabu všeobecne uvedený medián trvania
následného sledovania (približne 4 roky). Keďže pacienti mohli byť prestavení z jednej dávky na druhú, medián trvania následného sledovania je rôzny v prípadoch, keď sa spôsob použitia golimumabu charakterizoval prostredníctvom dávky (približne 2 roky pre 50 mg dávku a približne
3 roky pre 100 mg dávku).
Popis vybraných nežiaducich reakciíInfekcieV kontrolovanom období pivotných skúšaní bola najčastejšou nežiaducou reakciou infekcia horných dýchacích ciest, ktorá sa hlásila u 12,6 % pacientov liečených golimumabom (incidencia na
100 pacientskych rokov: 60,8; 95% IS: 55,0; 67,1) v porovnaní s 11,0 % pacientov v kontrolnej skupine (incidencia na 100 pacientskych rokov: 54,5; 95% IS: 46,1; 64,0). Počas kontrolovaných
a nekontrolovaných častí štúdií s mediánom sledovania približne 4 roky bola incidencia infekcií horných dýchacích ciest u pacientov liečených golimumabom 34,9 udalostí na 100 pacientskych rokov; 95% IS: 33,8; 36,0.
V kontrolovanom období pivotných skúšaní sa pozorovali infekcie u 23,0 % pacientov liečených golimumabom (incidencia na 100 pacientskych rokov: 132,0; 95% IS: 123,3; 141,1), v porovnaní s 20,2 % u pacientov v kontrolnej skupine (incidencia na 100 pacientskych rokov: 122,3; 95% IS:
109,5; 136,2). Počas kontrolovaných a nekontrolovaných častí klinických štúdií s mediánom
sledovania približne 4 roky bola incidencia infekcií u pacientov liečených golimumabom 81,1 udalostí na 100 pacientskych rokov; 95% IS: 79,5; 82,8.
V kontrolovanom období skúšaní RA, PsA, AS a nr-axiálnej SpA sa pozorovali závažné infekcie u 1,2 % pacientov liečených golimumabom a u 1,2 % pacientov v kontrolnej liečenej skupine. Incidencia závažných infekcií na 100 pacientskych rokov následného sledovania v kontrolovanom období skúšaní RA, PsA, AS a nr-axiálnej SpA bola 7,3; 95% IS: 4,6; 11,1 v skupine so 100 mg golimumabu, 2,9; 95% IS: 1,2; 6,0 v skupine s 50 mg golimumabu a 3,6; 95% IS: 1,5; 7,0 v skupine s placebom. V kontrolovanom období skúšaní zameraných na nasadenie golimumabu pri UC sa závažné infekcie pozorovali u 0,8 % pacientov liečených golimumabom v porovnaní s 1,5 % pacientov s kontrolnou liečbou. Závažné infekcie pozorované u pacientov liečených golimumabom zahŕňali tuberkulózu, bakteriálne infekcie vrátane sepsy a pneumónie, invazívne mykotické infekcie a iné oportúnne infekcie. Niektoré z týchto infekcií boli fatálne. Počas kontrolovaných
a nekontrolovaných častí pivotných skúšaní s mediánom sledovania až do 3 rokov bola u pacientov, ktorí dostávali 100 mg golimumabu, pozorovaná vyššia incidencia závažných infekcií vrátane oportúnnych infekcií a tuberkulózy v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu. Incidencia všetkých závažných infekcií na 100 pacientskych rokov bola 4,1; 95% IS: 3,6; 4,5
u pacientov, ktorí dostávali 100 mg golimumabu a 2,5; 95% IS: 2,0; 3,1 u pacientov, ktorí dostávali
50 mg golimumabu.
Malignity
Lymfóm
Počas pivotných skúšaní bola incidencia lymfómu u pacientov liečených golimumabom vyššia, než sa očakáva v bežnej populácii. Počas kontrolovaných a nekontrolovaných častí týchto skúšaní
s mediánom sledovania až do 3 rokov bola vyššia incidencia lymfómu pozorovaná u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu. Lymfóm
sa diagnostikoval u 11 osôb (1 v skupinách liečených s 50 mg golimumabu a 10 v skupinách liečených so 100 mg golimumabu) s incidenciou (95% IS) na 100 pacientskych rokov následného sledovania
0,03 (0,00; 0,15) udalostí pre golimumab 50 mg a 0,13 (0,06; 0,24) udalostí pre golimumab 100 mg
a 0,00 (0,00; 0,57) udalostí pre placebo. Väčšina lymfómov sa vyskytla v štúdii GO-AFTER, v ktorej boli zaradení pacienti s predošlou expozíciou anti-TNF liečivám, ktorí mali dlhšie trvajúce
a refraktérnejšie ochorenie (pozri časť 4.4).
Malignity iné než lymfóm
V kontrolovaných obdobiach pivotných skúšaní a počas približne 4 rokov následného sledovania bola podobná incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) medzi golimumabom a kontrolnými skupinami. Počas približne 4 rokov následného sledovania bola incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) podobná všeobecnej populácii.
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov bol nemelanómový karcinóm kože diagnostikovaný u 5 osôb v skupine dostávajúcej placebo,
10 s golimumabom 50 mg a 31 s golimumabom 100 mg s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,36 (0,26; 0,49) pre kombinovaný golimumab a 0,87 (0,28; 2,04) pre
placebo.
V kontrolovanom a nekontrolovanom období pivotných skúšaní s mediánom sledovania až do 3 rokov boli malignity okrem melanómového, nemelanómového karcinómu kože a lymfómu diagnostikované
u 5 osôb v skupine dostávajúcej placebo, 21 s golimumabom 50 mg a 34 s golimumabom 100 mg
s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,48 (0,36; 0,62) pre kombinovaný golimumab a 0,87 (0,28; 2,04) pre placebo (pozri časť 4.4).
Prípady hlásené v klinických štúdiách s astmou
V prieskumnej klinickej štúdii bol pacientom so závažnou perzistujúcou astmou podávaný golimumab v záťažovej dávke (150 % určenej terapeutickej dávky) subkutánne v týždni 0 a následne v dávkach
200 mg golimumabu, 100 mg golimumabu alebo 50 mg golimumabu každé 4 týždne subkutánne až do
52. týždňa. Hlásilo sa osem malignít v kombinovanej skupine liečenej golimumabom (n = 230)
a žiadna v skupine dostávajúcej placebo (n = 79). Lymfóm sa hlásil u 1 pacienta, nemelanómový karcinóm kože u 2 pacientov a iné malignity u 5 pacientov. U žiadneho typu malignity nevzniklo špecifické zoskupovanie.
Počas placebom kontrolovanej časti štúdie bola incidencia (95% IS) všetkých malignít na
100 pacientskych rokov v následnom sledovaní 3,19 (1,38; 6,28) v skupine s golimumabom. V tejto štúdii bola incidencia (95% IS) na 100 pacientskych rokov v následnom sledovaní u osôb liečených
golimumabom 0,40 (0,01; 2,20) pre lymfóm, 0,79 (0,10; 2,86) pre nemelanómové karcinómy kože a 1,99 (0,64; 4,63) pre iné malignity. U osôb s placebom bola incidencia (95% IS) na
100 pacientskych rokov v následnom sledovaní týchto malignít 0,00 (0,00; 2,94). Význam tohto nálezu nie je známy.
Neurologické udalosti
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov sa pozorovala väčšia incidencia demyelinizácie u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu (pozri časť 4.4).
Zvýšenia hepatálnych enzýmov
V kontrolovanom období pivotných skúšaní RA a PsA sa vyskytlo mierne zvýšenie ALT (> 1
a < 3 x horná hranica normálu (ULN)) v štúdiách s RA a PsA v podobnej miere u pacientov s golimumabom a u kontrolných pacientov (22,1 % k 27,4 % pacientov). V štúdiách s AS
a nr-axiálnou SpA malo mierne zvýšenie ALT viac pacientov liečených golimumabom (26,9 %) než
pacienti v kontrolnej skupine (10,6 %). V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní RA a PsA s mediánom sledovania približne 5 rokov bola v štúdiách s RA a PsA incidencia mierneho zvýšenia ALT podobná u pacientov liečených golimumabom a u kontrolných pacientov.
V kontrolovanom období pivotných skúšaní zameraných na nasadenie golimumabu pri UC sa vyskytli mierne zvýšenia ALT (> 1 a < 3 x ULN) v podobnej miere u pacientov liečených golimumabom
(8,0 %) a u pacientov v kontrolnej skupine (6,9 %). V kontrolovaných a nekontrolovaných obdobiach
pivotných skúšaní UC s mediánom sledovania približne 2 roky bol podiel pacientov s miernymi zvýšeniami ALT 24,7 % u pacientov dostávajúcich golimumab počas udržiavacej časti štúdie UC.
Zvýšenie ALT ≥ 5 x ULN bolo v kontrolovanom období pivotných skúšaní RA a AS menej časté a pozorovalo sa častejšie u pacientov liečených golimumabom (0,4 % a 0,9 %) než u kontrolných pacientov (0,0 %). Táto tendencia sa nepozorovala v skupine s PsA. V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní RA, PsA a AS s mediánom sledovania 5 rokov bola incidencia zvýšenia ALT ≥ 5 x ULN podobná u pacientov liečených golimumabom aj u kontrolných
pacientov. Vo všeobecnosti boli tieto zvýšenia asymptomatické a abnormality poklesli alebo vymizli, či už pri pokračovaní liečby alebo po ukončení podávania golimumabu alebo po modifikácii súbežne podávaných liekov. V kontrolovaných a nekontrolovaných obdobiach štúdie nr-axiálnej SpA (až do 1
roka) sa nehlásili žiadne prípady. V kontrolovaných obdobiach pivotných skúšaní zameraných na nasadenie golimumabu pri UC sa vyskytli zvýšenia ALT ≥ 5 x ULN v podobnej miere u pacientov
liečených golimumabom (0,3 %) a u kontrolných pacientov (1,0 %). V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní UC s mediánom sledovania približne 2 roky bol podiel pacientov so zvýšeniami ALT ≥ 5 x ULN 0,8 % u pacientov dostávajúcich golimumab počas
udržiavacej časti štúdie UC.
Počas pivotných skúšaní RA, PsA, AS a nr-axiálnej SpA sa u jedného pacienta v skúšaní RA liečeného golimumabom s existujúcimi hepatálnymi abnormalitami a zamenenými liekmi vyvinula fatálna neinfekčná hepatitída so žltačkou. Úloha golimumabu ako prispievajúceho alebo zhoršujúceho činiteľa nemôže byť vylúčená.
Reakcie v mieste podania
V kontrolovaných obdobiach pivotných skúšaní malo 5,4 % pacientov liečených golimumabom reakciu v mieste podania v porovnaní s 2,0 % kontrolných pacientov. Prítomnosť protilátok proti
golimumabu môže zvýšiť riziko reakcií v mieste podania. Väčšina reakcií v mieste podania bola
mierna alebo stredne závažná a najčastejšie sa manifestovala ako erytém v mieste podania. Reakcie v mieste podania všeobecne nemusia nutne viesť k ukončeniu liečby.
V kontrolovaných klinických skúšaniach fázy IIb a/alebo III s RA, PsA, AS, nr-axiálnou SpA, závažnou perzistujúcou astmou a v skúšaniach fázy II/III s UC sa u žiadneho pacienta liečeného golimumabom neobjavili anafylaktické reakcie.
Autoimunitné protilátkyV kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní boli počas 1 roka následného sledovania nanovo ANA-pozitívne 3,5 % pacientov liečených golimumabom a 2,3 % kontrolných pacientov (s titrom 1:160 alebo vyšším). Frekvencia protilátok anti-dsDNA za 1 rok následného sledovania bola 1,1 % u pacientov, ktorí boli na začiatku anti-dsDNA negatívni.
Pediatrická populáciaPolyartikulárna juvenilná idiopatická artritída (pJIA)Bezpečnosť golimumabu sa skúmala v štúdii fázy III so 173 pacientmi s pJIA vo veku 2 až 17 rokov. Priemerná doba sledovania bola približne 2 roky. V tejto štúdii boli typ a frekvencia hlásených nežiaducich udalostí vo všeobecnosti podobné tým pozorovaným v štúdiách u dospelých s RA.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinickej štúdii boli jednorazovo intravenózne podané dávky až do 10 mg/kg bez toxicity limitujúcej dávku. V prípade predávkovania sa odporúča sledovanie pacienta na akékoľvek prejavy a príznaky nežiaducich účinkov a okamžite sa má začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNF-α), ATC kód: L04AB06
Mechanizmus účinkuGolimumab je ľudská monoklonálna protilátka, ktorá vytvára stabilné komplexy s vysokou afinitou so solubilnými aj transmembránovými bioaktívnymi formami ľudského TNF-α, čím zabraňuje naviazanie
TNF-α na jeho receptory.
Farmakodynamické účinkyNaviazaním golimumabu na ľudský TNF sa preukázala neutralizácia expresie adhezívnych molekúl na povrchu bunky indukovaná TNF-α – E-selektínu, adhezívnej molekuly cievnych buniek (VCAM)-1
a intercelulárnej adhezívnej molekuly (ICAM)-1 ľudských endotelových buniek.
In vitro golimumab
tiež inhiboval TNF indukovanú sekréciu interleukínu (IL)-6 a IL-8 a faktoru stimulujúceho kolónie granulocytov a makrofágov (GM-CSF) ľudských endotelových buniek.
V porovnaní so skupinami s placebom sa pozorovalo relatívne zlepšenie hladín C-reaktívneho proteínu (CRP) a liečba Simponi viedla v porovnaní s kontrolnou liečbou k významnému zníženiu sérových hladín IL-6, ICAM-1, matrixovej metaloproteinázy (MMP)-3 a vaskulárneho endotelového rastového faktoru (VEGF) oproti východiskovým hodnotám. Došlo aj ku zníženiu hladín TNF-a u pacientov
s RA a AS a hladiny IL-8 sa znížili u pacientov s PsA. Tieto zmeny sa pozorovali pri prvom hodnotení
(4. týždeň) po úvodnom podaní Simponi a všeobecne pretrvávali až do 24. týždňa.
Klinická účinnosť
Reumatoidná artritída
Účinnosť Simponi sa preukázala v troch multicentrických randomizovaných dvojito zaslepených, placebom kontrolovaných štúdiách s vyše 1 500 pacientmi vo veku ≥ 18 rokov so stredne závažnou
a závažnou aktívnou RA, diagnostikovanou podľa kritérií American College of Rheumatology (ACR) najmenej 3 mesiace pred skríningom. Pacienti mali najmenej 4 opuchnuté a 4 bolestivé kĺby. Simponi alebo placebo sa podávali subkutánne každé 4 týždne.
V GO-FORWARD sa hodnotilo 444 pacientov, ktorí mali aktívnu RA napriek stabilnej dávke MTX aspoň 15 mg/týždeň a ktorí sa predtým neliečili žiadnym anti-TNF liečivom. Pacienti boli randomizovaní a dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. Po 24. týždni boli pacienti, ktorí dostávali placebo + MTX, prevedení na Simponi 50 mg + MTX. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia.
V GO-AFTER sa hodnotilo 445 pacientov, ktorí sa už predtým liečili jednou alebo viacerými anti- TNF liečivami adalimumabom, etanerceptom alebo infliximabom. Pacienti boli randomizovaní
a dostávali placebo, Simponi 50 mg alebo Simponi 100 mg. Počas tejto štúdie mohli pacienti
pokračovať v už prebiehajúcej liečbe DMARD s MTX, sulfasalazínom (SSZ) a/alebo hydroxychlorochinom (HCQ). Stanovené dôvody na ukončenie predchádzajúcej anti-TNF liečby boli nedostatočná účinnosť (58 %), intolerancia (13 %) a/alebo iné dôvody než bezpečnosť alebo účinnosť (29 %, zväčša finančné dôvody).
V GO-BEFORE sa hodnotilo 637 pacientov s aktívnou RA, ktorí sa predtým neliečili MTX ani anti- TNF liečivom. Pacienti boli randomizovaní tak, aby dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia, v ktorom pacienti dostávajúci placebo + MTX s aspoň 1 citlivým alebo opuchnutým kĺbom, boli prestavení na Simponi 50 mg + MTX.
Primárnymi spoločnými koncovými ukazovateľmi v GO-FORWARD boli stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni a zlepšenie v 24. týždni v dotazníku hodnotiacom zdravie (Health Assessment Questionnaire, HAQ) oproti východiskovej hodnote.
V GO-AFTER bolo primárnym koncovým ukazovateľom stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni. V GO-BEFORE boli primárnymi spoločnými koncovými
ukazovateľmi stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 50 v 24. týždni a zmena
skóre podľa Sharpa modifikovanom van der Heijdeovou (vdH-S) v 52. týždni oproti východiskovej hodnote. Okrem primárnych koncových ukazovateľov sa vykonali ďalšie hodnotenia vplyvu liečby Simponi na prejavy a príznaky artritídy, rádiografickú odpoveď, telesnú funkciu a kvalitu života spojenú so zdravím.
Celkovo sa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg so súbežným MTX až do 104. týždňa v GO-FORWARD
a GO-BEFORE a až do 24. týždňa v GO-AFTER. Podľa dizajnu každej z RA štúdií mohli byť pacienti v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznaky
Kľúčové výsledky ACR pre dávku Simponi 50 mg v 14., 24. a 52. týždni GO-FORWARD,
GO-AFTER a GO-BEFORE sú zobrazené v tabuľke 2 a sú popísané nižšie. Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po úvodnej aplikácii Simponi.
V skupine 89 osôb randomizovaných na Simponi 50 mg + MTX v GO-FORWARD bolo stále na tejto liečbe 48 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 40 pacientov, odpoveď ACR 50
u 33 pacientov a odpoveď ACR 70 u 24 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
V GO-AFTER bolo percento pacientov, ktorí dosiahli odpoveď ACR 20, vyššie u pacientov dostávajúcich Simponi než u pacientov dostávajúcich placebo, bez ohľadu na hlásený dôvod ukončenia jednej alebo viacerých predchádzajúcich terapií anti-TNF.
Tabuľka 2
Kľúčové výsledky účinnosti z kontrolovaných častí GO-FORWARD, GO-AFTER
a GO-BEFORE
GO-FORWARD Aktívna RA napriek MTX
Simponi
GO-AFTER Aktívna RA, predtým liečená jedným alebo viacerými anti-TNF liečivami
GO-BEFORE Aktívna RA, neliečená MTX
Simponi
Placebo
+ MTX
50 mg
+
MTX Placebo
Simponi
50 mg
Placebo
+ MTX
50 mg
+ MTX
na 133 89 150 147 160 159
Respondenti, % pacientov
ACR 20
14. týždeň 33 % 55 %* 18 % 35 %* NA NA
24. týždeň 28 % 60 %* 16 % 31 %
p = 0,002
49 % 62 %
52. týždeň NA NA NA NA 52 % 60 %
ACR 50
14. týždeň 10 % 35 %* 7 % 15 %
p = 0,021
NA NA
24. týždeň 14 % 37 %* 4 % 16 %* 29 % 40 %
52. týždeň NA NA NA NA 36 % 42 %
ACR 70
14. týždeň 4 % 14 %
p = 0,008
2 % 10 %
p = 0,005
NA NA
24. týždeň 5 % 20 %* 2 % 9 % p = 0,009 16 % 24 %
52. týždeň NA NA NA NA 22 % 28 %
a n vyjadruje randomizovaných pacientov; aktuálny počet pacientov hodnotiteľných pre každý koncový ukazovateľ sa môže líšiť podľa časového bodu.
* p ≤ 0,001
NA: Neaplikovateľné
V GO-BEFORE nebola primárna analýza v 24. týždni u pacientov so stredne závažnou až závažnou
reumatoidnou artritídou (kombinované skupiny Simponi 50 a 100 mg + MTX vs. samotný MTX pre ACR50) štatisticky signifikantná (p = 0,053). V 52. týždni bolo v celkovej populácii celkovo vyššie percento pacientov, ktorí dosiahli odpoveď ACR, v skupine so Simponi 50 mg + MTX, ale
v porovnaní so samotným MTX nebolo signifikantne odlišné (pozri tabuľku 2). Ďalšie analýzy sa vykonali v podskupinách reprezentujúcich indikovanú populáciu pacientov so závažnou aktívnou
a progresívnou RA. V indikovanej populácii bol preukázaný celkovo väčší účinok Simponi 50 mg + MTX oproti samotnému MTX v porovnaní s celkovou populáciou.
V GO-FORWARD a GO-AFTER sa v každom dopredu určenom časovom bode pozorovali klinicky významné a štatisticky signifikantné odpovede na stupnici aktivity ochorenia (Disease Activity Scale, DAS) 28, v 14. týždni a v 24. týždni (p ≤ 0,001). V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 od
104. týždňa až do 256. týždňa podobné.
V GO-BEFORE bola meraná výrazná klinická odpoveď, definovaná ako udržanie odpovede ACR 70 nepretržite počas 6-mesačného obdobia. V 52. týždni dosiahlo výraznú klinickú odpoveď 15 % pacientov v skupine so Simponi 50 mg + MTX v porovnaní so 7 % pacientov v skupine s placebom +
MTX (p = 0,018). V skupine 159 osôb randomizovaných na Simponi 50 mg + MTX bolo stále na tejto liečbe 96 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 85 pacientov, odpoveď ACR 50
u 66 pacientov a odpoveď ACR 70 u 53 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
Rádiografická odpoveď:V GO-BEFORE bola na stanovenie stupňa štrukturálneho poškodenia použitá zmena vo vdH-S skóre oproti východiskovej hodnote, čo je kombinované skóre štrukturálneho poškodenia, ktoré
rádiograficky stanovuje počet a veľkosť erózií kĺbov a stupeň zúženia kĺbovej štrbiny (joint space
narrowing, JSN) na rukách/zápästiach a nohách. Kľúčové výsledky pre dávku Simponi 50 mg v 52. týždni sú uvedené v tabuľke 3.
Počet pacientov bez nových erózií alebo so zmenou oproti východiskovej hodnote v celkovom vdH-S skóre ≤ 0 bol signifikantne vyšší v skupine liečenej Simponi ako v kontrolnej skupine (p = 0,003). Rádiografické účinky pozorované v 52. týždni boli udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli rádiografické účinky od 104. týždňa až do 256. týždňa podobné.
Tabuľka 3 Priemerné (štandardná odchýlka) rádiografické zmeny oproti východiskovej hodnote v celkovom vdH-S skóre v 52. týždni v celkovej populácii v GO-BEFOREPlacebo + MTX Simponi 50 mg + MTX
Priemerné (štandardná odchýlka) rádiografické zmeny oproti východiskovej hodnote v celkovom vdH-S skóre v 52. týždni v celkovej populácii v GO-BEFOREPlacebo + MTX Simponi 50 mg + MTXn
a 160 159Celkové skóreVýchodisková hodnota 19,7 (35,4) 18,7 (32,4) Zmena oproti východiskovej hodnote 1,4 (4,6) 0,7 (5,2)*
Skóre erózieVýchodisková hodnota 11,3 (18,6) 10,8 (17,4) Zmena oproti východiskovej hodnote 0,7 (2,8) 0,5 (2,1)
Skóre JSNVýchodisková hodnota 8,4 (17,8) 7,9 (16,1) Zmena oproti východiskovej hodnote 0,6 (2,3) 0,2 (2,0)** a n vyjadruje randomizovaných pacientov
* p = 0,015
** p = 0,044
Telesná funkcia a kvalita života spojená so zdravímTelesná funkcia a neschopnosť sa posudzovali ako samostatný koncový ukazovateľ v GO-FORWARD
a GO-AFTER používajúc index neschopnosti HAQ DI. V týchto štúdiách preukázal Simponi klinicky významné a štatisticky signifikantné zlepšenie v HAQ DI od základného stavu oproti kontrole v 24.
týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku
štúdie, sa udržalo zlepšenie v HAQ DI až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi bolo zlepšenie v HAQ DI od 104. týždňa až do 256. týždňa podobné.
V GO-FORWARD sa preukázali klinicky významné a štatisticky signifikantné zlepšenia kvality života spojenej so zdravím, merané pomocou skóre telesných komponentov SF-36 u pacientov liečených Simponi oproti placebu v 24. týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, bolo udržané zlepšenie telesných komponentov SF-36 až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi bolo zlepšenie telesných komponentov SF-36 od 104. týždňa až do 256. týždňa podobné. V GO-FORWARD
a GO-AFTER sa pozorovali štatisticky signifikantné zlepšenia únavy, merané škálou únavy-funkčné posúdenie liečby chronickej choroby (FACIT-F).
Psoriatická artritída
Bezpečnosť a účinnosť Simponi bola hodnotená v multicentrickej randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-REVEAL) so 405 dospelými pacientmi s aktívnou PsA (≥ 3 opuchnuté kĺby a ≥ 3 bolestivé kĺby) napriek liečbe nesteroidovými antiflogistikami (NSAID) alebo DMARD. Pacienti v tejto štúdii mali diagnózu PsA najmenej 6 mesiacov a mali aspoň miernu psoriatickú chorobu. Boli zaradení pacienti s každým podtypom psoriatickej artritídy, vrátane polyartikulárnej artritídy bez reumatoidných uzlíkov (43 %), asymetrickej periférnej artritídy (30 %), distálnej artritídy interfalangeálnych kĺbov (DIP) (15 %), spondylitídy s periférnou artritídou (11 %)
a arthritis mutilans (1 %). Predchádzajúca liečba anti-TNF liečivom nebola prípustná. Simponi alebo placebo sa podávali subkutánne každé 4 týždne. Pacientom bolo náhodne pridelené placebo, Simponi
50 mg alebo Simponi 100 mg. Po 24. týždni boli pacienti, ktorí dostávali placebo, prevedení na Simponi 50 mg. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia. Približne štyridsaťosem percent pacientov pokračovalo na stabilnej dávke metotrexátu (≤ 25 mg/týždeň).
Spoločnými primárnymi koncovými ukazovateľmi boli percento pacientov, ktorí dosiahli odpoveď
ACR 20 v 14. týždni a zmena v celkovom vdH-S skóre modifikovanom pre PsA v 24. týždni oproti východiskovej hodnote.
Celkovo sa až do 104. týždňa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg. Podľa dizajnu štúdie mohli byť pacienti
v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznaky
Kľúčové výsledky pre dávku 50 mg v 14. a 24. týždni sú zobrazené v tabuľke 4 a opísané nižšie.
Tabuľka 4
Kľúčové výsledky účinnosti z GO-REVEAL
Placebo
Simponi
50 mg*
na 113 146
Respondenti, % pacientov
ACR 20
AC
R 50
AC
R 70
PASI
b
75
c
14. týždeň
9 % 51 %24. týždeň 12 % 52 %

14. týždeň 2 % 30 %
24. týždeň 4 % 32 %
14. týždeň 1 % 12 %
24. týždeň 1 % 19 %
14. týždeň 3 % 40 %
24. týždeň 1 % 56 %
* p < 0,05 pre všetky porovnávania;
a n vyjadruje randomizovaných pacientov; aktuálny počet hodnotiteľných pacientov sa môže pre každý výsledný ukazovateľ líšiť podľa časového bodu.
b Oblasť postihnutá psoriázou a stupeň závažnosti
c Založené na podskupine pacientov so vstupným postihnutím BSA ≥ 3 %, 79 pacientov
(69,9 %) zo skupiny s placebom a 109 pacientov (74,3 %) zo skupiny so Simponi 50 mg.
Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po prvom podaní Simponi. Podobné odpovede
ACR 20 sa pozorovali v 14. týždni u pacientov s polyartikulárnou artritídou bez reumatoidných uzlíkov a pri asymetrickej periférnej artritíde, podtyp PsA. Počet pacientov s inými podtypmi PsA bol
príliš malý na zmysluplné vyhodnotenie. Odpovede pozorované v skupinách liečených Simponi boli podobné u pacientov užívajúcich a neužívajúcich súčasne metotrexát. V skupine 146 pacientov
randomizovaných na Simponi 50 mg bolo stále na tejto liečbe 70 pacientov v 104. týždni. Z týchto
70 pacientov bola odpoveď ACR 20 u 64 pacientov, odpoveď ACR 50 u 46 pacientov a odpoveď
ACR 70 u 31 pacientov. V skupine pacientov zostávajúcich v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede ACR 20/50/70.
Štatisticky významné odpovede v DAS 28 sa pozorovali aj v 14. a 24. týždni (p < 0,05).
V skupine pacientov liečených Simponi sa v 24. týždni pozorovali zlepšenia parametrov periférnej aktivity, charakteristických pre psoriatickú artritídu (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída). Liečba Simponi viedla k výraznému zlepšeniu telesných funkcií hodnotenej podľa HAQ DI, ako aj k signifikantnému zlepšeniu kvality života spojenej so zdravím ako bolo namerané pomocou sumárneho skóre telesných a mentálnych údajov podľa SF-36. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 a HAQ DI udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 a HAQ DI od 104. týždňa až do
256. týždňa podobné.
Rádiografická odpoveď:
Štrukturálne poškodenie oboch rúk a nôh sa rádiograficky hodnotilo zmenou vo východiskovej hodnote oproti vdH-S skóre modifikovanom pre PsA pridaním distálnych interfalangeálnych (DIP)
kĺbov rúk.
Liečba Simponi 50 mg znížila v 24. týždni rýchlosť progresie poškodenia periférnych kĺbov
v porovnaní s podávaním placeba podľa zmeny v celkovom modifikovanom vdH-S skóre oproti východiskovej hodnote (priemer ± SD skóre bolo 0,27 ± 1,3 v skupine s placebom v porovnaní
s -0,16 ± 1,3 v skupine so Simponi; p = 0,011). Zo 146 pacientov, ktorí boli randomizovaní na
Simponi 50 mg, boli RTG údaje dostupné u 126 pacientov v 52. týždni, z ktorých u 77 % sa nepreukázala žiadna progresia v porovnaní s východiskovou hodnotou. V 104. týždni boli RTG údaje
dostupné u 114 pacientov a u 77 % sa nepreukázala žiadna progresia oproti východiskovej hodnote.
V skupine pacientov zostávajúcich v štúdii a liečených Simponi v období od 104. týždňa až do 256. týždňa podobný pomer pacientov nepreukázal žiadnu progresiu oproti východiskovej hodnote.
Axiálna spondyloartritída
Ankylozujúca spondylitída
Bezpečnosť a účinnosť Simponi sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-RAISE) s 356 dospelými pacientmi s aktívnou ankylozujúcou spondylitídou (definovanou ako Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥ 4 a VAS ≥ 4 pri celkovej bolesti chrbtice na škále 0 až 10 cm). Pacienti zaradení do tejto štúdie mali aktívne ochorenie napriek prebiehajúcej alebo predchádzajúcej liečbe NSAID alebo DMARD
a predtým sa neliečili anti-TNF liečbou. Simponi alebo placebo sa podávali subkutánne každé
4 týždne. Pacientom bolo náhodne pridelené placebo, Simponi 50 mg a Simponi 100 mg a mali povolené pokračovať v súbežnej liečbe DMARD (MTX, SSZ a/alebo HCQ). Primárnym koncovým
ukazovateľom bolo percento pacientov, ktorí dosiahli v Ankylosing Spondylitis Assessment Study
Group (ASAS) 20 odpoveď v 14. týždni. Údaje o placebom kontrolovanej účinnosti boli zozbierané a analyzované v 24. týždni.
Kľúčové výsledky pre dávku 50 mg sú zobrazené v tabuľke 5 a popísané nižšie. Celkovo sa až do 24. týždňa nepozorovali žiadne klinicky významné rozdiely v hodnotách účinnosti medzi dávkovými režimami Simponi 50 mg a 100 mg. Podľa dizajnu štúdie mohli byť pacienti v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Tabuľka 5
 Kľúčové výsledky účinnosti v GO-RAISE
Kľúčové výsledky účinnosti v GO-RAISE
Placebo
Simponi
50 mg*
na 78 138
Respondenti, % pacientov
ASAS 20
14. týždeň 22 % 59 %
ASA
S 40
ASA
S 5/6
24. týždeň 23 % 56 %
14. týždeň 15 % 45 %
24. týždeň 15 % 44 %
14. týždeň 8 % 50 %
24. týždeň 13 % 49 %
* p ≤ 0,001 pre všetky porovnania
a n vyjadruje randomizovaných pacientov; aktuálny počet hodnotiteľných pacientov sa môže pre každý výsledný ukazovateľ líšiť podľa časového bodu.
V skupine pacientov zostávajúcich v štúdii a liečených Simponi bola časť pacientov s ASAS 20
a ASAS 40 odpoveďou od 24. týždňa až do 256. týždňa podobná.
Štatisticky signifikantné odpovede v BASDAI 50, 70 a 90 (p ≤ 0,017) sa tiež pozorovali v 14. a 24. týždni. Zlepšenie v kľúčových hodnotách aktivity choroby sa pozorovalo v prvom hodnotení (4. týždeň) po prvom podaní Simponi a pretrvávalo až do 24. týždňa. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa v BASDAI od 24. týždňa až do 256. týždňa pozoroval podobný pomer zmeny oproti východiskovej hodnote. Konzistentná účinnosť sa pozorovala bez ohľadu na použitie
DMARD (MTX, sulfasalazín a/alebo hydroxychlorochin), stav antigénu HLA-B27 alebo
východiskové hodnoty CRP, ako bolo zhrnuté v ASAS 20 odpovediach v 14. týždni.
Liečba Simponi vyústila do signifikantného zlepšenia telesnej funkcie ako bolo posúdené v BASFI zmenami v 14. a 24. týždni oproti východiskovej hodnote. Kvalita života spojená so zdravím hodnotená podľa skóre fyzických komponentov SF-36 sa tiež signifikantne zlepšila v 14. a 24. týždni. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli zlepšenia telesnej funkcie
a kvality života spojenej so zdravím od 24. týždňa až do 256. týždňa podobné.
Axiálna spondyloartritída bez rádiografického dôkazuBezpečnosť a účinnosť Simponi sa hodnotili v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-AHEAD) u 197 dospelých pacientov so závažnou aktívnou
nr-axiálnou SpA (definovaní ako tí pacienti, ktorí spĺňajú kritériá klasifikácie axiálnej
spondyloartritídy podľa ASAS, ale nespĺňajú upravené kritériá New York pre AS). Pacienti zaradení do tejto štúdie mali aktívne ochorenie (definované ako BASDAI ≥ 4 a celková bolesť chrbta ≥ 4 na vizuálnej analógovej stupnici (VAS), každé na stupnici 0-10 cm) napriek prebiehajúcej alebo predchádzajúcej liečbe NSAID a ktorí sa v minulosti neliečili žiadnymi biologickými liečivami vrátane anti-TNF liečby. Pacienti boli náhodne priradení na placebo alebo Simponi podávané v dávke
50 mg subkutánne každé 4 týždne. V 16. týždni pacienti vstúpili do otvoreného obdobia, v ktorom všetci pacienti dostávali Simponi v dávke 50 mg podávaných subkutánne každé 4 týždne počas 48 týždňov s hodnoteniami účinnosti vykonanými v 52. týždni a následnými hodnoteniami bezpečnosti
v 60. týždni. Približne 93 % pacientov, ktorí dostávali Simponi na začiatku otvoreného predĺženia (16. týždeň), pokračovalo v liečbe do konca štúdie (52. týždeň). Analýzy sa vykonali v populácii všetkých
liečených pacientov (All treated, AT, N = 197) a aj v populácii s objektívnymi prejavmi zápalu (Objective Signs of Inflammation, OSI, N = 158, definovanej na základe zvýšenej hladiny CRP a/alebo dôkazu sakroileitídy na MR na začiatku). V 16. týždni sa získali a analyzovali placebom
kontrolované údaje o účinnosti. Primárnym koncovým ukazovateľom bol podiel pacientov, ktorí dosiahli ASAS 20 odpoveď v 16. týždni. Kľúčové výsledky sú uvedené v tabuľke 6 a uvedené nižšie.
Tabuľka 6Kľúčové výsledky účinnosti z GO-AHEAD v 16. týždniZlepšenia v prejavoch a príznakoch Populácia všetkých liečených pacientov (AT)
Populácia s objektívnymi prejavmi zápalu (OSI)
Placebo Simponi 50 mg Placebo Simponi 50 mg na 100 97 80 78
Respondenti, % pacientov
ASAS 20 40 % 71 %** 38 % 77 %**
ASAS 40 23 % 57 %** 23 % 60 %** ASAS 5/6 23 % 54 %** 23 % 63 %** Parciálna remisia ASAS 18 % 33 %* 19 % 35 %* ASDAS-C b < 1,3 13 % 33 %* 16 % 35 %* BASDAI 50 30 % 58 %** 29 % 59 %**
Inhibícia zápalu v sakroiliakálnych (SI) kĺboch na základe merania pomocou MRPlacebo Simponi 50 mg Placebo Simponi 50 mg n C 87 74 69 61
Priemerná zmena v MR
skóre pre sakroiliakálne
kĺby SPARCCd -0,9 -5,3** -1,2 -6,4**
a n odráža randomizovaných a liečených pacientov
b Skóre C-reaktívneho proteínu pre aktivitu ochorenia ankylozujúcej spondylitídy (AT-placebo, N = 90; AT-Simponi
50 mg, N = 88; OSI-placebo, N = 71; OSI-Simponi 50 mg, N = 71)
c n odráža počet pacientov s údajmi z MR na začiatku a v 16. týždni
d SPARCC (Kanadské združenie pre výskum spondyloartritídy)
** p < 0,0001 pri porovnaniach Simponi
vs. placebo
* p < 0,05 pri porovnaniach Simponi
vs. placebo
U pacientov liečených Simponi v dávke 50 mg sa v 16. týždni v porovnaní s placebom preukázali
štatisticky významné zlepšenia prejavov a príznakov závažnej aktívnej nr-axiálnej SpA (tabuľka 6). Zlepšenia sa pozorovali pri prvom hodnotení (4. týždeň) po podaní úvodnej dávky Simponi. Pri skóre SPARCC sa na základe merania pomocou MR v 16. týždni preukázali štatisticky významné zmiernenia zápalu SI kĺbov u pacientov liečených Simponi v dávke 50 mg v porovnaní s placebom (tabuľka 6). Pri bolesti hodnotenej pomocou VAS pre celkovú bolesť chrbta a bolesť chrbta v noci
a pri aktivite ochorenia hodnotenej pomocou ASDAS-C sa tiež preukázalo štatisticky významné zlepšenie od začiatku do 16. týždňa u pacientov liečených Simponi v dávke 50 mg v porovnaní
s placebom (p < 0,0001).
U pacientov liečených Simponi v dávke 50 mg sa v porovnaní s pacientmi dostávajúcimi placebo preukázali štatisticky významné zlepšenia v spinálnej mobilite na základe hodnotenia podľa BASMI (
Bath Ankylosing Spondylitis Metrology Index) a v telesnej funkcii na základe hodnotenia podľa BASFI (p < 0,0001). U pacientov liečených Simponi sa objavilo významne viac zlepšení v kvalite života súvisiacej so zdravotným stavom na základe ASQoL, EQ-5D a v telesných a mentálnych zložkách SF-36 a objavilo sa u nich významne viac zlepšení v produktivite na základe zhodnotenia väčších znížení v celkovom narušení práce a narušení aktivity na základe hodnotenia dotazníkom WPAI ako u pacientov dostávajúcich placebo.
Pri všetkých koncových ukazovateľoch uvedených vyššie sa štatisticky významné výsledky preukázali aj v populácii OSI v 16. týždni.
V oboch populáciách, AT a OSI, zlepšenia prejavov a príznakov, spinálnej mobility, telesnej funkcie, kvality života a produktivity pozorované v 16. týždni u pacientov liečených Simponi v dávke 50 mg pokračovali aj u tých pacientov, ktorí zostali v štúdii až do 52. týždňa.
Ulcerózna kolitídaÚčinnosť Simponi sa hodnotila v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách u dospelých pacientov.
Štúdia zameraná na nasadenie lieku (PURSUIT-indukcia) hodnotila pacientov so stredne závažnou až závažnou aktívnou ulceróznou kolitídou (skóre 6 až 12 podľa Maya; subskóre ≥ 2 podľa endoskopie), ktorí nemali dostatočnú odpoveď na konvenčné liečby alebo ktorí netolerovali konvenčné liečby alebo boli závislí od kortikosteroidov. V časti štúdie na potvrdenie dávky bolo 761 pacientov randomizovaných na užívanie buď 400 mg Simponi s.c. v 0. týždni a 200 mg v 2. týždni, 200 mg Simponi s.c. v 0. týždni a 100 mg v 2. týždni alebo placebo s.c. v 0. a 2. týždni. Súbežné stabilné dávky perorálnych aminosalicylátov, kortikosteroidov a/alebo imunomodulátorov boli povolené.
V tejto štúdii sa hodnotila účinnosť Simponi v 6. týždni.
Výsledky štúdie zameranej na udržiavaciu liečbu (PURSUIT-udržiavanie) boli založené na hodnotení
456 pacientov, ktorí dosiahli klinickú odpoveď z predchádzajúceho nasadenia liečby Simponi. Pacienti boli randomizovaní na užívanie Simponi 50 mg, Simponi 100 mg alebo placebo, podávané subkutánne
každé 4 týždne. Súbežné stabilné dávky perorálnych aminosalicylátov a/alebo imunomodulátorov boli
povolené. Na začiatku štúdie zameranej na udržiavaciu liečbu sa dávky kortikosteroidov museli znížiť. V tejto štúdii sa hodnotila účinnosť Simponi v 54. týždni. Pacienti, ktorí dokončili štúdiu zameranú na udržiavaciu liečbu v 54. týždni, pokračovali v liečbe v predĺžení štúdie s hodnotením účinnosti v 216. týždni. Hodnotenie účinnosti pri predĺžení štúdie sa zakladalo na zmenách v používaní kortikosteroidov, na celkovom hodnotení lekárov (Physician’s Global Assessment, PGA) týkajúceho
sa aktivity ochorenia a na zlepšení kvality života meranej prostredníctvom dotazníka zápalového ochorenia čriev (Inflammatory Bowel Disease Questionnaire, IBQD).
Tabuľka 7
Kľúčové výsledky účinnosti zo štúdií PURSUIT-indukcia a PURSUIT-udržiavanie
PURSUIT-indukcia
Percentuálne vyjadrenie pacientov
Placebo
N = 251
Simponi
200/100 mg
N = 253
Pacienti s klinickou odpoveďou v 6. týždnia
Pacienti s klinickou remisiou v 6. týždnib
Pacienti s hojením sliznice v 6. týždnic
30 % 51 %**
6 % 18 %**
29 % 42 %*
PURSUIT-udržiavanie
 Placebo
d
Placebo
d
N = 154
Simponi50 mgN = 151
Simponi100 mgN = 151
Percentuálne vyjadrenie pacientov
Udržiavanie odpovede (Pacienti s klinickou odpoveďou v 54. týždni)e
Trvalá remisia (Pacienti
s klinickou remisiou v 30. a aj
54. týždni)f
N = počet pacientov
** p ≤ 0,001
* p ≤ 0,01
31 % 47 %* 50 %**
16 % 23 %g 28 %*
a definované ako pokles skóre podľa Maya o ≥ 30 % a ≥ 3 body z východiskovej hodnoty, sprevádzaný poklesom v subskóre rektálneho krvácania o ≥ 1 alebo subskóre rektálneho
krvácania 0 alebo 1.
b definované ako skóre podľa Maya ≤ 2 body bez žiadneho individuálneho subskóre > 1
c definované ako 0 alebo 1 v subskóre podľa endoskopie pri skóre podľa Maya.
d len nasadenie liečby Simponi.
e U pacientov sa každé 4 týždne hodnotila aktivita ochorenia UC pomocou parciálneho skóre podľa Maya (strata odpovede sa potvrdila pomocou endoskopie). Preto pri každom hodnotení
v 54. týždni boli pacienti s udržanou odpoveďou v stave kontinuálnej klinickej odpovede.
f Na dosiahnutie trvalej remisie musel byť pacient v remisii v 30. a aj 54. týždni (bez prejavu straty odpovede kedykoľvek v 54. týždni).
g U pacientov s telesnou hmotnosťou nižšou ako 80 kg sa vo väčšej časti pacientov, ktorí dostávali
50 mg udržiavaciu liečbu, preukázala trvalá klinická remisia v porovnaní s pacientmi, ktorí dostávali placebo.
U viacerých pacientov liečených Simponi, sa preukázalo pretrvávajúce hojenie sliznice (pacienti
s hojením sliznice v 30. a aj 54. týždni) v skupine s 50 mg (42 %, nominálna hodnota p < 0,05)
a v skupine so 100 mg (42 %, nominálna hodnota p < 0,005) v porovnaní s pacientmi v skupine s placebom (27 %).
Spomedzi 54 % pacientov (247/456), ktorí súbežne dostávali kortikosteroidy na začiatku štúdie PURSUIT-udržiavanie, bol podiel pacientov, u ktorých sa udržala klinická odpoveď v 54. týždni a ktorí v 54. týždni súbežne nedostávali kortikosteroidy, väčší v skupine s 50 mg (38 %, 30/78)
a v skupine so 100 mg (30 %, 25/82) v porovnaní so skupinou s placebom (21 %, 18/87). Podiel pacientov, u ktorých sa kortikosteroidy vysadili do 54. týždňa, bol väčší v skupine s 50 mg (41 %,
32/78) a v skupine so 100 mg (33 %, 27/82) v porovnaní so skupinou s placebom (22 %, 19/87). Spomedzi pacientov, ktorí vstúpili do predĺženia štúdie, bol podiel osôb, ktorí nepotrebovali liečbu
kortikosteroidmi, všeobecne udržaný až do 216. týždňa.
Pacientom, ktorí v štúdiách PURSUIT-indukcia nedosiahli klinickú odpoveď v 6. týždni, bola v štúdii PURSUIT-udržiavanie podávaná dávka 100 mg každé 4 týždne. V 14. týždni 28 % z týchto pacientov dosiahlo odpoveď definovanú parciálnymi skóre podľa Maya (zníženú o ≥ 3 body v porovnaní so začiatkom indukcie). V 54. týždni boli klinické výsledky pozorované u týchto pacientov podobné klinickým výsledkom hláseným u pacientov, ktorí dosiahli klinickú odpoveď v 6. týždni.
V 6. týždni Simponi výrazne zlepšil kvalitu života, na základe merania zmeny v špecifickom meraní ochorenia, v dotazníku zápalového ochorenia čriev (inflammatory bowel disease questionnaire, IBDQ). Medzi pacientmi, ktorí dostávali udržiavaciu liečbu Simponi, sa zlepšenie kvality života merané pomocou IBDQ v 54. týždni udržalo.
Približne 63 % pacientov dostávajúcich Simponi na začiatku predĺženia štúdie (56. týždeň)
pokračovalo v liečbe do konca štúdie (posledné podanie golimumabu v 212. týždni).
Imunogenita
Počas štúdií fázy III s RA, PsA a AS boli až do 52. týždňa použitím metódy enzýmovej imunoanalýzy (EIA) zistené protilátky proti golimumabu u 5 % (105/2 062) pacientov liečených golimumabom a kde sa testovali, boli takmer všetky protilátky neutralizujúce in vitro. Podobný pomer sa pozoroval vo všetkých reumatologických indikáciách. Súbežná liečba s MTX mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez MTX (približne 3 % [41/1 235] oproti 8 % [64/827] v uvedenom poradí).
Pri nr-axiálnej SpA sa protilátky proti golimumabu zistili použitím metódy EIA do 52. týždňa u 7 % (14/193) pacientov liečených golimumabom.
V štúdiách fázy II a III s UC boli až do 54. týždňa použitím metódy EIA zistené protilátky proti golimumabu u 3 % (26/946) pacientov liečených golimumabom. Šesťdesiatosem percent (21/31) pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky. Súbežná liečba
s imunomodulátormi (azatioprin, 6-merkaptopurín a MTX ) mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez
imunomodulátorov (1 % (4/308) oproti 3 % (22/638) v uvedenom poradí). Z pacientov, ktorí pokračovali v predĺžení štúdie a mali hodnotiteľné vzorky v 228. týždni, sa preukázali protilátky proti golimumabu u 4 % (23/604) pacientov liečených golimumabom. Osemdesiatdva percent (18/22)
pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky.
Na stanovenie protilátok proti golimumabu sa v štúdii pJIA použila metóda drug-tolerant EIA. Pri použití metódy drug-tolerant EIA v porovnaní s metódou EIA sa očakávalo zistenie väčšieho výskytu protilátok proti golimumabu z dôvodu vyššej citlivosti a lepšej tolerancie lieku. V štúdii fázy III pJIA sa počas 48. týždňa použitím metódy drug-tolerant EIA zistili protilátky proti golimumabu u 40 % (69/172) detí liečených golimumabom, z ktorých väčšina mala titer nižší ako 1:1 000. Vplyv na sérové koncentrácie golimumabu sa pozoroval pri titroch > 1:100, zatiaľ čo vplyv na účinnosť sa nepozoroval do titrov > 1:1 000, aj keď počet detí s titrom > 1:1 000 bol nízky (N = 8). U detí, u ktorých sa preukázali protilátky proti golimumabu malo 39 % (25/65) neutralizujúce protilátky. Keďže boli prítomné nízke titre protilátok, vyšší výskyt protilátok pri použití metódy drug-tolerant EIA nemal
zjavný vplyv na hladiny liečiva, účinnosť a bezpečnosť a preto nepredstavuje žiadny nový bezpečnostný signál.
Prítomnosť protilátok proti golimumabu môže zvýšiť riziko reakcií v mieste injekcie (pozri časť 4.4). Malý počet pacientov pozitívnych na protilátky proti golimumabu limituje schopnosť vyvodiť definitívne závery, týkajúce sa vzťahu medzi protilátkami proti golimumabu a klinickou účinnosťou alebo bezpečnostné opatrenia.
Pretože analýzy imunogenity sú špecifické pre daný liek a daný test, porovnanie frekvencie výskytu protilátok s frekvenciami pri ostatných liekoch nie je vhodné.
Pediatrická populáciaPolyartikulárna juvenilná idiopatická artritídaBezpečnosť a účinnosť Simponi sa hodnotili v randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii s ukončením liečby skúšaným liekom (GO-KIDS) u 173 detí (vo veku 2 až
17 rokov) s aktívnou pJIA s najmenej 5 aktívnymi kĺbmi a neadekvátnou odpoveďou na MTX. Do
štúdie boli zaradené deti s polyartikulárnym priebehom JIA (polyartritída s negatívnym alebo pozitívnym reumatoidným faktorom, rozšírená oligoartritída, juvenilná psoriatická artritída alebo
systémová JIA bez systémových príznakov v súčasnosti). Východiskový medián počtu aktívnych
kĺbov bol 12 a medián CRP bol 0,17 mg/dl.
Časť 1 štúdie pozostávala zo 16-týždňovej otvorenej fázy, v ktorej 173 zaradených detí dostávalo každé 4 týždne Simponi v dávke 30 mg/m2 (maximálne 50 mg) subkutánne a MTX. 154 detí, ktoré dosiahli odpoveď Ped ACR 30 v 16. týždni vstúpilo do časti 2 štúdie, randomizovanej fázy
s ukončením liečby skúšaným liekom a dostávalo Simponi v dávke 30 mg/m2 (maximálne
50 mg) + MTX alebo placebo + MTX každé 4 týždne. Po vzplanutí ochorenia dostávali deti Simponi v dávke 30 mg/m2 (maximálne 50 mg) + MTX. V 48. týždni deti vstúpili do dlhodobého predĺženia štúdie.
U detí v tejto štúdii sa preukázali odpovede Ped ACR 30, 50, 70, a 90 od 4. týždňa.
V 16. týždni malo 87 % detí odpoveď Ped ACR 30, 79 % odpoveď Ped ACR 50, 66 % odpoveď Ped ACR 70 a 36 % odpoveď Ped ACR 90. V 16. týždni malo 34 % detí neaktívne ochorenie, definované ako všetko z nasledujúcich: žiadny kĺb s aktívnou artritídou; bez horúčky, vyrážky, serozitídy, splenomegálie, hepatomegálie alebo generalizovanej lymfadenopatie pravdepodobne spojenej s JIA; bez aktívnej uveitídy; normálne hodnoty FW (< 20 mm/hodina) alebo CRP (< 1,0 mg/dl); celkové hodnotenie aktivity ochorenia lekárom (≤ 5 mm na VAS); dĺžka trvania rannej stuhnutosti < 15 minút.
V 16. týždni všetky zložky Ped ACR preukázali klinicky relevantné zlepšenie oproti východiskovým hodnotám (pozri tabuľku 8).
Tabuľka 8 Zlepšenia oproti východiskovým hodnotám všetkých zložiek Ped ACR v 16. týždniaMedián zlepšenia v percentách
Zlepšenia oproti východiskovým hodnotám všetkých zložiek Ped ACR v 16. týždniaMedián zlepšenia v percentách Simponi 30 mg/m2 nb = 173
Celkové hodnotenie ochorenia lekármi
(VASc 0-10 cm)
Celkové hodnotenie celkovej pohody pacientom/rodičom (VAS 0-10 cm)
88 %
67 %
Počet aktívnych kĺbov 92 % Počet kĺbov s obmedzeným rozsahom pohybu 80 % Telesné funkcie podľa CHAQd 50 % FW (mm/h)e 33 %

a východisková hodnota = týždeň 0
b “n” odráža zaradených pacientov
c VAS: vizuálna analógová stupnica
d CHAQ (Child Health Assessment Questionaire): dotazník hodnotiaci zdravie dieťaťa
e FW (mm/h): rýchlosť sedimentácie erytrocytov (milimetrov za hodinu)
Primárny koncový ukazovateľ, podiel detí, ktoré dosiahli odpoveď Ped ACR 30 v 16. týždni
a u ktorých medzi 16. a 48. týždňom nedošlo k vzplanutiu ochorenia, sa nedosiahol. U väčšiny detí nedošlo k vzplanutiu ochorenia medzi 16. a 48. týždňom (59 % v skupine Simponi + MTX a 53 % v skupine placebo + MTX; hodnota p = 0,41).
Analýzy primárneho koncového ukazovateľa vo vopred definovaných podskupinách podľa východiskových hladín CRP (≥ 1 mg/dl oproti < 1 mg/dl) u osôb s východiskovou hladinou
CRP ≥ 1 mg/dl preukázali vyššie miery vzplanutia ochorenia v skupine dostávajúcej placebo + MTX
oproti skupine so Simponi + MTX (87 % oproti 40 % p = 0,0068).
V 48. týždni dosiahlo odpoveď Ped ACR 30 53 % detí v skupine Simponi + MTX a 55 % detí
v skupine placebo + MTX a neaktívne ochorenie 40 % detí v skupine Simponi + MTX a 28 % detí v skupine placebo + MTX.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so Simponi v jednej alebo vo viacerých podskupinách pediatrickej populácie pre ulceróznu kolitídu (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2. Farmakokinetické vlastnostiAbsorpciaPo jednorazovej subkutánnej aplikácii golimumabu zdravým osobám alebo pacientom s RA bol priemerný čas na dosiahnutie maximálnych koncentrácií v sére (Tmax) od 2 do 6 dní. Subkutánna injekcia 50 mg golimumabu zdravým osobám priniesla priemernú hodnotu ± smerodajná odchýlka maximálnych koncentrácií (Cmax) v sére 3,1 ± 1,4 μg/ml.
Po jednorazovej subkutánnej injekcii 100 mg bola absorpcia golimumabu v hornej časti ramena, na bruchu a na stehne podobná, s priemernou absolútnou biologickou dostupnosťou 51 %. Keďže golimumab preukázal približnú dávkovo úmernú FK po subkutánnom podaní, dá sa očakávať, že absolútna biologická dostupnosť po dávke 50 mg alebo 200 mg golimumabu bude podobná.
DistribúciaPo jednorazovom i.v. podaní bol priemerný distribučný objem 115 ± 19 ml/kg.
ElimináciaSystémový klírens golimumabu bol odhadnutý na 6,9 ± 2,0 ml/deň/kg. Hodnota terminálneho polčasu u zdravých osôb bola odhadnutá na približne 12 ± 3 dni a podobné hodnoty sa pozorovali u pacientov s RA, PsA, AS alebo UC.
U pacientov s RA, PsA alebo AS, ktorí dostávali 50 mg golimumabu každé 4 týždne, sa dosiahol rovnovážny stav koncentrácií v sére v 12. týždni. Pri súbežnom podávaní MTX priniesla liečba
s 50 mg golimumabu subkutánne každé 4 týždne priemerné (± smerodajná odchýlka) minimálne
sérové koncentrácie v rovnovážnom stave približne 0,6 ± 0,4 μg/ml u pacientov s RA s aktívnou RA napriek liečbe MTX a približne 0,5 ± 0,4 μg/ml u pacientov s aktívnou PsA a približne 0,8 ± 0,4 μg/ml u pacientov s AS. Priemerné minimálne sérové koncentrácie golimumabu v rovnovážnom stave
u pacientov s nr-axiálnou SpA boli podobné koncentráciám, ktoré sa pozorovali u pacientov s AS po subkutánnom podaní 50 mg golimumabu každé 4 týždne.
Pacienti s RA, PsA alebo AS, ktorí nedostávali súbežne MTX, mali približne o 30 % nižšie hodnoty minimálnych koncentrácií golimumabu v rovnovážnom stave než tí, ktorí dostávali golimumab
s MTX. U obmedzeného počtu pacientov s RA liečených subkutánnym golimumabom počas 6- mesačného obdobia súbežné používanie MTX znížilo zdanlivý klírens golimumabu o približne 36 %. Populačná farmakokinetická analýza však naznačila, že súbežné používanie NSAID, perorálnych kortikosteroidov alebo sulfasalazínu nemalo vplyv na zdanlivý klírens golimumabu.
U pacientov s UC sa po indukčných dávkach 200 mg golimumabu v 0. týždni a 100 mg golimumabu v 2. týždni a udržiavacích dávkach 50 mg alebo 100 mg golimumabu subkutánne následne každé
4 týždne dosiahli sérové koncentrácie golimumabu v rovnovážnom stave približne 14 týždňov po
začiatku liečby. Liečba 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne počas udržiavania viedla k priemernej minimálnej sérovej koncentrácii v rovnovážnom stave približne
0,9 ± 0,5 mg/ml a 1,8 ± 1,1 mg/ml.
Súbežné užívanie imunomodulátorov u pacientov s UC liečených 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne nemalo významný vplyv na minimálne hladiny golimumabu
v rovnovážnom stave.
Pacienti, u ktorých sa vyvinuli protilátky proti golimumabu, mali v rovnovážnom stave všeobecne nízke minimálne sérové koncentrácie golimumabu (pozri časť 5.1).
LinearitaGolimumab vykazoval približne dávkovo úmernú farmakokinetiku u pacientov s RA v rozsahu dávok
0,1 mg až 10,0 mg/kg po jednorazovej intravenóznej dávke. Po jednorazovej subkutánnej dávke
u zdravých osôb sa v rozmedzí dávok 50 mg až 400 mg pozorovala farmakokinetika, ktorá bola tiež približne proporcionálna k dávke.
Vplyv telesnej hmotnosti na farmakokinetikuZistil sa trend k vyšším hodnotám zdanlivého klírensu golimumabu s narastajúcou telesnou hmotnosťou (pozri časť 4.2).
Pediatrická populáciaFarmakokinetika golimumabu sa stanovila u 173 detí s pJIA s vekovým rozpätím od 2 do 17 rokov.
V štúdii pJIA mali deti, ktoré dostávali subkutánne golimumab v dávke 30 mg/m2 (maximálne 50 mg)
každé 4 týždne, hodnoty mediánu minimálnych koncentrácií golimumabu v rovnovážnom stave, ktoré boli podobné v rôznych vekových skupinách a tiež podobné alebo trochu vyššie ako tie, pozorované
u dospelých pacientov s RA, ktorí dostávali 50 mg golimumabu každé 4 týždne.
Farmakokinetické/farmakodynamické modelovanie populácie a simulácia u detí s pJIA potvrdili vzťah medzi expozíciami golimumabu v sére a klinickou účinnosťou a podporili dávkovací režim 50 mg golimumabu každé 4 týždne u detí s pJIA s telesnou hmotnosťou najmenej 40 kg, pri ktorom sa dosahujú expozície podobné tým, ktoré sa ukázali ako účinné u dospelých.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
S golimumabom sa nevykonali žiadne štúdie mutagenity, štúdie fertility na zvieratách ani dlhodobé štúdie karcinogenity.
V štúdiách fertility a všeobecných reprodukčných funkcií myší, pri použití analogickej protilátky selektívne inhibujúcej funkčnú aktivitu myšieho TNFα, sa znížilo množstvo gravidných myší. Nie je známe, či bol tento nález spôsobený vplyvom na samce a/alebo samice. V štúdii vývinovej toxicity vykonanej na myšiach pri aplikácii rovnakej analógnej protilátky u myší a pri použití golimumabu
u opíc druhu Cynomolgus neboli zaznamenané žiadne dôkazy maternálnej toxicity, embryotoxicity alebo teratogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sorbitol (E420) Histidín
Monohydrát histidíniumchloridu
Polysorbát 80
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
24 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Naplnené pero alebo naplnenú injekčnú striekačku uchovávajte v škatuli na ochranu pred svetlom.
Simponi sa môže uchovávať pri teplotách do maximálne 25 °C počas jedného obdobia až do 30 dní, nesmie však presiahnuť pôvodný dátum exspirácie vytlačený na škatuli. Nový dátum exspirácie sa musí napísať na škatuľu (až do 30 dní od dátumu vybratia z chladničky).
Ak sa Simponi začne uchovávať pri izbovej teplote, nesmie sa vrátiť späť do chladničky. Ak sa
Simponi nepoužije počas 30 dní uchovávania pri izbovej teplote, musí sa zlikvidovať.
6.5 Druh obalu a obsah balenia
Simponi 50 mg injekčný roztok naplnený v injekčnom pere
0,5 ml roztoku naplneného v injekčnej striekačke (sklo typu 1) s napevno nasadenou ihlou (nerezová oceľ) a krytom ihly (kaučuk obsahujúci latex) v naplnenom pere. Simponi je dostupný v baleniach
obsahujúcich 1 naplnené pero a viacpočetných baleniach obsahujúcich 3 (3 balenia po 1) naplnené perá.
Simponi 50 mg injekčný roztok naplnený v injekčnej striekačke
0,5 ml roztoku naplneného v injekčnej striekačke (sklo typu 1) s napevno nasadenou ihlou (nerezová oceľ) a krytom ihly (kaučuk obsahujúci latex). Simponi je dostupný v baleniach obsahujúcich
1 naplnenú injekčnú striekačku a viacpočetných baleniach obsahujúcich 3 (3 balenia po 1) naplnené
injekčné striekačky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Simponi sa dodáva ako jednorazové naplnené pero, nazývané SmartJect alebo ako jednorazová naplnená injekčná striekačka. Každé balenie obsahuje návod na použitie, ktorý úplne popisuje spôsob používania pera alebo injekčnej striekačky. Po vybratí naplneného pera alebo naplnenej injekčnej striekačky z chladničky sa má naplnené pero alebo naplnená injekčná striekačka nechať 30 minút dosiahnuť izbovú teplotu pred podaním Simponi. Perom alebo injekčnou striekačkou sa nesmie triasť.
Roztok je číry až mierne opaleskujúci, bezfarebný až svetložltý a môže obsahovať malé množstvo priehľadných alebo bielych čiastočiek proteínov. Tento vzhľad nie je ničím nezvyčajným pre roztoky
s obsahom proteínov. Simponi sa nesmie použiť pri zmene farby roztoku, zakalení alebo ak obsahuje viditeľné cudzie častice.
Podrobné inštrukcie o spôsobe prípravy a aplikácie Simponi v naplnenom pere alebo naplnenej injekčnej striekačke sú súčasťou písomnej informácie pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/09/546/001 1 naplnené injekčné pero
EU/1/09/546/002 3 naplnené injekčné perá
EU/1/09/546/003 1 naplnená injekčná striekačka
EU/1/09/546/004 3 naplnené injekčné striekačky
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 1. októbra 2009
Dátum posledného predĺženia registrácie: 19. júna 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Simponi 100 mg injekčný roztok naplnený v injekčnom pere
Simponi 100 mg injekčný roztok naplnený v injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Simponi 100 mg injekčný roztok naplnený v injekčnom pere
Každé 1 ml naplnené pero obsahuje 100 mg golimumabu*.
Simponi 100 mg injekčný roztok naplnený v injekčnej striekačke
Každá 1 ml naplnená striekačka obsahuje 100 mg golimumabu*.
* Ľudská monoklonálna protilátka IgG1κ získaná z bunkovej línie hybridómu myši technológiou rekombinantnej DNA.
Pomocná látka so známym účinkom
Každé naplnené pero obsahuje 41 mg sorbitolu v 100 mg dávke.
Každá naplnená injekčná striekačka obsahuje 41 mg sorbitolu v 100 mg dávke.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok naplnený v injekčnom pere (injekcia), SmartJect Injekčný roztok naplnený v injekčnej striekačke (injekcia) Roztok je číry až mierne opaleskujúci, bezfarebný až svetložltý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidná artritída (RA)
Simponi je v kombinácii s metotrexátom (MTX) indikovaný na:
· liečbu stredne závažnej až závažnej aktívnej reumatoidnej artritídy dospelým, u ktorých sa nedosiahla dostatočná odpoveď na liečbu ochorenie modifikujúcimi antireumatikami (disease-modifying anti-rheumatic drug, DMARD) vrátane MTX.
· liečbu závažnej aktívnej a progresívnej reumatoidnej artritídy dospelým ešte neliečeným MTX.
Preukázalo sa, že Simponi v kombinácii s MTX znižuje rýchlosť progresie poškodenia kĺbov hodnotenú RTG a zlepšuje telesnú funkciu.
Informácie týkajúce sa polyartikulárnej juvenilnej idiopatickej artritídy pozri v súhrne charakteristických vlastností lieku Simponi 50 mg.
Psoriatická artritída (PsA)
Simponi, samostatne alebo v kombinácii s MTX, je indikovaný na liečbu aktívnej a progresívnej psoriatickej artritídy dospelým pacientom, u ktorých sa nedosiahla dostatočná odpoveď na liečbu
DMARD. Preukázalo sa, že Simponi znižuje rýchlosť progresie poškodenia periférnych kĺbov
hodnotenú RTG u pacientov s polyartikulárnymi symetrickými podtypmi ochorenia (pozri časť 5.1)
a zlepšuje telesnú funkciu.
Axiálna spondyloartritída
Ankylozujúca spondylitída (AS)
Simponi je indikovaný na liečbu závažnej, aktívnej ankylozujúcej spondylitídy dospelým, ktorí dostatočne neodpovedali na konvenčnú liečbu.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axiálna SpA)
Simponi je indikovaný na liečbu závažnej, aktívnej axiálnej spondyloartritídy bez rádiografického dôkazu dospelým s objektívnymi prejavmi zápalu na základe indikácie zvýšenou hladinou C-
reaktívneho proteínu (CRP) a/alebo dôkazom pri zobrazení magnetickou rezonanciou (MR),
s nedostatočnou odpoveďou na nesteroidové protizápalové liečivá (NSAID) alebo tým, ktorí tieto liečivá netolerujú.
Ulcerózna kolitída (UC)
Simponi je indikovaný na liečbu stredne závažnej až závažnej aktívnej ulceróznej kolitídy dospelým pacientom s nedostatočnou odpoveďou na konvenčnú liečbu vrátane kortikosteroidov a 6- merkaptopurínu (6-MP) alebo azatioprinu (AZA), alebo pacientom, ktorí takéto liečby netolerujú alebo sú u nich zdravotne kontraindikované.
4.2 Dávkovanie a spôsob podávania
Liečbu majú iniciovať a dohliadať na ňu iba lekári špecialisti skúsení v diagnostike a liečbe reumatoidnej artritídy, psoriatickej artritídy, ankylozujúcej spondylitídy, axiálnej spondyloartritídy bez rádiografického dôkazu alebo ulceróznej kolitídy. Pacienti, ktorí sa liečia Simponi, majú dostať kartu
s pripomienkami pre pacienta.
Dávkovanie
Reumatoidná artritída
Simponi 50 mg sa podáva raz mesačne, vždy v rovnaký deň v mesiaci. Simponi sa má podávať súbežne s MTX.
Psoriatická artritída, ankylozujúca spondylitída alebo axiálna spondyloartritída bez rádiografického dôkazu
Simponi 50 mg sa podáva raz mesačne, vždy v rovnaký deň v mesiaci.
Pre všetky z vyššie uvedených indikácií dostupné údaje naznačujú, že sa klinická odpoveď dosiahne zvyčajne v priebehu 12. až 14. týždňa liečby (po 3-4 dávkach). U pacientov, u ktorých sa počas tohto obdobia nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Pacienti s telesnou hmotnosťou vyššou ako 100 kg
Pre všetky z vyššie uvedených indikácií sa u pacientov s RA, PsA, AS alebo nr-axiálnou SpA
s telesnou hmotnosťou vyššou ako 100 kg, ktorí po 3 alebo 4 dávkach nedosiahli primeranú klinickú odpoveď, môže zvážiť zvýšenie dávky na 100 mg golimumabu raz mesačne, pričom treba vziať do
úvahy zvýšené riziko niektorých závažných nežiaducich reakcií pri dávke 100 mg v porovnaní
s dávkou 50 mg (pozri časť 4.8). U pacientov, u ktorých sa po podaní 3 až 4 ďalších dávok po 100 mg nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Ulcerózna kolitída
Pacienti s telesnou hmotnosťou nižšou ako 80 kg
Simponi sa podáva ako 200 mg úvodná dávka, po ktorej nasleduje 100 mg v 2. týždni. Pacienti, ktorí majú dostatočnú odpoveď, majú dostať 50 mg v 6. týždni a následne každé 4 týždne. U pacientov,
ktorí nemajú dostatočnú odpoveď, môže byť prospešné pokračovanie so 100 mg v 6. týždni a následne
každé 4 týždne(pozri časť 5.1).
Pacienti s telesnou hmotnosťou vyššou ako 80 kg alebo rovnou 80 kg
Simponi sa podáva ako 200 mg úvodná dávka, po ktorej nasleduje 100 mg v 2. týždni, potom 100 mg každé 4 týždne (pozri časť 5.1).
Počas udržiavacej liečby sa dávka kortikosteroidov môže znižovať v súlade s usmerneniami pre klinickú prax.
Dostupné údaje naznačujú, že sa klinická odpoveď zvyčajne dosiahne v priebehu 12. – 14. týždňa liečby (po 4 dávkach). U pacientov, u ktorých sa počas tohto obdobia nepotvrdil liečebný prínos, sa má zvážiť pokračovanie v liečbe.
Vynechanie dávky
V prípade, že si pacient zabudne podať Simponi v plánovanom termíne, vynechaná dávka sa má podať hneď, ako si pacient spomenie. Pacienti majú byť poučení o tom, aby si nepodali dvojnásobnú dávku
ako náhradu za vynechanú dávku.
Ďalšia dávka sa má podať podľa nasledujúcich pokynov:
· ak sa podanie oneskorí o menej ako 2 týždne, pacient si má podať vynechanú dávku a pokračovať podľa pôvodnej schémy.
· ak sa podanie oneskorí o viac ako 2 týždne, pacient si má podať vynechanú dávku a má sa stanoviť nová schéma, počínajúc dňom podania tejto injekcie.
Osobitné skupiny pacientov
Staršie osoby (≥ 65 rokov)
U starších osôb sa úprava dávky nevyžaduje.
Porucha funkcie obličiek a pečene
Simponi sa neskúmal v týchto skupinách pacientov. Odporúčanú dávku nie je možné stanoviť.
Pediatrická populácia
Simponi 100 mg sa neodporúča používať u detí mladších ako 18 rokov.
Spôsob podávania
Simponi je na subkutánne použitie. Po dôkladnom oboznámení sa s technikou subkutánnej aplikácie si ho môžu pacienti podať sami, ak to ich lekár uzná za vhodné, s následným lekárskym dohľadom podľa potreby. Pacienti majú byť poučení o podaní celej dávky Simponi podľa podrobných inštrukcií
o použití, ktoré sú súčasťou písomnej informácie pre používateľa. Ak sa vyžaduje podanie viacerých injekcií, injekcie sa majú podať do rôznych miest na tele.
Návod na použitie, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza (TBC) alebo iné závažné infekcie ako je sepsa a oportúnne infekcie (pozri časť
4.4).
Stredne závažné alebo závažné zlyhávanie srdca (NYHA trieda III/IV) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Na zlepšenie sledovateľnosti biologických liekov sa má jasne zaznamenať názov a číslo šarže podaného lieku.
Infekcie
Pacienti musia byť pred liečbou, počas liečby a po liečbe golimumabom dôkladne sledovaní, či sa u nich neobjaví infekcia vrátane tuberkulózy. Pretože eliminácia golimumabu môže trvať až
5 mesiacov, sledovanie má pokračovať počas celého obdobia. Ak sa u pacienta rozvinie závažná infekcia alebo sepsa, ďalšia liečba golimumabom sa nesmie podať (pozri časť 4.3).
Golimumab sa nemá podávať pacientom s klinicky závažnou aktívnou infekciou. Opatrnosť je potrebná, keď sa zvažuje použitie golimumabu u pacientov s chronickou infekciou alebo s anamnézou opakovaných infekcií. Pacientov treba poučiť a podľa možnosti sa vyhýbať potenciálnym rizikovým faktorom infekcií.
Pacienti užívajúci TNF-blokátory sú náchylnejší na závažné infekcie.
U pacientov, ktorým bol podaný golimumab, sa hlásili bakteriálne infekcie (vrátane sepsy
a pneumónie), mykobakteriálne infekcie (vrátane TBC), invazívne mykotické a oportúnne infekcie, vrátane smrteľných prípadov. Niektoré z týchto závažných infekcií sa vyskytli u pacientov so súbežnou imunosupresívnou liečbou, ktorá ich, okrem vlastného ochorenia, môže urobiť náchylnejšími na vznik infekcií. Pacienti, u ktorých sa vyvinula nová infekcia počas liečby golimumabom, sa majú starostlivo monitorovať a podstúpiť kompletné diagnostické vyšetrenie. Podávanie golimumabu sa má prerušiť, ak sa u pacienta vyvinie nová ťažká infekcia alebo sepsa a má sa začať vhodná antimikrobiálna alebo antimykotická liečba, pokým nie je infekcia pod kontrolou.
U pacientov, ktorí žili alebo cestovali do oblastí, kde sú endemické invazívne mykotické infekcie, ako je histoplazmóza, kokcidioidomykóza alebo blastomykóza, sa majú pred začatím liečby golimumabom starostlivo zvážiť prínosy a riziká liečby golimumabom. U rizikových pacientov liečených golimumabom, u ktorých sa vyvinie závažné systémové ochorenie, sa má zvážiť podozrenie na invazívnu mykotickú infekciu. Ak je to možné, diagnostika a podanie empirickej antimykotickej
liečby sa má u týchto pacientov vykonať po konzultácii s lekárom so skúsenosťami v starostlivosti o pacientov s invazívnymi mykotickými infekciami.
Tuberkulóza
U pacientov používajúcich golimumab sa hlásili prípady tuberkulózy. Je potrebné zdôrazniť, že vo väčšine týchto hlásení bola tuberkulóza extrapulmonálna, prebiehajúca buď ako lokálne alebo ako diseminované ochorenie.
Pred začatím liečby golimumabom sa musia všetci pacienti vyhodnotiť na aktívnu i neaktívnu („latentnú“) tuberkulózu. Toto vyhodnotenie má zahŕňať detailnú anamnézu s osobnou anamnézou tuberkulózy alebo možného predchádzajúceho kontaktu s tuberkulózou a predchádzajúcu a/alebo súčasnú imunosupresívnu liečbu. U všetkých pacientov sa majú urobiť vhodné skríningové vyšetrenia, t.j. tuberkulínový kožný test alebo krvné vyšetrenie a RTG hrudníka (možno aplikovať miestne odporúčania). Odporúča sa, aby sa vykonanie týchto testov zaznamenalo do karty s pripomienkami pre pacienta. Upozorňujeme predpisujúcich lekárov na riziko falošne negatívnych výsledkov tuberkulínových kožných testov, najmä u pacientov, ktorí sú ťažko chorí alebo imunokompromitovaní.
Ak sa diagnostikuje aktívna tuberkulóza, liečba golimumabom sa nesmie začať (pozri časť 4.3). Ak je podozrenie na latentnú tuberkulózu, treba to konzultovať s lekárom s odbornosťou v liečbe
tuberkulózy. Vo všetkých situáciách popísaných nižšie sa má veľmi starostlivo zvážiť vyváženie pomeru prínosu/rizika liečby golimumabom.
Ak sa diagnostikuje neaktívna („latentná“) tuberkulóza, musí sa, predtým ako sa iniciuje liečba golimumabom, začať antituberkulózna liečba latentnej tuberkulózy, a to v súlade s miestnymi odporúčaniami.
U pacientov, ktorí majú niekoľko rizikových faktorov alebo významné rizikové faktory pre vznik tuberkulózy a majú negatívny test na latentnú tuberkulózu, sa musí pred nasadením golimumabu zvážiť antituberkulózna liečba. Použitie antituberkulóznej liečby sa má tiež zvážiť pred nasadením golimumabu u pacientov s latentnou alebo aktívnou tuberkulózou v anamnéze, u ktorých nie je možné potvrdiť adekvátny priebeh liečby.
U pacientov liečených golimumabom počas a po liečbe latentnej tuberkulózy sa vyskytli prípady aktívnej tuberkulózy. Pacienti liečení golimumabom sa majú starostlivo sledovať na prejavy
a príznaky aktívnej tuberkulózy, vrátane pacientov s negatívnym testom na latentnú tuberkulózu,
pacientov, ktorí sú na latentnú tuberkulózu liečení, alebo pacientov, ktorí boli predtým liečení na tuberkulózu.
Všetci pacienti majú byť informovaní o tom, že musia vyhľadať lekársku pomoc, ak sa u nich počas liečby alebo po liečbe golimumabom objavia prejavy/príznaky, ktoré poukazujú na tuberkulózu (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, zvýšená teplota).
Reaktivácia vírusovej hepatitídy B
U pacientov liečených TNF-antagonistom vrátane golimumabu, ktorí sú chronickými nositeľmi tohto vírusu (t.j. pozitívny povrchový antigén), sa objavila reaktivácia hepatitídy B. Niektoré prípady mali
fatálne následky.
Pred začatím liečby golimumabom majú byť pacienti vyšetrení na infekciu HBV. Pacientom s pozitívnym výsledkom vyšetrenia na infekciu HBV sa odporúča konzultácia s lekárom
s odbornosťou na liečbu hepatitídy B.
Nositelia HBV, u ktorých je potrebná liečba golimumabom, sa majú starostlivo sledovať na prejavy
a príznaky aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby. Nie sú dostupné dostatočné údaje o liečbe pacientov, ktorí sú nositeľmi HBV a dostávajú antivírusovú liečbu v kombinácii s liečbou TNF-antagonistom, aby sa zabránilo reaktivácii HBV. U pacientov, u ktorých došlo k reaktivácii HBV, je potrebné golimumab ukončiť a má sa začať účinná antivírusová liečba
s príslušnou podpornou liečbou.
Malignity a lymfoproliferatívne poruchy
Potenciálna úloha liečby blokujúcej TNF v rozvoji malignít nie je známa. Podľa súčasných vedomostí nemožno u pacientov liečených TNF-antagonistom vylúčiť riziko vzniku lymfómov, leukémie alebo iných malignít. Keď sa zvažuje liečba blokujúca TNF u pacientov s malignitou v anamnéze alebo pri zvažovaní pokračovania liečby u pacientov, u ktorých sa vyvinula malignita, je potrebná opatrnosť.
Malignity u detí a dospievajúcich
Malignity, niektoré fatálne, sa hlásili u detí, dospievajúcich a mladých dospelých (do veku 22 rokov)
liečených liečivami blokujúcimi TNF (začatie liečby vo veku ≤ 18 rokov) v období po uvedení lieku na trh. Približne polovica týchto prípadov boli lymfómy. Ďalšie prípady predstavovali rôzne druhy
iných malignít a zahŕňali zriedkavé malignity zvyčajne súvisiace s imunosupresiou. Riziko rozvoja
malignít u detí a dospievajúcich liečených TNF-blokátormi nie je možné vylúčiť.
Lymfóm a leukémia
V kontrolovaných častiach klinických skúšaní všetkých liečiv blokujúcich TNF vrátane golimumabu, sa pozorovalo viac prípadov lymfómu u pacientov, ktorí dostávali anti-TNF liečbu, v porovnaní
s pacientmi v kontrolnej skupine. V priebehu fázy IIb a fázy III klinických skúšaní Simponi pri RA, PsA a AS, bola incidencia lymfómu u pacientov liečených golimumabom vyššia, ako sa očakáva
v bežnej populácii. U pacientov liečených golimumabom sa hlásili prípady leukémie. U pacientov
s reumatoidnou artritídou s dlhotrvajúcim, vysoko aktívnym zápalovým ochorením je zvýšené základné riziko vzniku lymfómov a leukémie, čo komplikuje odhad rizika.
U pacientov liečených inými liečivami blokujúcimi TNF sa po ich uvedení na trh hlásili zriedkavé prípady hepatosplenického lymfómu T-buniek (HSTCL) (pozri časť 4.8). Tento zriedkavý typ lymfómu T-buniek má veľmi agresívny priebeh ochorenia a je zvyčajne smrteľný. Väčšina prípadov sa objavila u dospievajúcich a mladých dospelých mužov, z ktorých takmer všetci užívali súbežnú liečbu azatioprinom (AZA) alebo 6-merkaptopurínom (6-MP) na zápalové ochorenie čreva. Možné riziko pri kombinácii AZA alebo 6-MP a golimumabu sa má starostlivo zvážiť. Riziko vzniku hepatosplenického lymfómu T-buniek u pacientov liečených blokátormi TNF nie je možné vylúčiť.
Malignity iné ako lymfóm
V kontrolovaných častiach klinických skúšaní Simponi fázy IIb a fázy III s RA, PsA, AS a UC, bola incidencia nelymfómových malignít (okrem nemelanómového karcinómu kože) podobná po golimumabe a v kontrolných skupinách.
Dysplázia/karcinóm hrubého čreva
Nie je známe, či má liečba golimumabom vplyv na riziko vzniku dysplázie alebo rakoviny hrubého čreva. Všetci pacienti s ulceróznou kolitídou, u ktorých je zvýšené riziko dysplázie alebo rakoviny
hrubého čreva (napríklad pacienti s dlhodobo pretrvávajúcou ulceróznou kolitídou alebo primárnou sklerotizujúcou cholangitídou) alebo s dyspláziou alebo karcinómom hrubého čreva v anamnéze, majú
byť pred liečbou a počas celého priebehu ich ochorenia v pravidelných intervaloch vyšetrovaní na prítomnosť dysplázie. Toto vyšetrenie má zahŕňať kolonoskopiu a biopsie podľa miestnych odporúčaní. U pacientov s novo diagnostikovanou dyspláziou, ktorí sa liečia golimumabom, sa musia
pozorne prehodnotiť riziká a prínosy pre konkrétneho pacienta a má sa zvážiť, či sa má v liečbe pokračovať.
V prieskumnom klinickom skúšaní, ktoré hodnotilo používanie golimumabu u pacientov so závažnou perzistujúcou astmou, bolo hlásených viac malignít u pacientov liečených golimumabom v porovnaní s pacientmi v kontrolnej skupine (pozri časť 4.8). Význam tohto nálezu nie je známy.
V prieskumnom klinickom skúšaní, ktoré hodnotilo používanie iného anti-TNF liečiva, infliximabu, u pacientov s mierne závažnou až závažnou chronickou obštrukčnou chorobou pľúc (CHOCHP), sa hlásilo viac malignít u pacientov liečených infliximabom, prevažne v pľúcach alebo na hlave a krku, v porovnaní s pacientmi v kontrolnej skupine. Všetci pacienti mali v anamnéze intenzívne fajčenie. Z tohto dôvodu je potrebná opatrnosť pri používaní akéhokoľvek TNF-antagonistu u pacientov
s CHOCHP, tak ako aj u pacientov so zvýšeným rizikom vývoja malignity z dôvodu intenzívneho fajčenia.
Rakovina kože
U pacientov liečených blokátormi TNF vrátane golimumabu sa hlásil výskyt melanómu a karcinómu z Merkelových buniek (pozri časť 4.8). Odporúča sa vykonávať pravidelné vyšetrenie kože, najmä
u pacientov s rizikovými faktormi pre vznik rakoviny kože.
Kongestívne zlyhávanie srdca (congestive heart failure, CHF)
Pri TNF-blokátoroch vrátane golimumabu boli hlásené prípady zhoršenia kongestívneho zlyhávania srdca (CHF) a nový vznik CHF. Niektoré prípady mali fatálne následky. V klinickom skúšaní s iným
TNF-antagonistom sa pozorovalo zhoršenie kongestívneho zlyhávania srdca a zvýšenie mortality
z dôvodu CHF. Golimumab sa u pacientov s CHF neskúmal. Golimumab sa má použiť s opatrnosťou u pacientov s miernym zlyhávaním srdca (NYHA trieda I/II). Pacienti, u ktorých sa rozvinú nové alebo sa zhoršia príznaky zlyhávania srdca, sa majú dôkladne sledovať a podávanie golimumabu sa musí ukončiť (pozri časť 4.3).
Neurologické udalosti
Použitie liečiv blokujúcich TNF vrátane golimumabu bolo spojené s prípadmi nového vzniku alebo exacerbácie klinických príznakov a/alebo rádiografickým dôkazom demyelinizačných porúch
centrálneho nervového systému vrátane roztrúsenej sklerózy a periférnych demyelinizačných porúch. U pacientov s existujúcimi alebo nedávno diagnostikovanými demyelinizačnými poruchami sa majú
pred začatím liečby golimumabom starostlivo zvážiť prínosy a riziká anti-TNF liečby. Ak sa tieto poruchy vyvinú, má sa zvážiť ukončenie liečby golimumabom (pozri časť 4.8).
Chirurgickývýkon
Skúsenosti o bezpečnosti liečby golimumabom u pacientov, ktorí sa podrobili chirurgickému výkonu vrátane artroplastiky, sú obmedzené. Ak sa plánuje chirurgický výkon, má sa zohľadniť dlhý
biologický polčas. Pacient, ktorý počas liečby golimumabom potrebuje chirurgický výkon, sa má
pozorne sledovať z dôvodu infekcií a majú sa vykonať náležité opatrenia.
Imunosupresia
Existuje možnosť, že liečivá blokujúce TNF vrátane golimumabu ovplyvňujú obranyschopnosť organizmu voči infekciám a malignitám, keďže TNF sprostredkúva zápalový proces a moduluje bunkové imunitné odpovede.
Autoimunitné procesy
Relatívny deficit TNFα spôsobený anti-TNF liečbou môže viesť k rozvoju autoimunitného procesu. Ak sa u pacienta po liečbe golimumabom vyvinú príznaky pripomínajúce lupusu podobný syndróm a ak má pacient pozitívne protilátky proti dvojvláknovej DNA, liečba golimumabom sa má ukončiť (pozri časť 4.8).
Hematologické reakcie
U pacientov dostávajúcich TNF-blokátory vrátane golimumabu boli hlásené pancytopénia, leukopénia, neutropénia, agranulocytóza, aplastická anémia a trombocytopénia. Všetkým pacientom sa má
odporučiť okamžite vyhľadať lekársku starostlivosť, ak sa rozvinú prejavy a príznaky naznačujúce
dyskrázie krvi (napr. pretrvávajúca horúčka, tvorba modrín, krvácanie, bledosť). U pacientov
s potvrdenými signifikantnými hematologickými abnormalitami sa má zvážiť ukončenie liečby golimumabom.
Súbežné podávanie TNF-antagonistov a anakinry
V klinických štúdiách so súbežným používaním anakinry a iného liečiva blokujúceho TNF, etanerceptu, sa pozorovali závažné infekcie a neutropénia, bez prídavného klinického prínosu. Kvôli charakteru nežiaducich udalostí, pozorovaných pri tejto kombinovanej liečbe, podobné toxicity môžu tiež vyústiť z kombinácie s anakinrou a inými liečivami blokujúcimi TNF. Kombinácia golimumabu
a anakinry sa neodporúča.
Súbežné podávanie TNF-antagonistov a abataceptu
V klinických štúdiách sa súbežné podávanie TNF-antagonistov a abataceptu spájalo so zvýšeným rizikom infekcií, vrátane závažných infekcií, v porovnaní s TNF-antagonistami samotnými, bez zvýšeného klinického prínosu. Kombinácia golimumabu a abataceptu sa neodporúča.
Súbežné podávanie s inými biologickými liečivami
Existujú nedostatočné informácie týkajúce sa súbežného používania golimumabu s inými biologickými liečivami používanými na liečbu rovnakých ochorení ako golimumab. Súbežné používanie golimumabu s týmito biologickými liečivami sa neodporúča vzhľadom na možnosť zvýšeného rizika vzniku infekcie a iných potenciálnych farmakologických interakcií.
Zámena jednotlivých biologických DMARDs
Pri prechode z jedného biologického liečiva na iné sa má postupovať s opatrnosťou a pacienti majú byť naďalej monitorovaní, pretože prekrývanie biologického účinku môže ďalej zvyšovať riziko
vzniku nežiaducich udalostí vrátane infekcie.
Očkovania/infekčné látky na terapeutické účely
Pacienti liečení golimumabom môžu byť súčasne očkovaní, s výnimkou živých vakcín (pozri časti 4.5
a 4.6). U pacientov, ktorí dostávajú anti-TNF liečbu, sú dostupné obmedzené údaje týkajúce sa odpovede na očkovanie živými vakcínami alebo sekundárneho prenosu infekcie živými vakcínami.
Použitie živých vakcín môže viesť k vzniku klinických infekcií vrátane diseminovaných infekcií.
Iné použitia infekčných látok na terapeutické účely, ako sú napr. živé atenuované baktérie (napr. BCG na instiláciu do močového mechúra na liečbu rakoviny) môžu viesť ku vzniku klinických infekcií vrátane diseminovaných infekcií. Odporúča sa, aby sa infekčné látky na terapeutické účely nepodávali súbežne s golimumabom.
Alergické reakcie
Po uvedení lieku na trh boli po podaní golimumabu hlásené závažné systémové hypersenzitívne reakcie (vrátane anafylaktickej reakcie). Niektoré z týchto reakcií sa vyskytli po prvom podaní
golimumabu. Ak sa objaví anafylaktická reakcia alebo iné závažné alergické reakcie, podávanie golimumabu sa má okamžite ukončiť a má sa iniciovať príslušná liečba.
Precitlivenosť na latex
Ochranný kryt ihly na naplnenom pere alebo naplnenej injekčnej striekačke je vyrobený zo suchého prírodného kaučuku, ktorý obsahuje latex a môže vyvolať alergické reakcie u osôb precitlivených na
latex.
Osobitné skupiny pacientov
Staršie osoby (≥ 65 rokov)
Vo fáze III štúdií s RA, PsA, AS a UC nebol pozorovaný výrazný rozdiel v nežiaducich udalostiach
(adverse event, AE), v závažných nežiaducich udalostiach (serious adverse event, SAE) a v závažných infekciách u pacientov vo veku 65 rokov alebo starších, ktorí dostávali golimumab v porovnaní
s mladšími pacientmi. Je však potrebná opatrnosť v liečbe starších osôb a zvláštna pozornosť sa musí
venovať vzhľadom k výskytu infekcií. V štúdii nr-axiálnej SpA neboli žiadni pacienti vo veku
45 rokov a starší.
Porucha funkcie obličiek a pečene
Špecifické štúdie golimumabu u pacientov s poruchou funkcie obličiek alebo pečene sa nevykonali. Golimumab sa má používať s opatrnosťou u osôb s poruchou funkcie pečene (pozri časť 4.2).
Pomocné látky
Simponi obsahuje sorbitol (E420). U pacientov s dedičnou neznášanlivosťou fruktózy sa musí vziať do úvahy aditívny účinok súbežne podávaných liekov obsahujúcich sorbitol (alebo fruktózu) a príjem sorbitolu (alebo fruktózy) v strave (pozri časť 2).
Možnosť chýb pri podávaní lieku
Simponi je registrovaný v silách 50 mg a 100 mg na subkutánne podanie. Je dôležité, aby sa použila správna sila na podanie správnej dávky, ako je to indikované v dávkovaní (pozri časť 4.2). Má sa dbať
na to, aby sa poskytla správna sila, aby sa zaistilo, že u pacientov nedôjde k poddávkovaniu alebo predávkovaniu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Súbežné používanie s inými biologickými liečivami
Kombinácia golimumabu s inými biologickými liečivami používanými na liečbu rovnakých ochorení ako golimumab vrátane anakinry a abataceptu sa neodporúča (pozri časť 4.4).
Živé vakcíny/infekčné látky na terapeutické účely
Živé vakcíny sa nemajú podávať súbežne s golimumabom (pozri časti 4.4 a 4.6).
Infekčné látky na terapeutické účely sa nemajú podávať súbežne s golimumabom (pozri časť 4.4). Metotrexát
I napriek tomu, že súbežné použitie MTX vyvoláva u pacientov s RA, PsA a AS vyššie minimálne
koncentrácie golimumabu v rovnovážnom stave, údaje nenaznačujú, že je potrebná úprava dávky či už golimumabu alebo MTX (pozri časť 5.2).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať vhodnú antikoncepciu na zabránenie gravidity a pokračovať v jej používaní najmenej 6 mesiacov po poslednej liečbe golimumabom.
Gravidita
Nie sú dostatočné údaje o použití golimumabu u gravidných žien. Z dôvodu jeho inhibície TNF môže golimumab podávaný počas gravidity ovplyvniť normálne imunitné odpovede novorodenca. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky s ohľadom na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3). Použitie golimumabu
u gravidných žien sa neodporúča; golimumab sa môže podať gravidnej žene len ak je to jednoznačne nevyhnutné.
Golimumab prechádza placentou. Po liečbe monoklonálnou protilátkou blokujúcou TNF počas gravidity sa počas 6 mesiacov zistili protilátky v sére dojčaťa narodeného liečenej matke. V dôsledku toho môžu mať tieto dojčatá zvýšené riziko infekcie. Podanie živých vakcín dojčatám vystaveným golimumabu in utero sa neodporúča po dobu 6 mesiacov po matkinej poslednej injekcii golimumabu počas gravidity (pozri časti 4.4 a 4.5).
Dojčenie
Nie je známe, či sa golimumab vylučuje do ľudského mlieka alebo či sa po užití systémovo absorbuje. Ukázalo sa, že golimumab prestupuje do materského mlieka opíc, a pretože ľudské imunoglobulíny prestupujú do materského mlieka, ženy nesmú dojčiť v priebehu liečby a najmenej 6 mesiacov po liečbe golimumabom.
Fertilita
S golimumabom sa nevykonali žiadne štúdie fertility na zvieratách. Štúdia fertility na myšiach pri použití analogickej protilátky selektívne inhibujúcej funkčnú aktivitu myšieho TNFα nepreukázala
žiadne významné účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Simponi má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní Simponi sa však môže objaviť závrat (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V kontrolovanom období pivotných skúšaní RA, PsA, AS, nr-axiálnej SpA a UC bola najčastejšou nežiaducou reakciou (adverse reaction, AR) infekcia horných dýchacích ciest, ktorá sa hlásila
u 12,6 % pacientov liečených golimumabom v porovnaní s 11,0 % pacientov v kontrolnej skupine. Najzávažnejšie AR, ktoré boli hlásené pri golimumabe, zahŕňajú závažné infekcie (vrátane sepsy,
pneumónie, tuberkulózy, invazívnych mykotických a oportúnnych infekcií), demyelinizačné poruchy, reaktiváciu HBV, CHF, autoimunitné procesy (lupusu podobný syndróm), hematologické reakcie, závažnú systémovú hypersenzitivitu (vrátane anafylaktickej reakcie), vaskulitídu, lymfóm a leukémiu
(pozri časť 4.4).
Tabuľkový zoznam nežiaducich reakcií
AR pozorované v klinických štúdiách a celosvetovo hlásené po uvedení golimumabu na trh sú uvedené v tabuľke 1. V rámci stanovených tried orgánových systémov sú AR vymenované pod
hlavičkami frekvencií výskytu s použitím nasledovnej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100
až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000); neznáme (nedá sa odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
Tabuľka 1Tabuľkový zoznam AR
Veľmi časté: Infekcie horných dýchacích ciest (nazofaryngitída, faryngitída, laryngitída a rinitída)
Časté: Bakteriálne infekcie (ako je flegmóna), infekcie dolných dýchacích ciest (ako je pneumónia), vírusové infekcie (ako je chrípka a herpes), bronchitída, sinusitída, povrchové mykotické infekcie, absces
Menej časté: Sepsa vrátane septického šoku, pyelonefritída
Zriedkavé: Tuberkulóza, oportúnne infekcie (ako sú invazívne mykotické infekcie [histoplazmóza, kokcidioidomykóza, pneumocystóza], bakteriálna, atypická mykobakteriálna a protozoálna infekcia), reaktivácia hepatitídy B, bakteriálna artritída, infekčná burzitída
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov
Menej časté: Neoplazmy (ako sú karcinóm kože, skvamocelulárny karcinóm a melanocytový névus)
Zriedkavé: Lymfóm, leukémia, melanóm, karcinóm z Merkelových buniek
Neznáme: Hepatosplenický lymfóm T-buniek*
Poruchy krvi a lymfatického systému
Časté: Leukopénia (vrátane neutropénie), anémia
Menej časté: Trombocytopénia, pancytopénia
Zriedkavé: Aplastická anémia, agranulocytóza
Poruchy imunitného systému
Časté: Alergické reakcie (bronchospazmus, hypersenzitivita, urtikária), pozitívne autoprotilátky
Zriedkavé: Závažné systémové hypersenzitívne reakcie (vrátane anafylaktickej reakcie), vaskulitída (systémová), sarkoidóza
Poruchy endokrinného systému
Menej časté: Poruchy štítnej žľazy (ako sú hypotyreoidizmus, hypertyreoidizmus a struma)
Poruchy metabolizmu a výživy
Menej časté: Zvýšenie glukózy v krvi, zvýšenie lipidov
Psychické poruchy
Časté: Depresia, insomnia
Poruchy nervového systému
Časté: Závrat, bolesť hlavy, parestézia
Menej časté: Poruchy rovnováhy
Zriedkavé: Demyelinizačné poruchy (centrálne a periférne), poruchy chuti
Poruchy oka
Menej časté: Poruchy zraku (ako je zahmlené videnie a zníženie zrakovej ostrosti), konjunktivitída, alergia oka (ako je svrbenie
a podráždenie)
Poruchy srdca a srdcovej činnosti
Menej časté: Arytmia, ischemické poruchy koronárnych artérií
Zriedkavé: Kongestívne zlyhávanie srdca (nové alebo zhoršujúce sa)
Poruchy ciev
Časté: Hypertenzia
Menej časté: Trombóza (ako je hĺbková venózna alebo aorty), sčervenenie
Zriedkavé: Raynaudov fenomén
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté: Astma a pridružené príznaky (ako sú sipot a bronchiálna hyperaktivita)
Menej časté: Intersticiálna choroba pľúc
Poruchy gastrointestinálneho traktu
Časté: Dyspepsia, gastrointestinálna a abdominálna bolesť, nauzea, gastrointestinálne zápalové poruchy (ako sú gastritída a kolitída), stomatitída
Menej časté: Zápcha, gastroezofágová refluxová choroba
Poruchy pečene a žlčových ciest
Časté: Zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza
Menej časté: Cholelitiáza, poruchy pečene
Poruchy kože a podkožného tkaniva
Časté: Svrbenie, vyrážka, alopécia, dermatitída
Menej časté: Bulózne kožné reakcie, psoriáza (nová alebo zhoršená psoriáza, palmárna/plantárna a pustulárna), urtikária
Zriedkavé: Lichenoidné reakcie, odlupovanie kože, vaskulitída (kutánna)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Zriedkavé: Lupusu podobný syndróm
Poruchy obličiek a močových ciest
Zriedkavé: Poruchy močového mechúra, poruchy obličiek
Poruchy reprodukčného systému a prsníkov
Menej časté: Poruchy prsníkov, menštruačné poruchy
Celkové poruchy a reakcie v mieste podania
Časté: Pyrexia, asténia, reakcie v mieste podania injekcie (ako sú erytém, urtikária, zdurenie, bolesť, modrina, svrbenie, podráždenie
a parestézia v mieste vpichu), diskomfort na hrudníku
Zriedkavé: Zhoršené hojenie
Úrazy, otravy a komplikácie liečebného postupu
Časté: Zlomeniny kostí
* pozorované u iných liečiv blokujúcich TNF
V celej tejto časti je pre všetky spôsoby použitia golimumabu všeobecne uvedený medián trvania
následného sledovania (približne 4 roky). Keďže pacienti mohli byť prestavení z jednej dávky na druhú, medián trvania následného sledovania je rôzny v prípadoch, keď sa spôsob použitia
golimumabu charakterizoval prostredníctvom dávky (približne 2 roky pre 50mg dávku a približne
3 roky pre 100mg dávku).
Popis vybraných nežiaducich reakciíInfekcieV kontrolovanom období pivotných skúšaní bola najčastejšou nežiaducou reakciou infekcia horných dýchacích ciest, ktorá sa hlásila u 12,6 % pacientov liečených golimumabom (incidencia na
100 pacientskych rokov: 60,8; 95% IS: 55,0; 67,1) v porovnaní s 11,0 % pacientov v kontrolnej skupine (incidencia na 100 pacientskych rokov: 54,5; 95% IS: 46,1; 64,0). Počas kontrolovaných a nekontrolovaných častí štúdií s mediánom sledovania približne 4 roky bola incidencia infekcií
horných dýchacích ciest u pacientov liečených golimumabom 34,9 udalostí na 100 pacientskych rokov; 95% IS: 33,8; 36,0.
V kontrolovanom období pivotných skúšaní sa pozorovali infekcie u 23,0 % pacientov liečených golimumabom (incidencia na 100 pacientskych rokov: 132,0; 95% IS: 123,3; 141,1), v porovnaní s 20,2 % u pacientov v kontrolnej skupine (incidencia na 100 pacientskych rokov: 122,3; 95% IS:
109,5; 136,2). Počas kontrolovaných a nekontrolovaných častí klinických štúdií s mediánom sledovania približne 4 roky bola incidencia infekcií u pacientov liečených golimumabom 81,1 udalostí
na 100 pacientskych rokov; 95% IS: 79,5; 82,8.
V kontrolovanom období skúšaní RA, PsA, AS a nr-axiálnej SpA sa pozorovali závažné infekcie u 1,2 % pacientov liečených golimumabom a u 1,2 % pacientov v kontrolnej liečenej skupine. Incidencia závažných infekcií na 100 pacientskych rokov následného sledovania v kontrolovanom období skúšaní RA, PsA, AS a nr-axiálnej SpA bola 7,3; 95% IS: 4,6; 11,1 v skupine so 100 mg golimumabu, 2,9; 95% IS: 1,2; 6,0 v skupine s 50 mg golimumabu a 3,6; 95% IS: 1,5; 7,0 v skupine s placebom. V kontrolovanom období skúšaní zameraných na nasadenie golimumabu pri UC sa závažné infekcie pozorovali u 0,8 % pacientov liečených golimumabom v porovnaní s 1,5 % pacientov s kontrolnou liečbou. Závažné infekcie pozorované u pacientov liečených golimumabom zahŕňali tuberkulózu, bakteriálne infekcie vrátane sepsy a pneumónie, invazívne mykotické infekcie a iné oportúnne infekcie. Niektoré z týchto infekcií boli fatálne. Počas kontrolovaných
a nekontrolovaných častí pivotných skúšaní s mediánom sledovania až do 3 rokov bola u pacientov, ktorí dostávali 100 mg golimumabu, pozorovaná vyššia incidencia závažných infekcií vrátane
oportúnnych infekcií a tuberkulózy v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu. Incidencia všetkých závažných infekcií na 100 pacientskych rokov bola 4,1; 95% IS: 3,6; 4,5
u pacientov, ktorí dostávali 100 mg golimumabu a 2,5; 95% IS: 2,0; 3,1 u pacientov, ktorí dostávali
50 mg golimumabu.
Malignity
Lymfóm
Počas pivotných skúšaní bola incidencia lymfómu u pacientov liečených golimumabom vyššia, než sa očakáva v bežnej populácii. Počas kontrolovaných a nekontrolovaných častí týchto skúšaní
s mediánom sledovania až do 3 rokov bola vyššia incidencia lymfómu pozorovaná u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu. Lymfóm
sa diagnostikoval u 11 osôb (1 v skupinách liečených s 50 mg golimumabu a 10 v skupinách liečených
so 100 mg golimumabu) s incidenciou (95% IS) na 100 pacientskych rokov následného sledovania
0,03 (0,00; 0,15) udalostí pre golimumab 50 mg a 0,13 (0,06; 0,24) udalostí pre golimumab 100 mg
a 0,00 (0,00; 0,57) udalostí pre placebo. Väčšina lymfómov sa vyskytla v štúdii GO-AFTER, v ktorej boli zaradení pacienti s predošlou expozíciou anti-TNF liečivám, ktorí mali dlhšie trvajúce
a refraktérnejšie ochorenie (pozri časť 4.4).
Malignity iné než lymfóm
V kontrolovaných obdobiach pivotných skúšaní a počas približne 4 rokov následného sledovania bola podobná incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) medzi golimumabom a kontrolnými skupinami. Počas približne 4 rokov následného sledovania bola incidencia nelymfómových malignít (vylúčený nemelanómový karcinóm kože) podobná všeobecnej populácii.
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov bol nemelanómový karcinóm kože diagnostikovaný u 5 osôb v skupine dostávajúcej placebo,
10 s golimumabom 50 mg a 31 s golimumabom 100 mg s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,36 (0,26; 0,49) pre kombinovaný golimumab a 0,87 (0,28; 2,04) pre
placebo.
V kontrolovanom a nekontrolovanom období pivotných skúšaní s mediánom sledovania až do 3 rokov boli malignity okrem melanómového, nemelanómového karcinómu kože a lymfómu diagnostikované
u 5 osôb v skupine dostávajúcej placebo, 21 s golimumabom 50 mg a 34 s golimumabom 100 mg
s incidenciou (95% IS) na 100 pacientskych rokov v následnom sledovaní 0,48 (0,36; 0,62) pre kombinovaný golimumab a 0,87 (0,28; 2,04) pre placebo (pozri časť 4.4).
Prípady hlásené v klinických štúdiách s astmou
V prieskumnej klinickej štúdii bol pacientom so závažnou perzistujúcou astmou podávaný golimumab v záťažovej dávke (150 % určenej terapeutickej dávky) subkutánne v týždni 0 a následne v dávkach
200 mg golimumabu, 100 mg golimumabu alebo 50 mg golimumabu každé 4 týždne subkutánne až do
52. týždňa. Hlásených bolo osem malignít v kombinovanej skupine liečenej golimumabom (n = 230) a žiadna v skupine dostávajúcej placebo (n = 79). Lymfóm sa hlásil u 1 pacienta, nemelanómový karcinóm kože u 2 pacientov a iné malignity u 5 pacientov. U žiadneho typu malignity nevzniklo špecifické zoskupovanie.
Počas placebom kontrolovanej časti štúdie bola incidencia (95% IS) všetkých malignít na
100 pacientskych rokov v následnom sledovaní 3,19 (1,38; 6,28) v skupine s golimumabom. V tejto štúdii bola incidencia (95% IS) na 100 pacientskych rokov v následnom sledovaní u osôb liečených
golimumabom 0,40 (0,01; 2,20) pre lymfóm, 0,79 (0,10; 2,86) pre nemelanómové karcinómy kože a 1,99 (0,64; 4,63) pre iné malignity. U osôb s placebom bola incidencia (95% IS) na
100 pacientskych rokov v následnom sledovaní týchto malignít 0,00 (0,00; 2,94). Význam tohto nálezu nie je známy.
Neurologické udalosti
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní s mediánom sledovania až do
3 rokov sa pozorovala väčšia incidencia demyelinizácie u pacientov, ktorí dostávali 100 mg golimumabu v porovnaní s pacientmi, ktorí dostávali 50 mg golimumabu (pozri časť 4.4).
Zvýšenia hepatálnych enzýmov
V kontrolovanom období pivotných skúšaní RA a PsA sa vyskytlo mierne zvýšenie ALT (> 1
a < 3 x horná hranica normálu (ULN)) v štúdiách s RA a PsA v podobnej miere u pacientov s golimumabom a u kontrolných pacientov (22,1 % k 27,4 % pacientov). V štúdiách s AS
a nr-axiálnou SpA malo mierne zvýšenie ALT viac pacientov liečených golimumabom (26,9 %) než
pacienti v kontrolnej skupine (10,6 %). V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní RA a PsA s mediánom sledovania približne 5 rokov bola v štúdiách s RA a PsA incidencia mierneho zvýšenia ALT podobná u pacientov liečených golimumabom a u kontrolných pacientov.
V kontrolovanom období pivotných skúšaní zameraných na nasadenie golimumabu pri UC sa vyskytli mierne zvýšenia ALT (> 1 a < 3 x ULN) v podobnej miere u pacientov liečených golimumabom
(8,0 %) a u pacientov v kontrolnej skupine (6,9 %). V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní UC s mediánom sledovania približne 2 roky bol podiel pacientov s miernymi zvýšeniami ALT 24,7 % u pacientov dostávajúcich golimumab počas udržiavacej časti štúdie UC.
Zvýšenie ALT ≥ 5 x ULN bolo v kontrolovanom období pivotných skúšaní RA a AS menej časté a pozorovalo sa častejšie u pacientov liečených golimumabom (0,4 % a 0,9 %) než u kontrolných pacientov (0,0 %). Táto tendencia sa nepozorovala v skupine s PsA. V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní RA, PsA a AS s mediánom sledovania 5 rokov bola incidencia zvýšenia ALT ≥ 5 x ULN podobná u pacientov liečených golimumabom aj u kontrolných
pacientov. Vo všeobecnosti boli tieto zvýšenia asymptomatické a abnormality poklesli alebo vymizli,
či už pri pokračovaní liečby alebo po ukončení podávania golimumabu alebo po modifikácii súbežne podávaných liekov. V kontrolovaných a nekontrolovaných obdobiach štúdie nr-axiálnej SpA (až do
1 roka) sa nehlásili žiadne prípady. V kontrolovaných obdobiach pivotných skúšaní zameraných na nasadenie golimumabu pri UC sa vyskytli zvýšenia ALT ≥ 5 x ULN v podobnej miere u pacientov liečených golimumabom (0,3 %) a u kontrolných pacientov (1,0 %). V kontrolovaných
a nekontrolovaných obdobiach pivotných skúšaní UC s mediánom sledovania približne 2 roky bol podiel pacientov so zvýšeniami ALT ≥ 5 x ULN 0,8 % u pacientov dostávajúcich golimumab počas
udržiavacej časti štúdie UC.
Počas pivotných skúšaní RA, PsA, AS a nr-axiálnej SpA sa u jedného pacienta v skúšaní RA liečeného golimumabom s existujúcimi hepatálnymi abnormalitami a zamenenými liekmi vyvinula fatálna neinfekčná hepatitída so žltačkou. Úloha golimumabu ako prispievajúceho alebo zhoršujúceho činiteľa nemôže byť vylúčená.
Reakcie v mieste podania
V kontrolovaných obdobiach pivotných skúšaní malo 5,4 % pacientov liečených golimumabom reakciu v mieste podania v porovnaní s 2,0 % kontrolných pacientov. Prítomnosť protilátok proti golimumabu môže zvýšiť riziko reakcií v mieste podania. Väčšina reakcií v mieste podania bola mierna alebo stredne závažná a najčastejšie sa manifestovala ako erytém v mieste podania. Reakcie v mieste podania všeobecne nemusia nutne viesť k ukončeniu liečby.
V kontrolovaných klinických skúšaniach fázy IIb a/alebo III s RA, PsA, AS, nr-axiálnou SpA, závažnou perzistujúcou astmou a v skúšaniach fázy II/III s UC sa u žiadneho pacienta liečeného golimumabom neobjavili anafylaktické reakcie.
Autoimunitné protilátky
V kontrolovaných a nekontrolovaných obdobiach pivotných skúšaní boli počas 1 roka následného sledovania nanovo ANA-pozitívne 3,5 % pacientov liečených golimumabom a 2,3 % kontrolných
pacientov (s titrom 1:160 alebo vyšším). Frekvencia protilátok anti-dsDNA za 1 rok následného
sledovania bola 1,1 % u pacientov, ktorí boli na začiatku anti-dsDNA negatívni.
H
lásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinickej štúdii boli jednorazovo intravenózne podané dávky až do 10 mg/kg bez toxicity limitujúcej dávku. V prípade predávkovania sa odporúča sledovanie pacienta na akékoľvek prejavy a príznaky nežiaducich účinkov a okamžite sa má začať vhodná symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNF-α), ATC kód: L04AB06
Mechanizmus účinkuGolimumab je ľudská monoklonálna protilátka, ktorá vytvára stabilné komplexy s vysokou afinitou so solubilnými aj transmembránovými bioaktívnymi formami ľudského TNF-α, čím zabraňuje naviazanie TNF-α na jeho receptory.
Farmakodynamické účinkyNaviazaním golimumabu na ľudský TNF sa preukázala neutralizácia expresie adhezívnych molekúl na povrchu bunky indukovaná TNF-α – E-selektínu, adhezívnej molekuly cievnych buniek (VCAM)-1
a intercelulárnej adhezívnej molekuly (ICAM)-1 ľudských endotelových buniek.
In vitro golimumab tiež inhiboval TNF indukovanú sekréciu interleukínu (IL)-6 a IL-8 a faktoru stimulujúceho kolónie granulocytov a makrofágov (GM-CSF) ľudských endotelových buniek.
V porovnaní so skupinami s placebom sa pozorovalo relatívne zlepšenie hladín C-reaktívneho proteínu (CRP) a liečba Simponi viedla v porovnaní s kontrolnou liečbou k významnému zníženiu sérových hladín IL-6, ICAM-1, matrixovej metaloproteinázy (MMP)-3 a vaskulárneho endotelového rastového faktoru (VEGF) oproti východiskovým hodnotám. Došlo aj ku zníženiu hladín TNF-a u pacientov
s RA a AS a hladiny IL-8 sa znížili u pacientov s PsA. Tieto zmeny sa pozorovali pri prvom hodnotení
(4. týždeň) po úvodnom podaní Simponi a všeobecne pretrvávali až do 24. týždňa.
Klinická účinnosťReumatoidná artritídaÚčinnosť Simponi sa preukázala v troch multicentrických randomizovaných dvojito zaslepených, placebom kontrolovaných štúdiách s vyše 1 500 pacientmi vo veku ≥ 18 rokov so stredne závažnou
a závažnou aktívnou RA, diagnostikovanou podľa kritérií American College of Rheumatology (ACR)
najmenej 3 mesiace pred skríningom. Pacienti mali najmenej 4 opuchnuté a 4 bolestivé kĺby. Simponi alebo placebo sa podávali subkutánne každé 4 týždne.
V GO-FORWARD sa hodnotilo 444 pacientov, ktorí mali aktívnu RA napriek stabilnej dávke MTX aspoň 15 mg/týždeň a ktorí sa predtým neliečili žiadnym anti-TNF liečivom. Pacienti boli randomizovaní a dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. Po 24. týždni boli pacienti, ktorí dostávali placebo + MTX, prevedení na Simponi 50 mg + MTX. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia.
V GO-AFTER sa hodnotilo 445 pacientov, ktorí sa už predtým liečili jednou alebo viacerými anti- TNF liečivami adalimumabom, etanerceptom alebo infliximabom. Pacienti boli randomizovaní
a dostávali placebo, Simponi 50 mg alebo Simponi 100 mg. Počas tejto štúdie mohli pacienti pokračovať v už prebiehajúcej liečbe DMARD s MTX, sulfasalazínom (SSZ) a/alebo
hydroxychlorochinom (HCQ). Stanovené dôvody na ukončenie predchádzajúcej anti-TNF liečby boli nedostatočná účinnosť (58 %), intolerancia (13 %) a/alebo iné dôvody než bezpečnosť alebo účinnosť (29 %, zväčša finančné dôvody).
V GO-BEFORE sa hodnotilo 637 pacientov s aktívnou RA, ktorí sa predtým neliečili MTX ani anti- TNF liečivom. Pacienti boli randomizovaní tak, aby dostávali placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX alebo Simponi 100 mg + placebo. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia, v ktorom pacienti dostávajúci placebo + MTX s aspoň 1 citlivým alebo opuchnutým kĺbom, boli prestavení na Simponi 50 mg + MTX.
Primárnymi spoločnými koncovými ukazovateľmi v GO-FORWARD boli stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni a zlepšenie v 24. týždni v dotazníku hodnotiacom zdravie (Health Assessment Questionnaire, HAQ) oproti východiskovej hodnote.
V GO-AFTER bolo primárnym koncovým ukazovateľom stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 20 v 14. týždni. V GO-BEFORE boli primárnymi spoločnými koncovými
ukazovateľmi stanovenie percenta pacientov, ktorí dosiahli odpoveď ACR 50 v 24. týždni a zmena skóre podľa Sharpa modifikovanom van der Heijdeovou (vdH-S) v 52. týždni oproti východiskovej hodnote. Okrem primárnych koncových ukazovateľov sa vykonali ďalšie hodnotenia vplyvu liečby
Simponi na prejavy a príznaky artritídy, rádiografickú odpoveď, telesnú funkciu a kvalitu života spojenú so zdravím.
Celkovo sa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg so súbežným MTX až do 104. týždňa v GO-FORWARD
a GO-BEFORE a až do 24. týždňa v GO-AFTER. Podľa dizajnu každej z RA štúdií mohli byť pacienti v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznaky
Kľúčové výsledky ACR pre dávku Simponi 50 mg v 14., 24. a 52. týždni GO-FORWARD,
GO-AFTER a GO-BEFORE sú zobrazené v tabuľke 2 a sú popísané nižšie. Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po úvodnej aplikácii Simponi.
V skupine 89 osôb randomizovaných na Simponi 50 mg + MTX v GO-FORWARD bolo stále na tejto liečbe 48 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 40 pacientov, odpoveď ACR 50 u 33 pacientov a odpoveď ACR 70 u 24 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
V GO-AFTER bolo percento pacientov, ktorí dosiahli odpoveď ACR 20, vyššie u pacientov dostávajúcich Simponi než u pacientov dostávajúcich placebo, bez ohľadu na hlásený dôvod ukončenia jednej alebo viacerých predchádzajúcich terapií anti-TNF.
Tabuľka 2
Kľúčové výsledky účinnosti z kontrolovaných častí GO-FORWARD, GO-AFTER
a GO-BEFORE
GO-FORWARD Aktívna RA napriek MTX
Simponi
GO-AFTER Aktívna RA, predtým liečená jedným alebo viacerými anti-TNF liečivami
GO-BEFORE Aktívna RA, neliečená MTX
Simponi
Placebo
+ MTX
50 mg
+
MTX Placebo
Simponi
50 mg
Placebo
+ MTX
50 mg
+ MTX
na 133 89 150 147 160 159
Respondenti, % pacientov
ACR 20
14. týždeň 33 % 55 %* 18 % 35 %* NA NA
24. týždeň 28 % 60 %* 16 % 31 %
p = 0,002
49 % 62 %
52. týždeň NA NA NA NA 52 % 60 %
ACR 50
14. týždeň 10 % 35 %* 7 % 15 %
p = 0,021
NA NA
24. týždeň 14 % 37 %* 4 % 16 %* 29 % 40 %
52. týždeň NA NA NA NA 36 % 42 %
ACR 70
14. týždeň 4 % 14 %
p = 0,008
2 % 10 %
p = 0,005
NA NA
24. týždeň 5 % 20 %* 2 % 9 % p = 0,009 16 % 24 %
52. týždeň NA NA NA NA 22 % 28 %
a n vyjadruje randomizovaných pacientov; aktuálny počet pacientov hodnotiteľných pre každý koncový ukazovateľ sa môže líšiť podľa časového bodu.
* p ≤ 0,001
NA: Neaplikovateľné
V GO-BEFORE nebola primárna analýza v 24. týždni u pacientov so stredne závažnou až závažnou
reumatoidnou artritídou (kombinované skupiny Simponi 50 a 100 mg + MTX vs. samotný MTX pre ACR50) štatisticky signifikantná (p = 0,053). V 52. týždni bolo v celkovej populácii celkovo vyššie percento pacientov, ktorí dosiahli odpoveď ACR, v skupine so Simponi 50 mg + MTX, ale
v porovnaní so samotným MTX nebolo signifikantne odlišné (pozri tabuľku 2). Ďalšie analýzy sa vykonali v podskupinách reprezentujúcich indikovanú populáciu pacientov so závažnou aktívnou
a progresívnou RA. V indikovanej populácii bol preukázaný celkovo väčší účinok Simponi 50 mg + MTX oproti samotnému MTX v porovnaní s celkovou populáciou.
V GO-FORWARD a GO-AFTER sa v každom dopredu určenom časovom bode pozorovali klinicky významné a štatisticky signifikantné odpovede na stupnici aktivity ochorenia (Disease Activity Scale, DAS) 28, v 14. týždni a v 24. týždni (p ≤ 0,001). V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 od
104. týždňa až do 256. týždňa podobné.
V GO-BEFORE bola meraná výrazná klinická odpoveď, definovaná ako udržanie odpovede ACR 70 nepretržite počas 6-mesačného obdobia. V 52. týždni dosiahlo výraznú klinickú odpoveď 15 % pacientov v skupine so Simponi 50 mg + MTX v porovnaní so 7 % pacientov v skupine s placebom + MTX (p = 0,018). V skupine 159 osôb randomizovaných na Simponi 50 mg + MTX bolo stále na tejto liečbe 96 osôb v 104. týždni. Z tých bola odpoveď ACR 20 u 85 pacientov, odpoveď ACR 50
u 66 pacientov a odpoveď ACR 70 u 53 pacientov v 104. týždni. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede
ACR 20/50/70.
Rádiografická odpoveď:
V GO-BEFORE bola na stanovenie stupňa štrukturálneho poškodenia použitá zmena vo vdH-S skóre oproti východiskovej hodnote, čo je kombinované skóre štrukturálneho poškodenia, ktoré rádiograficky stanovuje počet a veľkosť erózií kĺbov a stupeň zúženia kĺbovej štrbiny (joint space narrowing, JSN) na rukách/zápästiach a nohách. Kľúčové výsledky pre dávku Simponi 50 mg v 52. týždni sú uvedené v tabuľke 3.
Počet pacientov bez nových erózií alebo so zmenou oproti východiskovej hodnote v celkovom vdH-S skóre ≤ 0 bol signifikantne vyšší v skupine liečenej Simponi ako v kontrolnej skupine (p = 0,003). Rádiografické účinky pozorované v 52. týždni boli udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli rádiografické účinky od 104. týždňa až do 256. týždňa podobné.
Tabuľka 3
Priemerné (štandardná odchýlka) rádiografické zmeny oproti východiskovej hodnote v celkovom vdH-S skóre v 52. týždni v celkovej populácii v GO-BEFORE
Placebo + MTX Simponi 50 mg + MTX
n
a 160 159Celkové skóreVýchodisková hodnota 19,7 (35,4) 18,7 (32,4) Zmena oproti východiskovej hodnote 1,4 (4,6) 0,7 (5,2)*
Skóre erózieVýchodisková hodnota 11,3 (18,6) 10,8 (17,4) Zmena oproti východiskovej hodnote 0,7 (2,8) 0,5 (2,1)
Skóre JSNVýchodisková hodnota 8,4 (17,8) 7,9 (16,1) Zmena oproti východiskovej hodnote 0,6 (2,3) 0,2 (2,0)** a n vyjadruje randomizovaných pacientov
* p = 0,015
** p = 0,044
Telesná funkcia a kvalita života spojená so zdravímTelesná funkcia a neschopnosť sa posudzovali ako samostatný koncový ukazovateľ v GO-FORWARD
a GO-AFTER používajúc index neschopnosti HAQ DI. V týchto štúdiách preukázal Simponi klinicky významné a štatisticky signifikantné zlepšenie v HAQ DI od základného stavu oproti kontrole v 24. týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, sa udržalo zlepšenie v HAQ DI až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii
a liečených Simponi bolo zlepšenie v HAQ DI od 104. týždňa až do 256. týždňa podobné.
V GO-FORWARD sa preukázali klinicky významné a štatisticky signifikantné zlepšenia kvality života spojenej so zdravím, merané pomocou skóre telesných komponentov SF-36 u pacientov liečených Simponi oproti placebu v 24. týždni. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, bolo udržané zlepšenie telesných komponentov SF-36 až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi bolo zlepšenie telesných komponentov SF-36 od 104. týždňa až do 256. týždňa podobné. V GO-FORWARD
a GO-AFTER sa pozorovali štatisticky signifikantné zlepšenia únavy, merané škálou únavy-funkčné posúdenie liečby chronickej choroby (FACIT-F).
Psoriatická artritídaBezpečnosť a účinnosť Simponi bola hodnotená v multicentrickej randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-REVEAL) so 405 dospelými pacientmi s aktívnou PsA (≥ 3
opuchnuté kĺby a ≥ 3 bolestivé kĺby) napriek liečbe nesteroidovými antiflogistikami (NSAID) alebo
DMARD. Pacienti v tejto štúdii mali diagnózu PsA najmenej 6 mesiacov a mali aspoň miernu psoriatickú chorobu. Boli zaradení pacienti s každým podtypom psoriatickej artritídy, vrátane
polyartikulárnej artritídy bez reumatoidných uzlíkov (43 %), asymetrickej periférnej artritídy (30 %),
distálnej artritídy interfalangeálnych kĺbov (DIP) (15 %), spondylitídy s periférnou artritídou (11 %) a arthritis mutilans (1 %). Predchádzajúca liečba anti-TNF liečivom nebola prípustná. Simponi alebo placebo sa podávali subkutánne každé 4 týždne. Pacientom bolo náhodne pridelené placebo, Simponi
50 mg alebo Simponi 100 mg. Po 24. týždni boli pacienti, ktorí dostávali placebo, prevedení na
Simponi 50 mg. V 52. týždni pacienti vstúpili do nezaslepeného dlhodobého predĺženia. Približne štyridsaťosem percent pacientov pokračovalo na stabilnej dávke metotrexátu (≤ 25 mg/týždeň).
Spoločnými primárnymi koncovými ukazovateľmi boli percento pacientov, ktorí dosiahli odpoveď
ACR 20 v 14. týždni a zmena v celkovom vdH-S skóre modifikovanom pre PsA v 24. týždni oproti východiskovej hodnote.
Celkovo sa až do 104. týždňa nepozorovali žiadne klinicky významné rozdiely v mierach účinnosti medzi dávkovacími režimami Simponi 50 mg a 100 mg. Podľa dizajnu štúdie mohli byť pacienti
v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Prejavy a príznaky
Kľúčové výsledky pre dávku 50 mg v 14. a 24. týždni sú zobrazené v tabuľke 4 a opísané nižšie.
Tabuľka 4
Kľúčové výsledky účinnosti z GO-REVEAL
Placebo
Simponi
50 mg*
na 113 146
Respondenti, % pacientov
ACR 20
AC
R 50
AC
R 70
PASI
b
75
c
14. týždeň 9 % 51 %
24. týždeň 12 % 52 %
14. týždeň 2 % 30 %
24. týždeň 4 % 32 %
14. týždeň 1 % 12 %
24. týždeň 1 % 19 %
14. týždeň 3 % 40 %
24. týždeň 1 % 56 %
* p < 0,05 pre všetky porovnávania;
a n vyjadruje randomizovaných pacientov; aktuálny počet hodnotiteľných pacientov sa môže pre každý výsledný ukazovateľ líšiť podľa časového bodu.
b Oblasť postihnutá psoriázou a stupeň závažnosti
c Založené na podskupine pacientov so vstupným postihnutím BSA ≥ 3 %, 79 pacientov
(69,9 %) zo skupiny s placebom a 109 pacientov (74,3 %) zo skupiny so Simponi 50 mg.
Odpovede sa pozorovali v prvom hodnotení (4. týždeň) po prvom podaní Simponi. Podobné odpovede
ACR 20 sa pozorovali v 14. týždni u pacientov s polyartikulárnou artritídou bez reumatoidných uzlíkov a pri asymetrickej periférnej artritíde, podtyp PsA. Počet pacientov s inými podtypmi PsA bol
príliš malý na zmysluplné vyhodnotenie. Odpovede pozorované v skupinách liečených Simponi boli
podobné u pacientov užívajúcich a neužívajúcich súčasne metotrexát. V skupine 146 pacientov randomizovaných na Simponi 50 mg bolo stále na tejto liečbe 70 pacientov v 104. týždni. Z týchto
70 pacientov bola odpoveď ACR 20 u 64 pacientov, odpoveď ACR 50 u 46 pacientov a odpoveď ACR 70 u 31 pacientov. V skupine pacientov zostávajúcich v štúdii a liečených Simponi sa od 104. týždňa až do 256. týždňa pozoroval podobný pomer odpovede ACR 20/50/70.
Štatisticky významné odpovede v DAS 28 sa pozorovali aj v 14. a 24. týždni (p < 0,05).
V skupine pacientov liečených Simponi sa v 24. týždni pozorovali zlepšenia parametrov periférnej aktivity, charakteristických pre psoriatickú artritídu (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída). Liečba Simponi viedla k výraznému zlepšeniu telesných funkcií hodnotenej podľa HAQ DI, ako aj k signifikantnému zlepšeniu kvality života spojenej so zdravím ako bolo namerané pomocou sumárneho skóre telesných a mentálnych údajov podľa SF-36. V skupine pacientov zostávajúcich na liečbe Simponi, na ktorú boli randomizovaní na začiatku štúdie, boli odpovede v DAS 28 a HAQ DI udržané až do 104. týždňa. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli odpovede v DAS 28 a HAQ DI od 104. týždňa až do
256. týždňa podobné.
Rádiografická odpoveď:
Štrukturálne poškodenie oboch rúk a nôh sa rádiograficky hodnotilo zmenou vo východiskovej hodnote oproti vdH-S skóre modifikovanom pre PsA pridaním distálnych interfalangeálnych (DIP)
kĺbov rúk.
Liečba Simponi 50 mg znížila v 24. týždni rýchlosť progresie poškodenia periférnych kĺbov
v porovnaní s podávaním placeba podľa zmeny v celkovom modifikovanom vdH-S skóre oproti
východiskovej hodnote (priemer ± SD skóre bolo 0,27 ± 1,3 v skupine s placebom v porovnaní s -0,16 ± 1,3 v skupine so Simponi; p = 0,011). Zo 146 pacientov, ktorí boli randomizovaní na Simponi 50 mg, boli RTG údaje dostupné u 126 pacientov v 52. týždni, z ktorých u 77 % sa nepreukázala žiadna progresia v porovnaní s východiskovou hodnotou. V 104. týždni boli RTG údaje dostupné u 114 pacientov a u 77 % sa nepreukázala žiadna progresia oproti východiskovej hodnote.
V skupine pacientov zostávajúcich v štúdii a liečených Simponi v období od 104. týždňa až do 256. týždňa podobný pomer pacientov nepreukázal žiadnu progresiu oproti východiskovej hodnote.
Axiálna spondyloartritída
Ankylozujúca spondylitída
Bezpečnosť a účinnosť Simponi sa hodnotila v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-RAISE) s 356 dospelými pacientmi s aktívnou ankylozujúcou spondylitídou (definovanou ako Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥ 4 a VAS ≥ 4 pri celkovej bolesti chrbtice na škále 0 až 10 cm). Pacienti zaradení do tejto štúdie mali aktívne ochorenie napriek prebiehajúcej alebo predchádzajúcej liečbe NSAID alebo DMARD
a predtým sa neliečili anti-TNF liečbou. Simponi alebo placebo sa podávali subkutánne každé
4 týždne. Pacientom bolo náhodne pridelené placebo, Simponi 50 mg a Simponi 100 mg a mali povolené pokračovať v súbežnej liečbe DMARD (MTX, SSZ a/alebo HCQ). Primárnym koncovým
ukazovateľom bolo percento pacientov, ktorí dosiahli v Ankylosing Spondylitis Assessment Study
Group (ASAS) 20 odpoveď v 14. týždni. Údaje o placebom kontrolovanej účinnosti boli zozbierané a analyzované v 24. týždni.
Kľúčové výsledky pre dávku 50 mg sú zobrazené v tabuľke 5 a popísané nižšie. Celkovo sa až do 24. týždňa nepozorovali žiadne klinicky významné rozdiely v hodnotách účinnosti medzi dávkovými režimami Simponi 50 mg a 100 mg. Podľa dizajnu štúdie mohli byť pacienti v dlhodobom predĺžení po zvážení lekára štúdie prestavení na 50 mg alebo 100 mg dávky Simponi.
Tabuľka 5
Kľúčové výsledky účinnosti v GO-RAISE
Placebo
Simponi
50 mg*
na 78 138
Respondenti, % pacientov
ASAS 20
ASA
S 40
ASA
S 5/6
14. týždeň 22 % 59 %
24. týždeň 23 % 56 %
14. týždeň 15 % 45 %
24. týždeň 15 % 44 %
14. týždeň 8 % 50 %
24. týždeň 13 % 49 %
* p ≤ 0,001 pre všetky porovnania
a n vyjadruje randomizovaných pacientov; aktuálny počet hodnotiteľných pacientov sa môže pre každý výsledný ukazovateľ líšiť podľa časového bodu.
V skupine pacientov zostávajúcich v štúdii a liečených Simponi bola časť pacientov s ASAS 20
a ASAS 40 odpoveďou od 24. týždňa až do 256. týždňa podobná.
Štatisticky signifikantné odpovede v BASDAI 50, 70 a 90 (p ≤ 0,017) sa tiež pozorovali v 14. a 24. týždni. Zlepšenie v kľúčových hodnotách aktivity choroby sa pozorovalo v prvom hodnotení (4. týždeň) po prvom podaní Simponi a pretrvávalo až do 24. týždňa. V skupine pacientov zostávajúcich
v štúdii a liečených Simponi sa v BASDAI od 24. týždňa až do 256. týždňa pozoroval podobný pomer zmeny oproti východiskovej hodnote. Konzistentná účinnosť sa pozorovala bez ohľadu na použitie DMARD (MTX, sulfasalazín a/alebo hydroxychlorochin), stav antigénu HLA-B27 alebo východiskové hodnoty CRP, ako bolo zhrnuté v ASAS 20 odpovediach v 14. týždni.
Liečba Simponi vyústila do signifikantného zlepšenia telesnej funkcie ako bolo posúdené v BASFI zmenami v 14. a 24. týždni oproti východiskovej hodnote. Kvalita života spojená so zdravím hodnotená podľa skóre fyzických komponentov SF-36 sa tiež signifikantne zlepšila v 14. a 24. týždni. V skupine pacientov zostávajúcich v štúdii a liečených Simponi boli zlepšenia telesnej funkcie
a kvality života spojenej so zdravím od 24. týždňa až do 256. týždňa podobné.
Axiálna spondyloartritída bez rádiografického dôkazuBezpečnosť a účinnosť Simponi sa hodnotili v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (GO-AHEAD) u 197 dospelých pacientov so závažnou aktívnou
nr-axiálnou SpA (definovaní ako tí pacienti, ktorí spĺňajú kritériá klasifikácie axiálnej
spondyloartritídy podľa ASAS, ale nespĺňajú upravené kritériá New York pre AS). Pacienti zaradení do tejto štúdie mali aktívne ochorenie (definované ako BASDAI ≥ 4 a celková bolesť chrbta ≥ 4 na vizuálnej analógovej stupnici (VAS), každé na stupnici 0-10 cm) napriek prebiehajúcej alebo predchádzajúcej liečbe NSAID a ktorí sa v minulosti neliečili žiadnymi biologickými liečivami vrátane anti-TNF liečby. Pacienti boli náhodne priradení na placebo alebo Simponi podávané v dávke
50 mg subkutánne každé 4 týždne. V 16. týždni pacienti vstúpili do otvoreného obdobia, v ktorom všetci pacienti dostávali Simponi v dávke 50 mg podávaných subkutánne každé 4 týždne počas
48 týždňov s hodnoteniami účinnosti vykonanými v 52. týždni a následnými hodnoteniami
bezpečnosti v 60. týždni. Približne 93 % pacientov, ktorí dostávali Simponi na začiatku otvoreného predĺženia (16. týždeň), pokračovalo v liečbe do konca štúdie (52. týždeň). Analýzy sa vykonali
v populácii všetkých liečených pacientov (All treated, AT, N = 197) a aj v populácii s objektívnymi prejavmi zápalu (Objective Signs of Inflammation, OSI, N = 158, definovanej na základe zvýšenej hladiny CRP a/alebo dôkazu sakroileitídy na MR na začiatku). V 16. týždni sa získali a analyzovali
placebom kontrolované údaje o účinnosti. Primárnym koncovým ukazovateľom bol podiel pacientov, ktorí dosiahli ASAS 20 odpoveď v 16. týždni. Kľúčové výsledky sú uvedené v tabuľke 6 a uvedené
nižšie.
Tabuľka 6Kľúčové výsledky účinnosti z GO-AHEAD v 16. týždniZlepšenia v prejavoch a príznakoch Populácia všetkých liečených pacientov (AT)
Populácia s objektívnymi prejavmi zápalu (OSI)
Placebo Simponi 50 mg Placebo Simponi 50 mg na 100 97 80 78
Respondenti, % pacientov
ASAS 20 40 % 71 %** 38 % 77 %** ASAS 40 23 % 57 %** 23 % 60 %** ASAS 5/6 23 % 54 %** 23 % 63 %** Parciálna remisia ASAS 18 % 33 %* 19 % 35 %* ASDAS-C b < 1,3 13 % 33 %* 16 % 35 %* BASDAI 50 30 % 58 %** 29 % 59 %**
Inhibícia zápalu v sakroiliakálnych (SI) kĺboch na základe merania pomocou MR
Placebo Simponi 50 mg Placebo Simponi 50 mg n C 87 74 69 61
Priemerná zmena v MR
skóre pre sakroiliakálne
kĺby SPARCCd -0,9 -5,3** -1,2 -6,4**
a n odráža randomizovaných a liečených pacientov
b Skóre C-reaktívneho proteínu pre aktivitu ochorenia ankylozujúcej spondylitídy (AT-placebo, N = 90; AT-Simponi
50 mg, N = 88; OSI-placebo, N = 71; OSI-Simponi 50 mg, N = 71)
c n odráža počet pacientov s údajmi z MR na začiatku a v 16. týždni
d SPARCC (Kanadské združenie pre výskum spondyloartritídy)
** p < 0,0001 pri porovnaniach Simponi vs. placebo
* p < 0,05 pri porovnaniach Simponi vs. placebo
U pacientov liečených Simponi v dávke 50 mg sa v 16. týždni v porovnaní s placebom preukázali
štatisticky významné zlepšenia prejavov a príznakov závažnej aktívnej nr-axiálnej SpA (tabuľka 6).
Zlepšenia sa pozorovali pri prvom hodnotení (4. týždeň) po podaní úvodnej dávky Simponi. Pri skóre SPARCC sa na základe merania pomocou MR v 16. týždni preukázali štatisticky významné zmiernenia zápalu SI kĺbov u pacientov liečených Simponi v dávke 50 mg v porovnaní s placebom (tabuľka 6). Pri bolesti hodnotenej pomocou VAS pre celkovú bolesť chrbta a bolesť chrbta v noci
a pri aktivite ochorenia hodnotenej pomocou ASDAS-C sa tiež preukázalo štatisticky významné zlepšenie od začiatku do 16. týždňa u pacientov liečených Simponi v dávke 50 mg v porovnaní
s placebom (p < 0,0001).
U pacientov liečených Simponi v dávke 50 mg sa v porovnaní s pacientmi dostávajúcimi placebo preukázali štatisticky významné zlepšenia v spinálnej mobilite na základe hodnotenia podľa BASMI (Bath Ankylosing Spondylitis Metrology Index) a v telesnej funkcii na základe hodnotenia podľa BASFI (p < 0,0001). U pacientov liečených Simponi sa objavilo významne viac zlepšení v kvalite života súvisiacej so zdravotným stavom na základe ASQoL, EQ-5D a v telesných a mentálnych zložkách SF-36 a objavilo sa u nich významne viac zlepšení v produktivite na základe zhodnotenia väčších znížení v celkovom narušení práce a narušení aktivity na základe hodnotenia dotazníkom WPAI ako u pacientov dostávajúcich placebo.
Pri všetkých koncových ukazovateľoch uvedených vyššie sa štatisticky významné výsledky preukázali aj v populácii OSI v 16. týždni.
V oboch populáciách, AT a OSI, zlepšenia prejavov a príznakov, spinálnej mobility, telesnej funkcie, kvality života a produktivity pozorované v 16. týždni u pacientov liečených Simponi v dávke 50 mg pokračovali aj u tých pacientov, ktorí zostali v štúdii až do 52. týždňa.
Ulcerózna kolitída
Účinnosť Simponi sa hodnotila v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách u dospelých pacientov.
Štúdia zameraná na nasadenie lieku (PURSUIT-indukcia) hodnotila pacientov so stredne závažnou až závažnou aktívnou ulceróznou kolitídou (skóre 6 až 12 podľa Maya; subskóre ≥ 2 podľa endoskopie), ktorí nemali dostatočnú odpoveď na konvenčné liečby alebo ktorí netolerovali konvenčné liečby alebo boli závislí od kortikosteroidov. V časti štúdie na potvrdenie dávky bolo 761 pacientov randomizovaných na užívanie buď 400 mg Simponi s.c. v 0. týždni a 200 mg v 2. týždni, 200 mg Simponi s.c. v 0. týždni a 100 mg v 2. týždni alebo placebo s.c. v 0. a 2. týždni. Súbežné stabilné dávky perorálnych aminosalicylátov, kortikosteroidov a/alebo imunomodulátorov boli povolené.
V tejto štúdii sa hodnotila účinnosť Simponi v 6. týždni.
Výsledky štúdie zameranej na udržiavaciu liečbu (PURSUIT-udržiavanie) boli založené na hodnotení
456 pacientov, ktorí dosiahli klinickú odpoveď z predchádzajúceho nasadenia liečby Simponi. Pacienti boli randomizovaní na užívanie Simponi 50 mg, Simponi 100 mg alebo placebo, podávané subkutánne
každé 4 týždne. Súbežné stabilné dávky perorálnych aminosalicylátov a/alebo imunomodulátorov boli povolené. Na začiatku štúdie zameranej na udržiavaciu liečbu sa dávky kortikosteroidov museli znížiť.
V tejto štúdii sa hodnotila účinnosť Simponi v 54. týždni. Pacienti, ktorí dokončili štúdiu zameranú na udržiavaciu liečbu v 54. týždni, pokračovali v liečbe v predĺžení štúdie s hodnotením účinnosti v 216. týždni. Hodnotenie účinnosti pri predĺžení štúdie sa zakladalo na zmenách v používaní
kortikosteroidov, na celkovom hodnotení lekárov (Physician’s Global Assessment, PGA) týkajúceho sa aktivity ochorenia a na zlepšení kvality života meranej prostredníctvom dotazníka zápalového
ochorenia čriev (Inflammatory Bowel Disease Questionnaire, IBQD).
Tabuľka 7
Kľúčové výsledky účinnosti zo štúdií PURSUIT-indukcia a PURSUIT-udržiavanie
PURSUIT-indukcia
Percentuálne vyjadrenie pacientov
Placebo
N = 251
Simponi
200/100 mg
N = 253
Pacienti s klinickou odpoveďou v 6. týždnia
Pacienti s klinickou remisiou v 6. týždnib
Pacienti s hojením sliznice v 6. týždnic'
30 % 51 %**
6 % 18 %**
29 % 42 %*
PURSUIT-udržiavanie
Placebo
d
N = 154
Simponi
50 mg
N = 151
Simponi
100 mg
N = 151
Percentuálne vyjadrenie pacientov
Udržiavanie odpovede (Pacienti s klinickou odpoveďou v 54. týždni)e
Trvalá remisia (Pacienti
s klinickou remisiou v 30. a aj
54. týždni)f
N = počet pacientov
** p ≤ 0,001
* p ≤ 0,01
31 % 47 %* 50 %**
16 % 23 %g 28 %*
a definované ako pokles skóre podľa Maya o ≥ 30 % a ≥ 3 body z východiskovej hodnoty, sprevádzaný poklesom v subskóre rektálneho krvácania o ≥ 1 alebo subskóre rektálneho
krvácania 0 alebo 1.
b definované ako skóre podľa Maya ≤ 2 body bez žiadneho individuálneho subskóre > 1
c definované ako 0 alebo 1 v subskóre podľa endoskopie pri skóre podľa Maya.
d len nasadenie liečby Simponi.
e U pacientov sa každé 4 týždne hodnotila aktivita ochorenia UC pomocou parciálneho skóre podľa Maya (strata odpovede sa potvrdila pomocou endoskopie). Preto pri každom hodnotení
v 54. týždni boli pacienti s udržanou odpoveďou v stave kontinuálnej klinickej odpovede.
f Na dosiahnutie trvalej remisie musel byť pacient v remisii v 30. a aj 54. týždni (bez prejavu straty odpovede kedykoľvek v 54. týždni).
g U pacientov s telesnou hmotnosťou nižšou ako 80 kg sa vo väčšej časti pacientov, ktorí dostávali
50 mg udržiavaciu liečbu, preukázala trvalá klinická remisia v porovnaní s pacientmi, ktorí dostávali placebo.
U viacerých pacientov liečených Simponi, sa preukázalo pretrvávajúce hojenie sliznice (pacienti
s hojením sliznice v 30. a aj 54. týždni) v skupine s 50 mg (42 %, nominálna hodnota p < 0,05)
a v skupine so 100 mg (42 %, nominálna hodnota p < 0,005) v porovnaní s pacientmi v skupine s placebom (27 %).
Spomedzi 54 % pacientov (247/456), ktorí súbežne dostávali kortikosteroidy na začiatku štúdie PURSUIT-udržiavanie, bol podiel pacientov, u ktorých sa udržala klinická odpoveď v 54. týždni a ktorí v 54. týždni súbežne nedostávali kortikosteroidy, väčší v skupine s 50 mg (38 %, 30/78)
a v skupine so 100 mg (30 %, 25/82) v porovnaní so skupinou s placebom (21 %, 18/87). Podiel pacientov, u ktorých sa kortikosteroidy vysadili do 54. týždňa, bol väčší v skupine s 50 mg (41 %,
32/78) a v skupine so 100 mg (33 %, 27/82) v porovnaní so skupinou s placebom (22 %, 19/87).
Spomedzi pacientov, ktorí vstúpili do predĺženia štúdie, bol podiel osôb, ktorí nepotrebovali liečbu kortikosteroidmi, všeobecne udržaný až do 216. týždňa.
Pacientom, ktorí v štúdiách PURSUIT-indukcia nedosiahli klinickú odpoveď v 6. týždni, bola v štúdii
PURSUIT-udržiavanie podávaná dávka 100 mg každé 4 týždne. V 14. týždni 28 % z týchto pacientov
dosiahlo odpoveď definovanú parciálnymi skóre podľa Maya (zníženú o ≥ 3 body v porovnaní so začiatkom indukcie). V 54. týždni boli klinické výsledky pozorované u týchto pacientov podobné klinickým výsledkom hláseným u pacientov, ktorí dosiahli klinickú odpoveď v 6. týždni.
V 6. týždni Simponi výrazne zlepšil kvalitu života, na základe merania zmeny v špecifickom meraní ochorenia, v dotazníku zápalového ochorenia čriev (inflammatory bowel disease questionnaire, IBDQ). Medzi pacientmi, ktorí dostávali udržiavaciu liečbu Simponi, sa zlepšenie kvality života merané pomocou IBDQ v 54. týždni udržalo.
Približne 63 % pacientov dostávajúcich Simponi na začiatku predĺženia štúdie (56. týždeň)
pokračovalo v liečbe do konca štúdie (posledné podanie golimumabu v 212. týždni).
Imunogenita
Počas štúdií fázy III s RA, PsA a AS boli až do 52. týždňa zistené protilátky proti golimumabu u 5 % (105/2 062) pacientov liečených golimumabom a kde sa testovali, boli takmer všetky protilátky neutralizujúce in vitro. Podobný pomer sa pozoroval vo všetkých reumatologických indikáciách. Súbežná liečba s MTX mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez MTX (približne 3 % [41/1 235] oproti 8 % [64/827]
v uvedenom poradí).
Pri nr-axiálnej SpA sa protilátky proti golimumabu zistili do 52. týždňa u 7 % (14/193) pacientov liečených golimumabom.
V štúdiách fázy II a III štúdií s UC boli až do 54. týždňa zistené protilátky proti golimumabu u 3 % (26/946) pacientov liečených golimumabom. Šesťdesiatosem percent (21/31) pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky. Súbežná liečba s imunomodulátormi (azatioprin,
6-merkaptopurín a MTX ) mala za následok nižšie zastúpenie pacientov s protilátkami proti golimumabu, než u pacientov užívajúcich golimumab bez imunomodulátorov (1 % (4/308) oproti
3 % (22/638) v uvedenom poradí). Z pacientov, ktorí pokračovali v predĺžení štúdie a mali
hodnotiteľné vzorky v 228. týždni, sa preukázali protilátky proti golimumabu u 4 % (23/604)
pacientov liečených golimumabom. Osemdesiatdva percent (18/22) pacientov s pozitívnymi protilátkami malo in vitro neutralizujúce protilátky.
Prítomnosť protilátok proti golimumabu môže zvýšiť riziko reakcií v mieste injekcie (pozri časť 4.4). Malý počet pacientov pozitívnych na protilátky proti golimumabu limituje schopnosť vyvodiť definitívne závery, týkajúce sa vzťahu medzi protilátkami proti golimumabu a klinickou účinnosťou alebo bezpečnostné opatrenia.
Pretože analýzy imunogenity sú špecifické pre daný liek a daný test, porovnanie frekvencie výskytu protilátok s prekvenciami pri ostatných liekoch nie je vhodné.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so Simponi v jednej alebo viacerých podskupinách pediatrickej populácie pre ulceróznu kolitídu (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2. Farmakokinetické vlastnosti
Absorpcia
Po jednorazovej subkutánnej aplikácii golimumabu zdravým osobám alebo pacientom s RA bol priemerný čas na dosiahnutie maximálnych koncentrácií v sére (Tmax) od 2 do 6 dní. Subkutánna injekcia 50 mg golimumabu zdravým osobám priniesla priemernú hodnotu ± smerodajná odchýlka maximálnych koncentrácií (Cmax) v sére 3,1 ± 1,4 μg/ml.
Po jednorazovej subkutánnej injekcii 100 mg bola absorpcia golimumabu v hornej časti ramena, na bruchu a na stehne podobná, s priemernou absolútnou biologickou dostupnosťou 51 %. Keďže
golimumab preukázal približnú dávkovo úmernú FK po subkutánnom podaní, dá sa očakávať, že absolútna biologická dostupnosť po dávke 50 mg alebo 200 mg golimumabu bude podobná.
Distribúcia
Po jednorazovom i.v. podaní bol priemerný distribučný objem 115 ± 19 ml/kg.
Eliminácia
Systémový klírens golimumabu bol odhadnutý na 6,9 ± 2,0 ml/deň/kg. Hodnota terminálneho polčasu u zdravých osôb bola odhadnutá na približne 12 ± 3 dni a podobné hodnoty sa pozorovali u pacientov
s RA, PsA, AS alebo UC.
U pacientov s RA, PsA alebo AS, ktorí dostávali 50 mg golimumabu každé 4 týždne, sa dosiahol rovnovážny stav koncentrácií v sére v 12. týždni. Pri súbežnom podávaní MTX priniesla liečba
s 50 mg golimumabu subkutánne každé 4 týždne priemerné (± smerodajná odchýlka) minimálne sérové koncentrácie v rovnovážnom stave približne 0,6 ± 0,4 μg/ml u pacientov s RA s aktívnou RA
napriek liečbe MTX a približne 0,5 ± 0,4 μg/ml u pacientov s aktívnou PsA a približne 0,8 ± 0,4 μg/ml u pacientov s AS. Priemerné minimálne sérové koncentrácie golimumabu v rovnovážnom stave
u pacientov s nr-axiálnou SpA boli podobné koncentráciám, ktoré sa pozorovali u pacientov s AS po
subkutánnom podaní 50 mg golimumabu každé 4 týždne.
Pacienti s RA, PsA alebo AS, ktorí nedostávali súbežne MTX, mali približne o 30 % nižšie hodnoty minimálnych koncentrácií golimumabu v rovnovážnom stave než tí, ktorí dostávali golimumab
s MTX. U obmedzeného počtu pacientov s RA liečených subkutánnym golimumabom počas 6-
mesačného obdobia súbežné používanie MTX znížilo zdanlivý klírens golimumabu o približne 36 %. Populačná farmakokinetická analýza však naznačila, že súbežné používanie NSAID, perorálnych kortikosteroidov alebo sulfasalazínu nemalo vplyv na zdanlivý klírens golimumabu.
U pacientov s UC sa po indukčných dávkach 200 mg golimumabu v 0. týždni a 100 mg golimumabu v 2. týždni a udržiavacích dávkach 50 mg alebo 100 mg golimumabu subkutánne následne každé
4 týždne dosiahli sérové koncentrácie golimumabu v rovnovážnom stave približne 14 týždňov po
začiatku liečby. Liečba 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne počas udržiavania viedla k priemernej minimálnej sérovej koncentrácii v rovnovážnom stave približne
0,9 ± 0,5 mg/ml a 1,8 ± 1,1 mg/ml.
Súbežné užívanie imunomodulátorov u pacientov s UC liečených 50 mg alebo 100 mg golimumabu subkutánne každé 4 týždne nemalo významný vplyv na minimálne hladiny golimumabu
v rovnovážnom stave.
Pacienti, u ktorých sa vyvinuli protilátky proti golimumabu, mali v rovnovážnom stave všeobecne nízke minimálne sérové koncentrácie golimumabu (pozri časť 5.1).
Linearita
Golimumab vykazoval približne dávkovo úmernú farmakokinetiku u pacientov s RA v rozsahu dávok
0,1 mg až 10,0 mg/kg po jednorazovej intravenóznej dávke. Po jednorazovej subkutánnej dávke
u zdravých osôb sa v rozmedzí dávok 50 mg až 400 mg pozorovala farmakokinetika, ktorá bola tiež približne proporcionálna k dávke.
Vplyv telesnej hmotnosti na farmakokinetiku
Zistil sa trend k vyšším hodnotám zdanlivého klírensu golimumabu s narastajúcou telesnou hmotnosťou (pozri časť 4.2).
5.3. Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
S golimumabom sa nevykonali žiadne štúdie mutagenity, štúdie fertility na zvieratách ani dlhodobé štúdie karcinogenity.
V štúdiách fertility a všeobecných reprodukčných funkcií myší, pri použití analogickej protilátky selektívne inhibujúcej funkčnú aktivitu myšieho TNFα, sa znížilo množstvo gravidných myší. Nie je známe, či bol tento nález spôsobený vplyvom na samce a/alebo samice. V stúdii vývinovej toxicity vykonanej na myšiach pri aplikácii rovnakej analógnej protilátky u myší a pri použití golimumabu
u opíc druhu Cynomolgus neboli zaznamenané žiadne dôkazy maternálnej toxicity, embryotoxicity alebo teratogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sorbitol (E420) Histidín
Monohydrát histidíniumchloridu
Polysorbát 80
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
24 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Naplnené pero alebo naplnenú injekčnú striekačku uchovávajte v škatuli na ochranu pred svetlom. Simponi sa môže uchovávať pri teplotách do maximálne 25 °C počas jedného obdobia až do 30 dní,
nesmie však presiahnuť pôvodný dátum exspirácie vytlačený na škatuli. Nový dátum exspirácie sa musí napísať na škatuľu (až do 30 dní od dátumu vybratia z chladničky).
Ak sa Simponi začne uchovávať pri izbovej teplote, nesmie sa vrátiť späť do chladničky. Ak sa
Simponi nepoužije počas 30 dní uchovávania pri izbovej teplote, musí sa zlikvidovať.
6.5 Druh obalu a obsah balenia
Simponi 100 mg injekčný roztok naplnený v injekčnom pere
1 ml roztoku naplneného v injekčnej striekačke (sklo typu 1) s napevno nasadenou ihlou (nerezová oceľ) a krytom ihly (kaučuk obsahujúci latex) v naplnenom pere. Simponi je dostupný v baleniach
obsahujúcich 1 naplnené pero a viacpočetných baleniach obsahujúcich 3 (3 balenia po 1) naplnené
perá.
Simponi 100 mg injekčný roztok naplnený v injekčnej striekačke
1 ml roztoku naplneného v injekčnej striekačke (sklo typu 1) s napevno nasadenou ihlou (nerezová oceľ) a krytom ihly (kaučuk obsahujúci latex). Simponi je dostupný v baleniach obsahujúcich
1 naplnenú injekčnú striekačku a viacpočetných baleniach obsahujúcich 3 (3 balenia po 1) naplnené injekčné striekačky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Simponi sa dodáva ako jednorazové naplnené pero, nazývané SmartJect alebo ako jednorazová naplnená injekčná striekačka. Každé balenie obsahuje návod na použitie, ktorý úplne popisuje spôsob používania pera alebo injekčnej striekačky. Po vybratí naplneného pera alebo naplnenej injekčnej striekačky z chladničky sa má naplnené pero alebo naplnená injekčná striekačka nechať 30 minút dosiahnuť izbovú teplotu pred podaním Simponi. Perom alebo injekčnou striekačkou sa nesmie triasť.
Roztok je číry až mierne opaleskujúci, bezfarebný až svetložltý a môže obsahovať malé množstvo priehľadných alebo bielych čiastočiek proteínov. Tento vzhľad nie je ničím nezvyčajným pre roztoky s obsahom proteínov. Simponi sa nesmie použiť pri zmene farby roztoku, zakalení alebo ak obsahuje viditeľné cudzie častice.
Podrobné inštrukcie o spôsobe prípravy a aplikácie Simponi v naplnenom pere alebo naplnenej injekčnej striekačke sú súčasťou písomnej informácie pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/09/546/005 1 naplnené injekčné pero
EU/1/09/546/006 3 naplnené injekčné perá
EU/1/09/546/007 1 naplnená injekčná striekačka
EU/1/09/546/008 3 naplnené injekčné striekačky
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 1. októbra 2009
Dátum posledného predĺženia registrácie: 19. júna 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Návod na použitie
Simponi 45 mg/0,45 ml
injekčný roztok naplnený v injekčnom pere, VarioJect
Na použitie u detí a dospievajúcich
JEDNORAZOVÉ POUŽITIE
 Poznajte svoju dávku
Poznajte svoju dávkuMiesto vyššie použite na zaznamenanie vašej predpísanej dávky. Ak si nie ste istý svojou dávkou, overte si to u svojho lekára.
DôležitéAk sa váš lekár rozhodne, že si budete môcť injekcie Simponi podať doma sami alebo ich vám bude
podávať ošetrovateľ, musíte byť zaškolený na správny spôsob prípravy a podania injekcie Simponi.
Predtým, ako použijete naplnené pero Simponi a vždy, keď dostanete nové naplnené pero, prečítajte si tento návod na použitie. Môže obsahovať nové informácie.
Pozorne si prečítajte aj písomnú informáciu predtým, ako si podáte injekciu. Tento návod na použitie nenahrádza rozhovor s lekárom týkajúci sa vášho zdravotného stavu alebo vašej liečby.
Ak ste neboli zaškolený alebo ak máte akékoľvek otázky, kontaktujte, vášho lekára, zdravotnú sestru alebo lekárnika.
 Informácie o uchovávaní
Informácie o uchovávaníUchovávajte v chladničke pri teplote 2 °C až 8 °C.
Môže sa uchovávať pri izbovej teplote (až do 25 °C) počas jedného obdobia až do 30 dní, nesmie však presiahnuť pôvodný dátum exspirácie. Nový dátum exspirácie zahŕňajúci deň/mesiac/rok napíšte na zadnú stranu škatule (najviac 30 dní po vybratí lieku z chladničky). Nevracajte tento liek späť do chladničky, ak dosiahol izbovú teplotu.
Naplnené pero Simponi a všetky lieky uchovávajte mimo dohľadu a dosahu detí. PrehľadNaplnené pero je injekčné pero, pomocou ktorého si môžete
ručne nastaviť konkrétnu, predpísanú
dávku. Každé naplnené pero môže dodať 0,1 ml až 0,45 ml (čo zodpovedá 10 mg až 45 mg golimumabu) v čiastkach po 0,05 ml.
Predtým, ako začnete používať toto naplnené pero, musíte vedieť ako:
· odstrániť vzduchové bublinky,
· nastaviť predpísanú dávku,
· podať injekciu
stlačením piestu rukou, takisto ako pri injekčnej striekačke.
Naplnené pero sa má použiť len raz. Po použití naplnené pero zlikvidujte.
Nepokúšajte sa použiť akékoľvek zvyšné množstvo lieku v naplnenom pere.
Nedávajte naplnené pero nikomu inému.
Liek
netraste.
Potrebujete pomoc?Zavolajte svojmu lekárovi, zdravotnej sestre alebo lekárnikovi, aby ste sa s nimi porozprávali o akýchkoľvek otázkach, ktoré môžete mať.
Ak potrebujte ďalšiu pomoc, pozrite si kontaktné údaje miestneho zástupcu v písomnej informácii.
Plánujte v predstihuSkontrolujte škatuľuSkontrolujte dátum exspirácie („EXP“) vytlačený alebo napísaný na zadnej strane škatule.
Nepoužívajte v prípade, že dátum exspirácie vypršal.
Injekciu
nepodávajte, ak je perforácia na škatuli porušená. Zavolajte lekárovi alebo lekárnikovi ohľadom nového naplneného pera.
Naplnené pero vyberte zo škatuleNechajte naplnené pero
pri izbovej teplote počas najmenej 30 minút mimo dosahu detí.
Nezohrievajte ho žiadnym iným spôsobom.
Budete potrebovať tieto pomôcky:·
1 alkoholový tampón·
1 vatový tampón alebo
gázový štvorček·
1 adhezívny obväz·
1 nádobu na ostré predmety (pozri krok 3)
Vaše naplnené pero
Špička
Tenká skrytá injekčná ihla
 Kontrolné okienkoOranžový prúžok na nastavenie peraOznačenia zodpovedajúce dávkam
Kontrolné okienkoOranžový prúžok na nastavenie peraOznačenia zodpovedajúce dávkam
Kryt* Neodstraňujte ho predtým, ako je to uvedené v návode.
Oranžový
chránič ihly
DÔLEŽITÉ:
Nestláčajte oranžový chránič ihly pred podaním injekcie. Chránič sa uzamkne a vaša dávka nebude podaná.
Naplnené pero nedvíhajte z kože počas podávania injekcie. Oranžový chránič ihly sa uzamkne a nebude vám podaná celá dávka.
Zárez na zvolenie
dávky Piest
* NEBEZPEČENSTVO UDUSENIA! Uchovávajte mimo dosahu detí.
 1. Pripravte si svoju injekciuVyberte si miesto podania injekcie
1. Pripravte si svoju injekciuVyberte si miesto podania injekcieVyberte si z nasledovných miest na podanie vašej injekcie:
·
Predná strana stehien (odporúčané)
· Spodná časť brucha
Nepoužívajte v oblasti 5 centimetrov okolo vášho pupku.
· Zadná strana hornej časti ramien (ak vám injekciu podáva váš ošetrovateľ) Pre každú injekciu si vyberte iné miesto v rámci vašej uprednostňovanej oblasti.
Injekciu nepodávajte do kože, ktorá je citlivá, podliata krvou, červená, šupinatá, tvrdá alebo do
miest, kde sa nachádzajú jazvy.
 Očistite miesto podania injekcie
Očistite miesto podania injekcie
Dôkladne si umyte ruky mydlom a teplou vodou.

Zvolené miesto na podanie injekcie potrite alkoholovým tampónom a nechajte oschnúť. Po vyčistení sa
nedotýkajte miesta na podanie injekcie, nefénujte ho ani naň nefúkajte.
Skontrolujte roztokNaplnené pero vyberte zo škatule.
Skontrolujte roztok cez kontrolné okienko. Má byť číry až mierne opaleskujúci (lesknúci sa ako perla)
a bezfarebný až svetložltý a môže obsahovať malé množstvo priehľadných alebo bielych čiastočiek bielkovín. Môžete tiež vidieť jednu alebo viac vzduchových bubliniek. To je normálne.
 Injekciu nepodávajte,
Injekciu nepodávajte, ak má roztok nesprávnu farbu, je zakalený, alebo obsahuje veľké častice. Ak si nie ste istý, zavolajte lekárovi alebo lekárnikovi pre nové naplnené pero.
Poklepaním posuňte vzduchové bublinky do hornej časti
Naplnené pero držte v zvislej polohe s modrým krytom smerujúcim nahor.

Naplnené pero jemne poklepte prstami v blízkosti kontrolného okienka. Týmto sa všetky vzduchové bublinky dostanú do hornej časti.
Odstráňte krytNaplnené pero držte v zvislej polohe, potom otočte a vytiahnite kryt, aby ste ho odstránili.
DÔLEŽITÉ: Nestláčajte oranžový chránič ihly pred podaním injekcie. Chránič sa uzamkne a vaša dávka nebude podaná.
Po odstránení krytu podajte injekciu do 5 minút.Kryt
nedávajte späť, môže sa tým poškodiť skrytá injekčná ihla. Naplnené pero
nepoužívajte, ak vám spadne, keď kryt nie je nasadený. Zavolajte lekárovi alebo lekárnikovi pre nové naplnené pero.
Oranžový
p
rúžok na nastavenie pera
POTOM
 Odstráňte vzduchové bublinky*
Odstráňte vzduchové bublinky*
Naplnené pero držte v zvislej polohe.
Palcom jemne zatlačte piest, kým sa nezastaví. Roztok vystriekne von. To je normálne.
Oranžový prúžok na nastavenie pera zmizne.*Odstránenie vzduchových bubliniek pomôže zaistiť podanie správnej dávky.Po odstránení vzduchových bubliniek môžete v kontrolnom okienku vidieť čiarku. To je normálne.
2. Podajte Simponi pomocou naplneného pera
Zárez na
zvolenie
 dávky
Nastavte predpísanú dávku
dávky
Nastavte predpísanú dávku
Otáčajte piestom dovtedy, kým sa označenie pre vašu predpísanú dávku nezarovná so zárezom na zvolenie dávky. Naplnené pero je teraz pripravené na použitie.
Zvoliteľné dávky:0,1 ml
0,15 ml
0,2 ml
0,25 ml
0,3 ml
0,35 ml
0,4 ml
0,45 ml
POTOMVpichnite ihlu a pridržte na miesteDÔLEŽITÉ: Nedvíhajte naplnené pero z kože počas podávania injekcie. Oranžový chránič ihly sa uzamkne a nebude vám podaná celá dávka.
Nestláčajte piest počas zavádzania injekčnej ihly.
Zatlačte a pridržte špičku naplneného pera oproti koži tak, aby sa oranžový chránič ihly nadvihol, až kým sa nezastaví. Stále bude vidieť oranžovú farbu.
POTOM
Podajte injekciu Simponi
Naplnené pero držte stlačené oproti koži. Jemne zatlačte na piest, kým sa nezastaví.
Ak je nastavená nízka dávka, piest sa pohne len o malý kúsok.Dávku, ktorú podáte, si môžete skontrolovať pohľadom na zárez so zvolenou dávkou.

Naplnené pero
ešte nedvíhajte.
Držte ho stlačené a potom zdvihnite.Naplnené pero držte stlačené oproti koži počas približne 5 sekúnd.
Je to normálne, ak budete vidieť nejaké množstvo lieku v kontrolnom okienku. Naplnené pero zdvihnite z kože.
Oranžový chránič ihly sa predĺži a uzamkne.
 3. Po podaní injekcie
Použité naplnené pero vyhoďte.
3. Po podaní injekcie
Použité naplnené pero vyhoďte.
Použité naplnené pero ihneď po podaní vyhoďte do nádoby na ostré predmety.

Keď je nádoba plná, zabezpečte jej likvidáciu podľa pokynov svojho lekára alebo zdravotnej sestry.
Skontrolujte miesto podania injekcieV mieste podania injekcie sa môže objaviť malé množstvo krvi alebo tekutiny. Pritlačte vatový tampón alebo gázový štvorček na kožu, kým sa krvácanie nezastaví. Miesto podania injekcie
nešúchajte.Ak je to potrebné, miesto podania injekcie prekryte obväzom. Podanie vašej injekcie je dokončené!
NÁVO
D NA POUŽITIE
Ak si chcete sami podať Simponi, musíte byť zaškolený zdravotníckym pracovníkom na prípravu injekcie a jej podanie sebe samému. Ak ste neboli zaškolený, požiadajte o to svojho lekára, zdravotnú sestru alebo lekárnika a naplánujte si s nimi termín školenia.
V tomto návode:
1. Príprava naplneného pera na použitie
2. Výber a príprava miesta podania
3. Podávanie lieku
4. Po podaní injekcie
Nižšie uvedená schéma (pozri obrázok 1) znázorňuje, ako vyzerá naplnené pero „SmartJect“.
Kryt

Obrázok 1
1. Príprava naplneného pera na použitie
· Naplnené pero nikdy netraste.
· Neodstraňujte kryt z naplneného pera skôr, ako bezprostredne pred podaním injekcie.
Skontrolujte počet naplnených pier
Skontrolujte naplnené perá, aby ste sa uistili,
· že je počet naplnených pier a síl správny.
o Ak je vaša dávka 50 mg, dostanete jedno 50 mg naplnené pero
o Ak je vaša dávka 100 mg, dostanete dve 50 mg naplnené perá a bude potrebné, aby ste si
podali dve injekcie. Na tieto injekcie si vyberte dve odlišné miesta (napr. jedna injekcia
do pravého stehna a druhá injekcia do ľavého stehna) a injekcie si podajte hneď po sebe.
o Ak je vaša dávka 200 mg, dostanete štyri 50 mg naplnené perá a bude potrebné, aby ste si podali štyri injekcie. Na tieto injekcie si vyberte odlišné miesta a injekcie si podajte hneď po sebe.
Skontrolujte dátum exspirácie
· Skontrolujte dátum exspirácie vytlačený alebo napísaný na škatuli.
· Skontrolujte dátum exspirácie na naplnenom pere (vyznačený skratkou „EXP“).
· V prípade, že dátum exspirácie vypršal, naplnené pero nepoužívajte. Vytlačený dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci. Požiadajte, prosím, lekára alebo lekárnika o pomoc.
Skontrolujte bezpečnostnú plombu· Skontrolujte bezpečnostnú plombu okolo krytu naplneného pera.
· Ak je bezpečnostná plomba porušená, naplnené pero nepoužívajte. Vyhľadajte, prosím, lekára alebo lekárnika.
Počkajte 30 minút, aby naplnené pero dosiahlo izbovú teplotu· Na zabezpečenie správneho podania injekcie nechajte naplnené pero pri izbovej teplote vybraté z papierovej škatule 30 minút mimo dosahu detí.
· Naplnené pero žiadnym iným spôsobom nezohrievajte (napríklad nezohrievajte ho v mikrovlnnej rúre ani v horúcej vode).
· Kryt naplneného pera neodstraňujte, kým pero nedosiahne izbovú teplotu.
Pripravte si zvyšné pomôcky· Kým čakáte, môžete si pripraviť zvyšné pomôcky, ako sú alkoholový tampón, vata alebo gáza a nádoba na ostré predmety.
Skontrolujte roztok vo vnútri naplneného pera· Cez kontrolné okienko sa uistite, či roztok v naplnenom pere je číry až mierne opaleskujúci (lesknúci sa ako perla) a bezfarebný až svetložltý. Roztok môžete použiť, ak obsahuje malé množstvo malých priehľadných alebo bielych čiastočiek bielkovín.
· Môžete tiež pozorovať vzduchovú bublinu, čo je normálne.
· Nepoužívajte naplnené pero, ak má roztok nesprávnu farbu, je zakalený alebo obsahuje väčšie častice. Ak sa tak stane, poraďte sa so svojím lekárom alebo lekárnikom.
2. Výber a príprava miesta podania (pozri obrázok 2)· Zvyčajne si budete liek podávať spredu do strednej časti stehna.
· Môžete tiež použiť brušnú oblasť (abdómen) pod pupkom, s výnimkou plochy približne 5 cm priamo pod pupkom.
· Nepodávajte do miest, kde je koža citlivá, otlačená, červená, šupinatá, tvrdá alebo kde je koža s jazvami či striami.
· Ak sa na jedno podanie vyžaduje viacero injekcií, injekcie sa majú podať do rôznych miest na tele.
Obrázok 2
Výber miesta podania pre ošetrovateľov, ak si injekciu nepodávate sami (pozri obrázok 3)· Ak vám injekciu podáva ošetrovateľ, môže tiež použiť hornú vonkajšiu časť ramena.
· Opäť sa môžu použiť všetky spomenuté miesta nezávisle od typu alebo veľkosti vášho tela.
Obrázok 3
Príprava miesta podania· Dôkladne si umyte ruky mydlom a teplou vodou.
· Potrite miesto podania alkoholovým tampónom.
· Pred podaním nechajte miesto vpichu oschnúť. Vyčistené miesto neovievajte, ani naň nefúkajte.
· Nedotýkajte sa tohto miesta pred podaním injekcie.
3. Podávanie lieku· Kryt sa nesmie odstrániť, pokiaľ nie ste pripravený na podanie injekcie.
· Liek sa musí podať v priebehu 5 minút po odstránení krytu.
Odstráňte kryt (pozri obrázok 4)· Keď ste pripravený na injekciu, jemne otočte krytom, aby ste porušili bezpečnostnú plombu.
· Odstráňte kryt a po podaní injekcie ho vyhoďte.
· Kryt nedávajte naspäť, pretože môže poškodiť ihlu vo vnútri naplneného pera.
· Naplnené pero nepoužívajte, ak spadne bez nasadeného krytu. Ak sa tak stane, prosím, kontaktujte svojho lekára alebo lekárnika.
Obrázok 4
Naplnené pero pevne pritlačte na kožu (pozri obrázky 5 a 6)· Naplnené pero držte pohodlne v ruke. Tlačidlo teraz
NESTLÁČAJTE.
· Vyberiete si z dvoch metód vpichu. Odporúčaný je vpich bez vytvorenia kožnej riasy (obrázok 5a). Ak však chcete, môžete si vytvoriť kožnú riasu na spevnenie miesta vpichu (obrázok 5b).
· Pod uhlom 90 stupňov zatlačte otvorený koniec naplneného pera pevne proti koži, kým bezpečnostná manžeta úplne neskĺzne do priehľadného krytu (obrázok 6).
Obrázok 5a Obrázok 5b
Obrázok 6
Stlačte tlačidlo, aby sa injekcia podala (pozri obrázok 7)·
Naplnené pero stále pevne pritláčajte kolmo na kožu a prstami alebo palcom stlačte vyvýšenú časť tlačidla. Tlačidlo nebudete môcť stlačiť, ak naplnené pero nie je
pevnepritlačené na kožu a bezpečnostná manžeta neskĺzne do priehľadného krytu.
· Po stlačení zostane tlačidlo zatlačené a už nemusíte vyvíjať ďalší tlak.

Obrázok 7
·
Budete počuť hlasný „klik“- neľakajte sa. Prvý „klik“ znamená, že ihla je vpichnutá a začalo sa podávanie. V tomto momente môžete aj nemusíte pocítiť pichnutie ihly.
Zatiaľ naplnené pero nevyťahujte z kože. Ak naplnené pero z kože vytiahnete, nemusíte dostať celú dávku lieku.Naplnené pero držte do druhého „kliku“ (pozri obrázok 8)·
Naplnené pero ďalej pevne držte kolmo na koži, pokiaľ nebudete počuť druhý „klik“.Toto zvyčajne trvá 3-6 sekúnd, no môže to trvať aj 15 sekúnd, kým budete počuť druhý„klik“.· Druhý „klik“ znamená, že podanie injekcie bolo ukončené a ihla sa automaticky vsunula naspäť do naplneného pera. Ak máte sluchové ťažkosti, po prvom stlačení tlačidla počítajte 15 sekúnd
a potom zdvihnite naplnené pero z miesta podania injekcie.
· Zdvihnite naplnené pero z miesta podania injekcie.
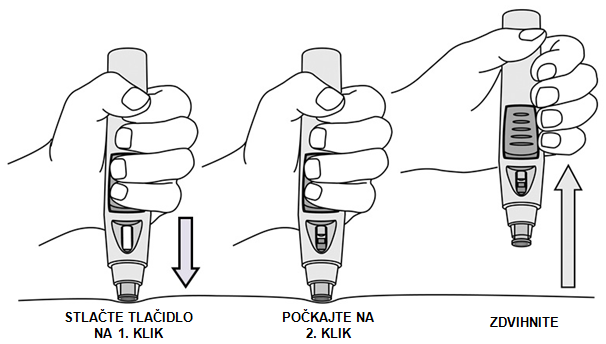
Obrázok 8
4. Po podaní injekciePoužite vatový tampón alebo gázu· V mieste podania injekcie sa môže objaviť malé množstvo krvi alebo tekutiny. Je to normálne.
· Pritlačte vatový tampón alebo gázu na miesto podania injekcie na 10 sekúnd.
· Ak je potrebné, miesto podania injekcie môžete prekryť náplasťou.
· Kožu nešúchajte.
Skontrolujte okienko – žltý indikátor potvrdzuje správne podanie (pozri obrázok 9)· Žltý indikátor je spojený s piestom naplneného pera. Ak sa žltý indikátor v okienku nezobrazuje, piest sa dostatočne neposunul a injekcia sa nepodala.
· Žltý indikátor vyplní približne polovicu kontrolného okienka. Je to normálne.
· Ak v okienku nie je viditeľný žltý indikátor alebo ak si myslíte, že ste nedostali celú dávku, povedzte to svojmu lekárovi alebo lekárnikovi. Nepodávajte si druhú dávku bez porady so
svojím lekárom.
Obrázok 9
N
aplnené pero vyhoďte (pozri obrázok 10)
· Použité pero hneď vyhoďte do nádoby na ostré predmety. Keď je nádoba plná, zabezpečte jej likvidáciu podľa pokynov svojho lekára alebo zdravotnej sestry.
Ak máte pocit, že niečo s podaním injekcie nebolo v poriadku alebo si nie ste istý, povedzte to svojmu lekárovi alebo lekárnikovi.
Obrázok 10
Písomná informácia pre používateľa
Simponi 50 mg injekčný roztok naplnený v injekčnej striekačke
golimumab
Pozorne si prečítajte celú písomnú informáciu predtým, ako začnete používať tento liek, pretože obsahuje pre vás dôležité informácie.
- Túto písomnú informáciu si uschovajte. Možno bude potrebné, aby ste si ju znovu prečítali.
- Ak máte akékoľvek ďalšie otázky, obráťte sa na svojho lekára, lekárnika alebo zdravotnú sestru.
- Tento liek bol predpísaný iba vám. Nedávajte ho nikomu inému. Môže mu uškodiť, dokonca aj vtedy, ak má rovnaké prejavy ochorenia ako vy.
- Ak sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára, lekárnika alebo
zdravotnú sestru. To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej informácii. Pozri časť 4.
Váš lekár vám tiež dá kartu s pripomienkami pre pacienta, ktorá obsahuje dôležité informácie o bezpečnosti, ktoré je potrebné si uvedomiť pred a počas liečby Simponi.
V tejto písomnej informácii sa dozviete:
1. Čo je Simponi a na čo sa používa
2. Čo potrebujete vedieť predtým, ako použijete Simponi
3. Ako používať Simponi
4. Možné vedľajšie účinky
5. Ako uchovávať Simponi
6. Obsah balenia a ďalšie informácie
1. Čo je Simponi a na čo sa používa
Simponi obsahuje liečivo nazývané golimumab.
Simponi patrí do skupiny liekov nazývaných „TNF-blokátory“. Používa sa u dospelých na liečbu nasledujúcich zápalových ochorení:
· Reumatoidná artritída
· Psoriatická artritída
· Axiálna spondyloartritída vrátane ankylozujúcej spondylitídy a axiálnej spondyloartritídy bez rádiografického dôkazu
· Ulcerózna kolitída
U detí vo veku 2 rokov a starších sa Simponi používa na liečbu polyartikulárnej juvenilnej idiopatickej artritídy.
Simponi pôsobí blokovaním účinku bielkoviny nazývanej „tumor nekrotizujúci faktor alfa“ (TNF-α). Táto bielkovina sa podieľa na zápalových procesoch v tele a jej blokovanie môže znížiť zápal vo vašom tele.
Reumatoidná artritída
Reumatoidná artritída je zápalové ochorenie kĺbov. Ak máte aktívnu reumatoidnú artritídu, najprv vám
dajú iné lieky. Ak na tieto lieky nebudete dostatočne dobre reagovať, môžu vám podať Simponi, ktorý budete používať v kombinácii s iným liekom nazývaným metotrexát na:
· zmiernenie prejavov a príznakov vašej choroby,
· spomalenie poškodenia vašich kostí a kĺbov,
· zlepšenie vašich telesných funkcií.
Psoriatická artritída
Psoriatická artritída je zápalové ochorenie kĺbov, ktoré je zvyčajne sprevádzané psoriázou, zápalovým ochorením kože. Ak máte aktívnu psoriatickú artritídu, najprv vám dajú iné lieky. Ak na tieto lieky
nebudete dostatočne dobre reagovať, môžu vám podať Simponi na:
· zmiernenie prejavov a príznakov vašej choroby,
· spomalenie poškodenia vašich kostí a kĺbov,
· zlepšenie vašich telesných funkcií.
Ankylozujúca spondylitída a axiálna spondyloartritída bez rádiografického dôkazu Ankylozujúca spondylitída a axiálna spondyloartritída bez rádiografického dôkazu sú zápalové ochorenia chrbtice. Ak máte ankylozujúcu spondylitídu alebo axiálnu spondyloartritídu bez rádiografického dôkazu, najprv vám dajú iné lieky. Ak na tieto lieky nebudete dostatočne dobre reagovať, môžu vám podať Simponi na:
· zmiernenie prejavov a príznakov vašej choroby,
· zlepšenie vašich telesných funkcií.
Ulcerózna kolitída
Ulcerózna kolitída je zápalové ochorenie čreva. Ak máte ulceróznu kolitídu, najprv vám dajú iné
lieky. Ak na tieto lieky nebudete dostatočne dobre reagovať, na liečbu vášho ochorenia dostanete
Simponi.
Polyartikulárna juvenilná idiopatická artritída
Polyartikulárna juvenilná idiopatická artritída je zápalové ochorenie, ktoré spôsobuje bolesť a opuch
kĺbov u detí. Ak máte polyartikulárnu juvenilnú idiopatickú artritídu, najprv vám podajú iné lieky. Ak nebudete na tieto lieky dostatočne dobre reagovať, na liečbu ochorenia vám podajú Simponi
v kombinácii s metotrexátom.
2. Čo potrebujete vedieť predtým, ako použijete Simponi
Nepoužívajte Simponi:
· ak ste alergický (precitlivený) na golimumab alebo na ktorúkoľvek z ďalších zložiek tohto lieku
(uvedených v časti 6),
· ak máte tuberkulózu (TBC) alebo akúkoľvek inú závažnú infekciu,
· ak máte stredne závažné alebo závažné zlyhávanie srdca.
Ak si nie ste istý, či sa vás niečo z vyššie uvedeného týka, opýtajte sa svojho lekára, lekárnika alebo zdravotnej sestry pred použitím Simponi.
Upozornenia a opatrenia
Predtým, ako začnete používať Simponi, obráťte sa na svojho lekára, lekárnika alebo zdravotnú sestru.
Infekcie
Okamžite povedzte svojmu lekárovi, ak máte alebo sa u vás počas liečby Simponi alebo po jej ukončení objavia akékoľvek príznaky infekcie. Príznaky infekcie zahŕňajú horúčku, kašeľ, dýchavičnosť, príznaky podobné chrípke, hnačku, rany, ťažkosti so zubami alebo pocit pálenia pri močení.
· Počas používania Simponi môžete ľahšie dostať infekcie.
· Infekcie sa môžu vyvíjať rýchlejšie a môžu byť závažnejšie. Okrem toho sa môžu znovu objaviť infekcie prekonané v minulosti.
Tuberkulóza (TBC)
Okamžite povedzte svojmu lekárovi, ak sa u vás objavia príznaky TBC v priebehu liečby alebo po liečbe. Príznaky TBC zahŕňajú pretrvávajúci kašeľ, úbytok telesnej hmotnosti, únavu,
horúčku alebo nočné potenie.
· U pacientov liečených Simponi sa hlásili prípady TBC, v zriedkavých prípadoch aj
u pacientov liečených na TBC liekmi. Váš lekár vás vyšetrí, aby zistil, či máte TBC. Váš lekár bude zaznamenávať tieto vyšetrenia do vašej karty s pripomienkami pre pacienta.
· Je veľmi dôležité, aby ste povedali svojmu lekárovi, ak ste niekedy mali TBC alebo ste boli v bezprostrednom kontakte s niekým, kto mal alebo má TBC.
· Ak sa vášmu lekárovi zdá, že vám hrozí riziko TBC, môže vás pred použitím Simponi liečiť liekmi na TBC.
Vírusová hepatitída B (HBV)
· Pred podaním Simponi povedzte svojmu lekárovi, ak ste nositeľom alebo ak máte alebo ste mali HBV.
· Povedzte svojmu lekárovi, ak si myslíte, že môžete mať riziko spojené s prenosom HBV.
· Váš lekár vás má vyšetriť na HBV.
· Liečba TNF-blokátormi, ako je Simponi, môže vyvolať reaktiváciu HBV u pacientov, ktorí sú nositeľmi tohto vírusu, čo môže byť v niektorých prípadoch život ohrozujúce.
Invazívne hubové infekcie
Okamžite povedzte svojmu lekárovi, ak ste žili alebo cestovali do oblastí, kde sú bežné infekcie spôsobené špecifickými druhmi húb (nazývané histoplazmóza, kokcidioidomykóza alebo
blastomykóza), ktoré môžu zasiahnuť pľúca alebo iné časti tela. Spýtajte sa svojho lekára, ak
neviete, či sú tieto hubové infekcie bežné v oblasti, kde ste žili alebo kam ste cestovali.
Rakovinaa lymfóm
Povedzte svojmu lekárovi, ak vám niekedy diagnostikovali lymfóm (typ rakoviny krvi) alebo akúkoľvek inú rakovinu, pred použitím Simponi.
· Ak používate Simponi alebo iné TNF-blokátory, môže sa zvýšiť vaše riziko rozvoja lymfómu alebo inej rakoviny.
· Pacienti so závažnou reumatoidnou artritídou a inými zápalovými ochoreniami, ktorí majú toto ochorenie dlhú dobu, môžu mať vyššie než priemerné riziko vzniku lymfómu.
· U detí a dospievajúcich pacientov, ktorí dostávali TNF-blokátory, sa objavili prípady rakoviny vrátane neobvyklých typov, ktoré niekedy viedli k smrti.
· V zriedkavých prípadoch sa u pacientov, ktorí používali iné blokátory TNF, pozoroval osobitný a závažný typ lymfómu nazývaný hepatosplenický lymfóm T-buniek. Väčšina týchto pacientov
boli dospievajúci alebo mladí dospelí muži. Tento typ rakoviny sa zvyčajne končil smrťou.
Takmer všetci títo pacienti tiež užívali liečivá známe ako azatioprin alebo 6-merkaptopurín. Ak so Simponi užívate azatioprin alebo 6-merkaptopurín, povedzte to svojmu lekárovi.
· Pacienti so závažnou pretrvávajúcou astmou, chronickou obštrukčnou chorobou pľúc
(CHOCHP) alebo silní fajčiari môžu mať zvýšené riziko rakoviny pri liečbe Simponi. Ak máte závažnú pretrvávajúcu astmu, CHOCHP alebo ste silným fajčiarom, prediskutujte so
svojím lekárom, či je liečba TNF-blokátormi pre vás vhodná.
· U niektorých pacientov liečených golimumabom sa objavili určité typy rakoviny kože. Ak sa počas liečby alebo po liečbe objavia akékoľvek zmeny vo vzhľade pokožky alebo výrastky na koži, povedzte to svojmu lekárovi.
Zlyhávanie srdca
Ak sa u vás vyskytnú nové alebo sa zhoršia príznaky zlyhávania srdca, okamžite to povedzte svojmu lekárovi. Príznaky zlyhávania srdca zahŕňajú dýchavičnosť alebo opuch chodidiel.
· Pri TNF-blokátoroch vrátane Simponi sa hlásilo nové a zhoršujúce sa kongestívne zlyhávanie srdca. Niektorí z týchto pacientov zomreli.
· Ak máte mierne zlyhávanie srdca a ste liečený Simponi, musíte byť pozorne sledovaný vaším lekárom.
Ochorenie nervového systému
Ak vám niekedy diagnostikovali alebo ste mali príznaky demyelinizačného ochorenia, ako je skleróza multiplex, okamžite to povedzte svojmu lekárovi. Príznaky môžu zahŕňať zmeny vášho zraku, slabosť
vašich rúk alebo nôh, či necitlivosť alebo mravčenie v ktorejkoľvek časti vášho tela. Váš lekár rozhodne, či máte používať Simponi.
Chirurgické zákroky alebo zákroky na zuboch
· Povedzte svojmu lekárovi, ak sa chystáte podstúpiť akékoľvek chirurgické zákroky alebo zákroky na zuboch.
· Povedzte svojmu chirurgovi alebo zubárovi, ktorý vykonáva zákrok, že sa liečite Simponi a ukážte mu vašu kartu s pripomienkami pre pacienta.
Autoimunitné ochorenie
Povedzte svojmu lekárovi, ak sa u vás vyvinú príznaky ochorenia nazývaného lupus. Príznaky zahŕňajú pretrvávajúcu vyrážku, horúčku, bolesť kĺbov a únavu.
· V zriedkavých prípadoch sa u ľudí, liečených TNF-blokátormi, rozvinul lupus.
Ochorenie krvi
U niektorých pacientov môže telo prestať vytvárať dostatok krviniek, ktoré pomáhajú vášmu telu bojovať proti infekciám alebo pomáhajú zastaviť krvácanie. Ihneď povedzte svojmu lekárovi, ak sa
u vás rozvinie horúčka, ktorá neustupuje, ak sa vám rýchlo tvoria modriny, ľahko sa spúšťa krvácanie
alebo ak vyzeráte veľmi bledo. Váš lekár sa môže rozhodnúť liečbu ukončiť.
Ak si nie ste istý, či sa vás niečo z vyššie uvedeného týka, pred použitím Simponi sa opýtajte svojho lekára alebo lekárnika.
Očkovania
Povedzte svojmu lekárovi, ak ste boli očkovaný alebo sa chystáte na očkovanie.
· Počas používania Simponi nesmiete byť očkovaný určitými (živými) vakcínami.
· Niektoré očkovania môžu spôsobiť infekcie. Ak ste dostávali Simponi počas tehotenstva, vaše dieťa môže mať vyššie riziko vzniku takejto infekcie približne až do šiestich mesiacov od poslednej dávky, ktorú ste dostali počas tehotenstva. Je dôležité, aby ste povedali lekárom
svojho dieťaťa a ďalším zdravotníckym pracovníkom o vašom používaní Simponi, aby mohli
rozhodnúť, kedy má vaše dieťa dostať ktorúkoľvek vakcínu.
Porozprávajte sa s lekárom vášho dieťaťa o jeho očkovaniach. Ak je to možné, vaše dieťa má mať pred použitím Simponi absolvované všetky očkovania.
Infekčné látky na liečebné účely
Ak ste v nedávnej minulosti dostali alebo je u vás naplánované, že dostanete liečbu infekčnou látkou na liečebné účely (ako je napr. podanie BCG používané na liečbu rakoviny), porozprávajte sa so
svojím lekárom.
Alergické reakcie
Okamžite povedzte svojmu lekárovi, ak sa u vás po liečbe Simponi vyvinú príznaky alergickej reakcie. Príznaky alergickej reakcie môžu zahŕňať opuch tváre, pier, úst alebo hrdla, ktoré môžu spôsobiť
ťažkosti s prehĺtaním alebo s dýchaním, kožnú vyrážku, žihľavku, opuch rúk, chodidiel alebo členkov.
· Niektoré z týchto reakcií môžu byť závažné alebo, zriedkavo, život ohrozujúce.
· Niektoré z týchto reakcií sa vyskytli po prvom podaní Simponi.
Deti
Simponi sa neodporúča pre deti s polyartikulárnou juvenilnou idiopatickou artritídou mladšie ako
2 roky, pretože sa v tejto vekovej skupine neskúmal.
Iné lieky a Simponi
· Ak teraz používate, alebo ste v poslednom čase používali, či práve budete používať ďalšie lieky vrátane akýchkoľvek iných liekov na liečbu reumatoidnej artritídy, polyartikulárnej juvenilnej
idiopatickej artritídy, psoriatickej artritídy, ankylozujúcej spondylitídy, axiálnej
spondyloartritídy bez rádiografického dôkazu alebo ulceróznej kolitídy, povedzte to svojmu lekárovi alebo lekárnikovi.
· Nesmiete používať Simponi s liekmi obsahujúcimi liečivo anakinra alebo abatacept. Tieto lieky sa používajú pri liečbe reumatických chorôb.
· Ak užívate akékoľvek iné lieky, ktoré ovplyvňujú váš imunitný systém, povedzte to svojmu lekárovi alebo lekárnikovi.
· Počas používania Simponi nesmiete byť očkovaný určitými (živými) vakcínami.
Ak si nie ste istý, či sa vás niečo z vyššie uvedeného týka, pred použitím Simponi sa opýtajte svojho lekára alebo lekárnika.
Tehotenstvo a dojčenie
Pred použitím Simponi povedzte lekárovi ak:
· Ste tehotná alebo plánujete otehotnieť počas používania Simponi. Účinky tohto lieku nie sú u tehotných žien známe. Používanie Simponi u tehotných žien sa neodporúča. Ak sa liečite Simponi, musíte zabrániť otehotneniu používaním vhodnej antikoncepcie počas liečby
a najmenej 6 mesiacov po poslednej injekcii Simponi.
· Pred začatím dojčenia musí byť vaša posledná liečba Simponi ukončená najmenej pred
6 mesiacmi. Ak budete liečená Simponi, musíte prestať dojčiť.
· Ak ste dostávali Simponi počas tehotenstva, vaše dieťa môže mať vyššie riziko vzniku infekcie.
Je dôležité, aby ste povedali lekárom svojho dieťaťa a ďalším zdravotníckym pracovníkom
o vašom používaní Simponi predtým, ako dieťa dostane ktorúkoľvek vakcínu (viac informácií pozri v časti o očkovaní).
Ak ste tehotná alebo dojčíte, ak si myslíte, že ste tehotná alebo ak plánujete otehotnieť, poraďte sa so svojím lekárom alebo lekárnikom predtým, ako začnete používať tento liek.
Vedenie vozidiel a obsluha strojov
Simponi má malý vplyv na schopnosť viesť vozidlá a obsluhovať nástroje či stroje. Po použití
Simponi sa však môže objaviť závrat. Ak sa to stane, neveďte vozidlá, nepoužívajte žiadne nástroje ani neobsluhujte stroje.
Simponi obsahuje latex a sorbitol
Precitlivenosť na latex
Časť naplnenej injekčnej striekačky, kryt ihly, obsahuje latex. Pretože latex môže vyvolať závažné alergické reakcie, pred použitím Simponi povedzte svojmu lekárovi, ak ste vy alebo váš ošetrovateľ alergický na latex.
Neznášanlivosť sorbitolu
Tento liek obsahuje 20,5 mg sorbitolu (E420) v každej naplnenej injekčnej striekačke.
3. Ako používať Simponi
Vždy používajte tento liek presne tak, ako vám povedal váš lekár alebo lekárnik. Ak si nie ste niečím istý, overte si to u svojho lekára alebo lekárnika.
Aké je dávkovanie Simponi
Reumatoidná artritída, psoriatická artritída a axiálna spondyloartritída vrátane ankylozujúcej
spondylitídy a axiálnej spondyloartritídy bez rádiografického dôkazu:
· Odporúčaná dávka je 50 mg (obsah 1 naplnenej injekčnej striekačky), podaná raz mesačne, vždy v rovnaký deň v mesiaci.
· Pred podaním štvrtej dávky sa porozprávajte so svojím lekárom. Váš lekár rozhodne, či máte pokračovať v liečbe Simponi.
o Ak je vaša telesná hmotnosť vyššia ako 100 kg, dávka sa môže zvýšiť na 100 mg (obsah
2 naplnených injekčných striekačiek), podaná raz mesačne, vždy v rovnaký deň
v mesiaci.
Polyartikulárna juvenilná idiopatická artritída
· Pre pacientov s telesnou hmotnosťou nejmenej 40 kg je odporúčaná dávka 50 mg, podaná raz mesačne, vždy v rovnaký deň v mesiaci. Pre pacientov s telesnou hmotnosťou nižšou ako 40 kg je dostupné 45 mg/0,45 ml naplnené pero. Váš lekár vám povie správnu dávku.
· Pred podaním štvrtej dávky sa porozprávajte s vaším lekárom. Váš lekár rozhodne, či máte pokračovať v liečbe Simponi.
Ulcerózna kolitída
· Tabuľka nižšie uvádza spôsob, akým budete tento liek zvyčajne používať.
Úvodná liečba Úvodná dávka 200 mg (obsah 4 naplnených injekčných striekačiek), po ktorej o 2 týždne neskôr nasleduje 100 mg (obsah 2 naplnených injekčných striekačiek).

Udržiavacia liečba · U pacientov s telesnou hmotnosťou nižšou ako 80 kg, 50 mg (obsah 1 naplnenej injekčnej striekačky) 4 týždne po vašej poslednej liečbe, následne potom každé 4 týždne. Na základe toho, ako dobre u vás Simponi účinkuje, sa váš lekár môže rozhodnúť predpísať vám 100 mg (obsah 2 naplnených striekačiek).
· U pacientov s telesnou hmotnosťou 80 kg alebo viac, 100 mg (obsah 2 naplnených injekčných striekačiek) 4 týždne po vašej poslednej liečbe, následne potom každé 4 týždne.
Ako sa Simponi podáva· Simponi sa podáva injekciou pod kožu (subkutánne).
· Na začiatku vám Simponi podá váš lekár alebo zdravotná sestra. Vy a váš lekár sa však môžete rozhodnúť, že si budete podávať Simponi sami. V tomto prípade absolvujete školenie, ako si
sami podať Simponi.
Opýtajte sa vášho lekára, ak máte akékoľvek otázky o samopodávaní injekcie. Na konci tejto písomnej informácie nájdete podrobný „Návod na použitie“.
Ak použijete viac Simponi, ako máteAk ste použili alebo ste dostali príliš veľa Simponi (buď ste si podali príliš veľa jednorazovo alebo
príliš častým podávaním), okamžite o tom informujte svojho lekára alebo lekárnika. Majte vždy pri sebe túto písomnú informáciu a vonkajšiu škatuľu, aj keď je prázdna.
Ak zabudnete použiť SimponiAk zabudnete použiť Simponi v plánovanom termíne, podajte si zabudnutú dávku hneď, ako si
spomeniete.
Nepoužívajte dvojnásobnú dávku, aby ste nahradili vynechanú dávku. Kedy si podať ďalšiu dávku:
· Ak sa oneskoríte s podaním o menej ako 2 týždne, podajte si zabudnutú dávku hneď, ako si
spomeniete a zostaňte pri pôvodnej schéme.
· Ak sa oneskoríte s podaním o viac ako 2 týždne, podajte si zabudnutú dávku hneď, ako si spomeniete a spýtajte sa lekára alebo lekárnika, kedy si máte podať ďalšiu dávku.
Ak si nie ste istý, čo máte robiť, opýtajte sa svojho lekára alebo lekárnika.
Ak prestanete používať SimponiPredtým, ako sa rozhodnete liečbu Simponi ukončiť, poraďte sa najprv so svojím lekárom alebo
lekárnikom.
Ak máte akékoľvek ďalšie otázky, týkajúce sa použitia tohto lieku, opýtajte sa svojho lekára, lekárnika alebo zdravotnej sestry.
4. Možné vedľajšie účinky
Tak ako všetky lieky, aj tento liek môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého. U niektorých pacientov sa môžu vyskytnúť závažné vedľajšie účinky vyžadujúce liečbu. Riziko niektorých vedľajších účinkov je väčšie pri dávke 100 mg v porovnaní s dávkou 50 mg. Vedľajšie účinky sa môžu objaviť aj niekoľko mesiacov po podaní poslednej dávky.
Okamžite povedzte svojmu lekárovi, ak si všimnete ktorýkoľvek z nasledujúcich závažných vedľajších účinkov Simponi, ktoré zahŕňajú:
· alergické reakcie, ktoré môžu byť závažné alebo zriedkavo život ohrozujúce (zriedkavé).
Príznaky alergickej reakcie môžu zahŕňať opuch tváre, pier, úst alebo hrdla, ktoré môžu
spôsobiť ťažkosti s prehĺtaním alebo s dýchaním, kožnú vyrážku, žihľavku, opuch rúk, chodidiel alebo členkov. Niektoré z týchto reakcií sa vyskytli po prvom podaní Simponi.
· závažné infekcie (vrátane TBC, bakteriálnych infekcií zahŕňajúcich závažné infekcie krvi a zápal pľúc, závažných hubových infekcií a iných oportúnnych infekcií) (časté). Príznaky infekcie môžu zahŕňať horúčku, únavu, (pretrvávajúci) kašeľ, dýchavičnosť, príznaky podobné chrípke, úbytok telesnej hmotnosti, nočné potenie, hnačku, rany, problémy so zubami a pocit pálenia pri močení.
· reaktiváciu vírusu hepatitídy B, ak ste nositeľom alebo ste mali predtým hepatitídu B (zriedkavé). Príznaky môžu zahŕňať zožltnutie kože a očí, tmavohnedo sfarbený moč, bolesť brucha na pravej strane, horúčku, pocit nevoľnosti, nevoľnosť a pocit veľkej únavy.
· ochorenie nervového systému, ako je skleróza multiplex (zriedkavé). Príznaky ochorenia nervového systému môžu zahŕňať zmeny vášho zraku, slabosť vašich rúk alebo nôh, či necitlivosť alebo mravčenie v ktorejkoľvek časti vášho tela.
· rakovinu lymfatických uzlín (lymfóm) (zriedkavé). Príznaky lymfómu môžu zahŕňať opuch lymfatických uzlín, úbytok telesnej hmotnosti alebo horúčku.
· zlyhávanie srdca (zriedkavé). Príznaky zlyhávania srdca môžu zahŕňať dýchavičnosť alebo opuch chodidiel.
· prejavy ochorení imunitného systému nazývaných:
- lupus (zriedkavé). Príznaky môžu zahŕňať bolesť kĺbov alebo vyrážku na lícach alebo
rukách, ktorá je citlivá na slnečné žiarenie.
- sarkoidóza (zriedkavé). Príznaky môžu zahŕňať pretrvávajúci kašeľ, dýchavičnosť, bolesť na hrudi, horúčku, opuch lymfatických uzlín, úbytok telesnej hmotnosti, vyrážky
na koži a rozmazané videnie.
· opuch malých krvných ciev (vaskulitída) (zriedkavé). Príznaky môžu zahŕňať horúčku, bolesť hlavy, úbytok telesnej hmotnosti, nočné potenie, vyrážku a problémy s nervami ako sú necitlivosť a mravčenie.
· rakovina kože (menej časté). Príznaky rakoviny kože môžu zahŕňať zmeny vo vzhľade vašej kože alebo výrastky na koži.
· ochorenie krvi (časté). Príznaky ochorenia krvi môžu zahŕňať horúčku, ktorá neustupuje, ľahkú tvorbu modrín alebo ľahko sa spúšťajúce krvácanie alebo ak vyzeráte veľmi bledo.
· rakovina krvi (leukémia) (zriedkavé). Príznaky leukémie môžu zahŕňať horúčku, pocit únavy, časté infekcie, ľahkú tvorbu modrín a nočné potenie.
Okamžite povedzte svojmu lekárovi, ak si všimnete ktorýkoľvek z vyššie uvedených príznakov.
Pri podávaní Simponi sa pozorovali nasledujúce ďalšie vedľajšie účinky:
Veľmi časté vedľajšie účinky (môžu postihovať viac ako 1 z 10 osôb):
· Infekcie horných dýchacích ciest, bolesť hrdla alebo zachrípnutie, nádcha
Časté vedľajšie účinky (môžu postihovať menej ako 1 z 10 osôb):
· Nezvyčajné výsledky pečeňových vyšetrení (zvýšené hladiny pečeňových enzýmov), zistené pri vyšetrení krvi vaším lekárom
· Pocit závratu
· Bolesť hlavy
· Pocit necitlivosti alebo mravčenia
· Povrchové hubové infekcie
· Hnisavé ložisko
· Bakteriálne infekcie (ako je flegmóna)
· Nízky počet červených krviniek
· Nízky počet bielych krviniek
· Pozitívne krvné vyšetrenie na lupus
· Alergické reakcie
· Poruchy trávenia
· Bolesť žalúdka
· Pocit nevoľnosti (nauzea)
· Chrípka
· Zápal priedušiek
· Infekcia dutín
· Jednoduchý opar
· Vysoký krvný tlak
· Horúčka
· Astma, dýchavičnosť, sipot
· Poruchy žalúdka a čriev, ktoré zahŕňajú zápal sliznice žalúdka a hrubého čreva, ktorý môže spôsobiť horúčku
· Bolesť a vriedky v ústach
· Reakcie v mieste podania injekcie (zahŕňajúce sčervenenie, tvrdosť, bolesť, modrinu, svrbenie, pocit brnenia a podráždenie)
· Vypadávanie vlasov
· Vyrážka a svrbenie pokožky
· Ťažkosti so spaním
· Depresia
· Pocit slabosti
· Zlomeniny kostí
· Nepríjemný pocit v hrudníku
Menej časté vedľajšie účinky (môžu postihovať menej ako 1 zo 100 osôb):
· Infekcia obličiek
· Rakoviny vrátane karcinómu kože a nenádorové výrastky alebo hrčky vrátane materských znamienok na koži
· Kožné pľuzgiere
· Závažná infekcia v celom tele (sepsa), niekedy zahŕňajúca nízky krvný tlak (septický šok)
· Psoriáza (vrátane na dlani ruky a/alebo na chodidle nôh a/alebo vo forme kožných pľuzgierov)
· Nízky počet krvných doštičiek
· Združený nízky počet krvných doštičiek, červených a bielych krviniek
· Poruchy štítnej žľazy
· Zvýšenie hladiny cukru v krvi
· Zvýšenie hladiny cholesterolu v krvi
· Poruchy rovnováhy
· Poruchy videnia
· Zápal oka (konjunktivitída)
· Alergia oka
· Pocit nepravidelného tlkotu srdca
· Zúženie krvných ciev v srdci
· Krvné zrazeniny
· Návaly tepla
· Zápcha
· Dlhodobé zápalové ochorenie pľúc
· Spätný návrat kyselín (reflux)
· Žlčové kamene
· Poruchy pečene
· Choroby prsníkov
· Poruchy menštruácie
Zriedkavé vedľajšie účinky (môžu postihovať menej ako 1 z 1 000 osôb):· Zlyhanie kostnej drene pri tvorbe krvných buniek
· Závažne znížený počet bielych krviniek
· Infekcia kĺbov alebo okolitého tkaniva
· Zhoršené hojenie
· Zápal krvných ciev vo vnútorných orgánoch
· Leukémia
· Melanóm (typ rakoviny kože)
· Nádor z Merkelových buniek (typ rakoviny kože)
· Lichenoidné reakcie (svrbiaca červeno-purpurová kožná vyrážka a/alebo vláknité bielosivé čiary na slizniciach)
· Šupinatá, odlupujúca sa koža
· Poruchy obranyschopnosti, ktoré môžu ovplyvniť pľúca, kožu a lymfatické uzliny (najčastejšie sa prejavujúce ako sarkoidóza)
· Bolesť a zmena sfarbenia prstov na rukách alebo na nohách
· Poruchy chuti
· Poruchy močového mechúra
· Poruchy obličiek
· Zápal krvných ciev v koži, čo vedie k vyrážke
Vedľajšie účinky, ktorých častosť nie je známa:· Zriedkavý typ rakoviny krvi postihujúci najmä mladých ľudí (hepatosplenický lymfóm T- buniek)
Hlásenie vedľajších účinkovAk sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára, lekárnika alebo zdravotnú sestru. To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej

informácii. Vedľajšie účinky môžete hlásiť aj priamo na národné centrum hlásenia uvedené
v
Prílohe V. Hlásením vedľajších účinkov môžete prispieť k získaniu ďalších informácií o bezpečnosti tohto lieku.
5. Ako uchovávať Simponi· Tento liek uchovávajte mimo dohľadu a dosahu detí.
· Nepoužívajte tento liek po dátume exspirácie, ktorý je uvedený na štítku a na škatuli po „EXP“.
Dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci.
· Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
· Naplnenú injekčnú striekačku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
· Tento liek sa môže uchovávať aj mimo chladničky pri teplotách do maximálne 25 °C počas jedného obdobia až do 30 dní, nesmie však presiahnuť pôvodný dátum exspirácie, ktorý je
vytlačený na škatuli. Nový dátum exspirácie zahŕňajúci deň/mesiac/rok napíšte na škatuľu
(najviac 30 dní po vybratí lieku z chladničky). Nevracajte tento liek späť do chladničky, ak dosiahol izbovú teplotu. Zlikvidujte tento liek, ak sa nepoužil do konca nového dátumu exspirácie alebo do dátumu exspirácie vytlačeného na škatuli, podľa toho, ktorý je skôr.
· Nepoužívajte tento liek, ak spozorujete, že roztok nie je bezfarebný až svetložltej farby, je zakalený alebo obsahuje cudzie častice.
· Nelikvidujte lieky odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.
6. Obsah balenia a ďalšie informácie
Čo Simponi obsahuje
Liečivo je golimumab. Jedna 0,5 ml naplnená injekčná striekačka obsahuje 50 mg golimumabu.
Ďalšie zložky sú: sorbitol (E420), histidín, monohydrát histidíniumchloridu, polysorbát 80 a voda na injekcie. Ďalšie informácie o sorbitole (E420), pozri časť 2.
Ako vyzerá Simponi a obsah balenia
Simponi sa dodáva ako injekčný roztok naplnený v injekčnej striekačke na jedno použitie. Simponi je
dostupný v baleniach obsahujúcich 1 naplnenú injekčnú striekačku a viacpočetných baleniach obsahujúcich 3 (3 balenia po 1) naplnené injekčné striekačky. Na trh nemusia byť uvedené všetky veľkosti balenia.
Roztok je číry až mierne opaleskujúci (lesknúci sa ako perla), bezfarebný až svetložltý a môže obsahovať malé množstvo malých priehľadných alebo bielych čiastočiek bielkovín. Simponi sa nesmie použiť pri zmene farby roztoku, zakalení alebo pri obsahu viditeľných cudzích častíc v ňom.
Držiteľ rozhodnutia o registrácii a výrobca
Janssen Biologics B.V.
Einsteinweg 101
2333 CB Leiden
Holandsko
Ak potrebujete akúkoľvek informáciu o tomto lieku, kontaktujte miestneho zástupcu držiteľa rozhodnutia o registrácii:
France
MSD France
Tél: + 33 (0) 1 80 46 40 40
PortugalMerck Sharp & Dohme, Lda
Tel: +351 21 4465700
inform_pt@merck.com
Ísland
Vistor hf.
Sími: + 354 535 7000
Slovenská republikaMerck Sharp & Dohme, s. r. o. Tel: +421 2 58282010
dpoc_czechslovak@merck.com
Italia
MSD Italia S.r.l. Tel: +39 06 361911
medicalinformation.it@merck.com
Suomi/FinlandMSD Finland Oy
Puh/Tel: +358 (0)9 804 650
info@msd.fi
Κύπρος
Merck Sharp & Dohme Cyprus Limited Τηλ.: 800 00 673 (+357 22866700)
cyprus_info@merck.com
SverigeMerck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488
medicinskinfo@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: + 371 67364224
msd_lv@merck.com
United KingdomMerck Sharp & Dohme Limited Tel: +44 (0) 1992 467272
medicalinformationuk@merck.com
Táto písomná informácia bola naposledy aktualizovaná v
NÁVOD NA POUŽITIE
Ak si chcete sami podať Simponi, musíte byť zaškolený zdravotníckym pracovníkom na prípravu injekcie a jej podanie sebe samému. Ak ste neboli zaškolený, požiadajte o to svojho lekára, zdravotnú sestru alebo lekárnika a naplánujte si s nimi termín školenia.
V tomto návode:
1. Príprava naplnenej injekčnej striekačky na použitie
2. Výber a príprava miesta podania
3. Podávanie lieku
4. Po podaní injekcie
Nižšie uvedená schéma (pozri obrázok 1) znázorňuje, ako vyzerá naplnená injekčná striekačka.
Obrázok 1
1. Príprava naplnenej injekčnej striekačky na použitieNaplnenú injekčnú striekačku držte za telo naplnenej injekčnej striekačky· Striekačku nedržte za hlavicu piestu, piest, ochranné krídla ihly alebo kryt ihly.
· Nikdy neťahajte piest.
· Naplnenú injekčnú striekačku nikdy netraste.
· Neodstraňujte kryt ihly z naplnenej injekčnej striekačky skôr, ako je to uvedené v návode.
· Nedotýkajte sa aktivačných spôn chrániča ihly (znázornených hviezdičkami * na obrázku 1), aby ste zabránili predčasnému zakrytiu ihly chráničom.
Skontrolujte počet naplnených injekčných striekačiekSkontrolujte naplnené injekčné striekačky, aby ste sa uistili,
· že je počet naplnených injekčných striekačiek a síl správny.
o Ak je vaša dávka 50 mg, dostanete jednu 50 mg naplnenú injekčnú striekačku
o Ak je vaša dávka 100 mg, dostanete dve 50 mg naplnené injekčné striekačky a bude
potrebné, aby ste si podali dve injekcie. Na tieto injekcie si vyberte dve odlišné miesta
(napr. jedna injekcia do pravého stehna a druhá injekcia do ľavého stehna) a injekcie si podajte hneď po sebe.
o Ak je vaša dávka 200 mg, dostanete štyri 50 mg naplnené injekčné striekačky a bude potrebné, aby ste si podali štyri injekcie. Na tieto injekcie si vyberte odlišné miesta
a injekcie si podajte hneď po sebe.
Skontrolujte dátum exspirácie (pozri obrázok 2)· Skontrolujte dátum exspirácie vytlačený alebo napísaný na škatuli.
· Skontrolujte dátum exspirácie (vyznačený skratkou „EXP“) na štítku pohľadom cez kontrolné okienko, ktoré je umiestnené na tele naplnenej injekčnej striekačky.
· Ak cez kontrolné okienko neuvidíte dátum exspirácie, držte naplnenú injekčnú striekačku za jej telo a otáčajte krytom ihly, kým sa vám v kontrolnom okienku neznázorní dátum exspirácie.
V prípade, že dátum exspirácie vypršal, naplnenú injekčnú striekačku nepoužívajte. Vytlačený dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci. Požiadajte, prosím, lekára alebo lekárnika
o pomoc.
Obrázok 2
Počkajte 30 minút, aby naplnená injekčná striekačka dosiahla izbovú teplotu· Na zabezpečenie správneho podania injekcie, nechajte naplnenú injekčnú striekačku pri izbovej teplote 30 minút mimo papierovej škatule a mimo dosahu detí.
Naplnenú injekčnú striekačku žiadnym iným spôsobom nezohrievajte (napríklad nezohrievajte ju
v mikrovlnnej rúre alebo v horúcej vode).
Kryt ihly naplnenej injekčnej striekačky neodstraňujte, kým striekačka nedosiahne izbovú teplotu.
Pripravte si zvyšné pomôckyKým čakáte, môžete si pripraviť zvyšné pomôcky, ako sú alkoholový tampón, vata alebo gáza
a nádoba na ostré predmety.
Skontrolujte roztok vo vnútri naplnenej injekčnej striekačky· Držte naplnenú injekčnú striekačku za jej telo tak, aby zakrytá ihla smerovala nadol.
· Cez kontrolné okienko naplnenej injekčnej striekačky sa uistite, či roztok v striekačke je číry až mierne opaleskujúci (lesknúci sa ako perla) a bezfarebný až svetložltý. Roztok môžete použiť,
ak obsahuje malé množstvo malých priehľadných alebo bielych čiastočiek bielkovín.
· Ak cez kontrolné okienko roztok neuvidíte, držte naplnenú injekčnú striekačku za jej telo a otáčajte krytom ihly, kým sa roztok neobjaví v kontrolnom okienku (pozri obrázok 2).
Nepoužívajte naplnenú injekčnú striekačku, ak má roztok nesprávnu farbu, je zakalený alebo obsahuje väčšie častice. Ak sa tak stane, poraďte sa so svojím lekárom alebo lekárnikom.
2. Výber a príprava miesta podania (pozri obrázok 3)· Zvyčajne si budete liek podávať spredu do strednej časti stehna.
· Môžete tiež použiť brušnú oblasť (abdómen) pod pupkom, s výnimkou plochy približne 5 cm priamo pod pupkom.
· Nepodávajte do miest, kde je koža citlivá, otlačená, červená, šupinatá, tvrdá alebo kde je koža s jazvami či striami.
· Ak sa na jedno podanie vyžaduje viacero injekcií, injekcie sa majú podať do rôznych miest na tele.
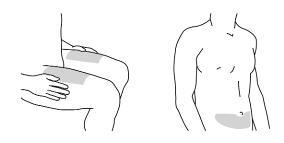
Obrázok 3
Výber miesta podania pre ošetrovateľov (pozri obrázok 4)· Ak vám injekciu podáva ošetrovateľ, môže tiež použiť hornú vonkajšiu časť ramena.
· Opäť sa môžu použiť všetky spomenuté miesta, nezávisle od typu alebo veľkosti vášho tela.
Obrázok 4
Príprava miesta podania· Dôkladne si umyte ruky mydlom a teplou vodou.
· Potrite miesto podania alkoholovým tampónom.
· Pred podaním nechajte miesto vpichu oschnúť. Vyčistené miesto neovievajte, ani naň nefúkajte. Nedotýkajte sa tohto miesta pred podaním injekcie.
3. Podávanie liekuKryt ihly sa nesmie odstrániť, pokiaľ nie ste pripravený na podanie injekcie. Liek sa musí podať v priebehu 5 minút po odstránení krytu ihly.
Pri odstraňovaní krytu ihly sa nedotýkajte piestu.
Odstráňte kryt ihly (pozri obrázok 5)· Ak ste pripravený na podanie injekcie, chyťte telo naplnenej injekčnej striekačky do jednej ruky.
· Odstráňte kryt ihly a po podaní injekcie ho vyhoďte. Kým odstraňujete kryt ihly, nedotýkajte sa piestu.
· V naplnenej striekačke môžete pozorovať vzduchovú bublinu alebo malé množstvo roztoku na konci ihly. Oba sú bežné javy a nemusia byť odstránené.
· Injekciu si podajte ihneď po odstránení krytu ihly.
Nedotýkajte sa ihly a nedotýkajte sa ihlou žiadnych predmetov.
Naplnenú injekčnú striekačku nepoužívajte, ak vám spadne, keď kryt ihly nie je nasadený. Ak sa tak stane, kontaktujte, prosím, svojho lekára alebo lekárnika.
Obrázok 5
Poloha naplnenej injekčnej striekačky pri podaní· Telo naplnenej injekčnej striekačky chyťte prostredníkom a ukazovákom jednej ruky a palec priložte na hlavicu piestu. Druhou rukou si jemne uštipnite očistené miesto podania. Pevne
držte.
Nikdy neťahajte za piest.
Podanie lieku· Ihlu držte približne v 45-stupňovom uhle voči pokožke. Jednoduchým a rýchlym pohybom vpichnite ihlu pod kožu najviac ako je to možné (pozri obrázok 6).
Obrázok 6
· Zatlačte na hlavicu piestu, až kým sa hlavica piestu nedostane medzi ochranné krídla a podajte si tým celý obsah lieku (pozri obrázok 7).
Obrázok 7
· Keď je piest striekačky zatlačený, ako je najviac možné, ešte chvíľu vyvíjajte tlak palcom na hlavicu piestu. Až potom vytiahnite ihlu z kože (pozri obrázok 8).
Obrázok 8
· Pomaly uvoľnite palec z hlavice piestu, aby ste umožnili prázdnej naplnenej injekčnej
striekačke vysunúť sa nahor, kým celá ihla nebude pokrytá chráničom ihly, ako je to znázornené na obrázku 9:
Obrázok 9
4. Po podaní injekciePoužite vatový tampón alebo gázu· V mieste podania injekcie sa môže objaviť malé množstvo krvi alebo tekutiny. Je to normálne.
· Pritlačte vatový tampón alebo gázu na miesto podania injekcie na 10 sekúnd.
· Ak je potrebné, miesto podania injekcie môžete prekryť náplasťou. Kožu nešúchajte.
Naplnenú injekčnú striekačku vyhoďte (pozri obrázok 10)· Použitú naplnenú injekčnú striekačku ihneď vyhoďte do nádoby na ostré predmety. Zabezpečte likvidáciu nádoby podľa pokynov svojho lekára alebo zdravotnej sestry.
Nepokúšajte sa znovu nasadiť kryt ihly.
Pre vašu bezpečnosť a zdravie a pre bezpečnosť iných nikdy opätovne nepoužívajte naplnenú injekčnú striekačku.
Ak máte pocit, že niečo s podaním injekcie nebolo v poriadku alebo si nie ste istý, povedzte to svojmu lekárovi alebo lekárnikovi.

Obrázok 10
NÁVOD NA POUŽITIE
Ak si chcete sami podať Simponi, musíte byť zaškolený zdravotníckym pracovníkom na prípravu injekcie a jej podanie sebe samému. Ak ste neboli zaškolený, požiadajte o to svojho lekára, zdravotnú sestru alebo lekárnika a naplánujte si s nimi termín školenia.
V tomto návode:
1. Príprava naplneného pera na použitie
2. Výber a príprava miesta podania
3. Podávanie lieku
4. Po podaní injekcie
Nižšie uvedená schéma (pozri obrázok 1) znázorňuje, ako vyzerá naplnené pero „SmartJect“.
 Kryt
KrytObrázok 1
1. Príprava naplneného pera na použitie· Naplnené pero nikdy netraste.
· Neodstraňujte kryt z naplneného pera skôr, ako bezprostredne pred podaním injekcie.
Skontrolujte počet naplnených pierSkontrolujte naplnené perá, aby ste sa uistili,
· že je počet naplnených pier a síl správny.
o Ak je vaša dávka 100 mg, dostanete jedno 100 mg naplnené pero
o Ak je vaša dávka 200 mg, dostanete dve 100 mg naplnené perá a bude potrebné, aby ste si
podali dve injekcie. Na tieto injekcie si vyberte odlišné miesta a injekcie si podajte hneď
po sebe.
Skontrolujte dátum exspirácie· Skontrolujte dátum exspirácie vytlačený alebo napísaný na škatuli.
· Skontrolujte dátum exspirácie na naplnenom pere (vyznačený skratkou „EXP“).
· V prípade, že dátum exspirácie vypršal, naplnené pero nepoužívajte. Vytlačený dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci. Požiadajte, prosím, lekára alebo
lekárnika o pomoc.
Skontrolujte bezpečnostnú plombu
· Skontrolujte bezpečnostnú plombu okolo krytu naplneného pera.
· Ak je bezpečnostná plomba porušená, naplnené pero nepoužívajte. Vyhľadajte, prosím, lekára alebo lekárnika.
Počkajte 30 minút, aby naplnené pero dosiahlo izbovú teplotu· Na zabezpečenie správneho podania injekcie nechajte naplnené pero pri izbovej teplote vybraté z papierovej škatule 30 minút mimo dosahu detí.
· Naplnené pero žiadnym iným spôsobom nezohrievajte (napríklad nezohrievajte ho v mikrovlnnej rúre ani v horúcej vode).
· Kryt naplneného pera neodstraňujte, kým pero nedosiahne izbovú teplotu.
Pripravte si zvyšné pomôcky· Kým čakáte, môžete si pripraviť zvyšné pomôcky, ako sú alkoholový tampón, vata alebo gáza a nádoba na ostré predmety.
Skontrolujte roztok vo vnútri naplneného pera· Cez kontrolné okienko sa uistite, či roztok v naplnenom pere je číry až mierne opaleskujúci
(lesknúci sa ako perla) a bezfarebný až svetložltý. Roztok môžete použiť, ak obsahuje malé množstvo malých priehľadných alebo bielych čiastočiek bielkovín.
· Môžete tiež pozorovať vzduchovú bublinu, čo je normálne.
· Nepoužívajte naplnené pero, ak má roztok nesprávnu farbu, je zakalený alebo obsahuje väčšie častice. Ak sa tak stane, poraďte sa so svojím lekárom alebo lekárnikom.
2. Výber a príprava miesta podania (pozri obrázok 2)· Zvyčajne si budete liek podávať spredu do strednej časti stehna.
· Môžete tiež použiť brušnú oblasť (abdómen) pod pupkom, s výnimkou plochy približne 5 cm priamo pod pupkom.
· Nepodávajte do miest, kde je koža citlivá, otlačená, červená, šupinatá, tvrdá alebo kde je koža s jazvami či striami.
· Ak sa na jedno podanie vyžaduje viacero injekcií, injekcie sa majú podať do rôznych miest na tele.
Obrázok 2
Výber miesta podania pre ošetrovateľov, ak si injekciu nepodávate sami (pozri obrázok 3)· Ak vám injekciu podáva ošetrovateľ, môže tiež použiť hornú vonkajšiu časť ramena.
· Opäť sa môžu použiť všetky spomenuté miesta nezávisle od typu alebo veľkosti vášho tela.
Obrázok 3
Príprava miesta podania· Dôkladne si umyte ruky mydlom a teplou vodou.
· Potrite miesto podania alkoholovým tampónom.
· Pred podaním nechajte miesto vpichu oschnúť. Vyčistené miesto neovievajte, ani naň nefúkajte.
· Nedotýkajte sa tohto miesta pred podaním injekcie.
3. Podávanie lieku· Kryt sa nesmie odstrániť, pokiaľ nie ste pripravený na podanie injekcie.
· Liek sa musí podať v priebehu 5 minút po odstránení krytu.
Odstráňte kryt (pozri obrázok 4)· Keď ste pripravený na injekciu, jemne otočte krytom, aby ste porušili bezpečnostnú plombu.
· Odstráňte kryt a po podaní injekcie ho vyhoďte.
· Kryt nedávajte naspäť, pretože môže poškodiť ihlu vo vnútri naplneného pera.
· Naplnené pero nepoužívajte, ak spadne bez nasadeného krytu. Ak sa tak stane, prosím, kontaktujte svojho lekára alebo lekárnika.
Obrázok 4
Naplnené pero pevne pritlačte na kožu (pozri obrázky 5 a 6)· Naplnené pero držte pohodlne v ruke. Tlačidlo teraz
NESTLÁČAJTE.
· Vyberiete si z dvoch metód vpichu. Odporúčaný je vpich bez vytvorenia kožnej riasy (obrázok 5a). Ak však chcete, môžete si vytvoriť kožnú riasu na spevnenie miesta vpichu (obrázok 5b).
· Pod uhlom 90 stupňov zatlačte otvorený koniec naplneného pera pevne proti koži, kým bezpečnostná manžeta úplne neskĺzne do priehľadného krytu (obrázok 6).
Obrázok 5a Obrázok 5b
Obrázok 6
Stlačte tlačidlo, aby sa injekcia podala (pozri obrázok 7)·
Naplnené pero stále pevne pritláčajte kolmo na kožu a prstami alebo palcom stlačte vyvýšenú časť tlačidla. Tlačidlo nebudete môcť stlačiť, ak naplnené pero nie je
pevnepritlačené na kožu a bezpečnostná manžeta neskĺzne do priehľadného krytu.
· Po stlačení zostane tlačidlo zatlačené a už nemusíte vyvíjať ďalší tlak.
Obrázok 7
·
Budete počuť hlasný „klik“- neľakajte sa. Prvý „klik“ znamená, že ihla je vpichnutá a začalo sa podávanie. V tomto momente môžete aj nemusíte pocítiť pichnutie ihly.
Zatiaľ naplnené pero nevyťahujte z kože. Ak naplnené pero z kože vytiahnete, nemusíte dostať celú dávku lieku.Naplnené pero držte do druhého „kliku“ (pozri obrázok 8)·
Naplnené pero ďalej pevne držte kolmo na koži, pokiaľ nebudete počuť druhý „klik“.Toto zvyčajne trvá 3-6 sekúnd, no môže to trvať aj 15 sekúnd, kým budete počuť druhý„klik“.· Druhý „klik“ znamená, že podanie injekcie bolo ukončené a ihla sa automaticky vsunula naspäť do naplneného pera. Ak máte sluchové ťažkosti, po prvom stlačení tlačidla počítajte 15 sekúnd
a potom zdvihnite naplnené pero z miesta podania injekcie.
· Zdvihnite naplnené pero z miesta podania injekcie.
Obrázok 8
4. Po podaní injekciePoužite vatový tampón alebo gázu· V mieste podania injekcie sa môže objaviť malé množstvo krvi alebo tekutiny. Je to normálne.
· Pritlačte vatový tampón alebo gázu na miesto podania injekcie na 10 sekúnd.
· Ak je potrebné, miesto podania injekcie môžete prekryť náplasťou.
· Kožu nešúchajte.
Skontrolujte okienko – žltý indikátor potvrdzuje správne podanie (pozri obrázok 9)· Žltý indikátor je spojený s piestom naplneného pera. Ak sa žltý indikátor v okienku nezobrazuje, piest sa dostatočne neposunul a injekcia sa nepodala.
· Žltý indikátor vyplní približne polovicu kontrolného okienka. Je to normálne.
· Ak v okienku nie je viditeľný žltý indikátor alebo ak si myslíte, že ste nedostali celú dávku, povedzte to svojmu lekárovi alebo lekárnikovi. Nepodávajte si druhú dávku bez porady so
svojím lekárom.
Obrázok 9
N
aplnené pero vyhoďte (pozri obrázok 10)
· Použité pero hneď vyhoďte do nádoby na ostré predmety. Keď je nádoba plná, zabezpečte jej likvidáciu podľa pokynov svojho lekára alebo zdravotnej sestry.
Ak máte pocit, že niečo s podaním injekcie nebolo v poriadku alebo si nie ste istý, povedzte to svojmu lekárovi alebo lekárnikovi.
Obrázok 10
NÁVOD NA POUŽITIE
Ak si chcete sami podať Simponi, musíte byť zaškolený zdravotníckym pracovníkom na prípravu injekcie a jej podanie sebe samému. Ak ste neboli zaškolený, požiadajte o to svojho lekára, zdravotnú sestru alebo lekárnika a naplánujte si s nimi termín školenia.
V tomto návode:
1. Príprava naplnenej injekčnej striekačky na použitie
2. Výber a príprava miesta podania
3. Podávanie lieku
4. Po podaní injekcie
Nižšie uvedená schéma (pozri obrázok 1) znázorňuje, ako vyzerá naplnená injekčná striekačka.
Obrázok 1
1. Príprava naplnenej injekčnej striekačky na použitieNaplnenú injekčnú striekačku držte za telo naplnenej injekčnej striekačky· Striekačku nedržte za hlavicu piestu, piest, ochranné krídla ihly alebo kryt ihly.
· Nikdy neťahajte piest.
· Naplnenú injekčnú striekačku nikdy netraste.
· Neodstraňujte kryt ihly z naplnenej injekčnej striekačky skôr, ako je to uvedené v návode.
· Nedotýkajte sa aktivačných spôn chrániča ihly (znázornených hviezdičkami * na obrázku 1), aby ste zabránili predčasnému zakrytiu ihly chráničom.
Skontrolujte počet naplnených injekčných striekačiekSkontrolujte naplnené injekčné striekačky, aby ste sa uistili,
· že je počet naplnených injekčných striekačiek a síl správny.
o Ak je vaša dávka 100 mg, dostanete jednu 100 mg naplnenú injekčnú striekačku.
o Ak je vaša dávka 200 mg, dostanete dve 100 mg naplnené injekčné striekačky a bude
potrebné, aby ste si podali dve injekcie. Na tieto injekcie si vyberte odlišné miesta
a injekcie si podajte hneď po sebe.
Skontrolujte dátum exspirácie (pozri obrázok 2)· Skontrolujte dátum exspirácie vytlačený alebo napísaný na škatuli.
· Skontrolujte dátum exspirácie (vyznačený skratkou „EXP“) na štítku pohľadom cez kontrolné okienko, ktoré je umiestnené na tele naplnenej injekčnej striekačky.
· Ak cez kontrolné okienko neuvidíte dátum exspirácie, držte naplnenú injekčnú striekačku za jej telo a otáčajte krytom ihly, kým sa vám v kontrolnom okienku neznázorní dátum exspirácie.
V prípade, že dátum exspirácie vypršal, naplnenú injekčnú striekačku nepoužívajte. Vytlačený dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci. Požiadajte, prosím, lekára alebo lekárnika
o pomoc.
Obrázok 2
Počkajte 30 minút, aby naplnená injekčná striekačka dosiahla izbovú teplotu· Na zabezpečenie správneho podania injekcie, nechajte naplnenú injekčnú striekačku pri izbovej teplote 30 minút mimo papierovej škatule a mimo dosahu detí.
Naplnenú injekčnú striekačku žiadnym iným spôsobom nezohrievajte (napríklad nezohrievajte ju
v mikrovlnnej rúre alebo v horúcej vode).
Kryt ihly naplnenej injekčnej striekačky neodstraňujte, kým striekačka nedosiahne izbovú teplotu.
Pripravte si zvyšné pomôckyKým čakáte, môžete si pripraviť zvyšné pomôcky, ako sú alkoholový tampón, vata alebo gáza
a nádoba na ostré predmety.
Skontrolujte roztok vo vnútri naplnenej injekčnej striekačky· Držte naplnenú injekčnú striekačku za jej telo tak, aby zakrytá ihla smerovala nadol.
· Cez kontrolné okienko naplnenej injekčnej striekačky sa uistite, či roztok v striekačke je číry až mierne opaleskujúci (lesknúci sa ako perla) a bezfarebný až svetložltý. Roztok môžete použiť,
ak obsahuje malé množstvo malých priehľadných alebo bielych čiastočiek bielkovín.
· Ak cez kontrolné okienko roztok neuvidíte, držte naplnenú injekčnú striekačku za jej telo a otáčajte krytom ihly, kým sa roztok neobjaví v kontrolnom okienku (pozri obrázok 2).
Nepoužívajte naplnenú injekčnú striekačku, ak má roztok nesprávnu farbu, je zakalený alebo obsahuje väčšie častice. Ak sa tak stane, poraďte sa so svojím lekárom alebo lekárnikom.
2. Výber a príprava miesta podania (pozri obrázok 3)· Zvyčajne si budete liek podávať spredu do strednej časti stehna.
· Môžete tiež použiť brušnú oblasť (abdómen) pod pupkom, s výnimkou plochy približne 5 cm priamo pod pupkom.
· Nepodávajte do miest, kde je koža citlivá, otlačená, červená, šupinatá, tvrdá alebo kde je koža s jazvami či striami.
· Ak sa na jedno podanie vyžaduje viacero injekcií, injekcie sa majú podať do rôznych miest na tele.
Obrázok 3
Výber miesta podania pre ošetrovateľov (pozri obrázok 4)
· Ak vám injekciu podáva ošetrovateľ, môže tiež použiť hornú vonkajšiu časť ramena.
· Opäť sa môžu použiť všetky spomenuté miesta, nezávisle od typu alebo veľkosti vášho tela.
Obrázok 4
Príprava miesta podania· Dôkladne si umyte ruky mydlom a teplou vodou.
· Potrite miesto podania alkoholovým tampónom.
· Pred podaním nechajte miesto vpichu oschnúť. Vyčistené miesto neovievajte, ani naň nefúkajte. Nedotýkajte sa tohto miesta pred podaním injekcie.
3. Podávanie liekuKryt ihly sa nesmie odstrániť, pokiaľ nie ste pripravený na podanie injekcie. Liek sa musí podať v priebehu 5 minút po odstránení krytu ihly.
Pri odstraňovaní krytu ihly sa nedotýkajte piestu.
Odstráňte kryt ihly (pozri obrázok 5)· Ak ste pripravený na podanie injekcie, chyťte telo naplnenej injekčnej striekačky do jednej ruky.
· Odstráňte kryt ihly a po podaní injekcie ho vyhoďte. Kým odstraňujete kryt ihly, nedotýkajte sa piestu.
· V naplnenej striekačke môžete pozorovať vzduchovú bublinu alebo malé množstvo roztoku na konci ihly. Oba sú bežné javy a nemusia byť odstránené.
· Injekciu si podajte ihneď po odstránení krytu ihly.
Nedotýkajte sa ihly a nedotýkajte sa ihlou žiadnych predmetov.
Naplnenú injekčnú striekačku nepoužívajte, ak vám spadne, keď kryt ihly nie je nasadený. Ak sa tak stane, kontaktujte, prosím, svojho lekára alebo lekárnika.
Obrázok 5
Poloha naplnenej injekčnej striekačky pri podaní
· Telo naplnenej injekčnej striekačky chyťte prostredníkom a ukazovákom jednej ruky a palec priložte na hlavicu piestu. Druhou rukou si jemne uštipnite očistené miesto podania. Pevne
držte.
Nikdy neťahajte za piest.
Podanie lieku· Ihlu držte približne v 45-stupňovom uhle voči pokožke. Jednoduchým a rýchlym pohybom vpichnite ihlu pod kožu najviac ako je to možné (pozri obrázok 6).
Obrázok 6
· Zatlačte na hlavicu piestu, až kým sa hlavica piestu nedostane medzi ochranné krídla a podajte si tým celý obsah lieku (pozri obrázok 7).
Obrázok 7
· Keď je piest striekačky zatlačený, ako je najviac možné, ešte chvíľu vyvíjajte tlak palcom na hlavicu piestu. Až potom vytiahnite ihlu z kože (pozri obrázok 8).
Obrázok 8
· Pomaly uvoľnite palec z hlavice piestu, aby ste umožnili prázdnej naplnenej injekčnej
striekačke vysunúť sa nahor, kým celá ihla nebude pokrytá chráničom ihly, ako je to znázornené na obrázku 9:

Obrázok 9
4. Po podaní injekciePoužite vatový tampón alebo gázu· V mieste podania injekcie sa môže objaviť malé množstvo krvi alebo tekutiny. Je to normálne.
· Pritlačte vatový tampón alebo gázu na miesto podania injekcie na 10 sekúnd.
· Ak je potrebné, miesto podania injekcie môžete prekryť náplasťou. Kožu nešúchajte.
Naplnenú injekčnú striekačku vyhoďte (pozri obrázok 10)· Použitú naplnenú injekčnú striekačku ihneď vyhoďte do nádoby na ostré predmety. Zabezpečte likvidáciu nádoby podľa pokynov svojho lekára alebo zdravotnej sestry.
Nepokúšajte sa znovu nasadiť kryt ihly.
Pre vašu bezpečnosť a zdravie a pre bezpečnosť iných nikdy opätovne nepoužívajte naplnenú injekčnú striekačku.
Ak máte pocit, že niečo s podaním injekcie nebolo v poriadku alebo si nie ste istý, povedzte to svojmu lekárovi alebo lekárnikovi.
Obrázok 10