
systémov
V
e
ľ
m
i časté Časté Menej časté
Infekcie a nákazy Infekcie horných dýchacích ciest
Poruchy gastrointestinálneho traktu
Poruchy kože a podkožného tkaniva Poruchy nervového systému
Laboratórne a funkčné
vyšetrenia
Celulitída, pneumónia, jednoduchý opar v oblasti úst, pásový opar
Bolesť brucha, ulcerácia v ústnej dutine, gastritída
Vyrážka, pruritus,
urtikária
Bolesť hlavy, závraty
Zvýšené hodnoty pečeňových transamináz, zvýšenie telesnej hmotnosti, zvýšené hodnoty celkového bilirubínu*
Divertikulitída
Stomatitída, žalúdočné vredy
Poruchy ciev Hypertenzia
Poruchy krvi a lymfatického systému Poruchy metabolizmu a výživy
Celkové poruchy a reakcie v mieste podania
Hypercholeste- rolémia*
Leukopénia, neutropénia
Periférny edém, reakcie
z precitlivenosti
Hypertriglyceridémia
Poruchy oka Konjunktivitída
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy obličiek
a močových ciest Poruchy endokrinného systému
Kašeľ, dyspnoe
Nefrolitiáza
Hypotyreoidizmus
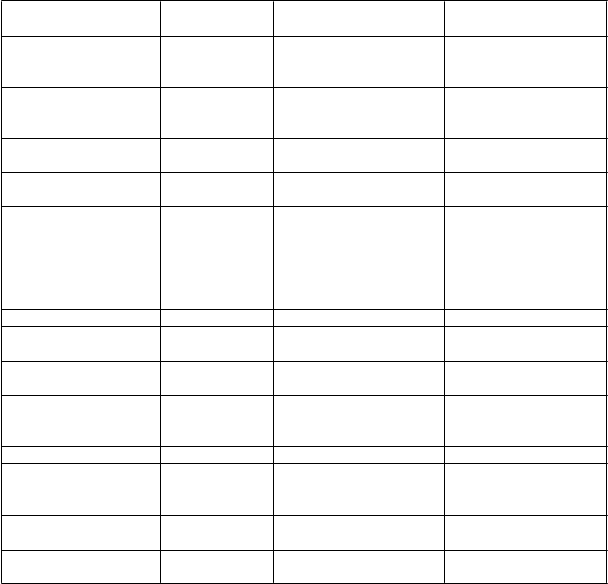
*zahŕňa zvýšenia zozbierané ako časť rut inného laborat órneho pozorovania (pozri t ext nižšie).
Inf ekcieV 6 mesačných kontrolovaných štúdiách bola miera výskytu všetkých infekcií hlásených pri liečbe tocilizumabom 8 mg/kg spolu s DMARDs 127 udalostí na 100 pacientorokov oproti 112 udalostiam na 100 pacientorokov v skupine s placebom v kombinácii s DMARDs. V súbore dlhodobej expozície bol celkový výskyt infekcií pri liečbe RoActemrou 108 udalostí na 100 pacientorokov.
V 6 mesačných kontrolovaných klinických štúdiách bola miera výskytu závažných infekcií pri liečbe tocilizumabom 8 mg/kg v kombinácii s DMARDs 5,3 udalosťami na 100 pacientorokov expozície oproti 3,9 udalostiam na 100 pacientorokov expozície v skupine s placebom v kombinácii s DMARDs. V štúdii monoterapie bola miera výskytu závažných infekcií 3,6 udalostí na 100 pacientorokov expozície v skupine liečenej tocilizumabom a 1,5 udalostí na 100 pacientorokov expozície v skupine liečenej MTX.
V súbore dlhodobej expozície bol celkový výskyt závažných infekcií (bakteriálne, vírusové
a mykotické) 4,7 udalostí na 100 pacientorokov. K hláseným závažným infekciám, z ktorých niektoré mali fatálne následky, patrila aktívna tuberkulóza, ktorá môže byť prítomná s vnútropľúcnym alebo mimopľúcnym ochorením, invazívne pľúcne infekcie, vrátene kandidózy, aspergilózy kokcidioidomykózy a
Pneumocystis jiroveci, pneumónia, celulitída, pásový opar, gastroenteritída, divertikulitída, sepsa a bakteriálna artritída. Boli hlásené prípady oportúnnych infekcií.
I
ntersticiálna choroba pľúc
Zhoršená funkcia pľúc môže zvýšiť riziko vzniku infekcií. Po uvedení lieku na trh boli hlásené
prípady intersticiálnej choroby pľúc (vrátane pneumonitídy a pľúcnej fibrózy), z ktorých niektoré boli
fatálne.
Gastrointestinálne perf orácie
Pri liečbe tocilizumabom počas 6-mesačných kontrolovaných klinických štúdií bol celkový výskyt gastrointestinálnych perforácií 0,26 udalostí na 100 pacientorokov. V dlhodobej expozícií bol celkový výskyt gastrointestinálnych perforácií 0,28 udalostí na 100 pacientorokov. Hlásenia gastrointestinálnej perforácie pri liečbe tocilizumabom boli primárne hlásené ako komplikácie divertikulitídy, zahŕňajúce generalizovanú purulentnú peritonitídu, perforáciu dolnej časti gastrointestinálneho traktu, fistulu
a absces.
Reakcie na inf úziu
V 6 mesačných kontrolovaných klinických štúdiách boli nežiaduce účinky súvisiace s podávaním infúzie (vybrané udalosti vyskytujúce sa počas podávania infúzie alebo v priebehu 24 hodín od podania infúzie) hlásené u 6,9 % pacientov v skupine liečenej tocilizumabom 8 mg/kg v kombinácii s DMARDs a u 5,1 % pacientov v skupine s placebom spolu s DMARDs. Udalosti hlásené počas podávania infúzie boli predovšetkým epizódy hypertenzie; udalosti hlásené v priebehu 24 hodín od ukončenia podávania infúzie boli bolesť hlavy a kožné reakcie (vyrážka, urtikária). Tieto udalosti neboli pre liečbu limitujúce.
Miera výskytu anafylaktických reakcií (vyskytujúcich sa celkovo u 8/4 009 pacientov, 0,2 %) bola niekoľkonásobne vyššia pri dávke 4 mg/kg oproti dávke 8 mg/kg. Klinicky významné reakcie
z precitlivenosti súvisiace s liečbou tocilizumabom a vyžadujúce ukončenie liečby boli hlásené
u celkovo 56 z 4 009 pacientov (1,4 %) liečených tocilizumabom počas kontrolovaných a otvorených klinických štúdií. Tieto reakcie sa zvyčajne pozorovali počas podávania druhej až piatej infúzie tocilizumabu (pozri časť 4.4). Po registrácii lieku bola hlásená fatálna anafylaxia počas liečby tocilizumabom (pozri časť 4.4).
Imunogenicita
V 6 mesačných kontrolovaných klinických štúdiách bolo celkovo 2 876 pacientov vyšetrených na protilátky proti tocilizumabu. Zo 46 pacientov (1,6 %), u ktorých sa vytvorili protilátky proti tocilizumabu, došlo u 6 k významnej reakcii z precitlivenosti, ktorá u 5 z nich viedla k trvalému ukončeniu liečby. U tridsiatich pacientov (1,1 % sa vytvorili neutralizujúce protilátky.
Hematologické odchýlky: Neutrof ily
V 6 mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu neutrofilov pod 1 x 109/l u
3,4 % pacientov liečených tocilizumabom 8 mg/kg spolu s DMARDs oproti < 0,1 % pacientov s
placebom spolu s DMARDs. Približne u polovice pacientov, u ktorých ANC klesol na < 1 x 109/l,
došlo k tomuto poklesu v priebehu 8 týždňov po začatí liečby. Pokles pod 0,5 x 109/l bol hlásený u 0,3 % pacientov liečených tocilizumabom 8 mg/kg spolu s DMARDs. Boli hlásené infekcie
s neutropéniou.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt poklesu počtu neutrofilov rovnaký ako sa pozoroval počas 6 mesačných kontrolovaných klinických štúdií.
Krvné doštičky
V 6 mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu trombocytov pod
100 x 103/µl u 1,7 % pacientov liečených tocilizumabom 8 mg/kg spolu s DMARDs oproti < 1 %
pacientov s placebom spolu s DMARDs. Tieto poklesy sa vyskytli bez asociácie s krvácaním.
Počas dvojito zaslepenej kontrolnej fázy a pri dlhodobej expozícii zostával charakter a výskyt poklesu počtu krvných doštičiek rovnaký ako sa pozoroval počas 6 mesačných kontrolovaných klinických štúdií.
V post marketingovom sledovaní sa vyskytli veľmi zriedkavo prípady pancytopénie.
Zvýšenie pečeňových transamináz
V 6 mesačných kontrolovaných klinických štúdiách bolo prechodné zvýšenie hodnôt
ALT/AST > 3 x ULN u 2,1 % pacientov liečených tocilizumabom 8 mg/kg oproti 4,9 % pacientov
liečených MTX, a u 6,5 % pacientov liečených 8 mg/kg tocilizumabu v kombinácii s DMARDs oproti
1,5 % pacientov s placebom v kombinácii s DMARDs.
Pridanie potenciálne hepatotoxických liekov (napr. MTX) k tocilizumabu podávanému v monoterapii viedlo k zvýšenému výskytu vyšších hodnôt. Zvyšovanie hladín ALT/AST > 5 násobok ULN sa pozorovalo u 0,7 % pacientov liečených tocilizumabom v monoterapii a u 1,4 % pacientov liečených tocilizumabom v kombinácii s DMARDs, pričom väčšina z nich liečbu tocilizumabom trvalo ukončila. Tieto vzostupy neboli spojené s klinicky významným zvýšením hodnôt priameho (konjugovaného) bilirubínu, ani nesúviseli s klinickými príznakmi hepatitídy či poruchy funkcie pečene. Počas dvojito zaslepeného kontrolovaného obdobia, incidencia zvýšenia nepriameho bilirubínu vyššia ako horný
limit normy sledované ako rutinný laboratórny parameter, je 6,2 % u pacientov liečených tocilizumabom v dávke 8 mg/kg + DMARD. Celkovo 5,8 % pacientov malo zvýšený nepriamy bilirubín od > 1 do 2 x ULN a 0,4 % pacientov malo zvýšenie > 2 x ULN.
Počas dvojito zaslepenej kontrolnej fázy a pri dlhodobej expozícii zostával charakter a výskyt
prípadov zvýšenia ALT/AST rovnaký ako sa pozoroval počas 6 mesačných kontrolovaných klinických
štúdií.
Hodnoty lipidových parametrov
Zvýšenie hodnôt lipidových parametrov, ako je napríklad celkový cholesterol, triglyceridy,
LDL-cholesterol a/alebo HDL-cholesterol, bolo počas 6-mesačných kontrolovaných štúdií hlásené
často. Rutinným laboratórnym sledovaním sa zistilo, že približne u 24 % pacientov, ktorí v klinických štúdiách dostávali RoActemru, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu na
≥ 6,2 mmol/l, pričom u 15 % pacientov došlo k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l.
Zvýšené lipidové parametre odpovedali na liečbu hypolipidemikami.
Počas dvojito zaslepenej kontrolnej fázy a pri dlhodobej expozícii zostával charakter a výskyt prípadov zvýšenia lipidových parametrov rovnaký ako sa pozoroval počas 6 mesačných kontrolovaných klinických štúdií.
Malignity
Klinické údaje nie sú dostatočné na zhodnotenie rizika možného výskytu malignity po expozícii
tocilizumabu. Hodnotenie dlhodobej bezpečnosti naďalej prebieha.
Kožné reakcie
V postmarketingovom sledovaní sa vyskytli veľmi zriedkavé hlásenia Stevensovho-Johnsonovho syndrómu.
Pediatrická populácia
Bezpečnosť tocilizumabu v pediatrickej populácii je uvedená v častiach o pJIA a sJIA nižšie. Vo
všeobecnosti boli ADRs u pacientov s pJIA a sJIA podobného typu ako tie, ktoré boli pozorované
u pacientov s RA, pozri časť 4.8.
ADRs pozorované u pacientov s pJIA a sJIA liečených tocilizumabom sú popísané nižšie a sú uvedené v tabuľke 2 podľa triedy orgánových systémov a kategórií frekvencie, ktoré sú definované s použitím nasledujúceho pravidla: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10) alebo menej časté (≥ 1/1 000
až < 1/100).
Tabuľka 2. Súhrn ADRs vyskytujúcich sa u pacientov so sJIA alebo pJIA liečených tocilizumabom
v monoterapii alebo v kombinácii s MTX
TOS Preferovaný termín Frek vencia
Infekcie a nákazy Veľmi časté
Časté Menej časté
Infekcie horných dýchacích ciest
pJIA, sJIA
Nazofaryngitída pJIA, sJIA
Poruchy gastrointestinálneho traktu
Nauzea pJIA Hnačka pJIA, sJIA
Celkové poruchy a reakcie v mieste podania
Reakcie na infúziu pJIA1, sJIA2
Poruchy nervového systému
Bolesť hlavy pJIA sJIA Laboratórne a funkčné vyšetrenia
Zvýšené hodnoty pečeňových transamináz Pokles počtu neutrofilov
Znížený počet krvných doštičiek
Zvýšené hodnoty cholesterolu
pJIA
sJIA pJIA
sJIA pJIA
sJIA pJIA
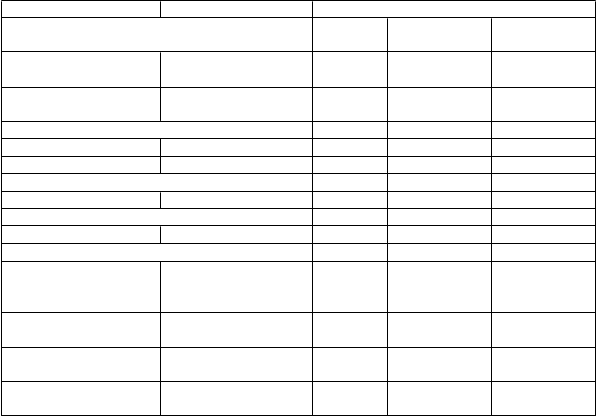
1. Reakcie na infúziu u pacient ov s pJIA zahŕňali bolesť hlavy, nauzeu a hypot enziu a nielen t iet o reakcie
2. Reakcie na infúziu u pacient ov so sJIA pacient ov zahŕňali vyrážku, urt ikáriu, hnačku, ťažkost i v epigast riu, art ralgiu a bolesť hlavy a nielen t iet o reakcie
Pacienti s pJIABezpečnosť tocilizumabu pri pJIA sa skúmala u 188 pacientov vo veku 2 až 17 rokov. Celková expozícia pacientov bola 184,4 pacientorokov. Frekvenciu ADRs u pJIA pacientov je možné nájsť v tabuľke 2. Typy ADRs u pacientov s pJIA boli podobné typom, ktoré sa pozorovali u pacientov
s RA a sJIA, pozri časť 4.8. Pri porovnaní s dospelou populáciou s RA boli prípady nazofaryngitídy, bolesti hlavy, nauzey a zníženého počtu neutrofilov hlásené častejšie v populácii s pJIA. Prípady zvýšeného cholesterolu boli hlásené menej častejšie v pJIA populácii ako u dospelej populácie s RA.
Inf ekcieVýskyt infekcií v populácii vystavenej tocilizumabu bola 163,7 na 100 pacientorokov. Najčastejšie pozorované prípady boli nazofaryngitída a infekcie horných dýchacích ciest. Výskyt závažných infekcií bola číselne vyšší u pacientov s telesnou hmotnosťou < 30 kg, ktorí sa liečili dávkou 10 mg/kg tocilizumabu (12,2 na 100 pacientorokov) v porovnaní s pacientmi s telesnou hmotnosťou ≥ 30 kg, ktorí sa liečili dávkou 8 mg/kg tocilizumabu (4,0 na 100 pacientorokov). Incidencia infekcií vedúcich
k prerušeniam dávky bola tiež číselne vyššia u pacientov s telesnou hmotnosťou < 30 kg, ktorí sa liečili dávkou 10 mg/kg tocilizumabu (21,4 %) v porovnaní s pacientmi s telesnou hmotnosťou
≥ 30 kg, ktorí sa liečili dávkou 8 mg/kg tocilizumabu (7,6 %).
Reakcie na inf úziuU pacientov s pJIA sa reakcie na infúziu definovali ako všetky udalosti, ktoré nastali v priebehu infúzie alebo počas 24 hodín od infúzie. V populácii vystavenej tocilizumabu sa u 11 pacientov
(5,9 %) vyskytli reakcie na infúziu počas infúzie a u 38 pacientov (20,2 %) sa vyskytla udalosť počas
24 hodín od infúzie. Najčastejšie udalosti, ktoré sa vyskytli v priebehu infúzie, boli bolesť hlavy, nevoľnosť a hypotenzia a počas 24 hodín od infúzie to bol závrat a hypotenzia. Vo všeobecnosti sa počas infúzie alebo počas 24 hodín od infúzie pozorovali nežiaduce reakcie na liek s podobným charakterom ako tie, ktoré sa pozorovali u pacientov s RA a sJIA, pozri časť 4.8.
Nezaznamenali sa žiadne klinicky významné hypersenzitívne reakcie súvisiace s tocilizumabom
a ktoré by vyžadovali vysadenie liečby.
Imunogenicita
U jedného pacienta v skupine s 10 mg/kg a < 30 kg sa vyvinuli pozitívne protilátky proti tocilizumabu bez rozvoja hypersenzitívnej reakcie a ten bol následne vyradený zo štúdie.
Neutrof ily
Počas rutinného laboratórneho vyšetrenia v populácii vystavenej tocilizumabu sa zaznamenal pokles
v počte neutrofilov pod 1 x 109/l u 3,7 % pacientov.
Krvné doštičky
Počas rutinného laboratórneho vyšetrenia v populácii vystavenej tocilizumabu malo 1 % pacientov pokles v počte krvných doštičiek pod 50 x 109/l bez sprievodných krvácavých udalostí.
Zvýšenie pečeňových transamináz
Počas rutinného laboratórneho vyšetrenia v populácii vystavenej tocilizumabu sa zvýšenie ALT alebo
AST ≥ 3 x ULN objavilo u 3,7 % a < 1 % pacientov.
Hodnoty lipidových parametrov
Počas rutinného laboratórneho vyšetrenia v populácii vystavenej tocilizumabu sa zvýšenie celkového cholesterolu > 1,5 - 2 x ULN objavilo u jedného pacienta (0,5 %) a zvýšenie LDL > 1,5-2 x ULN
u jedného pacienta (0,5 %).
Pacienti so sJIA
Bezpečnosť tocilizumabu pri sJIA bola skúmaná u 112 pacientov vo veku od 2 do 17 rokov. V 12 týždňovej dvojito zaslepenej, kontrolovanej fáze dostalo 75 pacientov liečbu tocilizumabom (8 mg/kg alebo 12 mg/kg na základe telesnej hmotnosti). Po 12 týždňoch alebo v čase prechodu na tocilizumab pre zhoršenie choroby boli pacienti liečení v pokračujúcej otvorenej nadstavbovej fáze štúdie.
Vo všeobecnosti bol typ ADRs u pacientov so sJIA podobný ako u pacientov s RA, pozri časť 4.8. Frekvenciu ADRs u sJIA pacientov je možné nájsť v tabuľke 2. Pri porovnaní s dospelou populáciou s RA sa u pacientov so sJIA vyskytovala nazofaryngitída, pokles počtu neutrofilov, zvýšenie pečeňových transamináz a hnačka s vyššou frekvenciou. Prípady zvýšeného cholesterolu boli hlásené menej častejšie v sJIA populácii ako u dospelej populácie s RA.
Inf ekcie
V 12-týždňovej kontrolovanej fáze bol výskyt všetkých infekcií v skupine s tocilizumabom 344,7 na
100 pacientorokov a v skupine s placebom 287,0 na 100 pacientorokov. V pokračujúcej otvorenej nadstavbovej fáze (časť II) ostal celkový výskyt infekcií podobný, a to 306,6 na 100 pacientorokov.
V 12-týždňovej kontrolovanej fáze bol výskyt závažných infekcií v skupine s tocilizumabom 11,5 na
100 pacientorokov. Po jednom roku v pokračujúcej otvorenej nadstavbovej fáze ostal celkový výskyt závažných infekcií stabilný, a to 11,3 na 100 pacientorokov. Hlásené závažné nežiaduce infekcie boli podobné ako tie u pacientov s RA, navyše varicella a otitis media.
Reakcie na inf úziu
Reakcie súvisiace s infúziou sú definované ako všetky nežiaduce účinky objavujúce sa počas infúzie alebo v rámci 24 hodín po nej. V 12-týždňovej kontrolovanej fáze zaznamenali 4 % pacientov zo skupiny s tocilizumabom udalosti, ktoré sa objavili počas infúzie. Jeden nežiaduci účinok (angioedém) bol považovaný za závažný a život ohrozujúci a liečba pacienta v štúdii bola ukončená.
V 12 týždňovej kontrolovanej fáze zaznamenalo 16 % pacientov zo skupiny s tocilizumabom a 5,4 % pacientov zo skupiny s placebom udalosť v rámci 24 hodín po infúzii. V skupine s tocilizumabom tieto nežiaduce účinky zahŕňali (avšak nielen tieto) vyrážku, urtikáriu, hnačku, nepohodlie v epigastriu, bolesť kĺbov a bolesť hlavy. Jeden z týchto nežiaducich účinkov, žihľavka, bol považovaný za
závažný.
Klinicky významné reakcie z precitlivenosti spojené s liečbou tocilizumabom, ktoré vyžadovali ukončenie liečby, boli zaznamenané u 1 zo 112 pacientov (<1 %) liečených tocilizumabom počas celej kontrolovanej fázy a vrátane otvorenej fázy klinickej štúdie.
ImunogenicitaNa začiatku štúdie boli všetci 112 pacienti testovaní na antitocilizumabové protilátky. U dvoch pacientov boli zistené antitocilizumabové protilátky, pričom jeden z týchto pacientov mal reakciu z precitlivenosti vedúcu k ukončeniu liečby. Incidencia antitocilizumabových protilátok môže byť podhodnotená vzhľadom na interferenciu tocilizumabu s rozborom a vyššou koncentráciou lieku, ktorá je pozorovaná u detí v porovnaní s dospelými.
Neutrof ilyPočas rutinného laboratórneho vyšetrenia v 12-týždňovej kontrolovanej fáze sa pokles v počte neutrofilov pod 1 x 109/l objavil u 7 % pacientov v skupine s tocilizumabom a žiadny pokles nebol
zaznamenaný v skupine s placebom.
V pokračujúcej otvorenej nadstavbovej fáze sa pokles v počte neutrofilov pod 1 x 109/l objavil u 15%
pacientov v skupine s tocilizumabom.
Krvné doštičkyPočas rutinného laboratórneho vyšetrenia v 12-týždňovej kontrolovanej fáze mali 3% pacientov
v skupine s placebom a 1% v skupine s tocilizumabom pokles v počte krvných doštičiek pod
100 x 109/l.
V pokračujúcej otvorenej nadstavbovej fáze sa pokles v počte krvných doštičiek pod 100 x 109/l
objavil u 3% pacientov v skupine s tocilizumabom, bez pridružených krvácaní.
Zvýšenie pečeňových transaminázPočas rutinného laboratórneho vyšetrenia v 12-týždňovej kontrolovanej fáze sa zvýšenie ALT alebo
AST ≥ 3 x ULN objavilo u 5% a 3% pacientov v skupine s tocilizumabom a 0% v skupine s placebom.
V pokračujúcej otvorenej nadstavbovej fáze sa zvýšenie ALT alebo AST ≥ 3 x ULN objavilo u 12% a
4% pacientov v skupine s tocilizumabom.
Imunoglobulín G
Hladiny IgG klesajú počas liečby. Pokles na dolnú hranicu normy sa vyskytol u 15 pacientov
v niektorom období štúdie.
Hodnoty lipidových parametrovPočas rutinného laboratórneho vyšetrenia v 12-týždňovej kontrolovanej fáze sa zvýšenie celkového
cholesterolu > 1,5 x ULN až 2 x ULN objavilo u 1,5 % pacientov v skupine s tocilizumabom
a u nikoho v skupine s placebom. Zvýšenie LDL > 1,5 x ULN až 2 x ULN sa objavilo u 1,9 %
pacientov v skupine s tocilizumabom a u 0 % v skupine s placebom.
V pokračujúcej otvorenej nadstavbovej fáze ostali charakter a incidencia zvýšení lipidových parametrov rovnaké ako v 12-týždňovej kontrolovanej fáze.
H
l
á
s
e
nie podozrení
na
nežiaduce
r
e
a
kcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovanieK dispozícii sú obmedzené údaje o predávkovaní RoActemrou. Hlásený bol jeden prípad náhodného predávkovania, pri ktorom pacient s mnohopočetným myelómom dostal jednorazovú dávku 40 mg/kg. Nepozorovali sa žiadne nežiaduce reakcie.
U zdravých dobrovoľníkov, ktorí dostali jednorazovú dávku do 28 mg/kg, sa nepozorovali žiadne závažné nežiaduce reakcie, hoci došlo k výskytu neutropénie limitujúcej dávku.
Pediatrick á populáciaV pediatrickej populácii sa nepozoroval žiaden prípad predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu; ATC kód: L04AC07.
MechanizmusúčinkuTocilizumab sa špecificky viaže na rozpustný aj na membránovo-viazaný receptor pre IL-6 (sIL-6R a
mIL-6R). Dokázalo sa, že tocilizumab inhibuje prenos signálu sprostredkovaný sIL-6R a mIL-6R.
IL-6 je pleiotropný prozápalový cytokín, ktorý produkujú rôzne typy buniek, vrátane T- a B-buniek, monocytov a fibroblastov. IL-6 sa zúčastňuje na rôznorodých fyziologických procesoch, ako je aktivácia T-buniek, indukcia sekrécie imunoglobulínov, indukcia syntézy proteínov akútnej fázy
v pečeni a stimulácia krvotvorby. IL-6 sa podieľa na patogenéze ochorení, medzi ktoré patria zápalové
ochorenia, osteoporóza a neoplázie.
PacientisRAFarmakodynamickéúčinkyV klinických štúdiách s tocilizumabom sa pozoroval rýchly pokles hodnôt CRP, sedimentácie erytrocytov (ESR) a sérového amyloidu A (SAA). V zhode s účinkom na reaktanty akútnej fázy bola liečba tocilizumabom spojená s poklesom počtu trombocytov na hodnoty v rámci referenčného rozpätia. Pozorovalo sa zvýšenie hladín hemoglobínu, ktoré tocilizumab vyvoláva tým, že znižuje IL-6 navodené účinky na tvorbu hepcidínu, čím sa zvyšuje dostupnosť železa. U pacientov liečených tocilizumabom sa pokles hladín CRP na hodnoty v rámci referenčného rozpätia pozoroval už od
2. týždňa a počas trvania liečby sa tento pokles udržal.
U zdravých dobrovoľníkov, ktorým sa podával tocilizumab v dávkach od 2 do 28 mg/kg, klesal absolútny počet neutrofilov k najnižším hladinám 3 až 5 dní po podaní. Potom sa počet neutrofilov vrátil k východiskovým hodnotám v závislosti na dávke. Pacienti s reumatoidnou artritídou po podaní tocilizumabu vykazovali podobný charakter absolútneho počtu neutrofilov (pozri časť 4.8).
Klinická účinnosťabezpečnosťÚčinnosť tocilizumabu na zmiernenie prejavov a príznakov RA sa hodnotila v piatich randomizovaných, dvojito zaslepených, multicentrických štúdiách. Do štúdií I - V boli zaradení pacienti vo veku ≥ 18 rokov s aktívnou RA diagnostikovanou podľa kritérií American College of Rheumatology (ACR), ktorí mali pred začiatkom liečby minimálne osem bolestivých a šesť opuchnutých kĺbov.
V štúdii I sa tocilizumab podával intravenózne raz za štyri týždne v monoterapii. V štúdiách II, III a V sa tocilizumab podával intravenózne raz za štyri týždne v kombinácii s MTX, kontrolnú skupinu tvorilo placebo v kombinácii s MTX. V štúdii IV sa tocilizumab podával intravenózne raz za 4 týždne v kombinácii s inými DMARDs, kontrolnú skupinu tvorilo placebo v kombinácii s inými DMARDs.
Primárny cieľový ukazovateľ pre každú z piatich štúdií bol podiel pacientov, ktorí dosiahli odpoveď
ACR 20 v 24. týždni.
Štúdia I hodnotila 673 pacientov, ktorí sa v priebehu šiestich mesiacov pred randomizáciou neliečili MTX a ktorí neprerušili predchádzajúcu liečbu MTX kvôli klinicky významným toxickým účinkom alebo nedostatočnej odpovedi na liečbu. Väčšina (67 %) pacientov sa MTX predtým neliečila. Dávky
8 mg/kg tocilizumabu sa podávali raz za štyri týždne v monoterapii. Porovnávacia skupina dostávala MTX raz týždenne (dávka titrovaná od 7,5 mg na maximálne 20 mg týždenne počas osemtýždňového obdobia).
Štúdia II, dvojročná štúdia s plánovanou analýzou v týždni 24., 52. a v týždni 104, hodnotila 1 196 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky 4 alebo 8 mg/kg tocilizumabu alebo placeba sa podávali raz za štyri týždne v zaslepenej fáze liečby trvajúcej 52 týždňov v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne). Po týždni 52 mohli všetci pacienti pokračovať v otvorenej fáze liečby s tocilizumabom v dávke 8 mg/kg. Z pacientov, ktorí dokončili štúdiu a ktorí boli pôvodne randomizovaní do skupiny s placebom + MTX, v 2. roku
86 % pacientov pokračovalo v otvorenej fáze liečby tocilizumabom v dávke 8 mg/kg. Primárny cieľový ukazovateľ v 24. týždni bol podiel pacientov, ktorí dosiahli odpoveď ACR 20. V 52. a 104. týždni boli prevencia poškodenia kĺbu a zlepšenie fyzických funkcií pridružené ako primárne cieľové ukazovatele.
Štúdia III hodnotila 623 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky
4 alebo 8 mg/kg tocilizumabu alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Štúdia IV hodnotila 1 220 pacientov, ktorí nedosiahli dostatočnú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viaceré DMARDs. Dávky 8 mg/kg tocilizumabu alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou DMARDs.
Štúdia V hodnotila 499 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na liečbu jedným alebo viacerými inhibítormi TNF, alebo ktorí takúto liečbu netolerovali. Liečba inhibítorom TNF sa pred randomizáciou ukončila. Dávky 4 alebo 8 mg/kg tocilizumabu alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Klinická odpoveď
Vo všetkých štúdiách mali pacienti liečení tocilizumabom 8 mg/kg štatisticky významne vyššiu mieru odpovede ACR 20, 50, 70 po šiestich mesiacoch oproti kontrolnej skupine (tabuľka 3). V štúdii I sa preukázala vyššia účinnosť tocilizumabu 8 mg/kg oproti aktívnej porovnávacej látke - MTX.
Účinok liečby bol u pacientov podobný nezávisle od prítomnosti reumatoidného faktora, veku, pohlavia, rasy, počtu predchádzajúcich terapií a stavu ochorenia. Účinok nastúpil rýchlo (už v
2. týždni) a stupeň odpovede sa počas liečby neustále zlepšoval. V prebiehajúcich predĺžených
otvorených štúdiách I-V sa počas viac ako 3 rokov pozorovali neustále pretrvávajúce odpovede.
Vo všetkých štúdiách sa u pacientov liečených tocilizumabom 8 mg/kg oproti pacientom s placebom a MTX alebo inými DMARDs zaznamenalo významné zlepšenie vo všetkých jednotlivých zložkách odpovede ACR zahŕňajúcich: počet bolestivých a opuchnutých kĺbov; celkové hodnotenie pacientmi a lekárom; skóre indexu funkčnej neschopnosti; hodnotenie bolesti a CRP.
Pacienti v štúdiách I - V mali pred začiatkom liečby priemerné skóre aktivity ochorenia (DAS28)
6,5 - 6,8. U pacientov liečených tocilizumabom sa v porovnaní s pacientmi v kontrolnej skupine (1,3-
2,1) pozorovalo významné zníženie DAS28 oproti východiskovej hodnote (priemerné zlepšenie) o
3,1 - 3,4. Podiel pacientov, ktorí v 24. týždni dosiahli klinickú remisiu ochorenia podľa DAS28
(DAS28 < 2,6), bol významne vyšší u pacientov liečených tocilizumabom (28 - 34 %) v porovnaní s 1 - 12 % pacientov v kontrolnej skupine. V štúdii II dosiahlo 65 % pacientov DAS28 < 2,6 v 104. týždni, v porovnaní so 48 % pacientov v 52. týždni a s 33 % pacientov v 24. týždni.
V súhrnnej analýze štúdií II, III a IV bol podiel pacientov, ktorí dosiahli odpoveď ACR 20, 50 a 70
významne vyšší (59 % oproti 50 %, 37 % oproti 27 %, 18 % oproti 11 % v uvedenom poradí) v skupine liečenej tocilizumabom 8 mg/kg v kombinácii s DMARDs oproti skupine liečenej tocilizumabom 4 mg/kg v kombinácii s DMARDs (p < 0,03). Podobne bol aj podiel pacientov, ktorí dosiahli remisiu ochorenia podľa DAS28 (DAS28 < 2,6), významne vyšší (31 % oproti 16 %)
u pacientov liečených tocilizumabom 8 mg/kg v kombinácii s DMARDs než u pacientov liečených tocilizumabom 4 mg/kg v kombinácii s DMARDs (p < 0,0001).
Tabuľka 3.Odpovede ACR v placebom/MTX/DMARDs kontrolovaných štúdiách (% pacientov)
Š
t
ú
d
i a I AMBITIO N
Š
t
ú
d
i a II LITHE
Š
t
ú
d
i a III OPTION
Š
t
ú
d
i a IV TO W ARD
Š
t
ú
d
i a V RADIATE
T ýž-
deň
TC Z
8 m g/k g
MTX TC Z
8 m g/k g
+ MTX
PBO + MTX
TC Z
8 m g/k g
+ MTX
PBO
+ MTX
TC Z
8 m g/k g
+
DMARD
PBO + DMARD
TC Z
8 m g/k g
+ MTX
PBO + MTX
N =
286
N =
284
N =
398
N =
393
N =
205
A
C R 20
N =
204
N =
803
N =
413
N =
170
N =
158
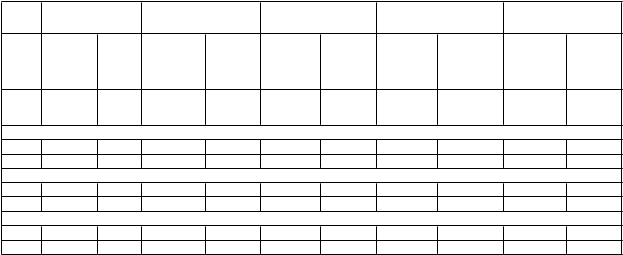
24 70 %*** 52 % 56 %*** 27 % 59 %*** 26 % 61 %*** 24 % 50 %*** 10 %
52 56 %*** 25 %
AC R 5024 44 %** 33 % 32 %*** 10 % 44 %*** 11 % 38 %*** 9 % 29 %*** 4 %
52 36 %*** 10 %
AC R 7024 28 %** 15 % 13 %*** 2 % 22 %*** 2 % 21 %*** 3 % 12 %** 1 %
52 20 %*** 4 %
TCZ - Tocilizum ab MTX - Metotrexát PBO - PlaceboDMARD - Antireum atikum m odifikujúce priebeh choroby** - p < 0,01, TCZ oproti PBO + MTX/DMARD*** - p < 0,0001, TCZ oproti PBO + MTX/DMARDVýznamná klinická odpoveďPo 2 rokoch liečby tocilizumabom s MTX dosiahlo 14 % pacientov významnú klinickú odpoveď
(udržanie ACR70 odpovede počas 24 týždňov alebo dlhšie).
Rádiograf ická odpoveďV štúdii II sa u pacientov s nedostatočnou odpoveďou na MTX hodnotila inhibícia štrukturálneho poškodenia kĺbov rádiograficky a vyjadrila sa ako zmena v modifikovanom Sharpovom skóre a jeho zložkách - skóre erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených tocilizumabom sa oproti kontrolnej skupine preukázala inhibícia štrukturálneho poškodenia kĺbov s významne nižšou rádiografickou progresiou ochorenia (tabuľka 4).
V otvorenej predĺženej fáze štúdie II bola inhibícia progresie štrukturálneho poškodenie kĺbu v skupine s tocilizumabom a MTX udržiavaná i v druhom roku liečby. Stredná zmena od východiskových hodnôt bola v 104. týždni v celkovom Sharp-Genatovom skóre významne nižšia u pacientov randomizovaných do skupiny s tocilizumabom v dávke 8 mg/kg a MTX (p < 0,0001) v porovnaní s pacientami, ktorí boli randomizovaní do skupiny s placebom a MTX.
T
abuľka 4. Rádiografické priemerné zmeny počas 52 týždňov v štúdii II
P
B
O + MTX (+TCZ od 24. týždňa) N = 393
T
C
Z 8 mg/k g + MTX N = 398
Celkové
Sharpovo-Genantovo
skóre
1,13 0,29*

Skóre erózie 0,71 0,17* Skóre JSN 0,42 0,12**
PBO - PlaceboMTX - MetotrexátTCZ - Tocilizum abJSN - Zúženie kĺbovej štrbiny* - p ≤ 0,0001, TCZ oproti PBO + MTX** - p < 0,005, TCZ oproti PBO + MTXPo 1 roku liečby tocilizumabom a MTX 85 % pacientov (N=348) nevykazovalo žiadnu progresiu štrukturálneho poškodenia kĺbov ako je definované v celkovom Sharpovom skóre 0 alebo menej,
v porovnaní so 67 % pacientov v skupine s placebom a MTX (n=290) (p ≤ 0,001). Tieto výsledky pretrvávali i po 2 rokoch liečby (83 %, n=353). Deväťdesiat tri percent (93 %; n=271) pacientov nevykazovalo žiadnu progresiu medzi 52. a 104. týždňom.
Zdravotné výsledky a výsledky týkajúce sa kvality životaPacienti liečení tocilizumabom hlásili zlepšenie vo všetkých výsledkoch hlásených pacientmi
(dotazník hodnotiaci zdravie a index funkčnej neschopnosti, - HAQ-DI), skrátený formulár 36
a dotazník funkčného hodnotenia liečby chronického ochorenia. U pacientov liečených RoActemrou sa oproti pacientom liečeným DMARDs pozorovalo štatisticky významné zlepšenie skóre HAQ-DI.
V priebehu otvorenej fázy štúdie II bolo udržanie zlepšenia fyzických funkcií až počas 2 rokov. V 52.
týždni bola stredná zmena v HAQ-DI - 0,58 v skupine s tocilizumabom 8 mg/kg a MTX v porovnaní
s 0,39 v skupine s placebom a MTX. Stredná zmena HAQ-DI bola v skupine s tocilizumabom 8 mg/kg a MTX udržovaná aj 104. týždni (-0,61).
Hladiny hemoglobínuPri liečbe tocilizumabom sa oproti liečbe DMARDs v 24. týždni pozorovalo štatisticky významné zlepšenie hladín hemoglobínu (p < 0,0001). Priemerné hodnoty hladín hemoglobínu sa zvýšili do
2. týždňa a udržali sa v referenčnom rozpätí až do 24. týždňa.
Tocilizumab verzus adalimumab v monoterapiiŠtúdia VI (WA19924), 24-týždňová dvojito zaslepená štúdia, ktorá porovnávala monoterapiu tocilizumabom s monoterapiou adalimumabom, hodnotila 326 pacientov s RA, ktorí netolerovali MTX alebo kde pokračovanie v liečbe MTX sa považovalo za nevhodné (vrátane nedostatočných respondérov na MTX). Pacienti v skupine s tocilizumabom dostávali intravenóznu (i.v.) infúziu tocilizumabu (8 mg/kg) každé 4 týždne a subkutánne (s.c.) injekciu s placebom každé 2 týždne. Pacienti v skupine s adalimumabom dostávali s.c. injekciu adalimumabu (40 mg) každé 2 týždne plus i.v. infúziu s placebom každé 4 týždne.
Pozoroval sa štatistický významný superiórny účinok liečby v prospech tocilizumabu v porovnaní s adalimumabom pri kontrole aktivity ochorenia od východiskovej hodnoty po 24. týždeň pre
primárny cieľový ukazovateľ zmenu DAS28 a pre všetky sekundárne cieľové ukazovatele (tabuľka 5).
T
abuľka 5. Výsledky účinnosti pre štúdiu VI (WA19924)
AD
A + placebo
 (
i
.
v
)
N = 162
T
o
c
ili
z
u
m
a
b +
p
l
a
ce
b
o (s .c.)
N = 163
(
i
.
v
)
N = 162
T
o
c
ili
z
u
m
a
b +
p
l
a
ce
b
o (s .c.)
N = 163 p-hodnota
(a)
P
r
i
m
á
r
n
y cieľový ukazovateľ – Priemerná zmena od východis kovej hodnoty v 24. týždni
DA
S
2
8 (upravený priemer) -1,8 -3,3
R
o
z
d
i
e
l v upravenom priemere (95 % IC)
-
1
,
5 (-1,8, -1,1) < 0,0001
S
e
k
un
d
á
r
n
e cieľové ukazovatele – Percento res pondérov v 24. týždni
(
b)
DAS28 < 2,6, n (%) 17 (10,5) 65 (39,9) < 0,0001
DAS28 ≤ 3,2, n (%) 32 (19,8) 84 (51,5) < 0,0001
ACR20 odpoveď, n (%) 80 (49,4) 106 (65,0) 0,0038
ACR50 odpoveď, n (%) 45 (27,8) 77 (47,2) 0,0002
ACR70 odpoveď, n (%) 29 (17,9) 53 (32,5) 0,0023
ap hodnota je upravená vzhľadom na oblasť a trvanie RA pre všetky cieľové ukazovatele a tiež východisková hodnota pre všetky pokračujúce cieľové ukazovatele.
b Neodpovedajúci na liečbu použití pre chýbajúce údaje. Multidisciplinárna kontrola použitím Bonferroni-Holm procedúry
Celkový klinický profil nežiaducich udalostí bol podobný pri tocilizumabe a adalimumabe. Podiel pacientov so závažnými nežiaducimi udalosťami bol medzi liečebnými skupinami (tocilizumab
11,7 % oproti adalimumabu 9,9 %). Nežiaduce účinky v skupine s tocilizumabom odpovedali známemu bezpečnostnému profilu tocilizumabu a nežiaduce účinky boli hlásené s podobnou frekvenciou v porovnaní s tabuľkou 1. Vyššia incidencia infekcií a infestácií bola hlásená v skupine
s tocilizumabom (48 % oproti 42 %), a to bez rozdielu v incidencii závažných infekcií (3,1 %). Obidve skúmané liečby indukovali rovnaké zmeny v laboratórnych bezpečnostných parametroch (poklesy počtu neutrofilov a krvných doštičiek, zvýšenie ALT, AST a lipidov), veľkosť zmien a frekvencie výrazných abnormalít však bola vyššia pri tocilizumabe v porovnaní s adalimumabom. U štyroch
(2,5 %) pacientov v skupine s tocilizumabom a dvoch (1,2 %) pacientov v skupine s adalimumabom sa vyskytli poklesy počtu neutrofilov 3. alebo 4. stupňa CTC. U jedenástich (6,8 %) pacientov v skupine
s tocilizumabom a piatich (3,1 %) pacientov v skupine s adalimumabom sa vyskytlo zvýšenie ALT 2. alebo vyššieho stupňa CTC. Priemerné zvýšenie LDL od východiskovej hodnoty bolo 0,64 mmol/l (25 mg/dl) u pacientov v skupine s tocilizumabom a 0,19 mmol/l (7 mg/dl) u pacientov v skupine s adalimumabom. Bezpečnosť pozorovaná v skupine s tocilizumabom sa zhodovala so známym bezpečnostných profilom tocilizumabu a nepozorovali sa žiadne nové alebo neočakávané nežiaduce liekové reakcie (pozri tabuľku 1).
Včasná RA, bez predchádzajúcej liečby MTX
Štúdia VII (WA19926), 2-ročná štúdia s plánovanou primárnou analýzou v 52. týždni hodnotila
1 162 dospelých pacientov so stredne ťažkou až ťažkou, aktívnou včasnou RA (priemerné trvanie ochorenia ≤ 6 mesiacov), ktorí neboli doteraz liečení MTX. Približne 20 % pacientov podstúpilo predchádzajúcu liečbu DMARDs inými ako MTX. Táto štúdia hodnotila účinnosť kombinovanej liečby i.v. tocilizumabom 4 alebo 8 mg/kg raz za 4 týždne/MTX, i.v. tocilizumabu 8 mg/kg
v monoterapii a MTX v monoterapii v zmierňovaní prejavov a príznakov a v spomaľovaní rýchlosti
progresie poškodenia kĺbov počas 104 týždňov. Primárny cieľový ukazovateľ bol podiel pacientov, ktorí v 24. týždni dosiahli remisiu ochorenia podľa DAS28 (DAS28 < 2,6). Významne vyšší podiel pacientov v skupine s tocilizumabom 8 mg/kg + MTX a v skupine s tocilizumabom v monoterapii dosiahol primárny cieľový ukazovateľ v porovnaní so samotným MTX. V skupine s tocilizumabom
8 mg/kg + MTX sa tiež preukázali štatisticky významné výsledky v porovnaní s kľúčovými
sekundárnymi cieľovými ukazovateľmi. V porovnaní so samotným MTX sa v skupine monoterapie tocilizumabom 8 mg/kg pozorovali percentuálne vyššie odpovede vo všetkých sekundárnych cieľových ukazovateľoch vrátane rádiografických cieľových ukazovateľov. V tejto štúdii sa tiež analyzovala remisia podľa kritérií ACR/EULAR (Boolean and Index) ako vopred špecifikované exploratívne cieľové ukazovatele, s vyššími odpoveďami pozorovanými v skupinách s tocilizumabom. Výsledky zo štúdie VII sú uvedené v tabuľke 6.
T
abuľka 6: Výsledky účinnosti v štúdii VII (WA19926) u pacientov so včasnou RA, bez
predchádzajúcej liečby MTX
T
C Z 8 m g/k g + MTX
N
=
290
T
C Z 8 m g/k g +
p
l ace bo
N
=
292
T
C Z 4 m g/k g + MTX
N
=
288
P
l ace bo + MTX
N
=
28
7
Remisia podľa DAS28
Pri m árn y ci eľový u kazovateľ
24. t ýždeň n (%) 130 (44,8)*** 113 (38,7)*** 92 (31,9) 43 (15,0)
Kľúčové s e kundárn e ci eľové u kazovatele
Remisia podľa DAS28
52. t ýždeň ACR, n (%) 142 (49,0)*** 115 (39,4) 98 (34,0) 56 (19,5)
24. t ýždeň ACR20, n (%) 216 (74,5)* 205 (70,2) 212 (73,6) 187 (65,2)
ACR50, n (%) 165 (56,9)** 139 (47,6) 138 (47,9) 124 (43,2)
ACR70, n (%) 112 (38,6)** 88 (30,1) 100 (34,7) 73 (25,4)
52. t ýždeň ACR20, n (%) 195 (67,2)* 184 (63,0) 181 (62,8) 164 (57,1) ACR50, n (%) 162 (55,9)** 144 (49,3) 151 (52,4) 117 (40,8) ACR70, n (%) 125 (43,1)** 105 (36,0) 107 (37,2) 83 (28,9)
HAQ-DI (upravená priemerná zmena oprot i východiskovej hodnot e)
52. t ýždeň -0,81* -0,67 -0,75 -0,64
Rádi ografické ci eľové u kazovatele (pri em erná z m ena oproti vých odisk ove j h odn ote)
52. t ýždeň mT SS 0,08*** 0,26 0,42 1,14
Skóre erózie 0,05** 0,15 0,25 0,63
JSN 0,03 0,11 0,17 0,51
Rádiograficky bez progresie n (%) (zmena oprot i východiskovej hodnot e v mT SS ≤ 0)
226 (83)‡ 226 (82)‡ 211 (79) 194 (73)
E
x
p
l oratí vn e ci eľové u kazovatele
24. t ýždeň: remisia podľa ACR/EULAR - Boolean, n (%)
47 (18,4)
‡ 38 (14,2) 43 (16,7)
25 (10,0)
remisia podľa ACR/EULAR - Index, n (%) 73 (28,5) ‡ 60 (22,6) 58 (22,6) 41 (16,4)
52. t ýždeň: remisia podľa ACR/EULAR - Boolean, n (%)
59 (25,7) ‡ 43 (18,7)
48 (21,1)
34 (15,5)

remisia podľa ACR/EULAR - Index, n (%) 83 (36,1)
‡ 69 (30,0) 66 (29,3) 49 (22,4)
mT SS - modified T ot al Sharp Score (celkové Sharpove skóre modifikované van der Heijdom) JSN - skóre Joint space narrowing (zúženie kĺbovej št rbiny )
Všet ky porovnania účinnost i vs. placebo + MT X. ***p ≤ 0,0001; **p < 0,001; *p < 0,05;
‡p-hodnot a < 0,05 vs. placebo + MT X, ale cieľový ukazovat eľ bol explorat ívny (nezahrnut ý v hierarchii št at istického t est ovania, a pret o nebol kont rolovaný na mult iplicit u)
Pediatrická populáciaPacienti so sJIAKlinická účinnosť
Účinnosť tocilizumabu pri liečbe aktívnej sJIA bola hodnotená v 12-týždňovej randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii s paralelnými skupinami a s dvoma ramenami.
Pacienti zaradení do klinického skúšania mali celkové trvanie choroby najmenej 6 mesiacov a aktívnu chorobu, ale bez akútneho vzplanutia vyžadujúceho dávku kortikosteroidov vyššiu ako ekvivalent prednizónu 0,5 mg/kg. Účinnosť na liečbu syndrómu aktivácie makrofágov nebola skúmaná.
Pacienti (liečení s MTX alebo bez neho) boli randomizovaní (tocilizumab:placebo = 2:1) do jednej
z dvoch liečených skupín, 75 pacientov dostávalo infúzie tocilizumabu každé dva týždne, buď
8 mg/kg pre pacientov s telesnou hmotnosťou ≥ 30 kg alebo 12 mg/kg pre pacientov s hmotnosťou
<30 kg a 37 pacientov dostávalo každé dva týždne infúzie s placebom. Postupné znižovanie dávok
kortikosteroidov bolo povolené od šiesteho týždňa u pacientov, ktorí dosiahli JIA ACR70 odpoveď. Po 12-týždňoch alebo v čase ukončenia kvôli zhoršeniu choroby boli pacienti liečení v otvorenej fáze dávkou zodpovedajúcou telesnej hmotnosti.
Klinická odpoveď
Primárny cieľový ukazovateľ bol podiel pacientov, ktorí dosiahli aspoň 30 % zlepšenie skóre JIA
ACR (odpoveď JIA ACR30) v 12. týždni a neprítomnosť horúčky (bez zaznamenania teploty ≥
37,5 °C počas predošlých 7 dní). Osemdesiat päť percent (64/75) pacientov liečených tocilizumabom a 24,3 % (9/37) pacientov liečených placebom dosiahlo tento cieľový ukazovateľ. Tieto percentá boli vysoko signifikantne odlišné (p<0,0001).
Percentá pacientov, ktorí dosiahli odpoveď JIA ACR 30, 50, 70 a 90 sú uvedené v tabuľke 7.
T
abuľka 7. Odpoveď JIA ACR v 12. týždni (% pacientov)
O
dp
oveď tocilizumab
N = 75
P
l
acebo
N = 37
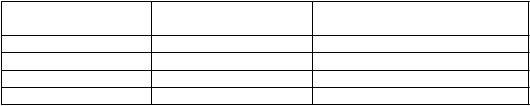
JIA ACR 30 90,7 %1 24,3 %
JIA ACR 50 85,3 %1 10,8 % JIA ACR 70 70,7 %1 8,1 % JIA ACR 90 37,3 %1 5,4 %
1
p<0,0001, tocilizum ab oproti placebuSystémové účinkyZ pacientov liečených tocilizumabom bolo 85 % takých, ktorí mali na začiatku štúdie horúčku kvôli sJIA a v 12. týždni boli bez horúčky (bez zaznamenania teploty ≥ 37,5 °C počas predošlých 14 dní) oproti 21 % pacientov s placebom (p<0,0001).
Upravenou priemernou zmenou vo VAS bolesti po 12 týždňoch liečby tocilizumabom bolo zníženie
o 41 bodov na škále 0 – 100 oproti zníženiu o 1 bod u pacientov s placebom (p<0,0001).
Postupné znižovanie dávok kortikosteroidovPacientom, ktorí dosiahli odpoveď JIA ACR70 bolo povolené zníženie dávky kortikosteroidov. Sedemnásť (24 %) pacientov liečených tocilizumabom oproti 1 (3 %) pacientovi s placebom bolo schopných znížiť svoju dávku kortikosteroidov o aspoň 20 % bez zaznamenania následného vzplanutia JIA ACR30 alebo objavenia sa systémových príznakov do 12. týždňa (p=0,028). Znižovanie kortikosteroidov pokračovalo, pričom 44 pacientov v 44. týždni úplne prestalo užívať kortikosteroidy pri súčasnom zachovaní odpovede JIA ACR.
Zdravotné výsledky a výsledky týkajúce sa kvality životaV 12. týždni bol pomer pacientov liečených tocilizumabom s minimálnym klinicky významným zlepšením v Detskom dotazníku hodnotenia zdravia – indexe neschopnosti (definovanom ako individuálne zníženie skóre ≥0,13) signifikantne vyšší ako u pacientov liečených placebom, 77 % oproti 19 % (p<0,0001).
Laboratórne parametrePäťdesiat zo sedemdesiat päť (67 %) pacientov liečených tocilizumabom malo na začiatku štúdie hemoglobín <LLN. Štyridsať (80 %) z týchto pacientov zaznamenalo zvýšenie hemoglobínu na
úroveň v rámci normálnych hodnôt v 12. týždni, v porovnaní s iba 2 z 29 (7 %) pacientov liečených placebom s hemoglobínom na pôvodnej hodnote (p<0,0001).
Pacienti s pJIA
Klinická účinnosť
Účinnosť tocilizumabu sa hodnotila v štúdii WA19977 s tromi časťami vrátane otvoreného rozšírenia u detí s aktívnou pJIA. Časť I pozostávala zo 16-týždnovej, aktívnej, začiatočnej etapy liečby tocilizumabom (n = 188), po ktorej nasledovala časť II, 24-týždňové, randomizované, dvojito zaslepené, placebom kontrolované obdobie ukončenia liečby (n = 163), a po nej časť III, 64-týždňové otvorené obdobie. V časti I vhodní pacienti ≥ 30 kg dostávali tocilizumab v dávke 8 mg/kg intravenózne 4 dávky každé 4 týždne. Pacienti < 30 kg boli randomizovaní 1:1 a dostávali buď
8 mg/kg alebo 10 mg/kg tocilizumabu i.v. každé 4 týždne 4 dávky. Pacienti, ktorí dokončili časť I štúdie a dosiahli aspoň JIA ACR30 odpoveď v 16. týždni v porovnaní s východiskovou hodnotou boli vhodní vstúpiť do zaslepeného obdobia s ukončenou liečbou (časť II). V časti II boli pacienti randomizovaní na tocilizumab (rovnakú dávku dostávali v časti I) alebo na placebo v pomere 1:1, boli stratifikovaní podľa súčasného použitia MTX a súčasného použitia kortikosteroidov. Každý pacient pokračoval v štúdii v časti II až do 40. týždňa alebo kým pacient dosiahol JIA ACR30 kritériá vzplanutia (oproti 16. týždňu) a bol vhodný pre ukončenie liečby tocilizumabom (rovnakú dávku dostali v časti I) .
Klinická odpoveď
Primárnym koncovým ukazovateľom bol podiel pacientov s JIA ACR30 vzplanutím v 40. týždni vzhľadom k 16. týždňu. Štyridsaťosem percent (48,1 %, 39/81) pacientov liečených placebom malo vzplanutie v porovnaní s 25,6 % (21/82) pacientov liečených tocilizumabom. Tieto podiely boli štatisticky významne rozdielne (p = 0,0024).
V závere časti I boli JIA ACR 30/50/70/90 odpovede 89,4 %; 83,0 %; 62,2 % a 26,1 %; v uvedenom
poradí.
V priebehu fázy vysadenia liečby (časť II) percento pacientov, ktorí dosiahli JIA ACR odpovede 30,
50 a 70 v 40. týždni v porovnaní s východiskovými hodnotami je uvedené v tabuľke 8. V tejto štatistickej analýze pacienti, u ktorých došlo počas časti II k vzplanutiu ochorenia (a prešli do tocilizumabovej skupiny) alebo ktorí odstúpili zo štúdie, boli klasifikovaní ako neodpovedajúci na liečbu. Ďalšie analýzy JIA ACR odpovedí, vzhľadom na pozorované údaje v 40.týždni, bez ohľadu na stav vzplanutia, ukázali, že do týždňa 40 91,5 % pacientov, ktorí dostávali kontinuálnu liečbu tocilizumabom dosiahli JIAACR 30 alebo vyššiu.
Tabuľka 8. Pozorovaný výskyt JIA ACR odpovede v 40. týždni v porovnaní s východiskovými
hodnotami (percento pacientov)
V
ýsk yt odpovede tocilizumab
N=82
placebo
N=81

ACR 30 74,4%* 54,3%*
ACR 50 73,2%* 51,9%*
ACR 70 64,6%* 42,0%*
*
p < 0,01, tocilizum ab oproti placebuPočet aktívnych kĺbov bol signifikantne nižší v porovnaní s východiskovými hodnotami u pacientov liečených tocilizumabom v porovnaní s placebom (upravené priemery zmien -14,3 oproti -11,4,
p = 0,0435). Celkové hodnotenie aktivity ochorenia lekárom, merané na škále 0-100 mm, ukázalo výrazné zníženie aktivity ochorenia u tocilizumabu v porovnaní s placebom (upravené priemery zmien
-45,2 mm oproti -35,2 mm, p = 0,0031).
Upravená priemerná zmena bolesti podľa VAS po 40. týždňoch liečby tocilizumabom bola 32,4 mm, na škále 0 - 100 mm, v porovnaní so znížením 22,3 mm u pacientov dostávajúcich placebo (výrazne štatisticky významné; p = 0,0076).
Výskyt ACR odpovedí bol početne nižší u pacientov s predchádzajúcou biologickou liečbou ako je uvedené v tabuľke 9 nižšie.
Tabuľka 9. Počet a podiel pacientov so vzplanutím ochorenia JIA ACR30 a podiel pacientov s odpoveďou JIA ACR30/50/70/90 v 40.týždni pri predchádzajúcej biologickej liečbe (OITT populácia – časť II štúdie)Placebo Všetci s tocilizumabom Biologick á liečba Áno (N =
23) Nie (N =
58) Áno (N =
27) Nie (N =
55) Vzplanutie JIA ACR30 18 (78,3) 21 (36,2) 12 (44,4) 9 (16,4) Odpoveď JIA ACR30 6 (26,1) 38 (65,5) 15 (55,6) 46 (83,6) Odpoveď JIA ACR50 5 (21,7) 37 (63,8) 14 (51,9) 46 (83,6) Odpoveď JIA ACR70 2 (8,7) 32 (55,2) 13 (48,1) 40 (72,7)
Odpoveď JIA ACR90 2 (8,7) 17 (29,3) 5 (18,5) 32 (58,2)
Pacienti randomizovaní na liečbu tocilizumabom mali menej vzplanutí ochorenia ACR30 a vyššie
celkové odpovede ACR ako pacienti, ktorí dostávali placebo bez ohľadu na predchádzajúcu biologickú liečbu.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdii pre RoActemru vo všetkých vekových podskupinách detí a dospievajúcich s reumatoidnou artritídou a udelila odklad z povinnosti predložiť výsledky štúdií s RoActemrou v jednej alebo vo viacerých vekových podskupinách detí a dospievajúcich s juvenilnou idiopatickou artritídou. Pre informácie o použití
u detí a dospievajúcich, pozri časť 4.2.
5.2 Farmak ok inetické vlastnostiPacienti s RAIntravenóznepoužitieFarmakokinetika tocilizumabu sa stanovila za použitia populačnej farmakokinetickej analýzy údajov z databázy zloženej z 3 552 pacientov s RA liečených dávkou 4 alebo 8 mg/kg tocilizumabu podávanou raz za 4 týždne formou jednu hodinu trvajúcej infúzie alebo so 162 mg tocilizumabu podávaného subkutánne buď raz týždenne alebo každý druhý týždeň počas 24 týždňov.
Nasledujúce parametre (predpokladaný priemer ± SD, štandardná odchýlka) sa odhadli pre dávku
8 mg/kg tocilizumabu podávanú raz za 4 týždne: plocha pod krivkou pre plazmatickú koncentráciu
(AUC) v rovnovážnom stave = 38 000 ± 13 000 h µg/ml, minimálna koncentrácia
(Cmin) =15,9± 13,1 µg/ml a maximálna koncentrácia (Cmax) = 182 ± 50,4 µg/ml a pomer kumulácie v hodnote 1,32 pri AUC a1,09 pri Cmax bol nízky. Pomer kumulácie bol vyšší pri Cmin (2,49), čo sa očakávalo na základe prispenia nelineárneho klírensu pri nižších koncentráciách. Rovnovážny stav sa dosiahol po podaní prvej dávky pri hodnote Cmax, po 8 týždňoch pri hodnote AUC a po 20 týždňoch pri hodnote Cmin. AUC, Cmin a Cmax tocilizumabu vzrástlo so stúpajúcou telesnou hmotnosťou. Pri telesnej hmotnosti ≥ 100 kg bol predpovedaný priemer (± SD) AUC tocilizumabu v rovnovážnom stave50 000 ± 16 800 μg•h/ml, Cmin tocilizumabu 24,4 ± 17,5 μg/ml a Cmax tocilizumabu
226 ± 50,3 μg/ml, čo sú vyššie hodnoty, než hodnoty pri priemernej expozícii v súbore pacientov (t.j.
celková hmotnosť všetkých pacientov) ako je uvedené vyššie. Krivka odpovede na dávku sa pri
tocilizumabe pri vyšších expozíciách splošťuje, čo vedie k nižšiemu nárastu účinnosti pre každé ďalšie zvýšenie koncentrácie tocilizumabu, takže u pacientov liečených tocilizumabom dávkou > 800 mg
nedochádza už k žiadnemu zmysluplnému zvýšeniu účinnosti. Preto sa neodporúčajú dávky, ktoré
presahujú 800 mg na infúziu (pozri časť 4.2).
Distribúcia
U pacientov s RA bol distribučný objem centrálneho kompartmentu 3,72, distribučný objem periférneho kompartmentu bol 3,35, čo malo za následok distribučný objem 7,07 v rovnovážnom stave.
Vylučovanie
Po intravenóznom podaní dávky podlieha tocilizumab dvojfázovému vylučovaniu z cirkulácie. Celkový klírens tocilizumabu bol závislý od koncentrácie a je súčtom lineárneho a nelineárneho klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze na
9,5 ml/h. Od koncentrácie závislý nelineárny klírens zohráva hlavnú úlohu pri nízkych koncentráciách tocilizumabu. Keď je cesta nelineárneho klírensu nasýtená, pri vyšších koncentráciách tocilizumabu je klírens určovaný hlavne lineárnym klírensom.
t1/2 tocilizumabu bol závislý od koncentrácie. V rovnovážnom stave sa po dávke 8 mg/kg podávanej raz za 4 týždne efektívny t1/2 znižoval so znižujúcimi sa koncentráciami v rámci dávkovacieho intervalu od 18 dní do 6 dní.
Linearita
Farmakokinetické parametre tocilizumabu sa postupom času nezmenili. Pri dávkach 4 a 8 mg/kg podávaných raz za 4 týždne sa pozorovalo vyššie ako dávke úmerné zvýšenie hodnoty AUC a Cmin. Hodnota Cmax sa zvyšovala úmerne dávke. V rovnovážnom stave bola pri dávke 8 mg/kg predpokladaná hodnota AUC 3,2-násobne a hodnota Cmin 30-násobne vyššia než pri dávke 4 mg/kg.
Osobitné skupiny pacientov
Porucha f unkcie obličiek: Štúdia vplyvu poruchy funkcie obličiek na farmakokinetiku tocilizumabu sa neuskutočnila. Väčšina pacientov zaradených do populačnej farmakokinetickej analýzy mala
normálnu funkciu obličiek alebo miernu poruchu funkcie obličiek. Mierna porucha funkcie obličiek (klírens kreatinínu podľa Cockcrofta-Gaulta < 80 ml/min a ≥ 50 ml/min) nemala vplyv na farmakokinetiku tocilizumabu.
Porucha f unkcie pečene: Štúdia vplyvu poruchy funkcie pečene na farmakokinetiku tocilizumabu sa neuskutočnila.
Vek, pohlavie a etnická príslušnosť: Populačné farmakokinetické analýzy u dospelých pacientov s RA
preukázali, že vek, pohlavie a etnická príslušnosť nemajú vplyv na farmakokinetiku tocilizumabu.
Pacienti so sJIA:
Farmakokinetika tocilizumabu bola stanovená pomocou populačnej farmakokinetickej analýzy databázy 75 pacientov so sJIA liečených s 8 mg/kg (pacienti s hmotnosťou ≥ 30 kg) alebo 12 mg/kg (pacienti s hmotnosťou <30 kg) podávanými každé dva týždne. Predpokladané priemerné (± SD) AUC2týždne, Cmax a Cmin tocilizumabu boli 32200 ± 9960 μg•h/ml, 245 ± 57,2 µg/ml a 57,5 ±
23,3 µg/ml. Akumulačný pomer Cmin (12. týždeň / 2. týždeň) bol 3,2 ± 1,3. Cmin pre tocilizumab sa stabilizoval po 12. týždni. Priemerné predpokladané parametre expozície tocilizumabu boli podobné v oboch skupinách telesnej hmotnosti.
U pacientov so sJIA bol centrálny distribučný objem 35 ml/kg a periférny distribučný objem 60 ml/kg, čo viedlo k distribučnému objemu pri nasýtenom stave 95 ml/kg. Odhadovaný lineárny klírens ako parameter populačnej farmakokinetickej analýzy bol 0,142 ml/h/kg.
Biologický polčas tocilizumabu u pacientov so sJIA je v 12. týždni až 23 dní pre obe hmotnostné kategórie ( 8 mg/kg pre telesnú hmotnosť ≥ 30 kg alebo 12 mg/kg pre telesnú hmotnosť <30 kg).
Pacienti s pJIA:
Farmakokinetika tocilizumabu bola stanovená pomocou populačnej farmakokinetickej analýzy databázy 188 pacientov s pJIA.
Nasledovné parametre sú platné pre dávku 8 mg/kg tocilizumabu (pacienti s telesnou hmotnosťou
≥ 30 kg) podávanú raz za 4 týždne. Predpokladané priemerné (± SD) AUC4týždne, Cmax a Cmin
tocilizumabu boli 29 500 ± 8 660 μg•h/ml, 182 ± 37 µg/ml a 7,49 ± 8,20 µg/ml, v uvedenom poradí.
Nasledovné parametre sú platné pre dávku 10 mg/kg tocilizumabu (pacienti s telesnou hmotnosťou
< 30 kg) podávanú raz za 4 týždne. Predpokladané priemerné (± SD) AUC4týždne, Cmax a Cmin
tocilizumabu boli 23 200 ± 6 100 μg•h/ml, 175 ± 32 µg/ml a 2,35 ± 3,59 µg/ml, v uvedenom poradí.
Akumulačné pomery boli 1,05 a 1,16 pre AUC4týždne, a 1,43 a 2,22 pre Cmin pre dávku 10 mg/kg (telesná hmotnosť < 30 kg) a 8 mg/kg (telesná hmotnosť ≥ 30 kg). Nepozorovala sa žiadna kumulácie pre Cmax.
U pacientov s pJIA bol centrálny distribučný objem 50 ml/kg, periférny distribučný objem bol
53 ml/kg, čo viedlo k distribučnému objemu v rovnovážnom stave 103 ml/kg. Odhadovaný lineárny
klírens ako parameter populačnej farmakokinetickej analýzy bol 0,146 ml/h/kg.
Biologický polčas tocilizumabu u pacientov s pJIA je 16 dní pre obe hmotnostné kategórie (8 mg/kg pre telesnú hmotnosť ≥ 30 kg alebo 10 mg/kg pre telesnú hmotnosť <30 kg) počas dávkového intervalu v rovnovážnom stave.
5.3 Predk linick é údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
Štúdie karcinogenity sa neuskutočnili pretože IG1 monoklonálne protilátky sa nepovažujú za látky s vlastným karcinogénnym potenciálom.
Dostupné predklinické údaje preukázali vplyv IL-6 na progresiu zhubných nádorov a na rezistenciu rôznych typov nádorov na apoptózu. Tieto údaje nepoukazujú na významné riziko pre vznik
a progresiu rakoviny počas liečby tocilizumabom. Okrem toho sa v 6-mesačných štúdiách chronickej toxicity na opiciach rodu Cynomolgus, ani u myší s deficitom IL-6 proliferatívne lézie nepozorovali.
Dostupné predklinické údaje nepotvrdili, že liečba tocilizumabom má vplyv na fertilitu. V štúdii chronickej toxicity na opiciach rodu Cynomolgus sa nepozorovali účinky na endokrinne aktívne orgány a na orgány reprodukčného systému a u myší s deficitom IL-6 nedošlo k poškodeniu reprodukčnej výkonnosti. Zistilo sa, že tocilizumab podávaný opiciam rodu Cynomolgus počas skorej fázy gestácie nemal priamy ani nepriamy škodlivý vplyv na graviditu alebo embryofetálny vývoj. Pozorovalo sa však mierne zvýšenie potratov/embryofetálnej úmrtnosti pri vysokej systémovej expozícii (> 100-násobok expozície dosiahnutej u ľudí) v skupine liečenej vysokou dávkou
50 mg/kg/deň oproti skupine liečenej placebom a inými nízkymi dávkami. Hoci IL-6 zrejme nie je rozhodujúcim cytokínom pre rast plodu alebo imunologickú kontrolu rozhrania materských
a fetálnych tkanív, súvislosť tohto zistenia s tocilizumabom nie je možné vylúčiť.
Liečba myšacím analógom na juvenilných myšiach nevykazovala toxicitu. Konkrétne, nebolo prítomné žiadne narušenie rastu kostí, imunitných funkcií a sexuálneho dozrievania
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Polysorbát 80
Dodekahydrát hydrogenfosforečnanu sodného Dihydrát dihydrogenfosforečnanu sodného Voda na injekciu
6.2 Ink ompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka: 30 mesiacov
Nariedený liek: Po nariedení je pripravený infúzny roztok fyzikálne a chemicky stabilný v injekčnom roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %) po dobu 24 hodín pri teplote 30 °C.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania pred použitím je zodpovedný používateľ a za normálnych podmienok by nemali presiahnuť 24 hodín pri teplote 2 - 8 °C, pokiaľ sa zriedenie neudialo
v kontrolovaných a overených aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Injekčnú liekovku (injekčné liekovky) uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčné liekovky uchovávajte v škatuli na ochranu pred svetlom.
Podmienky na uchovávanie nariedeného lieku pozri v časti 6.3.
6.5 Druh obalu a obsah balenia
RoActemra sa dodáva v injekčnej liekovke (sklo typu I) s uzáverom (butylkaučuk) s obsahom 4 ml,
10 ml alebo 20 ml koncentrátu. Veľkosti balenia po 1 a 4 injekčných liekovkách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návodnanariedeniepredpodaním
Parenterálne lieky sa majú pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc
alebo zmenu sfarbenia. Nariediť sa môžu iba roztoky, ktoré sú číre až opaleskujúce, bezfarebné až svetložlté a bez viditeľných častíc.
Pacientis RA
Za aseptických podmienok odoberte zo 100 ml infúzneho vaku objem sterilného, nepyrogénneho,
0,9 % injekčného roztoku chloridu sodného (9 mg/ml), ktorý sa rovná objemu koncentrátu RoActemry potrebného na dávku pre pacienta. Potrebné množstvo koncentrátu RoActemry (0,4 ml/kg) sa má odobrať z injekčnej liekovky a preniesť do 100 ml infúzneho vaku. Konečný objem má byť 100 ml. Roztok premiešajte tak, že infúzny vak jemne prevrátite, aby sa predišlo speneniu.
P
oužitie u detí a dospievajúcichPacientisosJIAaPJIA≥30 kgZa aseptických podmienok odoberte zo 100 ml infúzneho vaku objem sterilného, nepyrogénneho injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), ktorý sa rovná objemu koncentrátu RoActemry potrebného na dávku pre pacienta. Potrebné množstvo koncentrátu RoActemry
(0,4 ml/kg) sa má odobrať z injekčnej liekovky a preniesť do 100 ml infúzneho vaku. Konečný objem má byť 100 ml. Roztok premiešajte tak, že infúzny vak jemne prevrátite, aby sa predišlo speneniu.
PacientisosJIA<30 kgZa aseptických podmienok odoberte z 50 ml infúzneho vaku objem sterilného, nepyrogénneho, 0,9 % injekčného roztoku chloridu sodného (9 mg/ml), ktorý sa rovná objemu koncentrátu RoActemry potrebného na dávku pre pacienta. Potrebné množstvo koncentrátu RoActemry
(0,6 ml/kg) sa má odobrať z injekčnej liekovky a preniesť do 50 ml infúzneho vaku. Konečný objem má byť 50 ml. Roztok premiešajte tak, že infúzny vak jemne prevrátite, aby sa predišlo speneniu.
PacientispJIA<30 kgZa aseptických podmienok odoberte z 50 ml infúzneho vaku objem sterilného, nepyrogénneho, 0,9 % injekčného roztoku chloridu sodného (9 mg/ml), ktorý sa rovná objemu koncentrátu RoActemry potrebného na dávku pre pacienta. Potrebné množstvo koncentrátu RoActemry (
0,5 ml/kg) sa má odobrať z injekčnej liekovky a preniesť do 50 ml infúzneho vaku. Konečný objem má byť 50 ml. Roztok premiešajte tak, že infúzny vak jemne prevrátite, aby sa predišlo speneniu.
RoActemra je určená iba na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLAEU/1/08/492/001
EU/1/08/492/002
EU/1/08/492/003
EU/1/08/492/004
EU/1/08/492/005
EU/1/08/492/006
9. DÁTUM PRVEJ REGISTRÁCIE/ DÁTUM POSLEDNÉHO PREDĹŽENIADátum prvej registrácie: 16. január 2009
Dátum posledného predĺženia registrácie: 25. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.
1. NÁZOV LIEKU
RoActemra 162 mg injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá naplnená injekčná striekačka obsahuje 162 mg tocilizumabu v 0,9 ml.
Tocilizumab je rekombinantná humanizovaná antihumánna monoklonálna protilátka podtriedy imunoglobulínu G1 (IgG1) namierená proti solubilným a membránovo viazaným receptorom pre interleukín 6.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia). Bezfarebný až svetložltkastý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutick é indik ácie
RoActemra v kombinácii s metotrexátom (MTX) je indikovaná na
• liečbu ťažkej, aktívnej a progresívnej reumatoidnej artritídy (RA) u dospelých, ktorí neboli doteraz liečení MTX.
• liečbu stredne ťažkej až ťažkej aktívnej RA u dospelých pacientov, ktorí na predchádzajúcu liečbu jedným alebo viacerými antireumatikami modifikujúcimi priebeh choroby (DMARDs), alebo inhibítormi tumor nekrotizujúceho faktora (TNF) buď neodpovedali dostatočne, alebo ju netolerovali.
U týchto pacientov sa RoActemra môže podávať v monoterapii v prípade intolerancie MTX, alebo keď je pokračujúca liečba MTX nevhodná.
Dokázalo sa, že RoActemra spomaľuje progresiu poškodenia kĺbov meranú RTG vyšetrením
a zlepšuje fyzické funkcie, keď sa podáva v kombinácii s metotrexátom.
RoActemra v kombinácii s metotrexátom (MTX) je indikovaná na liečbu polyartikulárnej juvenilnej idiopatickej artritídy (pJIA; s pozitívnym alebo negatívnym reumatoidným faktorom a pretrvávajúcou oligoatritídou) u pacientov vo veku 2 rokov a starších, ktorí nedostatočne odpovedali na predošlú liečbu MTX.
RoActemra sa môže podávať v monoterapii v prípade intolerancie MTX, alebo keď je pokračujúca liečba MTX nevhodná.
RoActemra je indikovaná na liečbu obrovskobunkovej arteritídy (OBA) u dospelých pacientov.
4.2 Dávk ovanie a spôsob podávania
Subkutánna forma tocilizumabu sa podáva naplnenou injekčnou striekačkou na jednorazové použitie vybavenej bezpečnostným krytom ihly (pre-filled syringe (PFS) + needle safety device (NSD)). Liečbu majú začať zdravotnícki pracovníci, ktorí majú skúsenosti s diagnostikou a liečbou RA, pJIA a
/ alebo OBA. Prvá injekcia sa má podať pod dohľadom kvalifikovaného zdravotníckeho pracovníka.
Pacient si môže sám injekčne podávať RoActemru len vtedy, keď lekár rozhodne, že je to vhodné
a keď pacient súhlasí s lekárskymi prehliadkami vykonávanými podľa potreby a bol zaškolený
v správnej injekčnej technike.
Pacienti, ktorí prechádzajú z liečby i.v. podávaným tocilizumabom na subkutánne podávanie, si majú prvú s.c. dávku podať v čase ďalšej plánovanej i.v. dávky pod dohľadom kvalifikovaného zdravotníckeho pracovníka.
Všetkým pacientom, ktorí sú liečení RoActemrou, sa má poskytnúť karta pre pacienta.
Je potrebné zhodnotiť vhodnosť pacienta alebo rodiča/opatrovateľa na subkutánne podávanie lieku v domácom prostredí a pacienti alebo rodič/opatrovateľ musia byť poučení pred podaním ďalšej dávky, že ak sa objavia príznaky alergickej reakcie, musia o tom informovať zdravotníckeho pracovníka. Pacienti musia vyhľadať okamžitú lekársku pomoc, ak sa budú vyvíjať príznaky závažných alergických reakcií (pozri časť 4.4).
Dávkovanie
Subkutánna forma RoActemry nie je určená na intravenózne podávanie.
RA
Odporúčané dávkovanie je 162 mg raz za týždeň subkutánne.
K dispozícii sú obmedzené informácie týkajúce sa prestavenia pacientov z intravenóznej formy RoActemry na subkutánnu formu RoActemry vo fixnej dávke. Je potrebné dodržiavať interval podávania raz za týždeň.
Pacienti, ktorí prechádzajú z intravenóznej na subkutánnu formu, si majú podať svoju prvú subkutánnu dávku namiesto ďalšej plánovanej intravenóznej dávky pod dohľadom kvalifikovaného
zdravotníckeho pracovníka.
OBA
Odporúčané dávkovanie je 162 mg raz za týždeň subkutánne v kombinácii s postupným znižovaním dávky glukokortikoidov. Po ukončení podávania glukokortikoidov sa môže RoActemra podávať samostatne.
RoActemra v monoterapii sa nemá podávať na liečbu akútnych relapsov (pozri časť 4.4).
Vzhľadom na chronický charakter OBA sa musí liečba dlhšia ako 52 týždňov usmerňovať podľa prejavov (aktivity) ochorenia, zváženia lekára a voľby pacienta.
RA a OBA
Úpravy dávky kvôli laboratórnymodchýlkam(pozričasť4.4).
• Odchýlky hodnôt pečeňových enzýmov
Laboratórna hodnota Opatrenie
> 1- až 3-násobok
hornej hranice
referenčného rozpätia (ULN)
Úprava dávky súbežne podávaných DMARDs (RA) alebo
imunomodulačných látok (OBA) , ak je to vhodné.
Pri pretrvávajúcich vzostupoch v tomto rozpätí znížte frekvenciu podávania RoActemry na injekciu podanú každý druhý týždeň alebo prerušte podávanie RoActemry, kým nedôjde k normalizácii hodnôt alanínaminotransferázy (ALT) alebo aspartátaminotransferázy (AST).
Liečbu znovu začnite injekciou podanou každý týždeň alebo každý druhý týždeň, ak je to klinicky vhodné.
> 3- až 5-násobok
ULN
Prerušte podávanie RoActemry, pokým nebude hodnota < 3-násobok ULN a
postupujte podľa vyššie uvedených odporúčaní pre > 1- až 3-násobok ULN.
Pri pretrvávajúcich vzostupoch > 3-násobok ULN (potvrdených opakovaným vyšetrením, pozri časť 4.4) ukončite liečbu RoActemrou.
> 5-násobok ULN Ukončite liečbu RoActemrou.
• Nízky absolútny počet neutrofilov (ANC, absolute neutrophil count)
U pacientov, ktorí neboli doteraz liečení RoActemrou a majú absolútny počet neutrofilov (ANC) nižší
ako 2 x 109/l, sa neodporúča začať liečbu.
Laboratórna hodnota
(bunky x 109/l)
Opatrenie

ANC > 1 Udržiavajte dávku.
ANC 0,5 až 1 Prerušte podávanie RoActemry.
Keď sa ANC zvýši na > 1 x 109/l, liečbu RoActemrou znovu začnite injekciou podanou každý druhý týždeň a zvýšte frekvenciu podávania na injekciu podanú každý týždeň, ak je to klinicky vhodné.
ANC < 0,5 Ukončite liečbu RoActemrou.
• Nízky počet trombocytov
Laboratórna hodnota
(bunky x 103/μl)
Opatrenie

50 až 100 Prerušte podávanie RoActemry.
Keď bude počet trombocytov > 100 x 103/μl, liečbu RoActemrou znovu začnite injekciou podanou každý druhý týždeň a zvýšte frekvenciu podávania na injekciu podanú na každý týždeň, ak je to klinicky vhodné.
< 50 Ukončite liečbu RoActemrou.
VynechanádávkaAk pacient vynechá subkutánnu injekciu RoActemry, ktorú si podáva raz za týždeň, do 7 dní
od plánovanej dávky, treba ho poučiť, aby si vynechanú dávku podal v deň, v ktorý je plánovaná
ďalšia dávka. Ak pacient vynechá subkutánnu injekciu RoActemry, ktorú si podáva každý druhý týždeň, do 7 dní od plánovanej dávky, treba ho poučiť, aby si vynechanú dávku podal ihneď a ďalšiu dávku v deň, v ktorý je plánovaná ďalšia dávka.
Osobitné skupiny pacientovStarší pacientiU pacientov vo veku 65 rokov a starších nie je potrebná úprava dávky.
Porucha f unkcie obličiek:U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávky. U pacientov s ťažkou poruchou funkcie obličiek sa RoActemra neskúmala (pozri časť 5.2). U týchto pacientov sa má starostlivo monitorovať funkcia obličiek.
Porucha f unkcie pečene:U pacientov s poruchou funkcie pečene sa RoActemra neskúmala. Preto nie je možné poskytnúť
odporúčania na úpravu dávkovania.
Pediatrickí pacientiBezpečnosť a účinnosť subkutánnej formy RoActemry u detí od narodenia do menej ako 2 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Pacienti s pJIA:Zmena dávky má byť založená iba na konzistentnej zmene telesnej hmotnosti pacienta v čase.
Odporúčané dávkovanie pre pacientov starších ako 2 roky je 162 mg subkutánne raz za 2 týždne u pacientov s telesnou hmotnosťou vyššou alebo rovnou 30 kg alebo 162 mg subkutánne
raz za 3 týždne u pacientov s telesnou hmotnosťou nižšou ako 30 kg.
Ak je to vhodné, dávka súbežne podávaného MTX a/alebo iných liekov sa má upraviť alebo podávanie sa má zastaviť a dávkovanie tocilizumabu sa má prerušiť až do zhodnotenia klinického stavu. Pretože je veľa pridružených chorobných stavov pri pJIA, ktoré môžu ovplyvniť laboratórne hodnoty, rozhodnutie o ukončení používania tocilizumabu pre laboratórne odchýlky má byť založené na lekárskom zhodnotení každého individuálneho pacienta.
• Odchýlky hodnôt pečeňových enzýmov
Laboratórna hodnota
Opatrenie
> 1- až 3-násobok
ULN
> 3- až 5-násobok
ULN
Úprava dávky súbežne podávaného MTX, ak je to vhodné
Pri pretrvávajúcich vzostupoch v tomto rozpätí prerušte podávanie RoActemry, kým nedôjde k normalizácii hodnôt ALT alebo AST.
Upravte dávku súbežne podávaného MTX, ak je to vhodné

Prerušte podávanie RoActemry, pokým nebude hodnota
< 3-násobok ULN a postupujte podľa vyššie uvedených odporúčaní pre > 1- až 3-násobok ULN
> 5-násobok
ULN
Ukončite liečbu RoActemrou.
Rozhodnutie o ukončení používania RoActemry u pacientov
s pJIA pre laboratórne odchýlky má byť založené na lekárskom zhodnotení každého individuálneho pacienta.
• Nízky absolútny počet neutrofilov (ANC)
Laboratórna hodnota(bunk y x 109/l)ANC > 1 Udržiavajte dávku
 Opatrenie
Opatrenie
ANC 0,5 až 1 Prerušte podávanie RoActemry
Keď sa ANC zvýši na > 1 x 109/l, liečbu RoActemrou znovu začnite
ANC < 0,5 Ukončite liečbu RoActemrou
Rozhodnutie o ukončení používania RoActemry u pacientov
s pJIA pre laboratórne odchýlky má byť založené na lekárskom zhodnotení každého individuálneho pacienta.
• Nízky počet trombocytov
Laboratórna hodnota
(bunk y x 103/µ l)
Opatrenie
50 až 100 Úprava dávky súbežne podávaného MTX, ak je to vhodné
Prerušte podávanie RoActemry
Keď bude počet trombocytov > 100 x 103/µl, liečbu

RoActemrou znovu začnite.
< 50 Ukončite liečbu RoActemrou.
Rozhodnutie o ukončení používania RoActemry u pacientov
s pJIA pre laboratórne odchýlky má byť založené na lekárskom zhodnotení každého individuálneho pacienta.
Zníženie dávky tocilizumabu kvôli laboratórnym odchýlkam sa u pacientov s pJIA neskúmalo.
Bezpečnosť a účinnosť subkutánnej formy RoActemry u detí s ochoreniami inými ako pJIA neboli stanovené.
Dostupné údaje z intravenózneho podávania naznačujú, že klinické zlepšenie bolo pozorované do 12
týždňov od začiatku liečby RoActemrou. Pokračovanie liečby sa má starostlivo zvážiť u pacientov, u ktorých sa neprejavilo zlepšenie v rámci tohto časového obdobia.
Vynechaná dávkaAk pacient s pJIA vynechá subkutánnu injekciu RoActemry do 7 dní od plánovanej dávky, vynechanú
dávku si má podať hneď, ako si spomenie, a ďalšiu dávku si má podať v pôvodne plánovanom čase.
Ak pacient vynechá subkutánnu injekciu RoActemry o viac ako 7 dní od plánovanej dávky alebo si nie
je istý, kedy si má podať injekciu RoActemry, má zatelefonovať svojmu lekárovi alebo lekárnikovi.
SpôsobpodávaniaRoActemra je na subkutánne použitie.
Po náležitej inštruktáži o injekčnej technike si pacienti môžu sami injekčne podávať RoActemru, ak ich lekár rozhodne, že je to vhodné. Celý obsah (0,9 ml) naplnenej injekčnej striekačky sa má podať formou subkutánnej injekcie. Odporúčané miesta vpichu (brucho, stehno, horná časť ramena) sa majú striedať a injekcie sa nikdy nemajú podať do materských znamienok, jaziev alebo do miest, na ktorých je koža citlivá, podliata krvou, červená, stvrdnutá alebo porušená.
Naplnenou injekčnou striekačkou sa nemá triasť.
Úplné pokyny na podanie RoActemry v naplnenej injekčnej striekačke sú poskytnuté v písomnej informácii pre používateľa, pozri časť 6.6.
4.3 Kontraindik áciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívne, závažné infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
V záujme zlepšenia sledovateľnosti biologických liekov sa musí v zdravotnej dokumentácii pacienta
j
asne zaznamenať obchodný názov a číslo šarže podaného lieku.
I
nf ekcie
Závažné a niekedy fatálne infekcie boli hlásené u pacientov, ktorí dostávali imunosupresívne látky vrátane RoActemry (pozri časť 4.8). Liečba RoActemrou sa nesmie začať u pacientov s aktívnymi infekciami (pozri časť 4.3). Ak u pacienta dôjde ku vzniku závažnej infekcie, podávanie RoActemry sa má prerušiť, kým sa infekcia nevylieči (pozri časť 4.8). Zdravotnícki pracovníci majú postupovať
opatrne, ak uvažujú o použití RoActemry u pacientov s anamnézou opakujúcich sa alebo chronických infekcií alebo so základnými ochoreniami (napr. divertikulitída, diabetes a intersticiálna choroba pľúc), ktoré ich predisponujú ku vzniku infekcií.
Pacientov, ktorí na stredne ťažkú až ťažkú RA, pJIA alebo OBA, užívajú imunosupresívne látky ako je RoActemra, sa odporúča pozorne sledovať, aby sa včas odhalila závažná infekcia, keďže prejavy
a príznaky akútneho zápalu môžu byť zmiernené, z dôvodu potlačenia reaktantov akútnej fázy. Pri vyšetrovaní pacienta na možnú infekciu sa má vziať do úvahy vplyv tocilizumabu na C-reaktívny proteín (CRP), neutrofily a prejavy a príznaky infekcie. Pacientov treba poučiť, že keď sa u nich objavia akékoľvek príznaky svedčiace o infekcii, majú sa ihneď skontaktovať so zdravotníckym pracovníkom, aby sa zaistilo rýchle vyšetrenie a náležitá liečba.
Tuberkulóza
Tak ako sa odporúča pre iné typy biologickej liečby, aj pred začatím liečby RoActemrou majú všetci
pacienti podstúpiť skríningové vyšetrenie na latentnú tuberkulóznu (TB) infekciu. Pacienti s latentnou TB sa pred začatím liečby RoActemrou majú liečiť štandardnou antimykobakteriálnou terapiou. Lekári, ktorí predpisujú RoActemru, majú mať na mysli riziko falošne negatívnych výsledkov tuberkulinových kožných testov a krvného testu interferón-gamma TB, zvlášť u pacientov, ktorí sú vážne chorí alebo so zníženou imunitou.
Pacienti majú byť upozornení, aby vyhľadali lekársku pomoc, ak sa u nich v priebehu liečby RoActemrou alebo po jej ukončení vyskytnú príznaky/symptómy (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, mierna horúčka) svedčiace o tuberkulóznej infekcii.
Reaktivácia vírusu
Reaktivácia vírusu (napr. vírusu hepatitídy B) sa zaznamenala pri biologickej liečbe RA. Z klinických štúdií s RoActemrou boli vylúčení pacienti, ktorí mali pozitívny skríning na hepatitídu.
Komplikácie divertikulitídy
Prípady perforácie divertikulu ako komplikácie divertikulitídy boli u pacientov liečených RoActemrou
hlásené menej často (pozri časť 4.8). RoActemra sa má u pacientov s ulceráciou čriev alebo divertikulitídou v anamnéze používať opatrne. Pacientov s príznakmi, ktoré by mohli svedčiť
o komplikovanej divertikulitíde, ako sú bolesti brucha, krvácanie a/alebo nevysvetliteľná zmena
vo vyprázdňovaní stolice spolu s horúčkou, je potrebné promptne vyšetriť, aby sa včas rozpoznala divertikulitída, ktorá môže byť spojená s perforáciou gastrointestinálneho traktu.
Reakcie z precitlivenosti
Boli hlásené závažné reakcie z precitlivenosti, vrátane anafylaxie, súvisiace s podávaním RoActemry (pozri časť 4.8). Takéto reakcie môžu byť závažnejšie a potenciálne fatálne u pacientov, ktorí mali reakcie z precitlivenosti počas predchádzajúcej liečby tocilizumabom, aj keď dostali premedikáciu kortikosteroidmi a antihistaminikami. V prípade výskytu anafylaktickej reakcie alebo inej závažnej reakcie z precitlivenosti sa má podávanie RoActemry okamžite ukončiť, začať náležitá liečba a liečba tocilizumabom sa má navždy skončiť.
A
k
tí
v
ne ochorenie pečene a porucha f unkcie pečene
Liečba RoActemrou, najmä keď sa podáva súbežne s MTX, môže byť spojená so zvýšením pečeňových transamináz. Ak sa uvažuje o liečbe pacientov s aktívnym ochorením pečene alebo poruchou funkcie pečene, vyžaduje sa opatrnosť (pozri časti 4.2 a 4.8).
Zvýšenie pečeňových transamináz
V klinických štúdiách sa pri liečbe RoActemrou často hlásili prechodné alebo sporadické, mierne a stredne závažné zvýšenia pečeňových transamináz bez progresie do poškodenia pečene (pozri časť 4.8). Vyššia frekvencia týchto zvýšení pečeňových transamináz sa pozorovala vtedy, keď sa
v kombinácii s RoActemrou užívali potenciálne hepatotoxické liečivá (napr. MTX). Keď je klinicky indikované, majú sa zvážiť ďalšie vyšetrenia funkcie pečene, vrátane bilirubínu.
Keď sa uvažuje o začatí liečby RoActemrou u pacientov s hodnotami ALT alebo AST zvýšenými na > 1,5-násobok ULN, je nutná opatrnosť. U pacientov s východiskovými hodnotami ALT alebo AST > 5-násobok ULN sa liečba neodporúča.
U pacientov s RA a OBA sa hladiny ALT a AST majú skontrolovať raz za 4 až 8 týždňov počas prvých 6 mesiacov liečby a následne raz za 12 týždňov. Odporúčané úpravy dávky na základe transamináz pozri v časti 4.2. Pri vzostupoch hodnôt ALT alebo AST na > 3- až 5-násobok ULN sa má liečba RoActemrou prerušiť.
U pacientov s pJIA sa hladiny ALT a AST majú skontrolovať v čase podania druhej injekcie
a následne v súlade so správnou klinickou praxou (pozri časť 4.2).
Hematologické odchýlky
Po liečbe tocilizumabom v dávke 8 mg/kg v kombinácii s MTX sa vyskytoval pokles počtu neutrofilov a trombocytov (pozri časť 4.8). U pacientov, ktorí boli predtým liečení inhibítorom TNF, môže existovať zvýšené riziko neutropénie.
U pacientov, ktorí neboli doteraz liečení RoActemrou, sa neodporúča začať liečbu, ak je ANC nižší ako 2 x 109/l. Keď sa uvažuje o začatí liečby RoActemrou u pacientov s nízkym počtom trombocytov (t.j. počet trombocytov pod 100 x 103/μl), vyžaduje sa opatrnosť. U pacientov, u ktorých je
ANC < 0,5 x 109/l alebo počet trombocytov je < 50 x 103/μl, sa neodporúča pokračovať v liečbe.
Ťažká neutropénia môže byť spojená so zvýšeným rizikom závažných infekcií, i keď doteraz nie je jasné spojenie medzi zníženým počtom neutrofilov a výskytom závažných infekcií v klinických skúšaniach s RoActemrou.
U pacientov s RA a OBA sa má počet neutrofilov a trombocytov skontrolovať po 4 až 8 týždňoch od začatia liečby a následne v súlade so štandardnou klinickou praxou. Odporúčané úpravy dávky na základe počtu ANC a neutrofilov pozri v časti 4.2.
U pacientov s pJIA sa má počet neutrofilov a trombocytov skontrolovať v čase podania druhej injekcie
a následne v súlade so správnou klinickou praxou (pozri časť 4.2).
Hodnoty lipidov
U pacientov liečených tocilizumabom sa pozorovalo zvýšenie hodnôt lipidových parametrov, vrátane hladiny celkového cholesterolu, lipoproteínov s nízkou hustotou (LDL), lipoproteínov s vysokou hustotou (HDL) a triglyceridov (pozri časť 4.8). U väčšiny pacientov nedošlo k zvýšeniu aterogénneho indexu a zvýšenie celkového cholesterolu odpovedalo na liečbu hypolipidemikami.
U všetkých pacientov sa má hodnotenie lipidových parametrov vykonať po 4 až 8 týždňoch od začatia liečby RoActemrou. Pacienti sa majú liečiť v súlade s národnými klinickými odporúčaniami pre liečbu hyperlipidémií.
N
e
urologické poruchy
Lekári majú venovať zvýšenú pozornosť príznakom, ktoré by mohli svedčiť o vzniku centrálnych demyelinizačných porúch. V súčasnosti nie je známe, či RoActemra môže vyvolať centrálnu demyelinizáciu.
Malignita
Riziko vzniku malignity je u pacientov s RA zvýšené. Imunomodulačné lieky môžu riziko vzniku malignity zvyšovať.
Očkovania
Súbežne s RoActemrou sa nemajú podávať živé a živé oslabené očkovacie látky, keďže klinická bezpečnosť nebola stanovená. V randomizovanej otvorenej štúdii, dosahovali dospelí pacienti s RA, ktorí dostávali liečbu RoActemrou a MTX, účinnú odpoveď na 23-valentnú pneumokokovú polysacharidovú vakcínu ako aj na vakcínu obsahujúcu tetanický toxoid, čo bolo porovnateľné
s odpoveďou pozorovanou u pacientov, ktorí dostávali liečbu MTX v monoterapii.
Odporúča sa, aby všetci pacienti, a obzvlášť pediatrickí alebo starší pacienti, boli podrobení aktuálne platným imunizáciám v súlade s aktuálnymi odporúčaniami imunizácie ešte pred začatím liečby RoActemrou. Interval medzi podaním živých očkovacích látok a začatím liečby RoActemrou má byť v súlade s aktuálnymi odporúčaniami imunizácie ohľadne imunosupresívnych látok.
Kardiovaskulárne riziko
Pacientom s RA hrozí zvýšené riziko kardiovaskulárnych porúch; rizikové faktory (napr. hypertenzia, hyperlipidémia) je potrebné korigovať v rámci bežnej štandardnej zdravotnej starostlivosti.
Kombinácia s inhibítormi TNF
Nie sú skúsenosti s použitím RoActemry s inhibítormi TNF ani inými biologickými liekmi na liečbu pacientov s RA. RoActemra sa neodporúča používať spolu s inými biologickými liekmi.
OBA
RoActemra v monoterapii sa nesmie podávať na liečbu akútnych relapsov, keďže účinnosť pre tento
stav nebola stanovená. Glukokortikoidy sa majú podávať podľa zváženia lekára a praktických odporúčaní.
4.5 Liek ové a iné interakcie
Interakčné štúdie sa uskutočnili iba u dospelých.
Súbežné podanie jednorazovej dávky RoActemry 10 mg/kg s MTX v dávke 10 - 25 mg podávanej jedenkrát týždenne nemalo klinicky významný vplyv na expozíciu MTX.
Populačné farmakokinetické analýzy nezistili žiaden vplyv MTX, nesteroidných protizápalových liekov (NSAIDs) alebo kortikosteroidov na klírens RoActemry u pacientov s RA. U pacientov s OBA nebol pozorovaný žiaden účinok kumulatívnej dávky kortikosteroidov na expozíciu RoActemry.
Účinkom cytokínov, ako je napr. IL-6, ktoré stimulujú chronický zápal, dochádza k potlačeniu expresie pečeňových enzýmov CYP450. Pri začatí liečby silne účinným inhibítorom cytokínov, ako je RoActemra, preto môže dôjsť k obnoveniu expresie enzýmov CYP450.
Štúdie in vitro na kultivovaných ľudských hepatocytoch preukázali, že IL-6 spôsobuje zníženie expresie enzýmov CYP1A2, CYP2C9, CYP2C19 a CYP3A4. RoActemra normalizuje expresiu týchto enzýmov.
V štúdii s pacientami s RA boli hladiny simvastatínu (CYP3A4) znížené o 57 % jeden týždeň po jednotlivej dávke tocilizumabu na hodnotu podobnú alebo mierne vyššiu, ako sa pozorovala u zdravých ľudí.
Pri začatí alebo ukončení liečby tocilizumabom sa majú sledovať pacienti, ktorí užívajú individuálne upravované dávky liekov metabolizovaných prostredníctvom CYP450 3A4, 1A2 alebo 2C9 (napr. metylprednizolón, dexametazón (možnosť vzniku abstinenčného syndrómu v dôsledku vysadenia perorálnych glukokortikoidov), atorvastatín, blokátory kalciového kanála, teofylín, w arfarín, fenprokumón, fenytoín, cyklosporín alebo benzodiazepíny), keďže na udržanie ich terapeutického účinku môže byť potrebné zvýšenie dávok. Vzhľadom na dlhý eliminačný polčas (t1/2) môže vplyv tocilizumabu na aktivitu enzýmov CYP450 pretrvávať niekoľko týždňov po ukončení liečby.
4.6 Fertilita, gravidita a lak tácia
Ženy vofertilnomveku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a 3 mesiace po ukončení liečby.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití RoActemry u gravidných žien. Štúdia na zvieratách preukázala zvýšené riziko spontánneho potratu/embryofetálneho úmrtia pri podávaní vysokej dávky (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí.
RoActemra sa môže používať počas gravidity iba v nevyhnutných prípadoch.
DojčenieNie je známe, či sa tocilizumab vylučuje do ľudského materského mlieka. Vylučovanie RoActemry do mlieka sa u zvierat neskúmalo. Pri rozhodovaní o tom, či pokračovať v dojčení/ukončiť dojčenie, alebo či pokračovať v liečbe/ukončiť liečbu RoActemrou sa má brať do úvahy prínos dojčenia pre dieťa a prínos liečby RoActemrou pre ženu.
Fertilita
Dostupné predklinické údaje nenaznačujú vplyv liečby RoActemrou na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
RoActemra má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť 4.8, závraty).
4.8 Nežiaduce účink y
Súhrnbezpečnostnéhoprofilu
Bezpečnostný profil bol stanovený na základe 4 510 pacientov vystavených RoActemre v klinických štúdiách; väčšina týchto pacientov sa zúčastnila štúdií RA (n=4 009), kým zvyšné údaje pochádzajú zo štúdií OBA (n=149), pJIA (n=240) a sJIA (n=112). Bezpečnostný profil RoActemry zostáva v týchto indikáciách podobný a nediferencovaný.
Najčastejšie hlásené nežiaduce reakcie na liek (Adverse Drug Reactions, ADRs) boli infekcie horných dýchacích ciest, nazofaryngitída, bolesť hlavy, hypertenzia a zvýšené hodnoty ALT.
Najvážnejšie ADRs boli závažné infekcie, komplikácie divertikulitídy a reakcie z precitlivenosti. Súhrnnežiaducichreakciívtabuľke
ADRs vymenované v tabuľke 1 sú uvedené podľa triedy orgánových systémov a kategórií frekvencie, ktoré sú definované s použitím nasledujúceho pravidla: veľmi časté (≥ 1/10); časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (>1/10 000 až < 1/1 000) alebo veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
T
abuľka 1. Súhrn ADRs vyskytujúcich sa u pacientov liečených RoActemrou
T
r
i
e
d
a orgánových
s
ystémov [A1] Poruchy krvi a lymfatického systému Poruchy endokrinného systému
Veľmi časté Časté Menej časté
Leukopénia, neutropénia
Hypotyreoidizmus
Poruchy oka Konjunktivitída
Poruchy gastrointestinálneho traktu
Celkové poruchy a reakcie v mieste podania
Reakcia
v mieste vpichu
Bolesť brucha, ulcerácia
v ústnej dutine, gastritída
Periférny edém, reakcia
z precitlivenosti
Stomatitída, žalúdočné vredy
Infekcie a nákazy Infekcie horných dýchacích ciest
Laboratórne a funkčné vyšetrenia
Celulitída, pneumónia, jednoduchý opar v oblasti úst, pásový opar
Zvýšené hodnoty pečeňových transamináz, zvýšenie telesnej hmotnosti, zvýšené hodnoty celkového bilirubínu*
Divertikulitída
Poruchy metabolizmu a výživy
Poruchy nervového systému
Poruchy obličiek
a močových ciest
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy kože a
podkožného tkaniva
Hypercholeste-
rolémia*
Bolesť hlavy, závraty
Kašeľ, dyspnoe
Vyrážka, pruritus, urtikária
Hypertriglyceridémia
Nefrolitiáza
Poruchy kože a podkožného tkaniva

Poruchy ciev Hypertenzia
*Zahŕňa zvýšenia zozbierané ako časť rut inného laborat órneho pozorovania (pozri t ext nižšie).
RA
Intravenózne použitieBezpečnosť RoActemry bola skúmaná v 5 dvojito-zaslepených skúšaniach fázy III a v ich predĺžených fázach.
Populácia
všetkých kontrolných pacientov zahŕňala všetkých pacientov z dvojito-zaslepených fáz všetkých hlavných štúdií od randomizácie až po prvú zmenu režimu liečby alebo do dosiahnutia druhého roka. V 4 štúdiách bolo kontrolné obdobie 6 mesiacov a v 1 štúdii trvalo až 2 roky. V dvojito- zaslepených kontrolných štúdiách dostávalo 774 pacientov RoActemru 4 mg/kg v kombinácii s MTX,
1 870 pacientov dostávalo tocilizumab 8 mg/kg v kombinácii s MTX alebo inými DMARDs
a 288 pacientov dostávalo tocilizumab 8 mg/kg v monoterapii.
Populácia
všetkých exponovaných pacientov zahŕňala všetkých pacientov zo štúdií, ktorí dostali aspoň jednu dávku RoActemry buď v kontrolnom dvojito- zaslepenom období alebo v otvorenej predĺženej fáze. Z celkového počtu 4009 pacientov dostávalo 3577 pacientov liečbu po dobu najmenej 6 mesiacov, 3296 po dobu najmenej jedného roka; 2806 dostávalo liečbu po dobu aspoň 2 roky a 1222 pacientov počas 3 rokov.
P
opis vybraných nežiaducich reakcií
Inf ekcie
V 6-mesačných kontrolovaných štúdiách bola miera výskytu všetkých infekcií hlásených pri liečbe
RoActemrou 8 mg/kg spolu s DMARDs 127 udalostí na 100 pacientorokov oproti 112 udalostiam na
100 pacientorokov v skupine s placebom v kombinácii s DMARDs. V súbore dlhodobej expozície bol celkový výskyt infekcií pri liečbe RoActemrou 108 udalostí na 100 pacientorokov.
V 6-mesačných kontrolovaných klinických štúdiách bola miera výskytu závažných infekcií pri liečbe
RoActemrou 8 mg/kg v kombinácii s DMARDs 5,3 udalosťami na 100 pacientorokov expozície oproti
3,9 udalostiam na 100 pacientorokov expozície v skupine s placebom v kombinácii s DMARDs. V štúdii monoterapie bola miera výskytu závažných infekcií 3,6 udalostí na 100 pacientorokov expozície v skupine liečenej RoActemrou a 1,5 udalostí na 100 pacientorokov expozície v skupine liečenej MTX.
V súbore celkovej expozície bol celkový výskyt závažných infekcií 4,7 udalosti na 100 pacientorokov. K hláseným závažným infekciám, z ktorých niektoré mali fatálne následky patrila pneumónia, celulitída, pásový opar, gastroenteritída, divertikulitída, sepsa a bakteriálna artritída. Boli hlásené aj prípady oportúnnych infekcií.
Intersticiálna choroba pľúc
Zhoršená funkcia pľúc môže zvýšiť riziko vzniku infekcií. Po uvedení lieku na trh boli hlásené prípady intersticiálnej choroby pľúc (vrátane pneumonitídy a pľúcnej fibrózy), z ktorých niektoré boli fatálne.
Gastrointestinálne perf orácie
Pri liečbe tocilizumabom počas 6-mesačných kontrolovaných klinických štúdií bol celkový výskyt gastrointestinálnych perforácií 0,26 udalostí na 100 pacientorokov. V dlhodobej expozícií bol celkový výskyt gastrointestinálnych perforácií 0,28 udalostí na 100 pacientorokov. Hlásenia gastrointestinálnej perforácie pri liečbe RoActemrou boli primárne hlásené ako komplikácie divertikulitídy, zahŕňajúce generalizovanú purulentnú peritonitídu, perforáciu dolnej časti gastrointestinálneho traktu, fistulu
a absces.
Reakcie na inf úziu
V 6-mesačných kontrolovaných klinických štúdiách boli nežiaduce účinky súvisiace s podávaním
infúzie (vybrané udalosti vyskytujúce sa počas podávania infúzie alebo v priebehu 24 hodín od podania infúzie) hlásené u 6,9 % pacientov v skupine liečenej tocilizumabom 8 mg/kg
v kombinácii s DMARDs a u 5,1 % pacientov v skupine s placebom spolu s DMARDs. Udalosti hlásené počas podávania infúzie boli predovšetkým epizódy hypertenzie; udalosti hlásené v priebehu
24 hodín od ukončenia podávania infúzie boli bolesť hlavy a kožné reakcie (vyrážka, urtikária). Tieto udalosti neboli pre liečbu limitujúce.
Miera výskytu anafylaktických reakcií (vyskytujúcich sa celkovo u 6/3 778 ; 0,2 % pacientov) bola niekoľkonásobne vyššia pri dávke 4 mg/kg oproti dávke 8 mg/kg. Klinicky významné reakcie
z precitlivenosti súvisiace s liečbou RoActemrou a vyžadujúce ukončenie liečby boli hlásené
u celkovo 13 z 3 778 pacientov (0,3 %) liečených RoActemrou počas kontrolovaných a otvorených
klinických štúdií. Tieto reakcie sa zvyčajne pozorovali počas podávania druhej až piatej infúzie tocilizumabu (pozri časť 4.4). Po registrácii lieku bola hlásená fatálna anafylaxia počas liečby intravenózne podávanou RoActemrou (pozri časť 4.4).
Imunogenicita
V 6-mesačných kontrolovaných klinických štúdiách bolo celkovo 2 876 pacientov vyšetrených na protilátky proti RoActemre. Zo 46 pacientov (1,6 %), u ktorých sa vytvorili protilátky proti RoActemre, došlo u 6 k významnej reakcii z precitlivenosti, ktorá u 5 z nich viedla k trvalému ukončeniu liečby. U tridsiatich pacientov (1,1 %) sa vytvorili neutralizujúce protilátky.
H
e
m
atologické odchýlky:
N
e
utrof ily
V 6-mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu neutrofilov pod 1 x 109/l
u 3,4 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs oproti < 0,1 % pacientov
s placebom spolu s DMARDs. Približne u polovice pacientov, u ktorých ANC klesol na < 1 x 109/l,
došlo k tomuto poklesu v priebehu 8 týždňov po začatí liečby. Pokles pod 0,5 x 109/l bol hlásený u 0,3 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs. Boli hlásené infekcie
s neutropéniou.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt poklesu počtu neutrofilov rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
Krvné doštičky
V 6-mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu trombocytov pod
100 x 103/µl u 1,7 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs oproti < 1 %
pacientov s placebom spolu s DMARDs. Tieto poklesy sa vyskytli bez asociácie s krvácaním.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt poklesu počtu krvných doštičiek rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
V postmarketingovom sledovaní sa vyskytli veľmi zriedkavo prípady pancytopénie.
Zvýšenie pečeňových transamináz
V 6-mesačných kontrolovaných klinických štúdiách bolo prechodné zvýšenie hodnôt
ALT/AST > 3 x ULN u 2,1 % pacientov liečených RoActemrou 8 mg/kg oproti 4,9 % pacientov liečených MTX, a u 6,5 % pacientov liečených 8 mg/kg RoActemry v kombinácii s DMARDs oproti
1,5 % pacientov s placebom v kombinácii s DMARDs.
Pridanie potenciálne hepatotoxických liekov (napr. MTX) k tocilizumabu podávanému v monoterapii viedlo k zvýšenému výskytu vyšších hodnôt. Zvyšovanie hladín ALT/AST > 5-násobok ULN sa pozorovalo u 0,7 % pacientov liečených RoActemrou v monoterapii a u 1,4 % pacientov liečených tocilizumabom v kombinácii s DMARDs, pričom väčšina z nich liečbu tocilizumabom trvalo ukončila. Tieto vzostupy neboli spojené s klinicky významným zvýšením hodnôt priameho (konjugovaného) bilirubínu, ani nesúviseli s klinickými príznakmi hepatitídy či poruchy funkcie pečene. Počas dvojito zaslepeného kontrolovaného obdobia sa hodnoty nepriameho bilirubínu, sledovaného ako rutinný laboratórny parameter, vyššie ako horná hranica referenčného rozpätia vyskytli u 6,2 % pacientov liečených RoActemrou v dávke 8 mg/kg + DMARD. U celkovo 5,8 % pacientov došlo k zvýšeniu hodnoty nepriameho bilirubínu na > 1-násobok až 2-násobok ULN a 0,4 % pacientov malo zvýšenie
na > 2-násobok ULN.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt prípadov zvýšenia ALT/AST rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
Hodnoty lipidových parametrov
Zvýšenie hodnôt lipidových parametrov, ako je napríklad celkový cholesterol, triglyceridy,
LDL-cholesterol a/alebo HDL-cholesterol, bolo počas 6-mesačných kontrolovaných štúdií hlásené často. Rutinným laboratórnym sledovaním sa zistilo, že približne u 24 % pacientov, ktorí v klinických štúdiách dostávali RoActemru, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu
na ≥ 6,2 mmol/l, pričom u 15 % pacientov došlo k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l.
Zvýšené lipidové parametre odpovedali na liečbu hypolipidemikami.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt prípadov zvýšenia lipidových parametrov rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
M
alignity
Klinické údaje nie sú dostatočné na zhodnotenie rizika možného výskytu malignity po expozícii
tocilizumabu. Hodnotenie dlhodobej bezpečnosti naďalej prebieha.
Kožné reakcie
V postmarketingovom sledovaní sa vyskytli veľmi zriedkavé hlásenia Stevensovho-Johnsonovho syndrómu.
Subkutánnepoužitie
RA
Bezpečnosť subkutánne podávanej RoActemry pri RA sa hodnotila v dvojito zaslepenej, kontrolovanej, multicentrickej štúdii, SC-I. SC-I bola štúdia noninferiority, ktorá porovnávala účinnosť a bezpečnosť RoActemry v dávke 162 mg podávanej raz za týždeň s intravenóznou dávkou
8 mg/kg u 1 262 pacientov s RA. Všetkým pacientom boli v rámci základnej liečby podávané nebiologické DMARDs. Bezpečnosť a imunogenicita pozorované pri subkutánne podávanej RoActemre sa zhodovali so známym bezpečnostných profilom intravenózne podávanej RoActemry a nepozorovali sa žiadne nové alebo neočakávané nežiaduce reakcie na liek (pozri tabuľku 1). Vyšší
výskyt reakcií v mieste vpichu sa pozoroval v skupine so subkutánnym tocilizumabom v porovnaní so
subkutánnymi injekciami placeba v skupinách s intravenóznym podávaním.
Reakcie v mieste podania injekcie
V štúdii SC-I bol počas 6-mesačného kontrolovaného obdobia výskyt reakcií v mieste vpichu 10,1 % (64/631) pri subkutánnom tocilizumabe a 2,4 % (15/631) pri subkutánnych injekciách placeba (skupina s intravenóznym podávaním) podávaných raz za týždeň. Tieto reakcie v mieste vpichu (vrátane erytému, pruritu, bolesti a hematómu) boli mierne až stredne závažné. Väčšina z nich ustúpila bez potreby akejkoľvek liečby a žiadna nevyžadovala ukončenie podávania lieku.
Imunogenicita
V štúdii SC-I bolo celkovo 625 pacientov liečených RoActemrou v dávke 162 mg podávanej
raz za týždeň vyšetrených na protilátky proti RoActemre v 6-mesačnom kontrolovanom období. U piatich pacientov (0,8 %) sa zistila pozitivita protilátok proti RoActemre; u všetkých z nich sa vytvorili neutralizujúce protilátky proti RoActemre. U jedného pacienta sa zistila pozitivita protilátok izotypu IgE (0,2 %).
V štúdii SC-II bolo celkovo 434 pacientov liečených tocilizumabom v dávke 162 mg podávanej každý druhý týždeň vyšetrených na protilátky proti RoActemre v 6-mesačnom kontrolovanom období.
U siedmich pacientov (1,6 %) sa zistila pozitivita protilátok proti RoActemre: u šiestich z nich (1,4 %) sa vytvorili neutralizujúce protilátky proti RoActemre. U štyroch pacientov sa zistila pozitivita protilátok izotypu IgE (0,9 %).
Nepozorovala sa žiadna korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami.
Hematologické odchýlky:
Neutrof ily
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s tocilizumabom, SC-I, došlo k poklesu počtu neutrofilov pod 1 x 109/l u 2,9 % pacientov liečených subkutánnou dávkou podávanou raz za týždeň.
Nezistila sa žiadna jasná súvislosť medzi poklesom počtu neutrofilov pod 1 x 109/l a výskytom závažných infekcií.
Krvné doštičky
Počas rutinného laboratórneho sledovania v 6-mesačnom klinickom skúšaní s tocilizumabom, SC-I, nedošlo u žiadneho z pacientov liečených s.c. dávkou podávanou raz za týždeň k poklesu počtu krvných doštičiek na ≤ 50 x 103/μl.
Z
v
ý
š
e
nie pečeňových transamináz
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s tocilizumabom, SC-I, došlo k vzostup hodnôt ALT a AST na ≥ 3-násobok ULN u 6,5 % a 1,4 %, v uvedenom poradí, pacientov liečených subkutánnou dávkou podávanou raz za týždeň.
Hodnoty lipidových parametrov
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, SC-I, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu na > 6,2 mmol/l
(240 mg/dl) u 19 % pacientov liečených subkutánnou dávkou podávanou raz za týždeň a u 9 % došlo
k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l (160 mg/dl).
Subkutánnepoužitie
OBA
Bezpečnosť subkutánne podávanej RoActemry sa skúmala vo fáze III jednej štúdie (WA28119), ktorej sa zúčastnilo 251 pacientov s OBA. Počas 12-mesačnej, dvojito zaslepenej, placebom kontrolovanej fázy tejto štúdie, bolo celkové trvanie v pacientorokoch pri RoActemre u populácie s celkovou expozíciou 138,5 pacientorokov. Celkový bezpečnostný profil pozorovaný v liečebných skupinách
s RoActemrou bol zhodný so známym bezpečnostným profilom RoActemry (pozri tabuľku 1).
Inf ekcie
Miera výskytu udalostí infekcií/závažných infekcií bola vyrovnaná medzi skupinou s RoActemrou podávanou raz za týždeň (200,2/9,7 udalostí na 100 pacientorokov) a skupinou s placebom
v kombinácii s postupným znižovaním dávky prednizónu počas 26 týždňov (156,0/4,2 udalosti na 100 pacientorokov) a skupinou s placebom v kombinácii s postupným znižovaním dávky počas 52 týždňov (210,2/12,5 udalostí na 100 pacientorokov).
Reakcie v mieste podania injekcie
V skupine, v ktorej sa RoActemra podávala subkutánne raz za týždeň, celkovo 6 % (6/100) pacientov hlásilo nežiaduce reakcie v mieste podania subkutánnej injekcie. Žiadna reakcia v mieste podania injekcie nebola hlásená ako závažná nežiaduca udalosť, alebo žiadna nevyžadovala ukončenie liečby.
Imunogenicita
V skupine s RoActemrou podávanou subkutánne raz za týždeň, sa u jedného pacienta (1,1 %, 1/95) zistila pozitivita neutralizujúcich protilátok proti RoActemre, nie však protilátok izotypu IgE. U tohto pacienta nebola pozorovaná reakcia z precitlivenosti, ani reakcia v mieste podania injekcie.
Hematologické odchýlky:
Neutrof ily
Počas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní s RoActemrou sa pozoroval pokles počtu neutrofilov pod 1x109/l u 4% pacientov v skupine
so subkutánne podávanou RoActemrou raz za týždeň. Tento pokles sa nepozoroval v žiadnej skupine s placebom a s postupným znižovaním dávky prednizónu.
Trombocyty
Počas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou sa u 1 pacienta (1%, 1/100) v skupine so subkutánne podávanou Roactemrou raz za týždeň pozoroval jeden výskyt prechodného poklesu počtu tromobcytov na <100 × 103 /μl bez súvisiacich prípadov krvácania. Pokles počtu trombocytov pod počet 100 × 103 /μl nebol pozorovaný v žiadnej skupine s placebom a s postupným znižovaním dávky prednizónu.
Zvýšenie pečeňových transamináz
Počas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, sa pozoroval vzostup hodnôt ALT na > 3-násobok ULN u 3 % pacientov v skupine so subkutánne podávanou RoActemrou raz za týždeň v porovnaní s 2 % pacientov v skupine s placebom v kombinácii s postupným znižovaním dávky prednizónu počas 52 týždňov a 0 % pacientov v skupine s placebom v kombinácii s postupným znižovaním dávky prednizónu počas 26 týždňov. Vzostup
hodnôt AST na > 3 ULN sa pozoroval u 1 % pacientov v skupine s RoActemrou podávanou subkutánne raz za týždeň v porovnaní s 0 % pacientov v oboch skupinách s placebom s postupným znižovaním dávky prednizónu.
Hodnoty lipidových parametrovPočas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu na > 6,2 mmol/l (240 mg/dl)
u 34 % pacientov liečených RoActemrou podávanou subkutánne raz za týždeň a u 15 % došlo
k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l (160 mg/dl)
SubkutánnaformaRoActemrypJIABezpečnostný profil subkutánnej formy RoActemry sa hodnotil aj u 52 pediatrických pacientov
s pJIA. Celková expozícia RoActemre u všetkých pacientov s pJIA vystavených tocilizumabu bola
184,4 pacientoroka v prípade i.v. formy tocilizumabu a 50,4 pacientoroka v prípade s.c. formy tocilizumabu. Bezpečnostný profil pozorovaný u pacientov s pJIA sa vo všeobecnosti zhodoval so známym bezpečnostným profilom RoActemry s výnimkou reakcií v mieste podania injekcie
(injection site reactions, ISR) (pozri tabuľku 1). U pacientov s pJIA bol výskyt ISR po s.c. podávaných injekciách RoActemry vyšší v porovnaní s dospelými s RA.
Inf ekcieV štúdii so s.c. formou RoActemry bol výskyt infekcií u pacientov s pJIA liečených s.c. podávanou RoActemrou porovnateľný s výskytom infekcií zisteným u pacientov s pJIA liečených i.v. podávanou RoActemrou.
Reakcie v mieste podania injekcieCelkovo 28,8 % (15/52) pacientov s pJIA malo ISR po s.c. podávanej RoActemre. Tieto ISR sa vyskytli u 44 % pacientov s telesnou hmotnosťou ≥ 30 kg v porovnaní so 14,8 % pacientov s telesnou hmotnosťou nižšou ako 30 kg. Najčastejšími ISR boli erytém, opuch, hematóm, bolesť a pruritus
v mieste podania injekcie. Všetky hlásené ISR boli nezávažnými udalosťami 1. stupňa a žiadna ISR
nevyžadovala predčasné ukončenie liečby pacienta alebo prerušenie podávania dávok.
ImunogenicitaV štúdii so s.c. formou sa u 5,8 % [3/52] pacientov zistila pozitivita neutralizujúcich protilátok proti tocilizumabu bez toho, že by u nich vznikla závažná alebo klinicky významná reakcia
z precitlivenosti. Jeden z týchto 3 pacientov bol následne vyradený zo štúdie. Nepozorovala sa žiadna korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami.
Laboratórne odchýlkyPočas rutinného laboratórneho sledovania v populácii všetkých pacientov vystavených RoActemre došlo k poklesu počtu neutrofilov pod 1 × 109/l u 15,4 % pacientov liečených s.c. podávanou RoActemrou. K vzostupu hodnôt ALT a AST na ≥ 3-násobok ULN došlo, v uvedenom poradí,
u 9,6% a 3,8 % pacientov liečených s.c. podávanou RoActemrou. U žiadneho pacienta liečeného s.c. podávanou RoActemrou nedošlo k poklesu počtu trombocytov na ≤ 50 × 103/μl.
Hodnoty lipidových parametrovV štúdii so s.c. formou sa vzostup hodnoty LDL-cholesterolu na ≥ 130 mg/dl zistil u 14,3 % pacientov a vzostup hodnoty celkového cholesterolu na ≥ 200 mg/dl u 12,8 % pacientov, a to na ktorejkoľvek
z kontrolných návštev (post-baseline) vykonaných v priebehu skúšanej liečby.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávk ovanie
K dispozícii sú obmedzené údaje o predávkovaní RoActemrou. Hlásený bol jeden prípad náhodného predávkovania, pri ktorom pacient s mnohopočetným myelómom dostal jednorazovú intravenózne podanú dávku 40 mg/kg. Nepozorovali sa žiadne nežiaduce reakcie.
U zdravých dobrovoľníkov, ktorí dostali jednorazovú dávku do 28 mg/kg, sa nepozorovali žiadne závažné nežiaduce reakcie, hoci došlo k výskytu neutropénie limitujúcej dávku.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmak odynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu; ATC kód: L04AC07. Mechanizmusúčinku
RoActemra sa špecificky viaže na rozpustný aj na membránovo-viazaný receptor pre IL-6 (sIL-6R a
mIL-6R). Dokázalo sa, že tocilizumab inhibuje prenos signálu sprostredkovaný sIL-6R a mIL-6R.
IL-6 je pleiotropný prozápalový cytokín, ktorý produkujú rôzne typy buniek, vrátane T- a B-buniek, monocytov a fibroblastov. IL-6 sa zúčastňuje na rôznorodých fyziologických procesoch, ako je aktivácia T-buniek, indukcia sekrécie imunoglobulínov, indukcia syntézy proteínov akútnej fázy
v pečeni a stimulácia krvotvorby. IL-6 sa podieľa na patogenéze ochorení, medzi ktoré patria zápalové ochorenia, osteoporóza a neoplázie.
Farmakodynamickéúčinky
V klinických štúdiách s RoActemrou sa pozoroval rýchly pokles hodnôt CRP, sedimentácie erytrocytov (ESR) a sérového amyloidu A (SAA). V zhode s účinkom na reaktanty akútnej fázy bola liečba RoActemrou spojená s poklesom počtu trombocytov na hodnoty v rámci referenčného rozpätia. Pozorovalo sa zvýšenie hladín hemoglobínu, ktoré RoActemra vyvoláva tým, že znižuje IL-6 navodené účinky na tvorbu hepcidínu, čím sa zvyšuje dostupnosť železa. U pacientov liečených RoActemrou sa pokles hladín CRP na hodnoty v rámci referenčného rozpätia pozoroval už od
2. týždňa a počas trvania liečby sa tento pokles udržal.
V klinickej štúdii OBA WA28119 sa pozoroval podobný rýchly pokles hodnôt CRP a sedimentácie erytrocytov (ESR) s miernym zvýšením priemernej koncentrácie korpuskulárneho hemoglobínu. U zdravých dobrovoľníkov, ktorým sa podávala RoActemra v dávkach od 2 do 28 mg/kg intravenózne a od 81 do 162 mg subkutánne, klesal absolútny počet neutrofilov k najnižším hladinám 2 až 5 dní
po podaní. Potom sa počet neutrofilov vrátil k východiskovým hodnotám v závislosti od dávky. Pacienti po podaní RoActemry vykazujú porovnateľný (oproti zdravým osobám) pokles absolútneho počtu neutrofilov (pozri časť 4.8).
RA
Intravenóznepoužitie
Klinická účinnosť
Účinnosť tocilizumabu na zmiernenie prejavov a príznakov RA sa hodnotila v piatich
randomizovaných, dvojito zaslepených, multicentrických štúdiách. Do štúdií I - V boli zaradení pacienti vo veku ≥ 18 rokov s aktívnou RA diagnostikovanou podľa kritérií American College of Rheumatology (ACR), ktorí mali pred začiatkom liečby minimálne osem bolestivých a šesť opuchnutých kĺbov.
V štúdii I sa RoActemra podávala intravenózne raz za štyri týždne v monoterapii. V štúdiách II, III
a V sa RoActemra podávala intravenózne raz za štyri týždne v kombinácii s MTX, kontrolnú skupinu tvorilo placebo v kombinácii s MTX. V štúdii IV sa RoActemra podávala intravenózne raz za 4 týždne v kombinácii s inými DMARDs, kontrolnú skupinu tvorilo placebo v kombinácii s inými DMARDs. Primárny cieľový ukazovateľ pre každú z piatich štúdií bol podiel pacientov, ktorí dosiahli odpoveď ACR 20 v 24. týždni.
Štúdia I hodnotila 673 pacientov, ktorí sa v priebehu šiestich mesiacov pred randomizáciou neliečili MTX a ktorí neprerušili predchádzajúcu liečbu MTX kvôli klinicky významným toxickým účinkom alebo nedostatočnej odpovedi na liečbu. Väčšina (67 %) pacientov sa MTX predtým neliečila. Dávky
8 mg/kg RoActemry sa podávali raz za štyri týždne v monoterapii. Porovnávacia skupina dostávala MTX raz týždenne (dávka titrovaná od 7,5 mg na maximálne 20 mg týždenne počas osemtýždňového obdobia).
Štúdia II, dvojročná štúdia s plánovanou analýzou v týždni 24., 52. a v týždni 104, hodnotila
1 196 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky 4 alebo 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v zaslepenej fáze liečby trvajúcej 52 týždňov v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne). Po týždni 52 mohli všetci
pacienti pokračovať v otvorenej fáze liečby s RoActemrou v dávke 8 mg/kg. Z pacientov, ktorí dokončili štúdiu a ktorí boli pôvodne randomizovaní do skupiny s placebom + MTX, v 2. roku 86 % pacientov pokračovalo v otvorenej fáze liečby RoActemrou v dávke 8 mg/kg. Primárny cieľový ukazovateľ v 24. týždni bol podiel pacientov, ktorí dosiahli odpoveď ACR 20. V 52. a 104. týždni boli prevencia poškodenia kĺbu a zlepšenie fyzických funkcií pridružené ako primárne cieľové
ukazovatele.
Štúdia III hodnotila 623 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky
4 alebo 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Štúdia IV hodnotila 1 220 pacientov, ktorí nedosiahli dostatočnú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viaceré DMARDs. Dávky 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou DMARDs.
Štúdia V hodnotila 499 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na liečbu jedným alebo viacerými inhibítormi TNF, alebo ktorí takúto liečbu netolerovali. Liečba inhibítorom TNF sa pred randomizáciou ukončila. Dávky 4 alebo 8 mg/kg tocilizumabu alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Klinická odpoveď
Vo všetkých štúdiách mali pacienti liečení RoActemrou 8 mg/kg štatisticky významne vyššiu mieru odpovede ACR 20, 50, 70 po šiestich mesiacoch oproti kontrolnej skupine (tabuľka 3). V štúdii I sa preukázala vyššia účinnosť RoActemry 8 mg/kg oproti aktívnej porovnávacej látke - MTX.
Účinok liečby bol u pacientov podobný nezávisle od prítomnosti reumatoidného faktora, veku, pohlavia, rasy, počtu predchádzajúcich terapií a stavu ochorenia. Účinok nastúpil rýchlo
(už v 2. týždni) a stupeň odpovede sa počas liečby neustále zlepšoval. V prebiehajúcich predĺžených otvorených štúdiách I - V sa počas viac ako 3 rokov pozorovali neustále pretrvávajúce odpovede.
Vo všetkých štúdiách sa u pacientov liečených RoActemrou 8 mg/kg oproti pacientom s placebom
a MTX alebo inými DMARDs zaznamenalo významné zlepšenie vo všetkých jednotlivých zložkách odpovede ACR zahŕňajúcich: počet bolestivých a opuchnutých kĺbov; celkové hodnotenie pacientmi a lekárom; skóre indexu funkčnej neschopnosti; hodnotenie bolesti a CRP.
Pacienti v štúdiách I - V mali pred začiatkom liečby priemerné skóre aktivity ochorenia (DAS28)
6,5 - 6,8. U pacientov liečených tocilizumabom sa v porovnaní s pacientmi v kontrolnej skupine
(1,3 - 2,1) pozorovalo významné zníženie DAS28 oproti východiskovej hodnote (priemerné zlepšenie) o 3,1 - 3,4. Podiel pacientov, ktorí v 24. týždni dosiahli klinickú remisiu ochorenia podľa DAS28 (DAS28 < 2,6), bol významne vyšší u pacientov liečených tocilizumabom (28 - 34 %) v porovnaní
s 1 - 12 % pacientov v kontrolnej skupine. V štúdii II dosiahlo 65 % pacientov DAS28 < 2,6
v 104. týždni, v porovnaní so 48 % pacientov v 52. týždni a s 33 % pacientov v 24. týždni.
V súhrnnej analýze štúdií II, III a IV bol podiel pacientov, ktorí dosiahli odpoveď ACR 20, 50 a 70
významne vyšší (59 % oproti 50 %, 37 % oproti 27 %, 18 % oproti 11 % v uvedenom poradí) v skupine liečenej tocilizumabom 8 mg/kg v kombinácii s DMARDs oproti skupine liečenej tocilizumabom 4 mg/kg v kombinácii s DMARDs (p < 0,03). Podobne bol aj podiel pacientov, ktorí dosiahli remisiu ochorenia podľa DAS28 (DAS28 < 2,6), významne vyšší (31 % oproti 16 %)
u pacientov liečených RoActemrou 8 mg/kg v kombinácii s DMARDs než u pacientov liečených
RoActemrou 4 mg/kg v kombinácii s DMARDs (p < 0,0001).
Tabuľka 2.Odpovede ACR v placebom/MTX/DMARDs kontrolovaných štúdiách (% pacientov)
Š
t
ú
d
i a I AMBITIO N
Š
t
ú
d
i a II LITHE
Š tú di a III OPTION
Š
t
ú
d
i a IV TO W ARD
Š
t
ú
d
i a V RADIATE
T ýž-
deň
TC Z
8 m g/k g
MTX TC Z
8 m g/k g
+ MTX
PBO + MTX
TC Z
8 m g/k g
+ MTX
PBO
+ MTX
TC Z
8 m g/k g
+ DMARD
PBO + DMARD
TC Z
8 m g/k g
+ MTX
PBO + MTX
N =
286
N =
284
N =
398
N =
393
N =
205
A
C R 20
N =
204
N =
803
N =
413
N =
170
N =
158

24 70 %*** 52 % 56 %*** 27 % 59 %*** 26 % 61 %*** 24 % 50 %*** 10 %
52 56 %*** 25 %
AC R 5024 44 %** 33 % 32 %*** 10 % 44 %*** 11 % 38 %*** 9 % 29 %*** 4 %
52 36 %*** 10 %
AC R 7024 28 %** 15 % 13 %*** 2 % 22 %*** 2 % 21 %*** 3 % 12 %** 1 %
52 20 %*** 4 %
TCZ - Tocilizum ab MTX - Metotrexát PBO - PlaceboDMARD - Antireum atikum m odifikujúce priebeh choroby** - p < 0,01, TCZ oproti PBO + MTX/DMARD*** - p < 0,0001, TCZ oproti PBO + MTX/DMARDVýznamná klinická odpoveďPo 2 rokoch liečby tocilizumabom s MTX dosiahlo 14 % pacientov významnú klinickú odpoveď
(udržanie ACR70 odpovede počas 24 týždňov alebo dlhšie).
Rádiograf ická odpoveďV štúdii II sa u pacientov s nedostatočnou odpoveďou na MTX hodnotila inhibícia štrukturálneho
poškodenia kĺbov rádiograficky a vyjadrila sa ako zmena v modifikovanom Sharpovom skóre a jeho zložkách - skóre erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených RoActemrou sa oproti kontrolnej skupine preukázala inhibícia štrukturálneho poškodenia kĺbov s významne nižšou rádiografickou progresiou ochorenia (tabuľka 3).
V otvorenej predĺženej fáze štúdie II bola inhibícia progresie štrukturálneho poškodenie kĺbu
v skupine s tocilizumabom a MTX udržiavaná i v druhom roku liečby. Stredná zmena
od východiskových hodnôt bola v 104. týždni v celkovom Sharpovom-Genantovom skóre významne nižšia u pacientov randomizovaných do skupiny s RoActemrou v dávke 8 mg/kg a MTX (p < 0,0001) v porovnaní s pacientami, ktorí boli randomizovaní do skupiny s placebom a MTX.
T
abuľka 3. Rádiografické priemerné zmeny počas 52 týždňov v štúdii II
P
B
O + MTX (+TCZ od 24. týždňa) N = 393
T
C
Z 8 mg/k g + MTX N = 398
Celkové
Sharpovo-Genantovo
skóre
1,13 0,29*

Skóre erózie 0,71 0,17* Skóre JSN 0,42 0,12**
PBO - PlaceboMTX - MetotrexátTCZ - Tocilizum abJSN - Zúženie kĺbovej štrbiny* - p ≤ 0,0001, TCZ oproti PBO + MTX** - p < 0,005, TCZ oproti PBO + MTXPo 1 roku liečby tocilizumabom a MTX 85 % pacientov (n = 348) nevykazovalo žiadnu progresiu štrukturálneho poškodenia kĺbov ako je definované v celkovom Sharpovom skóre 0 alebo menej,
v porovnaní so 67 % pacientov v skupine s placebom a MTX (n = 290) (p ≤ 0,001). Tieto výsledky
pretrvávali i po 2 rokoch liečby (83 %, n = 353). Deväťdesiat tri percent (93 %; n = 271) pacientov nevykazovalo žiadnu progresiu medzi 52. a 104. týždňom.
Zdravotné výsledky a výsledky týkajúce sa kvality životaPacienti liečení RoActemrou hlásili zlepšenie vo všetkých výsledkoch hlásených pacientmi (dotazník
hodnotiaci zdravie a index funkčnej neschopnosti, - HAQ-DI), skrátený formulár 36 a dotazník funkčného hodnotenia liečby chronického ochorenia. U pacientov liečených RoActemrou sa oproti pacientom liečeným DMARDs pozorovalo štatisticky významné zlepšenie skóre HAQ-DI.
V priebehu otvorenej fázy štúdie II bolo udržanie zlepšenia fyzických funkcií až počas 2 rokov. V 52. týždni bola stredná zmena v HAQ-DI -0,58 v skupine s RoActemrou 8 mg/kg a MTX
v porovnaní s -0,39 v skupine s placebom a MTX. Stredná zmena HAQ-DI bola v skupine s RoActemrou 8 mg/kg a MTX udržovaná aj 104. týždni (-0,61).
Hladiny hemoglobínuPri liečbe RoActemrou sa oproti liečbe DMARDs v 24. týždni pozorovalo štatisticky významné zlepšenie hladín hemoglobínu (p < 0,0001). Priemerné hodnoty hladín hemoglobínu sa zvýšili do 2. týždňa a udržali sa v referenčnom rozpätí až do 24. týždňa.
Tocilizumab verzus adalimumab v monoterapiiŠtúdia VI (WA19924), 24-týždňová dvojito zaslepená štúdia, ktorá porovnávala monoterapiu RoActemrou s monoterapiou adalimumabom, hodnotila 326 pacientov s RA, ktorí netolerovali MTX alebo kde pokračovanie v liečbe MTX sa považovalo za nevhodné (vrátane nedostatočných respondérov na MTX). Pacienti v skupine s RoActemrou dostávali intravenóznu (i.v.) infúziu RoActemry (8 mg/kg) každé 4 týždne a subkutánne (s.c.) injekciu s placebom každé 2 týždne. Pacienti v skupine s adalimumabom dostávali s.c. injekciu adalimumabu (40 mg) každé 2 týždne plus i.v. infúziu s placebom každé 4 týždne.
Pozoroval sa štatistický významný superiórny účinok liečby v prospech RoActemry v porovnaní
s adalimumabom pri kontrole aktivity ochorenia od východiskovej hodnoty po 24. týždeň
pre primárny cieľový ukazovateľ zmenu DAS28 a pre všetky sekundárne cieľové ukazovatele
(tabuľka 4).
T
abuľka 4. Výsledky účinnosti pre štúdiu VI (WA19924)
AD
A + placebo
 (
i
.
v)
N = 162
T
o
c
ili
z
u
m
a
b +
p
l
a
ce
b
o (s .c.)
N = 163
(
i
.
v)
N = 162
T
o
c
ili
z
u
m
a
b +
p
l
a
ce
b
o (s .c.)
N = 163 p-hodnota
(a)
P
r
i
m
á
r
n
y cieľový ukazovateľ – Priemerná zmena od východis kovej hodnoty v 24. Týždni
DA
S
2
8 (upravený priemer) -1,8 -3,3
R
o
z
d
i
e
l v upravenom priemere (95 % IS)
-
1
,
5 (-1,8, -1,1) < 0,0001
S
e
k
un
d
á
r
n
e cieľové ukazovatele – Percento res pondérov v 24. týždni
(
b)
DAS28 < 2,6, n (%) 17 (10,5) 65 (39,9) < 0,0001
DAS28 ≤ 3,2, n (%) 32 (19,8) 84 (51,5) < 0,0001
ACR20 odpoveď, n (%) 80 (49,4) 106 (65,0) 0,0038
ACR50 odpoveď, n (%) 45 (27,8) 77 (47,2) 0,0002
ACR70 odpoveď, n (%) 29 (17,9) 53 (32,5) 0,0023
ap hodnota je upravená vzhľadom na oblasť a trvanie RA pre všetky cieľové ukazovatele a tiež východisková hodnota
pre všetky pokračujúce cieľové ukazovatele.
b Neodpovedajúci na liečbu použití pre chýbajúce údaje. Multidisciplinárna kontrola použitím Bonferroni-Holm procedúry
Celkový klinický profil nežiaducich udalostí bol podobný pri tocilizumabe a adalimumabe. Podiel pacientov so závažnými nežiaducimi udalosťami bol medzi liečebnými skupinami (RoActemra 11,7 % oproti adalimumabu 9,9 %). Nežiaduce účinky v skupine s tocilizumabom odpovedali známemu bezpečnostnému profilu RoActemry a nežiaduce účinky boli hlásené s podobnou frekvenciou
v porovnaní s tabuľkou 1. Vyššia incidencia infekcií a infestácií bola hlásená v skupine s RoActemrou
(48 % oproti 42 %), a to bez rozdielu v incidencii závažných infekcií (3,1 %). Obidve skúmané liečby indukovali rovnaké zmeny v laboratórnych bezpečnostných parametroch (poklesy počtu neutrofilov
a krvných doštičiek, zvýšenie ALT, AST a lipidov), veľkosť zmien a frekvencie výrazných abnormalít však bola vyššia pri RoActemre v porovnaní s adalimumabom. U štyroch (2,5 %) pacientov v skupine s RoActemrou a dvoch (1,2 %) pacientov v skupine s adalimumabom sa vyskytli poklesy počtu neutrofilov 3. alebo 4. stupňa CTC. U jedenástich (6,8 %) pacientov v skupine s RoActemrou a piatich (3,1 %) pacientov v skupine s adalimumabom sa vyskytlo zvýšenie ALT 2. alebo vyššieho
stupňa CTC. Priemerné zvýšenie LDL od východiskovej hodnoty bolo 0,64 mmol/l (25 mg/dl)
u pacientov v skupine s RoActemrou a 0,19 mmol/l (7 mg/dl) u pacientov v skupine s adalimumabom. Bezpečnosť pozorovaná v skupine s tocilizumabom sa zhodovala so známym bezpečnostných profilom RoActemry a nepozorovali sa žiadne nové alebo neočakávané nežiaduce liekové reakcie (pozri tabuľku 1).
Subkutánnepoužitie
Klinická účinnosť
Účinnosť subkutánne podávanej RoActemry v zmierňovaní prejavov a príznakov RA a rádiografická
odpoveď sa hodnotili v dvoch randomizovaných, dvojito zaslepených, kontrolovaných, multicentrických štúdiách. V štúdii I (SC-I) sa vyžadovalo, aby mali pacienti vek > 18 rokov, stredne ťažkú až ťažkú aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali pred začiatkom liečby minimálne 4 bolestivé a 4 opuchnuté kĺby. Všetkým pacientom boli v rámci základnej liečby podávané nebiologické DMARDs. V štúdii II (SC-II) sa vyžadovalo, aby pacienti mali vek > 18 rokov, stredne ťažkú až ťažkú aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali pred začiatkom liečby minimálne 8 bolestivých a 6 opuchnutých kĺbov.
Po prechode z dávky 8 mg/kg podávanej intravenózne raz za 4 týždne na dávku 162 mg podávanú subkutánne raz za týždeň dôjde u pacienta k zmene expozície. Rozsah tejto zmeny sa líši v závislosti od telesnej hmotnosti pacienta (zvýšenie expozície u pacientov s nízkou telesnou hmotnosťou
a zníženie expozície u pacientov s vysokou telesnou hmotnosťou), ale klinický výsledok sa zhoduje s klinickým výsledkom pozorovaným u pacientov liečených intravenóznou formou lieku.
K
li
nická odpoveď
Štúdia SC-I hodnotila pacientov so stredne ťažkou až ťažkou aktívnou RA, ktorí nedosiahli dostatočnú klinickú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viac DMARDs, pričom približne 20 % malo v anamnéze nedostatočnú odpoveď na liečbu najmenej jedným inhibítorom TNF. V štúdii SC-I bolo 1 262 pacientov randomizovaných v pomere 1:1 do skupiny s RoActemrou podávanou subkutánne v dávke 162 mg každý týždeň, alebo do skupiny s RoActemrou podávanou intravenózne v dávke 8 mg/kg každé štyri týždne v kombinácii s nebiologickým(i) DMARD(s). Primárny cieľový ukazovateľ v štúdii bol rozdiel v podiele pacientov, ktorí dosiahli odpoveď ACR20
v 24. týždni. Výsledky zo štúdie SC-I sú uvedené v tabuľke 5.
Tabuľka 5. Odpovede ACR v štúdii SC-I (% pacientov) v 24. týždni
SC-Ia
TCZ s .c. 162 mg
každý týždeň
+ DMARD
N = 558
TCZ i.v. 8 mg/kg
+ DMARD
N = 537

ACR20 24. týždeň 69,4 % 73,4 %
Vážený rozdiel (95 % IS) -4,0 (-9,2; 1,2)
ACR50 24. týždeň 47,0 % 48,6 %
Vážený rozdiel (95 % IS) -1,8 (-7,5; 4,0)
ACR70 24. týždeň 24,0 % 27,9 %
Vážený rozdiel (95 % IS) -3,8 (-9,0; 1,3)
T CZ = t ocilizumab
a = populácia podľa prot okolu
Pacienti v štúdii SC-I mali pred začiatkom liečby priemerné skóre aktivity ochorenia (DAS28) 6,6
v skupine so subkutánnym tocilizumabom a 6,7 v skupine s intravenóznym tocilizumabom. V 24. týždni sa v oboch liečebných skupinách pozorovalo významné zníženie DAS28 oproti východiskovej hodnote (priemerné zlepšenie) o 3,5 a porovnateľný podiel pacientov dosiahol klinickú remisiu ochorenia podľa DAS28 (DAS28 < 2,6) v skupine so subkutánnym tocilizumabom (38,4 %)
a v skupine s intravenóznym tocilizumabom (36,9 %).
Rádiograf ická odpoveďRádiografická odpoveď subkutánne podávanej RoActemry sa hodnotila v dvojito zaslepenej, kontrolovanej, multicentrickej štúdii s pacientmi s aktívnou RA (SC-II). Štúdia SC-II hodnotila pacientov so stredne ťažkou až ťažkou aktívnou RA, ktorí nedosiahli dostatočnú klinickú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viac DMARDs, pričom približne 20 %
malo v anamnéze nedostatočnú odpoveď na liečbu najmenej jedným inhibítorom TNF. Vyžadovalo sa, aby pacienti mali vek > 18 rokov a aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali
pred začiatkom liečby minimálne 8 bolestivých a 6 opuchnutých kĺbov. V štúdii SC-II bolo
656 pacientov randomizovaných v pomere 2:1 do skupiny s RoActemrou podávanou subkutánne
v dávke 162 mg každý druhý týždeň, alebo do skupiny s placebom, v kombinácii s nebiologickým(i)
DMARD(s).
V štúdii SC-II sa inhibícia štrukturálneho poškodenia kĺbov hodnotila rádiograficky a vyjadrila sa ako zmena oproti východiskovej hodnote v priemernom celkovom Sharpovom skóre modifikovanom
van der Heijdom (mTSS). V 24. týždni sa preukázala inhibícia štrukturálneho poškodenia kĺbov
s významne nižšou rádiografickou progresiou ochorenia u pacientov, ktorým bola subkutánne podávaná RoActemra, v porovnaní s pacientmi, ktorým bolo podávané placebo (priemerné mTSS 0,62 oproti 1,23, p = 0,0149 (van Elteren)). Tieto výsledky sa zhodujú s výsledkami pozorovanými u pacientov liečených intravenóznym tocilizumabom.
V štúdii SC-II sa v 24. týždni dosiahla odpoveď ACR20 u 60,9 %, ACR50 u 39,8 % a ACR70 u 19,7 % pacientov liečených RoActemrou podávanou subkutánne každý druhý týždeň oproti odpovedi ACR20 dosiahnutej u 31,5 %, ACR50 u 12,3 % a ACR70 u 5,0 % pacientov, ktorým bolo
podávané placebo. Pacienti mali pred začiatkom liečby priemerné DAS28 6,7 v skupine
so subkutánnym tocilizumabom a 6,6 v skupine s placebom. V 24. týždni sa pozorovalo významné zníženie DAS28 oproti východiskovej hodnote o 3,1 v skupine so subkutánnym tocilizumabom a
o 1,7 v skupine s placebom a DAS28 < 2,6 sa pozorovalo u 32,0 % pacientov v skupine so subkutánnym tocilizumabom a u 4,0 % pacientov v skupine s placebom.
Zdravotné výsledky a výsledky týkajúce sa kvality života
V štúdii SC-I sa skóre HAQ-DI od začiatku štúdie po 24. týždeň znížilo priemerne o 0,6 v skupine
so subkutánnym tocilizumabom aj v skupine s intravenóznym tocilizumabom. Podiel pacientov, ktorí
dosiahli klinicky významné zlepšenie skóre HAQ-DI v 24. týždni (zmena oproti východiskovému skóre o ≥ 0,3 jednotky), bol tiež porovnateľný v skupine so subkutánnym tocilizumabom (65,2 %) oproti skupine s intravenóznym tocilizumabom (67,4 %), pričom vážený rozdiel v podieloch bol
-2,3 % (95 % IS -8,1; 3,4). Pokiaľ ide o SF-36, v 24. týždni bola priemerná zmena oproti východiskovej hodnote v skóre mentálneho komponentu 6,22 v skupine so subkutánnym tocilizumabom a 6,54 v skupine s intravenóznym tocilizumabom a zmena v skóre fyzického komponentu bola tiež podobná, a to 9,49 v skupine so subkutánnym tocilizumabom a 9,65 v v skupine s intravenóznym tocilizumabom.
V štúdii SC-II bolo priemerné zníženie skóre HAQ-DI od začiatku štúdie po 24. týždeň významne väčšie u pacientov liečených tocilizumabom podávaným subkutánne každý druhý týždeň (0,4)
v porovnaní s pacientmi, ktorým bolo podávané placebo (0,3). Podiel pacientov, ktorí dosiahli klinicky významné zlepšenie skóre HAQ-DI v 24. týždni (zmena oproti východiskovému skóre
o ≥ 0,3 jednotky), bol vyšší u pacientov liečených tocilizumabom podávaným subkutánne každý druhý týždeň (58 %) v porovnaní s pacientmi, ktorým bolo podávané placebo (46,8 %). Zmena v SF-36 (priemerná zmena v skóre mentálneho a fyzického komponentu) bola významne väčšia v skupine
so subkutánnym tocilizumabom (6,5 a 5,3) v porovnaní so skupinou s placebom (3,8 a 2,9).
OBA
Subkutánne použitie
Klinická účinnosť
Klinická štúdia WA28119 bola randomizovaná, multicentrická, dvojito zaslepená, placebom kontrolovaná štúdia superiority fázy III, ktorá skúmala účinnosť a bezpečnosť RoActemry u pacientov s OBA.
Do štúdie bolo zaradených dvestopäťdesiatjeden (251) pacientov s novým nástupom alebo relapsom OBA, ktorí boli rozdelení do štyroch liečebných skupín. Táto štúdia sa skladala z 52-týždňového zaslepeného obdobia (1. časť), po ktorom nasledovalo 104-týždňové otvorené predĺženie (2. časť). Účelom 2. časti bolo popísať dlhodobú bezpečnosť a udržanie účinnosti po 52 týždňoch liečby RoActemrou, preskúmať mieru výskytu relapsu ochorenia a potrebu liečby RoActemrou nad rámec 52 týždňov, a pochopiť potenciálnu dlhodobú účinnosť RoActemry bez potreby podávania steroidov (steroidy šetriaci účinok).
Dve subkutánne dávky RoActemry (162 mg každý týždeň a 162 mg každý druhý týždeň) sa porovnávali s dvomi rozdielnymi placebom kontrolovanými skupinami randomizovanými v pomere
2:1:1:1.
Všetkým pacientom boli v rámci základnej liečby podávané glukokortikoidy (prednizón). Každá liečebná skupina s RoActemrou a jedna z liečebných skupín s placebom nasledovala po vopred špecifikovanom režime postupného znižovania dávky prednizónu v priebehu 26 týždňov, kým druhá skupina s placebom nasledovala po vopred špecifikovanom režime postupného znižovania prednizónu počas 52 týždňov, čo viac zodpovedá štandardnej klinickej praxi.
Trvanie liečby glukokortikoidmi počas fázy skríningu a pred začatím podávania RoActemry (alebo placeba) bolo podobné vo všetkých 4 skupinách liečby (pozri tabuľku 6).
T
abuľka 6. Trvanie liečby kortikosteroidmi počas obdobia skríningu v štúdii WA28119
P
l
ac
e bo + 26 týždňov pos tupne
z
n
i
ž
o
va
n
e j dávk y pre dnizónu
N
=
5
0
P
l
ac
e bo + 52 týždňov pos tupne
z
n
i
ž
o
va
n
e j dávk y pre dnizónu
 N
=
5
1
R
o
A
c
t
e m ra 162m g s .c. týžde nne + 26 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k aždý druhý týžde ň +
2
6 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
4
9
N
=
5
1
R
o
A
c
t
e m ra 162m g s .c. týžde nne + 26 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k aždý druhý týžde ň +
2
6 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
4
9
Trvanie (dni)
Priemer (SD) 35,7 (11,5) 36,3 (12,5) 35,6 (13,2) 37,4 (14,4) Medián 42,0 41,0 41,0 42,0
Min - Max 6 - 63 12 – 82 1 - 87 9 - 87
Primárny cieľový ukazovateľ účinnosti hodnotený na základe pomeru pacientov, ktorí dosiahli trvalú
remisiu ochorenia bez steroidov v 52 týždni liečby RoActemrou plus 26-týždňovým obdobím postupného znižovania dávky prednizónu v porovnaní so skupinou s placebom plus 26-týždňové obdobie postupného znižovania dávky prednizónu, bol dosiahnutý (tabuľka 7).
Kľúčový sekundárny cieľový ukazovateľ účinnosti, ktorý sa rovnako zakladal na pomere pacientov, ktorí dosiahli pretrvávajúcu remisiu ochorenia v 52. týždni, porovnávajúci skupinu s tocilizumabom plus 26 týždňov postupného znižovania dávky prednizónu a skupinu s placebom plus 52 týždňov postupného znižovania dávky prednizónu, bol rovnako dosiahnutý (tabuľka 7).
Štatisticky významný superiórny účinok liečby sa pozoroval v prospech RoActemry oproti placebu v dosiahnutí pretrvávajúcej remisie bez steroidov v 52. týždni liečby RoActemrou plus 26-týždňové obdobie postupného znižovania dávky prednizónu v porovnaní so skupinou s placebom plus 26- týždňové obdobie postupného znižovania dávky prednizónu a so skupinou s placebom plus 52- týždňové obdobie postupného znižovania dávky prednizónu.
Percento pacientov, ktorí dosiahli pertrvávajúcu remisiu ochorenia v 52. týždni liečby, je znázornená v tabuľke 7.
Sekundárne cieľové ukazovatele
Pri posudzovaní času do nástupu prvého vzplanutia OBA sa pozorovalo výrazne menšie riziko vzplanutia ochorenia v skupine s RoActemrou podávanou subkutánne raz za týždeň v porovnaní so skupinou s placebom plus 26 týždňov postupného znižovania prednizónu a so skupinou s placebom plus 52 týždňov postupného znižovania prednizónu, a v skupine s RoActemrou podávanou subkutánne každý druhý týždeň, v porovnaní so skupinou s placebom plus 26 týždňov postupného znižovania prednizónu (porovnávané pri hladine významnosti 0,01). RoActemra podávaná subkutánne raz za týždeň rovnako preukázala klinicky významný pokles rizika vzplanutia ochorenia v porovnaní so skupinou s placebom plus 26 týždňov prednizónu u pacientov, ktorí vstúpili do skúšania
s relapsujúcim OBA, rovnako ako aj u pacientov s novým nástupom ochorenia (tabuľka 7).
Kumulatívna dávka glukokortikoidov
Kumulatívna dávka prednizónu bola v 52. týždni výrazne nižšia v obidvoch skupinách s RoActemrou
v porovnaní s dvoma skupinami s placebom (tabuľka 6). V osobitnej analýze pacientov, ktorí dostávali prednizón ako emergentnú liečbu pri vzplanutí OBA počas prvých 52 týždňov, sa veľkosť kumulatívnej dávky prednizónu významne líšila. Priemerné dávky u pacientov, ktorým bol podávaná emergentná liečba, boli v skupine s RoActemrou podávanou raz za týždeň v dávke 3 129,75 mg
a v skupine s RoActemrou podávanou každý druhý týždeň v dávke 3 847 mg. V oboch prípadoch bola táto dávka významne nižšia ako v skupinách s placebom plus 26 týždňov postupného znižovania
prednizónu, kde bola 4 023,5 mg, a v skupine s placebom plus 52 týždňov postupného znižovania prednizónu, kde bola 5 389,5 mg.
Tabuľka 7. Výsledky účinnosti zo štúdie WA28119
P
r
i m árn y ci eľový u k azovateľ
P
l ace bo + 26 týž dň ov pos tu pn e
z n i ž ovan e j
d
á
v
k y
p
r
e dn i z ón u
N
=
5
0
P
l ace bo + 52
t
ý
ž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
5
1
R
o
A
c
t
e m ra 162m g s .c. raz z a týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k až dý dru h ý týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y pre dn i z ón u
N
=
4
9
****P ret rvávajúca remisia (skupiny s t ocilizumabom vs placebo+26)
P acient i, kt orí odpovedali na liečbu v 52. t ýždni, n (%)
7 (14 %) 9 (17,6 %) 56 (56 %) 26 (53,1 %)
Neupravený rozdiel v proporciách
(99,5% IS)
Kľú čový s e k u ndárny ci eľový u k azovateľ
N/A N/A 42 %*
(18,00 ; 66,00)
39,06 %*
(12,46 ; 65,66)
P ret rvávajúca remisia (skupiny s t ocilizumabom vs placebo+52)
P acient i, kt orí odpovedali na liečbu v
52. t ýždni, n (%)
7 (14
%)
9 (17,6 %) 56 (56 %) 26 (53,1 %)
Neupravený rozdiel v proporciách
(99,5% IS)
N/A N/A 38,35 %* (17,89 ; 58,81)
35,41 %** (10,41 ; 60,41)
Ď
a
l š i e s e kundárne ci eľové u kazovatel e
Čas do nást upu prvého vzplanut ia OBA¹ (skupiny s t ocilizumabom vs placebo+26)
HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (skupiny s t ocilizumabom vs placebo+52)
HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s relapsom ochorenia; skupiny s
t ocilizumabom vs placebo +26) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s relapsom ochorenia; skupiny s
t ocilizumabom vs placebo + 52) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s novým nást upom ochorenia; skupiny s t ocilizumabom vs placebo +26) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s novým nást upom ochorenia; skupiny s t ocilizumabom vs placebo + 52) HR (99% IS)
Kumulat ívna dávka glukokort ikoidov (mg)
medián v 52. t ýždni (skupiny s
t ocilizumabom vs placebo+262)
medián v 52. t ýždni (skupiny s
t ocilizumabom vs placebo +522)
N/A N/A
N/A N/A
N/A N/A
3296,00
N/A

N/A N/A
N/A N/A
N/A N/A
N/A
3817,50
0,23 * (0,11 ; 0,46)
0,39**
(0,18 ; 0,82)
0,23***
(0,09 ; 0,61)
0,36
(0,13 ; 1,00)
0,25***
(0,09 ; 0,70)
0,44
(0,14 ; 1,32)
1862,00*
1862,00*
0,28 ** (0,12 ; 0,66)
0,48
(0,20 ; 1,16)
0,42
(0,14 ; 1,28)
0,67
(0,21 ; 2,10)
0,20***
(0,05 ; 0,76)
0,35
(0,09 ; 1,42)
1862,00*
1862,00*
P
l ace bo + 26 týž dň ov pos tu pn e
z n i ž ovan e j
d
á
v
k y
p
r
e dn i z ón u
N
=
5
0
P
l ace bo + 52
t
ý
ž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
5
1
R
o
A
c
t
e m ra 162m g s .c. raz z a týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k až dý dru h ý týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y pre dn i z ón u
N
=
4
9
E
x
p
l oratí vn e ci eľové u kazovatele
Ročná miera relapsu ochorenia, 52.
t ýždeň§
P riemer (SD)
* p < 0,0001
1,74
(2,18)
1,30
(1,84)
0,41
(0,78)
0,67
(1,10)

** p < 0,005 (prah významnost i primárnych a kľúčových sekundárnych testov superiorit y)
*** popisná p hodnot a
< 0,005
****
vz pl anu ti e: re cidí va pre javov al e bo prí znakov O BA a/al ebo ES R ≥30 mm/h –pot rebné zvýšenie dávky prednizónu
Re m is ia: abs e n ci a vz pl an u ti a a n ávrat C RP do norm al i z ovaných hodnôtPre trvávajú ca re m isi a: re mis ia od 12- 52. týž dňa- pacienti musia dodržiavať prot okolom st anovené post upné znižovanie dávky prednizónu
¹ analýzy času (v dňoch) medzi klinickou remisiou a prvým vzplanut ím ochorenia
2 p hodnot y sú st anovené na základe Van Elt erenovej analýzy neparamet rických údajov
§ št at ist ická analýza sa neuskut očnila
N/A= nehodí sa
HR = pomer rizika
IS = int erval spoľahlivost i
Výsledky týkajúce sa kvality životaV klinickej štúdii WA28119 boli výsledky skóre SF-36 (skrátený formulár) rozdelené na fyzické (PCS) a mentálne (MCS) komponenty. Stredná zmena vo fyzických komponentoch východiskového stavu do 52. týždňa bola vyššia (preukázalo sa výraznejšie zlepšenie) v skupinách s RoActemrou podávanou raz za týždeň [4,10] a každý druhý týždeň [2,76], ako v oboch skupinách s placebom
[placebo + 26 týždňov; -0,28, placebo + 52 týždňov; -1,49], hoci štatisticky významný rozdiel (p=0,0024) sa pozoroval len v porovnaní skupiny s RoActemrou podávanou raz za týždeň plus 26 týždňov postupného znižovania prednizónu a skupiny s placebom plus 52 týždňov postupného znižovania prednizónu (5,59; 99% IS: 8,6; 10,32). V prípade mentálnych komponentov bola stredná zmena z východiskového stavu do 52. týždňa vyššia v skupinách s RoActemrou podávanou raz za týždeň [7,28] a každé dva týždne [6,12], ako v skupine s placebom plus 52 týždňov postupného znižovania dávky prednizónu [2,84] (hoci rozdiely neboli štatisticky významné [týždenná hodnota p=0,0252 pri podávaní raz za týždeň, p=0.1468 pri podávaní každý druhý týždeň]) a podobná ako
v skupine s placebom plus 26 týždňov postupného znižovania dávky prednizónu [6,67].
Celkové hodnotenie aktivity ochorenia pacientom sa posudzovalo na vizuálnej analógovej škále (VAS, visual analogue scale) 0-100 mm. Stredná zmena v celkovej VAS pacienta bola z východiskového stavu do 52. týždňa nižšia (preukázalo sa výraznejšie zlepšenie) v oboch skupinách s RoActemrou, podávanou raz za týždeň [-19,0] a každé dva týždne. [-25,3], ako v oboch skupinách s placebom
[placebo + 26 týždňov -3,4, placebo + 52 týždňov -7,2], hoci len v skupine s RoActemrou podávanou každé dva týždne plus 26 týždňov postupného znižovania dávky prednizónu sa preukázal štatisticky významný rozdiel oproti skupine s placebom [placebo + 26 týždňov p=0,0059, a placebo + 52 týždňov p=0,0081].
Skóre zmeny v stave únavy z východiskového stavu do 52. týždňa- FACIT (Funkčné hodnotenie liečby chronického ochorenia) sa vypočítalo pre všetky skupiny. Skóre strednej zmeny [SD] bolo nasledovné: RoActemra podávaná raz za týždeň + 26 týždňov 5,61 [10,115], RoActemra podávaná každý druhý týždeň + 26 týždňov 1,81 [8,836], placebo + 26 týždňov 0,26 [10,702], a placebo + 52 týždňov -1,63 [6,753].
Zmeny v skóre EQ5D z východiskového stavu do 52. týždňa boli nasledovné: RoActemra podávaná
raz za týždeň + 26 týždňov 0,10 [0,198], RoActemra podávaná každý druhý týždeň + 26 týždňov 0,05
[0,215], placebo + 26 týždňov 0,07 [0,293], a placebo + 52 týždňov -0,02 [0,159].
Vyššie skóre signalizuje zlepšenie v oboch hodnoteniach, FACIT-únava aj EQ5D. PacientispJIA
Uskutočnila sa 52-týždňová, otvorená, multicentrická štúdia skúmajúca farmakokinetiku (FK) - farmakodynamiku (FD) a bezpečnosť u pediatrických pacientov s pJIA vo veku od 1 do 17 rokov, ktorej cieľom bolo určiť vhodnú subkutánnu dávku RoActemry, pri ktorej sa dosiahne FK/FD profil a bezpečnostný profil, ktoré sú porovnateľné s tými, ktoré sa dosiahnu pri režime s i.v. dávkou.
Vhodným pacientom sa tocilizumab podával v dávke určenej podľa telesnej hmotnosti (TH), pričom pacientom s telesnou hmotnosťou ≥ 30 kg (n = 25) sa RoActemra podávala v dávke 162 mg
raz za 2 týždne (Q2W) a pacientom s telesnou hmotnosťou nižšou ako 30 kg (n = 27) sa RoActemra podávala v dávke 162 mg raz za 3 týždne (Q3W) počas 52 týždňov. Títo 52 pacienti zahŕňali
37 (71 %) pacientov, ktorí dovtedy neboli liečení RoActemrou, a 15 (29%) pacientov, ktorí boli liečení i.v. podávanou RoActemrou a pri zaradení do štúdie prešli na s.c. podávanú RoActemru.
Režimy so s.c. podávanou RoActemrou, v dávke 162 mg Q3W určenej pre pacientov s telesnou hmotnosťou nižšou ako 30 kg a v dávke 162 mg Q2W určenej pre pacientov s telesnou hmotnosťou ≥ 30 kg, poskytujú FK expozíciu a FD odpovede podporujúce dosiahnutie podobných výsledkov účinnosti a bezpečnosti ako pri režimoch s i.v. podávanou RoActemrou schválených
pre pacientov s pJIA.
Výsledky exploračnej analýzy účinnosti ukázali, že s.c. podávaná RoActemra zlepšila priemerné skóre
JADAS-71 (Juvenile Arthritis Disease Activity Score) u pacientov dovtedy neliečených RoActemrou
a udržala medián skóre JADAS-71 u pacientov, ktorí prešli z liečby i.v. podávanou RoActemrou
na s.c. podávanú RoActemru, počas celej doby trvania štúdie u pacientov v obidvoch hmotnostných skupinách (TH nižšia ako 30 kg a TH ≥ 30 kg).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s RoActemrou
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe chronickej idiopatickej artritídy (vrátane reumatoidnej artritídy, ankylozujúcej spondylitídy, psoriatickej artritídy a juvenilnej idiopatickej artritídy). Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmak ok inetické vlastnosti
Farmakokinetiku RoActemry charakterizuje nelineárna eliminácia, ktorá je kombináciou lineárneho klírensu a Michaelisa-Mentenovej eliminácie. Nelineárna časť eliminácie RoActemry vedie k zvýšenej expozícii, ktorá je vyššia ako dávke úmerná. Farmakokinetické parametre RoActemry sa postupom času nemenia. Nakoľko je celkový klírens závislý od koncentrácií RoActemry v sére, aj polčas eliminácie RoActemry je závislý od koncentrácií a jeho dĺžka je rôzna v závislosti od hladiny koncentrácie v sére. Populačná farmakokinetická analýza u všetkých skúmaných populácií pacientov zatiaľ nepreukázala žiaden vzťah medzi zdanlivým klírensom a prítomnosťou protilátok proti lieku.
Intravenóznepoužitie
Farmakokinetika RoActemry sa stanovila za použitia populačnej farmakokinetickej analýzy údajov
z databázy zloženej z 3 552 pacientov s RA liečených dávkou 4 alebo 8 mg/kg RoActemry podávanou raz za 4 týždne formou jednu hodinu trvajúcej infúzie alebo so 162 mg tocilizumabu podávaného subkutánne buď raz týždenne alebo každý druhý týždeň počas 24 týždňov.
Nasledujúce parametre (predpokladaný priemer ± SD, štandardná odchýlka) sa odhadli pre dávku
8 mg/kg RoActemry podávanú raz za 4 týždne: plocha pod krivkou pre plazmatickú koncentráciu
(AUC) v rovnovážnom stave = 38 000 ± 13 000 h•µg/ml, minimálna koncentrácia (Cmin) =15,9 ± 13,1
µg/ml a maximálna koncentrácia (Cmax) = 182 ± 50,4 µg/ml a pomer kumulácie v hodnote 1,32 pri
AUC a1,09 pri Cmax bol nízky. Pomer kumulácie bol vyšší pri Cmin (2,49), čo sa očakávalo na základe
prispenia nelineárneho klírensu pri nižších koncentráciách. Rovnovážny stav sa dosiahol po podaní prvej dávky pri hodnote Cmax, po 8 týždňoch pri hodnote AUC a po 20 týždňoch pri hodnote Cmin. AUC, Cmin a Cmax RoActemry vzrástlo so stúpajúcou telesnou hmotnosťou. Pri telesnej hmotnosti ≥
100 kg bol predpovedaný priemer (± SD) AUC RoActemry v rovnovážnom stave
50 000 ± 16 800 μg•h/ml, Cmin tocilizumabu 24,4 ± 7,5 μg/ml a Cmax tocilizumabu226 ± 50,3 μg/ml, čo sú vyššie hodnoty, než hodnoty pri priemernej expozícii v súbore pacientov (t.j. celková hmotnosť všetkých pacientov) ako je uvedené vyššie. Krivka odpovede na dávku sa pri tocilizumabe pri vyšších expozíciách splošťuje, čo vedie k nižšiemu nárastu účinnosti pre každé ďalšie zvýšenie koncentrácie RoActemry, takže u pacientov liečených RoActemrou dávkou > 800 mg nedochádza už k žiadnemu zmysluplnému zvýšeniu účinnosti. Preto sa neodporúčajú dávky RoActemry, ktoré presahujú 800 mg na infúziu (pozri časť 4.2).
Farmakokinetika RoActemry u pacientov s pJIA bola charakterizovaná pomocou populačnej farmakokinetickej analýzy zahŕňajúcej 237 pacientov, ktorí boli liečení dávkou 8 mg/kg i.v.
raz za 4 týždne (pacienti s telesnou hmotnosťou ≥ 30 kg), 10 mg/kg i.v. raz za 4 týždne (pacienti s telesnou hmotnosťou nižšou ako 30 kg), 162 mg s.c. raz za 2 týždne (pacienti s telesnou hmotnosťou ≥ 30 kg) alebo 162 mg s.c. raz za 3 týždne (pacienti s telesnou hmotnosťou nižšou ako
30 kg).
Tabuľka 8. Predpokladaný priemer ± SD PK parametrov v rovnovážnom stave po i.v. alebo s.c. podávaní u pacientov s pJIA
i.v. s .c.
F
K parameter
R
o
A
c
t
e
m
r
y
8 mg/kg Q4W
≥ 30 kg
1
0 mg/kg Q4W
m
e
n
e
j ako 30 kg
16
2 mg Q2W
≥ 30 kg
16
2 mg Q3W
m
e
n
e
j ako 30 kg
Cm ax (µg/ml) 183 ± 42,3 168 ± 24,8 29,4 ± 13,5 75,5 ± 24,1
Cm in (µg/ml) 6,55 ± 7,93 1,47 ± 2,44 11,8 ± 7,08 18,4 ± 12,9
Cavg (µg/ml) 42,2 ± 13,4 31,6 ± 7,84 21,7 ± 10,4 45,5 ± 19,8
Kumulácia z hľadiska
Cm ax
Kumulácia z hľadiska
Cm in
Kumulácia z hľadiska
Cavg alebo AUCτ *
1,04 1,01 1,72 1,32
2,22 1,43 3,58 2,08
1,16 1,05 2,04 1,46

*τ = 4 t ýždne pri režime s i.v. dávkou, 2 t ýždne alebo 3 t ýždne pri režimoch so s.c. dávkou
Po i.v. podávaní sa približne 90 % rovnovážneho stavu dosiahlo do 12. týždňa pri dávke 10 mg/kg
(TH < 30 kg) a do 16. týždňa pri dávke 8 mg/kg (TH ≥ 30 kg). Po s.c. podávaní sa približne
90 % rovnovážneho stavu dosiahlo do 12. týždňa pri režime so s.c. dávkou 162 mg Q2W aj pri režime so s.c. dávkou 162 mg Q3W.
DistribúciaU pacientov s RA bol distribučný objem centrálneho kompartmentu 3,72 l, distribučný objem
periférneho kompartmentu bol 3,35 l, čo malo za následok distribučný objem 7,07 l v rovnovážnom stave.
ElimináciaPo intravenóznom podaní dávky podlieha RoActemra dvojfázovému vylučovaniu z cirkulácie.
Celkový klírens RoActemry bol závislý od koncentrácie a je súčtom lineárneho a nelineárneho klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze na 9,5 ml/h. Od koncentrácie závislý nelineárny klírens zohráva hlavnú úlohu pri nízkych
koncentráciách RoActemry. Keď je cesta nelineárneho klírensu nasýtená, pri vyšších koncentráciách
RoActemry je klírens určovaný hlavne lineárnym klírensom.
t1/2 RoActemry bol závislý od koncentrácie. V rovnovážnom stave sa po dávke 8 mg/kg podávanej raz
za 4 týždne efektívny t1/2 znižoval so znižujúcimi sa koncentráciami v rámci dávkovacieho intervalu
od 18 dní do 6 dní.
Linearita
Farmakokinetické parametre RoActemry sa postupom času nezmenili. Pri dávkach 4 a 8 mg/kg podávaných raz za 4 týždne sa pozorovalo vyššie ako dávke úmerné zvýšenie hodnoty AUC a Cmin. Hodnota Cmax sa zvyšovala úmerne dávke. V rovnovážnom stave bola pri dávke 8 mg/kg predpokladaná hodnota AUC 3,2-násobne a hodnota Cmin 30-násobne vyššia než pri dávke 4 mg/kg.
Subkutánnepoužitie
Farmakokinetika RoActemry sa stanovila za použitia populačnej farmakokinetickej analýzy údajov
z databázy zloženej z 3 552 pacientov s RA liečených dávkou 162 mg podávanou subkutánne každý týždeň, dávkou 162 mg podávanou subkutánne každý druhý týždeň a dávkou 4 alebo 8 mg/kg podávanou intravenózne raz za 4 týždne počas 24 týždňov.
Farmakokinetické parametre RoActemry sa postupom času nezmenili. Pri dávke 162 mg podávanej každý týždeň bol predpokladaný priemer (± SD) AUC1. týždeň tocilizumabu v rovnovážnom stave
7 970 ± 3 432 µg•h/ml, Cmin RoActemry 43,0 ± 19,8 µg/ml a Cmax RoActemry 49,8 ± 21,0 µg/ml. Pomer kumulácie bol pri AUC 6,32, pri Cmin 6,30 a pri Cmax 5,27. Rovnovážny stav sa pri AUC, Cmin a Cmax dosiahol po 12 týždňoch.
Pri dávke 162 mg podávanej každý druhý týždeň bol predpokladaný priemer (± SD) AUC2. týždeň
RoActemry v rovnovážnom stave 3 430 ± 2 660 µg•h/ml, Cmin RoActemry 5,7 ± 6,8 µg/ml a
Cmax RoActemry 13,2 ± 8,8 µg/ml. Pomer kumulácie bol pri AUC 2,67, pri Cmin 6,02 a pri Cmax 2,12.
Rovnovážny stav sa pri AUC a Cmin dosiahol po 12 týždňoch a pri Cmax po 10 týždňoch.
Absorpcia
Po subkutánnom podávaní pacientom s RA bol čas do dosiahnutia maximálnej sérovej koncentrácie
RoActemry (tmax) 2,8 dňa. Biologická dostupnosť s.c. formy bola 79 %.
Po s.c. podávaní pacientom s pJIA bol polčas absorpcie približne 2 dní a biologická dostupnosť s.c. formy u pacientov s pJIA bola 96 %.
Distribúcia
U pediatrických pacientov s pJIA bol distribučný objem centrálneho kompartmentu 1,97 l; distribučný objem periférneho kompartmentu 2,03 l; čo malo za následok distribučný objem 4,0 l v rovnovážnom stave.
Eliminácia
Pri subkutánnom podávaní u pacientov s RA je efektívny t1/2 po dosiahnutí rovnovážneho stavu až 13 dní pri dávke 162 mg podávanej každý týždeň a 5 dní pri dávke 162 mg podávanej každý druhý týždeň. Populačné farmakokinetické analýzy u pacientov s pJIA preukázali vplyv veľkosti tela na lineárny klírens, z čoho vyplýva, že sa má vziať do úvahy dávkovanie odvodené od telesnej hmotnosti (pozri tabuľku 8).
Po subkutánnom podávaní je efektívny t1/2 RoActemry u pacientov s pJIA až 10 dní u pacientov
s hmotnosťou < 30 kg (pri režime so s.c. dávkou 162 mg Q3W) a až 7 dní u pacientov ≥ 30 kg (pri režime so s.c. dávkou 162 mg Q2W) počas dávkovacieho intervalu po dosiahnutí rovnovážneho stavu.
Po intravenóznom podaní podlieha tocilizumab dvojfázovému vylučovaniu z cirkulácie. Celkový klírens tocilizumabu bol závislý od koncentrácie a predstavoval súčet lineárneho a nelineráneho klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze na
6,25 ml/hod. Nelineárny klírens závislý od koncentrácie zohrával významnú úlohu pri nízkych koncentráciách tocilizumabu. Po dosiahnutí saturácie nelineárneho klírensu pri vyšších koncentráciách tocilizumabu je klírens určovaný predovšetkým lineárnym klírensom.
OBA
SubkutánnepoužitieFarmakokinetika RoActemry u pacientov s OBA sa stanovila za použitia populačného farmakokinetického modelu analýzy údajov z databázy zloženej zo 149 pacientov s OBA liečených dávkou 162 mg podávanou subkutánne každý týždeň alebo dávkou 162 mg podávanou subkutánne každý druhý týždeň. Tento vyvinutý model mal zhodnú štruktúru so štruktúrou populačného PK modelu, ktorý bol vyvinutý skoršie na základe údajov zo štúdie pacientov s RA (pozri tabuľku 9).
Tabuľka 9. Predpokladaný priemer ± SD PK paramentrov v rovnovážnom stave po subkutánnompodávaní u pacientov s OBA Subkutánne podávanie
Subkutánne podávanie
P
K parametre RoActemry
16
2 mg každý druhý týždeň
16
2 mg každý týždeň
Cm ax (µg/ml) 19,3 ± 12,8 73 ± 30,4
Cm in (µg/ml) 11,1 ± 10,3 68,1± 29,5
Cavg (µg/ml) 16,2 ± 11,8 71,3 ± 30,1
Kumulácia Cm ax 2,18 8,88
Kumulácia Cm in 5,61 9,59
Kumulácia Cavg alebo AUCτ 2,81 10,91
Profil rovnovážneho stavu po podávaní RoActemry každý týždeň bol takmer plochý, s veľmi malými
rozdielmi medzi minimálnymi a maximálnymi hodnotami, zatiaľ čo pri dávke RoActemry podávanej každý druhý týždeň sa pozorovali významné rozdiely. Približne 90 % rovnovážneho stavu (AUCτ) sa dosiahlo do 14. týždňa v skupine s RoActemrou podávanou každý druhý týždeň a do 17. týždňa pri skupine s RoActemrou podávanou každý týždeň.
Na základe súčasnej PK charakteristiky minimálne koncentrácie RoActermy v rovnovážnom stave sú o 50 % vyššie v tejto populácii, v porovnamí s priemernými koncentráciami uvedenými vo veľkom súbore údajov získaných od populácie s RA. Tieto rozdiely sa objavujú z neznámych dôvodov. Rozdiely v PK nesprevádzajú významné rozdiely v PD parametroch, a preto klinický význam týchto údajov nie je známy.
U pacientov s OBA bola pozorovaná vyššia expozícia u pacientov s nižšou telesnou hmotnosťou. V režime s dávkou 162 mg podávanou každý týždeň bol rovnovážny stav Cavg o 51 % vyšší u pacientov s telesnou hmotnosťou < 60 kg, oproti pacientom s telesnou hmotnosťou od 60 do 100 kg. V režime s dávkou 162 mg podávanou každý druhý týždeň bol rovnovážny stav Cavg o 129 % vyšší u pacientov s telesnou hmotnosťou < 60 kg, oproti pacientom s telesnou hmotnosťou od 60 do 100 kg. Údaje o pacientoch s telesnou hmotnosťou > 100 kg sú obmedzené (n=7).
Absorpcia
Po subkutánnom podávaní u pacientov s OBA bol absorpčný t½ približne 4 dni. Biologická
dostupnosť v prípade subkutánnej formy bola 0,8. Medián hodnôt Tmax bol 3 dni po podávaní
RoActemry každý týždeň a 4,5 dňa po podávaní tocilizumabu každý druhý týždeň.
Distribúcia
U pacientov s OBA bol distribučný objem centrálneho kompartmentu 4,09 l, distribučný objem
periférneho kompartmentu bol 3,37 l, čo malo za následok distribučný objem 7,46 l v rovnovážnom stave.
E
li
m
i
nácia
Celkový klírens RoActemry bol závislý od koncentrácie a je súčtom lineárneho a nelineárneho
klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze a u pacientov s OBA bol 6,7ml/h.
U pacientov s OBA, v rovnovážnom stave, sa t½ účinnosti RoActemry pohyboval v rozpätí od 18,3 do
18,9 dňa pri režime 162 mg podávaných každý týždeň, a v rozpätí od 4,2 do 7,9 dňa pri režime 162 mg
podávaných každý druhý týždeň. Pri vysokých koncentráciách v sére, v ktorých je celkový klírens RoActemry určovaný najmä lineárnym klírensom, bol t½ účinnosti približne 32 dní odvodený z odhadov populačných parametrov.
Osobitnéskupinypacientov
Porucha f unkcie obličiek: Štúdia vplyvu poruchy funkcie obličiek na farmakokinetiku RoActemry sa neuskutočnila. Väčšina pacientov v RA a OBA štúdiách zaradených do populačnej farmakokinetickej analýzy mala normálnu funkciu obličiek alebo miernu poruchu funkcie obličiek. Mierna porucha funkcie obličiek (odhadovaný klírens kreatinínu podľa Cockroftovho a Gaultovho vzorca) nemala vplyv na farmakokinetiku RoActemry.
Približne jedna tretina pacientov v štúdii OBA mala stredne ťažkú poruchu funkcie obličiek na začiatku liečby (odhadovaný klírens kreatínu 30-59 ml/min). U týchto pacientov nebol pozorovaný žiaden vplyv na expozíciu RoActemry.
U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávky.
Porucha f unkcie pečene: Štúdia vplyvu poruchy funkcie pečene na farmakokinetiku RoActemry sa neuskutočnila.
Vek, pohlavie a etnická príslušnosť: Populačné farmakokinetické analýzy u dospelých pacientov s RA a OBA preukázali, že vek, pohlavie a etnická príslušnosť nemajú vplyv na farmakokinetiku RoActemry.
Výsledky populačnej PK analýzy u pacientov s pJIA potvrdili, že veľkosť tela je jediný kovariát, ktorý má zreteľný účinok na farmakokinetiku RoActemry vrátane eliminácie a absorpcie, z čoho vyplýva, že sa má vziať do úvahy dávkovanie odvodené od telesnej hmotnosti (pozri tabuľku 8).
5.3 Predk linick é údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Štúdie karcinogenity sa neuskutočnili, pretože IG1 monoklonálne protilátky sa nepovažujú za látky s vlastným karcinogénnym potenciálom.
Dostupné predklinické údaje preukázali vplyv IL-6 na progresiu zhubných nádorov a na rezistenciu rôznych typov nádorov na apoptózu. Tieto údaje nepoukazujú na významné riziko pre vznik
a progresiu rakoviny počas liečby RoActemrou. Okrem toho sa v 6-mesačných štúdiách chronickej toxicity na opiciach rodu Cynomolgus, ani u myší s deficitom IL-6 proliferatívne lézie nepozorovali.
Dostupné predklinické údaje nepotvrdili, že liečba tocilizumabom má vplyv na fertilitu. V štúdii chronickej toxicity na opiciach rodu Cynomolgus sa nepozorovali účinky na endokrinne aktívne orgány a na orgány reprodukčného systému a u myší s deficitom IL-6 nedošlo k poškodeniu reprodukčnej výkonnosti. Zistilo sa, že RoActemra podávaná opiciam rodu Cynomolgus počas skorej fázy gestácie nemal priamy ani nepriamy škodlivý vplyv na graviditu alebo embryofetálny vývoj.
Pozorovalo sa však mierne zvýšenie potratov/embryofetálnej úmrtnosti pri vysokej systémovej expozícii (> 100-násobok expozície dosiahnutej u ľudí) v skupine liečenej vysokou dávkou
50 mg/kg/deň oproti skupine liečenej placebom a inými nízkymi dávkami. Hoci IL-6 zrejme nie je
rozhodujúcim cytokínom pre rast plodu alebo imunologickú kontrolu rozhrania materských
a fetálnych tkanív, súvislosť tohto zistenia s RoActemrou nie je možné vylúčiť.
Liečba myšacím analógom na juvenilných myšiach nevykazovala toxicitu. Konkrétne, nebolo prítomné žiadne narušenie rastu kostí, imunitných funkcií a sexuálneho dozrievania.
Predklinický bezpečnostný profil RoActemry u opíc rodu Cynomolgus nepoukazuje na rozdiel medzi intravenóznyn a subkutánynm spôsobom podávania.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidíniumchloridu
arginín arginíniumchlorid metionín polysorbát 80 voda na injekciu
6.2 Ink ompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
24 mesiacov.
Po vybratí z chladničky sa RoActemra musí podať do 8 hodín a má sa uchovávať pri teplote do 30 °C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Naplnené injekčné striekačky uchovávajte v škatuli na ochranu pred svetlom a vlhkosťou .
6.5 Druh obalu a obsah balenia
0,9 ml roztok v naplnenej injekčnej striekačke (sklo typu I) so vsadenou ihlou. Injekčná striekačka je uzatvorená pevným krytom ihly (elastomérové tesnenie s polypropylénovým plášťom) a gumovou zátkou (butylkaučuk s povlakom z fluorovanej živice).
Balenie po 4 naplnených injekčných striekačkách a spoločné balenie s obsahom 12 (3 balenia po
4ks) naplnených injekčných striekačiek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
RoActemra sa dodáva v naplnenej injekčnej striekačke na jednorazové použitie vybavenej bezpečnostným krytom ihly. Po vybratí naplnenej injekčnej striekačky z chladničky sa má nechať naplnená injekčná striekačka dosiahnuť izbovú teplotu (18°C až 28°C) tak, že sa počká 25 až 30 minút pred injekčným podaním RoActemry. Naplnenou injekčnou striekačkou sa nemá triasť. Po odstránení krytu ihly sa musí injekcia začať podávať do 5 minút, aby sa zabránilo vysušeniu lieku a zablokovaniu
ihly. Ak sa naplnená injekčná striekačka nepoužije do 5 minút, musí sa vyhodiť do nádoby odolnej proti prepichnutiu a použiť novú naplnenú injekčnú striekačku.
Ak sa nedá po vpichnutí ihly stlačiť piest, naplnenú injekčnúj striekačku musíte vyhodiť do nádoby odolnej proti prepichnutiu a použiť novú naplnenú injekčnú striekačku.
Nepoužívajte, ak je liek zakalený alebo obsahuje čiastočky, ak má inú farbu než bezfarebnú až svetložltkastú, alebo ak ktorákoľvek časť naplnenej injekčnej striekačky javí známky poškodenia.
Úplné pokyny na podanie RoActemry v naplnenej injekčnej striekačke sú poskytnuté v písomnej informácii pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/08/492/007
EU/1/08/492/008
9. DÁTUM PRVEJ REGISTRÁCIE/DÁTUM POSLEDNÉHO PREDĹŽENIADátum prvej registrácie: 16. január 2009
Dátum posledného predĺženia registrácie: 25. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.
1. NÁZOV LIEKU
RoActemra 162 mg injekčný roztok v naplnenom pere.
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené pero obsahuje 162 mg RoActemry (tocilizumabu) v 0,9 ml.
RoActemra je rekombinantná humanizovaná antihumánna monoklonálna protilátka podtriedy imunoglobulínu G1 (IgG1) namierená proti solubilným a membránovo viazaným receptorom pre interleukín 6.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok v naplnenom pere (ACTPen).
Bezfarebný až svetložltkastý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutick é indik ácie
RoActemra v kombinácii s metotrexátom (MTX) je indikovaná na
• liečbu ťažkej, aktívnej a progresívnej reumatoidnej artritídy (RA) u dospelých, ktorí neboli doteraz liečení MTX.
• liečbu stredne ťažkej až ťažkej aktívnej RA u dospelých pacientov, ktorí na predchádzajúcu liečbu jedným alebo viacerými antireumatikami modifikujúcimi priebeh choroby (DMARDs), alebo inhibítormi tumor nekrotizujúceho faktora (TNF) buď neodpovedali dostatočne, alebo ju netolerovali.
U týchto pacientov sa RoActemra môže podávať v monoterapii v prípade intolerancie MTX, alebo keď je pokračujúca liečba MTX nevhodná.
Dokázalo sa, že RoActemra spomaľuje progresiu poškodenia kĺbov meranú RTG vyšetrením
a zlepšuje fyzické funkcie, keď sa podáva v kombinácii s metotrexátom.
RoActemra je indikovaná na liečbu obrovskobunkovej arteritídy (OBA) u dospelých pacientov.
4.2 Dávk ovanie a spôsob podávania
Liečbu majú začať zdravotnícki pracovníci, ktorí majú skúsenosti s diagnostikou a liečbou RA
a/alebo OBA. Všetkým pacientom, ktorí sú liečení RoActemrou, sa má poskytnúť karta pre pacienta. Je potrebné zhodnotiť vhodnosť pacienta na subkutánne podávanie lieku v domácom prostredí
a pacienti musia byť poučení, že ak sa u nich objavia príznaky alergickej reakcie, musia o tom informovať zdravotníckeho pracovníka predtým, ako si podajú ďalšiu dávku. Pacienti musia vyhľadať okamžitú lekársku pomoc, ak u nich vzniknú príznaky závažných alergických reakcií (pozri časť 4.4).
Dávkovanie
Subkutánna forma RoActemry nie je určená na intravenózne podávanie.
RA
Odporúčané dávkovanie je 162 mg raz za týždeň subkutánne.
K dispozícii sú obmedzené informácie týkajúce sa prestavenia pacientov z intravenóznej formy RoActemry na subkutánnu formu RoActemry vo fixnej dávke. Je potrebné dodržiavať interval podávania raz za týždeň.
Pacienti, ktorí prechádzajú z intravenóznej na subkutánnu formu, si majú podať svoju prvú subkutánnu dávku namiesto ďalšej plánovanej intravenóznej dávky pod dohľadom kvalifikovaného
zdravotníckeho pracovníka.
OBA
Odporúčané dávkovanie je 162 mg raz za týždeň subkutánne v kombinácii s postupným znižovaním dávky glukokortikoidov. Po ukončení podávania glukokortikoidov sa môže RoActemra podávať samostatne.
RoActemra v monoterapii sa nemá podávať na liečbu akútnych relapsov (pozri časť 4.4).
Vzhľadom na chronický charakter OBA sa musí liečba dlhšia ako 52 týždňov usmerňovať podľa prejavov (aktivity) ochorenia, zváženia lekára a voľby pacienta.
RA a OBA
Úpravydávkykvôlilaboratórnymodchýlkam(pozričasť4.4).
• Odchýlky hodnôt pečeňových enzýmov
Laboratórna hodnota Opatrenie
> 1- až 3-násobok
hornej hranice referenčného rozpätia (ULN)
Úprava dávky súbežne podávaných DMARDs (RA) alebo
imunomodulačných látok (OBA) , ak je to vhodné.
Pri pretrvávajúcich vzostupoch v tomto rozpätí znížte frekvenciu podávania RoActemry na injekciu podanú každý druhý týždeň alebo prerušte podávanie RoActemry, kým nedôjde k normalizácii hodnôt alanínaminotransferázy (ALT) alebo aspartátaminotransferázy (AST).
Liečbu znovu začnite injekciou podanou každý týždeň alebo každý druhý týždeň, ak je to klinicky vhodné.
> 3- až 5-násobok
ULN
Prerušte podávanie RoActemry, pokým nebude hodnota < 3-násobok ULN a
postupujte podľa vyššie uvedených odporúčaní pre > 1- až 3-násobok ULN.
Pri pretrvávajúcich vzostupoch > 3-násobok ULN (potvrdených opakovaným vyšetrením, pozri časť 4.4) ukončite liečbu RoActemrou.
> 5-násobok ULN Ukončite liečbu RoActemrou.
• Nízky absolútny počet neutrofilov (ANC, absolute neutrophil count)
U pacientov, ktorí neboli doteraz liečení RoActemrou a majú absolútny počet neutrofilov (ANC) nižší
ako 2 x 109/l, sa neodporúča začať liečbu.
Laboratórna hodnota
(bunky x 109/l)
Opatrenie
ANC > 1 Udržiavajte dávku.
ANC 0,5 až 1 Prerušte podávanie RoActemry.
Keď sa ANC zvýši na > 1 x 109/l, liečbu RoActemrou znovu začnite injekciou podanou každý druhý týždeň a zvýšte frekvenciu podávania na injekciu podanú každý týždeň, ak je to klinicky vhodné.
ANC < 0,5 Ukončite liečbu RoActemrou.
'
• Nízky počet trombocytov
Laboratórna hodnota
(bunky x 103/μl)
Opatrenie
50 až 100 Prerušte podávanie RoActemry.
Keď bude počet trombocytov > 100 x 103/μl, liečbu RoActemrou znovu začnite injekciou podanou každý druhý týždeň a zvýšte frekvenciu podávania na injekciu podanú na každý týždeň, ak je to klinicky vhodné.
< 50 Ukončite liečbu RoActemrou.
VynechanádávkaAk pacient vynechá subkutánnu injekciu RoActemry, ktorú si podáva raz za týždeň, do 7 dní
od plánovanej dávky, treba ho poučiť, aby si vynechanú dávku podal v deň, v ktorý je plánovaná ďalšia dávka. Ak pacient vynechá subkutánnu injekciu RoActemry, ktorú si podáva každý druhý týždeň, do 7 dní od plánovanej dávky, treba ho poučiť, aby si vynechanú dávku podal ihneď a ďalšiu dávku v deň, v ktorý je plánovaná ďalšia dávka.
Osobitné skupiny pacientovStarší pacientiU pacientov vo veku 65 rokov a starších nie je potrebná úprava dávky.
Porucha f unkcie obličiek:U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávky. U pacientov s ťažkou poruchou funkcie obličiek sa RoActemra neskúmala (pozri časť 5.2). U týchto pacientov sa má starostlivo monitorovať funkcia obličiek.
Porucha f unkcie pečene:U pacientov s poruchou funkcie pečene sa RoActemra neskúmala. Preto nie je možné poskytnúť odporúčania na úpravu dávkovania.
Pediatrickí pacientiBezpečnosť a účinnosť subkutánnej formy RoActemry u detí od narodenia do menej ako 2 rokov
neboli stanovené. K dispozícii nie sú žiadne údaje.
S
pôsob
podávania
RoActemra je na subkutánne použitie.
Po náležitej inštruktáži o injekčnej technike si pacienti môžu sami injekčne podávať RoActemru, ak ich lekár rozhodne, že je to vhodné. Celý obsah (0,9 ml) naplneného pera sa má podať formou subkutánnej injekcie. Odporúčané miesta vpichu (brucho, stehno, horná časť ramena) sa majú striedať a injekcie sa nikdy nemajú podať do materských znamienok, jaziev alebo do miest, na ktorých je koža citlivá, podliata krvou, červená, stvrdnutá alebo porušená.
Naplneným perom sa nemá triasť.
Úplné pokyny na podanie RoActemry v naplnenom pere sú poskytnuté v písomnej informácii pre používateľa, pozri časť 6.6.
4.3 Kontraindik ácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívne, závažné infekcie (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
V záujme zlepšenia sledovateľnosti biologických liekov sa musí v zdravotnej dokumentácii pacienta jasne zaznamenať obchodný názov a číslo šarže podaného lieku.
Inf ekcie
Závažné a niekedy fatálne infekcie boli hlásené u pacientov, ktorí dostávali imunosupresívne látky vrátane RoActemry (pozri časť 4.8 Nežiaduce účinky). Liečba RoActemrou sa nesmie začať
u pacientov s aktívnymi infekciami (pozri časť 4.3). Ak u pacienta dôjde ku vzniku závažnej infekcie,
podávanie RoActemry sa má prerušiť, kým sa infekcia nevylieči (pozri časť 4.8). Zdravotnícki pracovníci majú postupovať opatrne, ak uvažujú o použití RoActemry u pacientov s anamnézou opakujúcich sa alebo chronických infekcií alebo so základnými ochoreniami (napr. divertikulitída, diabetes a intersticiálna choroba pľúc), ktoré ich predisponujú ku vzniku infekcií.
Pacientov, ktorí na stredne ťažkú až ťažkú RA alebo OBA užívajú imunosupresívne látky ako je RoActemra, sa odporúča pozorne sledovať, aby sa včas odhalila závažná infekcia, keďže prejavy a príznaky akútneho zápalu môžu byť zmiernené, z dôvodu potlačenia reaktantov akútnej fázy.
Pri vyšetrovaní pacienta na možnú infekciu sa má vziať do úvahy vplyv RoActemry na C-reaktívny proteín (CRP), neutrofily a prejavy a príznaky infekcie. Pacientov treba poučiť, že keď sa u nich objavia akékoľvek príznaky svedčiace o infekcii, majú sa ihneď skontaktovať so zdravotníckym pracovníkom, aby sa zaistilo rýchle vyšetrenie a náležitá liečba.
Tuberkulóza
Tak ako sa odporúča pre iné typy biologickej liečby, aj pred začatím liečby RoActemrou majú všetci pacienti podstúpiť skríningové vyšetrenie na latentnú tuberkulóznu (TB) infekciu. Pacienti s latentnou TB sa pred začatím liečby RoActemrou majú liečiť štandardnou antimykobakteriálnou terapiou. Lekári, ktorí predpisujú RoActemru, majú mať na mysli riziko falošne negatívnych výsledkov tuberkulinových kožných testov a krvného testu interferón-gamma TB, zvlášť u pacientov, ktorí sú vážne chorí alebo so zníženou imunitou.
Pacienti majú byť upozornení, aby vyhľadali lekársku pomoc, ak sa u nich v priebehu liečby RoActemrou alebo po jej ukončení vyskytnú príznaky/symptómy (napr. pretrvávajúci kašeľ, chradnutie/úbytok telesnej hmotnosti, mierna horúčka) svedčiace o tuberkulóznej infekcii.
R
e
aktivácia vírusu
Reaktivácia vírusu (napr. vírusu hepatitídy B) sa zaznamenala pri biologickej liečbe RA. Z klinických štúdií s RoActemrou boli vylúčení pacienti, ktorí mali pozitívny skríning na hepatitídu.
Komplikácie divertikulitídy
Prípady perforácie divertikulu ako komplikácie divertikulitídy boli u pacientov liečených RoActemrou hlásené menej často (pozri časť 4.8). RoActemra sa má u pacientov s ulceráciou čriev alebo divertikulitídou v anamnéze používať opatrne. Pacientov s príznakmi, ktoré by mohli svedčiť
o komplikovanej divertikulitíde, ako sú bolesti brucha, krvácanie a/alebo nevysvetliteľná zmena
vo vyprázdňovaní stolice spolu s horúčkou, je potrebné promptne vyšetriť, aby sa včas rozpoznala divertikulitída, ktorá môže byť spojená s perforáciou gastrointestinálneho traktu.
Reakcie z precitlivenosti
Boli hlásené závažné reakcie z precitlivenosti, vrátane anafylaxie, súvisiace s podávaním RoActemry
(pozri časť 4.8). Takéto reakcie môžu byť závažnejšie a potenciálne fatálne u pacientov, ktorí mali reakcie z precitlivenosti počas predchádzajúcej liečby RoActemrou, aj keď dostali premedikáciu kortikosteroidmi a antihistaminikami. V prípade výskytu anafylaktickej reakcie alebo inej závažnej reakcie z precitlivenosti sa má podávanie RoActemry okamžite ukončiť, začať náležitá liečba a liečba RoActemrou sa má navždy skončiť.
Aktívne ochorenie pečene a porucha f unkcie pečene
Liečba RoActemrou, najmä keď sa podáva súbežne s MTX, môže byť spojená so zvýšením pečeňových transamináz. Ak sa uvažuje o liečbe pacientov s aktívnym ochorením pečene alebo poruchou funkcie pečene, vyžaduje sa opatrnosť (pozri časti 4.2 a 4.8).
Zvýšenie pečeňových transamináz
V klinických štúdiách sa pri liečbe RoActemrou často hlásili prechodné alebo sporadické, mierne a stredne závažné zvýšenia pečeňových transamináz bez progresie do poškodenia pečene (pozri časť 4.8). Vyššia frekvencia týchto zvýšení pečeňových transamináz sa pozorovala vtedy, keď sa
v kombinácii s RoActemrou užívali potenciálne hepatotoxické liečivá (napr. MTX). Keď je klinicky
indikované, majú sa zvážiť ďalšie vyšetrenia funkcie pečene, vrátane bilirubínu.
Keď sa uvažuje o začatí liečby RoActemrou u pacientov s hodnotami ALT alebo AST zvýšenými na > 1,5-násobok ULN, je nutná opatrnosť. U pacientov s východiskovými hodnotami ALT alebo AST > 5-násobok ULN sa liečba neodporúča.
U pacientov s RA a OBA sa hladiny ALT a AST majú skontrolovať raz za 4 až 8 týždňov počas prvých 6 mesiacov liečby a následne raz za 12 týždňov. Pre odporúčané úpravy dávky na základe transamináz pozri časť 4.2. Pri vzostupoch hodnôt ALT alebo AST na > 3- až 5-násobok ULN sa má liečba RoActemrou prerušiť.
Hematologické odchýlky
Po liečbe RoActemrou v dávke 8 mg/kg v kombinácii s MTX sa vyskytoval pokles počtu neutrofilov a trombocytov (pozri časť 4.8). U pacientov, ktorí boli predtým liečení inhibítorom TNF, môže existovať zvýšené riziko neutropénie.
U pacientov, ktorí neboli doteraz liečení RoActemrou, sa neodporúča začať liečbu, ak je ANC nižší ako 2 x 109/l. Keď sa uvažuje o začatí liečby RoActemrou u pacientov s nízkym počtom trombocytov (t. j. počet trombocytov pod 100 x 103/μl), vyžaduje sa opatrnosť. U pacientov, u ktorých je
ANC < 0,5 x 109/l alebo počet trombocytov je < 50 x 103/μl, sa neodporúča pokračovať v liečbe.
Ťažká neutropénia môže byť spojená so zvýšeným rizikom závažných infekcií, i keď doteraz nie je jasné spojenie medzi zníženým počtom neutrofilov a výskytom závažných infekcií v klinických skúšaniach s RoActemrou.
U pacientov s RA a OBA sa má počet neutrofilov a trombocytov skontrolovať po 4 až 8 týždňoch od začatia liečby a následne v súlade so štandardnou klinickou praxou. Odporúčané úpravy dávky na základe počtu ANC a neutrofilov pozri v časti 4.2.
Hodnoty lipidov
U pacientov liečených RoActemrou sa pozorovalo zvýšenie hodnôt lipidových parametrov, vrátane hladiny celkového cholesterolu, lipoproteínov s nízkou hustotou (LDL), lipoproteínov s vysokou hustotou (HDL) a triglyceridov (pozri časť 4.8). U väčšiny pacientov nedošlo k zvýšeniu aterogénneho indexu a zvýšenie celkového cholesterolu odpovedalo na liečbu hypolipidemikami.
U pacientov s RA a OBA sa má hodnotenie lipidových parametrov vykonať po 4 až 8 týždňoch
od začatia liečby RoActemrou. Pacienti sa majú liečiť v súlade s národnými klinickými odporúčaniami pre liečbu hyperlipidémií.
Neurologické poruchy
Lekári majú venovať zvýšenú pozornosť príznakom, ktoré by mohli svedčiť o vzniku centrálnych demyelinizačných porúch. V súčasnosti nie je známe, či RoActemra môže vyvolať centrálnu demyelinizáciu.
Malignita
Riziko vzniku malignity je u pacientov s RA zvýšené. Imunomodulačné lieky môžu riziko vzniku malignity zvyšovať.
Očkovania
Súbežne s RoActemrou sa nemajú podávať živé a živé oslabené očkovacie látky, keďže klinická
bezpečnosť nebola stanovená. V randomizovanej otvorenej štúdii, dosahovali dospelí pacienti s RA, ktorí dostávali liečbu RoActemrou a MTX, účinnú odpoveď na 23-valentnú pneumokokovú polysacharidovú vakcínu ako aj na vakcínu obsahujúcu tetanický toxoid, čo bolo porovnateľné
s odpoveďou pozorovanou u pacientov, ktorí dostávali liečbu MTX v monoterapii. Odporúča sa, aby všetci pacienti, a obzvlášť starší pacienti, boli podrobení aktuálne platným imunizáciám v súlade
s aktuálnymi odporúčaniami imunizácie ešte pred začatím liečby RoActemrou. Interval medzi podaním živých očkovacích látok a začatím liečby RoActemrou má byť v súlade s aktuálnymi odporúčaniami imunizácie ohľadne imunosupresívnych látok.
Kardiovaskulárne riziko
Pacientom s RA hrozí zvýšené riziko kardiovaskulárnych porúch; rizikové faktory (napr. hypertenzia, hyperlipidémia) je potrebné korigovať v rámci bežnej štandardnej zdravotnej starostlivosti.
Kombinácia s inhibítormi TNF
Nie sú skúsenosti s použitím RoActemry s inhibítormi TNF ani inými biologickými liekmi na liečbu pacientov s RA. RoActemra sa neodporúča používať spolu s inými biologickými liekmi.
OBA
RoActemra v monoterapii sa nesmie podávať na liečbu akútnych relapsov, keďže účinnosť pre tento stav nebola stanovená. Glukokortikoidy sa majú podávať podľa zváženia lekára a praktických odporúčaní.
4.5 Liek ové a iné interakcie
Interakčné štúdie sa uskutočnili iba u dospelých.
Súbežné podanie jednorazovej dávky RoActemry 10 mg/kg s MTX v dávke 10 - 25 mg podávanej jedenkrát týždenne nemalo klinicky významný vplyv na expozíciu MTX.
Populačné farmakokinetické analýzy nezistili žiaden vplyv MTX, nesteroidných protizápalových liekov (NSAIDs) alebo kortikosteroidov na klírens RoActemry u pacientov s RA. U pacientov s OBA nebol pozorovaný žiaden účinok kumulatívnej dávky kortikosteroidov na expozíciu RoActemry.
Účinkom cytokínov, ako je napr. IL-6, ktoré stimulujú chronický zápal, dochádza k potlačeniu expresie pečeňových enzýmov CYP450. Pri začatí liečby silne účinným inhibítorom cytokínov, ako je RoActemra, preto môže dôjsť k obnoveniu expresie enzýmov CYP450.
Štúdie in vitro na kultivovaných ľudských hepatocytoch preukázali, že IL-6 spôsobuje zníženie expresie enzýmov CYP1A2, CYP2C9, CYP2C19 a CYP3A4. RoActemra normalizuje expresiu týchto enzýmov.
V štúdii s pacientmi s RA boli hladiny simvastatínu (CYP3A4) znížené o 57 % jeden týždeň po jednotlivej dávke RoActemry na hodnotu podobnú alebo mierne vyššiu, ako sa pozorovala u zdravých ľudí.
Pri začatí alebo ukončení liečby RoActemrou sa majú sledovať pacienti, ktorí užívajú individuálne upravované dávky liekov metabolizovaných prostredníctvom CYP450 3A4, 1A2 alebo 2C9
(napr. metylprednizolón, dexametazón (možnosť vzniku abstinenčného syndrómu v dôsledku vysadenia perorálnych glukokortikoidov), atorvastatín, blokátory kalciového kanála, teofylín, w arfarín, fenprokumón, fenytoín, cyklosporín alebo benzodiazepíny), keďže na udržanie ich terapeutického účinku môže byť potrebné zvýšenie dávok. Vzhľadom na dlhý eliminačný polčas (t1/2) môže vplyv RoActemry na aktivitu enzýmov CYP450 pretrvávať niekoľko týždňov po ukončení liečby.
4.6 Fertilita, gravidita a lak tácia
Ženy vofertilnomveku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a 3 mesiace po ukončení liečby.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití RoActemry u gravidných žien. Štúdia na zvieratách
preukázala zvýšené riziko spontánneho potratu/embryofetálneho úmrtia pri podávaní vysokej dávky
(pozri časť 5.3). Nie je známe potenciálne riziko u ľudí.
RoActemra sa môže používať počas gravidity iba v nevyhnutných prípadoch. Dojčenie
Nie je známe, či sa RoActemra vylučuje do ľudského materského mlieka. Vylučovanie RoActemry do mlieka sa u zvierat neskúmalo. Pri rozhodovaní o tom, či pokračovať v dojčení/ukončiť dojčenie, alebo či pokračovať v liečbe/ukončiť liečbu RoActemrou sa má brať do úvahy prínos dojčenia
pre dieťa a prínos liečby RoActemrou pre ženu.
Fertilita
Dostupné predklinické údaje nenaznačujú vplyv liečby RoActemrou na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
RoActemra má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť 4.8, závraty).
4.8 Nežiaduce účink y
Súhrnbezpečnostnéhoprofilu
Bezpečnostný profil bol stanovený na základe 4 158 pacientov vystavených RoActemre v klinických štúdiách; väčšina týchto pacientov sa zúčastnila štúdií RA (n=4 009), kým zvyšné údaje pochádzajú zo štúdií OBA (n=149). Bezpečnostný profil RoActemry zostáva v týchto indikáciách podobný
a nediferencovaný.
Najčastejšie hlásené nežiaduce reakcie na liek (Adverse Drug Reactions, ADRs) boli infekcie horných dýchacích ciest, nazofaryngitída, bolesť hlavy, hypertenzia a zvýšené hodnoty ALT.
Najvážnejšie ADRs boli závažné infekcie, komplikácie divertikulitídy a reakcie z precitlivenosti. Súhrnnežiaducichreakciívtabuľke
ADRs vymenované v tabuľke 1 sú uvedené podľa triedy orgánových systémov a kategórií frekvencie,
ktoré sú definované s použitím nasledujúceho pravidla: veľmi časté (≥ 1/10); časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (>1/10 000 až < 1/1 000) alebo veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1. Súhrn ADRs vyskytujúcich sa u pacientov liečených RoActemrou
T
r
i
e
d
a orgánových
s
ystémov [A1] Poruchy krvi a lymfatického systému Poruchy endokrinného systému
Veľmi časté Časté Menej časté
Leukopénia, neutropénia
Hypotyreoidizmus
Poruchy oka Konjunktivitída
Poruchy gastrointestinálneho traktu
Celkové poruchy a reakcie v mieste podania
Infekcie a nákazy Infekcie horných dýchacích ciest
Laboratórne a funkčné vyšetrenia
Bolesť brucha, ulcerácia
v ústnej dutine, gastritída
Periférny edém, reakcia z precitlivenosti, reakcia v mieste vpichu Celulitída, pneumónia,
jednoduchý opar v oblasti úst, pásový opar
Zvýšené hodnoty pečeňových transamináz, zvýšenie telesnej hmotnosti, zvýšené hodnoty celkového bilirubínu*
Stomatitída, žalúdočné vredy
Divertikulitída
Poruchy metabolizmu a výživy
Poruchy nervového systému
Poruchy obličiek
a močových ciest Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy kože a
podkožného tkaniva
Hypercholeste-
rolémia*
Bolesť hlavy, závraty
Kašeľ, dyspnoe
Vyrážka, pruritus, urtikária
Hypertriglyceridémia
Nefrolitiáza
Poruchy kože a podkožného tkaniva
Poruchy ciev Hypertenzia
*Zahŕňa zvýšenia zozbierané ako časť rut inného laborat órneho pozorovania (pozri t ext nižšie).
RAIntravenózne použitieBezpečnosť RoActemry bola skúmaná v 4 placebom kontrolovaných štúdiách (štúdie II, III, IV a V), v 1 MTX kontrolovanej štúdii (štúdia I) a v ich predĺžených fázach (pozri časť 5.1).
V 4 štúdiách bolo dvojito-zaslepené kontrolované obdobie 6 mesiacov (štúdie I, III, IV a V) a v jednej štúdii trvalo až 2 roky (štúdia II). V týchto dvojito zaslepených, kontrolovaných štúdiách dostávalo
774 pacientov RoActemru 4 mg/kg v kombinácii s MTX, 1 870 pacientov dostávalo RoActemru
8 mg/kg v kombinácii s MTX alebo inými DMARDs a 288 pacientov dostávalo RoActemru 8 mg/kg v monoterapii.
Populácia dlhodobej expozície zahŕňa všetkých pacientov, ktorí dostali aspoň jednu dávku RoActemry buď v dvojito zaslepenom kontrolovanom období alebo v otvorenej predĺženej fáze týchto štúdií.
Z celkovo 4 009 pacientov v tomto súbore dostávalo 3 577 pacientov liečbu aspoň počas 6 mesiacov,
3 296 aspoň počas 1 roku, 2 806 dostávalo liečbu aspoň počas 2 rokov a 1 222 počas 3 rokov.
Popis vybraných nežiaducich reakcií
Inf ekcie
V 6-mesačných kontrolovaných štúdiách bola miera výskytu všetkých infekcií hlásených pri liečbe
RoActemrou 8 mg/kg spolu s DMARDs 127 udalostí na 100 pacientorokov oproti 112 udalostiam
na 100 pacientorokov v skupine s placebom v kombinácii s DMARDs. V súbore dlhodobej expozície bol celkový výskyt infekcií pri liečbe RoActemrou 108 udalostí na 100 pacientorokov.
V 6-mesačných kontrolovaných klinických štúdiách bola miera výskytu závažných infekcií pri liečbe
RoActemrou 8 mg/kg v kombinácii s DMARDs 5,3 udalostí na 100 pacientorokov expozície oproti
3,9 udalostiam na 100 pacientorokov expozície v skupine s placebom v kombinácii s DMARDs. V štúdii monoterapie bola miera výskytu závažných infekcií 3,6 udalostí na 100 pacientorokov expozície v skupine liečenej RoActemrou a 1,5 udalosti na 100 pacientorokov expozície v skupine liečenej MTX.
V súbore dlhodobej expozície bol celkový výskyt závažných infekcií (bakteriálne, vírusové
a mykotické) 4,7 udalostí na 100 pacientorokov. K hláseným závažným infekciám, z ktorých niektoré mali fatálne následky, patrila aktívna tuberkulóza, ktorá môže byť prítomná s vnútropľúcnym alebo mimopľúcnym ochorením, invazívne pľúcne infekcie, vrátene kandidózy, aspergilózy kokcidioidomykózy a Pneumocystis jiroveci, pneumónia, celulitída, pásový opar, gastroenteritída, divertikulitída, sepsa a bakteriálna artritída. Boli hlásené prípady oportúnnych infekcií.
Intersticiálna choroba pľúc
Zhoršená funkcia pľúc môže zvýšiť riziko vzniku infekcií. Po uvedení lieku na trh boli hlásené
prípady intersticiálnej choroby pľúc (vrátane pneumonitídy a pľúcnej fibrózy), z ktorých niektoré boli
fatálne.
Gastrointestinálne perf orácie
Pri liečbe RoActemrou počas 6-mesačných kontrolovaných klinických štúdií bol celkový výskyt gastrointestinálnych perforácií 0,26 udalosti na 100 pacientorokov. V dlhodobej expozícií bol celkový výskyt gastrointestinálnych perforácií 0,28 udalosti na 100 pacientorokov. Hlásenia gastrointestinálnej perforácie pri liečbe RoActemrou boli primárne hlásené ako komplikácie divertikulitídy, zahŕňajúce generalizovanú purulentnú peritonitídu, perforáciu dolnej časti gastrointestinálneho traktu, fistulu
a absces.
Reakcie na inf úziu
V 6-mesačných kontrolovaných klinických štúdiách boli nežiaduce účinky súvisiace s podávaním infúzie (vybrané udalosti vyskytujúce sa počas podávania infúzie alebo v priebehu 24 hodín
od podania infúzie) hlásené u 6,9 % pacientov v skupine liečenej RoActemrou 8 mg/kg v kombinácii s DMARDs a u 5,1 % pacientov v skupine s placebom spolu s DMARDs. Udalosti hlásené počas podávania infúzie boli predovšetkým epizódy hypertenzie; udalosti hlásené v priebehu 24 hodín
od ukončenia podávania infúzie boli bolesť hlavy a kožné reakcie (vyrážka, urtikária). Tieto udalosti neboli pre liečbu limitujúce.
Miera výskytu anafylaktických reakcií (vyskytujúcich sa celkovo u 8/4 009 pacientov, 0,2 %) bola niekoľkonásobne vyššia pri dávke 4 mg/kg oproti dávke 8 mg/kg. Klinicky významné reakcie
z precitlivenosti súvisiace s liečbou RoActemrou a vyžadujúce ukončenie liečby boli hlásené
u celkovo 56 z 4 009 pacientov (1,4 %) liečených RoActemrou počas kontrolovaných a otvorených klinických štúdií. Tieto reakcie sa zvyčajne pozorovali počas podávania druhej až piatej infúzie RoActemry (pozri časť 4.4). Po registrácii lieku bola hlásená fatálna anafylaxia počas liečby intravenózne podávanou RoActemrou (pozri časť 4.4).
Imunogenicita
V 6-mesačných kontrolovaných klinických štúdiách bolo celkovo 2 876 pacientov vyšetrených na protilátky proti RoActemre. Zo 46 pacientov (1,6 %), u ktorých sa vytvorili protilátky proti RoActemre, došlo u 6 k významnej reakcii z precitlivenosti, ktorá u 5 z nich viedla k trvalému ukončeniu liečby. U tridsiatich pacientov (1,1 %) sa vytvorili neutralizujúce protilátky.
Hematologické odchýlky:
Neutrof ily
V 6-mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu neutrofilov pod 1 x 109/l
u 3,4 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs oproti < 0,1 % pacientov
s placebom spolu s DMARDs. Približne u polovice pacientov, u ktorých ANC klesol na < 1 x 109/l,
došlo k tomuto poklesu v priebehu 8 týždňov po začatí liečby. Pokles pod 0,5 x 109/l bol hlásený u 0,3 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs. Boli hlásené infekcie
s neutropéniou.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter
a výskyt poklesu počtu neutrofilov rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
Krvné doštičky
V 6-mesačných kontrolovaných klinických štúdiách došlo k poklesu počtu trombocytov
pod 100 x 103/μl u 1,7 % pacientov liečených RoActemrou 8 mg/kg spolu s DMARDs oproti
< 1 % pacientov s placebom spolu s DMARDs. Tieto poklesy sa vyskytli bez asociácie s krvácaním.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter a výskyt poklesu počtu krvných doštičiek rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
V postmarketingovom sledovaní sa vyskytli veľmi zriedkavo prípady pancytopénie.
Zvýšenie pečeňových transamináz
V 6-mesačných kontrolovaných klinických štúdiách bolo prechodné zvýšenie hodnôt
ALT/AST > 3 x ULN u 2,1 % pacientov liečených RoActemrou 8 mg/kg oproti 4,9 % pacientov liečených MTX, a u 6,5 % pacientov liečených 8 mg/kg RoActemry v kombinácii s DMARDs oproti
1,5 % pacientov s placebom v kombinácii s DMARDs.
Pridanie potenciálne hepatotoxických liekov (napr. MTX) k RoActemre podávanej v monoterapii viedlo k zvýšenému výskytu vyšších hodnôt. Zvyšovanie hladín ALT/AST > 5-násobok ULN sa pozorovalo u 0,7 % pacientov liečených RoActemrou v monoterapii a u 1,4 % pacientov liečených RoActemrou v kombinácii s DMARDs, pričom väčšina z nich liečbu RoActemrou trvalo ukončila. Tieto vzostupy neboli spojené s klinicky významným zvýšením hodnôt priameho (konjugovaného) bilirubínu, ani nesúviseli s klinickými príznakmi hepatitídy či poruchy funkcie pečene. Počas dvojito zaslepeného kontrolovaného obdobia sa hodnoty nepriameho bilirubínu, sledovaného ako rutinný laboratórny parameter, vyššie ako horná hranica referenčného rozpätia vyskytli u 6,2 % pacientov liečených RoActemrou v dávke 8 mg/kg + DMARD. U celkovo 5,8 % pacientov došlo k zvýšeniu hodnoty nepriameho bilirubínu na > 1-násobok až 2-násobok ULN a 0,4 % pacientov malo zvýšenie na > 2-násobok ULN.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter
a výskyt prípadov zvýšenia ALT/AST rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných
klinických štúdií.
H
odnoty lipidových parametrov
Zvýšenie hodnôt lipidových parametrov, ako je napríklad celkový cholesterol, triglyceridy,
LDL-cholesterol a/alebo HDL-cholesterol, bolo počas 6-mesačných kontrolovaných štúdií hlásené často. Rutinným laboratórnym sledovaním sa zistilo, že približne u 24 % pacientov, ktorí v klinických štúdiách dostávali RoActemru, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu
na ≥ 6,2 mmol/l, pričom u 15 % pacientov došlo k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l.
Zvýšené lipidové parametre odpovedali na liečbu hypolipidemikami.
Počas dvojito zaslepeného kontrolovaného obdobia a pri dlhodobej expozícii zostával charakter
a výskyt prípadov zvýšenia lipidových parametrov rovnaký ako sa pozoroval počas 6-mesačných kontrolovaných klinických štúdií.
Malignity
Klinické údaje nie sú dostatočné na zhodnotenie rizika možného výskytu malignity po expozícii
RoActemre. Hodnotenie dlhodobej bezpečnosti naďalej prebieha.
Kožné reakcie
V postmarketingovom sledovaní sa vyskytli veľmi zriedkavé hlásenia Stevensovho-Johnsonovho syndrómu.
Subkutánnepoužitie
RA
Bezpečnosť subkutánne podávanej RoActemry pri RA sa hodnotila v dvojito zaslepenej, kontrolovanej, multicentrickej štúdii, SC-I. SC-I bola štúdia noninferiority, ktorá porovnávala účinnosť a bezpečnosť RoActemry v dávke 162 mg podávanej raz za týždeň s intravenóznou dávkou
8 mg/kg u 1 262 pacientov s RA. Všetkým pacientom boli v rámci základnej liečby podávané nebiologické DMARDs. Bezpečnosť a imunogenicita pozorované pri subkutánne podávanej RoActemre sa zhodovali so známym bezpečnostných profilom intravenózne podávanej RoActemry a nepozorovali sa žiadne nové alebo neočakávané nežiaduce reakcie na liek (pozri tabuľku 1). Vyšší výskyt reakcií v mieste vpichu sa pozoroval v skupine so subkutánnym tocilizumabom v porovnaní so subkutánnymi injekciami placeba v skupinách s intravenóznym podávaním.
Reakcie v mieste podania injekcie
V štúdii SC-I bol počas 6-mesačného kontrolovaného obdobia výskyt reakcií v mieste vpichu 10,1 %
(64/631) pri subkutánnej RoActemre a 2,4 % (15/631) pri subkutánnych injekciách placeba (skupina s intravenóznym podávaním) podávaných raz za týždeň. Tieto reakcie v mieste vpichu (vrátane erytému, pruritu, bolesti a hematómu) boli mierne až stredne závažné. Väčšina z nich ustúpila bez potreby akejkoľvek liečby a žiadna nevyžadovala ukončenie podávania lieku.
Imunogenicita
V štúdii SC-I bolo celkovo 625 pacientov liečených RoActemrou v dávke 162 mg podávanej
raz za týždeň vyšetrených na protilátky proti RoActemre v 6-mesačnom kontrolovanom období. U piatich pacientov (0,8 %) sa zistila pozitivita protilátok proti RoActemre; u všetkých z nich sa vytvorili neutralizujúce protilátky proti RoActemre. U jedného pacienta sa zistila pozitivita protilátok izotypu IgE (0,2 %).
V štúdii SC-II bolo celkovo 434 pacientov liečených RoActemrou v dávke 162 mg podávanej každý druhý týždeň vyšetrených na protilátky proti RoActemre v 6-mesačnom kontrolovanom období.
U siedmich pacientov (1,6 %) sa zistila pozitivita protilátok proti RoActemre: u šiestich z nich (1,4 %) sa vytvorili neutralizujúce protilátky proti RoActemre. U štyroch pacientov sa zistila pozitivita protilátok izotypu IgE (0,9 %).
Nepozorovala sa žiadna korelácia medzi tvorbou protilátok a klinickou odpoveďou alebo nežiaducimi udalosťami.
H
e
m
atologické odchýlky: Neutrof ily
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, SC-I, došlo k poklesu počtu neutrofilov pod 1 x 109/l u 2,9 % pacientov liečených subkutánnou dávkou podávanou raz za týždeň.
Nezistila sa žiadna jasná súvislosť medzi poklesom počtu neutrofilov pod 1 x 109/l a výskytom závažných infekcií.
Krvné doštičky
Počas rutinného laboratórneho sledovania v 6-mesačnom klinickom skúšaní s RoActemrou, SC-I, nedošlo u žiadneho z pacientov liečených s.c. dávkou podávanou raz za týždeň k poklesu počtu krvných doštičiek na ≤ 50 x 103/μl.
Zvýšenie pečeňových transamináz
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, SC-I, došlo k vzostup hodnôt ALT a AST na ≥ 3-násobok ULN u 6,5 % a 1,4 %, v uvedenom poradí, pacientov liečených subkutánnou dávkou podávanou raz za týždeň.
Hodnoty lipidových parametrov
Počas rutinného laboratórneho sledovania v 6-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, SC-I, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu na > 6,2 mmol/l
(240 mg/dl) u 19 % pacientov liečených subkutánnou dávkou podávanou raz za týždeň a u 9 % došlo
k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l (160 mg/dl).
Subkutánnepoužitie
OBA
Bezpečnosť subkutánne podávanej RoActemry sa skúmala vo fáze III jednej štúdie (WA28119), ktorej sa zúčastnilo 251 pacientov s OBA. Počas 12-mesačnej, dvojito zaslepenej, placebom kontrolovanej fázy tejto štúdie, bolo celkové trvanie v pacientorokoch pri RoActemre u populácie s celkovou expozíciou 138,5 pacientorokov. Celkový bezpečnostný profil pozorovaný v liečebných skupinách
s RoActemrou bol zhodný so známym bezpečnostným profilom RoActemry (pozri tabuľku 1).
Inf ekcie
Miera výskytu udalostí infekcií/závažných infekcií bola vyrovnaná medzi skupinou s RoActemrou
podávanou raz za týždeň (200,2/9,7 udalostí na 100 pacientorokov) a skupinou s placebom
v kombinácii s postupným znižovaním dávky prednizónu počas 26 týždňov (156,0/4,2 udalostí
na 100 pacientorokov) a skupinou s placebom v kombinácii s postupným znižovaním dávky počas
52 týždňov (210,2/12,5 udalostí na 100 pacientorokov).
Reakcie v mieste podania injekcie
V skupine, v ktorej sa RoActemra podávala subkutánne raz za týždeň, celkovo 6 % (6/100) pacientov hlásilo nežiaduce reakcie v mieste podania subkutánnej injekcie. Žiadna reakcia v mieste podania injekcie nebola hlásená ako závažná nežiaduca udalosť, alebo žiadna nevyžadovala ukončenie liečby.
Imunogenicita
V skupine s RoActemrou podávanou subkutánne raz za týždeň, sa u jedného pacienta (1,1 %, 1/95) zistila pozitivita neutralizujúcich protilátok proti RoActemre, nie však protilátok izotypu IgE. U tohto pacienta nebola pozorovaná reakcia z precitlivenosti, ani reakcia v mieste podania injekcie.
Hematologické odchýlky:
Neutrof ily
Počas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní s RoActemrou sa pozoroval pokles počtu neutrofilov pod 1 x 109/l u 4 % pacientov v skupine
so subkutánne podávanou RoActemrou raz za týždeň. Tento pokles sa nepozoroval v žiadnej skupine s placebom a s postupným znižovaním dávky prednizónu.
T
r
ombocyty
Počas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní s RoActemrou sa u 1 pacienta (1 %, 1/100) v skupine so subkutánne podávanou Roactemrou
raz za týždeň pozoroval jeden výskyt prechodného poklesu počtu trombocytov na < 100 x 103/μl bez súvisiacich prípadov krvácania. Pokles počtu trombocytov pod počet 100 x103/μl nebol pozorovaný v žiadnej skupine s placebom a s postupným znižovaním dávky prednizónu.
Zvýšenie pečeňových transaminázPočas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, sa pozoroval vzostup hodnôt ALT na > 3-násobok ULN u 3 % pacientov v skupine so subkutánne podávanou RoActemrou raz za týždeň v porovnaní s 2 % pacientov v skupine
s placebom v kombinácii s postupným znižovaním dávky prednizónu počas 52 týždňov
a 0 % pacientov v skupine s placebom v kombinácii s postupným znižovaním dávky prednizónu počas
26 týždňov. Vzostup hodnôt AST na > 3-násobok ULN sa pozoroval u 1 % pacientov v skupine
s RoActemrou podávanou subkutánne raz za týždeň v porovnaní s 0 % pacientov v oboch skupinách s placebom s postupným znižovaním dávky prednizónu.
Hodnoty lipidových parametrovPočas rutinného laboratórneho sledovania v 12-mesačnom kontrolovanom klinickom skúšaní
s RoActemrou, došlo k trvalému zvýšeniu hodnôt celkového cholesterolu na > 6,2 mmol/l (240 mg/dl)
u 34 % pacientov liečených RoActemrou podávanou subkutánne raz za týždeň a u 15 % došlo
k trvalému zvýšeniu hodnôt LDL na ≥ 4,1 mmol/l (160 mg/dl)
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávk ovanieK dispozícii sú obmedzené údaje o predávkovaní RoActemrou. Hlásený bol jeden prípad náhodného predávkovania, pri ktorom pacient s mnohopočetným myelómom dostal jednorazovú intravenózne podanú dávku 40 mg/kg. Nepozorovali sa žiadne nežiaduce reakcie.
U zdravých dobrovoľníkov, ktorí dostali jednorazovú dávku do 28 mg/kg, sa nepozorovali žiadne závažné nežiaduce reakcie, hoci došlo k výskytu neutropénie limitujúcej dávku.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmak odynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínu; ATC kód: L04AC07.
MechanizmusúčinkuRoActemra sa špecificky viaže na rozpustný aj na membránovo-viazaný receptor pre IL-6 (sIL-6R
a mIL-6R). Dokázalo sa, že RoActemra inhibuje prenos signálu sprostredkovaný sIL-6R a mIL-6R. IL-6 je pleiotropný prozápalový cytokín, ktorý produkujú rôzne typy buniek, vrátane T- a B-buniek, monocytov a fibroblastov. IL-6 sa zúčastňuje na rôznorodých fyziologických procesoch, ako je aktivácia T-buniek, indukcia sekrécie imunoglobulínov, indukcia syntézy proteínov akútnej fázy
v pečeni a stimulácia krvotvorby. IL-6 sa podieľa na patogenéze ochorení, medzi ktoré patria zápalové ochorenia, osteoporóza a neoplázie.
FarmakodynamickéúčinkyV klinických štúdiách RA s RoActemrou sa pozoroval rýchly pokles hodnôt CRP, sedimentácie erytrocytov (ESR) a sérového amyloidu A (SAA). V zhode s účinkom na reaktanty akútnej fázy bola liečba RoActemrou spojená s poklesom počtu trombocytov na hodnoty v rámci referenčného rozpätia.
Pozorovalo sa zvýšenie hladín hemoglobínu, ktoré RoActemra vyvoláva tým, že znižuje IL-6 navodené účinky na tvorbu hepcidínu, čím sa zvyšuje dostupnosť železa. U pacientov liečených RoActemrou sa pokles hladín CRP na hodnoty v rámci referenčného rozpätia pozoroval už od
2. týždňa a počas trvania liečby sa tento pokles udržal.
V klinickej štúdii OBA WA28119 sa pozoroval podobný rýchly pokles hodnôt CRP a sedimentácie erytrocytov (ESR) s miernym zvýšením priemernej koncentrácie korpuskulárneho hemoglobínu.
U zdravých dobrovoľníkov, ktorým sa podávala RoActemra v dávkach od 2 do 28 mg/kg intravenózne
a od 81 do 162 mg subkutánne, klesal absolútny počet neutrofilov k najnižším hladinám 2 až 5 dní
po podaní. Potom sa počet neutrofilov vrátil k východiskovým hodnotám v závislosti od dávky. Pacienti s RA a OBA po podaní RoActemry vykazujú porovnateľný (oproti zdravým osobám) pokles absolútneho počtu neutrofilov (pozri časť 4.8).
RA
Intravenóznepoužitie
Klinická účinnosť
Účinnosť RoActemry na zmiernenie prejavov a príznakov RA sa hodnotila v piatich randomizovaných, dvojito zaslepených, multicentrických štúdiách. Do štúdií I - V boli zaradení pacienti vo veku ≥ 18 rokov s aktívnou RA diagnostikovanou podľa kritérií American College of Rheumatology (ACR), ktorí mali pred začiatkom liečby minimálne osem bolestivých a šesť opuchnutých kĺbov.
V štúdii I sa RoActemra podávala intravenózne raz za štyri týždne v monoterapii. V štúdiách II, III
a V sa RoActemra podávala intravenózne raz za štyri týždne v kombinácii s MTX, kontrolnú skupinu tvorilo placebo v kombinácii s MTX. V štúdii IV sa RoActemra podávala intravenózne raz za 4 týždne v kombinácii s inými DMARDs, kontrolnú skupinu tvorilo placebo v kombinácii s inými DMARDs. Primárny cieľový ukazovateľ pre každú z piatich štúdií bol podiel pacientov, ktorí dosiahli odpoveď ACR 20 v 24. týždni.
Štúdia I hodnotila 673 pacientov, ktorí sa v priebehu šiestich mesiacov pred randomizáciou neliečili MTX a ktorí neprerušili predchádzajúcu liečbu MTX kvôli klinicky významným toxickým účinkom alebo nedostatočnej odpovedi na liečbu. Väčšina (67 %) pacientov sa MTX predtým neliečila. Dávky
8 mg/kg RoActemry sa podávali raz za štyri týždne v monoterapii. Porovnávacia skupina dostávala MTX raz týždenne (dávka titrovaná od 7,5 mg na maximálne 20 mg týždenne počas osemtýždňového obdobia).
Štúdia II, dvojročná štúdia s plánovanou analýzou v týždni 24., 52. a v týždni 104, hodnotila
1 196 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky 4 alebo 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v zaslepenej fáze liečby trvajúcej 52 týždňov v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne). Po týždni 52 mohli všetci
pacienti pokračovať v otvorenej fáze liečby s RoActemrou v dávke 8 mg/kg. Z pacientov, ktorí
dokončili štúdiu a ktorí boli pôvodne randomizovaní do skupiny s placebom + MTX, v 2. roku
86 % pacientov pokračovalo v otvorenej fáze liečby RoActemrou v dávke 8 mg/kg. Primárny cieľový ukazovateľ v 24. týždni bol podiel pacientov, ktorí dosiahli odpoveď ACR 20. V 52. a 104. týždni boli prevencia poškodenia kĺbu a zlepšenie fyzických funkcií pridružené ako primárne cieľové
ukazovatele.
Štúdia III hodnotila 623 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na MTX. Dávky
4 alebo 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Štúdia IV hodnotila 1 220 pacientov, ktorí nedosiahli dostatočnú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viaceré DMARDs. Dávky 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou DMARDs.
Štúdia V hodnotila 499 pacientov, ktorí nedosiahli dostatočnú klinickú odpoveď na liečbu jedným alebo viacerými inhibítormi TNF, alebo ktorí takúto liečbu netolerovali. Liečba inhibítorom TNF sa
pred randomizáciou ukončila. Dávky 4 alebo 8 mg/kg RoActemry alebo placeba sa podávali raz za štyri týždne v kombinácii so stabilnou dávkou MTX (10 mg až 25 mg týždenne).
Klinická odpoveď
Vo všetkých štúdiách mali pacienti liečení RoActemrou 8 mg/kg štatisticky významne vyššiu mieru odpovede ACR 20, 50, 70 po šiestich mesiacoch oproti kontrolnej skupine (tabuľka 2). V štúdii I sa preukázala vyššia účinnosť RoActemry 8 mg/kg oproti aktívnej porovnávacej látke - MTX.
Účinok liečby bol u pacientov podobný nezávisle od prítomnosti reumatoidného faktora, veku, pohlavia, rasy, počtu predchádzajúcich terapií a stavu ochorenia. Účinok nastúpil rýchlo
(už v 2. týždni) a stupeň odpovede sa počas liečby neustále zlepšoval. V prebiehajúcich predĺžených otvorených štúdiách I - V sa počas viac ako 3 rokov pozorovali neustále pretrvávajúce odpovede.
Vo všetkých štúdiách sa u pacientov liečených RoActemrou 8 mg/kg oproti pacientom s placebom
a MTX alebo inými DMARDs zaznamenalo významné zlepšenie vo všetkých jednotlivých zložkách odpovede ACR zahŕňajúcich: počet bolestivých a opuchnutých kĺbov; celkové hodnotenie pacientmi a lekárom; skóre indexu funkčnej neschopnosti; hodnotenie bolesti a CRP.
Pacienti v štúdiách I - V mali pred začiatkom liečby priemerné skóre aktivity ochorenia (DAS28)
6,5 - 6,8. U pacientov liečených RoActemrou sa v porovnaní s pacientmi v kontrolnej skupine
(1,3 - 2,1) pozorovalo významné zníženie DAS28 oproti východiskovej hodnote (priemerné zlepšenie) o 3,1 - 3,4. Podiel pacientov, ktorí v 24. týždni dosiahli klinickú remisiu ochorenia podľa DAS28 (DAS28 < 2,6), bol významne vyšší u pacientov liečených RoActemrou (28 - 34 %) v porovnaní
s 1 - 12 % pacientov v kontrolnej skupine. V štúdii II dosiahlo 65 % pacientov DAS28 < 2,6
v 104. týždni, v porovnaní so 48 % pacientov v 52. týždni a s 33 % pacientov v 24. týždni.
V súhrnnej analýze štúdií II, III a IV bol podiel pacientov, ktorí dosiahli odpoveď ACR 20, 50 a 70
významne vyšší (59 % oproti 50 %, 37 % oproti 27 %, 18 % oproti 11 % v uvedenom poradí) v skupine liečenej RoActemrou 8 mg/kg v kombinácii s DMARDs oproti skupine liečenej RoActemrou 4 mg/kg v kombinácii s DMARDs (p < 0,03). Podobne bol aj podiel pacientov, ktorí dosiahli remisiu ochorenia podľa DAS28 (DAS28 < 2,6), významne vyšší (31 % oproti 16 %)
u pacientov liečených RoActemrou 8 mg/kg v kombinácii s DMARDs než u pacientov liečených
RoActemrou 4 mg/kg v kombinácii s DMARDs (p < 0,0001).
Tabuľka 2.Odpovede ACR v placebom/MTX/DMARDs kontrolovaných štúdiách (% pacientov)
Š
t
ú
d
i a I AMBITIO N
Š
t
ú
d
i a II LITHE
Š tú di a III OPTION
Š
t
ú
d
i a IV TO W ARD
Š
t
ú
d
i a V RADIATE
T ýž-
deň
TC Z
8 m g/k g
MTX TC Z
8 m g/k g
+ MTX
PBO + MTX
TC Z
8 m g/k g
+ MTX
PBO
+ MTX
TC Z
8 m g/k g
+ DMARD
PBO + DMARD
TC Z
8 m g/k g
+ MTX
PBO + MTX
N =
286
N =
284
N =
398
N =
393
N =
205
A
C R 20
N =
204
N =
803
N =
413
N =
170
N =
158
24 70 %*** 52 % 56 %*** 27 % 59 %*** 26 % 61 %*** 24 % 50 %*** 10 %
52 56 %*** 25 %
AC R 5024 44 %** 33 % 32 %*** 10 % 44 %*** 11 % 38 %*** 9 % 29 %*** 4 %
52 36 %*** 10 %
AC R 7024 28 %** 15 % 13 %*** 2 % 22 %*** 2 % 21 %*** 3 % 12 %** 1 %
52 20 %*** 4 %
TCZ - Tocilizum ab MTX - Metotrexát PBO - PlaceboDMARD - Antireum atikum m odifikujúce priebeh choroby** - p < 0,01, TCZ oproti PBO + MTX/DMARD*** - p < 0,0001, TCZ oproti PBO + MTX/DMARD
V
ý
z
namná klinická odpoveď
Po 2 rokoch liečby RoActemrou s MTX dosiahlo 14 % pacientov významnú klinickú odpoveď
(udržanie ACR70 odpovede počas 24 týždňov alebo dlhšie).
Rádiograf ická odpoveď
V štúdii II sa u pacientov s nedostatočnou odpoveďou na MTX hodnotila inhibícia štrukturálneho poškodenia kĺbov rádiograficky a vyjadrila sa ako zmena v modifikovanom Sharpovom skóre a jeho zložkách - skóre erózie a skóre zúženia kĺbovej štrbiny. U pacientov liečených RoActemrou sa oproti kontrolnej skupine preukázala inhibícia štrukturálneho poškodenia kĺbov s významne nižšou rádiografickou progresiou ochorenia (tabuľka 3).
V otvorenej predĺženej fáze štúdie II bola inhibícia progresie štrukturálneho poškodenie kĺbu
v skupine s RoActemrou a MTX udržiavaná i v druhom roku liečby. Stredná zmena
od východiskových hodnôt bola v 104. týždni v celkovom Sharpovom-Genantovom skóre významne
nižšia u pacientov randomizovaných do skupiny s RoActemrou v dávke 8 mg/kg a MTX (p < 0,0001)
v porovnaní s pacientami, ktorí boli randomizovaní do skupiny s placebom a MTX.
Tabuľka 3. Rádiografické priemerné zmeny počas 52 týždňov v štúdii II
P
B
O + MTX (+TCZ od 24. týždňa) N = 393
T
C
Z 8 mg/k g + MTX N = 398
Celkové
Sharpovo-Genantovo
skóre
1,13 0,29*
Skóre erózie 0,71 0,17* Skóre JSN 0,42 0,12**
PBO - PlaceboMTX - MetotrexátTCZ - Tocilizum abJSN - Zúženie kĺbovej štrbiny* - p ≤ 0,0001, TCZ oproti PBO + MTX** - p < 0,005, TCZ oproti PBO + MTXPo 1 roku liečby RoActemrou a MTX 85 % pacientov (n = 348) nevykazovalo žiadnu progresiu štrukturálneho poškodenia kĺbov ako je definované v celkovom Sharpovom skóre 0 alebo menej,
v porovnaní so 67 % pacientov v skupine s placebom a MTX (n = 290) (p ≤ 0,001). Tieto výsledky pretrvávali i po 2 rokoch liečby (83 %, n = 353). Deväťdesiat tri percent (93 %; n = 271) pacientov nevykazovalo žiadnu progresiu medzi 52. a 104. týždňom.
Zdravotné výsledky a výsledky týkajúce sa kvality životaPacienti liečení RoActemrou hlásili zlepšenie vo všetkých výsledkoch hlásených pacientmi (dotazník hodnotiaci zdravie a index funkčnej neschopnosti - HAQ-DI), skrátený formulár 36 a dotazník funkčného hodnotenia liečby chronického ochorenia. U pacientov liečených RoActemrou sa oproti pacientom liečeným DMARDs pozorovalo štatisticky významné zlepšenie skóre HAQ-DI.
V priebehu otvorenej fázy štúdie II bolo udržanie zlepšenia fyzických funkcií až počas 2 rokov.
V 52. týždni bola stredná zmena v HAQ-DI -0,58 v skupine s RoActemrou 8 mg/kg a MTX
v porovnaní s -0,39 v skupine s placebom a MTX. Stredná zmena HAQ-DI bola v skupine s RoActemrou 8 mg/kg a MTX udržovaná aj v 104. týždni (-0,61).
Hladiny hemoglobínuPri liečbe RoActemrou sa oproti liečbe DMARDs v 24. týždni pozorovalo štatisticky významné
zlepšenie hladín hemoglobínu (p < 0,0001). Priemerné hodnoty hladín hemoglobínu sa zvýšili do 2. týždňa a udržali sa v referenčnom rozpätí až do 24. týždňa.
Tocilizumab verzus adalimumab v monoterapiiŠtúdia VI (WA19924), 24-týždňová dvojito zaslepená štúdia, ktorá porovnávala monoterapiu
RoActemrou s monoterapiou adalimumabom, hodnotila 326 pacientov s RA, ktorí netolerovali MTX
alebo kde pokračovanie v liečbe MTX sa považovalo za nevhodné (vrátane nedostatočných
respondérov na MTX). Pacienti v skupine s RoActemrou dostávali intravenóznu (i.v.) infúziu RoActemry (8 mg/kg) každé 4 týždne a subkutánne (s.c.) injekciu s placebom každé 2 týždne. Pacienti v skupine s adalimumabom dostávali s.c. injekciu adalimumabu (40 mg) každé 2 týždne plus i.v. infúziu s placebom každé 4 týždne.
Pozoroval sa štatistický významný superiórny účinok liečby v prospech RoActemry v porovnaní s adalimumabom pri kontrole aktivity ochorenia od východiskovej hodnoty po 24. týždeň
pre primárny cieľový ukazovateľ zmenu DAS28 a pre všetky sekundárne cieľové ukazovatele
(tabuľka 4).
Tabuľka 4. Výsledky účinnosti pre štúdiu VI (WA19924)
AD
A + placebo
(
i
.
v)
N = 162
T
o
c
ili
z
u
m
a
b +
p
l
a
ce
b
o (s .c.)
N = 163 p-hodnota
(a)
P
r
i
m
á
r
n
y cieľový ukazovateľ – Priemerná zmena od východis kovej hodnoty v 24. týždni
DA
S
2
8 (upravený priemer) -1,8 -3,3
R
o
z
d
i
e
l v upravenom priemere (95 % IS)
-
1
,
5 (-1,8, -1,1) < 0,0001
S
e
k
un
d
á
r
n
e cieľové ukazovatele – Percento res pondérov v 24. týždni
(
b)
DAS28 < 2,6, n (%) 17 (10,5) 65 (39,9) < 0,0001
DAS28 ≤ 3,2, n (%) 32 (19,8) 84 (51,5) < 0,0001
ACR20 odpoveď, n (%) 80 (49,4) 106 (65,0) 0,0038
ACR50 odpoveď, n (%) 45 (27,8) 77 (47,2) 0,0002
ACR70 odpoveď, n (%) 29 (17,9) 53 (32,5) 0,0023
ap hodnota je upravená vzhľadom na oblasť a trvanie RA pre všetky cieľové ukazovatele a tiež východisková hodnota
pre všetky pokračujúce cieľové ukazovatele.
b Neodpovedajúci na liečbu použití pre chýbajúce údaje. Multidisciplinárna kontrola použitím Bonferroni-Holm procedúry
Celkový klinický profil nežiaducich udalostí bol podobný pri RoActemre a adalimumabe. Podiel pacientov so závažnými nežiaducimi udalosťami bol medzi liečebnými skupinami (RoActemra 11,7 % oproti adalimumabu 9,9 %). Nežiaduce účinky v skupine s RoActemrou odpovedali známemu bezpečnostnému profilu RoActemry a nežiaduce účinky boli hlásené s podobnou frekvenciou
v porovnaní s tabuľkou 1. Vyššia incidencia infekcií a infestácií bola hlásená v skupine s RoActemrou (48 % oproti 42 %), a to bez rozdielu v incidencii závažných infekcií (3,1 %). Obidve skúmané liečby indukovali rovnaké zmeny v laboratórnych bezpečnostných parametroch (poklesy počtu neutrofilov
a krvných doštičiek, zvýšenie ALT, AST a lipidov), veľkosť zmien a frekvencie výrazných abnormalít však bola vyššia pri RoActemre v porovnaní s adalimumabom. U štyroch (2,5 %) pacientov v skupine s RoActemrou a dvoch (1,2 %) pacientov v skupine s adalimumabom sa vyskytli poklesy počtu neutrofilov 3. alebo 4. stupňa CTC. U jedenástich (6,8 %) pacientov v skupine s RoActemrou a piatich (3,1 %) pacientov v skupine s adalimumabom sa vyskytlo zvýšenie ALT 2. alebo vyššieho
stupňa CTC. Priemerné zvýšenie LDL od východiskovej hodnoty bolo 0,64 mmol/l (25 mg/dl)
u pacientov v skupine s RoActemrou a 0,19 mmol/l (7 mg/dl) u pacientov v skupine s adalimumabom. Bezpečnosť pozorovaná v skupine s RoActemrou sa zhodovala so známym bezpečnostných profilom RoActemry a nepozorovali sa žiadne nové alebo neočakávané nežiaduce liekové reakcie (pozri tabuľku 1).
Subkutánnepoužitie
Klinická účinnosť
Účinnosť subkutánne podávanej RoActemry v zmierňovaní prejavov a príznakov RA a rádiografická odpoveď sa hodnotili v dvoch randomizovaných, dvojito zaslepených, kontrolovaných, multicentrických štúdiách. V štúdii I (SC-I) sa vyžadovalo, aby mali pacienti vek > 18 rokov, stredne ťažkú až ťažkú aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali pred začiatkom liečby minimálne 4 bolestivé a 4 opuchnuté kĺby. Všetkým pacientom boli v rámci základnej liečby podávané
nebiologické DMARDs. V štúdii II (SC-II) sa vyžadovalo, aby pacienti mali vek > 18 rokov, stredne ťažkú až ťažkú aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali pred začiatkom liečby minimálne 8 bolestivých a 6 opuchnutých kĺbov.
Po prechode z dávky 8 mg/kg podávanej intravenózne raz za 4 týždne na dávku 162 mg podávanú subkutánne raz za týždeň dôjde u pacienta k zmene expozície. Rozsah tejto zmeny sa líši v závislosti od telesnej hmotnosti pacienta (zvýšenie expozície u pacientov s nízkou telesnou hmotnosťou
a zníženie expozície u pacientov s vysokou telesnou hmotnosťou), ale klinický výsledok sa zhoduje s klinickým výsledkom pozorovaným u pacientov liečených intravenóznou formou lieku.
Klinická odpoveď
Štúdia SC-I hodnotila pacientov so stredne ťažkou až ťažkou aktívnou RA, ktorí nedosiahli dostatočnú klinickú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viac DMARDs, pričom približne 20 % malo v anamnéze nedostatočnú odpoveď na liečbu najmenej jedným inhibítorom TNF. V štúdii SC-I bolo 1 262 pacientov randomizovaných v pomere 1:1 do skupiny s RoActemrou podávanou subkutánne v dávke 162 mg každý týždeň, alebo do skupiny s RoActemrou podávanou intravenózne v dávke 8 mg/kg každé štyri týždne v kombinácii s nebiologickým(i) DMARD(s). Primárny cieľový ukazovateľ v štúdii bol rozdiel v podiele pacientov, ktorí dosiahli odpoveď ACR20
v 24. týždni. Výsledky zo štúdie SC-I sú uvedené v tabuľke 5.
Tabuľka 5. Odpovede ACR v štúdii SC-I (% pacientov) v 24. týždni
SC-Ia
TCZ s .c. 162 mg
každý týždeň
+ DMARD
N = 558
TCZ i.v. 8 mg/kg
+ DMARD
N = 537
ACR20 24. týždeň 69,4 % 73,4 %
Vážený rozdiel (95% IS) -4,0 (-9,2; 1,2)
ACR50 24. týždeň 47,0 % 48,6 %
Vážený rozdiel (95% IS) -1,8 (-7,5; 4,0)
ACR70 24. týždeň 24,0 % 27,9 %
Vážený rozdiel (95% IS) -3,8 (-9,0; 1,3)
T CZ = t ocilizumab
a = populácia podľa prot okolu
Pacienti v štúdii SC-I mali pred začiatkom liečby priemerné skóre aktivity ochorenia (DAS28) 6,6
v skupine so subkutánnym tocilizumabom a 6,7 v skupine s intravenóznym tocilizumabom. V 24. týždni sa v oboch liečebných skupinách pozorovalo významné zníženie DAS28 oproti východiskovej hodnote (priemerné zlepšenie) o 3,5 a porovnateľný podiel pacientov dosiahol klinickú remisiu ochorenia podľa DAS28 (DAS28 < 2,6) v skupine so subkutánnou RoActemrou (38,4 %)
a v skupine s intravenóznou RoActemrou (36,9 %).
Rádiograf ická odpoveďRádiografická odpoveď subkutánne podávanej RoActemry sa hodnotila v dvojito zaslepenej,
kontrolovanej, multicentrickej štúdii s pacientmi s aktívnou RA (SC-II). Štúdia SC-II hodnotila pacientov so stredne ťažkou až ťažkou aktívnou RA, ktorí nedosiahli dostatočnú klinickú odpoveď na existujúcu reumatologickú liečbu zahŕňajúcu jedno alebo viac DMARDs, pričom približne 20 %
malo v anamnéze nedostatočnú odpoveď na liečbu najmenej jedným inhibítorom TNF. Vyžadovalo sa, aby pacienti mali vek > 18 rokov a aktívnu RA diagnostikovanú podľa kritérií ACR a aby mali
pred začiatkom liečby minimálne 8 bolestivých a 6 opuchnutých kĺbov. V štúdii SC-II bolo
656 pacientov randomizovaných v pomere 2:1 do skupiny s RoActemrou podávanou subkutánne
v dávke 162 mg každý druhý týždeň, alebo do skupiny s placebom, v kombinácii s nebiologickým(i) DMARD(s).
V štúdii SC-II sa inhibícia štrukturálneho poškodenia kĺbov hodnotila rádiograficky a vyjadrila sa ako zmena oproti východiskovej hodnote v priemernom celkovom Sharpovom skóre modifikovanom
van der Heijdom (mTSS). V 24. týždni sa preukázala inhibícia štrukturálneho poškodenia kĺbov
s významne nižšou rádiografickou progresiou ochorenia u pacientov, ktorým bola subkutánne podávaná RoActemra, v porovnaní s pacientmi, ktorým bolo podávané placebo (priemerné mTSS 0,62 oproti 1,23; p = 0,0149 (van Elteren)). Tieto výsledky sa zhodujú s výsledkami pozorovanými
u pacientov liečených intravenóznou RoActemrou.
V štúdii SC-II sa v 24. týždni dosiahla odpoveď ACR20 u 60,9 %, ACR50 u 39,8 % a ACR70 u 19,7 % pacientov liečených RoActemrou podávanou subkutánne každý druhý týždeň oproti odpovedi ACR20 dosiahnutej u 31,5 %, ACR50 u 12,3 % a ACR70 u 5,0 % pacientov, ktorým bolo podávané placebo. Pacienti mali pred začiatkom liečby priemerné DAS28 6,7 v skupine
so subkutánnou RoActemrou a 6,6 v skupine s placebom. V 24. týždni sa pozorovalo významné zníženie DAS28 oproti východiskovej hodnote o 3,1 v skupine so subkutánnou RoActemrou
a o 1,7 v skupine s placebom a DAS28 < 2,6 sa pozorovalo u 32,0 % pacientov v skupine so subkutánnou RoActemrou a u 4,0 % pacientov v skupine s placebom.
Zdravotné výsledky a výsledky týkajúce sa kvality života
V štúdii SC-I sa skóre HAQ-DI od začiatku štúdie po 24. týždeň znížilo priemerne o 0,6 v skupine
so subkutánnou RoActemrou aj v skupine s intravenóznou RoActemrou. Podiel pacientov, ktorí dosiahli klinicky významné zlepšenie skóre HAQ-DI v 24. týždni (zmena oproti východiskovému skóre o ≥ 0,3 jednotky), bol tiež porovnateľný v skupine so subkutánnou RoActemrou (65,2 %) oproti skupine s intravenóznou RoActemrou (67,4 %), pričom vážený rozdiel v podieloch bol -2,3 %
(95 % IS -8,1; 3,4). Pokiaľ ide o SF-36, v 24. týždni bola priemerná zmena oproti východiskovej hodnote v skóre mentálneho komponentu 6,22 v skupine so subkutánnou RoActemrou
a 6,54 v skupine s intravenóznou RoActemrou a zmena v skóre fyzického komponentu bola tiež podobná, a to 9,49 v skupine so subkutánnou RoActemrou a 9,65 v v skupine s intravenóznou RoActemrou.
V štúdii SC-II bolo priemerné zníženie skóre HAQ-DI od začiatku štúdie po 24. týždeň významne väčšie u pacientov liečených RoActemrou podávanou subkutánne každý druhý týždeň (0,4)
v porovnaní s pacientmi, ktorým bolo podávané placebo (0,3). Podiel pacientov, ktorí dosiahli klinicky významné zlepšenie skóre HAQ-DI v 24. týždni (zmena oproti východiskovému skóre
o ≥ 0,3 jednotky), bol vyšší u pacientov liečených RoActemrou podávanou subkutánne každý druhý týždeň (58 %) v porovnaní s pacientmi, ktorým bolo podávané placebo (46,8 %). Zmena v SF-36 (priemerná zmena v skóre mentálneho a fyzického komponentu) bola významne väčšia v skupine
so subkutánnou RoActemrou (6,5 a 5,3) v porovnaní so skupinou s placebom (3,8 a 2,9).
OBA
Subkutánnepoužitie
Klinická účinnosť
Klinická štúdia WA28119 bola randomizovaná, multicentrická, dvojito zaslepená, placebom kontrolovaná štúdia superiority fázy III, ktorá skúmala účinnosť a bezpečnosť RoActemry u pacientov s OBA.
Do štúdie bolo zaradených dvestopäťdesiatjeden (251) pacientov s novým nástupom alebo relapsom OBA, ktorí boli rozdelení do štyroch liečebných skupín. Táto štúdia sa skladala z 52-týždňového zaslepeného obdobia (1. časť), po ktorom nasledovalo 104-týždňové otvorené predĺženie (2. časť). Účelom 2. časti bolo popísať dlhodobú bezpečnosť a udržanie účinnosti po 52 týždňoch liečby RoActemrou, preskúmať mieru výskytu relapsu ochorenia a potrebu liečby RoActemrou nad rámec
52 týždňov, a pochopiť potenciálnu dlhodobú účinnosť RoActemry bez potreby podávania steroidov
(steroidy šetriaci účinok).
Dve subkutánne dávky RoActemry (162 mg každý týždeň a 162 mg každý druhý týždeň) sa porovnávali s dvomi rozdielnymi placebom kontrolovanými skupinami randomizovanými v pomere
2:1:1:1.
Všetkým pacientom boli v rámci základnej liečby podávané glukokortikoidy (prednizón). Každá liečebná skupina s RoActemrou a jedna z liečebných skupín s placebom nasledovala po vopred špecifikovanom režime postupného znižovania dávky prednizónu v priebehu 26 týždňov, kým druhá skupina s placebom nasledovala po vopred špecifikovanom režime postupného znižovania prednizónu počas 52 týždňov, čo viac zodpovedá štandardnej klinickej praxi.
Trvanie liečby glukokortikoidmi počas fázy skríningu a pred začatím podávania RoActemry (alebo placeba) bolo podobné vo všetkých 4 skupinách liečby (pozri tabuľku 6).
Tabuľka 6. Trvanie liečby kortikosteroidmi počas obdobia skríningu v štúdii WA28119
P
l
ac
e bo + 26 týždňov pos tupne
z
n
i
ž
o
va
n
e j dávk y pre dnizónu
N
=
5
0
P
l
ac
e bo + 52 týždňov pos tupne
z
n
i
ž
o
va
n
e j dávk y pre dnizónu
 N
=
5
1
R
o
A
c
t
e m ra 162 m g s .c. raz týžde nne +
2
6 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k aždý druhý týžde ň + 26 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
4
9
N
=
5
1
R
o
A
c
t
e m ra 162 m g s .c. raz týžde nne +
2
6 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
100
R
o
A
c
t
e m ra 162 m g s .c. k aždý druhý týžde ň + 26 týždňov pos tupne znižovane j dávk y pre dnizónu
N
=
4
9
Trvanie (dni)
Priemer (SD) 35,7 (11,5) 36,3 (12,5) 35,6 (13,2) 37,4 (14,4) Medián 42,0 41,0 41,0 42,0
Min - Max 6 - 63 12 – 82 1 - 87 9 - 87
Primárny cieľový ukazovateľ účinnosti hodnotený na základe pomeru pacientov, ktorí dosiahli trvalú
remisiu ochorenia bez steroidov v 52. týždni liečby RoActemrou plus 26-týždňovým obdobím postupného znižovania dávky prednizónu v porovnaní so skupinou s placebom plus 26-týždňové obdobie postupného znižovania dávky prednizónu, bol dosiahnutý (tabuľka 7).
Kľúčový sekundárny cieľový ukazovateľ účinnosti, ktorý sa rovnako zakladal na pomere pacientov, ktorí dosiahli pretrvávajúcu remisiu ochorenia v 52. týždni, porovnávajúci skupinu s tocilizumabom plus 26 týždňov postupného znižovania dávky prednizónu a skupinu s placebom plus 52 týždňov postupného znižovania dávky prednizónu, bol rovnako dosiahnutý (tabuľka 7).
Štatisticky významný superiórny účinok liečby sa pozoroval v prospech RoActemry oproti placebu v dosiahnutí pretrvávajúcej remisie bez steroidov v 52. týždni liečby RoActemrou plus 26-týždňové obdobie postupného znižovania dávky prednizónu v porovnaní so skupinou s placebom
plus 26-týždňové obdobie postupného znižovania dávky prednizónu a so skupinou s placebom plus 52-týždňové obdobie postupného znižovania dávky prednizónu.
Percento pacientov, ktorí dosiahli pertrvávajúcu remisiu ochorenia v 52. týždni liečby, je znázornená
v tabuľke 7.
Sekundárne cieľové ukazovatele
Pri posudzovaní času do nástupu prvého vzplanutia OBA sa pozorovalo výrazne menšie riziko vzplanutia ochorenia v skupine s RoActemrou podávanou subkutánne raz za týždeň v porovnaní
so skupinou s placebom plus 26 týždňov postupného znižovania prednizónu a so skupinou s placebom plus 52 týždňov postupného znižovania prednizónu, a v skupine s RoActemrou podávanou subkutánne každý druhý týždeň, v porovnaní so skupinou s placebom plus 26 týždňov postupného znižovania prednizónu (porovnávané pri hladine významnosti 0,01). RoActemra podávaná subkutánne raz za týždeň rovnako preukázala klinicky významný pokles rizika vzplanutia ochorenia v porovnaní
so skupinou s placebom plus 26 týždňov prednizónu u pacientov, ktorí vstúpili do skúšania s relapsujúcim OBA, rovnako ako aj u pacientov s novým nástupom ochorenia (tabuľka 7).
K
umulatívna dávka glukokortikoidov
Kumulatívna dávka prednizónu bola v 52. týždni výrazne nižšia v obidvoch skupinách s RoActemrou
v porovnaní s dvoma skupinami s placebom (tabuľka 6). V osobitnej analýze pacientov, ktorí dostávali prednizón ako emergentnú liečbu pri vzplanutí OBA počas prvých 52 týždňov, sa veľkosť kumulatívnej dávky prednizónu významne líšila. Priemerné dávky u pacientov, ktorým bola podávaná emergentná liečba, boli v skupine s RoActemrou podávanou raz za týždeň v dávke 3 129,75 mg
a v skupine s RoActemrou podávanou každý druhý týždeň v dávke 3 847 mg. V oboch prípadoch bola táto dávka významne nižšia ako v skupinách s placebom plus 26 týždňov postupného znižovania prednizónu, kde bola 4 023,5 mg, a v skupine s placebom plus 52 týždňov postupného znižovania prednizónu, kde bola 5 389,5 mg.
T
abuľka 7. Výsledky účinnosti zo štúdie WA28119
P
l ace bo + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y pre dn i z ón u
N
=
5
0
P
l ace bo + 52
t
ý
ž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
5
1
R
o
A
c
t
e m ra 162 m g s .c. raz z a týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
100
R
o
A
c
t
e m ra
16
2 m g s .c.
k
a
ž dý druhý týž de ň + 26 týž dň ov
p
o
s tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
4
9
P
r
i m árn y ci eľový u k azovateľ
****P ret rvávajúca remisia (skupiny s t ocilizumabom vs placebo+26)
P acient i, kt orí odpovedali na liečbu v 52. t ýždni,
n (%)
7 (14 %) 9 (17,6 %) 56 (56 %) 26 (53,1 %)
Neupravený rozdiel v proporciách
(99,5% IS)
N/A N/A 42 %* (18,00 ; 66,00)
39,06 %* (12,46 ; 65,66)
K
ľ
ú čový s e k u ndárny ci eľový u k azovateľ
P ret rvávajúca remisia (skupiny s t ocilizumabom vs placebo+52)
P acient i, kt orí odpovedali na liečbu v 52. t ýždni,
n (%)
7 (14 %) 9 (17,6 %) 56 (56 %) 26 (53,1 %)
Neupravený rozdiel v proporciách
(99,5% IS)
N/A N/A 38,35 %* (17,89 ; 58,81)
35,41 %** (10,41 ; 60,41)
P
l ace bo + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y pre dn i z ón u
N
=
5
0
P
l ace bo + 52
t
ý
ž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
5
1
R
o
A
c
t
e m ra 162 m g s .c. raz z a týž de ň + 26 týž dň ov pos tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
100
R
o
A
c
t
e m ra
16
2 m g s .c.
k
a
ž dý druhý týž de ň + 26 týž dň ov
p
o
s tu pn e z n i ž ovan e j dávk y
p
r
e dn i z ón u
N
=
4
9
Ď
a
l š i e s e kundárne ci eľové u kazovatel e
Čas do nást upu prvého vzplanut ia OBA¹ (skupiny s t ocilizumabom vs placebo+26)
HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (skupiny s t ocilizumabom vs placebo+52)
HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s relapsom ochorenia; skupiny s t ocilizumabom vs placebo +26) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s relapsom ochorenia; skupiny s t ocilizumabom vs placebo + 52) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s novým nást upom ochorenia; skupiny s
t ocilizumabom vs placebo +26) HR (99% IS)
Čas do nást upu prvého vzplanut ia OBA ¹ (pacient i s novým nást upom ochorenia; skupiny s
t ocilizumabom vs placebo + 52) HR (99% IS)
N/A N/A
N/A N/A N/A
N/A
N/A N/A
N/A N/A N/A
N/A
0,23 * (0,11 ; 0,46)
0,39** (0,18 ; 0,82)
0,23*** (0,09 ; 0,61)
0,36
(0,13 ; 1,00)
0,25***
(0,09 ; 0,70)
0,44
(0,14 ; 1,32)
0,28 ** (0,12 ; 0,66)
0,48
(0,20 ; 1,16)
0,42 (0,14 ; 1,28)
0,67
(0,21 ; 2,10)
0,20***
(0,05 ; 0,76)
0,35
(0,09 ; 1,42)
Kumulat ívna dávka glukokort ikoidov (mg)
medián v 52. t ýždni (skupiny s t ocilizumabom vs placebo+262)
medián v 52. t ýždni (skupiny s t ocilizumabom vs placebo +522)
Expl oratí vn e ci eľové u kazovatele
Ročná miera relapsu ochorenia, 52. t ýždeň§
P riemer (SD)
* p < 0,0001
3296,00
N/A
1,74
(2,18)
N/A
3817,50
1,30 (1,84)
1862,00*
1862,00*
0,41
(0,78)
1862,00*
1862,00*
0,67
(1,10)
** p < 0,005 (prah významnost i primárnych a kľúčových sekundárnych t est ov superiorit y)
*** popisná p hodnot a
< 0,005**** vzplanutie:recidíva prejavov alebo príznakov OBAa/alebo ESR ≥30 mm/h –pot rebné zvýšenie dávky prednizónu
Re m i s i a: abs e n ci a vz pl an u ti a a n ávrat C RP do n orm al i z ovan ých h odn ôtPre trvávajú ca re m isi a: re mis ia od 12.- 52. týž dňa- pacienti musia dodržiavať prot okolom st anovené post upné znižovanie
dávky prednizónu
¹ analýzy času (v dňoch) medzi klinickou remisiou a prvým vzplanut ím ochorenia
2 p hodnot y sú st anovené na základe Van Elt erenovej analýzy neparamet rických údajov
§ št at ist ická analýza sa neuskut očnila
N/A= nehodí sa
HR = pomer rizika
IS = int erval spoľahlivost i
Výsledky týkajúce sa kvality životaV klinickej štúdii WA28119 boli výsledky skóre SF-36 (skrátený formulár) rozdelené na fyzické
(PCS) a mentálne (MCS) komponenty. Stredná zmena vo fyzických komponentoch východiskového
stavu do 52. týždňa bola vyššia (preukázalo sa výraznejšie zlepšenie) v skupinách s RoActemrou podávanou raz za týždeň [4,10] a každý druhý týždeň [2,76], ako v oboch skupinách s placebom
[placebo + 26 týždňov; -0,28, placebo + 52 týždňov; -1,49], hoci štatisticky významný rozdiel
(p=0,0024) sa pozoroval len v porovnaní skupiny s RoActemrou podávanou raz za týždeň plus
26 týždňov postupného znižovania prednizónu a skupiny s placebom plus 52 týždňov postupného znižovania prednizónu (5,59; 99 % IS: 8,6; 10,32). V prípade mentálnych komponentov bola stredná zmena z východiskového stavu do 52. týždňa vyššia v skupinách s RoActemrou podávanou raz za týždeň [7,28] a každé dva týždne [6,12], ako v skupine s placebom plus 52 týždňov postupného znižovania dávky prednizónu [2,84] (hoci rozdiely neboli štatisticky významné [týždenná hodnota p=0,0252 pri podávaní raz za týždeň, p=0,1468 pri podávaní každý druhý týždeň]) a podobná ako
v skupine s placebom plus 26 týždňov postupného znižovania dávky prednizónu [6,67].
Celkové hodnotenie aktivity ochorenia pacientom sa posudzovalo na vizuálnej analógovej škále (VAS, visual analogue scale) 0-100 mm. Stredná zmena v celkovej VAS pacienta bola z východiskového stavu do 52. týždňa nižšia (preukázalo sa výraznejšie zlepšenie) v oboch skupinách s RoActemrou, podávanou raz za týždeň [-19,0] a každé dva týždne. [-25,3], ako v oboch skupinách s placebom
[placebo + 26 týždňov -3,4, placebo + 52 týždňov -7,2], hoci len v skupine s RoActemrou podávanou každé dva týždne plus 26 týždňov postupného znižovania dávky prednizónu sa preukázal štatisticky významný rozdiel oproti skupine s placebom [placebo + 26 týždňov p=0,0059, a placebo + 52 týždňov p=0,0081].
Skóre zmeny v stave únavy z východiskového stavu do 52. týždňa - FACIT (Funkčné hodnotenie liečby chronického ochorenia) sa vypočítalo pre všetky skupiny. Skóre strednej zmeny [SD] bolo nasledovné: RoActemra podávaná raz za týždeň + 26 týždňov 5,61 [10,115], RoActemra podávaná každý druhý týždeň + 26 týždňov 1,81 [8,836], placebo + 26 týždňov 0,26 [10,702], a placebo +
52 týždňov -1,63 [6,753].
Zmeny v skóre EQ5D z východiskového stavu do 52. týždňa boli nasledovné: RoActemra podávaná raz za týždeň + 26 týždňov 0,10 [0,198], RoActemra podávaná každý druhý týždeň + 26 týždňov
0,05 [0,215], placebo + 26 týždňov 0,07 [0,293], a placebo + 52 týždňov -0,02 [0,159].
Vyššie skóre signalizuje zlepšenie v oboch hodnoteniach, FACIT-únava aj EQ5D.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s RoActemrou
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe chronickej idiopatickej
artritídy (vrátane reumatoidnej artritídy, ankylozujúcej spondylitídy, psoriatickej artritídy a juvenilnej idiopatickej artritídy). Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmak ok inetické vlastnosti
Farmakokinetiku RoActemry charakterizuje nelineárna eliminácia, ktorá je kombináciou lineárneho klírensu a Michaelisa-Mentenovej eliminácie. Nelineárna časť eliminácie RoActemry vedie k zvýšenej expozícii, ktorá je vyššia ako dávke úmerná. Farmakokinetické parametre RoActemry sa postupom času nemenia. Nakoľko je celkový klírens závislý od koncentrácií RoActemry v sére, aj polčas eliminácie RoActemry je závislý od koncentrácií a jeho dĺžka je rôzna v závislosti od hladiny koncentrácie v sére. Populačná farmakokinetická analýza u všetkých skúmaných populácií pacientov zatiaľ nepreukázala žiaden vzťah medzi zdanlivým klírensom a prítomnosťou protilátok proti lieku.
RA
Intravenóznepoužitie
Farmakokinetika RoActemry sa stanovila za použitia populačnej farmakokinetickej analýzy údajov
z databázy zloženej z 3 552 pacientov s RA liečených dávkou 4 alebo 8 mg/kg RoActemry podávanou raz za 4 týždne formou jednu hodinu trvajúcej infúzie alebo so 162 mg RoActemry podávanej subkutánne buď raz týždenne alebo každý druhý týždeň počas 24 týždňov.
Nasledujúce parametre (predpokladaný priemer ± SD, štandardná odchýlka) sa odhadli pre dávku
8 mg/kg RoActemry podávanú raz za 4 týždne: plocha pod krivkou pre plazmatickú koncentráciu
(AUC) v rovnovážnom stave = 38 000 ± 13 000 h•µg/ml, minimálna koncentrácia
(Cmin) =15,9 ± 13,1 µg/ml a maximálna koncentrácia (Cmax) = 182 ± 50,4 µg/ml a pomer kumulácie v hodnote 1,32 pri AUC a 1,09 pri Cmax bol nízky. Pomer kumulácie bol vyšší pri Cmin (2,49), čo sa očakávalo na základe prispenia nelineárneho klírensu pri nižších koncentráciách. Rovnovážny stav sa dosiahol po podaní prvej dávky pri hodnote Cmax, po 8 týždňoch pri hodnote AUC a po 20 týždňoch pri hodnote Cmin. AUC, Cmin a Cmax RoActemry vzrástlo so stúpajúcou telesnou hmotnosťou.
Pri telesnej hmotnosti ≥ 100 kg bol predpovedaný priemer (± SD) AUC RoActemry v rovnovážnom
stave 50 000 ± 16 800 μg•h/ml, Cmin RoActemry 24,4 ± 7,5 μg/ml a Cmax RoActemry
226 ± 50,3 μg/ml, čo sú vyššie hodnoty, než hodnoty pri priemernej expozícii v súbore pacientov
(t. j. celková hmotnosť všetkých pacientov) ako je uvedené vyššie. Krivka odpovede na dávku sa pri RoActemre pri vyšších expozíciách splošťuje, čo vedie k nižšiemu nárastu účinnosti pre každé ďalšie zvýšenie koncentrácie RoActemry, takže u pacientov liečených RoActemrou dávkou > 800 mg nedochádza už k žiadnemu zmysluplnému zvýšeniu účinnosti. Preto sa neodporúčajú dávky RoActemry, ktoré presahujú 800 mg na infúziu (pozri časť 4.2).
Distribúcia
U pacientov s RA bol distribučný objem centrálneho kompartmentu 3,72, distribučný objem periférneho kompartmentu bol 3,35, čo malo za následok distribučný objem 7,07 v rovnovážnom stave.
Eliminácia
Po intravenóznom podaní dávky podlieha RoActemra dvojfázovému vylučovaniu z cirkulácie. Celkový klírens RoActemry bol závislý od koncentrácie a je súčtom lineárneho a nelineárneho klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze na 9,5 ml/h. Od koncentrácie závislý nelineárny klírens zohráva hlavnú úlohu pri nízkych
koncentráciách RoActemry. Keď je cesta nelineárneho klírensu nasýtená, pri vyšších koncentráciách
RoActemry je klírens určovaný hlavne lineárnym klírensom.
t1/2 RoActemry bol závislý od koncentrácie. V rovnovážnom stave sa po dávke 8 mg/kg podávanej raz
za 4 týždne efektívny t1/2 znižoval so znižujúcimi sa koncentráciami v rámci dávkovacieho intervalu
od 18 dní do 6 dní.
Linearita
Farmakokinetické parametre RoActemry sa postupom času nezmenili. Pri dávkach 4 a 8 mg/kg podávaných raz za 4 týždne sa pozorovalo vyššie ako dávke úmerné zvýšenie hodnoty AUC a Cmin. Hodnota Cmax sa zvyšovala úmerne dávke. V rovnovážnom stave bola pri dávke 8 mg/kg predpokladaná hodnota AUC 3,2-násobne a hodnota Cmin 30-násobne vyššia než pri dávke 4 mg/kg.
Subkutánnepoužitie
Farmakokinetika RoActemry sa stanovila za použitia populačnej farmakokinetickej analýzy údajov
z databázy zloženej z 3 552 pacientov s RA liečených dávkou 162 mg podávanou subkutánne každý
týždeň, dávkou 162 mg podávanou subkutánne každý druhý týždeň a dávkou 4 alebo 8 mg/kg
podávanou intravenózne raz za 4 týždne počas 24 týždňov.
Farmakokinetické parametre RoActemry sa postupom času nezmenili. Pri dávke 162 mg podávanej každý týždeň bol predpokladaný priemer (± SD) AUC1. týždeň RoActemry v rovnovážnom stave
7 970 ± 3 432 µg•h/ml, Cmin RoActemry 43,0 ± 19,8 µg/ml a Cmax RoActemry 49,8 ± 21,0 µg/ml. Pomer kumulácie bol pri AUC 6,32, pri Cmin 6,30 a pri Cmax 5,27. Rovnovážny stav sa pri AUC, Cmin a Cmax dosiahol po 12 týždňoch.
Pri dávke 162 mg podávanej každý druhý týždeň bol predpokladaný priemer (± SD) AUC2. týždeň
RoActemry v rovnovážnom stave 3 430 ± 2 660 µg•h/ml, Cmin RoActemry 5,7 ± 6,8 µg/ml
a Cmax RoActemry 13,2 ± 8,8 µg/ml. Pomer kumulácie bol pri AUC 2,67, pri Cmin 6,02 a pri Cmax 2,12.
Rovnovážny stav sa pri AUC a Cmin dosiahol po 12 týždňoch a pri Cmax po 10 týždňoch.
A
bsorpcia
Po subkutánnom podávaní pacientom s RA bol čas do dosiahnutia maximálnej sérovej koncentrácie
RoActemry (tmax) 2,8 dňa. Biologická dostupnosť s.c. formy bola 79 %.
ElimináciaPri subkutánnom podávaní pacientom s RA je po dosiahnutí rovnovážneho stavu zdanlivý t1/2 závislý od koncentrácie až 12 dní pri dávke 162 mg podávanej každý týždeň a 5 dní pri dávke 162 mg podávanej každý druhý týždeň.
OBASubkutánnepoužitieFarmakokinetika RoActemry u pacientov s OBA sa stanovila za použitia populačného farmakokinetického modelu analýzy údajov z databázy zloženej zo 149 pacientov s OBA liečených dávkou 162 mg podávanou subkutánne každý týždeň alebo dávkou 162 mg podávanou subkutánne každý druhý týždeň. Tento vyvinutý model mal zhodnú štruktúru so štruktúrou populačného PK modelu, ktorý bol vyvinutý skoršie na základe údajov zo štúdie pacientov s RA (pozri tabuľku 8).
Tabuľka 8. Predpokladaný priemer ± SD PK paramentrov v rovnovážnom stave po subkutánnom podávaní u pacientov s OBASubkutánne podávanie
P
K parametre tocilizumabu
16
2 mg každý druhý týždeň
16
2 mg každý týždeň
Cm ax (µg/ml) 19,3 ± 12,8 73 ± 30,4
Ctrough (µg/ml) 11,1 ± 10,3 68,1± 29,5
Cm ean (µg/ml) 16,2 ± 11,8 71,3 ± 30,1
Kumulácia Cm ax 2,18 8,88
Kumulácia Ctrough 5,61 9,59
Kumulácia Cm ean alebo AUCτ 2,81 10,91
Profil rovnovážneho stavu po podávaní RoActemry každý týždeň bol takmer plochý, s veľmi malými
rozdielmi medzi minimálnymi a maximálnymi hodnotami, zatiaľ čo pri dávke RoActemry podávanej každý druhý týždeň sa pozorovali významné rozdiely. Približne 90 % rovnovážneho stavu (AUCτ) sa dosiahlo do 14. týždňa v skupine s RoActemrou podávanou každý druhý týždeň a do 17. týždňa
pri skupine s RoActemrou podávanou každý týždeň.
Na základe súčasnej PK charakteristiky minimálne koncentrácie RoActermy v rovnovážnom stave sú o 50 % vyššie v tejto populácii, v porovnamí s priemernými koncentráciami uvedenými vo veľkom súbore údajov získaných od populácie s RA. Tieto rozdiely sa objavujú z neznámych dôvodov. Rozdiely v PK nesprevádzajú významné rozdiely v PD parametroch, a preto klinický význam týchto údajov nie je známy.
U pacientov s OBA bola pozorovaná vyššia expozícia u pacientov s nižšou telesnou hmotnosťou.
V režime s dávkou 162 mg podávanou každý týždeň bol rovnovážny stav Cavg o 51 % vyšší
u pacientov s telesnou hmotnosťou < 60 kg, oproti pacientom s telesnou hmotnosťou od 60 do 100 kg.
V režime s dávkou 162 mg podávanou každý druhý týždeň bol rovnovážny stav Cavg o 129 % vyšší
u pacientov s telesnou hmotnosťou < 60 kg, oproti pacientom s telesnou hmotnosťou od 60 do 100 kg.
Údaje o pacientoch s telesnou hmotnosťou > 100 kg sú obmedzené (n=7).
A
bsorpcia
Po subkutánnom podávaní u pacientov s OBA bol absorpčný t½ približne 4 dni. Biologická
dostupnosť v prípade subkutánnej formy bola 0,8. Medián hodnôt Tmax bol 3 dni po podávaní
RoActemry každý týždeň a 4,5 dňa po podávaní tocilizumabu každý druhý týždeň.
Distribúcia
U pacientov s OBA bol distribučný objem centrálneho kompartmentu 4,09 l, distribučný objem
periférneho kompartmentu bol 3,37 l, čo malo za následok distribučný objem 7,46 l v rovnovážnom stave.
Eliminácia
Celkový klírens RoActemry bol závislý od koncentrácie a je súčtom lineárneho a nelineárneho
klírensu. Lineárny klírens bol odhadnutý ako parameter v populačnej farmakokinetickej analýze
a u pacientov s OBA bol 6,7 ml/h.
U pacientov s OBA, v rovnovážnom stave, sa t½ účinnosti RoActemry pohyboval v rozpätí
od 18,3 do 18,9 dňa pri režime 162 mg podávaných každý týždeň, a v rozpätí od 4,2 do 7,9 dňa
pri režime 162 mg podávaných každý druhý týždeň. Pri vysokých koncentráciách v sére, v ktorých je celkový klírens RoActemry určovaný najmä lineárnym klírensom, bol t½ účinnosti približne 32 dní odvodený z odhadov populačných parametrov.
Osobitnéskupinypacientov
Porucha f unkcie obličiek: Štúdia vplyvu poruchy funkcie obličiek na farmakokinetiku RoActemry sa neuskutočnila. Väčšina pacientov v RA a OBA štúdiách zaradených do populačnej farmakokinetickej analýzy mala normálnu funkciu obličiek alebo miernu poruchu funkcie obličiek. Mierna porucha funkcie obličiek (odhadovaný klírens kreatinínu podľa Cockroftovho a Gaultovho vzorca) nemala vplyv na farmakokinetiku RoActemry.
Približne jedna tretina pacientov v štúdii OBA mala stredne ťažkú poruchu funkcie obličiek na začiatku liečby (odhadovaný klírens kreatínu 30 -59 ml/min). U týchto pacientov nebol pozorovaný žiaden vplyv na expozíciu RoActemry.
U pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava dávky.
Porucha f unkcie pečene: Štúdia vplyvu poruchy funkcie pečene na farmakokinetiku RoActemry sa neuskutočnila.
Vek, pohlavie a etnická príslušnosť: Populačné farmakokinetické analýzy u dospelých pacientov s RA a OBA preukázali, že vek, pohlavie a etnická príslušnosť nemajú vplyv na farmakokinetiku RoActemry.
5.3 Predk linick é údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Štúdie karcinogenity sa neuskutočnili, pretože IG1 monoklonálne protilátky sa nepovažujú za látky s vlastným karcinogénnym potenciálom.
Dostupné predklinické údaje preukázali vplyv IL-6 na progresiu zhubných nádorov a na rezistenciu rôznych typov nádorov na apoptózu. Tieto údaje nepoukazujú na významné riziko pre vznik
a progresiu rakoviny počas liečby RoActemrou. Okrem toho sa v 6-mesačných štúdiách chronickej
toxicity na opiciach rodu Cynomolgus, ani u myší s deficitom IL-6 proliferatívne lézie nepozorovali.
Dostupné predklinické údaje nepotvrdili, že liečba RoActemrou má vplyv na fertilitu. V štúdii chronickej toxicity na opiciach rodu Cynomolgus sa nepozorovali účinky na endokrinne aktívne orgány a na orgány reprodukčného systému a u myší s deficitom IL-6 nedošlo k poškodeniu reprodukčnej výkonnosti. Zistilo sa, že RoActemra podávaná opiciam rodu Cynomolgus počas skorej fázy gestácie nemala priamy ani nepriamy škodlivý vplyv na graviditu alebo embryofetálny vývoj. Pozorovalo sa však mierne zvýšenie potratov/embryofetálnej úmrtnosti pri vysokej systémovej expozícii (> 100-násobok expozície dosiahnutej u ľudí) v skupine liečenej vysokou dávkou
50 mg/kg/deň oproti skupine liečenej placebom a inými nízkymi dávkami. Hoci IL-6 zrejme nie je
rozhodujúcim cytokínom pre rast plodu alebo imunologickú kontrolu rozhrania materských
a fetálnych tkanív, súvislosť tohto zistenia s RoActemrou nie je možné vylúčiť.
Liečba myšacím analógom na juvenilných myšiach nevykazovala toxicitu. Konkrétne, nebolo prítomné žiadne narušenie rastu kostí, imunitných funkcií a sexuálneho dozrievania.
Predklinický bezpečnostný profil RoActemry u opíc rodu Cynomolgus nepoukazuje na rozdiel medzi intravenóznyn a subkutánynm spôsobom podávania.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidíniumchloridu
arginín arginíniumchlorid metionín polysorbát 80 voda na injekciu
6.2 Ink ompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
24 mesiacov.
Po vybratí z chladničky sa RoActemra musí podať do 8 hodín a má sa uchovávať pri teplote do 30 °C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke. Naplnené perá uchovávajte v škatuli na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah balenia
0,9 ml roztok v naplnenej injekčnej striekačke (sklo typu I) so vsadenou ihlou, ktorá je zabudovaná do naplneného pera. Injekčná striekačka je uzatvorená pevným krytom ihly (elastomérové tesnenie s polypropylénovým plášťom) a gumovou zátkou (butylkaučuk s povlakom z fluorovanej živice).
Balenie po 4 naplnených perách a spoločné balenie s obsahom 12 (3 balenia po 4k s) naplnených pier. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
RoActemra sa dodáva v naplnenom pere na jednorazové použitie. Po vybratí naplneného pera
z chladničky sa má nechať naplnené pero dosiahnuť izbovú teplotu (18°C až 28°C) tak, že sa počká
45 minút pred injekčným podaním RoActemry. Naplneným perom sa nemá triasť. Po odstránení viečka sa musí injekcia začať podávať do 3 minút, aby sa zabránilo vysušeniu lieku a zablokovaniu ihly. Ak sa naplnené pero nepoužije do 3 minút, musí sa vyhodiť do nádoby odolnej proti prepichnutiu a použiť nové naplnené pero.
Ak sa fialový indikátor po stlačení aktivačného tlačidla neposúva, naplnené pero musíte vyhodiť
do nádoby odolnej proti prepichnutiu.
Nepok úšajte sa znovu použiť toto naplnené pero. Neopakujte podanie injekcie pomocou ďalšieho naplneného pera. Zatelefonujte svojmu lekárovi a poraďte sa
s ním.
Nepoužívajte, ak je liek zakalený alebo obsahuje čiastočky, ak má inú farbu než bezfarebnú až svetložltkastú, alebo ak ktorákoľvek časť naplneného pera javí známky poškodenia.
Úplné pokyny na podanie RoActemry v naplnenom pere sú poskytnuté v písomnej informácii pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/08/492/009
EU/1/08/492/010
9. DÁTUM PRVEJ REGISTRÁCIE/DÁTUM POSLEDNÉHO PREDĹŽENIADátum prvej registrácie: 16. január 2009
Dátum posledného predĺženia registrácie: 25. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.