
zlepšenia sa pozorovali do jedného týždňa po ukončení podávania aPCC a prerušení profylaxie Hemlibrou. Toto rýchle zlepšenie sa líši od zvyčajného klinického priebehu pozorovaného pri atypickom
hemolyticko-uremickom syndróme a klasických trombotických mikroangiopatiách, akou je napríklad trombotická trombocytopenická purpura (pozri časť 4.8). U jedného pacienta sa po odznení TMA opätovne začala profylaxia Hemlibrou a jej pokračovanie bolo pre neho bezpečné.
Pacientov, ktorí dostávajú profylaxiu Hemlibrou, je potrebné sledovať kvôli vzniku TMA, keď sa im podáva aPCC. Lekár má ihneď ukončiť podávanie aPCC a prerušiť liečbu Hemlibrou, ak sa zistia klinické príznaky a/alebo laboratórne nálezy zodpovedajúce TMA, a pri jej liečbe sa má riadiť klinickým stavom pacienta. Lekári a pacienti/opatrovatelia majú v každom individuálnom prípade zvážiť prínosy a riziká opätovného začatia profylaxie Hemlibrou po úplnom odznení TMA. V prípade, že je „bypassový“ prípravok indikovaný u pacienta, ktorý dostáva profylaxiu Hemlibrou, pozrite si odporúčania na dávkovanie „bypassových“ prípravkov uvedené nižšie.
Pri liečbe pacientov, ktorí majú vysoké riziko vzniku TMA (napr. majú TMA v predchádzajúcej anamnéze alebo v rodinnej anamnéze) alebo pacientov, ktorí sú súbežne liečení liekmi, o ktorých je známe, že sú rizikovým faktorom vzniku TMA (napr. cyklosporín, chinidín, takrolimus), sa má postupovať s opatrnosťou.
Tromboembólia súvisiaca s Hemlibrou a aPCC
V klinickej štúdii boli hlásené závažné trombotické príhody u pacientov, ktorí dostávali profylaxiu
Hemlibrou, keď im bol počas 24 hodín alebo dlhšie podávaný aPCC v priemernom kumulatívnom množstve > 100 U/kg/24 hodín (pozri časť 4.8). Žiadny prípad nevyžadoval antikoagulačnú liečbu. Po ukončení podávania aPCC a prerušení profylaxie Hemlibrou sa známky zlepšenia alebo odznenie príhody pozorovali do jedného mesiaca (pozri časť 4.8). U jedného pacienta sa po odznení trombotickej príhody opätovne začala profylaxia Hemlibrou a jej pokračovanie bolo pre neho bezpečné.
Pacientov, ktorí dostávajú profylaxiu Hemlibrou, je potrebné sledovať kvôli vzniku tromboembolizmu, keď sa im podáva aPCC. Lekár má ihneď ukončiť podávanie aPCC a prerušiť liečbu Hemlibrou, ak sa zistia klinické príznaky, nálezy zobrazovacích a/alebo laboratórnych vyšetrení zodpovedajúce trombotickým príhodam, a pri ich liečbe sa má riadiť klinickým stavom pacienta. Lekári a pacienti/opatrovatelia majú v každom individuálnom prípade zvážiť prínosy a riziká opätovného začatia profylaxie Hemlibrou po úplnom odznení trombotickej príhody. V prípade, že je
„bypassový“ prípravok indikovaný u pacienta, ktorý dostáva profylaxiu Hemlibrou, pozrite si
odporúčania na dávkovanie „bypassových“ prípravkov uvedené nižšie.
Odporúčania na používanie „bypassových“ prípravkov u pacientov, ktorí dostávajú profylaxiu
Hemlibrou
Liečba „bypassovými“ prípravkami sa má ukončiť deň pred začiatkom liečby Hemlibrou.
Lekári majú so všetkými pacientmi a/alebo opatrovateľmi prediskutovať presnú dávku a schému podávania „bypassových“ prípravkov, ak sú potrebné počas profylaktickej liečby Hemlibrou.
Hemlibra zvyšuje u pacienta koagulačný potenciál. Preto môže byť potrebná nižšia dávka
„bypassového“ prípravku ako je dávka, ktorá sa používa bez profylaxie Hemlibrou. Dávkovanie a trvanie liečby „bypassovými“ prípravkami bude závisieť od miesta a rozsahu krvácania
a od klinického stavu pacienta. Je potrebné vyhnúť sa použitiu aPCC, ak sú k dispozícii iné možnosti/alternatívy liečby. Ak je aPCC indikovaný u pacienta, ktorý dostáva profylaxiu Hemlibrou, úvodná dávka nemá prekročiť 50 U/kg a odporúča sa laboratórne monitorovanie (ktoré zahŕňa, ale neobmedzuje sa na monitorovanie funkcie obličiek, testy na stanovenie počtu trombocytov
a vyšetrenie na prítomnosť trombózy). Ak sa krvácanie nezvládne úvodnou dávkou aPCC do 50 U/kg,
ďalšie dávky aPCC sa majú podávať pod vedením alebo dohľadom lekára a má sa zvážiť laboratórne monitorovanie na odhalenie TMA alebo tromboembolizmu a skontrolovanie krvácania pred opakovaným podávaním. Celková dávka aPCC nemá prekročiť 100 U/kg v prvých 24 hodinách liečby. Ošetrujúci lekári musia starostlivo zvážiť riziko vzniku TMA a tromboembolizmu oproti riziku krvácania, keď uvažujú o liečbe aPCC v dávke prevyšujúcej maximálnych 100 U/kg v prvých
24 hodinách.
V klinických štúdiách sa nepozorovali žiadne prípady TMA ani trombotických príhod, keď sa
u pacientov, ktorí dostávali profylaxiu Hemlibrou, používal iba rFVIIa.
Odporúčania na dávkovanie „bypassových“ prípravkov sa majú dodržiavať počas aspoň 6 mesiacov
po ukončení profylaxie Hemlibrou (pozri časť 5.2).
Imunogenicita
Počas klinických štúdií sa menej často pozorovala tvorba neutralizačných protilátok proti
emicizumabu s klesajúcou koncentráciou emicizumabu, ktorá viedla k strate účinnosti (pozri časti 4.8 a 5.1). Pacienti s klinickými prejavmi straty účinnosti (napr. zvýšenie výskytu epizód akútneho krvácania [„breakthrough bleeding“, t. j. spontánne vnútorné krvácanie alebo krvácanie spôsobené úrazom či zranením, ktoré sa vyskytne napriek tomu, že pacient dostáva profylaxiu]) sa majú okamžite vyšetriť, aby sa stanovila príčina, a v prípade podozrenia na neutralizačné protilátky proti
emicizumabu sa majú zvážiť iné terapeutické možnosti.
Vplyv emicizumabu na koagulačné testy
Emicizumab nahrádza pôsobenie chýbajúceho aktivovaného faktora VIII (FVIIIa) ako kofaktora
v tenázovom komplexe. Laboratórne koagulačné testy založené na vnútornej ceste aktivácie koagulácie, zahŕňajúce aktivovaný čas zrážania (activated clotting time, ACT), aktivovaný parciálny tromboplastínový čas (aPTT), merajú celkový čas zrážania (t. j. čas potrebný na vytvorenie koagula) vrátane času potrebného na to, aby došlo k aktivácii FVIII na FVIIIa pôsobením trombínu. Výsledkom týchto testov založených na vnútornej ceste bude nadmerne skrátený čas zrážania pri podávaní emicizumabu, ktorý nevyžaduje aktiváciu trombínom. Nadmerne skrátený čas zrážania po aktivácii vnútornej cesty následne naruší všetky vyšetrenia aktivity jednotlivých koagulačných faktorov
na princípe aPTT, napríklad stanovenie funkčnej aktivity FVIII jednofázovou metódou (pozri časť 4.4, tabuľka 1). Vyšetrenia aktivity jednotlivých koagulačných faktorov, pri ktorých sa využívajú chromogénne alebo imunologické metódy, však nie sú ovplyvnené emicizumabom a môžu sa používať na hodnotenie koagulačných parametrov počas liečby, pričom je potrebné vziať do úvahy ďalej uvedené špecifické aspekty týkajúce sa chromogénnych testov na stanovenie aktivity FVIII.
Pri výrobe chromogénnych testov na stanovenie aktivity FVIII sa môžu použiť buď ľudské alebo bovinné (hovädzie) koagulačné proteíny. Testy obsahujúce ľudské koagulačné faktory reagujú na emicizumab, ale môžu nadhodnotiť klinický hemostatický potenciál emicizumabu. Naopak,
testy obsahujúce bovinné koagulačné faktory nie sú citlivé na emicizumab (nie je nameraná žiadna aktivita) a môžu sa použiť na monitorovanie aktivity endogénneho alebo infúziou podaného FVIII alebo na zistenie prítomnosti inhibítorov FVIII.
Emicizumab zostáva aktívny v prítomnosti inhibítora FVIII, a preto spôsobí falošne negatívny výsledok koagulačného Bethesda testu, ktorý sa používa na kvantifikáciu inhibítora FVIII. Namiesto tohto testu sa môže použiť chromogénny Bethesda test, pri ktorom sa na stanovenie aktivity FVIII využíva chromogénny test s bovinnými koagulačnými faktormi, ktorý nie je citlivý na emicizumab.
Tieto dva farmakodynamické markery (t. j. aPTT a aktivita FVIII) neodzrkadľujú skutočný hemostatický účinok emicizumabu v podmienkach in vivo (aPTT je nadmerne skrátený a nameraná aktivita FVIII môže byť nadhodnotená), ale naznačujú pravdepodobný relatívny prokoagulačný účinok emicizumabu.
Je možné zhrnúť, že výsledky laboratórnych koagulačných testov založených na vnútornej ceste sa u pacientov liečených Hemlibrou nemajú používať na sledovanie jej účinku, na určenie dávky substitučnej liečby či antikoagulačnej liečby, ani na meranie titra inhibítora FVIII. Pri používaní laboratórnych koagulačných testov založených na vnútornej ceste sa má postupovať s opatrnosťou, pretože nesprávna interpretácia ich výsledkov môže viesť k nedostatočnému liečeniu pacientov,
u ktorých sa vyskytujú krvácavé epizódy, čo môže potenciálne viesť k závažnému alebo život ohrozujúcemu krvácaniu.
V tabuľke 1 sú uvedené laboratórne testy, ktoré sú alebo nie sú ovplyvnené emicizumabom.
Vzhľadom na dlhý polčas emicizimabu môže tento vplyv na koagulačné testy pretrvávať až
6 mesiacov po podaní poslednej dávky (pozri časť 5.2).
Tabuľka 1 Výsledky koagulačných testov, ktoré sú alebo nie sú ovplyvnené emicizumabom
Výsledky, ktoré sú ovplyvnené emicizumabom
|
Výsledky, ktoré nie sú ovplyvnené emicizumabom
|
- Aktivovaný parciálny tromboplastínový
čas (aPTT)
- (Koagulačný) Bethesda test na stanovenie
titra inhibítora FVIII
- Jednofázové metódy stanovenia aktivity jednotlivých koagulačných faktorov na princípe aPTT
- Test rezistencie na aktivovaný proteín C (APC-R) na princípe aPTT
- Aktivovaný čas zrážania (ACT)
|
- (Chromogénny) Bethesda test (s
bovinnými koagulačnými faktormi) na stanovenie titra inhibítora FVIII
- Trombínový čas (TT)
- Jednofázové metódy stanovenia aktivity jednotlivých koagulačných faktorov na princípe protrombínového času (PT)
- Chromogénne testy na stanovenie aktivity jednotlivých koagulačných faktorov iných ako FVIII1
- Imunologické testy (napr. ELISA,
turbidimetrické metódy)
- Genetické testy zamerané na koagulačné faktory (napr. faktor V Leiden, alela 20210 protrombínového génu)
|
1 Dôležité aspekty týkajúce sa chromogénnych testov na stanovenie aktivity FVIII, pozri časť 4.4.
Pediatrická populáciaK dispozícii nie sú žiadne údaje týkajúce sa detí vo veku < 1 rok. Vyvíjajúci sa hemostatický systém
u novorodencov a dojčiat je dynamický a rozvíjajúci sa a pri hodnotení pomeru prínosu a rizika liečby vrátane možného rizika vzniku trombózy (napr. trombóza súvisiaca so zavedením centrálneho žilového katétra) je potrebné vziať do úvahy relatívne koncentrácie prokoagulačných
a antikoagulačných proteínov u týchto pacientov.
4.5 Liekové a iné interakcieS emicizumabom sa neuskutočnili žiadne adekvátne alebo dobre kontrolované štúdie interakcií.
Klinické skúsenosti naznačujú existenciu liekovej interakcie medzi emicizumabom a aPCC (pozri
časti 4.4 a 4.8).
Na základe predklinických experimentov existuje možnosť hyperkoagulability pri použití rFVIIa alebo FVIII s emicizumabom, a preto môže byť dávka FVIIa alebo FVIII potrebná na dosiahnutie hemostázy nižšia ako dávka používaná bez profylaxie Hemlibrou.
V prípade trombotických komplikácií má lekár zvážiť prerušenie liečby rFVIIa alebo FVIII a prerušiť profylaxiu Hemlibrou podľa klinických indikácií. Ďalšia liečba má byť prispôsobená individuálnym klinickým okolnostiam.
● Rozhodnutie o úpravách dávkovania má brať do úvahy polčas rozpadu liekov; konkrétne,
prerušenie emicizumabu nemusí mať okamžitý účinok.
● Monitorovanie pomocou chromogénneho testu FVIII môže viesť k podaniu koagulačných faktorov a môže sa zvážiť testovanie trombofilných vlastností.
K dispozícii sú obmedzené skúsenosti so súbežným podávaním antifibrinolytík a aPCC alebo rFVIIa u pacientov, ktorí dostávajú profylaxiu Hemlibrou. Je však potrebné zohľadniť možnosť vzniku trombotických príhod, keď sa systémové antifibrinolytiká používajú v kombinácii s aPCC alebo rFVIIa u pacientov, ktorí dostávajú emicizumab.
4
.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/Antikoncepcia
Ženy vo fertilnom veku, ktoré sú liečené Hemlibrou, musia používať účinnú antikoncepciu počas
liečby Hemlibrou a počas aspoň 6 mesiacov po jej ukončení (pozri časť 5.2).
Gravidita
K dispozícii nie sú žiadne klinické štúdie skúmajúce použitie emicizumabu u gravidných žien.
S Hemlibrou sa neuskutočnili reprodukčné štúdie na zvieratách. Nie je známe, či emicizumab môže spôsobiť poškodenie plodu, keď sa podáva gravidnej žene alebo či môže ovplyvniť reprodukčnú schopnosť. Hemlibra sa má používať počas gravidity, iba ak potenciálny prínos pre matku prevažuje nad potenciálnym rizikom pre plod, pričom je potrebné vziať do úvahy, že počas gravidity
a v popôrodnom období je riziko trombózy zvýšené, a že niektoré komplikácie gravidity sú spájané so zvýšeným rizikom diseminovanej intravaskulárnej koagulácie (DIK).
Dojčenie
Nie je známe, či sa emicizumab vylučuje do ľudského mlieka. Neuskutočnili sa žiadne štúdie
hodnotiace vplyv emicizumabu na tvorbu mlieka alebo na jeho prítomnosť v materskom mlieku. Je známe, že ľudský IgG je prítomný v ľudskom mlieku. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu Hemlibrou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej
toxicity (pozri časť 5.3). K dispozícii nie sú žiadne údaje týkajúce sa fertility ľudí. Účinok
emicizumabu na mužskú a ženskú fertilitu preto nie je známy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Hemlibra nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Celkový bezpečnostný profil Hemlibry je založený na údajoch z klinických štúdií a zo sledovania po
uvedení lieku na trh. Najzávažnejšími nežiaducimi reakciami na liek (adverse drug reactions, ADR) hlásenými v klinických štúdiách s Hemlibrou boli trombotická mikroangiopatia (TMA) a trombotické príhody, ktoré zahŕňali trombózu kavernózneho sínusu (cavernous sinus thrombosis, CST) a trombózu povrchových žíl vyskytujúcu sa súčasne s nekrózou kože (pozri nižšie a časť 4.4).
Najčastejšími ADR hlásenými u ≥ 10% pacientov liečených aspoň jednou dávkou Hemlibry boli:
reakcie v mieste podania injekcie (19,4%), artralgia (14,2%) a bolesť hlavy (14,0%).
Celkovo traja pacienti (0,7%), ktorí v klinických štúdiách dostávali profylaxiu Hemlibrou, predčasne ukončili liečbu z dôvodu ADR, ktorými boli TMA, nekróza kože vyskytujúca sa súčasne s povrchovou tromboflebitídou a bolesť hlavy.
Tabuľkový zoznam nežiaducich reakcií na liekNasledujúce nežiaduce reakcie na liek (ADR) sú založené na údajoch zo sledovania po uvedení lieku
na trh a na kombinovaných údajoch z piatich klinických štúdií fázy III (štúdie u dospelých
a dospievajúcich [BH29884 - HAVEN 1, BH30071 - HAVEN 3 a BO39182 - HAVEN 4], štúdia
u pacientov všetkých vekových kategórií [BO41423 - HAVEN 6] a štúdia u pediatrických pacientov
[BH29992 - HAVEN 2]), v ktorých celkovo 444 pacientov s hemofíliou A dostalo aspoň jednu dávku Hemlibry ako rutinnú profylaxiu (pozri časť 5.1). Tristosedem (69,1%) účastníkov klinickej štúdie bolo dospelých (z ktorých dvaja boli ženského pohlavia), 61 (13,7%) dospievajúcich (vo veku
≥ 12 až < 18 rokov), 71 (16,0%) detí (vo veku ≥ 2 až < 12 rokov) a päť (1,1%) dojčiat a batoliat
(vo veku 1 mesiac až < 2 roky). Medián trvania expozície v štúdiách bol 32 týždňov (rozmedzie:
0,1 až 94,3 týždňa).
ADR zistené v klinických štúdiách fázy III a zo sledovania po uvedení lieku na trh sú uvedené podľa triedy orgánových systémov MedDRA (tabuľka 2). Zodpovedajúce kategórie frekvencie výskytu uvedené pri každej ADR sú založené na nasledujúcej konvencii: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Tabuľka 2 Súhrn nežiaducich reakcií na liek založený na kombinovaných údajoch z klinických štúdií HAVEN s Hemlibrou a zo sledovania po uvedení lieku na trhTrieda orgánových systémov (System
Organ Class, SOC)
| Nežiaduce reakcie
(uprednostnený výraz, MedDRA)
| Frekvencia
|
Poruchy krvi a lymfatického systému
| Trombotická mikroangiopatia
| Menej časté
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté
|
Poruchy ciev
| Povrchová tromboflebitída
| Menej časté
|
Trombóza kavernózneho sínusua
| Menej časté
|
Poruchy gastrointestinálneho traktu
| Hnačka
| Časté
|
Poruchy kože a podkožného tkaniva
| Nekróza kože
| Menej časté
|
Angioedém
| Menej časté
|
Urtikária
| Časté
|
Vyrážka
| Časté
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Artralgia
| Veľmi časté
|
Myalgia
| Časté
|
Celkové poruchy a reakcie v mieste podania
| Reakcia v mieste podania injekcie
| Veľmi časté
|
Pyrexia
| Časté
|
Zníženie terapeutickej odpovedeb
| Menej časté
|
a Poruchy ciev je sekundárna SOC pre trombózu kavernózneho sínusu.
b Strata účinnosti (zníženie terapeutickej odpovede) sa prejavuje ako zvýšenie výskytu akútneho krvácania a bolo
hlásené pri neutralizačných protilátkach proti emicizumabu s klesajúcou koncentráciou emicizumabu (pozri Opis
vybraných nežiaducich reakcií na liek a časti 4.4 a 5.1).
|
Opis vybraných nežiaducich reakcií na liek
Trombotická mikroangiopatia
V súhrnne hodnotených klinických štúdií fázy III boli prípady TMA hlásené u menej ako
1% pacientov (3/444) a špecificky u 9,7% pacientov (3/31), ktorí dostali aspoň jednu dávku aPCC
počas liečby emicizumabom. Všetky 3 prípady TMA sa vyskytli, keď bol aPCC podávaný počas
24 hodín alebo dlhšie v priemernom kumulatívnom množstve > 100 U/kg/24 hodín počas profylaktickej liečby (pozri časť 4.4). U pacientov sa zistila trombocytopénia, mikroangiopatická hemolytická anémia a akútne poškodenie obličiek, bez závažne zníženej aktivity proteázy ADAMTS13. U jedného pacienta sa po odznení TMA opätovne začala profylaxia Hemlibrou bez toho, že by došlo k jej recidíve.
Trombotické príhodyV súhrnne hodnotených klinických štúdií fázy III boli závažné trombotické príhody hlásené u menej
ako 1% pacientov (2/444) a špecificky u 6,5% pacientov (2/31), ktorí dostali aspoň jednu dávku aPCC počas liečby emicizumabom. Obidve závažné trombotické príhody sa vyskytli, keď bol aPCC podávaný počas 24 hodín alebo dlhšie v priemernom kumulatívnom množstve > 100 U/kg/24 hodín počas profylaktickej liečby. U jedného pacienta sa po odznení trombotickej príhody opätovne začala profylaxia Hemlibrou bez toho, že by došlo k jej recidíve (pozri časť 4.4).
Charakterizácia interakcie medzi emicizumabom a liečbou aPCC v pivotných klinických štúdiáchZaznamenalo sa 82 prípadov liečby aPCC* u pacientov, ktorí dostávali profylaxiu Hemlibrou, pričom
v ôsmich (10%) z týchto prípadov bol aPCC podávaný počas 24 hodín alebo dlhšie v priemernom kumulatívnom množstve > 100 U/kg/24 hodín; dva z týchto ôsmich prípadov sa spájali
s trombotickými príhodami a tri z týchto ôsmich prípadov sa spájali s TMA (tabuľka 3). So zvyšnými prípadmi liečby aPCC sa nespájala žiadna TMA ani trombotické príhody. V 68% všetkých prípadov liečby aPCC bola podaná iba jedna infúzia < 100 U/kg.
Tabuľka 3 Charakterizácia liečby aPCC* v súhrnne hodnotených klinických štúdiách fázy III Dĺžka trvania liečby aPCC
| Priemerné kumulatívne množstvo aPCC počas 24 hodín
(U/kg/24 hodín)
|
< 50
| 50 - 100
| > 100
|
< 24 hodín
| 9
| 47
| 13
|
24 - 48 hodín
| 0
| 3
| 1b
|
> 48 hodín
| 1
| 1
| 7a,a,a,b
|
* Prípad liečby aPCC je definovaný ako všetky dávky aPCC, ktoré pacient dostal, z akéhokoľvek
dôvodu, až kým nedošlo k 36-hodinovej prestávke v liečbe. Zahŕňa to všetky prípady liečby aPCC okrem tých, ktoré sa vyskytli v prvých 7 dňoch a tých, ktoré sa vyskytli 30 dní po ukončení podávania Hemlibry.
a Trombotická mikroangiopatia
b Trombotická príhoda
Reakcie v mieste podania injekcieV súhrnne hodnotených klinických štúdiách fázy III boli veľmi často (19,4%) hlásené reakcie v mieste
podania injekcie (injection site reactions, ISR). Všetky ISR pozorované v klinických štúdiách
s Hemlibrou boli hlásené ako nezávažné a ich intenzita bola mierna až stredne závažná a 94,9% z nich odznelo bez potreby liečby. Najčastejšie hlásenými príznakmi ISR boli erytém v mieste podania injekcie (10,6%), bolesť v mieste podania injekcie (4,1%), pruritus v mieste podania injekcie (2,9%)
a opuch v mieste podania injekcie (2,7%).
Imunogenicita
V súhrnne hodnotených klinických štúdií fázy III s Hemlibrou bola tvorba neutralizačných protilátok
proti emicizumabu spojená s klesajúcou koncentráciou emicizumabu menej častá (pozri časť 5.1). U jedného pacienta, u ktorého sa vytvorili neutralizačné protilátky proti emicizumabu s klesajúcou koncentráciou emicizumabu, došlo k strate účinnosti (prejavujúcej sa akútnym krvácaním) po piatich týždňoch liečby a liečba Hemlibrou bola u neho neskôr ukončená (pozri časti 4.4 a 5.1).
Pediatrická populáciaSkúmaná pediatrická populácia pozostávala celkovo zo 137 pacientov, ktorí zahŕňali 5 (3,6%) dojčiat
a batoliat (vo veku od 1 mesiaca do menej ako 2 rokov), 71 (51,8%) detí (vo veku od 2 do menej ako
12 rokov) a 61 (44,5%) dospievajúcich (vo veku od 12 do menej ako 18 rokov).
Bezpečnostný profil Hemlibry bol medzi dojčatami, deťmi, dospievajúcimi a dospelými celkovo zhodný.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s predávkovaním Hemlibrou sú obmedzené.
PríznakyNáhodné predávkovanie môže mať za následok hyperkoagulabilitu.
LiečbaPacienti, ktorí si náhodne podajú nadmernú dávku, majú ihneď vyhľadať lekára a majú byť pozorne
sledovaní.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká, iné systémové hemostatiká; ATC kód: B02BX06
Mechanizmus účinkuEmicizumab je humanizovaná monoklonálna modifikovaná imunoglobulín G4 (IgG4) protilátka
s bišpecifickou štruktúrou protilátky.
Emicizumab premosťuje priestor medzi aktivovaným faktorom IX a faktorom X, aby sa obnovila funkcia chýbajúceho FVIIIa, ktorý je potrebný na účinnú hemostázu.
Emicizumab nie je štrukturálne príbuzný ani sekvenčne homológny s FVIII, a preto neindukuje ani nepodporuje vznik priameho inhibítora FVIII.
Farmakodynamické účinkyProfylaktická liečba Hemlibrou skracuje aPTT a zvyšuje nameranú aktivitu FVIII (pri použití
chromogénneho testu s ľudskými koagulačnými faktormi). Tieto dva farmakodynamické markery
neodzrkadľujú skutočný hemostatický účinok emicizumabu v podmienkach in vivo (aPTT je nadmerne skrátený a nameraná aktivita FVIII môže byť nadhodnotená), ale naznačujú pravdepodobný relatívny prokoagulačný účinok emicizumabu.
Klinická účinnosť a bezpečnosť
Účinnosť Hemlibry v rutinnej profylaxii u pacientov s hemofíliou A sa hodnotila v piatich klinických
štúdiách (tri štúdie u dospelých a dospievajúcich pacientov s hemofíliou A a s inhibítorom FVIII alebo bez neho [HAVEN 1, HAVEN 3 a HAVEN 4], štúdia u pediatrických pacientov
s hemofíliou A a inhibítorom FVIII [HAVEN 2] a štúdia u pacientov všetkých vekových kategórií s ľahkou alebo stredne ťažkou hemofíliou A bez inhibítora FVIII [HAVEN 6]).
Klinické štúdie u dospelých a dospievajúcich pacientov s hemofíliou A a s inhibítorom FVIII alebo bezneho
Pacienti (vo veku ≥ 12 rokov a s telesnou hmotnosťou > 40 kg) s hemofíliou A a bez inhibítora FVIII
(štúdia BH30071 - HAVEN 3)
Štúdia HAVEN 3 bola randomizovaná, multicentrická, otvorená klinická štúdia fázy III
u 152 dospelých mužov a dospievajúcich chlapcov (vo veku ≥ 12 rokov a s telesnou
hmotnosťou > 40 kg) s ťažkou hemofíliou A a bez inhibítora FVIII, ktorí v predchádzajúcom období
dostávali buď epizodickú („on demand“, t. j. liečbu akútneho krvácania až do jeho zastavenia),
alebo profylaktickú liečbu FVIII. Pacienti dostávali subkutánne podávanú Hemlibru v dávke 3 mg/kg raz za týždeň počas prvých štyroch týždňov, po ktorých nasledovalo podávanie buď 1,5 mg/kg
raz za týždeň (skupina A a skupina D), alebo 3 mg/kg raz za dva týždne (skupina B), alebo nedostávali žiadnu profylaxiu (skupina C). Pacienti v skupine C mohli prejsť na profylaxiu Hemlibrou (3 mg/kg
raz za dva týždne) po absolvovaní aspoň 24 týždňov bez profylaxie. V skupine A a v skupine B bola po 24 týždňoch profylaxie Hemlibrou povolená titrácia dávky smerom nahor na 3 mg/kg raz za týždeň u pacientov, u ktorých sa vyskytli dve alebo viacej epizód krvácania spĺňajúcich vopred definované
kritériá (t. j. epizódy spontánneho a klinicky významného krvácania vyskytujúce sa pri koncentráciách
emicizumabu v rovnovážnom stave). U pacientov v skupine D mohla byť dávka titrovaná smerom nahor po výskyte druhej epizódy spĺňajúcej vopred definované kritériá. V čase primárnej analýzy bola titrácia udržiavacej dávky smerom nahor vykonaná u piatich pacientov.
Osemdesiatdeväť pacientov, ktorí v predchádzajúcom období dostávali epizodickú („on demand“) liečbu FVIII, bolo randomizovaných v pomere 2:2:1 na podávanie Hemlibry buď raz za týždeň (skupina A; N = 36), raz za dva týždne (skupina B; N = 35) alebo na nepodávanie profylaxie
(skupina C; N = 18), so stratifikáciou podľa výskytu epizód krvácania v predchádzajúcich 24 týždňoch
(< 9 alebo ≥ 9). Šesťdesiattri pacientov, ktorí boli v predchádzajúcom období profylakticky
liečení FVIII, bolo zaradených do skupiny D dostávajúcej Hemlibru (1,5 mg/kg raz za týždeň).
Primárnym cieľom štúdie bolo vyhodnotiť u pacientov, ktorí v predchádzajúcom období dostávali epizodickú liečbu FVIII, účinnosť profylaxie Hemlibrou podávanou raz za týždeň (skupina A) alebo raz za dva týždne (skupina B) v porovnaní s nepodávaním profylaxie (skupina C) na základe počtu epizód krvácania vyžadujúcich liečbu koagulačnými faktormi (pozri tabuľku 4). Ďalšie ciele
štúdie zahŕňali vyhodnotenie randomizovaného porovnania skupiny A alebo skupiny B a skupiny C z hľadiska účinnosti profylaxie Hemlibrou v znižovaní počtu všetkých epizód krvácania, epizód spontánneho krvácania, epizód krvácania do kĺbov a epizód krvácania do cieľového (t. j. iba jedného) kĺbu (pozri tabuľku 4), ako aj zhodnotenie preferencie liečby zo strany pacientov pomocou preferenčného dotazníka.
Účinnosť profylaxie Hemlibrou sa porovnala aj s predchádzajúcou profylaktickou liečbou FVIII (skupina D) u pacientov, ktorí sa pred zaradením do štúdie zúčastnili na neintervenčnej štúdii (non-interventional study, NIS) (pozri tabuľku 5). Do tohto porovnania boli zahrnutí iba pacienti
z NIS, pretože údaje o krvácavých epizódach a o liečbe boli zozbierané na rovnakej úrovni granularity
(detailnosti) údajov ako bola úroveň granularity použitá v štúdii HAVEN 3. NIS je observačná štúdia,
ktorej hlavným cieľom je získať podrobné klinické údaje o krvácavých epizódach a používaní liekov na hemofíliu u pacientov s hemofíliou A mimo rámca intervenčnej klinickej štúdie.
Pacienti (vo veku ≥ 12 rokov) s hemofíliou A a inhibítorom FVIII (štúdia BH29884 - HAVEN 1)
Štúdia HAVEN 1 bola randomizovaná, multicentrická, otvorená klinická štúdia u 109 dospievajúcich chlapcov (vo veku ≥ 12 rokov) a dospelých mužov s hemofíliou A a inhibítorom FVIII, ktorí
v predchádzajúcom období dostávali buď epizodickú alebo profylaktickú liečbu „bypassovými“ prípravkami (aPCC a rFVIIa). V tejto štúdii dostávali pacienti profylaxiu Hemlibrou podávanou raz za týždeň (skupiny A, C a D) - 3 mg/kg raz za týždeň počas štyroch týždňov, po ktorých nasledovalo podávanie 1,5 mg/kg raz za týždeň - alebo nedostávali žiadnu profylaxiu (skupina B). Pacienti randomizovaní do skupiny B mohli prejsť na profylaxiu Hemlibrou po absolvovaní aspoň
24 týždňov bez profylaxie. Po 24 týždňoch profylaxie Hemlibrou bola povolená titrácia dávky smerom nahor na 3 mg/kg raz za týždeň u pacientov, u ktorých sa vyskytli dve alebo viacej epizód krvácania spĺňajúcich vopred definované kritériá (t. j. epizódy spontánneho a potvrdeného klinicky významného krvácania vyskytujúce sa pri koncentráciách emicizumabu v rovnovážnom stave). V čase primárnej analýzy bola titrácia udržiavacej dávky na 3 mg/kg raz za týždeň vykonaná u dvoch pacientov.
Päťdesiattri pacientov, ktorí v predchádzajúcom období dostávali epizodickú („on demand“) liečbu
„bypassovými“ prípravkami, bolo randomizovaných v pomere 2:1 na podávanie profylaxie Hemlibrou (skupina A) alebo na nepodávanie profylaxie (skupina B), so stratifikáciou podľa výskytu epizód krvácania v predchádzajúcich 24 týždňoch (< 9 alebo ≥ 9).
Štyridsaťdeväť pacientov, ktorí boli v predchádzajúcom období profylakticky liečení „bypassovými“ prípravkami, bolo zaradených do skupiny C dostávajúcej profylaxiu Hemlibrou. Sedem pacientov, ktorí v predchádzajúcom období dostávali epizodickú („on demand“) liečbu „bypassovými“ prípravkami a ktorí sa pred zaradením do štúdie zúčastnili na NIS, ale nemohli byť zaradení
do štúdie HAVEN 1 pred skompletizovaním skupín A a B, bolo zaradených do skupiny D
dostávajúcej profylaxiu Hemlibrou.
Primárnym cieľom štúdie bolo vyhodnotiť u pacientov, ktorí v predchádzajúcom období dostávali epizodickú („on demand“) liečbu „bypassovými“ prípravkami, vplyv profylaktickej liečby Hemlibrou podávanou raz za týždeň v porovnaní s nepodávaním profylaxie (skupina A v porovnaní
so skupinou B) na počet epizód krvácania vyžadujúcich liečbu koagulačnými faktormi v priebehu času (minimálne 24 týždňov alebo do dátumu ukončenia účasti na štúdii) (pozri tabuľku 6). Ďalšími sekundárnymi cieľmi randomizovaného porovnania skupín A a B boli účinnosť profylaxie Hemlibrou podávanou raz za týždeň v znižovaní počtu všetkých epizód krvácania, epizód spontánneho krvácania, epizód krvácania do kĺbov a epizód krvácania do cieľového (t. j. iba jedného) kĺbu (pozri tabuľku 6), ako aj hodnotenie kvality života súvisiacej so zdravím (health-related quality of life, HRQoL)
a zdravotného stavu pacientov (pozri tabuľky 10 a 11). Priemerná doba expozície (+ štandardná odchýlka (standard deviation, SD)) u všetkých pacientov v štúdii bola 21,38 týždňa (12,01). Pokiaľ ide o jednotlivé liečebné skupiny, priemerná doba expozície (+ SD) bola 28,86 týždňa (8,37) v skupine A,
8,79 týždňa (3,62) v skupine B, 21,56 týždňa (11,85) v skupine C a 7,08 týždňa (3,89) v skupine D. Jeden pacient v skupine A ukončil účasť na štúdii pred začatím profylaxie Hemlibrou.
Štúdia hodnotila aj účinnosť profylaxie Hemlibrou podávanou raz za týždeň v porovnaní
s predchádzajúcou epizodickou („on demand“) a profylaktickou liečbou „bypassovými“ prípravkami
(samostatné porovnania) u pacientov, ktorí sa pred zaradením do štúdie zúčastnili na NIS
(skupina A a skupina C v uvedenom poradí) (pozri tabuľku 7).
Pacienti (vo veku ≥ 12 rokov) s hemofíliou A a s inhibítorom FVIII alebo bez neho (štúdia
BO39182 - HAVEN 4)
Hemlibra bola skúmaná v multicentrickej klinickej štúdii fázy III s jednou skupinou u 41 dospelých mužov a dospievajúcich chlapcov (vo veku ≥ 12 rokov a s telesnou hmotnosťou > 40 kg), ktorí majú hemofíliu A s inhibítorom FVIII alebo ťažkú hemofíliu A bez inhibítora FVIII, ktorí
v predchádzajúcom období dostávali buď epizodickú („on demand“) alebo profylaktickú liečbu
„bypassovými“ prípravkami alebo FVIII. Pacienti dostávali profylaxiu Hemlibrou - 3 mg/kg
raz za týždeň počas štyroch týždňov, po ktorej nasledovalo podávanie 6 mg/kg raz za štyri týždne. Primárnym cieľom štúdie bolo vyhodnotiť účinnosť profylaxie Hemlibrou podávanou raz za štyri týždne pri udržiavaní adekvátnej kontroly krvácania na základe liečených epizód krvácania. Ďalším cieľom bolo vyhodnotiť klinickú účinnosť profylaxie Hemlibrou z hľadiska všetkých epizód krvácania, liečených epizód spontánneho krvácania, liečených epizód krvácania do kĺbov a liečených
epizód krvácania do cieľového (t. j. iba jedného) kĺbu (pozri tabuľku 8). Hodnotila sa aj preferencia
liečby zo strany pacientov pomocou preferenčného dotazníka.
Pacienti (všetkých vekových kategórií) s ľahkou alebo stredne ťažkou hemofíliou A
bez inhibítora FVIII (štúdia BO41423 - HAVEN 6)
Štúdia HAVEN 6 bola multicentrická, otvorená klinická štúdia fázy III s jednou skupinou
u 71 pacientov (všetkých vekových kategórií) liečených emicizumabom, s ľahkou (n = 20 [28,2%]) alebo stredne ťažkou (n = 51 [71,8%]) hemofíliou A bez inhibítora FVIII, u ktorých bola indikovaná profylaxia na základe rozhodnutia skúšajúceho lekára. Väčšina pacientov boli muži (69 pacientov
[97,2%]) a 2 boli ženy (2,8%). Pri vstupe do štúdie 34 pacienti (47,9%) dostávali epizodickú
(„on demand“) liečbu a 37 pacienti (52,1%) dostávali profylaktickú liečbu FVIII. Pacientom bola subkutánne podávaná Hemlibra v dávke 3 mg/kg raz za týždeň počas prvých štyroch týždňov,
po ktorých nasledovala jedna z nasledujúcich schém podávania udržiavacej dávky, podľa preferencie zo strany pacienta, od 5. týždňa: 1,5 mg/kg raz za týždeň (n = 24 [33,8%]), 3 mg/kg raz za dva týždne (n = 39 [54,9%]) alebo 6 mg/kg raz za štyri týždne (n = 8 [11,3%]). Po 24 týždňoch bola povolená
titrácia dávky smerom nahor na 3 mg/kg raz za týždeň u pacientov, u ktorých sa vyskytli dve
alebo viacej epizód krvácania spĺňajúcich vopred definované kritériá (t. j. epizódy spontánneho
a klinicky významného krvácania vyskytujúce sa pri koncentráciách emicizumabu v rovnovážnom stave). V čase predbežnej analýzy nebola titrácia udržiavacej dávky smerom nahor vykonaná
u žiadneho pacienta.
Primárnym cieľom účinnosti štúdie bolo vyhodnotiť účinnosť profylaxie Hemlibrou na základe počtu epizód krvácania vyžadujúcich liečbu koagulačnými faktormi v priebehu času (t. j. výskyt liečených epizód krvácania, pozri tabuľku 9). Ďalšími cieľmi boli vyhodnotiť účinnosť profylaxie Hemlibrou
na základe počtu všetkých epizód krvácania, epizód spontánneho krvácania, epizód krvácania
do kĺbov a epizód krvácania do cieľového (t. j. iba jedného) kĺbu v priebehu času, ako aj vyhodnotenie kvality života súvisiacej so zdravím (health-related quality of life, HRQoL) prostredníctvom pacientmi vyplneného dotazníka CATCH (Comprehensive Assessment Tool of Challenges in Haemophilia,
t. j. komplexný nástroj na hodnotenie výziev pri hemofílii) v priebehu času.
Výsledky účinnosti
HAVEN 3
Výsledky účinnosti profylaxie Hemlibrou v porovnaní s nepodávaním profylaxie z hľadiska výskytu liečených epizód krvácania, všetkých epizód krvácania, liečených epizód spontánneho krvácania, liečených epizód krvácania do kĺbov a liečených epizód krvácania do cieľového (t. j. iba jedného) kĺbu sú uvedené v tabuľke 4.
Tabuľka 4 Štúdia HAVEN 3: Ročný výskyt epizód krvácania v skupine s profylaxiou Hemlibrou v porovnaní so skupinou bez profylaxie u pacientov vo veku ≥ 12 rokov a bez inhibítora FVIII
Cieľový ukazovateľ
|
Skupina C: žiadna profylaxia (N = 18)
|
Skupina A: Hemlibra
1
,5 mg/kg
raz za týždeň
(N = 36)
|
Skupina B: Hemlibra
3 mg/kg
raz za 2 týždne
(N = 35)
|
Liečené epizódy krvácania
|
ABR (95% IS)
|
38,2 (22,9; 63,8)
|
1,5 (0,9; 2,5)
|
1,3 (0,8; 2,3)
|
% zníženie (RR), p-hodnota
|
NA
|
96% (0,04),
< 0,0001
|
97% (0,03), < 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
0,0 (0,0; 18,5)
|
55,6 (38,1; 72,1)
|
60,0 (42,1; 76,1)
|
Medián ABR (IQR)
|
40,4 (25,3; 56,7)
|
0 (0; 2,5)
|
0 (0; 1,9)
|
Všetky epizódy krvácania
|
ABR (95% IS)
|
47,6 (28,5; 79,6)
|
2,5 (1,6; 3,9)
|
2,6 (1,6; 4,3)
|
% zníženie (RR), p-hodnota
|
NA
|
95% (0,05), < 0,0001
|
94% (0,06),
< 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
0 (0,0:18,5)
|
50 (32,9; 67,1)
|
40 (23,9; 57,9)
|
Liečené epizódy spontánneho krvácania
|
ABR (95% IS)
|
15,6 (7,6; 31,9)
|
1,0 (0,5; 1,9)
|
0,3 (0,1; 0,8)
|
% zníženie (RR), p-hodnota
|
NA
|
94% (0,06), < 0,0001
|
98% (0,02),
< 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
22,2 (6,4; 47,6)
|
66,7 (49,0; 81,4)
|
88,6 (73,3; 96,8)
|
Cieľový ukazovateľ
|
Skupina C: žiadna profylaxia (N = 18)
|
Skupina A: Hemlibra
1
,5 mg/kg
raz za týždeň
(N = 36)
|
Skupina B: Hemlibra
3 mg/kg
raz za 2 týždne
(N = 35)
|
Liečené epizódy krvácania do kĺbov
|
ABR (95% IS)
|
26,5 (14,67; 47,79)
|
1,1 (0,59; 1,89)
|
0,9 (0,44; 1,67)
|
% zníženie (RR), p-hodnota
|
NA
|
96% (0,04), < 0,0001
|
97% (0,03),
< 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
0 (0; 18,5)
|
58,3 (40,8; 74,5)
|
74,3 (56,7; 87,5)
|
Liečené epizódy krvácania do cieľového kĺbu
|
ABR (95% IS)
|
13,0 (5,2; 32,3)
|
0,6 (0,3; 1,4)
|
0,7 (0,3; 1,6)
|
% zníženie (RR), p-hodnota
|
NA
|
95% (0,05), < 0,0001
|
95% (0,05),
< 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
27,8 (9,7; 53,5)
|
69,4 (51,9; 83,7)
|
77,1 (59,9; 89,6)
|
Pomer výskytu (rate ratio) a interval spoľahlivosti (IS) vychádzajú z modelu negatívne binomickej regresie
(NBR) a p-hodnota zo stratifikovaného Waldovho testu, pri porovnávaní výskytu epizód krvácania medzi špecifikovanými skupinami.
Skupina C: zahŕňa iba obdobie bez podávania profylaxie.
Definície epizód krvácania sú prevzaté na základe kritérií ISTH (International Society on Thrombosis and
Haemostasis).
Liečené epizódy krvácania = epizódy krvácania liečené FVIII.
Všetky epizódy krvácania = epizódy krvácania liečené a neliečené FVIII.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas 4 týždňov.
ABR = ročný výskyt epizód krvácania (Annualised Bleed Rate); IS = interval spoľahlivosti; RR = pomer výskytu (rate ratio); IQR = interkvartilové rozpätie (interquartile range), 25. percentil až 75. percentil. NA = Neuplatňuje sa
|
V klinickej štúdii HAVEN 3 sa v intraindividuálnej analýze zistilo, že profylaxia Hemlibrou viedla
k štatisticky signifikantnému (p < 0,0001) zníženiu (68%) výskytu epizód krvácania z hľadiska liečených epizód krvácania v porovnaní s údajmi o predchádzajúcej profylaxii FVIII zozbieranými v NIS pred zaradením do štúdie (pozri tabuľku 5).
Tabuľka 5 Štúdia HAVEN 3: Intraindividuálne porovnanie ročného výskytu epizód krvácania
(liečené epizódy krvácania) pri profylaxii Hemlibrou v porovnaní
s predchádzajúcou profylaxiou FVIII
Cieľový ukazovateľ
|
Skupina DNIS:
Predchádzajúca profylaxia FVIII
(N = 48)
|
Skupina D: Hemlibra 1,5 mg/kg raz za týždeň
(N = 48)
|
Medián obdobia účinnosti
(týždne)
|
30,1
|
33,7
|
Liečené epizódy krvácania
|
ABR (95% IS)
|
4,8 (3,2; 7,1)
|
1,5 (1; 2,3)
|
% zníženie (RR), p-hodnota
|
68% (0,32), < 0,0001
|
% pacientov s nulovým počtom
epizód krvácania (95% IS)
|
39,6 (25,8; 54,7)
|
54,2 (39,2; 68,6)
|
Medián ABR (IQR)
|
1,8 (0; 7,6)
|
0 (0; 2,1)
|
Pomer výskytu (rate ratio) a interval spoľahlivosti (IS) vychádzajú z modelu negatívne binomickej regresie
(NBR) a p-hodnota zo stratifikovaného Waldovho testu, pri porovnávaní ABR medzi špecifikovanými skupinami.
Intraindividuálne porovnávacie údaje z NIS. Zahrnutí sú iba pacienti, ktorí sa zúčastnili na NIS a na štúdii
HAVEN 3.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou.
Liečené epizódy krvácania = epizódy krvácania liečené FVIII. Definície epizód krvácania sú prevzaté na základe kritérií ISTH. ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; RR = pomer výskytu;
IQR = interkvartilové rozpätie, 25. percentil až 75. percentil.
Hoci sa pri profylaxii emicizumabom pozorovala vyššia adhézia ako pri predchádzajúcej profylaxii FVIII, nebol identifikovaný žiadny rozdiel v ABR u pacientov so zhodnými dávkami ≥ 80% alebo < 80% profylaxie FVIII podľa štandardných požiadaviek (údaje sa majú interpretovať s opatrnosťou kvôli malej veľkosti vzorky).
Kvôli krátkému polčasu FVIII sa po ukončení liečby nepredpokladá žiadny zostatkový účinok.
Iba prvých päť dávok emicizumabu sa muselo podať pod dozorom, aby sa zaistila zbehlosť v oblasti bezpečnosti a techniky vpichu injekcie. Podobne ako pri profylaxii FVIII bolo povolené podávanie v domácom prostredí pre všetky nasledujúce dávky emicizumabu.
Všetci pacienti boli liečení odborníkmi na hemofíliu, ktorí potvrdili, že primeraná FVIII-profylaxia bola
podávaná pacientom zahrnutým do porovnania medzi pacientmi, podporujúc ekvivalentnú bežnú profylaktickú
starostlivosť naprieč miestami a pacientmi.
|
HAVEN 1
Výsledky účinnosti profylaxie Hemlibrou v porovnaní s nepodávaním profylaxie z hľadiska výskytu liečených epizód krvácania, všetkých epizód krvácania, liečených epizód spontánneho krvácania, liečených epizód krvácania do kĺbov a liečených epizód krvácania do cieľového (t. j. iba jedného) kĺbu sú uvedené v tabuľke 6.
Tabuľka 6 Štúdia HAVEN 1: Ročný výskyt epizód krvácania v skupine s profylaxiou Hemlibrou v porovnaní so skupinou bez profylaxie u pacientov vo veku ≥ 12 rokov a s inhibítorom FVIII
Cieľový ukazovateľ
|
Skupina B: žiadna profylaxia
|
Skupina A: Hemlibra
1
,5 mg/kg raz za týždeň
|
|
N = 18
|
N = 35
|
Liečené epizódy krvácania
|
ABR (95% IS)
|
23,3 (12,33; 43,89)
|
2,9 (1,69; 5,02)
|
% zníženie (RR), p-hodnota
|
87% (0,13), < 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
5,6 (0,1; 27,3)
|
62,9 (44,9; 78,5)
|
Medián ABR (IQR)
|
18,8 (12,97; 35,08)
|
0 (0; 3,73)
|
Všetky epizódy krvácania
|
ABR (95% IS)
|
28,3 (16,79; 47,76)
|
5,5 (3,58; 8,60)
|
% zníženie (RR), p-hodnota
|
80% (0,20), < 0,0001
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
5,6 (0,1; 27,3)
|
37,1 (21,5; 55,1)
|
Liečené epizódy spontánneho krvácania
|
ABR (95% IS)
|
16,8 (9,94; 28,30)
|
1,3 (0,73; 2,19)
|
% zníženie (RR), p-hodnota
|
92% (0,08), < 0,0001
|
% pacientov s nulovým počtom epizód
krvácania (95% IS)
|
11,1 (1,4; 34,7)
|
68,6 (50,7; 83,1)
|
Liečené epizódy krvácania do kĺbov
|
ABR (95% IS)
|
6,7 (1,99; 22,42)
|
0,8 (0,26; 2,20)
|
% zníženie (RR), p-hodnota
|
89% (0,11), 0,0050
|
% pacientov s nulovým počtom epizód krvácania (95% IS)
|
50,0 (26,0; 74,0)
|
85,7 (69,7; 95,2)
|
Liečené epizódy krvácania do cieľového kĺbu
|
ABR (95% IS)
|
3,0 (0,96; 9,13)
|
0,1 (0,03; 0,58)
|
% zníženie (RR), p-hodnota
|
95% (0,05), 0,0002
|
% pacientov s nulovým počtom epizód
krvácania (95% IS)
|
50,0 (26,0; 74,0)
|
94,3 (80,8; 99,3)
|
Pomer výskytu (rate ratio) a interval spoľahlivosti (IS) vychádzajú z modelu negatívne binomickej regresie (NBR) a p-hodnota zo stratifikovaného Waldovho testu, pri porovnávaní výskytu epizód krvácania medzi špecifikovanými skupinami.
Skupina B: zahŕňa iba obdobie bez podávania profylaxie. Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
Liečené epizódy krvácania = epizódy krvácania liečené „bypassovými“ prípravkami.
Všetky epizódy krvácania = epizódy krvácania liečené a neliečené „bypassovými“ prípravkami.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas
4 týždňov.
ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; RR = pomer výskytu; IQR = interkvartilové rozpätie, 25. percentil až 75. percentil.
|
V štúdii HAVEN 1 sa v intraindividuálnej analýze zistilo, že profylaxia Hemlibrou viedla k štatisticky
signifikantnému (p = 0,0003) a klinicky významnému zníženiu (79%) výskytu epizód krvácania z hľadiska liečených epizód krvácania v porovnaní s údajmi o predchádzajúcej profylaxii
„bypassovými“ prípravkami zozbieranými v NIS pred zaradením do štúdie (pozri tabuľku 7).
Tabuľka 7 Štúdia HAVEN 1: Intraindividuálne porovnanie ročného výskytu epizód krvácania
(liečené epizódy krvácania) pri profylaxii Hemlibrou v porovnaní
s predchádzajúcou profylaxiou „bypassovými“ prípravkami (pacienti z NIS)
Cieľový ukazovateľ
|
Skupina C
NI
S
: predchádzajúca profylaxia „bypassovými“ prípravkami
|
Skupina C: Hemlibra
1
,5 mg/kg raz za týždeň
|
|
N = 24
|
N = 24
|
Liečené epizódy krvácania
|
ABR (95% IS)
|
15,7 (11,08; 22,29)
|
3,3 (1,33; 8,08)
|
% pacientov s nulovým
počtom epizód krvácania
(95% IS)
|
12,5 (2,7; 32,4)
|
70,8 (48,9; 87,4)
|
Medián ABR (IQR)
|
12,0 (5,73; 24,22)
|
0,0 (0,00; 2,23)
|
% zníženie (RR), p-hodnota
|
79% (0,21), 0,0003
|
Pomer výskytu (rate ratio) a interval spoľahlivosti (IS) vychádzajú z modelu negatívne binomickej regresie
(NBR) a p-hodnota zo stratifikovaného Waldovho testu, pri porovnávaní ABR medzi špecifikovanými skupinami.
Intraindividuálne porovnávacie údaje z NIS.
Zahrnutí sú iba pacienti, ktorí sa zúčastnili na NIS a na štúdii HAVEN 1.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou.
Liečené epizódy krvácania = epizódy krvácania liečené „bypassovými“ prípravkami. Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; RR = pomer výskytu; IQR = interkvartilové
rozpätie, 25. percentil až 75. percentil.
Hoci sa pri profylaxii emicizumabom pozorovala vyššia adhézia ako pri predchádzajúcej profylaxii bypassovými prípravkami (BPA), nebol identifikovaný žiadny rozdiel v ABR u pacientov so zhodnými dávkami ≥ 80% alebo < 80% profylaxie BPA podľa štandardných požiadaviek (údaje sa majú interpretovať s opatrnosťou kvôli malej veľkosti vzorky).
Kvôli krátkému polčasu bypassových prípravkov sa po ukončení liečby nepredpokladá žiadny zostatkový účinok.
Iba prvých päť dávok emicizumabu sa muselo podať pod dozorom, aby sa zaistila zbehlosť v oblasti bezpečnosti a techniky vpichu injekcie. Podobne ako pri profylaxii BPA bolo povolené podávanie v domácom prostredí pre všetky nasledujúce dávky emicizumabu.
|
HAVEN 4
Výsledky primárnej analýzy účinnosti profylaxie Hemlibrou podávanou raz za štyri týždne z hľadiska výskytu liečených epizód krvácania, všetkých epizód krvácania, liečených epizód spontánneho krvácania, liečených epizód krvácania do kĺbov a liečených epizód krvácania do cieľového (t. j. iba jedného) kĺbu sú uvedené v tabuľke 8. Účinnosť sa vyhodnotila u 41 pacientov vo veku ≥ 12 rokov, pričom medián obdobia pozorovania bol 25,6 týždňa (rozmedzie: 24,1 - 29,4).
Tabuľka 8 Štúdia HAVEN 4: Ročný výskyt epizód krvácania pri profylaxii Hemlibrou u pacientov vo veku ≥ 12 rokov a s inhibítorom FVIII alebo bez neho
|
Hemlibra 6 mg/kg Q4W
|
Cieľové ukazovatele
|
a
ABR (95% IS)
|
b
Medián ABR (IQR)
|
% pacientov
s nulovým počtom epizód krvácania (95% IS)
|
N
|
41
|
41
|
41
|
Liečené epizódy
krvácania
|
2,4 (1,4; 4,3)
|
0,0 (0,0; 2,1)
|
56,1 (39,7; 71,5)
|
Všetky epizódy krvácania
|
4,5 (3,1; 6,6)
|
2,1 (0,0; 5,9)
|
29,3 (16,1; 45,5)
|
Liečené epizódy
spontánneho krvácania
|
0,6 (0,3; 1,5)
|
0,0 (0,0; 0,0)
|
82,9 (67,9; 92,8)
|
Liečené epizódy krvácania do kĺbov
|
1,7 (0,8; 3,7)
|
0,0 (0,0; 1,9)
|
70,7 (54,5; 83,9)
|
Liečené epizódy
krvácania do cieľového kĺbu
|
1,0 (0,3; 3,3)
|
0,0 (0,0; 0,0)
|
85,4 (70,8; 94,4)
|
a Vypočítané pomocou modelu negatívne binomickej regresie (NBR).
b Vypočítaný ABR.
Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
Liečené epizódy krvácania: epizódy krvácania liečené FVIII alebo rFVIIa.
Všetky epizódy krvácania: epizódy krvácania liečené a neliečené FVIII alebo rFVIIa.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas 4 týždňov. ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; RR = pomer výskytu; IQR = interkvartilové rozpätie, 25. percentil až 75. percentil; Q4W = profylaxia dávkou podávanou raz za štyri týždne.
|
HAVEN 6 (predbežná analýza)
Účinnosť sa vyhodnotila u 51 pacientov so stredne ťažkou hemofíliou A vo veku od 2 do 56 rokov, pričom medián obdobia pozorovania bol 30,4 týždňa (rozsah: 17,4 - 61,7). Výsledky predbežnej analýzy účinnosti profylaxie Hemlibrou u pacientov so stredne ťažkou hemofíliou A (pozri časť 4.1) z hľadiska výskytu liečených epizód krvácania, všetkých epizód krvácania, liečených epizód spontánneho krvácania, liečených epizód krvácania do kĺbov a liečených epizód krvácania
do cieľového (t. j. iba jedného) kĺbu sú uvedené v tabuľke 9.
Tabuľka 9 HAVEN 6: Ročný výskyt epizód krvácania pri profylaxii Hemlibrou u pacientov so stredne ťažkou hemofíliou A bez inhibítora FVIII
|
c
Hemlibra 1,5 mg/kg QW, 3 mg/kg Q2W alebo 6 mg/kg Q4W
|
Cieľové ukazovatele
|
a
ABR (95% IS)
|
b
Medián ABR (IQR)
|
% pacientov
s nulovým počtom epizód krvácania (95% IS)
|
N
|
51
|
51
|
51
|
Liečené epizódy
krvácania
|
0,9 [0,43;1,89]
|
0,0 [0,00; 0,00]
|
78,4 [64,7; 88,7]
|
Všetky epizódy krvácania
|
2,6 [1,81; 3,81]
|
1,7 [0,00; 3,90]
|
43,1 [29,3; 57,8]
|
Liečené epizódy
spontánneho krvácania
|
0,1 [0,03; 0,30]
|
0,0 [0,00; 0,00]
|
94,1 [83,8; 98,8]
|
Liečené epizódy krvácania do kĺbov
|
0,3 [0,10; 0,84]
|
0,0 [0,00; 0,00]
|
90,2 [78,6; 96,7]
|
Liečené epizódy krvácania do cieľového
kĺbu
|
0,1 [0,02; 0,26]
|
0,0 [0,00; 0,00]
|
96,1 [86,5; 99,5]
|
a Vypočítané pomocou modelu negatívne binomickej regresie (NBR)
b Vypočítaný ABR
Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
Liečené epizódy krvácania: epizódy krvácania liečené FVIII.
Všetky epizódy krvácania: epizódy krvácania liečené a neliečené FVIII.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas 4 týždňov. ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; IQR = interkvartilové rozpätie, 25. percentil až 75. percentil; QW = profylaxia dávkou podávanou raz za týždeň; Q2W = profylaxia dávkou podávanou
raz za dva týždne; Q4W = profylaxia dávkou podávanou raz za štyri týždne.
C 1,5 mg/kg QW (n = 16); 3 mg/kg Q2W (n = 30); 6 mg/kg Q4W (n = 5).
|
Hodnotenie výsledkov týkajúcich sa zdravia
Klinické štúdie HAVEN hodnotili kvalitu života súvisiacu so zdravím (health-related quality of life,
HRQoL) a zdravotný stav pomocou nástrojov na hodnotenie klinických výsledkov. V štúdiách
HAVEN 1 a HAVEN 2 sa použil špecifický dotazník kvality života pri hemofílii
(
Haemophilia-specific Quality of Life, Haem-A-QoL) určený pre dospelých (≥ 18-ročných) a jeho verzia určená pre dospievajúcich (Haemo-QoL-SF, pre 8-ročných až < 18-ročných), v uvedenom poradí, pričom cieľovými ukazovateľmi definovanými protokolom boli skóre fyzického zdravia
(t. j. bolestivé opuchy, prítomnosť bolesti kĺbov, bolesť pri pohybe, ťažkosti s dlhšou chôdzou
a potreba dlhšieho času na prípravu) a celkové skóre (súhrn všetkých skóre). V štúdii HAVEN 2 sa okrem toho použil upravený špecifický dotazník kvality života pri hemofílii určený pre pacientov
s inhibítorom (dotazník Adapted InhibQoL), ktorý zahŕňa aspekty záťaže opatrovateľa, s cieľom získať opatrovateľmi hlásenú HRQoL u pediatrických pacientov vo veku < 12 rokov. Štúdia HAVEN 6 hodnotila HRQoL u dospelých a pediatrických pacientov, ako aj u opatrovateľov
pediatrických pacientov pomocou dotazníka CATCH (
Comprehensive Assessment Tool of Challenges in Haemophilia, t. j. komplexný nástroj na hodnotenie výziev pri hemofílii). Preskúmané boli domény vnímania rizika a vplyvu hemofílie na každodenné aktivity, sociálne aktivity, reakreačné aktivity
a na prácu/školu, ako aj miera zaoberania sa („preoccupation“) hemofíliou a záťaž spôsobená liečbou.
Zmena zdravotného stavu sa hodnotila na základe preskúmania skóre indexu užitočnosti (index utility score, IUS) a hodnôt vizuálnej analógovej škály (visual analog scale, VAS), ktoré sú súčasťou dotazníka
EuroQoL Five-Dimension-Five Levels Questionnaire (EQ-5D-5L, t. j. dotazník hodnotiaci zdravotný stav pacienta v piatich dimenziách a na piatich úrovniach vnímania závažnosti zdravotných problémov, ktorý vypracovala skupina EuroQoL).
Výsledky týkajúce sa zdravia zo štúdie HAVEN 1
V tejto štúdii východiskové celkové skóre (priemer = 41,14 a 44,58 v uvedenom poradí) a skóre škály hodnotiacej fyzické zdravie (priemer = 52,41 a 57,19 v uvedenom poradí) boli medzi skupinou
s profylaxiou Hemlibrou a skupinou bez profylaxie podobné. Tabuľka 10 poskytuje zhrnutie porovnania celkového skóre Haem-A-QoL a skóre škály hodnotiacej fyzické zdravie po 24 týždňoch liečby, po úprave vzhľadom na východiskové hodnoty, medzi skupinou s profylaxiou Hemlibrou (skupina A) a skupinou bez profylaxie (skupina B). Pri hodnotení v 25. týždni sa pri profylaxii Hemlibrou podávanou raz za týždeň v porovnaní s nepodávaním profylaxie preukázalo štatisticky signifikantné a klinicky významné zlepšenie vopred špecifikovaných cieľových ukazovateľov, ktorými bolo Haem-A-QoL skóre škály hodnotiacej fyzické zdravie.
Tabuľka 10 Štúdia HAVEN 1: Zmena v Haem-A-QoL fyzické zdravie a celkové skóre pri profylaxii Hemlibrou v porovnaní so žiadnou profylaxiou u pacientov vo veku ≥ 18 rokov s inhibítorom FVIII
Skóre Haem-A-QoL v 25. týždni
|
Skupina B: žiadna profylaxia
(N = 14)
|
'
Skupina A: Hemlibra
1,5 mg/kg raz za týždeň
(N = 25)
|
Skóre fyzického zdravia (rozmedzie 0 až 100)
|
Upravený priemer
| 54,17
| 32,61
|
Rozdiel v upravených priemeroch (95% IS)
| 21,55 (7,89; 35,22)
|
p-hodnota
| 0,0029
|
Celkové skóre (rozmedzie 0 až 100)
|
Upravený priemer
| 43,21
| 29,2
|
Rozdiel v upravených priemeroch (95% IS)
| 14,01 (5,56; 22,45)
|
Skupina B: zahŕňa iba obdobie bez podávania profylaxie.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou. Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas 4 týždňov. Skóre Haem-A-QoL sa pohybuje v rozmedzí od 0 do 100; nižšie skóre vyjadruje lepšiu HRQoL.
Klinicky významný rozdiel: celkové skóre: 7 bodov; fyzické zdravie: 10 bodov.
Analýzy sú založené na údajoch od jednotlivcov, ktorí poskytli odpovede v hodnoteniach na začiatku aj v 25. týždni.
|
| | | | |
Výsledky týkajúce sa zdravotného stavu zo štúdie HAVEN 1Tabuľka 11 poskytuje zhrnutie porovnania skóre IUS a hodnôt VAS dotazníka EQ-5D-5L
po 24 týždňoch liečby, po úprave vzhľadom na východiskové hodnoty, medzi skupinou s profylaxiou
Hemlibrou (skupina A) a skupinou bez profylaxie (skupina B).
Tabuľka 11 Štúdia HAVEN 1: Skóre EQ-5D-5L u pacientov vo veku ≥ 12 rokov v 25. týždni
Skóre EQ-5D-5L v 25. týždni
|
Skupina B: žiadna profylaxia
(N = 16)
|
Skupina A: Hemlibra
1
,5 mg/kg raz za týždeň
(N = 29)
|
Vizuálna analógová škála
|
Upravený priemer
|
74,36
|
84,08
|
Rozdiel v upravených priemeroch (95% IS)
|
-9,72 (-17,62; -1,82)
|
Skóre indexu užitočnosti (Index Utility Score)
|
Upravený priemer
|
0,65
|
0,81
|
Rozdiel v upravených priemeroch (95% IS)
|
-0,16 (-0,25; -0,07)
|
Skupina B: zahŕňa iba obdobie bez podávania profylaxie.
V prípade pacientov, ktorých dávka bola titrovaná smerom nahor, sú zahrnuté iba údaje, ktoré sa získali pred titráciou.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas
4 týždňov.
Vyššie skóre vyjadruje lepšiu kvalitu života.
Klinicky významný rozdiel: VAS: 7 bodov; skóre indexu užitočnosti: 0,07 bodu.
Analýzy sú založené na údajoch od jednotlivcov, ktorí poskytli odpovede v hodnoteniach na začiatku aj v
25. týždni.
|
|
|
|
|
|
|
Výsledky týkajúce sa zdravia zo štúdie HAVEN 6
V štúdii HAVEN 6 sa HRQoL u pacientov všetkých vekových kategórií so stredne ťažkou hemofíliou A hodnotila v 25. týždni na základe dotazníka CATCH. Dotazník CATCH (verzia 1.0) je validný nástroj, ktorý hodnotí vplyv hemofílie a jej liečby. Existujú rôzne verzie dotazníka
pre dospelých pacientov, pediatrických pacientov a opatrovateľov pediatrických pacientov. Kvalita života súvisiaca so zdravím zostala počas profylaxie Hemlibrou vo všeobecnosti stabilná a v doméne dotazníka CATCH zameranej na záťaž spôsobenú liečbou sa pozorovalo zlepšenie naprieč celými skupinami respondentov.
Pediatrická populáciaPediatrickí pacienti (vo veku < 12 rokov alebo 12 až 17 rokov a s telesnou hmotnosťou < 40 kg)s hemofíliou A a inhibítorom FVIII (štúdia BH29992 - HAVEN 2)Profylaxia Hemlibrou podávanou raz za týždeň sa hodnotila v multicentrickej, otvorenej klinickej štúdii s jednou skupinou u pediatrických pacientov (vo veku < 12 rokov alebo 12 až 17 rokov
a s telesnou hmotnosťou < 40 kg) s hemofíliou A a inhibítorom FVIII. Pacienti dostávali profylaxiu Hemlibrou v dávke 3 mg/kg podávanej raz za týždeň počas prvých 4 týždňov, po ktorých nasledovala dávka 1,5 mg/kg podávaná raz za týždeň.
Štúdia hodnotila farmakokinetiku (FK), bezpečnosť a účinnosť vrátane účinnosti profylaxie Hemlibrou podávanou raz za týždeň v porovnaní s predchádzajúcou epizodickou a profylaktickou liečbou „bypassovými“ prípravkami u pacientov, ktorí sa pred zaradením do štúdie zúčastnili na NIS (intraindividuálne porovnanie).
Výsledky účinnosti
HAVEN 2 (predbežná analýza)
V čase predbežnej analýzy bola účinnosť vyhodnotená u 59 pacientov, ktorí boli vo veku < 12 rokov a dostávali profylaxiu Hemlibrou podávanou raz za týždeň počas aspoň 12 týždňov, vrátane štyroch pacientov vo veku < 2 roky, 17 pacientov vo veku 2 až < 6 rokov, 38 pacientov vo veku
6 až < 12 rokov. Vypočítaný bol ročný výskyt epizód krvácania a percento pacientov s nulovým
počtom epizód krvácania (pozri tabuľku 12). U týchto pacientov bol medián obdobia pozorovania
29,6 týždňa (rozmedzie: 18,4 až 63,0 týždňov).
Tabuľka 12 Štúdia HAVEN 2: Prehľad účinnosti (predbežná analýza)Cieľový ukazovateľ
| aABR (95% IS)
bN = 59
| Medián ABR (IQR)
bN = 59
| % pacientov s nulovým počtom epizód krvácania (95% IS)
bN = 59
|
Liečené epizódy
krvácania
| 0,3 (0,1; 0,5)
| 0 (0; 0)
| 86,4 (75; 94)
|
Všetky epizódy krvácania
| 3,8 (2,2; 6,5)
| 0 (0; 3,4)
| 55,9 (42,4; 68,8)
|
Liečené epizódy spontánneho krvácania
| 0 (0; 0,2)
| 0 (0; 0)
| 98,3 (90,9; 100)
|
Liečené epizódy
krvácania do kĺbov
| 0,2 (0,1; 0,4)
| 0 (0; 0)
| 89,8 (79,2; 96,2)
|
Liečené epizódy krvácania do cieľového kĺbu
| 0,1 (0; 0,7)
| 0 (0; 0)
| 96,6 (88,3; 99,6)
|
ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; IQR = interkvartilové rozpätie,
25. percentil až 75. percentil.
a Vypočítané pomocou modelu negatívne binomickej regresie (NBR).
b Údaje o účinnosti od liečených pacientov vo veku < 12 rokov, ktorí boli v štúdii HAVEN 2 liečení aspoň
12 týždňov (N = 59), pretože štúdia sa zamerala hlavne na preskúmanie efektu liečby na základe veku.
c Vypočítaný ABR
Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
Liečené epizódy krvácania: epizódy krvácania liečené „bypassovými“ prípravkami.
Všetky epizódy krvácania: epizódy krvácania liečené a neliečené „bypassovými“ prípravkami.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas
4 týždňov.
|
V intraindividuálnej analýze sa zistilo, že profylaxia Hemlibrou podávanou raz za týždeň viedla
ku klinicky významnému zníženiu (98%) výskytu liečených epizód krvácania u 18 pediatrických pacientov, ktorí dostávali profylaxiu Hemlibrou aspoň 12 týždňov, v porovnaní s údajmi o výskyte epizód krvácania zozbieranými v NIS pred zaradením do štúdie (tabuľka 13).
Tabuľka 13 Štúdia HAVEN 2: Intraindividuálne porovnanie ročného výskytu epizód krvácania (liečené epizódy krvácania) pri profylaxii Hemlibrou v porovnaní s predchádzajúcou profylaxiou „bypassovým“ prípravkom
Cieľový ukazovateľ
|
Predchádzajúca liečba
„
bypassovým“ prípravkom* (N = 18)
|
Profylaxia Hemlibrou
(N = 18)
|
Liečené epizódy krvácania
|
ABR (95% IS)
|
19,8 (15,3; 25 7)
|
0,4 (0,15; 0,88)
|
% zníženie (RR)
|
98%
(0,02)
|
% pacientov s nulovým počtom
epizód krvácania (95% IS)
|
5,6 (0,1; 27,3)
|
77,8 (52,4; 93,6)
|
Medián ABR (IQR)
|
16,2 (11,49; 25,78)
|
0 (0; 0)
|
* Predchádzajúca profylaktická liečba u 15 z 18 pacientov; predchádzajúca epizodická („on demand“) liečba
u 3 osôb.
Pomer výskytu (rate ratio) a interval spoľahlivosti (IS) vychádzajú z modelu negatívne binomickej regresie (NBR) a p-hodnota zo stratifikovaného Waldovho testu, pri porovnávaní ABR medzi špecifikovanými skupinami.
Intraindividuálne porovnávacie údaje z NIS.
Zahrnutí sú iba pacienti, ktorí sa zúčastnili na NIS a na štúdii HAVEN 2. Definície epizód krvácania sú prevzaté na základe kritérií ISTH.
Liečené epizódy krvácania: epizódy krvácania liečené „bypassovými“ prípravkami.
Pacienti vystavení emicizumabu začali liečbu nasycovacou dávkou 3 mg/kg/týždeň podávanou počas
4 týždňov.
ABR = ročný výskyt epizód krvácania; IS = interval spoľahlivosti; RR = pomer výskytu; IQR = interkvartilové rozpätie, 25. percentil až 75. percentil.
Hoci sa pri profylaxii emicizumabom pozorovala vyššia adhézia ako pri predchádzajúcej profylaxii bypassovými prípravkami (BPA), nebol identifikovaný žiadny rozdiel v ABR u pacientov so zhodnými dávkami ≥ 80% alebo < 80% profylaxie BPA podľa štandardných požiadaviek (údaje sa majú interpretovať s opatrnosťou kvôli malej veľkosti vzorky).
Kvôli krátkému polčasu bypassových prípravkov sa po ukončení liečby nepredpokladá žiadny zostatkový účinok.
Iba prvých päť dávok emicizumabu sa muselo podať pod dozorom, aby sa zaistila zbehlosť v oblasti
bezpečnosti a techniky vpichu injekcie. Podobne ako pri profylaxii BPA bolo povolené podávanie v domácom prostredí pre všetky nasledujúce dávky emicizumabu.
|
Hodnotenie výsledkov týkajúcich sa zdravia u pediatrických pacientov
Výsledky týkajúce sa zdravia zo štúdie HAVEN 2
V štúdii HAVEN 2 sa v 25. týždni hodnotila HRQoL u pacientov vo veku ≥ 8 rokov až < 12 rokov
na základe dotazníka Haemo-QoL-SF určeného pre deti (pozri tabuľku 14). Dotazník Haemo-QoL-SF je validný a spoľahlivý nástroj na hodnotenie HRQoL. HRQoL u pacientov vo veku < 12 rokov sa hodnotila v 25. týždni aj na základe upraveného špecifického dotazníka kvality života pri hemofílii určeného pre pacientov s inhibítorom (dotazník Adapted InhibQoL), ktorý zahŕňa aspekty záťaže opatrovateľa, vyplneného opatrovateľmi (pozri tabuľku 14). Dotazník Adapted InhibQoL je validný
a spoľahlivý nástroj na hodnotenie HRQoL.
Tabuľka 14 Štúdia HAVEN 2: Zmena skóre fyzického zdravia v 25. týždni v porovnaní s východiskovým skóre u pacientov (vo veku < 12 rokov) po profylaktickej liečbe Hemlibrou, hlásená pacientmi a opatrovateľmi
|
Haemo-QoL-SF
|
Skóre fyzického zdravia (rozmedzie 0 až 100)
a
|
Priemerné východiskové skóre (95% IS) (n = 18)
|
29,5 (16,4 - 42,7)
|
Priemerná zmena v porovnaní s východiskovým skóre
(95% IS) (n = 15)
|
-21,7 (-37,1 - -6,3)
|
|
|
Dotazník Adapted InhibQoL zahŕňajúci aspekty záťaže opatrovateľa
|
Skóre fyzického zdravia (rozmedzie 0 až 100)
a
|
Priemerné východiskové skóre (95% IS) (n = 54)
|
37,2 (31,5 - 42,8)
|
Priemerná zmena v porovnaní s východiskovým skóre
(95% IS) (n = 43)
|
-32,4 (-38,6 - -26,2)
|
|
a Nižšie skóre (negatívna zmena skóre) vyjadrujú lepšie fungovanie.
Analýzy sú založené na údajoch od jednotlivcov, ktorí poskytli odpovede v hodnoteniach na začiatku
aj v 25. týždni.
|
K dispozícii sú obmedzené skúsenosti s použitím „bypassového“ prípravku alebo FVIII počas operácií
a procedúr. O použití „bypassového“ prípravku alebo FVIII počas operácií a procedúr rozhodol skúšajúci lekár.
V prípade, že sa u pacientov dostávajúcich profylaxiu emicizumabom vyskytne akútne krvácanie, majú byť liečení dostupnými terapiami. Odporúčania na používanie „bypassových“ prípravkov, pozri časť 4.4.
ImunogenicitaTak ako pri všetkých terapeutických proteínoch, u pacientov liečených emicizumabom existuje
možnosť vzniku imunitnej reakcie. V súhrnne hodnotených klinických štúdií bolo celkovo
739 pacientov testovaných na prítomnosť protilátok proti emicizumabu. U 36 pacientov (4,9%) sa zistila pozitivita protilátok proti emicizumabu. U 19 pacientov (2,6%) boli protilátky proti emicizumabu neutralizačné v podmienkach
in vitro. U 15 z týchto 19 pacientov nemali neutralizačné protilátky proti emicizumabu klinicky významný vplyv na farmakokinetiku alebo účinnosť Hemlibry, kým u štyroch pacientov (0,5%) bola pozorovaná znížená plazmatická koncentrácia emicizumabu.
U jedného pacienta (0,1%) s neutralizačnými protilátkami proti emicizumabu a zníženou plazmatickou koncentráciou emicizumabu došlo k strate účinnosti po piatich týždňoch liečby a liečba Hemlibrou
bola u neho ukončená. Celkovo bol bezpečnostný profil Hemlibry podobný medzi pacientmi
s protilátkami proti emicizumabu (vrátane neutralizačných protilátok) a pacientmi bez nich (pozri
časti 4.4 a 4.8).
Populácia starších pacientovPoužitie Hemlibry u pacientov vo veku 65 rokov a starších s hemofíliou A podporujú štúdie
HAVEN 1, HAVEN 3, HAVEN 4 a HAVEN 6. Na základe obmedzených údajov nie sú dôkazy
svedčiace o rozdiele v účinnosti alebo bezpečnosti u pacientov vo veku 65 rokov alebo starších.
5
.2 Farmakokinetické vlastnosti
Farmakokinetika emicizumabu sa stanovila pomocou nonkompartmentovej analýzy u zdravých osôb a pomocou populačnej farmakokinetickej analýzy údajov z databázy zahŕňajúcej 389 pacientov
s hemofíliou A.
AbsorpciaPo subkutánnom podaní pacientom s hemofíliou A bol počas absorpcie 1,6 dňa.
U pacientov s hemofíliou A sa po opakovanom subkutánnom podávaní 3 mg/kg raz za týždeň počas prvých 4 týždňov dosiahla priemerná (± SD) minimálna (trough, t. j. nameraná na konci dávkovacieho intervalu tesne pred podaním ďalšej dávky) plazmatická koncentrácia emicizumabu 52,6 ± 13,6 µg/ml v 5. týždni.
Predpokladané priemerné hodnoty (± SD) Ctrough a Cmax a pomery Cmax/Ctrough v rovnovážnom stave pri odporúčaných udržiavacích dávkach 1,5 mg/kg raz za týždeň, 3 mg/kg raz za dva týždne alebo
6 mg/kg raz za štyri týždne sú uvedené v tabuľke 15.
Tabuľka 15 Priemerné (± SD) koncentrácie emicizumabu v rovnovážnom stave
| Udržiavacia dávka
|
Parametre
| 1,5 mg/kg raz za týždeň
| 3 mg/kg raz za dva týždne
| 6 mg/kg raz za štyri týždne
|
Cmax, ss (µg/ml)
| 54,9 ± 15,9
| 58,1 ± 16,5
| 66,8 ± 17,7
|
Cavg, ss (µg/ml)
| 53,5 ± 15,7
| 53,5 ± 15,7
| 53,5 ± 15,7
|
Ctrough, ss (µg/ml)
| 51,1 ± 15,3
| 46,7 ± 16,9
| 38,3 ± 14,3
|
Pomer Cmax/Ctrough
| 1,08 ± 0,03
| 1,26 ± 0,12
| 1,85 ± 0,46
|
Cavg, ss = priemerná koncentrácia v rovnovážnom stave; Cmax, ss = maximálna plazmatická koncentrácia v rovnovážnom stave ; Ctrough, ss = minimálna koncentrácia v rovnovážnom stave; QW = raz za týždeň; Q2W = raz za dva týždne; Q4W = raz za štyri týždne. Farmakokinetické parametre odvodené
z populačného FK modelu.
|
Po podávaní dávky raz za týždeň (3 mg/kg/týždeň počas 4 týždňov, po ktorých nasledovalo podávanie
1,5 mg/kg/týždeň), boli u dospelých/dospievajúcich (vo veku ≥ 12 rokov) a u detí (vo veku
< 12 rokov) pozorované podobné FK profily (pozri graf 1).
Graf 1: Priemerná (± 95% IS) plazmatická koncentrácia emicizumabu oproti časovým profilom u pacientov vo veku ≥ 12 rokov (štúdie HAVEN 1 a HAVEN 3) v porovnaní s pacientmi vo veku < 12 rokov (štúdia HAVEN 2)
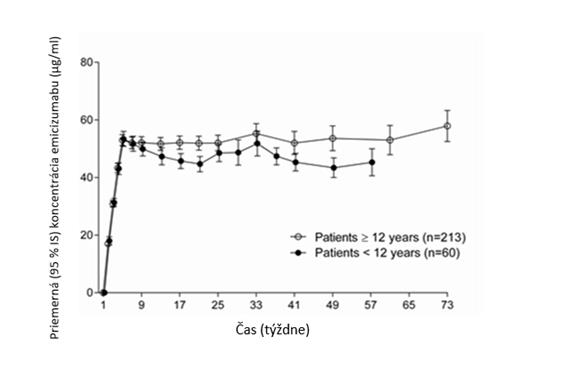
U zdravých osôb bola absolútna biologická dostupnosť po subkutánnom podaní 1 mg/kg
medzi 80,4% a 93,1% v závislosti od miesta podania injekcie. Podobné farmakokinetické profily sa pozorovali po subkutánnom podaní do brucha, hornej časti ramena a do stehna. Emicizumab sa môže podávať striedavo do týchto anatomických miest (pozri časť 4.2).
DistribúciaPo jednorazovej intravenóznej dávke 0,25 mg/kg emicizumabu podanej zdravým osobám bol
distribučný objem v rovnovážnom stave 106 ml/kg (t. j. 7,4 l u 70 kg dospelého).
Po opakovanom subkutánnom podávaní dávok emicizumabu pacientom s hemofíliou A bol zdanlivý
distribučný objem (V/F), odhadnutý v populačnej FK analýze, 10,4 l.
BiotransformáciaMetabolizmus emicizumabu sa neskúmal. IgG protilátky sú primárne katabolizované proteolýzou
v lyzozómoch a potom sú vylučované z organizmu alebo sú v organizme znovu použité.
ElimináciaPo intravenóznom podaní 0,25 mg/kg zdravým osobám bol celkový klírens emicizumabu
3,26 ml/kg/deň (t. j. 0,228 l/deň u 70 kg dospelého) a priemerný koncový polčas bol 26,7 dňa.
Po jednorazovej subkutánnej injekcii podanej zdravým osobám bol eliminačný polčas približne
4 až 5 týždňov.
Po opakovanom podávaní subkutánnych injekcií pacientom s hemofíliou A bol zdanlivý klírens
0,272 l/deň a zdanlivý eliminačný polčas bol 26,8 dňa.
Dávková linearita
U pacientov s hemofíliou A po prvej dávke Hemlibry vykazoval emicizumab dávkovo úmernú
farmakokinetiku v rozmedzí dávky od 0,3 do 6 mg/kg. Expozícia (Cavg, ss) vo viacerých dávkach je porovnateľná medzi dávkou 1,5 mg/kg každý týždeň, 3 mg/kg každé 2 týždne a 6 mg/kg každé 4 týždne.
Osobitné skupiny pacientov
Pediatrická populácia
Vplyv veku na farmakokinetiku emicizumabu sa hodnotil v populačnej farmakokinetickej analýze, ktorá zahŕňala 5 dojčiat (vo veku ≥ 1 mesiac až < 2 roky), 55 detí (mladších ako 12 rokov)
a 50 dospievajúcich (vo veku od 12 do < 18 rokov) s hemofíliou A.
Vek neovplyvnil farmakokinetiku emicizumabu u pediatrických pacientov.
Staršie osoby
Vplyv veku na farmakokinetiku emicizumabu sa hodnotil v populačnej farmakokinetickej analýze, ktorá zahŕňala trinásť osôb vo veku 65 rokov a starších (žiadna osoba nebola staršia ako 77 rokov). Relatívna biologická dostupnosť sa znižovala so starším vekom, ale nepozorovali sa žiadne klinicky významné rozdiely vo farmakokinetike emicizumabu medzi osobami < 65 rokov
a osobami ≥ 65 rokov.
Rasa
Populačné farmakokinetické analýzy u pacientov s hemofíliou A ukázali, že rasa neovplyvňuje farmakokinetiku emicizumabu. Nie je potrebná žiadna úprava dávky v súvislosti s týmto demografickým faktorom.
Pohlavie
Údaje u pacientov ženského pohlavia sú príliš obmezené na vyvodenie záveru.
Porucha funkcie obličiek
Neuskutočnili sa žiadne štúdie zamerané na zistenie vplyvu poruchy funkcie obličiek
na farmakokinetiku emicizumabu.
Väčšina pacientov s hemofíliou A, ktorí boli zahrnutí do populačnej farmakokinetickej analýzy, mala normálnu funkciu obličiek (N = 332; klírens kreatinínu [CLcr] ≥ 90 ml/min) alebo miernu poruchu funkcie obličiek (N = 27; CLcr 60 - 89 ml/min). Mierna porucha funkcie obličiek neovplyvnila farmakokinetiku emicizumabu. K dispozícii sú obmedzené údaje o použití Hemlibry u pacientov
so stredne závažnou poruchou funkcie obličiek (iba 2 pacienti s CLcr 30 - 59 ml/min) a žiadne údaje
u pacientov so závažnou poruchou funkcie obličiek. Vplyv stredne ťažkej a závažnej poruchy funkcie
obličiek na farmakokinetiku emicizumabu nemožno vyvodiť.
Emicizumab je monoklonálna protilátka, ktorá podlieha katabolizmu a nie renálnej exkrécii, a preto sa nepredpokladá, že u pacientov s poruchou funkcie obličiek je potrebná zmena dávky.
Porucha funkcie pečene
Neuskutočnili sa žiadne štúdie zamerané na zistenie vplyvu poruchy funkcie pečene
na farmakokinetiku emicizumabu. Väčšina pacientov s hemofíliou A, ktorí boli zahrnutí do populačnej farmakokinetickej analýzy, mala normálnu funkciu pečene (hodnoty bilirubínu a AST ≤ horná hranica referenčného rozpätia (upper limit of normal, ULN), N = 300) alebo miernu poruchu funkcie pečene (hodnota bilirubínu ≤ ULN a hodnota AST > ULN alebo hodnota bilirubínu od 1- do 1,5-násobku
ULN a akákoľvek hodnota AST, N = 51). Iba 6 pacientov malo stredne závažnú poruchu funkcie pečene (hodnota bilirubínu > 1,5-násobok ULN a ≤ 3-násobok ULN a akákoľvek hodnota AST). Mierna porucha funkcie pečene neovplyvnila farmakokinetiku emicizumabu (pozri časť 4.2). Bezpečnosť a účinnosť emicizumabu neboli špecificky skúmané u pacientov s poruchou funkcie pečene. V klinických štúdiách boli zahrnutí pacienti s miernou a stredne závažnou poruchou funkcie pečene. K dispozícii nie sú žiadne údaje o použití Hemlibry u pacientov so závažnou poruchou funkcie pečene.
Emicizumab je monoklonálna protilátka, ktorá podlieha katabolizmu a nie hepatálnemu metabolizmu, a preto sa nepredpokladá, že u pacientov s poruchou funkcie pečene je potrebná zmena dávky.
Iné osobitné skupiny pacientov
Modelovanie ukazuje, že menej časté dávkovanie u pacientov s hypoalbuminémiou a nízkou telesnou hmotnosťou v dôsledku veku, vedie k nižším expozíciám emicizumabu; simulácie naznačujú, že títo pacienti budú stále profitovať z klinicky významnej kontroly krvácania. V klinických štúdiách neboli zaradení žiadni pacienti s takýmito charakteristikami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe štúdií akútnej toxicity a toxicity po opakovanom podávaní, ktoré zahŕňali cieľové ukazovale farmakologickej bezpečnosti a cieľové ukazovatele reprodukčnej toxicity, neodhalili žiadne osobitné riziko pre ľudí.
Fertilita
Emicizumab nespôsobil žiadne zmeny v reprodukčných orgánoch samcov a samíc opíc rodu
Cynomolgus, keď sa podával až do najvyššej testovanej dávky 30 mg/kg/týždeň (zodpovedajúca
11-násobku expozície dosiahnutej u ľudí po podávaní najvyššej dávky 3 mg/kg/týždeň, na základe
AUC).
Teratogenita
K dispozícii nie sú žiadne údaje týkajúce sa potenciálnych nežiaducich účinkov emicizumabu
na embryo-fetálny vývin.
Reakcie v mieste podania injekcie
U zvierat sa po subkutánnej injekcii zaznamenali reverzibilné krvácanie, perivaskulárna infiltrácia
mononukleárnymi bunkami, degenerácia/nekróza podkožia a opuch endotelu v podkoží.
6
. FARMACEUTICKÉ INFORMÁCIE
6
.1 Zoznam pomocných látok
L-arginín
L-histidín
Kyselina L-asparágová
Poloxamér 188
Voda na injekcie
6.2 Inkompatibility
Nepozorovali sa žiadne inkompatibility medzi Hemlibrou a polypropylénovými alebo polykarbonátovými injekčnými striekačkami a ihlami z nehrdzavejúcej ocele.
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
Hemlibra 30 mg/mlinjekčnýroztok
2 roky.
Hemlibra 150 mg/ml injekčný roztok
2 roky.
Po vybratí z chladničky sa neotvorené injekčné liekovky môžu uchovávať pri izbovej teplote
(do 30 °C) najviac 7 dní.
Po uchovávaní pri izbovej teplote sa neotvorené injekčné liekovky môžu vrátiť späť do chladničky.
Ak sa uchovávajú mimo chladničky a potom sa do nej vrátia, celková súhrnná doba uchovávania
mimo chladničky nesmie byť dlhšia ako 7 dní. Injekčné liekovky sa nikdy nesmú vystavovať teplotám nad 30 °C. Injekčné liekovky, ktoré boli uchovávané pri izbovej teplote viac ako 7 dní alebo ktoré boli vystavené teplotám nad 30 ºC, sa musia zlikvidovať.
Prepichnutá injekčná liekovka a naplnená injekčná striekačka
Z mikrobiologického hľadiska sa má liek prenesený z injekčnej liekovky do injekčnej striekačky
použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky jeho uchovávania je zodpovedný
používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuľke na ochranu pre svetlom.
Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6
.5 Druh obalu a obsah balenia
Hemlibra 30 mg/ml injekčný roztok
Injekčná liekovka s objemom 3 ml vyrobená z číreho skla typu I, s butylkaučukovou zátkou
potiahnutou fluoro-živicovou vrstvou a so zalisovanou hliníkovou obrubou s plastovým odlamovacím viečkom. Každá injekčná liekovka obsahuje 30 mg emicizumabu v 1 ml injekčného roztoku. Každá škatuľka obsahuje jednu injekčnú liekovku.
Hemlibra 150 mg/ml injekčný roztok
Injekčná liekovka s objemom 3 ml vyrobená z číreho skla typu I, s butylkaučukovou zátkou
potiahnutou fluoro-živicovou vrstvou a so zalisovanou hliníkovou obrubou s plastovým odlamovacím viečkom. Každá injekčná liekovka obsahuje 60 mg emicizumabu v 0,4 ml injekčného roztoku. Každá škatuľka obsahuje jednu injekčnú liekovku.
Injekčná liekovka s objemom 3 ml vyrobená z číreho skla typu I, s butylkaučukovou zátkou potiahnutou fluoro-živicovou vrstvou a so zalisovanou hliníkovou obrubou s plastovým odlamovacím viečkom. Každá injekčná liekovka obsahuje 105 mg emicizumabu v 0,7 ml injekčného roztoku. Každá škatuľka obsahuje jednu injekčnú liekovku.
Injekčná liekovka s objemom 3 ml vyrobená z číreho skla typu I, s butylkaučukovou zátkou potiahnutou fluoro-živicovou vrstvou a so zalisovanou hliníkovou obrubou s plastovým odlamovacím viečkom. Každá injekčná liekovka obsahuje 150 mg emicizumabu v 1 ml injekčného roztoku. Každá škatuľka obsahuje jednu injekčnú liekovku.
Injekčná liekovka s objemom 3 ml vyrobená z číreho skla typu I, s butylkaučukovou zátkou potiahnutou fluoro-živicovou vrstvou a so zalisovanou hliníkovou obrubou s plastovým odlamovacím viečkom. Každá injekčná liekovka obsahuje 300 mg emicizumabu v 2 ml injekčného roztoku. Každá škatuľka obsahuje jednu injekčnú liekovku.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Roztok Hemlibry je sterilný roztok bez obsahu konzervačných látok, ktorý je pripravený na podanie subkutánnou injekciou a ktorý nie je potrebné riediť.
Hemlibra sa má pred podaním zrakom skontrolovať na prítomnosť tuhých častíc alebo zmenu farby. Hemlibra je bezfarebný až svetložltý roztok. Roztok sa má zlikvidivať, ak sú v ňom viditeľné cudzorodé častice alebo ak má zmenenú farbu.
Injekčnou liekovkou netraste.
Injekčné liekovky s injekčným roztokom Hemlibry sú určené iba na jednorazové použitie.
Injekčná striekačka, prenosová ihla a injekčná ihla sú potrebné na odobratie roztoku Hemlibry
z injekčnej liekovky a na jeho podanie subkutánnou injekciou.
Odporúčané charakteristiky uvedených pomôcok, pozri, prosím, nižšie:
Injekčná striekačka s objemom 1 ml sa má použiť na podanie injekčného roztoku Hemlibry s objemom do 1 ml, zatiaľ čo injekčná striekačka s objemom 2 až 3 ml sa má použiť na podanie injekčného
roztoku s objemom väčším ako 1 ml a neprevyšujúcim 2 ml.
Pokyny na zaobchádzanie pri kombinovaní obsahu injekčných liekoviek v injekčnej striekačke, pozri
„Návod na použitie“ Hemlibry. Na dosiahnutie predpísanej dávky v jednej injekčnej striekačke sa
nesmie kombinovať obsah injekčných liekoviek s rôznymi koncentráciami Hemlibry (30 mg/ml a 150 mg/ml).
Injekčná striekačka s objemom 1 ml
Kritériá: Priehľadná polypropylénová alebo polykarbonátová injekčná striekačka s hrotom so závitom
typu Luer-lock, so stupnicou delenou po 0,01 ml.
Injekčná striekačka s objemom 2 až 3 ml
Kritériá: Priehľadná polypropylénová alebo polykarbonátová injekčná striekačka s hrotom so závitom
typu Luer-lock, so stupnicou delenou po 0,1 ml.
Prenosová ihla s filtrom
Kritéria pre prenosovú ihlu s filtrom: Ihla z nehrdzavejúcej ocele s kónusom so závitom typu
Luer-lock, veľkosť 18 G, dĺžka 35 mm (1½″), obsahujúca 5 µm filter a najlepšie s tupo zrezaným hrotom.
Injekčná ihla
Kritériá: Ihla z nehrdzavejúcej ocele s kónusom so závitom typu Luer-lock, veľkosť 26 G
(akceptovateľné rozmedzie 25 - 27 G), dĺžka najlepšie 9 mm (3/8″) alebo maximálne 13 mm (½″), najlepšie s bezpečnostným mechanizmom ihly.
Ďalšie informácie týkajúce sa podania, pozri, prosím, časť 4.2 a písomnú informáciu pre používateľa
(časť 7 Návod na použitie).
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLA
EU/1/18/1271/001 (30 mg/1 ml) EU/1/18/1271/002 (60 mg/0,4 ml) EU/1/18/1271/003 (105 mg/0,7 ml) EU/1/18/1271/004 (150 mg/1 ml) EU/1/18/1271/005 (300 mg/2 ml)
9
. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 23. februára 2018
Dátum posledného predĺženia registrácie: 15. septembra 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.