
bezpečnostnéhoprofilu
Reumatoidnáartritída
Cimzia bola skúmaná u 4 049 pacientov s reumatoidnou artritídou v kontrolovaných a nezaslepených štúdiách po dobu až 92 mesiacov. Údaje v tabuľke 1 vychádzajú v prvom rade z placebom
kontrolovaných štúdií, ktoré zahŕňali 2 965 pacientov používajúcich Cimziu a 1 137 pacientov
užívajúcich placebo počas kontrolovaného obdobia.
V placebom kontrolovaných štúdiách mali pacienti používajúci Cimziu približne 4-krát dlhšie trvanie expozície v porovnaní s pacientmi v placebovej skupine. Tento rozdiel v expozícii je predovšetkým spôsobený tým, že u pacientov užívajúcich placebo bola vyššia pravdepodobnosť skorého prerušenia štúdie. Okrem toho, v štúdiách RA-I a RA-II bolo prerušenie štúdie povinné pre pacientov bez odpovede v 16. týždni, z ktorých väčšina užívala placebo.
Počas kontrolovaných štúdií bol podiel pacientov, ktorí prerušili liečbu z dôvodu nežiaducich udalostí,
4,4 % u pacientov liečených Cimziou a 2,7 % u pacientov liečených placebom.
Najčastejšie nežiaduce reakcie patrili do triedy orgánových systémov Infekcie a nákazy, hlásené u
14,4 % pacientov liečených Cimziou a 8,0 % pacientov liečených placebom, do triedy Celkové poruchy a reakcie v mieste podania, hlásené u 8,8 % pacientov liečených Cimziou a 7,4 % pacientov
liečených placebom a do triedy Poruchy kože a podkožného tkaniva, hlásené u 7,0 % pacientov
liečených Cimziou a u 2,4 % pacientov liečených placebom.
Axiálnaspondylartritída
Cimzia sa skúmala u 325 pacientov s aktívnou axiálnou spondylartritídou v AS001 klinickej štúdii počas až 4 rokov, ktorá zahŕňala 24 týždňov trvajúcu, placebom kontrolovanú fázu, po ktorej nasledovala 24 týdňov trvajúca zaslepená fáza a 156 týždňov trvajúca otvorená fáza. Bezpečnostný profil u pacientov s axiálnou spondylartritídou liečených Cimziou sa zhodoval s bezpečnostným profilom u reumatoidnej artritídy a s predchádzajúcimi skúsenosťami s Cimziou.
P
soriatická
artritída
Cimzia sa skúmala u 409 pacientov s psoriatickou artritídou v PsA001 klinickej štúdii počas až
4 rokov, ktorá zahŕňala 24 týždňov trvajúcu, placebom kontrolovanú fázu, po ktorej nasledovala 24
týždňov trvajúca zaslepená fáza a 168 týždňov trvajúca otvorená fáza. Bezpečnostný profil
u pacientov s psoriatickou artritídou liečených Cimziou bol zhodný s bezpečnostným profilom pri reumatoidnej artritíde a s predchádzajúcimi skúsenosťami s Cimziou.
Zoznam nežiaducich reakcií v tabuľke
Nežiaduce reakcie hlásené v klinických štúdiách s reumatoidnou artritídou a prípady po uvedení lieku
na trh s minimálne možnou súvislosťou s Cimziou sú vymenované v tabuľke 1 nižšie, podľa frekvencie a triedy orgánových systémov. Frekvencie kategórií sú definované nasledovne: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000), neznáme (nie je možné odhadnúť z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1 Nežiaduce reakcie v klinických štúdiách a po uvedení lieku na trh
Trieda orgánových systémov Frekvencia Nežiaduce reakcie
Infekcie a nákazy Časté bakteriálne infekcie (vrátane abscesu), vírusové infekcie (vrátane herpesu zoster, papilomavírusu, chrípky)
Menej časté sepsa (vrátane multiorgánového zlyhania, septického šoku), tuberkulóza (vrátane miliárneho, diseminovaného a extrapulmonálneho ochorenia), mykotické infekcie (vrátane oportúnnych)
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Poruchy krvi a lymfatického systému
Menej časté malignity krvi a lymfatického systému (vrátane lymfómu a leukémie), tumory solídnych orgánov, nemelanómové karcinómy kože, prekancerózne lézie (vrátane orálnej leukoplakie, névu z melanocytov), benígne tumory a cysty (vrátane papilómu kože)
Zriedkavé gastrointestinálne tumory, melanóm
Neznáme karcinóm z Merkelových buniek*
Časté eozinofilné poruchy, leukopénia (vrátane neutropénie, lymfopénie)
Menej časté anémia, lymfadenopatia, trombocytopénia, trombocytóza
Zriedkavé pancytopénia, splenomegália, erytrocytóza, abnormálna morfológia bielych krviniek
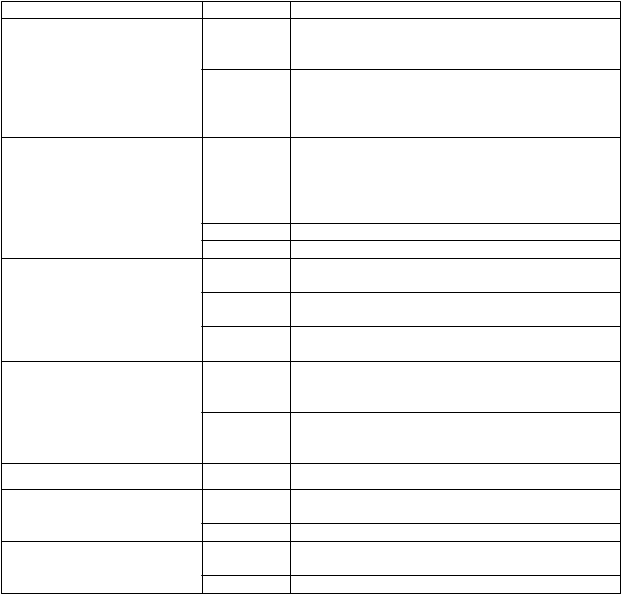
Poruchy imunitného systému Menej časté vaskulitídy, lupus erythematosus, precitlivenosť na lieky (vrátane anafylaktického šoku), alergické poruchy, pozitívne autoprotilátky
Zriedkavé angioneurotický edém, sarkoidóza, sérová choroba, panikulitída (vrátane nodózneho erytému), zhoršenie príznakov dermatomyozitídy**
Poruchy endokrinného systému Zriedkavé poruchy štítnej žľazy
Poruchy metabolizmu a výživy Menej časté nerovnováha elektrolytov, dyslipidémia, poruchy chuti do jedla, zmeny telesnej hmotnosti
Zriedkavé hemosideróza
Psychické poruchy Menej časté úzkosť a poruchy nálady (vrátane súvisiacich symptómov)
Zriedkavé pokus o samovraždu, delírium, duševná porucha
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Poruchy nervového systému Časté bolesti hlavy (vrátane migrény), zmyslové anomálie
Menej časté periférne neuropatie, závrat, tremor
Zriedkavé záchvat, zápal kraniálneho nervu, porucha koordinácie alebo rovnováhy
Neznáme skleróza multiplex*, Guillainov-Barrého syndróm* Poruchy oka Menej časté porucha zraku (vrátane zhoršeného videnia), zápal
oka a očného viečka, porucha slzenia
Poruchy ucha a labyrintu Menej časté tinnitus, vertigo
Poruchy srdca a srdcovej činnosti
Menej časté kardiomyopatie (vrátane srdcového zlyhania), ischemické koronárne arteriálne poruchy, arytmie (vrátane atriálnej fibrilácie), palpitácie
Zriedkavé perikarditída, atrioventrikulárna blokáda
Poruchy ciev Časté hypertenzia
Menej časté hemorágia alebo krvácanie (na ktoromkoľvek mieste), hyperkoagulácia (vrátane tromboflebitídy, pľúcnej embólie), synkopa, edém (vrátane periférneho, faciálneho), ekchymózy (vrátane hematómu, petechií)
Zriedkavé cerebrovaskulárna príhoda, arterioskleróza, Raynaudov fenomén, livedo reticularis, telangiektázia
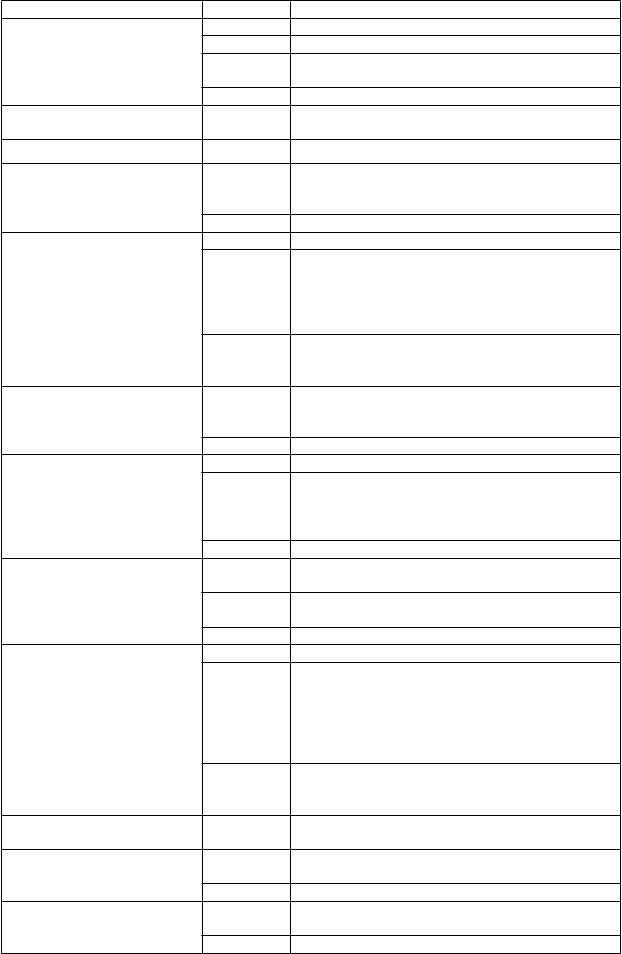
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Poruchy obličiek a močových ciest
Poruchy reprodukčného systému a prsníkov
Menej časté astma a s ňou súvisiace symptómy, pleurálny
výpotok a symptómy, kongescia a zápal respiračného traktu, kašeľ
Zriedkavé intersticiálna pľúcna choroba, pneumonitída
Časté nevoľnosť
Menej časté ascites, gastrointestinálna ulcerácia a perforácia, zápal gastrointestinálneho traktu (na ktoromkoľvek mieste), stomatitída, dyspepsia, abdominálna distenzia, orofaryngeálna suchosť
Zriedkavé odynofágia, hypermotilita
Časté hepatitída (vrátane zvýšenej hladiny pečeňových enzýmov)
Menej časté hepatopatia (vrátane cirhózy), cholestáza, zvýšená hladina bilirubínu v krvi
Zriedkavé cholelitiáza
Časté vyrážka
Menej časté alopécia, nový nástup alebo zhoršenie psoriázy
(vrátane palmoplantárnej pustulóznej psoriázy)
a s ňou súvisiace ochorenia, dermatitída a ekzém, porucha potnej žľazy, kožný vred, fotosenzitivita, akné, zmeny sfarbenia kože, suchá koža, poruchy nechtov a nechtového lôžka
Zriedkavé exfoliácia a deskvamácia kože, bulózne ochorenia, porucha štruktúry vlasov, Stevens-Johnsonov syndróm**, multiformný erytém**
Menej časté svalové poruchy, zvýšená hladina kreatínfosfokinázy v krvi
Menej časté porucha funkcie obličiek, krv v moči, symptómy močového mechúra a močovej rúry
Zriedkavé nefropatia (vrátane nefritídy)
Menej časté poruchy menštruačného cyklu a krvácania maternice
(vrátane amenorey), poruchy prsníkov
Zriedkavé sexuálna dysfunkcia
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Celkové poruchy a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Časté pyrexia, bolesť (na ktoromkoľvek mieste), asténia, pruritus (na ktoromkoľvek mieste), reakcie v mieste vpichu
Menej časté zimnica, ochorenie podobné chrípke, zmena vnímania teploty, nočné potenie, návaly tepla
Zriedkavé fistula (na akomkoľvek mieste)
Menej časté zvýšená hladina alkalickej fosfatázy v krvi, predĺženie času koagulácie
Zriedkavé zvýšená hladina kyseliny močovej v krvi
Menej časté kožné poranenia, zhoršené hojenie
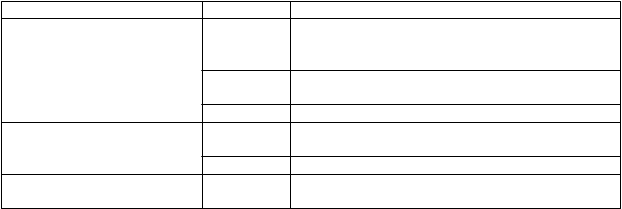
*Tieto udalosti sa týkali triedy antagonistov TNF, ale výskyt v súvislosti s certolizumab pegolom nie je známy.
**Tieto udalosti sa týkali triedy antagonistov TNF.
V iných indikáciách boli v súvislosti s Cimziou pozorované menej často ďalšie nasledujúce nežiaduce reakcie: gastrointestinálna stenóza a obštrukcie, zhoršenie celkového zdravotného stavu, spontánny potrat a azoospermia.
Popis vybraných nežiaducichreakciíInfekcieV placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou bola miera incidencie nových prípadov infekcií u všetkých pacientov liečených Cimziou 1,03 na pacientorok a u pacientov
liečených placebom 0,92 na pacientorok. Z infekcií boli zastúpené predovšetkým infekcie horných
dýchacích ciest, infekcie močového traktu a infekcie dolných dýchacích ciest a herpetické vírusové infekcie (pozri časti 4.3 a 4.4).
V placebom kontrolovaných klinických štúdiách bolo v skupinách liečených Cimziou (0,07 na pacientorok; pri všetkých dávkach) viac nových prípadov závažnej infekcie v porovnaní s placebom (0,02 na pacientorok). Medzi najčastejšie závažné infekcie patrili pneumónia, tuberkulózne infekcie. Medzi závažné infekcie patrili aj invazívne oportúnne infekcie (napr. pneumocystóza, mykotická ezofagitída, nokardióza a diseminovaný herpes zoster). Neexistuje dôkaz o zvýšenom riziku infekcií pri nepretržitej expozícii v priebehu času (pozri časť 4.4).
MalignityalymfoproliferatívneporuchyS výnimkou nemelanómových nádorov kože bolo pozorovaných 121 malignít vrátane 5 prípadov lymfómu v klinických RA štúdiách s Cimziou, v ktorých bolo liečených celkovo 4 049 pacientov, čo
predstavuje 9 277 pacientorokov. V klinických štúdiách s reumatoidnou artritídou sa v súvislosti
s Cimziou vyskytovali prípady lymfómu v rozsahu incidencie 0,05 na 100 pacientorokov a melanómu v rozsahu incidencie 0,08 na 100 pacientorokov (pozri časť 4.4). V klinickej štúdii fázy III
s psoriatickou artritídou sa pozoroval aj jeden prípad lymfómu.
AutoimunitaZ pacientov, ktorí boli na začiatku ANP negatívni, malo v pivotných štúdiách pozitívne ANP titre
16,7 % pacientov liečených Cimziou v porovnaní s 12,0 % pacientov v placebovej skupine. V rámci pacientov, ktorí mali na začiatku negatívne protilátky anti-dsDNA, u 2,2 % pacientov liečených Cimziou sa vyskytli pozitívne titre protilátok anti-dsDNA v porovnaní s 1,0 % pacientov v placebovej skupine. V obidvoch placebom kontrolovaných a nezaslepených následných klinických štúdiách s reumatoidnou artritídou boli menej často hlásené prípady syndrómu podobného lupusu. Zriedkavo boli hlásené iné imunologicky sprostredkované ochorenia; príčinná súvislosť s Cimziou nie je známa. Vplyv dlhodobej liečby Cimziou na vývoj autoimunitných ochorení nie je známy.
R
eakcie
v
m
i
este
p
odania
injekcie
V placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou sa reakcie v mieste podania injekcie vyskytovali u 5,8 % pacientov liečených Cimziou, ako je erytém, svrbenie, hematóm, bolesť, opuch alebo podliatina, v porovnaní s 4,8 % pacientov liečených placebom. Bolesť v mieste podania injekcie sa pozorovala u 1,5 % pacientov liečených Cimziou, pričom ani v jeden prípad nemal za následok prerušenie liečby.
ZvýšeniekreatínfosfokinázyFrekvencia zvýšenia kreatínfosfokinázy (CPK) bola celkovo vyššia u pacientov s axSpA v porovnaní s RA populáciou. Frekvencia bola zvýšená u pacientov liečených placebom (2,8 % oproti 0,4 %
v populácii s axSpA a RA, v uvedenom poradí), aj u pacientov liečených Cimziou (4,7 % oproti 0,8 % v populácii s axSpA a RA, v uvedenom poradí). Zvýšenie CPK v štúdii axSpA bolo väčšinou mierne až stredne závažné, prechodnej povahy a neznámeho klinického významu, pričom ani v jednom
prípade toto zvýšenie neviedlo k vysadeniu liečby.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePočas klinických štúdií sa nepozorovala žiadna toxicita limitujúca dávku. Podávané boli viacnásobné dávky až do 800 mg subkutánne a 20 mg/kg intravenózne. V prípade predávkovania sa odporúča
u pacientov starostlivé sledovanie akýchkoľvek nežiaducich reakcií alebo účinku a okamžité začatie vhodnej symptomatickej liečby.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNFα), ATC kód: L04AB05
Mechanizmus účinkuCimzia má vysokú afinitu k ľudskému TNFα a viaže sa s disociačnou konštantou (KD) 90 pM. TNFα
je kľúčovým prozápalovým cytokínom s najdôležitejšou úlohou pri zápalových procesoch. Cimzia selektívne neutralizuje TNFα (IC90 4 ng/ml pre inhibíciu ľudského TNFα v
in vitro hodnotení L929
fibrosarkómovej cytotoxicity u myší), ale neneutralizuje lymfotoxín α (TNFβ).
Ukázalo sa, že Cimzia neutralizuje rozpustný ľudský TNFα spojený s membránami, spôsobom závislým od dávky. Inkubácia monocytov s Cimziou viedla k inhibícii lipopolysacharidmi (LPS) indukovanej produkcie TNFα a IL1β v ľudských monocytoch, ktorá bola závislá od dávky.
Cimzia neobsahuje oblasť fragmentu schopného kryštalizácie (Fc), ktorá je zvyčajne súčasťou kompletnej protilátky a preto neviaže komplement ani nevyvoláva od protilátky závislú bunkami sprostredkovanú cytotoxicitu
in vitro. Neindukuje apoptózu
in vitro v ľudských monocytoch ani lymfocytoch pochádzajúcich z periférnej krvi, ani degranuláciu neutrofilov.
K
l
i
n
i
cká účinnosť
Reumatoidnáartritída
Účinnosť a bezpečnosť Cimzie boli hodnotené v 2 randomizovaných, placebom kontrolovaných, dvojito zaslepených klinických štúdiách u pacientov vo veku ≥ 18 rokov s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR (American College of Rheumatology) kritérií, RA-I (RAPID
1) a RA-II (RAPID 2). Každý pacient mal 9 alebo viac opuchnutých a citlivých kĺbov a trpel aktívnou
RA minimálne 6 mesiacov pred začiatkom štúdie. V obidvoch štúdiách bola Cimzia podávaná subkutánne v kombinácii s perorálnym MTX počas minimálne 6 mesiacov v stabilných dávkach
minimálne 10 mg týždenne po dobu 2 mesiacov. S používaním Cimzie v kombinácii s DMARD
okrem MTX nie sú žiadne skúsenosti.
Účinnosť a bezpečnosť Cimzie boli hodnotené u dospelých pacientov doteraz neliečených liekmi zo skupiny DMARD (tzv. DMARD-naivní pacienti) s aktívnou reumatoidnou artritídou
v randomizovanej, placebom kontrolovanej, dvojito zaslepenej klinickej štúdii (C-EARLY). V štúdii
C-EARLY boli pacienti vo veku ≥ 18 rokov, ktorí mali ≥ 4 opuchnuté a bolestivé kĺby a diagnóza stredne závažnej až závažnej progresívnej reumatoidnej artritídy u nich musela byť stanovená
v priebehu 1 roka (podľa definície klasifikačných kritérií ACR/EULAR (European League Against
Rheumatism) z roku 2010). Priemerný čas od diagnózy bol u pacientov vo východiskovom bode 2,9 mesiaca a neboli doteraz liečení pomocou DMARD (vrátane MTX). V ramene s Cimziou a v ramene s placebom bola liečba MTX začatá od 0. týždňa (10 mg/týždeň), dávka bola titrovaná až do maximálne tolerovanej dávky v 8. týždni (povolených min. 15 mg/týždeň, max. 25 mg/týždeň)
a udržiavaná v priebehu štúdie (priemerná dávka MTX po 8. týždni bola pre placebo 22,3 mg/týždeň, pre Cimziu 21,1 mg/týždeň).
Tabuľka 2 Popis klinickej štúdie
Č
íslo št
údie
P
očty pacientov
A
ktívna schéma dávkovania
C
i
ele štúdie
RA-I (52 týždňov)
RA-II (24 týždňov)
C-EARLY (do 52. týždňa
982 400 mg (v 0.,2.,4. týždni) s MTX
200 mg alebo 400 mg
každé 2 týždne s MTX
619 400 mg (v 0.,2.,4. týždni) s MTX
200 mg alebo 400 mg každé 2 týždne s MTX
879 400 mg (v 0., 2., 4. týždni)

200 mg každé
2 týždne s MTX
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Súbežne primárne koncové ukazovatele: ACR 20 v 24. týždni a zmena
z východiskovej hodnoty mTSS v 52. týždni
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Primárny výsledný ukazovateľ: ACR 20
v 24. týždni.
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia
u DMARD-naivných pacientov. Primárny výsledný ukazovateľ: podiel subjektov s pretrvávajúcou remisiou* v 52.
týždni
mTSS: modifikované TSS (Total Sharp Score)
*Pretrvávajúca remisia v 52. týždni je definovaná ako DAS28[ESR] < 2,6 v 40. týždni aj v 52. týždni.
Prejavy a príznaky
Výsledky klinických štúdií RA-I a RA-II sú uvedené v tabuľke 3. V oboch klinický štúdiách sa v porovnaní s placebom dosiahli štatisticky významne vyššie odpovede ACR 20 od 1. týždňa
a ACR 50 od 2. týždňa. Odpovede pretrvávali až do 52. týždňa (RA-I) a 24. týždňa (RA-II). Zo 783
pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo 52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie. Z nich 427 pacientov ukončilo 2-ročnú nezaslepenú následnú štúdiu a tak mali celkovú expozíciu Cimzii spolu 148 týždňov. Pozorovaná miera odpovede ACR 20 v tomto časovom bode bola 91 %. Zníženie (RA-I)
z východiskovej hodnoty DAS28 (ESR) bolo tiež v porovnaní s placebom významne vyššie
(p < 0,001) v 52. týždni (RA-I) a 24. týždni (RA-II) a pretrvávalo počas 2 rokov v nezaslepenej predĺženej štúdii RA-I.
T
abuľka 3 ACR odpoveď v klinických štúdiách RA-I a RA-II
Štúdia RA-I
Štúdia RA-II
K
ombinácia s metotrexátom
(
24 a 52 týždňov)
K
ombinácia s metotrexátom
(
24 týždňov)
Odpoveď Placebo + MTX N = 199
ACR 20
Cimzia
200 mg + MTX
každé 2 týždne
N = 393
Placebo + MTX N = 127
Cimzia
200 mg + MTX
každé 2 týždne
N = 246
24. týždeň 14 % 59 %** 9 % 57 %**
52. týždeň 13 % 53 %** N/A N/A
ACR 50
24. týždeň 8 % 37 %** 3 % 33 %**
52. týždeň 8 % 38 %** N/A N/A
ACR 70
24. týždeň 3 % 21 %** 1 % 16 %*
52. týždeň 4 % 21 %** N/A N/A
Hlavná klinická odpoveďa.
1 % 13 %**
Cimzia verzus placebo: * p ≤ 0,01, ** p < 0,001
a. Hlavná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 pri každom hodnotení počas nepretržitého 6-mesačného obdobia.
p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
Percentuálna odpoveď založená na počte pacientov, ktorí prispeli údajmi (n) pre tento koncový a časový ukazovateľ, ktorý sa môže odlišovať od N.
Štúdia C-EARLY splnila svoje primárne a kľúčové sekundárne výsledné ukazovatele. Kľúčové výsledky zo štúdie sú uvedené v tabuľke 4.
Tabuľka 4 Štúdia C-EARLY: percento pacientov s trvajúcou remisiou a trvajúcou nízkou aktivitou ochorenia v 52. týždni
Odpoveď Placebo + MTX N= 213
Cimzia 200 mg + MTX N= 655
P
r
etrvávajúca remisia*
(DAS28(ESR) <2,6 v
40. týždni aj 52. týždni)
Pretrvávajúca nízka aktivita ochorenia
(DAS28(ESR) ≤3,2 v 40. týždni aj v 52. týždni)
15,0 % 28,9%**
28,6 % 43,8%**

*Primárny výsledný ukazovateľ štúdie C-EARLY (do 52. týždňa)
Zostava úplnej analýzy, prirátanie nonrespondérov pre chýbajúce hodnoty.
**Cimzia+MTX vs placebo+MTX: p<0,001
p-hodnota bola odhadnutá z logistického regresného modelu s faktormi pre liečbu, miesto a dobu od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4 mesiace)
U pacientov v skupine Cimzia+MTX došlo k výraznejšej redukcii od východiskového stavu
v DAS 28 (ESR) v porovnaní so skupinou placebo+MTX, ktorá bola pozorovaná už od 2. týždňa a pokračovala do 52. týždňa (p<0,001 pri každej kontrole). Hodnotenia remisie (DAS 28 (ESR)
<2,6), stavu nízkej aktivity ochorenia (DAS 28 (ESR) ≤3,2) a ACR 50 a ACR 70 podľa kontrol
ukázali, že liečba Cimzia+MTX viedla k rýchlejšej a výraznejšej odpovedi ako liečba
PBO+MTX. Tieto výsledky u DMARD-naivných subjektov pretrvávali počas 52 týždňov liečby.
R
ádiografická odpoveď
V RA-I bolo štrukturálne poškodenie kĺbu hodnotené rádiograficky v 52. týždni a vyjadrené ako
zmena mTSS a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (JSN – joint space narrowing score) v porovnaní s východiskovými hodnotami. Pacienti liečení Cimziou preukázali významne menšiu rádiografickú progresiu ako pacienti užívajúci placebo v 24. týždni a 52. týždni (pozri tabuľku
5). V placebovej skupine v 52. týždni bolo 52 % pacientov bez rádiografickej progresie (mTSS ≤ 0,0)
v porovnaní so 69 % v skupine liečenej Cimziou v dávke 200 mg.
Tabuľka 5 Zmeny v priebehu 12 mesiacov v RA-I
m
T
SS
P
l
acebo + MTX N = 199
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX N = 393
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX – Placebo + MTX
Stredná hodnota rozdielu
52. týždeň 2,8 (7,8) 0,4 (5,7) -2,4
Skóre erózie
52. týždeň 1,5 (4,3) 0,1 (2,5) -1,4
JSN skóre
52. týždeň 1,4 (5,0) 0,4 (4,2) -1,0
p-hodnoty v prípade obidvoch – mTSS aj skóre erózie – boli < 0,001 a v prípade JSN skóre boli
≤ 0,01. ANCOVA zodpovedala hodnotenej zmene od východiskovej hodnoty pre každé meranie s oblasťou a liečbou ako faktormi a východiskovým hodnotením ako kovariátom.
Zo 783 pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo
52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie.
V podskupine 449 pacientov, ktorí ukončili minimálne 2-ročnú liečbu Cimziou (RA-I a nezaslepená predĺžená štúdia), sa preukázala pretrvávajúca inhibícia progresie štrukturálneho poškodenia
s hodnotiteľnými údajmi v časovom bode 2 roky.
V štúdii C-EARLY viedla Cimzia+MTX k inhibícii rádiografickej progresie v porovnaní
s placebom+MTX v 52. týždni (pozri tabuľku 6). V skupine placebo+MTX u 49,7 % pacientov nedošlo k žiadnej rádiografickej progresii (zmena mTSS ≤0,5) v 52. týždni, v porovnaní s 70,3 % v skupine Cimzia+MTX (p<0,001).
Tabuľka 6 Rádiografické zmeny v 52. týždni v štúdii C-EARLY
Placebo + MTX
N= 163
Priemer (SD)
Cimzia 200 mg + MTX
N = 528
Priemer (SD)
Cimzia 200 mg + MTX – Placebo + MTX Rozdiel*
m
T
SS
týždeň 52
Skóre erózie
týždeň 52
Skóre JSN
týždeň 52
1,8 (4,3) 0,2 (3,2)** -0,978 (-1,005, -0,500)
1,1 (3,0) 0,1 (2,1)** -0,500 (-0,508, -0,366)
0,7 (2,3) 0,1 (1,7)** 0,000 (0,000, 0,000)
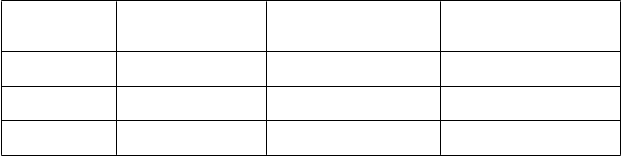

Rádiografická zostava s lineárnou extrapoláciou.
* Hodges-Lehmannov bodový odhad posunu a 95 % asymptotický (Moses) interval spoľahlivosti.
**Cimzia+MTX vs. placebo+MTX p<0,001. p-hodnota bola odhadnutá z ANCOVA modelu hodnotenia liečby, oblasti a času od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4
mesiace) ako faktory a hodnoty vo východiskovom stave ako kovariancia.
Odpoveď fyzickej funkcie a následky týkajúce sa zdraviaU pacientov liečených Cimziou v RA-I a RA-II bolo už od 1. týždňa až do konca štúdií, v porovnaní s placebom, hlásené významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI
(Health Assessment Questionnaire – Disability Index) a únavy (vyčerpanosti) hlásenej podľa škály
FAS (Fatigue Assessment Scale). V obidvoch klinických štúdiách hlásili pacienti liečení Cimziou významne väčšie zlepšenia v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a v skóre všetkých domén. Zlepšenia fyzickej funkcie a HRQoL pretrvávali
v priebehu 2 rokov v nezaslepenej predĺženej štúdii RA-I. Pacienti liečení Cimziou hlásili, v porovnaní s placebom, štatisticky významné zlepšenia v prieskume produktivity práce (Work Productivity Survey).
V štúdii C-EARLY pacienti liečení kombináciou Cimzia+MTX hlásili významné zlepšenie bolesti
v 52. týždni v porovnaní s kombináciou placebo+MTX, hodnotenej pomocou Hodnotenia artritickej bolesti pacientom (Patient Assessment of Arthritis Pain = PAAP) - 48,5 vs - 44,0 (priemer najmenších štvorcov) (p<0,05).
Klinická štúdia DoseFlex
Účinnosť a bezpečnosť 2 dávkovacích režimov Cimzie (200 mg každé 2 týždne a 400 mg každé 4 týždne) oproti placebu sa hodnotili v 18-týždňovej, nezaslepenej, predrandomizačnej a 16-týždňovej randomizovanej, dvojito-zaslepenej, placebom kontrolovanej klinickej štúdii u dospelých pacientov
s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR kritérií, ktorí nemali dostatočnú odpoveď na MTX.
Pacienti dostávali nárazové dávky 400 mg Cimzie v 0., 2. a 4. týždni, po ktorých nasledovala dávka
200 mg Cimzie každé 2 týždne počas začiatočného nezaslepeného obdobia. Respondéri (ACR 20
dosiahnuté) v 16. týždni boli randomizovaní v 18. týždni na Cimziu v dávke 200 mg každé 2 týždne, Cimziu v dávke 400 mg každé 4 týždne alebo placebo v kombinácii s MTX počas ďalších 16 týždňov (celková dĺžka štúdie: 34 týždňov). Tieto 3 skupiny boli dobre vyvážené čo sa týka klinickej odpovede po aktívnej vstupnej fáze (ACR 20: 83-84 % v 18. týždni).
Primárnym koncovým ukazovateľom štúdie bola miera ACR 20 respondérov v 34. týždni. Výsledky v v 34. týždni sú uvedené v tabuľke 7. Obidva dávkovacie režimy Cimzie preukázali pretrvávajúcu klinickú odpoveď a boli štatisticky významné v porovnaní s placebom v 34. týždni. Koncový ukazovateľ ACR 20 sa dosiahol pre oba režimy Cimzie, 200 mg každé 2 týždne a 400 mg každé 4 týždne.
Tabuľka 7 ACR odpoveď v klinickej štúdii DoseFlex v 34. týždni
L
i
ečebný režim v 0. až
16. týždni
C
im
z
i
a 400 mg + MTX v 0., 2. a 4. týždni, po ktorých nasledovala Cimzia v dávke 200 mg + MTX každé 2 týždne
R
andomizovaný, dvojito- zaslepený liečebný režim v 18. až 34. týždni
AC
R 20
p-hodnota*
ACR 50
p-hodnota*
ACR 70
p-hodnota*
N/A: neaplikovateľné
Placebo + MTX
N=69
45 % N/A
30 % N/A
16 % N/A
Cimzia
200 mg + MTX
každé 2 týždne
N=70
67 %
0,009
50 %
0,020
30 %
0,052
Cimzia
400 mg + MTX
každé 4 týždne
N=69
65 %
0,017
52 %
0,010
38 %
0,005
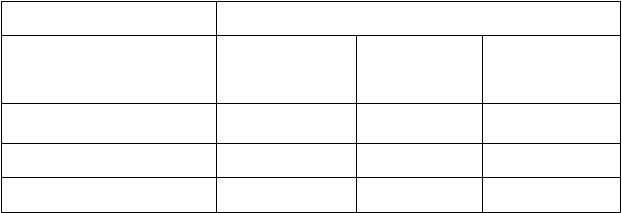
* p-hodnoty Waldovho testu pre porovnania Cimzie v dávke 200 mg oproti placebu a Cimzie v dávke
400 mg oproti placebu sú odhadované na základe logistickej regresie s faktormi pre liečbu.
AxiálnaspondylartritídaÚčinnosť a bezpečnosť Cimzie boli hodnotené v jednej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AS001) u 325 pacientov vo veku ≥ 18 rokov s aktívnou
axiálnou spondylartritídou s nástupom v dospelosti trvajúcou najmenej 3 mesiace definovanou
klasifikačnými kritériami spoločnosti ASAS (Assessment of Spondyloarthritis International Society). Celková populácia s axiálnou spondylartritídou zahŕňala subpopulácie s röntgenologickým dôkazom
ankylozujúcej spondylitídy (AS) a bez neho (axiálna spondylartritída bez röntgenologického dôkazu;
[nr-axSpA]). Pacienti mali aktívne ochorenie definované indexom aktivity ochorenia ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥ 4, bolesť chrbtice ≥ 4 na číselnej stupnici NRS (Numerical Rating Scale) s rozsahom 0 až 10 a zvýšené CRP alebo súčasný dôkaz sakroilitídy pri vyšetrení magnetickou rezonanciou (MR). Pacienti museli mať prejavy
neznášanlivosti alebo museli mať nedostatočnú odpoveď aspoň na jedno NSA. Celkovo 16 %
pacientov malo v anamnéze liečbu antagonistom TBF. Pacienti boli liečení nárazovou dávkou Cimzie
400 mg v 0., 2. a 4. týždni (u oboch liečebných skupín) alebo placebom, po ktorom nasledovala dávka
Cimzie buď 200 mg každé 2 týždne alebo 400 mg každé 4 týždne alebo placebo. 87,7 % pacientov dostávalo súčasne NSA. Primárnym výsledným ukazovateľom účinnosti bola miera odpovede ASAS20 v 12. týždni.
Po 24 týždňovej, dvojito zaslepenej, placebom kontrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 156 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 204 týždňov. Všetci
pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
199 účastníkov klinického skúšania (61,2 % z randomizovaných účastníkov) ukončilo štúdiu do týždňa 204.
Kľúčové výsledky účinnosti
V klinickej štúdii AS001 dosiahlo ASAS20 odpoveď v 12. týždni 58 % pacientov liečených Cimziou v dávke 200 mg každé 2 týždne a 64 % pacientov liečených Cimziou v dávke 400 mg každé 4 týždne v porovnaní s 38 % pacientmi užívajúcimi placebo (p < 0,01). V celkovej populácii bol podiel ASAS20 respondérov klinicky významný a významne vyšší pre liečebnú skupinu s Cimziou v dávke
200 mg každé 2 týždne a s Cimziou 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej kontrole od 1. týždňa až do 24. týždňa (p ≤ 0,001 pri každej kontrole). V 12. týždni a 24. týždni
bolo percento osôb s ASAS40 odpoveďou vyššie v skupinách liečených Cimziou v porovnaní s
placebom.
Podobné výsledky sa dosiahli u oboch subpopulácií s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu. U žien neboli ASAS20 odpovede štatisticky významne odlišné od placeba až do časového bodu po 12. týždni.
Zlepšenie ASAS 5/6, čiastočná remisia a BASDAI-50 boli štatisticky významné v 12. týždni a v
24. týždni a pretrvávali až do 48. týždňa v celkovej populácii rovnako ako v subpopuláciách. Kľúčové výsledky účinnosti z klinickej štúdie AS001 sú uvedené v tabuľke 8.
U pacientov, ktorí zostali v štúdii, sa zlepšenie vo všetkých vyššie uvedených kľúčových výsledkoch účinnosti zachovalo do týždňa 204 v celkovej populácii, rovnako ako v subpopuláciách.
T
abuľka 8 Kľúčové výsledky účinnosti v klinickej štúdii AS001 (percento pacientov)
Parametre
Ankylozujúca spondylitída
Axiálna spondylartritída bez röntgenologického dôkazu
Axiálna spondylartritída Celková populácia
A
SAS20
(b
,c
)
12. týždeň
24. týždeň
ASAS40(c,d)
12. týždeň
24. týždeň
ASAS 5/6(c,d)
12. týždeň
24. týždeň
Čiastočná remisia(c,d)
12. týždeň
24. týždeň
BASDAI 50(c,d)
12. týždeň
24. týždeň
Placebo
N=57
37%
33%
19%
16%
9%
5%
2%
7%
11%
16%
Cimzia všetky režimy dávkovania (a) N=121
60%*
69%**
45%**
53%**
42%**
40%**
20%**
28%**
41%**
49%**
Placebo
N=50
40%
24%
16%
14%
8%
4%
6%
10%
16%
20%
Cimzia všetky režimy
dávkovania (a)
N=97
61%*
68%**
47%**
51%**
44%**
45%**
29%**
33%**
49%**
57%**
Placebo
N=107
38%
29%
18%
15%
8%
5%
4%
9%
13%
18%
Cimzia všetky režimy
dávkovania (a)
N=218
61%**
68%**
46%**
52%**
43%**
42%**
24%**
30%**
45%**
52%**
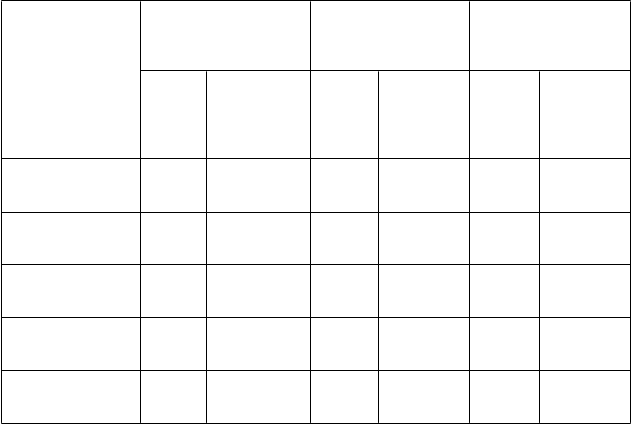
(a) Cimzia všetky režimy dávkovania = údaje pre Cimziu v dávke 200 mg podávanej každé 2 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni plus Cimzia 400 mg podávaná
každé 4 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni
(b) Výsledky sú z randomizovaného súboru
(c) p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
(d) Kompletný analyzovaný súbor
NA = nie je k dispozícii
* p ≤ 0,05, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
Mobilita chrbticeMobilita chrbtice sa hodnotila v dvojito zaslepenej, placebom kontrolovanej fáze pomocou BASMI v niekoľkých časových bodoch, vrátane východiskového stavu, v 12. týždni a v 24. týždni. Klinicky významné a štatisticky významné rozdiely u pacientov liečených Cimziou v porovnaní s pacientmi liečenými placebom sa preukázali pri každej kontrole od východiskového stavu. Rozdiel od placeba má tendenciu byť väčší u subpopulácie nr-axSpA ako u AS, čo môže byť dôsledkom menšieho chronického štrukturálneho poškodenia u pacientov s nr-axSpA.
Zlepšenie lineárneho skóre BASMI dosiahnuté v 24. týždni sa u pacientov, ktorí zostali v štúdii, udržalo až do týždňa 204.
O
dpoveď fyzickej funkcie a následky týkajúce sa zdravia
V klinickej štúdii AS001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotené pomocou BASFI a zlepšenie bolesti hodnotené NRS stupnicou celkovej a nočnej bolesti chrbtice v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie únavy (vyčerpanosti) hlásené pomocou položky BASDAI pre únavu a významné zlepšenie kvality života súvisiacej so zdravím merané dotazníkom Kvality života pri ankylozujúcej spondylitíde (ASQoL) a zlepšenie v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a v skóre všetkých domén v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie produktivity v práci a v domácnosti súvisiacej s axiálnou spondylartritídou, ktoré bolo hlásené v prieskume produktivity práce v porovnaní s placebom.
U pacientov, ktorí zostali v štúdii, bolo zlepšenie všetkých vyššie uvedených výsledkov do značnej miery zachované až do týždňa 204.
Inhibícia zápalu pri magnetickej rezonancii (MR)
V zobrazovacej subštúdii zahŕňajúcej 153 pacientov sa hodnotili prejavy zápalu hodnotené MR v
12. týždni a boli vyjadrené ako zmena skóre SPARCC (Spondyloarthritis Research Consortium of Canada) oproti východiskovému stavu pre krížovo-driekové kĺby a skóre ASspiMRI-a v berlínskej modifikácii pre chrbticu. Významná inhibícia zápalových príznakov u krížovo-driekových kĺbov aj chrbtice sa pozorovala u pacientov liečených Cimziou (všetky dávkové skupiny) v 12. týždni, a to v celkovej populácii s axiálnou spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu.
U pacientov, ktorí zostali v štúdii a u ktorých boli k dispozícii východiskové hodnoty aj hodnoty z týždňa 204, bola do značnej miery zachovaná inhibícia zápalových príznakov až do týždňa 204
v krížovo-driekových kĺboch (n=72) aj chrbtici (n=82), a to v celkovej populácii s axiálnou spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou
bez röntgenologického dôkazu.
Psoriatickáartritída
Účinnosť a bezpečnosť Cimzie sa hodnotili v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii (PsA001) u 409 pacientov vo veku ≥ 18 rokov s aktívnou
psoriatickou artritídou s nástupom v dospelosti trvajúcou najmenej 6 mesiacov definovanou klasifikačnými kritériami pre psoriatickú artritídu (Classification Criteria for Psoriatic Arthritis -
CASPAR). Pacienti mali ≥ 3 opuchnuté a bolestivé kĺby a zvýšené hodnoty markerov akútnej fázy. Pacienti mali tiež aktívne psoriatické kožné lézie alebo zdokumentovanú psoriázu v anamnéze a zlyhala u nich jedna alebo viac terapií DMARD. Predchádzajúca liečba jedným antagonistom TNF
bola povolená a 20 % pacientov bolo predtým liečených TNF-antagonistom. Pacienti dostávali nárazovú dávku Cimzie 400 mg v 0., 2. a 4. týždni (v oboch liečebných skupinách) alebo placebo, po
ktorej nasledovalo podávanie 200 mg Cimzie každé 2 týždne alebo 400 mg Cimzie každé 4 týždne alebo placebo každé 2 týždne. 72,6 % pacientov súbežne užívalo NSA a 70,2 % konvenčné DMARD. Štúdia mala dva primárne koncové ukazovatele: percento pacientov, ktorí dosiahli ACR 20 odpoveď
v 12. týždni a zmena modifikovaného skóre mTSS (Total Sharp Score) v 24. týždni od východiskovej hodnoty. Účinnosť a bezpečnosť Cimzie u pacientov s PsA, u ktorých prevládali symptómy
sakroilitídy alebo axiálnej spondylartritídy, neboli analyzované samostatne.
Po 24 týždňovej dvojito zaslepenej, placebom kotrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 168 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 216 týždňov. Všetci
pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
264 účastníkov klinického skúšania (64,5 %) ukončilo štúdiu až v týždni 216.
ACR odpoveď
Pacienti liečení Cimziou mali štatisticky významne vyššiu mieru ACR 20 odpovede v 12. týždni
a v 24. týždni v porovnaní s pacientmi, ktorým bolo podávané placebo (p < 0,001). Percento ACR 20 respondérov bolo klinicky významné v liečebných skupinách s Cimziou v dávke 200 mg každé 2 týždne a s Cimziou v dávke 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej návšteve od začiatku až do 24. týždňa (nominálne p ≤ 0,001 pri každej návšteve). Pacienti liečení Cimziou mali tiež významné zlepšenie miery odpovede ACR 50 a 70. V 12. a 24. týždni sa u pacientov liečených Cimziou pozorovalo zlepšenie v parametroch periférnej aktivity psoriatickej artritídy (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída)
(nominálna p-hodnota p < 0,01). Kľúčové výsledky účinnosti z klinickej štúdie PsA001 sú uvedené v tabuľke 9.
Tabuľka 9 Kľúčové výsledky účinnosti v klinickej štúdii PsA001 (percento pacientov)
O
dpoveď Placebo
N
=136
C
im
z
i
a
(
a)
200 mg každé 2 týždne
N
=138
C
im
z
i
a
(b
)
400 mg každé 4 týždne
N
=135
ACR
20
12. týždeň
24. týždeň
ACR5012. týždeň
24. týždeň
ACR7012. týždeň
24. týždeň
24 %
24 %
11 %
13 %
3 %
4 %
58 %**
64 %**
36 %**
44 %**
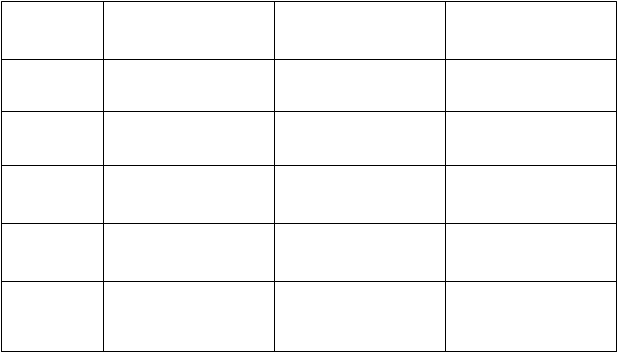
25 %**
28 %**
52 %**
56 %**
33 %**
40 %**
13 %*
24 %**
O
dpoveď Placebo
N
=86
C
im
z
i
a
(
a)
200 mg každé 2 týždne N=90
C
im
z
i
a
(b
)
400 mg každé 4 týždne N=76
P
A
SI 75 (c)
12. týždeň
24. týždeň
48. týždeň
14 %
15 % N/A
47 %***
62 %***
67 %
47 %***
61 %***
62 %
(a) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 2 týždne (b) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 4 týždne (c) U osôb s minimálne 3% psoriázy BSA na začiatku
* p < 0,01, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
*** p < 0,001 (nominálne), Cimzia oproti placebu
Výsledky sú z randomizovaného súboru. Rozdiel liečby: Cimzia 200 mg - placebo, Cimzia 400 mg - placebo (a zodpovedajúci 95 % interval spoľahlivosti a p-hodnota) boli posudzované pomocou štandardného obojstranného Waldovho testu asymptotických štandardných chýb. U pacientov, ktorí prerušili liečbu alebo u nich chýbajú údaje, sa použil systém „Non-responder Imputation„ (NRI).
Spomedzi 273 pacientov, ktorí boli na začiatku randomizovaní na Cimziu v dávke 200 mg každé 2
týždne a Cimziu v dávke 400 mg každé 4 týždne, zostalo na tejto liečbe 237 (86,8 %) pacientov až do
48. týždňa. Zo 138 pacientov randomizovaných na Cimziu v dávke 200 mg každé 2 týždne dosiahlo
92, 68 a 48 pacientov odpoveď ACR 20/50/70 v 48. týždni, v uvedenom poradí. Zo 135 pacientov randomizovaných na Cimziu v dávke 400 mg každé 4 týždne dosiahlo 89, 62 a 41 pacientov odpoveď
ACR 20/50/70, v uvedenom poradí.
Medzi pacientmi, ktorí zostali v štúdii, sa miera odpovede ACR 20, 50 a 70 zachovala až do týždňa
216. To platilo aj v prípade ďalších parametrov periférnej aktivity (napr. počet opuchnutých kĺbov, počet bolestivých /citlivých kĺbov, daktylitída a entezitída).
Rádiografická odpoveď
V klinickej štúdii PsA001 bola inhibícia progresie štrukturálneho poškodenia hodnotená rádiograficky
a vyjadrená ako zmena mTSS a jeho zložiek, skóre erózie (ES) a skóre zúženia kĺbovej štrbiny (JSN) v
24. týždni v porovnaní s východiskovými hodnotami. Skóre mTSS bolo upravené pre psoriatickú artritídu pridaním distálnych interfalangeálnych kĺbov ruky. Liečba Cimziou inhibovala rádiografickú
progresiu v porovnaní s liečbou placebom v 24. týždni meranú ako zmena celkového skóre mTSS (LS
priemerné [±SE] skóre oproti východiskovému stavu 0,28 [±0,07] v placebovej skupine v porovnaní s
0,06 [±0,06] v skupine so všetkými dávkami Cimzie, p = 0,007). Inhibícia rádiografickej progresie sa udržiavala pri liečbe Cimziou až do 48. týždňa v podskupine pacientov s vyšším rizikom
rádiografickej progresie (pacienti s východiskovým skóre mTSS > 6). U pacientov, ktorí sa zúčastnili štúdie, sa inhibícia rádiografickej progresie naďalej udržala až do týždňa 216.
Odpoveď fyzickej funkcie a následky týkajúce sa zdravia
V klinickej štúdii PsA001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI (Health Assessment Questionnaire – Disability Index), bolesti
hodnotenej podľa Hodnotenia artritickej bolesti pacientom PAAP a únavy (vyčerpanosti), čo sa
preukázalo podľa Škály hodnotenia únavy (FAS - Fatigue Assessment Scale) v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie kvality života súvisiacej so zdravím merané
dotazníkom Kvality života pri psoriatickej artritíde (PsAQoL) a SF-36 fyzickej a duševnej zložky a
produktivity súvisiacej s psoriatickou artritídou v zamestnaní a v domácnosti hlásenej v v prieskume produktivity práce v porovnaní s placebom. Zlepšenie vo všetkých vyššie uvedených výsledkoch pretrvávalo až do týždňa 216.
Imunogenicita
Reumatoidnáartritída
Celkový podiel pacientov s detekovateľnými protilátkami proti Cimzii pri aspoň 1 príležitosti bol
9,6 % v placebom kontrolovaných RA štúdiách. Približne u jednej tretiny pacientov s pozitívnymi protilátkami boli zistené in vitro neutralizujúce protilátky. U pacientov liečených súbežne s imunosupresívami (MTX) sa protilátky tvorili v menšej miere ako u pacientov, ktorí na začiatku neužívali imunosupresíva. Tvorba protilátok bola spojená s nižšou plazmatickou koncentráciou lieku a u niektorých pacientov s nižšou účinnosťou.
V 2 dlhodobých (až 5 rokov expozície) otvorených štúdiách celkový podiel pacientov
s detekovateľnými protilátkami proti Cimzii pri aspoň jednej príležitosti bol 13 % (8,4 % z celkového počtu pacientov malo prechodnú tvorbu protilátok a ďalších 4,7 % pacientov malo trvalú tvorbu protilátok). Celkové percento pacientov, ktorí boli pozitívni na tvorbu protilátok a mali trvale zníženú plazmatickú koncentráciu lieku, bolo odhadované na 9,1 %. Podobne, ako v placebom kontrolovaných štúdiách, bola tvorba protilátok u niektorých pacientov spojená s nižšou účinnosťou.
Farmakodynamický model založený na údajoch z III. fázy klinickej štúdie predpokladá, že približne u 15 % pacientov sa pri odporúčanom dávkovaní (200 mg každé 2 týždne po začiatočnej dávke), bez súbežnej liečby s MTX, vyvinú protilátky v priebehu 6 mesiacov. Tento počet sa znižuje so zvyšujúcimi sa dávkami súbežnej liečby s MTX. Tieto údaje sú primerane v súlade s pozorovanými údajmi.
Axiálnaspondylartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 4,4 % vo fáze III placebom kontrolovanej štúdie u pacientov s axiálnou spondylartritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až počas 192 týždňov) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 9,6 % (4,8 % malo prechodnú tvorbu protilátok, ďalších
4,8 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo na
6,8 %.
Psoriatickáartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 11,7 % vo fáze III placebom kontrolovanej štúdie u pacientov s psoriatickou artritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až do 4 rokov expozície) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 17,3 % (8,7 % malo prechodnú tvorbu protilátok, ďalších
8,7 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo
na 11,5 %.
V
šetky
i
ndikácie
Údaje vyjadrujú podiel pacientov, u ktorých bola vyšetrením ELISA stanovená prítomnosť protilátok proti Cimzii a do veľkej miery závisia od senzitivity a špecificity tohto vyšetrenia.
Okrem toho môže byť pozorovaný výskyt protilátok v hodnotení ovplyvnený viacerými faktormi, vrátane manipulácie so vzorkou, načasovania odberu vzorky, súbežne podávaných liekov a
základného ochorenia. Z týchto dôvodov nie je vhodné porovnávať výskyt protilátok proti Cimzii s výskytom protilátok proti iným antagonistom TNF.
5.2 Farmakokinetické vlastnosti
Plazmatické koncentrácie certolizumab pegolu boli značne úmerné dávke. Farmakokinetika pozorovaná u pacientov s reumatoidnou artritídou zodpovedala farmakokinetike pozorovanej u zdravých jedincov.
Absorpcia
Po subkutánnom podaní sa maximálne plazmatické koncentrácie certolizumab pegolu dosiahli v rozmedzí 54 a 171 hodín od podania injekcie. Biologická dostupnosť (F) certolizumab pegolu po subkutánnom podaní, v porovnaní s intravenóznym podaním, je približne 80 % (rozmedzie 76 % až
88%).
Distribúcia
Vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou bol zdanlivý distribučný objem (V/F) odhadovaný na 8,01 l.
Biotransformáciaaeliminácia
PEGyláciou, kovalentnou väzbou PEG polymérov na peptidy, sa odďaľuje eliminácia týchto látok z krvného obehu viacerými mechanizmami, vrátane zníženého renálneho klírensu, zníženej proteolýzy a zníženej imunogenicity. Certolizumab pegol je preto Fab' fragmentom protilátky konjugovanej
s PEG, aby predĺžil terminálny plazmatický eliminačný polčas Fab' fragmentu na hodnotu porovnateľnú s kompletnou molekulou protilátky. Polčas terminálnej eliminačnej fázy (t1/2) bol pre všetky testované dávky približne 14 dní.
Klírens po subkutánnom podaní bol odhadovaný na 21,0 ml/h vo farmakokinetickej analýze populácie s reumatoidnou artritídou, s interindividuálnou variabilitou 30,8 % (CV) a intraindividuálnou variabilitou 22,0 %. Prítomnosť protilátok proti certolizumab pegolu viedla k približne trojnásobnému nárastu klírensu. V porovnaní s osobou s hmotnosťou 70 kg je klírens o 29 % nižší u individuálnych pacientov s RA vážiacich 40 kg a o 38 % vyšší u pacientov vážiacich 120 kg.
Fab' fragment sa skladá z bielkovinových zložiek a predpokladá sa, že je odbúravaný na peptidy
a aminokyseliny proteolýzou. Dekonjugovaná PEG zložka sa rýchlo eliminuje z plazmy a vylučuje sa renálne v neznámej miere.
Osobitnépopulácie
Porucha funkcie obličiek
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili účinok poruchy funkcie obličiek na farmakokinetiku certolizumab pegolu alebo jeho PEG frakcie. Farmakokinetická analýza populácie
pacientov s ľahkou poruchou renálnej funkcie nepreukázala žiadny vplyv klírensu kreatinínu. Na to,
aby bolo možné odporučiť dávkovanie pri stredne ťažkej a ťažkej poruche funkcie obličiek, nie je dostatok informácií. Predpokladá sa, že farmakokinetika PEG frakcie certolizumab pegolu závisí od funkcie obličiek, avšak u pacientov s poruchou funkcie obličiek sa nehodnotila.
Porucha funkcie pečene
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili vplyv poruchy funkcie pečene na farmakokinetiku certolizumab pegolu.
Starší pacienti (≥ 65 rokov)
Neboli vykonané špecifické klinické štúdie u starších pacientov. Avšak vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou sa nepozoroval žiadny vplyv veku, 78 pacientov
(13,2 % populácie) bolo vo veku 65 rokov alebo viac, pričom najstarší pacient mal 83 rokov.
Pohlavie
Pohlavie nemalo žiadny vplyv na farmakokinetiku certolizumab pegolu. Pretože sa klírens znižuje so znižujúcou sa telesnou hmotnosťou, ženy zvyčajne môžu dosiahnuť o niečo vyššiu systémovú
expozíciu certolizumab pegolu.
Farmakokinetický/farmakodynamickývzťah
Na základe údajov z II. a III. fázy klinickej štúdie bol stanovený vzťah expozícia - odpoveď populácie medzi priemernou plazmatickou koncentráciou certolizumab pegolu počas dávkovacieho intervalu
(Cavg) a účinnosťou (definícia ACR 20 odpovede). Charakteristická Cavg, ktorá tvorí polovicu maximálnej pravdepodobnosti odpovede ACR 20 (EC50), bola 17 µg/ml (95 % CI: 10-23 µg/ml).
5.3 Predklinické údaje o bezpečnosti
Pivotné predklinické štúdie bezpečnosti boli vykonané u opíc Cynomolgus. U potkanov a opíc sa, pri dávkach vyšších ako sú dávky u ľudí, histopatologicky zistila celulárna vakuolizácia prítomná najmä v makrofágoch viacerých orgánov (v lymfatických uzlinách, v miestach vpichu, v slezine, nadobličkách, maternici, cervixe, cievovkovej spleti mozgu a v epitelových bunkách cievovkovej spleti). Je pravdepodobné, že tento nález bol spôsobený bunkovým vychytávaním PEG komponentu. In vitro funkčné štúdie ľudských vakuolizovaných makrofágov naznačili, že všetky skúmané funkcie zostali zachované. Štúdie s potkanmi naznačili, že > 90 % podaného PEG sa eliminovalo počas 3 mesiacov
po jednorazovej dávke, pričom hlavnou cestou exkrécie bolo vylúčenie močom.
Certolizumab pegol nereaguje skrížene s TNF hlodavcov. Preto boli reprodukčné toxikologické štúdie vykonané s homologickou skupinou rozpoznávajúcou TNF potkanov. Význam týchto údajov pri hodnotení rizika u ľudí môže byť obmedzený. Neboli pozorované žiadne nežiaduce účinky na zdravie matiek alebo fertilitu samíc, embryo-fetálne, peri- a postnatálne reprodukčné indexy u potkanov, ktorým bol po pretrvávajúcej supresii TNFα podávaný PEGylovaný Fab' fragment anti-rat TNFα (cTN3 PF). U samcov potkanov bola pozorovaná znížená motilita spermií a tendencia k poklesu počtu spermií.
Štúdie distribúcie preukázali, že prestup cTN3 PF placentou a materským mliekom do cirkulácie plodu a novorodenca je zanedbateľný. Certolizumab pegol sa neviaže na ľudský neonatálny Fc receptor (FcRn). Údaje z modelu ľudského placentárneho transferu s uzatvoreným okruhom ex vivo naznačujú nízky alebo zanedbateľný transfer do fetálneho kompartmentu. Okrem toho experimenty
s transcytózou sprostredkovanou FcRn v bunkách transfektovaných ľudským FcRn poukázali na zanedbateľný transfer (pozri časť 4.6).
V predklinických štúdiách sa nepreukázali žiadne mutagénne ani klastogénne účinky. Štúdie na karcinogenicitu sa neuskutočnili s certolizumab pegolom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
octan sodný chlorid sodný voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Jednomililitrová naplnená injekčná striekačka (zo skla typu I) s plunžerovým uzáverom (z bromobutylovej gumy), s obsahom 200 mg certolizumab pegolu.
Veľkosť balenia: 2 naplnené injekčné striekačky a 2 liehové tampóny.
Multibalenie obsahujúce 6 (3 balenia po 2) naplnených injekčných striekačiek a 6 (3 balenia po 2)
liehových tampónov.
Multibalenie obsahujúce 10 (5 balení po 2) naplnených injekčných striekačiek a 10 (5 balení po 2)
liehových tampónov.
Veľkosť balenia: 2 naplnené injekčné striekačky s chráničom ihly a 2 liehové tampóny (len na používanie zdravotníckymi pracovníkmi).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Obsiahle pokyny na prípravu a podanie Cimzie v naplnenej striekačke sú uvedené v písomnej informácii pre používateľa.
Tento liek je len na jednorazové použitie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
UCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/09/544/001
EU/1/09/544/002
EU/1/09/544/003
EU/1/09/544/004
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 1. október 2009
Dátum posledného predĺženia registrácie: 16. máj 2014
10. DÁTUM REVÍZIE TEXTU
{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Cimzia 200 mg injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každé naplnené pero obsahuje 200 mg certolizumab pegolu v jednom ml.
Certolizumab pegol je Fab' fragment rekombinantnej, humanizovanej protilátky proti tumor nekrotizujúcemu faktoru alfa (TNFα) získaný z Escherichia coli a konjugovaný s polyetylénglykolom (PEG).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia).
Číry až opalizujúci, bezfarebný až žltý roztok. pH roztoku je približne 4,7.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidnáartritída
Cimzia, v kombinácii s metotrexátom (MTX), je indikovaná na:
- liečbu stredne závažnej až závažnej aktívnej reumatoidnej artritídy (RA) u dospelých pacientov, keď je odpoveď na antireumatiká modifikujúce ochorenie (DMARD –Disease-Modifying Antirheumatic Drugs), vrátane MTX, nedostatočná. Cimzia sa môže podávať v monoterapii
v prípade intolerancie MTX, alebo keď pokračovanie v liečbe MTX nie je vhodné.
- liečbu závažnej, aktívnej a progresívnej formy RA u dospelých, doteraz neliečených MTX alebo inými DMARD.
Preukázalo sa, že Cimzia znižuje mieru progresie poškodenia kĺbov meranú röntgenologicky a zlepšuje fyzickú funkciu, keď sa podáva v kombinácii s MTX.
Axiálnaspondylartritída
Cimzia je indikovaná na liečbu dospelých pacientov so závažnou aktívnou axiálnou spondylartritídou, ktorú tvorí:
Ankylozujúca spondylitída (AS)
Dospelí pacienti so závažnou aktívnou ankylozujúcou spondylitídou, u ktorých nedošlo k primeranej odpovedi na nesteroidné antiflogistiká (NSA) alebo ktorí ich netolerujú.
Axiálna spondylartritída bez röntgenového dôkazu AS
Dospelí pacienti so závažnou aktívnou axiálnou spondylartritídou bez röntgenového dôkazu AS, avšak s objektívnymi prejavmi zápalu a to zvýšeným C-reaktívnym proteínom (CRP) a/alebo podľa vyšetrenia magnetickou rezonanciou (MR), u ktorých nedošlo k primeranej odpovedi na NSA alebo ktorí ich netolerujú.
P
soriatická
artritída
Cimzia, v kombinácii s MTX, je indikovaná na liečbu aktívnej psoriatickej artritídy u dospelých, keď je odpoveď na liečbu DMARD nedostatočná.
Cimzia sa môže podávať v monoterapii v prípade intolerancie metotrexátu, alebo keď pokračovanie v liečbe metotrexátom nie je vhodné.
Podrobnosti o terapeutických účinkoch, pozri časť 5.1.
4.2 Dávkovanie a spôsob podávania
Liečbu má začať lekár - špecialista so skúsenosťami v diagnostike a liečbe ochorení, na ktoré je indikovaná Cimzia, ktorý bude dohliadať na priebeh liečby. Pacienti majú dostať špeciálnu kartičku s upozornením.
Dávkovanie
Nárazovádávka
Odporúčaná začiatočná dávka Cimzie pre dospelých pacientov je 400 mg (podávaná vo forme dvoch
200 mg subkutánnych injekcií) v 0., 2. a 4. týždni. Ak je to vhodné, s podávaním metotrexátu sa má pri reumatoidnej artritíde a psoriatickej artritíde pokračovať počas liečby Cimziou.
Udržiavaciadávka
Reumatoidná artritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s reumatoidnou artritídou 200 mg každé 2 týždne. Po potvrdení klinickej odpovede sa môže zvážiť alternatívne
udržiavacie dávkovanie 400 mg každé 4 týždne. Ak je to vhodné, s podávaním MTX sa má
pokračovať počas liečby Cimziou.
Axiálna spondylartritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s axiálnou spondylartritídou 200 mg každé 2 týždne alebo 400 mg každé 4 týždne.
Psoriatická artritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s psoriatickou artritídou 200 mg každé 2 týždne. Po potvrdení klinickej odpovede sa môže zvážiť alternatívne
udržiavacie dávkovanie 400 mg každé 4 týždne. Ak je to vhodné, s podávaním MTX sa má
pokračovať počas liečby Cimziou.
Pre indikácie uvedené vyššie dostupné údaje ukazujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby. Pokračovanie v liečbe sa má starostlivo prehodnotiť u pacientov, u ktorých sa nepreukázal žiadny prospech z liečby počas prvých 12 týždňov liečby.
Vynechaniedávky
Pacientom, ktorí vynechali dávku, treba odporučiť, aby si aplikovali injekčne ďalšiu dávku Cimzie, hneď ako si spomenú a potom pokračovali v injekčnej aplikácii nasledujúcich dávok tak, ako boli
poučení.
Osobitnéskupinypacientov
Pediatrická populácia (< 18 rokov)
Bezpečnosť a účinnosť Cimzie u detí a dospievajúcich vo veku do 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Starší pacienti ( ≥ 65 rokov)
Nie je potrebná žiadna úprava dávky. Farmakokinetické analýzy populácie nepreukázali žiadny vplyv veku (pozri časť 5.2).
P
orucha funkcie obličiek a pečene
V týchto populáciách pacientov sa Cimzia neskúmala. Nie je možné poskytnúť žiadne odporúčania pre dávkovanie (pozri časť 5.2).
Spôsobpodávania
Celkový obsah (1 ml) naplneného pera sa má podávať len vo forme subkutánnej injekcie. Medzi vhodné miesta pre injekčnú aplikáciu patrí stehno alebo brucho.
Po náležitom vyškolení v injekčnej technike si môžu pacienti, ak to ich lekár považuje za vhodné, sami aplikovať injekcie pomocou naplneného pera, s lekárskou kontrolou podľa potreby. Lekár by mal s pacientom prediskutovať, ktorá forma podania injekcie je najvhodnejšia.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza alebo iné závažné infekcie, ako sú sepsa alebo oportúnne infekcie (pozri časť
4.4).
Stredne závažné až závažné srdcové zlyhanie (NYHA trieda III/IV) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Infekcie
Pred liečbou Cimziou, počas nej a po jej skončení sa musia u pacientov starostlivo sledovať prejavy a príznaky infekcií, vrátane tuberkulózy. Pretože eliminácia certolizumab pegolu môže trvať až do
5 mesiacov, sledovanie má pokračovať aj počas tohto obdobia (pozri časť 4.3).
Liečba Cimziou sa nesmie začať u pacientov s klinicky významnou aktívnou infekciou, vrátane chronických alebo lokalizovaných infekcií, pokiaľ nie je infekcia zvládnutá (pozri časť 4.3).
Pacientov, u ktorých sa objavila nová infekcia v priebehu liečby Cimziou, je potrebné starostlivo sledovať. Ak sa u pacienta vyskytne nová závažná infekcia, podávanie Cimzie sa má prerušiť, pokiaľ nie je táto infekcia zvládnutá. Lekári majú byť opatrní pri zvažovaní použitia Cimzie u pacientov
s opakovanou alebo oportúnnou infekciou v anamnéze alebo s prebiehajúcimi ochoreniami, ktoré môžu pacientov predisponovať na infekcie, vrátane súbežného používania imunosupresívnych liekov.
U pacientov s reumatoidnou artritídou sa nemusia prejaviť typické príznaky infekcie, vrátane horúčky, z dôvodu ich ochorenia a súbežne podávaných liekov. Preto je skoré odhalenie akejkoľvek infekcie, predovšetkým atypických klinických prejavov závažnej infekcie, veľmi dôležité, aby sa minimalizovalo oneskorenie diagnostiky a začiatku liečby.
U pacientov, ktorí používajú Cimziu, boli hlásené závažné infekcie, vrátane sepsy a tuberkulózy (vrátane miliárneho, diseminovaného a mimopľúcneho ochorenia) a oportúnne infekcie (napr. histoplazmóza, nokardia, kandidóza). Niektoré z týchto udalostí boli fatálne.
Tuberkulóza
Pred začiatkom liečby Cimziou sa musia všetci pacienti vyšetriť na aktívnu alebo inaktívnu (latentnú)
tuberkulóznu infekciu. Toto vyšetrenie má obsahovať podrobnú lekársku anamnézu u pacientov
s tuberkulózou v osobnej anamnéze, s možnou predchádzajúcou expozíciou iným jedincom s aktívnou tuberkulózou a s predchádzajúcim a/alebo súčasným používaním imunosupresívnej liečby. U všetkých pacientov sa majú urobiť príslušné skríningové vyšetrenia, napr. tuberkulínový kožný test a röntgen hrudníka (je potrebné postupovať podľa miestnych odporúčaní). Odporúča sa, aby sa vykonanie týchto vyšetrení zaznamenalo v kartičke s upozornením pre pacienta. Predpisujúcich lekárov to upozorní na riziko falošne negatívnych výsledkov tuberkulínového kožného testu, najmä u pacientov, ktorí sú závažne chorí alebo majú oslabený imunitný systém.
V prípade, že je aktívna tuberkulóza diagnostikovaná pred liečbou alebo počas nej, liečba Cimziou sa nesmie začať a musí sa prerušiť (pozri časť 4.3).
Pri podozrení na inaktívnu („latentnú“) tuberkulózu sa treba poradiť s lekárom s kvalifikáciou v liečbe tuberkulózy. Vo všetkých prípadoch popísaných nižšie sa má veľmi dôkladne zvážiť pomer prínosu
a rizika liečby Cimziou.
Ak sa stanoví diagnóza latentnej tuberkulózy, musí sa pred začiatkom liečby Cimziou začať príslušná antituberkulózna liečba v súlade s miestnymi odporúčaniami.
Použitie antituberkulóznej liečby sa má zvážiť tiež pred začatím liečby Cimziou u pacientov
s latentnou alebo aktívnou tuberkulózou v predchádzajúcej anamnéze, u ktorých nie je možné potvrdiť adekvátny priebeh liečby a u pacientov s významnými rizikovými faktormi tuberkulózy, napriek
negatívnemu vyšetreniu na latentnú tuberkulózu. Biologické testy pre skríning tuberkulózy sa majú zvážiť pred začiatkom liečby Cimziou, ak existuje možnosť výskytu latentnej tuberkulóznej infekcie,
bez ohľadu na BCG vakcináciu.
Napriek predchádzajúcej alebo súbežnej profylaktickej antituberkulóznej liečbe sa u pacientov liečených antagonistami TNF, vrátane Cimzie, vyskytli prípady aktívnej tuberkulózy. U niektorých pacientov úspešne liečených na aktívnu tuberkulózu sa opätovne vyvinula tuberkulóza počas liečby Cimziou.
Pacientov je potrebné poučiť, aby vyhľadali lekársku pomoc v prípade, že sa u nich počas liečby Cimziou alebo po jej skončení objavia prejavy/príznaky pripomínajúce tuberkulóznu infekciu (napr. pretrvávajúci kašeľ, chudnutie/zníženie hmotnosti, horúčka nízkeho stupňa, ľahostajnosť).
ReaktiváciavírusuhepatitídytypuB(HBV)
U pacientov liečených antagonistom TNF vrátane certolizumab pegolu, ktorí sú chronickými nosičmi tohto vírusu (t.j. pozitívny povrchový antigén) sa objavila reaktivácia hepatitídy B. Niektoré prípady mali fatálny následok.
Pred začatím liečby Cimziou sa majú pacienti vyšetriť na infekciu HBV. Pacientom s pozitívnym testom na infekciu HBV sa odporúča konzultácia s lekárom so skúsenosťami v liečbe hepatitídy B.
U nosičov HBV, ktorí si vyžadujú liečbu Cimziou, je potrebné počas liečby a niekoľko mesiacov po skončení liečby starostlivo sledovať výskyt znakov a príznakov aktívnej infekcie HBV. Nie sú dostupné dostatočné údaje o liečbe pacientov, ktorí sú nosičmi HBV a dostávajú antivírusovú liečbu v kombinácii s liečbou TNF-antagonistom na prevenciu reaktivácie HBV. U pacientov, u ktorých sa vyvinula reaktivácia HBV, je potrebné ukončiť podávanie Cimzie a má sa začať účinná antivírusová liečba s príslušnou podpornou liečbou.
Malignityalymfoproliferatívneporuchy
Potenciálna úloha liečby antagonistom TNF v rozvoji malignít nie je známa. Pri zvažovaní liečby antagonistom TNF u pacientov s malignitou v anamnéze alebo pri zvažovaní pokračovania v liečbe u pacientov, u ktorých sa rozvinula malignita, je potrebná opatrnosť.
Pri súčasných znalostiach nie je možné vylúčiť možné riziko rozvoja lymfómov, leukémie alebo iných malignít u pacientov liečených s antagonistom TNF.
V klinických štúdiách s Cimziou a inými antagonistami TNF bolo hlásených viac prípadov lymfómov a iných malignít medzi pacientmi užívajúcimi antagonisty TNF ako u kontrolných pacientov užívajúcich placebo (pozri časť 4.8). Po uvedení lieku na trh sa u pacientov liečených antagonistom TNF zaznamenali prípady leukémie. Existuje zvýšené riziko lymfómu a leukémie v pozadí
u pacientov s reumatoidnou artritídou s dlhotrvajúcim, vysoko aktívnym, zápalovým ochorením, ktoré komplikuje odhad rizika.
Neboli vykonané žiadne štúdie, ktoré zahŕňajú pacientov s anamnézou malignity alebo štúdie,
v ktorých liečba pokračovala u pacientov, u ktorých sa rozvinula malignita počas liečby Cimziou.
Kožné karcinómy
U pacientov liečených antagonistami TNF vrátane certolizumab pegolu sa zaznamenal melanóm
a karcinóm z Merkelových buniek (pozri časť 4.8). Odporúča sa pravidelné vyšetrenie kože, hlavne u pacientov s rizikovými faktormi pre kožný karcinóm.
Malignita u detí
U detí, dospievajúcich a mladých dospelých (do 22 rokov) liečených antagonistami TNF (začiatok liečby vo veku ≤ 18 rokov) sa po uvedení lieku na trh zaznamenali malignity, niekedy fatálne. Približne polovica prípadov boli lymfómy. Ďalšie prípady predstavovali rôzne druhy rozličných malignít a patrili medzi ne zriedkavé malignity zvyčajne súvisiace s imunosupresiou. Riziko rozvoja malignít u detí a dospievajúcich liečených antagonistami TNF nemožno vylúčiť.
U pacientov liečených antagonistami TNF sa po uvedení lieku na trh zaznamenali prípady hepatosplenického T-bunkového lymfómu (hepatosplenic T-cell lymphoma - HSTCL). Tento zriedkavý typ T-bunkového lymfómu má veľmi agresívny priebeh ochorenia a zvyčajne je smrteľný. Väčšina hlásených prípadov s antagonistami TNF sa vyskytovala u dospievajúcich a mladých dospelých mužov s Crohnovou chorobou alebo ulceróznou kolitídou. Takmer všetci títo pacienti užívali liečbu imunosupresívami azatioprinom a/alebo 6-merkaptopurínom súbežne s antagonistom TNF počas diagnózy alebo pred ňou. Riziko vývoja hepatosplenického T-bunkového lymfómu
u pacientov liečených Cimziou nemožno vylúčiť.
Chronickáobštrukčnáchorobapľúc(CHOCHP)
V prieskumnej klinickej štúdii hodnotiacej používanie iného antagonistu TNF, infliximabu, bolo
u pacientov so stredne závažnou až závažnou chronickou obštrukčnou chorobou pľúc (CHOCHP) liečených infliximabom hlásených viac malignít, najmä v pľúcach alebo v hlave a krku, v porovnaní s kontrolnými pacientmi. Všetci pacienti mali silné fajčenie v anamnéze. Preto pri používaní ktoréhokoľvek antagonistu TNF u pacientov so CHOCHP je potrebná opatrnosť, rovnako ako
u pacientov so zvýšeným rizikom malignít z dôvodu silného fajčenia.
Kongestívnesrdcovézlyhanie
Cimzia je kontraindikovaná v prípade stredne závažného alebo závažného srdcového zlyhania (pozri časť 4.3). V klinickej štúdii s iným antagonistom TNF sa pozorovalo zhoršenie kongestívneho
srdcového zlyhania a zvýšená mortalita v dôsledku kongestívneho srdcového zlyhania. Rovnako boli
prípady kongestívneho srdcového zlyhania hlásené aj u pacientov s reumatoidnou artritídou liečených Cimziou. U pacientov s miernym srdcovým zlyhaním (I/II trieda NYHA) sa má Cimzia používať opatrne. U pacientov, u ktorých sa objavia nové príznaky kongestívneho srdcového zlyhania alebo sa existujúce príznaky zhoršia, sa liečba Cimziou musí prerušiť.
Hematologickéreakcie
Hlásenia o pancytopénii, vrátane aplastickej anémie, boli pri antagonistoch TNF zriedkavé.
V súvislosti s Cimziou boli hlásené nežiaduce reakcie hematologického systému, vrátane medicínsky významnej cytopénie (napr. leukopénia, pancytopénia, trombocytopénia) (pozri časť 4.8). Je potrebné
upozorniť všetkých pacientov, aby okamžite vyhľadali lekársku pomoc, ak sa u nich počas liečby
Cimziou objavia prejavy a príznaky pripomínajúce krvnú dyskráziu alebo infekciu (napr. pretrvávajúca horúčka, podliatiny, krvácanie, bledosť). U pacientov s potvrdenými významnými hematologickými abnormalitami sa má zvážiť prerušenie liečby Cimziou.
Neurologickéudalosti
Používanie antagonistov TNF bolo v zriedkavých prípadoch spájané s novým nástupom alebo exacerbáciou klinických symptómov a/alebo rádiograficky potvrdeným demyelinizačným ochorením, vrátane sklerózy multiplex. U pacientov s preexistujúcim demyelizačným ochorením alebo s jeho nedávnym nástupom je potrebné pred začatím liečby Cimziou starostlivo zvážiť prínosy a riziká liečby
antagonistom TNF. U pacientov liečených Cimziou boli hlásené zriedkavé prípady neurologických porúch, vrátane záchvatovej poruchy, neuritídy a periférnej neuropatie.
Precitlivenosť
Po podaní Cimzie boli zriedkavo hlásené závažné reakcie z precitlivenosti. Niektoré z týchto reakcií sa vyskytli po prvom podaní Cimzie. Ak sa vyskytnú závažné reakcie, podávanie Cimzie sa má okamžite
prerušiť a začať príslušná liečba.
Údaje o používaní Cimzie u pacientov, u ktorých sa vyskytla závažná reakcia precitlivenosti na iného antagonistu TNF sú obmedzené, u týchto pacientov je potrebná opatrnosť.
Imunosupresia
Pretože tumor nekrotizujúci faktor (TNF) sprostredkováva zápal a modifikuje bunkové imunitné odpovede, pre antagonisty TNF, vrátane Cimzie, existuje možnosť, že spôsobia imunosupresiu,
pričom ovplyvnia ochranu hostiteľa proti infekciám a malignitám.
Autoimunita
Liečba Cimziou môže vyvolať tvorbu antinukleárnych protilátok (ANP) a menej často môže viesť k vzniku syndrómu, ktorý je podobný lupusu (pozri časť 4.8). Vplyv dlhodobej liečby Cimziou na
rozvoj autoimunitných ochorení nie je známy. Ak sa po liečbe Cimziou u pacienta objavia symptómy
pripomínajúce syndróm podobný lupusu, liečba sa musí prerušiť. Cimzia sa neskúmala špeciálne v populácii pacientov s lupusom (pozri časť 4.8).
Vakcinácie
Pacienti liečení Cimziou môžu byť očkovaní, s výnimkou očkovaní živými vakcínami. Nie sú dostupné žiadne údaje o odpovedi na očkovanie živými vakcínami alebo na sekundárny prenos
infekcie živými vakcínami u pacientov liečených Cimziou. Živé vakcíny sa nemajú podávať súbežne s
Cimziou.
V placebom kontrolovanej klinickej štúdii u pacientov s reumatoidnou artritídou sa pozorovala rovnaká protilátková odpoveď medzi liečebnými skupinami s Cimziou a s placebom pri podávaní pneumokokovej polysacharidovej vakcíny a chrípkovej vakcíny súbežne s Cimziou. Pacienti liečení Cimziou a súbežne metotrexátom mali nižšiu humorálnu odpoveď v porovnaní s pacientmi liečenými samotnou Cimziou. Klinický význam tohto nie je známy.
Súbežnépoužívaniesinýmibiologickýmilátkami
Závažné infekcie a neutropénia boli hlásené v klinických štúdiách so súbežným používaním anakinry
(antagonista interleukínu-1) alebo abataceptu (modulátor CD28) a iných antagonistov TNF, etanerceptu, ktoré však nepredstavovali žiadny ďalší prínos v porovnaní s liečbou samotným antagonistom TNF. Vzhľadom na povahu nežiaducich udalostí pozorovaných pri kombinácii iného antagonistu TNF s liečbou abataceptom alebo anakinrou môže kombinácia anakinry alebo abataceptu a iných antagonistov TNF viesť k podobným toxicitám. Preto sa používanie certolizumab pegolu v kombinácii s anakinrou alebo abataceptom neodporúča (pozri časť 4.5).
Chirurgickývýkon
U pacientov liečených Cimziou existujú s chirurgickými výkonmi obmedzené skúsenosti týkajúce sa bezpečnosti. Ak je plánovaný chirurgický výkon, je potrebné vziať do úvahy 14-dňový polčas certolizumab pegolu. Pacienta, u ktorého je nutný chirurgický výkon počas liečby Cimziou, je potrebné starostlivo sledovať kvôli výskytu infekcie a vykonať vhodné opatrenia.
Vyšetrenieaktivovanéhoparciálnehotromboplastínovéhočasu(aPTT)
U pacientov liečených Cimziou bola pozorovaná interferencia s niektorými koagulačnými vyšetreniami. Cimzia môže spôsobiť falošne zvýšené výsledky vyšetrenia aPTT u pacientov bez
koagulačných anomálií. Tento vplyv bol pozorovaný pri teste na lupus antikoagulans (PTT-LA)
a štandardných automatizovaných testoch aktivovaného parciálneho tromboplastínovéhu času (STA-PTT - Standard Target Activated Partial Thromboplastin time) od firmy Diagnostica Stago a testoch ”HemosIL APTT-SP liquid' a 'HemosIL lyophilized silica“ od firmy Instrumention
Laboratories. Rovnako môžu byť ovplyvnené aj iné aPTT vyšetrenia. Neexistuje dôkaz o tom, že liečba Cimziou má vplyv na koaguláciu in vivo. Po podaní Cimzie pacientom sa má interpretácii abnormálnych výsledkov koagulačných vyšetrení venovať zvýšená pozornosť. Nebola pozorovaná interferencia s vyšetrením trombínového času (TT) a protrombínového času (PT).
Staršípacienti
V klinických štúdiách bol u pacientov vo veku ≥ 65 rokov zjavne vyšší výskyt infekcií ako u mladších pacientov, hoci skúsenosti sú obmedzené. Opatrnosť je potrebná pri liečbe starších pacientov
a mimoriadnu opatrnosť je potrebné venovať výskytu infekcií.
4.5 Liekové a iné interakcie
Súbežná liečba s metotrexátom, kortikosteroidmi, nesteroidnými aniflogistikami (NSAID – nonsteroidal anti-inflammatory drugs) a analgetikami nepreukázala žiadny vplyv na farmakokinetiku certolizumab pegolu vychádzajúc z farmakokinetickej analýzy populácie.
Kombinácia certolizumab pegolu a anakinry alebo abataceptu sa neodporúča (pozri časť 4.4). Súbežné podávanie Cimzie s metotrexátom nemá žiadny významný vplyv na farmakokinetiku
metotrexátu. Pri porovnaní jednotlivých štúdií sa farmakokinetika certolizumab pegolu javila ako
porovnateľná s farmakokinetikou pozorovanou predtým u zdravých jedincov.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
U žien vo fertilnom veku je potrebné zvážiť použitie primeranej antikoncepcie. U žien, ktoré plánujú otehotnieť, vzhľadom na rýchlosť eliminácie lieku možno zvážiť pokračujúce používanie antikoncepcie počas 5 mesiacov po poslednej dávke Cimzie (pozri časť 5.2), ale je tiež potrebné vziať do úvahy potrebu liečby žien (pozri ďalej).
Gravidita
Údaje z vyše 500 prospektívne zozbieraných gravidít, ktoré boli vystavené účinkom Cimzie so známymi výsledkami gravidity, vrátane viac ako 400 gravidít vystavených Cimzii počas prvého
trimestra, nepoukazujú na malformačný účinok Cimzie. Dostupné klinické skúsenosti sú však príliš
obmedzené na vyvodenie záveru s odôvodnenou istotou, že s podávaním Cimzie počas gravidity nie je spojené zvýšené riziko.
Štúdie na zvieratách, v ktorých sa používal anti-rat TNFα hlodavcov, neodhalili žiadny dôkaz o poškodení fertility alebo poškodení plodu. Tieto informácie sú však nedostatočné čo sa týka reprodukčnej toxicity u ľudí (pozri časť 5.3). Z dôvodu jej inhibície TNFα by Cimzia podávaná počas gravidity mohla ovplyvňovať normálnu imunitnú odpoveď u novorodenca.
Cimzia sa má používať počas gravidity len v prípade, že je to klinicky potrebné.
Predklinické štúdie naznačujú nízku alebo zanedbateľnú úroveň placentárneho transferu homológneho
Fab-fragmentu certolizumab pegolu (bez Fc oblasti) (pozri časť 5.3).
V klinickej štúdii bolo 16 žien liečených certolizumab pegolom (200 mg každé 2 týždne alebo 400 mg každé 4 týždne) počas gravidity. Plazmatické koncentrácie certolizumab pegolu namerané u 14 dojčiat pri narodení boli pod hranicou kvantifikácie (BLQ - Below the Limit of Quantification) pri 13 vzorkách; jedna bola 0,042 µg/ml s plazmatickým pomerom dojča/matka pri narodení 0,09 %. Vo
4. týždni a v 8. týždni boli všetky koncentrácie u dojčiat BLQ. Klinický význam nízkych hladín certolizumab pegolu pre dojčatá nie je známy. Odporúča sa počkať minimálne 5 mesiacov po podaní poslednej dávky Cimzie matke počas gravidity pred podaním živej alebo živej oslabenej vakcíny (napr. vakcína BCG), ak prínos vakcinácie zjavne neprevyšuje teoretické riziko podania živej alebo živej oslabenej vakcíny dojčatám.
D
ojčenie
V klinickej štúdii bol u 17 dojčiacich žien liečených Cimziou pozorovaný minimálny transfer certolizumab pegolu z plazmy do materského mlieka. Percentuálny podiel materskej dávky certolizumab pegolu, ktorá prechádza na dojča počas 24 hodín, bol odhadnutý na 0,04 % až 0,30 %. Okrem toho, keďže certolizumab pegol je proteín, ktorý sa po perorálnom podaní rozkladá
v gastrointestinálnom trakte, predpokladá sa, že absolútna biologická dostupnosť bude u dojčeného dieťaťa veľmi nízka.
Cimzia sa preto môže používať počas dojčenia. Fertilita
Boli pozorované účinky na mieru motility spermií a tendencia zníženého počtu spermií u samcov
hlodavcov, ktoré však nemali dopad na ich fertilitu (pozri časť 5.3).
V klinickej štúdii na zhodnotenie účinku certolizumab pegolu na parametre kvality semena bolo 20 zdravých mužov randomizovaných na aplikáciu jednorazovej subkutánnej dávky 400 mg certolizumab pegolu alebo placeba. Počas 14-týždňového sledovania sa nepozorovali žiadne účinky liečby certolizumab pegolom na parametre kvality semena v porovnaní s placebom.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cimzia môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní Cimzie sa môže objaviť závrat (vrátane vertiga, poruchy videnia a vyčerpanosti) (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Reumatoidnáartritída
Cimzia bola skúmaná u 4 049 pacientov s reumatoidnou artritídou v kontrolovaných a nezaslepených štúdiách po dobu až 92 mesiacov. Údaje v tabuľke 1 vychádzajú v prvom rade z placebom
kontrolovaných štúdií, ktoré zahŕňali 2 965 pacientov používajúcich Cimziu a 1 137 pacientov
užívajúcich placebo počas kontrolovaného obdobia.
V placebom kontrolovaných štúdiách mali pacienti používajúci Cimziu približne 4-krát dlhšie trvanie expozície v porovnaní s pacientmi v placebovej skupine. Tento rozdiel v expozícii je predovšetkým spôsobený tým, že u pacientov užívajúcich placebo bola vyššia pravdepodobnosť skorého prerušenia štúdie. Okrem toho, v štúdiách RA-I a RA-II bolo prerušenie štúdie povinné pre pacientov bez odpovede v 16. týždni, z ktorých väčšina užívala placebo.
Počas kontrolovaných štúdií bol podiel pacientov, ktorí prerušili liečbu z dôvodu nežiaducich udalostí,
4,4 % u pacientov liečených Cimziou a 2,7 % u pacientov liečených placebom.
Najčastejšie nežiaduce reakcie patrili do triedy orgánových systémov Infekcie a nákazy, hlásené u
14,4 % pacientov liečených Cimziou a 8,0 % pacientov liečených placebom, do triedy Celkové poruchy a reakcie v mieste podania, hlásené u 8,8 % pacientov liečených Cimziou a 7,4 % pacientov
liečených placebom a do triedy Poruchy kože a podkožného tkaniva, hlásené u 7,0 % pacientov
liečených Cimziou a u 2,4 % pacientov liečených placebom.
Axiálnaspondylartritída
Cimzia sa skúmala u 325 pacientov s aktívnou axiálnou spondylartritídou v AS001 klinickej štúdii počas až 4 rokov, ktorá zahŕňala 24 týždňov trvajúcu placebom kontrolovanú fázu, po ktorej nasledovala 24 týdňov trvajúca zaslepená fáza a 156 týždňov trvajúca otvorená fáza. Bezpečnostný profil u pacientov s axiálnou spondylartritídou liečených Cimziou sa zhodoval s bezpečnostným profilom u reumatoidnej artritídy a s predchádzajúcimi skúsenosťami s Cimziou.
P
soriatická
artritída
Cimzia sa skúmala u 409 pacientov s psoriatickou artritídou v PsA001 klinickej štúdii počas až
4 rokov, ktorá zahŕňala 24 týždňov trvajúcu placebom kontrolovanú fázu, po ktorej nasledovala
24 týždňov trvajúca zaslepená fáza a 168 týždňov trvajúca otvorená fáza. Bezpečnostný profil
u pacientov s psoriatickou artritídou liečených Cimziou bol zhodný s bezpečnostným profilom pri reumatoidnej artritíde a s predchádzajúcimi skúsenosťami s Cimziou.
Zoznam nežiaducich reakcií v tabuľke
Nežiaduce reakcie hlásené v klinických štúdiách s reumatoidnou artritídou a prípady po uvedení lieku
na trh s minimálne možnou súvislosťou s Cimziou sú vymenované v tabuľke 1 nižšie, podľa frekvencie a triedy orgánových systémov. Frekvencie kategórií sú definované nasledovne: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000), neznáme (nie je možné odhadnúť z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1 Nežiaduce reakcie v klinických štúdiách a po uvedení lieku na trh
Trieda orgánových systémov Frekvencia Nežiaduce reakcie
Infekcie a nákazy Časté bakteriálne infekcie (vrátane abscesu), vírusové infekcie (vrátane herpesu zoster, papilomavírusu, chrípky)
Menej časté sepsa (vrátane multiorgánového zlyhania, septického šoku), tuberkulóza (vrátane miliárneho, diseminovaného a extrapulmonálneho ochorenia), mykotické infekcie (vrátane oportúnnych)
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Poruchy krvi a lymfatického systému
Menej časté malignity krvi a lymfatického systému (vrátane lymfómu a leukémie), tumory solídnych orgánov, nemelanómové karcinómy kože, prekancerózne lézie (vrátane orálnej leukoplakie, névu z melanocytov), benígne tumory a cysty (vrátane papilómu kože)
Zriedkavé gastrointestinálne tumory, melanóm
Neznáme karcinóm z Merkelových buniek*
Časté eozinofilné poruchy, leukopénia (vrátane neutropénie, lymfopénie)
Menej časté anémia, lymfadenopatia, trombocytopénia, trombocytóza
Zriedkavé pancytopénia, splenomegália, erytrocytóza, abnormálna morfológia bielych krviniek

Poruchy imunitného systému Menej časté vaskulitídy, lupus erythematosus, precitlivenosť na lieky (vrátane anafylaktického šoku), alergické poruchy, pozitívne autoprotilátky
Zriedkavé angioneurotický edém, sarkoidóza, sérová choroba, panikulitída (vrátane nodózneho erytému), zhoršenie príznakov dermatomyozitídy**
Poruchy endokrinného systému Zriedkavé poruchy štítnej žľazy
Poruchy metabolizmu a výživy Menej časté nerovnováha elektrolytov, dyslipidémia, poruchy chuti do jedla, zmeny telesnej hmotnosti
Zriedkavé hemosideróza
Psychické poruchy Menej časté úzkosť a poruchy nálady (vrátane súvisiacich symptómov)
Zriedkavé pokus o samovraždu, delírium, duševná porucha
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Poruchy nervového systému Časté bolesti hlavy (vrátane migrény), zmyslové anomálie
Menej časté periférne neuropatie, závrat, tremor
Zriedkavé záchvat, zápal kraniálneho nervu, porucha koordinácie alebo rovnováhy
Neznáme skleróza multiplex*, Guillainov-Barrého syndróm* Poruchy oka Menej časté porucha zraku (vrátane zhoršeného videnia), zápal
oka a očného viečka, porucha slzenia
Poruchy ucha a labyrintu Menej časté tinnitus, vertigo
Poruchy srdca a srdcovej činnosti
Menej časté kardiomyopatie (vrátane srdcového zlyhania), ischemické koronárne arteriálne poruchy, arytmie (vrátane atriálnej fibrilácie), palpitácie
Zriedkavé perikarditída, atrioventrikulárna blokáda
Poruchy ciev Časté Hypertenzia
Menej časté hemorágia alebo krvácanie (na ktoromkoľvek mieste), hyperkoagulácia (vrátane tromboflebitídy, pľúcnej embólie), synkopa, edém (vrátane periférneho, faciálneho), ekchymózy (vrátane hematómu, petechií)
Zriedkavé cerebrovaskulárna príhoda, arterioskleróza, Raynaudov fenomén, livedo reticularis, telangiektázia
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Poruchy obličiek a močových ciest
Poruchy reprodukčného systému a prsníkov
Menej časté astma a s ňou súvisiace symptómy, pleurálny
výpotok a symptómy, kongescia a zápal respiračného traktu, kašeľ
Zriedkavé intersticiálna pľúcna choroba, pneumonitída
Časté Nevoľnosť
Menej časté ascites, gastrointestinálna ulcerácia a perforácia, zápal gastrointestinálneho traktu (na ktoromkoľvek mieste), stomatitída, dyspepsia, abdominálna distenzia, orofaryngeálna suchosť
Zriedkavé odynofágia, hypermotilita
Časté hepatitída (vrátane zvýšenej hladiny pečeňových enzýmov)
Menej časté hepatopatia (vrátane cirhózy), cholestáza, zvýšená hladina bilirubínu v krvi
Zriedkavé Cholelitiáza
Časté Vyrážka
Menej časté alopécia, nový nástup alebo zhoršenie psoriázy
(vrátane palmoplantárnej pustulóznej psoriázy)
a s ňou súvisiace ochorenia, dermatitída a ekzém, porucha potnej žľazy, kožný vred, fotosenzitivita, akné, zmeny sfarbenia kože, suchá koža, poruchy nechtov a nechtového lôžka
Zriedkavé exfoliácia a deskvamácia kože, bulózne ochorenia, porucha štruktúry vlasov, Stevens-Johnsonov syndróm**, multiformný erytém**
Menej časté svalové poruchy, zvýšená hladina kreatínfosfokinázy v krvi
Menej časté porucha funkcie obličiek, krv v moči, symptómy močového mechúra a močovej rúry
Zriedkavé nefropatia (vrátane nefritídy)
Menej časté poruchy menštruačného cyklu a krvácania maternice
(vrátane amenorey), poruchy prsníkov
Zriedkavé sexuálna dysfunkcia
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Celkové poruchy a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Časté pyrexia, bolesť (na ktoromkoľvek mieste), asténia, pruritus (na ktoromkoľvek mieste), reakcie v mieste vpichu
Menej časté zimnica, ochorenie podobné chrípke, zmena vnímania teploty, nočné potenie, návaly tepla
Zriedkavé fistula (na akomkoľvek mieste)
Menej časté zvýšená hladina alkalickej fosfatázy v krvi, predĺženie času koagulácie
Zriedkavé zvýšená hladina kyseliny močovej v krvi
Menej časté kožné poranenia, zhoršené hojenie
*Tieto udalosti sa týkali triedy antagonistov TNF, ale výskyt v súvislosti s certolizumab pegolom nie je známy.
**Tieto udalosti sa týkali triedy antagonistov TNF.
V iných indikáciách boli v súvislosti s Cimziou pozorované menej často ďalšie nasledujúce nežiaduce reakcie: gastrointestinálna stenóza a obštrukcie, zhoršenie celkového zdravotného stavu, spontánny potrat a azoospermia.
Popis vybraných nežiaducichreakciíInfekcieV placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou bola miera incidencie nových prípadov infekcií u všetkých pacientov liečených Cimziou 1,03 na pacientorok a u pacientov
liečených placebom 0,92 na pacientorok. Z infekcií boli zastúpené predovšetkým infekcie horných
dýchacích ciest, infekcie močového traktu a infekcie dolných dýchacích ciest a herpetické vírusové infekcie (pozri časti 4.3 a 4.4).
V placebom kontrolovaných klinických štúdiách bolo v skupinách liečených Cimziou (0,07 na pacientorok; pri všetkých dávkach) viac nových prípadov závažnej infekcie v porovnaní s placebom (0,02 na pacientorok). Medzi najčastejšie závažné infekcie patrili pneumónia, tuberkulózne infekcie. Medzi závažné infekcie patrili aj invazívne oportúnne infekcie (napr. pneumocystóza, mykotická ezofagitída, nokardióza a diseminovaný herpes zoster). Neexistuje dôkaz o zvýšenom riziku infekcií pri nepretržitej expozícii v priebehu času (pozri časť 4.4).
MalignityalymfoproliferatívneporuchyS výnimkou nemelanómových nádorov kože bolo pozorovaných 121 malignít vrátane 5 prípadov lymfómu v klinických RA štúdiách s Cimziou, v ktorých bolo liečených celkovo 4 049 pacientov, čo
predstavuje 9 277 pacientorokov. V klinických štúdiách s reumatoidnou artritídou sa v súvislosti
s Cimziou vyskytovali prípady lymfómu v rozsahu incidencie 0,05 na 100 pacientorokov a melanómu v rozsahu incidencie 0,08 na 100 pacientorokov (pozri časť 4.4). V klinickej štúdii fázy III
s psoriatickou artritídou sa pozoroval aj jeden prípad lymfómu.
AutoimunitaZ pacientov, ktorí boli na začiatku ANP negatívni, malo v pivotných štúdiách pozitívne ANP titre
16,7 % pacientov liečených Cimziou v porovnaní s 12,0 % pacientov v placebovej skupine. V rámci pacientov, ktorí mali na začiatku negatívne protilátky anti-dsDNA, u 2,2 % pacientov liečených Cimziou sa vyskytli pozitívne titre protilátok anti-dsDNA v porovnaní s 1,0 % pacientov v placebovej skupine. V obidvoch placebom kontrolovaných a nezaslepených následných klinických štúdiách s reumatoidnou artritídou boli menej často hlásené prípady syndrómu podobného lupusu. Zriedkavo boli hlásené iné imunologicky sprostredkované ochorenia; príčinná súvislosť s Cimziou nie je známa. Vplyv dlhodobej liečby Cimziou na vývoj autoimunitných ochorení nie je známy.
R
eakcie
v
m
i
este
p
odania
injekcie
V placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou sa reakcie v mieste podania injekcie vyskytovali u 5,8 % pacientov liečených Cimziou, ako je erytém, svrbenie, hematóm, bolesť, opuch alebo podliatina, v porovnaní s 4,8 % pacientov liečených placebom. Bolesť v mieste podania injekcie sa pozorovala u 1,5 % pacientov liečených Cimziou, pričom ani v jeden prípad nemal za následok prerušenie liečby.
ZvýšeniekreatínfosfokinázyFrekvencia zvýšenia kreatínfosfokinázy (CPK) bola celkovo vyššia u pacientov s axSpA v porovnaní s RA populáciou. Frekvencia bola zvýšená u pacientov liečených placebom (2,8 % oproti 0,4 %
v populácii s axSpA a RA, v uvedenom poradí), aj u pacientov liečených Cimziou (4,7 % oproti 0,8 % v populácii s axSpA a RA, v uvedenom poradí). Zvýšenie CPK v štúdii axSpA bolo väčšinou mierne až stredne závažné, prechodnej povahy a neznámeho klinického významu, pričom ani v jednom
prípade toto zvýšenie neviedlo k vysadeniu liečby.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePočas klinických štúdií sa nepozorovala žiadna toxicita limitujúca dávku. Podávané boli viacnásobné dávky až do 800 mg subkutánne a 20 mg/kg intravenózne. V prípade predávkovania sa odporúča
u pacientov starostlivé sledovanie akýchkoľvek nežiaducich reakcií alebo účinku a okamžité začatie
vhodnej symptomatickej liečby.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNFα), ATC kód: L04AB05
Mechanizmus účinkuCimzia má vysokú afinitu k ľudskému TNFα a viaže sa s disociačnou konštantou (KD) 90 pM. TNFα
je kľúčovým prozápalovým cytokínom s najdôležitejšou úlohou pri zápalových procesoch. Cimzia selektívne neutralizuje TNFα (IC90 4 ng/ml pre inhibíciu ľudského TNFα v
in vitro hodnotení L929
fibrosarkómovej cytotoxicity u myší), ale neneutralizuje lymfotoxín α (TNFβ).
Ukázalo sa, že Cimzia neutralizuje rozpustný ľudský TNFα spojený s membránami, spôsobom závislým od dávky. Inkubácia monocytov s Cimziou viedla k inhibícii lipopolysacharidmi (LPS) indukovanej produkcie TNFα a IL1β v ľudských monocytoch, ktorá bola závislá od dávky.
Cimzia neobsahuje oblasť fragmentu schopného kryštalizácie (Fc), ktorá je zvyčajne súčasťou kompletnej protilátky a preto neviaže komplement ani nevyvoláva od protilátky závislú bunkami sprostredkovanú cytotoxicitu
in vitro. Neindukuje apoptózu
in vitro v ľudských monocytoch ani lymfocytoch pochádzajúcich z periférnej krvi, ani degranuláciu neutrofilov.
Klinická účinnosťReumatoidnáartritídaÚčinnosť a bezpečnosť Cimzie boli hodnotené v 2 randomizovaných, placebom kontrolovaných, dvojito zaslepených klinických štúdiách u pacientov vo veku ≥ 18 rokov s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR (American College of Rheumatology) kritérií, RA-I (RAPID
1) a RA-II (RAPID 2). Každý pacient mal 9 alebo viac opuchnutých a citlivých kĺbov a trpel aktívnou RA minimálne 6 mesiacov pred začiatkom štúdie. V obidvoch štúdiách bola Cimzia podávaná subkutánne v kombinácii s perorálnym MTX počas minimálne 6 mesiacov v stabilných dávkach minimálne 10 mg týždenne po dobu 2 mesiacov. S používaním Cimzie v kombinácii s DMARD okrem MTX nie sú žiadne skúsenosti.
Účinnosť a bezpečnosť Cimzie boli hodnotené u dospelých pacientov doteraz neliečených liekmi zo skupiny DMARD (tzv. DMARD-naivní pacienti) s aktívnou reumatoidnou artritídou
v randomizovanej, placebom kontrolovanej, dvojito zaslepenej klinickej štúdii (C-EARLY). V štúdii
C-EARLY boli pacienti vo veku ≥ 18 rokov, ktorí mali ≥ 4 opuchnuté a bolestivé kĺby a diagnóza stredne závažnej až závažnej progresívnej reumatoidnej artritídy u nich musela byť stanovená
v priebehu 1 roka (podľa definície klasifikačných kritérií ACR/EULAR (European League Against
Rheumatism) z roku 2010). Priemerný čas od diagnózy bol u pacientov vo východiskovom bode 2,9
mesiaca a neboli doteraz liečení pomocou DMARD (vrátane MTX). V ramene s Cimziou a v ramene s placebom bola liečba MTX začatá od 0. týždňa (10 mg/týždeň), dávka bola titrovaná až do maximálne tolerovanej dávky v 8. týždni (povolených min. 15 mg/týždeň, max. 25 mg/týždeň)
a udržiavaná v priebehu štúdie (priemerná dávka MTX po 8. týždni bola pre placebo 22,3 mg/týždeň, pre Cimziu 21,1 mg/týždeň).
Tabuľka 2 Popis klinickej štúdie
Č
íslo št
údie
P
očty pacientov
A
ktívna schéma dávkovania
C
i
ele štúdie
RA-I (52 týždňov)
RA-II (24 týždňov)
C-EARLY (do 52. týždňa
982 400 mg (v 0.,2.,4. týždni) s MTX
200 mg alebo 400 mg každé 2 týždne s MTX
619 400 mg (v 0.,2.,4. týždni) s MTX

200 mg alebo 400 mg
každé 2 týždne s MTX
879 400 mg (v 0., 2., 4. týždni)
200 mg každé
2 týždne s MTX
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Súbežne primárne koncové ukazovatele: ACR 20 v 24. týždni a zmena
z východiskovej hodnoty mTSS v 52. týždni
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Primárny výsledný ukazovateľ: ACR 20
v 24. týždni.
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia
u DMARD-naivných pacientov.
Primárny výsledný ukazovateľ: podiel subjektov s pretrvávajúcou remisiou* v 52. týždni
mTSS: modifikované TSS (Total Sharp Score)
*Pretrvávajúca remisia v 52. týždni je definovaná ako DAS28[ESR] < 2,6 v 40. týždni aj v 52. týždni.
Prejavy a príznaky
Výsledky klinických štúdií RA-I a RA-II sú uvedené v tabuľke 3. V oboch klinický štúdiách sa v porovnaní s placebom dosiahli štatisticky významne vyššie odpovede ACR 20 od 1. týždňa
a ACR 50 od 2. týždňa. Odpovede pretrvávali až do 52. týždňa (RA-I) a 24. týždňa (RA-II). Zo 783
pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo 52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie. Z nich 427 pacientov
ukončilo 2-ročnú nezaslepenú následnú štúdiu a tak mali celkovú expozíciu Cimzii spolu 148 týždňov.
Pozorovaná miera odpovede ACR 20 v tomto časovom bode bola 91 %. Zníženie (RA-I)
z východiskovej hodnoty DAS28 (ESR) bolo tiež v porovnaní s placebom významne vyššie
(p < 0,001) v 52. týždni (RA-I) a 24. týždni (RA-II) a pretrvávalo počas 2 rokov v nezaslepenej predĺženej štúdii RA-I.
T
abuľka 3 ACR odpoveď v klinických štúdiách RA-I a RA-II Štúdia RA-I
Štúdia RA-II
K
ombinácia s metotrexátom
(
24 a 52 týždňov)
K
ombinácia s metotrexátom
(
24 týždňov)
Odpoveď Placebo + MTX N = 199
ACR 20
Cimzia
200 mg + MTX
každé 2 týždne
N = 393
Placebo + MTX N = 127
Cimzia
200 mg + MTX
každé 2 týždne
N = 246
24. týždeň 14 % 59 %** 9 % 57 %**
52. týždeň 13 % 53 %** N/A N/A
ACR 50
24. týždeň 8 % 37 %** 3 % 33 %**
52. týždeň 8 % 38 %** N/A N/A
ACR 70
24. týždeň 3 % 21 %** 1 % 16 %*
52. týždeň 4 % 21 %** N/A N/A
Hlavná klinická odpoveďa.
1 % 13 %**
Cimzia verzus placebo: * p ≤ 0,01, ** p < 0,001
a. Hlavná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 pri každom hodnotení počas nepretržitého 6-mesačného obdobia.
p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
Percentuálna odpoveď založená na počte pacientov, ktorí prispeli údajmi (n) pre tento koncový a časový ukazovateľ, ktorý sa môže odlišovať od N.
Štúdia C-EARLY splnila svoje primárne a kľúčové sekundárne výsledné ukazovatele. Kľúčové výsledky zo štúdie sú uvedené v tabuľke 4.
Tabuľka 4 Štúdia C-EARLY: percento pacientov s trvajúcou remisiou a trvajúcou nízkou aktivitou ochorenia v 52. týždni
Odpoveď Placebo + MTX N= 213
Cimzia 200 mg + MTX N= 655
P
r
etrvávajúca remisia*
(DAS28(ESR) <2,6 v
40. týždni aj 52. týždni)
Pretrvávajúca nízka aktivita ochorenia
(DAS28(ESR) ≤3,2 v 40. týždni aj v 52. týždni)
15,0 % 28,9%**
28,6 % 43,8%**


*Primárny výsledný ukazovateľ štúdie C-EARLY (do 52. týždňa)
Zostava úplnej analýzy, prirátanie nonrespondérov pre chýbajúce hodnoty.
**Cimzia+MTX vs placebo+MTX: p<0,001
p-hodnota bola odhadnutá z logistického regresného modelu s faktormi pre liečbu, miesto a dobu od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4 mesiace)
U pacientov v skupine Cimzia+MTX došlo k výraznejšej redukcii od východiskového stavu
v DAS 28 (ESR) v porovnaní so skupinou placebo+MTX, ktorá bola pozorovaná už od 2. týždňa a pokračovala do 52. týždňa (p<0,001 pri každej kontrole). Hodnotenia remisie (DAS 28 (ESR)
<2,6), stavu nízkej aktivity ochorenia (DAS 28 (ESR) ≤3,2) a ACR 50 a ACR 70 podľa kontrol ukázali, že liečba Cimzia+MTX viedla k rýchlejšej a výraznejšej odpovedi ako liečba
PBO+MTX. Tieto výsledky u DMARD-naivných subjektov pretrvávali počas 52 týždňov liečby.
R
ádiografická odpoveď
V RA-I bolo štrukturálne poškodenie kĺbu hodnotené rádiograficky v 52. týždni a vyjadrené ako
zmena mTSS a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (JSN – joint space narrowing score) v porovnaní s východiskovými hodnotami. Pacienti liečení Cimziou preukázali významne
menšiu rádiografickú progresiu ako pacienti užívajúci placebo v 24. týždni a 52. týždni (pozri tabuľku
5). V placebovej skupine v 52. týždni bolo 52 % pacientov bez rádiografickej progresie (mTSS ≤ 0,0)
v porovnaní so 69 % v skupine liečenej Cimziou v dávke 200 mg.
a.
Tabuľka 5 Zmeny v priebehu 12 mesiacov v RA-I
m
T
SS
P
l
acebo + MTX N = 199
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX N = 393
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX – Placebo + MTX
Stredná hodnota rozdielu
52. týždeň 2,8 (7,8) 0,4 (5,7) -2,4
Skóre erózie
52. týždeň 1,5 (4,3) 0,1 (2,5) -1,4
JSN skóre
52. týždeň 1,4 (5,0) 0,4 (4,2) -1,0
p-hodnoty v prípade obidvoch – mTSS aj skóre erózie – boli < 0,001 a v prípade JSN skóre boli
≤ 0,01. ANCOVA zodpovedala hodnotenej zmene od východiskovej hodnoty pre každé meranie s oblasťou a liečbou ako faktormi a východiskovým hodnotením ako kovariátom.
Zo 783 pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo
52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie.
V podskupine 449 pacientov, ktorí ukončili minimálne 2-ročnú liečbu Cimziou (RA-I a nezaslepená predĺžená štúdia), sa preukázala pretrvávajúca inhibícia progresie štrukturálneho poškodenia
s hodnotiteľnými údajmi v časovom bode 2 roky.
V štúdii C-EARLY viedla Cimzia+MTX k inhibícii rádiografickej progresie v porovnaní
s placebom+MTX v 52. týždni (pozri tabuľku 6). V skupine placebo+MTX u 49,7 % pacientov nedošlo k žiadnej rádiografickej progresii (zmena mTSS ≤0,5) v 52. týždni, v porovnaní s 70,3 %
v skupine Cimzia+MTX (p<0,001).
Tabuľka 6 Rádiografické zmeny v 52. týždni v štúdii C-EARLY
P
l
acebo + MTX
N= 163
Priemer (SD)
Cimzia 200 mg + MTX
N = 528
Priemer (SD)
Cimzia 200 mg + MTX – Placebo + MTX Rozdiel*
m
T
SS
týždeň 52
Skóre erózie
týždeň 52
Skóre JSN
týždeň 52
1,8 (4,3) 0,2 (3,2)** -0,978 (-1,005, -0,500)
1,1 (3,0) 0,1 (2,1)** -0,500 (-0,508, -0,366)
0,7 (2,3) 0,1 (1,7)** 0,000 (0,000, 0,000)
Rádiografická zostava s lineárnou extrapoláciou.
* Hodges-Lehmannov bodový odhad posunu a 95 % asymptotický (Moses) interval spoľahlivosti.
**Cimzia+MTX vs. placebo+MTX p<0,001. p-hodnota bola odhadnutá z ANCOVA modelu hodnotenia liečby, oblasti a času od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4 mesiace) ako faktory a hodnoty vo východiskovom stave ako kovariancia.
Odpoveď fyzickej funkcie a následky týkajúce sa zdraviaU pacientov liečených Cimziou v RA-I a RA-II bolo už od 1. týždňa až do konca štúdií, v porovnaní s placebom, hlásené významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI (Health Assessment Questionnaire – Disability Index) a únavy (vyčerpanosti) hlásenej podľa škály FAS (Fatigue Assessment Scale). V obidvoch klinických štúdiách hlásili pacienti liečení Cimziou významne väčšie zlepšenia v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a v skóre všetkých domén. Zlepšenia fyzickej funkcie a HRQoL pretrvávali
v priebehu 2 rokov v nezaslepenej predĺženej štúdii RA-I. Pacienti liečení Cimziou hlásili, v porovnaní
s placebom, štatisticky významné zlepšenia v prieskume produktivity práce (Work Productivity
Survey).
V štúdii C-EARLY pacienti liečení kombináciou Cimzia+MTX hlásili významné zlepšenie bolesti
v 52. týždni v porovnaní s kombináciou placebo+MTX, hodnotenej pomocou Hodnotenia artritickej bolesti pacientom (Patient Assessment of Arthritis Pain = PAAP) - 48,5 vs - 44,0 (priemer najmenších
štvorcov) (p<0,05).
Klinická štúdia DoseFlex
Účinnosť a bezpečnosť 2 dávkovacích režimov Cimzie (200 mg každé 2 týždne a 400 mg každé 4
týždne) oproti placebu sa hodnotili v 18-týždňovej, nezaslepenej, predrandomizačnej a 16-týždňovej randomizovanej, dvojito-zaslepenej, placebom kontrolovanej klinickej štúdii u dospelých pacientov
s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR kritérií, ktorí nemali dostatočnú
odpoveď na MTX.
Pacienti dostávali nárazové dávky 400 mg Cimzie v 0., 2. a 4. týždni, po ktorých nasledovala dávka
200 mg Cimzie každé 2 týždne počas začiatočného nezaslepeného obdobia. Respondéri (ACR 20
dosiahnuté) v 16. týždni boli randomizovaní v 18. týždni na Cimziu v dávke 200 mg každé 2 týždne, Cimziu v dávke 400 mg každé 4 týždne alebo placebo v kombinácii s MTX počas ďalších 16 týždňov
(celková dĺžka štúdie: 34 týždňov). Tieto 3 skupiny boli dobre vyvážené čo sa týka klinickej odpovede
po aktívnej vstupnej fáze (ACR 20: 83-84 % v 18. týždni).
Primárnym koncovým ukazovateľom štúdie bola miera ACR 20 respondérov v 34. týždni. Výsledky v v 34. týždni sú uvedené v tabuľke 7. Obidva dávkovacie režimy Cimzie preukázali pretrvávajúcu klinickú odpoveď a boli štatisticky významné v porovnaní s placebom v 34. týždni. Koncový ukazovateľ ACR 20 sa dosiahol pre oba režimy Cimzie, 200 mg každé 2 týždne a 400 mg každé 4 týždne.
Tabuľka 7 ACR odpoveď v klinickej štúdii DoseFlex v 34. týždni
L
i
ečebný režim v 0. až
16. týždni
C
im
z
i
a 400 mg + MTX v 0., 2. a 4. týždni, po ktorých nasledovala Cimzia v dávke 200 mg + MTX každé 2 týždne
R
andomizovaný, dvojito- zaslepený liečebný režim v 18. až 34. týždni
AC
R 20
p-hodnota*
ACR 50
p-hodnota*
ACR 70
p-hodnota*
N/A: neaplikovateľné
Placebo + MTX
N=69
45 % N/A
30 % N/A
16 % N/A
Cimzia
200 mg + MTX každé 2 týždne N=70
67 %
0,009
50 %
0,020
30 %
0,052
Cimzia
400 mg + MTX každé 4 týždne N=69
65 %
0,017
52 %
0,010
38 %
0,005
* p-hodnoty Waldovho testu pre porovnania Cimzie v dávke 200 mg oproti placebu a Cimzie v dávke
400 mg oproti placebu sú odhadované na základe logistickej regresie s faktormi pre liečbu.
AxiálnaspondylartritídaÚčinnosť a bezpečnosť Cimzie boli hodnotené v jednej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AS001) u 325 pacientov vo veku ≥ 18 rokov s aktívnou axiálnou spondylartritídou s nástupom v dospelosti trvajúcou najmenej 3 mesiace definovanou klasifikačnými kritériami spoločnosti ASAS (Assessment of Spondyloarthritis International Society). Celková populácia s axiálnou spondylartritídou zahŕňala subpopulácie s röntgenologickým dôkazom ankylozujúcej spondylitídy (AS) a bez neho (axiálna spondylartritída bez röntgenologického dôkazu;
[nr-axSpA]). Pacienti mali aktívne ochorenie definované indexom aktivity ochorenia ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥ 4, bolesť chrbtice ≥ 4
na číselnej stupnici NRS (Numerical Rating Scale) s rozsahom 0 až 10 a zvýšené CRP alebo súčasný
dôkaz sakroilitídy pri vyšetrení magnetickou rezonanciou (MR). Pacienti museli mať prejavy neznášanlivosti alebo museli mať nedostatočnú odpoveď aspoň na jedno NSA. Celkovo 16 %
pacientov malo v anamnéze liečbu antagonistom TBF. Pacienti boli liečení nárazovou dávkou Cimzie
400 mg v 0., 2. a 4. týždni (u oboch liečebných skupín) alebo placebom, po ktorom nasledovala dávka Cimzie buď 200 mg každé 2 týždne alebo 400 mg každé 4 týždne alebo placebo. 87,7 % pacientov dostávalo súčasne NSA. Primárnym výsledným ukazovateľom účinnosti bola miera odpovede ASAS20 v 12. týždni.
Po 24 týždňovej, dvojito zaslepenej, placebom kontrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 156 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 204 týždňov. Všetci pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
199 účastníkov klinického skúšania (61,2 % z randomizovaných účastníkov) ukončilo štúdiu do týždňa 204.
Kľúčové výsledky účinnosti
V klinickej štúdii AS001 dosiahlo ASAS20 odpoveď v 12. týždni 58 % pacientov liečených Cimziou v dávke 200 mg každé 2 týždne a 64 % pacientov liečených Cimziou v dávke 400 mg každé 4 týždne
v porovnaní s 38 % pacientmi užívajúcimi placebo (p < 0,01). V celkovej populácii bol podiel
ASAS20 respondérov klinicky významný a významne vyšší pre liečebnú skupinu s Cimziou v dávke
200 mg každé 2 týždne a s Cimziou 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej kontrole od 1. týždňa až do 24. týždňa (p ≤ 0,001 pri každej kontrole). V 12. týždni a 24. týždni
bolo percento osôb s ASAS40 odpoveďou vyššie v skupinách liečených Cimziou v porovnaní s placebom.
Podobné výsledky sa dosiahli u oboch subpopulácií s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu. U žien neboli ASAS20 odpovede štatisticky významne odlišné od placeba až do časového bodu po 12. týždni.
Zlepšenie ASAS 5/6, čiastočná remisia a BASDAI-50 boli štatisticky významné v 12. týždni a v
24. týždni a pretrvávali až do 48. týždňa v celkovej populácii rovnako ako v subpopuláciách. Kľúčové výsledky účinnosti z klinickej štúdie AS001 sú uvedené v tabuľke 8.
U pacientov, ktorí zostali v štúdii, sa zlepšenie vo všetkých vyššie uvedených kľúčových výsledkoch účinnosti zachovalo do týždňa 204 v celkovej populácii, rovnako ako v subpopuláciách.
T
abuľka 8 Kľúčové výsledky účinnosti v klinickej štúdii AS001 (percento pacientov)
Parametre
Ankylozujúca spondylitída
Axiálna spondylartritída bez röntgenologického dôkazu
Axiálna spondylartritída Celková populácia
A
SAS20
(b
,c
)
12. týždeň
24. týždeň
ASAS40(c,d)
12. týždeň
24. týždeň
ASAS 5/6(c,d)
12. týždeň
24. týždeň
Čiastočná remisia(c,d)
12. týždeň
24. týždeň
BASDAI 50(c,d)
12. týždeň
24. týždeň
Placebo
N=57
37%
33%
19%
16%
9%
5%
2%
7%
11%
16%
Cimzia všetky režimy dávkovania (a)
N=121
60%*
69%**
45%**
53%**
42%**
40%**
20%**
28%**
41%**
49%**
Placebo
N=50
40%
24%
16%
14%
8%
4%
6%
10%
16%
20%
Cimzia všetky režimy
dávkovania (a)
N=97
61%*
68%**
47%**
51%**
44%**
45%**
29%**
33%**
49%**
57%**
Placebo
N=107
38%
29%
18%
15%
8%
5%
4%
9%
13%
18%
Cimzia všetky režimy
dávkovania (a)
N=218
61%**
68%**
46%**
52%**
43%**
42%**
24%**
30%**
45%**
52%**

(a) Cimzia všetky režimy dávkovania = údaje pre Cimziu v dávke 200 mg podávanej každé 2 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni plus Cimzia 400 mg podávaná každé 4 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni
(b) Výsledky sú z randomizovaného súboru
(c) p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
(d) Kompletný analyzovaný súbor
NA = nie je k dispozícii
* p ≤ 0,05, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
Mobilita chrbticeMobilita chrbtice sa hodnotila v dvojito zaslepenej, placebom kontrolovanej fáze pomocou BASMI
v niekoľkých časových bodoch, vrátane východiskového stavu, v 12. týždni a v 24. týždni. Klinicky významné a štatisticky významné rozdiely u pacientov liečených Cimziou v porovnaní s pacientmi
liečenými placebom sa preukázali pri každej kontrole od východiskového stavu. Rozdiel od placeba
má tendenciu byť väčší u subpopulácie nr-axSpA ako u AS, čo môže byť dôsledkom menšieho chronického štrukturálneho poškodenia u pacientov s nr-axSpA.
Zlepšenie lineárneho skóre BASMI dosiahnuté v 24. týždni sa u pacientov, ktorí zostali v štúdii,
udržalo až do týždňa 204.
Odpoveď fyzickej funkcie a následky týkajúce sa zdraviaV klinickej štúdii AS001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotené pomocou BASFI a zlepšenie bolesti hodnotené NRS stupnicou celkovej a nočnej bolesti
chrbtice v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie únavy
(vyčerpanosti) hlásené pomocou položky BASDAI pre únavu a významné zlepšenie kvality života súvisiacej so zdravím merané dotazníkom Kvality života pri ankylozujúcej spondylitíde (ASQoL) a
zlepšenie v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a
v skóre všetkých domén v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie
produktivity v práci a v domácnosti súvisiacej s axiálnou spondylartritídou, ktoré bolo hlásené v prieskume produktivity práce v porovnaní s placebom.
U pacientov, ktorí zostali v štúdii, bolo zlepšenie všetkých vyššie uvedených výsledkov do značnej
miery zachované až do týždňa 204.
Inhibícia zápalu pri magnetickej rezonancii (MR)
V zobrazovacej subštúdii zahŕňajúcej 153 pacientov sa hodnotili prejavy zápalu hodnotené MR
v 12. týždni a boli vyjadrené ako zmena skóre SPARCC (Spondyloarthritis Research Consortium of
Canada) oproti východiskovému stavu pre krížovo-driekové kĺby a skóre ASspiMRI-a v berlínskej modifikácii pre chrbticu. Významná inhibícia zápalových príznakov u krížovo-driekových kĺbov aj chrbtice sa pozorovala u pacientov liečených Cimziou (všetky dávkové skupiny) v 12. týždni, a to
v celkovej populácii s axiálnou spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu.
U pacientov, ktorí zostali v štúdii a u ktorých boli k dispozícii východiskové hodnoty aj hodnoty z týždňa 204, bola do značnej miery zachovaná inhibícia zápalových príznakov až do týždňa 204
v krížovo-driekových kĺboch (n=72) aj chrbtici (n=82), a to v celkovej populácii s axiálnou spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu.
Psoriatickáartritída
Účinnosť a bezpečnosť Cimzie sa hodnotili v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii (PsA001) u 409 pacientov vo veku ≥ 18 rokov s aktívnou psoriatickou artritídou s nástupom v dospelosti trvajúcou najmenej 6 mesiacov definovanou klasifikačnými kritériami pre psoriatickú artritídu (Classification Criteria for Psoriatic Arthritis - CASPAR). Pacienti mali ≥ 3 opuchnuté a bolestivé kĺby a zvýšené hodnoty markerov akútnej fázy. Pacienti mali tiež aktívne psoriatické kožné lézie alebo zdokumentovanú psoriázu v anamnéze a zlyhala u nich jedna alebo viac terapií DMARD. Predchádzajúca liečba jedným antagonistom TNF bola povolená a 20 % pacientov bolo predtým liečených TNF-antagonistom. Pacienti dostávali nárazovú dávku Cimzie 400 mg v 0., 2. a 4. týždni (v oboch liečebných skupinách) alebo placebo, po ktorej nasledovalo podávanie 200 mg Cimzie každé 2 týždne alebo 400 mg Cimzie každé 4 týždne alebo placebo každé 2 týždne. 72,6 % pacientov súbežne užívalo NSA a 70,2 % konvenčné DMARD. Štúdia mala dva primárne koncové ukazovatele: percento pacientov, ktorí dosiahli ACR 20 odpoveď
v 12. týždni a zmena modifikovaného skóre mTSS (Total Sharp Score) v 24. týždni od východiskovej hodnoty. Účinnosť a bezpečnosť Cimzie u pacientov s PsA, u ktorých prevládali symptómy sakroilitídy alebo axiálnej spondylartritídy, neboli analyzované samostatne.
Po 24 týždňovej dvojito zaslepenej, placebom kotrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 168 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 216 týždňov. Všetci
pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
264 účastníkov klinického skúšania (64,5 %) ukončilo štúdiu až v týždni 216.
ACR odpoveď
Pacienti liečení Cimziou mali štatisticky významne vyššiu mieru ACR 20 odpovede v 12. týždni
a v 24. týždni v porovnaní s pacientmi, ktorým bolo podávané placebo (p < 0,001). Percento ACR 20 respondérov bolo klinicky významné v liečebných skupinách s Cimziou v dávke 200 mg každé 2 týždne a s Cimziou v dávke 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej návšteve od začiatku až do 24. týždňa (nominálne p ≤ 0,001 pri každej návšteve). Pacienti liečení Cimziou mali tiež významné zlepšenie miery odpovede ACR 50 a 70. V 12. a 24. týždni sa
u pacientov liečených Cimziou pozorovalo zlepšenie v parametroch periférnej aktivity psoriatickej artritídy (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída) (nominálna p-hodnota p < 0,01).
Kľúčové výsledky účinnosti z klinickej štúdie PsA001 sú uvedené v tabuľke 9.
T
abuľka 9 Kľúčové výsledky účinnosti v klinickej štúdii PsA001 (percento pacientov)
O
dpoveď Placebo
N
=136
C
im
z
i
a
(
a)
200 mg každé 2 týždne
N
=138
C
im
z
i
a
(b
)
400 mg každé 4 týždne
N
=135
ACR
20
12. týždeň
24. týždeň
ACR5012. týždeň
24. týždeň
ACR7012. týždeň
24. týždeň
24 %
24 %
11 %
13 %
3 %
4 %
58 %**
64 %**
36 %**
44 %**

25 %**
28 %**
52 %**
56 %**
33 %**
40 %**
13 %*
24 %**
O
dpoveď Placebo
N
=86
C
im
z
i
a
(
a)
200 mg každé 2 týždne N=90
C
im
z
i
a
(b
)
400 mg každé 4 týždne N=76
P
A
SI 75 (c)
12. týždeň
24. týždeň
48. týždeň
14 %
15 % N/A
47 %***
62 %***
67 %
47 %***
61 %***
62 %
(a) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 2 týždne (b) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 4 týždne (c) U osôb s minimálne 3% psoriázy BSA na začiatku
* p < 0,01, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
*** p < 0,001 (nominálne), Cimzia oproti placebu
Výsledky sú z randomizovaného súboru. Rozdiel liečby: Cimzia 200 mg - placebo, Cimzia 400 mg - placebo (a zodpovedajúci 95 % interval spoľahlivosti a p-hodnota) boli posudzované pomocou štandardného obojstranného Waldovho testu asymptotických štandardných chýb. U pacientov, ktorí prerušili liečbu alebo u nich chýbajú údaje, sa použil systém „Non-responder Imputation„ (NRI).
Spomedzi 273 pacientov, ktorí boli na začiatku randomizovaní na Cimziu v dávke 200 mg každé 2
týždne a Cimziu v dávke 400 mg každé 4 týždne, zostalo na tejto liečbe 237 (86,8 %) pacientov až do
48. týždňa. Zo 138 pacientov randomizovaných na Cimziu v dávke 200 mg každé 2 týždne dosiahlo
92, 68 a 48 pacientov odpoveď ACR 20/50/70 v 48. týždni, v uvedenom poradí. Zo 135 pacientov randomizovaných na Cimziu v dávke 400 mg každé 4 týždne dosiahlo 89, 62 a 41 pacientov odpoveď
ACR 20/50/70, v uvedenom poradí.
Medzi pacientmi, ktorí zostali v štúdii, sa miera odpovede ACR 20, 50 a 70 zachovala až do týždňa
216. To platilo aj v prípade ďalších parametrov periférnej aktivity (napr. počet opuchnutých kĺbov, počet bolestivých /citlivých kĺbov, daktylitída a entezitída).
Rádiografická odpoveď
V klinickej štúdii PsA001 bola inhibícia progresie štrukturálneho poškodenia hodnotená rádiograficky
a vyjadrená ako zmena mTSS a jeho zložiek, skóre erózie (ES) a skóre zúženia kĺbovej štrbiny (JSN) v
24. týždni v porovnaní s východiskovými hodnotami. Skóre mTSS bolo upravené pre psoriatickú artritídu pridaním distálnych interfalangeálnych kĺbov ruky. Liečba Cimziou inhibovala rádiografickú progresiu v porovnaní s liečbou placebom v 24. týždni meranú ako zmena celkového skóre mTSS (LS priemerné [±SE] skóre oproti východiskovému stavu 0,28 [±0,07] v placebovej skupine v porovnaní s
0,06 [±0,06] v skupine so všetkými dávkami Cimzie, p = 0,007). Inhibícia rádiografickej progresie sa udržiavala pri liečbe Cimziou až do 48. týždňa v podskupine pacientov s vyšším rizikom
rádiografickej progresie (pacienti s východiskovým skóre mTSS > 6). U pacientov, ktorí sa zúčastnili
štúdie, sa inhibícia rádiografickej progresie naďalej udržala až do týždňa 216.
O
dpoveď fyzickej funkcie a následky týkajúce sa zdravia
V klinickej štúdii PsA001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI (Health Assessment Questionnaire – Disability Index), bolesti hodnotenej podľa Hodnotenia artritickej bolesti pacientom PAAP a únavy (vyčerpanosti), čo sa preukázalo podľa Škály hodnotenia únavy (FAS - Fatigue Assessment Scale) v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie kvality života súvisiacej so zdravím merané dotazníkom Kvality života pri psoriatickej artritíde (PsAQoL) a SF-36 fyzickej a duševnej zložky a produktivity súvisiacej s psoriatickou artritídou v zamestnaní a v domácnosti hlásenej v v prieskume produktivity práce v porovnaní s placebom. Zlepšenie vo všetkých vyššie uvedených výsledkoch pretrvávalo až do týždňa 216.
Imunogenicita
Reumatoidnáartritída
Celkový podiel pacientov s detekovateľnými protilátkami proti Cimzii pri aspoň 1 príležitosti bol
9,6 % v placebom kontrolovaných RA štúdiách. Približne u jednej tretiny pacientov s pozitívnymi protilátkami boli zistené in vitro neutralizujúce protilátky. U pacientov liečených súbežne s imunosupresívami (MTX) sa protilátky tvorili v menšej miere ako u pacientov, ktorí na začiatku neužívali imunosupresíva. Tvorba protilátok bola spojená s nižšou plazmatickou koncentráciou lieku a u niektorých pacientov s nižšou účinnosťou.
V 2 dlhodobých (až 5 rokov expozície) otvorených štúdiách celkový podiel pacientov
s detekovateľnými protilátkami proti Cimzii pri aspoň jednej príležitosti bol 13 % (8,4 % z celkového počtu pacientov malo prechodnú tvorbu protilátok a ďalších 4,7 % pacientov malo trvalú tvorbu
protilátok). Celkové percento pacientov, ktorí boli pozitívni na tvorbu protilátok a mali trvale zníženú
plazmatickú koncentráciu lieku, bolo odhadované na 9,1 %. Podobne, ako v placebom kontrolovaných štúdiách, bola tvorba protilátok u niektorých pacientov spojená s nižšou účinnosťou.
Farmakodynamický model založený na údajoch z III. fázy klinickej štúdie predpokladá, že približne u 15 % pacientov sa pri odporúčanom dávkovaní (200 mg každé 2 týždne po začiatočnej dávke), bez súbežnej liečby s MTX, vyvinú protilátky v priebehu 6 mesiacov. Tento počet sa znižuje so zvyšujúcimi sa dávkami súbežnej liečby s MTX. Tieto údaje sú primerane v súlade s pozorovanými údajmi.
Axiálnaspondylartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 4,4 % vo fáze III placebom kontrolovanej štúdie u pacientov s axiálnou spondylartritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až počas 192 týždňov) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 9,6 % (4,8 % malo prechodnú tvorbu protilátok, ďalších
4,8 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo na 6,8 %.
Psoriatickáartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 11,7 % vo fáze III placebom kontrolovanej štúdie u pacientov s psoriatickou artritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až do 4 rokov expozície) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 17,3 % (8,7 % malo prechodnú tvorbu protilátok, ďalších
8,7 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo
na 11,5 %.
V
šetky
i
ndikácie
Údaje vyjadrujú podiel pacientov, u ktorých bola vyšetrením ELISA stanovená prítomnosť protilátok proti Cimzii a do veľkej miery závisia od senzitivity a špecificity tohto vyšetrenia.
Okrem toho môže byť pozorovaný výskyt protilátok v hodnotení ovplyvnený viacerými faktormi, vrátane manipulácie so vzorkou, načasovania odberu vzorky, súbežne podávaných liekov a
základného ochorenia. Z týchto dôvodov nie je vhodné porovnávať výskyt protilátok proti Cimzii s výskytom protilátok proti iným antagonistom TNF.
5.2 Farmakokinetické vlastnosti
Plazmatické koncentrácie certolizumab pegolu boli značne úmerné dávke. Farmakokinetika pozorovaná u pacientov s reumatoidnou artritídou zodpovedala farmakokinetike pozorovanej u zdravých jedincov.
Absorpcia
Po subkutánnom podaní sa maximálne plazmatické koncentrácie certolizumab pegolu dosiahli v rozmedzí 54 a 171 hodín od podania injekcie. Biologická dostupnosť (F) certolizumab pegolu po subkutánnom podaní, v porovnaní s intravenóznym podaním, je približne 80 % (rozmedzie 76 % až
88%).
Distribúcia
Vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou bol zdanlivý distribučný objem (V/F) odhadovaný na 8,01 l.
Biotransformáciaaeliminácia
PEGyláciou, kovalentnou väzbou PEG polymérov na peptidy, sa odďaľuje eliminácia týchto látok z krvného obehu viacerými mechanizmami, vrátane zníženého renálneho klírensu, zníženej proteolýzy a zníženej imunogenicity. Certolizumab pegol je preto Fab' fragmentom protilátky konjugovanej
s PEG, aby predĺžil terminálny plazmatický eliminačný polčas Fab' fragmentu na hodnotu porovnateľnú s kompletnou molekulou protilátky. Polčas terminálnej eliminačnej fázy (t1/2) bol pre všetky testované dávky približne 14 dní.
Klírens po subkutánnom podaní bol odhadovaný na 21,0 ml/h vo farmakokinetickej analýze populácie s reumatoidnou artritídou, s interindividuálnou variabilitou 30,8 % (CV) a intraindividuálnou variabilitou 22,0 %. Prítomnosť protilátok proti certolizumab pegolu viedla k približne trojnásobnému nárastu klírensu. V porovnaní s osobou s hmotnosťou 70 kg je klírens o 29 % nižší u individuálnych pacientov s RA vážiacich 40 kg a o 38 % vyšší u pacientov vážiacich 120 kg.
Fab' fragment sa skladá z bielkovinových zložiek a predpokladá sa, že je odbúravaný na peptidy
a aminokyseliny proteolýzou. Dekonjugovaná PEG zložka sa rýchlo eliminuje z plazmy a vylučuje sa renálne v neznámej miere.
Osobitnépopulácie
Porucha funkcie obličiek
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili účinok poruchy funkcie obličiek na farmakokinetiku certolizumab pegolu alebo jeho PEG frakcie. Farmakokinetická analýza populácie
pacientov s ľahkou poruchou renálnej funkcie nepreukázala žiadny vplyv klírensu kreatinínu. Na to,
aby bolo možné odporučiť dávkovanie pri stredne ťažkej a ťažkej poruche funkcie obličiek, nie je dostatok informácií. Predpokladá sa, že farmakokinetika PEG frakcie certolizumab pegolu závisí od funkcie obličiek, avšak u pacientov s poruchou funkcie obličiek sa nehodnotila.
Porucha funkcie pečene
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili vplyv poruchy funkcie pečene na farmakokinetiku certolizumab pegolu.
Starší pacienti (≥ 65 rokov)
Neboli vykonané špecifické klinické štúdie u starších pacientov. Avšak vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou sa nepozoroval žiadny vplyv veku, 78 pacientov
(13,2 % populácie) bolo vo veku 65 rokov alebo viac, pričom najstarší pacient mal 83 rokov.
Pohlavie
Pohlavie nemalo žiadny vplyv na farmakokinetiku certolizumab pegolu. Pretože sa klírens znižuje so znižujúcou sa telesnou hmotnosťou, ženy zvyčajne môžu dosiahnuť o niečo vyššiu systémovú
expozíciu certolizumab pegolu.
Farmakokinetický/farmakodynamickývzťah
Na základe údajov z II. a III. fázy klinickej štúdie bol stanovený vzťah expozícia - odpoveď populácie medzi priemernou plazmatickou koncentráciou certolizumab pegolu počas dávkovacieho intervalu
(Cavg) a účinnosťou (definícia ACR 20 odpovede). Charakteristická Cavg, ktorá tvorí polovicu maximálnej pravdepodobnosti odpovede ACR 20 (EC50), bola 17 µg/ml (95 % CI: 10-23 µg/ml).
5.3 Predklinické údaje o bezpečnosti
Pivotné predklinické štúdie bezpečnosti boli vykonané u opíc Cynomolgus. U potkanov a opíc sa, pri dávkach vyšších ako sú dávky u ľudí, histopatologicky zistila celulárna vakuolizácia prítomná najmä v makrofágoch viacerých orgánov (v lymfatických uzlinách, v miestach vpichu, v slezine, nadobličkách, maternici, cervixe, cievovkovej spleti mozgu a v epitelových bunkách cievovkovej spleti). Je pravdepodobné, že tento nález bol spôsobený bunkovým vychytávaním PEG komponentu. In vitro funkčné štúdie ľudských vakuolizovaných makrofágov naznačili, že všetky skúmané funkcie zostali zachované. Štúdie s potkanmi naznačili, že > 90 % podaného PEG sa eliminovalo počas 3 mesiacov
po jednorazovej dávke, pričom hlavnou cestou exkrécie bolo vylúčenie močom.
Certolizumab pegol nereaguje skrížene s TNF hlodavcov. Preto boli reprodukčné toxikologické štúdie vykonané s homologickou skupinou rozpoznávajúcou TNF potkanov. Význam týchto údajov pri hodnotení rizika u ľudí môže byť obmedzený. Neboli pozorované žiadne nežiaduce účinky na zdravie matiek alebo fertilitu samíc, embryo-fetálne, peri- a postnatálne reprodukčné indexy u potkanov, ktorým bol po pretrvávajúcej supresii TNFα podávaný PEGylovaný Fab' fragment anti-rat TNFα (cTN3 PF). U samcov potkanov bola pozorovaná znížená motilita spermií a tendencia k poklesu počtu spermií.
Štúdie distribúcie preukázali, že prestup cTN3 PF placentou a materským mliekom do cirkulácie plodu a novorodenca je zanedbateľný. Certolizumab pegol sa neviaže na ľudský neonatálny Fc receptor (FcRn). Údaje z modelu ľudského placentárneho transferu s uzatvoreným okruhom ex vivo naznačujú nízky alebo zanedbateľný transfer do fetálneho kompartmentu. Okrem toho experimenty
s transcytózou sprostredkovanou FcRn v bunkách transfektovaných ľudským FcRn poukázali na zanedbateľný transfer (pozri časť 4.6).
V predklinických štúdiách sa nepreukázali žiadne mutagénne ani klastogénne účinky. Štúdie na karcinogenicitu sa neuskutočnili s certolizumab pegolom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
octan sodný chlorid sodný voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Naplnené pero uchovávajte vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah baleniaJednomililitrové naplnené pero (AutoClicks) obsahujúce naplnenú injekčnú striekačku (zo skla typu I)
s plunžerovým uzáverom (z bromobutylovej gumy), s obsahom 200 mg certolizumab pegolu.
Veľkosť balenia: 2 naplnené perá a 2 liehové tampóny.
Multibalenie obsahujúce 6 (3 balenia po 2) naplnených pier a 6 (3 balenia po 2) liehových tampónov. Multibalenie obsahujúce 10 (5 balení po 2) naplnených pier a 10 (5 balení po 2) liehových tampónov.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomObsiahle pokyny na prípravu a podanie Cimzie v naplnenom pere sú uvedené v písomnej informácii pre používateľa.
Tento liek je len na jednorazové použitie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/09/544/005
EU/1/09/544/006
EU/1/09/544/007
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 1. október 2009
Dátum posledného predĺženia registrácie: 16. máj 2014
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky:
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Cimzia 200 mg injekčný roztok v náplni do dávkovacieho zariadenia
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá náplň do dávkovacieho zariadenia obsahuje 200 mg certolizumab pegolu v jednom ml. Certolizumab pegol je Fab' fragment rekombinantnej, humanizovanej protilátky proti tumor
nekrotizujúcemu faktoru alfa (TNFα) získaný z Escherichia coli a konjugovaný s polyetylénglykolom
(PEG).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (injekcia).
Číry až opalizujúci, bezfarebný až žltý roztok. pH roztoku je približne 4,7.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Reumatoidnáartritída
Cimzia, v kombinácii s metotrexátom (MTX), je indikovaná na:
- liečbu stredne závažnej až závažnej aktívnej reumatoidnej artritídy (RA) u dospelýchpacientov, keď je odpoveď na antireumatiká modifikujúce ochorenie (DMARD –Disease-Modifying Antirheumatic Drugs), vrátane MTX, nedostatočná. Cimzia sa môže podávať v monoterapii
v prípade intolerancie MTX, alebo keď pokračovanie v liečbe MTX nie je vhodné.
- liečbu závažnej, aktívnej a progresívnej formy RA u dospelých, doteraz neliečených MTX alebo inými DMARD.
Preukázalo sa, že Cimzia znižuje mieru progresie poškodenia kĺbov meranú röntgenologicky a zlepšuje fyzickú funkciu, keď sa podáva v kombinácii s MTX.
Axiálnaspondylartritída
Cimzia je indikovaná na liečbu dospelých pacientov so závažnou aktívnou axiálnou spondylartritídou, ktorú tvorí:
Ankylozujúca spondylitída (AS)
Dospelí pacienti so závažnou aktívnou ankylozujúcou spondylitídou, u ktorých nedošlo k primeranej odpovedi na nesteroidné antiflogistiká (NSA) alebo ktorí ich netolerujú.
Axiálna spondylartritída bez röntgenového dôkazu AS
Dospelí pacienti so závažnou aktívnou axiálnou spondylartritídou bez röntgenového dôkazu AS, avšak s objektívnymi prejavmi zápalu a to zvýšeným C-reaktívnym proteínom (CRP) a/alebo podľa
vyšetrenia magnetickou rezonanciou (MR), u ktorých nedošlo k primeranej odpovedi na NSA alebo
ktorí ich netolerujú.
P
soriatická
artritída
Cimzia, v kombinácii s MTX, je indikovaná na liečbu aktívnej psoriatickej artritídy u dospelých, keď je odpoveď na liečbu DMARD nedostatočná.
Cimzia sa môže podávať v monoterapii v prípade intolerancie metotrexátu, alebo keď pokračovanie v liečbe metotrexátom nie je vhodné.
Podrobnosti o terapeutických účinkoch, pozri časť 5.1.
4.2 Dávkovanie a spôsob podávania
Liečbu má začať lekár - špecialista so skúsenosťami v diagnostike a liečbe ochorení, na ktoré je indikovaná Cimzia, ktorý bude dohliadať na priebeh liečby. Pacienti majú dostať špeciálnu kartičku s upozornením.
Dávkovanie
Nárazovádávka
Odporúčaná začiatočná dávka Cimzie pre dospelých pacientov je 400 mg (podávaná vo forme dvoch
200 mg subkutánnych injekcií) v 0., 2. a 4. týždni. Ak je to vhodné, s podávaním metotrexátu sa má pri reumatoidnej artritíde a psoriatickej artritíde pokračovať počas liečby Cimziou.
Udržiavaciadávka
Reumatoidná artritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s reumatoidnou artritídou 200 mg každé 2 týždne. Po potvrdení klinickej odpovede sa môže zvážiť alternatívne
udržiavacie dávkovanie 400 mg každé 4 týždne. Ak je to vhodné, s podávaním MTX sa má
pokračovať počas liečby Cimziou.
Axiálna spondylartritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s axiálnou spondylartritídou 200 mg každé 2 týždne alebo 400 mg každé 4 týždne.
Psoriatická artritída
Po začiatočnej dávke je odporúčaná udržiavacia dávka Cimzie u dospelých pacientov s psoriatickou artritídou 200 mg každé 2 týždne. Po potvrdení klinickej odpovede sa môže zvážiť alternatívne
udržiavacie dávkovanie 400 mg každé 4 týždne. Ak je to vhodné, s podávaním MTX sa má
pokračovať počas liečby Cimziou.
Pre indikácie uvedené vyššie dostupné údaje ukazujú, že klinická odpoveď sa zvyčajne dosiahne v priebehu 12 týždňov liečby. Pokračovanie v liečbe sa má starostlivo prehodnotiť u pacientov, u ktorých sa nepreukázal žiadny prospech z liečby počas prvých 12 týždňov liečby.
Vynechaniedávky
Pacientom, ktorí vynechali dávku, treba odporučiť, aby si aplikovali injekčne ďalšiu dávku Cimzie, hneď ako si spomenú a potom pokračovali v injekčnej aplikácii nasledujúcich dávok tak, ako boli
poučení.
Osobitnéskupinypacientov
Pediatrická populácia (< 18 rokov)
Bezpečnosť a účinnosť Cimzie u detí a dospievajúcich vo veku do 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Starší pacienti ( ≥ 65 rokov)
Nie je potrebná žiadna úprava dávky. Farmakokinetické analýzy populácie nepreukázali žiadny vplyv veku (pozri časť 5.2).
P
orucha funkcie obličiek a pečene
V týchto populáciách pacientov sa Cimzia neskúmala. Nie je možné poskytnúť žiadne odporúčania pre dávkovanie (pozri časť 5.2).
Spôsobpodávania
Celý obsah (1 ml) náplne do dávkovacieho zariadenia sa podáva pomocou elektromechanického injekčného zariadenia ava určeného len na subkutánnu injekciu. K vhodným miestam pre aplikáciu injekcie patria stehno alebo brucho.
Cimzia injekčný roztok v náplni do dávkovacieho zariadenia je určená pre jednorazové použitie elektromechanickým injekčným zariadením ava. Po príslušnom zaškolení v technike injekcie si pacienti môžu sami aplikovať injekciu pomocou elektromechanického injekčného zariadenia
s jednorazovou náplňou do dávkovacieho zariadenia, ak sa ich lekár rozhodne, že je to vhodné a
s následným lekárskym dohľadom podľa potreby. Lekár by mal s pacientom prediskutovať, ktorá forma podania injekcie je najvhodnejšia.
Pri podávaní sa majú dodržiavať pokyny na použitie uvedené na konci písomnej informácie pre používateľa a v Návode na použitie elektromechanického injekčného zariadenia ava.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Aktívna tuberkulóza alebo iné závažné infekcie, ako sú sepsa alebo oportúnne infekcie (pozri časť
4.4).
Stredne závažné až závažné srdcové zlyhanie (NYHA trieda III/IV) (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Infekcie
Pred liečbou Cimziou, počas nej a po jej skončení sa musia u pacientov starostlivo sledovať prejavy a príznaky infekcií, vrátane tuberkulózy. Pretože eliminácia certolizumab pegolu môže trvať až do
5 mesiacov, sledovanie má pokračovať aj počas tohto obdobia (pozri časť 4.3).
Liečba Cimziou sa nesmie začať u pacientov s klinicky významnou aktívnou infekciou, vrátane chronických alebo lokalizovaných infekcií, pokiaľ nie je infekcia zvládnutá (pozri časť 4.3).
Pacientov, u ktorých sa objavila nová infekcia v priebehu liečby Cimziou, je potrebné starostlivo sledovať. Ak sa u pacienta vyskytne nová závažná infekcia, podávanie Cimzie sa má prerušiť, pokiaľ nie je táto infekcia zvládnutá. Lekári majú byť opatrní pri zvažovaní použitia Cimzie u pacientov
s opakovanou alebo oportúnnou infekciou v anamnéze alebo s prebiehajúcimi ochoreniami, ktoré môžu pacientov predisponovať na infekcie, vrátane súbežného používania imunosupresívnych liekov.
U pacientov s reumatoidnou artritídou sa nemusia prejaviť typické príznaky infekcie, vrátane horúčky, z dôvodu ich ochorenia a súbežne podávaných liekov. Preto je skoré odhalenie akejkoľvek infekcie, predovšetkým atypických klinických prejavov závažnej infekcie, veľmi dôležité, aby sa minimalizovalo oneskorenie diagnostiky a začiatku liečby.
U pacientov, ktorí používajú Cimziu, boli hlásené závažné infekcie, vrátane sepsy a tuberkulózy (vrátane miliárneho, diseminovaného a mimopľúcneho ochorenia) a oportúnne infekcie (napr. histoplazmóza, nokardia, kandidóza). Niektoré z týchto udalostí boli fatálne.
Tuberkulóza
Pred začiatkom liečby Cimziou sa musia všetci pacienti vyšetriť na aktívnu alebo inaktívnu (latentnú)
tuberkulóznu infekciu. Toto vyšetrenie má obsahovať podrobnú lekársku anamnézu u pacientov
s tuberkulózou v osobnej anamnéze, s možnou predchádzajúcou expozíciou iným jedincom s aktívnou
tuberkulózou a s predchádzajúcim a/alebo súčasným používaním imunosupresívnej liečby. U všetkých pacientov sa majú urobiť príslušné skríningové vyšetrenia, napr. tuberkulínový kožný test a röntgen hrudníka (je potrebné postupovať podľa miestnych odporúčaní). Odporúča sa, aby sa vykonanie týchto vyšetrení zaznamenalo v kartičke s upozornením pre pacienta. Predpisujúcich lekárov to upozorní na riziko falošne negatívnych výsledkov tuberkulínového kožného testu, najmä u pacientov, ktorí sú závažne chorí alebo majú oslabený imunitný systém.'
V prípade, že je aktívna tuberkulóza diagnostikovaná pred liečbou alebo počas nej, liečba Cimziou sa nesmie začať a musí sa prerušiť (pozri časť 4.3).
Pri podozrení na inaktívnu („latentnú“) tuberkulózu sa treba poradiť s lekárom s kvalifikáciou v liečbe tuberkulózy. Vo všetkých prípadoch popísaných nižšie sa má veľmi dôkladne zvážiť pomer prínosu
a rizika liečby Cimziou.
Ak sa stanoví diagnóza latentnej tuberkulózy, musí sa pred začiatkom liečby Cimziou začať príslušná antituberkulózna liečba v súlade s miestnymi odporúčaniami.
Použitie antituberkulóznej liečby sa má zvážiť tiež pred začatím liečby Cimziou u pacientov
s latentnou alebo aktívnou tuberkulózou v predchádzajúcej anamnéze, u ktorých nie je možné potvrdiť adekvátny priebeh liečby a u pacientov s významnými rizikovými faktormi tuberkulózy, napriek negatívnemu vyšetreniu na latentnú tuberkulózu. Biologické testy pre skríning tuberkulózy sa majú zvážiť pred začiatkom liečby Cimziou, ak existuje možnosť výskytu latentnej tuberkulóznej infekcie, bez ohľadu na BCG vakcináciu.
Napriek predchádzajúcej alebo súbežnej profylaktickej antituberkulóznej liečbe sa u pacientov liečených antagonistami TNF, vrátane Cimzie, vyskytli prípady aktívnej tuberkulózy. U niektorých pacientov úspešne liečených na aktívnu tuberkulózu sa opätovne vyvinula tuberkulóza počas liečby Cimziou.
Pacientov je potrebné poučiť, aby vyhľadali lekársku pomoc v prípade, že sa u nich počas liečby Cimziou alebo po jej skončení objavia prejavy/príznaky pripomínajúce tuberkulóznu infekciu (napr. pretrvávajúci kašeľ, chudnutie/zníženie hmotnosti, horúčka nízkeho stupňa, ľahostajnosť).
ReaktiváciavírusuhepatitídytypuB(HBV)
U pacientov liečených antagonistom TNF vrátane certolizumab pegolu, ktorí sú chronickými nosičmi tohto vírusu (t.j. pozitívny povrchový antigén) sa objavila reaktivácia hepatitídy B. Niektoré prípady
mali fatálny následok.
Pred začatím liečby Cimziou sa majú pacienti vyšetriť na infekciu HBV. Pacientom s pozitívnym testom na infekciu HBV sa odporúča konzultácia s lekárom so skúsenosťami v liečbe hepatitídy B.
U nosičov HBV, ktorí si vyžadujú liečbu Cimziou, je potrebné počas liečby a niekoľko mesiacov po skončení liečby starostlivo sledovať výskyt znakov a príznakov aktívnej infekcie HBV. Nie sú dostupné dostatočné údaje o liečbe pacientov, ktorí sú nosičmi HBV a dostávajú antivírusovú liečbu v kombinácii s liečbou TNF-antagonistom na prevenciu reaktivácie HBV. U pacientov, u ktorých sa vyvinula reaktivácia HBV, je potrebné ukončiť podávanie Cimzie a má sa začať účinná antivírusová liečba s príslušnou podpornou liečbou.
Malignityalymfoproliferatívneporuchy
Potenciálna úloha liečby antagonistom TNF v rozvoji malignít nie je známa. Pri zvažovaní liečby antagonistom TNF u pacientov s malignitou v anamnéze alebo pri zvažovaní pokračovania v liečbe
u pacientov, u ktorých sa rozvinula malignita, je potrebná opatrnosť.
Pri súčasných znalostiach nie je možné vylúčiť možné riziko rozvoja lymfómov, leukémie alebo iných malignít u pacientov liečených s antagonistom TNF.
V klinických štúdiách s Cimziou a inými antagonistami TNF bolo hlásených viac prípadov lymfómov a iných malignít medzi pacientmi užívajúcimi antagonisty TNF ako u kontrolných pacientov užívajúcich placebo (pozri časť 4.8). Po uvedení lieku na trh sa u pacientov liečených antagonistom TNF zaznamenali prípady leukémie. Existuje zvýšené riziko lymfómu a leukémie v pozadí
u pacientov s reumatoidnou artritídou s dlhotrvajúcim, vysoko aktívnym, zápalovým ochorením, ktoré komplikuje odhad rizika.
Neboli vykonané žiadne štúdie, ktoré zahŕňajú pacientov s anamnézou malignity alebo štúdie,
v ktorých liečba pokračovala u pacientov, u ktorých sa rozvinula malignita počas liečby Cimziou.
Kožné karcinómy
U pacientov liečených antagonistami TNF vrátane certolizumab pegolu sa zaznamenal melanóm
a karcinóm z Merkelových buniek (pozri časť 4.8). Odporúča sa pravidelné vyšetrenie kože, hlavne u pacientov s rizikovými faktormi pre kožný karcinóm.
Malignita u detí
U detí, dospievajúcich a mladých dospelých (do 22 rokov) liečených antagonistami TNF (začiatok liečby vo veku ≤ 18 rokov) sa po uvedení lieku na trh zaznamenali malignity, niekedy fatálne.
Približne polovica prípadov boli lymfómy. Ďalšie prípady predstavovali rôzne druhy rozličných malignít a patrili medzi ne zriedkavé malignity zvyčajne súvisiace s imunosupresiou. Riziko rozvoja
malignít u detí a dospievajúcich liečených antagonistami TNF nemožno vylúčiť.
U pacientov liečených antagonistami TNF sa po uvedení lieku na trh zaznamenali prípady hepatosplenického T-bunkového lymfómu (hepatosplenic T-cell lymphoma - HSTCL). Tento zriedkavý typ T-bunkového lymfómu má veľmi agresívny priebeh ochorenia a zvyčajne je smrteľný. Väčšina hlásených prípadov s antagonistami TNF sa vyskytovala u dospievajúcich a mladých dospelých mužov s Crohnovou chorobou alebo ulceróznou kolitídou. Takmer všetci títo pacienti užívali liečbu imunosupresívami azatioprinom a/alebo 6-merkaptopurínom súbežne s antagonistom TNF počas diagnózy alebo pred ňou. Riziko vývoja hepatosplenického T-bunkového lymfómu
u pacientov liečených Cimziou nemožno vylúčiť.
Chronickáobštrukčnáchorobapľúc(CHOCHP)
V prieskumnej klinickej štúdii hodnotiacej používanie iného antagonistu TNF, infliximabu, bolo
u pacientov so stredne závažnou až závažnou chronickou obštrukčnou chorobou pľúc (CHOCHP)
liečených infliximabom hlásených viac malignít, najmä v pľúcach alebo v hlave a krku, v porovnaní s kontrolnými pacientmi. Všetci pacienti mali silné fajčenie v anamnéze. Preto pri používaní
ktoréhokoľvek antagonistu TNF u pacientov so CHOCHP je potrebná opatrnosť, rovnako ako
u pacientov so zvýšeným rizikom malignít z dôvodu silného fajčenia.
Kongestívnesrdcovézlyhanie
Cimzia je kontraindikovaná v prípade stredne závažného alebo závažného srdcového zlyhania (pozri časť 4.3). V klinickej štúdii s iným antagonistom TNF sa pozorovalo zhoršenie kongestívneho srdcového zlyhania a zvýšená mortalita v dôsledku kongestívneho srdcového zlyhania. Rovnako boli prípady kongestívneho srdcového zlyhania hlásené aj u pacientov s reumatoidnou artritídou liečených Cimziou. U pacientov s miernym srdcovým zlyhaním (I/II trieda NYHA) sa má Cimzia používať opatrne. U pacientov, u ktorých sa objavia nové príznaky kongestívneho srdcového zlyhania alebo sa existujúce príznaky zhoršia, sa liečba Cimziou musí prerušiť.
Hematologickéreakcie
Hlásenia o pancytopénii, vrátane aplastickej anémie, boli pri antagonistoch TNF zriedkavé.
V súvislosti s Cimziou boli hlásené nežiaduce reakcie hematologického systému, vrátane medicínsky významnej cytopénie (napr. leukopénia, pancytopénia, trombocytopénia) (pozri časť 4.8). Je potrebné upozorniť všetkých pacientov, aby okamžite vyhľadali lekársku pomoc, ak sa u nich počas liečby Cimziou objavia prejavy a príznaky pripomínajúce krvnú dyskráziu alebo infekciu (napr. pretrvávajúca horúčka, podliatiny, krvácanie, bledosť). U pacientov s potvrdenými významnými hematologickými abnormalitami sa má zvážiť prerušenie liečby Cimziou.
N
eurologické
udalosti
Používanie antagonistov TNF bolo v zriedkavých prípadoch spájané s novým nástupom alebo exacerbáciou klinických symptómov a/alebo rádiograficky potvrdeným demyelinizačným ochorením, vrátane sklerózy multiplex. U pacientov s preexistujúcim demyelizačným ochorením alebo s jeho nedávnym nástupom je potrebné pred začatím liečby Cimziou starostlivo zvážiť prínosy a riziká liečby antagonistom TNF. U pacientov liečených Cimziou boli hlásené zriedkavé prípady neurologických porúch, vrátane záchvatovej poruchy, neuritídy a periférnej neuropatie.
Precitlivenosť
Po podaní Cimzie boli zriedkavo hlásené závažné reakcie z precitlivenosti. Niektoré z týchto reakcií sa vyskytli po prvom podaní Cimzie. Ak sa vyskytnú závažné reakcie, podávanie Cimzie sa má okamžite
prerušiť a začať príslušná liečba.
Údaje o používaní Cimzie u pacientov, u ktorých sa vyskytla závažná reakcia precitlivenosti na iného antagonistu TNF sú obmedzené, u týchto pacientov je potrebná opatrnosť.
Imunosupresia
Pretože tumor nekrotizujúci faktor (TNF) sprostredkováva zápal a modifikuje bunkové imunitné odpovede, pre antagonisty TNF, vrátane Cimzie, existuje možnosť, že spôsobia imunosupresiu,
pričom ovplyvnia ochranu hostiteľa proti infekciám a malignitám.
Autoimunita
Liečba Cimziou môže vyvolať tvorbu antinukleárnych protilátok (ANP) a menej často môže viesť k vzniku syndrómu, ktorý je podobný lupusu (pozri časť 4.8). Vplyv dlhodobej liečby Cimziou na
rozvoj autoimunitných ochorení nie je známy. Ak sa po liečbe Cimziou u pacienta objavia symptómy
pripomínajúce syndróm podobný lupusu, liečba sa musí prerušiť. Cimzia sa neskúmala špeciálne v populácii pacientov s lupusom (pozri časť 4.8).
Vakcinácie
Pacienti liečení Cimziou môžu byť očkovaní, s výnimkou očkovaní živými vakcínami. Nie sú dostupné žiadne údaje o odpovedi na očkovanie živými vakcínami alebo na sekundárny prenos
infekcie živými vakcínami u pacientov liečených Cimziou. Živé vakcíny sa nemajú podávať súbežne s
Cimziou.
V placebom kontrolovanej klinickej štúdii u pacientov s reumatoidnou artritídou sa pozorovala rovnaká protilátková odpoveď medzi liečebnými skupinami s Cimziou a s placebom pri podávaní pneumokokovej polysacharidovej vakcíny a chrípkovej vakcíny súbežne s Cimziou. Pacienti liečení Cimziou a súbežne metotrexátom mali nižšiu humorálnu odpoveď v porovnaní s pacientmi liečenými samotnou Cimziou. Klinický význam tohto nie je známy.
Súbežnépoužívaniesinýmibiologickýmilátkami
Závažné infekcie a neutropénia boli hlásené v klinických štúdiách so súbežným používaním anakinry
(antagonista interleukínu-1) alebo abataceptu (modulátor CD28) a iných antagonistov TNF, etanerceptu, ktoré však nepredstavovali žiadny ďalší prínos v porovnaní s liečbou samotným antagonistom TNF. Vzhľadom na povahu nežiaducich udalostí pozorovaných pri kombinácii iného antagonistu TNF s liečbou abataceptom alebo anakinrou môže kombinácia anakinry alebo abataceptu a iných antagonistov TNF viesť k podobným toxicitám. Preto sa používanie certolizumab pegolu v kombinácii s anakinrou alebo abataceptom neodporúča (pozri časť 4.5).
Chirurgickývýkon
U pacientov liečených Cimziou existujú s chirurgickými výkonmi obmedzené skúsenosti týkajúce sa bezpečnosti. Ak je plánovaný chirurgický výkon, je potrebné vziať do úvahy 14-dňový polčas certolizumab pegolu. Pacienta, u ktorého je nutný chirurgický výkon počas liečby Cimziou, je potrebné starostlivo sledovať kvôli výskytu infekcie a vykonať vhodné opatrenia.
V
y
š
etrenie
aktivovaného
parciálneho
tromboplastínového
času
(aPTT)
U pacientov liečených Cimziou bola pozorovaná interferencia s niektorými koagulačnými vyšetreniami. Cimzia môže spôsobiť falošne zvýšené výsledky vyšetrenia aPTT u pacientov bez koagulačných anomálií. Tento vplyv bol pozorovaný pri teste na lupus antikoagulans (PTT-LA) a štandardných automatizovaných testoch aktivovaného parciálneho tromboplastínovéhu času
(STA-PTT - Standard Target Activated Partial Thromboplastin time) od firmy Diagnostica Stago a testoch ”HemosIL APTT-SP liquid' a 'HemosIL lyophilized silica“ od firmy Instrumention Laboratories. Rovnako môžu byť ovplyvnené aj iné aPTT vyšetrenia. Neexistuje dôkaz o tom, že liečba Cimziou má vplyv na koaguláciu in vivo. Po podaní Cimzie pacientom sa má interpretácii abnormálnych výsledkov koagulačných vyšetrení venovať zvýšená pozornosť. Nebola pozorovaná interferencia s vyšetrením trombínového času (TT) a protrombínového času (PT).
Staršípacienti
V klinických štúdiách bol u pacientov vo veku ≥ 65 rokov zjavne vyšší výskyt infekcií ako u mladších pacientov, hoci skúsenosti sú obmedzené. Opatrnosť je potrebná pri liečbe starších pacientov
a mimoriadnu opatrnosť je potrebné venovať výskytu infekcií.
4.5 Liekové a iné interakcie
Súbežná liečba s metotrexátom, kortikosteroidmi, nesteroidnými aniflogistikami (NSAID – nonsteroidal anti-inflammatory drugs) a analgetikami nepreukázala žiadny vplyv na farmakokinetiku certolizumab pegolu vychádzajúc z farmakokinetickej analýzy populácie.
Kombinácia certolizumab pegolu a anakinry alebo abataceptu sa neodporúča (pozri časť 4.4). Súbežné podávanie Cimzie s metotrexátom nemá žiadny významný vplyv na farmakokinetiku
metotrexátu. Pri porovnaní jednotlivých štúdií sa farmakokinetika certolizumab pegolu javila ako
porovnateľná s farmakokinetikou pozorovanou predtým u zdravých jedincov.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
U žien vo fertilnom veku je potrebné zvážiť použitie primeranej antikoncepcie. U žien, ktoré plánujú otehotnieť, vzhľadom na rýchlosť eliminácie lieku možno zvážiť pokračujúce používanie antikoncepcie počas 5 mesiacov po poslednej dávke Cimzie (pozri časť 5.2), ale je tiež potrebné vziať do úvahy potrebu liečby žien (pozri ďalej).
Gravidita
Údaje z vyše 500 prospektívne zozbieraných gravidít, ktoré boli vystavené účinkom Cimzie so známymi výsledkami gravidity, vrátane viac ako 400 gravidít vystavených Cimzii počas prvého
trimestra, nepoukazujú na malformačný účinok Cimzie. Dostupné klinické skúsenosti sú však príliš obmedzené na vyvodenie záveru s odôvodnenou istotou, že s podávaním Cimzie počas gravidity nie je
spojené zvýšené riziko.
Štúdie na zvieratách, v ktorých sa používal anti-rat TNFα hlodavcov, neodhalili žiadny dôkaz o poškodení fertility alebo poškodení plodu. Tieto informácie sú však nedostatočné čo sa týka reprodukčnej toxicity u ľudí (pozri časť 5.3). Z dôvodu jej inhibície TNFα by Cimzia podávaná počas gravidity mohla ovplyvňovať normálnu imunitnú odpoveď u novorodenca.
Cimzia sa má používať počas gravidity len v prípade, že je to klinicky potrebné.
Predklinické štúdie naznačujú nízku alebo zanedbateľnú úroveň placentárneho transferu homológneho
Fab-fragmentu certolizumab pegolu (bez Fc oblasti) (pozri časť 5.3).
V klinickej štúdii bolo 16 žien liečených certolizumab pegolom (200 mg každé 2 týždne alebo 400 mg každé 4 týždne) počas gravidity. Plazmatické koncentrácie certolizumab pegolu namerané u 14 dojčiat pri narodení boli pod hranicou kvantifikácie (BLQ - Below the Limit of Quantification) pri 13 vzorkách; jedna bola 0,042 µg/ml s plazmatickým pomerom dojča/matka pri narodení 0,09 %. Vo
4. týždni a v 8. týždni boli všetky koncentrácie u dojčiat BLQ. Klinický význam nízkych hladín certolizumab pegolu pre dojčatá nie je známy. Odporúča sa počkať minimálne 5 mesiacov po podaní poslednej dávky Cimzie matke počas gravidity pred podaním živej alebo živej oslabenej vakcíny (napr. vakcína BCG), ak prínos vakcinácie zjavne neprevyšuje teoretické riziko podania živej alebo živej oslabenej vakcíny dojčatám.
Dojčenie
V klinickej štúdii bol u 17 dojčiacich žien liečených Cimziou pozorovaný minimálny transfer certolizumab pegolu z plazmy do materského mlieka. Percentuálny podiel materskej dávky
certolizumab pegolu, ktorá prechádza na dojča počas 24 hodín, bol odhadnutý na 0,04 % až 0,30 %.
Okrem toho, keďže certolizumab pegol je proteín, ktorý sa po perorálnom podaní rozkladá v gastrointestinálnom trakte, predpokladá sa, že absolútna biologická dostupnosť bude u dojčeného dieťaťa veľmi nízka.
Cimzia sa preto môže používať počas dojčenia. Fertilita
Boli pozorované účinky na mieru motility spermií a tendencia zníženého počtu spermií u samcov
hlodavcov, ktoré však nemali dopad na ich fertilitu (pozri časť 5.3).
V klinickej štúdii na zhodnotenie účinku certolizumab pegolu na parametre kvality semena bolo 20 zdravých mužov randomizovaných na aplikáciu jednorazovej subkutánnej dávky 400 mg certolizumab pegolu alebo placeba. Počas 14-týždňového sledovania sa nepozorovali žiadne účinky liečby certolizumab pegolom na parametre kvality semena v porovnaní s placebom.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cimzia môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní Cimzie sa môže objaviť závrat (vrátane vertiga, poruchy videnia a vyčerpanosti) (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Reumatoidnáartritída
Cimzia bola skúmaná u 4 049 pacientov s reumatoidnou artritídou v kontrolovaných a nezaslepených štúdiách po dobu až 92 mesiacov. Údaje v tabuľke 1 vychádzajú v prvom rade z placebom kontrolovaných štúdií, ktoré zahŕňali 2 965 pacientov používajúcich Cimziu a 1 137 pacientov užívajúcich placebo počas kontrolovaného obdobia.
V placebom kontrolovaných štúdiách mali pacienti používajúci Cimziu približne 4-krát dlhšie trvanie expozície v porovnaní s pacientmi v placebovej skupine. Tento rozdiel v expozícii je predovšetkým spôsobený tým, že u pacientov užívajúcich placebo bola vyššia pravdepodobnosť skorého prerušenia štúdie. Okrem toho, v štúdiách RA-I a RA-II bolo prerušenie štúdie povinné pre pacientov bez odpovede v 16. týždni, z ktorých väčšina užívala placebo.
Počas kontrolovaných štúdií bol podiel pacientov, ktorí prerušili liečbu z dôvodu nežiaducich udalostí,
4,4 % u pacientov liečených Cimziou a 2,7 % u pacientov liečených placebom.
Najčastejšie nežiaduce reakcie patrili do triedy orgánových systémov Infekcie a nákazy, hlásené u
14,4 % pacientov liečených Cimziou a 8,0 % pacientov liečených placebom, do triedy Celkové poruchy a reakcie v mieste podania, hlásené u 8,8 % pacientov liečených Cimziou a 7,4 % pacientov liečených placebom a do triedy Poruchy kože a podkožného tkaniva, hlásené u 7,0 % pacientov liečených Cimziou a u 2,4 % pacientov liečených placebom.
A
xiálna
spondylartritída
Cimzia sa skúmala u 325 pacientov s aktívnou axiálnou spondylartritídou v AS001 klinickej štúdii počas až 4 rokov, ktorá zahŕňala 24 týždňov trvajúcu, placebom kontrolovanú fázu, po ktorej nasledovala 24 týdňov trvajúca zaslepená fáza a 156 týždňov trvajúca otvorená fáza. Bezpečnostný profil u pacientov s axiálnou spondylartritídou liečených Cimziou sa zhodoval s bezpečnostným profilom u reumatoidnej artritídy a s predchádzajúcimi skúsenosťami s Cimziou.
Psoriatickáartritída
Cimzia sa skúmala u 409 pacientov s psoriatickou artritídou v PsA001 klinickej štúdii počas až
4 rokov, ktorá zahŕňala 24 týždňov trvajúcu, placebom kontrolovanú fázu, po ktorej nasledovala 24
týždňov trvajúca zaslepená fáza a 168 týždňov trvajúca otvorená fáza. Bezpečnostný profil
u pacientov s psoriatickou artritídou liečených Cimziou bol zhodný s bezpečnostným profilom pri reumatoidnej artritíde a s predchádzajúcimi skúsenosťami s Cimziou.
Zoznam nežiaducich reakcií v tabuľke
Nežiaduce reakcie hlásené v klinických štúdiách s reumatoidnou artritídou a prípady po uvedení lieku
na trh s minimálne možnou súvislosťou s Cimziou sú vymenované v tabuľke 1 nižšie, podľa frekvencie a triedy orgánových systémov. Frekvencie kategórií sú definované nasledovne: veľmi časté
(≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000), neznáme (nie je možné odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1 Nežiaduce reakcie v klinických štúdiách a po uvedení lieku na trh
Trieda orgánových systémov Frekvencia Nežiaduce reakcie
Infekcie a nákazy Časté bakteriálne infekcie (vrátane abscesu), vírusové infekcie (vrátane herpesu zoster, papilomavírusu, chrípky)
Menej časté sepsa (vrátane multiorgánového zlyhania, septického šoku), tuberkulóza (vrátane miliárneho, diseminovaného a extrapulmonálneho ochorenia), mykotické infekcie (vrátane oportúnnych)
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Poruchy krvi a lymfatického systému
Menej časté malignity krvi a lymfatického systému (vrátane lymfómu a leukémie), tumory solídnych orgánov, nemelanómové karcinómy kože, prekancerózne lézie (vrátane orálnej leukoplakie, névu z melanocytov), benígne tumory a cysty (vrátane papilómu kože)
Zriedkavé gastrointestinálne tumory, melanóm
Neznáme karcinóm z Merkelových buniek*
Časté eozinofilné poruchy, leukopénia (vrátane neutropénie, lymfopénie)
Menej časté anémia, lymfadenopatia, trombocytopénia, trombocytóza
Zriedkavé pancytopénia, splenomegália, erytrocytóza, abnormálna morfológia bielych krviniek
Poruchy imunitného systému Menej časté vaskulitídy, lupus erythematosus, precitlivenosť na lieky (vrátane anafylaktického šoku), alergické poruchy, pozitívne autoprotilátky
Zriedkavé angioneurotický edém, sarkoidóza, sérová choroba, panikulitída (vrátane nodózneho erytému), zhoršenie príznakov dermatomyozitídy**
Poruchy endokrinného systému Zriedkavé poruchy štítnej žľazy
Poruchy metabolizmu a výživy Menej časté nerovnováha elektrolytov, dyslipidémia, poruchy chuti do jedla, zmeny telesnej hmotnosti
Zriedkavé hemosideróza
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Psychické poruchy Menej časté úzkosť a poruchy nálady (vrátane súvisiacich symptómov)
Zriedkavé pokus o samovraždu, delírium, duševná porucha
Poruchy nervového systému Časté bolesti hlavy (vrátane migrény), zmyslové anomálie
Menej časté periférne neuropatie, závrat, tremor
Zriedkavé záchvat, zápal kraniálneho nervu, porucha koordinácie alebo rovnováhy
Neznáme skleróza multiplex*, Guillainov-Barrého syndróm* Poruchy oka Menej časté porucha zraku (vrátane zhoršeného videnia), zápal
oka a očného viečka, porucha slzenia
Poruchy ucha a labyrintu Menej časté tinnitus, vertigo
Poruchy srdca a srdcovej činnosti
Menej časté kardiomyopatie (vrátane srdcového zlyhania), ischemické koronárne arteriálne poruchy, arytmie (vrátane atriálnej fibrilácie), palpitácie
Zriedkavé perikarditída, atrioventrikulárna blokáda
Poruchy ciev Časté hypertenzia
Menej časté hemorágia alebo krvácanie (na ktoromkoľvek mieste), hyperkoagulácia (vrátane tromboflebitídy, pľúcnej embólie), synkopa, edém (vrátane periférneho, faciálneho), ekchymózy (vrátane hematómu, petechií)
Zriedkavé cerebrovaskulárna príhoda, arterioskleróza, Raynaudov fenomén, livedo reticularis, telangiektázia
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Poruchy obličiek a močových ciest
Menej časté astma a s ňou súvisiace symptómy, pleurálny
výpotok a symptómy, kongescia a zápal respiračného traktu, kašeľ
Zriedkavé intersticiálna pľúcna choroba, pneumonitída
Časté nevoľnosť
Menej časté ascites, gastrointestinálna ulcerácia a perforácia, zápal gastrointestinálneho traktu (na ktoromkoľvek mieste), stomatitída, dyspepsia, abdominálna distenzia, orofaryngeálna suchosť
Zriedkavé odynofágia, hypermotilita
Časté hepatitída (vrátane zvýšenej hladiny pečeňových enzýmov)
Menej časté hepatopatia (vrátane cirhózy), cholestáza, zvýšená hladina bilirubínu v krvi
Zriedkavé cholelitiáza
Časté vyrážka
Menej časté alopécia, nový nástup alebo zhoršenie psoriázy
(vrátane palmoplantárnej pustulóznej psoriázy)
a s ňou súvisiace ochorenia, dermatitída a ekzém, porucha potnej žľazy, kožný vred, fotosenzitivita,
akné, zmeny sfarbenia kože, suchá koža, poruchy
nechtov a nechtového lôžka
Zriedkavé exfoliácia a deskvamácia kože, bulózne ochorenia, porucha štruktúry vlasov, Stevens-Johnsonov syndróm**, multiformný erytém**
Menej časté svalové poruchy, zvýšená hladina kreatínfosfokinázy v krvi
Menej časté porucha funkcie obličiek, krv v moči, symptómy močového mechúra a močovej rúry
Zriedkavé nefropatia (vrátane nefritídy)
T
rieda orgánových systémov Frekvencia Nežiaduce reakcie
Poruchy reprodukčného systému a prsníkov
Celkové poruchy a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Menej časté poruchy menštruačného cyklu a krvácania maternice
(vrátane amenorey), poruchy prsníkov
Zriedkavé sexuálna dysfunkcia
Časté pyrexia, bolesť (na ktoromkoľvek mieste), asténia, pruritus (na ktoromkoľvek mieste), reakcie v mieste vpichu
Menej časté zimnica, ochorenie podobné chrípke, zmena vnímania teploty, nočné potenie, návaly tepla
Zriedkavé fistula (na akomkoľvek mieste)
Menej časté zvýšená hladina alkalickej fosfatázy v krvi, predĺženie času koagulácie
Zriedkavé zvýšená hladina kyseliny močovej v krvi
Menej časté kožné poranenia, zhoršené hojenie
*Tieto udalosti sa týkali triedy antagonistov TNF, ale výskyt v súvislosti s certolizumab pegolom nie je známy.
**Tieto udalosti sa týkali triedy antagonistov TNF.
V iných indikáciách boli v súvislosti s Cimziou pozorované menej často ďalšie nasledujúce nežiaduce reakcie: gastrointestinálna stenóza a obštrukcie, zhoršenie celkového zdravotného stavu, spontánny potrat a azoospermia.
Popis vybraných nežiaducichreakciíInfekcieV placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou bola miera incidencie nových prípadov infekcií u všetkých pacientov liečených Cimziou 1,03 na pacientorok a u pacientov liečených placebom 0,92 na pacientorok. Z infekcií boli zastúpené predovšetkým infekcie horných dýchacích ciest, infekcie močového traktu a infekcie dolných dýchacích ciest a herpetické vírusové infekcie (pozri časti 4.3 a 4.4).
V placebom kontrolovaných klinických štúdiách bolo v skupinách liečených Cimziou (0,07 na pacientorok; pri všetkých dávkach) viac nových prípadov závažnej infekcie v porovnaní s placebom (0,02 na pacientorok). Medzi najčastejšie závažné infekcie patrili pneumónia, tuberkulózne infekcie. Medzi závažné infekcie patrili aj invazívne oportúnne infekcie (napr. pneumocystóza, mykotická ezofagitída, nokardióza a diseminovaný herpes zoster). Neexistuje dôkaz o zvýšenom riziku infekcií pri nepretržitej expozícii v priebehu času (pozri časť 4.4).
MalignityalymfoproliferatívneporuchyS výnimkou nemelanómových nádorov kože bolo pozorovaných 121 malignít vrátane 5 prípadov lymfómu v klinických RA štúdiách s Cimziou, v ktorých bolo liečených celkovo 4 049 pacientov, čo
predstavuje 9 277 pacientorokov. V klinických štúdiách s reumatoidnou artritídou sa v súvislosti
s Cimziou vyskytovali prípady lymfómu v rozsahu incidencie 0,05 na 100 pacientorokov a melanómu v rozsahu incidencie 0,08 na 100 pacientorokov (pozri časť 4.4). V klinickej štúdii fázy III
s psoriatickou artritídou sa pozoroval aj jeden prípad lymfómu.
AutoimunitaZ pacientov, ktorí boli na začiatku ANP negatívni, malo v pivotných štúdiách pozitívne ANP titre
16,7 % pacientov liečených Cimziou v porovnaní s 12,0 % pacientov v placebovej skupine. V rámci pacientov, ktorí mali na začiatku negatívne protilátky anti-dsDNA, u 2,2 % pacientov liečených
Cimziou sa vyskytli pozitívne titre protilátok anti-dsDNA v porovnaní s 1,0 % pacientov v placebovej
skupine. V obidvoch placebom kontrolovaných a nezaslepených následných klinických štúdiách s reumatoidnou artritídou boli menej často hlásené prípady syndrómu podobného lupusu. Zriedkavo boli
hlásené iné imunologicky sprostredkované ochorenia; príčinná súvislosť s Cimziou nie je známa.
Vplyv dlhodobej liečby Cimziou na vývoj autoimunitných ochorení nie je známy.
R
eakcie
v
m
i
este
p
odania
injekcie
V placebom kontrolovaných klinických štúdiách s reumatoidnou artritídou sa reakcie v mieste podania injekcie vyskytovali u 5,8 % pacientov liečených Cimziou, ako je erytém, svrbenie, hematóm, bolesť, opuch alebo podliatina, v porovnaní s 4,8 % pacientov liečených placebom. Bolesť v mieste podania injekcie sa pozorovala u 1,5 % pacientov liečených Cimziou, pričom ani v jeden prípad nemal za následok prerušenie liečby.
ZvýšeniekreatínfosfokinázyFrekvencia zvýšenia kreatínfosfokinázy (CPK) bola celkovo vyššia u pacientov s axSpA v porovnaní s RA populáciou. Frekvencia bola zvýšená u pacientov liečených placebom (2,8 % oproti 0,4 %
v populácii s axSpA a RA, v uvedenom poradí), aj u pacientov liečených Cimziou (4,7 % oproti 0,8 % v populácii s axSpA a RA, v uvedenom poradí). Zvýšenie CPK v štúdii axSpA bolo väčšinou mierne až stredne závažné, prechodnej povahy a neznámeho klinického významu, pričom ani v jednom
prípade toto zvýšenie neviedlo k vysadeniu liečby.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePočas klinických štúdií sa nepozorovala žiadna toxicita limitujúca dávku. Podávané boli viacnásobné dávky až do 800 mg subkutánne a 20 mg/kg intravenózne. V prípade predávkovania sa odporúča
u pacientov starostlivé sledovanie akýchkoľvek nežiaducich reakcií alebo účinku a okamžité začatie
vhodnej symptomatickej liečby.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory tumor nekrotizujúceho alfa faktora (TNFα), ATC kód: L04AB05
Mechanizmus účinkuCimzia má vysokú afinitu k ľudskému TNFα a viaže sa s disociačnou konštantou (KD) 90 pM. TNFα
je kľúčovým prozápalovým cytokínom s najdôležitejšou úlohou pri zápalových procesoch. Cimzia selektívne neutralizuje TNFα (IC90 4 ng/ml pre inhibíciu ľudského TNFα v
in vitro hodnotení L929
fibrosarkómovej cytotoxicity u myší), ale neneutralizuje lymfotoxín α (TNFβ).
Ukázalo sa, že Cimzia neutralizuje rozpustný ľudský TNFα spojený s membránami, spôsobom závislým od dávky. Inkubácia monocytov s Cimziou viedla k inhibícii lipopolysacharidmi (LPS) indukovanej produkcie TNFα a IL1β v ľudských monocytoch, ktorá bola závislá od dávky.
Cimzia neobsahuje oblasť fragmentu schopného kryštalizácie (Fc), ktorá je zvyčajne súčasťou kompletnej protilátky a preto neviaže komplement ani nevyvoláva od protilátky závislú bunkami sprostredkovanú cytotoxicitu
in vitro. Neindukuje apoptózu
in vitro v ľudských monocytoch ani lymfocytoch pochádzajúcich z periférnej krvi, ani degranuláciu neutrofilov.
Klinická účinnosťReumatoidnáartritídaÚčinnosť a bezpečnosť Cimzie boli hodnotené v 2 randomizovaných, placebom kontrolovaných, dvojito zaslepených klinických štúdiách u pacientov vo veku ≥ 18 rokov s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR (American College of Rheumatology) kritérií, RA-I (RAPID
1) a RA-II (RAPID 2). Každý pacient mal 9 alebo viac opuchnutých a citlivých kĺbov a trpel aktívnou RA minimálne 6 mesiacov pred začiatkom štúdie. V obidvoch štúdiách bola Cimzia podávaná subkutánne v kombinácii s perorálnym MTX počas minimálne 6 mesiacov v stabilných dávkach minimálne 10 mg týždenne po dobu 2 mesiacov. S používaním Cimzie v kombinácii s DMARD okrem MTX nie sú žiadne skúsenosti.
Účinnosť a bezpečnosť Cimzie boli hodnotené u dospelých pacientov doteraz neliečených liekmi zo skupiny DMARD (tzv. DMARD-naivní pacienti) s aktívnou reumatoidnou artritídou
v randomizovanej, placebom kontrolovanej, dvojito zaslepenej klinickej štúdii (C-EARLY). V štúdii
C-EARLY boli pacienti vo veku ≥ 18 rokov, ktorí mali ≥ 4 opuchnuté a bolestivé kĺby a diagnóza stredne závažnej až závažnej progresívnej reumatoidnej artritídy u nich musela byť stanovená
v priebehu 1 roka (podľa definície klasifikačných kritérií ACR/EULAR (European League Against
Rheumatism) z roku 2010). Priemerný čas od diagnózy bol u pacientov vo východiskovom bode 2,9
mesiaca a neboli doteraz liečení pomocou DMARD (vrátane MTX). V ramene s Cimziou a v ramene s placebom bola liečba MTX začatá od 0. týždňa (10 mg/týždeň), dávka bola titrovaná až do maximálne tolerovanej dávky v 8. týždni (povolených min. 15 mg/týždeň, max. 25 mg/týždeň)
a udržiavaná v priebehu štúdie (priemerná dávka MTX po 8. týždni bola pre placebo 22,3 mg/týždeň, pre Cimziu 21,1 mg/týždeň).
Tabuľka 2 Popis klinickej štúdie
Č
íslo št
údie
P
očty pacientov
A
ktívna schéma dávkovania
C
i
ele štúdie
RA-I (52 týždňov)
RA-II (24 týždňov)
C-EARLY (do 52. týždňa
982 400 mg (v 0.,2.,4. týždni) s MTX
200 mg alebo 400 mg každé 2 týždne s MTX
619 400 mg (v 0.,2.,4. týždni) s MTX
200 mg alebo 400 mg
každé 2 týždne s MTX
879 400 mg (v 0., 2., 4. týždni)
200 mg každé
2 týždne s MTX
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Súbežne primárne koncové ukazovatele: ACR 20 v 24. týždni a zmena
z východiskovej hodnoty mTSS v 52. týždni
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia. Primárny výsledný ukazovateľ: ACR 20
v 24. týždni.
Vyhodnotenie liečby prejavov a príznakov a inhibície štrukturálneho poškodenia
u DMARD-naivných pacientov.
Primárny výsledný ukazovateľ: podiel subjektov s pretrvávajúcou remisiou* v 52. týždni
mTSS: modifikované TSS (Total Sharp Score)
*Pretrvávajúca remisia v 52. týždni je definovaná ako DAS28[ESR] < 2,6 v 40. týždni aj v 52. týždni.
Prejavy a príznaky
Výsledky klinických štúdií RA-I a RA-II sú uvedené v tabuľke 3. V oboch klinický štúdiách sa v porovnaní s placebom dosiahli štatisticky významne vyššie odpovede ACR 20 od 1. týždňa
a ACR 50 od 2. týždňa. Odpovede pretrvávali až do 52. týždňa (RA-I) a 24. týždňa (RA-II). Zo 783
pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo 52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie. Z nich 427 pacientov
ukončilo 2-ročnú nezaslepenú následnú štúdiu a tak mali celkovú expozíciu Cimzii spolu 148 týždňov.
Pozorovaná miera odpovede ACR 20 v tomto časovom bode bola 91 %. Zníženie (RA-I)
z východiskovej hodnoty DAS28 (ESR) bolo tiež v porovnaní s placebom významne vyššie
(p < 0,001) v 52. týždni (RA-I) a 24. týždni (RA-II) a pretrvávalo počas 2 rokov v nezaslepenej predĺženej štúdii RA-I.
T
abuľka 3 ACR odpoveď v klinických štúdiách RA-I a RA-II Štúdia RA-I
Štúdia RA-II
K
ombinácia s metotrexátom
(
24 a 52 týždňov)
K
ombinácia s metotrexátom
(
24 týždňov)
Odpoveď Placebo + MTX N = 199
ACR 20
Cimzia
200 mg + MTX
každé 2 týždne
N = 393
Placebo + MTX N = 127
Cimzia
200 mg + MTX
každé 2 týždne
N = 246
24. týždeň 14 % 59 %** 9 % 57 %**
52. týždeň 13 % 53 %** N/A N/A
ACR 50
24. týždeň 8 % 37 %** 3 % 33 %**
52. týždeň 8 % 38 %** N/A N/A
ACR 70
24. týždeň 3 % 21 %** 1 % 16 %*
52. týždeň 4 % 21 %** N/A N/A
Hlavná klinická odpoveďa.
1 % 13 %**
Cimzia verzus placebo: * p ≤ 0,01, ** p < 0,001
a. Hlavná klinická odpoveď je definovaná ako dosiahnutie odpovede ACR 70 pri každom hodnotení počas nepretržitého 6-mesačného obdobia.
p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
Percentuálna odpoveď založená na počte pacientov, ktorí prispeli údajmi (n) pre tento koncový a časový ukazovateľ, ktorý sa môže odlišovať od N.
Štúdia C-EARLY splnila svoje primárne a kľúčové sekundárne výsledné ukazovatele. Kľúčové výsledky zo štúdie sú uvedené v tabuľke 4.
Tabuľka 4 Štúdia C-EARLY: percento pacientov s trvajúcou remisiou a trvajúcou nízkou aktivitou ochorenia v 52. týždni
Odpoveď Placebo + MTX N= 213
Cimzia 200 mg + MTX N= 655
P
r
etrvávajúca remisia*
(DAS28(ESR) <2,6 v
40. týždni aj 52. týždni)
Pretrvávajúca nízka aktivita ochorenia
(DAS28(ESR) ≤3,2 v 40. týždni aj v 52. týždni)
15,0 % 28,9%**
28,6 % 43,8%**
*Primárny výsledný ukazovateľ štúdie C-EARLY (do 52. týždňa)
Zostava úplnej analýzy, prirátanie nonrespondérov pre chýbajúce hodnoty.
**Cimzia+MTX vs placebo+MTX: p<0,001
p-hodnota bola odhadnutá z logistického regresného modelu s faktormi pre liečbu, miesto a dobu od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4 mesiace)
U pacientov v skupine Cimzia+MTX došlo k výraznejšej redukcii od východiskového stavu
v DAS 28 (ESR) v porovnaní so skupinou placebo+MTX, ktorá bola pozorovaná už od 2. týždňa a pokračovala do 52. týždňa (p<0,001 pri každej kontrole). Hodnotenia remisie (DAS 28 (ESR)
<2,6), stavu nízkej aktivity ochorenia (DAS 28 (ESR) ≤3,2) a ACR 50 a ACR 70 podľa kontrol ukázali, že liečba Cimzia+MTX viedla k rýchlejšej a výraznejšej odpovedi ako liečba
PBO+MTX. Tieto výsledky u DMARD-naivných subjektov pretrvávali počas 52 týždňov liečby.
R
ádiografická odpoveď
V RA-I bolo štrukturálne poškodenie kĺbu hodnotené rádiograficky v 52. týždni a vyjadrené ako
zmena mTSS a jeho zložiek, skóre erózie a skóre zúženia kĺbovej štrbiny (JSN – joint space narrowing score) v porovnaní s východiskovými hodnotami. Pacienti liečení Cimziou preukázali významne
menšiu rádiografickú progresiu ako pacienti užívajúci placebo v 24. týždni a 52. týždni (pozri tabuľku
5). V placebovej skupine v 52. týždni bolo 52 % pacientov bez rádiografickej progresie (mTSS ≤ 0,0)
v porovnaní so 69 % v skupine liečenej Cimziou v dávke 200 mg.
Tabuľka 5 Zmeny v priebehu 12 mesiacov v RA-I
m
T
SS
P
l
acebo + MTX N = 199
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX N = 393
Stredná hodnota (SD)
C
im
z
i
a 200 mg + MTX – Placebo + MTX
Stredná hodnota rozdielu
52. týždeň 2,8 (7,8) 0,4 (5,7) -2,4
Skóre erózie
52. týždeň 1,5 (4,3) 0,1 (2,5) -1,4
JSN skóre
52. týždeň 1,4 (5,0) 0,4 (4,2) -1,0
p-hodnoty v prípade obidvoch – mTSS aj skóre erózie – boli < 0,001 a v prípade JSN skóre boli
≤ 0,01. ANCOVA zodpovedala hodnotenej zmene od východiskovej hodnoty pre každé meranie s oblasťou a liečbou ako faktormi a východiskovým hodnotením ako kovariátom.
Zo 783 pacientov, ktorí boli na začiatku randomizovaní k aktívnej liečbe v RA-I, 508 ukončilo
52-týždňovú placebom kontrolovanú liečbu a zúčastnilo sa nezaslepeného predĺženia štúdie.
V podskupine 449 pacientov, ktorí ukončili minimálne 2-ročnú liečbu Cimziou (RA-I a nezaslepená predĺžená štúdia), sa preukázala pretrvávajúca inhibícia progresie štrukturálneho poškodenia
s hodnotiteľnými údajmi v časovom bode 2 roky.
V štúdii C-EARLY viedla Cimzia+MTX k inhibícii rádiografickej progresie v porovnaní
s placebom+MTX v 52. týždni (pozri tabuľku 6). V skupine placebo+MTX u 49,7 % pacientov nedošlo k žiadnej rádiografickej progresii (zmena mTSS ≤0,5) v 52. týždni, v porovnaní s 70,3 %
v skupine Cimzia+MTX (p<0,001).
Tabuľka 6 Rádiografické zmeny v 52. týždni v štúdii C-EARLY
P
l
acebo + MTX
N= 163
Priemer (SD)
Cimzia 200 mg + MTX
N = 528
Priemer (SD)
Cimzia 200 mg + MTX – Placebo + MTX Rozdiel*
m
T
SS
týždeň 52
Skóre erózie
týždeň 52
Skóre JSN
týždeň 52
1,8 (4,3) 0,2 (3,2)** -0,978 (-1,005, -0,500)
1,1 (3,0) 0,1 (2,1)** -0,500 (-0,508, -0,366)
0,7 (2,3) 0,1 (1,7)** 0,000 (0,000, 0,000)
Rádiografická zostava s lineárnou extrapoláciou.
* Hodges-Lehmannov bodový odhad posunu a 95 % asymptotický (Moses) interval spoľahlivosti.
**Cimzia+MTX vs. placebo+MTX p<0,001. p-hodnota bola odhadnutá z ANCOVA modelu hodnotenia liečby, oblasti a času od diagnózy RA vo východiskovom stave (≤4 mesiace vs >4 mesiace) ako faktory a hodnoty vo východiskovom stave ako kovariancia.
Odpoveď fyzickej funkcie a následky týkajúce sa zdraviaU pacientov liečených Cimziou v RA-I a RA-II bolo už od 1. týždňa až do konca štúdií, v porovnaní s placebom, hlásené významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI (Health Assessment Questionnaire – Disability Index) a únavy (vyčerpanosti) hlásenej podľa škály FAS (Fatigue Assessment Scale). V obidvoch klinických štúdiách hlásili pacienti liečení Cimziou významne väčšie zlepšenia v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a v skóre všetkých domén. Zlepšenia fyzickej funkcie a HRQoL pretrvávali
v priebehu 2 rokov v nezaslepenej predĺženej štúdii RA-I. Pacienti liečení Cimziou hlásili, v porovnaní
s placebom, štatisticky významné zlepšenia v prieskume produktivity práce (Work Productivity
Survey).
V štúdii C-EARLY pacienti liečení kombináciou Cimzia+MTX hlásili významné zlepšenie bolesti
v 52. týždni v porovnaní s kombináciou placebo+MTX, hodnotenej pomocou Hodnotenia artritickej bolesti pacientom (Patient Assessment of Arthritis Pain = PAAP) - 48,5 vs - 44,0 (priemer najmenších
štvorcov) (p<0,05).
Klinická štúdia DoseFlex
Účinnosť a bezpečnosť 2 dávkovacích režimov Cimzie (200 mg každé 2 týždne a 400 mg každé 4
týždne) oproti placebu sa hodnotili v 18-týždňovej, nezaslepenej, predrandomizačnej a 16-týždňovej randomizovanej, dvojito-zaslepenej, placebom kontrolovanej klinickej štúdii u dospelých pacientov
s aktívnou reumatoidnou artritídou diagnostikovanou podľa ACR kritérií, ktorí nemali dostatočnú
odpoveď na MTX.
Pacienti dostávali nárazové dávky 400 mg Cimzie v 0., 2. a 4. týždni, po ktorých nasledovala dávka
200 mg Cimzie každé 2 týždne počas začiatočného nezaslepeného obdobia. Respondéri (ACR 20
dosiahnuté) v 16. týždni boli randomizovaní v 18. týždni na Cimziu v dávke 200 mg každé 2 týždne, Cimziu v dávke 400 mg každé 4 týždne alebo placebo v kombinácii s MTX počas ďalších 16 týždňov
(celková dĺžka štúdie: 34 týždňov). Tieto 3 skupiny boli dobre vyvážené čo sa týka klinickej odpovede
po aktívnej vstupnej fáze (ACR 20: 83-84 % v 18. týždni).
Primárnym koncovým ukazovateľom štúdie bola miera ACR 20 respondérov v 34. týždni. Výsledky v v 34. týždni sú uvedené v tabuľke 7. Obidva dávkovacie režimy Cimzie preukázali pretrvávajúcu klinickú odpoveď a boli štatisticky významné v porovnaní s placebom v 34. týždni. Koncový ukazovateľ ACR 20 sa dosiahol pre oba režimy Cimzie, 200 mg každé 2 týždne a 400 mg každé 4 týždne.
Tabuľka 7 ACR odpoveď v klinickej štúdii DoseFlex v 34. týždni
L
i
ečebný režim v 0. až
16. týždni
C
im
z
i
a 400 mg + MTX v 0., 2. a 4. týždni, po ktorých nasledovala Cimzia v dávke 200 mg + MTX každé 2 týždne
R
andomizovaný, dvojito- zaslepený liečebný režim v 18. až 34. týždni
AC
R 20
p-hodnota*
ACR 50
p-hodnota*
ACR 70
p-hodnota*
N/A: neaplikovateľné
Placebo + MTX
N=69
45 % N/A
30 % N/A
16 % N/A
Cimzia
200 mg + MTX každé 2 týždne N=70
67 %
0,009
50 %
0,020
30 %
0,052
Cimzia
400 mg + MTX každé 4 týždne N=69
65 %
0,017
52 %
0,010
38 %
0,005
* p-hodnoty Waldovho testu pre porovnania Cimzie v dávke 200 mg oproti placebu a Cimzie v dávke
400 mg oproti placebu sú odhadované na základe logistickej regresie s faktormi pre liečbu.
AxiálnaspondylartritídaÚčinnosť a bezpečnosť Cimzie boli hodnotené v jednej multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (AS001) u 325 pacientov vo veku ≥ 18 rokov s aktívnou axiálnou spondylartritídou s nástupom v dospelosti trvajúcou najmenej 3 mesiace definovanou klasifikačnými kritériami spoločnosti ASAS (Assessment of Spondyloarthritis International Society). Celková populácia s axiálnou spondylartritídou zahŕňala subpopulácie s röntgenologickým dôkazom ankylozujúcej spondylitídy (AS) a bez neho (axiálna spondylartritída bez röntgenologického dôkazu;
[nr-axSpA]). Pacienti mali aktívne ochorenie definované indexom aktivity ochorenia ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Disease Activity Index - BASDAI) ≥ 4, bolesť chrbtice ≥ 4
na číselnej stupnici NRS (Numerical Rating Scale) s rozsahom 0 až 10 a zvýšené CRP alebo súčasný
dôkaz sakroilitídy pri vyšetrení magnetickou rezonanciou (MR). Pacienti museli mať prejavy neznášanlivosti alebo museli mať nedostatočnú odpoveď aspoň na jedno NSA. Celkovo 16 %
pacientov malo v anamnéze liečbu antagonistom TBF. Pacienti boli liečení nárazovou dávkou Cimzie
400 mg v 0., 2. a 4. týždni (u oboch liečebných skupín) alebo placebom, po ktorom nasledovala dávka Cimzie buď 200 mg každé 2 týždne alebo 400 mg každé 4 týždne alebo placebo. 87,7 % pacientov dostávalo súčasne NSA. Primárnym výsledným ukazovateľom účinnosti bola miera odpovede ASAS20 v 12. týždni.
Po 24 týždňovej, dvojito zaslepenej, placebom kontrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 156 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 204 týždňov. Všetci pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
199 účastníkov klinického skúšania (61,2 % z randomizovaných účastníkov) ukončilo štúdiu do týždňa 204.
Kľúčové výsledky účinnosti
V klinickej štúdii AS001 dosiahlo ASAS20 odpoveď v 12. týždni 58 % pacientov liečených Cimziou v dávke 200 mg každé 2 týždne a 64 % pacientov liečených Cimziou v dávke 400 mg každé 4 týždne
v porovnaní s 38 % pacientmi užívajúcimi placebo (p < 0,01). V celkovej populácii bol podiel
ASAS20 respondérov klinicky významný a významne vyšší pre liečebnú skupinu s Cimziou v dávke
200 mg každé 2 týždne a s Cimziou 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej kontrole od 1. týždňa až do 24. týždňa (p ≤ 0,001 pri každej kontrole). V 12. týždni a 24. týždni
bolo percento osôb s ASAS40 odpoveďou vyššie v skupinách liečených Cimziou v porovnaní s placebom.
Podobné výsledky sa dosiahli u oboch subpopulácií s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu. U žien neboli ASAS20 odpovede štatisticky významne odlišné od placeba až do časového bodu po 12. týždni.
Zlepšenie ASAS 5/6, čiastočná remisia a BASDAI-50 boli štatisticky významné v 12. týždni a v
24. týždni a pretrvávali až do 48. týždňa v celkovej populácii rovnako ako v subpopuláciách. Kľúčové výsledky účinnosti z klinickej štúdie AS001 sú uvedené v tabuľke 8.
U pacientov, ktorí zostali v štúdii, sa zlepšenie vo všetkých vyššie uvedených kľúčových výsledkoch účinnosti zachovalo do týždňa 204 v celkovej populácii, rovnako ako v subpopuláciách.
T
abuľka 8 Kľúčové výsledky účinnosti v klinickej štúdii AS001 (percento pacientov)
Parametre
Ankylozujúca spondylitída
Axiálna spondylartritída bez röntgenologického dôkazu
Axiálna spondylartritída Celková populácia
A
SAS20
(b
,c
)
12. týždeň
24. týždeň
ASAS40(c,d)
12. týždeň
24. týždeň
ASAS 5/6(c,d)
12. týždeň
24. týždeň Čiastočná remisia(c,d)
12. týždeň
24. týždeň
BASDAI 50(c,d)
12. týždeň
24. týždeň
Placebo
N=57
37%
33%
19%
16%
9%
5%
2%
7%
11%
16%
Cimzia všetky režimy dávkovania (a) N=121
60%*
69%**
45%**
53%**
42%**
40%**
20%**
28%**
41%**
49%**
Placebo
N=50
40%
24%
16%
14%
8%
4%
6%
10%
16%
20%
Cimzia všetky režimy dávkovania (a)
N=97
61%*
68%**
47%**
51%**
44%**
45%**
29%**
33%**
49%**
57%**
Placebo
N=107
38%
29%
18%
15%
8%
5%
4%
9%
13%
18%
Cimzia všetky režimy dávkovania (a)
N=218
61%**
68%**
46%**
52%**
43%**
42%**
24%**
30%**
45%**
52%**

(a) Cimzia všetky režimy dávkovania = údaje pre Cimziu v dávke 200 mg podávanej každé 2 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni plus Cimzia 400 mg podávaná
každé 4 týždne, ktorej predchádzala nárazová dávka 400 mg v 0., 2. a 4. týždni
(b) Výsledky sú z randomizovaného súboru
(c) p-hodnoty Waldovho testu sú uvedené na porovnanie terapií použitím logistickej regresie s faktormi pre liečbu a oblasť.
(d) Kompletný analyzovaný súbor
NA = nie je k dispozícii
* p ≤ 0,05, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
Mobilita chrbticeMobilita chrbtice sa hodnotila v dvojito zaslepenej, placebom kontrolovanej fáze pomocou BASMI
v niekoľkých časových bodoch, vrátane východiskového stavu, v 12. týždni a v 24. týždni. Klinicky významné a štatisticky významné rozdiely u pacientov liečených Cimziou v porovnaní s pacientmi liečenými placebom sa preukázali pri každej kontrole od východiskového stavu. Rozdiel od placeba má tendenciu byť väčší u subpopulácie nr-axSpA ako u AS, čo môže byť dôsledkom menšieho chronického štrukturálneho poškodenia u pacientov s nr-axSpA.
Zlepšenie lineárneho skóre BASMI dosiahnuté v 24. týždni sa u pacientov, ktorí zostali v štúdii, udržalo až do týždňa 204.
Odpoveď fyzickej funkcie a následky týkajúce sa zdraviaV klinickej štúdii AS001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotené pomocou BASFI a zlepšenie bolesti hodnotené NRS stupnicou celkovej a nočnej bolesti
chrbtice v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie únavy
(vyčerpanosti) hlásené pomocou položky BASDAI pre únavu a významné zlepšenie kvality života súvisiacej so zdravím merané dotazníkom Kvality života pri ankylozujúcej spondylitíde (ASQoL) a zlepšenie v SF-36 súhrnov fyzickej a duševnej zložky (Physical and Mental Component Summaries) a v skóre všetkých domén v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie
produktivity v práci a v domácnosti súvisiacej s axiálnou spondylartritídou, ktoré bolo hlásené v prieskume produktivity práce v porovnaní s placebom.
U pacientov, ktorí zostali v štúdii, bolo zlepšenie všetkých vyššie uvedených výsledkov do značnej
miery zachované až do týždňa 204.
Inhibícia zápalu pri magnetickej rezonancii (MR)
V zobrazovacej subštúdii zahŕňajúcej 153 pacientov sa hodnotili prejavy zápalu hodnotené MR v
12. týždni a boli vyjadrené ako zmena skóre SPARCC (Spondyloarthritis Research Consortium of
Canada) oproti východiskovému stavu pre krížovo-driekové kĺby a skóre ASspiMRI-a v berlínskej modifikácii pre chrbticu. Významná inhibícia zápalových príznakov u krížovo-driekových kĺbov aj chrbtice sa pozorovala u pacientov liečených Cimziou (všetky dávkové skupiny) v 12. týždni, a to v celkovej populácii s axiálnou spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu.
U pacientov, ktorí zostali v štúdii a u ktorých boli k dispozícii východiskové hodnoty aj hodnoty z týždňa 204, bola do značnej miery zachovaná inhibícia zápalových príznakov až do týždňa 204 v krížovo-driekových kĺboch (n=72) aj chrbtici (n=82), a to v celkovej populácii s axiálnou
spondylartritídou, ako aj v subpopulácii s ankylozujúcou spondylitídou a s axiálnou spondylartritídou bez röntgenologického dôkazu.
Psoriatickáartritída
Účinnosť a bezpečnosť Cimzie sa hodnotili v multicentrickej, randomizovanej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii (PsA001) u 409 pacientov vo veku ≥ 18 rokov s aktívnou psoriatickou artritídou s nástupom v dospelosti trvajúcou najmenej 6 mesiacov definovanou klasifikačnými kritériami pre psoriatickú artritídu (Classification Criteria for Psoriatic Arthritis - CASPAR). Pacienti mali ≥ 3 opuchnuté a bolestivé kĺby a zvýšené hodnoty markerov akútnej fázy. Pacienti mali tiež aktívne psoriatické kožné lézie alebo zdokumentovanú psoriázu v anamnéze a zlyhala u nich jedna alebo viac terapií DMARD. Predchádzajúca liečba jedným antagonistom TNF bola povolená a 20 % pacientov bolo predtým liečených TNF-antagonistom. Pacienti dostávali nárazovú dávku Cimzie 400 mg v 0., 2. a 4. týždni (v oboch liečebných skupinách) alebo placebo, po ktorej nasledovalo podávanie 200 mg Cimzie každé 2 týždne alebo 400 mg Cimzie každé 4 týždne alebo placebo každé 2 týždne. 72,6 % pacientov súbežne užívalo NSA a 70,2 % konvenčné DMARD. Štúdia mala dva primárne koncové ukazovatele: percento pacientov, ktorí dosiahli ACR 20 odpoveď
v 12. týždni a zmena modifikovaného skóre mTSS (Total Sharp Score) v 24. týždni od východiskovej hodnoty. Účinnosť a bezpečnosť Cimzie u pacientov s PsA, u ktorých prevládali symptómy sakroilitídy alebo axiálnej spondylartritídy, neboli analyzované samostatne.
Po 24 týždňovej dvojito zaslepenej, placebom kotrolovanej fáze štúdie nasledovala 24 týždňová zaslepená fáza a 168 týždňová otvorená fáza. Maximálna dĺžka trvania štúdie bola 216 týždňov. Všetci
pacienti dostávali Cimziu ako v zaslepenej fáze, tak aj v následnej otvorenej fáze. Celkom
264 účastníkov klinického skúšania (64,5 %) ukončilo štúdiu až v týždni 216.
ACR odpoveď
Pacienti liečení Cimziou mali štatisticky významne vyššiu mieru ACR 20 odpovede v 12. týždni
a v 24. týždni v porovnaní s pacientmi, ktorým bolo podávané placebo (p < 0,001). Percento ACR 20 respondérov bolo klinicky významné v liečebných skupinách s Cimziou v dávke 200 mg každé 2 týždne a s Cimziou v dávke 400 mg každé 4 týždne v porovnaní s placebovou skupinou pri každej návšteve od začiatku až do 24. týždňa (nominálne p ≤ 0,001 pri každej návšteve). Pacienti liečení Cimziou mali tiež významné zlepšenie miery odpovede ACR 50 a 70. V 12. a 24. týždni sa u pacientov liečených Cimziou pozorovalo zlepšenie v parametroch periférnej aktivity psoriatickej artritídy (napr. počet opuchnutých kĺbov, počet bolestivých/citlivých kĺbov, daktylitída a entezitída) (nominálna p-hodnota p < 0,01). Kľúčové výsledky účinnosti z klinickej štúdie PsA001 sú uvedené v tabuľke 9.
T
abuľka 9 Kľúčové výsledky účinnosti v klinickej štúdii PsA001 (percento pacientov)
O
dpoveď Placebo
N
=136
C
im
z
i
a
(
a)
200 mg každé 2 týždne
N
=138
C
im
z
i
a
(b
)
400 mg každé 4 týždne
N
=135
ACR
20
12. týždeň
24. týždeň
ACR50
12. týždeň
24. týždeň
ACR70
12. týždeň
24. týždeň
24 %
24 %
11 %
13 %
3 %
4 %
58 %**
64 %**
36 %**
44 %**
25 %**
28 %**
52 %**
56 %**
33 %**
40 %**
13 %*
24 %**
O
dpoveď Placebo
N
=86
C
im
z
i
a
(
a)
200 mg každé 2 týždne N=90
C
im
z
i
a
(b
)
400 mg každé 4 týždne N=76
P
A
SI 75 (c)
12. týždeň
24. týždeň
48. týždeň
14 %
15 % N/A
47 %***
62 %***
67 %
47 %***
61 %***
62 %
(a) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 2 týždne (b) Po nárazovej dávke 400 mg v 0., 2. a 4. týždni sa Cimzia podávala každé 4 týždne (c) U osôb s minimálne 3% psoriázy BSA na začiatku
* p < 0,01, Cimzia oproti placebu
** p < 0,001, Cimzia oproti placebu
*** p < 0,001 (nominálne), Cimzia oproti placebu
Výsledky sú z randomizovaného súboru. Rozdiel liečby: Cimzia 200 mg - placebo, Cimzia 400 mg - placebo (a zodpovedajúci 95 % interval spoľahlivosti a p-hodnota) boli posudzované pomocou štandardného obojstranného Waldovho testu asymptotických štandardných chýb. U pacientov, ktorí prerušili liečbu alebo u nich chýbajú údaje, sa použil systém „Non-responder Imputation„ (NRI).
Spomedzi 273 pacientov, ktorí boli na začiatku randomizovaní na Cimziu v dávke 200 mg každé 2
týždne a Cimziu v dávke 400 mg každé 4 týždne, zostalo na tejto liečbe 237 (86,8 %) pacientov až do
48. týždňa. Zo 138 pacientov randomizovaných na Cimziu v dávke 200 mg každé 2 týždne dosiahlo
92, 68 a 48 pacientov odpoveď ACR 20/50/70 v 48. týždni, v uvedenom poradí. Zo 135 pacientov randomizovaných na Cimziu v dávke 400 mg každé 4 týždne dosiahlo 89, 62 a 41 pacientov odpoveď
ACR 20/50/70, v uvedenom poradí.
Medzi pacientmi, ktorí zostali v štúdii, sa miera odpovede ACR 20, 50 a 70 zachovala až do týždňa
216. To platilo aj v prípade ďalších parametrov periférnej aktivity (napr. počet opuchnutých kĺbov, počet bolestivých /citlivých kĺbov, daktylitída a entezitída).
Rádiografická odpoveďV klinickej štúdii PsA001 bola inhibícia progresie štrukturálneho poškodenia hodnotená rádiograficky
a vyjadrená ako zmena mTSS a jeho zložiek, skóre erózie (ES) a skóre zúženia kĺbovej štrbiny (JSN) v
24. týždni v porovnaní s východiskovými hodnotami. Skóre mTSS bolo upravené pre psoriatickú artritídu pridaním distálnych interfalangeálnych kĺbov ruky. Liečba Cimziou inhibovala rádiografickú progresiu v porovnaní s liečbou placebom v 24. týždni meranú ako zmena celkového skóre mTSS (LS priemerné [±SE] skóre oproti východiskovému stavu 0,28 [±0,07] v placebovej skupine v porovnaní s
0,06 [±0,06] v skupine so všetkými dávkami Cimzie, p = 0,007). Inhibícia rádiografickej progresie sa udržiavala pri liečbe Cimziou až do 48. týždňa v podskupine pacientov s vyšším rizikom
rádiografickej progresie (pacienti s východiskovým skóre mTSS > 6). U pacientov, ktorí sa
zúčastnili štúdie, sa inhibícia rádiografickej progresie naďalej udržala až do týždňa 216.
O
dpoveď fyzickej funkcie a následky týkajúce sa zdravia
V klinickej štúdii PsA001 hlásili pacienti liečení Cimziou významné zlepšenie fyzickej funkcie hodnotenej podľa dotazníka HAQ-DI (Health Assessment Questionnaire – Disability Index), bolesti hodnotenej podľa Hodnotenia artritickej bolesti pacientom PAAP a únavy (vyčerpanosti), čo sa preukázalo podľa Škály hodnotenia únavy (FAS - Fatigue Assessment Scale) v porovnaní s placebom. Pacienti liečení Cimziou hlásili významné zlepšenie kvality života súvisiacej so zdravím merané dotazníkom Kvality života pri psoriatickej artritíde (PsAQoL) a SF-36 fyzickej a duševnej zložky a produktivity súvisiacej s psoriatickou artritídou v zamestnaní a v domácnosti hlásenej v v prieskume produktivity práce v porovnaní s placebom. Zlepšenie vo všetkých vyššie uvedených výsledkoch pretrvávalo až do týždňa 216.
Imunogenicita
Reumatoidnáartritída
Celkový podiel pacientov s detekovateľnými protilátkami proti Cimzii pri aspoň 1 príležitosti bol
9,6 % v placebom kontrolovaných RA štúdiách. Približne u jednej tretiny pacientov s pozitívnymi protilátkami boli zistené in vitro neutralizujúce protilátky. U pacientov liečených súbežne s imunosupresívami (MTX) sa protilátky tvorili v menšej miere ako u pacientov, ktorí na začiatku neužívali imunosupresíva. Tvorba protilátok bola spojená s nižšou plazmatickou koncentráciou lieku a u niektorých pacientov s nižšou účinnosťou.
V 2 dlhodobých (až 5 rokov expozície) otvorených štúdiách celkový podiel pacientov
s detekovateľnými protilátkami proti Cimzii pri aspoň jednej príležitosti bol 13 % (8,4 % z celkového počtu pacientov malo prechodnú tvorbu protilátok a ďalších 4,7 % pacientov malo trvalú tvorbu
protilátok). Celkové percento pacientov, ktorí boli pozitívni na tvorbu protilátok a mali trvale zníženú
plazmatickú koncentráciu lieku, bolo odhadované na 9,1 %. Podobne, ako v placebom kontrolovaných štúdiách, bola tvorba protilátok u niektorých pacientov spojená s nižšou účinnosťou.
Farmakodynamický model založený na údajoch z III. fázy klinickej štúdie predpokladá, že približne u 15 % pacientov sa pri odporúčanom dávkovaní (200 mg každé 2 týždne po začiatočnej dávke), bez súbežnej liečby s MTX, vyvinú protilátky v priebehu 6 mesiacov. Tento počet sa znižuje so zvyšujúcimi sa dávkami súbežnej liečby s MTX. Tieto údaje sú primerane v súlade s pozorovanými údajmi.
Axiálnaspondylartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 4,4 % vo fáze III placebom kontrolovanej štúdie u pacientov s axiálnou spondylartritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až počas 192 týždňov) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 9,6 % (4,8 % malo prechodnú tvorbu protilátok, ďalších
4,8 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo na
6,8 %.
Psoriatickáartritída
Celkové percento pacientov s protilátkami proti Cimzii zistenými aspoň pri jednej príležitosti až do
24. týždňa bolo 11,7 % vo fáze III placebom kontrolovanej štúdie u pacientov s psoriatickou artritídou. Tvorba protilátok bola spojená so zníženou plazmatickou koncentráciou liečiva.
V priebehu celej štúdie (až do 4 rokov expozície) bolo celkové percento pacientov s protilátkami proti
Cimzii zistenými aspoň pri jednej príležitosti 17,3 % (8,7 % malo prechodnú tvorbu protilátok, ďalších
8,7 % malo pretrvávajúcu tvorbu protilátok proti Cimzii). Celkové percento pacientov, ktorí mali pozitívne protilátky s pretrvávajúcou zníženou plazmatickou koncentráciou liečiva sa odhadovalo na 11,5 %.
V
šetky
i
ndikácie
Údaje vyjadrujú podiel pacientov, u ktorých bola vyšetrením ELISA stanovená prítomnosť protilátok proti Cimzii a do veľkej miery závisia od senzitivity a špecificity tohto vyšetrenia.
Okrem toho môže byť pozorovaný výskyt protilátok v hodnotení ovplyvnený viacerými faktormi, vrátane manipulácie so vzorkou, načasovania odberu vzorky, súbežne podávaných liekov a
základného ochorenia. Z týchto dôvodov nie je vhodné porovnávať výskyt protilátok proti Cimzii s výskytom protilátok proti iným antagonistom TNF.
5.2 Farmakokinetické vlastnosti
Plazmatické koncentrácie certolizumab pegolu boli značne úmerné dávke. Farmakokinetika pozorovaná u pacientov s reumatoidnou artritídou zodpovedala farmakokinetike pozorovanej u zdravých jedincov.
Absorpcia
Po subkutánnom podaní sa maximálne plazmatické koncentrácie certolizumab pegolu dosiahli v rozmedzí 54 a 171 hodín od podania injekcie. Biologická dostupnosť (F) certolizumab pegolu po subkutánnom podaní, v porovnaní s intravenóznym podaním, je približne 80 % (rozmedzie 76 % až
88%).
Distribúcia
Vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou bol zdanlivý distribučný objem (V/F) odhadovaný na 8,01 l.
Biotransformáciaaeliminácia
PEGyláciou, kovalentnou väzbou PEG polymérov na peptidy, sa odďaľuje eliminácia týchto látok z krvného obehu viacerými mechanizmami, vrátane zníženého renálneho klírensu, zníženej proteolýzy a zníženej imunogenicity. Certolizumab pegol je preto Fab' fragmentom protilátky konjugovanej
s PEG, aby predĺžil terminálny plazmatický eliminačný polčas Fab' fragmentu na hodnotu porovnateľnú s kompletnou molekulou protilátky. Polčas terminálnej eliminačnej fázy (t1/2) bol pre všetky testované dávky približne 14 dní.
Klírens po subkutánnom podaní bol odhadovaný na 21,0 ml/h vo farmakokinetickej analýze populácie s reumatoidnou artritídou, s interindividuálnou variabilitou 30,8 % (CV) a intraindividuálnou variabilitou 22,0 %. Prítomnosť protilátok proti certolizumab pegolu viedla k približne trojnásobnému nárastu klírensu. V porovnaní s osobou s hmotnosťou 70 kg je klírens o 29 % nižší u individuálnych pacientov s RA vážiacich 40 kg a o 38 % vyšší u pacientov vážiacich 120 kg.
Fab' fragment sa skladá z bielkovinových zložiek a predpokladá sa, že je odbúravaný na peptidy
a aminokyseliny proteolýzou. Dekonjugovaná PEG zložka sa rýchlo eliminuje z plazmy a vylučuje sa renálne v neznámej miere.
Osobitnépopulácie
Porucha funkcie obličiek
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili účinok poruchy funkcie obličiek na farmakokinetiku certolizumab pegolu alebo jeho PEG frakcie. Farmakokinetická analýza populácie
pacientov s ľahkou poruchou renálnej funkcie nepreukázala žiadny vplyv klírensu kreatinínu. Na to,
aby bolo možné odporučiť dávkovanie pri stredne ťažkej a ťažkej poruche funkcie obličiek, nie je dostatok informácií. Predpokladá sa, že farmakokinetika PEG frakcie certolizumab pegolu závisí od funkcie obličiek, avšak u pacientov s poruchou funkcie obličiek sa nehodnotila.
Porucha funkcie pečene
Neboli vykonané špecifické klinické štúdie, ktoré by hodnotili vplyv poruchy funkcie pečene na farmakokinetiku certolizumab pegolu.
Starší pacienti (≥ 65 rokov)
Neboli vykonané špecifické klinické štúdie u starších pacientov. Avšak vo farmakokinetickej analýze populácie pacientov s reumatoidnou artritídou sa nepozoroval žiadny vplyv veku, 78 pacientov
(13,2 % populácie) bolo vo veku 65 rokov alebo viac, pričom najstarší pacient mal 83 rokov.
Pohlavie
Pohlavie nemalo žiadny vplyv na farmakokinetiku certolizumab pegolu. Pretože sa klírens znižuje so znižujúcou sa telesnou hmotnosťou, ženy zvyčajne môžu dosiahnuť o niečo vyššiu systémovú
expozíciu certolizumab pegolu.
Farmakokinetický/farmakodynamickývzťah
Na základe údajov z II. a III. fázy klinickej štúdie bol stanovený vzťah expozícia - odpoveď populácie medzi priemernou plazmatickou koncentráciou certolizumab pegolu počas dávkovacieho intervalu
(Cavg) a účinnosťou (definícia ACR 20 odpovede). Charakteristická Cavg, ktorá tvorí polovicu maximálnej pravdepodobnosti odpovede ACR 20 (EC50), bola 17 µg/ml (95 % CI: 10-23 µg/ml).
5.3 Predklinické údaje o bezpečnosti
Pivotné predklinické štúdie bezpečnosti boli vykonané u opíc Cynomolgus. U potkanov a opíc sa, pri dávkach vyšších ako sú dávky u ľudí, histopatologicky zistila celulárna vakuolizácia prítomná najmä v makrofágoch viacerých orgánov (v lymfatických uzlinách, v miestach vpichu, v slezine, nadobličkách, maternici, cervixe, cievovkovej spleti mozgu a v epitelových bunkách cievovkovej spleti). Je pravdepodobné, že tento nález bol spôsobený bunkovým vychytávaním PEG komponentu. In vitro funkčné štúdie ľudských vakuolizovaných makrofágov naznačili, že všetky skúmané funkcie zostali zachované. Štúdie s potkanmi naznačili, že > 90 % podaného PEG sa eliminovalo počas 3 mesiacov
po jednorazovej dávke, pričom hlavnou cestou exkrécie bolo vylúčenie močom.
Certolizumab pegol nereaguje skrížene s TNF hlodavcov. Preto boli reprodukčné toxikologické štúdie vykonané s homologickou skupinou rozpoznávajúcou TNF potkanov. Význam týchto údajov pri hodnotení rizika u ľudí môže byť obmedzený. Neboli pozorované žiadne nežiaduce účinky na zdravie matiek alebo fertilitu samíc, embryo-fetálne, peri- a postnatálne reprodukčné indexy u potkanov, ktorým bol po pretrvávajúcej supresii TNFα podávaný PEGylovaný Fab' fragment anti-rat TNFα (cTN3 PF). U samcov potkanov bola pozorovaná znížená motilita spermií a tendencia k poklesu počtu spermií.
Štúdie distribúcie preukázali, že prestup cTN3 PF placentou a materským mliekom do cirkulácie plodu a novorodenca je zanedbateľný. Certolizumab pegol sa neviaže na ľudský neonatálny Fc receptor (FcRn). Údaje z modelu ľudského placentárneho transferu s uzatvoreným okruhom ex vivo naznačujú nízky alebo zanedbateľný transfer do fetálneho kompartmentu. Okrem toho experimenty
s transcytózou sprostredkovanou FcRn v bunkách transfektovaných ľudským FcRn poukázali na zanedbateľný transfer (pozri časť 4.6).
V predklinických štúdiách sa nepreukázali žiadne mutagénne ani klastogénne účinky. Štúdie na karcinogenicitu sa neuskutočnili s certolizumab pegolom.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
octan sodný chlorid sodný voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Náplň do dávkovacieho zariadenia uchovávajte vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Jednomililitrová náplň do dávkovacieho zariadenia obsahuje naplnenú injekčnú striekačku (zo skla typu I) s plunžerovým uzáverom (z bromobutylovej gumy). Naplnená injekčná striekačka obsahuje
200 mg certolizumab pegolu.
Veľkosť balenia: 2 náplne do dávkovacieho zariadenia a 2 liehové tampóny.
Multibalenie obsahujúce 6 (3 balenia po 2) náplní do dávkovacieho zariadenia a 6 (3 balenia po 2)
liehových tampónov.
Multibalenie obsahujúce 10 (5 balení po 2) náplní do dávkovacieho zariadenia a 10 (5 balení po 2)
liehových tampónov.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Podrobné pokyny na prípravu a podanie Cimzie v náplni do dávkovacieho zariadenia sú uvedené v písomnej informácii pre používateľa a v Návode na použitie elektromechanického injekčného zariadenia ava.
Tento liek je určený len na jednorazové použitie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
UCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/09/544/008
EU/1/09/544/009
EU/1/09/544/010
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 1. október 2009
Dátum posledného predĺženia registrácie: 16. máj 2014
10. DÁTUM REVÍZIE TEXTU
{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky:
http://www.ema.europa.eu.