
nózne
Liek Xydalba musí byť vopred rozpustený a následne doriedený pred podaním vo forme intravenóznej infúzie v časovom rozpätí 30 minút. Návod na rozpustenie a nariedenie liečiva pred podávaním sa nachádza v časti 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Hypersenzitívna reakcia
Podávanie lieku Xydalba sa má zvážiť u pacientov, u ktorých je známa precitlivenosť na iné
glykopeptidy, pretože by mohlo dôjsť ku ich krížovému znásobeniu. Ak sa dostaví alergická reakcia na liek Xydalba, je potrebné jeho podávanie prerušiť a nasadiť vhodnú liečbu alergickej reakcie.
Hnačka súvisiaca s Clostridiumdifficile
Výskyt kolitídy súvisiacej s antibakteriálnym účinkom a pseudomembránnej kolitídy je hlásený
v súvislosti s použitím takmer všetkých antibiotík a môže sa vyskytovať v rozsahu od mierneho po život ohrozujúci stav. Preto je potrebné brať do úvahy túto diagnózu u pacientov, u ktorých sa
vyskytne hnačka počas alebo následne po užívaní dalbavancínu (pozri časť 4.8). V takom prípade je potrebné zvážiť prerušenie podávania dalbavancínu a použitie podporných prostriedkov spolu
s nasadením špecifickej liečby v súvislosti s Clostridium difficile. Takíto pacienti nesmú byť nikdy liečení liečivami, ktoré potláčajú peristaltiku.
R
eak
c
i
e v súvislosti s infúziou
Liek Xydalba má byť podávaný vo forme intravenóznej infúzie v časovom rozpätí 30 minút, aby sa
minimalizovali riziká reakcií s súvislosti s infúziou. Rýchla intravenózna infúzia glykopeptidových antibakteriálnych látok môže spôsobiť reakcie pripomínajúce syndróm červeného človeka (“Red-Man Syndrome”), vrátane sčervenania hornej časti tela, žihľavky, svrbenia a/alebo vyrážky. Zastavenie
alebo spomalenie infúzie môže mať za následok vymiznutie reakcií.
Nedostatočná činnosť obličiek
Množstvo informácií o účinnosti a bezpečnosti dalbavancínu u pacientov s klírensom kreatinínu
< 30 ml/min je obmedzené. Na základe simulácií je potrebné upraviť dávku u pacientov s chronickou nedostatočnou činnosťou obličiek, ktorých klírens kreatinínu je < 30 ml/min a ktorí nedostávajú pravidelnú hemodialýzu (pozri časti 4.2 a 5.2).
Zmiešané infekcie
V prípade zmiešaných infekcií, kde je podozrenie na prítomnosť gram-negatívnych baktérií, je
potrebná aj liečba pomocou vhodných antibakteriálnych látok, ktoré sú účinné na gram-negatívne baktérie (pozri časť 5.1).
Rezistentné organizmy
Používanie antibiotík môže podporiť premnoženie voči rezistentným organizmom. Ak počas liečby
dôjde k superinfekcii, je potrebné vykonať potrebné opatrenia.
Obmedzené množstvo klinických údajov
K dispozícii je len obmedzené množstvo údajov týkajúcich sa bezpečnosti a účinnosti dalbavancínu
v prípade, že sú podané viac ako dve dávky (v odstupe jedného týždňa). Pri hlavných testoch
s ABSSSI sa typy liečených infekcií obmedzovali na celulitídu/eryzipel (ružu), abscesy a infikované rany. Nie sú skúsenosti s dalbavancínom pri liečbe pacientov s vážne oslabeným imunitným
systémom.
4.5 Liekové a iné interakcie
Výsledky in vitro receptorovej skríningovej štúdie neindikujú pravdepodobnosť interakcie s inými terapeutickými cieľmi alebo potenciál pre klinicky relevantné farmakodynamické interakcie
(pozri časť 5.1).
Klinické štúdie ohľadom interakcie dalbavancínu s inými liečivami neboli uskutočnené. Potenciálneúčinkyinýchliekovnafarmakokinetikudalbavancínu.
Dalbavancín nie je metabolizovaný CYP (cytochróm P-450) enzýmami in vitro, preto spoločné
podávanie CYP induktorov alebo inhibítorov s veľkou pravdepodobnosťou neovplyvňuje
farmakokinetiku dalbavancínu.
Nie je známe, či dalbavancín je substrátom pre hepatickú absorpciu a efluxné transportéry. Súbežné podávanie s inhibítormi týchto transportérov môže zvýšiť účinnosť pôsobenia dalbavancínu. Príkladmi takýchto transportných inhibítorov sú posilnené inhibítory proteázy, verapamil, chinidín, itrakonazol, klaritromycín a cyklosporín.
Potenciáldalbavancínuovplyvňovaťfarmakokinetikuinýchliekov.
Očakáva sa nízky interakčný potenciál dalbavancínu na liečivá metabolizované CYP enzýmami, pretože táto látka nie je ani inhibítor, ani induktor CYP enzýmov in vitro. Nie sú dostupné údaje o dalbavancíne ako inhibítore CYP2C8.
Nie je známe, či je dalbavancín inhibítorom transportérov. Zvýšená expozícia transportným substrátom citlivých na inhibovanú aktivitu transportéru, ako sú statíny a digoxín, sa v kombinácii
s dalbavancínom nedajú vylúčiť.
4.6 Fertilita, gravidita a laktácia
Gravidita
Údaje týkajúce sa užívania dalbavancínu tehotnými ženami nie sú k dispozícii. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3).
Podávanie lieku Xydalba sa počas gravidity neodporúča, pokiaľ to nie je jasne nevyhnutné. Laktácia
Nie je známe, či sa dalbavancín vylučuje do materského mlieka u ľudí. Avšak, keďže dalbavancín sa
vylučuje do mlieka u dojčiacich potkanov, je možné, že sa vylučuje do ľudského materského mlieka. Dalbavancín sa perorálne nevstrebáva dobre; avšak napriek tomu nie je možné vylúčiť, že má vplyv na gastrointestinálnu flóru alebo ústnu flóru dojčeného dieťaťa. Rozhodnutie, či pokračovať
v dojčení/ukončiť dojčenie alebo pokračovať/prerušiť liečbu liekom Xydalba sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na zvieratách preukázali zníženú fertilitu (pozri časť 5.3). Potenciálny riziko pre ľudí nie je
známe.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Xydalba môže mierne ovplyvňovať schopnosť viesť vozidlá a obsluhovať stroje, pretože u malého počtu pacientov boli hlásené závraty (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrnný bezpečnostný profil
Vo fáze 2 / 3 klinických štúdií 1 778 pacientov užívalo dalbavancín. Najčastejšími nežiaduci
reakciami, ku ktorým došlo u ≥ 1 % pacientov liečených dalbavancínom, boli nevoľnosť (2,8 %), hnačka (2,5 %), bolesť hlavy (1,5 %), zvýšená gama-glutamyl transferáza (1,1 %), vyrážka (1,0 %) a vracanie (1,0 %) a boli vo všeobecnosti mierne alebo stredne závažné.
Prehľadný zoznam nežiaducich reakcií
Počas fázy 2/3 klinických štúdií dalbavancínu boli zistené nasledovné nežiaduce reakcie. Nežiaduce
reakcie sú klasifikované podľa triedenia orgánových systémov a frekvencie. Kategórie na základe frekvencie sú odvodené na základe nasledovných kritérií: veľmi časté (≥1/10), časté (≥1/100 až
<1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000).
Triedenie orgánových
s
ystémov (System Organ
C
l
ass, SOC)
|
Č
asté
|
Me
n
e
j časté
|
Z
rie
dk
avé
|
I
n
fekcie a nákazy
|
|
Vulvovaginálna mykotická infekcia,
infekcia močových ciest, plesňová infekcia, kolitída spôsobená Clostridium difficile, kandidóza ústnej dutiny
|
|
P
oruchy krvi
a lymfatického systému
|
|
Anémia, trombocytóza, eozinofília,
leukopénia, neutropénia
|
|
P
oruchy imunitného
s
ystému
|
|
|
Anafylaktoidná
reakcia
|
P
oruchy metabolizmu
a výživy
|
|
Znížená chuť do jedla
|
|
D
u
š
e
vné poruchy
|
|
Nespavosť
|
|
P
oruchy nervového systému
|
Bolesť hlavy
|
Dysgeúzia, závraty
|
|
P
oruchy ciev
|
|
Sčervenanie, flebitída
|
|
R
e
s
p
ir
ačné, torakálne
a mediastinálne poruchy
|
|
Kašeľ
|
Bronchospazmus
|
P
oruchy
gastrointenstinálneho traktu
|
Nevoľnosť,
hnačka, vracanie
|
Zápcha, bolesť brucha, dyspepsia,
abdominálny diskomfort
|
|
P
oruchy kože a podkožného
tkaniva
|
Vyrážka
|
Svrbenie, žihľavka
|
|
P
oruchy reprodukčného
s
ystému a prsníkov
|
|
Vulvovaginálne svrbenie
|
|
B
ež
n
é ochorenia a stav
m
i
e
s
ta podania lieku
|
|
Reakcie v súvislosti s infúziou
|
|
S
kú
m
ané účinky
|
Zvýšená gama-
glutamyl
transferáza
|
Zvýšená krvná laktát dehydrogenáza,
zvýšená alanín aminotransferáza,
zvýšená aspartát aminotransferáza, zvýšená kyselina močová v krvi, abnormálna funkcia pečene, transaminázy zvýšené, krvná alkalická fosfatáza, zvýšený počet krvných doštičiek, zvýšená telesná teplota, zvýšený hepatický enzým
|
|
Pop
i
s
vy
b
r
an
ý
c
h
n
e
ž
i
a
d
u
c
i
c
h
reakcií
Ve
d
ľajšie účinky súvisiace s touto skupinou liekov
Ototoxicita sa spájala s použitím glykopeptidu (vankomycín a teikoplanín); pacienti, ktorí dostávajú sprievodnú terapiu ototoxickou látkou, ako je aminoglykozid, môžu byť vystavení zvýšenému riziku.
Hlásenie podozrení nanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
PríloheV.
4.9 PredávkovanieŽiadne špecifické informácie, týkajúce sa liečenia predávkovania dalbavancínom nie sú k dispozícii, pretože dávku obmedzujúca toxicita v klinických štúdiách nebola pozorovaná. V štúdiách Fázy 1 dostávali zdraví dobrovoľníci jednotlivé dávky do výšky až 1 500 mg a kumulatívne dávky až do
4 500 mg v období až do 8 týždňov bez toho, aby boli pozorované akékoľvek prejavy toxicity alebo preukázané laboratórne výsledky vyvolávajúce klinické obavy.
Liečba predávkovania dalbavancínom by mala pozostávať z pozorovania a všeobecnej podpornej starostlivosti. Hoci nie sú k dispozícii žiadne informácie týkajúce sa špecificky použitia hemodialýzy na liečbu predávkovania, je treba podotknúť, že v štúdii Fázy 1 u pacientov s poruchou funkcie obličiek bolo po 3 hodinách hemodialýzy odstránených menej ako 6% dávky dalbavancínu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antibiotiká na systémové použitie, glykopeptidové antibiotiká, ATC kód: J01XA04.
Mechanizmus účinku
Dalbavancín je baktericídny lipoglykopeptid.
Mechanizmus jeho účinku na citlivé Gram-pozitívne baktérie spočíva v inhibícii syntézy bunkových stien naviazaním na koncový D-alanyl-D-alanín kmeňového peptidu vo vznikajúcej bunkovej stene peptidoglykánu, pričom sa zabráni krížovej väzbe (transpeptidácii a transglykozylácii) disacharidových podjednotiek, čím nastane smrť bakteriálnej bunky.
Mechanizmus rezistencie
VšetkyGram-negatívnebaktériesúinherentnerezistentnévočidalbavancínu.
Rezistencia na dalbavancín u Staphylococcus spp. a Enterococcus spp. je spôsobená VanA,
genotypom, ktorý spôsobuje modifikáciu cieľového peptidu vo vznikajúcej bunkovej stene. Na základe štúdií in vitro aktivita dalbavancínu nie je ovplyvnená inými triedami génov rezistentných na vankomycín.
Hodnoty MIC (Minimum inhibitory concentration) pre dalbavancín sú vyššie pre stafylokoky intermediálne rezistentné na vankomycín (vancomycin-intermediate staphylococci, VISA) než pre kmene úplne citlivé na vankomycín. Ak izoláty s vyšším MIC dalbavancínu reprezentujú stabilné fenotypy a sú v korelácii s rezistenciou na ostatné glykopeptidy, potom by pravdepodobný mechanizmus spočíval vo zvýšení počtu glykopeptidových cieľov vo vznikajúcom peptidoglykáne.
Skrížená rezistencia medzi dalbavancínom a inými triedami antibiotík nebola in vitro pozorovaná. Meticilínová rezistencia nemá vplyv na aktivitu dalbavancínu.
Interakcie s inýmiantibakteriálnymilátkami
V in vitro štúdiách nebolo pozorované žiadne protikladné pôsobenie dalbavancínu a iných bežne
používaných antibiotík (napr. cefepím, ceftazidím, ceftriaxón, imipenem, meropenem, amikacín, aztreonám, ciprofloxacín, piperacilín/tazobaktám a trimetoprim/sulfametoxazol), keď bol testovaný voči 12 druhom Gram-negatívnych patogénov (pozri časť 4.5).
Koncové body testovania citlivosti
Koncové body Minimálnej inhibičnej koncentrácie (MIC) určené Európskou komisiou pre testovanie
antimikrobiálnej citlivosti (European Committee on Antimicrobial Susceptibility Testing, (EUCAST))
sú:
• Stafylococcus spp.: Citlivé ≤ 0,125 mg/l; Rezistentné > 0,125 mg/l,
• Beta-hemolytické streptokoky skupín A, B, C, G: Citlivé ≤ 0,125 mg/l; Rezistentné > 0,125 mg/l,
• Viridujúce streptokoky (iba zo skupiny Streptococcus anginosus): Citlivé ≤ 0,125 mg/l; Rezistentné > 0,125 mg/l.
PK/PD vzťah
Baktericídna aktivita voči stafylokokom in vitro závisí od času pri sérových koncentráciách
dalbavancínu podobným tým, ktoré boli získané pri odporúčanej dávke u ľudí. In vivo PK/PD vzťah
dalbavancínu ku S. aureus bol skúmaný použitím neutropenického modelu animálnej infekcie, ktorá preukázala, že čistý úbytok log10 kolóniotvorných jednotiek (colony-forming units, (CFU)) bol najväčší, keď sa väčšie dávky podávali menej často.
Klinická účinnosť voči špecifickýmpatogénom
Účinnosť bola dokázaná v klinických štúdiách voči patogénom uvádzaným pre ABSSSI, ktoré boli
citlivé na dalbavancín in vitro:
• Staphylococcus aureus,
• Streptococcus pyogenes,
• Streptococcus agalactiae,
• Streptococcus dysgalactiae,
• Streptococcus anginosus skupina (vrátane S. anginosus, S. intermedius a S. constellatus).
Antibakteriálna aktivita voči ostatnýmrelevantnýmpatogénom
Klinická účinnosť nebola stanovená voči nasledovným patogénom, aj keď in vitro štúdie naznačujú, že
by mohli byť citlivé na dalbavancín v prípade absencie získaného mechanizmu rezistencie:
• Streptokoky skupiny G
• Clostridium perfringens,
• Peptostreptococcus spp.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Xydalba v
jednej alebo viacerých podskupinách pediatrickej populácie pri ABSSSI (pozri časti 4.2 a 5.2 týkajúce sa informácií o pediatrickom použití).
5.2 Farmakokinetické vlastnosti
Farmakokinetika dalbavancínu bola popísaná u zdravých ľudí, pacientov a u osobitných skupín populácie. Systémová expozícia dalbavancínu je závislá od dávky po podaní jednotlivých dávok
v rozsahu od 140 do 1 120 mg, indikujúca lineárnu farmakokinetiku dalbavancínu. Nebolo pozorované žiadne akumulovanie dalbavancínu po niekoľkonásobnej intravenóznej infúzii podávanej raz týždenne po dobu až 8 týždňov (1 000 mg 1. deň, po čom nasledovalo až 7 týždenných dávok po 500 mg)
u zdravých dospelých.
Bezprostredne po 30-minútovej intravenóznej infúzii 1 000 mg dalbavancínu na 1. deň bola priemerná hodnota (±SD) plazmatickej Cmax 278 (± 53) μg/ml a AUC (dni 1-7) bola 10 577 μg•h /ml. Na deň 8, následne po 30-minútovej intravenóznej infúzii 500 mg dalbavancínu, bola priemerná hodnota
Cmax 166 (±43) μg/ml a priemerná celková AUC (dni 1-14) bola 20 473 μg•h /ml.
Priemerný polčas konečnej eliminácie (t1/2) bol 372 (rozsah 333 až 405) hodín. Farmakokinetika dalbavancínu je najlepšie popísaná pomocou trojdielneho modelu (α a β distribučné fázy, po ktorých nasleduje konečná eliminačná fáza). Distribučný polčas (t1/2β), ktorý predstavuje zásadnú súčasť klinicky relevantnej závislosti koncentrácie od času, sa tak pohyboval v rozsahu 5 až 7 dní a je konzistentný s dávkovaním raz týždenne.
Pri adekvátne senzitívnej analýze a dôkladnom vzorkovaní bola odhadovaná konečná eliminačná fáza určená ako 16 dní.
Obr. 1: Priemerné plazmatické koncentrácie dalbavancínu v závislosti od času u zdravých subjektov (n=10)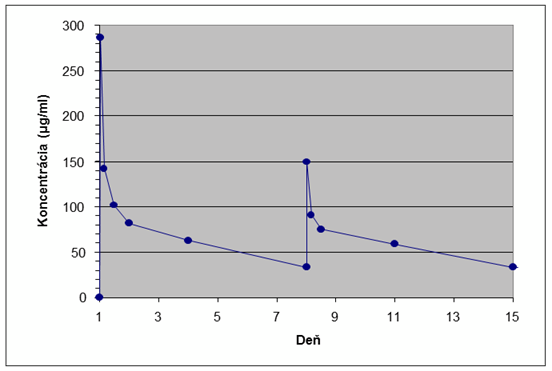 Distribúcia
DistribúciaDoba vylučovania a distribučný objem v rovnovážnom stave sú porovnateľné medzi zdravými'
a pacientami s infekciou. Distribučný objem v rovnovážnom stave bol podobný, ako objem extracelulárnej kvapaliny. Dalbavancín sa reverzibilne viaže na proteíny ľudskej plazmy, primárne na albumín. Proteínová väzba dalbavancínu v plazme je 93% a nemení sa ani v závislosti od koncentrácie lieku ani v prípade renálnej poruchy alebo poruchy pečene. Následne po jednorazovej intravenóznej dávke 1 000 mg u zdravých dobrovoľníkov predstavovala hodnota AUC v tekutine pľuzgiera na koži obsahovala (viazaný aj neviazaný dalbavancín) približne 60% AUC v plazme v deň 7 po podaní
dávky.
BiotransformáciaV ľudskej plazme neboli pozorované metabolity v signifikantných množstvách. Metabolity hydroxy-
dalbavancín a manózyl aglykón boli pozorované v moči (< 25 % podanej dávky). Metabolické cesty zodpovedné za tvorbu metabolitov neboli identifikované; avšak kvôli relatívne malému príspevku metabolizmu ku celkovému vylúčeniu dalbavancínu sa interakcie liek-liek v dôsledku inhibície alebo indukcie metabolizmu dalbavancínu neočakávajú. Hydroxy-dalbavancín a manózyl aglykón vykazujú značne nižšiu antibakteriálnu aktivitu v porovnaní s dalbavancínom.
ElimináciaPo podaní jednej 1 000 mg dávky zdravým jedincom, v priemere 19 % až 33 % podanej dávky
dalbavancínu bolo vylúčených močom vo forme dalbavancínu a 8 % až 12 % vo forme metabolitu hydroxy-dalbavancínu. Približne 20 % podanej dávky sa vylúčilo stolicou.
Osobitné skupiny pacientovPorucha funkcie obličiekFarmakokinetika dalbavancínu sa hodnotila u 28 pacientov s rôznym stupňom poškodenia funkcie obličiek a u 15 porovnávacích kontrolných subjektov s normálnou funkciou obličiek. Následne po
podaní jednorazovej dávky 500 mg alebo 1 000 mg dalbavancínu sa priemerný plazmatický klírens
(CLT) znížil o 11 %, 35 %, a 47 % u pacientov s miernym (CLCR 50 - 79 ml/min), stredným
(CLCR 30 – 49 ml/min) a závažným (CLCR < 30 ml/min) poškodením funkcie obličiek v tomto poradí
v porovnaní so subjektami s normálnou funkciou obličiek. Priemerná AUC u pacientov s klírensom
kreatinínu < 30 ml/min bola približne 2 – násobne vyššia. Klinický význam zníženia priemernej plazmatickej CLT a s tým spojené zvýšenie AUC0-∞ pozorované v týchto farmakokinetických štúdiách dalbavancínu nebolo u pacientov so závažným poškodením funkcie obličiek potvrdené. Farmakokinetika dalbavancínu u pacientov v konečnom štádiu renálneho ochorenia, ktorí dostávali pravidelne prebiehajúcu renálnu dialýzu (3-krát/týždeň) bola podobná tej, aká bola pozorovaná
u pacientov s miernym až stredným poškodením funkcie obličiek a menej ako 6% podanej dávky sa vylúči po 3 hodinách hemodialýzy. Pokyny týkajúce sa dávkovania u pacientov s poškodenou funkciou obličiek sa nachádzajú v časti 4.2.
Porucha funkcie pečene
Farmakokinetika dalbavancínu sa hodnotila u 17 pacientov s mierne, stredne alebo závažne poškodenou funkciou pečene a porovnávala sa s 9 priradenými zdravými subjektami s normálnou funkciou pečene. Priemerné AUC sa nemenilo u pacientov s miernym poškodením funkcie pečene v porovnaní so subjektami s normálnou funkciou pečene, priemerná AUC sa znížil o 28 % a 31 %,
v tomto poradí, u pacientov so stredným a závažným poškodením funkcie pečene. Príčina a klinický význam zníženého účinku u subjektov so stredným a závažným poškodením funkcie pečene nie sú známe. Pokyny týkajúce sa dávkovania u pacientov s poškodením funkcie pečene sa nachádzajú
v časti 4.2.
Pohlavie
Neboli pozorované žiadne klinicky významné rozdiely vo farmakokinetike dalbavancínu týkajúce sa pohlavia, a to ani u zdravých subjektov, ani u pacientov s infekciami. Nie sú odporúčané žiadne úpravy dávkovania na základe pohlavia.
Starší ľudia
Farmakokinetika dalbavancínu sa s vekom výrazne nemenila; preto nie je potrebné upravovať dávkovanie v závislosti od veku (pozri časť 4.2.). Skúsenosti s dalbavancínom u starších ľudí sú limitované: 220 pacientov vo veku > 75 rokov bolo zaradených do fázy 2/3 klinických štúdií, z ktorých
127 dostalo dalbavancín. Do klinických štúdií boli zaradení pacienti až do veku 93 rokov.
Pediatrická populácia
Bezpečnosť a účinnosť lieku Xydalba u detí vo veku od narodenia až po < 18 rokov nebola stanovená. Celkovo 10 pediatrických pacientov vo veku od 12 do 16 rokov, ktorí mali zápalové infekcie, dostalo
jednorazovú dávku dalbavancínu buď 1 000 mg (telesná hmotnosť ≥ 60 kg) alebo 15 mg/kg (telesná hmotnosť < 60 kg).
Priemerné plazmatické expozície dalbavancínu, založené na AUCinf (17 495 µg•h/ml a
16 248 µg •h/ml) a Cmax (212 µg/ml a 191 µg/ml) boli podobné, keď bola podaná dávka 1 000 mg pediatrickým pacientom (12-16 ročným) s hmotnosťou > 60 kg ( 61,9 – 105,2 kg) alebo ako 15 mg/kg pediatrickým pacientom s hmotnosťou < 60 kg (47,9-58,9 kg). Zjavná konečná eliminácia t½ bola podobná pre dávky dalbavancínu 1 000 mg a 15 mg/kg, so strednými hodnotami 227 a 202 hodín,
v tomto poradí. Bezpečnostný profil dalbavancínu u pacientov vo veku medzi 12 a 16 rokom v tejto štúdii bol konzistentný s bezpečnostným profilom pozorovaným u dospelých, ktorým bol podaný dalbavancín.
5
.
3 Predklinické bezpečnostné údaje
Toxicita dalbavancínu bola hodnotená po jeho každodennom intravenóznom podávaní po dobu
3 mesiacov potkanom a psom. Toxicita v závislosti od dávky bola stanovená na základe chemických rozborov séra a histologických nálezov poškodenia obličiek a pečene, znížených parametrov červených krviniek a podráždenia miesta vpichu. Iba u psov sa reakcia na podanie infúzie v súvislosti s dávkou prejavila vo forme opuchnutia pokožky a/alebo sčervenania (nie v súvislosti s miestom vpichu injekcie), zblednutia sliznice, slinenia, vracania, sedácie a mierneho poklesu krvného tlaku a zvýšenia srdcového tepu. Tieto reakcie na podanie infúzie boli prechodné (odzneli do 1 hodiny po podaní dávky) a boli pripisované vylučovaniu histamínu. Profil toxicity dalbavancínu u mladých potkanov sa zhodoval s tým, ktorý bol predtým pozorovaný u dospelých potkanov pri rovnakej veľkosti dávky (mg/kg /deň).
Štúdie reprodukčnej toxicity u potkanov a králikov nepreukázali žiadne prejavy teratogenného účinku. U potkanov pri vystavení účinkom približne 3-násobkom klinických dávok bola pozorovaná znížená fertilita a zvýšený výskyt embryonálnej letality, zníženie hmotnosti plodu a kostrového skostnatenia
a zvýšenej neonatálnej úmrtnosti. U králikov dochádzalo k potratom v súvislosti s materskou toxicitou pri vystavení dávkam nižším ako je terapeutický rozsah u ľudí.
Štúdie dlhodobej karcinogenity neboli vykonané. Dalbavancín nebol mutagénny ani klastogénny v sérii in vitro a in vivo testoch genotoxicity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol (E421) Monohydrát laktózy
Kyselina chlorovodíková (na úpravu pH) Hydroxid sodný (na úpravu pH)
6.2 Inkompatibilita
Roztoky chloridu sodného môžu spôsobiť vyzrážanie a nesmú sa používať na rozpúšťanie ani riedenie
(pozri časť 6.6).
Toto liečivo sa nesmie zmiešavať so žiadnymi inými liečivami alebo intravenóznymi roztokmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Doba použiteľnosti
Suchý prášok: 3 roky
Chemická a fyzikálna stabilita pre použiteľnosť lieku Xydalba bola preukázaná pre rozpustený koncentrát, ako aj pre nariedený roztok po dobu 48 hodín pri teplote 25 °C alebo nižšej. Celková stabilita pre použiteľnosť od okamihu rozpustenia po podanie nemá presiahnuť 48 hodín.
Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa nepoužije okamžite, doba skladovania a podmienky pred použitím sú na osobnej zodpovednosti používateľa a za normálnych okolností by nemali presiahnuť 24 hodín pri teplote 2 až 8 °C, pokiaľ sa rozpúšťanie nevykonalo za kontrolovaných a potvrdených aseptických podmienok. Nezmrazujte.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne špeciálne podmienky na uchovávanie.
Podmienky uchovávania po rozpustení a nariedení tohto lieku sa nachádzajú v časti 6.3.
6.5 Druh obalu a obsah balenia
Jednorazová sklenená liekovka typu I s objemom 48 ml s elastomérickou zátkou a zeleným odklápacím uzáverom.
Každé balenie obsahuje 1 liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie
Liek Xydalba musí byť pre podanie vo forme infúzie rozpustený v sterilnej vode pre injekcie a následne nariedený infúznym roztokom glukózy o koncentrácii 50 mg/ml (5 %).
Liekovky lieku Xydalba sú len na jednorazové použitie. Návodnarozpúšťanieariedenie
Pri rozpúšťaní a narieďovaní lieku Xydalba sa musia používať aseptické techniky.
1. Obsah každej liekovky musí byť rozpustený pomalým pridávaním 25 ml vody pre injekcie.
2. Nepretrepávať. Aby ste zabránili tvorbe peny, striedajte jemný krútivý pohyb a obracanie liekovky, kým sa jej obsah úplne nerozpustí. Rozpúšťanie môže trvať až 5 minút.
3. Rozpustený koncentrát obsahuje 20 mg/ml dalbavancínu.
4. Rozpustený koncentrát musí byť číry, bezfarebný až žltý roztok bez akýchkoľvek viditeľných častíc.
5. Rozpustený koncentrát sa musí ďalej nariediť roztokom glukózy o koncentrácii 50 mg/ml
(5 %) na použitie vo forme infúzie.
6. Príslušný objem 20 mg/ml koncentrátu sa musí preniesť do intravenózneho vrecúška alebo fľaše obsahujúcej infúzny roztok glukózy 50 mg/ml (5 %). Napríklad: 25 ml koncentrátu obsahuje 500 mg dalbavancínu.
7. Po nariedení roztoku na infúziu musí mať finálnu koncentráciu 1 až 5 mg/ml dalbavancínu.
8. Infúzny roztok musí byť číry, bezfarebný až žltý roztok bez akýchkoľvek viditeľných častíc.
9. Ak sa spozoruje výskyt čiastočiek alebo zmena sfarbenia, roztok musí byť zlikvidovaný.
Xydalba sa nesmie zmiešavať so žiadnymi inými liečivami alebo intravenóznymi roztokmi. Roztoky chloridu sodného môžu spôsobiť vyzrážanie a NESMÚ sa používať na rozpúšťanie ani riedenie. Kompatibilita rozpusteného koncentrátu lieku Xydalba bola stanovená len s infúznym roztokom glukózy o koncentrácii 50 mg/ml (5 %).
Likvidácia
Akékoľvek množstvo nespotrebovaného rozpusteného liečiva musí byť zlikvidované.
Nepoužitý liek alebo odpad vzniknutý z lieku majú byť zlikvidované podľa platných legislatívnych požiadaviek danej krajiny.
7
. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Durata Therapeutics International B.V. Spaces Zuidas II,
Barbara Strozzilaan 101,
1083HN Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/14/986/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. február 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.