
hnológiou v bunkách geneticky upravenej ľudskej embryonálnej obličky (HEK) 293F. Počas výrobného procesu a ani do finálneho lieku sa nepridávajú žiadne materiály zo zvierat a ľudí.
Pomocná látka so známym účinkom
7,35 mg sodíka na ml rekonštituovaného roztoku (18,4 mg sodíka na injekčnú liekovku).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok a rozpúšťadlo na injekčný roztok. Prášok: biely až takmer biely sypký prášok. Rozpúšťadlo: číra, bezfarebná kvapalina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba a prevencia krvácania u pacientov s hemofíliou typu A (vrodený nedostatok faktora VIII). Vihuma sa môže používať u všetkých vekových skupín.
4.2 Dávkovanie a spôsob podávania
Liečba musí byť pod dohľadom lekára, ktorý má skúsenosti s liečbou hemofílie. Monitorovanie liečby
V priebehu liečby sa odporúča vhodným spôsobom zisťovať hladiny faktora VIII kvôli stanoveniu
dávky, ktorá sa má podávať, a frekvencii opakovaných infúzií. Jednotliví pacienti sa môžu líšiť na základe ich reakcie na faktor VIII, preukázaných rôznych polčasov rozpadu a zotavení. Dávka podľa
telesnej hmotnosti môže vyžadovať úpravu u podvýživených alebo obéznych pacientov. Predovšetkým
v prípade veľkých chirurgických zákrokov je nevyhnutné presné monitorovanie substitučnej terapie pomocou koagulačnej analýzy (plazmatickej aktivity faktora VIII).
Pri používaní jednostupňovej analýzy zrážania na základe tromboplastínového času (aPTT,
z anglického výrazu activated parcial tromboplastine time) in vitro na stanovenie aktivity faktora VIII
vo vzorkách krvi pacientov môžu byť výsledné aktivity faktora VIII v plazme výrazne ovplyvnené typom reagencie v aPTT a referenčným štandardom použitým pri analýze. Taktiež sa môžu vyskytnúť výrazné odlišnosti medzi výsledkom analýzy pomocou jednostupňovej analýzy zrážania na základe
aPTT a pomocou chromogénneho spôsobu stanovenia podľa Ph. Eur. Je to dôležité obzvlášť pri výmene laboratória a/alebo reagencií používaných v analýze.
DávkovanieDávka a trvanie substitučnej terapie závisí od závažnosti nedostatku faktora VIII, od miesta a rozsahu krvácania a od klinického stavu pacienta.
Počet podávaných jednotiek faktora VIII sa vyjadruje v medzinárodných jednotkách (IU), čo súvisí s aktuálnym štandardom pre koncentráty podľa WHO pre lieky s faktorom VIII. Aktivita faktora VIII v plazme sa vyjadruje buď ako percento (v pomere k normálnej ľudskej plazme) alebo pokiaľ možno v medzinárodných jednotkách (v pomere k medzinárodnej norme pre faktor VIII v plazme).
Jedna medzinárodná jednotka (IU) aktivity faktora VIII zodpovedá množstvu faktora VIII v jednom ml normálnej ľudskej plazmy.
Ošetrenie na základe potrebyVýpočet potrebnej dávky faktora VIII vychádza z empirického zistenia, že 1 medzinárodná jednotka
(IU) faktora VIII na kg telesnej hmotnosti zvyšuje plazmatickú aktivitu faktora VIII o približne 2 %
normálnej aktivity alebo 2 IU/dl. Potrebná dávka sa stanoví pomocou nasledovného vzorca:
Potrebné jednotky = telesná hmotnosť (kg) x potrebné zvýšenie faktora VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl)
Očakávané zvýšenie faktora VIII (% normálu) =
2 x podávané IU telesná hmotnosť (kg)
Množstvo, ktoré sa má podať, a frekvencia podávania musí byť vždy orientovaná na klinickú účinnosť
v danom individuálnom prípade.

V prípade nasledujúcich krvácaní aktivita faktora VIII nesmie klesnúť pod uvedenú úroveň plazmatickej aktivity (v % normálu alebo IU/dl) v zodpovedajúcom období. Ako pomôcku dávkovania pri epizódach krvácania a chirurgickej operácii je možné použiť nasledovnú tabuľku.
Stupeň krvácania/
 t
yp chirurgického postupu
t
yp chirurgického postupu
 Kr
v
ácanie
P
ožadovaná úroveň
f
aktora VIII (%) (IU/dl)
F
r
ekvencia dávok (hodiny)/trvanie terapie (dni)
Kr
v
ácanie
P
ožadovaná úroveň
f
aktora VIII (%) (IU/dl)
F
r
ekvencia dávok (hodiny)/trvanie terapie (dni)
Počiatočná hemartróza, svalové krvácanie alebo krvácanie v ústach

Rozsiahlejšia hemartróza, svalové krvácanie alebo hematóm
20 – 40 Opakujte každých 12 až 24 hodín.
Najmenej 1 deň, kým krvácanie, ktoré sa prejavuje bolesťou, neprestane
alebo kým sa nedosiahne zahojenie.
30 – 60 Opakujte infúziu každých 12 až 24 hodín 3 až 4 dni alebo viac, kým bolesť a akútna porucha neustúpia.
Život ohrozujúce krvácania 60 – 100 Opakujte infúziu každých 8 až 24
hodín, kým sa ohrozenie neodstráni.
C
hirurgická operácia
Menšia operácia

vrátane vytrhnutia zuba
30 – 60 Každých 24 hodín, najmenej 1 deň,
kým sa nedosiahne zahojenie.
Stupeň krvácania/
t
yp chirurgického postupu
P
ožadovaná úroveň faktora VIII (%) (IU/dl)
F
r
ekvencia dávok (hodiny)/trvanie terapie (dni)
Väčšia operácia 80 – 100
(pred operáciou a po operácii)

Opakujte infúziu každých 8 – 24 hodín až do primeraného zahojenia rany, potom terapia najmenej ďalších 7 dní na udržanie aktivity faktora VIII na úrovni 30 % až 60 % (IU/dl).
Prevencia
Na dlhodobú prevenciu pred krvácaním u pacientov so silnou hemofíliou typu A sú zvyčajné dávky 20
až 40 IU faktora VIII na kg telesnej hmotnosti v intervaloch 2 až 3 dní. V niektorých prípadoch, najmä
u mladších pacientov, však môžu byť potrebné kratšie intervaly dávkovania alebo vyššie dávky.
Pediatrická populácia
Dávkovanie je rovnaké u dospelých aj u detí, avšak u detí môžu byť potrebné kratšie intervaly dávok alebo vyššie dávky. V súčasnosti dostupné údaje sú popísané v častiach 4.8, 5.1 a 5.2.
Spôsob podávania
Vihuma je na intravenózne použitie.
Odporúča sa podávať maximálne 4 ml za minútu.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Precitlivenosť
Rovnako ako v prípade akéhokoľvek intravenózneho proteínového lieku sú možné reakcie precitlivenosti alergického typu. Vihuma obsahuje stopy iných ľudských proteínov hostiteľských buniek než faktor VIII. Ak sa vyskytnú symptómy precitlivenosti, pacienti musia liek ihneď prestať používať a musia sa obrátiť na svojho lekára. Pacienti musia byť informovaní o skorých prejavoch reakcií precitlivenosti vrátane žihľavky, generalizovanej žihľavky, pocitu zvierania v hrudi, dýchavičnosti, hypotenzie a anafylaxie.
V prípade šoku je nutné vykonať štandardné lekárske ošetrenie šoku.
Inhibítory
Známou komplikáciou liečby jedincov s hemofíliou A je vznik neutralizujúcich protilátok (inhibítorov) faktora VIII. Tieto inhibítory sú zvyčajne imunoglobulíny IgG zamerané proti prokoagulačnej aktivite faktora VIII, ktoré sú kvantifikované v Bethesdových jednotkách (BU, z anglického výrazu Bethesda Units) na ml plazmy použitím modifikovanej skúšky. Riziko vzniku inhibítorov koreluje so
závažnosťou ochorenia, ako aj s expozíciou faktoru VIII, toto riziko býva najvyššie počas
prvých 20 dní expozície. V zriedkavých prípadoch môžu inhibítory vzniknúť po prvých 100 dňoch
expozície.
Boli pozorované prípady opakovaného výskytu inhibítorov (nízky titer) po prechode z jedného lieku s faktorom VIII na iný u predtým liečených pacientov s viac ako 100 dňami expozície, ktorí majú
v anamnéze vznik inhibítorov. Odporúča sa preto, aby všetci pacienti po prechode z jedného lieku na iný boli pozorne sledovaní na vznik inhibítorov.
Klinický význam tvorby inhibítorov bude závisieť od titra inhibítora, pričom menšie riziko nedostatočnej klinickej odpovede hrozí v prípade inhibítorov nízkeho titra, ktoré sú prítomné dočasne alebo zostávajú trvalo nízkeho titra, než v prípade vysokého titra inhibítorov.
Vo všeobecnosti všetci pacienti liečení liekmi s koagulačným faktorom VIII majú byť pomocou náležitých klinických pozorovaní a laboratórnych vyšetrení pozorne sledovaní na vznik inhibítorov. Ak sa očakávané hladiny aktivity faktora VIII v plazme nedosiahnu, alebo ak krvácanie nie je kontrolované vhodnou dávkou, má sa vykonať testovanie prítomnosti inhibítorov faktora VIII. U
pacientov s vysokými hladinami inhibítora, terapia faktorom VIII nemusí byť účinná a treba zvážiť iné možnosti liečby. Liečba takých pacientov má byť riadená lekármi, ktorí majú skúsenosti s liečbou hemofílie a s inhibítormi faktora VIII.
Kardiovaskulárne udalosti
U pacientov s existujúcimi kardiovaskulárnymi rizikovými faktormi môže substitučná liečba s FVIII
zvýšiť kardiovaskulárne riziko.
Komplikácie súvisiace s katétrom
Ak je potrebné zariadenie na centrálny žilový prístup (CVAD), treba vziať do úvahy komplikácie
súvisiace so zariadením CVAD vrátane lokálnych infekcií, bakterémie a trombózy v mieste zavedenia katétra.
Vždy, keď sa Vihuma podáva pacientovi, dôrazne sa odporúča zaznamenať názov a číslo výrobnej
šarže lieku, aby sa zachovalo prepojenie medzi pacientom a výrobnou šaržou lieku.
Pediatrická populácia
Uvedené upozornenia a opatrenia sa týkajú dospelých aj detí.
Informácie o pomocných látkach (obsah sodíka)
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na injekčnú liekovku.
V závislosti od telesnej hmotnosti a štúdie podávania liekov však môže pacient dostávať viac než
jednu liekovku. Tento fakt by sa mal brať do úvahy u pacientov s kontrolovanou diétou príjmu sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
S Vihumaom sa nevykonali reprodukčné štúdie na zvieratách.
Na základe zriedkavého výskytu hemofílie typu A u žien nie sú k dispozícii skúsenosti týkajúce sa
použitia faktora VIII počas tehotenstva a dojčenia. Preto by sa Vihuma mal používať počas gravidity a dojčenia iba vtedy, ak je na toto použitie jasne indikovaný. Nie sú k dispozícii žiadne údaje o fertilite.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Vihuma nemá vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
U prípravkov FVIII bola zriedkavo pozorovaná precitlivenosť alebo alergické reakcie (ktoré môžu zahŕňať angioedém, pálenie a štípanie v mieste podania infúzie, zimnicu, začervenanie, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, vyrážku, nepokoj, tachykardiu, zvieranie v hrudi, mravčenie, urtikáriu, vrátane generalizovanej urtikárie, zvracanie, dýchavičnosť), ktoré v niektorých prípadoch môžu prejsť do závažnej anafylaxie (vrátane šoku).
U pacientov s hemofíliou A, ktorí sú liečení pomocou faktora VIII vrátane Vihumau, môžu vzniknúť neutralizačné protilátky (inhibítory). Ak sa takéto inhibítory vyskytnú, stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované pracovisko zamerané na liečbu hemofílie.
Tabuľkovýzoznamnežiaducichreakcií
Počas klinických štúdií s Vihumaom u predtým liečených pediatrických (2 až 11 rokov, n = 58),
dospievajúcich (12 až 17 rokov, n = 3) a dospelých pacientov (n = 74) so závažnou hemofíliou typu A
bolo hlásených celkovo 8 nežiaducich reakcií na liek (ADR) (6 u dospelých, 2 u detí) u 5 pacientov (3
dospelí, 2 deti).
Tabuľka 1 uvedená nižšie je podľa triedy orgánových systémov podľa databázy MedDRA (SOC, z anglického výrazu system organ classification a preferovaných termínov).
Frekvencie boli posudzované podľa nasledovných konvencií: veľmi časté (≥ 1/10), časté (≥ 1/100 až <
1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až <1/1 000), veľmi zriedkavé (<
1/10 000), neznáme (z dostupných údajov).
V každej skupine frekvencií sú uvedené nežiaduce reakcie podľa klesajúcej závažnosti.
Tabuľka 1. Frekvencia výskytu nežiaducich reakcií na liek (ADR, z anglického výrazu adverse drug reactions) v klinických štúdiách
T
rieda orgánových systémov
podľa databázy MedDRA
N
ežiaduce reakcie Frekvencia
Poruchy krvi a lymfatického systému
Hemoragická anémia
Inhibícia faktora VIII
Menej časté*
Menej časté (PTP) #
Veľmi časté (PUP) #
Poruchy imunitného systému Precitlivenosť Časté*
Poruchy nervového systému Parestézia
Bolesť hlavy
Menej časté*
Poruchy ucha a labyrintu Závrat Menej časté*
Poruchy gastrointestinálneho traktu
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Sucho v ústach Menej časté*
Bolesť chrbta Menej časté*
Celkové poruchy a reakcie v mieste podania
Pyrexia
Zápal miesta vpichu
Bolesť v mieste vpichu
Časté*
Menej časté*
Laboratórne a funkčné vyšetrenia Pozitívny nález neneutralizačnej protilátky (u PTP)
Menej časté*
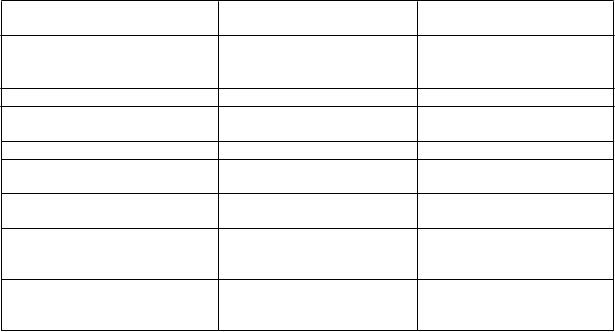
*Vypočítané ako pacienti s ADR na celkový počet 225 pacientov, ktorí sa zúčastnili skúšania, z ktorých
135 pacientov bolo predtým liečených (PTP) a 90 predtým neliečených pacientov (PUP).
# Frekvencia vychádza zo štúdií so všetkými liekmi FVIII, ktoré zahŕňali pacientov so závažnou hemofíliou A. PTP = predtým liečení pacienti (z anglického výrazu previously-treated patients), PUP = predtým neliečení pacienti (z anglického výrazu previously-untreated patients)
Popis vybraných nežiaducich reakciíU jedného dospelého pacienta bola zistená neneutralizačná protilátka faktora VIII (pozri Tabuľku 1). Vzorku testovalo centrálne laboratórium v ôsmich riedeniach. Výsledok bol pozitívny iba v
zrieďovacom pomere 1 a titer protilátky bol veľmi nízky. Inhibičná aktivita, meraná pomocou upraveného Bethesdovho testu, nebola u tohto pacienta zistená. Klinická účinnosť Vihumau a jeho
obnova
in vivo nebola u tohto pacienta ovplyvnená.
Pediatrická populáciaPredpokladá sa, že frekvencia, typ a závažnosť nežiaducich reakcií u detí je rovnaká ako u dospelých.
H
l
ásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeboli zaznamenané žiadne prípady predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antihemoragiká: krvný koagulačný faktor VIII, ATC kód: B02BD02.
Komplex faktora VIII/von Willebrandovho faktora tvoria dve molekuly (faktor VIII a von Willebrandov faktor) s odlišnými fyziologickými funkciami. Po infúzii pacientovi s hemofíliou sa faktor VIII naviaže na von Willebrandov faktor v krvnom obehu pacienta. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX a urýchľuje premenu faktora X na aktivovaný faktor X. Aktivovaný faktor X premieňa protrombín na trombín. Trombín potom premieňa fibrinogén na fibrín a tým sa môže vytvoriť krvná zrazenina. Hemofília typu A je pohlavne viazaná dedičná porucha zrážanlivosti krvi z dôvodu zníženej úrovne faktora VIII:C a spôsobuje profúzne krvácanie do kĺbov, svalov alebo vnútorných orgánov, buď spontánne alebo v dôsledku úrazovej alebo chirurgickej
traumy. Plazmatické úrovne faktora VIII sa zvyšujú substitučnou liečbou, čo dočasne umožňuje
korekciu nedostatku faktora VIII a korekciu tendencií krvácania.
Imunogenicita Vihumau bola posudzovaná v klinických štúdiách u 135 predtým liečených pacientov so závažnou hemofíliou typu A (74 dospelých a 61 pediatrických pacientov). U žiadneho z pacientov sa nevyvinuli inhibítory.
V klinickej štúdii u 32 dospelých pacientov so závažnou hemofíliou typu A bola priemerná spotreba
Vihumau na prevenciu 468,7 IU/kg/mesiac. Priemerná dávka na liečbu epizódy krvácania bola
33,0 IU/kg v prípade nečakaných krvácaní u pacientov, ktorí sa liečili preventívne. V ďalšej klinickej
štúdii sa 22 dospelých pacientov liečilo podľa potreby. Celkovo sa liečilo 986 epizód krvácania s
priemernou dávkou 30,9 IU/kg. Vo všeobecnosti si menšie krvácania vyžadovali o niečo nižšie a
silnejšie krvácania až trojnásobne vyššie priemerné dávky.
Pediatrická populáciaÚdaje sa získavali od 29 predtým liečených detí vo veku 2 až 5 rokov, 31 detí vo veku 6 až 12 rokov a jedného dospievajúceho vo veku 14 rokov. Medián dávky na preventívnu infúziu bol 37,8 IU/kg. Dvadsať pacientov používalo priemerné dávky viac ako 45 IU/kg. Medián spotreby Vihumau na prevenciu na mesiac bol 521,9 IU/kg. Na liečbu krvácania u detí bola potrebná vyššia priemerná dávka Vihumau (43,9 IU/kg) než u dospelých (33,0 IU/kg) a na liečbu stredných až silnejších krvácaní bola tiež potrebná vyššia priemerná dávka než na liečbu slabších krvácaní (78,2 IU/kg oproti 41,7 IU/kg).
U mladších detí boli vo všeobecnosti potrebné vyššie priemerné dávky (6 – 12 rokov: 43,9 IU/kg; 2 –
5 rokov: 52,6 IU/kg).
Aktuálne prebieha prospektívna otvorená klinická štúdia u PUP so závažnou hemofíliou A (<1 % FVIII:C).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Vihumaom v
jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe hemofílie typu A (vrodeného
nedostatku faktora VIII) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
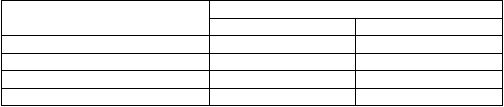 T
abuľka 2. Farmakokinetické parametre pre Vihuma (dávka: 50 IU/kg) u dospelých, predtým liečených pacientov (vek 18 – 65 rokov) so závažnou hemofíliou typu A (n = 20) Farmakokinetický parameter Chromogénny spôsob stanovenia
P
r
i
e
m
er ± SD Medián (rozsah)
T
abuľka 2. Farmakokinetické parametre pre Vihuma (dávka: 50 IU/kg) u dospelých, predtým liečených pacientov (vek 18 – 65 rokov) so závažnou hemofíliou typu A (n = 20) Farmakokinetický parameter Chromogénny spôsob stanovenia
P
r
i
e
m
er ± SD Medián (rozsah) AUC (hod*IU/ml) 22,6 ± 8,0 22,3 (8,4 – 38,1) T1/2 (hod) 14,7 ± 10,4 12,5 (5,4 – 55,6) IVR (%/IU/kg) 2,5 ± 0,4 2,5 (1,7 – 3,2)
CL (ml/hod/kg) 3,0 ± 1,2 2,7 (1,5-6,4) AUC = plocha pod krivkou (FVIII:C), T1/2 = terminálny polčas rozpadu, IVR = prírastková obnova
in vivo, CL = klírens, SD = Štandardná odchýlka
Tabuľka 3. Farmakokinetické parametre pre Vihuma (dávka: 50 IU/kg) u predtým liečenýchdetí vo veku 6 až 12 rokov so závažnou hemofíliou typu A (n = 12)Farmakokinetický parameter Chromogénny spôsob stanoveniaPriemer ± SD Medián (rozsah) AUC (hod*IU/ml) 13,2 ± 3,4 12,8 (7,8 – 19,1) T1/2 (hod) 10,0 ± 1,9 9,9 (7,6 – 14,1) IVR (%/IU/kg) 1,9 ± 0,4 1,9 (1,2 – 2,6)
CL (ml/hod/kg) 4,3 ± 1,2 4,2 (2,8 - 6,9) AUC = plocha pod krivkou (FVIII:C), T1/2 = terminálny polčas rozpadu, IVR = prírastková obnova
in vivo, CL = klírens, SD = Štandardná odchýlka
Tabuľka 4. Farmakokinetické parametre pre Vihuma (dávka: 50 IU/kg) u predtým liečených detí vo veku 2 až 5 rokov so závažnou hemofíliou typu A (n = 13)Farmakokinetický parameter Chromogénny spôsob stanoveniaPriemer ± SD Medián (rozsah)
detí vo veku 2 až 5 rokov so závažnou hemofíliou typu A (n = 13)Farmakokinetický parameter Chromogénny spôsob stanoveniaPriemer ± SD Medián (rozsah) AUC (hod*IU/ml) 11,7 ± 5,3 10,5 (4,9 – 23,8) T1/2 (hod) 9,5 ± 3,3 8,2 (4,3 – 17,3) IVR (%/IU/kg) 1,9 ± 0,3 1,8 (1,5 – 2,4)
CL (ml/hod/kg) 5,4 ± 2,4 5,1 ( 2,3 – 10,9) AUC = plocha pod krivkou (FVIII:C), T1/2 = terminálny polčas rozpadu,
IVR = prírastková obnova
in vivo, CL = klírens, SD = Štandardná odchýlka
Pediatrická populáciaAko je známe z literatúry, obnova a polčas rozpadu bol kratší u mladých detí než u dospelých a klírens vyšší, čo môže byť spôsobené čiastočne známym vyšším objemom plazmy na kilogram telesnej hmotnosti u mladších pacientov.
Podskupiny s upravenou hmotnosťouTabuľka 5. Farmakokinetické parametre pre Vihuma s upravenou hmotnosťou (dávka: 50IU/kg) u dospelých, predtým liečených pacientov (vek 18 – 65 rokov) so závažnou hemofíliou typu A (n = 20)
F
armakokinetický parameter
V
šetci
(
n=20)
N
ormálna hmotnosť (n=14)
Mierne adipózni
(
n=4)
A
dipózni
(
n=2)
'
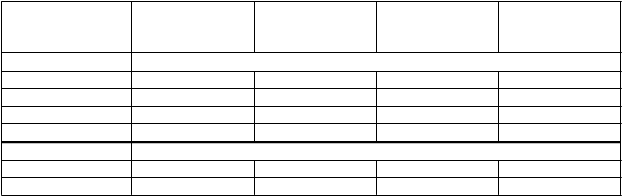 C
hromogénny spôsob stanovenia, Priemer ± SD
C
hromogénny spôsob stanovenia, Priemer ± SD
AUC (hod*IU/ml) 22,6 ± 8,0 20,4 ± 6,9 24,9 ± 8,9 33,5 ± 6,5
T1/2 (hod) 14,7 ± 10,4 14,7 ± 12,1 13,4 ± 5,9 17,2 ± 4,8
IVR (%/IU/kg) 2,5 ± 0,4 2,4 ± 0,4 2,7 ± 0,4 2,8 ± 0,3
CL (ml/hod/kg) 3,0 ± 1,2 3,2 ± 1,3 2,6 ± 1,0 1,8 ± 0,4
Chromogénny spôsob stanovenia, medián (rozsah)AUC (hod*IU/ml) 22,3 (8,4 – 38,1) 21,2 (8,4 – 32,6) 23,3 (17,4 – 35,5) 33,5 (28,9 – 38,1) T1/2 (hod) 12,5 (5,4 – 55,6) 12,3 (5,4 – 55,6) 11,2 (9,3 – 22,0) 17,2 (13,8 – 20,6)

IVR (%/IU/kg) 2,5 (1,7 – 3,2) 2,4 (1,7 – 3,1) 2,8 (2,3 – 3,2) 2,8 (2,6 – 3,0) CL (ml/hod/kg) 2,7 (1,5 – 6,4) 2,8 (1,7 – 6,4) 2,5 (1,6 – 3,7) 1,8 (1,5 – 2,0) Normálna hmotnosť: BMI 18,5 – 25 kg/m2, mierne adipózni: BMI 25 – 30 kg/m2, adipózni: BMI > 30 kg/m2, SD = Štandardná odchýlka
5.3 Predklinické údaje o bezpečnostiV predklinických štúdiách sa Vihuma používal na bezpečnú a účinnú obnovu hemostázy u psov s hemofíliou. Toxikologické štúdie preukázali, že lokálne intravenózne podávanie a systémová expozícia boli u laboratórnych zvierat (potkany a makaky dlhochvosté) dobre tolerované.
Osobitné štúdie s dlhodobým opakovaným podávaním ako štúdie reprodukčnej toxicity, chronickej toxicity a karcinogenity sa s Vihumaom nevykonali z dôvodu imunitnej reakcie na heterológne proteíny u všetkých neľudských cicavčích druhov.
Nevykonali sa žiadne štúdie mutagénneho potenciálu Vihumau.
Hodnotenia
ex vivo pomocou komerčnej testovacej súpravy na stanovenie reakcie T buniek na
proteínovú terapiu naznačujú nízke riziko imunogenicity.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokPrášok Sacharóza Chlorid sodný
Dihydrát chloridu vápenatého
Arginíniumchlorid
Dihydrát citronanu sodného
Poloxamér 188
RozpúšťadloVoda na injekciu
6.2 InkompatibilityNevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
Môžu sa používať iba injekčné súpravy, ktoré sú súčasťou balenia, pretože adsorpcia ľudského koagulačného faktora VIII na vnútorné povrchy niektorých injekčných zariadení môže spôsobiť neúspešnosť liečby.
6.3 Čas použiteľnostiNeotvorenáinjekčnáliekovka2 roky
Počas času použiteľnosti môže byť liek uchovávaný nepretržite pri izbovej teplote (do 25°C) počas obdobia nie dlhšie ako 1 mesiac. Ak bol liek už raz vybratý z chladničky, nesmie sa už do chladničky vrátiť. Zaznačte si, prosím, na krabičke dátum, kedy ste začali liek uchovávať pri izbovej teplote. Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Po rekonštitúciiPo rekonštitúcii bola preukázaná chemická a fyzikálna stabilita pre použitie počas 24 hodín pri skladovaní pri izbovej teplote.
Z mikrobiologického hľadiska sa liek musí použiť ihneď po rekonštitúcii. Ak sa nepoužije ihneď, za čas skladovania po otvorení a za podmienky pred použitím je zodpovedný používateľ. Rekonštituovaný roztok skladujte pri izbovej teplote. Neuchovávajte v chladničke po rekonštitúcii.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Uchovávanie pri izbovej teplote a podmienky uchovávania lieku po rekonštitúcii, pozri časť 6.3.
6.5 Druh obalu a obsah baleniaKaždé balenie obsahuje:
- 1 injekčnú liekovku s práškom s 250, 500, 1000, 2000, 2500, 3000 alebo 4000 IU simoctocog alfa v sklenenej injekčnej liekovke typu 1, uzatvorenej obalenou brómbutylovou zátkou
a utesnenej hliníkovým vyklápacím viečkom
- Rozpúšťadlo: 2,5 ml vody na injekciu v naplnenej injekčnej striekačke z borokremičitého skla
- 1 sterilný adaptér injekčnej liekovky na rekonštitúciu s 1 motýlikovou ihlou a 2 alkoholovými tampónmi
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPrášok rekonštituujte iba s rozpúšťadlom (2,5 ml vody na injekcie), ktoré je súčasťou balenia, pomocou dodávanej injekčnej súpravy. Injekčnú liekovku pozvoľna otáčajte, kým sa všetok prášok nerozpustí. Po rekonštitúcii naberte roztok späť do injekčnej striekačky.
Pred podávaním vizuálne skontrolujte, či v rekonštituovanom roztoku nie sú cudzorodé častice
a zafarbenie. Rekonštituovaný liek je číry, bezfarebný roztok, bez prítomnosti cudzorodých častíc,
ktorý má pH 6,5 až 7,5. Nepoužívajte roztoky, ktoré sú zakalené alebo majú usadeniny.
Pokyny na prípravu a podávanie1. Počkajte, kým injekčná striekačka s rozpúšťadlom (voda na injekciu) a prášok v uzatvorenej
injekčnej liekovke dosiahnu izbovú teplotu. Môžete ich podržať v rukách, kým nebudete cítiť, že sú rovnako teplé ako vaše ruky. Injekčnú liekovku a vopred naplnenú injekčnú striekačku nezohrievajte iným spôsobom. Táto teplota by sa mala zachovať počas rekonštitúcie.
2. Odstránením plastového vyklápacieho viečka z injekčnej liekovky s práškom odkryte stredové časti gumenej zátky. Neodstraňujte sivú zátku ani kovový krúžok okolo vrchu injekčnej
liekovky.
3. Utrite vrch injekčnej liekovky alkoholovým tampónom. Nechajte alkohol uschnúť.
4. Stiahnite papierový kryt z obalu adaptéra injekčnej liekovky. Adaptér z obalu nevyberajte.
5. Položte injekčnú liekovku s práškom na rovný povrch a chyťte ju. Zoberte obal adaptéra a umiestnite adaptér injekčnej liekovky na stred gumenej zátky injekčnej liekovky s práškom. Silno zatlačte obal adaptéra, kým hrot adaptéra neprejde cez gumenú zátku. Adaptér nakoniec zacvakne do injekčnej liekovky.
6. Stiahnite papierový kryt z obalu naplnenej injekčnej striekačky. Držte tyčinku piestu na konci a nedotýkajte sa drieku. Pripojte závitový koniec tyčinky piestu k piestu injekčnej striekačky
s rozpúšťadlom. Otáčajte piest v smere hodinových ručičiek, kým nebudete cítiť slabý odpor.
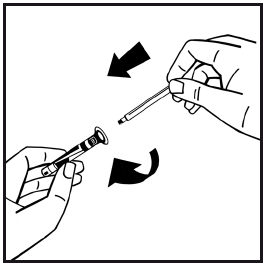
7. Odlomte ochranný plastový hrot od injekčnej striekačky s rozpúšťadlom prelomením perforácie viečka. Nedotýkajte sa vnútra viečka ani hrotu injekčnej striekačky. Ak sa roztok okamžite nepoužije, na uskladnenie uzavrite naplnenú injekčnú striekačku ochranným plastovým hrotom.
8. Odstráňte obal adaptéra a zahoďte ho.
9. Pevne spojte injekčnú striekačku s rozpúšťadlom s adaptérom injekčnej liekovky otočením v smere hodinových ručičiek, kým nebudete cítiť odpor.
10. Pomaly vstreknite všetko rozpúšťadlo do injekčnej liekovky s práškom zatlačením tyčinky
piestu.
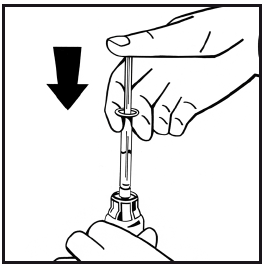
11. Bez odstránenia injekčnej striekačky rozpustite prášok jemným pohybom alebo viacnásobným krúžením s injekčnou liekovkou. Netrepte. Počkajte, kým sa všetok prášok úplne nerozpustí.
12. Pred podávaním vizuálne skontrolujte, či v konečnom roztoku nie sú žiadne častice. Roztok
musí byť číry a bezfarebný, prakticky bez viditeľných častíc. Nepoužívajte roztoky, ktoré sú
zakalené alebo majú usadeniny.
13. Obráťte injekčnú liekovku pripojenú k injekčnej striekačke hore dnom a pomaly vtiahnite konečný roztok do injekčnej striekačky. Do injekčnej striekačky musí prejsť celý obsah injekčnej liekovky.
14. Odpojte naplnenú injekčnú striekačku od adaptéra injekčnej liekovky otočením proti smeru hodinových ručičiek a prázdnu injekčnú liekovku zahoďte.
15. Roztok je teraz pripravený na okamžité použitie. Neuchovávajte v chladničke.
16. Očistite zvolené miesto vpichu jedným z alkoholových tampónov, ktoré sú súčasťou balenia.
17. Pripojte k injekčnej striekačke infúznu súpravu, ktorá je súčasťou balenia.
Vsuňte ihlu infúznej súpravy do zvolenej žily. Ak ste použili škrtidlo, aby bola žila lepšie
vidieť, pred začatím vstrekovania roztoku toto škrtidlo uvoľnite.
Do injekčnej striekačky nesmie prúdiť žiadna krv z dôvodu rizika vzniku fibrínových
zrazenín.
18. Roztok vstrekujte do žily pomalou rýchlosťou, nie rýchlejšie než 4 ml za minútu.
Ak použijete viac ako jednu injekčnú liekovku s práškom na jedno ošetrenie, môžete použiť tú istú injekčnú ihlu znovu. Adaptér injekčnej liekovky a injekčná striekačka sú určené len na jedno použitie.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIOctapharma AB
Lars Forssells gata 23
112 75 Stockholm
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1168/001
EU/1/16/1168/002
EU/1/16/1168/003
EU/1/16/1168/004
EU/1/16/1168/005
EU/1/16/1168/006
EU/1/16/1168/007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 13. februára 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.