aždých 6 hodín alebo 1 000 mg každých 12 hodín. Alebo 30 až 40 mg/kg/deň v 2 až 4 denných dávkach.
Pri bakteriálnej endokarditíde sa zvyčajne toleruje dávkovanie 1 000 mg vankomycínu intravenózne každých 12 hodín počas 4 týždňov, a to buď samostatne alebo v kombinácii s inými antibiotikami (gentamicín s rifampicínom, gentamicín, streptomycín). Enterokoková endokarditída sa lieči počas 6 týždňov vankomycínom v kombinácii s aminoglykozidom. Je potrebné si preštudovať oficiálne pokyny.
Deti vo veku od jedného mesiaca do 12 rokov:
Zvyčajné intravenózne dávkovanie je 10 mg/kg na dávku podávaných každých šesť hodín (celkové denné dávkovanie 40 mg/kg telesnej hmotnosti). Každá dávka sa má podávať minimálne počas 60 minút.
Novorodenci (narodení v termíne):Vo veku 0 – 7 dní: Počiatočná dávka je 15 mg/kg, po ktorej nasleduje 10 mg/kg každých 12 hodín.
Vo veku 7 – 30 dní: Počiatočná dávka je 15 mg/kg, po ktorej nasleduje 10 mg/kg každých 8 hodín.
Každá dávka sa má podávať počas 60 minút. U týchto pacientov môže byť potrebné dôkladné sledovanie sérových koncentrácií vankomycínu.
Gravidita:
Zaznamenalo sa, že na dosiahnutie terapeutických sérových koncentrácií u gravidných pacientok môžu byť potrebné významne vyššie dávky – pozri časť 4.6 Gravidita a laktácia.
Starší pacienti:
V dôsledku zníženej funkcie obličiek môže byť potrebné oveľa väčšie zníženie dávkovania ako sa očakáva (pozri nižšie).
Obézni pacienti:
Môže byť potrebná úprava zvyčajných denných dávok.
Pacienti s insuficienciou pečene:
Neexistuje dôkaz o tom, že sa u pacientov s insuficienciou pečene musí dávka znížiť.
Pacienti s poruchou funkcie obličiek::Dávkovanie sa má upraviť, aby sa zabránilo toxickým sérovým hladinám. U predčasne narodených detí a starších pacientov môže byť z dôvodu zníženej funkcie obličiek nevyhnutné väčšie zníženie dávkovania, ako sa očakáva. U týchto pacientov sa odporúča pravidelné sledovanie sérových hladín, pretože sa zaznamenala kumulácia, predovšetkým po dlhodobej liečbe.
Sérové koncentrácie vankomycínu sa môžu stanoviť pomocou mikrobiologickej analýzy, rádioimunoanalýzy, fluorescenčnej polarizačnej imunoanalýzy, fluorescenčnej imunoanalýzy alebo vysokotlakovej kvapalinovej chromatografie. Nasledujúci nomogram slúži ako návod pre úpravu dávky na základe hodnôt klírensu kreatinínu:
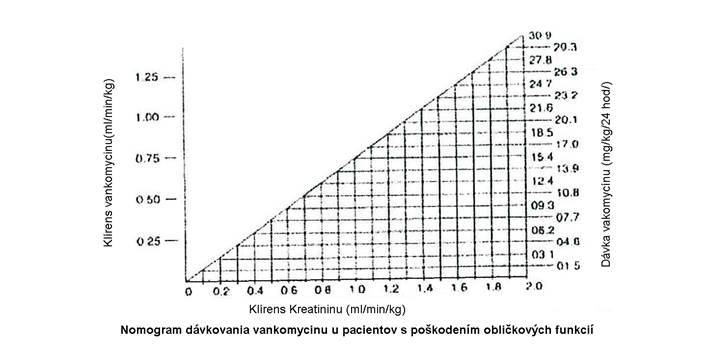
Nomogram neplatí pre funkčne anefrických pacientov na dialýze. Týmto pacientom sa má podať nárazová dávka 15 mg/kg telesnej hmotnosti na rýchle dosiahnutie terapeutických sérových hladín a na udržanie stabilných hladín je potrebná dávka 1,9 mg/kg/24 hodín. Keďže sú vhodné individuálne udržiavacie dávky 250 mg až 1 g, u pacientov s výraznou poruchou funkcie obličiek sa má podávať dávka radšej každých niekoľko dní ako každý deň. Pri anúrii sa odporúča dávka 1 g každých sedem až desať dní.
Ak je dostupná samotná sérová hladina kreatinínu, na výpočet klírensu kreatinínu sa môže použiť nasledujúci vzorec:
Muži:
Hmotnosť (kg) x (140 – vek (roky))72 x sérový kreatinín (mg/100 ml)
Ženy: 0,85 x hodnota vypočítaná podľa vyššie uvedeného vzorca.
Pokyny na prípravu roztokov, pozri časť 6.6.
Sledovanie sérových koncentrácií vankomycínu:Sérová koncentrácia vankomycínu sa má sledovať na druhý deň liečby bezprostredne pred ďalšou dávkou a jednu hodinu po infúzii. Terapeutické hladiny vankomycínu v krvi sa jednu hodinu po skončení infúzie majú pohybovať v rozmedzí 30 a 40 mg/l (maximálne 50 mg/l), maximálna hladina (krátko pred nasledujúcim podaním) sa má pohybovať v rozmedzí 5 a 10 mg/l.
Koncentrácie sa majú zvyčajne sledovať dvakrát alebo trikrát týždenne.
Dĺžka liečbyDĺžka liečby závisí od závažnosti infekcie rovnako ako od klinického a bakteriologického vývoja.
4.3 KontraindikáciePrecitlivenosť na vankomycín.
4.4 Osobitné upozornenia a opatrenia pri používaníRýchle bolusové podanie (napr. počas niekoľkých minút) môže byť spojené s výraznou hypotenziou, vrátane šoku a zriedkavo so zastavením srdca, reakciami podobnými histamínovým a makulopapulóznou alebo erytematóznou vyrážkou („syndróm červeného človeka“ alebo „syndróm červeného krku“). Vankomycín sa má podávať infúziou vo forme zriedeného roztoku počas viac ako 60 minút, aby sa zabránilo reakciám zapríčineným rýchlou infúziou. Zastavenie infúzie má zvyčajne za následok rýchly ústup týchto reakcií (pozri časť 4.2 Dávkovanie a spôsob podávania a časť 4.8 Nežiaduce účinky).
Z dôvodu svojej potenciálnej ototoxicity a nefrotoxicity sa má vankomycín u pacientov s insuficienciou obličiek používať opatrne a dávka sa má znížiť podľa stupňa poruchy funkcie obličiek. Riziko toxicity sa pri vysokých koncentráciách v krvi alebo pri dlhodobej liečbe výrazne zvyšuje. Je potrebné sledovať krvné hladiny a pravidelne vykonávať testy funkcie obličiek.
Podávaniu vankomycínu sa treba vyhnúť aj u pacientov s predchádzajúcou stratou sluchu. Ak sa používa u týchto pacientov, dávka sa má regulovať, ak je to možné, pravidelným stanovovaním hladiny liečiva v krvi. Hluchote môže predchádzať tinitus.
Staršie osoby sú citlivejšie na poškodenie sluchu. Skúsenosti s inými antibiotikami naznačujú, že hluchota môže postupovať napriek ukončeniu liečby.
Použitie u detí a dospievajúcich: U predčasne narodených novorodencov a malých detí môže byť vhodné skontrolovať požadované sérové koncentrácie vankomycínu. Súbežné podávanie vankomycínu a anestetík sa u detí spájalo s erytémom a sčervenaním podobným histamínovému.
Použitie u starších pacientov: Keď sa dávkovanie neupraví, prirodzený pokles glomerulárnej filtrácie so zvyšujúcim sa vekom môže viesť k zvýšeniu sérových koncentrácií vankomycínu (pozri „Dávkovanie a spôsob podávania“).
OpatreniaPri dlhodobom používaní je indikované pravidelné sledovanie hladín vankomycínu v krvi, predovšetkým u pacientov s dysfunkciou obličiek alebo poruchou sluchu rovnako ako pri súbežnom podávaní nefrotoxických alebo ototoxických látok (v uvedenom poradí).
Dávky sa majú titrovať na základe sérových hladín. Krvné hladiny sa majú sledovať a pravidelne sa majú vykonávať testy funkcie obličiek.
Pacienti s hraničnou funkciou obličiek a jedinci starší ako 60 rokov majú podstúpiť sériu vyšetrení sluchovej funkcie a krvných hladín vankomycínu. U všetkých pacientov, ktorým sa liek podáva, sa majú vykonávať pravidelné hematologické vyšetrenia, analýza moču a vyšetrenia funkcie obličiek.
Vankomycín veľmi dráždi tkanivo a vyvoláva nekrózu v mieste injekcie, keď sa podáva intramuskulárne – musí sa podávať intravenóznou infúziou. U veľkého počtu pacientov, ktorým sa podáva vankomycín, sa môže objaviť bolesť v mieste vpichu a tromboflebitída, ktoré sú občas závažné.
Frekvencia a závažnosť tromboflebitídy sa môže minimalizovať pomalým podávaním liečiva vo forme zriedeného roztoku (2,5 až 5,0 g/l) a obmieňaním miest podania infúzie.
Dlhodobé používanie vankomycínu môže mať za následok nadmerný rast necitlivých organizmov. Je nevyhnutné starostlivé sledovanie pacienta. Ak sa počas liečby vyskytne superinfekcia, treba urobiť vhodné opatrenia. V zriedkavých prípadoch sa zaznamenali hlásenia o pseudomembranóznej kolitíde zapríčinenej
C. difficile, ktorá sa vyvinula u pacientov, ktorým sa podával intravenózny vankomycín.
Vankomycín sa musí podávať opatrne pacientom so známou precitlivenosťou na teikoplanín, pretože sa zaznamenali prípady skríženej precitlivenosti.
4.5 Liekové a iné interakcieSúbežné podávanie vankomycínu s anestetikami sa spájalo s erytémom, sčervenaním podobným histamínovému a s anafylaktoidnými reakciami.
Zaznamenalo sa, že frekvencia udalostí súvisiacich s infúziou sa zvyšuje pri súbežnom podávaní s anestetikami. Udalosti súvisiace s infúziou možno minimalizovať podávaním vankomycínu vo forme 60‑minútovej infúzie pred indukciou anestézy.
Súbežné alebo sekvenčné systémové alebo topické používanie iných potenciálne
ototoxických, neurotoxických alebo nefrotoxických liečiv, ako amfotericín B, aminoglykozidy, bacitracín, polymyxín B, kolistín, viomycín alebo cisplatina, si v prípade indikácie vyžaduje starostlivé monitorovanie.
Pri súbežnom podávaní vankomycínu a neuromuskulárnych blokátorov existuje zvýšený potenciál neuromuskulárnej blokády.
4.6 Gravidita a laktáciaGravidita:Nie sú dostupné žiadne dostatočné skúsenosti týkajúce sa bezpečnosti podávania vankomycínu počas gravidity u ľudí. Reprodukčné toxikologické štúdie u zvierat nenaznačujú žiadne účinky na vývoj embrya, plodu alebo obdobie gestácie (pozri časť 5.3).
Vankomycín však prechádza placentou a potenciálne riziko embryonálnej a neonatálnej ototoxicity a nefrotoxicity nemožno vylúčiť. Preto sa má vankomycín počas gravidity podávať len ak je to jednoznačne nevyhnutné a po starostlivom zhodnotení rizika/prínosu.
Laktácia:Vankomycín sa vylučuje do ľudského mlieka, a preto sa má počas laktácie použiť len vtedy, ak iné antibiotiká neúčinkovali. Vankomycín sa má podávať dojčiacim matkám opatrne z dôvodu potenciálnych nežiaducich reakcií u dieťaťa (poruchy črevnej flóry s hnačkou, kolonizácia hubami podobnými kvasinkám a možno senzibilizácia).
Vzhľadom na dôležitosť tohto lieku pre dojčiacu matku sa má zvážiť rozhodnutie o skončení dojčenia.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeVankomycín má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkyV rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Nežiaduce reakcie uvedené nižšie sú definované pomocou nasledujúcich termínov MedDRA:
Veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100), zriedkavé (³ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (častosť nemožno odhadnúť z dostupných údajov).
Intravenózna infúzia:Najčastejšie nežiaduce reakcie v súvislosti s príliš rýchlou intravenóznou infúziou vankomycínu sú flebitída a pseudoalergické reakcie.
Poruchy krvi a lymfatického systému:
Zriedkavé (³ 1/10 000 až < 1/1 000): trombocytopénia, neutropénia, agranulocytózy, eozinofília.
Poruchy imunitného systému:
Zriedkavé (³ 1/10 000 až < 1/1 000): anafylaktické reakcie, reakcie z precitlivenosti.
Poruchy ucha a labyrintu:
Menej časté (³ 1/1 000 až < 1/100): prechodná alebo trvalá strata sluchu.
Zriedkavé (³ 1/10 000 až < 1/1 000): tinitus, závrat.
Poruchy srdca a srdcovej činnosti:
Veľmi zriedkavé (< 1/10 000): zastavenie srdca.
Poruchy ciev:
Časté (³ 1/100 až < 1/10): zníženie krvného tlaku.
Zriedkavé (³ 1/10 000 až < 1/1 000): vaskulitída.
Poruchy dýchacej sústavy, hrudníka a mediastína:
Časté (³ 1/100 až < 1/10): dyspnoe, chrčanie.
Poruchy gastrointestinálneho traktu:
Zriedkavé (³ 1/10 000 až < 1/1 000): nauzea.
Veľmi zriedkavé (< 1/10 000): pseudomembranózna enterokolitída.
Poruchy kože a podkožného tkaniva:
Časté (³ 1/100 až < 1/10): exantém a zápal sliznice, pruritus, urtikária.
Veľmi zriedkavé (< 1/10 000): exfoliatívna dermatitída, Stevensov‑Johnsonov syndróm, Lyellov syndróm, IgA lineárna bulózna dermatóza.
Poruchy obličiek a močových ciest:
Časté (³ 1/100 až < 1/10): insuficiencia obličiek, ktorá sa prejavuje predovšetkým zvýšeným sérovým kreatinínom.
Zriedkavé (³ 1/10 000 až < 1/1 000): intersticiálna nefritída, akútne zlyhávanie obličiek.
Celkové poruchy a reakcie v mieste podania:
Časté (³ 1/100 až < 1/10): flebitída, sčervenanie hornej časti tela a tváre.
Zriedkavé (³ 1/10 000 až < 1/1 000): lieková horúčka, triaška. Bolesť svalov na hrudi a na chrbte.
Udalosti súvisiace s infúziou:
Počas rýchlej infúzie alebo krátko po nej sa môžu vyskytnúť anafylaktoidné reakcie, vrátane hypotenzie, dyspnoe, urtikárie alebo pruritu. Môže sa vyskytnúť sčervenanie kože v hornej časti tela (syndróm červeného človeka), bolesť a kŕče svalov na hrudi alebo na chrbte.
Reakcie sa zmierňujú po skončení podávania, zvyčajne v rozmedzí 20 minút a 2 hodín. Vankomycín sa má podávať pomalou infúziou (dlhšie ako 60 minút – pozri časť 4.4).
Ototoxicita môže byť reverzibilná alebo trvalá a zaznamenala sa predovšetkým u predávkovaných pacientov, u pacientov s anamnézou zhoršeného sluchu a pri súbežnej liečbe ototoxickými liečivami, ako aminoglykozidy.
4.9 PredávkovanieOdporúča sa podporná starostlivosť s udržovaním glomerulárnej filtrácie. Vankomycín sa slabo odstraňuje z krvi hemodialýzou alebo peritoneálnou dialýzou. Zistilo sa, že hemoperfúzia so živicou Amberlite XAD‑4 má obmedzený prínos.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antibakteriálne látky na systémové použitie, glykopeptidové antibiotiká, ATC kód: J01X A01.
Spôsob účinku
Vankomycín je tricyklické glykopeptidové antibiotikum, ktoré inhibuje syntézu bunkovej steny u citlivých baktérií väzbou s vysokou afinitou k D-alanyl-D-alanínovému koncu jednotiek prekurzora bunkovej steny. Liečivo pôsobí baktericídne na deliace sa mikroorganizmy.
Vzťah FK/FD
Aktivita vankomycínu sa považuje za časovo závislú.
Mechanizmus rezistencie:
Získaná rezistencia na glykopeptidy je najčastejšia u enterokokov a je založená na získaní rozličných komplexov
van-génov, ktoré modifikujú D‑alanyl‑D‑alanínový cieľ na D‑alanyl‑D‑laktát alebo D‑alanyl‑D‑serín, ktoré vankomycín viažu slabo. U niektorých
van-génov sa zaznamenala skrížená rezistencia s teikoplanínom.
Van-gény sa zriedkavo zistili u
Staphylococcus aureus, u ktorého zmeny v štruktúre bunkovej steny mali za následok „strednú” citlivosť, ktorá je najčastejšie heterogénna.
Citlivosť:
Vankomycín je aktívny predovšetkým proti grampozitívnym baktériám, ako stafylokoky, streptokoky, enterokoky, pneumokoky a klostrídie a difteroidy. Gramnegatívne baktérie sú rezistentné.
Prevalencia získanej rezistencie sa pre vybrané kmene môže líšiť v závislosti od geografickej polohy a času a sú potrebné lokálne informácie o rezistencii, predovšetkým pri liečbe závažných infekcií. Keď je lokálna prevalencia rezistencie taká, že prospešnosť liečiva je prinajmenšom u niektorých typov infekcií sporná, má sa v prípade potreby vyhľadať pomoc odborníka.
Hraničné hodnotyOdporúčania EUCAST (Európsky výbor pre testovanie antimikrobiálnej citlivosti, European Committee on Antimicrobial Susceptibility testing)
| Citlivé
| Rezistentné
|
Staphylococcus spp.
| ≤ 2 mg/l
| > 2 mg/l
|
Enterococcus spp.
| ≤ 4 mg/l
| > 4 mg/l
|
Streptococcus spp
| ≤ 2 mg/l
| > 2 mg/l
|
Streptococcus pneumoniae
| ≤ 2 mg/l
| > 2 mg/l
|
Grampozitívne anaeróby
| ≤ 2 mg/l
| ≤ 2 mg/l
|
Hraničné hodnoty, ktoré sa nevzťahujú na druh*
| ≤ 2 mg/l
| > 4 mg/l
|
*Hraničné hodnoty, ktoré sa nevzťahujú na druh, sa zistili predovšetkým na základe údajov FK/FD a nezávisia od distribúcií MIC u konkrétnych druhov. Sú určené len na použitie pre druhy, pre ktoré sa nestanovili hraničné hodnoty pre konkrétne druhy a nie pre tie druhy, u ktorých sa testovanie citlivosti neodporúča.'
Triedy
|
Bežne citlivé druhy
|
Grampozitívne
Enterococcus faecalis
Staphylococcus aureus
Koaguláza-negatívny Staphylococcus
Streptococcus spp.
Streptococcus pneumoniae
Clostridium spp.
|
Druhy, pre ktoré môže byť problémom získaná rezistencia
|
Enterococcus faecium
|
Prirodzene rezistentné
|
Gramnegatívne baktérie
Chlamydia spp.
Mykobaktérie
Mycoplasma spp.
Rickettsia spp.
|
5.2 Farmakokinetické vlastnostiAbsorpciaVankomycín sa podáva intravenózne na liečbu systémových infekcií. V prípade pacientov s normálnou funkciou obličiek vyvolá intravenózna infúzia viacnásobných dávok 1 g vankomycínu (15 mg/kg) počas 60 minút približne priemerné plazmatické koncentrácie 50 – 60 µg/ml (ihneď po skončení infúzie), 20 – 25 µg/ml (2 hodiny po skončení infúzie) a 5 – 10 µg/ml (11 hodín po skončení infúzie). Intravenózna infúzia viacnásobných dávok 500 mg počas 30 minút vyvolá priemerné plazmatické koncentrácie 40 – 50 mg/l (ihneď po skončení infúzie), 19 – 20 mg/l (2 hodiny po skončení infúzie) a 10 – 11 mg/l (6 hodín po skončení infúzie). Plazmatické hladiny dosiahnuté po viacnásobných dávkach sú podobné hladinám, ktoré sa dosahujú po jednorazovej dávke.
V prípade perorálneho použitia sa vysoko polárny vankomycín prakticky neabsorbuje. Po perorálnom podaní sa v aktívnej forme objavuje v stolici, a preto je vhodným chemoterapeutikom pri psedomembranóznej kolitíde a stafylokokovej kolitíde.
DistribúciaPri sérových koncentráciách vankomycínu 10 mg/l až 100 mg/l je väzba liečiva na plazmatické proteíny približne 30 – 55 %, čo sa stanovilo ultrafiltráciou.
Po intravenóznom podaní vankomycíniumchloridu sa inhibičné koncentrácie zistili v pleurálnej, perikardiálnej, ascitickej a sinoviálnej tekutine, v moči a tekutine na peritoneálnu dialýzu a v tkanive predsieňového prívesku.
V nezapálených meningoch prestupuje vankomycín len v nízkej miere cez hematoencefalickú bariéru.
ElimináciaEliminačný polčas vankomycínu u pacientov s normálnou funkciou obličiek je 4 až 6 hodín. V priebehu prvých 24 hodín sa približne 80 % podanej dávky vankomycínu vylučuje močom prostredníctvom glomerulárnej filtrácie. Dysfunkcia obličiek spomaľuje vylučovanie vankomycínu. U anefrických pacientov je priemerný polčas 7,5 dní. Liečivo sa len vo veľmi malej miere metabolizuje. Približne 35 – 65 % intraperitoneálnej dávky vankomycínu podaného počas peritoneálnej dialýzy sa systémovo absorbuje počas šiestich hodín. Sérové koncentrácie približne 8 mg/liter sa dosahujú intraperitoneálnou injekciou dávky 30 mg vankomycínu/kg. Hoci sa vankomycín neeliminuje účinne hemodialýzou ani peritoneálnou dialýzou, zaznamenalo sa zvýšenie klírensu vankomycínu vplyvom hemoperfúzie a hemofiltrácie. Celkový systémový a renálny klírens vankomycínu môže byť znížený u osôb v pokročilom veku.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti a toxicity po opakovanom podaní neodhalili žiadne osobitné riziko pre ľudí.
Obmedzené údaje o mutagénnych účinkoch preukázali negatívne výsledky, dlhodobé štúdie u zvierat týkajúce sa karcinogénneho potenciálu nie sú k dispozícii. V štúdiách teratogenity, v ktorých potkany a králiky dostávali dávky približne zodpovedajúce dávke u ľudí na základe plochy povrchu tela (mg/m
2), sa nepozorovali žiadne priame ani nepriame teratogénne účinky.
Štúdie u zvierat týkajúce sa používania počas perinatálneho/postnatálneho obdobia a účinkov na fertilitu nie sú k dispozícii.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokŽiadne.
6.2 InkompatibilityRoztok vankomycínu má nízke pH, ktoré môže vyvolať chemickú alebo fyzikálnu nestabilitu, keď sa mieša s inými látkami. Miešaniu s alkalickými roztokmi sa treba vyhnúť. Každý parenterálny roztok sa má pred používaním vizuálne skontrolovať kvôli precipitácii a zmene sfarbenia.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnostiPrášok v pôvodnom obale:2 roky
Pripravený koncentrát:Pripravený koncentrát sa má bezprostredne po príprave ďalej zriediť.
Zriedený liek:Z mikrobiologického a fyzikálnochemického hľadiska sa má liek použiť ihneď.
6.4 Špeciálne upozornenia na uchovávaniePrášok v pôvodnom obale:Uchovávajte pri teplote do 25 °C.
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Pripravený koncentrát a zriedený liek:Podmienky na uchovávanie pripraveného koncentrátu a zriedeného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah baleniaBezfarebná 10 ml sklenená injekčná liekovka (typu 1) s chlórbutylovou zátkou (typu 1) potiahnutou silikónom a šedým hliníkovým/polypropylénovým vyklápacím viečkom.
Veľkosti balenia: 1 injekčná liekovka, 10 x injekčná liekovka.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomLiek sa musí pripraviť a vzniknutý koncentrát sa musí pred použitím zriediť.
Príprava koncentrátu:Rozpustite obsah každej 500 mg injekčnej liekovky v 10 ml sterilnej vody na injekciu.
Vzhľad pripraveného koncentrátu:Číry a bezfarebný roztok bez častíc.
Jeden ml pripraveného koncentrátu obsahuje 50 mg vankomycínu.
Podmienky uchovávania pripraveného koncentrátu, pozri časť 6.3.
Príprava finálneho zriedeného infúzneho roztoku:Pripravený koncentrát obsahujúci 50 mg vankomycínu/ml sa má bezprostredne po príprave ďalej riediť.
Vhodné roztoky na zriedenie sú:Injekčný roztok chloridu sodného 9 mg/ml (0,9 %), injekčný roztok glukózy 50 mg/ml (5 %), injekčný roztok chloridu sodného 9 mg/ml (0,9 %) a injekčný roztok glukózy 50 mg/ml (5 %) alebo Ringerov acetátový injekčný roztok.
Pred podaním sa majú pripravené a zriedené roztoky vizuálne skontrolovať, či neobsahujú častice a či nezmenili farbu. Má sa používať len číry a bezfarebný roztok bez častíc.
Intermitentná infúzia:Pripravený koncentrát obsahujúci 500 mg vankomycínu (50 mg/ml) sa musí bezprostredne po príprave ďalej zriediť s minimálne 100 ml roztoku na zriedenie.
Koncentrácia vankomycínu v infúznom roztoku nemá prekročiť 5 mg/ml.
Požadovaná dávka sa má podať pomalou intravenóznou infúziou s rýchlosťou maximálne 10 mg/minútu minimálne počas 60 minút alebo aj dlhšie.
Podmienky na uchovávanie zriedeného lieku, pozri časť 6.3.
LikvidáciaInjekčné liekovky sú určené len na jednorazové použitie. Nepoužitý liek sa musí zlikvidovať.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIFresenius Kabi s.r.o., Želetavská 1525/1, 140 00 Praha 4, Česká republika
8. REGISTRAČNÉ ČÍSLO15/0324/11-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 13.05.2011
10. DÁTUM REVÍZIE TEXTUNovember 2013