> 10 kg až ≤ 33 kg
| 15 mg/kg
| 1,5 ml/kg
| 49,5 ml
| 60 mg/kg, bez prekročenia 2 g
|
> 33 kg až ≤ 50 kg
| 15 mg/kg
| 1,5 ml/kg
| 75 ml
| 60 mg/kg, bez prekročenia 3 g
|
> 50 kg s ďalšími rizikovými faktormi pre hepatotoxicitu
| 1 g
| 100 ml
| 100 ml
| 3 g
|
> 50 kg bez ďalších rizikových faktorov pre hepatotoxicitu
| 1 g
| 100 ml
| 100 ml
| 4 g
|
*
Predčasne narodené deti: nie sú dostupné údaje o účinnosti a bezpečnosti pre predčasne narodené deti.
**
Maximálna denná dávka: maximálna denná dávka uvedená v tabuľke vyššie je pre pacientov, ktorí neužívajú iné lieky s obsahom paracetamolu a dávka sa má upraviť podľa užívania týchto liekov.
***
Pacienti s nižšou telesnou hmotnosťou potrebujú menší objem/množstvo.Minimálny interval medzi jednotlivými podaniami musí byť najmenej 4 hodiny. Počas 24 hodín sa môžu podať najviac 4 dávky.Pediatrická populáciaNie sú k dispozícii žiadne údaje o bezpečnosti a účinnosti pri predčasne narodených deťoch (pozri časť 5.2).Pacienti s poruchou funkcie obličiekMinimálny interval medzi jednotlivými dávkami u pacientov so závažnou poruchou funkcie obličiek musí byť najmenej 6 hodín.
Pri podávaní paracetamolu pacientom so závažnou poruchou funkcie obličiek (klírens kreatinínu ≤ 30 ml/min) sa odporúča zvýšiť minimálny interval medzi jednotlivými aplikáciami na 6 hodín (pozri časť 5.2).
U dospelých pacientov s hepatocelulárnou insuficienciou, chronickým alkoholizmom, chronickou malnutríciou (nízke zásoby glutatiónu v pečeni), dehydratáciou:
Maximálna denná dávka nesmie prekročiť 3 g (pozri časť 4.4).
Spôsob podávaniaOpatrenia pred zaobchádzaním alebo podaním liekuPacienti s telesnou hmotnosťou ≤ 10 kg:- Fľaša Paracetamolu Noridem sa nemá zavesiť ako infúzia kvôli malému objemu lieku, ktorý bude týmto pacientom podaný.
- Objem, ktorý bude podaný, má byť odobratý z fľaše a nariedený v 9 mg/ml (0,9 %) roztoku chloridu sodného alebo 50 mg/ml (5 %) roztokom glukózy až do jednej desatiny (jeden diel Paracetamolu Noridem do deviatich dielov roztoku) a má sa podávať po dobu 15 minút.
- 5 alebo 10 ml injekčná striekačka sa má použiť na odmeranie dávky primeranej pre telesnú hmotnosť dieťaťa a požadovaného objemu lieku. Nesmie sa však prekročiť 7,5ml na jednotlivú dávku.
- Používateľ sa má riadiť pokynmi pre dávkovanie, ktoré sú uvedené v písomnej informácii o lieku.
Na intravenózne použitie.
Iba na jednorazové použitie. Všetok nespotrebovaný roztok musí byť odborne zlikvidovaný.
Roztok paracetamolu sa podáva formou 15 minútovej intravenóznej infúzie.
Informácia k veľkosti balenia 50 ml:
50 ml Paracetamolu Noridem môže byť tiež nariedený 0,9 % roztokom chloridu sodného alebo 5 % roztokom glukózy až do jednej desatiny (jeden diel Paracetamolu Noridem do deviatich dielov roztoku). V tomto prípade sa má zriedený roztok použiť do 1 hodiny od jeho prípravy (vrátane času na podanie infúzie).
4.3 KontraindikácieParacetamol Noridem je kontraindikovaný:
- u pacientov s hypersenzitivitou na paracetamol alebo propacetamóliumchlorid (proliečivo paracetamolu) alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1,
- v prípade závažnej hepatocelulárnej insuficiencie.
4.4 Osobitné upozornenia a opatrenia pri používaní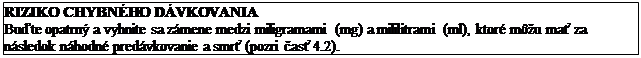
Odporúča sa použiť vhodnú perorálnu analgetickú liečbu, len čo bude možná táto cesta podania.
Aby sa zabránilo riziku predávkovania, je potrebné zistiť, či ďalšie podávané lieky neobsahujú paracetamol alebo propacetamol.
Dávky vyššie ako sú odporúčané dávky majú za následok riziko veľmi závažného poškodenia pečene. Klinické prejavy a symptómy poškodenia pečene (vrátane fulminantnej hepatitídy, zlyhania pečene, cholestatickej hepatitídy, cytolytickej hepatitídy) sa zvyčajne neprejavia skôr ako o dva dni a maximálne do 4–6 dní po podaní. Čo najskôr treba začať liečbu antidotom (pozri časť 4.9).
Tento liek obsahuje menej ako 1 mmol (23 mg) sodíka na 100 ml lieku, t.j. v podstate zanedbateľné množstvo sodíka.
Informácia pre fľaše s objemom 50 ml a 100 ml:
Rovnako ako pri všetkých infúznych roztokoch, je potrebné starostlivo sledovať pacienta, najmä na konci infúzie (pozri časť 4.2).
Paracetamol sa má používať mimoriadne opatrne za nasledujúcich okolností:
- hepatocelulárna insuficiencia,
- závažná renálna insuficiencia (klírens kreatinínu £ 30 ml/min), pozri časti 4.2 a 5.2,
- chronický alkoholizmus,
- chronická malnutrícia (nízke zásoby glutatiónu v pečeni),
- dehydratácia.
4.5 Liekové a iné interakcie- Probenecid vyvoláva takmer dvojnásobné zníženie klírensu paracetamolu inhibíciou jeho konjugácie s kyselinou glukurónovou. Pri súbežnom používaní s probenecidom sa má zvážiť zníženie dávky paracetamolu.
- Salicylamid môže predĺžiť eliminačný polčas paracetamolu.
- Je potrebné venovať pozornosť súbežnému užívaniu látok indukujúcich enzýmy (pozri časť 4.9)
- Súbežné používanie paracetamolu (4 g denne po dobu minimálne 4 dní) s perorálnymi antikoagulanciami môže vyvolať malé zmeny hodnôt INR. V tomto prípade sa majú častejšie monitorovať hodnoty INR počas súbežného podávania rovnako ako aj 1 týždeň po ukončení liečby paracetamolom.
Pediatrická populáciaInterakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktáciaGravidita:Klinická skúsenosť s intravenóznym podaním paracetamolu je obmedzená. Epidemiologické údaje pri použití perorálnych terapeutických dávok paracetamolu však nepreukazujú žiadne nežiaduce účinky na tehotenstvo ani na zdravie plodu/novorodenca.
Perspektívne údaje o tehotenstvách vystavených predávkovaniu nepreukázali zvýšené riziko malformácie.
Reprodukčné štúdie s intravenóznou formou paracetamolu neboli vykonané u zvierat. Štúdie s perorálnou formou však nepreukázali žiadnu malformáciu fetotoxických účinkov.
Paracetamol Noridem sa však má používať počas tehotenstva iba po starostlivom posúdení prínosov a rizík. V takomto prípade je nutné prísne dodržiavať odporúčané dávkovanie a trvanie liečby.
Dojčenie:Po perorálnom podaní sa paracetamol vylučuje v malých množstvách do materského mlieka. U dojčených detí sa nezaznamenali žiadne nežiaduce účinky.
Preto sa Paracetamol Noridem môže používať u dojčiacich žien.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeParacetamol Noridem nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkyPodobne ako u iných liekov obsahujúcich paracetamol sú nežiaduce reakcie zriedkavé (>1/10 000, < 1/1 000) alebo veľmi zriedkavé (< 1/10 000) a sú popísané nižšie:
Trieda orgánových systémov
| Zriedkavé
> 1/10 000, < 1/1 000
| Veľmi zriedkavé
< 1/10 000
|
Celkové poruchy a reakcie v mieste podania
| Malátnosť
| Reakcie z precitlivenosti
|
Poruchy srdca a srdcovej činnosti
| Hypotenzia
|
|
Poruchy pečene a žlčových ciest
|
| Zvýšené hladiny pečeňových transamináz
|
Poruchy krvi a lymfatického systému
|
| Trombocytopénia, leukopénia, neutropénia
|
Počas klinických skúšaní boli hlásené časté nežiaduce reakcie v mieste vpichu injekcie (bolesť a pálenie).
Veľmi zriedkavo boli hlásené prípady závažných kožných reakcií.
Boli hlásené veľmi zriedkavé prípady reakcií z precitlivenosti od jednoduchých kožných vyrážok alebo žihľavky až po anafylaktický šok, ktoré vyžadujú ukončenie liečby.
Boli hlásené prípady sčervenenia kože, návaly tepla, svrbenie a nezvyčajne rýchly tlkot srdca.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 PredávkovanieStarší pacienti, malé deti, pacienti s poruchami pečene, chronickým alkoholizmom, s chronickou malnutríciou a pacienti súbežne užívajúci lieky, ktoré spôsobujú indukciu enzýmov, sú vystavení mimoriadnemu riziku poškodenia pečene (vrátane fulminantnej hepatitídy, hepatálneho zlyhania, cholestatickej hepatitídy, cytolytickej hepatitídy). V takýchto prípadoch môže byť predávkovanie fatálne.
Príznaky
Príznaky sa zvyčajne objavia v priebehu prvých 24 hodín a zahŕňajú: nauzeu, vracanie, anorexiu, bledosť a bolesť brucha. Predávkovanie jednorazovou dávkou 7,5 g paracetamolu alebo viac u dospelých alebo jednorazovou dávkou 140 mg/kg telesnej hmotnosti u pediatrických pacientov má za následok nekrózu pečeňových buniek, ktorá môže zapríčiniť kompletnú a ireverzibilnú nekrózu a následne hepatocelulárnu insuficienciu, metabolickú acidózu a encefalopatiu. To následne môže viesť ku kómea a smrti. Súčasne sa pozorujú zvýšené hladiny pečeňových transamináz (AST, ALT), laktátdehydrogenázy a bilirubínu v kombinácii so zníženými hladinami protrombínu, ktoré sa môžu objaviť 12 až 48 hodín po podaní.
Klinické príznaky poškodenia pečene sú zvyčajne viditeľné najskôr po 2 dňoch a maximum dosiahnu po 4–6 dňoch.
Liečba- Okamžitá hospitalizácia.
- Pred začiatkom liečby a čo najskôr po predávkovaní sa má odobrať vzorka krvi na stanovenie plazmatických koncentrácií paracetamolu.
- Liečba zahŕňa podávanie antidota, N-acetylcysteínu (NAC), intravenózne alebo perorálne, ak je to možné v priebehu prvých 10 hodín. N-acetylcysteín má protektívny účinok pri podaní aj po viac ako 10 hodinách, v tomto prípade je však potrebná dlhšie trvajúca liečba.
- Symptomatická liečba.
- Na začiatku liečby a potom opakovane každých 24 hodín sa musia vykonať testy pečeňovej funkcie. Pečeňové transaminázy sa zvyčajne vrátia na normálnu hodnotu v priebehu jedného až dvoch týždňov s kompletným obnovením pečeňovej funkcie. U veľmi ťažkých prípadov môže byť však potrebná transplantácia pečene.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné analgetiká a antipyretiká, ATC kód: N02BE01
Presný analgetický a antipyretický spôsob účinku paracetamolu sa nestanovil. Pravdepodobné je centrálne i periférne pôsobenie.
Účinok Paracetamolu Noridem v úľave od bolesti nastupuje v priebehu 5–10 minút po podaní. Maximálny analgetický účinok sa dosiahne v priebehu 1 hodiny a analgézia zvyčajne pretrváva
4 – 6 hodín.
Paracetamol Noridem znižuje horúčku v priebehu 30 minút po podaní. Antipyretický účinok pretrváva minimálne 6 hodín.
5.2 Farmakokinetické vlastnostiDospelí:AbsorpciaPo jednorazovom a opakovanom podávaní počas 24 hodín je farmakokinetika paracetamolu lineárna až do 2 g.'
Biologická dostupnosť paracetamolu po infúzii 500 mg a 1 g Paracetamolu Noridem je podobná ako po podaní infúzie 1 g a 2 g propacetamolu (čo zodpovedá 500 mg a 1 g paracetamolu), v uvedenom poradí.
Maximálna plazmatická koncentrácia (C
max)paracetamolu pozorovaná na konci 15 minútovej intravenóznej infúzie 500 mg a 1 g Paracetamolu Noridem je približne 15 mg/ml a 30 mg/ml, v uvedenom poradí.
DistribúciaDistribučný objem paracetamolu je približne 1 l/kg.
Paracetamol sa neviaže vo výraznej miere na plazmatické bielkoviny.
Významné koncentrácie paracetamolu (približne 1,5 mg/ml) sa pozorovali v cerebrospinálnom moku dvadsať minút po podaní infúzie 1 g paracetamolu.
BiotransformáciaParacetamol sa metabolizuje hlavne v pečeni dvoma hlavnými metabolickými cestami: konjugáciou s kyselinou glukurónovou a s kyselinou sírovou. Druhá cesta je pri dávkach, ktoré prevyšujú terapeutickú dávku, rýchlo saturovaná. Malé množstvo (menej ako 4 %) sa metabolizuje cytochrómom P450 na reaktívny medziprodukt (N-acetylbenzochinónimín), ktorý je pri normálnom dávkovaní rýchlo detoxikovaný redukovaným glutatiónom a po konjugácii s cysteínom a kyselinou merkapturovou vylučovaný močom. V prípade závažného predávkovania je však množstvo tohto toxického metabolitu zvýšené.
ElimináciaMetabolity paracetamolu sú vylučované predovšetkým močom. 90 % podanej dávky sa vylúči v priebehu 24 hodín, hlavne vo forme glukuronidových (60–80 %) a sulfátových (20–30 %) konjugátov. Menej ako 5 % sa vylúči v nezmenenej forme. Plazmatický polčas je 2,7 hodín a celkový telesný klírens je 18 l/h.
Pediatrická populáciaFarmakokinetické parametre paracetamolu pozorované u dojčiat a detí sú podobné ako parametre pozorované u dospelých s výnimkou plazmatického polčasu, ktorý je mierne kratší (1,5 až 2 hodiny) ako u dospelých. U novorodencov je plazmatický polčas dlhší ako u dojčiat, t.j. okolo 3,5 hodiny. Novorodenci, dojčatá a deti do 10 rokov vylučujú významne menej glukuronidových a viac sulfátových konjugátov ako dospelí.
Tabuľka: Farmakokinetické hodnoty v závislosti od veku (štandardizovaný klírens, *CLstd/Foral (l.h-1 70 kg-1)Vek
| Telesná hmotnost (kg)
| CLstd/Foral (l.h-1 70 kg-1)
|
40 týždňov (vek po počatí)
3 mesiace po narodení (PNA)
6 mesiacov PNA
1 rok PNA
2 roky PNA
3 rokov PNA
8 rokov PNA
| 3,3
6
7,5
10
12
20
25
| 5,9
8,8
11,1
13,6
15,6
16,3
16,3
|
*CL
std je odhad CL populácie
Osobitné skupiny pacientov:Porucha funkcie obličiekPri závažnej poruche funkcie obličiek (klírens kreatinínu 10–30 ml/min) je eliminácia paracetamolu mierne oneskorená, eliminačný polčas sa pohybuje od 2 do 5,3 hodín. U glukuronidových a sulfátových konjugátov je rýchlosť eliminácie 3-krát nižšia u osôb so závažnou poruchou funkcie obličiek ako u zdravých jedincov. Preto pri podávaní paracetamolu pacientom so závažnou poruchou funkcie obličiek (klírens kreatinínu ≤ 30 ml/min) sa má predĺžiť minimálny interval medzi jednotlivými dávkami na 6 hodín (pozri časť 4.2 Dávkovanie a spôsob podávania).
Staršie osoby Farmakokinetika a metabolizmus paracetamolu je u starších jedincov nezmenená. U tejto skupiny pacientov nie je potrebná žiadna úprava dávky.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje neodhalili žiadne osobitné riziko pre ľudí okrem informácií uvedených v iných častiach SPC.
Štúdie lokálnej tolerancie infúzneho roztoku paracetamolu u potkanov a králikov preukázali dobrú znášanlivosť. Testy uskutočnené na morčatách nepreukázali oneskorenú kontaktnú hypersenzitivitu.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokManitol, hydrogenfosforečnan sodný, kyselina chlorovodíková (na úpravu pH), hydroxid sodný (na úpravu pH) a voda na injekcie
6.2 InkompatibilityParacetamol Noridem sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnostiFľaša pred otvorením: 2 rokyZ mikrobiologického hľadiska, pokiaľ spôsob otvorenia nevylučuje riziko mikrobiálnej kontaminácie, sa liek má použiť ihneď. Ak sa nepoužije ihneď, za čas použiteľnosti a podmienky pred použitím je zodpovedný používateľ.
Informácia k veľkosti balenia 50 ml:
Po nariedení roztokom chloridu sodného 9 mg/ml (0,9 %) alebo roztokom glukózy 50 mg/ml (5 %), sa roztok musí použiť ihneď. Ak však zriedený roztok nie je použitý ihneď, neuchovávajte ho dlhšie ako 1 hodinu (vrátane času na podanie infúzie).
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote do 30 °C.
Chráňte pred chladom alebo mrazom.
Vnútorný obal skladujte vo vonkajšom hliníkovom obale
Po otvorení vnútorného obalu sa má produkt okamžite použiť.
6.5 Druh obalu a obsah balenia 50 ml a 100 ml fľaše z polypropylénu, s tvarovaným plastovým viečkom, gumovým tesnením (typ II) a odtrhávacím krúžkom. Každá fľaša je umiestnená do metalického ochranného plastového obalu.
50ml a 100ml fľaše sú dostupné v baleniach po 1, 5, 10 a 12 fliaš.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomInformácia pre 50 ml a 100 ml fľaše:
Pred podaním liek vizuálne skontrolujte kvôli prítomnosti častíc a zmene farby.
Na jednorazové použitie. Všetok nepoužitý roztok sa má zlikvidovať.
Zriedený roztok sa má vizuálne skontrolovať a nesmie sa použiť, pokiaľ je zakalený (opaleskuje), sú v ňom viditeľné častice alebo zrazenina.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINoridem Enterprises Limited
Makariou & Evagorou
Mitsi Building 3, Office115
1065 Nikózia
Cyprus
8. REGISTRAČNÉ ČÍSLO07/0168/20-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU07/2020