
y v predpisovaní liečby vrátane
predávkovania a poddávkovania, je potrebné postupovať podľa pokynov pre výpočet dávky a úpravu dávky (pozri časť 4.2).
Predávkovanie môže viesť k nadmenému zvýšeniu počtu krvných doštičiek súvisiacemu
s trombotickými/tromboembolickými komplikáciami. Ak sa počet krvných doštičiek nadmerne zvýšil, vysaďte Nplate a sledujte počet krvných doštičiek. Liečbu liekom Nplate nasaďte znova v súlade
s odporúčaniami na dávkovanie a podanie. Poddávkovanie môže mať za následok nižší počet krvných
doštičiek, ako sa očakáva, a možnosť výskytu krvácania. U pacientov dostávajúcich Nplate sa má sledovať počet krvných doštičiek (pozri časti 4.2, 4.4 a 4.9).
Progresia existujúceho myelodysplastického syndrómu(MDS)
Pozitívny pomer prínosu a rizika pre romiplostím je stanovený len na liečbu trombocytopénie spojenej
s chronickou ITP a romiplostím nesmie byť použitý pri iných klinických stavoch spojených s trombocytopéniou.
Diagnóza ITP u dospelých a starších pacientov sa má potvrdiť vylúčením iných klinických príčin výskytu trombocytopénie, zvlášť diagnóza MDS musí byť vylúčená. Aspiráciu a biopsiu kostnej drene je zvyčajne potrebné urobiť v priebehu ochorenia a liečby, hlavne u pacientov vo veku nad 60 rokov,
u tých, ktorí majú systémové príznaky alebo abnormálne prejavy, ako je zvýšený počet nezrelých buniek v periférii.
V klinických skúšaniach, ktoré skúmali liečbu romiplostímom u pacientov s MDS, sa pozorovali prípady prechodných nárastov nezrelých buniek a zaznamenali sa prípady progresie MDS do akútnej myeloidnej leukémie (AML). V randomizovanom, placebom kontrolovanom klinickom skúšaní
u pacientov s MDS bola liečba romiplostímom predčasne ukončená z dôvodu numerickej prevahy progresie ochorenia do AML a zvýšenia počtu cirkulujúcich nezrelých buniek o viac ako 10 %
u pacientov liečených romiplostímom. Z pozorovaných prípadov progresie MDS do AML boli
pacienti s RAEB-1 podtypom MDS na začiatku náchylnejší na výskyt progresie ochorenia do AML
v porovnaní s nižším rizikom MDS.
Romiplostím sa nesmie používať na liečbu trombocytopénie spôsobenej MDS ani trombocytopénie zapríčinenej iným spôsobom, ako je ITP mimo klinických štúdií.
Strata odpovede na romiplostím
Pri strate odpovede alebo neschopnosti udržať odpoveď krvných doštičiek na liečbu romiplostímom
v rámci odporúčaného dávkovania sa majú hľadať príčinné faktory vrátane imunogenicity (pozri časť 4.8) a zvýšeného retikulínu kostnej drene (pozri časť vyššie).
Účinky romiplostímunačervenéabielekrvinky
Zmeny parametrov červených (zníženie) a bielych (zvýšenie) krviniek boli pozorované
v predklinických toxikologických štúdiách (potkan a opica), ako aj u ITP pacientov. U pacientov sa bez ohľadu na status splenektómie môžu súbežne vyskytnúť anémia a leukocytóza (v 4-týždňovom
časovom rozpätí), častejšie sa však pozorovali u pacientov po splenektómii. Monitorovanie týchto parametrov sa má zvážiť u pacientov liečených romiplostímom.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Možné interakcie romiplostímu so súčasne podávanými liekmi z dôvodu väzby na plazmatické bielkoviny zostávajú neznáme.
Lieky používané na liečbu ITP v kombinácii s romiplostímom v klinických skúšaniach zahŕňali kortikosteroidy, danazol a/alebo azatioprin, intravenózny imunoglobulín (IVIG) a anti-D imunoglobulín. Pri kombinovaní romiplostímu s inými liekmi na liečbu ITP sa má sledovať počet krvných doštičiek, aby počty krvných doštičiek neprekročili odporúčané rozmedzie (pozri časť 4.2).
Používanie kortikosteroidov, danazolu a azatioprinu sa môže znížiť alebo ukončiť, ak sa podávajú
v kombinácii s romiplostímom (pozri časť 5.1). Pri znižovaní alebo prerušení iných terapií ITP sa má sledovať počet krvných doštičiek, aby počty krvných doštičiek neklesli pod odporúčané rozmedzie
(pozri časť 4.2).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití romiplostímu u gravidných
žien.
Štúdie na zvieratách preukázali, že romiplostím prechádzal placentou a zvyšoval počet krvných doštičiek plodov. V štúdiách na zvieratách sa vyskytovali aj poimplantačná strata a mierne zvýšenie výskytu perinatálnej mortality mláďat (pozri časť 5.3).
Romiplostím sa neodporúča užívať počas gravidity a u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
Dojčenie
Nie je známe, či sa romiplostím a metabolity vylučujú do materského mlieka. Riziko u novorodencov
a dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu romiplostímom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
O fertilite nie sú k dispozícii žiadne údaje.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nplate má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. V klinických štúdiách sa u niektorých pacientov vyskytli mierne až stredne závažné prechodné závraty.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Na základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4
kontrolovaných a 5 nekontrolovaných klinických skúšaniach, bol celkový výskyt všetkých nežiaducich reakcií u pacientov liečených romiplostímom 91,5 % (248/271). Priemerné trvanie
expozície romiplostímu u tejto skúmanej populácie bolo 50 týždňov.
Najzávažnejšie nežiaduce reakcie, ktoré sa môžu objaviť v priebehu liečby liekom Nplate, sú: opätovný výskyt trombocytopénie a krvácania po vysadení liečby, zvýšenie retikulínu kostnej drene, trombotické/tromboembolické komplikácie, chyby pri predpisovaní liečby a progresia existujúceho MDS do AML. Najčastejšími pozorovanými nežiaducimi reakciami sú hypersenzitívne reakcie (zahŕňajúce prípady vyrážky, urtikárie a angioedému) a bolesť hlavy.
Zoznam nežiaducich reakcií zoradených dotabuľky
Frekvencie sú definované takto: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (častosť sa nedá odhadnúť z dostupných údajov). V rámci jednotlivých tried orgánových systémov podľa MedDRA a skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúceho výskytu.
T
rieda orgánových
systémov podľa
MedDRA
V
eľmi časté Časté Menej časté

Infekcie a nákazy infekcia horných dýchacích ciest
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
gastroenteritída chrípka,
lokalizovaná infekcia, nazofaryngitída
mnohonásobný myelóm, myelofibróza
T
rieda orgánových systémov podľa MedDRA
V
eľmi časté Časté Menej časté
Poruchy krvi
a lymfatického systému
Poruchy imunitného systému
Poruchy metabolizmu a výživy
porucha kostnej drene*, trombocytopénia*, anémia
hypersenzitivita** angioedém
aplastická anémia, zlyhanie kostnej drene, leukocytóza, splenomegália, trombocytémia, zvýšený počet krvných doštičiek,
abnormálny počet krvných doštičiek
intolerancia alkoholu, anorexia,
znížená chuť do jedla,
dehydratácia, dna
Psychické poruchy nespavosť depresia, abnormálne sny
Poruchy nervového systému
bolesť hlavy závrat, migréna, parestézia
klonus, dysgeúzia, hypestézia, hypogeúzia,
periférna neuropatia, trombóza priečnych
splavov
Poruchy oka konjunktiválna hemorágia, porucha akomodácie, slepota,
porucha oka, očný pruritus, zvýšená lakrimácia, papiloedém, poruchy videnia
Poruchy ucha a labyrintu Poruchy srdca
a srdcovej činnosti
vertigo
palpitácie infarkt myokardu, zvýšená frekvencia srdca
Poruchy ciev sčervenanie hlboká žilová trombóza, hypotenzia,
periférna embólia, periférna ischémia,
flebitída,
povrchová tromboflebitída, trombóza,
erytromelalgia
Poruchy dýchacej sústavy, hrudníka a mediastína
pľúcna embólia* kašeľ, rinorea,
suchosť v hrdle,
dyspnoe, kongescia nosa, bolestivé dýchanie
T
rieda orgánových systémov podľa MedDRA
V
eľmi časté Časté Menej časté
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest Poruchy kože
a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy obličiek a močových ciest Poruchy reprodukčného systému a prsníkov Celkové poruchy
a reakcie v mieste podania
nauzea, hnačka,
bolesť brucha,
zápcha, dyspepsia
pruritus, ekchymóza, vyrážka
artralgia, myalgia, svalové kŕče,
bolesť končatín, bolesť chrbta,
bolesť v kostiach
únava,
periférny edém, ochorenie podobné chrípke,
bolesť, asténia,
pyrexia, zimnica,
reakcia v mieste vpichu
vracanie,
rektálna hemorágia, zápach z úst, dysfágia,
gastro-ezofageálna refluxná choroba,
hematochézia,
hemorágia z úst, žalúdočný dyskomfort,
stomatitída,
zmena sfarbenia zubov trombóza portálnej vény, zvýšenie transaminázy alopécia,
fotosenzitívna reakcia,
akné,
kontaktná dermatitída, suchá koža,
ekzém,
erytém,
exfoliatívna vyrážka, abnormálny rast vlasov,
prurigo, purpura,
papulózna vyrážka, pruritická vyrážka, kožný nodulus,
abnormálny zápach kože, urtikária
napnuté svalstvo, svalová slabosť, bolesť ramena, svalové zášklby
prítomnosť bielkovín v moči
vaginálna hemorágia
hemorágia v mieste vpichu, bolesť na hrudi, podráždenosť,
nevoľnosť, edém tváre, pocit horúčavy, pocit nervozity
T
rieda orgánových systémov podľa MedDRA
Laboratórne a funkčné vyšetrenia
Veľmi časté Časté Menej časté
zvýšený krvný tlak, zvýšená laktátdehydrogenáza v krvi,
zvýšená telesná teplota, znížená hmotnosť,
zvýšená hmotnosť
Úrazy, otravy a komplikácie liečebného postupu
* pozri časť 4.4
kontúzia
** Hypersenzitívne reakcie vrátane prípadov vyrážky, urtikárie a angioedému
Opis vybraných nežiaducichreakciíOkrem toho sa za reakcie súvisiace s liečbou považovali aj udalosti uvedené nižšie.
TrombocytózaNa základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4 kontrolovaných a 5 nekontrolovaných klinických skúšaniach, boli hlásené 3 prípady trombocytózy, n = 271. V súvislosti so zvýšeným počtom krvných doštičiek u ktoréhokoľvek z týchto 3 pacientov neboli hlásené žiadne klinické následky.
Trombocytopénia po skončení liečbyNa základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4 kontrolovaných a 5 nekontrolovaných klinických skúšaniach, boli hlásené 4 prípady trombocytopénie po skončení liečby, n = 271 (pozri časť 4.4).
Progresia existujúceho myelodysplastického syndrómu (MDS)V randomizovanom, placebom kontrolovanom klinickom skúšaní u pacientov s MDS bola liečba romiplostímom predčasne ukončená z dôvodu numerického zvýšenia progresie MDS do AML
a prechodného zvýšenia počtu nezrelých buniek u pacientov liečených romiplostímom v porovnaní s placebom. Z pozorovaných prípadov progresie MDS do AML boli pacienti s RAEB-1 podtypom MDS na začiatku náchylnejší na výskyt progresie ochorenia do AML (pozri časť 4.4). Celkové
prežívanie bolo podobné placebu.
Zvýšenie retikulínu kostnej dreneV klinických skúšaniach bola liečba romiplostímom vysadená u 4 z 271 pacientov z dôvodu hromadenia retikulínu v kostnej dreni. U 6 ďalších pacientov sa retikulín pozoroval po biopsii kostnej drene (pozri časť 4.4).
ImunogenicitaV klinických skúšaniach sa u dospelých pacientov s ITP zisťovali protilátky proti romiplostímu. Kým 5,8 % osôb bolo pozitívnych na tvorbu viažucich protilátok proti romiplostímu a 3,9 % na
protilátky proti TPO, len 2 osoby (0,4 %) boli pozitívne na neutralizujúce protilátky proti romiplostímu, tieto protilátky však nereagovali skrížene s endogénnym TPO. Obe osoby mali
negatívny test neutralizujúcich protilátok proti romiplostímu 4 mesiace po skončení dávkovania. Incidencia preexistujúcich protilátok proti romiplostímu bola 8,0 % a protilátok proti TPO 5,4 %.
Rovnako ako pri všetkých terapeutických proteínoch existuje možnosť imunogenicity. Ak existuje podozrenie na tvorbu neutralizujúcich protilátok, kontaktujte miestneho zástupcu Držiteľa rozhodnutia o registrácii (pozri časť 6 písomnej informácie pre používateľa) kvôli vyšetreniu protilátok.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovaniePri potkanoch, ktorým sa podávala jednorazová dávka 1 000 µg/kg (100-násobok maximálnej klinickej dávky10 µg/kg), ani pri opiciach po opakovanom podávaní romiplostímu v dávke 500 µg/kg (50-násobok maximálnej klinickej dávky10 µg/kg) sa nepozorovali žiadne nežiaduce účinky.
V prípade predávkovania sa môžu počty krvných doštičiek nadmerne zvýšiť a môžu mať za následok trombotické/tromboembolické komplikácie. Ak sa počet krvných doštičiek nadmerne zvýši, ukončite podávanie Nplate a sledujte počty krvných doštičiek. Liečbu liekom Nplate začnite znova podľa odporúčaní na dávkovanie a podávanie (pozri časti 4.2 a 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antihemoragiká, iné systémové hemostatiká, ATC kód: B02BX04
Mechanizmus účinkuRomiplostím je Fc-peptidový fúzny proteín (peptilátka), ktorý signalizuje a aktivuje intracelulárne
transkripčné dráhy prostredníctvom TPO receptora (známeho aj ako cMpl) na zvýšenie tvorby krvných doštičiek. Peptilátková molekula sa skladá z Fc domény ľudského imunoglobulínu IgG1,
ktorej každá jednoreťazcová podjednotka sa kovalentne viaže C-terminálom na peptidový reťazec
obsahujúci 2 domény, ktoré sa viažu na TPO receptor.
Romiplostím nemá žiadnu sekvenciu aminokyselín homológnu s endogénnym TPO. V predklinických a klinických skúšaniach žiadne anti-romiplostímové protilátky nereagovali skrížene s endogénnym TPO.
Klinická účinnosť abezpečnosťBezpečnosť a účinnosť romiplostímu bola hodnotená v liečbe trvajúcej až 3 roky. V klinických
skúšaniach viedla liečba romiplostímom k zvýšeniu počtu krvných doštičiek, ktoré bolo závislé od dávky. Čas do dosiahnutia maximálneho účinku na počet krvných doštičiek je približne 10 – 14 dní
a nezávisí od dávky. Po jednorazovej subkutánnej dávke 1 až 10 µg/kg romiplostímu podanej pacientom s ITP bol maximálny počet krvných doštičiek 1,3- až 14,9-krát vyšší ako pôvodný počet
krvných doštičiek v priebehu 2 až 3 týždňov a medzi pacientmi bola odpoveď rôzna. Počty krvných doštičiek u pacientov s ITP, ktorí dostali 6 dávok 1 alebo 3 µg/kg romiplostímu raz týždenne, sa pohybovali v rozsahu 50 až 450 x 109/l u väčšiny pacientov. Z 271 pacientov, ktorí dostali romiplostím v klinických skúšaniach s ITP, bolo 55 (20 %) vo veku 65 rokov a viac a 27 (10 %) vo
veku 75 a viac. Medzi staršími a mladšími pacientmi sa v placebom kontrolovaných štúdiách nezistili žiadne celkové rozdiely v bezpečnosti ani účinnosti.
Výsledky z hlavných placebom kontrolovaných štúdií
Bezpečnosť a účinnosť romiplostímu bola hodnotená v dvoch placebom kontrolovaných, dvojito zaslepených štúdiách u dospelých s ITP, ktorí pred vstupom do štúdie absolvovali aspoň jednu liečbu a sú predstaviteľmi celého spektra takýchto pacientov s ITP.
Štúdia S1 (212) hodnotila pacientov, ktorí neabsolvovali splenektómiu a nedostatočne odpovedali alebo neznášali predchádzajúce spôsoby liečby. Pri vstupe do štúdie mali pacienti diagnostikovanú ITP približne 2 roky. Pacienti mali pred vstupom do štúdie medián 3 terapií ITP (rozsah 1 až 7). Predchádzajúce spôsoby liečby zahŕňali kortikosteroidy (90 % všetkých pacientov), imunoglobulíny (76 %), rituximab (29 %), cytotoxické liečby (21 %), danazol (11 %) a azatioprin (5 %). Pri vstupe do štúdie mali pacienti medián počtu krvných doštičiek 19 x 109/l.
Štúdia S2 (105) hodnotila pacientov, ktorí absolvovali splenektómiu a mali aj naďalej trombocytopéniu. Pri vstupe do štúdie mali pacienti diagnostikovanú ITP približne 8 rokov. Okrem splenektómie mali pacienti pred vstupom do štúdie medián 6 terapií ITP (rozsah 3 až 10). Predchádzajúce spôsoby liečby zahŕňali kortikosteroidy (98 % všetkých pacientov), imunoglobulíny (97 %), rituximab (71 %), danazol (37 %), cytotoxické liečby (68 %) a azatioprin (24 %). Pri vstupe do štúdie mali pacienti medián počtu krvných doštičiek 14 x 109/l.
Obidve štúdie mali rovnaký dizajn. Pacienti (≥ 18 rokov) boli randomizovaní v pomere 2 : 1, aby dostávali počiatočnú dávku romiplostímu 1 µg/kg alebo placebo. Pacienti dostávali subkutánne injekcie jedenkrát za týždeň počas 24 týždňov. Dávky boli upravené tak, aby sa udržali počty krvných doštičiek (50 až 200 x 109/l). V oboch štúdiách sa dokázala účinnosť zvýšením podielu pacientov,
ktorí dosiahli trvalú odpoveď krvných doštičiek. Medián priemernej týždennej dávky pre pacientov po splenektómii bol 3 µg/kg a pre pacientov, ktorí nepodstúpili splenektómiu, bol 2 µg/kg.
Významne vyšší podiel pacientov, ktorí dostávali romiplostím, dosiahol trvalú odpoveď krvných doštičiek v porovnaní s pacientmi, ktorí dostávali placebo v obidvoch štúdiách. Po prvých štyroch týždňoch štúdie romiplostím udržoval počty krvných doštičiek ≥ 50 x 109/l u 50 % až 70 % pacientov počas 6 mesiacov liečby v placebom kontrolovaných štúdiách. V placebovej skupine bolo 0 % až 7 % pacientov schopných dosiahnuť odpoveď krvných doštičiek počas 6 mesiacov liečby. Súhrn kľúčových cieľov účinnosti je uvedený nižšie.
Súhrn kľúčových výsledkov účinnosti z placebom kontrolovaných štúdií
Štúdia 1
pacienti bez splenektómie
Štúdia 2
pacienti po splenektómii
K
ombinované
štúdie 1 & 2
P
očet (%)
pacientov
romiplostím
(n = 41)
placebo
(n = 21)
romiplostím
(n = 42)
placebo
(n = 21)
romiplostím
(n = 83)
placebo
(n = 42)
s trvalou odpoveďou krvných doštičiek
a
25 (61 %) 1 (5 %) 16 (38 %) 0 (0 %) 41 (50 %) 1 (2 %)
(95 % CI) (45 %,
76 %)
(0 %,
24 %) (24 %, 54 %)
(0 %,
16 %) (38 %, 61 %)
(0 %,
13 %)
p-hodnota < 0,0001 0,0013 < 0,0001
Počet (%)
 pacientov
s celkovou
pacientov
s celkovou
36 (88 %) 3 (14 %) 33 (79 %) 0 (0 %) 69 (83 %) 3 (7 %)
odpoveďou krvných doštičiek
b
Štúdia 1 pacienti bez splenektómie
Štúdia 2
pacienti po splenektómii
K
ombinované štúdie 1 & 2
(95 % CI) (74 %,
96 %)
(3 %,
36 %) (63 %, 90 %)
(0 %,
16 %) (73 %, 91 %)
(2 %,
20 %)
p-hodnota < 0,0001 < 0,0001 < 0,0001
Priemerný počet
t
ýždňov
s odpoveďou krvných doštičiek
c
15 1 12 0 14 1
(SD) 3,5 7,5 7,9 0,5 7,8 2,5
p-hodnota < 0,0001 < 0,0001 < 0,0001
Počet (%)
pacientov,
ktorí potrebovali záchrannú liečbu
d
8 (20 %) 13 (62 %) 11 (26 %) 12 (57 %) 19 (23 %) 25 (60 %)
(95 % CI) (9 %, 35 %) (38 %,
82 %)
(14 %, 42 %) (34 %,
78 %)
(14 %, 33 %) (43 %,
74 %)
p-hodnota 0,001 0,0175 < 0,0001
Počet (%) pacientov s trvalou
odpoveďou krvných doštičiek pri stabilnej dávke
e
21 (51 %) 0 (0 %) 13 (31 %) 0 (0 %) 34 (41 %) 0 (0 %)
(95% CI) (35 %,
67 %)
(0 %,
16 %) (18 %, 47 %)
(0 %,
16 %) (30 %, 52 %) (0 %, 8 %)

p-hodnota 0,0001 0,0046 < 0,0001
a Trvalá odpoveď krvných doštičiek je definovaná ako týždenný počet krvných doštičiek ≥ 50 x 109/l 6- alebo viackrát počas 18 – 25 týždňov štúdie, pričom záchranná liečba nie je počas liečby nikdy
potrebná.
b Celková odpoveď krvných doštičiek je definovaná ako dosiahnutie trvalých alebo prechodných odpovedí krvných doštičiek. Prechodná odpoveď krvných doštičiek bola definovaná ako týždenný počet krvných doštičiek ≥ 50 x 109/l 4- alebo viackrát počas 2 – 25 týždňov štúdie bez trvalej
odpovede krvných doštičiek. Pacient nemusí mať týždennú odpoveď počas 8 týždňov po podaní akýchkoľvek záchranných liekov.
c Počet týždňov s odpoveďou krvných doštičiek je definovaný ako počet týždňov s počtami krvných doštičiek ≥ 50 x 109/l počas 2 – 25 týždňoch štúdie. Pacient nemusí mať týždennú odpoveď počas
8 týždňov po podaní akýchkoľvek záchranných liekov.
d Záchranná liečba je definovaná ako akákoľvek liečba podaná na zvýšenie počtov krvných doštičiek. Pacienti, ktorí potrebovali záchranné lieky, sa nepovažovali za pacientov s trvalou odpoveďou
krvných doštičiek. Záchranné liečby povolené v štúdii boli IVIG, transfúzie krvných doštičiek, anti-D
imunoglobulíny a kortikosteroidy.
e Stabilná dávka je definovaná ako dávka udržiavaná ±1 µg/kg počas posledných 8 týždňov liečby.
Výsledky štúdií v porovnaní so štandardnou starostlivosťou (ŠS) u pacientov bez splenektómie
Štúdia S3 (131) bola otvorené randomizované 52-týždňové klinické skúšanie s pacientmi, ktorí dostávali romiplostím alebo liečbu v rámci štandardnej starostlivosti (ŠS). V tejto štúdii sa hodnotili pacienti bez splenektómie s ITP a počtom krvných doštičiek < 50 x 109/l. Romiplostím bol podávaný
157 pacientom formou subkutánnej (s.c.) injekcie raz týždenne začnúc s dávkou 3 µg/kg, ktorá bola počas štúdie upravovaná v rozmedzí 1 – 10 µg/kg, aby sa počet krvných doštičiek udržal medzi 50
a 200 x 109/l. 77 pacientov dostávalo liečbu ŠS podľa štandardnej praxe zdravotníckeho zariadenia alebo podľa terapeutických odporúčaní.
Celková miera incidencie splenektómie bola 8,9 % (14 zo 157 pacientov) v skupine s romiplostímom v porovnaní s 36,4 % (28 zo 77 pacientov) v skupine so ŠS, pričom pomer pravdepodobnosti (odds ratio, OR) (romiplostím vs ŠS) bol 0,17 (95 % CI: 0,08; 0,35).
Celkový výskyt zlyhania liečby predstavoval 11,5 % (18 zo 157 pacientov) v skupine
s romiplostímom v porovnaní s 29,9 % (23 zo 77 pacientov) v skupine so ŠS, pričom pomer pravdepodobnosti (romiplostím vs ŠS) bol 0,31 (95 % CI: 0,15; 0,61).
Zo 157 pacientov randomizovaných do skupiny s romiplostímom traja pacienti nedostávali romiplostím. Medzi 154 pacientmi, ktorí dostávali romiplostím, bol medián celkovej expozície romiplostímu 52,0 týždňa a pohyboval sa od 2 do 53 týždňov. Najčastejšie užitá týždenná dávka bola medzi 3 – 5 µg/kg (t. j. 25. – 75. percentil; medián 3 µg/kg).
Zo 77 pacientov randomizovaných do skupiny so ŠS dvaja pacienti nedostávali žiadnu liečbu v rámci ŠS. Medzi 75 pacientmi, ktorí dostali najmenej jednu dávku liečby v rámci ŠS, bol medián celkovej expozície ŠS 51 týždňov a pohyboval sa od 0,4 do 52 týždňov.
Redukcia povolených súbežných spôsobov liečby ITP
V obidvoch placebom kontrolovaných, dvojito zaslepených štúdiách mohli pacienti, ktorí už dostávali liečbu ITP v konštantnej dávke, pokračovať v tejto liečbe počas štúdie (kortikosteroidy, danazol a/alebo azatioprin). Na začiatku štúdie dostávalo 21 pacientov bez splenektómie a 18 pacientov po splenektómii predchádzajúcu ITP liečbu (najmä kortikosteroidy). Všetci (100 %) pacienti po splenektómii, ktorí dostávali romiplostím, boli schopní znížiť dávku o viac ako 25 % alebo vysadiť súbežnú ITP liečbu do konca liečby v porovnaní so 17 % placebom liečených pacientov. 73 % pacientov bez splenektómie, ktorí dostávali romiplostím, boli schopní znížiť dávku o viac ako 25 % alebo vysadiť súbežnú ITP liečbu do konca štúdie v porovnaní s 50 % placebom liečených pacientov (pozri časť 4.5).
Krvácavé príhody
V celom klinickom ITP programe bol pozorovaný inverzný vzťah medzi krvácavými príhodami
a počtami krvných doštičiek. Všetky klinicky významné (≥ 3. stupeň) krvácavé príhody sa vyskytovali pri počtoch krvných doštičiek < 30 x 109/l. Všetky krvácavé príhody ≥ 2. stupeň sa vyskytovali pri počtoch krvných doštičiek < 50 x 109/l. Medzi skupinou pacientov liečenou liekom Nplate a skupinou liečenou placebom sa nezistili žiadne štatisticky významné rozdiely v celkovom výskyte krvácavých príhod.
V dvoch placebom kontrolovaných štúdiách 9 pacientov hlásilo krvácavú príhodu, ktorá sa považovala za závažnú (5 [6,0 %] romiplostím, 4 [9,8 %] placebo; pomer pravdepodobnosti [romiplostím/placebo]
= 0,59; 95 % CI = (0,15; 2,31)). Krvácavé príhody 2. alebo vyššieho stupňa boli hlásené u 15 %
pacientov liečených romiplostímom a 34 % pacientov liečených placebom (pomer pravdepodobnosti
[romiplostím/placebo] = 0,35; 95 % CI = (0,14; 0,85)).
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Nplate
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe imunitnej trombocytopénie
(idiopatická trombocytopenická purpura) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika romiplostímu zahŕňala elimináciu na cieľových štruktúrach, ktorá je pravdepodobne sprostredkovaná TPO receptormi na krvných doštičkách a iných bunkách trombopoetického radu, ako sú megakaryocyty.
Absorpcia
Po subkutánnom podaní 3 až 15 µg/kg romiplostímu sa dosiahli maximálne sérové hladiny
romiplostímu u ITP pacientov po 7 – 50 hodinách (medián 14 hodín). Sérové koncentrácie sa medzi pacientmi odlišovali a nekorelovali s podanou dávkou. Zdá sa, že sérové hladiny romiplostímu sú
v inverznom vzťahu s počtami krvných doštičiek.
Distribúcia
Distribučný objem romiplostímu po intravenóznom podaní romiplostímu klesal nelineárne zo 122;
78,8 na 48,2 ml/kg pre intravenózne dávky 0,3; 1,0 a 10 µg/kg u zdravých osôb. Tento nelineárny pokles distribučného objemu je v súlade s cieľovou väzbou romiplostímu (na megakaryocyty a krvné
doštičky), ktorá sa môže saturovať pri podávaní vysokých dávok.
Eliminácia
Polčas eliminácie romiplostímu u ITP pacientov sa pohyboval od 1 do 34 dní (medián 3,5 dňa).
Eliminácia sérového romiplostímu čiastočne závisí od TPO receptora krvných doštičiek. Ako následok podanej dávky sa u pacientov s vysokým počtom krvných doštičiek pozorovali nízke sérové
koncentrácie a naopak. V inom ITP klinickom skúšaní sa nepozorovala žiadna kumulácia sérových
koncentrácií po 6 dávkach romiplostímu raz týždenne (3 µg/kg).
Osobitné populácie
Farmakokinetika romiplostímu u pacientov s poruchou funkcie obličiek a pečene sa neskúmala. Zdá
sa, že farmakokinetika romiplostinu nie je klinicky signifikantne ovplyvnená vekom, hmotnosťou ani pohlavím.
5.3 Predklinické údaje o bezpečnosti
Uskutočnili sa toxikologické štúdie s viacnásobnou dávkou romiplostímu na potkanoch trvajúce
4 týždne a na opiciach trvajúce až 6 mesiacov. Vo všeobecnosti sa účinky pozorované počas týchto štúdií týkali trombopoetického účinku romiplostímu a boli podobné bez ohľadu na trvanie štúdie. Reakcie v mieste podania sa tiež týkali podania romiplostímu. Pri všetkých testovaných dávkach sa pozorovala myelofibróza v kostnej dreni potkanov. V týchto štúdiách sa myelofibróza pri zvieratách nepozorovala 4 týždne po skončení liečby, čo naznačuje reverzibilnosť.
V jednomesačných toxikologických štúdiách na potkanoch a opiciach sa pozorovalo mierne zníženie počtu červených krviniek, hematokritu a hemoglobínu. Pozoroval sa aj stimulačný účinok na tvorbu leukocytov, pretože počty neutrofilov, lymfocytov, monocytov a eozinofilov v periférnej krvi boli mierne zvýšené. V dlhšie trvajúcej chronickej štúdii s opicami sa nezistil žiadny účinok na erytroidné a leukocytové rady pri podávaní romiplostímu počas 6 mesiacov, kde sa podávanie romiplostímu
znížilo z trikrát týždenne na jedenkrát týždenne. Okrem toho v hlavných štúdiách fázy 3 romiplostím neovplyvňoval rady červených a bielych krviniek v porovnaní so subjektmi liečenými placebom.
Z dôvodu tvorby neutralizujúcich protilátok sa farmakodynamické účinky romiplostímu pri potkanoch často znižovali pri dlhodobom podávaní. Toxikokinetické štúdie nepreukázali žiadne interakcie protilátok s meranými koncentráciami. Hoci boli vysoké dávky testované v štúdiách na zvieratách, nemožno spoľahlivo určiť hranice bezpečnosti z dôvodu rozdielov medzi laboratórnymi zvieratami
a ľuďmi, čo sa týka citlivosti na farmakodynamický účinok romiplostímu a účinok neutralizujúcich protilátok.
Karcinogenéza
Karcinogénny potenciál romiplostímu sa nehodnotil. Preto riziko potenciálnej karcinogenicity
romiplostímu u ľudí zostáva neznáme.
Reprodukčná toxikológia
Vo všetkých vývojových štúdiách sa tvorili neutralizujúce protilátky, ktoré môžu inhibovať účinky
romiplostímu. V štúdiách embryofetálneho vývoja myší a potkanov boli pozorované zníženia telesnej hmotnosti matky iba pri myšiach. Pri myšiach sa dokázala zvýšená post-implantačná strata. V štúdii
prenatálneho a postnatálneho vývoja sa pri potkanoch zistilo predĺženie trvania gestácie a mierne
zvýšenie výskytu perinatálnej mortality mláďat. O romiplostíme je známe, že prechádza placentárnou bariérou potkanov, a môže sa prenášať z matky na vyvíjajúci sa plod a stimulovať tvorbu fetálnych
krvných doštičiek. Nepozoroval sa žiadny účinok romiplostímu na fertilitu potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
manitol (E421)
sacharóza
L-histidín
kyselina chlorovodíková (na úpravu pH)
polysorbát 20
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
5 rokov.
Po rekonštitúcii: Chemická a fyzikálna stabilita pri používaní sa stanovila na 24 hodín pri 25 °C a na
24 hodín pri 2 °C – 8 °C, ak je produkt chránený pred svetlom a uchovávaný v pôvodnej injekčnej liekovke.
Z mikrobiologického hľadiska sa liek musí použiť ihneď. Ak sa nepoužije ihneď, za čas použiteľnosti
a podmienky pred použitím je zodpovedný používateľ a nemali by byť za normálnych okolností dlhšie ako 24 hodín pri 25 °C alebo 24 hodín v chladničke (2 °C – 8 °C), chránené pred svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Môže sa dočasne vybrať z chladničky maximálne na dobu 24 hodín pri izbovej teplote (do 25 °C).
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
5 ml injekčná liekovka (z bezfarebného skla typu I) so zátkou (z chlorobutylovej gumy), s uzáverom
(z hliníka) a odklápacím viečkom (z polypropylénu).
Škatuľa obsahuje 1 alebo 4 injekčné liekovky s romiplostímom. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Nplate je sterilný liek, ale bez konzervačnej prísady, a je určený len na jednorazové použitie. Nplate sa má rekonštituovať podľa správnej aseptickej praxe.
Nplate 250 mikrogramovprášoknainjekčnýroztok
Nplate 250 mikrogramov prášok na injekčný roztok sa má rekonštituovať s 0,72 ml sterilnej vody na
injekcie, čím sa získa doručiteľný objem 0,5 ml. V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 250 µg romiplostímu (pozri tabuľku nižšie s obsahom injekčných liekoviek).
Nplate 500 mikrogramovprášoknainjekčnýroztok
Nplate 500 mikrogramov prášok na injekčný roztok sa má rekonštituovať s 1,2 ml sterilnej vody na
injekcie, čím sa získa doručiteľný objem 1 ml. V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 500 µg romiplostímu (pozri tabuľku nižšie s obsahom injekčných liekoviek).
Obsah injekčnej liekovky:
N
plate jednorazová injekčná liekovka
C
elkový obsah injekčnej liekovky s romiplostímom
O
bjem sterilnej vody na injekciu
D
oručiteľný liek a objem
V
ýsledná koncentrácia
250 µg 375 µg + 0,72 ml = 250 µg v 0,5 ml 500 µg/ml
500 µg 625 µg + 1,2 ml = 500 µg v 1 ml 500 µg/ml
Roztoky chloridu sodného alebo bakteriostatická voda sa nemajú používať pri rekonštitúcii lieku.
Do injekčnej liekovky sa má vstreknúť voda na injekcie. Obsah injekčnej liekovky sa môže rozpúšťať jemným krúžením a prevracaním.
Injekčnáliekovkasanemápretrepávaťanisilnomiešať. Rozpúšťanie Nplate trvá zvyčajne menej ako 2 minúty. Roztok pred podaním vizuálne skontrolujte, či neobsahuje častice a či nezmenil farbu. Rekonštituovaný roztok má byť číry a bezfarebný a nemá sa podávať, ak obsahuje častice a/alebo zmenil farbu.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Amgen Europe B.V. Minervum 7061
4817 ZK Breda
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/08/497/001
EU/1/08/497/003
EU/1/08/497/002
EU/1/08/497/004
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 4. február 2009
Dátum posledného predĺženia registrácie: 20. december 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.1. NÁZOV LIEKUNplate 250 mikrogramov prášok a rozpúšťadlo na injekčný roztok
Nplate 500 mikrogramov prášok a rozpúšťadlo na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIENplate 250 mikrogramovprášokarozpúšťadlonainjekčnýroztokKaždá injekčná liekovka obsahuje 250 µg romiplostímu. Po rekonštitúcii doručiteľný objem 0,5 ml
roztoku obsahuje 250 µg romiplostímu (500 µg/ml). V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 250 µg romiplostímu.
Nplate 500 mikrogramovprášokarozpúšťadlonainjekčnýroztokKaždá injekčná liekovka obsahuje 500 µg romiplostímu. Po rekonštitúcii doručiteľný objem 1 ml
roztoku obsahuje 500 µg romiplostímu (500 µg/ml). V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 500 µg romiplostímu.
Romiplostím je produkovaný bunkami
Escherichia coli (E. coli) technológiou rekombinantnej DNA. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPrášok a rozpúšťadlo na injekčný roztok (prášok na injekciu) Prášok je biely.
Rozpúšťadlo je číra bezfarebná tekutina.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieNplate je indikovaný dospelým pacientom s chronickou imunitnou (idiopatickou) trombocytopenickou purpurou (ITP), ktorí sú rezistentní alebo intolerantní voči iným terapiám (napr. kortikosteroidy, imunoglobulíny) (pozri časti 4.2 a 5.1).
4.2 Dávkovanie a spôsob podávaniaLiečba má prebiehať pod dohľadom lekára so skúsenosťami s liečbou hematologických ochorení.
DávkovanieNplate sa má podávať jedenkrát týždenne vo forme subkutánnej injekcie.
Počiatočná dávkaPočiatočná dávka romiplostímu je 1 µg/kg momentálnej telesnej hmotnosti.
Výpočet dávky
Počiatočná alebo následná
dávka podávaná jedenkrát týždenne:
Hmotnosť* v kg x Dávka v µg/kg = Individuálna dávka pre pacienta
v µg
Objem určený na podanie: Dávka v µg x _1ml_
500 µg = Množstvo určené na aplikáciu v ml
Príklad: Pacient s hmotnosťou 75 kg začína dávkou 1 µg/kg romiplostímu.
Individuálna dávka pre pacienta =
75 kg x 1 µg/kg = 75 µg
Zodpovedajúce množstvo roztoku Nplate určené na aplikáciu =
75 µg x _1ml_ = 0,15 ml
500 µg
*Pri výpočte dávky romiplostímu na začiatku liečby sa má vždy vychádzať z momentálnej telesnej hmotnosti. Ďalšie úpravy dávky vychádzajú len zo zmien v počte krvných doštičiek a uskutočňujú sa v prírastkoch o 1 µg/kg (pozri tabuľku nižšie).
Úpravy dávky
Na výpočet dávky na začiatku terapie sa má použiť momentálna telesná hmotnosť pacienta. Dávka romiplostímu jedenkrát týždenne sa má zvýšiť v prírastkoch o 1 µg/kg, až kým sa u pacienta nedosiahne počet krvných doštičiek ≥ 50 x 109/l. Množstvo krvných doštičiek sa má vyšetrovať raz za týždeň, až kým sa nedosiahne stabilný počet krvných doštičiek (≥ 50 x 109/l po dobu minimálne
4 týždňov bez úpravy dávky). Počet krvných doštičiek sa má následne vyšetrovať raz za mesiac. Maximálna dávka 10 µg/kg jedenkrát týždenne sa nemá prekročiť.
Dávku upravujte nasledujúcim spôsobom: Počet krvných doštičiek
(x 109/l) Postup
< 50 Zvýšte dávku podávanú jedenkrát týždenne o 1 µg/kg
> 150 počas dvoch
nasledujúcich týždňov Znížte dávku podávanú jedenkrát týždenne o 1 µg/kg
Nepodávajte, ďalej pokračujte vo vyšetrovaní počtu krvných doštičiek raz za týždeň
> 250
Pri poklese počtu krvných doštičiek na < 150 x 109/l pokračujte v podávaní jedenkrát týždenne v dávke zníženej o 1 µg/kg


Z dôvodu interindividuálnej rozličnej odpovede krvných doštičiek môže u niektorých pacientov po
znížení dávky alebo vysadení liečby náhle klesnúť počet krvných doštičiek pod 50 x 109/l. V týchto prípadoch sa majú zvážiť, ak je to klinicky vhodné, vyššie cut-off hladiny krvných doštičiek na
zníženie dávky (200 x 109/l) a na prerušenie liečby (400 x 109/l) podľa úsudku lekára.
Pri strate odpovede alebo neschopnosti udržať odpoveď krvných doštičiek na romiplostím v rámci odporúčaného dávkovania sa majú okamžite hľadať príčinné faktory (pozri časť 4.4, strata odpovede na romiplostím).
Ukončenie liečbyLiečba romiplostímom sa má ukončiť, ak sa po štyroch týždňoch liečby romiplostímom s najvyššou týždennou dávkou 10 µg/kg počet krvných doštičiek nezvýši na hodnotu, ktorá je dostatočná na zabránenie klinicky závažnému krvácaniu.
Pacientov treba pravidelne klinicky vyšetrovať a o pokračovaní v liečbe má ošetrujúci lekár rozhodnúť na základe individuálneho posúdenia; u pacientov, ktorí neabsolvovali splenektómiu, má ísť aj
o posúdenie z hľadiska splenektómie. Po skončení liečby je pravdepodobný opätovný výskyt trombocytopénie (pozri časť 4.4).
Starší pacienti (≥ 65 rokov)
U pacientov vo veku < 65 a ≥ 65 rokov sa nepozorovali žiadne celkové rozdiely v bezpečnosti alebo účinnosti (pozri časť 5.1). Hoci na základe týchto údajov nie je potrebná žiadna úprava dávkovania pre starších pacientov, odporúča sa opatrnosť, pretože v klinických štúdiách bol dosiaľ zahrnutý len malý počet starších pacientov.
Pediatrická populácia
Bezpečnosť a účinnosť romiplostímu u detí vo veku do 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Pacienti s poruchou funkcie pečene
Romiplostím sa nemá používať u pacientov so stredne závažnou až závažnou poruchou funkcie pečene (Childovo-Pughovo skóre ≥ 7), pokiaľ očakávaný prínos neprevýši identifikované riziko trombózy portálnej vény u pacientov s trombocytopéniou v súvislosti s insuficienciou pečene liečenou
agonistami trombopoetínu (TPO) (pozri časť 4.4).
Ak sa použitie romiplostímu považuje za nevyhnutné, je potrebné starostlivo sledovať počet krvných doštičiek, aby sa minimalizovalo riziko tromboembolických komplikácií.
Pacienti s poruchou funkcie obličiek
U týchto skupín pacientov sa neuskutočnili žiadne oficiálne klinické skúšania. Nplate sa má používať opatrne u týchto skupín pacientov.
Spôsob podávania
Na subkutánne použitie.
Po rekonštitúcii prášku sa injekčný roztok Nplate podáva subkutánne. Objem injekcie môže byť veľmi malý. Počas prípravy Nplate je potrebná opatrnosť pri výpočte dávky a rekonštitúcii so správnym objemom sterilnej vody na injekciu. Špeciálna opatrnosť je potrebná na zaistenie, že z injekčnej liekovky je natiahnutý správny objem Nplate na subkutánne podanie – je potrebné používať injekčnú striekačku s dielikmi po 0,01 ml.
Pacienti, ktorí majú stabilný počet krvných doštičiek ≥ 50 x 109/l počas minimálne 4 týždňov bez úpravy dávky, môžu si podľa zváženia dohliadajúceho lekára sami podávať injekčný roztok Nplate. Pacienti, ktorí sú schopní svojpomocne si podávať Nplate, majú absolvovať školenie na tieto procedúry.
Po prvých 4 týždňoch svojpomocného podávania by mal byť pacient počas rekonštitúcie a podávania Nplate opäť pod dohľadom. Môžu v tom pokračovať iba pacienti, ktorí preukážu schopnosť rekonštitúcie a svojpomocného podania lieku Nplate.
Pokyny na rekonštitúciu a podanie lieku, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 alebo na bielkoviny pochádzajúce z E. coli.
4.4 Osobitné upozornenia a opatrenia pri používaní
O
pätovný výskyt trombocytopénie a krvácaniaposkončeníliečby
Trombocytopénia sa pravdepodobne opätovne vyskytne po skončení liečby romiplostímom. Ak sa
preruší liečba romiplostímom v prítomnosti antikoagulancií alebo protidoštičkových látok, riziko krvácania je zvýšené. U pacientov sa má starostlivo sledovať zníženie počtu krvných doštičiek
a vhodnou liečbou zamedziť krvácaniu po skončení liečby romiplostímom. Po skončení liečby romiplostímom sa odporúča začať liečbu ITP podľa súčasných nariadení na liečbu. Vhodná liečba sa
môže skladať z prerušenia podávania antikoagulancií a/alebo protidoštičkovej liečby, výmeny antikoagulačnej liečby alebo podpornej doštičkovej liečby.
Zvýšenie retikulínu kostnejdrene
Zvýšenie retikulínu kostnej drene je pravdepodobne výsledkom stimulácie TPO receptora, ktorá vedie
k zvýšenému počtu megakaryocytov v kostnej dreni, čo môže následne uvoľniť cytokíny. Morfologické zmeny v periférnych krvných bunkách môžu naznačovať zvýšený retikulín, ktorý sa dá
zistiť biopsiou kostnej drene. Preto sa pred liečbou romiplostímom a počas nej odporúčajú vyšetrenia bunkových morfologických anomálií použitím periférneho krvného náteru a kompletného krvného
obrazu (CBC). Informácie o nárastoch retikulínu pozorovaných v klinických štúdiách s romiplostímom sú uvedené v časti 4.8.
Ak sa u pacientov zistí nedostatok účinnosti a anomálie periférneho krvného náteru, podávanie romiplostímu sa má ukončiť, treba vykonať lekársku prehliadku a zvážiť biopsiu kostnej drene
s vhodným ofarbením retikulínu. Ak je dostupný výsledok predchádzajúcej biopsie kostnej drene, je vhodné urobiť porovnanie. Ak je účinnosť zachovaná a zároveň sú u pacientov pozorované anomálie
periférneho krvného náteru, lekár má urobiť príslušné klinické posúdenie vrátane zváženia biopsie kostnej drene a znovu sa má prehodnotiť pomer prínosu a rizika pre romiplostím a pre alternatívne možnosti liečby ITP.
Trombotické/tromboembolické komplikácie
Počet krvných doštičiek nad normálne rozmedzie predstavuje riziko
trombotických/tromboembolických komplikácií. Výskyt trombotických/tromboembolických udalostí pozorovaných v klinických štúdiách bol 6,0 % pri romiplostíme a 3,6 % pri placebe. Opatrnosť je
potrebná pri podávaní romiplostímu pacientom so známymi rizikovými faktormi pre tromboembóliu
vrátane vrodených (napr. faktor V Leiden) alebo získaných rizikových faktorov (napr. deficit ATIII, antifosfolipidový syndróm), pokročilého veku, pacientov s dlhodobými obdobiami imobilizácie, malignít, kontraceptív a hormonálnej substitučnej liečby, chirurgického zákroku/traumy, obezity
a fajčenia, ale aj iných faktorov.
U pacientov s chronickým ochorením pečene užívajúcich romiplostím sa zaznamenali prípady tromboembolických príhod (TEE) vrátane trombózy portálnej vény. Romiplostím sa má používať opatrne u týchto skupín pacientov. Je potrebné riadiť sa pokynmi na úpravu dávky (pozri časť 4.2).
Chyby vpredpisovaníliečby
U pacientov liečených liekom Nplate sa zaznamenali chyby v predpisovaní liečby vrátane
predávkovania a poddávkovania, je potrebné postupovať podľa pokynov na výpočet dávky a úpravu dávky (pozri časť 4.2).
Predávkovanie môže viesť k nadmernému zvýšeniu počtu krvných doštičiek súvisiacemu
s trombotickými/tromboembolickými komplikáciami. Ak sa počet krvných doštičiek nadmerne zvýšil, vysaďte Nplate a sledujte počet krvných doštičiek. Liečbu liekom Nplate nasaďte znova v súlade
s odporúčaniami na dávkovanie a podanie. Poddávkovanie môže mať za následok nižší počet krvných
doštičiek, ako sa očakáva, a možnosť výskytu krvácania. U pacientov dostávajúcich Nplate sa má sledovať počet krvných doštičiek (pozri časti 4.2, 4.4 a 4.9).
Progresia existujúceho myelodysplastického syndrómu(MDS)
Pozitívny pomer prínosu a rizika pre romiplostím je stanovený len na liečbu trombocytopénie spojenej
s chronickou ITP a romiplostím nesmie byť použitý pri iných klinických stavoch spojených s trombocytopéniou.
Diagnóza ITP u dospelých a starších pacientov sa má potvrdiť vylúčením iných klinických príčin výskytu trombocytopénie, zvlášť diagnóza MDS musí byť vylúčená. Aspiráciu a biopsiu kostnej drene je zvyčajne potrebné urobiť v priebehu ochorenia a liečby, hlavne u pacientov vo veku nad 60 rokov,
u tých, ktorí majú systémové príznaky alebo abnormálne prejavy, ako je zvýšený počet nezrelých buniek v periférii.
V klinických skúšaniach, ktoré skúmali liečbu romiplostímom u pacientov s MDS, sa pozorovali prípady prechodných nárastov nezrelých buniek a zaznamenali sa prípady progresie MDS do akútnej myeloidnej leukémie (AML). V randomizovanom, placebom kontrolovanom klinickom skúšaní
u pacientov s MDS bola liečba romiplostímom predčasne ukončená z dôvodu numerickej prevahy progresie ochorenia do AML a zvýšenia počtu cirkulujúcich nezrelých buniek o viac ako 10 %
u pacientov liečených romiplostímom. Z pozorovaných prípadov progresie MDS do AML boli
pacienti s RAEB-1 podtypom MDS na začiatku náchylnejší na výskyt progresie ochorenia do AML
v porovnaní s nižším rizikom MDS.
Romiplostím sa nesmie používať na liečbu trombocytopénie spôsobenej MDS ani trombocytopénie zapríčinenej iným spôsobom ako je ITP mimo klinických štúdií.
Strata odpovede na romiplostím
Pri strate odpovede alebo neschopnosti udržať odpoveď krvných doštičiek na liečbu romiplostímom
v rámci odporúčaného dávkovania sa majú hľadať príčinné faktory vrátane imunogenicity (pozri časť 4.8) a zvýšeného retikulínu kostnej drene (pozri časť vyššie).
Účinky romiplostímunačervenéabielekrvinky
Zmeny parametrov červených (zníženie) a bielych (zvýšenie) krviniek boli pozorované v
predklinických toxikologických štúdiách (potkan a opica), ako aj u ITP pacientov. U pacientov sa bez ohľadu na status splenektómie môžu súbežne vyskytnúť anémia a leukocytóza (v 4-týždňovom časovom rozpätí), častejšie sa však pozorovali u pacientov po splenektómii. Monitorovanie týchto parametrov sa má zvážiť u pacientov liečených romiplostímom.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Možné interakcie romiplostímu so súčasne podávanými liekmi z dôvodu väzby na plazmatické bielkoviny zostávajú neznáme.
Lieky používané na liečbu ITP v kombinácii s romiplostímom v klinických skúšaniach zahŕňali kortikosteroidy, danazol a/alebo azatioprin, intravenózny imunoglobulín (IVIG) a anti-D imunoglobulín. Pri kombinovaní romiplostímu s inými liekmi na liečbu ITP sa má sledovať počet krvných doštičiek, aby počty krvných doštičiek neprekročili odporúčané rozmedzie (pozri časť 4.2).
Používanie kortikosteroidov, danazolu a azatioprinu sa môže znížiť alebo ukončiť, ak sa podávajú
v kombinácii s romiplostímom (pozri časť 5.1). Pri znižovaní alebo prerušení iných terapií ITP sa má sledovať počet krvných doštičiek, aby počty krvných doštičiek neklesli pod odporúčané rozmedzie
(pozri časť 4.2).
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití romiplostímu u gravidných
žien.
Štúdie na zvieratách preukázali, že romiplostím prechádzal placentou a zvyšoval počet krvných doštičiek plodov. V štúdiách na zvieratách sa vyskytovali aj poimplantačná strata a mierne zvýšenie výskytu perinatálnej mortality mláďat (pozri časť 5.3).
Romiplostím sa neodporúča užívať počas gravidity a u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
Dojčenie
Nie je známe, či sa romiplostím a metabolity vylučujú do materského mlieka. Riziko u novorodencov
a dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu romiplostímom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
O fertilite nie sú k dispozícii žiadne údaje.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Nplate má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. V klinických štúdiách sa u niektorých pacientov vyskytli mierne až stredne závažné prechodné závraty.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Na základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4
kontrolovaných a 5 nekontrolovaných klinických skúšaniach, bol celkový výskyt všetkých nežiaducich reakcií u pacientov liečených romiplostímom 91,5 % (248/271). Priemerné trvanie
expozície romiplostímu u tejto skúmanej populácie bolo 50 týždňov.
Najzávažnejšie nežiaduce reakcie, ktoré sa môžu objaviť v priebehu liečby liekom Nplate, sú: opätovný výskyt trombocytopénie a krvácania po vysadení liečby, zvýšenie retikulínu kostnej drene, trombotické/tromboembolické komplikácie, chyby pri predpisovaní liekov a progresia existujúceho MDS do AML. Najčastejšími pozorovanými nežiaducimi reakciami sú hypersenzitívne reakcie (zahŕňajúce prípady vyrážky, urtikárie a angioedému) a bolesť hlavy.
Zoznam nežiaducich reakcií zoradených dotabuľky
Frekvencie sú definované takto: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10) a menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (častosť sa nedá odhadnúť z dostupných údajov). V rámci jednotlivých tried orgánových systémov podľa MedDRA a skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúceho výskytu.
T
rieda orgánových systémov podľa MedDRA
V
eľmi časté Časté Menej časté
Infekcie a nákazy infekcia horných dýchacích ciest
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
gastroenteritída chrípka,
lokalizovaná infekcia, nazofaryngitída mnohonásobný myelóm, myelofibróza
Poruchy krvi
a lymfatického systému
Poruchy imunitného systému
Poruchy metabolizmu a výživy
porucha kostnej drene*, trombocytopénia*, anémia
hypersenzitivita** angioedém
aplastická anémia, zlyhanie kostnej drene, leukocytóza, splenomegália, trombocytémia, zvýšený počet krvných doštičiek,
abnormálny počet krvných doštičiek
intolerancia alkoholu, anorexia,
znížená chuť do jedla, dehydratácia,
dna
Psychické poruchy nespavosť depresia, abnormálne sny
Poruchy nervového systému
bolesť hlavy závrat, migréna, parestézia
klonus, dysgeúzia, hypestézia, hypogeúzia,
periférna neuropatia, trombóza priečnych splavov
Poruchy oka konjunktiválna hemorágia, porucha akomodácie, slepota,
porucha oka, očný pruritus,
zvýšená lakrimácia,
papiloedém, poruchy videnia,
Poruchy ucha a labyrintu vertigo
Poruchy srdca a srdcovej činnosti
palpitácie infarkt myokardu, zvýšená frekvencia srdca
Poruchy ciev sčervenanie hlboká žilová trombóza, hypotenzia,
periférna embólia, periférna ischémia,
flebitída,
povrchová tromboflebitída, trombóza,
erytromelalgia
T
rieda orgánových systémov podľa MedDRA
Poruchy dýchacej sústavy, hrudníka a mediastína
Veľmi časté Časté Menej časté
pľúcna embólia* kašeľ, rinorea,
suchosť v hrdle,
dyspnoe, kongescia nosa, bolestivé dýchanie

Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest Poruchy kože
a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy obličiek a močových ciest Poruchy reprodukčného systému a prsníkov
nauzea, hnačka,
bolesť brucha, zápcha,
dyspepsia
pruritus, ekchymóza, vyrážka
artralgia, myalgia, svalové kŕče,
bolesť končatín, bolesť chrbta, bolesť v kostiach
vracanie,
rektálna hemorágia, zápach z úst,
dysfágia,
gastro-ezofageálna refluxná choroba, hematochézia, hemorágia z úst, žalúdočný dyskomfort, stomatitída,
zmena sfarbenia zubov
trombóza portálnej vény, zvýšenie transaminázy alopécia,
fotosenzitívna reakcia,
akné,
kontaktná dermatitída, suchá koža
ekzém, erytém,
exfoliatívna vyrážka, abnormálny rast vlasov, prurigo,
purpura,
papulózna vyrážka, pruritická vyrážka,
kožný nodulus,
abnormálny zápach kože, urtikária
napnuté svalstvo, svalová slabosť, bolesť ramena, svalové zášklby
prítomnosť bielkovín v moči
vaginálna hemorágia
T
rieda orgánových systémov podľa MedDRA
V
eľmi časté Časté Menej časté
Celkové poruchy
a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
* pozri časť 4.4
únava,
periférny edém, ochorenie podobné chrípke,
bolesť, asténia,
pyrexia,
zimnica,
reakcia v mieste vpichu
kontúzia
hemorágia v mieste vpichu, bolesť na hrudi, podráždenosť,
nevoľnosť, edém tváre, pocit horúčavy, pocit nervozity
zvýšený krvný tlak, zvýšená laktátdehydrogenáza v krvi,
zvýšená telesná teplota, znížená hmotnosť,
zvýšená hmotnosť

** Hypersenzitívne reakcie vrátane prípadov vyrážky, urtikárie a angioedému
Opis vybraných nežiaducichreakciíOkrem toho sa za reakcie súvisiace s liečbou považovali aj udalosti uvedené nižšie.
TrombocytózaNa základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4 kontrolovaných a 5 nekontrolovaných klinických skúšaniach, boli hlásené 3 prípady trombocytózy, n = 271. V súvislosti so zvýšeným počtom krvných doštičiek u ktoréhokoľvek z týchto 3 pacientov neboli hlásené žiadne klinické následky.
Trombocytopénia po skončení liečbyNa základe analýzy všetkých dospelých pacientov s ITP, ktorí dostávali romiplostím v 4 kontrolovaných a 5 nekontrolovaných klinických skúšaniach, boli hlásené 4 prípady trombocytopénie po skončení liečby, n = 271 (pozri časť 4.4).
Progresia existujúceho myelodysplastického syndrómu (MDS)V randomizovanom, placebom kontrolovanom klinickom skúšaní u pacientov s MDS bola liečba romiplostímom predčasne ukončená z dôvodu numerického zvýšenia progresie MDS do AML
a prechodného zvýšenia počtu nezrelých buniek u pacientov liečených romiplostímom v porovnaní s placebom. Z pozorovaných prípadov progresie MDS do AML boli pacienti s RAEB-1 podtypom MDS na začiatku náchylnejší na výskyt progresie ochorenia do AML (pozri časť 4.4). Celkové
prežívanie bolo podobné placebu.
Zvýšenie retikulínu kostnej dreneV klinických skúšaniach bola liečba romiplostímom vysadená u 4 z 271 pacientov z dôvodu hromadenia retikulínu v kostnej dreni. U 6 ďalších pacientov sa retikulín pozoroval po biopsii kostnej drene (pozri časť 4.4).
I
m
unogenicita
V klinických skúšaniach sa u dospelých pacientov s ITP zisťovali protilátky proti romiplostímu. Kým 5,8 % osôb bolo pozitívnych na tvorbu viažucich protilátok proti romiplostímu a 3,9 % na
protilátky proti TPO, len 2 osoby (0,4 %) boli pozitívne na neutralizujúce protilátky proti romiplostímu, tieto protilátky však nereagovali skrížene s endogénnym TPO. Obe osoby mali
negatívny test neutralizujúcich protilátok proti romiplostímu 4 mesiace po skončení dávkovania.
Incidencia preexistujúcich protilátok proti romiplostímu bola 8,0 % a protilátok proti TPO 5,4 %.
Rovnako ako pri všetkých terapeutických proteínoch existuje možnosť imunogenicity. Ak existuje podozrenie na tvorbu neutralizujúcich protilátok, kontaktujte miestneho zástupcu Držiteľa rozhodnutia o registrácii (pozri časť 6 písomnej informácie pre používateľa) kvôli vyšetreniu protilátok.'
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovaniePri potkanoch, ktorým sa podávala jednorazová dávka 1 000 µg/kg (100-násobok maximálnej
klinickej dávky 10 µg/kg), ani pri opiciach po opakovanom podávaní romiplostímu v dávke 500 µg/kg
(50-násobok maximálnej klinickej dávky 10 µg/kg) sa nepozorovali žiadne nežiaduce účinky.
V prípade predávkovania sa môžu počty krvných doštičiek nadmerne zvýšiť a môžu mať za následok trombotické/tromboembolické komplikácie. Ak sa počet krvných doštičiek nadmerne zvýši, ukončite podávanie Nplate a sledujte počty krvných doštičiek. Liečbu liekom Nplate začnite znova podľa odporúčaní pre dávkovanie a podávanie (pozri časti 4.2 a 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antihemoragiká, iné systémové hemostatiká, ATC kód: B02BX04
Mechanizmus účinkuRomiplostím je Fc-peptidový fúzny proteín (peptilátka), ktorý signalizuje a aktivuje intracelulárne
transkripčné dráhy prostredníctvom TPO receptora (známeho aj ako cMpl) na zvýšenie tvorby krvných doštičiek. Peptilátková molekula sa skladá z Fc domény ľudského imunoglobulínu IgG1,
ktorej každá jednoreťazcová podjednotka sa kovalentne viaže C-terminálom na peptidový reťazec
obsahujúci 2 domény, ktoré sa viažu na TPO receptor.
Romiplostím nemá žiadnu sekvenciu aminokyselín homológnu s endogénnym TPO. V predklinických a klinických skúšaniach žiadne anti-romiplostímové protilátky nereagovali skrížene s endogénnym TPO.
Klinická účinnosť abezpečnosťBezpečnosť a účinnosť romiplostímu bola hodnotená v liečbe trvajúcej až 3 roky. V klinických
skúšaniach viedla liečba romiplostímom k zvýšeniu počtu krvných doštičiek, ktoré bolo závislé od
dávky. Čas do dosiahnutia maximálneho účinku na počet krvných doštičiek je približne 10 – 14 dní a nezávisí od dávky. Po jednorazovej subkutánnej dávke 1 až 10 µg/kg romiplostímu podanej pacientom s ITP bol maximálny počet krvných doštičiek 1,3- až 14,9-krát vyšší ako pôvodný počet krvných doštičiek v priebehu 2 až 3 týždňov a medzi pacientmi bola odpoveď rôzna. Počty krvných doštičiek u pacientov s ITP, ktorí dostali 6 dávok 1 alebo 3 µg/kg romiplostímu raz týždenne, sa pohybovali v rozsahu 50 až 450 x 109/l u väčšiny pacientov. Z 271 pacientov, ktorí dostali romiplostím v klinických skúšaniach s ITP, bolo 55 (20 %) vo veku 65 rokov a viac a 27 (10 %) vo veku 75 a viac. Medzi staršími a mladšími pacientmi sa v placebom kontrolovaných štúdiách nezistili žiadne celkové rozdiely v bezpečnosti ani účinnosti.
Výsledky z hlavných placebom kontrolovaných štúdií
Bezpečnosť a účinnosť romiplostímu bola hodnotená v dvoch placebom kontrolovaných, dvojito zaslepených štúdiách u dospelých s ITP, ktorí pred vstupom do štúdie absolvovali aspoň jednu liečbu a sú predstaviteľmi celého spektra takýchto pacientov s ITP.
Štúdia S1 (212) hodnotila pacientov, ktorí neabsolvovali splenektómiu a nedostatočne odpovedali alebo neznášali predchádzajúce spôsoby liečby. Pri vstupe do štúdie mali pacienti diagnostikovanú ITP približne 2 roky. Pacienti mali pred vstupom do štúdie medián 3 terapií ITP (rozsah 1 až 7). Predchádzajúce spôsoby liečby zahŕňali kortikosteroidy (90 % všetkých pacientov), imunoglobulíny (76 %), rituximab (29 %), cytotoxické liečby (21 %), danazol (11 %) a azatioprin (5 %). Pri vstupe do štúdie mali pacienti medián počtu krvných doštičiek 19 x 109/l.
Štúdia S2 (105) hodnotila pacientov, ktorí absolvovali splenektómiu a mali aj naďalej trombocytopéniu. Pri vstupe do štúdie mali pacienti diagnostikovanú ITP približne 8 rokov. Okrem splenektómie mali pacienti pred vstupom do štúdie medián 6 terapií ITP (rozsah 3 až 10). Predchádzajúce spôsoby liečby zahŕňali kortikosteroidy (98 % všetkých pacientov), imunoglobulíny (97 %), rituximab (71 %), danazol (37 %), cytotoxické liečby (68 %) a azatioprin (24 %). Pri vstupe do štúdie mali pacienti medián počtu krvných doštičiek 14 x 109/l.
Obidve štúdie mali rovnaký dizajn. Pacienti (≥ 18 rokov) boli randomizovaní v pomere 2 : 1, aby dostávali počiatočnú dávku romiplostímu 1 µg/kg alebo placebo. Pacienti dostávali subkutánne injekcie jedenkrát za týždeň počas 24 týždňov. Dávky boli upravené tak, aby sa udržali počty krvných doštičiek (50 až 200 x 109/l). V oboch štúdiách sa dokázala účinnosť zvýšením podielu pacientov,
ktorí dosiahli trvalú odpoveď krvných doštičiek. Medián priemernej týždennej dávky pre pacientov po splenektómii bol 3 µg/kg a pre pacientov, ktorí nepodstúpili splenektómiu, bol 2 µg/kg.
Významne vyšší podiel pacientov, ktorí dostávali romiplostím, dosiahol trvalú odpoveď krvných doštičiek v porovnaní s pacientmi, ktorí dostávali placebo v obidvoch štúdiách. Po prvých štyroch týždňoch štúdie romiplostím udržoval počty krvných doštičiek ≥ 50 x 109/l u 50 % až 70 % pacientov počas 6 mesiacov liečby v placebom kontrolovaných štúdiách. V placebovej skupine bolo 0 % až 7 % pacientov schopných dosiahnuť odpoveď krvných doštičiek počas 6 mesiacov liečby. Súhrn kľúčových cieľov účinnosti je uvedený nižšie.
Súhrn kľúčových výsledkov účinnosti z placebom kontrolovaných štúdií
Štúdia 1
pacienti bez splenektómie
Štúdia 2
pacienti po splenektómii
K
ombinované
štúdie 1 & 2
P
očet (%)
pacientov
romiplostím
(n = 41)
placebo
(n = 21)
romiplostím
(n = 42)
placebo
(n = 21)
romiplostím
(n = 83)
placebo
(n = 42)
s trvalou odpoveďou krvných doštičiek
a
25 (61 %) 1 (5 %) 16 (38 %) 0 (0 %) 41 (50 %) 1 (2 %)
(95 % CI) (45 %,
76 %)
(0 %,
24 %) (24 %, 54 %)
(0 %,
16 %) (38 %, 61 %)
(0 %,
13 %)
p-hodnota < 0,0001 0,0013 < 0,0001
Počet (%)
pacientov
s celkovou odpoveďou krvných doštičiek
b
36 (88 %) 3 (14 %) 33 (79 %) 0 (0 %) 69 (83 %) 3 (7 %)
(95 % CI) (74 %,
96 %)
(3 %,
36 %) (63 %, 90 %)
(0 %,
16 %) (73 %, 91 %)
(2 %,
20 %)
p-hodnota < 0,0001 < 0,0001 < 0,0001
Priemerný počet
t
ýždňov
s odpoveďou krvných doštičiek
c
15 1 12 0 14 1
(SD) 3,5 7,5 7,9 0,5 7,8 2,5
p-hodnota < 0,0001 < 0,0001 < 0,0001
Počet (%)
pacientov,
ktorí potrebovali záchrannú liečbu
d
8 (20 %) 13 (62 %) 11 (26 %) 12 (57 %) 19 (23 %) 25 (60 %)
(95 % CI) (9 %, 35 %) (38 %,
82 %)
(14 %, 42 %) (34 %,
78 %)
(14 %, 33 %) (43 %,
74 %)
p-hodnota 0,001 0,0175 < 0,0001
Počet (%) pacientov s trvalou
odpoveďou krvných
doštičiek pri stabilnej dávke
e
21 (51 %) 0 (0 %) 13 (31 %) 0 (0 %) 34 (41 %) 0 (0 %)
(95 % CI) (35 %,
67 %)
(0 %,
16 %) (18 %, 47 %)
(0 %,
16 %) (30 %, 52 %) (0 %, 8 %)
p-hodnota 0,0001 0,0046 < 0,0001
a Trvalá odpoveď krvných doštičiek je definovaná ako týždenný počet krvných doštičiek ≥ 50 x 109/l 6- alebo viackrát počas 18 – 25 týždňov štúdie, pričom záchranná liečba nie je nikdy potrebná počas
liečby.
Štúdia 1 pacienti bez splenektómie
Štúdia 2
pacienti po splenektómii
Kombinované štúdie 1 & 2

b Celková odpoveď krvných doštičiek je definovaná ako dosiahnutie trvalých alebo prechodných odpovedí krvných doštičiek. Prechodná odpoveď krvných doštičiek bola definovaná ako týždenný
počet krvných doštičiek ≥ 50 x 109/l 4- alebo viackrát počas 2 – 25 týždňov štúdie bez trvalej odpovede krvných doštičiek. Pacient nemusí mať týždennú odpoveď počas 8 týždňov po podaní
akýchkoľvek záchranných liekov.
c Počet týždňov s odpoveďou krvných doštičiek je definovaný ako počet týždňov s počtami krvných doštičiek ≥ 50 x 109/l počas 2 – 25 týždňoch štúdie. Pacient nemusí mať týždennú odpoveď počas
8 týždňov po podaní akýchkoľvek záchranných liekov.
d Záchranná liečba je definovaná ako akákoľvek liečba podaná na zvýšenie počtov krvných doštičiek. Pacienti, ktorí potrebovali záchranné lieky, sa nepovažovali za pacientov s trvalou odpoveďou
krvných doštičiek. Záchranné liečby povolené v štúdii boli IVIG, transfúzie krvných doštičiek, anti-D
imunoglobulíny a kortikosteroidy.
e Stabilná dávka je definovaná ako dávka udržiavaná ±1 µg/kg počas posledných 8 týždňov liečby.
Výsledky štúdií v porovnaní so štandardnou starostlivosťou (ŠS) u pacientov bez splenektómieŠtúdia S3 (131) bola otvorené randomizované 52-týždňové klinické skúšanie s pacientmi, ktorí dostávali romiplostím alebo liečbu v rámci štandardnej starostlivosti (ŠS). V tejto štúdii sa hodnotili pacienti bez splenektómie s ITP a počtom krvných doštičiek < 50 x 109/l. Romiplostím bol podávaný
157 pacientom formou subkutánnej (s.c.) injekcie raz týždenne začnúc s dávkou 3 µg/kg, ktorá bola počas štúdie upravovaná v rozmedzí 1 – 10 µg/kg, aby sa počet krvných doštičiek udržal medzi 50
a 200 x 109/l. 77 pacientov dostávalo liečbu ŠS podľa štandardnej praxe zdravotníckeho zariadenia alebo podľa terapeutických odporúčaní.
Celková miera incidencie splenektómie bola 8,9 % (14 zo 157 pacientov) v skupine s romiplostímom v porovnaní s 36,4 % (28 zo 77 pacientov) v skupine so ŠS, pričom pomer pravdepodobnosti (odds ratio, OR) (romiplostím vs ŠS) bol 0,17 (95 % CI: 0,08; 0,35).
Celkový výskyt zlyhania liečby predstavoval 11,5 % (18 zo 157 pacientov) v skupine
s romiplostímom v porovnaní s 29,9 % (23 zo 77 pacientov) v skupine so ŠS, pričom pomer pravdepodobnosti (romiplostím vs ŠS) bol 0,31 (95 % CI: 0,15; 0,61).
Zo 157 pacientov randomizovaných do skupiny s romiplostímom traja pacienti nedostávali romiplostím. Medzi 154 pacientmi, ktorí dostávali romiplostím, bol medián celkovej expozície romiplostímu 52,0 týždňa a pohyboval sa od 2 do 53 týždňov. Najčastejšie užitá týždenná dávka bola medzi 3 – 5 µg/kg (t. j. 25. – 75. percentil; medián 3 µg/kg).
Zo 77 pacientov randomizovaných do skupiny so ŠS dvaja pacienti nedostávali žiadnu liečbu v rámci ŠS. Medzi 75 pacientmi, ktorí dostali najmenej jednu dávku liečby v rámci ŠS, bol medián celkovej expozície ŠS 51 týždňov a pohyboval sa od 0,4 do 52 týždňov.
Redukcia povolených súbežných spôsobov liečby ITPV obidvoch placebom kontrolovaných, dvojito zaslepených štúdiách mohli pacienti, ktorí už dostávali liečbu ITP v konštantnej dávke, pokračovať v tejto liečbe počas štúdie (kortikosteroidy, danazol a/alebo azatioprin). Na začiatku štúdie dostávalo 21 pacientov bez splenektómie a 18 pacientov po splenektómii predchádzajúcu ITP liečbu (najmä kortikosteroidy). Všetci (100 %) pacienti po splenektómii, ktorí dostávali romiplostím, boli schopní znížiť dávku o viac ako 25 % alebo vysadiť súbežnú ITP liečbu do konca liečby v porovnaní so 17 % placebom liečených pacientov. 73 % pacientov bez splenektómie, ktorí dostávali romiplostím, boli schopní znížiť dávku o viac ako 25 % alebo vysadiť súbežnú ITP liečbu do konca štúdie v porovnaní s 50 % placebom liečených pacientov (pozri časť 4.5).
K
rvácavé príhody
V celom klinickom ITP programe bol pozorovaný inverzný vzťah medzi krvácavými príhodami
a počtami krvných doštičiek. Všetky klinicky významné (≥ 3. stupeň) krvácavé príhody sa vyskytovali pri počtoch krvných doštičiek < 30 x 109/l. Všetky krvácavé príhody ≥ 2. stupeň sa vyskytovali pri počtoch krvných doštičiek < 50 x 109/l. Medzi skupinou pacientov liečenou liekom Nplate a skupinou liečenou placebom sa nezistili žiadne štatisticky významné rozdiely v celkovom výskyte krvácavých príhod.
V dvoch placebom kontrolovaných štúdiách 9 pacientov hlásilo krvácavú príhodu, ktorá sa považovala za závažnú (5 [6,0 %] romiplostím, 4 [9,8 %] placebo; pomer pravdepodobnosti
[romiplostím/placebo] = 0,59; 95 % CI = (0,15; 2,31)). Krvácavé príhody 2. alebo vyššieho stupňa boli hlásené u 15 % pacientov liečených romiplostímom a 34 % pacientov liečených placebom (pomer
pravdepodobnosti [romiplostím/placebo] = 0,35; 95 % CI = (0,14; 0,85)).
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s liekom Nplate
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe imunitnej trombocytopénie
(idiopatická trombocytopenická purpura) (informácie o použití v pediatrickej populácii, pozri časť
4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika romiplostímu zahŕňala elimináciu na cieľových štruktúrach, ktorá je pravdepodobne sprostredkovaná TPO receptormi na krvných doštičkách a iných bunkách trombopoetického radu, ako sú megakaryocyty.
Absorpcia
Po subkutánnom podaní 3 až 15 µg/kg romiplostímu sa dosiahli maximálne sérové hladiny
romiplostímu u ITP pacientov po 7 – 50 hodinách (medián 14 hodín). Sérové koncentrácie sa medzi pacientmi odlišovali a nekorelovali s podanou dávkou. Zdá sa, že sérové hladiny romiplostímu sú
v inverznom vzťahu s počtami krvných doštičiek.
Distribúcia
Distribučný objem romiplostímu po intravenóznom podaní romiplostímu klesal nelineárne zo 122;
78,8 na 48,2 ml/kg pre intravenózne dávky 0,3; 1,0 a 10 µg/kg u zdravých osôb. Tento nelineárny pokles distribučného objemu je v súlade s cieľovou väzbou romiplostímu (na megakaryocyty a krvné doštičky), ktorá sa môže saturovať pri podávaní vysokých dávok.
Eliminácia
Polčas eliminácie romiplostímu u ITP pacientov sa pohyboval od 1 do 34 dní (medián 3,5 dňa).
Eliminácia sérového romiplostímu čiastočne závisí od TPO receptora krvných doštičiek. Ako následok podanej dávky sa u pacientov s vysokým počtom krvných doštičiek pozorovali nízke sérové koncentrácie a naopak. V inom ITP klinickom skúšaní sa nepozorovala žiadna kumulácia sérových koncentrácií po 6 dávkach romiplostímu raz týždenne (3 µg/kg).
Osobitné populácie
Farmakokinetika romiplostímu u pacientov s poruchou funkcie obličiek a pečene sa neskúmala. Zdá
sa, že farmakokinetika romiplostinu nie je klinicky signifikantne ovplyvnená vekom, hmotnosťou ani pohlavím.
5.3 Predklinické údaje o bezpečnosti
Uskutočnili sa toxikologické štúdie s viacnásobnou dávkou romiplostímu na potkanoch trvajúce
4 týždne a na opiciach trvajúce až 6 mesiacov. Vo všeobecnosti sa účinky pozorované počas týchto štúdií týkali trombopoetického účinku romiplostímu a boli podobné bez ohľadu na trvanie štúdie. Reakcie v mieste podania sa tiež týkali podania romiplostímu. Pri všetkých testovaných dávkach sa pozorovala myelofibróza v kostnej dreni potkanov. V týchto štúdiách sa myelofibróza nepozorovala pri zvieratách 4 týždne po skončení liečby, čo naznačuje reverzibilnosť.
V jednomesačných toxikologických štúdiách na potkanoch a opiciach sa pozorovalo mierne zníženie počtu červených krviniek, hematokritu a hemoglobínu. Pozoroval sa aj stimulačný účinok na tvorbu leukocytov, pretože počty neutrofilov, lymfocytov, monocytov a eozinofilov v periférnej krvi boli mierne zvýšené. V dlhšie trvajúcej chronickej štúdii s opicami sa nezistil žiadny účinok na erytroidné a leukocytové rady pri podávaní romiplostímu počas 6 mesiacov, kde sa podávanie romiplostímu znížilo z trikrát týždenne na jedenkrát týždenne. Okrem toho v hlavných štúdiách fázy 3 romiplostím neovplyvňoval rady červených a bielych krviniek v porovnaní so subjektmi liečenými placebom.
Z dôvodu tvorby neutralizujúcich protilátok sa farmakodynamické účinky romiplostímu pri potkanoch často znižovali pri dlhodobom podávaní. Toxikokinetické štúdie nepreukázali žiadne interakcie protilátok s meranými koncentráciami. Hoci boli vysoké dávky testované v štúdiách na zvieratách, nemožno spoľahlivo určiť hranice bezpečnosti z dôvodu rozdielov medzi laboratórnymi zvieratami
a ľuďmi, čo sa týka citlivosti na farmakodynamický účinok romiplostímu a účinok neutralizujúcich protilátok.
Karcinogenéza
Karcinogénny potenciál romiplostímu sa nehodnotil. Preto riziko potenciálnej karcinogenicity
romiplostímu u ľudí zostáva neznáme.
Reprodukčná toxikológia
Vo všetkých vývojových štúdiách sa tvorili neutralizujúce protilátky, ktoré môžu inhibovať účinky
romiplostímu. V štúdiách embryofetálneho vývoja myší a potkanov boli pozorované zníženia telesnej hmotnosti matky iba pri myšiach. Pri myšiach sa dokázala zvýšená post-implantačná strata. V štúdii
prenatálneho a postnatálneho vývoja sa pri potkanoch zistilo predĺženie trvania gestácie a mierne
zvýšenie výskytu perinatálnej mortality mláďat. O romiplostíme je známe, že prechádza placentárnou bariérou potkanov, a môže sa prenášať z matky na vyvíjajúci sa plod a stimulovať tvorbu fetálnych krvných doštičiek. Nepozoroval sa žiadny účinok romiplostímu na fertilitu potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
manitol (E421)
sacharóza
L-histidín
kyselina chlorovodíková (na úpravu pH)
polysorbát 20
Rozpúšťadlo:
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi, okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky.
Po rekonštitúcii: Chemická a fyzikálna stabilita pri používaní sa stanovila na 24 hodín pri 25 °C a na
24 hodín pri 2 °C – 8 °C, ak je produkt chránený pred svetlom a uchovávaný v pôvodnej injekčnej liekovke.
Z mikrobiologického hľadiska sa liek musí použiť ihneď. Ak sa nepoužije ihneď, za čas použiteľnosti
a podmienky pred použitím je zodpovedný používateľ a nemali by byť za normálnych okolností dlhšie ako 24 hodín pri 25 °C alebo 24 hodín v chladničke (2 °C – 8 °C), chránené pred svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Môže sa dočasne vybrať z chladničky maximálne na dobu 24 hodín pri izbovej teplote (do 25 °C).
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok:
5 ml injekčná liekovka (z bezfarebného skla typu I) so zátkou (z chlorobutylovej gumy), s uzáverom
(z hliníka) a odklápacím viečkom (z polypropylénu).
Rozpúšťadlo:
Nplate 250 mikrogramov prášok a rozpúšťadlo na injekčný roztok: Naplnená injekčná striekačka
(z bezfarebného skla typu I s bromobutylovým gumovým piestom), ktorá obsahuje 0.72 ml vody na injekciu na rekonštitúciu.
Nplate 500 mikrogramov prášok a rozpúšťadlo na injekčný roztok: Naplnená injekčná striekačka
(z bezfarebného skla typu I s bromobutylovým gumovým piestom), ktorá obsahuje 1,2 ml vody na injekciu na rekonštitúciu.
Veľkosť balenia:
Nplate 250 mikrogramov prášok a rozpúšťadlo na injekčný roztok:
Nplate je dodávaný ako 1 balenie alebo multibalenie obsahujúce 4 balenia. Každé balenie obsahuje:
1 injekčnú liekovku s 250 mikrogramami romiplostímu.
1 naplnenú injekčnú striekačku s obsahom 0,72 ml vody na injekciu na rekonštitúciu.
1 piestovú tyčinku pre naplnenú injekčnú striekačku.
1 sterilný adaptér injekčnej liekovky.
1 sterilnú 1 ml Luer lock injekčnú striekačku.
1 sterilnú bezpečnostnú ihlu.
4 liehové tampóny.
Nplate 500 mikrogramov prášok a rozpúšťadlo na injekčný roztok:
Nplate je dodávaný ako 1 balenie alebo multibalenie obsahujúce 4 balenia. Každé balenie obsahuje:
1 injekčnú liekovku s 500 mikrogramami romiplostímu.
1 naplnenú injekčnú striekačku s obsahom 1,2 ml vody na injekciu na rekonštitúciu.
1 piestovú tyčinku pre naplnenú injekčnú striekačku.
1 sterilný adaptér injekčnej liekovky.
1 sterilnú 1 ml Luer lock injekčnú striekačku.
1 sterilnú bezpečnostnú ihlu.
4 liehové tampóny.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Nplate je sterilný liek, ale bez konzervačnej prísady, a je určený len na jednorazové použitie. Nplate sa má rekonštituovať podľa správnej aseptickej praxe.
Nplate 250 mikrogramovprášokarozpúšťadlonainjekčnýroztok
Nplate 250 mikrogramov prášok na injekčný roztok sa má rekonštituovať s 0,72 ml sterilnej vody na
injekcie, čím sa získa doručiteľný objem 0,5 ml. V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 250 µg romiplostímu (pozri tabuľku nižšie s obsahom injekčných liekoviek).
Nplate 500 mikrogramovprášokarozpúšťadlonainjekčnýroztok
Nplate 500 mikrogramov prášok na injekčný roztok sa má rekonštituovať s 1,2 ml sterilnej vody na
injekcie, čím sa získa doručiteľný objem 1 ml. V každej injekčnej liekovke je nadbytok prášku, aby sa zabezpečilo dodanie 500 µg romiplostímu (pozri tabuľku nižšie s obsahom injekčných liekoviek).
Obsah injekčnej liekovky:
N
plate jednorazová injekčná liekovka
C
elkový obsah injekčnej liekovky s romiplostímom
O
bjem sterilnej vody na injekciu
D
oručiteľný liek a objem
V
ýsledná koncentrácia

250 µg 375 µg + 0,72 ml = 250 µg v 0,5 ml 500 µg/ml
500 µg 625 µg + 1,2 ml = 500 µg v 1 ml 500 µg/ml
Z mikrobiologického hľadiska sa liek musí použiť ihneď. Ak sa nepoužije ihneď, za čas použiteľnosti
a podmienky pred použitím je zodpovedný používateľ a nemali by byť za normálnych okolností dlhšie ako 24 hodín pri 25 °C alebo 24 hodín v chladničke (2 °C – 8 °C), chránené pred svetlom.
1. Odstráňte plastový kryt z injekčnej liekovky s práškom Nplate a priloženým liehovým tampónom očistite gumovú zátku.
2. Odtrhnite
papierovú podložku z adaptéra injekčnejliekovky, pričom adaptér injekčnej liekovky uchovajte v obale, a nasaďte adaptér injekčnej liekovky na injekčnú liekovku Nplate
. S injekčnouliekovkou položenou na stole zatlačte adaptér injekčnej liekovky dole do stredu injekčnej liekovky, až kým nie je pevne na mieste.
Poznámka: Nedotýkajte sa hrotu adaptéra injekčnej liekovky ani Luer locku, aby nedošlo ku kontaminácii lieku.3.
Odstráňte a zlikvidujte obal adaptéra injekčnej liekovky.
4. Nasaďte piestovú tyčinku na naplnenú injekčnú striekačku s vodou na injekciu otáčaním piestovej tyčinky v smere hodinových ručičiek na pieste injekčnej striekačky, až kým nepocítite slabý odpor.
5.
V jednej ruke držte naplnenú injekčnú striekačku s vodou na injekciu a druhou rukou ohnite hrot bieleho krytuz plastu smerom nadol. Tým sa zlomí spoj bielehoplastového krytu. Po zlomení spoja stiahnite kryt, aby ste oddelili sivý gumový uzáver od priehľadného plastového hrotu na injekčnej striekačke.6.
S injekčnou liekovkou položenou na stole nasaďte naplnenú injekčnú striekačku s vodou na injekciu na adaptér injekčnej liekovky: jednou rukou podržte vonkajší okraj adaptéra injekčnej liekovky a druhou rukou otáčajte hrot injekčnej striekačky v smere hodinových ručičiek na adaptéri, pokiaľ nepocítite slabý odpor.
7.
Veľmipomalyajemnevytlačtevšetkuvodudo injekčnej liekovky s práškom. Voda má stekať pomaly na prášok. JEMNE zakrúžte injekčnou liekovkou, až kým sa nerozpustí všetok prášok a kým nie je tekutina v injekčnej liekovke číra a bezfarebná.
Injekčnouliekovkounetraste.Poznámka: Z mikrobiologického hľadiska sa liek musí použiť ihneď po rekonštitúcii. Ak sa rekonštituovaný roztok nepoužije ihneď, injekčná striekačka sa nemá vyťahovať z adaptéra injekčnej liekovky na udržanie mikrobiologickej integrity.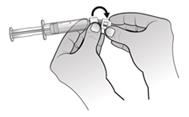
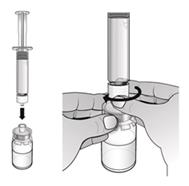 Poznámka:
Poznámka: Úplné rozpustenie prášku môže trvať až 2 minúty.
 P
r
edtým, ako budete pokračovať:
P
r
edtým, ako budete pokračovať:
Vizuálne
skontrolujte rekonštituovaný roztok, či neobsahuje častice a/alebo či nezmenil farbu. Rekonštituovaný roztok má byť číry a bezfarebný a nemá sa podávať, ak spozorujete častice a/alebo zmenu sfarbenia.
Pred odstránením injekčnej striekačky
sa uistite, že roztok je úplne rozpustený.
8. Odstráňte prázdnu naplnenú injekčnú striekačku z adaptéra
injekčnej liekovky.
9.
Z obalu vyberte 1 ml injekčnú striekačku na aplikáciu.Nasaďte 1 ml injekčnú striekačku na adaptér injekčnej liekovky
srekonštituovanýmroztokom otáčaním hrotu injekčnej striekačky na adaptéri injekčnej liekovky, až kým
nepocítite slabý odpor.
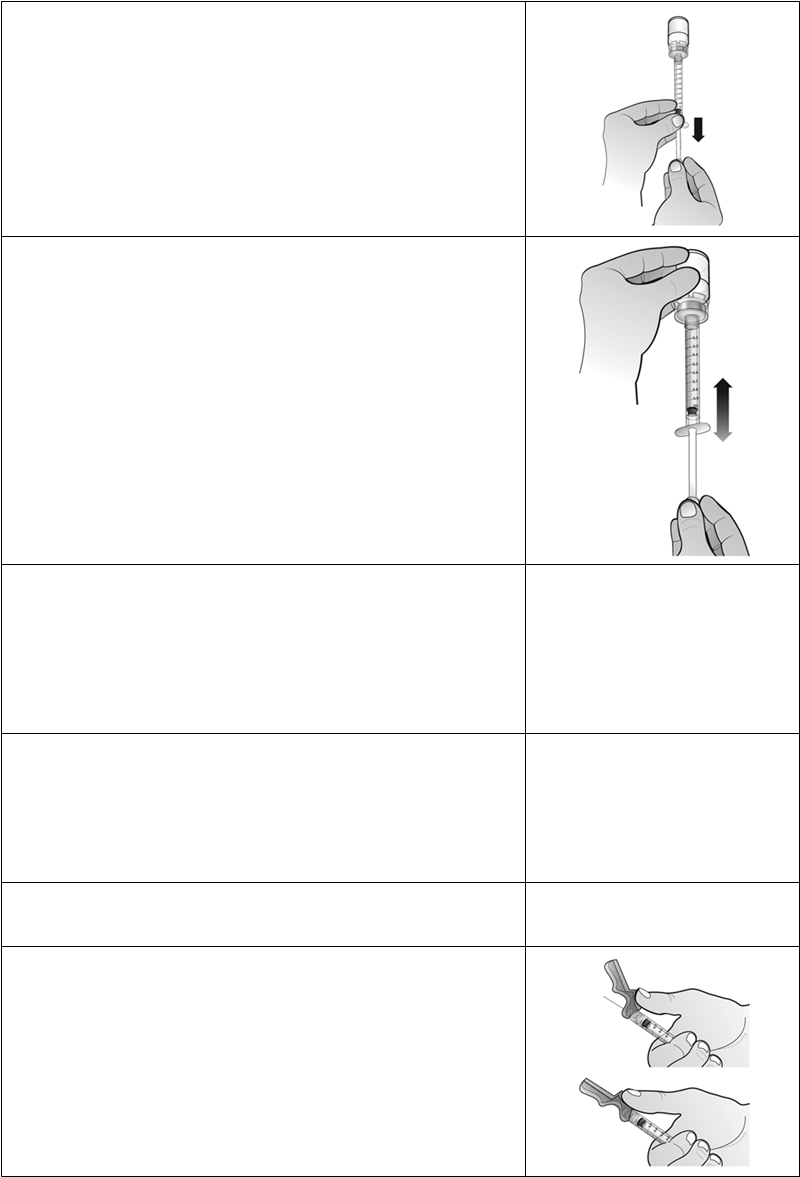
10.
Prevráťte zostavenú jednotku injekčná striekačka – injekčná liekovka hore dnom, aby bola injekčná liekovka s rekonštituovaným roztokom nad injekčnou striekačkou.
Natiahnite všetok roztok lieku do injekčnej striekačky na aplikáciu.
Uistite sa, že piest zostáva v injekčnej striekačke.
11.
Uistite sa, že v injekčnej striekačke na aplikáciu je
správnemnožstvo roztoku na dávku pre pacienta, a to tým, že nadbytočné množstvo roztoku vstreknete naspäť do injekčnej
liekovky.
Poznámka: Odstráňte všetky vzduchové bubliny z injekčnej striekačky, aby ste zaistili presné množstvo roztoku v injekčnej striekačke. 12.
Odkrúťte injekčnú striekačku na aplikáciu z adaptérainjekčnej liekovky.Nasaďte bezpečnostnú ihlu na naplnenú injekčnú striekačku na aplikáciu otáčaním ihly
v smere hodinových ručičiek do Luer lock hrotu injekčnej striekačky.
13. Pripravte miesto vpichu pomocou nového liehového tampónu.
Odklopte ružový bezpečnostný kryt smerom k injekčnej striekačke a bokom od ihly.
Odstráňte priehľadný ochranný kryt ihly z pripravenej ihly, a to tak, že v jednej ruke podržíte injekčnú striekačku a druhou rukou opatrne rovno odtiahnete ochranný kryt.
14.
Podajte subkutánnu injekciu podľa miestnych postupov a správnej aseptickej techniky
.15.
Po aplikácii aktivujte ružový bezpečnostný kryt stlačením
krytu dopredu pomocou tej istej ruky, pokiaľ nebudete počuť a/alebo cítiť kliknutie/zapadnutie.

16.
Okamžite zlikvidujte injekčnú striekačku a ihlu do schváleného kontajnera na ihly.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAmgen Europe B.V. Minervum 7061
4817 ZK Breda
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/08/497/005
EU/1/08/497/006
EU/1/08/497/007
EU/1/08/497/008
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 4. február 2009
Dátum posledného predĺženia registrácie: 20. december 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.