
ignifikantné
rozdiely medzi výsledkami testov získanými pomocou aPTT-na báze jednostupňového testu zrážavosti
a chromogénnym testom podľa Ph.Eur. To je dôležité predovšetkým pri zmene laboratória a/alebo činidiel použitých pri teste.
Dávkovanie
Dávka, interval dávkovania a trvanie substitučnej liečby závisia od závažnosti deficitu faktora VIII,
miesta a rozsahu krvácania, úrovne aktivity cieľového faktora VIII a od klinického stavu pacienta. Počet jednotiek podávaného faktora VIII je vyjadrený v medzinárodných jednotkách (IU), ktoré
zodpovedajú súčasnému WHO štandardu koncentrácie pre lieky obsahujúce faktor VIII. Aktivita faktora VIII v plazme je vyjadrená buď v percentách (porovnaním s normálnou hladinou v ľudskej
plazme), alebo v medzinárodných jednotkách na dl (porovnaním so súčasným medzinárodným štandardom pre faktor VIII v plazme).
Jedna medzinárodná jednotka (IU) aktivity faktora VIII zodpovedá množstvu faktora VIII v jednom
ml ľudskej plazmy.
L
i
ečba podľa potreby (on-demand) a liečba epizód krvácania
Výpočet požadovanej dávky faktora VIII vychádza z empirického zistenia, že 1 medzinárodná
jednotka (IU) faktora VIII na kg telesnej hmotnosti zvýši aktivitu plazmatického faktora VIII
o 2 IU/dl.
Požadovaná dávka sa určí použitím nasledujúceho vzorca:
Požadovaný počet jednotiek (IU) = telesná hmotnosť (kg) x požadovaný vzostup faktora VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Množstvo, ktoré sa má podať a frekvencia podávania sa majú vždy riadiť podľa klinickej účinnosti u jednotlivého pacienta.
Usmernenia k dávkovaniu Esperoctu pri liečbe podľa potreby a liečbe epizód krvácania sú uvedené v tabuľke 1. Úroveň aktivity faktora VIII v plazme má byť udržiavaná na opísaných úrovniach
v plazme alebo nad nimi (v IU na dl alebo % normálnej hodnoty). Na liečbu krvácania sa môže podať maximálna jednotlivá dávka Esperoctu v množstve 75 IU/kg a maximálna celková dávka
200 IU/kg/24 hodín.
Tabuľka 1 Usmernenia k liečbeepizódkrvácaniaEsperoctom
Stupeň krvácania Požadovaná hladina faktora VIII
 (I
U
/
d
l alebo % normálnej hodnoty)
(I
U
/
d
l alebo % normálnej hodnoty)a
Frekvencia dávok (hodiny)Trvanie liečby
Mierne
Začínajúca hemartróza, mierne krvácanie
do svalov alebo mierne
krvácanie do ústnej dutiny
Stredné
Rozsiahlejšia hemartróza, krvácanie do svalov, hematóm
Závažné alebo život ohrozujúce krvácanie
20-40 12-24 Do ukončenia krvácania
30-60 12-24 Do ukončenia krvácania
60-100 8-24 Do odstránenia ohrozenia
a Požadovaná dávka sa určí použitím nasledujúceho vzorca:
Požadovaný počet jednotiek (IU) = telesná hmotnosť (kg) x požadovaný vzostup faktora VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
Perioperačný manažment
Veľkosť dávky a intervaly dávkovania pri chirurgickom zákroku závisia od zákroku a od miestnych
postupov. Môže sa podať maximálna jednotlivá dávka Esperoctu 75 IU/kg a maximálna celková dávka
200 IU/kg/24 hodín.
Frekvencia dávok a trvanie liečby majú byť vždy individuálne prispôsobené podľa individuálnej klinickej odpovede.
V tabuľke 2 je uvedené všeobecne odporúčané dávkovanie Esperoctu na účely perioperačného manažmentu. Aktivitu faktora VIII je potrebné udržiavať v cieľovom rozpätí alebo nad ním.
Tabuľka 2 Usmernenie k dávkovaniu Esperoctu na účely perioperačného manažmentu
T
yp chirurgického zákroku
P
ožadovaná hladina faktora VIII (%) (IU/dl)a
Frekvencia dávok (hodiny) Trvanie liečby
Malý chirurgický zákrok
vrátane extrakcie zuba
30-60 Do jednej hodiny pred chirurgickým zákrokom
V prípade potreby zopakovať
po 24 hodinách
Jedna dávka alebo opakovaná injekcia každých 24 hodín najmenej 1 deň až do zahojenia
V
eľký chirurgický zákrok
80-100
(pred a po ope- rácii)
Do jednej hodiny
pred chirurgickým zákrokom s cieľom dosiahnuť aktivitu faktora VIII v cieľovom rozpätí
Opakovať každých 8 až
24 hodín s cieľom udržať aktivitu faktora VIII
v cieľovom rozpätí
Injekciu podľa potreby
opakovane podávať každých 8 až
24 hodín až do adekvátneho zahojenia rany
Kvôli udržaniu aktivity
faktora VIII na úrovni 30 % až
60 % (IU/dl) zvážte pokračovanie v liečbe v trvaní ďalších 7 dní
aPožadovaná dávka sa určí použitím nasledujúceho vzorca:
Požadovaný počet jednotiek (IU) = telesná hmotnosť (kg) x požadovaný vzostup faktora VIII (%) (IU/dl) x 0,5 (IU/kg na IU/dl).
ProfylaxiaOdporúčaná počiatočná dávka je 50 IU Esperoctu na kg telesnej hmotnosti každé 4 dni.
Maximálna jednorazová dávka je 75 IU/kg.
Úprava dávok a intervaly podávania sa majú zvážiť na základe dosiahnutých hladín faktora VIII
a individuálnej tendencie ku krvácaniu.
Pediatrická populáciaDávka u dospievajúcich (12 rokov a viac) je taká istá ako u dospelých. Dlhodobá bezpečnosť u detí do 12 rokov nebola stanovená.
Spôsob podávaniaEsperoct je určený na intravenózne použitie.
Esperoct sa má podávať intravenóznou injekciou (počas cca 2 minút) po rekonštitúcii prášku so 4 ml
dodaného rozpúšťadla (injekčného roztoku chloridu sodného 9 mg/ml (0,9 %)).
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Známa alergická reakcia na škrečiu bielkovinu.
4.4 Osobitné upozornenia a opatrenia pri používaníSledovateľnosťS cieľom zlepšiť sledovateľnosť biologických liekov, sa má jasne zaznamenať názov a číslo šarže
podávaného lieku.
Precitlivenosť
Pri používaní Esperoctu sa môžu vyskytnúť reakcie z precitlivenosti alergického typu. Liek obsahuje
stopy škrečích bielkovín, ktoré môžu u niektorých pacientov vyvolávať alergické reakcie. Ak sa objavia príznaky precitlivenosti, pacienti majú byť poučení, aby ihneď prerušili používanie tohto lieku a obrátili sa na svojho lekára. Pacienti majú byť informovaní o počiatočných prejavoch hypersenzitívnych reakcií vrátane žihľavky, generalizovanej urtikárie, zvierania hrudníka, sipotu, hypotenzie a anafylaxie.
V prípade šoku sa má dodržať štandardná medicínska liečba pri šoku. Inhibítory
Známou komplikáciou liečby v jednotlivcov s hemofíliou A je tvorba neutralizujúcich protilátok
(inhibítorov) proti faktoru VIII. Tieto inhibítory sú obyčajne IgG imunoglobulíny pôsobiace proti koagulačnej aktivite faktora VIII, ktorá je kvantifikovaná Bethesda jednotkami (BU) na ml plazmy pri
použití modifikovanej metódy. Riziko vzniku inhibítorov koreluje so závažnosťou ochorenia, ako aj
s expozíciou faktoru VIII. Toto riziko býva najvyššie počas prvých 50 dní expozície, ale pokračuje počas celého života, hoci nie je časté.
Klinický význam tvorby inhibítorov bude závisieť od titra inhibítora, pričom menšie riziko nedostatočnej klinickej odpovede hrozí v prípade inhibítorov nízkeho titra, než v prípade vysokého titra inhibítorov.
Všeobecne u všetkých pacientov liečených liekmi s koagulačným faktorom VIII sa má pozorne sledovať tvorba inhibítorov vhodným klinickým pozorovaním a laboratórnymi testami. Ak sa
nedosiahne očakávaná hladina aktivity faktora VIII v plazme alebo ak sa krvácanie nezvládne príslušnou dávkou, má sa robiť testovanie na prítomnosť inhibítora faktora VIII. U pacientov
s vysokou hladinou inhibítora nemusí byť liečba faktorom VIII účinná a má sa uvažovať o iných možnostiach liečby. Liečbu takýchto pacientov má viesť lekár so skúsenosťami s liečbou hemofílie a s inhibítormi faktora VIII.
Kardiovaskulárne udalosti
U pacientov s existujúcimi kardiovaskulárnymi rizikovými faktormi môže substitučná liečba
faktorom VIII zvýšiť kardiovaskulárne riziko.
Komplikácie spojené s katétrom
Ak je potrebný centrálny žilový katéter (CVAD), má sa brať do úvahy riziko komplikácií spojených
s CVAD vrátane lokálnych infekcií, bakterémie a trombózy v mieste katetrizácie.
Pediatrická populácia
Uvedené upozornenia a opatrenia platia pre dospelých a dospievajúcich (12-18 rokov).
Pomocné látky, ktorétrebavziaťdoúvahy
Tento liek obsahuje 30,5 mg sodíka na rekonštituovanú injekčnú liekovku, čo zodpovedá 1,5 % WHO
odporúčaného maximálneho denného príjmu 2,0 g sodíka pre dospelú osobu.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne interakcie liekov ľudského koagulačného faktora VIII (rDNA) s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Neuskutočnili sa žiadne reprodukčné štúdie s faktorom VIII na zvieratách. Vzhľadom na zriedkavý výskyt hemofílie A u žien, nie sú skúsenosti s použitím faktora VIII počas gravidity a dojčenia. Preto sa má faktor VIII použiť počas gravidity a laktácie, len ak je to jednoznačne indikované.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Esperoct nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Zriedkavo boli pozorované hypersenzitivita alebo alergické reakcie (ktoré môžu zahŕňať angioedém,
pálenie a štípanie v mieste podania infúzie, zimnicu, návaly horúčavy, generalizovanú urtikáriu, bolesť hlavy, žihľavku, hypotenziu, letargiu, nauzeu, nepokoj, tachykardiu, zvieranie hrudníka, brnenie, vracanie, sipot) a v niektorých prípadoch môžu viesť k závažnej anafylaxii (vrátane šoku).
Veľmi zriedkavo bola pozorovaná tvorba protilátok na škrečie bielkoviny spojená s hypersenzitívnymi reakciami.
U pacientov s hemofíliou A, ktorí sú liečení faktorom VIII vrátane Esperoctu, môžu vzniknúť neutralizačné protilátky (inhibítory). Ak sa objavia takéto inhibítory, stav sa bude prejavovať ako nedostatočná klinická odpoveď. V týchto prípadoch sa odporúča obrátiť sa na špecializované centrum pre liečbu hemofílie.
Tabuľkový zoznamnežiaducichreakcií
V tabuľke 3 sú uvedené frekvencie nežiaducich reakcií pozorované u 270 jednotlivých subjektov
v piatich prospektívnych, multicentrických klinických štúdiach na predtým liečených pacientoch
(PTP) so závažnou hemofíliou A (aktivita endogénneho faktora VIII <1 %) a bez inhibítorov v anamnéze. Kategórie nežiaducich reakcií sú v tabuľke 3 uvedené podľa klasifikácie triedy
orgánových systémov (SOC a preferované termíny početnosti podľa databázy MedDRA).
Frekvencie boli hodnotené podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000), neznáme (nedajú sa odhadnúť z dostupných údajov).
Tabuľka 3 Frekvencianežiaducichreakcií v klinickýchskúšaniachuPTP*
T
rieda orgánových systémov podľa
MedDRA
N
ežiaduce reakcie Frekvencia
Poruchy krvi a lymfatického systému Inhibícia faktora VIII Menej časté (PTP)** Poruchy imunitného systému Precitlivenosť Menej časté
Poruchy kože a podkožného tkaniva Vyrážka Erytém Svrbenie
Časté
Celkové poruchy a reakcie v mieste
podania
*PTP: predtým liečení pacienti.
Reakcie v mieste podania

injekcie***
Časté
**Frekvencia vychádza zo štúdií so všetkými liekmi s faktorom VIII, ktoré zahŕňali pacientov so závažnou hemofíliou A.
***Medzi preferované pojmy patrili reakcie v mieste podania injekcie: reakcia v mieste podania injekcie, hematóm v mieste prepichnutia cievy, reakcia v mieste podania infúzie, erytém v mieste podania injekcie, vyrážka v mieste podania injekcie, bolesť v mieste prepichnutia cievy a opuch v mieste podania injekcie.
Opisvybranýchnežiaducichreakcií
I
nhibítory faktora VIII
Jeden potvrdený prípad inhibítora faktora VIII sa vyskytol u 18-ročného predtým liečeného pacienta na profylaktickej liečbe Esperoctom. Tento pacient mal inverziu intrónu génu 22 faktora VIII a mal vysoké riziko vzniku inhibítorov faktora VIII.
Neexistujú žiadne indikácie zvýšeného rizika tvorby ihibítora faktora VIII pri liečbe Esperoctom
v porovnaní s inymi liekmi obsahujúcimi faktor VIII.
Protilátky proti liekuExistoval jeden prípad persistentných protilátok proti lieku súbežne s potvrdeným prípadom inhibítorov faktora VIII (pozri
Inhibítory faktora VIII). Traja pacienti mali prechodne pozitívne výsledky testu na protilátky proti lieku po podaní Esperoctu, ale nebola zistená žiadna spojitosť s nežiaducimi udalosťami.
Anti-PEG protilátkyTridsaťdva pacientov malo pre-existujúce anti-PEG protilátky pred podaním Esperoctu. Dvadsať z 32
pacientov bolo negatívnych na anti-PEG protilátky po podaní Esperoctu. U jedenástich pacientov prechodne vznikli anti-PEG protilátky s nízkym titrom. Nebola nájdená žiadna korelácia s nežiaducimi udalosťami.
Pediatrická populáciaMedzi predtým liečenými dospievajúcimi (12-18 rokov) a dospelými pacientmi nebol pozorovaný
žiadny rozdiel v bezpečnostnom profile.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieŽiadne príznaky predávkovania rekombinantným koagulačným faktorom VIII neboli hlásené.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká, koagulačný faktor VIII, ATC kód: B02BD02.
MechanizmusúčinkuTuroktokog alfa pegol je purifikovaný rekombinantný ľudský faktor VIII (rFVIII)
s polyetylénglykolom (PEG) s molekulovou hmotnosťou 40 kDa konjugovaným na proteín. PEG je spojený s O-viazaným glykánom v skrátenej B-doméne rFVIII (turoktokogu alfa). Podstata
mechanizmu účinku turoktokog alfa pegolu je v náhrade nedostatočného alebo neprítomného
faktora VIII u pacientov s hemofíliou A.
Pri aktivácii turoktokogu alfa pegolu trombínom na mieste poranenia dôjde k odštiepeniu B-domény s obsahom PEG zložky a regiónu a3, v dôsledku čoho dôjde k tvorbe rekombinantného faktora VIII
(rFVIIIa), ktorý má podobnú štruktúru ako prirodzený faktor VIIIa.
Komplex faktora VIII/von Willebrandovho faktora sa skladá z dvoch molekúl (faktor VIII a von
Willebrandov faktor) s rôznymi fyziologickými funkciami. Po podaní injekcie pacientovi s hemofíliou sa faktor VIII viaže na von Willebrandov faktor v krvnom obehu pacienta. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX, urýchľujúci konverziu faktora X na aktivovaný
faktor X. Aktivovaný faktor X konvertuje protrombín na trombín. Trombín potom konvertuje fibrinogén na fibrín a môže sa vytvoriť zrazenina. Hemofília A je vrodená porucha krvnej zrážavosti
viazaná na pohlavie, zapríčinená zníženou hladinou faktora VIII:C a dôsledkom je profúzne krvácanie do kĺbov, svalov alebo vnútorných orgánov, buď spontánne, alebo ako dôsledok úrazu alebo chirurgickej traumy. Pri substitučnej liečbe faktorom VIII sa plazmatické hladiny faktora VIII zvýšia, čím sa dočasne upraví deficit faktora a upraví sa tendencia ku krvácaniu.
Klinická účinnosť počasprofylaxiealiečbyepizódkrvácania
Klinická účinnosť Esperoctu v profylaxii a liečbe krvácania bola skúmaná v piatich prospektívnych,
multicentrických klinických štúdiách u 270 predtým liečených pacientov (PTP) so závažnou hemofíliou A.
Profylaxia u dospelých/dospievajúcich
Účinnosť Esperoctu pri profylaxii a liečbe krvácania sa hodnotila v otvorenom, nekontrolovanom
skúšaní na dospievajúcich a dospelých pacientoch so závažnou hemofíliou A vo veku 12 a viac rokov. Profylaktický účinok Esperoctu bol preukázaný u 175 pacientov pri dávkovaní 50 IU na kg telesnej hmotnosti podávané každé 4 dni alebo každé 3–4 dni (dvakrát týždenne). Odhadovaná priemerná
[medián] ročná frekvencia krvácania (ABR) u dospelých a dospievajúcich, ktorí dostávali Esperoct bola 1,18 (Interquartile range IQR: 0,00; 4,25), zatiaľ čo spontánna ABR bola 0,00 (IQR: 0,00; 1,82), traumatická ABR bola 0,00 (IQR: 0,00; 1,74) a kĺbová ABR bola 0,85 (IQR: 0,00; 2,84). Pri zahrnutí
imputácií (nahradenie chýbajúcich údajov u pacientov, ktorí boli vyradení, substituovanou hodnotou)
bola odhadovaná priemerná ABR pre všetky krvácania 3,70 (95% CI: 2,94; 4,66).
Zo 175 dospelých/dospievajúcich na profylaktickej liečbe nemalo 70 (40 %) žiadne krvácania. Priemerná ročná spotreba na profylaxiu bola 4641 IU/kg.
Je nutné poznamenať, že ročnú frekvenciu krvácania (ABR) nie je možné porovnať medzi rôznymi koncentráciami faktora a medzi rôznymi klinickými štúdiami.
Dospelí/dospievajúci s nízkou frekvenciou krvácania na úrovni 0-2 epizódy krvácania počas posledných 6 mesiacov, ktorí zároveň dostali najmenej 50 dávok Esperoctu, mali možnosť byť randomizovaní na profylaktickú liečbu každých 7 dní (75 IU/kg každých 7 dní) alebo každé 4 dni (50 IU/kg každé 4 dni). Celkovo 55 zo 120 spôsobilých pacientov sa rozhodlo pre randomizáciu (17 pre dávkovanie každé 4 dni a 38 pre 75 IU každých 7 dní). ABR randomizovaných pacientov bola
1,77 (0,59; 5,32) pri liečbe každé 4 dni a 3,57 (2,13; 6,00) pri profylaxii jedenkrát týždenne. Deväť
z týchto pacientov sa vrátilo späť na profylaxiu každé 4 dni počas randomizačnej fázy štúdie. Celkovo, vrátane všetkých predĺžených častí, sa 31 zo 61 pacientov na profylaxii každých 7 dní vrátilo späť na liečbu každé 4 dni.
Profylaxia u detí (do 12rokov)
Použitie Esperoctu u detí mladších ako 12 rokov nie je indikované (pozri časť 4.2, informácia
o pediatrickom použití).
Účinnosť a bezpečnosť Esperoctu pri profylaktickej liečbe krvácania sa hodnotila v otvorenom, nekontrolovanom skúšaní s jedným ramenom u 68 detí mladších ako 12 rokov, ktoré mali závažnú hemofíliu A. Profylaktický účinok Esperoctu bol preukázaný pri dávkovaní 60 IU na kg telesnej hmotnosti (50-75 IU/kg) dvakrát týždenne. Medián a odhadovaná priemerná ročná frekvencia krvácania u detí do 12 rokov, ktoré dostávali Esperoct dvakrát týždenne, bola 1,95 a 2,13 (95 % CI:
1,48; 3,06), zatiaľ čo spontánna ABR bola 0,00 a 0,58 (95 % CI: 0,24; 1,40), traumatická ABR bola
0,00 a 1,52 (95 % CI: 1,07; 2,17) a kĺbová ABR bola 0,00 a 1,03 (95 % CI: 0,59; 1,81). Zo 68 detí do
12 rokov na profylaktickej liečbe nemalo 29 (42,6 %) žiadne krvácania. Priemerná ročná spotreba na profylaxiu bola 6475 IU/kg.
Klinická účinnosť Esperoctuvliečbeepizódkrvácaniaapočasliečbypodľapotreby
Účinnosť Esperoctu v liečbe epizód krvácania bola preukázaná vo všetkých vekových skupinách.
Prevažná časť krvácaní liečených Esperoctom bola mierna/stredne závažná.
Celková miera úspešnosti pri liečbe krvácania bola 87,7 % a 94,4 % všetkých krvácaní liečených
1-2 injekciami.
U 12 pacientov starších ako 18 rokov bolo liečených liečbou podľa potreby s priemernou liečebnou dávkou 38,1 IU/kg liečených 1 126 prípadov krvácania s priemernou ročnou spotrebou 1457 IU/kg. Z celkového počtu 1 126 prípadov krvácania bolo 86,9 % účinne liečených 1 injekciou a 96,8 % bolo účinne liečených 1-2 injekciami Esperoctu.
Klinická účinnosť Esperoctupočasveľkýchchirurgickýchzákrokov
Esperoct bol účinný pri udržiavaní hemostázy počas veľkých chirurgických zákrokov s mierou
úspešnosti 95,6 % pri všetkých vykonaných veľkých chirurgických zákrokoch (43 zo 45 bol účinok ohodnotený ako „vynikajúci“ alebo „dobrý“).
5.2 Farmakokinetické vlastnosti
Celkovo sa u 86 pacientov (vrátane 24 pediatrických pacientov vo veku 0 až menej ako 12 rokov)
hodnotilo 129 farmakokinetických (PK) profilov jednotlivej dávky Esperoctu.
Všetky farmakokinetické štúdie s Esperoctom boli uskutočnené u pacientov so závažnou hemofíliou A (FVIII <1 %), ktorí boli už predtým liečení. Pacienti dostali jednotlivú dávku v množstve 50 IU/kg, pričom vzorky krvi im boli odobraté pred podaním dávky a v niekoľkých časových bodoch najneskôr do 96 hodín po podaní dávky.
Polčas Esperoctu u dospelých bol v porovnaní s liekmi s nemodifikovaným faktorom VIII 1,6-násobne dlhší.
Farmakokinetické parametre
Celkovo sa hodnotilo 108 farmakokinetických profilov jednotlivej dávky v množstve 50 IU/kg
Esperoctu u 69 pacientov. Farmakokinetické parametre jednotlivej dávky sú porovnateľné u malých detí (0 až menej ako 6 rokov) a starších detí (6 až menej ako 12 rokov) a u dospievajúcich
(12 až17 rokov) a dospelých (18 a viac rokov).
Očakávaný nárast zotavenia bol nižší, zatiaľ čo klírens upravený podľa hmotnosti bol u detí
v porovnaní s dospelými a dospievajúcimi vyšší. Vo všeobecnosti bol pozorovaný trend zvyšovania nárastu zotavenia a znižovania klírensu (ml/h/kg) podľa veku. Táto skutočnosť zodpovedá vyššiemu
objemu distribúcie na kilogram telesnej hmotnosti u detí v porovnaní s dospelými (tabuľka 4).
Farmakokinetické parametre jednotlivej dávky stanovené po 28 týždňoch profylaktickej liečby
Esperoctom boli konzistentné s úvodnými farmakokinetickými parametrami.
Farmakokinetické parametre jednotlivej dávky Esperoctu sú uvedené v tabuľke 4. Použitie Esperoctu u detí do 12 rokov nie je indikované.
Tabuľka 4 Farmakokinetické parametre jednotlivej dávky Esperoctu 50 IU/kg u detí, dospievajúcich a dospelých podľa veku za použitia chromogénneho testu
(geometrický priemer [CV %])
Farmakokinetický parameter
N= počet pacientov
0 až menej ako
6 rokov N=13
6 až menej ako
12 rokov N=11
12 až menej ako
18 rokov
N=3
18 rokov a viac
N=42
Počet profilov 13 11 5 79
IR (IU/dl) na
IU/kg)a
Maximálna aktivita faktora VIII
(IU/dl)a
1,80 (29) 1,99 (25) 2,79 (12) 2,63 (22)
101,2 (28) 119,6 (25) 133,2 (9) 134,4 (23)
t1/2 (hodiny) 13,6 (20) 14,2 (26) 15,8 (43) 19,9 (34)
AUCinf
(IU*hodina/dl)
2 147 (47) 2 503 (42) 3 100 (44) 3 686 (35)
CL (ml/hodina/kg) 2,6 (45) 2,4 (40) 1,5 (43) 1,4 (32) Vss (ml/kg) 44,2 (34) 41,2 (25) 33,4 (10) 37,7 (27) MRT (hodín) 17,0 (22) 17,3 (31) 21,7 (45) 25,2 (29)b
Skratky: AUC = plocha pod časovým profilom aktivity faktora VIII; t1/2 = terminálny polčas; MRT = priemerný čas zotrvania; CL = klírens;
Vss = distribučný objem v ustálenom stave; IR = nárast zotavenia.
a Nárast zotavenia a faktor VIII sa u pacientov vo veku 12 rokov a viac hodnotil 30 minút po podaní dávky a u detí do 12 rokov 60 minút
po podaní dávky (prvá vzorka).
b Výpočet na základe 67 profiloch.
Priemerná najnižšia aktivita faktora VIII v plazme v stabilnom stave počas profylaktickej liečby
Esperoctom davkovaným po 50 IU/kg každé 4 dni je 3,0 IU/dl (95% CI: 2,6; 3,4) u pacientov
12 rokov a starších.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe konvenčných farmakologických štúdií bezpečnosti a toxicity po opakovanom podaní neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokPrášokchlorid sodný
L-histidín sacharóza
polysorbát 80
L-metionín
dihydrát chloridu vápenatého hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
Rozpúšťadlochlorid sodný
voda na injekcie
6.2 InkompatibilityNevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi alebo rekonštituovať inými injekčnými roztokmi, ako je dodané rozpúšťadlo chloridu sodného.
Rekonštituovaný liek sa nemá podávať s inými liekmi v tých istých hadičkách alebo zásobníku.
6.3 Čas použiteľnostiInjekčná liekovka pred otvorením30 mesiacov pri teplote 2 °C – 8 °C, počas ktorých môže byť liek uchovávaný pri teplote do 30 °C
počas jedného nepretržitého obdobia, ktoré neprekračuje 12 mesiacov, nie však po dátume exspirácie
uvedenom na obale. Ak liek už raz bol vybratý z chladničky, nesmie sa už do chladničky vrátiť. Na škatuľke lieku si zaznačte dátum, kedy ste začali liek uchovávať pri izbovej teplote.
Po rekonštitúcii
Chemická a fyzikálna stabilita po rekonštitúcii bola dokázaná počas 24 hodín pri uchovávaní
v chladničke (2 °C – 8 °C) a počas 4 hodín pri uchovávaní pri izbovej teplote (do 30 °C).
Z mikrobiologického hľadiska sa tento liek má použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky uchovávania po rekonštitúcii a pred použitím nesú zodpovednosť používatelia
a neodporúča sa uchovávanie dlhší čas ako 4 hodiny pri izbovej teplote (do 30 °C) alebo 24 hodín v chladničke (2 °C – 8 °C), pokiaľ rekonštitúcia neprebehla za kontrolovaných a validovaných aseptických podmienok. Rekonštituovaný roztok sa má uchovávať v injekčnej liekovke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky uchovávania pri izbovej teplote a po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Každé balenie Esperoctu obsahuje:
– 1 sklenenú injekčnú liekovku (typu I) s práškom uzavretú chlórbutylovou gumenou zátkou, hliníkovým tesnením s plastovým uzáverom so západkou
– 1 sterilný adaptér injekčnej liekovky na rekonštitúciu
– 1 naplnenú injekčnú striekačku so 4 ml rozpúšťadla s uzáverom proti spätnému chodu
(polypropylén), gumeným piestom (brómbutyl) a gumeným krytom hrotu (brómbutyl).
– 1 nástavec piesta (polypropylén).
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie
Esperoct sa podáva intravenózne po rekonštitúcii prášku s rozpúšťadlom, ktoré sa dodáva v injekčnej striekačke. Po rekonštitúcii je roztok číra a bezfarebná tekutina bez viditeľných častíc. Rekonštituovaný liek je potrebné pred podávaním vizuálne skontrolovať, či neobsahuje častice a či nie je sfarbený. Roztok má byť číry a bezfarebný. Roztok nepoužite, ak je zakalený alebo obsahuje usadeniny.
Pokyny na rekonštitúciu lieku pred podávaním, pozri písomnú informáciu pre používateľa.
Rýchlosť podávania sa má určiť tak, aby vyhovovala pacientovi a podávanie trvalo približne 2 minúty. Budete potrebovať aj infúznu súpravu (motýlikovú ihlu s hadičkou), sterilné alkoholové tampóny,
gázové tampóny a náplasti. Tieto pomôcky nie sú súčasťou balenia Esperoctu.
Vždy používajte aseptickú techniku. Likvidácia
Po aplikácii injekčnú striekačku, infúznu súpravu a injekčnú liekovku s adaptérom zlikvidujte'
bezpečným spôsobom.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novo Nordisk A/S Novo Allé
DK-2880 Bagsværd
Dánsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/19/1374/001
EU/1/19/1374/002
EU/1/19/1374/003
EU/1/19/1374/004
EU/1/19/1374/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
 N
ávod na používanie Esperoctu
P
r
ed použitím Esperoctu si pozorne prečítajte tento návod.
N
ávod na používanie Esperoctu
P
r
ed použitím Esperoctu si pozorne prečítajte tento návod.
Esperoct sa dodáva ako prášok. Pred injekciou (podávaním) sa musí rekonštituovať (rozpustiť) rozpúšťadlom, ktoré sa dodáva v injekčnej striekačke. Rozpúšťadlo je injekčný roztok chloridu sodného 9 mg/ml (0,9 %). Rekonštituovaný liek si musíte podať do žily (intravenózna (i.v.) injekcia). Súčasti tohto balenia sú určené na rekonštitúciu a injekčné podávanie Esperoctu.
Budete tiež potrebovať:
• infúznu súpravu (motýlikovú ihlu s hadičkou)
• sterilné tampóny namočené v alkohole
• gázové tampóny a náplasti.
Tieto pomôcky nie sú súčasťou balenia Esperoctu.
Nepoužívajte túto súpravu bez riadneho školenia od svojho lekára alebo zdravotnej sestry. Vždy si umyte ruky a presvedčte sa, že priestor okolo vás je čistý.Keď pripravujete a podávate injekciu lieku priamo do žily, je dôležité
dodržiavať čistúa mikrobiologicky vyhovujúcu (aseptickú) techniku. Nesprávnou technikou sa môžu preniesť choroboplodné zárodky, ktoré môžu infikovať vašu krv.
Neotvárajte súpravu, kým nie ste pripravený na jej použitie.Nepoužite súpravu, ak spadla alebo je poškodená. Namiesto nej použite nové balenie.
Nepoužívajte súpravu po exspirácii. Namiesto nej použite nové balenie. Dátum exspirácie je uvedený na vonkajšom obale, na injekčnej liekovke, na adaptéri injekčnej liekovky a na naplnenej injekčnej striekačke.
Nepoužite súpravu, ak máte podozrenie, že je kontaminovaná. Namiesto nej použite nové balenie.
Nevyhadzujte žiadnu súčasť, kým ste si injekčne nepodali rekonštituovaný roztok. Súprava je určená len na jednorazové použitie.ObsahBalenie obsahuje:
• 1 injekčná liekovka s práškom Esperoct
• 1 adaptér injekčnej liekovky
• 1 naplnená injekčná striekačka s rozpúšťadlom
• 1 nástavec piesta (umiestnený pod injekčnou striekačkou)
P
r
e
h
ľ
ad
In
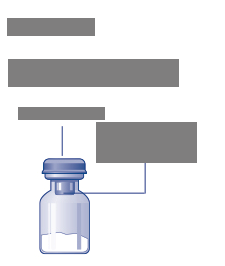
jekčná liekovka s práškom
Esperoct
Plastové viečko
Gumenná zátka (pod plastovým viečkom)
Adaptér injekčnej liekovky
Ochranný kryt
Hrot
(pod ochranným papierom)
Ochranný
papier
Naplnená injekčná striekačka s rozpúšťadlom
Hrot injekčnej
striekačky (pod krytom injekčnej striekačky)
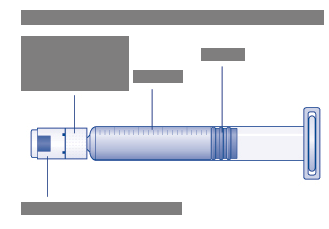
Stupnica
Piest

Kryt injekčnej striekačky
 Nástavec piesta
Nástavec piestaZávit Širší koniec

 1. Príprava injekčnej liekovky a injekčnej striekačky
A
1. Príprava injekčnej liekovky a injekčnej striekačky
A
•
Vyberte potrebný počet balení Esperoctu.
•
Skontrolujte dátum exspirácie.•
Skontrolujte názov, silu a farbu balenia, aby ste sa presvedčili, že obsahuje správny liek.
•
Umyte si ruky a riadne si ich usušte použitím čistého uteráku alebo voľným vysušením na vzduchu.
• Vyberte injekčnú liekovku, adaptér injekčnej liekovky a naplnenú injekčnú striekačku
zo škatuľky.
Nástavec piesta nechajte nedotknutý v škatuľke.•
Nechajte injekčnú liekovku a naplnenú injekčnú striekačku dosiahnuť izbovú teplotu. Môžete to urobiť ich podržaním v rukách, až kým necítite, že sú také teplé ako vaše ruky, pozri obrázok
A.Injekčnú liekovku a naplnenú injekčnú striekačku
nezohrievajte žiadnym iným spôsobom.
•
Odstráňte plastové viečko z injekčnej liekovky.
BAk je plastové viečko uvoľnené alebo chýba,injekčnú liekovku nepoužite.•
Očistite gumovú zátku sterilným alkoholovým tampónom a nechajte ju pred použitím pár sekúnd schnúť na vzduchu, aby ste zaistili, že je mikrobiologicky vyhovujúca.
Nedotýkajte sa gumovej zátky prstami, aby stenepreniesli baktérie. 2. Pripojenie adaptéra injekčnej liekovky C
2. Pripojenie adaptéra injekčnej liekovky C•
Odstráňte ochranný papier z adaptéra injekčnej liekovky.
Ak ochranný papier nie je úplne uzavretý alebo je roztrhnutý, nepoužite adaptér injekčnej liekovky.Nevyberajte adaptér injekčnej liekovkyz ochranného
krytu prstami.Ak sa dotknete hrotu adaptéra injekčnej liekovky, môžete prstami preniesť baktérie.
•
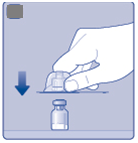
Položte injekčnú liekovku na rovný a pevný Dpovrch.•
Otočte ochranný kryt a zatlačte adaptér injekčnej liekovky na injekčnú liekovku.
Neodstraňujte adaptér z injekčnej liekovky, keď je už pripevnený.
• Jemne
pritlačte ochranný kryt palcom a
Eukazovákom ako je znázornené.
•
Odstráňte ochranný kryt z adaptéra injekčnej liekovky.
Nenadvihnite adaptér z injekčnej liekovky, keď odstraňujete ochranný kryt.
 3. Pripojenie nástavca piesta a injekčnej striekačky F
3. Pripojenie nástavca piesta a injekčnej striekačky F• Uchopte nástavec piesta za širší koniec a vyberte
ho zo škatuľky.
Nedotýkajte sa strán alebo závitunástavca piesta. Ak sa dotknete strán alebo závitu, môžete prstami preniesť baktérie.
•
Ihneď pripojte nástavec piesta k injekčnej striekačke, otáčaním v smere hodinových ručičiek, na piest vo vnútri naplnenej injekčnej striekačky, až kým nepocítite odpor.

•
Odstráňte kryt injekčnej striekačky z naplnenej
Ginjekčnej striekačky ohnutím dole, až kým sa
nezlomí perforácia.
Nedotýkajte sa hrotu injekčnej striekačky pod krytom injekčnej striekačky. Ak sa dotknete
hrotu injekčnej striekačky, môžete prstami preniesť baktérie.
Ak je kryt injekčnej striekačky uvoľnený alebochýba, naplnenú injekčnú striekačku nepoužite.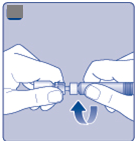
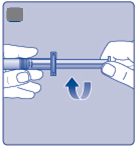
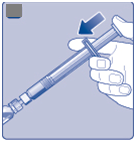
•
Naskrutkujte naplnenú injekčnú striekačku Hpevne na adaptér injekčnej liekovky, až kým
nepocítite odpor.
 4. Rekonštitúcia prášku rozpúšťadlom
I
4. Rekonštitúcia prášku rozpúšťadlom
I
•
Držte naplnenú injekčnú striekačku s injekčnou liekovkou naklonenou smerom dole.
•
Tlačte nástavec piesta, aby ste vstrekli všetko rozpúšťadlo do injekčnej liekovky.
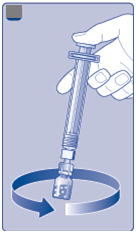
•
Držte nástavec piesta stlačený dole a jemne Jkrúžte injekčnou liekovkou, až sa rozpustí všetok
prášok.
Netraste injekčnou liekovkou, lebo to spôsobíspenenie.•
Skontrolujte rekonštituovaný roztok. Musí byť číry a bezfarebný a bez obsahu akýchkoľvek viditeľných častíc.
Ak spozorujete častice alebo zmenu jeho farby, nepoužite ho. Namiesto neho použite nové balenie.
Odporúča sa použiť Esperoct ihneď po rekonštitúcii.Ak nemôžete použiť rekonštituovaný roztok Esperoctu hneď, musíte ho použiť do 4 hodín, keď ho uchovávate pri izbovej teplote (do 30 °C) a do 24 hodín, keď ho uchovávate v chladničke (2 °C –
8 °C). Rekonštituovaný liek uchovávajte v injekčnej liekovke.
Neuchovávajte rekonštituovaný roztok v mrazničke, ani ho neuchovávajte v injekčnej striekačke.Chráňte rekonštituovaný roztok pred priamym svetlom. Ak potrebujete väčšiu dávku ako jedna injekčná liekovka, opakujte kroky
A až
J s ďalšími injekčnými liekovkami, adaptérmi injekčnej liekovky a naplnenými injekčnými striekačkami, až kým
nedosiahnete požadovanú dávku.
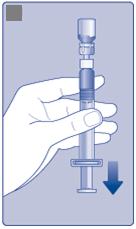
•
Držte nástavec piestu stlačený na doraz. K•
Otočte injekčnú striekačku s injekčnouliekovkou hore dnom.
•
Prestaňte tlačiť nástavec piesta a nechajte ho pohnúť sa samovoľne naspäť, kým rekonštituovaný roztok naplní injekčnú striekačku.
•
Ťahajte nástavec piesta jemne smerom dole, aby ste natiahli rekonštituovaný roztok do injekčnej striekačky.
•
Ak nepotrebujete použiť celý
 rekonštituovaný liek z injekčnej liekovky,
rekonštituovaný liek z injekčnej liekovky, na nabratie potrebnej dávky použite stupnicu na injekčnej striekačke, ako vám povedal lekár alebo zdravotná sestra.
Ak je v niektorom mieste vzduch
v injekčnej striekačke, vytlačte vzduch naspäť do injekčnej liekovky.
• Kým držíte injekčnú liekovku hore dnom,
klepkajte jemne na injekčnú striekačku, aby všetky bubliny vystúpili nahor.
•
Tlačte nástavec piesta pomaly, až kým všetky vzduchové bubliny nevytlačíte.
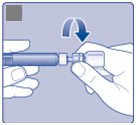
•
Odskrutkujte adaptér s injekčnou
Lliekovkou.
Nedotýkajte sa hrotu injekčnej striekačky. Ak sa dotknete špičky injekčnej striekačky, môžete prstami preniesť baktérie.
5. Injekčné podanie rekonštituovaného roztokuEsperoct je teraz pripravený na injekčné podanie do žily.
• Injekčne si podajte rekonštituovaný roztok podľa pokynov lekára alebo zdravotnej sestry.
• Injekciu podávajte pomaly počas približne 2 minút.
Nezmiešavajte Esperoct so žiadnymi inými intravenóznymi injekciami alebo liekmi.
Injekčné podávanie Esperoctu cez konektory bez ihly na intravenózne (i.v.) katétre Upozornenie: Naplnená injekčná striekačka je vyrobená zo skla a je navrhnutá tak, aby bola
kompatibilná so štandardnými luer-lock konektormi. Niektoré konektory bez ihly s vnútorným hrotom
nie sú kompatibilné s naplnenou injekčnou striekačkou. Táto inkompatibilita môže zabrániť podaniu lieku a spôsobiť poškodenie konektora bez ihly.
Injekčné podávanie roztoku cez centrálny žilový katéter (CVAD − central venous access device), ako je centrálny žilový katéter alebo podkožný port:
• Používajte čistú a mikrobiologicky vyhovujúcu (aseptickú) techniku. Po konzultácii s lekárom alebo zdravotnou sestrou postupujte podľa pokynov na správne používanie konektora a CVAD.
• Vstreknutie do CVAD môže vyžadovať použitie sterilnej 10 ml plastovej injekčnej striekačky
na nasatie rekonštituovaného roztoku. To by malo nasledovať hneď po kroku
J.
• Ak je potrebné hadičku CVAD prepláchnuť pred podaním alebo po podaní injekcie Esperoctu, použite injekčný roztok chloridu sodného 9 mg/ml (0,9 %).

 L
i
kvidácia
M
L
i
kvidácia
M
•
Po podaní injekcie bezpečne zlikvidujte všetok nepoužitý roztok Esperoctu, injekčnú striekačku s infúznou súpravou, injekčnú liekovku s adaptérom injekčnej liekovky a ďalšie odpadové materiály podľa pokynov lekárnika.
Nelikvidujte ho s bežným domovým odpadom.
Nerozoberajte súpravu pred likvidáciou.Nepoužívajte znova túto súpravu.