
vácania podľa PLATO
celkovo
Veľké + malé krvácania podľa PLATO
nesúvisiace s liečebným postupom
Veľké krvácania definované podľa kritérií
TIMI
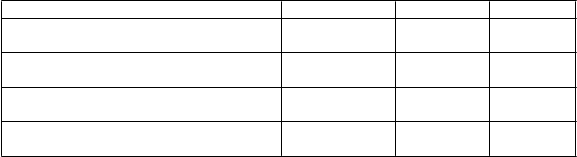
Veľké + malé krvácania definované podľa kritérií TIMI
Definície kategórií krvácania:16,1 14,6 0,0084
5,9 4,3 <0,0001
7,9 7,7 0,5669
11,4 10,9 0,3272
Veľké fatálne/ život ohrozujúce krvácanie: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o viac ako 50 g/l
alebo s transfúziou ≥ 4 jednotiek erytrocytov alebo fatálne alebo intrakraniálne alebo intraperikardiálne krvácanie s tamponádou srdca; alebo hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov alebo chirurgickú intervenciu.
Veľké iné: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50 g/l alebo s transfúziou 2 až 3 jednotiek erytrocytov; alebo významne vysiľujúce krvácanie.
Malé krvácanie: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
Veľké krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu > 50 g/l alebo intrakraniálne krvácanie.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50g/l.
*p-hodnota vypočítaná na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO, veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI alebo malého krvácania podľa kritérií TIMI (tabuľka 2). V porovnaní
s klopidogrelom sa však pri tikagrelore vyskytlo spolu viac veľkých a malých krvácaní podľa definície PLATO. Niekoľko pacientov v štúdii PLATO malo fatálne krvácania: 20 (0,2 %) pri tikagrelore a 23 (0,3 %) pri klopidogrele (pozri časť 4.4).
Vek, pohlavie, hmotnosť, rasa, geografické územie, súbežné ochorenia, súbežná liečba a anamnéza pacienta, vrátane prekonanej cievnej mozgovej príhody alebo prekonaného prechodného ischemického záchvatu, neboli prediktívne z hľadiska celkového výskytu krvácaní, ani z hľadiska výskytu veľkého krvácania podľa definície PLATO nesúvisiaceho s liečebným postupom. Preto pre žiaden podtyp krvácania nebola konkrétna skupina identifikovaná ako riziková.
Krvácanie súvisiace s CABG:
V štúdii PLATO sa u 42 % z 1 584 pacientov (12 % kohorty), ktorí podstúpili zákrok CABG, vyskytlo veľké fatálne/život ohrozujúce krvácanie podľa definície PLATO, bez rozdielu medzi liečebnými
skupinami. Fatálne krvácanie súvisiace s CABG sa vyskytlo v každej liečebnej skupine u 6 pacientov
(pozri časť 4.4).
Krvácanie nesúvisiace s CABG a krvácanie nesúvisiace s liečebným postupom:
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO nesúvisiaceho s CABG, ale výskyt veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI a výskyt veľkého a malého krvácania podľa kritérií TIMI bol častejší pri tikagrelore. Rovnako, po vylúčení všetkých krvácaní súvisiacich s liečebným postupom, sa viac krvácaní vyskytlo pri tikagrelore ako pri klopidogrele (tabuľka 2). Ukončenie liečby pre krvácanie nesúvisiace s liečebným postupom bolo častejšie pri tikagrelore (2,9 %) ako pri klopidogrele (1,2 %; p< 0,001).
Intrakraniálne krvácanie:
V skupine s tikagrelorom sa vyskytlo viac intrakraniálnych krvácaní nesúvisiacich s liečebným postupom (n = 27 krvácaní u 26 pacientov, 0,3 %) ako v skupine s klopidogrelom (n = 14 krvácaní,
0,2 %), z ktorých bolo fatálnych 11 v skupine s tikagrelorom a 1 v skupine s klopidogrelom.
V celkovom výskyte fatálnych krvácaní nebol žiadny rozdiel.
Zistenia týkajúce sa krvácania v štúdii PEGASUS
V tabuľke 3 sú uvedené celkové výsledky udalostí súvisiacich s krvácaním v štúdii PEGASUS.
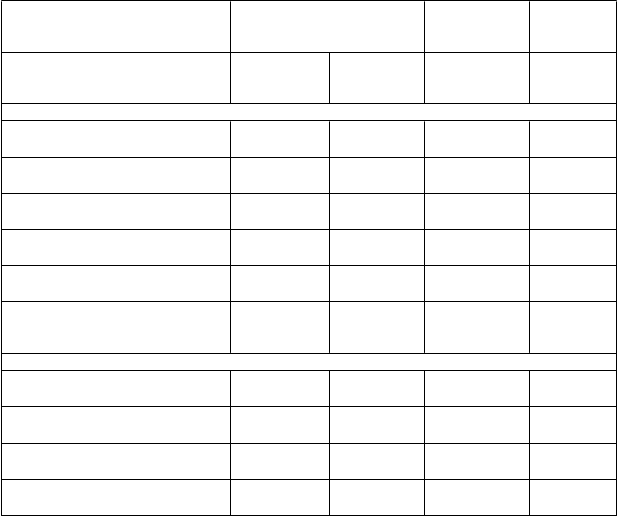
Tabuľka 3 – Analýza celkových krvácavých príhod, Kaplanov-Meierov odhad v 36. mesiaci
(štúdia PEGASUS)
tikagrelor 60 mg dvakrát
denne + ASA N = 6 958
Pomer
samotná ASA N = 6 996
Koncový ukazovateľ bezpečnosti KM %
Kategórie krvácania podľa kritérií TIMI
rizika
(95 % IS)
KM % p-hodnota
Veľké krvácania podľa TIMI 2,3 2,32 (1,68; 3,21)
Fatálne 0,3 1,00 (0,44; 2,27)
ICH 0,6 1,33 (0,77; 2,31)
1,1 < 0,0001
0,3 1,0000
0,5 0,3130
Iné veľké krvácania podľa
TIMI 1,6
Veľké alebo malé krvácania podľa
TIMI 3,4
Veľké alebo malé krvácania alebo
3,61
(2,31; 5,65) 0,5 < 0,0001
2,54
(1,93; 3,35) 1,4 < 0,0001
krvácania vyžadujúce lekársku starostlivosť podľa TIMI
16,6 2,64 (2,35; 2,97)
7,0 < 0,0001
Kategórie krvácania podľa definície PLATO
Veľké krvácania podľa PLATO 3,5 2,57 (1,95; 3,37)
Fatálne/život ohrozujúce 2,4 2,38 (1,73; 3,26)
1,4 < 0,0001
1,1 < 0,0001
Iné veľké krvácania podľa
PLATO 1,1
Veľké alebo malé krvácania podľa
PLATO 15,2
Definície kategórií krvácania:
3,37
(1,95; 5,83) 0,3 < 0,0001
2,71
(2,40; 3,08) 6,2 < 0,0001
 Veľké krvácanie podľa TIMI:
Veľké krvácanie podľa TIMI: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO klinicky zjavné prejavy hemorágie spojené s poklesom hemoglobínu (Hgb) ≥ 50 g/l, alebo 15 % pokles hematokritu (Hct) v prípade nedostupnosti údajov o Hgb.
Fatálne krvácanie: Krvácavá príhoda, ktorá viedla priamo k smrti v priebehu 7 dní.
ICH: Intrakraniálne krvácanie.
Iné veľké krvácanie podľa TIMI: Veľké non-fatálne krvácanie podľa TIMI iné než ICH.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l.
Krvácanie vyžadujúce lekársku starostlivosť podľa TIMI: Vyžadujúce zásah, ALEBO vedúce k hospitalizácii, ALEBO
vyžadujúce vyšetrenie.
Veľké fatálne/život ohrozujúce krvácanie podľa PLATO: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO intraperikardiálne krvácanie s tamponádou srdca, ALEBO s hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov/inotropík alebo chirurgickú intervenciu, ALEBO klinicky zjavné krvácanie s poklesom
hemoglobínu ˃ 50 g/l alebo s transfúziou ≥ 4 jednotiek erytrocytov.
Iné veľké krvácanie podľa PLATO: Významne vysiľujúce krvácanie, ALEBO klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l alebo s transfúziou 2 - 3 jednotiek erytrocytov.
Malé krvácanie podľa PLATO: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
V štúdii PEGASUS bol výskyt veľkého krvácania podľa kritérií TIMI pri tikagrelore v dávke 60 mg dvakrát denne vyšší ako pri samotnej ASA. Nepozorovalo sa zvýšené riziko fatálneho krvácania
a pozorovalo sa len menej významné zvýšenie výskytu intrakraniálneho krvácania v porovnaní
s liečbou samotnou ASA. V štúdii sa vyskytlo niekoľko fatálnych krvácavých príhod, 11 (0,3 %) pri
60 mg tikagreloru a 12 (0,3 %) pri liečbe samotnou ASA. Pozorované zvýšené riziko výskytu veľkého krvácania podľa kritérií TIMI pri 60 mg tikagreloru bolo zapríčinené predovšetkým vyššou frekvenciou výskytu iného veľkého krvácania podľa kritérií TIMI zastúpeného udalosťami
v gastrointestinálnej TOS.
Podobne zvýšený výskyt veľkého krvácania podľa kritérií TIMI bol pozorovaný aj pri kategóriách krvácania zahŕňajúcich veľké alebo malé krvácania podľa kritérií TIMI, veľké krvácania podľa definície PLATO a veľké alebo malé krvácania podľa definície PLATO (pozri tabuľku 3). Ukončenie liečby pre krvácanie bolo častejšie pri 60 mg tikagreloru (6,2 %) v porovnaní s liečbou samotnou ASA (1,5 %). Väčšina týchto krvácaní bola menej závažná (klasifikované ako krvácania vyžadujúce lekársku starostlivosť podľa kritérií TIMI), napr. epistaxa, tvorba krvných podliatin a hematómov.
Profil krvácania pri 60 mg tikagreloru bol pre udalosti veľkého krvácania podľa kritérií TIMI, veľkého alebo malého krvácania podľa kritérií TIMI a veľkého krvácania podľa definície PLATO konzistentný v rámci viacerých vopred definovaných podskupín (napr. podľa veku, pohlavia, hmotnosti, rasy, geografického regiónu, súbežných ochorení, súbežnej liečby a anamnézy).
Intrakraniálne krvácanie:
Pre 60 mg tikagreloru a liečbu samotnou ASA sa hlásili podobné miery výskytu spontánneho intrakraniálneho krvácania (n = 13, 0,2 % v oboch liečebných skupinách). Pri liečbe 60 mg tikagreloru (n = 15, 0,2 %) v porovnaní s liečbou samotnou ASA (n = 10, 0,1 %) sa preukázalo menej významné zvýšenie výskytu poúrazového intrakraniálneho krvácania a intrakraniálneho krvácania súvisiaceho
s liečebným postupom. Pri 60 mg tikagreloru sa vyskytlo 6 fatálnych prípadov intrakraniálneho krvácania a pri liečbe samotnou ASA 5 prípadov. Výskyt intrakraniálneho krvácania bol v oboch
liečebných skupinách nízky vzhľadom na to, že populácia štúdie sa vyznačovala významnou mierou
komorbidity a KV rizikových faktorov.
Dyspnoe
Pacienti liečení tikagrelorom hlásili dyspnoe, pocit sťaženého dýchania. V štúdii PLATO nežiaduce udalosti týkajúce sa dyspnoe (dyspnoe, kľudové dyspnoe, námahové dyspnoe, paroxyzmálne nočné dyspnoe a nočné dyspnoe) hlásili u 13,8 % pacientov liečených s tikagrelorom a u 7,8 % pacientov liečených s klopidogrelom. V štúdii PLATO u 2,2 % pacientov užívajúcich tikagrelor a u 0,6 % pacientov užívajúcich klopidogrel skúšajúci považovali dyspnoe za príčinu súvisiacu s liečbou a málo prípadov bolo závažných (0,14 % tikagrelor; 0,02 % klopidogrel) (pozri časť 4.4). Väčšina hlásených príznakov dyspnoe bola miernej až stredne ťažkej intenzity a väčšinou sa hlásili ako jedna epizóda krátko po začatí liečby.
V porovnaní s klopidogrelom bolo u pacientov s astmou/CHOCHP, ktorí boli liečení tikagrelorom, zvýšené riziko výskytu nezávažného dyspnoe (3,29 % v prípade tikagreloru oproti 0,53 % v prípade klopidogrelu) a závažného dyspnoe (0,38 % v prípade tikagreloru oproti 0,0 % v prípade klopidogrelu). V absolútnom vyjadrení bolo toto riziko vyššie ako v celej populácii pacientov v štúdii PLATO. Tikagrelor sa musí užívať opatrne u pacientov s anamnézou astmy a/alebo CHOCHP (pozri časť 4.4).
Približne 30 % všetkých prípadov dyspnoe sa upravilo v priebehu 7 dní. Štúdie PLATO sa zúčastnili pacienti s kongestívnym srdcovým zlyhaním, CHOCHP alebo astmou pred začatím skúšania; títo pacienti a starší pacienti hlásili najčastejšie dyspnoe. Kvôli dyspnoe sa liečba tikagrelorom vysadila u
0,9 % pacientov a liečba klopidogrelom u 0,1 % pacientov. Vyšší výskyt dyspnoe u tikagreloru nesúvisí s novým alebo zhoršujúcim sa srdcovým alebo pľúcnym ochorením (pozri časť 4.4). Tikagrelor nemá vplyv na výsledky testov funkcie pľúc.
V štúdii PEGASUS sa dyspnoe hlásilo u 14,2 % pacientov užívajúcich tikagrelor v dávke 60 mg dvakrát denne a u 5,5 % pacientov užívajúcich samotnú ASA. Rovnako ako v štúdii PLATO, väčšina hlásených prípadov dyspnoe bola miernej až stredne ťažkej intenzity (pozri časť 4.4). Pacienti, ktorí hlásili dýchavičnosť boli starší a mali na začiatku častejšie dýchavičnosť, CHOCHP alebo astmu.
Vyšetrenia
Zvýšenie kyseliny močovej: V štúdii PLATO došlo k zvýšeniu kyseliny močovej v sére nad hornú hranicu normálu u 22 % pacientov dostávajúcich tikagrelor, v porovnaní s 13 % pacientov dostávajúcich klopidogrel. V štúdii PEGASUS boli príslušné hodnoty 9,1 % pre 90 mg tikagreloru,
8,8 % pre 60 mg tikagreloru a 5,5 % pre placebo. Priemerná hladina kyseliny močovej v sére sa pri tikagrelore zvýšila približne o 15 % v porovnaní so zvýšením približne o 7,5 % pri klopidogrele a po
ukončení liečby sa znížila približne o 7 % v prípade tikagreloru, v prípade klopidogrelu sa však žiadne zníženie nezistilo. V štúdii PEGASUS sa zistilo reverzibilné zvýšenie priemerných hladín kyseliny močovej v sére o 6,3 % pre 90 mg tikagreloru a 5,6 % pre 60 mg tikagreloru, v porovnaní s 1,5 % znížením v skupine s placebom. V štúdii PLATO bola frekvencia dnovej artritídy 0,2 % pri tikagrelore oproti 0,1 % pri klopidogrele. V štúdii PEGASUS boli príslušné hodnoty frekvencie dny/dnovej artritídy 1,6 % pre 90 mg tikagreloru, 1,5 % pre 60 mg tikagreloru a 1,1 % pre placebo.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieTikagrelor je v jednorazových dávkach až do 900 mg dobre znášaný. Gastrointestinálna toxicita bola určujúcim faktorom v štúdii zameranej na zvyšovanie jednorazovej dávky. Iné klinicky významné nežiaduce reakcie, ktoré sa môžu vyskytnúť pri predávkovaní, zahŕňajú dyspnoe a ventrikulárne pauzy (pozri časť 4.8).
V prípade predávkovania sa môžu objaviť vyššie uvedené potenciálne nežiaduce reakcie a je potrebné zvážiť monitorovanie EKG.
V súčasnosti nie je známe žiadne antidotum na zvrátenie účinkov tikagreloru a nepredpokladá sa, že by bol tikagrelor dialyzovateľný (pozri časť 4.4). Liečba predávkovania sa má riadiť štandardnou lekárskou praxou na miestnej úrovni. Predpokladaným účinkom nadmerných dávok tikagreloru je predĺžené trvanie rizika krvácania, ktoré súvisí s inhibíciou krvných doštičiek. Transfúzia krvných doštičiek pravdepodobne nepredstavuje klinický prínos pre pacientov s krvácaním (pozri časť 4.4). Ak dôjde ku krvácaniu, je potrebné prijať ďalšie príslušné podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiagreganciá trombocytov okrem heparínu, ATC kód: B01AC24
MechanizmusúčinkuBrilique obsahuje tikagrelor patriaci do chemickej skupiny cyklopentyltriazolopyrimidínov (CPTP), ktorý je perorálnym, priamo pôsobiacim, selektívnym antagonistom, reverzibilne sa viažucim na receptor P2Y12, ktorý zabraňuje aktivácii a agregácii krvných doštičiek sprostredkovanej ADP
a závislej na P2Y12. Tikagrelor nezabraňuje väzbe ADP, ale po naviazaní na receptor P2Y12 zabraňuje
signálnej transdukcii indukovanej ADP. Vzhľadom na to, že krvné doštičky sa podieľajú na vzniku
a/alebo vývoji trombotických komplikácií aterosklerotických ochorení, preukázalo sa, že inhibícia funkcie krvných doštičiek znižuje riziko kardiovaskulárnych príhod, ako je smrť, IM alebo cievna mozgová príhoda.
Tikagrelor tiež zvyšuje lokálne hladiny endogénneho adenozínu inhibíciou rovnovážnych nukleozidových transportérov-1 (ENT-1).
Zistilo sa, že tikagrelor u zdravých dobrovoľníkov a pacientov s AKS zvýrazňuje nasledujúce účinky indukované adenozínom: vazodilatácia (merané zvýšením koronárneho prietoku krvi u zdravých dobrovoľníkov a pacientov s AKS; bolesť hlavy), inhibícia funkcie krvných doštičiek (v plnej ľudskej krvi
in vitro) a dyspnoe. Súvislosť medzi pozorovanými zvýšeniami adenozínu a klinickými výsledkami (napr. morbidita-mortalita) však nebola jasne vysvetlená.
Farmakodynamické
účinky
Nástup
účinku
Tikagrelor u pacientov so stabilnou koronárnou artériovou chorobou (CAD) užívajúcich ASA
vykazuje rýchly nástup farmakologického účinku, čo sa preukázalo priemernou inhibíciou agregácie krvných doštičiek (IPA) tikagrelorom po 0,5 hodiny od podania nárazovej dávky 180 mg približne
41 %, s maximálnym účinkom na IPA 89 % po 2 – 4 hodinách od podania dávky a tento účinok pretrvával 2 – 8 hodín. U 90 % pacientov bol finálny rozsah IPA po 2 hodinách od podania dávky
> 70 %.
Odznievanieúčinku
Pri plánovanom zákroku CABG existuje zvýšené riziko krvácania pre tikagrelor oproti klopidogrelu, pokiaľ je liečba vysadená v kratšej dobe ako 96 hodín pred zákrokom.
Údajetýkajúcesaprechodunainúliečbu
Prechod z liečby klopidogrelom v dávke 75 mg na tikagrelor v dávke 90 mg dvakrát denne má za následok absolútne zvýšenie IPA o 26,4 % a prechod z liečby tikagrelorom na klopidogrel má za následok absolútne zníženie IPA o 24,5 %. Pacientov možno prestaviť z liečby klopidogrelom na tikagrelor bez akéhokoľvek prerušenia protidoštičkového účinku (pozri časť 4.2).
Klinickáúčinnosťabezpečnosť
Klinický dôkaz účinnosti a bezpečnosti tikagreloru je odvodený z dvoch skúšaní fázy 3:
· Štúdia PLATO [PLATelet Inhibition and Patient Outcomes], porovnávajúca tikagrelor oproti klopidogrelu, oba podávané v kombinácii s ASA a ďalšou štandardnou liečbou.
· Štúdia PEGASUS TIMI-54 [PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients], porovnávajúca tikagrelor v kombinácii s ASA oproti liečbe samotnou ASA.
Štúdia PLATO(akútnykoronárnysyndróm)
Do štúdie PLATO bolo zahrnutých 18 624 pacientov, u ktorých v priebehu ostatných 24 hodín došlo
k nástupu príznakov nestabilnej anginy pectoris (unstable angina, UA), infarktu myokardu bez elevácie ST segmentu (NSTEMI) alebo infarktu myokardu s eleváciou ST segmentu (STEMI), a ktorí
spočiatku dostávali medikamentóznu liečbu alebo sa u nich vykonala perkutánna koronárna intervencia (PCI) alebo CABG.
Klinická účinnosť
Pri dennom podávaní ASA preukázal tikagrelor v dávke 90 mg dvakrát denne v prevencii výskytu združeného koncového ukazovateľa KV smrti, IM alebo cievnej mozgovej príhody, lepší účinok ako
klopidogrel v dávke 75 mg denne, na tomto rozdiele sa podieľal najmä výskyt KV smrti a IM. Pacienti
dostali 300 mg nárazovú dávku klopidogrelu (prípadne 600 mg v prípade PCI) alebo 180 mg tikagreloru.
Výsledok liečby sa prejavil skoro (zníženie absolútneho rizika [ARR] 0,6 % a zníženie relatívneho rizika [RRR] 12 % po 30 dňoch) a účinok liečby ostal rovnaký po celé obdobie 12 mesiacov, ARR/rok bolo 1,9 % a RRR 16 %. To naznačuje, že je vhodné liečiť pacientov tikagrelorom 90 mg dvakrát denne až 12 mesiacov (pozri časť 4.2). Liečbou 54 pacientov s AKS tikagrelorom namiesto klopidogrelu sa zabráni 1 aterotrombotickej príhode; liečbou 91 pacientov tikagrelorom namiesto klopidogrelu sa zabráni 1 KV smrti (pozri obrázok 1 a tabuľku 4).
Účinok liečby tikagrelorom sa popri klopidogrele ukazuje ako konzistentný v rámci rôznych podskupín pacientov, vrátane členenia podľa hmotnosti, pohlavia, diabetu v anamnéze, prechodného ischemického záchvatu alebo nehemoragickej cievnej mozgovej príhody alebo revaskularizácie, súbežnej liečby zahŕňajúcej liečbu heparínmi, inhibítormi GpIIb/IIIa a inhibítormi protónovej pumpy (pozri časť 4.5), podľa diagnózy, u ktorej sa sledovali koncové ukazovatele (STEMI, NSTEMI alebo UA) a podľa zámeru liečebného postupu pri randomizácii (invazívny alebo medikamentózny).
Pozorovala sa málo významná súčinnosť liečby a geografického územia, pričom podľa pomeru rizika (HR) pre primárny koncový ukazovateľ zo svetového hľadiska vychádza priaznivejšie účinok tikagreloru s výnimkou Severnej Ameriky, kde vychádza priaznivejšie účinok klopidogrelu, ktorá predstavovala približne 10 % celkovej skúmanej populácie (p-hodnota interakcie = 0,045). Výskumné analýzy poukázali na možnú súvislosť s dávkou ASA tak, že znížená účinnosť tikagreloru sa pozorovala pri zvyšujúcich sa dávkach ASA. Pri dlhodobom podávaní ASA s tikagrelorom má byť rozmedzie dennej dávky 75 - 150 mg (pozri časť 4.2 a 4.4).
Obrázok 1 vyjadruje odhad rizika prvého výskytu akejkoľvek udalosti zahrnutej do združeného koncového ukazovateľa účinnosti.
Obrázok 1 Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IM
a cievnej mozgovej príhody (štúdia PLATO)

0 6 0 1 2 0 1 8 0 2 4 0 3 0 0 3 6 0
Počet v ohroizení í
Dn i o d ra nd omizácie
T 9 3 3 3
C 9 2 9 1
8 6 2 8

8 5 2 1
8 4 6 0
8 3 6 2
8 2 1 9
8 1 2 4
6 7 4 3
6 6 5 0
5 1 6 1
5 0 9 6
4 1 4 7
4 0 7 4
Tikagrelor v porovnaní s klopidogrelom znížil výskyt primárneho združeného koncového ukazovateľa
rovnako u pacientov s UA/NSTEMI ako aj u pacientov so STEMI (tabuľka 4). Liečbu liekom Brilique
90 mg dvakrát denne spolu s nízkou dávkou ASA možno teda použiť u pacientov s AKS (nestabilná angina pectoris, infarkt myokardu bez elevácie ST segmentu [NSTEMI] alebo infarkt myokardu
s eleváciou ST segmentu [STEMI]); vrátane pacientov, ktorí dostávajú medikamentóznu liečbu
a pacientov, u ktorých sa vykonala perkutánna koronárna intervencia (PCI) alebo koronárny artériový by-pass (CABG).
Tabuľka 4 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PLATO)
tikagrelor
90 mg dvakrát
denne
(%
pacientov
s udalosťou) N = 9 333
klopidogrel
75 mg jedenkrát
denne
(% pacientov s udalosťou)
N = 9 291
ARR
a
(%/rok)
RRR
a
(%)
(95 % IS)
p
-
hodnota
KV smrť/IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
9,3 10,9 1,9 16 (8; 23) 0,0003
So zámerom
invazívneho zákroku 8,5 10,0 1,7 16 (6; 25) 0,0025
So zámerom
medikamentóznej liečby
11,3 13,2 2,3 15 (0,3; 27) 0,0444d

KV smrť 3,8 4,8 1,1 21 (9; 31) 0,0013
IM (s vylúčením
tichého IM)b 5,4 6,4 1,1 16 (5; 25) 0,0045
Cievna mozgová
príhoda 1,3 1,1 −0,2 −17 (−52; 9) 0,2249
Úmrtnosť zo všetkých príčin, IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
KV smrť, IM celkovo, cievna mozgová príhoda, SRI, RI, TIA alebo iná ATEc
Úmrtnosť zo
9,7 11,5 2,1 16 (8; 23) 0,0001
13,8 15,7 2,1 12 (5; 19) 0,0006
d
všetkých príčin 4,3 5,4 1,4 22 (11; 31) 0,0003
Definitívna d
trombóza stentu 1,2 1,7 0,6 32 (8; 49) 0,0123
aARR = zníženie absolútneho rizika; RRR = zníženie relatívneho rizika = (1 − pomer rizika) x 100 %. Hodnoty s negatívnym
RRR naznačujú zvýšenie relatívneho rizika.
bS vylúčením tichého IM.
cSRI = ťažká rekurentná ischémia (serious recurrent ischaemia); RI = rekurentná ischémia (recurrent ischaemia); TIA =
prechodný ischemický záchvat (transient ischaemic attack); ATE = arteriálna trombotická príhoda. IM celkovo zahŕňa tichý
IM, s dátumom, kedy bol zistený.
dNominálna hladina významnosti (significance value); všetky ostatné sú formálne štatisticky významné vopred definovaným hierarchickým testovaním.
Genetická podštúdia štúdie PLATO
Genotypizáciou CYP2C19 a ABCB1 u 10 285 pacientov v štúdii PLATO sa zistili súvislosti medzi
skupinami genotypu a výsledkami štúdie PLATO. Lepší účinok tikagreloru oproti klopidogrelu
v znížení výskytu závažných KV príhod nebol významne ovplyvnený genotypom CYP2C19 alebo ABCB1 pacienta. Podobne ako v celej štúdii PLATO sa celkový výskyt veľkých krvácaní podľa definície PLATO pri tikagrelore a klopidogrele neodlišoval, bez ohľadu na genotyp CYP2C19 alebo ABCB1. Výskyt veľkých krvácaní podľa definície PLATO nesúvisiacich s CABG bol pri tikagrelore
v porovnaní s klopidogrelom zvýšený u pacientov s jednou alebo viacerými nefunkčnými alelami
CYP2C19, ale podobný ako pri klopidogrele u pacientov bez nefunkčnej alely.
Združený ukazovateľ účinnosti a bezpečnosti
Združený ukazovateľ účinnosti a bezpečnosti (KV smrť, IM, cievna mozgová príhoda alebo veľké krvácanie podľa definície PLATO celkovo) naznačuje, že klinický prínos účinnosti tikagreloru
v porovnaní s klopidogrelom nie je kompenzovaný udalosťami súvisiacimi s veľkým krvácaním (ARR
1,4 %, RRR 8 %, HR 0,92; p = 0,0257) v priebehu 12 mesiacov od AKS.
Klinická bezpečnosť
Podštúdia s Holterovým monitorovaním:
Na sledovanie výskytu ventrikulárnych páuz a iných epizód arytmií počas štúdie PLATO skúšajúci vykonali Holterovo monitorovanie u podskupiny takmer 3 000 pacientov, z ktorých približne 2 000
malo záznam aj v akútnej fáze ich AKS a aj po jednom mesiaci. Primárnym sledovaným
ukazovateľom bol výskyt ventrikulárnych páuz ≥ 3 sekundy. Ventrikulárne pauzy sa u pacientov v akútnej fáze vyskytovali častejšie pri tikagrelore (6,0 %) ako pri klopidogrele (3,5 %); a po 1 mesiaci to bolo 2,2 % pri tikagrelore a 1,6 % pri klopidogrele (pozri časť 4.4). Nárast výskytu ventrikulárnych páuz v akútnej fáze AKS bol v skupine s tikagrelorom výraznejší u pacientov
s chronickým srdcovým zlyhávaním (CHF) v anamnéze (9,2 % oproti 5,4 % u pacientov bez CHF
v anamnéze; v skupine s klopidogrelom to bolo 4,0 % u pacientov s CHF v anamnéze oproti 3,6 %
u pacientov bez CHF v anamnéze). Tento rozdiel sa po 1 mesiaci nevyskytoval: 2,0 % oproti 2,1 % pri tikagrelore pre pacientov s a bez CHF v anamnéze a 3,8 % oproti 1,4 % pri klopidogrele. Tento rozdiel
nesúvisel so žiadnymi nežiaducimi klinickými dôsledkami (vrátane zavedenia kardiostimulátora)
v tejto populácii pacientov.
Štúdia PEGASUS (anamnézainfarktumyokardu)
Štúdia PEGASUS TIMI-54 bola medzinárodná, multicentrická, randomizovaná, dvojito zaslepená,
placebom kontrolovaná štúdia s paralelnou skupinou a s dĺžkou určenou počtom udalostí potrebných pre štatistické vyhodnotenie (event-driven), zahŕňajúca 21 162 pacientov, ktorá hodnotila prevenciu
aterotrombotických príhod pri tikagrelore podávanom v 2 dávkach (buď 90 mg dvakrát denne alebo
60 mg dvakrát denne) v kombinácii s nízkou dávkou ASA (75 – 150 mg), v porovnaní s liečbou samotnou ASA u pacientov s IM v anamnéze a ďalšími rizikovými faktormi aterotrombózy.
Pacienti boli vhodní na zaradenie do štúdie, ak boli vo veku 50 rokov alebo starší, mali IM
v anamnéze (1 až 3 roky pred randomizáciou) a mali aspoň jeden z nasledujúcich rizikových faktorov aterotrombózy: vek ≥ 65 rokov, diabetes mellitus vyžadujúci liečbu, druhý predchádzajúci IM,
potvrdenú CAD postihujúcu viaceré cievy alebo chronickú dysfunkciu obličiek v non-terminálnom
štádiu.
Pacienti neboli vhodní na zaradenie do štúdie, ak sa u nich počas obdobia štúdie plánoval použiť antagonista receptorov P2Y12, dipyridamol, cilostazol alebo antikoagulačná liečba; ak mali krvácavú poruchu alebo ischemickú cievnu mozgovú príhodu alebo intrakraniálne krvácanie v anamnéze, nádor centrálneho nervového systému alebo abnormalitu intrakraniálnych ciev; ak mali v priebehu predchádzajúcich 6 mesiacov gastrointestinálne krvácanie alebo veľký chirurgický zákrok v priebehu predchádzajúcich 30 dní.
Klinická účinnosť
Obrázok 2 – Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IM
a cievnej mozgovej príhody (štúdia PEGASUS)

Tikagrelor 60 mg bd - - - - Placebo
N 7045 7067
Pacienti s udalosťami 487 (6.9%) 578 (8.2%)
KM% v 36 mesiaci 7.8% 9.0%
Pomer rizika (95% CI) 0.84 (0.74, 0.95)
 p
p-hodnota 0.0043
Počet v riziku
Ti 60 mg
Placebo
Dni od randomizácie
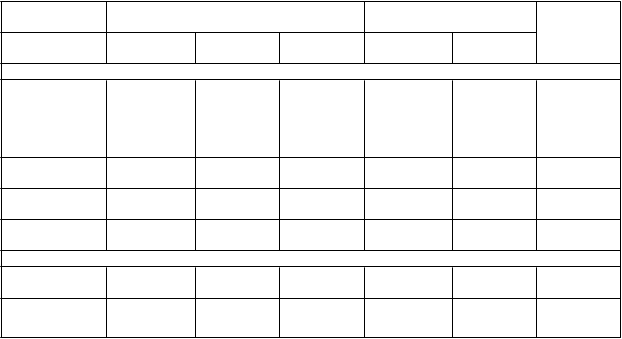
Tabuľka 5 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PEGASUS)
tikagrelor 60 mg dvakrát denne + ASA
N = 7 045
sa
m
otn
á ASA
N = 7 067
p
-
hodnota
Charakteristika
Pacienti
s udalosťami
Primárny koncový ukazovateľ
Združený pre
KV
KM % HR
(95 % IS)
Pacienti
s udalosťami KM %
smrť/IM/cievnu mozgovú príhodu
487 (6,9 %) 7,8 % 0,84 (0,74; 0,95)
578 (8,2 %) 9,0 % 0,0043 (s)
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
IM 285 (4,0 %) 4,5 % 0,84 (0,72; 0,98)
210 (3,0 %) 3,4 % 0,0676
338 (4,8 %) 5,2 % 0,0314
Cievna mozgová
príhoda 91 (1,3 %) 1,5 %
Sekundárny koncový ukazovateľ
0,75
(0,57; 0,98) 122 (1,7 %) 1,9 % 0,0337
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
210 (3,0 %) 3,4 % -
Úmrtnosť zo
všetkých príčin 289 (4,1 %) 4,7 %
0,89 326 (4,6 %) 5,2 % - (0,76; 1,04)
Pomer rizika (HR) a
p-hodnota sú vypočítané osobitne pre tikagrelor oproti liečbe samotnou ASA na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou.
Percentá KM vypočítané po 36 mesiacoch.
Poznámka: počty prvých udalostí pre zložky KV smrť, IM a cievna mozgová príhoda sú skutočné počty prvých udalostí pre každú zo zložiek a nesčítavajú sa do počtu udalostí v združenom koncovom ukazovateli.
(s) Predstavuje štatistickú významnosť (significance).
IS = interval spoľahlivosti; KV = kardiovaskulárna; HR = pomer rizika; KM = Kaplanov-Meierov odhad; IM = infarkt myokardu;
N = počet pacientov.
Oba režimy s tikagrelorom, 60 mg dvakrát denne a 90 mg dvakrát denne, v kombinácii s ASA preukázali lepší účinok v prevencii aterotrombotických príhod v porovnaní so samotnou ASA (združený koncový ukazovateľ: KV smrť, IM a cievna mozgová príhoda), s konzistentným účinkom liečby počas celého obdobia štúdie a dosiahnutím RRR 16 % a ARR 1,27 % pre 60 mg tikagreloru
a RRR 15 % a ARR 1,19 % pre 90 mg tikagreloru.
Hoci bol profil účinnosti 90 mg a 60 mg dávky podobný, bolo dokázané, že nižšia dávka je lepšie znášaná a má lepší bezpečnostný profil v súvislosti s rizikom krvácania a dyspnoe. Preto sa na prevenciu aterotrombotických príhod (KV smrť, IM a cievna mozgová príhoda) u pacientov s IM
v anamnéze a vysokým rizikom aterotrombotickej príhody odporúča iba Brilique 60 mg dvakrát denne podávaný súbežne s ASA.
V porovnaní so samotnou ASA, tikagrelor v dávke 60 mg dvakrát denne významne znížil primárny združený koncový ukazovateľ KV smrti, IM a cievnej mozgovej príhody. Každá zo zložiek prispela k zníženiu primárneho združeného koncového ukazovateľa (RRR KV smrti 17 %, RRR IM 16 %
a RRR cievnej mozgovej príhody 25 %).
RRR pre združený koncový ukazovateľ od 1. do 360. dňa (RRR 17 %) a od 361. dňa ďalej (RRR
16 %) bolo podobné. K dispozícii sú obmedzené údaje o účinnosti a bezpečnosti tikagreloru za 3 roky predĺženej liečby.
Nebol nájdený žiadny dôkaz o prínose (neprišlo k zníženiu primárneho koncového ukazovateľa zloženého z KV úmrtia, infarktu myokardu a cievnej mozgovej príhody, ale zvýšeniu závažného krvácania), pri užívaní tikagrelor 60 mg dvakrát denne u klinicky stabilných pacientov > 2 roky od infarktu myokardu alebo viac ako jeden rok po ukončení predchádzajúcej liečby inhibítorom receptora ADP (pozri tiež časť 4.2).
Klinická bezpečnosť
Počet prerušení liečby s tikagrelorom 60 mg kvôli krvácaniu a dýchavičnosti bola vyššia u pacientov
> 75 rokov (42 %) ako u mladších pacientov (rozmedzie: 23-31 %), s rozdielom v porovnaní s placebom vyšším ako 10 % (42 % vs. 29 %), u pacientov > 75 rokov.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Brilique vo všetkých podskupinách pediatrickej populácie s akútnym koronárnym syndrómom (AKS) a
infarktom myokardu (IM) v anamnéze (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika tikagreloru je lineárna a expozícia tikagreloru a aktívnemu metabolitu
(AR-C124910XX) je približne úmerná dávke až po dávku 1 260 mg.
Absorpcia
K absorpcii tikagreloru dochádza rýchlo, s mediánom tmax približne 1,5 hodiny. Tvorba hlavného cirkulujúceho metabolitu AR-C124910XX (tiež aktívneho) z tikagreloru je rýchla s mediánom tmax približne 2,5 hodín. Po perorálnom podaní jednorazovej dávky tikagreloru 90 mg nalačno zdravým dobrovoľníkom je Cmax 529 ng/ml a AUC 3 451 ng*h/ml. Pomer metabolit/pôvodné liečivo pre Cmax je
0,28 a pre AUC 0,42. Farmakokinetika tikagreloru a AR-C124910XX u pacientov s IM v anamnéze
bola vo všeobecnosti podobná farmakokinetike v populácii s AKS. Na základe populačnej farmakokinetickej analýzy štúdie PEGASUS bol medián Cmax tikagreloru v rovnovážnom stave
391 ng/ml a AUC 3 801 ng*h/ml pre dávku 60 mg tikagreloru. Pre dávku 90 mg tikagreloru bola Cmax
v rovnovážnom stave 627 ng/ml a AUC 6 255 ng*h/ml.
Priemerná absolútna biologická dostupnosť tikagreloru sa odhadla na 36 %. Príjem potravy s vysokým obsahom tukov mal za následok zvýšenie AUC tikagreloru o 21 % a zníženie Cmax aktívneho metabolitu o 22 %, na Cmax tikagreloru a na AUC aktívneho metabolitu však nemal žiadny vplyv. Tieto malé zmeny sa považujú za klinicky minimálne významné, preto sa tikagrelor môže užívať s jedlom alebo bez jedla. Tikagrelor ako aj jeho aktívny metabolit sú substrátmi P-gp.
Tikagrelor, vo forme rozdrvených tabliet zmiešaných s vodou, podávaný perorálne alebo nazogastrickou sondou do žalúdka má porovnateľnú biologickú dostupnosť s celými tabletami
s ohľadom na AUC a Cmax tikagreloru a aktívneho metabolitu. Úvodná expozícia (0,5 a 1 hodinu po podaní dávky) po podaní rozdrvených tabliet tikagreloru zmiešaných s vodou bola vyššia v porovnaní
s celými tabletami, pričom neskôr (o 2 až 48 hodín) bol koncentračný profil vo všeobecnosti identický.
Distribúcia
Distribučný objem tikagreloru v rovnovážnom stave je 87,5 l. Tikagrelor a aktívny metabolit sa vo veľkej miere viaže na bielkoviny ľudskej plazmy (> 99,0 %).
Biotransformácia
CYP3A4 je hlavným enzýmom zodpovedným za metabolizmus tikagreloru a tvorbu aktívneho metabolitu a ich interakcie s inými substrátmi CYP3A sú v rozmedzí od aktivácie po inhibíciu.
Hlavným metabolitom tikagreloru je AR-C124910XX, ktorý je na základe dôkazu jeho väzby na doštičkový receptor P2Y12 pre ADP v podmienkach in vitro tiež aktívny. Systémová expozícia aktívnemu metabolitu predstavuje približne 30 – 40 % systémovej expozície tikagreloru.
Eliminácia
Primárnou cestou eliminácie tikagreloru je metabolizácia v pečeni. V prípade podávania rádioizotopom značeného tikagreloru sa zachytí v priemere približne 84 % rádioizotopom značenej dávky (57,8 % v stolici, 26,5 % v moči). Detegované množstvá tikagreloru a aktívneho metabolitu v moči v obidvoch prípadoch predstavovali menej ako 1 % dávky. Primárnou cestou eliminácie
aktívneho metabolitu je s najväčšou pravdepodobnosťou biliárna sekrécia. Priemerná hodnota t1/2 pre tikagrelor bola približne 7 hodín a pre aktívny metabolit 8,5 hodín.
Osobitné skupinypacientov
Staršípacienti
U starších osôb (≥ 75 rokov) s AKS v porovnaní s mladšími pacientmi sa farmakokinetickou analýzou populácie zistilo zvýšenie expozície tikagreloru (približne 25 % pre Cmax aj AUC) a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné (pozri časť 4.2).
Pediatrickápopulácia
Tikagrelor sa neskúmal v pediatrickej populácii (pozri časti 4.2 a 5.1).
Pohlavie
U žien sa v porovnaní s mužmi zaznamenalo zvýšenie expozície tikagreloru a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné.
Poruchafunkcieobličiek
U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) v porovnaní s osobami s normálnou funkciou obličiek bola expozícia tikagreloru približne o 20 % nižšia
a expozícia aktívnemu metabolitu približne o 17 % vyššia (pozri časť 4.2).
Poruchafunkciepečene
Cmax tikagreloru bol o 12 % a AUC o 23 % vyššia u pacientov s miernou poruchou funkcie pečene v porovnaní s kontrolnými zdravými osobami, účinok tikagreloru na IPA bol však u oboch skupín podobný. U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Tikagrelor sa neskúmal u pacientov s ťažkou poruchou funkcie pečene a nie sú k dispozícii farmakokinetické údaje u pacientov so stredne ťažkou poruchou funkcie pečene. U pacientov, ktorí
mali stredne závažné alebo závažné východiskové zvýšenie jedného alebo viacerých vyšetrení funkcie pečene, boli plazmatické koncentrácie tikagreloru v priemere podobné alebo mierne vyššie
v porovnaní s pacientmi bez východiskových zvýšení. U pacientov so stredne ťažkou poruchou
funkcie pečene sa neodporúča žiadna úprava dávky (pozri časti 4.2 a 4.4).
Etnickápríslušnosť
Priemerná biologická dostupnosť je u pacientov ázijského pôvodu o 39 % vyššia ako u belochov. Biologická dostupnosť tikagreloru u pacientov, ktorí sami uviedli černošský pôvod bola o 18 % nižšia
ako u belochov, v klinických farmakologických štúdiách bola expozícia (Cmax a AUC) tikagreloru u Japoncov približne o 40 % (o 20 % po úprave podľa telesnej hmotnosti) vyššia ako u belochov. Expozícia u pacientov, ktorí sami uviedli hispánsky alebo latinskoamerický pôvod bola podobná ako u belochov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje týkajúce sa tikagreloru a jeho hlavného metabolitu nepreukázali neprijateľné riziko nežiaducich účinkov u ľudí na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po jednorazovom a opakovanom podávaní a genotoxického potenciálu.
Pri klinicky relevantných hladinách expozície sa pozorovalo gastrointestinálne podráždenie u niekoľkých druhov zvierat (pozri časť 4.8).
Pri vysokých dávkach tikagreloru sa u samíc potkanov preukázal zvýšený výskyt nádorov maternice (adenokarcinómov) a zvýšený výskyt adenómov pečene. Pravdepodobným mechanizmom vzniku nádorov maternice je hormonálna nerovnováha, ktorá u potkanov môže viesť k vzniku nádorov. Pravdepodobným mechanizmom vzniku adenómov pečene je indukcia enzýmov v pečeni špecifických pre hlodavce. Relevantnosť týchto zistení týkajúcich sa karcinogenity sa preto považuje za nepravdepodobnú pre ľudí.
U potkanov sa pozorovali menej významné vývinové anomálie pri dávkach toxických pre samicu (bezpečnostný pomer 5,1). U králikov sa pozorovalo mierne oneskorenie dozrievania pečene a vývoja skeletu plodu u samíc, ktorým boli podávané vysoké dávky, bez známok materskej toxicity (bezpečnostný pomer 4,5).
Štúdie na potkanoch a králikoch sa preukázali reprodukčnú toxicitu s mierne zníženým nárastom telesnej hmotnosti brezivých samíc a so zníženou životaschopnosťou a zníženou pôrodnou hmotnosťou mláďat s oneskoreným rastom. Tikagrelor spôsoboval nepravidelné cykly (najmä predĺžené cykly) u samíc potkanov, nemal však vplyv na celkovú fertilitu samcov a samíc potkanov. Farmakokinetické štúdie s rádioizotopom značeným tikagrelorom preukázali, že pôvodné liečivo
a jeho metabolity sa u potkanov vylučujú do materského mlieka (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety manitol (E421)
dihydrát hydrogenfosforečnanu vápenatého magnéziumstearát (E470b)
sodná soľ karboxymetylškrobu typu A
hydroxypropylcelulóza (E463)
Obaltablety
oxid titaničitý (E171)
čierny oxid železitý (E172)
červený oxid železitý (E172)
makrogol 400
hypromelóza (E464)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti3 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia· PVC-PVDC/Al priehľadný blister (so symbolmi slnko/mesiac) obsahujúci 10 tabliet; škatule s obsahom 60 tabliet (6 blistrov) a 180 tabliet (18 blistrov).
· PVC-PVDC/Al priehľadný kalendárový blister (so symbolmi slnko/mesiac) obsahujúci 14
tabliet; škatule s obsahom 14 tabliet (1 blister), 56 tabliet (4 blistre) a 168 tabliet (12 blistrov). Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/10/655/007-011
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 03. december 2010
Dátum posledného predĺženia registrácie: 17. júl 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Brilique 90 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 90 mg tikagreloru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta (tableta).
Okrúhle, bikonvexné, žlté tablety s označením „90“ nad „T“ na jednej strane a bez označenia na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Brilique podávaný spolu s kyselinou acetylsalicylovou (ASA) je indikovaný na prevenciu aterotrombotických príhod u dospelých pacientov s
- akútnym koronárnym syndrómom (AKS) alebo
- infarktom myokardu (IM) v anamnéze a vysokým rizikom vzniku aterotrombotickej príhody
(pozri časti 4.2 a 5.1).
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Pacienti užívajúci Brilique majú užívať aj nízku udržiavaciu dávku ASA 75 – 150 mg denne, pokiaľ to
nie je výslovne kontraindikované.
Akútnykoronárnysyndróm
Liečba liekom Brilique sa má začať s jednou 180 mg nárazovou dávkou (dve 90 mg tablety) a potom sa má pokračovať dávkou 90 mg dvakrát denne. Odporúča sa, aby liečba liekom Brilique 90 mg
dvakrát denne u pacientov s AKS trvala 12 mesiacov, pokiaľ nie je klinicky indikované ukončenie liečby (pozri časť 5.1).
Infarktmyokarduvanamnéze
Brilique 60 mg dvakrát denne je odporúčanou dávkou na predĺženú liečbu pacientov s IM v anamnéze aspoň jeden rok a vysokým rizikom aterotrombotickej príhody (pozri časť 5.1). Liečba by sa mala
začať bez prerušenia ako liečba nasledujúca po úvodnej jednoročnej liečbe liekom Brilique 90 mg
alebo liečbe iným inhibítorom receptora adenozíndifosfátu (ADP) u pacientov s AKS s vysokým rizikom aterotrombotickej príhody. Liečbu tiež možno začať až do 2 rokov po IM alebo v priebehu jedného roka po ukončení predchádzajúcej liečby inhibítorom receptora ADP. K dispozícii sú obmedzené údaje o účinnosti a bezpečnosti tikagreloru pri predĺženej liečbe presahujúcej 3 roky.
Ak je potrebné prestaviť pacientov na liečbu liekom Brilique, prvá dávka sa má podať 24 hodín po poslednej dávke predchádzajúceho protidoštičkového lieku.
Vynechaná
dávka
Tiež je potrebné sa vyvarovať vynechaniu liečby. Pacient, ktorý vynechá dávku lieku Brilique, má užiť iba jednu tabletu (svoju ďalšiu dávku) v obvyklom čase užitia ďalšej dávky.
Osobitnéskupinypacientov
Starší pacienti
U starších pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2). K dispozícii nie sú žiadne informácie týkajúce sa dialyzovaných pacientov a preto sa tikagrelor
u týchto pacientov neodporúča.
Porucha funkcie pečene
Tikagrelor sa neskúmal u pacientov s ťažkou poruchou funkcie pečene a jeho použitie u týchto pacientov je preto kontraindikované (pozri časť 4.3). K dispozícii sú len obmedzené údaje u pacientov
so stredne ťažkou poruchou funkcie pečene. Úprava dávky sa neodporúča, tikagrelor sa však má
používať s opatrnosťou (pozri časti 4.4 a 5.2). U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť tikagreloru u detí mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsobpodávania
Na perorálne použitie.
Brilique sa môže podávať s jedlom alebo bez jedla.
Pre pacientov, ktorí nie sú schopní prehltnúť tabletu (tablety) vcelku, sa tablety môžu rozdrviť na jemný prášok a zmiešať v pol pohári vody a ihneď vypiť. Pohár sa má opláchnuť ďalším pol pohárom vody a obsah sa má vypiť. Zmes sa môže podávať aj pomocou nazogastrickej sondy (CH8 alebo širšou). Po podaní zmesi je dôležité prepláchnuť nazogastrickú sondu vodou.
4.3 Kontraindikácie
· Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 (pozri časť 4.8).
· Aktívne patologické krvácanie.
· Intrakraniálne krvácanie v anamnéze (pozri časť 4.8).
· Ťažká porucha funkcie pečene (pozri časti 4.2, 4.4 a 5.2).
· Súbežné podávanie tikagreloru so silnými inhibítormi CYP3A4 (napr. ketokonazolom, klaritromycínom, nefazodónom, ritonavirom a atazanavirom) môže viesť k podstatnému
zvýšeniu expozície tikagreloru (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Rizikokrvácania
Pri použití tikagreloru u pacientov so známym zvýšeným rizikom krvácania sa má zvážiť prínos
z hľadiska prevencie aterotrombotických príhod (pozri časti 4.8 a 5.1). Pri klinickom opodstatnení sa má tikagrelor používať s opatrnosťou v nasledujúcich skupinách pacientov:
· Pacienti náchylní na krvácanie (napr. kvôli nedávnemu zraneniu, nedávnemu chirurgickému zákroku, poruchám koagulácie, aktívnemu alebo nedávnemu gastrointestinálnemu krvácaniu). Použitie tikagreloru je kontraindikované u pacientov s aktívnym patologickým krvácaním,
u pacientov s intrakraniálnym krvácaním v anamnéze a u pacientov s ťažkou poruchou funkcie
pečene (pozri časť 4.3).
· Pacienti súbežne liečení liekmi, ktoré môžu zvyšovať riziko krvácania (napr. nesteroidové protizápalové lieky (NSAID), perorálne antikoagulanciá a/alebo fibrinolytiká), v priebehu
24 hodín od podania tikagreloru.
Transfúzia krvných doštičiek nezvrátila protidoštičkový účinok tikagreloru u zdravých dobrovoľníkov a preto pravdepodobne nepredstavuje klinický prínos pre pacientov s krvácaním. Keďže podávaním tikagreloru spolu s dezmopresínom sa štandardizovaný čas krvácania neskrátil, nie je pravdepodobné, že by bol dezmopresín účinný pri zvládaní klinických krvácavých príhod (pozri časť 4.5).
Antifibrinolytická liečba (kyselinou aminokaprónovou alebo kyselinou tranexámovou) a/alebo liečba rekombinantným faktorom VIIa môže zvyšovať hemostázu. Tikagrelor možno znovu začať podávať po tom, ako bola príčina krvácania identifikovaná a je pod kontrolou.
Chirurgickýzákrok
Pacientov je potrebné poučiť, aby pred akýmkoľvek plánovaným chirurgickým zákrokom a pred užívaním akéhokoľvek nového lieku informovali lekárov a zubných lekárov, že užívajú tikagrelor.
V štúdii PLATO bol výskyt krvácania u pacientov, ktorí podstúpili koronárny artériový by –pass (coronary artery bypass grafting, CABG), vyšší pri tikagrelore ako pri klopidogrele pri jeho vysadení v priebehu 1 dňa pred chirurgickým zákrokom, ale výskyt veľkého krvácania pri vysadení liečby 2 alebo viac dní pred chirurgickým zákrokom bol podobný ako pri klopidogrele (pozri časť 4.8). Ak má pacient podstúpiť plánovaný chirurgický zákrok a protidoštičkový účinok nie je požadovaný, tikagrelor sa má vysadiť 7 dní pred chirurgickým zákrokom (pozri časť 5.1).
Pacientispredchádzajúcouischemickoucievnoumozgovoupríhodou
Pacienti s AKS s predchádzajúcou ischemickou cievnou mozgovou príhodou môžu byť liečení tikagrelorom až 12 mesiacov (štúdia PLATO).
Do štúdie PEGASUS neboli zahrnutí pacienti s IM v anamnéze a predchádzajúcou ischemickou cievnou mozgovou príhodou. Vzhľadom na chýbajúce údaje sa u týchto pacientov liečba presahujúca jeden rok neodporúča.
Poruchafunkciepečene
Použitie tikagreloru je kontraindikované u pacientov s ťažkou poruchou funkcie pečene (pozri časti
4.2 a 4.3). K dispozícii sú obmedzené skúsenosti s tikagrelorom u pacientov so stredne ťažkou poruchou funkcie pečene, u týchto pacientov sa preto odporúča opatrnosť (pozri časti 4.2 a 5.2).
Pacientisrizikombradykardickýchpríhod
Kvôli zisteniam väčšinou asymptomatických prípadov ventrikulárnej pauzy v skoršej klinickej štúdii boli z hlavných štúdií hodnotiacich bezpečnosť a účinnosť tikagreloru vyradení pacienti so zvýšeným rizikom bradykardických príhod (napr. pacienti bez kardiostimulátora so syndrómom chorého sínusového uzla, AV blokádou 2. alebo 3. stupňa alebo synkopou súvisiacou s bradykardiou). Vzhľadom na obmedzené klinické skúsenosti sa má preto tikagrelor u týchto pacientov používať
s opatrnosťou (pozri časť 5.1).
Okrem toho je potrebná opatrnosť aj pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že vyvolávajú bradykardiu. V štúdii PLATO sa však po súbežnom podaní s jedným alebo viacerými liekmi, o ktorých je známe, že vyvolávajú bradykardiu (napr. 96 % betablokátory, 33 % blokátory kalciových kanálov diltiazem a verapamil a 4 % digoxín), nepozorovali žiadne klinicky významné nežiaduce reakcie (pozri časť 4.5).
V priebehu podštúdie s Holterovým monitorovaním v štúdii PLATO sa ventrikulárne pauzy trvajúce
≥ 3 sekundy vyskytli u väčšieho počtu pacientov užívajúcich tikagrelor ako pacientov užívajúcich klopidogrel v akútnej fáze ich AKS. Nárast počtu ventrikulárnych páuz pri tikagrelore, odhalených
Holterovým monitorovaním, bol u pacientov s chronickým srdcovým zlyhávaním (CHF) vyšší ako
u celkového počtu pacientov v štúdii počas akútnej fázy AKS, avšak nie po 1 mesiaci liečby tikagrelorom alebo v porovnaní s klopidogrelom. Z tejto nerovnováhy však u tejto skupiny pacientov nevyplývali žiadne nežiaduce klinické dôsledky (vrátane synkopy alebo zavedenia kardiostimulátora) (pozri časť 5.1).
Dyspnoe
U pacientov liečených tikagrelorom sa hlásilo dyspnoe. Dyspnoe je zvyčajne miernej až stredne ťažkej intenzity a často ustúpi bez potreby ukončenia liečby. Absolútne riziko výskytu dyspnoe pri užívaní tikagreloru môže byť vyššie u pacientov s astmou/chronickou obštrukčnou chorobou pľúc (CHOCHP). Tikagrelor sa má používať opatrne u pacientov s astmou a/alebo CHOCHP v anamnéze. Mechanizmus nie je objasnený. Ak pacient hlási nové, dlhotrvajúce alebo zhoršené dyspnoe, dyspnoe sa má
dôkladne vyšetriť a pri neznášanlivosti sa má liečba tikagrelorom ukončiť. Pre ďalšie informácie pozri časť 4.8.
Zvýšeniekreatinínu
Počas liečby tikagrelorom sa môžu zvýšiť hladiny kreatinínu. Mechanizmus nie je objasnený. Funkcia obličiek sa má kontrolovať v súlade s bežnou lekárskou praxou. U pacientov s AKS sa tiež odporúča skontrolovať funkciu obličiek jeden mesiac po začatí liečby tikagrelorom s venovaním osobitnej pozornosti pacientom vo veku ≥ 75 rokov, pacientom so stredne ťažkou/ťažkou poruchou funkcie obličiek a pacientom súbežne liečeným blokátorom receptora angiotenzínu (ARB).
Zvýšeniekyselinymočovej
Počas liečby tikagrelorom sa môže objaviť hyperurikémia (pozri časť 4.8). U pacientov
s hyperurikémiou alebo dnovou artritídou v anamnéze sa odporúča opatrnosť. Ako preventívne opatrenie je potrebné zabrániť použitiu tikagreloru u pacientov s urátovou nefropatiou.
Iné
Na základe vzťahu zaznamenaného v štúdii PLATO medzi udržiavacou dávkou ASA a relatívnou účinnosťou tikagreloru v porovnaní s klopidogrelom sa súbežné podávanie tikagreloru a vysokej
udržiavacej dávky ASA (> 300 mg) neodporúča (pozri časť 5.1).
Predčasnéukončenieliečby
Predčasné ukončenie akejkoľvek protidoštičkovej liečby, vrátane lieku Brilique, môže viesť
k zvýšenému riziku kardiovaskulárnej (KV) smrti alebo IM v dôsledku základného ochorenia pacienta. Preto sa treba vyhnúť predčasnému ukončeniu liečby.
4.5 Liekové a iné interakcie
Tikagrelor je predovšetkým substrátom CYP3A4 a miernym inhibítorom CYP3A4. Tikagrelor je tiež substrátom P-glykoproteínu (P-gp) a slabým inhibítorom P-gp a môže zvyšovať expozíciu substrátom P-gp.
Účinky inýchliekovnatikagrelor
Liečivámetabolizované prostredníctvom CYP3A4
Inhibítory CYP3A4
· Silné inhibítory CYP3A4 – pri súbežnom podávaní ketokonazolu a tikagreloru sa Cmax tikagreloru zvýšil 2,4-násobne a AUC 7,3-násobne. Cmax aktívneho metabolitu sa znížil o 89 % a AUC o 56 %. Dá sa predpokladať, že účinky iných silných inhibítorov CYP3A4
(klaritromycín, nefazodón, ritonavir a atazanavir) sú podobné a preto je súbežné použitie silných inhibítorov CYP3A4 s tikagrelorom kontraindikované (pozri časť 4.3).
· Stredne silné inhibítory CYP3A4 – pri súbežnom podávaní diltiazemu a tikagreloru sa Cmax tikagreloru zvýšil o 69 % a AUC 2,7-násobne a v prípade aktívneho metabolitu došlo k zníženiu Cmax o 38 % a AUC ostala nezmenená. Tikagrelor nemal žiadny vplyv na hladiny diltiazemu
v plazme. Podobný účinok možno predpokladať aj u iných stredne silných inhibítorov CYP3A4
(napr. amprenavir, aprepitant, erytromycín a flukonazol) a tiež ich možno podávať súbežne s tikagrelorom.
Induktory CYP3A4
Pri súbežnom podávaní rifampicínu a tikagreloru sa Cmax tikagreloru znížil o 73 % a AUC o 86 %.
Cmax aktívneho metabolitu ostal nezmenený a AUC sa znížila o 46 %. Dá sa predpokladať, že aj ďalšie
induktory CYP3A4 (napr. fenytoín, karbamazepín a fenobarbital) znižujú expozíciu tikagreloru.
Súbežné podávanie tikagreloru so silnými induktormi CYP3A4 môže znižovať expozíciu a účinnosť tikagreloru, preto sa má zabrániť ich súbežnému použitiu s tikagrelorom.
Cyklosporín(inhibítorP-gpaCYP3A)
Pri súbežnom podávaní cyklosporínu (600 mg) a tikagreloru sa Cmax tikagreloru zvýšil 2,3-násobne
a AUC 2,8-násobne. V prítomnosti cyklosporínu sa AUC aktívneho metabolitu zvýšila o 32 % a Cmax
sa znížil o 15 %.
K dispozícii nie sú žiadne údaje týkajúce sa súbežného podávania tikagreloru a ďalších liečiv, ktoré sú tiež silnými inhibítormi P-gp a stredne silnými inhibítormi CYP3A4 (napr. verapamil, chinidín), ktoré takisto môžu zvyšovať expozíciu tikagreloru. Ak sa takejto kombinácii nie je možné vyhnúť, ich súbežné použitie si vyžaduje opatrnosť.
Iné
Klinické štúdie farmakologických interakcií preukázali, že súbežné podávanie tikagreloru
s heparínom, enoxaparínom a ASA alebo dezmopresínom v porovnaní s podávaním samotného tikagreloru nemalo žiadny vplyv na farmakokinetiku tikagreloru alebo jeho aktívneho metabolitu,
alebo na agregáciu krvných doštičiek indukovanú ADP. Ak je to klinicky indikované, lieky
ovplyvňujúce hemostázu sa majú v kombinácii s tikagrelorom používať opatrne.
Pri dennej konzumácii väčšieho množstva grapefruitovej šťavy (3 x 200 ml) sa pozorovalo 2-násobné zvýšenie expozície tikagreloru. Neočakáva sa, že rozsah tejto zvýšenej expozície bude klinicky významný pre väčšinu pacientov.
Účinky tikagreloru na inélieky
Liečivámetabolizované prostredníctvom CYP3A4
· Simvastatín – pri súbežnom podávaní tikagreloru a simvastatínu sa Cmax simvastatínu zvýšil o 81 % a AUC o 56 %, Cmax kyseliny simvastatínovej sa zvýšil o 64 % a AUC o 52 %,
v niektorých jednotlivých prípadoch boli zvýšenia 2- až 3-násobné. Súbežné podávanie tikagreloru so simvastatínom v dávkach vyšších ako 40 mg denne môže spôsobiť nežiaduce reakcie simvastatínu a potenciálny prínos tejto kombinácie je potrebné zvážiť. Simvastatín
nemal žiadny vplyv na hladiny tikagreloru v plazme. Tikagrelor môže mať podobný účinok na lovastatín. Súbežné použitie tikagreloru so simvastatínom alebo lovastatínom v dávkach vyšších
ako 40 mg sa neodporúča.
· Atorvastatín - pri súbežnom podávaní atorvastatínu a tikagreloru sa Cmax kyseliny atorvastatínovej zvýšil o 23 % a AUC o 36 %. Podobné zvýšenia AUC a Cmax sa pozorovali u všetkých metabolitov kyseliny atorvastatínovej. Tieto zvýšenia sa nepovažujú za klinicky významné.
· Podobný účinok na iné statíny metabolizované prostredníctvom CYP3A4 nemožno vylúčiť.
U pacientov dostávajúcich tikagrelor v štúdii PLATO, ktorí užívali rôzne statíny, nevznikli
v súvislosti s bezpečnosťou statínov žiadne obavy u 93 % pacientov v kohorte štúdie PLATO
užívajúcej tieto lieky.
Tikagrelor je miernym inhibítorom CYP3A4. Súbežné podávanie tikagreloru a substrátov CYP3A4 s úzkym terapeutickým indexom (napr. cisaprid alebo námeľové alkaloidy) sa neodporúča, nakoľko tikagrelor môže zvyšovať expozíciu týchto liekov.
SubstrátyP-gp(vrátanedigoxínu,cyklosporínu)
Pri súbežnom podávaní tikagreloru sa Cmax digoxínu zvýšil o 75 % a AUC o 28 %. Priemerné hladiny digoxínu pred podaním ďalšej dávky sa pri súbežnom podávaní s tikagrelorom zvýšili približne
o 30 %, v niektorých jednotlivých prípadoch maximálne až na 2-násobok. V prítomnosti digoxínu nebola ovplyvnená Cmax a AUC tikagreloru a jeho aktívneho metabolitu. Pri podávaní P-gp-
dependentných liečiv s úzkym terapeutickým indexom, ako je digoxín, súbežne s tikagrelorom sa preto odporúča náležité klinické a/alebo laboratórne monitorovanie.
Nebol pozorovaný žiadny vplyv tikagreloru na hladinu cyklosporínu v krvi. Vplyv tikagreloru na ďalšie substráty P-gp sa neskúmal.
LiečivámetabolizovanéCYP2C9
Súbežné podávanie tikagreloru a tolbutamidu neviedlo k zmenám plazmatických hladín žiadneho z liečiv, čo naznačuje, že tikagrelor nie je inhibítorom CYP2C9 a nie je pravdepodobné, že by
spôsoboval zmeny v metabolizme sprostredkovanom CYP2C9 u liečiv, akými sú warfarín a
tolbutamid.
Perorálnekontraceptíva
Pri súbežným podávaní tikagreloru a levonorgestrelu a etinylestradiolu sa expozícia etinylestradiolu zvýšila približne o 20 %, ale k zmenám vo farmakokinetike levonorgestrelu nedošlo. Pri súbežnom podávaní levonorgestrelu a etinylestradiolu s tikagrelorom sa nepredpokladá žiadny klinicky významný vplyv na účinnosť perorálnych kontraceptív.
Liečivá,ktorévyvolávajúbradykardiu
Pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že vyvolávajú bradykardiu, je potrebná opatrnosť vzhľadom na pozorované prípady zväčša asymptomatickej ventrikulárnej pauzy
a bradykardie (pozri časť 4.4). V štúdii PLATO sa však po súbežnom podaní s jedným alebo
viacerými liekmi, o ktorých je známe, že vyvolávajú bradykardiu (napr. 96 % betablokátory, 33 %
blokátory kalciových kanálov diltiazem a verapamil a 4 % digoxín) nepozorovali žiadne klinicky významné nežiaduce reakcie.
Ďalšiasúbežnáliečba
V klinických štúdiách sa tikagrelor bežne podával spolu s ASA, inhibítormi protónovej pumpy, statínmi, betablokátormi, inhibítormi enzýmu konvertujúceho angiotenzín (ACE) a s blokátormi receptorov angiotenzínu dlhodobo, ak si to súbežné ochorenia vyžadovali a tiež s heparínom, nízkomolekulovým heparínom a intravenóznymi inhibítormi GpIIb/IIIa pri krátkodobom podávaní (pozri časť 5.1). Nepozorovali sa žiadne klinicky významné nežiaduce interakcie s týmito liečivami.
Súbežné podávanie tikagreloru s heparínom, enoxaparínom alebo dezmopresínom nemalo žiadny vplyv na aktivovaný parciálny tromboplastínový čas (aPTT), aktivovaný koagulačný čas (ACT) alebo na testy faktora Xa. Pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že ovplyvňujú hemostázu, je však potrebné postupovať s opatrnosťou pre možné farmakodynamické interakcie.
Vzhľadom na hlásenia abnormalít kožného krvácania so SSRI (napr. paroxetín, sertralín a citalopram) sa pri súbežnom podávaní tikagreloru so SSRI odporúča opatrnosť, pretože to môže zvýšiť riziko krvácania.
4.6 Fertilita, gravidita a laktácia
Ženyvreprodukčnomveku
Ženy v reprodukčnom veku majú počas liečby tikagrelorom používať vhodné antikoncepčné metódy na zabránenie gravidity.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití tikagreloru u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Tikagrelor sa neodporúča užívať
počas gravidity.
Dojčenie
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie tikagreloru a jeho aktívnych metabolitov do mlieka (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť
vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu tikagrelorom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Tikagrelor nemal žiadny vplyv na fertilitu samcov alebo samíc zvierat (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tikagrelor nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Počas liečby tikagrelorom sa hlásil závrat a zmätenosť. Pacienti, u ktorých sa objavia tieto príznaky, majú byť preto pri vedení vozidiel alebo obsluhe strojov opatrní.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnostný profil tikagreloru sa hodnotil v dvoch rozsiahlych skúšaniach fázy 3 (PLATO
a PEGASUS), ktoré zahŕňali viac ako 39 000 pacientov (pozri časť 5.1).
V štúdii PLATO bol pri tikagrelore vyšší výskyt pacientov, ktorí ukončili liečbu pre nežiaduce udalosti, ako pri klopidogrele (7,4 % oproti 5,4 %). V štúdii PEGASUS bol pri tikagrelore vyšší výskyt pacientov, ktorí ukončili liečbu pre nežiaduce udalosti, v porovnaní s liečbou samotnou ASA (16,1 % pre 60 mg tikagreloru s ASA oproti 8,5 % pre liečbu samotnou ASA). Najčastejšie hlásenými nežiaducimi reakciami u pacientov liečených tikagrelorom boli krvácanie a dyspnoe (pozri časť 4.4).
Tabuľkovýzoznamnežiaducichreakcií
V klinických štúdiách s tikagrelorom alebo v období po uvedení lieku na trh sa zistili a hlásili nasledujúce nežiaduce reakcie (tabuľka 1).
Nežiaduce reakcie sú uvedené podľa tried orgánových systémov (TOS) MedDRA. V rámci každej
TOS sú nežiaduce reakcie usporiadané podľa kategórie frekvencie. Kategórie frekvencie sú
definované podľa nasledovných pravidiel: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme
(z dostupných údajov).
Tabuľka 1 – Nežiaduce reakcie podľa frekvencie a triedy orgánových systémov (TOS) TOS Veľmi časté Časté Menej časté
Benígne a malígne
nádory, vrátane nešpecifikovaných
novotvarov (cysty
a polypy) Poruchy krvi
a lymfatického
systému
Poruchy imunitného systému
Poruchy metabolizmu a výživy
krvácavá poruchab
hyperurikémiad dna/dnová artritída
krvácanie nádorua
precitlivenosť vrátane angioedémuc
Psychické poruchy zmätenosť
Poruchy nervového systému
závrat, synkopa, bolesť hlavy
intrakraniálne krvácanie
Poruchy oka krvácanie do okae
Poruchy ucha a labyrintu
vertigo krvácanie do ucha
 Poruchy ciev
Poruchy ciev hypotenzia
T
O
S Veľmi časté Časté Menej časté
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy obličiek a močových ciest Poruchy reprodukčného systému a prsníkov Laboratórne
a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
dyspnoe krvácanie do dýchacích ciestf
gastrointestinálne krvácanieg, hnačka, nauzea, dyspepsia, zápcha
podkožné alebo kožné krvácanieh, vyrážka, pruritus
krvácanie do močových ciestj
zvýšený kreatinín v krvid
krvácanie po zákroku, poúrazové krvácaniel
retroperitoneálne krvácanie
krvácanie do svalui
krvácanie do reprodukčného systémuk
a napr. krvácanie z nádoru močového mechúra, nádoru žalúdka, nádoru hrubého čreva
b napr. zvýšený sklon k tvorbe krvných podliatin, spontánny hematóm, hemoragická diatéza
c Identifikované v období po uvedení lieku na trh.
d Frekvencie odvodené z laboratórnych pozorovaní (Zvýšenia kyseliny močovej ˃ horná hranica normálu oproti východiskovej hodnote, pod alebo v rámci referenčného rozmedzia. Zvýšenia kreatinínu ˃ 50 % oproti východiskovej
hodnote.), a nie neprepočítané frekvencie hlásených nežiaducich udalostí.
e napr. krvácanie do spojovky, sietnice, vnútroočné krvácanie
f napr. epistaxa, hemoptýza
g napr. krvácanie ďasien, rektálne krvácanie, krvácanie žalúdkového vredu
h napr. ekchymóza, krvácanie do kože, petechie
i napr. hemartróza, krvácanie do svalu
j napr. hematúria, hemoragická cystitída
k napr. vaginálne krvácanie, hematospermia, postmenopauzálne krvácanie
l napr. kontúzia, poúrazový hematóm, poúrazové krvácanie
Popis vybraných nežiaducichreakcií
Krvácanie
Zistenia týkajúce sa krvácania v štúdii PLATO
V tabuľke 2 sú uvedené celkové výsledky miery krvácania v štúdii PLATO.
Tabuľka 2 – Analýza celkových krvácavých príhod, Kaplanov-Meierov odhad v 12.mesiaci
(štúdia PLATO)
tikagrelor
90 mg dvakrát denne
 N=9235
klopidogrel
N=9186 p-hodnota*
N=9235
klopidogrel
N=9186 p-hodnota*
Veľké krvácania podľa PLATO celkovo 11,6 11,2 0,4336
Veľké fatálne/život ohrozujúce krvácania podľa PLATO
Veľké krvácania podľa PLATO
nesúvisiace s CABG
5,8 5,8 0,6988
4,5 3,8 0,0264
Veľké krvácania podľa PLATO 3,1 2,3 0,0058
nesúvisiace s liečebným postupom Veľké + malé krvácania podľa PLATO celkovo
Veľké + malé krvácania podľa PLATO
nesúvisiace s liečebným postupom
Veľké krvácania definované podľa kritérií
TIMI
Veľké + malé krvácania definované podľa kritérií TIMI
Definície kategórií krvácania:16,1 14,6 0,0084
5,9 4,3 <0,0001
7,9 7,7 0,5669
11,4 10,9 0,3272
Veľké fatálne/ život ohrozujúce krvácanie: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o viac ako 50 g/l
alebo s transfúziou ≥ 4 jednotiek erytrocytov alebo fatálne alebo intrakraniálne alebo intraperikardiálne krvácanie s tamponádou srdca; alebo hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov alebo chirurgickú intervenciu.
Veľké iné: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50 g/l alebo s transfúziou 2 až 3 jednotiek erytrocytov; alebo významne vysiľujúce krvácanie.
Malé krvácanie: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
Veľké krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu > 50 g/l alebo intrakraniálne krvácanie.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50g/l.
*p-hodnota vypočítaná na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO, veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI alebo malého krvácania podľa kritérií TIMI (tabuľka 2). V porovnaní
s klopidogrelom sa však pri tikagrelore vyskytlo spolu viac veľkých a malých krvácaní podľa definície PLATO. Niekoľko pacientov v štúdii PLATO malo fatálne krvácania: 20 (0,2 %) pri tikagrelore a 23 (0,3 %) pri klopidogrele (pozri časť 4.4).
Vek, pohlavie, hmotnosť, rasa, geografické územie, súbežné ochorenia, súbežná liečba a anamnéza pacienta, vrátane prekonanej cievnej mozgovej príhody alebo prekonaného prechodného ischemického záchvatu, neboli prediktívne z hľadiska celkového výskytu krvácaní, ani z hľadiska výskytu veľkého krvácania podľa definície PLATO nesúvisiaceho s liečebným postupom. Preto pre žiaden podtyp krvácania nebola konkrétna skupina identifikovaná ako riziková.
Krvácanie súvisiace s CABG:
V štúdii PLATO sa u 42 % z 1 584 pacientov (12 % kohorty), ktorí podstúpili zákrok CABG, vyskytlo veľké fatálne/život ohrozujúce krvácanie podľa definície PLATO, bez rozdielu medzi liečebnými
skupinami. Fatálne krvácanie súvisiace s CABG sa vyskytlo v každej liečebnej skupine u 6 pacientov
(pozri časť 4.4).
Krvácanie nesúvisiace s CABG a krvácanie nesúvisiace s liečebným postupom:
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO nesúvisiaceho s CABG, ale výskyt veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI a výskyt veľkého a malého krvácania podľa kritérií TIMI bol častejší pri tikagrelore. Rovnako, po vylúčení všetkých krvácaní súvisiacich s liečebným postupom, sa viac krvácaní vyskytlo pri tikagrelore ako pri klopidogrele (tabuľka 2). Ukončenie liečby pre krvácanie nesúvisiace s liečebným postupom bolo častejšie pri tikagrelore (2,9 %) ako pri klopidogrele (1,2 %; p< 0,001).
Intrakraniálne krvácanie:
V skupine s tikagrelorom sa vyskytlo viac intrakraniálnych krvácaní nesúvisiacich s liečebným postupom (n = 27 krvácaní u 26 pacientov, 0,3 %) ako v skupine s klopidogrelom (n = 14 krvácaní,
0,2 %), z ktorých bolo fatálnych 11 v skupine s tikagrelorom a 1 v skupine s klopidogrelom.
V celkovom výskyte fatálnych krvácaní nebol žiadny rozdiel.
Zistenia týkajúce sa krvácania v štúdii PEGASUS
V tabuľke 3 sú uvedené celkové výsledky udalostí súvisiacich s krvácaním v štúdii PEGASUS.
Tabuľka 3 – Analýza celkových krvácavých príhod, Kaplanov-Meierov odhad v 36. mesiaci
(štúdia PEGASUS)
tikagrelor 60 mg dvakrát
denne + ASA N = 6 958
Pomer
samotná ASA N = 6 996
Koncový ukazovateľ bezpečnosti KM %
Kategórie krvácania podľa kritérií TIMI
rizika
(95 % IS)
KM % p-hodnota
Veľké krvácania podľa TIMI 2,3 2,32 (1,68; 3,21)
Fatálne 0,3 1,00 (0,44; 2,27)
ICH 0,6 1,33 (0,77; 2,31)
1,1 < 0,0001
0,3 1,0000
0,5 0,3130
Iné veľké krvácania podľa
TIMI 1,6
Veľké alebo malé krvácania podľa
TIMI 3,4
Veľké alebo malé krvácania alebo
3,61
(2,31; 5,65) 0,5 < 0,0001
2,54
(1,93; 3,35) 1,4 < 0,0001
krvácania vyžadujúce lekársku starostlivosť podľa TIMI
16,6 2,64 (2,35; 2,97)
7,0 < 0,0001
Kategórie krvácania podľa definície PLATO
Veľké krvácania podľa PLATO 3,5 2,57 (1,95; 3,37)
Fatálne/život ohrozujúce 2,4 2,38 (1,73; 3,26)
1,4 < 0,0001
1,1 < 0,0001
Iné veľké krvácania podľa
PLATO 1,1
Veľké alebo malé krvácania podľa
PLATO 15,2
Definície kategórií krvácania:
3,37
(1,95; 5,83) 0,3 < 0,0001
2,71
(2,40; 3,08) 6,2 < 0,0001
 Veľké krvácanie podľa TIMI:
Veľké krvácanie podľa TIMI: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO klinicky zjavné prejavy hemorágie spojené s poklesom hemoglobínu (Hgb) ≥ 50 g/l, alebo 15 % pokles hematokritu (Hct) v prípade nedostupnosti údajov o Hgb.
Fatálne krvácanie: Krvácavá príhoda, ktorá viedla priamo k smrti v priebehu 7 dní.
ICH: Intrakraniálne krvácanie.
Iné veľké krvácanie podľa TIMI: Veľké non-fatálne krvácanie podľa TIMI iné než ICH.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l.
Krvácanie vyžadujúce lekársku starostlivosť podľa TIMI: Vyžadujúce zásah, ALEBO vedúce k hospitalizácii, ALEBO
vyžadujúce vyšetrenie.
Veľké fatálne/život ohrozujúce krvácanie podľa PLATO: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO intraperikardiálne krvácanie s tamponádou srdca, ALEBO s hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov/inotropík alebo chirurgickú intervenciu, ALEBO klinicky zjavné krvácanie s poklesom
hemoglobínu ˃ 50 g/l alebo s transfúziou ≥ 4 jednotiek erytrocytov.
Iné veľké krvácanie podľa PLATO: Významne vysiľujúce krvácanie, ALEBO klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l alebo s transfúziou 2 - 3 jednotiek erytrocytov.
Malé krvácanie podľa PLATO: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
V štúdii PEGASUS bol výskyt veľkého krvácania podľa kritérií TIMI pri tikagrelore v dávke 60 mg dvakrát denne vyšší ako pri samotnej ASA. Nepozorovalo sa zvýšené riziko fatálneho krvácania
a pozorovalo sa len menej významné zvýšenie výskytu intrakraniálneho krvácania v porovnaní
s liečbou samotnou ASA. V štúdii sa vyskytlo niekoľko fatálnych krvácavých príhod, 11 (0,3 %) pri
60 mg tikagreloru a 12 (0,3 %) pri liečbe samotnou ASA. Pozorované zvýšené riziko výskytu veľkého krvácania podľa kritérií TIMI pri 60 mg tikagreloru bolo zapríčinené predovšetkým vyššou frekvenciou výskytu iného veľkého krvácania podľa kritérií TIMI zastúpeného udalosťami
v gastrointestinálnej TOS.
Podobne zvýšený výskyt veľkého krvácania podľa kritérií TIMI bol pozorovaný aj pri kategóriách krvácania zahŕňajúcich veľké alebo malé krvácania podľa kritérií TIMI, veľké krvácania podľa definície PLATO a veľké alebo malé krvácania podľa definície PLATO (pozri tabuľku 3). Ukončenie liečby pre krvácanie bolo častejšie pri 60 mg tikagreloru (6,2 %) v porovnaní s liečbou samotnou ASA (1,5 %). Väčšina týchto krvácaní bola menej závažná (klasifikované ako krvácania vyžadujúce lekársku starostlivosť podľa kritérií TIMI), napr. epistaxa, tvorba krvných podliatin a hematómov.
Profil krvácania pri 60 mg tikagreloru bol pre udalosti veľkého krvácania podľa kritérií TIMI, veľkého alebo malého krvácania podľa kritérií TIMI a veľkého krvácania podľa definície PLATO konzistentný v rámci viacerých vopred definovaných podskupín (napr. podľa veku, pohlavia, hmotnosti, rasy, geografického regiónu, súbežných ochorení, súbežnej liečby a anamnézy).
Intrakraniálne krvácanie:
Pre 60 mg tikagreloru a liečbu samotnou ASA sa hlásili podobné miery výskytu spontánneho intrakraniálneho krvácania (n = 13, 0,2 % v oboch liečebných skupinách). Pri liečbe 60 mg tikagreloru (n = 15, 0,2 %) v porovnaní s liečbou samotnou ASA (n = 10, 0,1 %) sa preukázalo menej významné zvýšenie výskytu poúrazového intrakraniálneho krvácania a intrakraniálneho krvácania súvisiaceho
s liečebným postupom. Pri 60 mg tikagreloru sa vyskytlo 6 fatálnych prípadov intrakraniálneho krvácania a pri liečbe samotnou ASA 5 prípadov. Výskyt intrakraniálneho krvácania bol v oboch
liečebných skupinách nízky vzhľadom na to, že populácia štúdie sa vyznačovala významnou mierou
komorbidity a KV rizikových faktorov.
Dyspnoe
Pacienti liečení Brilique hlásili dyspnoe, pocit sťaženého dýchania. V štúdii PLATO nežiaduce udalosti týkajúce sa dyspnoe (dyspnoe, kľudové dyspnoe, námahové dyspnoe, paroxyzmálne nočné dyspnoe a nočné dyspnoe) hlásili u 13,8 % pacientov liečených s tikagrelorom a u 7,8 % pacientov liečených s klopidogrelom. V štúdii PLATO u 2,2 % pacientov užívajúcich tikagrelor a u 0,6 % pacientov užívajúcich klopidogrel skúšajúci považovali dyspnoe za príčinu súvisiacu s liečbou a málo prípadov bolo závažných (0,14 % tikagrelor; 0,02 % klopidogrel) (pozri časť 4.4). Väčšina hlásených príznakov dyspnoe bola miernej až stredne ťažkej intenzity a väčšinou sa hlásili ako jedna epizóda krátko po začatí liečby.
V porovnaní s klopidogrelom bolo u pacientov s astmou/CHOCHP, ktorí boli liečení tikagrelorom, zvýšené riziko výskytu nezávažného dyspnoe (3,29 % v prípade tikagreloru oproti 0,53 % v prípade klopidogrelu) a závažného dyspnoe (0,38 % v prípade tikagreloru oproti 0,0 % v prípade klopidogrelu). V absolútnom vyjadrení bolo toto riziko vyššie ako v celej populácii pacientov v štúdii PLATO. Tikagrelor sa musí užívať opatrne u pacientov s anamnézou astmy a/alebo CHOCHP (pozri časť 4.4).
Približne 30 % všetkých prípadov dyspnoe sa upravilo v priebehu 7 dní. Štúdie PLATO sa zúčastnili pacienti s kongestívnym srdcovým zlyhaním, CHOCHP alebo astmou pred začatím skúšania; títo pacienti a starší pacienti hlásili najčastejšie dyspnoe. Kvôli dyspnoe sa liečba tikagrelorom vysadila u
0,9 % pacientov a liečba klopidogrelom u 0,1 % pacientov. Vyšší výskyt dyspnoe u tikagreloru nesúvisí s novým alebo zhoršujúcim sa srdcovým alebo pľúcnym ochorením (pozri časť 4.4). Tikagrelor nemá vplyv na výsledky testov funkcie pľúc.
V štúdii PEGASUS sa dyspnoe hlásilo u 14,2 % pacientov užívajúcich tikagrelor v dávke 60 mg dvakrát denne a u 5,5 % pacientov užívajúcich samotnú ASA. Rovnako ako v štúdii PLATO, väčšina hlásených prípadov dyspnoe bola miernej až stredne ťažkej intenzity (pozri časť 4.4). Pacienti, ktorí hlásili dýchavičnosť boli starší a mali na začiatku častejšie dýchavičnosť, CHOCHP alebo astmu.
Vyšetrenia
Zvýšenie kyseliny močovej: V štúdii PLATO došlo k zvýšeniu kyseliny močovej v sére nad hornú hranicu normálu u 22 % pacientov dostávajúcich tikagrelor, v porovnaní s 13 % pacientov dostávajúcich klopidogrel. V štúdii PEGASUS boli príslušné hodnoty 9,1 % pre 90 mg tikagreloru,
8,8 % pre 60 mg tikagreloru a 5,5 % pre placebo. Priemerná hladina kyseliny močovej v sére sa pri tikagrelore zvýšila približne o 15 % v porovnaní so zvýšením približne o 7,5 % pri klopidogrele a po
ukončení liečby sa znížila približne o 7 % v prípade tikagreloru, v prípade klopidogrelu sa však žiadne zníženie nezistilo. V štúdii PEGASUS sa zistilo reverzibilné zvýšenie priemerných hladín kyseliny močovej v sére o 6,3 % pre 90 mg tikagreloru a 5,6 % pre 60 mg tikagreloru, v porovnaní s 1,5 % znížením v skupine s placebom. V štúdii PLATO bola frekvencia dnovej artritídy 0,2 % pri tikagrelore oproti 0,1 % pri klopidogrele. V štúdii PEGASUS boli príslušné hodnoty frekvencie dny/dnovej artritídy 1,6 % pre 90 mg tikagreloru, 1,5 % pre 60 mg tikagreloru a 1,1 % pre placebo.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieTikagrelor je v jednorazových dávkach až do 900 mg dobre znášaný. Gastrointestinálna toxicita bola určujúcim faktorom v štúdii zameranej na zvyšovanie jednorazovej dávky. Iné klinicky významné nežiaduce reakcie, ktoré sa môžu vyskytnúť pri predávkovaní, zahŕňajú dyspnoe a ventrikulárne pauzy (pozri časť 4.8).
V prípade predávkovania sa môžu objaviť vyššie uvedené potenciálne nežiaduce reakcie a je potrebné zvážiť monitorovanie EKG.
V súčasnosti nie je známe žiadne antidotum na zvrátenie účinkov tikagreloru a nepredpokladá sa, že by bol tikagrelor dialyzovateľný (pozri časť 4.4). Liečba predávkovania sa má riadiť štandardnou lekárskou praxou na miestnej úrovni. Predpokladaným účinkom nadmerných dávok tikagreloru je predĺžené trvanie rizika krvácania, ktoré súvisí s inhibíciou krvných doštičiek. Transfúzia krvných doštičiek pravdepodobne nepredstavuje klinický prínos pre pacientov s krvácaním (pozri časť 4.4). Ak dôjde ku krvácaniu, je potrebné prijať ďalšie príslušné podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiagreganciá trombocytov okrem heparínu, ATC kód: B01AC24
MechanizmusúčinkuBrilique obsahuje tikagrelor patriaci do chemickej skupiny cyklopentyltriazolopyrimidínov (CPTP), ktorý je perorálnym, priamo pôsobiacim, selektívnym antagonistom, reverzibilne sa viažucim na
receptor P2Y12, ktorý zabraňuje aktivácii a agregácii krvných doštičiek sprostredkovanej ADP
a závislej na P2Y12. Tikagrelor nezabraňuje väzbe ADP, ale po naviazaní na receptor P2Y12 zabraňuje
signálnej transdukcii indukovanej ADP. Vzhľadom na to, že krvné doštičky sa podieľajú na vzniku
a/alebo vývoji trombotických komplikácií aterosklerotických ochorení, preukázalo sa, že inhibícia funkcie krvných doštičiek znižuje riziko kardiovaskulárnych príhod, ako je smrť, IM alebo cievna
mozgová príhoda.
Tikagrelor tiež zvyšuje lokálne hladiny endogénneho adenozínu inhibíciou rovnovážnych nukleozidových transportérov-1 (ENT-1).
Zistilo sa, že tikagrelor u zdravých dobrovoľníkov a pacientov s AKS zvýrazňuje nasledujúce účinky indukované adenozínom: vazodilatácia (merané zvýšením koronárneho prietoku krvi u zdravých dobrovoľníkov a pacientov s AKS; bolesť hlavy), inhibícia funkcie krvných doštičiek (v plnej ľudskej krvi
in vitro) a dyspnoe. Súvislosť medzi pozorovanými zvýšeniami adenozínu a klinickými výsledkami (napr. morbidita-mortalita) však nebola jasne vysvetlená.
Farmakodynamické
účinky
Nástup
účinku
Tikagrelor u pacientov so stabilnou koronárnou artériovou chorobou (CAD) užívajúcich ASA
vykazuje rýchly nástup farmakologického účinku, čo sa preukázalo priemernou inhibíciou agregácie krvných doštičiek (IPA) tikagrelorom po 0,5 hodiny od podania nárazovej dávky 180 mg približne
41 %, s maximálnym účinkom na IPA 89 % po 2 – 4 hodinách od podania dávky a tento účinok pretrvával 2 – 8 hodín. U 90 % pacientov bol finálny rozsah IPA po 2 hodinách od podania dávky
> 70 %.
Odznievanie účinku
Pri plánovanom zákroku CABG existuje zvýšené riziko krvácania pre tikagrelor oproti klopidogrelu, pokiaľ je liečba vysadená v kratšej dobe ako 96 hodín pred zákrokom.
Údajetýkajúcesa prechodunainúliečbu
Prechod z liečby klopidogrelom v dávke 75 mg na tikagrelor v dávke 90 mg dvakrát denne má za následok absolútne zvýšenie IPA o 26,4 % a prechod z liečby tikagrelorom na klopidogrel má za následok absolútne zníženie IPA o 24,5 %. Pacientov možno prestaviť z liečby klopidogrelom na tikagrelor bez akéhokoľvek prerušenia protidoštičkového účinku (pozri časť 4.2).
Klinickáúčinnosťabezpečnosť
Klinický dôkaz účinnosti a bezpečnosti tikagreloru je odvodený z dvoch skúšaní fázy 3:
· Štúdia PLATO [PLATelet Inhibition and Patient Outcomes], porovnávajúca tikagrelor oproti klopidogrelu, oba podávané v kombinácii s ASA a ďalšou štandardnou liečbou.
· Štúdia PEGASUS TIMI-54 [PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients], porovnávajúca tikagrelor v kombinácii s ASA oproti liečbe samotnou ASA.
Štúdia PLATO(akútnykoronárnysyndróm)
Do štúdie PLATO bolo zahrnutých 18 624 pacientov, u ktorých v priebehu ostatných 24 hodín došlo
k nástupu príznakov nestabilnej anginy pectoris (unstable angina, UA), infarktu myokardu bez elevácie ST segmentu (NSTEMI) alebo infarktu myokardu s eleváciou ST segmentu (STEMI), a ktorí
spočiatku dostávali medikamentóznu liečbu alebo sa u nich vykonala perkutánna koronárna intervencia (PCI) alebo CABG.
Klinická účinnosť
Pri dennom podávaní ASA preukázal tikagrelor v dávke 90 mg dvakrát denne v prevencii výskytu združeného koncového ukazovateľa KV smrti, IM alebo cievnej mozgovej príhody, lepší účinok ako
klopidogrel v dávke 75 mg denne, na tomto rozdiele sa podieľal najmä výskyt KV smrti a IM. Pacienti
dostali 300 mg nárazovú dávku klopidogrelu (prípadne 600 mg v prípade PCI) alebo 180 mg tikagreloru.
Výsledok liečby sa prejavil skoro (zníženie absolútneho rizika [ARR] 0,6 % a zníženie relatívneho rizika [RRR] 12 % po 30 dňoch) a účinok liečby ostal rovnaký po celé obdobie 12 mesiacov, ARR/rok bolo 1,9 % a RRR 16 %. To naznačuje, že je vhodné liečiť pacientov tikagrelorom 90 mg dvakrát denne až 12 mesiacov (pozri časť 4.2). Liečbou 54 pacientov s AKS tikagrelorom namiesto klopidogrelu sa zabráni 1 aterotrombotickej príhode; liečbou 91 pacientov tikagrelorom namiesto klopidogrelu sa zabráni 1 KV smrti (pozri obrázok 1 a tabuľku 4).
Účinok liečby tikagrelorom sa popri klopidogrele ukazuje ako konzistentný v rámci rôznych podskupín pacientov, vrátane členenia podľa hmotnosti, pohlavia, diabetu v anamnéze, prechodného ischemického záchvatu alebo nehemoragickej cievnej mozgovej príhody alebo revaskularizácie, súbežnej liečby zahŕňajúcej liečbu heparínmi, inhibítormi GpIIb/IIIa a inhibítormi protónovej pumpy (pozri časť 4.5), podľa diagnózy, u ktorej sa sledovali koncové ukazovatele (STEMI, NSTEMI alebo UA) a podľa zámeru liečebného postupu pri randomizácii (invazívny alebo medikamentózny).
Pozorovala sa málo významná súčinnosť liečby a geografického územia, pričom podľa pomeru rizika (HR) pre primárny koncový ukazovateľ zo svetového hľadiska vychádza priaznivejšie účinok tikagreloru s výnimkou Severnej Ameriky, kde vychádza priaznivejšie účinok klopidogrelu, ktorá predstavovala približne 10 % celkovej skúmanej populácie (p-hodnota interakcie = 0,045). Výskumné analýzy poukázali na možnú súvislosť s dávkou ASA tak, že znížená účinnosť tikagreloru sa pozorovala pri zvyšujúcich sa dávkach ASA. Pri dlhodobom podávaní ASA s tikagrelorom má byť rozmedzie dennej dávky 75 - 150 mg (pozri časť 4.2 a 4.4).
Obrázok 1 vyjadruje odhad rizika prvého výskytu akejkoľvek udalosti zahrnutej do združeného koncového ukazovateľa účinnosti.
Obrázok 1 Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IMa cievnej mozgovej príhody (štúdia PLATO)

0 6 0 1 2 0 1 8 0 2 4 0 3 0 0 3 6 0
Počet v ohroizení í
Dn i o d ra nd omizácie
T 9 3 3 3
C 9 2 9 1
8 6 2 8
8 5 2 1
8 4 6 0
8 3 6 2
8 2 1 9
8 1 2 4
6 7 4 3
6 6 5 0
5 1 6 1
5 0 9 6
4 1 4 7

4 0 7 4
Tikagrelor v porovnaní s klopidogrelom znížil výskyt primárneho združeného koncového ukazovateľa
rovnako u pacientov s UA/NSTEMI ako aj u pacientov so STEMI (tabuľka 4). Liečbu liekom Brilique
90 mg dvakrát denne spolu s nízkou dávkou ASA možno teda použiť u pacientov s AKS (nestabilná angina pectoris, infarkt myokardu bez elevácie ST segmentu [NSTEMI] alebo infarkt myokardu
s eleváciou ST segmentu [STEMI]); vrátane pacientov, ktorí dostávajú medikamentóznu liečbu
a pacientov, u ktorých sa vykonala perkutánna koronárna intervencia (PCI) alebo koronárny artériový by-pass (CABG).
Tabuľka 4 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PLATO)
tikagrelor
90 mg dvakrát
denne
(%
pacientov
s udalosťou) N = 9 333
klopidogrel
75 mg jedenkrát
denne
(% pacientov s udalosťou)
N = 9 291
ARR
a
(%/rok)
RRR
a
(%)
(95 % IS)
p
-
hodnota
KV smrť/IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
9,3 10,9 1,9 16 (8; 23) 0,0003
So zámerom
invazívneho zákroku 8,5 10,0 1,7 16 (6; 25) 0,0025
So zámerom
medikamentóznej liečby
11,3 13,2 2,3 15 (0,3; 27) 0,0444d

KV smrť 3,8 4,8 1,1 21 (9; 31) 0,0013
IM (s vylúčením
tichého IM)b 5,4 6,4 1,1 16 (5; 25) 0,0045
Cievna mozgová
príhoda 1,3 1,1 −0,2 −17 (−52; 9) 0,2249
Úmrtnosť zo všetkých príčin, IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
KV smrť, IM celkovo, cievna mozgová príhoda, SRI, RI, TIA alebo iná ATEc
Úmrtnosť zo
9,7 11,5 2,1 16 (8; 23) 0,0001
13,8 15,7 2,1 12 (5; 19) 0,0006
d
všetkých príčin 4,3 5,4 1,4 22 (11; 31) 0,0003
Definitívna d
trombóza stentu 1,2 1,7 0,6 32 (8; 49) 0,0123
aARR = zníženie absolútneho rizika; RRR = zníženie relatívneho rizika = (1 − pomer rizika) x 100 %. Hodnoty s negatívnym
RRR naznačujú zvýšenie relatívneho rizika.
bS vylúčením tichého IM.
cSRI = ťažká rekurentná ischémia (serious recurrent ischaemia); RI = rekurentná ischémia (recurrent ischaemia); TIA =
prechodný ischemický záchvat (transient ischaemic attack); ATE = arteriálna trombotická príhoda. IM celkovo zahŕňa tichý
IM, s dátumom, kedy bol zistený.
dNominálna hladina významnosti (significance value); všetky ostatné sú formálne štatisticky významné vopred definovaným hierarchickým testovaním.
Genetická podštúdia štúdie PLATO
Genotypizáciou CYP2C19 a ABCB1 u 10 285 pacientov v štúdii PLATO sa zistili súvislosti medzi
skupinami genotypu a výsledkami štúdie PLATO. Lepší účinok tikagreloru oproti klopidogrelu
v znížení výskytu závažných KV príhod nebol významne ovplyvnený genotypom CYP2C19 alebo ABCB1 pacienta. Podobne ako v celej štúdii PLATO sa celkový výskyt veľkých krvácaní podľa definície PLATO pri tikagrelore a klopidogrele neodlišoval, bez ohľadu na genotyp CYP2C19 alebo ABCB1. Výskyt veľkých krvácaní podľa definície PLATO nesúvisiacich s CABG bol pri tikagrelore
v porovnaní s klopidogrelom zvýšený u pacientov s jednou alebo viacerými nefunkčnými alelami
CYP2C19, ale podobný ako pri klopidogrele u pacientov bez nefunkčnej alely.
Združený ukazovateľ účinnosti a bezpečnosti
Združený ukazovateľ účinnosti a bezpečnosti (KV smrť, IM, cievna mozgová príhoda alebo veľké krvácanie podľa definície PLATO celkovo) naznačuje, že klinický prínos účinnosti tikagreloru
v porovnaní s klopidogrelom nie je kompenzovaný udalosťami súvisiacimi s veľkým krvácaním (ARR
1,4 %, RRR 8 %, HR 0,92; p = 0,0257) v priebehu 12 mesiacov od AKS.
Klinická bezpečnosť
Podštúdia s Holterovým monitorovaním:
Na sledovanie výskytu ventrikulárnych páuz a iných epizód arytmií počas štúdie PLATO skúšajúci vykonali Holterovo monitorovanie u podskupiny takmer 3 000 pacientov, z ktorých približne 2 000
malo záznam aj v akútnej fáze ich AKS a aj po jednom mesiaci. Primárnym sledovaným
ukazovateľom bol výskyt ventrikulárnych páuz ≥ 3 sekundy. Ventrikulárne pauzy sa u pacientov v akútnej fáze vyskytovali častejšie pri tikagrelore (6,0 %) ako pri klopidogrele (3,5 %); a po 1 mesiaci to bolo 2,2 % pri tikagrelore a 1,6 % pri klopidogrele (pozri časť 4.4). Nárast výskytu ventrikulárnych páuz v akútnej fáze AKS bol v skupine s tikagrelorom výraznejší u pacientov
s chronickým srdcovým zlyhávaním (CHF) v anamnéze (9,2 % oproti 5,4 % u pacientov bez CHF
v anamnéze; v skupine s klopidogrelom to bolo 4,0 % u pacientov s CHF v anamnéze oproti 3,6 %
u pacientov bez CHF v anamnéze). Tento rozdiel sa po 1 mesiaci nevyskytoval: 2,0 % oproti 2,1 % pri tikagrelore pre pacientov s a bez CHF v anamnéze a 3,8 % oproti 1,4 % pri klopidogrele. Tento rozdiel
nesúvisel so žiadnymi nežiaducimi klinickými dôsledkami (vrátane zavedenia kardiostimulátora)
v tejto populácii pacientov.
Štúdia PEGASUS (anamnézainfarktumyokardu)
Štúdia PEGASUS TIMI-54 bola medzinárodná, multicentrická, randomizovaná, dvojito zaslepená,
placebom kontrolovaná štúdia s paralelnou skupinou a s dĺžkou určenou počtom udalostí potrebných pre štatistické vyhodnotenie (event-driven), zahŕňajúca 21 162 pacientov, ktorá hodnotila prevenciu
aterotrombotických príhod pri tikagrelore podávanom v 2 dávkach (buď 90 mg dvakrát denne alebo
60 mg dvakrát denne) v kombinácii s nízkou dávkou ASA (75 – 150 mg), v porovnaní s liečbou samotnou ASA u pacientov s IM v anamnéze a ďalšími rizikovými faktormi aterotrombózy.
Pacienti boli vhodní na zaradenie do štúdie, ak boli vo veku 50 rokov alebo starší, mali IM
v anamnéze (1 až 3 roky pred randomizáciou) a mali aspoň jeden z nasledujúcich rizikových faktorov aterotrombózy: vek ≥ 65 rokov, diabetes mellitus vyžadujúci liečbu, druhý predchádzajúci IM,
potvrdenú CAD postihujúcu viaceré cievy alebo chronickú dysfunkciu obličiek v non-terminálnom
štádiu.
Pacienti neboli vhodní na zaradenie do štúdie, ak sa u nich počas obdobia štúdie plánoval použiť antagonista receptorov P2Y12, dipyridamol, cilostazol alebo antikoagulačná liečba; ak mali krvácavú poruchu alebo ischemickú cievnu mozgovú príhodu alebo intrakraniálne krvácanie v anamnéze, nádor centrálneho nervového systému alebo abnormalitu intrakraniálnych ciev; ak mali v priebehu predchádzajúcich 6 mesiacov gastrointestinálne krvácanie alebo veľký chirurgický zákrok v priebehu predchádzajúcich 30 dní.
Klinická účinnosť
Obrázok 2 – Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IM
a cievnej mozgovej príhody (štúdia PEGASUS)

Tikagrelor 60 mg bd - - - - Placebo
N 7045 7067
Pacienti s udalosťami 487 (6.9%) 578 (8.2%) KM% v 36 mesiaci 7.8% 9.0%
Pomer rizika (95% CI) 0.84 (0.74, 0.95)
 p
p-hodnota 0.0043
Počet v riziku
Ti 60 mg
Placebo
Dni od randomizácie
Tabuľka 5 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PEGASUS)
tikagrelor 60 mg dvakrát denne + ASA
N = 7 045
sa
m
otn
á ASA
N = 7 067
p
-
hodnota
Charakteristika
Pacienti
s udalosťami
Primárny koncový ukazovateľ
Združený pre
KV
KM % HR
(95 % IS)
Pacienti
s udalosťami KM %
smrť/IM/cievnu mozgovú príhodu
487 (6,9 %) 7,8 % 0,84 (0,74; 0,95)
578 (8,2 %) 9,0 % 0,0043 (s)
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
IM 285 (4,0 %) 4,5 % 0,84 (0,72; 0,98)
210 (3,0 %) 3,4 % 0,0676
338 (4,8 %) 5,2 % 0,0314
Cievna mozgová
príhoda 91 (1,3 %) 1,5 %
Sekundárny koncový ukazovateľ
0,75
(0,57; 0,98) 122 (1,7 %) 1,9 % 0,0337
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
210 (3,0 %) 3,4 % -
Úmrtnosť zo
všetkých príčin 289 (4,1 %) 4,7 %
0,89 326 (4,6 %) 5,2 % - (0,76; 1,04)

Pomer rizika (HR) a
p-hodnota sú vypočítané osobitne pre tikagrelor oproti liečbe samotnou ASA na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou.
Percentá KM vypočítané po 36 mesiacoch.
Poznámka: počty prvých udalostí pre zložky KV smrť, IM a cievna mozgová príhoda sú skutočné počty prvých udalostí pre každú zo zložiek a nesčítavajú sa do počtu udalostí v združenom koncovom ukazovateli.
(s) Predstavuje štatistickú významnosť (significance).
IS = interval spoľahlivosti; KV = kardiovaskulárna; HR = pomer rizika; KM = Kaplanov-Meierov odhad; IM = infarkt myokardu;
N = počet pacientov.
Oba režimy s tikagrelorom, 60 mg dvakrát denne a 90 mg dvakrát denne, v kombinácii s ASA preukázali lepší účinok v prevencii aterotrombotických príhod v porovnaní so samotnou ASA (združený koncový ukazovateľ: KV smrť, IM a cievna mozgová príhoda), s konzistentným účinkom liečby počas celého obdobia štúdie a dosiahnutím RRR 16 % a ARR 1,27 % pre 60 mg tikagreloru
a RRR 15 % a ARR 1,19 % pre 90 mg tikagreloru.
Hoci bol profil účinnosti 90 mg a 60 mg dávky podobný, bolo dokázané, že nižšia dávka je lepšie znášaná a má lepší bezpečnostný profil v súvislosti s rizikom krvácania a dyspnoe. Preto sa na prevenciu aterotrombotických príhod (KV smrť, IM a cievna mozgová príhoda) u pacientov s IM
v anamnéze a vysokým rizikom aterotrombotickej príhody odporúča iba Brilique 60 mg dvakrát denne podávaný súbežne s ASA.
V porovnaní so samotnou ASA, tikagrelor v dávke 60 mg dvakrát denne významne znížil primárny združený koncový ukazovateľ KV smrti, IM a cievnej mozgovej príhody. Každá zo zložiek prispela k zníženiu primárneho združeného koncového ukazovateľa (RRR KV smrti 17 %, RRR IM 16 %
a RRR cievnej mozgovej príhody 25 %).
RRR pre združený koncový ukazovateľ od 1. do 360. dňa (RRR 17 %) a od 361. dňa ďalej (RRR
16 %) bolo podobné. K dispozícii sú obmedzené údaje o účinnosti a bezpečnosti tikagreloru za 3 roky predĺženej liečby.
Nebol nájdený žiadny dôkaz o prínose (neprišlo k zníženiu primárneho koncového ukazovateľa zloženého z KV úmrtia, infarktu myokardu a cievnej mozgovej príhody, ale zvýšeniu závažného krvácania), pri užívaní tikagrelor 60 mg dvakrát denne u klinicky stabilných pacientov > 2 roky od infarktu myokardu alebo viac ako jeden rok po ukončení predchádzajúcej liečby inhibítorom receptora ADP (pozri tiež časť 4.2).
Klinická bezpečnosť
Počet prerušení liečby s tikagrelorom 60 mg kvôli krvácaniu a dýchavičnosti bola vyššia u pacientov
> 75 rokov (42 %) ako u mladších pacientov (rozmedzie: 23-31 %), s rozdielom v porovnaní s placebom vyšším ako 10 % (42 % vs. 29 %), u pacientov > 75 rokov.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Brilique vo všetkých podskupinách pediatrickej populácie s akútnym koronárnym syndrómom (AKS) a infarktom myokardu (IM) v anamnéze (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika tikagreloru je lineárna a expozícia tikagreloru a aktívnemu metabolitu
(AR-C124910XX) je približne úmerná dávke až po dávku 1 260 mg.
Absorpcia
K absorpcii tikagreloru dochádza rýchlo, s mediánom tmax približne 1,5 hodiny. Tvorba hlavného cirkulujúceho metabolitu AR-C124910XX (tiež aktívneho) z tikagreloru je rýchla s mediánom tmax približne 2,5 hodín. Po perorálnom podaní jednorazovej dávky tikagreloru 90 mg nalačno zdravým dobrovoľníkom je Cmax 529 ng/ml a AUC 3 451 ng*h/ml. Pomer metabolit/pôvodné liečivo pre Cmax je
0,28 a pre AUC 0,42. Farmakokinetika tikagreloru a AR-C124910XX u pacientov s IM v anamnéze
bola vo všeobecnosti podobná farmakokinetike v populácii s AKS. Na základe populačnej farmakokinetickej analýzy štúdie PEGASUS bol medián Cmax tikagreloru v rovnovážnom stave
391 ng/ml a AUC 3 801 ng*h/ml pre dávku 60 mg tikagreloru. Pre dávku 90 mg tikagreloru bola Cmax
v rovnovážnom stave 627 ng/ml a AUC 6 255 ng*h/ml.
Priemerná absolútna biologická dostupnosť tikagreloru sa odhadla na 36 %. Príjem potravy s vysokým obsahom tukov mal za následok zvýšenie AUC tikagreloru o 21 % a zníženie Cmax aktívneho metabolitu o 22 %, na Cmax tikagreloru a na AUC aktívneho metabolitu však nemal žiadny vplyv. Tieto malé zmeny sa považujú za klinicky minimálne významné, preto sa tikagrelor môže užívať s jedlom alebo bez jedla. Tikagrelor ako aj jeho aktívny metabolit sú substrátmi P-gp.
Tikagrelor, vo forme rozdrvených tabliet zmiešaných s vodou, podávaný perorálne alebo nazogastrickou sondou do žalúdka má porovnateľnú biologickú dostupnosť s celými tabletami
s ohľadom na AUC a Cmax tikagreloru a aktívneho metabolitu. Úvodná expozícia (0,5 a 1 hodinu po podaní dávky) po podaní rozdrvených tabliet tikagreloru zmiešaných s vodou bola vyššia v porovnaní
s celými tabletami, pričom neskôr (o 2 až 48 hodín) bol koncentračný profil vo všeobecnosti identický.
Distribúcia
Distribučný objem tikagreloru v rovnovážnom stave je 87,5 l. Tikagrelor a aktívny metabolit sa vo veľkej miere viaže na bielkoviny ľudskej plazmy (> 99,0 %).
Biotransformácia
CYP3A4 je hlavným enzýmom zodpovedným za metabolizmus tikagreloru a tvorbu aktívneho metabolitu a ich interakcie s inými substrátmi CYP3A sú v rozmedzí od aktivácie po inhibíciu.
Hlavným metabolitom tikagreloru je AR-C124910XX, ktorý je na základe dôkazu jeho väzby na doštičkový receptor P2Y12 pre ADP v podmienkach in vitro tiež aktívny. Systémová expozícia aktívnemu metabolitu predstavuje približne 30 – 40 % systémovej expozície tikagreloru.
Eliminácia
Primárnou cestou eliminácie tikagreloru je metabolizácia v pečeni. V prípade podávania rádioizotopom značeného tikagreloru sa zachytí v priemere približne 84 % rádioizotopom značenej
dávky (57,8 % v stolici, 26,5 % v moči). Detegované množstvá tikagreloru a aktívneho metabolitu v moči v obidvoch prípadoch predstavovali menej ako 1 % dávky. Primárnou cestou eliminácie
aktívneho metabolitu je s najväčšou pravdepodobnosťou biliárna sekrécia. Priemerná hodnota t1/2 pre tikagrelor bola približne 7 hodín a pre aktívny metabolit 8,5 hodín.
Osobitné skupinypacientov
Staršípacienti
U starších osôb (≥ 75 rokov) s AKS v porovnaní s mladšími pacientmi sa farmakokinetickou analýzou populácie zistilo zvýšenie expozície tikagreloru (približne 25 % pre Cmax aj AUC) a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné (pozri časť 4.2).
Pediatrickápopulácia
Tikagrelor sa neskúmal v pediatrickej populácii (pozri časti 4.2 a 5.1).
Pohlavie
U žien sa v porovnaní s mužmi zaznamenalo zvýšenie expozície tikagreloru a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné.
Poruchafunkcieobličiek
U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) v porovnaní s osobami s normálnou funkciou obličiek bola expozícia tikagreloru približne o 20 % nižšia
a expozícia aktívnemu metabolitu približne o 17 % vyššia (pozri časť 4.2).
Poruchafunkciepečene
Cmax tikagreloru bol o 12 % a AUC o 23 % vyššia u pacientov s miernou poruchou funkcie pečene v porovnaní s kontrolnými zdravými osobami, účinok tikagreloru na IPA bol však u oboch skupín
podobný. U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Tikagrelor sa neskúmal u pacientov s ťažkou poruchou funkcie pečene a nie sú k dispozícii farmakokinetické údaje u pacientov so stredne ťažkou poruchou funkcie pečene. U pacientov, ktorí mali stredne závažné alebo závažné východiskové zvýšenie jedného alebo viacerých vyšetrení funkcie pečene, boli plazmatické koncentrácie tikagreloru v priemere podobné alebo mierne vyššie
v porovnaní s pacientmi bez východiskových zvýšení. U pacientov so stredne ťažkou poruchou funkcie pečene sa neodporúča žiadna úprava dávky (pozri časti 4.2 a 4.4).
Etnickápríslušnosť
Priemerná biologická dostupnosť je u pacientov ázijského pôvodu o 39 % vyššia ako u belochov.
Biologická dostupnosť tikagreloru u pacientov, ktorí sami uviedli černošský pôvod bola o 18 % nižšia ako u belochov, v klinických farmakologických štúdiách bola expozícia (Cmax a AUC) tikagreloru
u Japoncov približne o 40 % (o 20 % po úprave podľa telesnej hmotnosti) vyššia ako u belochov.
Expozícia u pacientov, ktorí sami uviedli hispánsky alebo latinskoamerický pôvod bola podobná ako u belochov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje týkajúce sa tikagreloru a jeho hlavného metabolitu nepreukázali neprijateľné riziko nežiaducich účinkov u ľudí na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po jednorazovom a opakovanom podávaní a genotoxického potenciálu.
Pri klinicky relevantných hladinách expozície sa pozorovalo gastrointestinálne podráždenie u niekoľkých druhov zvierat (pozri časť 4.8).
Pri vysokých dávkach tikagreloru sa u samíc potkanov preukázal zvýšený výskyt nádorov maternice (adenokarcinómov) a zvýšený výskyt adenómov pečene. Pravdepodobným mechanizmom vzniku nádorov maternice je hormonálna nerovnováha, ktorá u potkanov môže viesť k vzniku nádorov. Pravdepodobným mechanizmom vzniku adenómov pečene je indukcia enzýmov v pečeni špecifických pre hlodavce. Relevantnosť týchto zistení týkajúcich sa karcinogenity sa preto považuje za nepravdepodobnú pre ľudí.
U potkanov sa pozorovali menej významné vývinové anomálie pri dávkach toxických pre samicu (bezpečnostný pomer 5,1). U králikov sa pozorovalo mierne oneskorenie dozrievania pečene a vývoja skeletu plodu u samíc, ktorým boli podávané vysoké dávky, bez známok materskej toxicity (bezpečnostný pomer 4,5).
Štúdie na potkanoch a králikoch sa preukázali reprodukčnú toxicitu s mierne zníženým nárastom telesnej hmotnosti brezivých samíc a so zníženou životaschopnosťou a zníženou pôrodnou hmotnosťou mláďat s oneskoreným rastom. Tikagrelor spôsoboval nepravidelné cykly (najmä predĺžené cykly) u samíc potkanov, nemal však vplyv na celkovú fertilitu samcov a samíc potkanov. Farmakokinetické štúdie s rádioizotopom značeným tikagrelorom preukázali, že pôvodné liečivo
a jeho metabolity sa u potkanov vylučujú do materského mlieka (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety manitol (E421)
dihydrát hydrogenfosforečnanu vápenatého
magnéziumstearát (E470b)
sodná soľ karboxymetylškrobu typu A
hydroxypropylcelulóza (E463)
Obal
tablety mastenec
oxid titaničitý (E171)
žltý oxid železitý (E172) makrogol 400 hypromelóza (E464)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
· PVC-PVDC/Al priehľadný blister (so symbolmi slnko/mesiac) obsahujúci 10 tabliet; škatule s obsahom 60 tabliet (6 blistrov) a 180 tabliet (18 blistrov).
· PVC-PVDC/Al priehľadný kalendárový blister (so symbolmi slnko/mesiac) obsahujúci 14
tabliet; škatule s obsahom 14 tabliet (1 blister), 56 tabliet (4 blistre) a 168 tabliet (12 blistrov).
· PVC-PVDC/hliníkový priehľadný blister s perforáciou umožňujúcou oddelenie jednotlivej dávky, obsahujúci 10 tabliet; škatule s obsahom 100 x 1 tabliet (10 blistrov).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/10/655/001-006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 03. december 2010
Dátum posledného predĺženia registrácie: 17. júl 2015
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.1. NÁZOV LIEKUBrilique 90 mg orodispergovateľné tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá orodispergovateľná tableta obsahuje 90 mg tikagreloru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAOrodispergovateľná tableta.
Okrúhle, ploché, biele až bledoružové orodispergovateľné tablety so skosenými hranami s označením
„90“ nad „TI“ na jednej strane a bez označenia na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieBrilique podávaný spolu s kyselinou acetylsalicylovou (ASA) je indikovaný na prevenciu aterotrombotických príhod u dospelých pacientov s
- akútnym koronárnym syndrómom (AKS) alebo
- infarktom myokardu (IM) v anamnéze a vysokým rizikom vzniku aterotrombotickej príhody
(pozri časti 4.2 a 5.1).
4.2 Dávkovanie a spôsob podávaniaDávkovaniePacienti užívajúci Brilique majú užívať aj nízku udržiavaciu dávku ASA 75 – 150 mg denne, pokiaľ to
nie je výslovne kontraindikované.
AkútnykoronárnysyndrómLiečba liekom Brilique sa má začať s jednou 180 mg nárazovou dávkou (dve 90 mg tablety) a potom sa má pokračovať dávkou 90 mg dvakrát denne. Odporúča sa, aby liečba liekom Brilique 90 mg
dvakrát denne u pacientov s AKS trvala 12 mesiacov, pokiaľ nie je klinicky indikované ukončenie liečby (pozri časť 5.1).
InfarktmyokarduvanamnézeBrilique 60 mg dvakrát denne je odporúčanou dávkou na predĺženú liečbu pacientov s IM v anamnéze aspoň jeden rok a vysokým rizikom aterotrombotickej príhody (pozri časť 5.1). Liečba by sa mala
začať bez prerušenia ako liečba nasledujúca po úvodnej jednoročnej liečbe liekom Brilique 90 mg
alebo liečbe iným inhibítorom receptora adenozíndifosfátu (ADP) u pacientov s AKS s vysokým rizikom aterotrombotickej príhody. Liečbu tiež možno začať až do 2 rokov po IM alebo v priebehu jedného roka po ukončení predchádzajúcej liečby inhibítorom receptora ADP. K dispozícii sú obmedzené údaje o účinnosti a bezpečnosti tikagreloru pri predĺženej liečbe presahujúcej 3 roky.
Ak je potrebné prestaviť pacientov na liečbu liekom Brilique, prvá dávka sa má podať 24 hodín po poslednej dávke predchádzajúceho protidoštičkového lieku.
Vynechaná
dávka
Tiež je potrebné sa vyvarovať vynechaniu liečby. Pacient, ktorý vynechá dávku lieku Brilique, má užiť iba jednu tabletu (svoju ďalšiu dávku) v obvyklom čase užitia ďalšej dávky.
Osobitnéskupinypacientov
Starší pacienti
U starších pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2). K dispozícii nie sú žiadne informácie týkajúce sa dialyzovaných pacientov a preto sa tikagrelor
u týchto pacientov neodporúča.
Porucha funkcie pečene
Tikagrelor sa neskúmal u pacientov s ťažkou poruchou funkcie pečene a jeho použitie u týchto pacientov je preto kontraindikované (pozri časť 4.3). K dispozícii sú len obmedzené údaje u pacientov
so stredne ťažkou poruchou funkcie pečene. Úprava dávky sa neodporúča, tikagrelor sa však má
používať s opatrnosťou (pozri časti 4.4 a 5.2). U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť tikagreloru u detí mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsobpodávania
Na perorálne použitie.
Brilique sa môže podávať s jedlom alebo bez jedla.
Orodispergovateľné tablety sa môžu používať ako alternatíva filmom obalených tabliet Brilique 90 mg pre pacientov, ktorí majú problémy s prehĺtaním tabliet vcelku alebo pre pacientov, ktorí dávajú prednosť orodispergovateľným tabletám. Tableta sa má položiť na jazyk, kde sa v slinách rýchlo rozpustí. Potom sa môže prehltnúť s vodou alebo bez vody (pozri časť 5.2). Tableta sa môže tiež rozpustiť vo vode a podávať nazogastrickou sondou (CH8 alebo širšou). Po podaní zmesi je dôležité prepláchnuť nazogastrickú sondu vodou. Orodispergovateľné tablety so silou 60 mg nie sú
k dispozícii.
4.3 Kontraindikácie
· Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 (pozri časť 4.8).
· Aktívne patologické krvácanie.
· Intrakraniálne krvácanie v anamnéze (pozri časť 4.8).
· Ťažká porucha funkcie pečene (pozri časti 4.2, 4.4 a 5.2).
· Súbežné podávanie tikagreloru so silnými inhibítormi CYP3A4 (napr. ketokonazolom, klaritromycínom, nefazodónom, ritonavirom a atazanavirom) môže viesť k podstatnému
zvýšeniu expozície tikagreloru (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Rizikokrvácania
Pri použití tikagreloru u pacientov so známym zvýšeným rizikom krvácania sa má zvážiť prínos
z hľadiska prevencie aterotrombotických príhod (pozri časti 4.8 a 5.1). Pri klinickom opodstatnení sa má tikagrelor používať s opatrnosťou v nasledujúcich skupinách pacientov:
· Pacienti náchylní na krvácanie (napr. kvôli nedávnemu zraneniu, nedávnemu chirurgickému zákroku, poruchám koagulácie, aktívnemu alebo nedávnemu gastrointestinálnemu krvácaniu). Použitie tikagreloru je kontraindikované u pacientov s aktívnym patologickým krvácaním,
u pacientov s intrakraniálnym krvácaním v anamnéze a u pacientov s ťažkou poruchou funkcie
pečene (pozri časť 4.3).
· Pacienti súbežne liečení liekmi, ktoré môžu zvyšovať riziko krvácania (napr. nesteroidové protizápalové lieky (NSAID), perorálne antikoagulanciá a/alebo fibrinolytiká), v priebehu
24 hodín od podania tikagreloru.
Transfúzia krvných doštičiek nezvrátila protidoštičkový účinok tikagreloru u zdravých dobrovoľníkov a preto pravdepodobne nepredstavuje klinický prínos pre pacientov s krvácaním. Keďže podávaním tikagreloru spolu s dezmopresínom sa štandardizovaný čas krvácania neskrátil, nie je pravdepodobné, že by bol dezmopresín účinný pri zvládaní klinických krvácavých príhod (pozri časť 4.5).
Antifibrinolytická liečba (kyselinou aminokaprónovou alebo kyselinou tranexámovou) a/alebo liečba rekombinantným faktorom VIIa môže zvyšovať hemostázu. Tikagrelor možno znovu začať podávať po tom, ako bola príčina krvácania identifikovaná a je pod kontrolou.
Chirurgickýzákrok
Pacientov je potrebné poučiť, aby pred akýmkoľvek plánovaným chirurgickým zákrokom a pred užívaním akéhokoľvek nového lieku informovali lekárov a zubných lekárov, že užívajú tikagrelor.
V štúdii PLATO bol výskyt krvácania u pacientov, ktorí podstúpili koronárny artériový by –pass (coronary artery bypass grafting, CABG), vyšší pri tikagrelore ako pri klopidogrele pri jeho vysadení v priebehu 1 dňa pred chirurgickým zákrokom, ale výskyt veľkého krvácania pri vysadení liečby 2 alebo viac dní pred chirurgickým zákrokom bol podobný ako pri klopidogrele (pozri časť 4.8). Ak má pacient podstúpiť plánovaný chirurgický zákrok a protidoštičkový účinok nie je požadovaný, tikagrelor sa má vysadiť 7 dní pred chirurgickým zákrokom (pozri časť 5.1).
Pacientispredchádzajúcouischemickoucievnoumozgovoupríhodou
Pacienti s AKS s predchádzajúcou ischemickou cievnou mozgovou príhodou môžu byť liečení tikagrelorom až 12 mesiacov (štúdia PLATO).
Do štúdie PEGASUS neboli zahrnutí pacienti s IM v anamnéze a predchádzajúcou ischemickou cievnou mozgovou príhodou. Vzhľadom na chýbajúce údaje sa u týchto pacientov liečba presahujúca jeden rok neodporúča.
Poruchafunkciepečene
Použitie tikagreloru je kontraindikované u pacientov s ťažkou poruchou funkcie pečene (pozri časti
4.2 a 4.3). K dispozícii sú obmedzené skúsenosti s tikagrelorom u pacientov so stredne ťažkou poruchou funkcie pečene, u týchto pacientov sa preto odporúča opatrnosť (pozri časti 4.2 a 5.2).
Pacientisrizikombradykardickýchpríhod
Kvôli zisteniam väčšinou asymptomatických prípadov ventrikulárnej pauzy v skoršej klinickej štúdii boli z hlavných štúdií hodnotiacich bezpečnosť a účinnosť tikagreloru vyradení pacienti so zvýšeným rizikom bradykardických príhod (napr. pacienti bez kardiostimulátora so syndrómom chorého sínusového uzla, AV blokádou 2. alebo 3. stupňa alebo synkopou súvisiacou s bradykardiou). Vzhľadom na obmedzené klinické skúsenosti sa má preto tikagrelor u týchto pacientov používať
s opatrnosťou (pozri časť 5.1).
Okrem toho je potrebná opatrnosť aj pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že vyvolávajú bradykardiu. V štúdii PLATO sa však po súbežnom podaní s jedným alebo viacerými liekmi, o ktorých je známe, že vyvolávajú bradykardiu (napr. 96 % betablokátory, 33 % blokátory kalciových kanálov diltiazem a verapamil a 4 % digoxín), nepozorovali žiadne klinicky významné nežiaduce reakcie (pozri časť 4.5).'
V priebehu podštúdie s Holterovým monitorovaním v štúdii PLATO sa ventrikulárne pauzy trvajúce
≥ 3 sekundy vyskytli u väčšieho počtu pacientov užívajúcich tikagrelor ako pacientov užívajúcich klopidogrel v akútnej fáze ich AKS. Nárast počtu ventrikulárnych páuz pri tikagrelore, odhalených Holterovým monitorovaním, bol u pacientov s chronickým srdcovým zlyhávaním (CHF) vyšší ako
u celkového počtu pacientov v štúdii počas akútnej fázy AKS, avšak nie po 1 mesiaci liečby tikagrelorom alebo v porovnaní s klopidogrelom. Z tejto nerovnováhy však u tejto skupiny pacientov
nevyplývali žiadne nežiaduce klinické dôsledky (vrátane synkopy alebo zavedenia kardiostimulátora) (pozri časť 5.1).
Dyspnoe
U pacientov liečených tikagrelorom sa hlásilo dyspnoe. Dyspnoe je zvyčajne miernej až stredne ťažkej intenzity a často ustúpi bez potreby ukončenia liečby. Absolútne riziko výskytu dyspnoe pri užívaní
tikagreloru môže byť vyššie u pacientov s astmou/chronickou obštrukčnou chorobou pľúc (CHOCHP).
Tikagrelor sa má používať opatrne u pacientov s astmou a/alebo CHOCHP v anamnéze. Mechanizmus nie je objasnený. Ak pacient hlási nové, dlhotrvajúce alebo zhoršené dyspnoe, dyspnoe sa má
dôkladne vyšetriť a pri neznášanlivosti sa má liečba tikagrelorom ukončiť. Pre ďalšie informácie pozri
časť 4.8.
Zvýšeniekreatinínu
Počas liečby tikagrelorom sa môžu zvýšiť hladiny kreatinínu. Mechanizmus nie je objasnený. Funkcia obličiek sa má kontrolovať v súlade s bežnou lekárskou praxou. U pacientov s AKS sa tiež odporúča skontrolovať funkciu obličiek jeden mesiac po začatí liečby tikagrelorom s venovaním osobitnej pozornosti pacientom vo veku ≥ 75 rokov, pacientom so stredne ťažkou/ťažkou poruchou funkcie obličiek a pacientom súbežne liečeným blokátorom receptora angiotenzínu (ARB).
Zvýšeniekyselinymočovej
Počas liečby tikagrelorom sa môže objaviť hyperurikémia (pozri časť 4.8). U pacientov
s hyperurikémiou alebo dnovou artritídou v anamnéze sa odporúča opatrnosť. Ako preventívne opatrenie je potrebné zabrániť použitiu tikagreloru u pacientov s urátovou nefropatiou.
Iné
Na základe vzťahu zaznamenaného v štúdii PLATO medzi udržiavacou dávkou ASA a relatívnou účinnosťou tikagreloru v porovnaní s klopidogrelom sa súbežné podávanie tikagreloru a vysokej udržiavacej dávky ASA (> 300 mg) neodporúča (pozri časť 5.1).
Predčasnéukončenieliečby
Predčasné ukončenie akejkoľvek protidoštičkovej liečby, vrátane lieku Brilique, môže viesť
k zvýšenému riziku kardiovaskulárnej (KV) smrti alebo IM v dôsledku základného ochorenia pacienta. Preto sa treba vyhnúť predčasnému ukončeniu liečby.
4.5 Liekové a iné interakcie
Tikagrelor je predovšetkým substrátom CYP3A4 a miernym inhibítorom CYP3A4. Tikagrelor je tiež substrátom P-glykoproteínu (P-gp) a slabým inhibítorom P-gp a môže zvyšovať expozíciu substrátom P-gp.
Účinky inýchliekovnatikagrelor
Liečivámetabolizované prostredníctvom CYP3A4
Inhibítory CYP3A4
· Silné inhibítory CYP3A4 – pri súbežnom podávaní ketokonazolu a tikagreloru sa Cmax tikagreloru zvýšil 2,4-násobne a AUC 7,3-násobne. Cmax aktívneho metabolitu sa znížil o 89 % a AUC o 56 %. Dá sa predpokladať, že účinky iných silných inhibítorov CYP3A4
(klaritromycín, nefazodón, ritonavir a atazanavir) sú podobné a preto je súbežné použitie silných inhibítorov CYP3A4 s tikagrelorom kontraindikované (pozri časť 4.3).
· Stredne silné inhibítory CYP3A4 – pri súbežnom podávaní diltiazemu a tikagreloru sa Cmax tikagreloru zvýšil o 69 % a AUC 2,7-násobne a v prípade aktívneho metabolitu došlo k zníženiu Cmax o 38 % a AUC ostala nezmenená. Tikagrelor nemal žiadny vplyv na hladiny diltiazemu
v plazme. Podobný účinok možno predpokladať aj u iných stredne silných inhibítorov CYP3A4
(napr. amprenavir, aprepitant, erytromycín a flukonazol) a tiež ich možno podávať súbežne s tikagrelorom.
Induktory CYP3A4
Pri súbežnom podávaní rifampicínu a tikagreloru sa Cmax tikagreloru znížil o 73 % a AUC o 86 %.
Cmax aktívneho metabolitu ostal nezmenený a AUC sa znížila o 46 %. Dá sa predpokladať, že aj ďalšie
induktory CYP3A4 (napr. fenytoín, karbamazepín a fenobarbital) znižujú expozíciu tikagreloru.
Súbežné podávanie tikagreloru so silnými induktormi CYP3A4 môže znižovať expozíciu a účinnosť tikagreloru, preto sa má zabrániť ich súbežnému použitiu s tikagrelorom.
Cyklosporín(inhibítorP-gpaCYP3A)
Pri súbežnom podávaní cyklosporínu (600 mg) a tikagreloru sa Cmax tikagreloru zvýšil 2,3-násobne
a AUC 2,8-násobne. V prítomnosti cyklosporínu sa AUC aktívneho metabolitu zvýšila o 32 % a Cmax
sa znížil o 15 %.
K dispozícii nie sú žiadne údaje týkajúce sa súbežného podávania tikagreloru a ďalších liečiv, ktoré sú tiež silnými inhibítormi P-gp a stredne silnými inhibítormi CYP3A4 (napr. verapamil, chinidín), ktoré takisto môžu zvyšovať expozíciu tikagreloru. Ak sa takejto kombinácii nie je možné vyhnúť, ich súbežné použitie si vyžaduje opatrnosť.
Iné
Klinické štúdie farmakologických interakcií preukázali, že súbežné podávanie tikagreloru
s heparínom, enoxaparínom a ASA alebo dezmopresínom v porovnaní s podávaním samotného tikagreloru nemalo žiadny vplyv na farmakokinetiku tikagreloru alebo jeho aktívneho metabolitu, alebo na agregáciu krvných doštičiek indukovanú ADP. Ak je to klinicky indikované, lieky ovplyvňujúce hemostázu sa majú v kombinácii s tikagrelorom používať opatrne.
Pri dennej konzumácii väčšieho množstva grapefruitovej šťavy (3 x 200 ml) sa pozorovalo 2-násobné zvýšenie expozície tikagreloru. Neočakáva sa, že rozsah tejto zvýšenej expozície bude klinicky významný pre väčšinu pacientov.
Účinky tikagreloru na inélieky
Liečivámetabolizované prostredníctvom CYP3A4
· Simvastatín – pri súbežnom podávaní tikagreloru a simvastatínu sa Cmax simvastatínu zvýšil o 81 % a AUC o 56 %, Cmax kyseliny simvastatínovej sa zvýšil o 64 % a AUC o 52 %,
v niektorých jednotlivých prípadoch boli zvýšenia 2- až 3-násobné. Súbežné podávanie
tikagreloru so simvastatínom v dávkach vyšších ako 40 mg denne môže spôsobiť nežiaduce reakcie simvastatínu a potenciálny prínos tejto kombinácie je potrebné zvážiť. Simvastatín
nemal žiadny vplyv na hladiny tikagreloru v plazme. Tikagrelor môže mať podobný účinok na
lovastatín. Súbežné použitie tikagreloru so simvastatínom alebo lovastatínom v dávkach vyšších ako 40 mg sa neodporúča.
· Atorvastatín - pri súbežnom podávaní atorvastatínu a tikagreloru sa Cmax kyseliny atorvastatínovej zvýšil o 23 % a AUC o 36 %. Podobné zvýšenia AUC a Cmax sa pozorovali u všetkých metabolitov kyseliny atorvastatínovej. Tieto zvýšenia sa nepovažujú za klinicky významné.
· Podobný účinok na iné statíny metabolizované prostredníctvom CYP3A4 nemožno vylúčiť.
U pacientov dostávajúcich tikagrelor v štúdii PLATO, ktorí užívali rôzne statíny, nevznikli
v súvislosti s bezpečnosťou statínov žiadne obavy u 93 % pacientov v kohorte štúdie PLATO
užívajúcej tieto lieky.
Tikagrelor je miernym inhibítorom CYP3A4. Súbežné podávanie tikagreloru a substrátov CYP3A4 s úzkym terapeutickým indexom (napr. cisaprid alebo námeľové alkaloidy) sa neodporúča, nakoľko tikagrelor môže zvyšovať expozíciu týchto liekov.
SubstrátyP-gp(vrátanedigoxínu,cyklosporínu)
Pri súbežnom podávaní tikagreloru sa Cmax digoxínu zvýšil o 75 % a AUC o 28 %. Priemerné hladiny digoxínu pred podaním ďalšej dávky sa pri súbežnom podávaní s tikagrelorom zvýšili približne
o 30 %, v niektorých jednotlivých prípadoch maximálne až na 2-násobok. V prítomnosti digoxínu
nebola ovplyvnená Cmax a AUC tikagreloru a jeho aktívneho metabolitu. Pri podávaní P-gp- dependentných liečiv s úzkym terapeutickým indexom, ako je digoxín, súbežne s tikagrelorom sa preto odporúča náležité klinické a/alebo laboratórne monitorovanie.
Nebol pozorovaný žiadny vplyv tikagreloru na hladinu cyklosporínu v krvi. Vplyv tikagreloru na ďalšie substráty P-gp sa neskúmal.
LiečivámetabolizovanéCYP2C9
Súbežné podávanie tikagreloru a tolbutamidu neviedlo k zmenám plazmatických hladín žiadneho z liečiv, čo naznačuje, že tikagrelor nie je inhibítorom CYP2C9 a nie je pravdepodobné, že by spôsoboval zmeny v metabolizme sprostredkovanom CYP2C9 u liečiv, akými sú warfarín a tolbutamid.
Perorálnekontraceptíva
Pri súbežným podávaní tikagreloru a levonorgestrelu a etinylestradiolu sa expozícia etinylestradiolu zvýšila približne o 20 %, ale k zmenám vo farmakokinetike levonorgestrelu nedošlo. Pri súbežnom
podávaní levonorgestrelu a etinylestradiolu s tikagrelorom sa nepredpokladá žiadny klinicky
významný vplyv na účinnosť perorálnych kontraceptív.
Liečivá,ktorévyvolávajúbradykardiu
Pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že vyvolávajú bradykardiu, je potrebná opatrnosť vzhľadom na pozorované prípady zväčša asymptomatickej ventrikulárnej pauzy a bradykardie (pozri časť 4.4). V štúdii PLATO sa však po súbežnom podaní s jedným alebo viacerými liekmi, o ktorých je známe, že vyvolávajú bradykardiu (napr. 96 % betablokátory, 33 % blokátory kalciových kanálov diltiazem a verapamil a 4 % digoxín) nepozorovali žiadne klinicky významné nežiaduce reakcie.
Ďalšiasúbežnáliečba
V klinických štúdiách sa tikagrelor bežne podával spolu s ASA, inhibítormi protónovej pumpy, statínmi, betablokátormi, inhibítormi enzýmu konvertujúceho angiotenzín (ACE) a s blokátormi
receptorov angiotenzínu dlhodobo, ak si to súbežné ochorenia vyžadovali a tiež s heparínom, nízkomolekulovým heparínom a intravenóznymi inhibítormi GpIIb/IIIa pri krátkodobom podávaní
(pozri časť 5.1). Nepozorovali sa žiadne klinicky významné nežiaduce interakcie s týmito liečivami.
Súbežné podávanie tikagreloru s heparínom, enoxaparínom alebo dezmopresínom nemalo žiadny vplyv na aktivovaný parciálny tromboplastínový čas (aPTT), aktivovaný koagulačný čas (ACT) alebo na testy faktora Xa. Pri súbežnom podávaní tikagreloru s liekmi, o ktorých je známe, že ovplyvňujú hemostázu, je však potrebné postupovať s opatrnosťou pre možné farmakodynamické interakcie.
Vzhľadom na hlásenia abnormalít kožného krvácania so SSRI (napr. paroxetín, sertralín a citalopram) sa pri súbežnom podávaní tikagreloru so SSRI odporúča opatrnosť, pretože to môže zvýšiť riziko krvácania.
4.6 Fertilita, gravidita a laktácia
Ženyvreprodukčnomveku
Ženy v reprodukčnom veku majú počas liečby tikagrelorom používať vhodné antikoncepčné metódy na zabránenie gravidity.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití tikagreloru u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Tikagrelor sa neodporúča užívať počas gravidity.
Dojčenie
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie tikagreloru a jeho aktívnych metabolitov do mlieka (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť
vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu tikagrelorom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Tikagrelor nemal žiadny vplyv na fertilitu samcov alebo samíc zvierat (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tikagrelor nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Počas liečby tikagrelorom sa hlásil závrat a zmätenosť. Pacienti, u ktorých sa objavia tieto príznaky, majú byť preto pri vedení vozidiel alebo obsluhe strojov opatrní.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnostný profil tikagreloru sa hodnotil v dvoch rozsiahlych skúšaniach fázy 3 (PLATO
a PEGASUS), ktoré zahŕňali viac ako 39 000 pacientov (pozri časť 5.1).
V štúdii PLATO bol pri tikagrelore vyšší výskyt pacientov, ktorí ukončili liečbu pre nežiaduce udalosti, ako pri klopidogrele (7,4 % oproti 5,4 %). V štúdii PEGASUS bol pri tikagrelore vyšší výskyt pacientov, ktorí ukončili liečbu pre nežiaduce udalosti, v porovnaní s liečbou samotnou ASA (16,1 % pre 60 mg tikagreloru s ASA oproti 8,5 % pre liečbu samotnou ASA). Najčastejšie hlásenými nežiaducimi reakciami u pacientov liečených tikagrelorom boli krvácanie a dyspnoe (pozri časť 4.4).
Tabuľkovýzoznamnežiaducichreakcií
V klinických štúdiách s tikagrelorom alebo v období po uvedení lieku na trh sa zistili a hlásili nasledujúce nežiaduce reakcie (tabuľka 1).
Nežiaduce reakcie sú uvedené podľa tried orgánových systémov (TOS) MedDRA. V rámci každej
TOS sú nežiaduce reakcie usporiadané podľa kategórie frekvencie. Kategórie frekvencie sú
definované podľa nasledovných pravidiel: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme
(z dostupných údajov).
Tabuľka 1 – Nežiaduce reakcie podľa frekvencie a triedy orgánových systémov (TOS) TOS Veľmi časté Časté Menej časté
Benígne a malígne
nádory, vrátane nešpecifikovaných
novotvarov (cysty
a polypy) Poruchy krvi
a lymfatického
systému
Poruchy imunitného systému
Poruchy metabolizmu a výživy
krvácavá poruchab
hyperurikémiad dna/dnová artritída
krvácanie nádorua
precitlivenosť vrátane angioedémuc
Psychické poruchy zmätenosť
Poruchy nervového systému
závrat, synkopa, bolesť hlavy
intrakraniálne krvácanie
Poruchy oka krvácanie do okae
Poruchy ucha a labyrintu
vertigo krvácanie do ucha
Poruchy ciev hypotenzia
T
O
S Veľmi časté Časté Menej časté
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho
traktu
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy obličiek a močových ciest Poruchy reprodukčného systému a prsníkov Laboratórne
a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
dyspnoe krvácanie do dýchacích ciestf
gastrointestinálne krvácanieg, hnačka, nauzea, dyspepsia, zápcha
podkožné alebo kožné krvácanieh, vyrážka, pruritus
krvácanie do močových ciestj
zvýšený kreatinín v krvid
krvácanie po zákroku, poúrazové krvácaniel
retroperitoneálne krvácanie
krvácanie do svalui
krvácanie do reprodukčného systémuk
a napr. krvácanie z nádoru močového mechúra, nádoru žalúdka, nádoru hrubého čreva
b napr. zvýšený sklon k tvorbe krvných podliatin, spontánny hematóm, hemoragická diatéza
c Identifikované v období po uvedení lieku na trh.
d Frekvencie odvodené z laboratórnych pozorovaní (Zvýšenia kyseliny močovej ˃ horná hranica normálu oproti východiskovej hodnote, pod alebo v rámci referenčného rozmedzia. Zvýšenia kreatinínu ˃ 50 % oproti východiskovej
hodnote.), a nie neprepočítané frekvencie hlásených nežiaducich udalostí.
e napr. krvácanie do spojovky, sietnice, vnútroočné krvácanie
f napr. epistaxa, hemoptýza
g napr. krvácanie ďasien, rektálne krvácanie, krvácanie žalúdkového vredu
h napr. ekchymóza, krvácanie do kože, petechie
i napr. hemartróza, krvácanie do svalu
j napr. hematúria, hemoragická cystitída
k napr. vaginálne krvácanie, hematospermia, postmenopauzálne krvácanie
l napr. kontúzia, poúrazový hematóm, poúrazové krvácanie
Popis vybraných nežiaducichreakcií
Krvácanie
Zistenia týkajúce sa krvácania v štúdii PLATO
V tabuľke 2 sú uvedené celkové výsledky miery krvácania v štúdii PLATO.
Tabuľka 2 – Analýza celkových krvácavých príhod, Kaplanov-Meierov odhad v 12.mesiaci
(štúdia PLATO)
tikagrelor
90 mg dvakrát denne
N=9235
klopidogrel
N=9186 p-hodnota*
Veľké krvácania podľa PLATO celkovo 11,6 11,2 0,4336
Veľké fatálne/život ohrozujúce krvácania podľa PLATO
Veľké krvácania podľa PLATO nesúvisiace s
CABG
5,8 5,8 0,6988
4,5 3,8 0,0264
Veľké krvácania podľa PLATO nesúvisiace s 3,1 2,3 0,0058
liečebným postupom
Veľké + malé krvácania podľa PLATO
celkovo
Veľké + malé krvácania podľa PLATO
nesúvisiace s liečebným postupom
Veľké krvácania definované podľa kritérií
TIMI
Veľké + malé krvácania definované podľa kritérií TIMI
Definície kategórií krvácania:16,1 14,6 0,0084
5,9 4,3 <0,0001
7,9 7,7 0,5669
11,4 10,9 0,3272
Veľké fatálne/ život ohrozujúce krvácanie: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o viac ako 50 g/l
alebo s transfúziou ≥ 4 jednotiek erytrocytov alebo fatálne alebo intrakraniálne alebo intraperikardiálne krvácanie s tamponádou srdca; alebo hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov alebo chirurgickú intervenciu.
Veľké iné: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50 g/l alebo s transfúziou 2 až 3 jednotiek erytrocytov; alebo významne vysiľujúce krvácanie.
Malé krvácanie: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
Veľké krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu > 50 g/l alebo intrakraniálne krvácanie.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie súvisiace s poklesom hemoglobínu o 30 až 50g/l.
*p-hodnota vypočítaná na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO, veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI alebo malého krvácania podľa kritérií TIMI (tabuľka 2). V porovnaní
s klopidogrelom sa však pri tikagrelore vyskytlo spolu viac veľkých a malých krvácaní podľa definície PLATO. Niekoľko pacientov v štúdii PLATO malo fatálne krvácania: 20 (0,2 %) pri tikagrelore a 23 (0,3 %) pri klopidogrele (pozri časť 4.4).
Vek, pohlavie, hmotnosť, rasa, geografické územie, súbežné ochorenia, súbežná liečba a anamnéza pacienta, vrátane prekonanej cievnej mozgovej príhody alebo prekonaného prechodného ischemického záchvatu, neboli prediktívne z hľadiska celkového výskytu krvácaní, ani z hľadiska výskytu veľkého krvácania podľa definície PLATO nesúvisiaceho s liečebným postupom. Preto pre žiaden podtyp krvácania nebola konkrétna skupina identifikovaná ako riziková.
Krvácanie súvisiace s CABG:
V štúdii PLATO sa u 42 % z 1 584 pacientov (12 % kohorty), ktorí podstúpili zákrok CABG, vyskytlo veľké fatálne/život ohrozujúce krvácanie podľa definície PLATO, bez rozdielu medzi liečebnými
skupinami. Fatálne krvácanie súvisiace s CABG sa vyskytlo v každej liečebnej skupine u 6 pacientov
(pozri časť 4.4).
Krvácanie nesúvisiace s CABG a krvácanie nesúvisiace s liečebným postupom:
Tikagrelor a klopidogrel sa neodlišovali vo výskyte veľkého fatálneho/život ohrozujúceho krvácania podľa definície PLATO nesúvisiaceho s CABG, ale výskyt veľkého krvácania podľa definície PLATO celkovo, veľkého krvácania podľa kritérií TIMI a výskyt veľkého a malého krvácania podľa kritérií TIMI bol častejší pri tikagrelore. Rovnako, po vylúčení všetkých krvácaní súvisiacich s liečebným postupom, sa viac krvácaní vyskytlo pri tikagrelore ako pri klopidogrele (tabuľka 2). Ukončenie liečby pre krvácanie nesúvisiace s liečebným postupom bolo častejšie pri tikagrelore (2,9 %) ako pri klopidogrele (1,2 %; p< 0,001).
Intrakraniálne krvácanie:
V skupine s tikagrelorom sa vyskytlo viac intrakraniálnych krvácaní nesúvisiacich s liečebným postupom (n = 27 krvácaní u 26 pacientov, 0,3 %) ako v skupine s klopidogrelom (n = 14 krvácaní,
0,2 %), z ktorých bolo fatálnych 11 v skupine s tikagrelorom a 1 v skupine s klopidogrelom.
V celkovom výskyte fatálnych krvácaní nebol žiadny rozdiel.
Zistenia týkajúce sa krvácania v štúdii PEGASUS
V tabuľke 3 sú uvedené celkové výsledky udalostí súvisiacich s krvácaním v štúdii PEGASUS.
Tabuľka 3 – Analýza celkových krvácavých príhod, Kaplanov-Meierov odhad v 36. mesiaci
(štúdia PEGASUS)
tikagrelor 60 mg dvakrát
denne + ASA N = 6 958
Pomer
samotná ASA N = 6 996
Koncový ukazovateľ bezpečnosti KM %
Kategórie krvácania podľa kritérií TIMI
rizika
(95 % IS)
KM % p-hodnota
Veľké krvácania podľa TIMI 2,3 2,32 (1,68; 3,21)
Fatálne 0,3 1,00 (0,44; 2,27)
ICH 0,6 1,33 (0,77; 2,31)
1,1 < 0,0001
0,3 1,0000
0,5 0,3130
Iné veľké krvácania podľa
TIMI 1,6
Veľké alebo malé krvácania podľa
TIMI 3,4
Veľké alebo malé krvácania alebo
3,61
(2,31; 5,65) 0,5 < 0,0001
2,54
(1,93; 3,35) 1,4 < 0,0001
krvácania vyžadujúce lekársku starostlivosť podľa TIMI
16,6 2,64 (2,35; 2,97)
7,0 < 0,0001
Kategórie krvácania podľa definície PLATO
Veľké krvácania podľa PLATO 3,5 2,57 (1,95; 3,37)
Fatálne/život ohrozujúce 2,4 2,38 (1,73; 3,26)
1,4 < 0,0001
1,1 < 0,0001
Iné veľké krvácania podľa
PLATO 1,1
Veľké alebo malé krvácania podľa
PLATO 15,2
Definície kategórií krvácania:
3,37
(1,95; 5,83) 0,3 < 0,0001
2,71
(2,40; 3,08) 6,2 < 0,0001
Veľké krvácanie podľa TIMI: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO klinicky zjavné prejavy hemorágie spojené s poklesom hemoglobínu (Hgb) ≥ 50 g/l, alebo 15 % pokles hematokritu (Hct) v prípade nedostupnosti údajov o Hgb.
Fatálne krvácanie: Krvácavá príhoda, ktorá viedla priamo k smrti v priebehu 7 dní.
ICH: Intrakraniálne krvácanie.
Iné veľké krvácanie podľa TIMI: Veľké non-fatálne krvácanie podľa TIMI iné než ICH.
Malé krvácanie podľa TIMI: Klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l.
Krvácanie vyžadujúce lekársku starostlivosť podľa TIMI: Vyžadujúce zásah, ALEBO vedúce k hospitalizácii, ALEBO
vyžadujúce vyšetrenie.
Veľké fatálne/život ohrozujúce krvácanie podľa PLATO: Fatálne krvácanie, ALEBO akékoľvek intrakraniálne krvácanie, ALEBO intraperikardiálne krvácanie s tamponádou srdca, ALEBO s hypovolemickým šokom alebo ťažkou hypotenziou vyžadujúcou si podanie vazopresorov/inotropík alebo chirurgickú intervenciu, ALEBO klinicky zjavné krvácanie s poklesom
hemoglobínu ˃ 50 g/l alebo s transfúziou ≥ 4 jednotiek erytrocytov.
Iné veľké krvácanie podľa PLATO: Významne vysiľujúce krvácanie, ALEBO klinicky zjavné krvácanie s poklesom hemoglobínu o 30 – 50 g/l alebo s transfúziou 2 - 3 jednotiek erytrocytov.
Malé krvácanie podľa PLATO: Vyžaduje si lekársky zásah na zastavenie alebo zvládnutie krvácania.
V štúdii PEGASUS bol výskyt veľkého krvácania podľa kritérií TIMI pri tikagrelore v dávke 60 mg dvakrát denne vyšší ako pri samotnej ASA. Nepozorovalo sa zvýšené riziko fatálneho krvácania
a pozorovalo sa len menej významné zvýšenie výskytu intrakraniálneho krvácania v porovnaní
s liečbou samotnou ASA. V štúdii sa vyskytlo niekoľko fatálnych krvácavých príhod, 11 (0,3 %) pri
60 mg tikagreloru a 12 (0,3 %) pri liečbe samotnou ASA. Pozorované zvýšené riziko výskytu veľkého krvácania podľa kritérií TIMI pri 60 mg tikagreloru bolo zapríčinené predovšetkým vyššou frekvenciou výskytu iného veľkého krvácania podľa kritérií TIMI zastúpeného udalosťami
v gastrointestinálnej TOS.
Podobne zvýšený výskyt veľkého krvácania podľa kritérií TIMI bol pozorovaný aj pri kategóriách krvácania zahŕňajúcich veľké alebo malé krvácania podľa kritérií TIMI, veľké krvácania podľa definície PLATO a veľké alebo malé krvácania podľa definície PLATO (pozri tabuľku 3). Ukončenie liečby pre krvácanie bolo častejšie pri 60 mg tikagreloru (6,2 %) v porovnaní s liečbou samotnou ASA (1,5 %). Väčšina týchto krvácaní bola menej závažná (klasifikované ako krvácania vyžadujúce lekársku starostlivosť podľa kritérií TIMI), napr. epistaxa, tvorba krvných podliatin a hematómov.
Profil krvácania pri 60 mg tikagreloru bol pre udalosti veľkého krvácania podľa kritérií TIMI, veľkého alebo malého krvácania podľa kritérií TIMI a veľkého krvácania podľa definície PLATO konzistentný v rámci viacerých vopred definovaných podskupín (napr. podľa veku, pohlavia, hmotnosti, rasy, geografického regiónu, súbežných ochorení, súbežnej liečby a anamnézy).
Intrakraniálne krvácanie:
Pre 60 mg tikagreloru a liečbu samotnou ASA sa hlásili podobné miery výskytu spontánneho intrakraniálneho krvácania (n = 13, 0,2 % v oboch liečebných skupinách). Pri liečbe 60 mg tikagreloru (n = 15, 0,2 %) v porovnaní s liečbou samotnou ASA (n = 10, 0,1 %) sa preukázalo menej významné zvýšenie výskytu poúrazového intrakraniálneho krvácania a intrakraniálneho krvácania súvisiaceho
s liečebným postupom. Pri 60 mg tikagreloru sa vyskytlo 6 fatálnych prípadov intrakraniálneho krvácania a pri liečbe samotnou ASA 5 prípadov. Výskyt intrakraniálneho krvácania bol v oboch
liečebných skupinách nízky vzhľadom na to, že populácia štúdie sa vyznačovala významnou mierou
komorbidity a KV rizikových faktorov.
Dyspnoe
Pacienti liečení tikagrelorom hlásili dyspnoe, pocit sťaženého dýchania. V štúdii PLATO nežiaduce udalosti týkajúce sa dyspnoe (dyspnoe, kľudové dyspnoe, námahové dyspnoe, paroxyzmálne nočné dyspnoe a nočné dyspnoe) hlásili u 13,8 % pacientov liečených s tikagrelorom a u 7,8 % pacientov liečených s klopidogrelom. V štúdii PLATO u 2,2 % pacientov užívajúcich tikagrelor a u 0,6 % pacientov užívajúcich klopidogrel skúšajúci považovali dyspnoe za príčinu súvisiacu s liečbou a málo prípadov bolo závažných (0,14 % tikagrelor; 0,02 % klopidogrel) (pozri časť 4.4). Väčšina hlásených príznakov dyspnoe bola miernej až stredne ťažkej intenzity a väčšinou sa hlásili ako jedna epizóda krátko po začatí liečby.
V porovnaní s klopidogrelom bolo u pacientov s astmou/CHOCHP, ktorí boli liečení tikagrelorom, zvýšené riziko výskytu nezávažného dyspnoe (3,29 % v prípade tikagreloru oproti 0,53 % v prípade klopidogrelu) a závažného dyspnoe (0,38 % v prípade tikagreloru oproti 0,0 % v prípade klopidogrelu). V absolútnom vyjadrení bolo toto riziko vyššie ako v celej populácii pacientov v štúdii PLATO. Tikagrelor sa musí užívať opatrne u pacientov s anamnézou astmy a/alebo CHOCHP (pozri časť 4.4).
Približne 30 % všetkých prípadov dyspnoe sa upravilo v priebehu 7 dní. Štúdie PLATO sa zúčastnili pacienti s kongestívnym srdcovým zlyhaním, CHOCHP alebo astmou pred začatím skúšania; títo pacienti a starší pacienti hlásili najčastejšie dyspnoe. Kvôli dyspnoe sa liečba tikagrelorom vysadila u
0,9 % pacientov a liečba klopidogrelom u 0,1 % pacientov. Vyšší výskyt dyspnoe u tikagreloru nesúvisí s novým alebo zhoršujúcim sa srdcovým alebo pľúcnym ochorením (pozri časť 4.4). Tikagrelor nemá vplyv na výsledky testov funkcie pľúc.
V štúdii PEGASUS sa dyspnoe hlásilo u 14,2 % pacientov užívajúcich tikagrelor v dávke 60 mg dvakrát denne a u 5,5 % pacientov užívajúcich samotnú ASA. Rovnako ako v štúdii PLATO, väčšina hlásených prípadov dyspnoe bola miernej až stredne ťažkej intenzity (pozri časť 4.4). Pacienti, ktorí hlásili dýchavičnosť boli starší a mali na začiatku častejšie dýchavičnosť, CHOCHP alebo astmu.
Vyšetrenia
Zvýšenie kyseliny močovej: V štúdii PLATO došlo k zvýšeniu kyseliny močovej v sére nad hornú hranicu normálu u 22 % pacientov dostávajúcich tikagrelor, v porovnaní s 13 % pacientov dostávajúcich klopidogrel. V štúdii PEGASUS boli príslušné hodnoty 9,1 % pre 90 mg tikagreloru,
8,8 % pre 60 mg tikagreloru a 5,5 % pre placebo. Priemerná hladina kyseliny močovej v sére sa pri tikagrelore zvýšila približne o 15 % v porovnaní so zvýšením približne o 7,5 % pri klopidogrele a po
ukončení liečby sa znížila približne o 7 % v prípade tikagreloru, v prípade klopidogrelu sa však žiadne zníženie nezistilo. V štúdii PEGASUS sa zistilo reverzibilné zvýšenie priemerných hladín kyseliny močovej v sére o 6,3 % pre 90 mg tikagreloru a 5,6 % pre 60 mg tikagreloru, v porovnaní s 1,5 % znížením v skupine s placebom. V štúdii PLATO bola frekvencia dnovej artritídy 0,2 % pri tikagrelore oproti 0,1 % pri klopidogrele. V štúdii PEGASUS boli príslušné hodnoty frekvencie dny/dnovej artritídy 1,6 % pre 90 mg tikagreloru, 1,5 % pre 60 mg tikagreloru a 1,1 % pre placebo.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieTikagrelor je v jednorazových dávkach až do 900 mg dobre znášaný. Gastrointestinálna toxicita bola určujúcim faktorom v štúdii zameranej na zvyšovanie jednorazovej dávky. Iné klinicky významné nežiaduce reakcie, ktoré sa môžu vyskytnúť pri predávkovaní, zahŕňajú dyspnoe a ventrikulárne pauzy (pozri časť 4.8).
V prípade predávkovania sa môžu objaviť vyššie uvedené potenciálne nežiaduce reakcie a je potrebné zvážiť monitorovanie EKG.
V súčasnosti nie je známe žiadne antidotum na zvrátenie účinkov tikagreloru a nepredpokladá sa, že by bol tikagrelor dialyzovateľný (pozri časť 4.4). Liečba predávkovania sa má riadiť štandardnou lekárskou praxou na miestnej úrovni. Predpokladaným účinkom nadmerných dávok tikagreloru je predĺžené trvanie rizika krvácania, ktoré súvisí s inhibíciou krvných doštičiek. Transfúzia krvných doštičiek pravdepodobne nepredstavuje klinický prínos pre pacientov s krvácaním (pozri časť 4.4). Ak dôjde ku krvácaniu, je potrebné prijať ďalšie príslušné podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antiagreganciá trombocytov okrem heparínu, ATC kód: B01AC24
MechanizmusúčinkuBrilique obsahuje tikagrelor patriaci do chemickej skupiny cyklopentyltriazolopyrimidínov (CPTP), ktorý je perorálnym, priamo pôsobiacim, selektívnym antagonistom, reverzibilne sa viažucim na
receptor P2Y12, ktorý zabraňuje aktivácii a agregácii krvných doštičiek sprostredkovanej ADP
a závislej na P2Y12. Tikagrelor nezabraňuje väzbe ADP, ale po naviazaní na receptor P2Y12 zabraňuje
signálnej transdukcii indukovanej ADP. Vzhľadom na to, že krvné doštičky sa podieľajú na vzniku
a/alebo vývoji trombotických komplikácií aterosklerotických ochorení, preukázalo sa, že inhibícia funkcie krvných doštičiek znižuje riziko kardiovaskulárnych príhod, ako je smrť, IM alebo cievna
mozgová príhoda.
Tikagrelor tiež zvyšuje lokálne hladiny endogénneho adenozínu inhibíciou rovnovážnych nukleozidových transportérov-1 (ENT-1).
Zistilo sa, že tikagrelor u zdravých dobrovoľníkov a pacientov s AKS zvýrazňuje nasledujúce účinky indukované adenozínom: vazodilatácia (merané zvýšením koronárneho prietoku krvi u zdravých dobrovoľníkov a pacientov s AKS; bolesť hlavy), inhibícia funkcie krvných doštičiek (v plnej ľudskej krvi
in vitro) a dyspnoe. Súvislosť medzi pozorovanými zvýšeniami adenozínu a klinickými výsledkami (napr. morbidita-mortalita) však nebola jasne vysvetlená.
Farmakodynamické
účinky
Nástup
účinku
Tikagrelor u pacientov so stabilnou koronárnou artériovou chorobou (CAD) užívajúcich ASA
vykazuje rýchly nástup farmakologického účinku, čo sa preukázalo priemernou inhibíciou agregácie krvných doštičiek (IPA) tikagrelorom po 0,5 hodiny od podania nárazovej dávky 180 mg približne
41 %, s maximálnym účinkom na IPA 89 % po 2 – 4 hodinách od podania dávky a tento účinok pretrvával 2 – 8 hodín. U 90 % pacientov bol finálny rozsah IPA po 2 hodinách od podania dávky
> 70 %.
Odznievanie účinku
Pri plánovanom zákroku CABG existuje zvýšené riziko krvácania pre tikagrelor oproti klopidogrelu, pokiaľ je liečba vysadená v kratšej dobe ako 96 hodín pred zákrokom.
Údajetýkajúcesa prechodunainúliečbu
Prechod z liečby klopidogrelom v dávke 75 mg na tikagrelor v dávke 90 mg dvakrát denne má za následok absolútne zvýšenie IPA o 26,4 % a prechod z liečby tikagrelorom na klopidogrel má za následok absolútne zníženie IPA o 24,5 %. Pacientov možno prestaviť z liečby klopidogrelom na tikagrelor bez akéhokoľvek prerušenia protidoštičkového účinku (pozri časť 4.2).
Klinickáúčinnosťabezpečnosť
Klinický dôkaz účinnosti a bezpečnosti tikagreloru je odvodený z dvoch skúšaní fázy 3:
· Štúdia PLATO [PLATelet Inhibition and Patient Outcomes], porovnávajúca tikagrelor oproti klopidogrelu, oba podávané v kombinácii s ASA a ďalšou štandardnou liečbou.
· Štúdia PEGASUS TIMI-54 [PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients], porovnávajúca tikagrelor v kombinácii s ASA oproti liečbe samotnou ASA.
Štúdia PLATO(akútnykoronárnysyndróm)
Do štúdie PLATO bolo zahrnutých 18 624 pacientov, u ktorých v priebehu ostatných 24 hodín došlo
k nástupu príznakov nestabilnej anginy pectoris (unstable angina, UA), infarktu myokardu bez elevácie ST segmentu (NSTEMI) alebo infarktu myokardu s eleváciou ST segmentu (STEMI), a ktorí
spočiatku dostávali medikamentóznu liečbu alebo sa u nich vykonala perkutánna koronárna intervencia (PCI) alebo CABG.
Klinická účinnosť
Pri dennom podávaní ASA preukázal tikagrelor v dávke 90 mg dvakrát denne v prevencii výskytu združeného koncového ukazovateľa KV smrti, IM alebo cievnej mozgovej príhody, lepší účinok ako
klopidogrel v dávke 75 mg denne, na tomto rozdiele sa podieľal najmä výskyt KV smrti a IM. Pacienti
dostali 300 mg nárazovú dávku klopidogrelu (prípadne 600 mg v prípade PCI) alebo 180 mg tikagreloru.
Výsledok liečby sa prejavil skoro (zníženie absolútneho rizika [ARR] 0,6 % a zníženie relatívneho rizika [RRR] 12 % po 30 dňoch) a účinok liečby ostal rovnaký po celé obdobie 12 mesiacov, ARR/rok bolo 1,9 % a RRR 16 %. To naznačuje, že je vhodné liečiť pacientov tikagrelorom 90 mg dvakrát denne až 12 mesiacov (pozri časť 4.2). Liečbou 54 pacientov s AKS tikagrelorom namiesto klopidogrelu sa zabráni 1 aterotrombotickej príhode; liečbou 91 pacientov tikagrelorom namiesto klopidogrelu sa zabráni 1 KV smrti (pozri obrázok 1 a tabuľku 4).
Účinok liečby tikagrelorom sa popri klopidogrele ukazuje ako konzistentný v rámci rôznych podskupín pacientov, vrátane členenia podľa hmotnosti, pohlavia, diabetu v anamnéze, prechodného ischemického záchvatu alebo nehemoragickej cievnej mozgovej príhody alebo revaskularizácie, súbežnej liečby zahŕňajúcej liečbu heparínmi, inhibítormi GpIIb/IIIa a inhibítormi protónovej pumpy (pozri časť 4.5), podľa diagnózy, u ktorej sa sledovali koncové ukazovatele (STEMI, NSTEMI alebo UA) a podľa zámeru liečebného postupu pri randomizácii (invazívny alebo medikamentózny).
Pozorovala sa málo významná súčinnosť liečby a geografického územia, pričom podľa pomeru rizika (HR) pre primárny koncový ukazovateľ zo svetového hľadiska vychádza priaznivejšie účinok tikagreloru s výnimkou Severnej Ameriky, kde vychádza priaznivejšie účinok klopidogrelu, ktorá predstavovala približne 10 % celkovej skúmanej populácie (p-hodnota interakcie = 0,045). Výskumné analýzy poukázali na možnú súvislosť s dávkou ASA tak, že znížená účinnosť tikagreloru sa pozorovala pri zvyšujúcich sa dávkach ASA. Pri dlhodobom podávaní ASA s tikagrelorom má byť rozmedzie dennej dávky 75 - 150 mg (pozri časť 4.2 a 4.4).
Obrázok 1 vyjadruje odhad rizika prvého výskytu akejkoľvek udalosti zahrnutej do združeného koncového ukazovateľa účinnosti.
Obrázok 1 Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IMa cievnej mozgovej príhody (štúdia PLATO)
0 6 0 1 2 0 1 8 0 2 4 0 3 0 0 3 6 0
Počet v ohroizení í
Dn i o d ra nd omizácie
T 9 3 3 3
C 9 2 9 1
8 6 2 8
8 5 2 1
8 4 6 0
8 3 6 2
8 2 1 9
8 1 2 4
6 7 4 3
6 6 5 0
5 1 6 1
5 0 9 6
4 1 4 7
4 0 7 4
Tikagrelor v porovnaní s klopidogrelom znížil výskyt primárneho združeného koncového ukazovateľa
rovnako u pacientov s UA/NSTEMI ako aj u pacientov so STEMI (tabuľka 4). Liečbu liekom Brilique
90 mg dvakrát denne spolu s nízkou dávkou ASA možno teda použiť u pacientov s AKS (nestabilná angina pectoris, infarkt myokardu bez elevácie ST segmentu [NSTEMI] alebo infarkt myokardu
s eleváciou ST segmentu [STEMI]); vrátane pacientov, ktorí dostávajú medikamentóznu liečbu
a pacientov, u ktorých sa vykonala perkutánna koronárna intervencia (PCI) alebo koronárny artériový by-pass (CABG).
Tabuľka 4 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PLATO)
tikagrelor
90 mg dvakrát
denne
(%
pacientov
s udalosťou) N = 9 333
klopidogrel
75 mg jedenkrát
denne
(% pacientov s udalosťou)
N = 9 291
ARR
a
(%/rok)
RRR
a
(%)
(95 % IS)
p
-
hodnota
KV smrť/IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
9,3 10,9 1,9 16 (8; 23) 0,0003
So zámerom
invazívneho zákroku 8,5 10,0 1,7 16 (6; 25) 0,0025
So zámerom
medikamentóznej liečby
11,3 13,2 2,3 15 (0,3; 27) 0,0444d
KV smrť 3,8 4,8 1,1 21 (9; 31) 0,0013
IM (s vylúčením
tichého IM)b 5,4 6,4 1,1 16 (5; 25) 0,0045
Cievna mozgová
príhoda 1,3 1,1 −0,2 −17 (−52; 9) 0,2249
Úmrtnosť zo všetkých príčin, IM
(s vylúčením tichého IM) alebo cievna mozgová príhoda
KV smrť, IM celkovo, cievna mozgová príhoda, SRI, RI, TIA alebo iná ATEc
Úmrtnosť zo
9,7 11,5 2,1 16 (8; 23) 0,0001
13,8 15,7 2,1 12 (5; 19) 0,0006
d
všetkých príčin 4,3 5,4 1,4 22 (11; 31) 0,0003
Definitívna d
trombóza stentu 1,2 1,7 0,6 32 (8; 49) 0,0123
aARR = zníženie absolútneho rizika; RRR = zníženie relatívneho rizika = (1 − pomer rizika) x 100 %. Hodnoty s negatívnym
RRR naznačujú zvýšenie relatívneho rizika.
bS vylúčením tichého IM.
cSRI = ťažká rekurentná ischémia (serious recurrent ischaemia); RI = rekurentná ischémia (recurrent ischaemia); TIA =
prechodný ischemický záchvat (transient ischaemic attack); ATE = arteriálna trombotická príhoda. IM celkovo zahŕňa tichý
IM, s dátumom, kedy bol zistený.
dNominálna hladina významnosti (significance value); všetky ostatné sú formálne štatisticky významné vopred definovaným hierarchickým testovaním.
Genetická podštúdia štúdie PLATO
Genotypizáciou CYP2C19 a ABCB1 u 10 285 pacientov v štúdii PLATO sa zistili súvislosti medzi
skupinami genotypu a výsledkami štúdie PLATO. Lepší účinok tikagreloru oproti klopidogrelu
v znížení výskytu závažných KV príhod nebol významne ovplyvnený genotypom CYP2C19 alebo ABCB1 pacienta. Podobne ako v celej štúdii PLATO sa celkový výskyt veľkých krvácaní podľa definície PLATO pri tikagrelore a klopidogrele neodlišoval, bez ohľadu na genotyp CYP2C19 alebo ABCB1. Výskyt veľkých krvácaní podľa definície PLATO nesúvisiacich s CABG bol pri tikagrelore
v porovnaní s klopidogrelom zvýšený u pacientov s jednou alebo viacerými nefunkčnými alelami
CYP2C19, ale podobný ako pri klopidogrele u pacientov bez nefunkčnej alely.
Združený ukazovateľ účinnosti a bezpečnosti
Združený ukazovateľ účinnosti a bezpečnosti (KV smrť, IM, cievna mozgová príhoda alebo veľké krvácanie podľa definície PLATO celkovo) naznačuje, že klinický prínos účinnosti tikagreloru
v porovnaní s klopidogrelom nie je kompenzovaný udalosťami súvisiacimi s veľkým krvácaním (ARR
1,4 %, RRR 8 %, HR 0,92; p = 0,0257) v priebehu 12 mesiacov od AKS.
Klinická bezpečnosť
Podštúdia s Holterovým monitorovaním:
Na sledovanie výskytu ventrikulárnych páuz a iných epizód arytmií počas štúdie PLATO skúšajúci vykonali Holterovo monitorovanie u podskupiny takmer 3 000 pacientov, z ktorých približne 2 000
malo záznam aj v akútnej fáze ich AKS a aj po jednom mesiaci. Primárnym sledovaným
ukazovateľom bol výskyt ventrikulárnych páuz ≥ 3 sekundy. Ventrikulárne pauzy sa u pacientov v akútnej fáze vyskytovali častejšie pri tikagrelore (6,0 %) ako pri klopidogrele (3,5 %); a po 1 mesiaci to bolo 2,2 % pri tikagrelore a 1,6 % pri klopidogrele (pozri časť 4.4). Nárast výskytu ventrikulárnych páuz v akútnej fáze AKS bol v skupine s tikagrelorom výraznejší u pacientov
s chronickým srdcovým zlyhávaním (CHF) v anamnéze (9,2 % oproti 5,4 % u pacientov bez CHF
v anamnéze; v skupine s klopidogrelom to bolo 4,0 % u pacientov s CHF v anamnéze oproti 3,6 %
u pacientov bez CHF v anamnéze). Tento rozdiel sa po 1 mesiaci nevyskytoval: 2,0 % oproti 2,1 % pri tikagrelore pre pacientov s a bez CHF v anamnéze a 3,8 % oproti 1,4 % pri klopidogrele. Tento rozdiel
nesúvisel so žiadnymi nežiaducimi klinickými dôsledkami (vrátane zavedenia kardiostimulátora)
v tejto populácii pacientov.
Štúdia PEGASUS (anamnézainfarktumyokardu)
Štúdia PEGASUS TIMI-54 bola medzinárodná, multicentrická, randomizovaná, dvojito zaslepená,
placebom kontrolovaná štúdia s paralelnou skupinou a s dĺžkou určenou počtom udalostí potrebných pre štatistické vyhodnotenie (event-driven), zahŕňajúca 21 162 pacientov, ktorá hodnotila prevenciu
aterotrombotických príhod pri tikagrelore podávanom v 2 dávkach (buď 90 mg dvakrát denne alebo
60 mg dvakrát denne) v kombinácii s nízkou dávkou ASA (75 – 150 mg), v porovnaní s liečbou samotnou ASA u pacientov s IM v anamnéze a ďalšími rizikovými faktormi aterotrombózy.
Pacienti boli vhodní na zaradenie do štúdie, ak boli vo veku 50 rokov alebo starší, mali IM
v anamnéze (1 až 3 roky pred randomizáciou) a mali aspoň jeden z nasledujúcich rizikových faktorov aterotrombózy: vek ≥ 65 rokov, diabetes mellitus vyžadujúci liečbu, druhý predchádzajúci IM,
potvrdenú CAD postihujúcu viaceré cievy alebo chronickú dysfunkciu obličiek v non-terminálnom
štádiu.
Pacienti neboli vhodní na zaradenie do štúdie, ak sa u nich počas obdobia štúdie plánoval použiť antagonista receptorov P2Y12, dipyridamol, cilostazol alebo antikoagulačná liečba; ak mali krvácavú poruchu alebo ischemickú cievnu mozgovú príhodu alebo intrakraniálne krvácanie v anamnéze, nádor centrálneho nervového systému alebo abnormalitu intrakraniálnych ciev; ak mali v priebehu predchádzajúcich 6 mesiacov gastrointestinálne krvácanie alebo veľký chirurgický zákrok v priebehu predchádzajúcich 30 dní.
Klinická účinnosť
Obrázok 2 – Analýza primárneho klinického združeného koncového ukazovateľa KV smrti, IM
a cievnej mozgovej príhody (štúdia PEGASUS)

Tikagrelor 60 mg bd - - - - Placebo
N 7045 7067
Pacienti s udalosťami 487 (6.9%) 578 (8.2%) KM% v 36 mesiaci 7.8% 9.0%
Pomer rizika (95% CI) 0.84 (0.74, 0.95)
p-hodnota 0.0043
Počet v riziku
Ti 60 mg
Placebo
Dni od randomizácie
Tabuľka 5 – Analýza primárnych a sekundárnych koncových ukazovateľov účinnosti (štúdia
PEGASUS)
tikagrelor 60 mg dvakrát denne + ASA
N = 7 045
sa
m
otn
á ASA
N = 7 067
p
-
hodnota
Charakteristika
Pacienti
s udalosťami
Primárny koncový ukazovateľ
Združený pre
KV
KM % HR
(95 % IS)
Pacienti
s udalosťami KM %
smrť/IM/cievnu mozgovú príhodu
487 (6,9 %) 7,8 % 0,84 (0,74; 0,95)
578 (8,2 %) 9,0 % 0,0043 (s)
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
IM 285 (4,0 %) 4,5 % 0,84 (0,72; 0,98)
210 (3,0 %) 3,4 % 0,0676
338 (4,8 %) 5,2 % 0,0314
Cievna mozgová
príhoda 91 (1,3 %) 1,5 %
Sekundárny koncový ukazovateľ
0,75
(0,57; 0,98) 122 (1,7 %) 1,9 % 0,0337
KV smrť 174 (2,5 %) 2,9 % 0,83 (0,68; 1,01)
210 (3,0 %) 3,4 % -
Úmrtnosť zo
všetkých príčin 289 (4,1 %) 4,7 %
0,89 326 (4,6 %) 5,2 % - (0,76; 1,04)
Pomer rizika (HR) a
p-hodnota sú vypočítané osobitne pre tikagrelor oproti liečbe samotnou ASA na základe Coxovho modelu proporcionálneho rizika s liečebnou skupinou ako jedinou vysvetľujúcou premennou.
Percentá KM vypočítané po 36 mesiacoch.
Poznámka: počty prvých udalostí pre zložky KV smrť, IM a cievna mozgová príhoda sú skutočné počty prvých udalostí pre každú zo zložiek a nesčítavajú sa do počtu udalostí v združenom koncovom ukazovateli.
(s) Predstavuje štatistickú významnosť (significance).
IS = interval spoľahlivosti; KV = kardiovaskulárna; HR = pomer rizika; KM = Kaplanov-Meierov odhad; IM = infarkt myokardu;
N = počet pacientov.
Oba režimy s tikagrelorom, 60 mg dvakrát denne a 90 mg dvakrát denne, v kombinácii s ASA preukázali lepší účinok v prevencii aterotrombotických príhod v porovnaní so samotnou ASA (združený koncový ukazovateľ: KV smrť, IM a cievna mozgová príhoda), s konzistentným účinkom liečby počas celého obdobia štúdie a dosiahnutím RRR 16 % a ARR 1,27 % pre 60 mg tikagreloru
a RRR 15 % a ARR 1,19 % pre 90 mg tikagreloru.
Hoci bol profil účinnosti 90 mg a 60 mg dávky podobný, bolo dokázané, že nižšia dávka je lepšie znášaná a má lepší bezpečnostný profil v súvislosti s rizikom krvácania a dyspnoe. Preto sa na prevenciu aterotrombotických príhod (KV smrť, IM a cievna mozgová príhoda) u pacientov s IM
v anamnéze a vysokým rizikom aterotrombotickej príhody odporúča iba Brilique 60 mg dvakrát denne podávaný súbežne s ASA.
V porovnaní so samotnou ASA, tikagrelor v dávke 60 mg dvakrát denne významne znížil primárny združený koncový ukazovateľ KV smrti, IM a cievnej mozgovej príhody. Každá zo zložiek prispela k zníženiu primárneho združeného koncového ukazovateľa (RRR KV smrti 17 %, RRR IM 16 %
a RRR cievnej mozgovej príhody 25 %).
RRR pre združený koncový ukazovateľ od 1. do 360. dňa (RRR 17 %) a od 361. dňa ďalej (RRR
16 %) bolo podobné. K dispozícii sú obmedzené údaje o účinnosti a bezpečnosti tikagreloru za 3 roky predĺženej liečby.
Nebol nájdený žiadny dôkaz o prínose (neprišlo k zníženiu primárneho koncového ukazovateľa zloženého z KV úmrtia, infarktu myokardu a cievnej mozgovej príhody, ale zvýšeniu závažného krvácania), pri užívaní tikagrelor 60 mg dvakrát denne u klinicky stabilných pacientov > 2 roky od infarktu myokardu alebo viac ako jeden rok po ukončení predchádzajúcej liečby inhibítorom receptora ADP (pozri tiež časť 4.2).
Klinická bezpečnosť
Počet prerušení liečby s tikagrelorom 60 mg kvôli krvácaniu a dýchavičnosti bola vyššia u pacientov
> 75 rokov (42 %) ako u mladších pacientov (rozmedzie: 23-31 %), s rozdielom v porovnaní s placebom vyšším ako 10 % (42 % vs. 29 %), u pacientov > 75 rokov.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Brilique vo všetkých podskupinách pediatrickej populácie s akútnym koronárnym syndrómom (AKS) a infarktom myokardu (IM) v anamnéze (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika tikagreloru je lineárna a expozícia tikagreloru a aktívnemu metabolitu
(AR-C124910XX) je približne úmerná dávke až po dávku 1 260 mg.
Absorpcia
K absorpcii tikagreloru dochádza rýchlo, s mediánom tmax približne 1,5 hodiny. Tvorba hlavného cirkulujúceho metabolitu AR-C124910XX (tiež aktívneho) z tikagreloru je rýchla s mediánom tmax približne 2,5 hodín. Po perorálnom podaní jednorazovej dávky tikagreloru 90 mg nalačno zdravým dobrovoľníkom je Cmax 529 ng/ml a AUC 3 451 ng*h/ml. Pomer metabolit/pôvodné liečivo pre Cmax je
0,28 a pre AUC 0,42. Farmakokinetika tikagreloru a AR-C124910XX u pacientov s IM v anamnéze
bola vo všeobecnosti podobná farmakokinetike v populácii s AKS. Na základe populačnej farmakokinetickej analýzy štúdie PEGASUS bol medián Cmax tikagreloru v rovnovážnom stave
391 ng/ml a AUC 3 801 ng*h/ml pre dávku 60 mg tikagreloru. Pre dávku 90 mg tikagreloru bola Cmax
v rovnovážnom stave 627 ng/ml a AUC 6 255 ng*h/ml.
Priemerná absolútna biologická dostupnosť tikagreloru sa odhadla na 36 %. Príjem potravy s vysokým obsahom tukov mal za následok zvýšenie AUC tikagreloru o 21 % a zníženie Cmax aktívneho metabolitu o 22 %, na Cmax tikagreloru a na AUC aktívneho metabolitu však nemal žiadny vplyv. Tieto malé zmeny sa považujú za klinicky minimálne významné, preto sa tikagrelor môže užívať s jedlom alebo bez jedla. Tikagrelor ako aj jeho aktívny metabolit sú substrátmi P-gp.
Orodispergovateľné tablety tikagreloru rozpustené v slinách a prehltnuté bez vody alebo suspendované vo vode a podávané nazogastrickou sondou do žalúdka boli bioekvivalentné s filmom obalenými tabletami vcelku (AUC a Cmax v rozmedzí 80-125 % pre tikagrelor a aktívny metabolit). Po rozpustení dispergovateľnej tablety v slinách a prehltnutí s vodou bola AUC tikagreloru rovnaká, zatiaľ čo Cmax bola približne o 15 % nižšia ako pre filmom obalenú tabletu. Pozorovaný malý rozdiel v Cmax pravdepodobne nemá klinický význam.
Distribúcia
Distribučný objem tikagreloru v rovnovážnom stave je 87,5 l. Tikagrelor a aktívny metabolit sa vo veľkej miere viaže na bielkoviny ľudskej plazmy (> 99,0 %).
Biotransformácia
CYP3A4 je hlavným enzýmom zodpovedným za metabolizmus tikagreloru a tvorbu aktívneho metabolitu a ich interakcie s inými substrátmi CYP3A sú v rozmedzí od aktivácie po inhibíciu.
Hlavným metabolitom tikagreloru je AR-C124910XX, ktorý je na základe dôkazu jeho väzby na doštičkový receptor P2Y12 pre ADP v podmienkach in vitro tiež aktívny. Systémová expozícia aktívnemu metabolitu predstavuje približne 30 – 40 % systémovej expozície tikagreloru.
Eliminácia
Primárnou cestou eliminácie tikagreloru je metabolizácia v pečeni. V prípade podávania rádioizotopom značeného tikagreloru sa zachytí v priemere približne 84 % rádioizotopom značenej
dávky (57,8 % v stolici, 26,5 % v moči). Detegované množstvá tikagreloru a aktívneho metabolitu
v moči v obidvoch prípadoch predstavovali menej ako 1 % dávky. Primárnou cestou eliminácie aktívneho metabolitu je s najväčšou pravdepodobnosťou biliárna sekrécia. Priemerná hodnota t1/2 pre tikagrelor bola približne 7 hodín a pre aktívny metabolit 8,5 hodín.
Osobitné skupinypacientov
Staršípacienti
U starších osôb (≥ 75 rokov) s AKS v porovnaní s mladšími pacientmi sa farmakokinetickou analýzou populácie zistilo zvýšenie expozície tikagreloru (približne 25 % pre Cmax aj AUC) a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné (pozri časť 4.2).
Pediatrickápopulácia
Tikagrelor sa neskúmal v pediatrickej populácii (pozri časti 4.2 a 5.1).
Pohlavie
U žien sa v porovnaní s mužmi zaznamenalo zvýšenie expozície tikagreloru a aktívnemu metabolitu. Tieto rozdiely sa nepovažujú za klinicky významné.
Poruchafunkcieobličiek
U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) v porovnaní s osobami s normálnou funkciou obličiek bola expozícia tikagreloru približne o 20 % nižšia
a expozícia aktívnemu metabolitu približne o 17 % vyššia (pozri časť 4.2).
Porucha
funkcie
pečene
Cmax tikagreloru bol o 12 % a AUC o 23 % vyššia u pacientov s miernou poruchou funkcie pečene v porovnaní s kontrolnými zdravými osobami, účinok tikagreloru na IPA bol však u oboch skupín podobný. U pacientov s miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Tikagrelor sa neskúmal u pacientov s ťažkou poruchou funkcie pečene a nie sú k dispozícii farmakokinetické údaje u pacientov so stredne ťažkou poruchou funkcie pečene. U pacientov, ktorí mali stredne závažné alebo závažné východiskové zvýšenie jedného alebo viacerých vyšetrení funkcie pečene, boli plazmatické koncentrácie tikagreloru v priemere podobné alebo mierne vyššie
v porovnaní s pacientmi bez východiskových zvýšení. U pacientov so stredne ťažkou poruchou funkcie pečene sa neodporúča žiadna úprava dávky (pozri časti 4.2 a 4.4).
Etnickápríslušnosť
Priemerná biologická dostupnosť je u pacientov ázijského pôvodu o 39 % vyššia ako u belochov.
Biologická dostupnosť tikagreloru u pacientov, ktorí sami uviedli černošský pôvod bola o 18 % nižšia ako u belochov, v klinických farmakologických štúdiách bola expozícia (Cmax a AUC) tikagreloru
u Japoncov približne o 40 % (o 20 % po úprave podľa telesnej hmotnosti) vyššia ako u belochov. Expozícia u pacientov, ktorí sami uviedli hispánsky alebo latinskoamerický pôvod bola podobná ako u belochov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje týkajúce sa tikagreloru a jeho hlavného metabolitu nepreukázali neprijateľné riziko nežiaducich účinkov u ľudí na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po jednorazovom a opakovanom podávaní a genotoxického potenciálu.
Pri klinicky relevantných hladinách expozície sa pozorovalo gastrointestinálne podráždenie u niekoľkých druhov zvierat (pozri časť 4.8).
Pri vysokých dávkach tikagreloru sa u samíc potkanov preukázal zvýšený výskyt nádorov maternice (adenokarcinómov) a zvýšený výskyt adenómov pečene. Pravdepodobným mechanizmom vzniku nádorov maternice je hormonálna nerovnováha, ktorá u potkanov môže viesť k vzniku nádorov. Pravdepodobným mechanizmom vzniku adenómov pečene je indukcia enzýmov v pečeni špecifických pre hlodavce. Relevantnosť týchto zistení týkajúcich sa karcinogenity sa preto považuje za nepravdepodobnú pre ľudí.
U potkanov sa pozorovali menej významné vývinové anomálie pri dávkach toxických pre samicu (bezpečnostný pomer 5,1). U králikov sa pozorovalo mierne oneskorenie dozrievania pečene a vývoja skeletu plodu u samíc, ktorým boli podávané vysoké dávky, bez známok materskej toxicity (bezpečnostný pomer 4,5).
Štúdie na potkanoch a králikoch sa preukázali reprodukčnú toxicitu s mierne zníženým nárastom telesnej hmotnosti brezivých samíc a so zníženou životaschopnosťou a zníženou pôrodnou hmotnosťou mláďat s oneskoreným rastom. Tikagrelor spôsoboval nepravidelné cykly (najmä predĺžené cykly) u samíc potkanov, nemal však vplyv na celkovú fertilitu samcov a samíc potkanov. Farmakokinetické štúdie s rádioizotopom značeným tikagrelorom preukázali, že pôvodné liečivo
a jeho metabolity sa u potkanov vylučujú do materského mlieka (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
manitol (E421)
mikrokryštalická celulóza (E460)
krospovidón (E1202)
xylitol (E967)
bezvodý hydrogenfosforečnan vápenatý (E341)
stearylfumarát sodný hydroxypropylcelulóza (E463) koloidný oxid kremičitý
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti3 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaAl/Al blister s perforáciou umožňujúcou oddelenie jednotlivej dávky s 8 alebo 10 tabletami; škatule s 10 x 1 tabletami (1 blister), škatule s 56 x 1 tabletami (7 blistrov) a škatule s 60 x 1 tabletami (6 blistrov).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/10/655/012-014
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 03. december 2010
Dátum posledného predĺženia registrácie: 17. júl 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.