
ivita lieku AFSTYLA je 7 400 – 16 000 IU/mg proteínu.
AFSTYLA je rekombinantný jednoreťazcový ľudský faktor VIII produkovaný v bunkách vaječníkov čínskych škrečkov (Chinese hamster ovary, CHO). Ide o stavbu, z ktorej bola odstránená podstatná časť B-domény, vyskytujúcej sa v prirodzenom faktore VIII s úplnou dĺžkou, a 4 aminokyseliny susediacej kyslej domény a3 (aminokyseliny 756 až 1 652 faktora VIII s úplnou dĺžkou) boli odstránené.
Novo vytvorené spojenie ťažkého a ľahkého reťazca faktora VIII vytvára novú N-glykozylovanú
oblasť. Keďže miesto štiepenia furínu prítomné v prirodzenom faktore VIII medzi B-doménou
a a3 doménou bolo odstránené, AFSTYLA sa prejavuje ako molekula jednoreťazcového faktora
VIII.
Pomocná látka so známymúčinkom:
AFSTYLA 250, 500 a 1000 IU (2,5 ml rozpúšťadla)
Každá injekčná liekovka obsahuje 17,5 mg (0,76 mmol) sodíka.
AFSTYLA 1500, 2000, 500 a 3000 IU (5 ml rozpúšťadla)
Každá injekčná liekovka obsahuje 35 mg (1,52 mmol) sodíka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok a rozpúšťadlo na injekčný roztok.
Biely až slabožltý prášok alebo drobivá hmota a číry a bezfarebný injekčný roztok.
pH: 6,6 – 7,3
Osmolalita: 500 – 600 mOsm/kg
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba a profylaxia krvácania u pacientov s hemofíliou A (vrodený nedostatok faktora VIII). AFSTYLA je indikovaná na liečbu všetkých vekových skupín.
4.2 Dávkovanie a spôsob podávania
Liečba musí byť pod dohľadom lekára, ktorý má skúsenosti s liečbou hemofílie.
Predtým neliečenípacienti
Bezpečnosť a účinnosť AFSTYLY u predtým neliečených pacientov nebola stanovená. K dispozícii nie sú žiadne údaje.
Monitorovanie
l
i
ečby
V priebehu liečby sa odporúča robiť príslušné stanovenie hladín faktora VIII, aby sa určila veľkosť dávky, ktorá sa má podať, a frekvencia podávania opakovaných injekcií. Odpoveď
jednotlivých pacientov na faktor VIII sa môže líšiť, čím sa prejavia rôzne biologické polčasy
a zlepšenia. Dávku odvodenú od telesnej hmotnosti môže byť potrebné upraviť u pacientov s nízkou hmotnosťou alebo u pacientov s nadváhou. Predovšetkým v prípade veľkých chirurgických zákrokov je nevyhnutné starostlivo sledovať substitučnú liečbu pomocou koagulačnej analýzy (aktivita faktora VIII v plazme).
Pri použití jednostupňového testu zrážania založeného na tromboplastínovom čase (aPTT) in vitro na stanovenie aktivity faktora VIII z krvných vzoriek pacientov môžu byť výsledky aktivity faktora VIII v plazme významne ovplyvnené typom činidla aPTT, aj referenčným štandardom použitým v teste. Okrem toho sa môžu vyskytnúť významné rozdiely medzi výsledkami
získanými pomocou jednostupňového testu zrážania (aPTT) a chromogénneho testu podľa
Európskeho liekopisu (European Pharmacopoeia, Ph. Eur). Táto skutočnosť je dôležitá najmä
vtedy, ak sa zmení laboratórium, ktoré test vykonáva, alebo sa zmenia činidlá použité v teste.
Aktivita faktora VIII v plazme sa má sledovať u pacientov liečených AFSTYLOU pomocou chromogénneho testu alebo jednostupňového testu zrážania na určovanie podávaných dávok a frekvencie opakovaných injekcií. Výsledok chromogénneho testu najpresnejšie odráža klinický hemostatický potenciál AFSTYLY a je uprednostňovaný. Výsledok jednostupňového testu zrážania podhodnocuje hladinu aktivity faktora VIII v porovnaní s výsledkom chromogénneho testu o približne 45 %. Pri použití jednostupňového testu zrážania na určenie hladiny aktivity faktora VIII u pacienta vynásobte výsledok konverzným faktorom 2.
Dávkovanie
Dávka a dĺžka substitučnej liečby závisia od závažnosti nedostatku faktora VIII, od miesta a rozsahu krvácania a od klinického stavu pacienta.
Počet jednotiek podávaného faktora VIII sa vyjadruje v medzinárodných jednotkách (International Units, IU), ktoré zodpovedajú aktuálnemu štandardu Svetovej zdravotníckej organizácie (World Health Organization, WHO) pre lieky s obsahom faktora VIII. Aktivita faktora VIII v plazme sa vyjadruje v percentách (v pomere k normálnej ľudskej plazme) alebo prednostne v medzinárodných jednotkách (v pomere k medzinárodnému štandardu pre faktor VIII v plazme).
Jedna medzinárodná jednotka (IU) aktivity faktora VIII zodpovedá množstvu faktora VIII
v jednom ml normálnej ľudskej plazmy.
Účinnosť sa stanovuje chromogénnym substrátovým testom.
Hladiny faktora VIII v plazme možno sledovať pomocou chromogénneho substrátového testu
alebo jednostupňového testu zrážania.
Liečba podľa potreby
Výpočet požadovanej dávky faktora VIII vychádza z empirického zistenia, že 1 medzinárodná jednotka (IU) faktora VIII na kg telesnej hmotnosti zvýši aktivitu faktora VIII v plazme o 2 IU/dl.
Potrebná dávka sa stanoví použitím nasledovného vzorca:
Dávka (IU) = telesná hmotnosť (kg) x požadované zvýšenie faktora VIII (IU/dl alebo % normálu)
x 0,5 (IU/kg na IU/dl)
Množstvo lieku, ktoré sa má podať, a frekvencia podávania sa majú vždy riadiť podľa klinickej účinnosti v jednotlivých prípadoch.
V prípade nasledovných hemoragických príhod by aktivita faktora VIII nemala v danom období klesnúť pod určenú hladinu plazmatickej aktivity (v % normálu alebo IU/dl). Nasledujúcu tabuľku možno použiť ako návod na dávkovanie pri epizódach krvácania alebo pri chirurgickom zákroku:
Stupeň krvácania/typ
chirurgického zákroku
Kr
v
ácanie
P
ožadovaná hladina
f
aktora VIII (%) (IU/dl)
F
r
ekvencia dávok (hodiny)/
t
rvanie liečby (dni)
Začínajúca hemartróza, krvácanie do svalov alebo ústnej dutiny
Rozsiahlejšie hemartrózy, krvácanie do svalov alebo hematóm
Život ohrozujúce krvácanie
Chirurgický zákrokMenší chirurgický zákrok vrátane extrakcie zuba
20 – 40 Injekciu opakujte každých 12 až
24 hodín. Aspoň 1 deň, kým sa krvácanie indikované bolesťou nezastaví, alebo do zahojenia.
30 – 60 Injekciu opakujte každých 12 –
24 hodín počas 3 – 4 dní alebo dlhšie, kým bolesť a akútna slabosť nevymiznú.
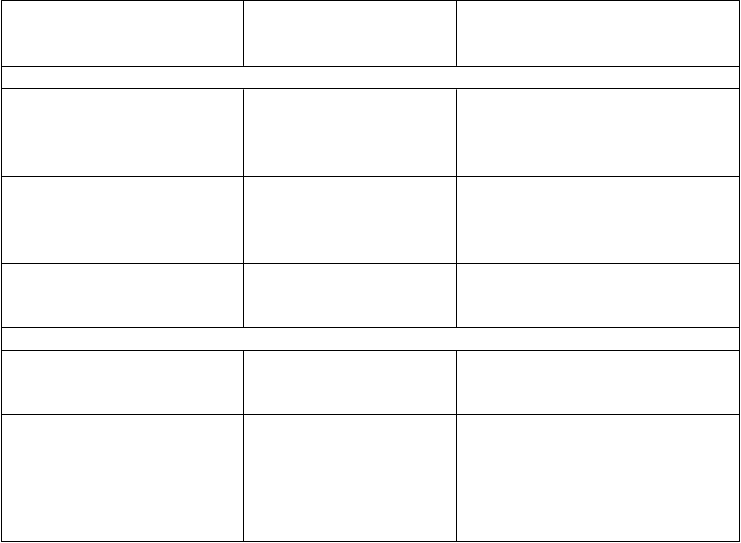
60 – 100 Opakujte injekciu
každých 8 až 24 hodín, až kým
ohrozenie nevymizne.
30 – 60 Injekciu podávajte
každých 24 hodín, najmenej 1 deň
až do zahojenia.
V
eľký
chirurgický zákrok 80 – 100
(pred a po operácii)
Opakujte injekciu
každých 8 až 24 hodín až do primeraného zahojenia rany, potom
pokračujte v liečbe najmenej ďalších
7 dní na udržanie aktivity faktora
VIII na 30 % až 60 % (IU/dl).
Profylaxia
Odporúčaný počiatočný režim dávkovania je 20 až 50 IU/kg AFSTYLY podávanej dvakrát až trikrát týždenne. Dávkovanie sa môže upraviť na základe odpovede pacienta.
Pediatrická populácia
Odporúčaný počiatočný režim dávkovania u detí (vo veku 0 až < 12 rokov) je 30 až 50 IU/kg
AFSTYLY podávanej dvakrát až trikrát týždenne. U detí mladších ako 12 rokov sa môžu
požadovať častejšie alebo vyššie dávky zohľadňujúce vyšší klírens v tejto vekovej skupine.
Pre dospievajúcich vo veku 12 rokov a starších sú odporúčania dávkovania rovnaké ako pre dospelých (pozri časť 5.2).
Starší pacienti
Klinické skúšania AFSTYLY nezahŕňajú pacientov starších ako 65 rokov.
Spôsob podávania
Intravenózne použitie.
Rekonštituovaný liek sa má podávať pomalou injekciou rýchlosťou, ktorá vyhovuje pacientovi,
ale maximálne 10 ml/min.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Známa alergická reakcia na proteíny škrečka.
4.4 Osobitné upozornenia a opatrenia pri používaní
Precitlivenosť
U AFSTYLY sa môže vyskytnúť precitlivenosť alergického typu. Liek obsahuje stopy proteínov
škrečka. Pacienti musia byť poučení, aby v prípade výskytu príznakov precitlivenosti okamžite prestali používať tento liek a kontaktovali svojho lekára. Pacienti majú byť informovaní o počiatočných
prejavoch reakcií z precitlivenosti vrátane žihľavky, generalizovanej žihľavky, pocitu tiesne na
hrudníku, sipotu, hypotenzie a anafylaxie.
U pacientov s predchádzajúcimi reakciami z precitlivenosti treba zvážiť premedikáciu.
V prípade šoku sa má použiť štandardná lekárska liečba šoku.
Inhibítory
Známou komplikáciou liečby u pacientov s hemofíliou A je vznik protilátok (inhibítorov)
neutralizujúcich faktor VIII. Tieto inhibítory sú zvyčajne imunoglobulíny IgG pôsobiace proti prokoagulačnej aktivite faktora VIII, ktoré sú kvantifikované v Bethesdových jednotkách (BU) na ml
plazmy pomocou modifikovanej metódy. Riziko vzniku inhibítorov koreluje s rozsahom expozície
faktoru VIII, toto riziko býva najvyššie počas prvých 20 dní expozície. V zriedkavých prípadoch sa môžu inhibítory tvoriť aj po prvých 100 dňoch expozície.
Boli pozorované prípady opakovaného vzniku inhibítorov (nízky titer) po prechode z jedného lieku s faktorom VIII na iný u predtým liečených pacientov s viac ako 100 dňami expozície, ktorí majú v anamnéze vznik inhibítorov. Odporúča sa preto, aby všetci pacienti po prechode z jedného lieku na iný boli pozorne sledovaní na vznik inhibítorov.
Vo všeobecnosti sa musí u všetkých pacientov liečených liekmi s obsahom koagulačného faktora VIII pozorne sledovať vznik inhibítorov pomocou príslušných klinických pozorovaní a laboratórnych testov. Ak sa očakávané hladiny aktivity faktora VIII v plazme nedosiahnu, alebo ak krvácanie nebude po podaní príslušnej dávky pod kontrolou, má sa vykonať testovanie na prítomnosť inhibítorov faktora VIII. U pacientov s vysokými hladinami inhibítora môže byť liečba faktorom VIII neúčinná a treba zvážiť iné možnosti liečby. Liečbu takých pacientov majú viesť lekári so skúsenosťami s liečbou hemofílie a s inhibítormi faktora VIII.
Monitoring laboratórnych testov
Pri použití jednostupňového testu zrážania na určenie hladiny aktivity faktora VIII u pacienta vynásobte výsledok konverzným faktorom (pozri časť 4.2).
Kardiovaskulárne príhody
U pacientov s existujúcimi kardiovaskulárnymi rizikovými faktormi môže substitučná liečba faktorom
VIII zvyšovať kardiovaskulárne riziko.
Komplikácie súvisiace s katétrom
Ak sa vyžaduje zariadenie na centrálny venózny prístup (Central Venous Access Device, CVAD), má sa zvážiť riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakterémie a trombózy v mieste zavedenia katétra.
Obsah sodíka
Tento liek obsahuje až 7 mg (0,3 mmol) sodíka na ml po rekonštitúcii. Má sa to vziať do úvahy u
pacientov na diéte s kontrolovaným príjmom sodíka.
Záznam o použití
Pri každom podaní AFSTYLY pacientovi sa dôrazne odporúča zaznamenať názov a číslo šarže
lieku, aby sa udržiavali záznamy o prepojení medzi pacientom a šaržou lieku.
Pediatrická populácia
Uvedené upozornenia a opatrenia sa vzťahujú na dospelých aj deti.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne interakcie liekov obsahujúcich ľudský koagulačný faktor VIII s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Neuskutočnili sa reprodukčné štúdie s faktorom VIII na zvieratách. Vzhľadom k zriedkavému výskytu hemofílie A u žien nie sú k dispozícii skúsenosti týkajúce sa použitia faktora VIII počas gravidity a dojčenia. Z toho dôvodu sa má faktor VIII používať počas gravidity a dojčenia iba ak je presne indikovaný.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
AFSTYLA nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Pri používaní liekov s obsahom faktora VIII boli zriedkavo pozorované reakcie z precitlivenosti alebo alergické reakcie (ktoré môžu zahŕňať angioedém, pálenie a pichanie v mieste vpichu, zimnicu, návaly
horúčavy, generalizovanú žihľavku, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, nepokoj,
tachykardiu, pocit tiesne na hrudníku, mravčenie, vracanie, sipot), ktoré v niektorých prípadoch môžu
viesť k závažnej anafylaxii (vrátane šoku).
U pacientov s hemofíliou A môžu vznikať neutralizačné protilátky (inhibítory) faktora VIII. Ak sa vyskytnú takéto inhibítory, stav sa prejaví nedostatočnou klinickou odpoveďou. V týchto prípadoch sa odporúča obrátiť na centrum špecializované na liečbu hemofílie.
Tabuľkovýzoznamnežiaducichreakcií
Tabuľka nižšie uvedená je podľa klasifikácie orgánových systémov MedDRA (SOC a frekvencie výskytu).
Frekvencie v nižšie uvedenej tabuľke boli zaznamenané v ukončených klinických štúdiách
u predtým liečených pacientov s ťažkou hemofíliou A.
Nežiaduce reakcie sú uvedené podľa nasledujúceho pravidla: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi
zriedkavé (<1/10 000), neznáme (z dostupných údajov).
T
rieda orgánových
s
ystémov podľa databázy
MedDRA
P
oruchy imunitného
N
ežiaduca reakcia Frekvencia
Časté
s
ystému Precitlivenosť
Poruchy nervového systému Závrat Časté
Parestézia Časté
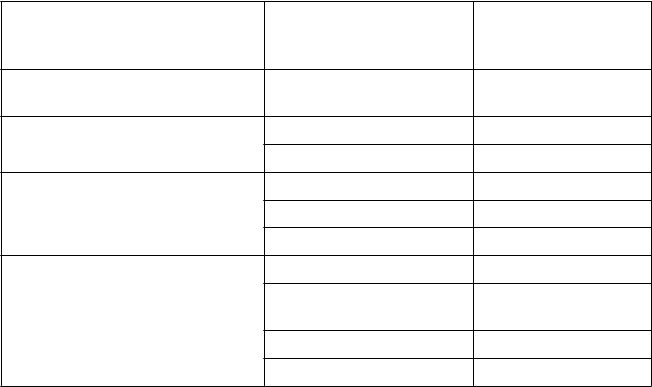 P
oruchy kože a podkožného tkaniva
C
elkové poruchy a reakcie
P
oruchy kože a podkožného tkaniva
C
elkové poruchy a reakcie
Vyrážky Časté Erytém Menej časté Pruritus Menej časté
Pyrexia Časté
v mieste podania
Reakcia v mieste vpichu
Menej časté
Zimnica Menej časté
Návaly tepla Menej časté
Pediatrická populácia
Neboli pozorované žiadne rozdiely v nežiaducich reakciách súvisiace s vekom medzi pediatrickými a dospelými pacientmi.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené
v
Prílohe V.4.9 PredávkovanieV , Pacient z dokončenej klinickej štúdii, ktorý dostal viac ako dvojnásobok predpísanej dávky AFSTYLY, pociťoval závrat, návaly tepla a svrbenie. Tieto neboli považované v súvislosti s AFSTYLOU, ale boli pravdepodobne vyvolané súčasným podaním analgetika.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antihemoragiká: Krvný koagulačný faktor VIII ATC kód: B02BD02
Mechanizmus účinkuAFSTYLA (INN: lonoktokog alfa) je rekombinantný ľudský proteín, ktorý nahrádza chýbajúci
koagulačný faktor VIII potrebný na efektívnu hemostázu.
AFSTYLA je jeden polypeptidový reťazec so skrátenou B-doménou, ktorá umožňuje
kovalentným mostíkom spojiť ťažké a ľahké reťazce faktora VIII. AFSTYLA prejavuje vyššiu VWF afinitu vo vzťahu k celej dĺžke rFVIII. VWF stabilizuje faktor VIII a chráni ho pred degradáciou. Aktivovaná AFSTYLA má aminokyselinovú sekvenciu identickú s endogénnym FVIIIa.
FarmakodynamickéúčinkyKomplex faktora VIII/von Willebrandovho faktora sa skladá z dvoch molekúl (faktor VIII a von
Willebrandov faktor) s rôznymi fyziologickými funkciami. Pri podávaní infúziou hemofilickému pacientovi sa faktor VIII viaže na von Willebrandov faktor v pacientovom krvnom obehu. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX, urýchľujúci konverziu faktora X na aktivovaný faktor X. Aktivovaný faktor X premieňa protrombín na trombín. Trombín potom premieňa fibrinogén na fibrín a môže sa vytvoriť krvná zrazenina.
Hemofília A je dedičná porucha zrážania krvi viazaná na chromozóm X, spôsobená zníženými hladinami faktora VIII a má za následok profúzne krvácanie do kĺbov, svalov alebo vnútorných orgánov, ktoré vzniká buď spontánne, alebo ako dôsledok úrazu alebo chirurgickej traumy. Substitučnou liečbou sa zvýšia hladiny faktora VIII v plazme, čím sa dočasne upraví deficit faktora a náchylnosť na krvácanie.
Klinická účinnosť abezpečnosťDospelí a dospievajúci vo veku 12 – 65 rokovŠtúdia 1001 určovala účinnosť a bezpečnosť v prevencii krvácania, v profylaxii, hemostatickej
účinnosti pri kontrole krvácania a počas perioperatívneho riadenia. Do štúdie bolo zaradených 175
predtým liečených pacientov (vo veku 12 až 65 rokov) s ťažkou hemofíliou A (bol prijatý 1 pacient
>60 rokov), ktorí boli pod účinkom jednoreťazcového rVIII celkom 14 306 dní. U žiadneho pacienta sa nevytvoril inhibítor ani sa nevyskytla anafylaktická reakcia.
Profylaxia: 146 pacientov bolo zaradených do skupiny podstupujúcej profylaktické liečebné režimy (medián ABR, 1,14 (medzikvartilový rozsah: 0,0; 4,2)), 79 (54 %) bolo zaradených do skupiny s liečebným režimom trikrát týždenne a 47 (32 %) do skupiny s liečebným režimom dvakrát týždenne. Pacienti v skupine s profylaxiou dvakrát a trikrát týždenne mali určenú strednú dávku 35 a 30 IU/kg na injekciu, respektíve s mediánom ročnej spotreby pre všetky režimy profylaxie 4283 IU/kg za rok.
Liečba krvácania: Z 848 udalostí krvácania pozorovaných v štúdii 1001, 93,5 % bolo liečených
podaním 2 alebo menej injekcií. Stredná hodnota dávky na liečbu epizódy krvácania bola 34,7 IU/kg.
Perioperačná liečba (chirurgická profylaxia): V štúdii 1001 sa celkovo vykonalo a vyhodnotilo
16 veľkých chirurgických zákrokov u 13 osôb. Hemostatická účinnosť jednoreťazcového rVIII v chirurgickej profylaxii bola hodnotená ako vynikajúca alebo dobrá u všetkých chirurgických zákrokov. Pediatrickí pacienti <18 rokov neboli zahrnutí do chirurgickej profylaxie.
Pediatrická populácia vo veku < 12 rokov
Do štúdie 3002 bolo zaradených celkovo 84 predtým liečených pacientov vo veku < 12 rokov (35 vo veku < 6 rokov a 49 vo veku 6 až < 12 rokov). Účastníci štúdie boli účinkom jednoreťazcového rVIII vystavovaní celkom 5 239 dní. U žiadneho z pacientov sa nepozorovala tvorba inhibítorov ani sa nevyskytla anafylaktická reakcia.
Individualizovaná profylaxia: Z 81 pacientov v skupine s profylaxiou (medián ABR 3,69 (medzikvartilový rozsah: 0,00; 7,20)), malo 43 pacientov (53 %) určený liečebný režim dvakrát týždenne a 25 pacientov (31 %) liečebný režim trikrát týždenne.
Pacientom s profylaxiou dvakrát a trikrát týždenne boli stanovené dávky s mediánom 35 a 32 IU/kg v
jednej injekcii, respektíve s priemernou ročnou spotrebou v rámci všetkých režimov profylaxie
4109 IU/kg za rok.
Liečba krvácania: Z 347 udalostí krvácania pozorovaných v štúdii 3002, 95,7% bolo liečených
podaním dvoch alebo menej injekcií. Stredná hodnota dávky na liečbu krvácania bola 27,6 IU/kg.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií
s AFSTYLOU u predtým neliečených pediatrických pacientov v liečbe dedičného deficitu faktora
VIII (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Dospelí
Farmakokinetika AFSTYLY bola hodnotená u 81 predtým liečených dospelých pacientov vo
veku 18 – 60 rokov s diagnostikovanou ťažkou hemofíliou A s < 1 % faktora VIII po intravenóznej injekcii 50 IU/kg.
Farmakokinetické parametre boli založené na aktivite faktora VIII v plazme meranej chromogénnym substrátovým testom (rozdiel v aktivite faktora VIII určenej jednostupňovým testom zrážania, pozri časť 4.2). Farmakokinetický profil získaný 3 až 6 mesiacov po
počiatočnom hodnotení farmakokinetiky bol porovnateľný s farmakokinetickým profilom
získaným po prvej dávke.
Farmakokinetické parametre po jednej injekcii 50 IU/kg AFSTYLY – Chromogénny substrátový test:
PK parametre
Jednoreťazcový rVIII
50 IU/kg
(
N = 81) Priemer (CV %)
Stredná hodnota (Min, Max)
IR (IU/dl)/(IU/kg) 2,00 (20,8)
1,99 (0,868, 2,90)
106 (18,1)
Cmax (IU/dl)
AUC0-inf (IU*h/dl)
t1/2 (h)
106 (62,4, 151)
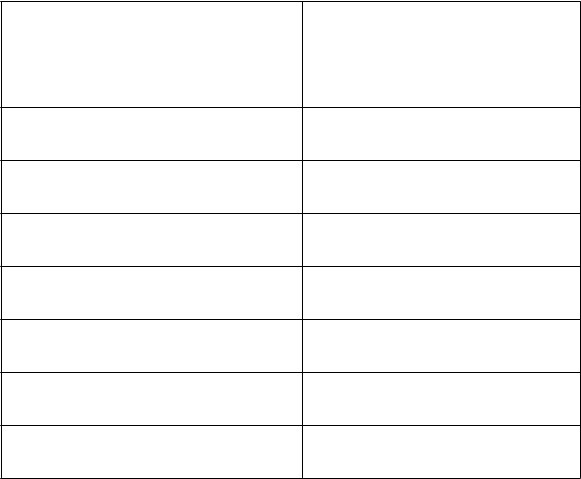
1 960 (33,1)
1 910 (932, 4 090)
14,2 (26,0)
13,7 (7,54, 23,9)
MRT (h) 20,4 (25,8)
20,2 (10,8, 35,1)
CL (ml/h/kg) 2,90 (34,4)
2,67 (1,26, 5,79)
55,2 (20,8)
Vss (ml/kg)
53,2 (32,4, 99,6)
IR = prírastková obnova zaznamenaná 30 minút po injekcii; Cmax = maximálna koncentrácia, AUC0-inf = plocha pod krivkou časového priebehu aktivity FVIII extrapolovaná na nekonečno; t1/2 = terminálny polčas; MRT = priemerná doba zdržania; CL = klírens upravený na základe telesnej hmotnosti s N = 80; Vss = distribučný objem v ustálenom stave upravený na základe telesnej hmotnosti. Hodnoty IR a Cmax boli korigované úvodnou hodnotou, kým ostatné parametre neboli korigované úvodnou hodnotou s N = 81.
Pediatrická populácia
Farmakokinetika AFSTYLY bola hodnotená u 10 predtým liečených dospievajúcich pacientov
(vo veku 12 až < 18 rokov) a 39 predtým liečených detí (vo veku 0 až < 12 rokov) po intravenóznej injekcii jednej dávky 50 IU/kg. Všetci pacienti mali diagnostikovanú ťažkú
hemofíliu A s < 1 % faktora VIII.
Farmakokinetické parametre boli založené na aktivite faktora VIII v plazme meranej chromogénnym substrátovým testom (rozdiel v aktivite faktora VIII určenej jednostupňovým testom zrážania, pozri časť 4.2).
Porovnanie farmakokinetických parametrov podľa vekovej kategórie po jednej injekcii 50 IU/kg
AFSTYLY – Chromogénny test:
PK parametre
0 až < 6 rokov
(
N = 20) Priemer (CV %) Stredná hodnota (Min, Max)
6 až < 12 rokov
(
N = 19) Priemer (CV %) Stredná hodnota (Min, Max)
12 až < 18 rokov
(
N = 10) Priemer (CV %) Stredná hodnota (Min, Max)
'
IR (IU/dl)/(IU/kg) 1,60 (21,1)
1,55 (1,18, 2,76)
80,2 (20,6)
1,66 (19,7)
1,69 (0,92, 2,35)
83,5 (19,5)
1,69 (24,8)
1,76 (0,88, 2,44)
89,7 (24,8)
Cmax (IU/dl)
AUC0-inf (IU*h/dl)
t1/2 (h)
78,6 (59,3, 138)
1 080 (31,0)
985 (561, 2 010)
10,4 (28,7)
10,1 (5,19, 17,8)
84,5 (46,4, 117)
1 170 (26,3)
1 120 (641, 1 810)
10,2 (19,4)
10,0 (6,92, 14,8)
92,4 (45,5, 131)
1 540 (36,5)
1 520 (683, 2 380)
14,3 (33,3)
13,5 (6,32, 23,8)
MRT (h) 12,4 (25,0)
13,0 (6,05, 17,9)
CL (ml/h/kg) 5,07 (29,6)
5,08 (2,52, 8,92)
71,0 (11,8)
12,3 (16,8)
12,8 (8,22, 16,0)
4,63 (29,5)
4,48 (2,79, 7,71)
67,1 (22,3)
20,0 (32,2)
18,6 (9,17, 31,7)
3,80 (46,9)
3,31 (2,10, 7,32)
68,5 (29,9)
Vss (ml/kg)
70,7 (57,3, 88,3)
64,9 (44,3, 111)
62,0 (45,9, 121)
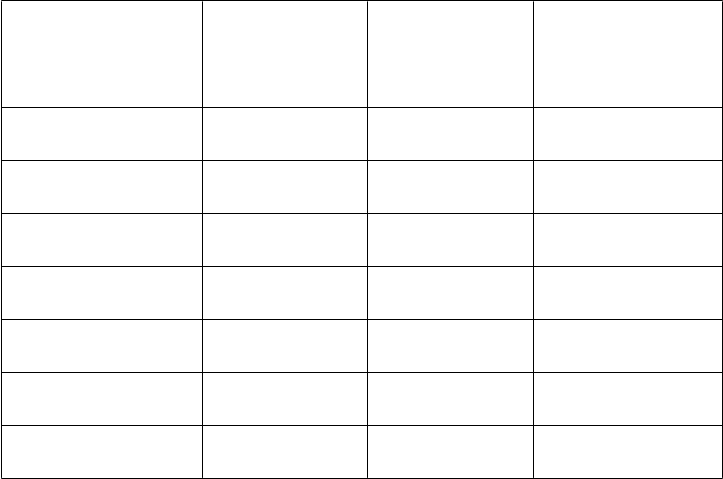
IR = prírastková obnova zaznamenaná 30 minút po injekcii u pacientov vo veku 12 až < 18 rokov a 60 minút po injekcii u pacientov vo veku 1 až < 12 rokov; Cmax = maximálna koncentrácia, AUC = plocha pod krivkou časového priebehu aktivity FVIII extrapolovaná na nekonečno; t1/2 = terminálny polčas; MRT = priemerná doba zdržania; CL = klírens upravený na základe telesnej hmotnosti; Vss = distribučný objem v ustálenom stave upravený na základe telesnej hmotnosti. Hodnoty IR a Cmax boli korigované úvodnou hodnotou, kým ostatné parametre neboli korigované úvodnou hodnotou.
5.3 Predklinické údaje o bezpečnostiNeklinické údaje získané na základe konvenčných farmakologických štúdií bezpečnosti, farmakológie,
toxicity po jednorazovom a opakovanom podaní, lokálnej tolerancie a trombogenicity neodhalili
žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokPrášok:L-histidín, polysorbát 80, dihydrát chloridu vápenatého, chlorid sodný, sacharóza
Rozpúšťadlo:Voda na injekciu
6.2 InkompatibilityNevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi alebo roztokmi okrem tých, ktoré sú uvedené v častiach 2. a 6.5.
6.3 Čas použiteľnosti3 roky.
Po rekonštitúcii bola preukázaná chemická a fyzikálna stabilita počas 48 hodín pri uchovávaní pri izbovej teplote (do 25 °C). Z mikrobiologického hľadiska sa má liek použiť okamžite. Ak sa liek nepoužije ihneď, za čas a podmienky uchovávania počas používania zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke. Injekčné liekovky uchovávajte vo vonkajšom obale na ochranu pred
svetlom.
AFSTYLU možno uchovávať pri izbovej teplote do 25 °C počas obdobia nepresahujúceho
3 mesiace do dátumu exspirácie vyznačeného na škatuľke a štítkoch injekčných liekoviek. Ak liek vyberiete z chladničky nesmiete ho vrátiť späť do chladničky. Prosím, zaznamenajte na krabičku
lieku začiatok uchovávania pri izbovej teplote.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
AFSTYLA 250 IUprášokarozpúšťadlonainjekčnýroztok
Prášok (250 IU) v 6 ml injekčnej liekovke (sklo typu I) so zátkou (guma), oranžovým diskom
(plast) a zeleným pruhovaným uzáverom (hliník).
2,5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast)
a uzáverom (hliník).
AFSTYLA500IUprášokarozpúšťadlonainjekčnýroztok
Prášok (500 IU) v 6 ml injekčnej liekovke (sklo typu I) so zátkou (guma), modrým diskom (plast)
a zeleným pruhovaným uzáverom (hliník).
2,5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast)
a uzáverom (hliník).
AFSTYLA1000IUprášokarozpúšťadlonainjekčnýroztok
Prášok (1000 IU) v 6 ml injekčnej liekovke (sklo typu I) so zátkou (guma), zeleným diskom
(plast) a uzáverom (hliník) so zelenými prúžkami.
2,5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast)
a uzáverom (hliník).
AFSTYLA1500IUprášokarozpúšťadlonainjekčnýroztok
Prášok (1500 IU) v 10 ml injekčnej liekovke (sklo typu I) so zátkou (guma), tyrkysovým diskom
(plast) a uzáverom (hliník) so zelenými prúžkami.
5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast) a uzáverom
(hliník).
AFSTYLA2000IUprášokarozpúšťadlonainjekčnýroztok
Prášok (2000 IU) v 10 ml injekčnej liekovke (sklo typu I) so zátkou (guma), purpurovým diskom
(plast) a uzáverom (hliník) so zelenými prúžkami.
5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast) a uzáverom
(hliník).
AFSTYLA 2500 IU prášokarozpúšťadlonainjekčnýroztok
Prášok (2500 IU) v 10 ml injekčnej liekovke (sklo typu I) so zátkou (guma), šedým diskom (plast)
a uzáverom (hliník) so zelenými prúžkami.
5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast) a uzáverom
(hliník).
A
FSTYLA
3000
IU
prášok
a
r
ozpúšťadlo
na
i
njekčný
roztok
Prášok (3000 IU) v 10 ml injekčnej liekovke (sklo typu I) so zátkou (guma), žltým diskom (plast)
a uzáverom (hliník) so zelenými prúžkami.
5 ml rozpúšťadla v injekčnej liekovke (sklo typu I) so zátkou (guma), diskom (plast) a uzáverom
(hliník).
VeľkosťbaleniaJedno balenie s 250, 500 alebo 1000 IU obsahuje:
1 injekčnú liekovku s práškom
1 injekčnú liekovku s 2,5 ml vody na injekciu
Jedna aplikačná súprava obsahuje:
1 filtračnú prenosovú súpravu 20/20
1 jednorazovú 5 ml injekčnú striekačku
1 súpravu na podanie do žily
2 tampóny namočené v alkohole
1 nesterilnú náplasť
Jedno balenie s 1500, 2000, 2500 alebo 3000 IU obsahuje:
1 injekčnú liekovku s práškom
1 injekčnú liekovku s 5 ml vody na injekciu
Jedna aplikačná súprava obsahuje:
1 filtračnú prenosovú súpravu 20/20
1 jednorazovú 10 ml injekčnú striekačku
1 súpravu na podanie do žily
2 tampóny namočené v alkohole
1 nesterilnú náplasť
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšeobecné pokyny- Roztok má byť takmer bezfarebný, číry alebo mierne opalescentný. Po filtrácii/natiahnutí do injekčnej striekačky (pozri nižšie) sa má rekonštituovaný liek pred podaním opticky skontrolovať na prítomnosť častíc alebo zmenu farby.
- Nepoužívajte roztoky, ktoré sú viditeľne zakalené alebo ktoré obsahujú vločky alebo častice.
- Rekonštitúcia a natiahnutie do injekčnej striekačky sa musia vykonávať za aseptických
podmienok.
Rekonštitúcia a podávanieNechajte rozpúšťadlo zohriať na izbovú teplotu. Pred otvorením Mix2Vial (balenie pomôcky) sa
presvedčte, či sú odstránené vyklápacie viečka z injekčných liekoviek s práškom a rozpúšťadlom
a zátky sú ošetrené antiseptickým roztokom a vysušené.
1. Otvorte Mix2Vial odlepením viečka.
Nevyberajte
súpravu Mix2Vial z blistra!
1
2. Umiestnite
injekčnú liekovku s rozpúšťadlom na rovný a čistý povrch a pevne ju držte. Uchopte Mix2Vial spolu s blistrom a zatlačte hrot na konci
modrého adaptéra
rovno nadol cez zátku injekčnej liekovky s rozpúšťadlom.
2
3. Opatrne odstráňte blister zo súpravy Mix2Vial podržaním za okraj a potiahnutím
zvislo nahor. Dbajte na to, aby ste vytiahli iba blistrový obal a nie súpravu Mix2Vial.
3
4. Umiestnite
injekčnú liekovku s práškom na rovný a pevný povrch. Injekčnú liekovku s rozpúšťadlom a pripojenou súpravou Mix2Vial otočte hore dnom a zatlačte hrot na konci
priesvitného adaptéra
rovno nadol cez zátku injekčnej liekovky s práškom. Rozpúšťadlo bude automaticky pretekať do injekčnej liekovky s práškom.
4
5. Jednou rukou uchopte časť súpravy Mix2Vial s liekom. Druhou rukou uchopte časť súpravy s rozpúšťadlom a opatrne odskrutkujte súpravu proti smeru hodinových ručičiek na dve časti.

Injekčnú liekovku s rozpúšťadlom a nasadeným
modrým adaptérom Mix2Vial zlikvidujte.
5
6. Jemne premiešajte injekčnú liekovku s liekom s nasadeným priesvitným adaptérom, až kým sa prášok úplne nerozpustí. Nepretrepávajte.
6
7. Natiahnite vzduch do prázdnej sterilnej injekčnej striekačky. Injekčnú liekovku s liekom držte zvislo a pripojte injekčnú striekačku ku konektoru Luer Lock súpravy Mix2Vial otáčaním v smere hodinových ručičiek. Vstreknite vzduch do liekovky s liekom.
7
Natiahnutie a podanie8. Počas držania stlačeného piestu striekačky prevráťte
súpravu hore dnom a natiahnite roztok do injekčnej striekačky pomalým spätným vyťahovaním piestu.

8
9. Teraz sa roztok premiestnil do injekčnej striekačky.
Pevne uchopte telo injekčnej striekačky (piest smeruje
smerom nadol) a odpojte priesvitný adaptér Mix2Vial
od injekčnej striekačky odskrutkovaním proti smeru hodinových ručičiek.

9
Na injekčné podanie AFSTYLY sa používa iba dodávaná aplikačná súprava, pretože môže dôjsť
k zlyhaniu liečby následkom adsorpcie faktora VIII na vnútorný povrch injekčnej súpravy.
Je potrebné dbať na to, aby sa do injekčnej striekačky s liekom nedostala žiadna krv, pretože hrozí, že by sa krv mohla zrážať v injekčnej striekačke a zrazenina fibrínu by mohla byť podaná pacientovi.
Roztok AFSTYLY sa nesmie riediť.
Rekonštituovaný roztok sa má podávať samostatnou injekčnou/infúznou linkou pomalou intravenóznou injekciou rýchlosťou, ktorá je pohodlná pre pacienta, maximálne 10 ml/min.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIICSL Behring GmbH
Emil-von-Behring-Str. 76
35041 Marburg
Nemecko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1158/001
EU/1/16/1158/002
EU/1/16/1158/003
EU/1/16/1158/004
EU/1/16/1158/005
EU/1/16/1158/006
EU/1/16/1158/007
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: {DD mesiac RRRR}
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry (EMA):
http://www.ema.europa.eu.