ZOLGENSMA sol inf 2x5,5 ml+8x8,3 ml (liek.inj.)

11,1 – 11,5
| 1,27 x 1015
| 63,3
|
11,6 – 12,0
| 1,32 x 1015
| 66,0
|
12,1 – 12,5
| 1,36 x 1015
| 68,8
|
12,6 – 13,0
| 1,44 x 1015
| 71,5
|
13,1 – 13,5
| 1,49 x 1015
| 74,3
|
13,6 – 14,0
| 1,54 x 1015
| 77,0
|
14,1 – 14,5
| 1,59 x 1015
| 79,8
|
14,6 – 15,0
| 1,65 x 1015
| 82,5
|
15,1 – 15,5
| 1,71 x 1015
| 85,3
|
15,6 – 16,0
| 1,76 x 1015
| 88,0
|
16,1 – 16,5
| 1,82 x 1015
| 90,8
|
16,6 – 17,0
| 1,87 x 1015
| 93,5
|
17,1 – 17,5
| 1,93 x 1015
| 96,3
|
Tabuľka 1 Odporúčané dávkovanie na základe telesnej hmotnosti pacienta
R
ozsah hmotnosti pacienta (kg)
|
D
ávka (vg)
|
C
elkový objem dávky
a
(
m
l
)
|
17,6 – 18,0
|
1,98 x 1015
|
99,0
|
18,1 – 18,5
|
2,04 x 1015
|
101,8
|
18,6 – 19,0
|
2,09 x 1015
|
104,5
|
19,1 – 19,5
|
2,15 x 1015
|
107,3
|
19,6 – 20,0
|
2,20 x 1015
|
110,0
|
20,1 – 20,5
|
2,26 x 1015
|
112,8
|
20,6 – 21,0
|
2,31 x 1015
|
115,5
|
a POZNÁMKA: Počet injekčných liekoviek v súprave a požadovaný počet súprav závisí od telesnej hmotnosti. Objem dávky sa vypočíta použitím hornej hranice rozsahu telesnej hmotnosti pacienta.
ImunomodulačnýrežimPo podaní onasemnogénu abeparvoveku sa prejaví imunitná odpoveď na kapsid adeno-asociovaného vírusového vektora sérotypu 9 (AAV9) (pozri časť 4.4). Môže to viesť k zvýšeniu hladiny pečeňových transamináz, zvýšeniu hladiny troponínu I alebo zníženiu počtu trombocytov (pozri časti 4.4 a 4.8). Na zníženie imunitnej odpovede sa odporúča imunomodulácia kortikosteroidmi. Ak je to možné, harmonogram očkovania pacienta sa má upraviť tak, aby umožnil súbežné podávanie kortikosteroidov pred infúziou a po infúzii onasemnogénu abeparvoveku (pozri časť 4.5).
Pred začatím imunomodulačného režimu a pred podaním onasemnogénu abeparvoveku musí byť pacient vyšetrený kvôli výskytu symptómov aktívneho infekčného ochorenia akéhokoľvek charakteru.
24 hodín pred infúziou onasemnogénu abeparvoveku sa odporúča začať imunomodulačný režim podľa nižšie uvedeného plánu (tabuľka 2). Odchýlky od týchto odporúčaní sú na rozhodnutí ošetrujúceho lekára (pozri časť 4.4).
Tabuľka 2 Imunomodulačný režim pred infúziou a po infúzii Pred infúziou
| 24 hodín pred podaním onasemnogénu
abeparvoveku
| Prednizolón perorálne 1 mg/kg/deň
(alebo ekvivalent)
|
Po infúzii
| 30 dní (vrátane dňa podania onasemnogénu
abeparvoveku)
| Prednizolón perorálne 1 mg/kg/deň
(alebo ekvivalent)
|
a potom počas 28 dní:
V prípade pacientov bez významných zistení (normálny výsledok klinického vyšetrenia, normálne hodnoty celkového bilirubínu
a hodnoty ALT a AST sú na konci 30-dňového obdobia nižšie ako dvojnásobok hornej
hranice normálu (ULN, upper limit of
normal)):
alebo
V prípade pacientov s abnormalitami funkcie pečene na konci 30-dňového obdobia: pokračovať, kým hodnoty AST a ALT nie sú nižšie ako dvojnásobok ULN a kým sa
výsledky všetkých ostatných vyšetrení nevrátia do normálneho rozsahu; potom nasleduje postupné znižovanie dávky počas 28 dní.
|
Postupné znižovanie dávky prednizolónu (alebo ekvivalentnu), napr. 2 týždne sa podáva prednizolón perorálne v dávke
0,5 mg/kg/deň a ďalšie 2 týždne sa
podáva v dávke 0,25 mg/kg/deň
Systémové kortikosteroidy (ekvivalentné perorálnemu prednizolónu 1 mg/kg/deň)
|
Najmenej 3 mesiace po infúzii onasemnogénu abeparvoveku je potrebné monitorovať hladinu pečeňových transamináz (pozri časť 4.4)
|
Ak pacienti neodpovedajú primerane na ekvivalent perorálneho prednizolónu 1 mg/kg/deň, poraďte sa
s odborníkom (odborníkmi).
Ak lekár namiesto prednizolónu použije iný kortikosteroid, je potrebné po 30 dňoch podľa potreby prijať podobné stanovisko a prístup pre znižovanie dávky.
Osobitné skupiny pacientov
Porucha funkcie obličiek
Bezpečnosť a účinnosť onasemnogénu abeparvoveku u pacientov s poruchou funkcie obličiek neboli stanovené a liečbu onasemnogénom abeparvovekom je potrebné dôkladne zvážiť. Nie je potrebné
zvažovať úpravu dávky.
Porucha funkcie pečene
Onasemnogén abeparvovek sa neskúmal u pacientov s poruchou funkcie pečene. Onasemnogén
abeparvovek sa nemá podať v infúzii, ak zvýšená hladina bilirubínu nie je spôsobená novorodeneckou žltačkou. U pacientov s poruchou funkcie pečene je potrebné dôkladne zvážiť liečbu onasemnogénom abeparvovekom (pozri časti 4.4 a 4.8). Nie je potrebné zvažovať úpravu dávky.
Genotyp 0SMN1/1SMN2
U pacientov s bialelickou mutáciou génu SMN1 a len s jednou kópiou SMN2 nie je potrebné zvažovať úpravu dávky (pozri časť 5.1).
Protilátky proti AAV9
U pacientov s východiskovými titrami protilátok proti AAV9 vyššími ako 1:50 nie je potrebné zvažovať úpravu dávky (pozri časťs 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť onasemnogénu abeparvoveku u predčasne narodených novorodencov pred
dosiahnutím gestačného veku donoseného dieťaťa neboli stanovené. K dispozícii nie sú žiadne údaje. Podanie onasemnogénu abeparvoveku je potrebné dôkladne zvážiť, pretože súbežná liečba kortikosteroidmi môže nežiaducim spôsobom ovplyvniť neurologický vývoj.
U pacientov vo veku 2 rokov a starších alebo s telesnou hmotnosťou vyššou ako 13,5 kg sú
obmedzené skúsenosti. Bezpečnosť a účinnosť onasemnogénu abeparvoveku u týchto pacientov neboli stanovené. Údaje, ktoré sú v súčasnosti k dispozícii, sú opísané v časti 5.1. Nie je potrebné zvažovať úpravu dávky (pozri tabuľku 1).
Spôsob podávania
Na intravenózne použitie.
Onasemnogén abeparvovek sa podáva ako jednorazová intravenózna infúzia. Má sa podať pomocou striekačkovej (injekčnej) pumpy ako pomalá infúzia v trvaní približne 60 minút. Nesmie sa podávať formou intravenóznej pretlakovej ani bolusovej injekcie.
V prípade zablokovania primárneho katétra sa odporúča zavedenie sekundárneho (tzv. záložného)
katétra. Po skončení infúzie je potrebné infúznu linku prepláchnuť fyziologickým roztokom. Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
Opatrenia, ktoré je potrebné vykonať pred zaobchádzaním s liekom alebo pred jeho podaním Tento liek obsahuje geneticky modifikovaný organizmus. Pri príprave a podávaní onasemnogénu abeparvoveku je potrebné používať osobné ochranné pomôcky (laboratórny plášť, ochranné okuliare
a rukavice) (pozri časť 6.6). Pokyny na prípravu, zaobchádzanie, náhodnú expozíciu a likvidáciu lieku vrátane správneho zaobchádzania s telesným odpadom, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo šarže podaného lieku.
ExistujúcaimunitaprotiAAV9
Po prirodzenej expozícii môže dochádzať k tvorbe protilátok proti AAV9. Uskutočnilo sa niekoľko štúdií skúmajúcich prevalenciu protilátok AAV9 v celkovej populácii, v ktorých sa v pediatrickej populácii preukázali nízke miery predchádzajúcej expozície AAV9. Pacientov je potrebné pred podaním infúzie onasemnogénu abeparvoveku vyšetriť z hľadiska prítomnosti protilátok AAV9. Opakované vyšetrenie možno uskutočniť, ak sú titre proitlátok AAV9 vyššie ako 1:50. Doteraz nie je známe, či a za akých podmienok sa môže onasemnogén abeparvovek bezpečne a účinne podať
v prítomnosti protilátok proti AAV9 s titrom vyšším ako 1:50 (pozri časti 4.2 a 5.1).
PokročiláSMA
Keďže SMA vedie k progresívnemu a nevratnému poškodeniu motorických neurónov, prínos onasemnogénu abeparvoveku u symptomatických pacientov závisí od stupňa záťaže ochorením v čase
liečby, pričom skoršia liečba vedie k potenciálne väčšiemu prínosu. Hoci pacienti s pokročilou
symptomatickou SMA nedosiahnu rovnaký vývoj hrubej motoriky ako zdravé deti v rovnakom veku, génová substitučná liečba môže byť pre nich klinicky prínosná v závislosti od progresie ochorenia
v čase liečby (pozri časť 5.1).
Ošetrujúci lekár si má byť vedomý, že prínos liečby je významne znížený u pacientov s výraznou svalovou slabosťou a respiračným zlyhaním, u pacientov na permanentnej ventilácii a u pacientov, ktorí nie sú schopní prehĺtať.
Profil prínosu a rizika onasemnogénu abeparvoveku u pacientov s pokročilou SMA, udržiavaných nažive pomocou permanentnej ventilácie a bez schopnosti prosperovať nie je stanovený.
Imunogenita
Imunitná odpoveď na kapsid adeno-asociovaného vírusového vektora sérotypu 9 (AAV9) sa objaví po infúzii onasemnogénu abeparvoveku vrátane tvorby protilátok proti kapsidu AAV9 (napriek imunomodulačnému režimu odporúčanému v časti 4.2), a imunitnej odpovede sprostredkovanej
T-bunkami.
V klinickom programe onasemnogénu abeparvoveku bola hlásená systémová imunitná odpoveď zahŕňajúca hepatotoxicitu sprostredkovanú imunitným systémom, čo môže vyžadovať úpravu imunomodulačného režimu vrátane dlhšieho trvania alebo zvýšenia dávky. Podrobné informácie o imunomodulačnom režime sú uvedené v časti 4.2 a v častiach “Poškodenie pečene”
a “Imunomodulačný režim” nižšie".
Poškodeniepečene
• Podanie vektora AAV môže viesť k zvýšeniu hladiny transamináz, čo môže byť závažné.
• Vyskytlo sa akútne závažné poškodenie pečene (pozri časť 4.8).
• Pacienti s existujúcou poruchou funkcie pečene alebo s akútnou vírusovou infekciou pečene môžu byť vystavení zvýšenému riziku poškodenia pečene.
• Pred infúziou je potrebné u všetkých pacientov vyhodnotiť funkciu pečene klinickým vyšetrením a laboratórnymi testami (napr. pečeňové aminotransferázy AST a ALT a celkový bilirubín (pozri časť 4.2)).
• Na zmiernenie potenciálneho zvýšenia hladiny transamináz je potrebné všetkým pacientom podať systémový kortikosteroid pred infúziou a po infúzii onasemnogénu abeparvoveku (pozri časť 4.2).
• Najmenej 3 mesiace po infúzii je potrebné monitorovať funkciu pečene.
• Je potrebné dôkladne zvážiť riziká a prínosy infúzie onasemnogénu abeparvoveku pri existujúcej poruche funkcie pečene oproti rizikám vyplývajúcim z toho, že pacient liečbu nedostane.
Hladinu AST/ALT/bilirubínu je potrebné kontrolovať jedenkrát týždenne počas 30 dní a následne každé dva týždne počas ďalších 60 dní po podaní onasemnogénu abeparvoveku, až do konca znižovania dávky (úplného vysadenia) kortikosteroidu alebo v prípade potreby dlhšie. O znižovaní dávky prednizolónu sa nemá uvažovať, ak hladina AST/ALT nie je nižšia ako dvojnásobok ULN.
Trombocytopénia
V klinických skúšaniach s onasemnogénom abeparvovekom sa pozorovalo prechodné zníženie počtu trombocytov, ktoré v niektorých prípadoch spĺňalo kritériá trombocytopénie. Vo väčšine prípadov sa
najnižšia hodnota počtu trombocytov pozorovala v prvom týždni po infúzii onasemnogénu
abeparvoveku. Pred infúziou onasemnogénu abeparvoveku je potrebné stanoviť počet trombocytov a potom ho pravidelne monitorovať; počas prvého mesiaca jedenkrát za týždeň a počas druhého
a tretieho mesiaca každý druhý týždeň, kým sa počet trombocytov nevráti na východiskovú úroveň.
Zvýšenáhladinatroponínu-I
Po infúzii onasemnogénu abeparvoveku sa pozorovalo zvýšenie hladiny srdcového troponínu-I. Zvýšená hladina troponínu-I zistená u niektorých pacientov môže naznačovať potenciálne poškodenie
tkaniva myokardu. Na základe týchto zistení a pozorovanej srdcovej toxicity u myší je potrebné pred
infúziou onasemnogénu abeparvoveku stanoviť hladinu troponínu-I a monitorovať ju najmenej
3 mesiace po infúzii onasemnogénu abeparvoveku alebo kým sa hladina nevráti k normálnemu referenčnému rozsahu u pacientov s SMA. V prípade potreby zvážte konzultáciu s kardiológom.
Imunomodulačnýrežim
Imunomodulačná liečba sa nemá začínať, ak sú prítomné aktívne infekcie, buď akútne (ako sú akútne respiračné infekcie alebo akútna hepatitída) alebo chronické, ktoré nie sú pod kontrolou (ako je chronická aktívna hepatitída B) (pozri časti 4.2 a 4.4).
Imunomodulačný režim (pozri časť 4.2) môže ovplyvniť aj imunitnú odpoveď na súbežné (respiračné) infekcie, čo môže viesť k závažnejšiemu klinickému priebehu súbežnej infekcie. Odporúča sa zvýšená opatrnosť, pokiaľ ide o načasovanie podania onasemnogénu abeparvoveku v prítomnosti prodromálneho alebo odznievajúceho (vírusového) ochorenia. Odporúča sa zvýšená pozornosť pri diagnostike a aktívnej liečbe (vírusovej) respiračnej infekcie. Odporúča sa aktuálna sezónna profylaktická liečba, ktorá zabráni infekciám spôsobeným respiračným syncyciálnym vírusom (RSV). Ak je to možné, harmonogram očkovania pacienta sa má upraviť tak, aby umožnil súbežné podávanie kortikosteroidov pred infúziou a po infúzii onasemnogénu abeparvoveku (pozri časť 4.5).
Ošetrujúci lekár si má byť vedomý možného rizika adrenálnej nedostatočnosti súvisiaceho
s dlhodobejšou liečbou kortikosteroidmi, čo môže ovplyvniť navrhnutý imunomodulačný režim.
Vylučovanie lieku
Dochádza k dočasnému vylučovaniu onasemnogénu abeparvoveku, najmä prostredníctvom telesného odpadu. Opatrovateľov a členov rodiny pacientov je potrebné informovať o nasledujúcich pokynoch na správne zaobchádzanie so stolicou pacienta:
• Vyžaduje sa dôkladná hygiena rúk, ak dôjde k priamemu kontaktu s telesným odpadom pacienta minimálne 1 mesiac po liečbe onasemnogénom abeparvovekom.
• Jednorazové plienky je potrebné uzavrieť do dvojvrstvových plastových vreciek a zlikvidovať pomocou domového odpadu.
Obsahsodíka
Tento liek obsahuje 14,6 mg sodíka na 1 ml, čo zodpovedá 0,23 % WHO odporúčaného maximálneho
denného príjmu 2 g sodíka pre dospelú osobu. Každá 5,5 ml injekčná liekovka obsahuje 25,3 mg
sodíka a každá 8,3 ml injekčná liekovka obsahuje 38,2 mg sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Skúsenosti s použitím onasemnogénu abeparvoveku u pacientov užívajúcich hepatotoxické lieky alebo používajúcich hepatotoxické látky sú obmedzené. Bezpečnosť onasemnogénu abeparvoveku u týchto pacientov nebola stanovená.
Skúsenosti s použitím súbežných liekov zacielených na 5q SMA sú obmedzené.
OčkovanieAk je to možné, harmonogram očkovania pacienta sa má upraviť tak, aby umožnil súbežné podávanie
kortikosteroidov pred infúziou a po infúzii onasemnogénu abeparvoveku (pozri časti 4.2 a 4.4). Odporúča sa sezónna profylaxia RSV (pozri časť 4.4). Živé vakcíny, ako napr. MMR a vakcína proti ovčím kiahňam, sa nemajú podávať pacientom užívajúcim dávku kortikosteroidov spôsobujúcu imunosupresiu (t. j. ≥ 2 týždne denná dávka 20 mg alebo 2 mg prednizónu alebo jeho ekvivalentu/kg telesnej hmotnosti).
4.6 Fertilita, gravidita a laktáciaNie sú k dispozícii údaje o použití počas gravidity alebo dojčenia u ľudí a štúdie fertility alebo reprodukcie na zvieratách sa neuskutočnili.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeOnasemnogén abeparvovek nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluNajčastejšie hlásenou nežiaducou reakciou po podaní lieku bolo prechodné zvýšenie hladiny pečeňových transamináz (12,4 %) a vracanie (8,2 %), pozri časť 4.4.
TabuľkovýzoznamnežiaducichreakciíNežiaduce reakcie identifikované pri použití onasemnogénu abeparvoveku u všetkých pacientov liečených intravenóznou infúziou, ktoré súvisia s liečbou, sú uvedené v tabuľke 3. Nežiaduce reakcie sú klasifikované podľa triedy orgánových systémov(SOC, system organ class) a frekvencie ich výskytu podľa databázy MedDRA. Kategórie frekvencií sú odvodené podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé
(≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci každej skupiny frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Nežiaduce reakcie podľa SOC/preferovanej terminológie a frekvencie databázy MedDRA
| Poruchy krvi a lymfatického systému
| Časté
| Trombocytopénia
| Poruchy gastrointestinálneho traktu
| Časté
| Vracanie
| Celkové poruchy a reakcie v mieste podania
| Časté
| Pyrexia
| Laboratórne a funkčné vyšetrenia
| Veľmi časté
| Zvýšená hladina transamináz
| Časté
| Zvýšená hladina aspartátaminotransferázy, zvýšená hladina alanínaminotransferázy, zvýšená hladina troponínu-I
|
|
|
Tabuľka 3 Tabuľkový zoznam nežiaducich reakcií onasemnogénu abeparvoveku
Popis vybraných nežiaducich reakcií
Poruchy pečene a žlčových ciest
Celkovo bola až u 12 % pacientov liečených odporúčanou dávkou hlásená zvýšená hladina transamináz na viac ako dvojnásobok ULN, ktorá súvisela so skúmaným liekom. Dvaja pacienti mali zvýšenie hladiny AST a ALT > 20 × ULN (jeden z týchto pacientov mal vírusovú infekciu). Títo pacienti boli klinicky asymptomatickí, nemali žltačku ani klinicky významné zvýšenie hladiny bilirubínu a nespĺňali kritériá Hyovho zákona. Zvýšená sérová hladina transamináz sa upravila pomocou liečby prednizolónom (pozri časti 4.2 a 4.4) a pacienti sa uzdravili bez klinických následkov.
Mimo klinických skúšaní bol hlásený prípad akútneho závažného poškodenia pečene pri použití onasemnogénu abeparvoveku, pričom pacient pokračoval v liečbe nusinersenom a pred liečbou onasemnogénom abeparvovekom mal zvýšenie hladiny AST a ALT > 3 × ULN. Pacient sa uzdravil pomocou ďalšej liečby steroidmi.
Prechodná trombocytopénia
V rôznych časových bodoch po podaní dávky sa pozorovalo prechodné zníženie priemerného počtu
trombocytov (4,1 %) oproti východiskovému počtu a tento pokles sa zvyčajne napravil do
dvoch týždňov. Zníženie počtu trombocytov bolo výraznejšie počas prvého týždňa liečby. Žiaden pacient nemal klinické príznaky súvisiace so zníženým počtom trombocytov (pozri časť 4.4).
Zvýšené hladiny troponínu-I
Po infúzii onasemnogénu abeparvoveku sa pozorovalo zvýšenie hladiny srdcového troponínu-I
(3,1 %) až do 0,2 µg/l. V programe klinických skúšaní sa po podaní onasemnogénu abeparvoveku
nepozorovali žiadne klinicky zjavné problémy so srdcom (pozri časť 4.4).
Imunogenita
V klinických štúdiách sa merali titre protilátok proti AAV9 pred génovou terapiu a po nej (pozri časť
4.4). Všetci pacienti, ktorí dostali onasemnogén abeparvovek, mali pred liečbou titre protilátok proti
AAV9 na úrovni 1:50 alebo menej. Priemerné zvýšenia od východiskovej hodnoty titra AAV9 sa pozorovali u všetkých pacientov vo všetkých časových bodoch okrem jedného, pre hladiny titra
protilátok proti peptidu AAV9, čo odzrkadľuje normálnu odpoveď na cudzí vírusový antigén.
U niektorých pacientov titre AAV9 presahovali úroveň kvantifikácie, väčšina týchto pacientov však nemala potenciálne klinicky významné nežiaduce reakcie. Preto nebol stanovený žiaden vzťah medzi vysokými titrami protilátok proti AAV9 a možnosťou nežiaducich reakcií alebo parametrami účinnosti.
V klinickej štúdii AVXS-101-CL-101 bolo vyšetrených 16 pacientov na titer protilátok proti AAV9:
13 pacientov malo titer nižší ako 1:50 a títo pacienti boli zaradení do štúdie; traja pacienti mali titer
vyšší ako 1:50, pričom dvaja z nich boli opätovne vyšetrení po ukončení dojčenia a ich zmeraný titer bol nižší ako 1:50 a obidvaja pacienti boli zaradení do štúdie. K dispozícii nie sú informácie o tom, či
sa má dojčenie obmedziť u matiek, ktoré môžu byť séropozitívne na protilátky proti AAV9. Všetci
pacienti mali pred liečbou onasemnogénom abeparvovekom titer protilátok proti AAV9 nižší alebo rovnajúci sa 1:50 a následne sa preukázalo očakávané zvýšenie titra protilátok proti AAV9 minimálne na úroveň 1:102 400 až po viac ako 1:819 200.
Detekcia tvorby protilátok do veľkej miery závisí od citlivosti a špecifickosti vyšetrenia. Okrem toho, pozorovaný výskyt pozitivity na protilátky (vrátane neutralizačnej protilátky) pri vyšetrení môže byť ovplyvnený niekoľkými faktormi vrátane metódy vyšetrenia, manipulácie so vzorkou, načasovaním odberu vzorky, súbežným podávaním liekov a základným ochorením.
U žiadneho z pacientov liečených onasemnogénom abeparvovekom sa nepreukázala imunitná odpoveď na transgén.
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii nie sú žiadne údaje z klinických štúdií o predávkovaní onasemnogénom abeparvovekom. Odporúča sa úprava dávky prednizolónu, pozorné klinické sledovanie a monitorovanie laboratórnych parametrov (vrátane klinickej chémie a hematológie) pre systémovú imunitnú odpoveď (pozri
časť 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá na poruchy muskuloskeletálnej sústavy, ATC kód: M09AX09
MechanizmusúčinkuOnasemnogén abeparvovek je génová terapia určená na zavedenie funkčnej kópie génu prežívania motorických neurónov (SMN1) do transdukovaných buniek na vyriešenie monogénnej hlavnej príčiny ochorenia. Očakáva sa, že zabezpečením alternatívneho zdroja expresie proteínu SMN v motorických neurónoch sa podporí prežitie a funkcia transdukovaných motorických neurónov.
Onasemnogén abeparvovek je nereplikujúci sa rekombinantný AAV vektor, ktorý využíva kapsid AAV9 na zavedenie stabilného plne funkčného ľudského transgénu SMN. Preukázala sa schopnosť kapsidu AAV9 prechádzať cez hematoencefalickú bariéru a transdukovať motorické neuróny. Gén SMN1 prítomný v onasemnogéne abeparvoveku je vytvorený tak, aby zotrvával ako DNA epizóm
v jadre transdukovaných buniek a očakáva sa, že bude dlhodobo stabilne exprimovaný
v postmitotických bunkách. Nie je známe, že vírus AAV9 spôsobuje u ľudí ochorenie. Transgén je zavedený do cieľových buniek ako samokomplementárna dvojvláknová molekula. Expresia transgénu je riadená konštitutívnym promótorom (cytomegalovírusom enhancovaný hybrid kuracieho β-aktínu), čo vedie ku kontinuálnej a pretrvávajúcej expresii proteínu SMN. Dôkaz mechanizmu účinku je podporený neklinickými štúdiami a údajmi biologickej distribúcie u ľudí.
Klinická účinnosť abezpečnosťŠtúdia AVXS-101-CL-303 vo fáze III u pacientov s SMA 1. typuAVXS-101-CL-303 (štúdia 303) je otvorená štúdia v 3. fáze s jednou skupinou skúmajúca intravenózne podanie onasemnogénu abeparvoveku v jednorazovej terapeutickej dávke
(1,1 × 1014 vg/kg). Do štúdie bolo zaradených dvadsaťdva pacientov s SMA 1. typu a 2 kópiami
SMN2. Vek pacientov pri podaní bol v rozsahu od 0,5 do 5,9 mesiacov. Z 22 zaradených pacientov traja pacienti ukončili štúdiu a z nich sa u dvoch vyskytla udalosť (úmrtie alebo permanentná
ventilácia), čo viedlo k 90,9 % (95 % IS: 79,7 %, 100,0 %) prežitiu bez udalostí (pacient je nažive
a bez permanentnej ventilácie) vo veku 14. mesiacov, pozri obrázok 1.
O
brázok 1 Čas (dni) do úmrtia alebo permanentnej ventilácie na základe súhrnných údajov zo štúdií s intravenózne podávaným onasemnogénom abeparvovekom (CL-101, CL-302, CL-303, kohorta s 2 kópiami v CL-304)
With Number of Subjects at Ris k
12 12 12 12 12 12 0
33 33 30 17 0
22 22 21 20 0
14 14 7 5 0
16 14 7 4 2 1 0
23 21 13 6 5 2 0
|
|
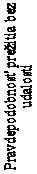
.0
.8
11
00
yy tt ii ll
bibi
obaoba 00
rr
PP
va 00
ll
va
vi vi
urur
SS 00
00
|
|
.6
.4
.2
.0
| + Cens ored
|
|
|
|
|
|
101/Coh 2
| 12 12
| 12
| 12
| 12
| 12
| 0
|
302
| 33 33
| 30
| 17
| 0
|
|
|
303
| 22 22
| 21
| 20
| 0
|
|
|
304/2 Copy
| 14 14
| 7
| 5
| 0
|
|
|
NeuroNext
| 16 14
| 7
| 4
| 2
| 1
| 0
|
PNCR
| 23 21
| 13
| 6
| 5
| 2
| 0
|
0
|
5
|
10
|
15
|
20
|
25
|
30
|
101/Coh 2
Vek ((mesiace)
S udy
101 Coh 2 302 303
304 2 Copy Neu oNex PNCR
|
|

Study
302 *
303
304/2 Copy NeuroNext PNCR
With number of subjects at risk
| S počtom ohrozených pacientov
|
Censored
| Cenzurované
|
Study
| Štúdia
|
101/Coh 2
| 101/Koh 2
|
304/2 Copy
| 304/2 kópie
|
PNCR = kohorta s prirodzeným vývojom v pediatrickom neuromuskulárnom klinickom výskume.
NeuroNext = kohorta s prirodzeným vývojom organizácie Network for Excellence in Neuroscience Clinical
Trials
* AVXS-101-CL-302 je prebiehajúca multicentrická, otvorená štúdia v 3. fáze s jednou skupinou s jednorazovou dávkou overujúca liečbu AVXS-101 (génová substitučná liečba) u pacientov s SMA 1. typu s 1 aleo 2 kópiami génu SMN2, ktorá je podobná štúdii AVXS-101-CL-303. Priemerný vek pacientov v štúdii bol k uzávierke údajov 31. decembra 2019 10,62 mesiaca (rozsah 1,8 až 15,4 mesiaca).
V prípade 14 pacientov v štúdii CL-303, ktorí dosiahli sledovaný míľnik – sedenie bez pomoci aspoň počas 30 sekúnd, bol medián veku, keď k tomu prvýkrát došlo, 12,5 mesiaca (rozsah 9,2 až
18,6 mesiaca). Trinásť pacientov dosiahlo míľnik sedenia bez pomoci aspoň počas 30 sekúnd pri
návšteve v 18. mesiaci (primárny parameter, p < 0,0001). Jeden pacient dosiahol míľnik sedenia bez pomoci počas 30 sekúnd vo veku 16 mesiacov, ale tento míľnik sa nepotvrdil pri návšteve
v 18 mesiaci. Dosiahnutie míľnikov vo vývine potvrdené videom u pacientov v štúdii CL-303 sú zhrnuté v tabuľke 4. Traja pacienti (13,6 %) nedosiahli žiadne motorické míľniky a šesť pacientov (27,2 %) dosiahlo pred poslednou návštevou v rámci štúdie v 18 mesiacoch veku ako maximálny
motorický míľnik ovládanie hlavy.
T
abuľka 4 Medián času do dosiahnutia motorických míľnikov zdokumentovaných videom
v štúdii 303
Míľnik zdokumentovaný videom
|
Počet pacientov, ktorí dosiahli míľnik
n/N (%)
|
Medián veku do dosiahnutia míľniku (mesiace)
|
95 % interval spoľahlivosti
|
Ovládanie hlavy
|
17/20 (85)
|
6,8
|
(4,77; 7,17)
|
Prevalenie z chrbta na boky
|
13/22 (59)
|
11,5
|
(7,77; 14,53)
|
Sedenie bez opory počas
30 sekúnd (Bayley)
|
14/22 (64)
|
12,5
|
(10,17; 15,20)
|
Sedenie bez opory aspoň počas 10 sekúnd (WHO)
|
14/22 (64)
|
13,9
|
(11,00; 16,17)
|
* U dvoch pacientov bolo pri vstupnej klinickej prehliadke hlásené ovládanie hlavy.
Jeden pacient (4,5 %) bol tiež schopný chodiť s pomocou vo veku 12,9 mesiaca. Na základe prirodzeného vývoja ochorenia by sa v prípade pacientov, ktorí spĺňali kritériá vstupu do štúdie, neočakávalo, že budú schopní sedieť bez opory.
Zlepšenia motorickej funkcie boli tiež pozorované na skóre CHOP INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders), pozri obrázok 2. Dvadsaťjeden pacientov (95,5 %) dosiahlo skóre CHOP-INTEND ≥ 40, štrnásť pacientov (64 %) dosiahlo skóre CHOP-
INTEND ≥ 50 a piati pacienti (23 %) dosiahli skóre CHOP-INTEND ≥ 60. Pacienti s neliečenou SMA
1. typu takmer nikdy nedosiahnu skóre CHOP-INTEND ≥ 40. Dosiahnutie motorického míľnika sa pozorovalo u niekoľkých pacientov napriek nemennému stavu skóre CHOP-INTEND. Medzi skóre CHOP-INTEND a dosiahnutím motorických míľnikov sa nepozorovala zjavná korelácia.
 Obrázok 2 Skóre CHOP-INTEND motorickej funkcie v štúdii 303
Obrázok 2 Skóre CHOP-INTEND motorickej funkcie v štúdii 303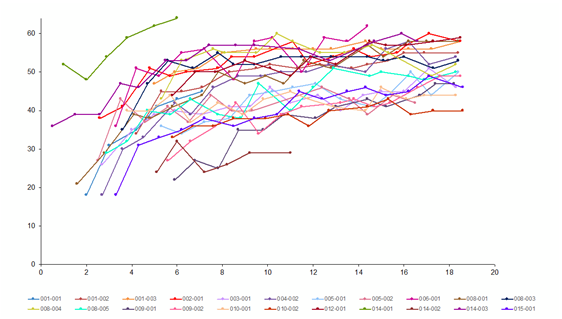
Vek (mesiace)
Štúdia AVXS-101-CL-101 vo fáze I u pacientov s SMA 1. typuVýsledky pozorované v štúdii 303 sú podporené štúdiou AVXS-101-CL-101 (štúdia v 1. fáze skúmajúca SMA 1. typu, štúdia 101), v ktorej sa onasemnogén abeparvovek podával ako jednorazová intravenózna infúzia u 12 pacientov s hmotnosťou 2,6 kg až 8,5 kg (vo veku 0,9 až 7,9 mesiaca).
Vo veku 14 mesiacov sa udalosť nevyskytla u žiadneho z liečených pacientov; t. j. prežili bez permanentnej ventilácie v porovnaní s 25 % pacientov v kohorte s prirodzeným vývojom. Na konci
štúdie (24 mesiacov po podaní dávky) sa udalosť nevyskytla u žiadneho z liečených pacientov v porovnaní s menej ako 8 % pacientov v štúdii skúmajúcej prirodzený vývoj ochorenia, pozri obrázok 1.
V 24. mesiaci sledovania po podaní dávky bolo 10 z 12 pacientov schopných sedieť bez pomoci
≥ 10 sekúnd, 9 pacientov bolo schopných sedieť bez pomoci ≥ 30 sekúnd a 2 pacienti boli schopní stáť a chodiť bez pomoci. Jeden z 12 pacientov pred dosiahnutím veku 24 mesiacov nedosiahol schopnosť ovládania hlavy ako maximálny motorický míľnik. Desať z 12 pacientov zo štúdie CL-101, ktorým bola podaná navrhnutá terapeutická dávka onasemnogénu abeparvoveku, bolo ďalej sledovaných
v dlhodobej štúdii (až 5,7 roka po podaní dávky) a u všetkých sa zachovali všetky predtým dosiahnuté míľniky, alebo dokonca dosiahli nové míľniky, napríklad sedenie s oporou, státie s pomocou
a samostatné chodenie. Štyria z 10 pacientov dostávali súbežnú liečbu nusinersenom v niektorom bode
počas dlhodobej štúdie. Zachovanie účinnosti a dosiahnutie míľnikov preto nemožno u všetkých pacientov pripísať výlučne onasemnogénu abeparvoveku. Míľnik státia s pomocou dosiahli nanovo
dvaja pacienti, ktorí nedostávali nusinersen.
Štúdia AVXS-101-CL-304 vo fáze III u pacientov s presymptomatickou SMA
Štúdia CL-304 je prebiehajúca, globálna, otvorená, multicentrická štúdia v 3. fáze s jednou skupinou a jednorazovou intravenóznou dávkou AVXS-101 u presymptomatických novorodencov do veku
6 týždňov s 2 (kohorta 1, n = 14) alebo 3 (kohorta 2, n = 15) kópiami SMN2.
Kohorta 1
V čase poslednej návštevy v rámci štúdie pred 31. decembrom 2019 boli liečení pacienti
s 2 kópiami SMN2 vo veku od 6 mesiacov do 18,6 mesiaca a do štúdie boli zapojení priemerne
10,5 mesiaca (rozsah: 5,1 až 18,0 mesiacov). Všetci pacienti boli nažive a bez permanentnej ventilácie.
Osem pacientov dosiahlo sedenie bez pomoci v trvaní aspoň 30 sekúnd vo veku od 6,4 do
11,8 mesiaca a 7 z týchto 8 pacientov (87,5 %) bolo schopných sedieť bez pomoci pred dosiahnutím veku 9,2 mesiaca, čo je 99. percentil pre vývoj tohto míľnika. Štyria pacienti (28,6 %) dosiahli míľnik
samostatnej chôdze. Dvanásť pacientov (85,7 %) dosiahlo k uzávierke údajov 31. decembra 2019
skóre CHOP-INTEND ≥ 60.
Kohorta 2
V čase poslednej návštevy v rámci štúdie pred 31. decembrom 2019 boli liečení pacienti s 3 kópiami
SMN2 vo veku 3,3 až 15,1 mesiaca a do štúdie boli zapojení priemerne 8,74 mesiaca (rozsah 2 až
13,9 mesiaca). Všetci pacienti boli nažive a bez permanentnej ventilácie. Desať z 15 pacientov bolo'
schopných sedieť bez pomoci aspoň 30 sekúnd, 4 pacienti boli schopní stáť bez pomoci aspoň
3 sekundy a 2 pacienti prešli aspoň päť krokov bez pomoci.
Dĺžka sledovania je príliš krátka na posúdenie vývoja pacientov s liečbou AVXS-101 v porovnaní s prirodzeným vývojom pacientov s 3 kópiami SMN2, ktorí majú heterogénne klinické prejavy.
V tomto momente preto nemožno vyvodiť definitívne závery o prínose v tejto populácii pacientov.
V klinických skúšaniach sa onasemnogén abeparvovek neskúmal u pacientov s bialelickou mutáciou génu SMN1 a len s jednou kópiou SMN2.
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s onasemnogénom abeparvovekom v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe spinálnej muskulárnej atrofie pre schválenú indikáciu (informácie o použití v pediatrickej populácii, pozri
časť 4.2).
5.2 Farmakokinetické vlastnosti
Uskutočnili sa štúdie vylučovania vektora onasemnogénu abeparvoveku, v ktorých sa hodnotilo množstvo vektora vylúčeného z tela slinami, močom a stolicou.
Onasemnogén abeparvovek bol po podaní infúzie detegovateľný vo vzorkách výlučkov. Onasemnogén abeparvovek sa vylučoval najmä stolicou a väčšina sa vylúčila počas 30 dní po podaní dávky. Koncentrácia onasemnogénu abeparvoveku v moči a slinách bola 0,1 % až 0,01 % pôvodnej koncentrácie v organizme v 1. deň po podaní infúzie a následne klesala.
Biologická distribúcia sa hodnotila u dvoch pacientov, ktorí zomreli 5,7 mesiaca a 1,7 mesiaca po infúzii onasemnogénu abeparvoveku v dávke 1,1 x 1014 vg/kg. V obidvoch prípadoch sa najvyššie hladiny vektorovej DNA zistili v pečeni. Vektorová DNA sa zistila aj v slezine, srdci, pankrease, inguinálnej lymfatickej uzline, kostrových svaloch, periférnych nervoch, obličkách, pľúcach, črevách, mieche, mozgu a týmuse. Imunofarbenie detegujúce proteín SMN preukázalo generalizovanú expresiu SMN v motorických neurónoch chrbtice, nervových a gliových bunkách mozgu, v srdci, pečeni, kostrových svaloch a v ďalších hodnotených tkanivách.
5.3 Predklinické údaje o bezpečnosti
Po intravenóznom podaní novonarodeným myšiam sa vektor a transgén distribuovali vo veľkej miere, pričom najvyššia expresia sa pozorovala v srdci a pečeni, a významná expresia sa pozorovala v mozgu a mieche. V pivotných 3-mesačných toxikologických štúdiách na myšiach boli hlavným cieľom orgánovej toxicity srdce a pečeň. Nálezy súvisiace s onasemnogénom abeparvovekom v srdcových komorách zahŕňali zápal, edém a fibrózu a boli závislé od dávky. V srdcových predsieňach sa pozoroval zápal, trombóza, degenerácia/nekróza myokardu a fibroplázia a boli závislé od dávky. Nálezy v pečeni zahŕňali hepatocelulárnu hypertrofiu, aktiváciu Kupfferových buniek a roztrúsenú hepatocelulárnu nekrózu. Pre onasemnogén abeparvovek nebola u myší identifikovaná hladina bez pozorovaného nežiaduceho účinku (NoAEL, no adverse effect level), keďže zápal/edém/fibróza srdcového svalu a zápal srdcových predsiení sa pozorovali pri najnižšej testovanej dávke
(1,5 × 1014 vg/kg). Táto dávka sa považuje za maximálnu tolerovanú dávku a predstavuje približne
1,4-násobok odporúčanej klinickej dávky. Mortalita súvisiaca s onasemnogénom abeparvovekom u väčšiny myší súvisela s trombózou srdcových predsiení, ktorá sa pozorovala pri
dávke 2,4 × 1014 vg/kg. Príčina úmrtí u ostatných zvierat sa neurčila, hoci sa v srdciach týchto zvierat zistila mikroskopická degenerácia/regenerácia.
S onasemnogénom abeparvovekom sa neuskutočnili štúdie genotoxicity, karcinogenity a reprodukčnej toxicity.
V toxikologickej štúdii uskutočnenej u mladých dospelých nehumánnych primátov viedlo podanie
(v Trendelenburgovej polohe) jednorazovej intratekálnej dávky 3 x 1013 vg/zviera (medián dávky
1,08 x 1013 vg/kg) onasemnogénu abeparvoveku bez liečby kortikosteroidmi k minimálnemu až výraznému zápalu mononukleárnych buniek (najmä lymfocytov) v niektorých dorzálnych koreňových
gangliách (na všetkých skúmaných úrovniach miechy) sprevádzanému neuronálnou satelitózou, neuronálnou nekrózou alebo úplnou stratou neurónov so zriedkavou mineralizáciou. Klinický význam
týchto zistení nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
trometamín chlorid horečnatý chlorid sodný poloxamér 188
kyselina chlorovodíková (na úpravu pH)
voda na injekcie
6.2 InkompatibilityNevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti1 rok
Po rozmrazeníPo rozmrazení sa liek nemá znova zmrazovať a má sa uchovávať v chlade pri teplote 2 °C až 8 °C
v pôvodnom obale počas 14 dní.
Po odobratí objemu dávky do injekčnej striekačky sa dávka musí podať formou infúzie do 8 hodín. Ak sa dávka nepodá do 8 hodín, zlikvidujte injekčnú striekačku obsahujúcu vektor.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte a prepravujte v mraze (≤ -60 °C).
Ihneď po prijatí uchovávajte v chladničke (2 °C – 8 °C). Uchovávajte v pôvodnom obale.
Podmienky na uchovávanie po rozmrazení lieku, pozri časť 6.3.
Na pôvodný obal je potrebné uviesť dátum prijatia predtým, ako sa liek uskladní v chladničke.
6.5 Druh obalu a obsah baleniaOnasemnogén abeparvovek sa dodáva v injekčnej liekovke (10 ml, polymér Crystal Zenith) so zátkou (20 mm, chlórbutylová guma), viečkom (hliníkové, odklápacie) a farebným krytom (plastový) v dvoch rôznych veľkostiach plniaceho objemu injekčnej liekovky (5,5 ml alebo 8,3 ml).
Dávka onasemnogénu abeparvoveku a presný počet injekčných liekoviek potrebných pre každého pacienta sa vypočíta podľa hmotnosti pacienta (pozri časť 4.2 a tabuľka 5 nižšie).
Tabuľka 5 Zostavy škatúľ/súprav
Hmotnosť pacienta
(kg)
|
5,5 ml injekčná
liekovkaa
|
8,3 ml injekčná
liekovkab
| Celkový počet injekčných liekoviek v škatuli
|
2,6 – 3,0
| 0
| 2
| 2
|
3,1 – 3,5
| 2
| 1
| 3
|
3,6 – 4,0
| 1
| 2
| 3
|
4,1 – 4,5
| 0
| 3
| 3
|
4,6 – 5,0
| 2
| 2
| 4
|
5,1 – 5,5
| 1
| 3
| 4
|
5,6 – 6,0
| 0
| 4
| 4
|
6,1 – 6,5
| 2
| 3
| 5
|
6,6 – 7,0
| 1
| 4
| 5
|
7,1 – 7,5
| 0
| 5
| 5
|
7,6 – 8,0
| 2
| 4
| 6
|
8,1 – 8,5
| 1
| 5
| 6
|
8,6 – 9,0
| 0
| 6
| 6
|
9,1 – 9,5
| 2
| 5
| 7
|
Hm
o
t
nosť pacienta
(
kg)
|
5,5 ml injekčná
li
ekovka
a
|
8,3 ml injekčná
li
ekovka
b
|
C
elkový počet injekčných liekoviek v škatuli
|
9,6 – 10,0
|
1
|
6
|
7
|
10,1 – 10,5
|
0
|
7
|
7
|
10,6 – 11,0
|
2
|
6
|
8
|
11,1 – 11,5
|
1
|
7
|
8
|
11,6 – 12,0
|
0
|
8
|
8
|
12,1 – 12,5
|
2
|
7
|
9
|
12,6 – 13,0
|
1
|
8
|
9
|
13,1 – 13,5
|
0
|
9
|
9
|
13,6 – 14,0
|
2
|
8
|
10
|
14,1 – 14,5
|
1
|
9
|
10
|
14,6 – 15,0
|
0
|
10
|
10
|
15,1 – 15,5
|
2
|
9
|
11
|
15,6 – 16,0
|
1
|
10
|
11
|
16,1 – 16,5
|
0
|
11
|
11
|
16,6 – 17,0
|
2
|
10
|
12
|
17,1 – 17,5
|
1
|
11
|
12
|
17,6 – 18,0
|
0
|
12
|
12
|
18,1 – 18,5
|
2
|
11
|
13
|
18,6 – 19,0
|
1
|
12
|
13
|
19,1 – 19,5
|
0
|
13
|
13
|
19,6 – 20,0
|
2
|
12
|
14
|
20,1 – 20,5
|
1
|
13
|
14
|
20,6 – 21,0
|
0
|
14
|
14
|
a Nominálna koncentrácia v injekčnej liekovke je 2 × 1013 vg/ml a obsahuje odoberateľný objem minimálne
5,5 ml.
b Nominálna koncentrácia v injekčnej liekovke je 2 × 1013 vg/ml a obsahuje odoberateľný objem minimálne
8,3 ml.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomTento liek obsahuje geneticky modifikované organizmy. Je potrebné dodržiavať príslušné opatrenia na zaobchádzanie, likvidáciu alebo náhodnú expozíciu onasemnogénu abeparvoveku:
• S injekčnou striekačkou s onasemnogénom abeparvovekom sa má zaobchádzať asepticky za sterilných podmienok.
• Pri zaobchádzaní alebo podávaní onasemnogénu abeparvoveku je potrebné používať osobné ochranné pomôcky (rukavice, ochranné okuliare, laboratórny plášť a ochranné rukávy). Ak má niekto z personálu porezanú alebo poškriabanú kožu, nemá pracovať s liekom.
• Rozliaty liek sa musí utrieť savým gázovým tampónom a zasiahnuté miesto sa musí vydezinfikovať bieliacim roztokom a následne alkoholovým tampónom. Všetok materiál
použitý pri čistení sa musí zabaliť do dvojvrstvového vrecka a zlikvidovať v súlade s národnými
požiadavkami pre zaobchádzanie s biologickým odpadom.
• Všetok materiál, ktorý sa mohol dostať do kontaktu s liekom (napr. injekčná liekovka, všetok materiál použitý na podanie injekcie vrátane sterilných rúšok a ihiel), sa musí zlikvidovať
v súlade s národnými požiadavkami pre zaobchádzanie s biologickým odpadom.
• Je potrebné vyhnúť sa náhodnej expozícii onasemnogénu abeparvoveku. V prípade expozície kože sa postihnuté miesto musí dôkladne umývať mydlom a vodou najmenej 15 minút.
V prípade zasiahnutia očí sa postihnuté miesto musí dôkladne vyplachovať vodou aspoň
15 minút.
Prijatiearozmrazenieinjekčnýchliekoviek
• Injekčné liekovky sa majú prepravovať v mraze (≤ -60 °C). Po prijatí sa injekčné liekovky majú ihneď uchovávať v chlade pri teplote 2 °C až 8 °C a v pôvodnom obale. Liečba
onasemnogénom abeparvovekom sa má začať do 14 dní po prijatí injekčných liekoviek.
• Injekčné liekovky sa musia pred použitím rozmraziť. Nepoužívajte liek, ak nie je rozmrazený.
• Pri baleniach škatúľ obsahujúcich do 9 injekčných liekoviek sa liek rozmrazí približne po
12 hodinách v chladničke. Pri baleniach škatúľ obsahujúcich do 14 injekčných liekoviek sa liek rozmrazí približne po 16 hodinách v chladničke. Alternatívne a v prípade okamžitého použitia
môže rozmrazovanie prebiehať pri izbovej teplote.
• Pri baleniach škatúľ obsahujúcich do 9 injekčných liekoviek dôjde k rozmrazeniu približne po
4 hodinách pri izbovej teplote (20 °C až 25 °C). Pri baleniach škatúľ obsahujúcich do
14 injekčných liekoviek dôjde k rozmrazeniu približne po 6 hodinách pri izbovej teplote (20 °C
až 25 °C).
• Pred natiahnutím objemu dávky do injekčnej striekačky premiešajte rozmrazený liek jemným krúživým pohybom. Liekovkou NETRASTE.
• Nepoužívajte tento liek, ak po rozmrazení zmrazeného lieku a pred jeho podaním spozorujete prítomnosť akýkoľvek častíc alebo zmenu zafarbenia.
• Liek sa nemá po rozmrazení znova zmrazovať.
• Liek sa má po rozmrazení podať čo najskôr. Po odobratí objemu dávky do injekčnej striekačky sa dávka musí podať formou infúzie do 8 hodín. Ak sa dávka nepodá do 8 hodín, zlikvidujte injekčnú liekovku obsahujúcu vektor.
Podanieonasemnogénu abeparvoveku pacientovi
• Na podanie onasemnogénu abeparvoveku odoberte do injekčnej striekačky celý objem dávky.
Pred podaním intravenóznej infúzie cez venózny katéter odstráňte z injekčnej striekačky všetok vzduch a pripravte infúzny vak.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami pre zaobchádzanie s biologickým odpadom.
Môže dochádzať k dočasnému vylučovaniu onasemnogénu abeparvoveku, najmä prostredníctvom telesného odpadu. Opatrovateľov a členov rodiny pacientov je potrebné informovať o nasledujúcich pokynoch na správne zaobchádzanie s telesnými tekutinami a stolicou pacienta:
• Je potrebná dôkladná hygiena rúk (nosenie ochranných rukavíc a následné dôkladné umytie rúk mydlom a teplou tečúcou vodou alebo dezinfekčným prostriedkom na ruky na báze alkoholu), ak dôjde k priamemu kontaktu s telesnými tekutinami a stolicou pacienta minimálne 1 mesiac po liečbe onasemnogénom abeparvovekom.
• Jednorazové plienky je potrebné uzavrieť do dvojvrstvových plastových vreciek a možno ich zlikvidovať pomocou domového odpadu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AveXis EU Limited
Block B, The Crescent Building
Northwood, Santry
Dublin 9
D09 C6X8
Írsko
8. REGISTRAČNÉ ČÍSLA
EU/1/20/1443/001
EU/1/20/1443/002
EU/1/20/1443/003
EU/1/20/1443/004
EU/1/20/1443/005
EU/1/20/1443/006
EU/1/20/1443/007
EU/1/20/1443/008
EU/1/20/1443/009
EU/1/20/1443/010
EU/1/20/1443/011
EU/1/20/1443/012
EU/1/20/1443/013
EU/1/20/1443/014
EU/1/20/1443/015
EU/1/20/1443/016
EU/1/20/1443/017
EU/1/20/1443/018
EU/1/20/1443/019
EU/1/20/1443/020
EU/1/20/1443/021
EU/1/20/1443/022
EU/1/20/1443/023
EU/1/20/1443/024
EU/1/20/1443/025
EU/1/20/1443/026
EU/1/20/1443/027
EU/1/20/1443/028
EU/1/20/1443/029
EU/1/20/1443/030
EU/1/20/1443/031
EU/1/20/1443/032
EU/1/20/1443/033
EU/1/20/1443/034
EU/1/20/1443/035
EU/1/20/1443/036
EU/1/20/1443/037
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.