
linické údaje (dostupné údaje týkajúce sa juvenilných zvierat, pozri časť 5.3).
Spôsob podávania
Volibris je na perorálne použitie. Odporúča sa, aby sa tableta prehltla vcelku a môže sa užívať s
jedlom alebo bez jedla. Odporúča sa tabletu nedeliť, nedrviť ani nežuť.
4.3 Kontraindikácie
Precitlivenosť na liečivo, na sóju alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Gravidita (pozri časť 4.6).
Ženy vo fertilnom veku, ktoré nepoužívajú spoľahlivú antikoncepciu (pozri časti 4.4 a 4.6).
Dojčenie (pozri časť 4.6).
Ťažká porucha funkcie pečene (s cirhózou alebo bez nej) (pozri časť 4.2).
Východiskové hodnoty pečeňových aminotransferáz (aspartátaminotransferázy (AST) a/alebo
alanínaminotransferázy (ALT)) >3xULN (pozri časti 4.2 a 4.4).
Idiopatická pľúcna fibróza (IPF) so sekundárnou pľúcnou artériovou hypertenziou alebo bez nej (pozri
časť 5.1).
4.4 Osobitné upozornenia a opatrenia pri používaní
Ambrisentan nebol skúmaný u dostatočného počtu pacientov, aby bolo možné stanoviť rovnováhu medzi prínosom a rizikom pri PAH funkčnej triedy I podľa SZO.
Účinnosť ambrisentanu v monoterapii nebola stanovená u pacientov s PAH funkčnej triedy IV podľa SZO. Ak dôjde k zhoršeniu klinického stavu, má sa zvážiť liečba, ktorá je odporúčaná pre ťažké štádium ochorenia (napr. epoprostenol).
Funkcia pečene
PAH je spojená s abnormalitami funkcie pečene. Pri používaní ambrisentanu boli pozorované prípady
zodpovedajúce autoimunitnej hepatitíde, vrátane možnej exacerbácie už existujúcej autoimunitnej
hepatitídy, poškodenia pečene a zvýšenia hodnôt pečeňových enzýmov potenciálne súvisiaceho
s liečbou (pozri časti 4.8 a 5.1). Pred začiatkom liečby ambrisentanom sa preto majú stanoviť hodnoty pečeňových aminotransferáz (ALT a AST) a liečba sa nemá začať u pacientov s východiskovými hodnotami ALT a/alebo AST >3xULN (pozri časť 4.3).
Pacienti majú byť sledovaní kvôli prejavom poškodenia pečene a odporúča sa kontrolovať hodnoty ALT a AST raz za mesiac. Ak u pacienta dôjde k trvalému, neobjasnenému, klinicky významnému zvýšeniu hodnôt ALT a/alebo AST, alebo ak je zvýšenie hodnôt ALT a/alebo AST sprevádzané znakmi alebo príznakmi poškodenia pečene (napr. žltačka), liečba ambrisentanom sa má ukončiť.
U pacientov bez klinických príznakov poškodenia pečene alebo bez žltačky sa opätovné začatie liečby
ambrisentanom môže zvážiť po normalizácii hodnôt pečeňových enzýmov. Odporúča sa vyšetrenie
u hepatológa.
Koncentrácia hemoglobínu
Používanie antagonistov endotelínových receptorov (ERA) vrátane ambrisentanu je spojené
so znížením koncentrácií hemoglobínu a hematokritu. Znížené koncentrácie boli väčšinou zistené počas prvých 4 týždňov liečby a potom sa hemoglobín zvyčajne stabilizoval. Priemerné poklesy
(v rozmedzí od 0,9 do 1,2 g/dl) koncentrácií hemoglobínu, v porovnaní s východiskovými
koncentráciami, pretrvávali počas až 4 rokov liečby ambrisentanom v dlhodobom otvorenom predĺžení
pivotných klinických štúdií fázy 3. V období po uvedení lieku na trh boli hlásené prípady anémie vyžadujúcej si transfúziu krvi (pozri časť 4.8).
Neodporúča sa začať liečbu ambrisentanom u pacientov s klinicky významnou anémiou. Počas liečby
ambrisentanom sa odporúča kontrolovať hladiny hemoglobínu a/alebo hematokritu, napríklad
po 1 mesiaci, 3 mesiacoch a potom v pravidelných intervaloch v súlade s klinickou praxou. Ak sa zistí klinicky významné zníženie hladín hemoglobínu alebo hematokritu a vylúčia sa iné príčiny, má sa zvážiť zníženie dávky alebo ukončenie liečby. Výskyt anémie bol zvýšený, keď sa ambrisentan podával v kombinácii s tadalafilom (15 % frekvencia výskytu tejto nežiaducej udalosti), v porovnaní
s výskytom anémie, keď sa ambrisentan a tadalafil podávali v monoterapii (7 % a 11 %, v uvedenom poradí).
Retencia tekutín
Pri používaní ERA vrátane ambrisentanu sa pozoroval periférny edém. Väčšina prípadov periférneho
edému v klinických štúdiách s ambrisentanom bola ľahkej až stredne ťažkej závažnosti, aj keď sa môže vyskytnúť s väčšou frekvenciou a závažnosťou u pacientov ≥ 65 rokov. V krátkodobých klinických štúdiách bol periférny edém hlásený častejšie pri podávaní 10 mg ambrisentanu (pozri časť 4.8).
Po uvedení lieku na trh boli hlásené prípady retencie tekutín vyskytujúcej sa v priebehu niekoľkých týždňov po začatí liečby ambrisentanom a v niektorých prípadoch liečba retencie tekutín alebo dekompenzovaného srdcového zlyhania vyžadovala podanie diuretika alebo hospitalizáciu. Ak pacienti trpia už existujúcim preťažením tekutinami, treba ho zvládnuť klinicky vhodným spôsobom pred začatím liečby ambrisentanom.
Ak počas liečby ambrisentanom vznikne klinicky významná retencia tekutín, ktorá je alebo nie je spojená s prírastkom telesnej hmotnosti, majú sa vykonať ďalšie vyšetrenia, aby sa určila príčina, napr. liečba ambrisentanom alebo už existujúce srdcové zlyhanie, a možná potreba špecifickej liečby alebo ukončenia liečby ambrisentanom. Výskyt periférneho edému bol zvýšený, keď sa ambrisentan podával v kombinácii s tadalafilom (45 % frekvencia výskytu tejto nežiaducej udalosti), v porovnaní s výskytom periférneho edému, keď sa ambrisentan a tadalafil podávali v monoterapii (38 % a 28 %,
v uvedenom poradí). Výskyt periférneho edému bol najvyšší v priebehu prvého mesiaca po začatí
liečby.
Ženy vo fertilnom veku
Liečba Volibrisom sa nesmie začať u žien vo fertilnom veku, pokiaľ výsledok tehotenského testu pred
začiatkom liečby nie je negatívny a pokiaľ nepoužívajú spoľahlivú antikoncepciu. V prípade akýchkoľvek pochybností ohľadom toho, aká antikoncepcia sa má odporučiť konkrétnej pacientke, sa
má zvážiť konzultácia s gynekológom. Počas liečby ambrisentanom sa odporúča vykonávať tehotenské testy v mesačných intervaloch (pozri časti 4.3 a 4.6).
Pľúcna venookluzívna choroba
Pri používaní vazodilatačných liekov, ako sú ERA, u pacientov s pľúcnou venookluzívnou chorobou
boli zaznamenané prípady pľúcneho edému. V dôsledku toho, ak u pacientov s PAH vznikne akútny pľúcny edém počas liečby ambrisentanom, je potrebné zvážiť možnosť pľúcnej venookluzívnej
choroby.
Súbežné podávanie s inými liekmi
Pacienti užívajúci ambrisentan majú byť starostlivo sledovaní pri začatí liečby rifampicínom (pozri
časti 4.5 a 5.2).
Pomocné látky
Volibris 2,5 mg, 5 mg a 10 mg filmom obalené tablety
Laktóza
Tento liek obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Lectín (sójový)
Tento liek obsahuje lecitín pochádzajúci zo sóje. Ak je pacient precitlivený na sóju, ambrisentan sa
nesmie použiť (pozri časť 4.3).
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v tablete, t. j. v podstate zanedbateľné
množstvo sodíka.
Volibris 5 mg a 10 mg filmom obalené tablety
Hlinitý lak červene allura AC
Tablety Volibrisu 5 mg a 10 mg obsahujú azofarbivo hlinitý lak červene allura AC (E129), ktoré môže
vyvolať alergické reakcie.
4.5 Liekové a iné interakcie
V predklinických štúdiách in vitro a in vivo ambrisentan v klinicky významných koncentráciách neinhiboval ani neindukoval enzýmy I. alebo II. fázy metabolizácie liečiv, čo svedčí o nízkej schopnosti ambrisentanu zmeniť profil liekov metabolizovaných týmito cestami.
Schopnosť ambrisentanu indukovať aktivitu CYP3A4 bola skúmaná u zdravých dobrovoľníkov a výsledky svedčia o nedostatočnom vplyve ambrisentanu na indukciu izoenzýmu CYP3A4.
C
yklosporín A
Súbežné podávanie ambrisentanu a cyklosporínu A v rovnovážnom stave viedlo u zdravých
dobrovoľníkov k 2-násobnému zvýšeniu expozície ambrisentanu. Toto môže byť spôsobené inhibíciou transportérov a metabolických enzýmov podieľajúcich sa na farmakokinetike ambrisentanu cyklosporínom A. Z toho dôvodu pri súbežnom podávaní s cyklosporínom A sa má dávka ambrisentanu u dospelých pacientov alebo u pediatrických pacientov ≥50 kg obmedziť na 5 mg jedenkrát denne; u pediatrických pacientov ≥20 až ˂50 kg sa má dávka obmedziť na 2,5 mg jedenkrát denne (pozri časť 4.2). Podávanie opakovaných dávok ambrisentanu nemalo vplyv na expozíciu cyklosporínu A a úprava dávky cyklosporínu A nie je potrebná.
Rifampicín
Súbežné podávanie rifampicínu (inhibítor transportného polypeptidu organických aniónov [OATP],
silný induktor CYP3A a 2C19 a induktor P-gp a uridíndifosfát glukuronozyltransferáz [UGTs]) bolo spojené s prechodným (približne 2-násobným) zvýšením expozície ambrisentanu po podaní úvodných
dávok zdravým dobrovoľníkom. Podávanie rifampicínu v rovnovážnom stave však na 8. deň nemalo
klinicky významný vplyv na expozíciu ambrisentanu. Pacienti užívajúci ambrisentan majú byť starostlivo sledovaní pri začatí liečby rifampicínom (pozri časti 4.4 a 5.2).
Inhibítory fosfodiesterázy
Súbežné podávanie ambrisentanu s inhibítorom fosfodiesterázy, buď so sildenafilom alebo tadalafilom
(oba sú substráty pre CYP3A4), zdravým dobrovoľníkom významne neovplyvnilo farmakokinetiku inhibítora fosfodiesterázy alebo ambrisentanu (pozri časť 5.2).
Iné cielené liečby PAH
Účinnosť a bezpečnosť ambrisentanu, keď sa podával súbežne s inými liekmi na PAH
(napr. s prostanoidmi a so stimulátormi rozpustnej guanylátcyklázy), sa špecificky nesledovali
v kontrolovaných klinických skúšaniach u pacientov s PAH (pozri časť 5.1). Na základe známych údajov o biotransformácii sa nepredpokladajú žiadne špecifické interakcie medzi ambrisentanom
a stimulátormi rozpustnej guanylátcyklázy ani s prostanoidmi (pozri časť 5.2). S týmito liekmi sa však neuskutočnili žiadne špecifické interakčné štúdie. V prípade ich súbežného podávania sa preto odporúča obozretnosť.
Perorálna antikoncepcia
V klinickej štúdii u zdravých dobrovoľníkov podávanie ambrisentanu v rovnovážnom stave v dávke
10 mg jedenkrát denne významne nezmenilo farmakokinetiku jednorazovej dávky etinylestradiolu a noretindrónu obsiahnutých v kombinovanej perorálnej antikoncepcii (pozri časť 5.2). Na základe tejto
farmakokinetickej štúdie sa neočakáva, že by ambrisentan významne ovplyvnil expozíciu
antikoncepcii založenej na estrogénoch alebo progestagéne.
Warfarín
V štúdii u zdravých dobrovoľníkov nemal ambrisentan žiadny vplyv na rovnovážne farmakokinetické
parametre a antikoagulačný účinok warfarínu (pozri časť 5.2). Warfarín taktiež nemal klinicky významný vplyv na farmakokinetiku ambrisentanu. Ambrisentan okrem toho nemal u pacientov žiadny celkový vplyv na týždennú dávku antikoagulancia warfarínového typu, na protrombínový čas
(Pothrombin Time, PT) a medzinárodný normalizovaný pomer (International Normalized Ratio, INR).
Ketokonazol
Podávanie ketokonazolu (silný inhibítor CYP3A4) v rovnovážnom stave neviedlo ku klinicky
významnému zvýšeniu expozície ambrisentanu (pozri časť 5.2).
V
plyv ambrisentanu na transportéry xenobiotík
Ambrisentan v klinicky významných koncentráciách nemá in vitro inhibičný vplyv na ľudské
transportéry vrátane P-glykoproteínu (Pgp), proteínu zodpovedného za rezistenciu pri rakovine prsníka (breast cancer resistance protein, BCRP), proteínu 2 súvisiaceho s mnohopočetnou liekovou rezistenciou (multi-drug resistance related protein 2, MRP2), transportnej pumpy solí žlčových kyselín (bile salt export pump, BSEP), transportných polypeptidov organických aniónov (organic anion transporting polypeptides, OATP1B1 a OATP1B3) a kontransportného polypeptidu sodíka
a taurocholátu (sodium-dependent taurocholate co-transporting polypeptide, NTCP). Ambrisentan je substrát pre Pgp-sprostredkovaný eflux.
Štúdie in vitro na potkaních hepatocytoch taktiež ukázali, že ambrisentan neindukoval expresiu proteínov Pgp, BSEP alebo MRP2.
Ambrisentan v rovnovážnom stave podávaný zdravým dobrovoľníkom nemal klinicky významný vplyv na farmakokinetiku jednorazovej dávky digoxínu, substrátu pre Pgp (pozri časť 5.2).
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Liečba ambrisentanom sa nesmie začať u žien vo fertilnom veku, pokiaľ výsledok tehotenského testu
pred začiatkom liečby nie je negatívny a pokiaľ nepoužívajú spoľahlivú antikoncepciu. Počas liečby
ambrisentanom sa odporúča vykonávať tehotenské testy v mesačných intervaloch.
Gravidita
Ambrisentan je kontraindikovaný počas gravidity (pozri časť 4.3). Štúdie na zvieratách preukázali, že
ambrisentan je teratogénny. Nie sú k dispozícii skúsenosti s použitím u ľudí.
Ženy liečené ambrisentanom musia byť informované o riziku poškodenia plodu a ak dôjde
k otehotneniu, má sa začať alternatívna liečba (pozri časti 4.3, 4.4 a 5.3).
Dojčenie
Nie je známe, či sa ambrisentan vylučuje do ľudského materského mlieka. Vylučovanie ambrisentanu
do mlieka sa u zvierat neskúmalo. Z tohto dôvodu je dojčenie kontraindikáciou u pacientok užívajúcich ambrisentan (pozri časť 4.3).
Mužská fertilita
Dlhodobé podávanie ERA, vrátane ambrisentanu, bolo u samcov spojené so vznikom atrofie
semenníkových tubulov (pozri časť 5.3). Hoci sa v štúdii ARIES-E nezistili jasné dôkazy o škodlivom účinku dlhodobej expozície ambrisentanu na počet spermií, dlhodobé podávanie ambrisentanu sa spájalo so zmenami v markeroch spermatogenézy. Pozoroval sa pokles plazmatickej koncentrácie inhibínu B a zvýšenie plazmatickej koncentrácie FSH. Vplyv na mužskú fertilitu nie je známy, ale nie je možné vylúčiť zhoršenie spermatogenézy. V klinických štúdiách nebolo dlhodobé podávanie ambrisentanu spojené so zmenou plazmatických hladín testosterónu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ambrisentan má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri posudzovaní pacientovej schopnosti vykonávať činnosti, ktoré vyžadujú úsudok, motorické alebo kognitívne zručnosti, treba mať na pamäti klinický stav pacienta a profil nežiaducich reakcií
na ambrisentan (akými sú hypotenzia, závraty, slabosť, únava) (pozri časť 4.8). Pred vedením vozidiel
alebo obsluhou strojov majú pacienti vedieť, ako by ich ambrisentan mohol ovplyvniť.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluNajčastejšie nežiaduce reakcie pozorované pri liečbe ambrisentanom boli periférny edém (37 %)
a bolesť hlavy (28 %). V krátkodobých klinických štúdiách sa liečba vyššou dávkou (10 mg) spájala s vyšším výskytom týchto nežiaducich reakcií a periférny edém bol zvyčajne závažnejší
u pacientov ≥ 65 rokov (pozri časť 4.4).
Závažné nežiaduce reakcie spojené s použitím ambrisentanu zahŕňajú anémiu (znížená koncentrácia hemoglobínu, znížený hematokrit) a hepatotoxicitu.
Pokles koncentrácie hemoglobínu a pokles hematokritu (10 %) sa spájal s ERA, vrátane ambrisentanu. Väčšina týchto znížení bola zistená počas prvých 4 týždňov liečby a hemoglobín sa vo všeobecnosti následne stabilizoval (pozri časť 4.4).
S ambrisentanom boli pozorované zvýšenia pečeňových enzýmov (2 %), poškodenie pečene
a autoimunitná hepatitída (vrátane vzplanutia základného ochorenia) (pozri časti 4.4 a 5.1).
Zoznam nežiaducich reakcií uvedený vtabuľkeFrekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000
až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme
(z dostupných údajov). U nežiaducich reakcií súvisiacich s dávkou sa kategória frekvencie vzťahuje na
vyššiu dávku ambrisentanu. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových
systémov
| Frekvencia
| Nežiaduca(e) reakcia(e)
|
Poruchy krvi a
lymfatického systému
| Veľmi časté
| Anémia (znížená koncentrácia hemoglobínu, znížený hematokrit)1
|
Poruchy imunitného systému
| Časté
| Reakcie z precitlivenosti (napr. angioedém, vyrážka, pruritus)
|
Poruchy nervového systému
| Veľmi časté
| Bolesť hlavy (vrátane bolesti hlavy v dôsledku sinusitídy, migrény)2,
závraty
|
Poruchy oka
| Časté
| Rozmazané videnie,
porucha zraku
|
Poruchy ucha a labyrintu
| Časté
| Tinnitus3
|
Menej časté
| Náhla strata sluchu3
|
Poruchy srdca a srdcovej
činnosti
| Veľmi časté
| Palpitácie
|
Časté
| Srdcové zlyhanie4
|
Poruchy ciev
| Veľmi časté
| Návaly tepla5
|
| Časté
| Hypotenzia,
synkopa
|
Poruchy dýchacej sústavy,
hrudníka a mediastína
| Veľmi časté
| Dyspnoe6,
kongescia v oblasti horných dýchacích ciest (napr. kongescia nosovej sliznice, kongescia sliznice prinosových dutín)7, nazofaryngitída7
|
Časté
| Epistaxa,
|
|
|
rinitída7, sinusitída7
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
|
Nauzea, hnačka, vracanie5
|
Časté
|
Bolesť brucha, zápcha
|
Poruchy pečene a žlčových
ciest
|
Časté
|
Zvýšené hladiny pečeňových transamináz
|
Menej časté
|
Poškodenie pečene (pozri časť 4.4),
autoimunitná hepatitída (pozri časť 4.4)
|
Poruchy kože
a podkožného tkaniva
|
Časté
|
Vyrážka8
|
Celkové porucha a reakcie v mieste podania
|
Veľmi časté
|
Periférny edém, retencia tekutín,
bolesť/nepríjemný pocit na hrudníku5,
únava
|
Časté
|
Slabosť
|
1 Pozri časť „
Popis vybraných nežiaducich reakcií“.
2 Frekvencia bolesti hlavy bola zvyčajne vyššia pri liečbe 10 mg dávkou ambrisentanu.
3 Prípady boli pozorované len v placebom kontrolovanej klinickej štúdii s ambrisentanom v kombinácii s tadalafilom.
4 Väčšina hlásených prípadov srdcového zlyhania bola spojená s retenciou tekutín.
5 Frekvencie boli pozorované v placebom kontrolovanej klinickej štúdii s ambrisentanom
v kombinácii s tadalafilom. Nižší výskyt bol pozorovaný s ambrisentanom v monoterapii.
6 Prípady zhoršujúceho sa dyspnoe nejasnej etiológie boli hlásené v krátkom čase po začatí liečby
ambrisentanom.
7 Počas liečby ambrisentanom súvisel výskyt kongescie nosovej sliznice s dávkou.
8 Vyrážka zahŕňa erytematóznu vyrážku, generalizovanú vyrážku, papulóznu vyrážku a pruritickú vyrážku.
Popis vybraných nežiaducich reakciíZnížená koncentrácia hemoglobínuV období po uvedení lieku na trh boli hlásené prípady anémie vyžadujúcej si transfúziu krvi (pozri
časť 4.4). Frekvencia zníženej koncentrácie hemoglobínu (anémia) bola vyššia pri liečbe 10 mg dávkou ambrisentanu. Vo všetkých 12-týždňových placebom kontrolovaných klinických štúdiách
fázy 3 došlo u pacientov v skupine s ambrisentanom k zníženiu priemernej koncentrácie hemoglobínu, ktoré bolo zistené už v 4. týždni (zníženie o 0,83 g/dl); priemerné zmeny oproti východiskovej koncentrácii sa zvyčajne stabilizovali počas nasledujúcich 8 týždňov. U celkovo 17 pacientov (6,5 %)
v skupine s ambrisentanom sa koncentrácia hemoglobínu znížila o ≥ 15 % oproti východiskovej
koncentrácii a klesla pod dolnú hranicu referenčného rozpätia.
Pediatrická populáciaBezpečnosť ambrisentanu u pediatrických pacientov s PAH vo veku od 8 rokov do menej ako
18 rokov bola hodnotená u 41 pacientov, ktorí boli liečení ambrisentanom 2,5 mg alebo 5 mg jedenkrát denne (nízkodávková skupina) alebo ambrisentanom 2,5 mg alebo 5 mg jedenkrát denne
titrovaným na 5 mg, 7,5 mg alebo 10 mg podľa telesnej hmotnosti (vysokodávková skupina)
v monoterapii alebo v kombinácii s iným liekom na PAH počas 24 týždňov vo fáze 2b otvoreného skúšania. Bezpečnosť bola ďalej hodnotená v následnej dlhodobej rozšírenej štúdii u 38 zo 41 jedincov. Pozorované nežiaduce reakcie, ktoré boli vyhodnotené ako súvisiace s ambrisentanom, boli konzistentné s tými, ktoré boli pozorované v kontrolovaných štúdiách u dospelých pacientov, najčastejšie sa vyskytujúce boli bolesť hlavy (15 %, 6/41 jedincov počas 24 týždňov fázy 2b otvoreného skúšania a 8 %, 3/38 jedincov počas dlhodobej rozšírenej štúdie) a kongescia nosovej sliznice (8 %, 3/41 jedincov počas 24 týždňov fázy 2b otvoreného skúšania).
H
l
ásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieU zdravých dobrovoľníkov bolo podanie jednorazovej dávky 50 a 100 mg (5- až 10-násobok maximálnej odporúčanej dávky) spojené s bolesťou hlavy, návalmi tepla, závratmi, nauzeou a kongesciou nosovej sliznice.
Vzhľadom na mechanizmus účinku by predávkovanie ambrisentanom mohlo potenciálne spôsobiť hypotenziu (pozri časť 5.3). V prípade výraznej hypotenzie môže byť potrebná aktívna kardiovaskulárna podpora. Nie je k dispozícii špecifické antidotum.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antihypertenzíva, iné antihypertenzíva, ATC kód: C02KX02
Mechanizmus účinkuAmbrisentan je perorálne účinný derivát kyseliny propiónovej a ERA so selektivitou pre receptor pre
endotelín A (ETA). Endotelín má významnú úlohu v patofyziológii PAH.
Ambrisentan je antagonista ETA (približne 4 000-násobne selektívnejší pre ETA ako pre ETB). Ambrisentan blokuje ETA receptorový podtyp, ktorý sa nachádza predovšetkým na bunkách hladkého svalstva ciev a srdcových myocytoch. To zabraňuje endotelínom sprostredkovanej aktivácii sekundárnych signálnych systémov, čo má za následok vazokonstrikciu a proliferáciu buniek hladkého svalstva. Očakáva sa, že vďaka selektivite ambrisentanu pre ETA receptor a nie pre ETB receptor sa zachová ETB receptorom sprostredkovaná tvorba vazodilatačných látok, oxidu dusnatého a prostacyklínu.
Klinická účinnosť abezpečnosťBoli vykonané dve randomizované, dvojito zaslepené, multicentrické, placebom kontrolované pivotné
štúdie fázy 3 (ARIES-1 a 2). Štúdia ARIES-1 zahŕňala 201 pacientov a porovnala 5 mg a 10 mg dávku ambrisentanu s placebom. Štúdia ARIES-2 zahŕňala 192 pacientov a porovnala 2,5 mg a 5 mg dávku
ambrisentanu s placebom. V oboch štúdiách bol ambrisentan pridaný k podporným/základným liekom
pacientov, čo mohlo zahŕňať podávanie v kombinácii s digoxínom, antikoagulanciami, diuretikami, kyslíkom a vazodilatanciami (blokátory kalciových kanálov, ACE inhibítory). Zaradení pacienti mali IPAH alebo PAH spojenú s ochorením spojivového tkaniva (PAH-CTD). Väčšina pacientov mala príznaky funkčnej triedy II (38,4 %) alebo III (55,0 %) podľa SZO. Pacienti s už existujúcim ochorením pečene (s cirhózou alebo s klinicky významne zvýšenými hodnotami aminotransferáz) a pacienti používajúci inú cielenú liečbu PAH (napr. prostanoidy) boli vylúčení. Hemodynamické parametre neboli v týchto štúdiách hodnotené.
Primárnym cieľovým ukazovateľom definovaným pre štúdie fázy 3 bolo zlepšenie záťažovej kapacity
hodnotené prostredníctvom zmeny oproti východiskovej hodnote v 6-minútovom teste chôdzou
(6-minute walk distance, 6MWD) v 12. týždni. V oboch štúdiách viedla liečba ambrisentanom k významnému zlepšeniu v 6MWD pri každej dávke ambrisentanu.
Na placebo korigované zlepšenie priemernej hodnoty v 6MWD v 12. týždni oproti východiskovej hodnote v skupine s 5 mg dávkou bolo 30,6 m (95 % IS: 2,9 až 58,3; p=0,008) v štúdii ARIES-1 a
59,4 m (95 % IS: 29,6 až 89,3; p<0,001) v štúdii ARIES-2. Na placebo korigované zlepšenie priemernej hodnoty v 6MWD v 12. týždni u pacientov v skupine s 10 mg dávkou v štúdii ARIES-1 bolo 51,4 m (95 % IS: 26,6 až 76,2; p<0,001).
Uskutočnila sa vopred špecifikovaná kombinovaná analýza štúdií fázy 3 (ARIES-C). Na placebo korigované priemerné zlepšenie v 6MWD bolo 44,6 m (95 % IS: 24,3 až 64,9; p<0,001) pri 5 mg dávke a 52,5 m (95 % IS: 28,8 až 76,2; p<0,001) pri 10 mg dávke.
V štúdii ARIES-2 ambrisentan (skupina s kombinovanou dávkou) významne spomalil čas do klinického zhoršenia PAH v porovnaní s placebom (p<0,001), hazard ratio preukázalo 80 % zníženie (95 % IS: 47 % až 92 %). Hodnotené premenné zahŕňali: úmrtie, transplantáciu pľúc, hospitalizáciu kvôli PAH, septostómiu predsiení, pridanie ďalších látok na liečbu PAH a kritériá pre predčasné ukončenie liečby. V skupine s kombinovanou dávkou sa pozorovalo štatisticky významné zvýšenie (3,41 ± 6,96) skóre škály hodnotiacej fyzické fungovanie v rámci dotazníka o zdraví SF-36
v porovnaní s placebom (-0,20 ± 8,14, p=0,005). Liečba ambrisentanom viedla k štatisticky významnému zlepšeniu skóre Borgovej škály hodnotiacej dýchavičnosť (Borg Dyspnea Index, BDI)
v 12. týždni (na placebo korigované skóre BDI v hodnote -1,1 (95 % IS: -1,8 až -0,4; p=0,019; skupina
s kombinovanou dávkou)).
Dlhodobé údaje
Pacienti, ktorí sa zúčastnili štúdie ARIES-1 a ARIES-2, mohli byť zaradení do dlhodobej otvorenej predĺženej štúdie ARIES-E (n=383). Kombinovaná priemerná expozícia bola približne
145 ± 80 týždňov a maximálna expozícia bola približne 259 týždňov. Hlavné primárne cieľové ukazovatele tejto štúdie boli výskyt a závažnosť nežiaducich udalostí súvisiacich s dlhodobou expozíciou ambrisentanu, vrátane sérových LFT (funkčných vyšetrení pečene). Zistenia ohľadom bezpečnosti pozorované pri dlhodobej expozícii ambrisentanu v tejto štúdii boli zvyčajne zhodné
s tými, ktoré sa pozorovali v 12-týždňových placebom kontrolovaných štúdiách.
Pravdepodobnosť prežitia zistená u jedincov užívajúcich ambrisentan (skupina s kombinovanou dávkou ambrisentanu) bola po 1 roku 93 %, po 2 rokoch 85 % a po 3 rokoch 79 %.
V otvorenej štúdii (AMB222) bol ambrisentan skúmaný u 36 pacientov za účelom zhodnotenia výskytu zvýšených sérových koncentrácií aminotransferáz u pacientov, ktorí prerušili predchádzajúcu liečbu iným ERA kvôli abnormalitám aminotransferáz. Počas priemerne 53 týždňov liečby ambrisentanom sa u žiadneho zo zaradených pacientov nepotvrdila hodnota sérovej ALT >3xULN, ktorá by vyžadovala trvalé ukončenie liečby. Počas tejto doby bola u päťdesiatich percent pacientov dávka ambrisentanu zvýšená z 5 mg na 10 mg.
Kumulatívny výskyt abnormalít sérových aminotransferáz >3xULN vo všetkých štúdiách fázy 2 a 3 (vrátane príslušných otvorených predĺžených štúdií) bol 17 zo 483 jedincov počas priemernej doby expozície 79,5 týždňa. To pri ambrisentane zodpovedá miere výskytu 2,3 udalosti na
100 pacientorokov expozície. V otvorenej dlhodobej predĺženej štúdii ARIES-E sa zistilo, že po
2 rokoch liečby je riziko vzniku vzostupov koncentrácií aminotransferáz na >3xULN u pacientov
liečených ambrisentanom 3,9 %.
Ďalšieklinickéinformácie
Zlepšenie hemodynamických parametrov sa pozorovalo u pacientov s PAH po 12 týždňoch (n = 29)
v štúdii fázy 2 (AMB220). Liečba ambrisentanom viedla k zvýšeniu priemerného srdcového indexu,
zníženiu priemerného tlaku v pľúcnych artériách a zníženiu priemernej pľúcnej cievnej rezistencie.
Pri liečbe ambrisentanom bol hlásený pokles systolického a diastolického krvného tlaku. V placebom kontrolovaných klinických skúšaniach trvajúcich 12 týždňov sa zistil priemerný pokles systolického krvného tlaku o 3 mmHg a diastolického krvného tlaku o 4,2 mmHg, keď sa porovnali hodnoty namerané na začiatku a na konci liečby. Priemerný pokles systolického a diastolického krvného tlaku pretrvával počas až 4 rokov liečby ambrisentanom v dlhodobej otvorenej predĺženej štúdii ARIES-E.
Počas interakčnej štúdie u zdravých dobrovoľníkov nebol pozorovaný klinicky významný vplyv na farmakokinetiku ambrisentanu alebo sildenafilu a táto kombinácia bola dobre znášaná. Počet pacientov, ktorí boli súbežne liečení ambrisentanom a sildenafilom, bol 22 pacientov (5,7 %) v štúdii ARIES-E a 17 pacientov (47 %) v štúdii AMB222. U týchto pacientov sa nezistili žiadne ďalšie bezpečnostné obavy.
Klinickáúčinnosťvkombinácii s tadalafilom
Uskutočnila sa multicentrická, dvojito zaslepená, aktívnymi komparátormi kontrolovaná štúdia fázy 3
typu „event-driven“ (dosiahnutie cieľa štúdie bolo podmienené výskytom vopred definovanej príhody), ktorá sledovala efekt liečby (AMB112565/AMBITION) s cieľom zhodnotiť účinnosť začiatočnej liečby kombináciou ambrisentanu a tadalafilu v porovnaní s monoterapiou
buď ambrisentanom, alebo tadalafilom, u 500 pacientov s PAH bez predchádzajúcej liečby, ktorí boli na uvedené liečby randomizovaní v pomere 2:1:1. Žiadni pacienti neužívali samotné placebo.
Primárna analýza porovnala skupinu s kombinovanou liečbou so súhrnne hodnotenými skupinami s monoterapiami. Uskutočnili sa aj podporné porovnania skupiny s kombinovanou liečbou
s jednotlivými skupinami s monoterapiami. Pacienti s významnou anémiou, s retenciou tekutín alebo so zriedkavými ochoreniami sietnice boli z účasti na štúdii vylúčení na základe kritérií stanovených skúšajúcimi lekármi. Z účasti na štúdii boli vylúčení aj pacienti, ktorí mali východiskové hodnoty
ALT a AST > 2xULN.
Pri vstupe do štúdie bolo 96 % pacientov bez predchádzajúcej špecifickej liečby PAH a medián času
od stanovenia diagnózy do zaradenia do štúdie bol 22 dní. Pacienti začali liečbu 5 mg ambrisentanu a 20 mg tadalafilu a v 4. týždni mali dávku tadalafilu titrovanú na 40 mg a v 8. týždni mali dávku
ambrisentanu titrovanú na 10 mg, pokiaľ nemali žiadne problémy so znášanlivosťou liečby. Medián trvania dvojito zaslepenej liečby bol pri kombinovanej liečbe dlhší ako 1,5 roku.
Primárnym cieľovým ukazovateľom bol čas do prvého výskytu príhody, ktorou bolo klinické zlyhanie
liečby definované ako:
- smrť alebo
- hospitalizácia z dôvodu zhoršujúcej sa PAH,
- progresia ochorenia,
- neuspokojivá dlhodobá klinická odpoveď na liečbu.
Priemerný vek všetkých pacientov bol 54 rokov (SD 15; vekové rozmedzie 18 - 75 rokov). Pri vstupe do štúdie boli pacienti vo FT II (31 %) a FT III (69 %) podľa SZO. Najčastejšou etiológiou v populácii tejto štúdie bola idiopatická alebo dedičná PAH (56 %), po nej nasledovala PAH zapríčinená poruchami spojivového tkaniva (37 %), PAH súvisiaca s liekmi a toxínmi (3 %), s korigovanou jednoduchou vrodenou srdcovou chybou (2 %) a s HIV (2 %). Pacienti vo FT II a III podľa SZO mali priemernú východiskovú hodnotu v 6MWD rovnú 353 m.
Cieľové ukazovatele efektu liečby
Kombinovaná liečba viedla v porovnaní so súhrnne hodnotenými skupinami s monoterapiami
k 50 % zníženiu rizika (hazard ratio [HR] 0,502; 95 % IS: 0,348 až 0,724; p = 0,0002) výskytu združeného cieľového ukazovateľa, ktorým bolo klinické zlyhanie liečby, a to až do hodnotenia na záverečnej návšteve [obrázok 1 a tabuľka 1]. Efekt liečby bol založený na 63 % znížení počtu hospitalizácií pri kombinovanej liečbe, dosiahol sa v krátkom čase a zostal zachovaný. Účinnosť kombinovanej liečby z hľadiska primárneho cieľového ukazovateľa sa preukázala aj v porovnaní s jednotlivými monoterapiami a naprieč podskupinami vytvorenými na základe veku, etnického pôvodu, geografickej oblasti, etiológie (IPAH/hPAH a PAH-CTD). Efekt liečby bol významný
u pacientov vo FC II aj u pacientov vo FC III.
Obrázok 1
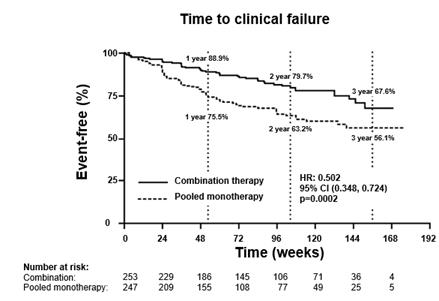
Čas do klinického zlyhania liečby (Time to Clinical Failure), Pacienti bez príhody (Event-Free) (%), Kombinovaná liečba (Combination therapy), Monoterapie - súhrnne (Pooled monotherapy); year (rok), Čas (týždne) (Time (weeks)), Počet pacientov v riziku (Number at risk), Kombinovaná liečba (Combination), Monoterapie - súhrnne (Pooled monotherapy)
Tabuľka 1
| Ambrisentan
+ tadalafil
(N = 253)
| Monoterapie - súhrnne
(N = 247)
| Ambrisentan v monoterapii
(N = 126)
| Tadalafil
v monoterapii
(N = 121)
|
Čas do prvej príhody klinického zlyhania liečby (formálne posúdenej)
|
Klinické zlyhanie liečby,
počet (%)
| 46 (18)
| 77 (31)
| 43 (34)
| 34 (28)
|
Hazard ratio (95 % IS)
|
| 0,502 (0,348; 0,724)
| 0,477 (0,314; 0,723)
| 0,528 (0,338; 0,827)
|
p-hodnota, log-rank test
|
| 0,0002
| 0,0004
| 0,0045
|
Zložky tvoriace prvú príhodu klinického zlyhania liečby (formálne posúdené)
|
Smrť (z akýchkoľvek príčin)
| 9 (4 %)
| 8 (3 %)
| 2 (2 %)
| 6 (5 %)
|
Hospitalizácia z dôvodu zhoršujúcej sa PAH
|
10 (4 %)
|
30 (12 %)
|
18 (14 %)
|
12 (10 %)
|
Progresia ochorenia
| 10 (4 %)
| 16 (6 %)
| 12 (10 %)
| 4 (3 %)
|
Neuspokojivá dlhodobá
klinická odpoveď na liečbu
|
17 (7 %)
|
23 (9 %)
|
11 (9 %)
|
12 (10 %)
|
Čas do prvej hospitalizácie z dôvodu zhoršujúcej sa PAH (formálne posúdenej)
|
Prvá hospitalizácia, počet
(%)
| 19 (8 %)
| 44 (18 %)
| 27 (21 %)
| 17 (14 %)
|
Hazard ratio (95 % IS)
|
| 0,372
| 0,323
| 0,442
|
p-hodnota, log-rank test
|
| 0,0002
| < 0,0001
| 0,0124
|
Sekundárna cieľové ukazovatele
Hodnotené boli sekundárne cieľové ukazovatele: Tabuľka 2
Sekundárna cieľové
ukazovatele (zmena
v 24. týždni v porovnaní
s východiskovým stavom)
| Ambrisentan
+ tadalafil
| Monoterapie - súhrnne
| Rozdiel a interval
spoľahlivosti
| p-hodnota
|
NT-proBNP (% zníženie)
|
-67,2
|
-50,4
| % rozdiel: -33,8;
95 % IS: -44,8; -20,7
|
p < 0,0001
|
% osôb, ktoré v 24. týždni
dosiahli uspokojivú
klinickú odpoveď na liečbu
|
39
|
29
|
Pomer šancí: 1,56;
95 % IS: 1,05; 2,32
|
p = 0,026
|
6MWD (m, medián zmeny)
|
49,0
|
23,8
| 22,75 m; 95 % IS:
12,00; 33,50
|
p < 0,0001
|
I
diopatická
pľúcna
f
i
bróza
Uskutočnila sa štúdia so 492 pacientmi (ambrisentan N = 329, placebo N = 163) s idiopatickou pľúcnou fibrózou (IPF), z ktorých 11 % malo sekundárnu pľúcnu hypertenziu (skupiny 3 podľa SZO), ktorá však bola predčasne ukončená, keď sa zistilo, že nie je možné splniť primárny cieľový ukazovateľ účinnosti (štúdia ARTEMIS-IPF). V skupine s ambrisentanom sa pozorovalo 90 prípadov (27 %) progresie IPF (vrátane hospitalizácie z dôvodu respiračných ťažkostí) alebo úmrtia v porovnaní s 28 prípadmi (17 %) v skupine s placebom. Ambrisentan je preto kontraindikovaný u pacientov s IPF so sekundárnou pľúcnou hypertenziou alebo bez nej (pozri časť 4.3).
Pediatrická populácia
Štúdia AMB112529
Bezpečnosť a znášanlivosť ambrisentanu jedenkrát denne počas 24 týždňov bola hodnotená
v otvorenej nekontrolovanej štúdii u 41 pediatrických pacientov s PAH vo veku od 8 do menej ako
18 rokov (medián: 13 rokov). Etiológia PAH bola idiopatická (n=26; 63 %), perzistujúca vrodená
PAH napriek chirurgickej korekcii (n=11; 27 %), sekundárna k ochoreniu spojivového tkaniva (n=1;
2 %) alebo familiárna (n=3; 7,3 %). Spomedzi 11 jedincov s vrodenou srdcovou chybou 9 mali defekty komorového septa, 2 mali defekty atriálneho septa a 1 mal perzistujúci otvorený ductus.'
Pacienti boli vo funkčnej triede II podľa SZO (n=32; 78 %) alebo v triede III (n=9; 22 %) na začiatku
liečby v štúdii. Na vstupe do štúdie boli pacienti liečení liekmi na PAH (najčastejšie PDE5i
v monoterapii [n=18; 44 %], kombinovanou liečbou PDE5i a prostanoidmi [n=8; 20 %] alebo prostanoidmi v monoterapii [n=1; 2 %] a počas štúdie pokračovali vo svojej liečbe PAH. Pacienti boli
rozdelení do dvoch dávkovacích skupín: ambrisentan 2,5 mg alebo 5 mg jedenkrát denne (nízka
dávka, n=21) a ambrisentan 2,5 mg alebo 5 mg jedenkrát denne titrované na 5 mg, 7,5 mg alebo 10 mg na základe telesnej hmotnosti (vysoká dávka, n=20). Spolu 20 pacientov z oboch dávkovacích skupín
bolo titrovaných v 2 týždňoch na základe klinickej odpovede a znášanlivosti; 37 pacientov ukončilo
štúdiu; 4 pacienti boli vyradení zo štúdie.
Nebol pozorovaný žiadny trend v závislosti účinku na dávke ambrisentanu na záťažovú kapacitu
(6MWD) ako hlavný parameter účinnosti. Stredná zmena oproti východiskovej hodnote v 24. týždni
v 6MWD z merania na začiatku a v 24. týždni bola +55,14 m (95 % IS: 4,32 až 105,95) u 18 pacientov v nízkodávkovej skupine a +26,25 m (95 % IS: -4,59 až 57,09) u 18 pacientov vo vysokodávkovej
skupine. Stredná zmena oproti východiskovej hodnote v 24. týždni v 6MWD spolu pre 36 pacientov
(obe dávky spojené) bola +40,69 m (95 % IS: 12,08 až 69,31). Tieto výsledky boli konzistentné
s tými, ktoré boli pozorované u dospelých. V 24. týždni 95 % pacientov v nízkodávkovej skupine
a 100 % pacientov vo vysokodávkovej skupine zostalo stabilných (funkčná trieda nezmenená alebo
zlepšená). Kaplanov-Meierov odhad prežitia nezávislý od udalosti pre zhoršujúcu sa PAH (úmrtie
[všetky príčiny], transplantácia pľúc alebo hospitalizácia pre zhoršenie PAH alebo pre zhoršenie súvisiace s PAH) v 24. týždni bol 86 % v nízkodávkovej skupine a 85 % vo vysokodávkovej skupine.
Hemodynamika bola meraná u 5 pacientov (nízkodávková skupina). Priemerné zvýšenie srdcového indexu oproti východiskovej hodnote bol +0,94 l/min/m2, priemerný pokles stredného pľúcneho arteriálneho tlaku bol -2,2 mmHg a stredný pokles PVR bol -277 dyn°s/cm5 (-3,46 mmHg/l/min).
U pediatrických pacientov s PAH, ktorí dostávali ambrisentan počas 24 týždňov, pokles geometrického priemeru NT-pro-BNP oproti východiskovej hodnote bol 31 % v nízkodávkovej skupine (2,5 a 5 mg) a 28 % vo vysokodávkovej skupine (5; 7,5 a 10 mg).
Štúdia AMB112588
Dlhodobé údaje boli získané od 38 zo 41 pacientov, ktorí boli liečení ambrisentanom v 24-týždňovej
randomizovanej štúdii. Priemerná doba expozície liečbe ambrisentanom bola 3,4 ± 1,8 roku (do
6,4 roku), pričom 63 % pacientov bolo liečených aspoň 3 roky a 42 % aspoň 4 roky. Pacienti mohli
dostávať ďalšiu liečbu PAH, ako bolo požadované v otvorenom rozšírení. Väčšina pacientov mala diagnostikovanú idiopatickú alebo dedičnú PAH (68 %). Celkovo 46 % pacientov zostalo vo funkčnej triede II podľa SZO. Kaplanove-Meierove odhady prežitia boli 94,42 % po 3 rokoch a 90,64 % po
4 rokoch od začiatku liečby. V rovnakých časových bodoch u 77,09 % a 73,24 % pacientov nedošlo k zhoršeniu PAH, pričom zhoršenie bolo definované ako úmrtie (všetky príčiny), zapísanie do
zoznamu na transplantáciu pľúc alebo atriálna septostómia, alebo zhoršenie PAH vedúce
k hospitalizácii, zmena dávky ambrisentanu, pridanie alebo zmena dávky existujúceho cieleného liečiva v liečbe PAH, nárast funkčnej triedy podľa SZO; pokles v 6MWD alebo prejavy/príznaky
pravostranného srdcového zlyhávania.
5.2 Farmakokinetické vlastnosti
Absorpcia
Ambrisentan sa u ľudí rýchlo absorbuje. Po perorálnom podaní sa maximálne plazmatické
koncentrácie (Cmax) ambrisentanu zvyčajne dosiahnu v priebehu približne 1,5 hodiny po podaní dávky spolu s jedlom ako aj nalačno. Cmax a plocha pod krivkou závislosti plazmatickej koncentrácie od času (AUC) sa zvyšujú úmerne dávke v rozmedzí terapeutických dávok. Rovnovážny stav sa zvyčajne dosiahne po 4 dňoch opakovaného podávania.
Štúdia skúmajúca vplyv jedla, v ktorej bol ambrisentan podávaný zdravým dobrovoľníkom nalačno a s jedlom s vysokým obsahom tuku poukázala na to, že hodnota Cmax sa znížila o 12 %, zatiaľ čo hodnota AUC zostala nezmenená. Tento pokles maximálnej koncentrácie nie je klinicky významný, a preto sa ambrisentan môže užívať s jedlom alebo bez jedla.
Distribúcia
Ambrisentan sa vo veľkej miere viaže na plazmatické bielkoviny. Väzba ambrisentanu na plazmatické
bielkoviny in vitro bola v priemere 98,8 % a nezávislá od koncentrácie v rozmedzí
0,2 - 20 mikrogramov/ml. Ambrisentan sa viaže predovšetkým na albumín (96,5 %) a v menšej miere na alfa1-kyslý glykoproteín.
Distribúcia ambrisentanu do červených krviniek je nízka a priemerný pomer krv:plazma je 0,57 u žien
a 0,61 u mužov.
Biotransformácia
Ambrisentan je nesulfónamidový ERA (je to derivát kyseliny propiónovej).
Ambrisentan je glukuronidovaný niekoľkými izoenzýmami UGT (UGT1A9S, UGT2B7S a UGT1A3S) na glukuronid ambrisentanu (13 %). Ambrisentan podlieha aj oxidačnému metabolizmu sprostredkovanému hlavne CYP3A4 a v menšej miere CYP3A5 a CYP2C19, pri ktorom vzniká
4-hydroxymetylambrisentan (21 %), ktorý je ďalej glukuronidovaný na glukuronid
4-hydroxymetylambrisentanu (5 %). Väzbová afinita 4-hydroxymetylambrisentanu k ľudskému
receptoru pre endotelín je 65-násobne nižšia ako u ambrisentanu. Z tohto dôvodu sa pri koncentráciách pozorovaných v plazme (približne 4 % oproti pôvodnému ambrisentanu) neočakáva, že sa
4-hydroxymetylambrisentan bude podieľať na farmakologickom účinku ambrisentanu.
Údaje in vitro ukazujú, že ambrisentan v koncentrácii 300 μmol/l spôsobil menej ako 50 % inhibíciu
UGT1A1, UGT1A6, UGT1A9, UGT2B7 (do 30 %) alebo enzýmov 1A2, 2A6, 2B6, 2C8, 2C9, 2C19,
2D6, 2E1 a 3A4 cytochrómu P450 (do 25 %). Ambrisentan v klinicky významných koncentráciách nemá in vitro žiaden inhibičný vplyv na ľudské transportéry vrátane Pgp, BCRP, MRP2, BSEP,
OATP1B1, OATP1B3 a NTCP. Okrem toho ambrisentan neindukoval expresiu proteínov MRP2, Pgp
alebo BSEP v potkaních hepatocytoch. Celkovo vzaté, údaje in vitro naznačujú, že sa nepredpokladá, že by ambrisentan v klinicky významných koncentráciách (plazmatická Cmax do 3,2 μmol/l) mal vplyv na UGT1A1, UGT1A6, UGT1A9, UGT2B7 alebo enzýmy 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6,
2E1, 3A4 cytochrómu P450 alebo na transport sprostredkovaný BSEP, BCRP, Pgp, MRP2, OATP1B1/3 alebo NTCP.
Vplyv ambrisentanu v rovnovážnom stave (10 mg jedenkrát denne) na farmakokinetiku a farmakodynamiku jednorazovej dávky warfarínu (25 mg), hodnotený pomocou PT a INR, bol skúmaný u 20 zdravých dobrovoľníkov. Ambrisentan nemal žiadny klinicky významný vplyv na farmakokinetiku alebo farmakodynamiku warfarínu. Podobne ani súbežné podávanie warfarínu nemalo vplyv na farmakokinetiku ambrisentanu (pozri časť 4.5).
Vplyv 7-dňového podávania sildenafilu (20 mg trikrát denne) na farmakokinetiku jednorazovej dávky ambrisentanu a vplyv 7-dňového podávania ambrisentanu (10 mg jedenkrát denne) na farmakokinetiku jednorazovej dávky sildenafilu bol skúmaný u 19 zdravých dobrovoľníkov. Po súbežnom podávaní ambrisentanu sa hodnota Cmax sildenafilu zvýšila o 13 %, avšak okrem toho sa nezistili žiadne ďalšie zmeny farmakokinetických parametrov sildenafilu, N-desmetylsildenafilu a ambrisentanu. Toto mierne zvýšenie hodnoty Cmax sildenafilu sa nepovažuje za klinicky významné (pozri časť 4.5).
Vplyv ambrisentanu v rovnovážnom stave (10 mg jedenkrát denne) na farmakokinetiku jednorazovej dávky tadalafilu a vplyv tadalafilu v rovnovážnom stave (40 mg jedenkrát denne) na farmakokinetiku jednorazovej dávky ambrisentanu bol skúmaný u 23 zdravých dobrovoľníkov. Ambrisentan nemal žiadny klinicky významný vplyv na farmakokinetiku tadalafilu. Podobne ani súbežné podávanie tadalafilu nemalo vplyv na farmakokinetiku ambrisentanu (pozri časť 4.5).
Vplyv opakovaného podávania ketokonazolu (400 mg jedenkrát denne) na farmakokinetiku jednorazovej 10 mg dávky ambrisentanu bol skúmaný u 16 zdravých dobrovoľníkov. Expozícia ambrisentanu sa zvýšila o 35 % pri hodnotení pomocou AUC(0-inf) a o 20 % pri hodnotení pomocou Cmax. Táto zmena expozície pravdepodobne nie je klinicky významná, a preto sa ambrisentan môže podávať súbežne s ketokonazolom.
Vplyv opakovaného podávania cyklosporínu A (100 - 150 mg dvakrát denne) na farmakokinetiku ambrisentanu (5 mg jedenkrát denne) v rovnovážnom stave a vplyv opakovaného podávania ambrisentanu (5 mg jedenkrát denne) na farmakokinetiku cyklosporínu A (100 - 150 mg dvakrát denne) v rovnovážnom stave bol skúmaný u zdravých dobrovoľníkov. Pri podávaní opakovaných dávok cyklosporínu A došlo k zvýšeniu hodnoty Cmax a AUC(0–t) ambrisentanu (48 % a 121 %, v tomto poradí). Na základe týchto zmien sa má pri súbežnom podávaní cyklosporínu A dávka ambrisentanu
u dospelých pacientov alebo u pediatrických pacientov s hmotnosťou ≥50 kg obmedziť na 5 mg jedenkrát denne; u pediatrických pacientov ≥20 až ˂50 kg sa má dávka obmedziť na 2,5 mg jedenkrát
denne (pozri časť 4.2). Podávanie opakovaných dávok ambrisentanu však nemalo klinicky významný vplyv na expozíciu cyklosporínu A a úprava dávky cyklosporínu A nie je potrebná.
Vplyv akútneho a opakovaného podávania rifampicínu (600 mg jedenkrát denne) na farmakokinetiku ambrisentanu (10 mg jedenkrát denne) v rovnovážnom stave bol sledovaný u zdravých dobrovoľníkov. Po podaní úvodných dávok rifampicínu sa pozorovalo prechodné zvýšenie hodnôt AUC(0–τ) ambrisentanu (o 121 % po prvej dávke a o 116 % po druhej dávke rifampicínu), pravdepodobne kvôli inhibícii OATP spôsobenej rifampicínom. Po podávaní opakovaných dávok rifampicínu sa však na 8. deň nezistil klinicky významný vplyv na expozíciu ambrisentanu. Pacienti užívajúci ambrisentan majú byť starostlivo sledovaní pri začatí liečby rifampicínom (pozri časti 4.4
a 4.5).
Vplyv opakovaného podávania ambrisentanu (10 mg) na farmakokinetiku jednorazovej dávky digoxínu bol skúmaný u 15 zdravých dobrovoľníkov. Podávanie opakovaných dávok ambrisentanu spôsobilo mierne zvýšenie AUC0-last a minimálnych (trough) koncentrácií digoxínu a zvýšenie Cmax digoxínu o 29 %. Zvýšenie expozície digoxínu pozorované po podávaní opakovaných dávok ambrisentanu sa nepovažovalo za klinicky významné a úprava dávky digoxínu nie je potrebná (pozri časť 4.5).
Vplyv 12-dňového podávania ambrisentanu (10 mg jedenkrát denne) na farmakokinetiku jednorazovej dávky perorálneho kontraceptíva obsahujúceho etinylestradiol (35 μg) a noretindrón (1 mg) bol skúmaný u zdravých dobrovoľníkov. Cmax a AUC(0–∞) boli mierne znížené pri etinylestradiole (o 8 % a
4 %) a mierne zvýšené pri noretindróne (o 13 % a 14 %). Tieto zmeny v expozícii etinylestradiolom alebo noretindrónom boli malé a pravdepodobne nie sú klinicky významné (pozri časť 4.5).
Eliminácia
Ambrisentan a jeho metabolity sa vylučujú predovšetkým žlčou po hepatálnom a/alebo
extrahepatálnom metabolizme. Po perorálnom podaní sa približne 22 % podanej dávky zistilo v moči, pričom 3,3 % tvoril ambrisentan v nezmenenej forme. Plazmatický eliminačný polčas u ľudí je v
rozmedzí od 13,6 do 16,5 hodín.
Osobitné skupiny pacientov
Dospelá populácia (pohlavie, vek)
Na základe výsledkov populačnej farmakokinetickej analýzy u zdravých dobrovoľníkov a pacientov s
PAH nebola farmakokinetika ambrisentanu významne ovplyvnená pohlavím alebo vekom (pozri
časť 4.2).
Pediatrická populácia
Dostupné sú obmedzené farmakokinetické údaje v pediatrickej populácii. Farmakokinetika bola skúmaná u pediatrických jedincov vo veku od 8 do menej ako 18 rokov v jednej klinickej štúdii
(AMB112529).
Farmakokinetika ambrisentanu po perorálnom podaní u jedincov s PAH vo veku od 8 do menej ako
18 rokov bola vo veľkej miere konzistentná s farmakokinetikou u dospelých po zohľadnení telesnej
hmotnosti. Modelové odvodené expozície v pediatrickej populácii v rovnovážnom stave (AUCss) boli pri nízkych dávkach a vysokých dávkach pre všetky hmotnostné skupiny v 5. a 95. percentile
historickej expozície u dospelých pri nízkej dávke (5 mg) alebo vysokej dávke (10 mg), v uvedenom
poradí.
Porucha funkcie obličiek
Ambrisentan nepodlieha významnému renálnemu metabolizmu alebo renálnemu klírensu (exkrécii). V
populačnej farmakokinetickej analýze sa zistilo, že klírens kreatinínu je štatisticky významný kovariant ovplyvňujúci perorálny klírens ambrisentanu. U pacientov so stredne ťažkou poruchou funkcie obličiek je rozsah poklesu perorálneho klírensu mierny (20-40 %), a preto pravdepodobne nie je klinicky významný. U pacientov s ťažkou poruchou funkcie obličiek je však potrebná opatrnosť (pozri časť 4.2).
Porucha funkcie pečene
Hlavné cesty metabolizmu ambrisentanu sú glukuronidácia a oxidácia, po ktorých nasleduje
vylučovanie žlčou, a preto je možné očakávať, že porucha funkcie pečene zvýši expozíciu (Cmax a
AUC) ambrisentanu. V populačnej farmakokinetickej analýze sa zistilo, že perorálny klírens je
znížený v dôsledku zvýšených hladín bilirubínu. Vplyv bilirubínu je však len mierne závažný (u pacienta s hladinou bilirubínu zvýšenou na 4,5 mg/dl by bol perorálny klírens ambrisentanu znížený o
približne 30 % v porovnaní s typickým pacientom s hladinou bilirubínu 0,6 mg/dl). Farmakokinetika
tohto dôvodu sa liečba ambrisentanom nemá začať u pacientov s ťažkou poruchou funkcie pečene alebo s klinicky významne zvýšenými hodnotami pečeňových aminotransferáz (>3xULN) (pozri časti 4.3 a 4.4).
5.3 Predklinické údaje o bezpečnosti
Vzhľadom na hlavný skupinový farmakologický účinok by podanie veľkej jednorazovej dávky ambrisentanu (t.j. predávkovanie) mohlo znížiť artériový tlak a mohlo by spôsobiť hypotenziu a príznaky súvisiace s vazodilatáciou.
Preukázalo sa, že ambrisentan nie je inhibítor transportu žlčových kyselín ani nevyvoláva zjavnú hepatotoxicitu.
Po dlhodobom podávaní boli u hlodavcov pozorované zápal a zmeny epitelu nosovej dutiny pri expozíciách nižších ako sú terapeutické hladiny u ľudí. Po dlhodobom podávaní vysokých dávok ambrisentanu boli u psov pozorované mierne zápalové reakcie pri expozíciách viac ako 20-násobne prevyšujúcich expozície zistené u pacientov.
U potkanov liečených ambrisentanom bola v nosovej dutine pozorovaná hyperplázia tkaniva nosovej kosti v oblasti nosových mušlí pri expozičných hladinách 3-násobne prevyšujúcich klinickú hodnotu AUC. Hyperplázia tkaniva nosovej kosti nebola pozorovaná pri podávaní ambrisentanu myšiam alebo psom. U potkanov je hyperplázia nosovej mušle známou reakciou na zápal nosovej sliznice, a to na základe skúseností s inými látkami.
Ambrisentan bol klastogénny, keď sa testoval vo vysokých koncentráciách v cicavčích bunkách in vitro. V bakteriálnych testoch a v dvoch štúdiách in vivo na hlodavcoch sa nepreukázali mutagénne ani genotoxické účinky ambrisentanu.
V 2-ročných štúdiách s perorálnym podávaním vykonaných na potkanoch a myšiach sa nepreukázal karcinogénny potenciál. U samcov potkanov došlo k malému zvýšeniu výskytu fibroadenómov
prsníka, benígneho nádoru, iba pri podávaní najvyššej dávky. Pri tejto dávke bola systémová expozícia ambrisentanu u samcov potkanov (na základe hodnoty AUC v rovnovážnom stave) 6-násobne vyššia
ako systémová expozícia dosiahnutá pri klinickej dávke 10 mg/deň.
Atrofia semenníkových tubulov, ktorá bola ojedinelo spojená s aspermiou, bola pozorovaná v štúdiách toxicity po opakovanom podávaní perorálnej dávky a v štúdiách fertility na samcoch potkanov a myší, a to bez stanovenia bezpečnostného rozpätia. Zmeny na semenníkoch neboli plne reverzibilné počas hodnotených období bez podávania dávky. Zmeny na semenníkoch však neboli pozorované v štúdiách na psoch, ktoré trvali až 39 týždňov, a to pri expozícii 35-násobne prevyšujúcej hodnotu AUC dosiahnutú u ľudí. U samcov potkanov nemal ambrisentan žiadne účinky na pohyblivosť spermií
pri všetkých skúšaných dávkach (až do 300 mg/kg/deň). Zaznamenal sa mierny (< 10 %) pokles percenta morfologicky normálnych spermií pri dávke 300 mg/kg/deň, ale nie pri dávke 100 mg/kg/deň (> 9-násobok klinickej expozície dosiahnutej pri dávke 10 mg/deň). Vplyv ambrisentanu na mužskú fertilitu nie je známy.
Zistilo sa, že ambrisentan mal teratogénne účinky u potkanov a králikov. Abnormality dolnej čeľuste, jazyka a/alebo podnebia boli pozorované pri všetkých skúšaných dávkach. V štúdii na potkanoch sa okrem toho preukázal zvýšený výskyt defektov interventrikulárneho septa, defektov v oblasti ciev trupu, abnormalít štítnej žľazy a týmu, osifikácie spodinovoklinovej kosti a výskyt umbilikálnej artérie lokalizovanej na ľavej strane močového mechúra namiesto na pravej strane. Teratogenita je suspektným skupinovým účinkom ERA.
Podávanie ambrisentanu samiciam potkanov v neskorom štádiu brezivosti a počas obdobia laktácie viedlo k nežiaducim účinkom na správanie samíc, k zníženému prežívaniu mláďat a k poruche reprodukčnej schopnosti potomkov (pri pitve sa zistili malé semenníky), a to pri expozícii 3-násobne prevyšujúcej hodnotu AUC dosiahnutú u ľudí po podaní maximálnej odporúčanej dávky.
U juvenilných potkanov, ktorým bol ambrisentan podávaný perorálne jedenkrát denne v postnatálnom období od 7. do 26., 36. alebo 62. dňa (čo zodpovedá novorodencom až obdobiu neskorého dospievania u ľudí), sa vyskytlo zníženie hmotnosti mozgu (-3 % až -8 %) bez morfologických alebo neurobehaviorálnych zmien po tom, ako boli pozorované abnormálne zvuky pri dýchaní, apnoe
a hypoxia. Tieto účinky sa vyskytli pri AUC hodnotách 1,8- až 7-násobne vyšších, ako je expozícia dosiahnutá pri 10 mg u pediatrických pacientov u ľudí. V inej štúdii, kde bol liek podávaný 5-
týždňovým potkanom (čo zodpovedá veku približne 8 rokov u ľudí), bolo zníženie hmotnosti mozgu
pozorované len pri veľmi vysokej dávke len u samcov. Dostupné predklinické údaje neumožňujú
pochopiť klinický význam tohto zistenia u detí mladších ako 8 rokov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
monohydrát laktózy
mikrokryštalická celulóza sodná soľ kroskarmelózy stearát horečnatý
Filmový obal
Volibris 2,5 mg filmom obalené tablety
polyvinylalkohol mastenec
oxid titaničitý (E171)
makrogol
lecitín (sójový) (E322)
Volibris 5 mg a 10 mg filmom obalené tablety
polyvinylalkohol mastenec
oxid titaničitý (E171)
makrogol
lecitín (sójový) (E322)
hlinitý lak červene allura AC (E129)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Volibris 2,5 mg filmom obalené tablety
2 roky
Volibris 5 mg a 10 mg filmom obalené tablety
5 rokov
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
V
olibris 2,5 mg filmom obalené tablety
Fľašky z nepriehľadného, bieleho polyetylénu s vysokou hustotou (HDPE) uzavreté uzávermi
z polypropylénu s detskou bezpečnostnou poistkou s polyetylénovým tepelne zapečatým prstencom.
Fľašky obsahujú 30 filmom obalených tabliet.
Volibris 5 mg a 10 mg filmom obalené tabletyPVC/PVDC/hliníkové fóliové blistre.
Veľkosti balenia obsahujúce blistre s jednotlivou dávkou po 10×1 alebo 30×1 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
8. REGISTRAČNÉ ČÍSLAVolibris 2,5 mg filmom obalené tabletyEU/1/08/451/005
Volibris 5 mg filmom obalené tabletyEU/1/08/451/001
EU/1/08/451/002
Volibris 10 mg filmom obalené tabletyEU/1/08/451/003
EU/1/08/451/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 21. apríla 2008
Dátum posledného predĺženia registrácie: 14. januára 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.