
kreatitídy. Pacientov je potrebné informovať o charakteristických príznakoch
akútnej pankreatitídy, ktorými sú pretrvávajúce intenzívne bolesti brucha, ktoré môžu vystreľovať do
oblasti chrbta. V prípade podozrenia na pankreatitídu je nutné ukončiť podávanie Vipdometu.
V prípade potvrdenia akútnej pankreatitídy sa podávanie Vipdometu nesmie obnoviť. U pacientov s pankreatitídou v minulosti je nutné postupovať opatrne.
Účinky napečeň
Po uvedení lieku na trh boli hlásené prípady dysfunkcie pečene vrátane zlyhania pečene. Kauzálny
súvis doteraz nebol stanovený. Pacienti musia byť dôkladne sledovaní vzhľadom na výskyt možných abnormalít pečene. U pacientov s podozrením na symptómy poškodenia pečene treba urýchlene
vykonať testy funkcie pečene. Ak sa zistí akákoľvek abnormalita a nestanoví sa žiadna alternatívna
etiológia, zvážte prerušenie liečby alogliptínom.
4.5 Liekové a iné interakcie
Súbežné podávanie 100 mg alogliptínu raz denne a 1000 mg metformín hydrochloridu dvakrát denne po dobu 6 dní nemalo u zdravých účastníkov žiadne klinicky významné účinky na farmakokinetické vlastnosti alogliptínu ani metformínu.
Špecifické farmakokinetické interakčné štúdie liečiv s Vipdometom neboli vykonané. Nasledujúca časť uvádza interakcie pozorované u jednotlivých zložiek Vipdometu (alogliptín/metformín), ako sú uvedené v ich príslušných súhrnoch charakteristických vlastností lieku.
Možné účinky iných liekovnaalogliptín
Alogliptín sa primárne vylučuje nezmenený v moči a jeho metabolizmus enzýmovým systémom
cytochrómu (CYP) P450 je zanedbateľný (pozri časť 5.2). Interakcie s inhibítormi CYP sa preto neočakávajú a neboli ani preukázané.
Výsledky klinických interakčných štúdií tiež nepreukázali klinicky významné účinky gemfibrozilu
(inhibítor CYP2C8/9), flukonazolu (inhibítor CYP2C9), ketokonazolu (inhibítor CYP3A4),
cyklosporínu (inhibítor P-glykoproteínu), voglibózy (inhibítor alfa-glukozidázy), digoxínu, metformínu, cimetidínu, pioglitazónu alebo atorvastatínu na farmakokinetické vlastnosti alogliptínu.
Možné účinky alogliptínu nainélieky
Štúdie in vitro ukazujú, že alogliptín neinhibuje ani neindukuje izoformy CYP 450 pri koncentráciách
dosiahnutých s odporúčanou dávkou 25 mg alogliptínu (pozri časť 5.2). Interakcie so substrátmi izoforiem CYP 450 sa preto neočakávajú a neboli ani preukázané. V štúdiách in vitro nebol alogliptín
potvrdený ani ako substrát, ani ako inhibítor kľúčových transportérov spojených s metabolizáciou liečiva v príslušnej obličke: organického aniónového transportéra-1, organického aniónového
transportéra-3 ani organického katiónového transportéra-2 (OCT2). Klinické údaje ďalej nepoukazujú na interakciu s inhibítormi ani substrátmi P-glykoproteínu.
Klinické štúdie alogliptínu nepotvrdili žiadne klinicky významné účinky na farmakokinetické vlastnosti kofeínu, (R)–warfarínu, pioglitazónu, glyburidu, tolbutamidu, (S)–warfarínu, dextrometorfánu, atorvastatínu, midazolamu, perorálnej antikoncepcie (noretisterónu a etinylestradiolu), digoxínu, fexofenadínu, metformínu ani cimetidínu, čo poskytuje in vivo dôkaz o slabej tendencii k vyvolaniu interakcií so substrátmi CYP1A2, CYP3A4, CYP2D6, CYP2C9, P- glykoproteínu a OCT2.
Alogliptín nemá u zdravých účastníkov pri súbežnom podaní s warfarínom žiadny účinok na protrombínový čas ani na medzinárodný normalizovaný pomer (INR).
Kombinácia alogliptínusinýmiantidiabetikami
Výsledky štúdií s metformínom, pioglitazónom (tiazolidíndiónom), voglibózou (inhibítor alfa-
glukozidázy) a glyburidom (sulfonylureou) nepreukázali žiadne klinicky významné farmakokinetické interakcie.
Interakcie s metformínom
Kombinácie,ktorésaneodporúčajú
Alkohol
Liečivo metformín spôsobuje zvýšené riziko vzniku laktátovej acidózy pri akútnej intoxikácii alkoholom (najmä pri hladovaní, podvýžive alebo poruchách funkcie pečene) (pozri časť 4.4). Je
potrebné vyhýbať sa požívaniu alkoholu a užívaniu liekov obsahujúcich alkohol.
Katiónové lieky
Pri katiónových liečivách, ktoré sú eliminované obličkovou tubulárnou sekréciou (napr. cimetidín), môže dochádzať k interakcii s metformínom spôsobenej kompetíciou o spoločné obličkové tubulárne
transportné systémy. Štúdia so siedmimi zdravými dobrovoľníkmi ukázala, že cimetidín (400 mg
dvakrát denne) zvyšoval systémovú expozíciu metformínu (AUC) o 50 % a Cmax o 81 %. Preto je potrebné zvážiť starostlivé monitorovanie hladiny glykémie, úpravu dávky v rámci odporúčaného dávkovania a zmeny liečby diabetu, keď sa súbežne podávajú katiónové lieky, ktoré sa eliminujú obličkovou tubulárnou sekréciou.
Jódované kontrastné látky
Intravaskulárne podanie jódovaných kontrastných látok môže viesť k zlyhaniu obličiek, čo môže mať za následok akumuláciu metformínu s rizikom vzniku laktátovej acidózy. Liečba Vipdometom sa
preto pred testom a počas testu musí ukončiť a nemá sa začať skôr ako 48 hodín po ňom a len vtedy, ak opätovné vyšetrenie obličiek potvrdilo ich normálnu funkciu (pozri časť 4.4).
K
ombinácie,
priktorýchsavyžadujúbezpečnostnéopatrenia
Lieky s vlastným hyperglykemizujúcim účinkom
Glukokortikoidy (podávané systémovo a lokálne), beta-2-agonisty a diuretiká (pozri tiež časť 4.4) majú vlastný hyperglykemizujúci účinok. Pacienta je o tomto potrebné informovať a častejšie u neho monitorovať hladinu glukózy v krvi, hlavne na začiatku liečby týmito liekmi. Ak je to potrebné, dávkovanie Vipdometu je možné upraviť počas súbežnej liečby s iným liekom a po jej ukončení.
Inhibítory ACE
Inhibítory enzýmu konvertujúceho angiotenzín (ACE) môžu znižovať hladinu glukózy v krvi. Ak je to potrebné, dávkovanie Vipdometu je možné upraviť počas súbežnej liečby s iným liekom a po jej
ukončení.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití Vipdometu u gravidných žien. Štúdie kombinovanej liečby
alogliptínom a metformínom na gravidných potkanoch preukázali reprodukčnú toxicitu u približne
5 až 20-násobku (pre metformín a alogliptín, v uvedenom poradí) expozície u ľudí pri odporúčanej dávke.
Vipdomet sa nesmie užívať počas gravidity.
Riziko spojenésalogliptínom
Nie sú k dispozícii údaje o použití alogliptínu počas gravidity. Štúdie na zvieratách nepreukázali
priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Riziko spojenésmetformínom
Obmedzené množstvo údajov o použití metformínu počas gravidity nepoukazuje na zvýšené riziko
kongenitálnych abnormalít. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity pri klinicky významných dávkach (pozri časť 5.3).
Laktácia
Neboli vykonané žiadne štúdie kombinácie liečiv Vipdometu na laktujúcich zvieratách. V štúdiách
jednotlivých liečiv sa alogliptín, ako aj metformín, vylučovali do mlieka laktujúcich potkanov. Nie je známe, či sa alogliptín vylučuje do ľudského mlieka. Metformín sa vylučuje do ľudského mlieka v
malých množstvách. Riziko u dojčiat nemôže byť vylúčené.
Rozhodnutie o prerušení dojčenia alebo o ukončení/prerušení liečby Vipdometom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinok Vipdometu na ľudskú fertilitu nebol študovaný. Štúdie alogliptínu alebo metformínu na
zvieratách nepreukázali žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Vipdomet nemá žiadny alebo má len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov treba upozorniť na riziko hypoglykémie, najmä pri jeho užívaní v kombinácii s inzulínom alebo pioglitazónom.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Klinické štúdie vykonané na podporu účinnosti a bezpečnosti Vipdometu obsahovali súbežné
podávanie alogliptínu a metformínu ako samostatných tabliet. Výsledky bioekvivalenčných štúdií však preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné s príslušnými dávkami
alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Poskytnuté informácie pochádzajú celkovo od 7150 pacientov s ochorením diabetes mellitus 2. typu vrátane 4201 pacientov liečených alogliptínom a metformínom, ktorí sa zúčastnili 3. etapy 7 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií. Tieto štúdie hodnotili účinky súbežne podávaného alogliptínu a metformínu na hodnotu glykémie a ich bezpečnosť ako úvodnej kombinovanej liečby, ako dvojkombinovanej liečby pacientov pôvodne liečených samotným metformínom a ako prídavnej liečby k tiazolidíndiónu alebo inzulínu.
Bezpečnostný profil súbežne podávaného alogliptínu a metformínu bol konzistentný s profilom jednotlivých zložiek, ako preukázali klinické skúšky alogliptínu a komplexné údaje dostupné pre metformín. Nasledujúca časť uvádza nežiaduce reakcie jednotlivých zložiek Vipdometu (alogliptín/metformín), ako sú uvedené v ich príslušných súhrnoch charakteristických vlastností lieku.
Alogliptín
Poskytnuté informácie pochádzajú celkovo od 9405 pacientov s ochorením diabetes mellitus 2. typu vrátane 3750 pacientov liečených 25 mg alogliptínu a 2476 pacientov liečených 12,5 mg alogliptínu, ktorí sa zúčastnili 2. etapy jednej alebo 3. etapy 12 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií. Okrem toho sa uskutočnila štúdia kardiovaskulárnych výsledkov
s 5380 pacientmi s ochorením diabetes mellitus 2. typu a nedávnym akútnym koronárnym syndrómom, z ktorých 2701 pacientov bolo randomizovaných do skupiny s alogliptínom
a 2679 pacientov do skupiny s placebom. Tieto štúdie hodnotili účinky alogliptínu na kontrolu
glykémie a jeho bezpečnosť ako monoterapie, ako úvodnej kombinovanej liečby s metformínom alebo tiazolidíndiónom a ako prídavnej liečby k metformínu alebo k sulfonylurey, alebo k tiazolidíndiónu
(s metformínom alebo sulfonylureou alebo bez nich), alebo k inzulínu (s metformínom alebo bez neho).
V súhrnnej analýze údajov z 13 štúdií boli celkové incidencie nežiaducich účinkov, závažných nežiaducich účinkov a nežiaducich účinkov vedúcich k ukončeniu liečby porovnateľné u pacientov liečených 25 mg alogliptínu, 12,5 mg alogliptínu, aktívnou kontrolou alebo placebom.
Najčastejšou nežiaducou reakciou u pacientov liečených 25 mg alogliptínu bola bolesť hlavy. Bezpečnosť alogliptínu v rámci starších pacientov (≥ 65 rokov) a mladších ako starších pacientov
(< 65 rokov) bola na podobnej úrovni.
Tabuľkový zoznamnežiaducichreakcií
Tieto nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a frekvencie. Frekvencie sú
definované ako veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1 /10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000); neznáme (nedá sa určiť z dostupných údajov).
Alogliptín
Počas súhrnných kontrolovaných pivotných klinických skúšok fázy 3 boli v prípade alogliptínu ako monoterapie a ako prídavnej kombinovanej liečby zahŕňajúcej 5659 pacientov pozorované nežiaduce reakcie, ktoré sú uvedené nižšie (Tabuľka 1).
T
abuľka 1: Nežiaduce reakcie pozorované pri súhrnných pivotných klinických skúškach fázy 3
T
rieda orgánových systémov Frekvencia nežiaducich reakcií
Nežiaduca reakcia
Infekcie a nákazy
Infekcia horných dýchacích ciest Časté
Zápal nosohltana Časté
Poruchy nervového systému
Bolesť hlavy Časté
Poruchy gastrointestinálneho traktu
Bolesť brucha Časté
Gastroezofageálny reflux Časté
Poruchy kože a podkožného tkaniva
Pruritus Časté
Vyrážka Časté
Alogliptín/metformín
Počas súhrnných kontrolovaných pivotných klinických skúšok fázy 3 boli v prípade alogliptínu ako prídavnej kombinovanej liečby s metformínom, zahŕňajúcej 7151 pacientov, pozorované nežiaduce reakcie, ktoré sú uvedené nižšie (Tabuľka 2).
Tabuľka 2: Nežiaduce reakcie pozorované pri kontrolovaných pivotných klinických skúškach fázy 3
T
rieda orgánových systémov
Nežiaduca reakcia
Infekcie a nákazy
Infekcia horných dýchacích ciest
Zápal nosohltana
Poruchy nervového systému
Frekvencia nežiaducich reakcií
Časté
Časté
Bolesť hlavy Časté
Poruchy gastrointestinálneho traktu
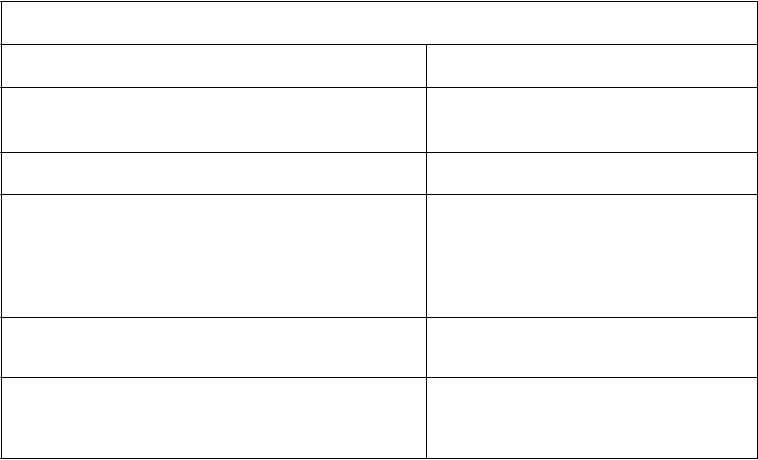
Gastroenteritída Bolesť brucha Hnačka Vracanie Gastritída
Gastroezofageálny reflux
Poruchy kože a podkožného tkanivaPruritus
Vyrážka
Časté Časté Časté Časté Časté Časté
Časté
Časté
Alogliptín
Skúsenosti
po
uvedení
na
trh
V Tabuľke 3 sú uvedené dodatočné nežiaduce reakcie, ktoré boli spontánne hlásené po uvedení lieku
na trh.
Tabuľka 3: Spontánne hlásené nežiaduce reakcie na alogliptín po jeho uvedení na trh
T
rieda orgánových systémov
Nežiaduca reakcia
Poruchy imunitného systému
Frekvencia nežiaducich reakcií
Precitlivenosť Neznáme
Poruchy gastrointestinálneho systému
Akútna pankreatitída Neznáme
Poruchy hepatobiliárneho systému
Dysfunkcia pečene vrátane zlyhania pečene Neznáme
Poruchy kože a podkožného tkaniva
Exfoliatívne kožné choroby vrátane
Stevensovho-Johnsonovho syndrómu
Multiformý erytém
Neznáme
Neznáme
Angioedém Neznáme
Urtikária Neznáme
Metformín
Údajezklinickýchskúšokaskúsenostipouvedenínatrh
V Tabuľke 4 sú uvedené dodatočné nežiaduce reakcie, ktoré boli hlásené v rámci klinických skúšok a
po uvedení lieku na trh.
Tabuľka 4: Frekvencia nežiaducich reakcií na metformín identifikovaných údajmi z klinických skúšok a skúsenosťami po uvedení na trh
T
rieda orgánových systémov
Nežiaduca reakcia
Poruchy metabolizmu a výživy
Frekvencia nežiaducich reakcií
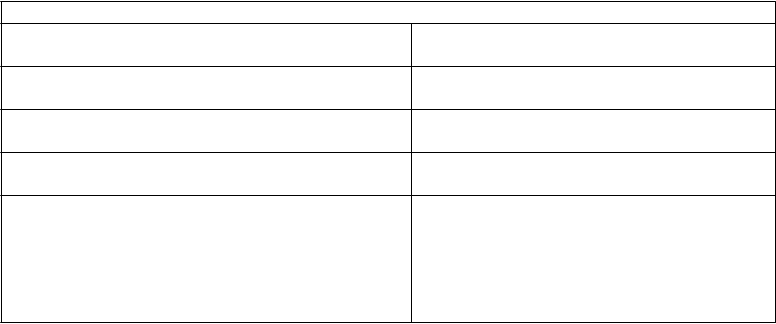
Laktátová acidóza Veľmi zriedkavé
Deficit vitamínu B12 Veľmi zriedkavé
Poruchy nervového systémuKovová chuť Časté
Poruchy gastrointestinálneho traktuBolesť brucha Veľmi časté
Hnačka Veľmi časté Strata chuti do jedla Veľmi časté Nevoľnosť Veľmi časté Zvracanie Veľmi časté
Poruchy pečene a žlčových ciestHepatitída Veľmi zriedkavé
Abnormality v testoch funkcie pečene Veľmi zriedkavé
Poruchy kože a podkožného tkanivaErytém Veľmi zriedkavé Pruritus Veľmi zriedkavé Urtikária Veľmi zriedkavé
PopisvybranýchnežiaducichreakciíLaktátová acidóza: 0,03 prípadov/1000 pacientorokov, pozri časť 4.4.
U pacientov dlhodobo liečených metformínom sa pozorovala znížená absorpcia vitamínu B12, ktorá vo všeobecnosti nemá klinický význam. Veľmi zriedkavo však môže viesť ku klinicky významnému deficitu vitamínu B12 (napr. megaloblastickej anémii).
Gastrointestinálne príznaky sa vyskytujú najčastejšie na začiatku liečby a vo väčšine prípadov spontánne vymiznú. Aby sa im zabránilo, odporúča sa užívať metformín v 2 denných dávkach počas jedla alebo po ňom.
Boli hlásené ojedinelé prípady hepatitídy alebo abnormalít v testoch funkcie pečene, ktoré ustúpili po vysadení metformínu.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie sú k dispozícii žiadne údaje týkajúce sa predávkovania Vipdometom.
AlogliptínNajvyššie dávky alogliptínu podané počas klinických skúšok boli jednorazové dávky 800 mg zdravým
účastníkom a 400 mg raz denne počas 14 dní pacientom s ochorením diabetes mellitus 2. typu (čo je ekvivalentné 32-násobku a 16-násobku odporúčanej celkovej dennej dávky 25 mg alogliptínu,
v uvedenom poradí).
MetformínVeľké predávkovanie metformínom alebo sprievodné riziká môžu viesť k laktátovej acidóze.
Laktátová acidóza je z medicínskeho hľadiska naliehavý prípad a musí sa liečiť v nemocnici.
Postup pri predávkovaníV prípade predávkovania je nutné vykonať vhodné podporné opatrenia v súlade s klinickým stavom
pacienta.
Hemodialýzou je možné odstrániť minimálne množstvá alogliptínu (počas 3-hodinovej hemodialýzy bolo odstránených približne 7 % tejto látky). Preto má hemodialýza pri predávkovaní len malý klinický prínos. Možnosť odstránenia alogliptínu peritoneálnou dialýzou nie je známa.
Najúčinnejšou metódou odstránenia laktátu a metformínu je hemodialýza.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antidiabetiká; biguanidy a sulfónamidy v kombinácii. ATC kód: A10BD13.
Mechanizmus účinku afarmakodynamickéúčinky
Vipdomet kombinuje dve hypoglykemicky účinné látky so vzájomne sa dopĺňajúcimi rozdielnymi
mechanizmami účinku na zlepšenie kontroly glykémie u pacientov s ochorením diabetes mellitus 2. typu: alogliptín, inhibítor dipeptidylpeptidázy-4 (DPP-4), a metformín, člen biguanidovej skupiny.
Alogliptín
Alogliptín je potentný a vysoko selektívny inhibítor DPP-4, > 10 000-krát selektívnejší pre DPP-4 ako
ostatné súvisiace enzýmy vrátane DPP-8 a DPP-9. DPP-4 je hlavný enzým vyvolávaný počas rapídnej degradácie inkretínových hormónov, glukagónu podobného peptidu 1 (GLP-1) a GIP (glukózo-
dependentný inzulínotropný polypeptid), ktoré sú uvoľňované črevami a ich hladiny stúpajú v rámci
odozvy na jedlo. GLP-1 a GIP zvyšujú biosyntézu inzulínu a sekréciu od pankreatických beta buniek, zatiaľ čo GLP-1 tiež inhibuje sekréciu glukagónu a tvorbu glukózy v pečeni. Alogliptín preto zlepšuje
glykemickú kontrolu pomocou glukózo-dependentného mechanizmu, nakoľko zlepšuje uvoľňovanie
inzulínu a pri zvýšených hladinách glukózy bráni zvyšovaniu hladín glukagónu.
Metformín
Metformín je biguanid s antidiabetickými účinkami, ktorý znižuje bazálnu aj postprandiálnu hladinu
glukózy v plazme. Nestimuluje sekréciu inzulínu, a preto nevyvoláva hypoglykémiu. Účinok metformínu môže mať 3 mechanizmy:
- zníženie tvorby glukózy v pečeni inhibíciou glukoneogenézy a glykogenolýzy.
- mierne zvyšovanie citlivosti na inzulín vo svaloch, zlepšovanie periférneho vychytávania a utilizácie glukózy.
- spomalenie intestinálnej absorpcie glukózy.
Metformín stimuluje intracelulárnu syntézu glykogénu účinkom na glykogénsyntázu. Taktiež zvyšuje transportnú kapacitu špecifických typov transportérov glukózy cez membrány (GLUT-1 a GLUT-4).
U ľudí má metformín priaznivé účinky na metabolizmus lipidov nezávisle od jeho pôsobenia na glykémiu. Ukázalo sa to pri terapeutických dávkach v kontrolovaných strednodobých alebo dlhodobých klinických štúdiách: metformín znižuje hladiny celkového cholesterolu, LDL cholesterolu a triglyceridov.
Klinická účinnosť
Klinické štúdie vykonané na podporu účinnosti Vipdometu obsahovali súbežné podávanie alogliptínu
a metformínu ako samostatných tabliet. Výsledky bioekvivalenčných štúdií však preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné s príslušnými dávkami alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Tieto štúdie hodnotili účinky súbežne podávaného alogliptínu a metformínu ako dvojkombinovanej liečby pacientov pôvodne liečených samotným metformínom a ako prídavnej liečby k tiazolidíndiónu alebo inzulínu.
Podanie 25 mg alogliptínu pacientom s ochorením diabetes mellitus 2. typu vyvolalo vrcholovú inhibíciu DPP-4 do 1 až 2 hodín a presiahlo 93 % po jednorazovej dávke 25 mg, ako aj po 14 dňoch podávania raz denne. Inhibícia DPP-4 zostala nad úrovňou 81 % 24 hodín po 14 dňoch podávania. Keď sa vypočítal priemer postprandiálnych koncentrácií glukózy 4 hodiny po raňajkách, obede
a večeri, výsledkom 14-dňovej liečby 25 mg alogliptínu bolo stredné, placebom kontrolované zníženie oproti prvej návšteve o -35,2 mg/dl.
Alogliptín v samostatnej dávke 25 mg, ako aj v kombinácii s 30 mg pioglitazónu, preukazoval významné zníženia postprandiálnej hladiny glukózy a postprandiálneho glukagónu, zatiaľ čo
významne zvyšoval postprandiálne hladiny aktívneho GLP-1 pri návšteve v týždni 16 v porovnaní s placebom (p < 0,05). Okrem toho vyvolal alogliptín v samostatnej dávke 25 mg, ako aj v kombinácii s
30 mg pioglitazónu, štatisticky významné (p < 0,001) zníženia celkových triglyceridov pri návšteve v
týždni 16 namerané podľa postprandiálnej prírastkovej zmeny AUC(0-8) z prvej návštevy v porovnaní s placebom.
Celkovo 7151 pacientov s ochorením diabetes mellitus 2. typu vrátane 4202 pacientov liečených alogliptínom a metformínom sa zúčastnilo 3. etapy 7 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií vykonaných za účelom hodnotenia účinkov súbežného podávania alogliptínu a metformínu na kontrolu glykémie a ich bezpečnosť. V týchto štúdiách bolo
696 pacientov liečených alogliptínom/metformínom vo veku ≥ 65 rokov.
Celkovo povedané, odporúčaná celková denná dávka 25 mg alogliptínu v kombinácii s metformínom zlepšila glykemickú kontrolu. Toto sa potvrdilo klinicky a štatisticky významnými zníženiami hladiny glykovaného hemoglobínu (HbA1c) a plazmatickej glukózy nalačno v porovnaní s kontrolou od prvej až po poslednú návštevu štúdie. Zníženia hladiny HbA1c boli podobné medzi rôznymi podskupinami vrátane ochorenia obličiek, veku, pohlavia a indexu telesnej hmotnosti, zatiaľ čo rozdiely medzi
rasami (napr. belosi a nebelosi) boli malé. Klinicky významné zníženia hladiny HbA1c v porovnaní s kontrolou boli tiež pozorované bez ohľadu na úvodnú základnú liečbu. Vyššia hladina HbA1c pri
prvej návšteve bola spojená s výraznejším znížením hladiny HbA1c. Účinky alogliptínu na telesnú
hmotnosť a lipidy boli vo všeobecnosti neutrálne.
Alogliptín ako prídavná liečba k metformínu
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe metformínom vo forme hydrochloridu (stredná hodnota dávky = 1847 mg) boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a plazmatickej glukózy nalačno pri návšteve v týždni 26 pri porovnaní s pridaním placeba (Tabuľka 5). Významne viac pacientov užívajúcich 25 mg alogliptínu (44,4 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (18,3 %) v týždni 26
(p < 0,001).
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe metformínom vo forme hydrochloridu (stredná hodnota dávky = 1835 mg) boli nepretržité zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c pri návšteve v týždni 52 a týždni 104. Pri návšteve v týždni 52 bolo zníženie hladiny HbA1c liečbou 25 mg alogliptínu spolu s metformínom (-0,76 %, tabuľka 6) podobné zníženiu vyvolanému liečbou glipizidom (stredná hodnota dávky = 5,2 mg) spolu s metformínom vo forme hydrochloridu (stredná hodnota dávky = 1824 mg, 0,73 %). Pri návšteve v týždni 104 bolo zníženie hladiny HbA1c liečbou 25 mg alogliptínu spolu s metformínom (-0,72 %, tabuľka 6) vyššie ako zníženie vyvolané liečbou glipizidom spolu s metformínom (-0,59 %). Stredná hodnota zmeny od prvej návštevy
v súvislosti s koncentráciou plazmatickej glukózy nalačno pri návšteve v týždni 52 pre 25 mg alogliptínu bola signifikantne vyššia ako pri liečbe glipizidom a metformínom (p < 0,001). V týždni 104, stredná hodnota zmeny od prvej návštevy v súvislosti s koncentráciou plazmatickej glukózy nalačno pre 25 mg alogliptínu a metformínu bola -3,2 mg/dl v porovnaní s 5,4 mg/dl pri liečbe glipizidom a metformínom. Viac pacientov užívajúcich 25 mg alogliptínu a metformínu (48,5 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi glipizid
a metformín (42,8 %) (p = 0,004).
Súbežné podávanie 12,5 mg alogliptínu a 1000 mg metformín hydrochloridu dvakrát denne viedlo k štatisticky významným zlepšeniam od prvej návštevy, čo sa týka hladiny HbA1c a plazmatickej glukózy nalačno pri návšteve v týždni 26 pri porovnaní s podávaním 12,5 mg samotného alogliptínu dvakrát denne alebo s podávaním 1000 mg samotného metformín hydrochloridu dvakrát denne. Významne viac pacientov užívajúcich 12,5 mg alogliptínu a 1000 mg metformín hydrochloridu dvakrát denne (59,5 %) dosiahlo cieľové hladiny HbA1c < 7,0 % v porovnaní s pacientmi užívajúcimi
12,5 mg samotného alogliptínu dvakrát denne (20,2 %, p < 0,001) alebo 1000 mg samotného metformín hydrochloridu dvakrát denne (34,3 %, p < 0,001) v týždni 26.
Alogliptín ako prídavná liečba k metformínu s tiazolidíndiónom
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe pioglitazónom (stredná hodnota
dávky = 35,0 mg, s metformínom alebo sulfonylureou alebo bez nich), boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a FPG pri návšteve v týždni 26 pri porovnaní
s pridaním placeba (Tabuľka 5). Klinicky významné zníženia hladiny HbA1c v porovnaní s placebom
boli tiež pozorované u 25 mg alogliptínu bez ohľadu na to, či pacienti súbežne užívali metformín alebo sulfonylureu. Významne viac pacientov užívajúcich 25 mg alogliptínu (49,2 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (34,0 %) v týždni 26 (p = 0,004).
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe 30 mg pioglitazónu v kombinácii
s metformín hydrochloridom (stredná hodnota dávky = 1867,9 mg) boli zlepšenia od prvej návštevy,
čo sa týka hladiny HbA1c pri návšteve v týždni 52, ktoré boli neinferiórne ako aj štatisticky superiórne zlepšenia vyvolané liečbou 45 mg pioglitazónu v kombinácii s metformín hydrochloridom (stredná hodnota dávky = 1847,6 mg, Tabuľka 6). Významné zníženia hladiny HbA1c pozorované u 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu boli konzistentné počas celého obdobia liečby v trvaní 52 týždňov v porovnaní so 45 mg pioglitazónu a metformínu (p < 0,001 vo všetkých časových bodoch). Okrem toho bola stredná hodnota zmeny od prvej návštevy v súvislosti s FPG pri návšteve v týždni 52 pre 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu významne väčšia ako zmena
pri liečbe 45 mg pioglitazónu a metformínu (p < 0,001). Významne viac pacientov užívajúcich 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu (33,2 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi 45 mg pioglitazónu a metformínu (21,3 %) v týždni 52 (p < 0,001).
Alogliptín ako prídavná liečba k metformínu s inzulínom
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe inzulínom (stredná hodnota
dávky = 56,5 IU, s metformínom alebo bez neho) boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a FPG pri návšteve v týždni 26 pri porovnaní s pridaním placeba
(Tabuľka 5). Klinicky významné zníženia hladiny HbA1c v porovnaní s placebom boli tiež pozorované u 25 mg alogliptínu bez ohľadu na to, či pacienti súbežne užívali metformín. Viac
pacientov užívajúcich 25 mg alogliptínu (7,8 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (0,8 %) v týždni 26.
T
abuľka 5: Zmena v hladine HbA1c (%) od prvej návštevy s 25 mg alogliptínu v týždni 26
v placebom kontrolovanej štúdii (FAS, LOCF)
Štúdia Stredná hodnota
H
bA1c pri prvej návšteve (%) (štandardná odchýlka (SD))
Stredná hodnota zmeny HbA1c od prvej návštevy (%)† (štandardná chyba priemeru (SE))
Placebom korigovaná zmena HbA1c od prvej návštevy (%)†
(2-stranný
95% interval spoľahlivosti (CI))
Placebom kontrolované štúdie prídavnej kombinovanej liečby
Alogliptín 25 mg raz denne s metformínom
(n = 203)
Alogliptín 25 mg raz denne so sulfonylureou
(n = 197)
Alogliptín 25 mg raz denne s tiazolidíndiónom ± metformín alebo sulfonylurea
(n = 195)
Alogliptín 25 mg raz denne s inzulínom ± metformín
(n = 126)
7,93 (0,799)
8,09 (0,898)
8,01 (0,837)
9,27 (1,127)
-0,59 (0,054)
-0,52 (0,058)
-0,80 (0,056)
-0,71 (0,078)
-0,48*
(-0,67, -0,30)
-0,53*
(-0,73, -0,33)
-0,61*
(-0,80, -0,41)
-0,59*
(-0,80, -0,37)
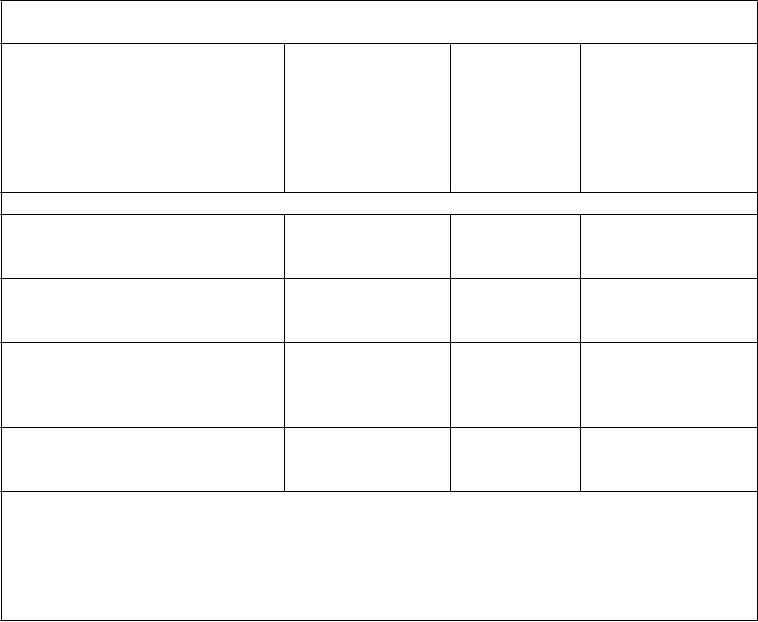
FAS = úplná populácia pacientov randomizovaných do liečby, ktorí prijali aspoň jednu dávku skúšaného lieku (full analysis set)
LOCF = použitie poslednej pozorovanej hodnoty na mieste chýbajúcej (last observation carried forward)
† Metóda najmenších štvorcov prispôsobená na stav pred antihyperglykemickou liečbou a hodnoty pri prvej návšteve
* p < 0.001 porovnané s placebom kontrolovanou liečbou alebo kombinovanou liečbou + placebo
T
abuľka 6: Zmena v hladine HbA1c (%) od prvej návštevy s 25 mg alogliptínu v aktívne kontrolovanej štúdii (PPS, LOCF)
Štúdia Stredná hodnota HbA1c pri prvej návšteve (%) (štandardná odchýlka
(
SD))
Štúdie prídavnej kombinovanej liečby
Alogliptín 25 mg raz denne s metformínom
v porovnaní so
sulfonylureou + metformín
Stredná hodnota zmeny HbA1c od
prvej návštevy (%)† (štandardná chyba
priemeru (SE))
Liečbou korigovaná zmena HbA1c od prvej návštevy (%)†
(1-stranný interval
spoľahlivosti (CI))
Zmena v týždni 52
(n = 382)
Zmena v týždni 104 (n = 382)
7,61 (0,526)
7,61 (0,526)
-0,76 (0,027)
-0,72 (0,037)
-0,03
(-nekonečno,
0,059)
-0,13*
(-nekonečno, -
0,006)
Alogliptín 25 mg raz denne
s tiazolidíndiónom +
metformín
v porovnaní s titrovaným tiazolidíndiónom + metformín
Zmena v týždni 26 (n = 303)
Zmena v týždni 52 (n = 303)
8,25 (0,820)
8,25 (0,820)
-0,89 (0,042)
-0,70 (0,048)
-0,47*
(-nekonečno, -0,35)
-0,42*
(-nekonečno, -0,28)
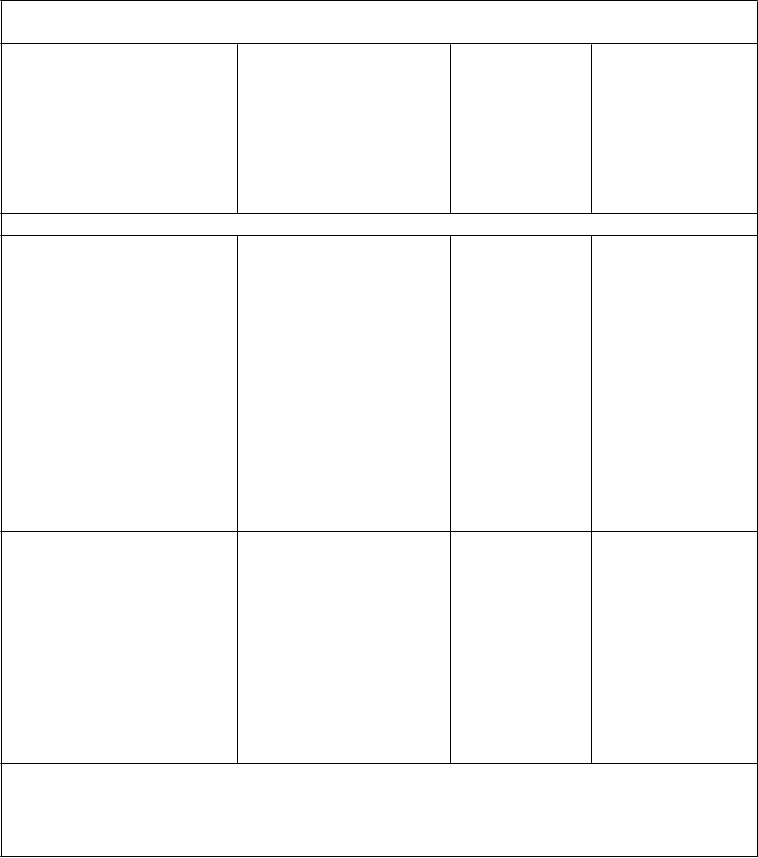
PPS = podskupina populácie pacientov spĺňajúcich kritériá protokolu (per protocol set)
LOCF = použitie poslednej pozorovanej hodnoty na mieste chýbajúcej (last observation carried forward)
* Štatisticky preukázaná neinferiorita a superiorita
† Metóda najmenších štvorcov prispôsobená na stav pred antihyperglykemickou liečbou a hodnoty pri prvej návšteve
Starší pacienti (≥ 65 rokov)Po kontrole účinnosti a bezpečnosti odporúčaných dávok alogliptínu a metformínu u podskupiny pacientov s ochorením diabetes mellitus 2. typu a vo veku ≥ 65 rokov sa zistila ich konzistentnosť
s profilom získaným u pacientov vo veku < 65 rokov.
Klinická bezpečnosťKardiovaskulárna bezpečnosťV súhrnnej analýze údajov z 13 štúdií boli celkové incidencie kardiovaskulárnej smrti, nefatálneho infarktu myokardu a nefatálnej mozgovej príhody porovnateľné u pacientov liečených 25 mg
alogliptínu, aktívnou kontrolou alebo placebom.
Okrem toho sa uskutočnila prospektívna randomizovaná štúdia kardiovaskulárnych výsledkov zameraná na bezpečnosť s 5380 pacientmi s vysokým základným kardiovaskulárnym rizikom, ktorej cieľom bolo preskúmať účinok alogliptínu v porovnaní s placebom (po pridaní k štandardnej
starostlivosti) na významné kardiovaskulárne nežiaduce udalosti (MACE) vrátane času do prvého výskytu akejkoľvek udalosti patriacej do skupiny zloženej z kardiovaskulárnej smrti, nefatálneho infarktu myokardu a nefatálnej mozgovej príhody u pacientov s nedávnou (15 až 90 dní) akútnou koronárnou udalosťou. Pri prvej návšteve mali pacienti stredný vek 61 rokov, stredné trvanie diabetu
9,2 roka a strednú hladinu HbA1c 8,0 %.


V štúdii sa preukázalo, že alogliptín nezvýšil riziko udalostí MACE v porovnaní s placebom [pomer rizika: 0,96; 1-stranný 99 % interval spoľahlivosti: 0 – 1,16]. V skupine s alogliptínom sa udalosť MACE vyskytla u 11,3 % pacientov v porovnaní s 11,8 % pacientov v skupine s placebom.
Tabuľka 7. Udalosti MACE hlásené v štúdii kardiovaskulárnych výsledkovPočet pacientov (%)Alogliptín25 mg Placebo
P
r
i
m
árny zložený koncový bod [prvá udalosť kardiovaskulárnej smrti, nefatálneho infarktu myokardu
a nefatálnej mozgovej príhody]
N = 2701 N = 2679
 305 (11,3) 316 (11,8)
305 (11,3) 316 (11,8)89 (3,3) 111 (4,1)
187 (6,9) 173 (6,5)
29 (1,1) 32 (1,2)
*Celkovo zomrelo (všetky príčiny) 153 pacientov (5,7 %) v skupine s alogliptínom a 173 pacientov (6,5 %) v skupine s placebom.
U 703 pacientov sa vyskytla udalosť zo sekundárnej zloženej skupiny koncových bodov MACE (prvá
udalosť kardiovaskulárnej smrti, nefatálneho infarktu myokardu, nefatálnej mozgovej príhody
a naliehavej revaskularizácie v dôsledku nestabilnej angíny). V skupine liečenej alogliptínom sa udalosť zo sekundárnej zloženej skupiny koncových bodov MACE vyskytla u 12,7 % (344) pacientov

v porovnaní s 13,4 % (359) pacientov v skupine s placebom [pomer rizika = 0,95; 1-stranný 99 %

interval spoľahlivosti: 0 – 1,14].
HypoglykémiaV súhrnnej analýze údajov z 12 štúdií bola celková incidencia akejkoľvek epizódy hypoglykémie nižšia u pacientov liečených 25 mg alogliptínu ako u pacientov liečených 12,5 mg alogliptínu, aktívnou kontrolou alebo placebom (3,6 %, 4,6 %, 12,9 % a 6,2 % v uvedenom poradí). Väčšina
týchto epizód mala miernu až stredne ťažkú intenzitu. Celková incidencia epizód ťažkej hypoglykémie bola porovnateľná u pacientov liečených 25 mg alogliptínu alebo 12,5 mg alogliptínu a nižšia ako
incidencia u pacientov liečených aktívnou kontrolou alebo placebom (0,1 %, 0,1 %, 0,4 % a 0,4 %
v uvedenom poradí). V prospektívnej randomizovanej kontrolovanej štúdii kardiovaskulárnych výsledkov skúšajúci uviedol, že incidencia udalostí hypoglykémie bola u pacientov užívajúcich
placebo (6,5 %) a pacientov užívajúcich alogliptín (6,7 %) ako doplnok štandardnej starostlivosti
podobná.
Počas klinickej skúšky alogliptínu ako monoterapie bola incidencia hypoglykémie podobná ako u placeba a nižšia ako u placeba v inej skúške alogliptínu ako prídavnej liečby k sulfonylurey.
Vyššia frekvencia hypoglykémie bola pozorovaná pri kombinovanej liečbe tromi liekmi s tiazolidíndiónom a metformínom a v kombinácii s inzulínom, ako pri iných inhibítoroch DPP-4.
Pacienti (≥ 65 rokov) s ochorením diabetes mellitus 2. typu sa považujú za náchylnejších na epizódy hypoglykémie ako pacienti vo veku < 65 rokov. V súhrnnej analýze údajov z 12 štúdií bola celková incidencia akejkoľvek hypoglykémie podobná u pacientov vo veku ≥ 65 rokov liečených 25 mg alogliptínu (3,8 %) ako incidencia u pacientov vo veku < 65 rokov (3,6 %).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Vipdomet vo
všetkých vekových podskupinách detí a dospievajúcich v liečbe ochorenia diabetes mellitus 2. typu
(pozri časť 4.2 pre informácie o používaní u detí a dospievajúcich).
5.2 Farmakokinetické vlastnosti
Výsledky bioekvivalenčných štúdií na zdravých účastníkoch preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné k príslušným dávkam alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Súbežné podávanie 100 mg alogliptínu raz denne a 1000 mg metformín hydrochloridu dvakrát denne po dobu 6 dní nemalo u zdravých účastníkov žiadne klinicky významné účinky na farmakokinetické vlastnosti alogliptínu ani metformínu.
Podávanie Vipdometu s jedlom neviedlo k zmene v celkovej expozícii (AUC) alogliptínu alebo metformínu. Stredné hodnoty vrcholových plazmatických koncentrácií alogliptínu a metformínu sa však znížili o 13 % a 28 %, v uvedenom poradí, ak sa Vipdomet podával s jedlom. U alogliptínu nedošlo k žiadnej zmene času dosiahnutia vrcholovej plazmatickej koncentrácie (Tmax), u metformínu však došlo k oneskoreniu Tmax o 1,5 hodiny. Klinická významnosť týchto zmien nie je pravdepodobná (pozri nižšie).
Vipdomet sa má užívať dvakrát denne z dôvodu farmakokinetických vlastností metformínovej zložky. Má sa tiež užívať s jedlom, aby sa znížili gastrointestinálne nežiaduce účinky spojené s metformínom (pozri časť 4.2).
Farmakokinetické vlastnosti Vipdometu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje (pozri časť 4.2).
Nasledujúca časť uvádza farmakokinetické vlastnosti jednotlivých zložiek Vipdometu
(alogliptín/metformín), ako sú uvedené v ich príslušných Súhrnoch charakteristických vlastností lieku.
Alogliptín
Farmakokinetické vlastnosti alogliptínu sú preukázateľne podobné u zdravých účastníkov
a u pacientov s ochorením diabetes mellitus 2. typu 2.
Absorpcia
Absolútna biodostupnosť alogliptínu je približne 100 %.
Podanie s jedlom s vysokým obsahom tuku nespôsobilo v celkovej a vrcholovej expozícii alogliptínu žiadne zmeny. Alogliptín sa preto môže podávať s jedlom alebo bez jedla.
Po podaní jednorazových perorálnych dávok až do 800 mg u zdravých účastníkov sa alogliptín rýchlo absorboval s výskytom vrcholových plazmatických koncentrácií 1 až 2 hodiny (stredná hodnota Tmax) po užití.
U zdravých účastníkov ani u pacientov s ochorením diabetes mellitus 2. typu nebola pozorovaná žiadna klinicky významná akumulácia po viacnásobnom podaní.
Celková a vrcholová expozícia alogliptínu sa primerane zvyšovala naprieč jednorazovými dávkami
6,25 mg až do 100 mg alogliptínu (pokrývajúcimi rozsah terapeutických dávok). Koeficient interindividuálnej variability (AUC) pre alogliptín bol nízky (17 %).
Distribúcia
Po podaní jednorazovej intravenóznej dávky 12,5 mg alogliptínu zdravým účastníkom bol distribučný
objem počas terminálnej fázy 417 l, čo indikuje dobrú distribúciu liečiva do tkanív.
Väzba alogliptínu na plazmatické bielkoviny je 20 – 30 %. Biotransformácia
Alogliptín nepodlieha rozsiahlemu metabolizmu – 60 – 70 % dávky sa vylúči močom ako nezmenený
liek. Po podaní perorálnej dávky [14C] alogliptínu boli zistené dva vedľajšie metabolity –
N-demetylovaný alogliptín, M-I (< 1 % pôvodnej látky) a N-acetylovaný alogliptín, M-II (< 6 % pôvodnej látky). M-I je aktívny metabolit a vysoko selektívny inhibítor DPP-4 podobný alogliptínu. M-II nevykazuje žiadnu inhibičnú aktivitu voči DPP-4 ani enzýmom súvisiacim s DPP. In vitro údaje indikujú, že CYP2D6 a CYP3A4 prispievajú k limitovanému metabolizmu alogliptínu.
In vitro štúdie dokazujú, že alogliptín pri koncentráciách dosiahnutých s odporúčanou dávkou 25 mg alogliptínu neindukuje CYP1A2, CYP2B6 a CYP2C9 a neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ani CYP3A4. Štúdie in vitro dokázali, že alogliptín mierne indukuje CYP3A4, avšak v štúdiách in vivo sa jeho indukovanie CYP3A4 nedokázalo.
V štúdiách in vitro sa alogliptín nepreukázal ako inhibítor týchto obličkových transportérov; OAT1, OAT3 a OCT2.
Alogliptín existuje prevažne ako (R)-enantiomér (> 99 %) a podlieha slabej alebo žiadnej chirálnej konverzii in vivo na (S)-enantiomér. (S)-enantiomér nie je pri terapeutických dávkach detekovateľný.
Eliminácia
Alogliptín bol eliminovaný so strednou hodnotou polčasu rozpadu (T1/2) približne 21 hodín.
Po podaní perorálnej dávky [14C] alogliptínu bolo 76 % celkovej rádioaktivity eliminovaných v moči a 13 % bolo zistených v stolici.
Priemerný renálny klírens alogliptínu (170 ml/min.) bol väčší ako priemerná odhadovaná rýchlosť glomerulárnej filtrácie (pribl. 120 ml/min.), čo naznačuje určitú aktívnu renálnu exkréciu.
Linearita
Celková expozícia (AUC(0-inf)) alogliptínu po podaní jednorazovej dávky bola podobná expozícii počas
intervalu jednej dávky (AUC(0-24)) po 6 dňoch dávkovania raz denne. Toto nenaznačuje časovú
závislosť v kinetike alogliptínu po viacnásobnom podaní.
Osobitné skupiny pacientov
Pacienti s poruchami funkcie obličiek
Jednorazová dávka 50 mg alogliptínu bola podaná 4 skupinám pacientov s rôznymi stupňami poruchy funkcie obličiek (klírens kreatinínu (CrCl) pomocou Cockcroftho a Gaultovho vzorca):
mierna (CrCl = > 50 až ≤ 80 ml/min.), stredne ťažká (CrCl = ≥ 30 až ≤ 50 ml/min.),
závažná (CrCl = < 30 ml/min.) a s ochorením obličiek v terminálnom štádiu na hemodialýze.
U pacientov s miernou poruchou funkcie obličiek bolo pozorované približne 1,7-násobné zvýšenie
AUC pre alogliptín. Avšak, keďže distribúcia hodnôt AUC pre alogliptín bola u týchto pacientov
v rámci rovnakého rozsahu ako u kontrolných účastníkov, úprava dávky alogliptínu u pacientov s miernou poruchou funkcie obličiek nie je nevyhnutná (pozri časť 4.2).
U pacientov so stredne ťažkou alebo závažnou poruchou funkcie obličiek, alebo s ochorením obličiek v terminálnom štádiu na hemodialýze bolo pozorované 2-násobné a 4-násobné zvýšenie v systémovej expozícii alogliptínu, v uvedenom poradí. (Pacienti s ochorením obličiek v terminálnom štádiu podstúpili hemodialýzu okamžite po dávkovaní alogliptínu. Na základe stredných hodnôt koncentrácií dialyzátu bolo počas 3-hodinovej hemodialýzy odstránených približne 7 % liečiva.) Preto sa za účelom zachovania systémových expozícií alogliptínu, ktoré sú podobné ako expozície pozorované u
pacientov s normálnou funkciou obličiek, odporúčajú nižšie dávky alogliptínu pre pacientov so stredne ťažkou alebo závažnou poruchou funkcie obličiek, alebo s ochorením obličiek v terminálnom štádiu
vyžadujúcim dialýzu (pozri vyššie uvedené a časť 4.2).
Pacienti s poruchami funkcie pečene
Celková expozícia alogliptínu bola u pacientov so stredne ťažkou poruchou funkcie pečene približne o 10 % nižšia a vrcholová expozícia bola približne o 8 % nižšia ako u kontrolných účastníkov.
Magnitúda týchto redukcií sa nepovažovala za klinicky významnú. U pacientov s miernou až stredne
ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre 5 až 9) preto nie je nutná úprava dávky alogliptínu. Použitie alogliptínu u pacientov so závažnou poruchou funkcie pečene (Childovo-
Pughovo skóre > 9) nebolo klinicky overené.
Vek, pohlavie, etnicita, telesná hmotnosť
Vek (65 – 81 rokov), pohlavie, etnicita (belosi, černosi a aziati) a telesná hmotnosť nemali na farmakokinetické vlastnosti alogliptínu žiadny klinicky významný účinok. Úprava dávky nie je nutná
(pozri časť 4.2).
Pediatrická populácia
Farmakokinetické vlastnosti alogliptínu u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. Nie sú k dispozícii žiadne údaje (pozri časť 4.2 a vyššie).
Metformín
Absorpcia
Po perorálnom podaní metformínu sa maximálna koncentrácia v plazme (Cmax) dosiahne približne za
2,5 hodiny (Tmax). Absolútna biodostupnosť 500 mg alebo 850 mg tabliet metformínu je približne 50 –
60 % u zdravých účastníkov. Neabsorbovaná frakcia nájdená po perorálnom podaní v stolici bola 20 –
30 %.
Po perorálnom podaní je absorpcia metformínu saturovateľná a neúplná. Predpokladá sa, že farmakokinetika absorpcie metformínu nie je lineárna.
Pri odporúčaných dávkach a dávkovacej schéme metformínu sa dosiahnu rovnovážne plazmatické koncentrácie metformínu do 24 – 48 hodín a všeobecne sú nižšie než 1 mikrogram/ml.
V kontrolovaných klinických skúškach maximálne hladiny metformínu v plazme (Cmax) neprekročili
4 mikrogramy/ml ani pri maximálnych dávkach.
Jedlo mierne spomaľuje a znižuje rozsah absorpcie metformínu. Po perorálnom podaní 850 mg tablety metformín hydrochloridu bola maximálna plazmatická koncentrácia o 40 % nižšia, AUC sa zmenšil
o 25 % a čas dosiahnutia vrcholovej plazmatickej koncentrácie (Tmax) sa predĺžil o 35 minút. Klinický význam týchto zistení nie je známy.
Distribúcia
Väzba na plazmatické bielkoviny je zanedbateľná. Metformín sa distribuuje do erytrocytov. Vrcholová
koncentrácia v krvi je nižšia ako v plazme a objavuje sa približne v rovnakom čase. Erytrocyty
predstavujú s najväčšou pravdepodobnosťou sekundárny kompartment distribúcie. Stredný distribučný objem (Vd) bol v rozmedzí 63 – 276 l.
Biotransformácia
Metformín sa vylučuje nezmenený močom. U ľudí sa nezistili žiadne metabolity.
Eliminácia
Obličkový klírens metformínu je > 400 ml/min., čo naznačuje, že metformín sa eliminuje
glomerulárnou filtráciou a tubulárnou sekréciou. Po perorálnom podaní je zdanlivý terminálny eliminačný polčas približne 6,5 hodiny.
Pri poruche funkcie obličiek sa znižuje obličkový klírens úmerne s klírensom kreatinínu a predlžuje sa tak eliminačný polčas, čo vedie k zvýšeniu hladiny metformínu v plazme.
Vipdomet
Osobitné skupiny pacientov
Pacienti s poruchami funkcie obličiek
Vzhľadom na obsah metformínu sa Vipdomet nesmie používať u pacientov s miernou až závažnou poruchou funkcie obličiek alebo s ochorením obličiek v terminálnom štádiu vyžadujúcim hemodialýzu
(pozri časť 4.2).
Pacienti s poruchami funkcie pečene
Vipdomet sa nemá používať u pacientov s poruchou funkcie pečene (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Súbežná liečba alogliptínom a metformínom nevyvolala žiadne nové poškodenia a neboli pozorované žiadne účinky na toxikokinetiku jednotlivých liečiv.
U potkanov sa nevyskytli žiadne fetálne abnormality po súbežnom podávaní pri rozpätiach expozície približne 28-násobku až 29-násobku pre alogliptín a 2-násobku až 2,5-násobku pre metformín pri maximálnej odporúčanej dávke pre ľudí 25 mg/denne a 2000 mg/denne, v uvedenom poradí. Táto kombinácia odhalila teratogenický potenciál u malého počtu plodov (mikroftalmia, malé vyklenutie oka a rázštep podnebia) pri vyšších dávkach metformínu (rozpätia expozície približne 20-násobku a
5 až 6-násobku maximálnej odporúčanej dávky pre ľudí pre alogliptín a metformín, v uvedenom poradí).
Nasledujúce údaje sú zistenia zo štúdií vykonaných samostatne s alogliptínom alebo metformínom. Alogliptín
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti a toxicity
neodhalili žiadne osobitné riziko pre ľudí.
Hladina bez pozorovaného nežiaduceho účinku (NOAEL) v štúdiách toxicity po opakovanom podávaní u potkanov a psov v trvaní až do 26 a 39 týždňov, v uvedenom poradí, preukázala rozpätia expozície, ktoré boli približne 147-násobkom a 227-násobkom, v uvedenom poradí, expozície u ľudí pri odporúčanej celkovej dennej dávke 25 mg alogliptínu.
Alogliptín nebol genotoxický v štandardnom rade in vitro a in vivo štúdií genotoxicity. Alogliptín nebol karcinogénny v 2-ročných štúdiách karcinogenity na potkanoch a myšiach.
V močovom mechúre potkaních samcov bola pozorovaná minimálna až slabá jednoduchá hyperplázia
prechodného epitelu, a to pri najnižšej použitej dávke (27-krát presahujúcej expozíciu u ľudí) bez vyvinutia jasnej NOEL (úroveň bez pozorovaného účinku).
Neboli pozorované žiadne nežiaduce účinky alogliptínu na fertilitu, reprodukčnú schopnosť ani raný embryonálny vývoj u potkanov až do systémovej expozície vysoko presahujúcej expozíciu u ľudí pri odporúčanej dávke. Hoci nedošlo k žiadnym negatívnym vplyvom na fertilitu, bolo pozorované nepatrné štatistické zvýšenie v počte abnormálnej spermy u samcov pri expozícii vysoko presahujúcej expozíciu u ľudí pri odporúčanej dávke.
U potkanov sa zaznamenal prestup alogliptínu cez placentu.
Alogliptín nebol teratogénny u potkanov ani u králikov so systémovou expozíciou pri hladinách NOAEL vysoko presahujúcou expozíciu u ľudí pri odporúčanej dávke. Vyššie dávky alogliptínu neboli teratogenické, ale spôsobovali toxicitu u matky a boli spojené s oneskorenou a/alebo nedostatočnou osifikáciou kostí a zníženou telesnou hmotnosťou plodu.
V štúdiách prenatálneho a postnatálneho vývoja u potkanov expozície vysoko presahujúce expozíciu
u ľudí pri odporúčanej dávke nepoškodili vyvíjajúce sa embryo ani negatívne neovplyvnili rast a vývoj potomstva. Vyššie dávky alogliptínu znižovali telesnú hmotnosť potomstva a vyvolali určité vývojové
zmeny považované za sekundárne v porovnaní s nízkou telesnou hmotnosťou.
Štúdie na laktujúcich potkanoch naznačujú, že alogliptín sa vylučuje do mlieka.
Po opakovanom podávaní v trvaní 4 a 8 týždňov sa u mladých potkanov nepozorovali žiadne účinky súvisiace s alogliptínom.
Metformín
Predklinické údaje pre metformín na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity
po opakovanom podaní, genotoxicity, karcinogénneho potenciálu a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Manitol
Mikrokryštalická celulóza
Povidón
Krospovidón
Magnéziumstearát
Filmový obal
Hypromelóza
Mastenec
Oxid titaničitý (E171) Oxid železitý žltý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPolychlorotrifluoroetylénové (PCTFE)/polyvinylchloridové (PVC) blistrové balenia s pretláčacou hliníkovou fóliou. Veľkosti balenia: 10, 14, 20, 28, 56, 60, 98, 112, 120, 180, 196, 196 (2x98 viacnásobné balenie) alebo 200 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITakeda Pharma A/S Dybendal Alle 10
2630 Taastrup
Dánsko
8. REGISTRAČNÉ ČÍSLOEU/1/13/843/001-012 , 025
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUVipdomet 12,5 mg/1000 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tableta obsahuje alogliptín benzoát zodpovedajúci 12,5 mg alogliptínu a 1000 mg metformín hydrochloridu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta (tableta).
Bledožlté, podlhovasté (približne 22,3 mm dlhé a 10,7 mm široké), bikonvexné, filmom obalené tablety s označením „12.5/1000“ vyrazeným na jednej strane a označením „322M“ vyrazeným na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieVipdomet je indikovaný na liečbu dospelých pacientov vo veku 18 a viac rokov s ochorením diabetes mellitus 2. typu:
• ako prídavný liek k diéte a k cvičeniu na zlepšenie kontroly glykémie u dospelých pacientov s neadekvátnou kontrolou svojej maximálnej tolerovanej dávky samotného metformínu alebo u pacientov, ktorí sa už liečia kombináciou alogliptínu a metformínu.
• v kombinácii s pioglitazónom (t.j. kombinovaná liečba tromi liekmi) ako prídavný liek k diéte a k cvičeniu u dospelých pacientov s neadekvátnou kontrolou svojej maximálnej tolerovanej dávky metformínu a pioglitazónu.
• v kombinácii s inzulínom (t.j. kombinovaná liečba tromi liekmi) ako prídavný liek k diéte a k cvičeniu na zlepšenie glykemickej kontroly u pacientov, keď inzulín v stabilnej dávke a samotný metformín neposkytujú adekvátnu kontrolu glykémie.
4.2 Dávkovanie a spôsob podávaniaDávkovaniePre rôzne dávkovacie režimy je Vipdomet k dispozícii vo filmom obalených tabletách
12,5 mg/850 mg a 12,5 mg/1000 mg.
Dospelí (≥ 18 rokov)Dávku Vipdometu je potrebné individualizovať na základe pacientovho aktuálneho liečebného režimu.
U pacientov s neadekvátnou kontrolou svojej maximálnej tolerovanej dávky samotného metformín hydrochloridu je odporúčaná dávka jedna tableta 12,5 mg/850 mg alebo 12,5 mg/1000 mg dvakrát denne, čo zodpovedá 25 mg alogliptínu plus 1700 alebo 2000 mg metformín hydrochloridu denne, v závislosti na dávke metformín hydrochloridu, ktorá sa už užíva.
U pacientov s dvojkombinovanou liečbou s neadekvátnou kontrolou s maximálnou tolerovanou dávkou metformínu a pioglitazónu sa má zachovať dávka pioglitazónu a Vipdomet sa má podávať súbežne. Alogliptín sa má užívať v dávke 12,5 mg dvakrát denne (celková denná dávka 25 mg) a metformín hydrochlorid v podobnej dávke (850 mg alebo 1000 mg dvakrát denne) k tomu, ktorý sa už užíva.
Pri používaní alogliptínu v kombinácii s metformínom a tiazolidíndiónom je nutné postupovať opatrne vzhľadom na zvýšené nebezpečenstvo hypoglykémie pozorovanej v súvislosti s touto trojkombinovanou liečbou (pozri časť 4.4). V prípade hypoglykémie zvážte možnosť zníženia dávky tiazolidíndiónu alebo metformínu.
U pacientov prechádzajúcich zo samostatných tabliet alogliptínu a metformínu (ako dvojkombinovanej liečby alebo ako súčasť kombinovanej liečby tromi liekmi spolu s inzulínom) sa má alogliptín aj metformín podávať v doteraz užívanej celkovej dennej dávke. Individuálnu dávku alogliptínu je potrebné rozdeliť na polovicu, keďže sa bude užívať dvakrát denne, kým dávkovanie metformínu je potrebné zachovať nezmenené.
U pacientov s neadekvátnou kontrolou s dvojkombinovanou liečbou s inzulínom a maximálnou tolerovanou dávkou metformínu má dávka Vipdometu zabezpečiť alogliptín v dávke 12,5 mg dvakrát denne (celková denná dávka 25 mg) a metformín v podobnej dávke, ako sa doteraz užíval.
S cieľom znížiť riziko hypoglykémie zvážte možnosť nižšej dávky inzulínu. Maximálna odporúčaná denná dávka 25 mg alogliptínu sa nesmie prekročiť.
Osobitné populácie
Starší pacienti (≥ 65 rokov)
Úprava dávky u starších pacientov nie je nutná. K možnosti podávania alogliptínu pacientom
v pokročilom veku však treba pristupovať konzervatívne vzhľadom k potenciálnemu zníženiu funkcie obličiek v rámci tejto populácie.
Pacienti s poruchami funkcie obličiek
U pacientov s miernou poruchou funkcie obličiek (klírens kreatinínu ≥ 60 ml/min.) nie je nutná úprava dávky Vipdometu (pozri časť 5.2).
Nakoľko Vipdomet obsahuje metformín, nesmie sa používať u pacientov so stredne ťažkou alebo závažnou poruchou funkcie obličiek, ani pri ochorení obličiek v terminálnom štádiu vyžadujúcim dialýzu (klírens kreatinínu < 60 ml/min.) (pozri časti 4.3, 4.4 a 5.2).
Pred začatím liečby Vipdometom a potom v pravidelných intervaloch sa odporúča dôkladné vyšetrenie funkcie obličiek (pozri časť 4.4).
Pacienti s poruchami funkcie pečene
Vipdomet sa nemá používať u pacientov s poruchami funkcie pečene (pozri časti 4.3 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Vipdometu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje.
Spôsob podania
Perorálne použitie.
Vipdomet sa má užívať dvakrát denne vzhľadom na farmakokinetické vlastnosti metformínovej
zložky. Aby sa znížili gastrointestinálne nežiaduce reakcie spojené s metformínom, liek sa má užívať s jedlom. Tablety sa majú prehltnúť celé a zapiť vodou.
V prípade vynechania dávky treba liek užiť hneď, ako si pacient spomenie. Dvojitá dávka lieku sa nesmie užiť naraz. V takomto prípade treba dávku lieku vynechať.
4.3 Kontraindikácie
• Precitlivenosť na liečivá alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 (pozri časť 4.8) alebo v minulosti vážna reakcia z precitlivenosti na akýkoľvek inhibítor dipeptidyl peptidázy 4 (DPP-4), vrátane anafylaktickej reakcie, anafylaktického šoku či angioedému (pozri časti 4.4 a 4.8).
• Diabetická ketoacidóza, diabetická pre-kóma
• Stredne ťažká a závažná porucha funkcie obličiek (klírens kreatinínu < 60 ml/min.; pozri časť 4.4)
• Akútne stavy s potenciálom zmeny funkcie obličiek, ako napr.:
o dehydratácia
o závažná infekcia
o šok
• Akútne alebo chronické ochorenie, ktoré môže spôsobiť tkanivovú hypoxiu (pozri časť 4.4),
ako napr.:
o zlyhanie srdca alebo dýchania
o nedávny infarkt myokardu
o šok
• Poruchy funkcie pečene (pozri časť 4.4)
• Akútna intoxikácia alkoholom, alkoholizmus (pozri časti 4.4 a 4.5)
4.4 Osobitné upozornenia a opatrenia pri používaní
Všeobecné
Vipdomet sa nemá používať u pacientov s ochorením diabetes mellitus 1. typu. Vipdomet nie je
náhradou inzulínu u pacientov vyžadujúcich inzulín.
Laktátová acidóza
Laktátová acidóza je veľmi zriedkavá, ale závažná (s vysokou úmrtnosťou, ak nie je liečená rýchlo)
metabolická komplikácia, ku ktorej môže dôjsť následkom akumulácie metformínu. Hlásené prípady laktátovej acidózy u pacientov liečených metformínom sa vyskytli prevažne u diabetických pacientov
so závažným zlyhávaním obličiek. Incidenciu laktátovej acidózy je možné znížiť a má byť znížená aj
posúdením iných sprievodných rizikových faktorov, ako je nedostatočne kontrolovaný diabetes,
ketóza, dlhšie trvajúce hladovanie, nadmerné požívanie alkoholu, poruchy funkcie pečene a akýkoľvek stav spojený s hypoxiou.
Diagnóza
Diagnózu laktátovej acidózy je nutné vziať do úvahy v prípade nešpecifických príznakov, ako sú napr. svalové kŕče a/alebo bolesti brucha a/alebo ťažká asténia. Laktátovú acidózu charakterizuje aj acidotické dyspnoe a hypotermia s následnou kómou. Diagnostické laboratórne nálezy sú znížená hodnota pH krvi, hladina laktátu v plazme nad 5 mmol/l, zvýšenie aniónového deficitu a pomeru laktát/pyruvát. Pri podozrení na metabolickú acidózu sa musí podávanie Vipdometu ukončiť a
pacienta je potrebné okamžite hospitalizovať (pozri časť 4.9).
Poruchy funkcie obličiek
Alogliptín a metformín sa vylučujú hlavne obličkami. Riziko laktátovej acidózy súvisiace
s metformínom sa zvyšuje so stupňom poruchy funkcie obličiek, a preto je pred začatím liečby a potom v pravidelných intervaloch nutné stanoviť koncentráciu kreatinínu v sére (a príslušnú odhadovanú rýchlosť glomerulárnej filtrácie alebo odhadovaný klírens):
• minimálne raz ročne u pacientov s normálnou funkciou obličiek
• minimálne dva až štyrikrát ročne u pacientov s hladinami kreatinínu v sére na hornom limite normálu a u starších pacientov
Zhoršená funkcia obličiek u starších pacientov býva častá a asymptomatická. Mimoriadna opatrnosť je potrebná v situáciách, keď môže nastať zhoršenie funkcie obličiek, napr. na začiatku
antihypertenzívnej alebo diuretickej liečby, alebo pri začatí liečby nesteroidným protizápalovým liekom (NSAID).
Vipdomet sa neodporúča podávať pacientom so stredne závažnou až závážnou poruchou funkcie obličiek a ochorením obličiek v terminálnom štádiu (klírens kreatinínu < 60 ml/min)(pozri časť 4.3).
Poruchy funkcie pečene
Použitie alogliptínu u pacientov so závažnou poruchou funkcie pečene (Childovo-Pughovo skóre > 9)
nebolo klinicky overené, a preto sa jeho použitie u takýchto pacientov neodporúča (pozri časti 4.2, 4.3
a 5.2).
Chirurgický zákrok
Keďže Vipdomet obsahuje metformín, liečba sa má prerušiť 48 hodín pred zvoleným chirurgickým
zákrokom v celkovej, spinálnej alebo epidurálnej anestézii. Liečba sa zvyčajne nemá obnoviť skôr ako
48 hodín po zákroku a len vtedy, ak opätovné vyšetrenie obličiek potvrdilo ich normálnu funkciu.
Podanie jódovaných kontrastnýchlátok
Intravaskulárne podanie jódovaných kontrastných látok pri rádiologických vyšetreniach môže
zapríčiniť zlyhanie obličiek, ktoré sa u pacientov užívajúcich metformín spájalo s laktátovou acidózou. Liečba Vipdometom sa preto pred testom a počas testu musí ukončiť a nemá sa začať skôr ako
48 hodín po ňom a len vtedy, ak opätovné vyšetrenie obličiek potvrdilo ich normálnu funkciu (pozri
časť 4.5).
Použitie s hypoglykemikami a liekmi spôsobujúcimi hypoglykémiu
O inzulíne je známe, že spôsobuje hypoglykémiu. Preto sa pri jeho použití v kombinácii s
Vipdometom odporúča zvážiť nižšiu dávku inzulínu, aby sa znížilo riziko hypoglykémie (pozri časť
4.2).
Vzhľadom k zvýšenému riziku hypoglykémie pri kombinovanej liečbe s pioglitazónom treba zvážiť nižšiu dávku pioglitazónu, aby sa znížilo riziko hypoglykémie, ak sú tieto lieky kombinované
s Vipdometom (pozri časť 4.2).
Klinicky neoverenékombinácie
Vipdomet sa nemá používať v kombinácii so sulfonylureou, keďže bezpečnosť a účinnosť tejto
kombinácie neboli doteraz úplne stanovené.
Zmena klinického stavupacientovspredtýmkontrolovanýmochorenímdiabetesmellitus2.typu
Keďže Vipdomet obsahuje metformín, akýkoľvek pacient s ochorením diabetes mellitus 2. typu dobre
kontrolovaný Vipdometom, u ktorého sa vyskytnú laboratórne abnormality alebo klinické ochorenie (obzvlášť neurčitá a nedostatočne definovaná choroba), sa má okamžite vyšetriť s podozrením na ketoacidózu alebo laktátovú acidózu. Toto vyšetrenie má zahŕňať stanovenie elektrolytov a ketónov
v sére, stanovenie glukózy v krvi a ak je indikované, aj stanovenie pH krvi a hladiny laktátu, pyruvátu a metformínu. V prípade výskytu akejkoľvek z týchto foriem acidózy sa musí liečba Vipdometom
okamžite ukončiť a musia sa prijať iné vhodné nápravné opatrenia.
Reakcie z precitlivenosti
Reakcie z precitlivenosti vrátane anafylaktických reakcií, angioedému a exfoliatívnych kožných
chorôb vrátane Stevensovho-Johnsonovho syndrómu a multiformného erytému boli pozorované u inhibítorov DPP-4 a spontánne hlásené pre alogliptín po jeho uvedení na trh. Klinické štúdie alogliptínu hlásia nízku incidenciu anafylaktických reakcií.
Akútna pankreatitída
Použitie inhibítorov DPP-4 sa spája s rizikom vyvolania akútnej pankreatitídy. Podľa súhrnnej analýzy
údajov z 13 štúdií boli počty hlásenej incidencie pankreatitídy u pacientov liečených 25 mg alogliptínu, 12,5 mg alogliptínu, aktívnou kontrolou alebo placebom 2, 1, 1 alebo 0 udalostí na
1000 pacientorokov, v uvedenom poradí. V štúdii kardiovaskulárnych výsledkov boli počty hlásenej
incidencie pankreatitídy u pacientov liečených alogliptínom alebo placebom 3 alebo 2 udalosti na
1000 pacientorokov, v uvedenom poradí. Po uvedení na trh došlo k spontánnemu hláseniu nežiaducich reakcií akútnej pankreatitídy. Pacientov je potrebné informovať o charakteristických príznakoch
akútnej pankreatitídy, ktorými sú pretrvávajúce intenzívne bolesti brucha, ktoré môžu vystreľovať do
oblasti chrbta. V prípade podozrenia na pankreatitídu je nutné ukončiť podávanie Vipdometu.
V prípade potvrdenia akútnej pankreatitídy sa podávanie Vipdometu nesmie obnoviť. U pacientov s pankreatitídou v minulosti je nutné postupovať opatrne.
Účinky na pečeň
Po uvedení lieku na trh boli hlásené prípady dysfunkcie pečene vrátane zlyhania pečene. Kauzálny
súvis doteraz nebol stanovený. Pacienti musia byť dôkladne sledovaní vzhľadom na výskyt možných abnormalít pečene. U pacientov s podozrením na symptómy poškodenia pečene treba urýchlene
vykonať testy funkcie pečene. Ak sa zistí akákoľvek abnormalita a nestanoví sa žiadna alternatívna
etiológia, zvážte prerušenie liečby alogliptínom.
4.5 Liekové a iné interakcie
Súbežné podávanie 100 mg alogliptínu raz denne a 1000 mg metformín hydrochloridu dvakrát denne po dobu 6 dní nemalo u zdravých účastníkov žiadne klinicky významné účinky na farmakokinetické vlastnosti alogliptínu ani metformínu.
Špecifické farmakokinetické interakčné štúdie liečiv s Vipdometom neboli vykonané. Nasledujúca časť uvádza interakcie pozorované u jednotlivých zložiek Vipdometu (alogliptín/metformín), ako sú uvedené v ich príslušných súhrnoch charakteristických vlastností lieku.
Možné účinky iných liekovnaalogliptín
Alogliptín sa primárne vylučuje nezmenený v moči a jeho metabolizmus enzýmovým systémom
cytochrómu (CYP) P450 je zanedbateľný (pozri časť 5.2). Interakcie s inhibítormi CYP sa preto neočakávajú a neboli ani preukázané.
Výsledky klinických interakčných štúdií tiež nepreukázali klinicky významné účinky gemfibrozilu
(inhibítor CYP2C8/9), flukonazolu (inhibítor CYP2C9), ketokonazolu (inhibítor CYP3A4),
cyklosporínu (inhibítor P-glykoproteínu), voglibózy (inhibítor alfa-glukozidázy), digoxínu, metformínu, cimetidínu, pioglitazónu alebo atorvastatínu na farmakokinetické vlastnosti alogliptínu.
Možné účinky alogliptínu nainélieky
Štúdie in vitro ukazujú, že alogliptín neinhibuje ani neindukuje izoformy CYP 450 pri koncentráciách
dosiahnutých s odporúčanou dávkou 25 mg alogliptínu (pozri časť 5.2). Interakcie so substrátmi izoforiem CYP 450 sa preto neočakávajú a neboli ani preukázané. V štúdiách in vitro nebol alogliptín
potvrdený ani ako substrát, ani ako inhibítor kľúčových transportérov spojených s metabolizáciou liečiva v príslušnej obličke: organického aniónového transportéra-1, organického aniónového
transportéra-3 ani organického katiónového transportéra-2 (OCT2). Klinické údaje ďalej nepoukazujú na interakciu s inhibítormi ani substrátmi P-glykoproteínu.
Klinické štúdie alogliptínu nepotvrdili žiadne klinicky významné účinky na farmakokinetické vlastnosti kofeínu, (R)–warfarínu, pioglitazónu, glyburidu, tolbutamidu, (S)–warfarínu, dextrometorfánu, atorvastatínu, midazolamu, perorálnej antikoncepcie (noretisterónu a etinylestradiolu), digoxínu, fexofenadínu, metformínu ani cimetidínu, čo poskytuje in vivo dôkaz o slabej tendencii k vyvolaniu interakcií so substrátmi CYP1A2, CYP3A4, CYP2D6, CYP2C9, P- glykoproteínu a OCT2.
Alogliptín nemá u zdravých účastníkov pri súbežnom podaní s warfarínom žiadny účinok na protrombínový čas ani na medzinárodný normalizovaný pomer (INR).
Kombinácia alogliptínusinýmiantidiabetikami
Výsledky štúdií s metformínom, pioglitazónom (tiazolidíndiónom), voglibózou (inhibítor alfa-
glukozidázy) a glyburidom (sulfonylureou) nepreukázali žiadne klinicky významné farmakokinetické interakcie.
Interakcie s metformínom
Kombinácie,ktorésaneodporúčajú
Alkohol
Liečivo metformín spôsobuje zvýšené riziko vzniku laktátovej acidózy pri akútnej intoxikácii alkoholom (najmä pri hladovaní, podvýžive alebo poruchách funkcie pečene) (pozri časť 4.4). Je
potrebné vyhýbať sa požívaniu alkoholu a užívaniu liekov obsahujúcich alkohol.
Katiónové lieky
Pri katiónových liečivách, ktoré sú eliminované obličkovou tubulárnou sekréciou (napr. cimetidín), môže dochádzať k interakcii s metformínom spôsobenej kompetíciou o spoločné obličkové tubulárne
transportné systémy. Štúdia so siedmimi zdravými dobrovoľníkmi ukázala, že cimetidín (400 mg
dvakrát denne) zvyšoval systémovú expozíciu metformínu (AUC) o 50 % a Cmax o 81 %. Preto je potrebné zvážiť starostlivé monitorovanie hladiny glykémie, úpravu dávky v rámci odporúčaného dávkovania a zmeny liečby diabetu, keď sa súbežne podávajú katiónové lieky, ktoré sa eliminujú obličkovou tubulárnou sekréciou.
Jódované kontrastné látky
Intravaskulárne podanie jódovaných kontrastných látok môže viesť k zlyhaniu obličiek, čo môže mať za následok akumuláciu metformínu s rizikom vzniku laktátovej acidózy. Liečba Vipdometom sa
preto pred testom a počas testu musí ukončiť a nemá sa začať skôr ako 48 hodín po ňom a len vtedy, ak opätovné vyšetrenie obličiek potvrdilo ich normálnu funkciu (pozri časť 4.4).
K
ombinácie,
priktorýchsavyžadujúbezpečnostnéopatrenia
Lieky s vlastným hyperglykemizujúcim účinkom
Glukokortikoidy (podávané systémovo a lokálne), beta-2-agonisty a diuretiká (pozri tiež časť 4.4) majú vlastný hyperglykemizujúci účinok. Pacienta je o tomto potrebné informovať a častejšie u neho monitorovať hladinu glukózy v krvi, hlavne na začiatku liečby týmito liekmi. Ak je to potrebné, dávkovanie Vipdometu je možné upraviť počas súbežnej liečby s iným liekom a po jej ukončení.
Inhibítory ACE
Inhibítory enzýmu konvertujúceho angiotenzín (ACE) môžu znižovať hladinu glukózy v krvi. Ak je to potrebné, dávkovanie Vipdometu je možné upraviť počas súbežnej liečby s iným liekom a po jej
ukončení.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití Vipdometu u gravidných žien. Štúdie kombinovanej liečby
alogliptínom a metformínom na gravidných potkanoch preukázali reprodukčnú toxicitu u približne
5 až 20-násobku (pre metformín a alogliptín, v uvedenom poradí) expozície u ľudí pri odporúčanej dávke.
Vipdomet sa nesmie užívať počas gravidity.
Riziko spojenésalogliptínom
Nie sú k dispozícii údaje o použití alogliptínu počas gravidity. Štúdie na zvieratách nepreukázali
priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Riziko spojenésmetformínom
Obmedzené množstvo údajov o použití metformínu počas gravidity nepoukazuje na zvýšené riziko
kongenitálnych abnormalít. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity pri klinicky významných dávkach (pozri časť 5.3).
Laktácia
Neboli vykonané žiadne štúdie kombinácie liečiv Vipdometu na laktujúcich zvieratách. V štúdiách
jednotlivých liečiv sa alogliptín, ako aj metformín, vylučovali do mlieka laktujúcich potkanov. Nie je známe, či sa alogliptín vylučuje do ľudského mlieka. Metformín sa vylučuje do ľudského mlieka v
malých množstvách. Riziko u dojčiat nemôže byť vylúčené.
Rozhodnutie o prerušení dojčenia alebo o ukončení/prerušení liečby Vipdometom, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinok Vipdometu na ľudskú fertilitu nebol študovaný. Štúdie alogliptínu alebo metformínu na
zvieratách nepreukázali žiadne nežiaduce účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Vipdomet nemá žiadny alebo má len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov treba upozorniť na riziko hypoglykémie, najmä pri jeho užívaní v kombinácii s inzulínom alebo pioglitazónom.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Klinické štúdie vykonané na podporu účinnosti a bezpečnosti Vipdometu obsahovali súbežné
podávanie alogliptínu a metformínu ako samostatných tabliet. Výsledky bioekvivalenčných štúdií však preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné s príslušnými dávkami
alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Poskytnuté informácie pochádzajú celkovo od 7150 pacientov s ochorením diabetes mellitus 2. typu vrátane 4201 pacientov liečených alogliptínom a metformínom, ktorí sa zúčastnili 3. etapy 7 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií. Tieto štúdie hodnotili účinky súbežne podávaného alogliptínu a metformínu na hodnotu glykémie a ich bezpečnosť ako úvodnej kombinovanej liečby, ako dvojkombinovanej liečby pacientov pôvodne liečených samotným metformínom a ako prídavnej liečby k tiazolidíndiónu alebo inzulínu.
Bezpečnostný profil súbežne podávaného alogliptínu a metformínu bol konzistentný s profilom jednotlivých zložiek, ako preukázali klinické skúšky alogliptínu a komplexné údaje dostupné pre metformín. Nasledujúca časť uvádza nežiaduce reakcie jednotlivých zložiek Vipdometu (alogliptín/metformín), ako sú uvedené v ich príslušných súhrnoch charakteristických vlastností lieku.
Alogliptín
Poskytnuté informácie pochádzajú celkovo od 9405 pacientov s ochorením diabetes mellitus 2. typu vrátane 3750 pacientov liečených 25 mg alogliptínu a 2476 pacientov liečených 12,5 mg alogliptínu, ktorí sa zúčastnili 2. etapy jednej alebo 3. etapy 12 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií. Okrem toho sa uskutočnila štúdia kardiovaskulárnych výsledkov
s 5380 pacientmi s ochorením diabetes mellitus 2. typu a nedávnym akútnym koronárnym syndrómom, z ktorých 2701 pacientov bolo randomizovaných do skupiny s alogliptínom
a 2679 pacientov do skupiny s placebom. Tieto štúdie hodnotili účinky alogliptínu na kontrolu
glykémie a jeho bezpečnosť ako monoterapie, ako úvodnej kombinovanej liečby s metformínom alebo tiazolidíndiónom a ako prídavnej liečby k metformínu alebo k sulfonylurey, alebo k tiazolidíndiónu
(s metformínom alebo sulfonylureou alebo bez nich), alebo k inzulínu (s metformínom alebo bez neho).
V súhrnnej analýze údajov z 13 štúdií boli celkové incidencie nežiaducich účinkov, závažných nežiaducich účinkov a nežiaducich účinkov vedúcich k ukončeniu liečby porovnateľné u pacientov liečených 25 mg alogliptínu, 12,5 mg alogliptínu, aktívnou kontrolou alebo placebom.
Najčastejšou nežiaducou reakciou u pacientov liečených 25 mg alogliptínu bola bolesť hlavy. Bezpečnosť alogliptínu v rámci starších pacientov (≥ 65 rokov) a mladších ako starších pacientov
(< 65 rokov) bola na podobnej úrovni.
Tabuľkový zoznamnežiaducichreakcií
Tieto nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a frekvencie. Frekvencie sú
definované ako veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1 /10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000); neznáme (nedá sa určiť z dostupných údajov).
Alogliptín
Počas súhrnných kontrolovaných pivotných klinických skúšok fázy 3 boli v prípade alogliptínu ako monoterapie a ako prídavnej kombinovanej liečby zahŕňajúcej 5659 pacientov pozorované nežiaduce reakcie, ktoré sú uvedené nižšie (Tabuľka 1).
T
abuľka 1: Nežiaduce reakcie pozorované pri súhrnných pivotných klinických skúškach fázy 3
T
rieda orgánových systémov Frekvencia nežiaducich reakcií
Nežiaduca reakcia
Infekcie a nákazy
Infekcia horných dýchacích ciest Časté
Zápal nosohltana Časté
Poruchy nervového systému
Bolesť hlavy Časté
Poruchy gastrointestinálneho traktu
Bolesť brucha Časté
Gastroezofageálny reflux Časté
Poruchy kože a podkožného tkaniva
Pruritus Časté
Vyrážka Časté
Alogliptín/metformín
Počas súhrnných kontrolovaných pivotných klinických skúšok fázy 3 boli v prípade alogliptínu ako prídavnej kombinovanej liečby s metformínom, zahŕňajúcej 7151 pacientov, pozorované nežiaduce reakcie, ktoré sú uvedené nižšie (Tabuľka 2).
Tabuľka 2: Nežiaduce reakcie pozorované pri kontrolovaných pivotných klinických skúškach fázy 3
T
rieda orgánových systémov
Nežiaduca reakcia
Infekcie a nákazy
Infekcia horných dýchacích ciest
Zápal nosohltana
Poruchy nervového systému
Frekvencia nežiaducich reakcií
Časté
Časté
Bolesť hlavy Časté
Poruchy gastrointestinálneho traktu
Gastroenteritída Bolesť brucha Hnačka Vracanie Gastritída
Gastroezofageálny reflux
Poruchy kože a podkožného tkaniva'
Pruritus
Vyrážka
Časté Časté Časté Časté Časté Časté
Časté
Časté
Alogliptín
Skúsenosti
po
uvedení
na
trh
V Tabuľke 3 sú uvedené dodatočné nežiaduce reakcie, ktoré boli spontánne hlásené po uvedení lieku
na trh.
Tabuľka 3: Spontánne hlásené nežiaduce reakcie na alogliptín po jeho uvedení na trh
T
rieda orgánových systémov
Nežiaduca reakcia
Poruchy imunitného systému
Frekvencia nežiaducich reakcií
Precitlivenosť Neznáme
Poruchy gastrointestinálneho systému
Akútna pankreatitída Neznáme
Poruchy hepatobiliárneho systému
Dysfunkcia pečene vrátane zlyhania pečene Neznáme
Poruchy kože a podkožného tkaniva
Exfoliatívne kožné choroby vrátane
Stevensovho-Johnsonovho syndrómu
Multiformný erytém
Neznáme
Neznáme
Angioedém Neznáme
Urtikária Neznáme
Metformín
Údajezklinickýchskúšokaskúsenostipouvedenínatrh
V Tabuľke 4 sú uvedené dodatočné nežiaduce reakcie, ktoré boli hlásené v rámci klinických skúšok a
po uvedení lieku na trh.
Tabuľka 4: Frekvencia nežiaducich reakcií na metformín identifikovaných údajmi z klinických skúšok a skúsenosťami po uvedení na trh
T
rieda orgánových systémov
Nežiaduca reakcia
Poruchy metabolizmu a výživy
Frekvencia nežiaducich reakcií
Laktátová acidóza Veľmi zriedkavé
Deficit vitamínu B12 Veľmi zriedkavé
Poruchy nervového systémuKovová chuť Časté
Poruchy gastrointestinálneho traktuBolesť brucha Veľmi časté
Hnačka Veľmi časté Strata chuti do jedla Veľmi časté Nevoľnosť Veľmi časté Zvracanie Veľmi časté
Poruchy pečene a žlčových ciestHepatitída Veľmi zriedkavé
Abnormality v testoch funkcie pečene Veľmi zriedkavé
Poruchy kože a podkožného tkanivaErytém Veľmi zriedkavé Pruritus Veľmi zriedkavé Urtikária Veľmi zriedkavé
PopisvybranýchnežiaducichreakciíLaktátová acidóza: 0,03 prípadov/1000 pacientorokov, pozri časť 4.4.
U pacientov dlhodobo liečených metformínom sa pozorovala znížená absorpcia vitamínu B12, ktorá vo všeobecnosti nemá klinický význam. Veľmi zriedkavo však môže viesť ku klinicky významnému deficitu vitamínu B12 (napr. megaloblastickej anémii).
Gastrointestinálne príznaky sa vyskytujú najčastejšie na začiatku liečby a vo väčšine prípadov spontánne vymiznú. Aby sa im zabránilo, odporúča sa užívať metformín v 2 denných dávkach počas jedla alebo po ňom.
Boli hlásené ojedinelé prípady hepatitídy alebo abnormalít v testoch funkcie pečene, ktoré ustúpili po vysadení metformínu.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieNie sú k dispozícii žiadne údaje týkajúce sa predávkovania Vipdometom.
AlogliptínNajvyššie dávky alogliptínu podané počas klinických skúšok boli jednorazové dávky 800 mg zdravým
účastníkom a 400 mg raz denne počas 14 dní pacientom s ochorením diabetes mellitus 2. typu (čo je ekvivalentné 32-násobku a 16-násobku odporúčanej celkovej dennej dávky 25 mg alogliptínu,
v uvedenom poradí).
MetformínVeľké predávkovanie metformínom alebo sprievodné riziká môžu viesť k laktátovej acidóze.
Laktátová acidóza je z medicínskeho hľadiska naliehavý prípad a musí sa liečiť v nemocnici.
Postup pri predávkovaníV prípade predávkovania je nutné vykonať vhodné podporné opatrenia v súlade s klinickým stavom
pacienta.
Hemodialýzou je možné odstrániť minimálne množstvá alogliptínu (počas 3-hodinovej hemodialýzy bolo odstránených približne 7 % tejto látky). Preto má hemodialýza pri predávkovaní len malý klinický prínos. Možnosť odstránenia alogliptínu peritoneálnou dialýzou nie je známa.
Najúčinnejšou metódou odstránenia laktátu a metformínu je hemodialýza.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antidiabetiká; biguanidy a sulfónamidy v kombinácii. ATC kód: A10BD13.
Mechanizmus účinku afarmakodynamickéúčinky
Vipdomet kombinuje dve hypoglykemicky účinné látky so vzájomne sa dopĺňajúcimi rozdielnymi
mechanizmami účinku na zlepšenie kontroly glykémie u pacientov s ochorením diabetes mellitus 2. typu: alogliptín, inhibítor dipeptidylpeptidázy-4 (DPP-4), a metformín, člen biguanidovej skupiny.
Alogliptín
Alogliptín je potentný a vysoko selektívny inhibítor DPP-4, > 10 000-krát selektívnejší pre DPP-4 ako
ostatné súvisiace enzýmy vrátane DPP-8 a DPP-9. DPP-4 je hlavný enzým vyvolávaný počas rapídnej degradácie inkretínových hormónov, glukagónu podobného peptidu 1 (GLP-1) a GIP (glukózo-
dependentný inzulínotropný polypeptid), ktoré sú uvoľňované črevami a ich hladiny stúpajú v rámci
odozvy na jedlo. GLP-1 a GIP zvyšujú biosyntézu inzulínu a sekréciu od pankreatických beta buniek, zatiaľ čo GLP-1 tiež inhibuje sekréciu glukagónu a tvorbu glukózy v pečeni. Alogliptín preto zlepšuje
glykemickú kontrolu pomocou glukózo-dependentného mechanizmu, nakoľko zlepšuje uvoľňovanie
inzulínu a pri zvýšených hladinách glukózy bráni zvyšovaniu hladín glukagónu.
Metformín
Metformín je biguanid s antidiabetickými účinkami, ktorý znižuje bazálnu aj postprandiálnu hladinu
glukózy v plazme. Nestimuluje sekréciu inzulínu, a preto nevyvoláva hypoglykémiu. Účinok metformínu môže mať 3 mechanizmy:
- zníženie tvorby glukózy v pečeni inhibíciou glukoneogenézy a glykogenolýzy.
- mierne zvyšovanie citlivosti na inzulín vo svaloch, zlepšovanie periférneho vychytávania a utilizácie glukózy.
- spomalenie intestinálnej absorpcie glukózy.
Metformín stimuluje intracelulárnu syntézu glykogénu účinkom na glykogénsyntázu. Taktiež zvyšuje transportnú kapacitu špecifických typov transportérov glukózy cez membrány (GLUT-1 a GLUT-4).
U ľudí má metformín priaznivé účinky na metabolizmus lipidov nezávisle od jeho pôsobenia na glykémiu. Ukázalo sa to pri terapeutických dávkach v kontrolovaných strednodobých alebo dlhodobých klinických štúdiách: metformín znižuje hladiny celkového cholesterolu, LDL cholesterolu a triglyceridov.
Klinická účinnosť
Klinické štúdie vykonané na podporu účinnosti Vipdometu obsahovali súbežné podávanie alogliptínu
a metformínu ako samostatných tabliet. Výsledky bioekvivalenčných štúdií však preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné s príslušnými dávkami alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Tieto štúdie hodnotili účinky súbežne podávaného alogliptínu a metformínu ako dvojkombinovanej liečby pacientov pôvodne liečených samotným metformínom a ako prídavnej liečby k tiazolidíndiónu alebo inzulínu.
Podanie 25 mg alogliptínu pacientom s ochorením diabetes mellitus 2. typu vyvolalo vrcholovú inhibíciu DPP-4 do 1 až 2 hodín a presiahlo 93 % po jednorazovej dávke 25 mg, ako aj po 14 dňoch podávania raz denne. Inhibícia DPP-4 zostala nad úrovňou 81 % 24 hodín po 14 dňoch podávania. Keď sa vypočítal priemer postprandiálnych koncentrácií glukózy 4 hodiny po raňajkách, obede
a večeri, výsledkom 14-dňovej liečby 25 mg alogliptínu bolo stredné, placebom kontrolované zníženie oproti prvej návšteve o -35,2 mg/dl.
Alogliptín v samostatnej dávke 25 mg, ako aj v kombinácii s 30 mg pioglitazónu, preukazoval významné zníženia postprandiálnej hladiny glukózy a postprandiálneho glukagónu, zatiaľ čo
významne zvyšoval postprandiálne hladiny aktívneho GLP-1 pri návšteve v týždni 16 v porovnaní s placebom (p < 0,05). Okrem toho vyvolal alogliptín v samostatnej dávke 25 mg, ako aj v kombinácii s
30 mg pioglitazónu, štatisticky významné (p < 0,001) zníženia celkových triglyceridov pri návšteve v
týždni 16 namerané podľa postprandiálnej prírastkovej zmeny AUC(0-8) z prvej návštevy v porovnaní s placebom.
Celkovo 7151 pacientov s ochorením diabetes mellitus 2. typu vrátane 4202 pacientov liečených alogliptínom a metformínom sa zúčastnilo 3. etapy 7 dvojito zaslepených, placebom alebo aktívne kontrolovaných klinických štúdií vykonaných za účelom hodnotenia účinkov súbežného podávania alogliptínu a metformínu na kontrolu glykémie a ich bezpečnosť. V týchto štúdiách bolo
696 pacientov liečených alogliptínom/metformínom vo veku ≥ 65 rokov.
Celkovo povedané, odporúčaná celková denná dávka 25 mg alogliptínu v kombinácii s metformínom zlepšila glykemickú kontrolu. Toto sa potvrdilo klinicky a štatisticky významnými zníženiami hladiny glykovaného hemoglobínu (HbA1c) a plazmatickej glukózy nalačno v porovnaní s kontrolou od prvej až po poslednú návštevu štúdie. Zníženia hladiny HbA1c boli podobné medzi rôznymi podskupinami vrátane ochorenia obličiek, veku, pohlavia a indexu telesnej hmotnosti, zatiaľ čo rozdiely medzi
rasami (napr. belosi a nebelosi) boli malé. Klinicky významné zníženia hladiny HbA1c v porovnaní s kontrolou boli tiež pozorované bez ohľadu na úvodnú základnú liečbu. Vyššia hladina HbA1c pri
prvej návšteve bola spojená s výraznejším znížením hladiny HbA1c. Účinky alogliptínu na telesnú
hmotnosť a lipidy boli vo všeobecnosti neutrálne.
Alogliptín ako prídavná liečba k metformínu
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe metformínom vo forme hydrochloridu (stredná hodnota dávky = 1847 mg) boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a plazmatickej glukózy nalačno pri návšteve v týždni 26 pri porovnaní s pridaním placeba (Tabuľka 5). Významne viac pacientov užívajúcich 25 mg alogliptínu (44,4 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (18,3 %) v týždni 26
(p < 0,001).
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe metformínom vo forme hydrochloridu (stredná hodnota dávky = 1835 mg) boli nepretržité zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c pri návšteve v týždni 52 a týždni 104. Pri návšteve v týždni 52 bolo zníženie hladiny HbA1c liečbou 25 mg alogliptínu spolu s metformínom (-0,76 %, tabuľka 6) podobné zníženiu vyvolanému liečbou glipizidom (stredná hodnota dávky = 5,2 mg) spolu s metformínom vo forme hydrochloridu (stredná hodnota dávky = 1824 mg, 0,73 %). Pri návšteve v týždni 104 bolo zníženie hladiny HbA1c liečbou 25 mg alogliptínu spolu s metformínom (-0,72 %, tabuľka 6) vyššie ako zníženie vyvolané liečbou glipizidom spolu s metformínom (-0,59 %). Stredná hodnota zmeny od prvej návštevy
v súvislosti s koncentráciou plazmatickej glukózy nalačno pri návšteve v týždni 52 pre 25 mg alogliprínu bola signifikantne vyššia ako pri liečbe glipizidom a metformínom (p < 0,001). V týždni
104, stredná hodnota zmeny od prvej návštevy v súvislosti s koncentráciou plazmatickej glukózy nalačno pre 25 mg alogliptínu a metformínu bola -3,2 mg/dl v porovnaní s 5,4 mg/dl pri liečbe glipizidom a metformínom. Viac pacientov užívajúcich 25 mg alogliptínu a metformínu (48,5 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi glipizid a metformín (42,8 %) (p = 0,004).
Súbežné podávanie 12,5 mg alogliptínu a 1000 mg metformín hydrochloridu dvakrát denne viedlo k štatisticky významným zlepšeniam od prvej návštevy, čo sa týka hladiny HbA1c a plazmatickej glukózy nalačno pri návšteve v týždni 26 pri porovnaní s podávaním 12,5 mg samotného alogliptínu dvakrát denne alebo s podávaním 1000 mg samotného metformín hydrochloridu dvakrát denne. Významne viac pacientov užívajúcich 12,5 mg alogliptínu a 1000 mg metformín hydrochloridu dvakrát denne (59,5 %) dosiahlo cieľové hladiny HbA1c < 7,0 % v porovnaní s pacientmi užívajúcimi
12,5 mg samotného alogliptínu dvakrát denne (20,2 %, p < 0,001) alebo 1000 mg samotného metformín hydrochloridu dvakrát denne (34,3 %, p < 0,001) v týždni 26.
Alogliptín ako prídavná liečba k metformínu s tiazolidíndiónom
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe pioglitazónom (stredná hodnota
dávky = 35,0 mg, s metformínom alebo sulfonylureou alebo bez nich), boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a FPG pri návšteve v týždni 26 pri porovnaní
s pridaním placeba (Tabuľka 5). Klinicky významné zníženia hladiny HbA1c v porovnaní s placebom
boli tiež pozorované u 25 mg alogliptínu bez ohľadu na to, či pacienti súbežne užívali metformín alebo sulfonylureu. Významne viac pacientov užívajúcich 25 mg alogliptínu (49,2 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (34,0 %) v týždni 26 (p = 0,004).
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe 30 mg pioglitazónu v kombinácii
s metformín hydrochloridom (stredná hodnota dávky = 1867,9 mg) boli zlepšenia od prvej návštevy,
čo sa týka hladiny HbA1c pri návšteve v týždni 52, ktoré boli neinferiórne ako aj štatisticky superiórne zlepšenia vyvolané liečbou 45 mg pioglitazónu v kombinácii s metformín hydrochloridom (stredná hodnota dávky = 1847,6 mg, Tabuľka 6). Významné zníženia hladiny HbA1c pozorované u 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu boli konzistentné počas celého obdobia liečby v trvaní 52 týždňov v porovnaní so 45 mg pioglitazónu a metformínu (p < 0,001 vo všetkých časových bodoch). Okrem toho bola stredná hodnota zmeny od prvej návštevy v súvislosti s FPG pri návšteve v týždni 52 pre 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu významne väčšia ako zmena
pri liečbe 45 mg pioglitazónu a metformínu (p < 0,001). Významne viac pacientov užívajúcich 25 mg alogliptínu plus 30 mg pioglitazónu a metformínu (33,2 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi 45 mg pioglitazónu a metformínu (21,3 %) v týždni 52 (p < 0,001).
Alogliptín ako prídavná liečba k metformínu s inzulínom
Výsledkom pridania 25 mg alogliptínu raz denne k liečbe inzulínom (stredná hodnota
dávky = 56,5 IU, s metformínom alebo bez neho) boli štatisticky významné zlepšenia od prvej návštevy, čo sa týka hladiny HbA1c a FPG pri návšteve v týždni 26 pri porovnaní s pridaním placeba
(Tabuľka 5). Klinicky významné zníženia hladiny HbA1c v porovnaní s placebom boli tiež pozorované u 25 mg alogliptínu bez ohľadu na to, či pacienti súbežne užívali metformín. Viac
pacientov užívajúcich 25 mg alogliptínu (7,8 %) dosiahlo cieľové hladiny HbA1c ≤ 7,0 % v porovnaní s pacientmi užívajúcimi placebo (0,8 %) v týždni 26.
T
abuľka 5: Zmena v hladine HbA1c (%) od prvej návštevy s 25 mg alogliptínu v týždni 26
v placebom kontrolovanej štúdii (FAS, LOCF)
Štúdia Stredná hodnota
H
bA1c pri prvej návšteve (%) (štandardná odchýlka (SD))
Stredná hodnota zmeny HbA1c od prvej návštevy (%)† (štandardná chyba priemeru (SE))
Placebom korigovaná zmena HbA1c od prvej návštevy (%)†
(2-stranný
95% interval spoľahlivosti (CI))
Placebom kontrolované štúdie prídavnej kombinovanej liečby
Alogliptín 25 mg raz denne s metformínom
(n = 203)
Alogliptín 25 mg raz denne so sulfonylureou
(n = 197)
Alogliptín 25 mg raz denne s tiazolidíndiónom ± metformín alebo sulfonylurea
(n = 195)
Alogliptín 25 mg raz denne s inzulínom ± metformín
(n = 126)
7,93 (0,799)
8,09 (0,898)
8,01 (0,837)
9,27 (1,127)
-0,59 (0,054)
-0,52 (0,058)
-0,80 (0,056)
-0,71 (0,078)
-0,48*
(-0,67, -0,30)
-0,53*
(-0,73, -0,33)
-0,61*
(-0,80, -0,41)
-0,59*
(-0,80, -0,37)
FAS = úplná populácia pacientov randomizovaných do liečby, ktorí prijali aspoň jednu dávku skúšaného lieku (full analysis set)
LOCF = použitie poslednej pozorovanej hodnoty na mieste chýbajúcej (last observation carried forward)
† Metóda najmenších štvorcov prispôsobená na stav pred antihyperglykemickou liečbou a hodnoty pri prvej návšteve
* p < 0.001 porovnané s placebom kontrolovanou liečbou alebo kombinovanou liečbou + placebo
T
abuľka 6: Zmena v hladine HbA1c (%) od prvej návštevy s 25 mg alogliptínu v aktívne kontrolovanej štúdii (PPS, LOCF)
Štúdia Stredná hodnota HbA1c pri prvej návšteve (%) (štandardná odchýlka
(
SD))
Štúdie prídavnej kombinovanej liečby
Alogliptín 25 mg raz denne s metformínom
v porovnaní so
sulfonylureou + metformín
Stredná hodnota zmeny HbA1c od
prvej návštevy (%)† (štandardná chyba
priemeru (SE))
Liečbou korigovaná zmena HbA1c od prvej návštevy (%)†
(1-stranný interval
spoľahlivosti (CI))
Zmena v týždni 52
(n = 382)
Zmena v týždni 104 (n = 382)
7,61 (0,526)
7,61 (0,526)
-0,76 (0,027)
-0,72 (0,037)
-0,03
(-nekonečno,
0,059)
-0,13*
(-nekonečno, -
0,006)
Alogliptín 25 mg raz denne
s tiazolidíndiónom +
metformín
v porovnaní s titrovaným tiazolidíndiónom + metformín
Zmena v týždni 26 (n = 303)
Zmena v týždni 52 (n = 303)
8,25 (0,820)
8,25 (0,820)
-0,89 (0,042)
-0,70 (0,048)
-0,47*
(-nekonečno, -0,35)
-0,42*
(-nekonečno, -0,28)
PPS = podskupina populácie pacientov spĺňajúcich kritériá protokolu (per protocol set)
LOCF = použitie poslednej pozorovanej hodnoty na mieste chýbajúcej (last observation carried forward)
* Štatisticky preukázaná neinferiorita a superiorita
† Metóda najmenších štvorcov prispôsobená na stav pred antihyperglykemickou liečbou a hodnoty pri prvej návšteve
Starší pacienti (≥ 65 rokov)Po kontrole účinnosti a bezpečnosti odporúčaných dávok alogliptínu a metformínu u podskupiny pacientov s ochorením diabetes mellitus 2. typu a vo veku ≥ 65 rokov sa zistila ich konzistentnosť
s profilom získaným u pacientov vo veku < 65 rokov.
Klinická bezpečnosťKardiovaskulárna bezpečnosťV súhrnnej analýze údajov z 13 štúdií boli celkové incidencie kardiovaskulárnej smrti, nefatálneho infarktu myokardu a nefatálnej mozgovej príhody porovnateľné u pacientov liečených 25 mg
alogliptínu, aktívnou kontrolou alebo placebom.
Okrem toho sa uskutočnila prospektívna randomizovaná štúdia kardiovaskulárnych výsledkov zameraná na bezpečnosť s 5380 pacientmi s vysokým základným kardiovaskulárnym rizikom, ktorej cieľom bolo preskúmať účinok alogliptínu v porovnaní s placebom (po pridaní k štandardnej
starostlivosti) na významné kardiovaskulárne nežiaduce udalosti (MACE) vrátane času do prvého výskytu akejkoľvek udalosti patriacej do skupiny zloženej z kardiovaskulárnej smrti, nefatálneho infarktu myokardu a nefatálnej mozgovej príhody u pacientov s nedávnou (15 až 90 dní) akútnou koronárnou udalosťou. Pri prvej návšteve mali pacienti stredný vek 61 rokov, stredné trvanie diabetu
9,2 roka a strednú hladinu HbA1c 8,0 %.
V štúdii sa preukázalo, že alogliptín nezvýšil riziko udalostí MACE v porovnaní s placebom [pomer rizika: 0,96; 1-stranný 99 % interval spoľahlivosti: 0 – 1,16]. V skupine s alogliptínom sa udalosť MACE vyskytla u 11,3 % pacientov v porovnaní s 11,8 % pacientov v skupine s placebom.
Tabuľka 7. Udalosti MACE hlásené v štúdii kardiovaskulárnych výsledkovPočet pacientov (%)Alogliptín25 mg Placebo
P
r
i
m
árny zložený koncový bod [prvá udalosť kardiovaskulárnej smrti, nefatálneho infarktu myokardu
a nefatálnej mozgovej príhody]
N = 2701 N = 2679
305 (11,3) 316 (11,8)89 (3,3) 111 (4,1)
187 (6,9) 173 (6,5)
29 (1,1) 32 (1,2)
*Celkovo zomrelo (všetky príčiny) 153 pacientov (5,7 %) v skupine s alogliptínom a 173 pacientov (6,5 %) v skupine s placebom.
U 703 pacientov sa vyskytla udalosť zo sekundárnej zloženej skupiny koncových bodov MACE (prvá
udalosť kardiovaskulárnej smrti, nefatálneho infarktu myokardu, nefatálnej mozgovej príhody
a naliehavej revaskularizácie v dôsledku nestabilnej angíny). V skupine liečenej alogliptínom sa udalosť zo sekundárnej zloženej skupiny koncových bodov MACE vyskytla u 12,7 % (344) pacientov
v porovnaní s 13,4 % (359) pacientov v skupine s placebom [pomer rizika = 0,95; 1-stranný 99 %
interval spoľahlivosti: 0 – 1,14].
HypoglykémiaV súhrnnej analýze údajov z 12 štúdií bola celková incidencia akejkoľvek epizódy hypoglykémie nižšia u pacientov liečených 25 mg alogliptínu ako u pacientov liečených 12,5 mg alogliptínu, aktívnou kontrolou alebo placebom (3,6 %, 4,6 %, 12,9 % a 6,2 % v uvedenom poradí). Väčšina
týchto epizód mala miernu až stredne ťažkú intenzitu. Celková incidencia epizód ťažkej hypoglykémie bola porovnateľná u pacientov liečených 25 mg alogliptínu alebo 12,5 mg alogliptínu a nižšia ako
incidencia u pacientov liečených aktívnou kontrolou alebo placebom (0,1 %, 0,1 %, 0,4 % a 0,4 %
v uvedenom poradí). V prospektívnej randomizovanej kontrolovanej štúdii kardiovaskulárnych výsledkov skúšajúci uviedol, že incidencia udalostí hypoglykémie bola u pacientov užívajúcich
placebo (6,5 %) a pacientov užívajúcich alogliptín (6,7 %) ako doplnok štandardnej starostlivosti
podobná.
Počas klinickej skúšky alogliptínu ako monoterapie bola incidencia hypoglykémie podobná ako u placeba a nižšia ako u placeba v inej skúške alogliptínu ako prídavnej liečby k sulfonylurey.
Vyššia frekvencia hypoglykémie bola pozorovaná pri kombinovanej liečbe tromi liekmi s tiazolidíndiónom a metformínom a v kombinácii s inzulínom, ako pri iných inhibítoroch DPP-4.
Pacienti (≥ 65 rokov) s ochorením diabetes mellitus 2. typu sa považujú za náchylnejších na epizódy hypoglykémie ako pacienti vo veku < 65 rokov. V súhrnnej analýze údajov z 12 štúdií bola celková
incidencia akejkoľvek hypoglykémie podobná u pacientov vo veku ≥ 65 rokov liečených 25 mg alogliptínu (3,8 %) ako incidencia u pacientov vo veku < 65 rokov (3,6 %).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Vipdomet vo
všetkých vekových podskupinách detí a dospievajúcich v liečbe ochorenia diabetes mellitus 2. typu
(pozri časť 4.2 pre informácie o používaní u detí a dospievajúcich).
5.2 Farmakokinetické vlastnosti
Výsledky bioekvivalenčných štúdií na zdravých účastníkoch preukázali, že filmom obalené tablety Vipdometu sú bioekvivalentné k príslušným dávkam alogliptínu a metformínu súbežne podávaných ako samostatné tablety.
Súbežné podávanie 100 mg alogliptínu raz denne a 1000 mg metformín hydrochloridu dvakrát denne po dobu 6 dní nemalo u zdravých účastníkov žiadne klinicky významné účinky na farmakokinetické vlastnosti alogliptínu ani metformínu.
Podávanie Vipdometu s jedlom neviedlo k zmene v celkovej expozícii (AUC) alogliptínu alebo metformínu. Stredné hodnoty vrcholových plazmatických koncentrácií alogliptínu a metformínu sa však znížili o 13 % a 28 %, v uvedenom poradí, ak sa Vipdomet podával s jedlom. U alogliptínu nedošlo k žiadnej zmene času dosiahnutia vrcholovej plazmatickej koncentrácie (Tmax), u metformínu však došlo k oneskoreniu Tmax o 1,5 hodiny. Klinická významnosť týchto zmien nie je pravdepodobná (pozri nižšie).
Vipdomet sa má užívať dvakrát denne z dôvodu farmakokinetických vlastností metformínovej zložky. Má sa tiež užívať s jedlom, aby sa znížili gastrointestinálne nežiaduce účinky spojené s metformínom (pozri časť 4.2).
Farmakokinetické vlastnosti Vipdometu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje (pozri časť 4.2).
Nasledujúca časť uvádza farmakokinetické vlastnosti jednotlivých zložiek Vipdometu
(alogliptín/metformín), ako sú uvedené v ich príslušných Súhrnoch charakteristických vlastností lieku.
Alogliptín
Farmakokinetické vlastnosti alogliptínu sú preukázateľne podobné u zdravých účastníkov
a u pacientov s ochorením diabetes mellitus 2. typu.
Absorpcia
Absolútna biodostupnosť alogliptínu je približne 100 %.
Podanie s jedlom s vysokým obsahom tuku nespôsobilo v celkovej a vrcholovej expozícii alogliptínu žiadne zmeny. Alogliptín sa preto môže podávať s jedlom alebo bez jedla.
Po podaní jednorazových perorálnych dávok až do 800 mg u zdravých účastníkov sa alogliptín rýchlo absorboval s výskytom vrcholových plazmatických koncentrácií 1 až 2 hodiny (stredná hodnota Tmax) po užití.
U zdravých účastníkov ani u pacientov s ochorením diabetes mellitus 2. typu nebola pozorovaná žiadna klinicky významná akumulácia po viacnásobnom podaní.
Celková a vrcholová expozícia alogliptínu sa primerane zvyšovala naprieč jednorazovými dávkami
6,25 mg až do 100 mg alogliptínu (pokrývajúcimi rozsah terapeutických dávok). Koeficient interindividuálnej variability (AUC) pre alogliptín bol nízky (17 %).
Distribúcia
Po podaní jednorazovej intravenóznej dávky 12,5 mg alogliptínu zdravým účastníkom bol distribučný
objem počas terminálnej fázy 417 l, čo indikuje dobrú distribúciu liečiva do tkanív.
Väzba alogliptínu na plazmatické bielkoviny je 20 – 30 %. Biotransformácia
Alogliptín nepodlieha rozsiahlemu metabolizmu – 60 – 70 % dávky sa vylúči močom ako nezmenený
liek.Po podaní perorálnej dávky [14C] alogliptínu boli zistené dva vedľajšie metabolity –
N-demetylovaný alogliptín, M-I (< 1 % pôvodnej látky) a N-acetylovaný alogliptín, M-II (< 6 % pôvodnej látky). M-I je aktívny metabolit a vysoko selektívny inhibítor DPP-4 podobný alogliptínu. M-II nevykazuje žiadnu inhibičnú aktivitu voči DPP-4 ani enzýmom súvisiacim s DPP. In vitro údaje indikujú, že CYP2D6 a CYP3A4 prispievajú k limitovanému metabolizmu alogliptínu.
In vitro štúdie dokazujú, že alogliptín pri koncentráciách dosiahnutých s odporúčanou dávkou 25 mg alogliptínu neindukuje CYP1A2, CYP2B6 a CYP2C9 a neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ani CYP3A4. Štúdie in vitro dokázali, že alogliptín mierne indukuje CYP3A4, avšak v štúdiách in vivo sa jeho indukovanie CYP3A4 nedokázalo.
V štúdiách in vitro sa alogliptín nepreukázal ako inhibítor týchto obličkových transportérov; OAT1, OAT3 a OCT2.
Alogliptín existuje prevažne ako (R)-enantiomér (> 99 %) a podlieha slabej alebo žiadnej chirálnej konverzii in vivo na (S)-enantiomér. (S)-enantiomér nie je pri terapeutických dávkach detekovateľný.
Eliminácia
Alogliptín bol eliminovaný so strednou hodnotou polčasu rozpadu (T1/2) približne 21 hodín.
Po podaní perorálnej dávky [14C] alogliptínu bolo 76 % celkovej rádioaktivity eliminovaných v moči a 13 % bolo zistených v stolici.
Priemerný renálny klírens alogliptínu (170 ml/min.) bol väčší ako priemerná odhadovaná rýchlosť glomerulárnej filtrácie (pribl. 120 ml/min.), čo naznačuje určitú aktívnu renálnu exkréciu.
Linearita
Celková expozícia (AUC(0-inf)) alogliptínu po podaní jednorazovej dávky bola podobná expozícii počas
intervalu jednej dávky (AUC(0-24)) po 6 dňoch dávkovania raz denne. Toto nenaznačuje časovú
závislosť v kinetike alogliptínu po viacnásobnom podaní.
Osobitné skupiny pacientov
Pacienti s poruchami funkcie obličiek
Jednorazová dávka 50 mg alogliptínu bola podaná 4 skupinám pacientov s rôznymi stupňami poruchy funkcie obličiek (klírens kreatinínu (CrCl) pomocou Cockcroftho a Gaultovho vzorca):
mierna (CrCl = > 50 až ≤ 80 ml/min.), stredne ťažká (CrCl = ≥ 30 až ≤ 50 ml/min.),
závažná (CrCl = < 30 ml/min.) a s ochorením obličiek v terminálnom štádiu na hemodialýze.
U pacientov s miernou poruchou funkcie obličiek bolo pozorované približne 1,7-násobné zvýšenie
AUC pre alogliptín. Avšak, keďže distribúcia hodnôt AUC pre alogliptín bola u týchto pacientov
v rámci rovnakého rozsahu ako u kontrolných účastníkov, úprava dávky alogliptínu u pacientov s miernou poruchou funkcie obličiek nie je nevyhnutná (pozri časť 4.2).
U pacientov so stredne ťažkou alebo závažnou poruchou funkcie obličiek, alebo s ochorením obličiek v terminálnom štádiu na hemodialýze bolo pozorované 2-násobné a 4-násobné zvýšenie v systémovej expozícii alogliptínu, v uvedenom poradí. (Pacienti s ochorením obličiek v terminálnom štádiu podstúpili hemodialýzu okamžite po dávkovaní alogliptínu. Na základe stredných hodnôt koncentrácií dialyzátu bolo počas 3-hodinovej hemodialýzy odstránených približne 7 % liečiva.) Preto sa za účelom zachovania systémových expozícií alogliptínu, ktoré sú podobné ako expozície pozorované u
pacientov s normálnou funkciou obličiek, odporúčajú nižšie dávky alogliptínu pre pacientov so stredne ťažkou alebo závažnou poruchou funkcie obličiek, alebo s ochorením obličiek v terminálnom štádiu
vyžadujúcim dialýzu (pozri vyššie uvedené a časť 4.2).
Pacienti s poruchami funkcie pečene
Celková expozícia alogliptínu bola u pacientov so stredne ťažkou poruchou funkcie pečene približne o 10 % nižšia a vrcholová expozícia bola približne o 8 % nižšia ako u kontrolných účastníkov.
Magnitúda týchto redukcií sa nepovažovala za klinicky významnú. U pacientov s miernou až stredne
ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre 5 až 9) preto nie je nutná úprava dávky alogliptínu. Použitie alogliptínu u pacientov so závažnou poruchou funkcie pečene (Childovo-
Pughovo skóre > 9) nebolo klinicky overené.
Vek, pohlavie, etnicita, telesná hmotnosť
Vek (65 – 81 rokov), pohlavie, etnicita (belosi, černosi a aziati) a telesná hmotnosť nemali na farmakokinetické vlastnosti alogliptínu žiadny klinicky významný účinok. Úprava dávky nie je nutná
(pozri časť 4.2).
Pediatrická populácia
Farmakokinetické vlastnosti alogliptínu u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. Nie sú k dispozícii žiadne údaje (pozri časť 4.2 a vyššie).
Metformín
Absorpcia
Po perorálnom podaní metformínu sa maximálna koncentrácia v plazme (Cmax) dosiahne približne za
2,5 hodiny (Tmax). Absolútna biodostupnosť 500 mg alebo 850 mg tabliet metformínu je približne 50 –
60 % u zdravých účastníkov. Neabsorbovaná frakcia nájdená po perorálnom podaní v stolici bola 20 –
30 %.
Po perorálnom podaní je absorpcia metformínu saturovateľná a neúplná. Predpokladá sa, že farmakokinetika absorpcie metformínu nie je lineárna.
Pri odporúčaných dávkach a dávkovacej schéme metformínu sa dosiahnu rovnovážne plazmatické koncentrácie metformínu do 24 – 48 hodín a všeobecne sú nižšie než 1 mikrogram/ml.
V kontrolovaných klinických skúškach maximálne hladiny metformínu v plazme (Cmax) neprekročili
4 mikrogramy/ml ani pri maximálnych dávkach.
Jedlo mierne spomaľuje a znižuje rozsah absorpcie metformínu. Po perorálnom podaní 850 mg tablety metformín hydrochloridu bola maximálna plazmatická koncentrácia o 40 % nižšia, AUC sa zmenšil
o 25 % a čas dosiahnutia vrcholovej plazmatickej koncentrácie (Tmax) sa predĺžil o 35 minút. Klinický význam týchto zistení nie je známy.
Distribúcia
Väzba na plazmatické bielkoviny je zanedbateľná. Metformín sa distribuuje do erytrocytov. Vrcholová
koncentrácia v krvi je nižšia ako v plazme a objavuje sa približne v rovnakom čase. Erytrocyty
predstavujú s najväčšou pravdepodobnosťou sekundárny kompartment distribúcie. Stredný distribučný objem (Vd) bol v rozmedzí 63 – 276 l.
Biotransformácia
Metformín sa vylučuje nezmenený močom. U ľudí sa nezistili žiadne metabolity.
Eliminácia
Obličkový klírens metformínu je > 400 ml/min., čo naznačuje, že metformín sa eliminuje
glomerulárnou filtráciou a tubulárnou sekréciou. Po perorálnom podaní je zdanlivý terminálny eliminačný polčas približne 6,5 hodiny.
Pri poruche funkcie obličiek sa znižuje obličkový klírens úmerne s klírensom kreatinínu a predlžuje sa tak eliminačný polčas, čo vedie k zvýšeniu hladiny metformínu v plazme.
Vipdomet
Osobitné skupiny pacientov
Pacienti s poruchami funkcie obličiek
Vzhľadom na obsah metformínu sa Vipdomet nesmie používať u pacientov s miernou až závažnou poruchou funkcie obličiek alebo s ochorením obličiek v terminálnom štádiu vyžadujúcim hemodialýzu
(pozri časť 4.2).
Pacienti s poruchami funkcie pečene
Vipdomet sa nemá používať u pacientov s poruchou funkcie pečene (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Súbežná liečba alogliptínom a metformínom nevyvolala žiadne nové poškodenia a neboli pozorované žiadne účinky na toxikokinetiku jednotlivých liečiv.
U potkanov sa nevyskytli žiadne fetálne abnormality po súbežnom podávaní pri rozpätiach expozície približne 28-násobku až 29-násobku pre alogliptín a 2-násobku až 2,5-násobku pre metformín pri maximálnej odporúčanej dávke pre ľudí 25 mg/denne a 2000 mg/denne, v uvedenom poradí. Táto kombinácia odhalila teratogenický potenciál u malého počtu plodov (mikroftalmia, malé vyklenutie oka a rázštep podnebia) pri vyšších dávkach metformínu (rozpätia expozície približne 20-násobku a
5 až 6-násobku maximálnej odporúčanej dávky pre ľudí pre alogliptín a metformín, v uvedenom poradí).
Nasledujúce údaje sú zistenia zo štúdií vykonaných samostatne s alogliptínom alebo metformínom. Alogliptín
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti a toxicity
neodhalili žiadne osobitné riziko pre ľudí.
Hladina bez pozorovaného nežiaduceho účinku (NOAEL) v štúdiách toxicity po opakovanom podávaní u potkanov a psov v trvaní až do 26 a 39 týždňov, v uvedenom poradí, preukázala rozpätia expozície, ktoré boli približne 147-násobkom a 227-násobkom, v uvedenom poradí, expozície u ľudí pri odporúčanej celkovej dennej dávke 25 mg alogliptínu.
Alogliptín nebol genotoxický v štandardnom rade in vitro a in vivo štúdií genotoxicity. Alogliptín nebol karcinogénny v 2-ročných štúdiách karcinogenity na potkanoch a myšiach.
V močovom mechúre potkaních samcov bola pozorovaná minimálna až slabá jednoduchá hyperplázia
prechodného epitelu, a to pri najnižšej použitej dávke (27-krát presahujúcej expozíciu u ľudí) bez vyvinutia jasnej NOEL (úroveň bez pozorovaného účinku).
Neboli pozorované žiadne nežiaduce účinky alogliptínu na fertilitu, reprodukčnú schopnosť ani raný embryonálny vývoj u potkanov až do systémovej expozície vysoko presahujúcej expozíciu u ľudí pri odporúčanej dávke. Hoci nedošlo k žiadnym negatívnym vplyvom na fertilitu, bolo pozorované nepatrné štatistické zvýšenie v počte abnormálnej spermy u samcov pri expozícii vysoko presahujúcej expozíciu u ľudí pri odporúčanej dávke.
U potkanov sa zaznamenal prestup alogliptínu cez placentu.
Alogliptín nebol teratogénny u potkanov ani u králikov so systémovou expozíciou pri hladinách NOAEL vysoko presahujúcou expozíciu u ľudí pri odporúčanej dávke. Vyššie dávky alogliptínu neboli teratogenické, ale spôsobovali toxicitu u matky a boli spojené s oneskorenou a/alebo nedostatočnou osifikáciou kostí a zníženou telesnou hmotnosťou plodu.
V štúdiách prenatálneho a postnatálneho vývoja u potkanov expozície vysoko presahujúce expozíciu
u ľudí pri odporúčanej dávke nepoškodili vyvíjajúce sa embryo ani negatívne neovplyvnili rast a vývoj potomstva. Vyššie dávky alogliptínu znižovali telesnú hmotnosť potomstva a vyvolali určité vývojové
zmeny považované za sekundárne v porovnaní s nízkou telesnou hmotnosťou.
Štúdie na laktujúcich potkanoch naznačujú, že alogliptín sa vylučuje do mlieka.
Po opakovanom podávaní v trvaní 4 a 8 týždňov sa u mladých potkanov nepozorovali žiadne účinky súvisiace s alogliptínom.
Metformín
Predklinické údaje pre metformín na základe obvyklých štúdií farmakologickej bezpečnosti, toxicity
po opakovanom podaní, genotoxicity, karcinogénneho potenciálu a reprodukčnej toxicity neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Manitol
Mikrokryštalická celulóza
Povidón
Krospovidón
Magnéziumstearát
Filmový obal
Hypromelóza
Mastenec
Oxid titaničitý (E171) Oxid železitý žltý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPolychlorotrifluoroetylénové (PCTFE)/polyvinylchloridové (PVC) blistrové balenia s pretláčacou hliníkovou fóliou. Veľkosti balenia: 10, 14, 20, 28, 56, 60, 98, 112, 120, 180, 196, 196 (2x98 viacnásobné balenie) alebo 200 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuNepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITakeda Pharma A/S Dybendal Alle 10
2630 Taastrup
Dánsko
8. REGISTRAČNÉ ČÍSLOEU/1/13843/013-024, 026
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. september 2013
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.