
.
Superinfekcia
V klinických skúšaniach u pacientov s komplikovanými intraabdominálnymi infekciami (cIAI) boli
poruchy hojenia operačnej rany spájané so superinfekciou. Pacienti s rozvíjajúcim sa zhoršeným hojením by mali byť sledovaní pre detekciu superinfekcie (pozri časť 4.8).
Pacienti, u ktorých sa vyvinuli superinfekcie, najmä nozokomiálna pneumónia, sa zdajú byť spojení so slabšími výsledkami. Pacientov je potrebné pozorne sledovať pre rozvoj superinfekcie. Ak sa po začatí liečby tigecyklínom identifikuje iné ohnisko infekcie ako cSSTI (complicated skin and soft tissue infection, cSSTI) alebo cIAI (complicated intra-abdominal infection, cIAI), je potrebné zvážiť začatie alternatívnej antibakteriálnej terapie, ktorá preukázala účinnosť v liečbe prítomnej infekcie (infekcií) špecifického typu.
Anafylaxia
Anafylaxia/anafylaktoidné reakcie, potenciálne život ohrozujúce, boli hlásené u tigecyklínu (pozri
časti 4.3 a 4.8).
Zlyhávanie pečene
U pacientov liečených tigecyklínom boli hlásené prípady poškodenia pečene s prevažne
cholestatickým obrazom vrátane niekoľkých prípadov zlyhávania pečene s fatálnym dôsledkom. Napriek tomu, že sa zlyhávanie pečene môže objaviť u pacientov liečených tigecyklínom kvôli základnému ochoreniu alebo súbežne používaným liekom, je potrebné zvážiť podiel tigecyklínu (pozri časť 4.8).
Antibiotiká triedytetracyklínu
Trieda antibiotík glycylcyklínu sa štruktúrne podobá antibiotikám triedy tetracyklínu. Tigecyklín môže
mať nežiaduce reakcie podobné antibiotikám triedy tetracyklínu. Tieto reakcie môžu zahŕňať fotosenzitivitu, pseudotumor mozgu, pankreatitídu a antianabolický účinok, ktorý viedol k zvýšeniu
dusíka močoviny v krvi (BUN), azotémii, acidóze a hyperfosfatémii (pozri časť 4.8).
Pankreatitída
Akútna pankreatitída, ktorá môže byť závažná, sa vyskytla (frekvencia: menej časté) v súvislosti
s liečbou tigecyklínom (pozri časť 4.8). Diagnóza akútnej pankreatitídy sa má brať do úvahy
u pacientov užívajúcich tigecyklín, u ktorých sa rozvinuli klinické príznaky, prejavy alebo laboratórne abnormality pripomínajúce akútnu pankreatitídu. Väčšina hlásených prípadov sa rozvinula po
minimálne jednom týždni liečby. Boli hlásené prípady u pacientov bez známych rizikových faktorov
pankreatitídy. Pacienti sa zvyčajne zotavia po vysadení tigecyklínu. V prípadoch podozrenia na rozvoj pankreatitídy sa má zvážiť ukončenie liečby tigecyklínom.
Základné ochorenia
Skúsenosti s použitím tigecyklínu na liečbu infekcií u pacientov so závažnými základnými
ochoreniami sú obmedzené.
V klinických skúšaniach s cSSTI bola najčastejším typom infekcie u pacientov liečených tigecyklínom celulitída (58,6 %) nasledovaná veľkými abscesmi (24,9 %). Pacienti so závažným základným ochorením, ako sú imunokompromitovaní pacienti, pacienti s infekciami dekubitov alebo pacienti, ktorí mali infekcie vyžadujúce si dlhšiu ako 14-dňovú liečbu (napríklad nekrotizujúca fasciitída)
neboli do štúdie zahrnutí. Štúdie sa zúčastnil limitovaný počet pacientov s pridruženými faktormi, ako sú diabetes (25,8 %), ochorenie periférnych ciev (10,4 %), intravenózne zneužívanie látok (4,0 %)
a HIV pozitívna infekcia (1,2 %). Limitované skúsenosti sú k dispozícii taktiež v liečbe pacientov so
súčasne sa vyskytujúcou bakteriémiou (3,4 %). Preto sa pri liečbe týchto pacientov odporúča postupovať opatrne. Výsledky veľkej štúdie u pacientov s infekciou diabetickej nohy poukázali na to, že tigecyklín bol menej účinný než komparátor, preto sa tigecyklín neodporúča používať u týchto pacientov (pozri časť 4.1).
V klinických skúšaniach s cIAI bola u pacientov liečených tigecyklínom najčastejším typom infekcií komplikovaná apendicitída (50,3 %) nasledovaná inými, menej často hlásenými diagnózami, ako sú komplikovaná cholecystitída (9,6 %), perforácia čreva (9,6 %), intraabdominálny absces (8,7 %), perforácia žalúdočných alebo duodenálnych vredov (8,3 %), peritonitída (6,2 %) a komplikovaná diventrikulitída (6,0 %). Z týchto pacientov malo 77,8 % chirurgicky zjavnú peritonitídu. Bol zahrnutý limitovaný počet pacientov so závažným základným ochorením, ako sú imunokompromitovaní pacienti, pacienti s APACHE II skóre > 15 (3,3 %) alebo s chirurgicky zjavnými mnohonásobnými intraabdominálnymi abscesmi (11,4 %). Limitované skúsenosti sú k dispozícii taktiež v liečbe pacientov so súčasne sa vyskytujúcou bakteriémiou (5,6 %). Preto sa pri liečbe týchto pacientov odporúča postupovať opatrne.
Je potrebné zvážiť použitie kombinovanej antibakteriálnej liečby vždy, ak sa tigecyklín podáva ťažko chorým pacientom s cIAI, ktorí majú sekundárne klinicky zjavnú intestinálnu perforáciu alebo pacientom so začínajúcou sepsou alebo septickým šokom (pozri časť 4.8).
Vplyv cholestázy na farmakokinetiku tigecyklínu sa náležite nestanovil. Biliárna exkrécia predstavuje približne 50 % celkovej exkrécie tigecyklínu. Preto je potrebné pacientov s cholestázou dôkladne monitorovať.
Ak sa tigecyklín podáva spolu s antikoagulačnými látkami, u sledovaných pacientov sa má stanoviť protrombínový čas alebo použiť iný vhodný antikoagulačný test (pozri časť 4.5).
Pseudomembranózna kolitída bola hlásená u takmer všetkých antibakteriálnych látok a jej závažnosť môže siahať od miernej až po život ohrozujúcu. Preto je dôležité zvážiť túto diagnózu u pacientov,
u ktorých sa počas alebo po podaní akejkoľvek antibakteriálnej látky objaví hnačka (pozri časť 4.8).
Použitie tigecyklínu môže viesť k premnoženiu necitlivých organizmov vrátane húb. Pacientov je potrebné počas liečby starostlivo sledovať (pozri časť 4.8).
Výsledky štúdií na potkanoch liečených tigecyklínom preukázali zmenu farby kostí. Tigecyklín môže súvisieť so stálou zmenou farby zubov u ľudí, ak sa použije v období vývoja zubov (pozri časť 4.8).
Pediatrická populácia
Klinické skúsenosti pri používaní tigecyklínu v liečbe infekcií pediatrických pacientov vo veku
8 rokov a starších sú veľmi obmedzené (pozri časti 4.8 a 5.1). V dôsledku toho sa má použiť u detí len v tých klinických situáciách, kedy nie je dostupná žiadna alternatívna antibakteriálna liečba.
Veľmi častými nežiaducimi reakciami u detí a dospievajúcich je nauzea a vracanie (pozri časť 4.8). Pozornosť sa má venovať možnej dehydratácii. Pediatrickým pacientom by sa mal tigecyklín podávať prednostne v 60 minút trvajúcej infúzii.
Podobne ako u dospelých, aj u detí sa často hlási bolesť brucha. Bolesť brucha môže poukazovať na pankreatitídu. Ak sa objaví pankreatitída, liečba tigecyklínom sa má vysadiť.
Pred začiatkom liečby tigecyklínom a pravidelne počas liečby sa majú monitorovať pečeňové testy, hemokoagulačné parametre, hematologické parametre, amyláza a lipáza.
Tygacil sa nemá používať u detí vo veku menej ako 8 rokov z dôvodu nedostatku údajov o bezpečnosti a účinnosti v tejto vekovej skupine a pretože tigecyklín môže byť spojený s trvalou zmenou farby zubov (pozri časti 4.2 a 4.8).
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Súbežné podávanie tigecyklínu a warfarínu (25 mg jednorazová dávka) zdravým osobám viedlo k poklesu klírensu R-warfarínu a S-warfarínu o 40 % a 23 % a k nárastu AUC o 68 % a 29 %. Mechanizmus týchto interakcií nie je ešte objasnený. Dostupné údaje nenasvedčujú, že tieto interakcie môžu viesť k významným zmenám INR. Keďže tigecyklín môže predlžovať protrombínový čas (PT)
a aktivovaný parciálny tromboplastínový čas (aPTT), majú sa v prípade súbežného podania tigecyklínu a antikoagulancií dôkladne sledovať zodpovedajúce koagulačné testy (pozri časť 4.4).
Warfarín neovplyvňoval farmakokinetický profil tigecyklínu.
Tigecyklín sa nemetabolizuje vo veľkej miere. Preto sa nepredpokladá vplyv liečiv inhibujúcich alebo indukujúcich aktivitu izoforiem CYP450 na klírens tigecyklínu. V podmienkach in vitro nie je tigecyklín kompetitívnym a ani ireverzibilným inhibítorom enzýmov CYP450 (pozri časť 5.2).
Pri podaní zdravým dospelým osobám v odporúčaných dávkach neovplyvňoval tigecyklín rýchlosť alebo mieru absorpcie alebo klírens digoxínu (0,5 mg nasledovaných 0,25 mg denne). Digoxín neovplyvňoval farmakokinetický profil tigecyklínu. Preto nie je potrebná úprava dávky pri podaní tigecyklínu s digoxínom.
V štúdiách in vitro sa nepozoroval žiadny antagonizmus medzi tigecyklínom a inými bežne používanými triedami antibiotík.
Súbežné používanie antibiotík s perorálnou antikoncepciou môže znižovať účinnosť perorálnej antikoncepcie.
Na základe in vitro štúdie tigecyklín je P-gp substrát. Súčasné podávanie P-gp inhibítorov (napr. ketokonazol alebo cyklosporín) alebo P-gp induktorov (napr. rifampicín) môže ovplyvniť farmakokinetiku tigecyklínu (pozri časť 5.2).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne alebo iba omedzené údaje o použití tigecyklínu u gravidných žien. Štúdie na
zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Ako je známe pre triedu tetracyklínových antibiotík, tigecyklín môže tiež indukovať tvorbu trvalých zubných porúch (zmena farby a poškodenie skloviny) a spomaliť proces osifikácie u plodov vystavených liečivu v maternici počas druhej polovice gravidity a u detí vo veku menej ako osem rokov. To je spôsobené hromadením v tkanivách s vysokým obratom vápnika a tvorbou chelátových komplexov s vápnikom (pozri časť 4.4). Tigecyklín sa nemá používať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu tigecyklínom.
Dojčenie
Nie je známe, či sa tigecyklín/metabolity vylučujú do ľudského mlieka. Dostupné
farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie tigecyklínu/metabolitov do mlieka (pozri časť 5.3). Riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť
dojčenie alebo či ukončiť/prerušiť liečbu tigecyklínom sa má urobiť po zvážení prínosu dojčenia pre
dieťa a prínosu liečby pre ženu.
Fertilita
Tigecyklín neovplyvňoval schopnosť párenia alebo fertilitu u potkanov pri expozíciách vyšších ako
4,7-násobok dennej dávky u ľudí založenej na AUC. U samíc potkanov sa pri expozíciách vyšších ako
4,7-násobok dennej dávky u ľudí založenej na AUC nezistili žiadne účinky na vaječníky alebo na estrálne cykly, ktoré by súviseli so zlúčeninou.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Môžu sa objaviť závraty, ktoré môžu mať vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Prehľad profilu bezpečnosti
Celkový počet cSSTI a cIAI pacientov liečených tigecyklínom v klinických štúdiách 3. a 4. fázy bol
2393.
V klinických skúšaniach boli najčastejšími akútnymi nežiaducimi reakciami súvisiacimi s liekom: reverzibilná nauzea (21 %) a vracanie (13 %). Tieto sa zvyčajne objavili skoro (počas 1. a 2. dňa liečby) a boli všeobecne mierne až stredne ťažké.
Nežiaduce reakcie hlásené u tigecyklínu vrátane klinických skúšaniach a skúseností po uvedení lieku na trh sú uvedené v tabuľke nižšie:
Tabuľkový zoznamnežiaducichreakcií
Trieda
orgánových systémov
Veľmi
časté
(
≥
1/10)
Časté ≥1/100 až
<1/10
Menej časté
≥1/1 000 až
<1/100
Neznáme (z dostupných
údajov)
Infekcie a nákazy sepsa/septický šok, pneumónia, absces, infekcie
Poruchy krvi a lymfatického systému
Poruchy imunitného systému Poruchy metabolizmu a výživy Poruchy
nervového systému
predĺžený aktivovaný parciálny tromboplastínový čas (aPTT), predĺžený protrombínový čas (PT)
hypoglykémia, hypoproteinémia
závraty
trombocytopénia, zvýšený medzinárodný normalizovaný pomer (INR)
hypofibrinogenémia
anafylaxia/anafylaktoidné reakcie* (pozri časti 4.3
a 4.4)
Poruchy ciev flebitída tromboflebitída
Poruchy gastrointestinálneh o traktu
Poruchy pečene a žlčových ciest
nauzea, vracanie, hnačka
bolesť brucha, dyspepsia, anorexia zvýšená hladina aspartát
aminotransferázy (AST) v sére a zvýšená hladina
alanín aminotransferázy
(ALT) v sére, hyperbilirubinémi a
akútna pankreatitída (pozri časť 4.4) žltačka, poškodenie pečene, prevažne cholestatické
zlyhávanie pečene* (pozri časť 4.4)
Poruchy kože a podkožného tkaniva
svrbenie, vyrážky závažné kožné reakcie, vrátane Stevens- Johnsonovho syndrómu*
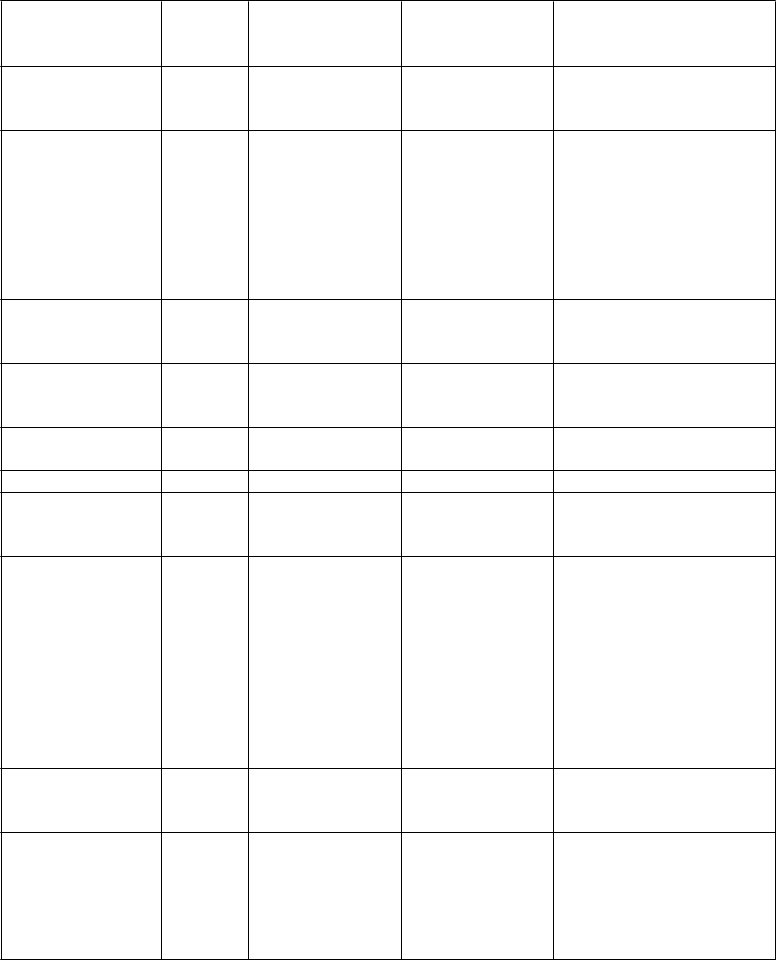
Celkové poruchy a reakcie v mieste podania
zhoršené hojenie, reakcia v mieste podania, bolesť hlavy
zápal v mieste podania, bolesť
v mieste podania,
opuch v mieste podania, flebitída v mieste podania
Trieda orgánových systémov Laboratórne a funkčné vyšetrenia
Veľmi časté (≥1/10)
Časté ≥1/100 až
<1/10
zvýšená hladina amylázy v sére, zvýšená hladina dusíka močoviny
v krvi (blood urea nitrogen, BUN)
Menej časté
≥1/1 000 až
<1/100
Neznáme (z dostupných údajov)

*Nežiaduca reakcia identifikovaná po uvedení lieku na trh
OpisvybranýchnežiaducichreakciíÚčinky triedy antibiotíkPseudomembranózna kolitída, ktorá môže mať rozsah závažnosti od miernej až po život ohrozujúcu
(pozri časť 4.4).
Nadmerný rast necitlivých organizmov vrátane húb (pozri časť 4.4).
Účinky triedy tetracyklínovAntibiotiká glycylcyklínovej triedy sú štruktúrne podobné antibiotikám tetracyklínovej triedy. Nežiaduce účinky tetracyklínovej triedy môžu zahŕňať fotosenzitivitu, pseudotumor cerebri,
pankreatitídu a antianabolický účinok, ktorý viedol ku zvýšeniu dusíka močoviny v krvi (BUN),
azotémii, acidóze a hyperfosfatémii (pozri časť 4.4).
Tigecyklín môže súvisieť so stálou zmenou farby zubov u ľudí, ak sa použije v období vývoja zubov
(pozri časť 4.4).
V cSSTI a cIAI klinických štúdiách 3. a 4. fázy boli závažné nežiaduce reakcie súvisiace s infekciami častejšie hlásené u osôb liečených tigecyklínom (7,1 %) v porovnaní s pacientmi liečenými komparátorom (5,3 %). Pozorovali sa signifikantné rozdiely v sepse/septickom šoku medzi tigecyklínom (2,2 %) a komparátorom (1,1 %).
Abnormality v hladinách AST a ALT sa u pacientov liečených tigecyklínom zaznamenali častejšie v období po ukončení liečby v porovnaní s pacientmi liečenými komparátorom, u ktorých sa zaznamenali častejšie počas liečby.
Vo všetkých klinických štúdiách 3. a 4. fázy (cSSTI a cIAI) sa úmrtie vyskytlo u 2,4 % (54/2216) pacientov, ktorým sa podával tigecyklín a u 1,7 % (37/2206) pacientov, ktorým sa podávali aktívne komparátory.
Pediatrická populáciaZ dvoch farmakokinetických štúdií boli dostupné veľmi obmedzené údaje o bezpečnosti (pozri časť
5.2). Nepozorovali sa žiadne nové alebo neočakávané situácie týkajúce sa bezpečnosti tigecyklínu v týchto štúdiách.
V otvorenej farmakokinetickej štúdii zvyšujúcej sa jednorazovej dávky sa bezpečnosť tigecyklínu skúmala u 25 detí vo veku 8 až 16 rokov, ktoré sa v nedávnej minulosti zotavili z infekcií. Profil nežiaducich reakcií tigecyklínu u týchto 25 jedincov bol vo všeobecnosti totožný s profilom
u dospelých.
Bezpečnosť tigecyklínu sa tiež skúmala v otvorenej farmakokinetickej štúdii so zvyšujúcimi sa viacnásobnými dávkami u 58 detí vo veku 8 až 11 rokov s cSTTI (n = 15), cIAI (n = 24) alebo s pneumóniou získanou v komunite (n = 19). Profil nežiaducich reakcií tigecyklínu u týchto
58 jedincov bol vo všeobecnosti totožný s profilom u dospelých, s výnimkou nauzey (48,3 %),
vracania (46,6 %) a zvýšenej aktivity lipázy v sére (6,9 %), ktoré boli pozorované častejšie ako u dospelých.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieNie sú k dispozícii špecifické informácie o liečbe predávkovania. Intravenózne podanie tigecyklínu formou jednorazovej dávky 300 mg počas 60 minút zdravým dobrovoľníkom viedlo k zvýšenému výskytu nauzey a vracania. Hemodialýzou sa podstatné množstvo tigecyklínu neodstraňuje.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antibakteriálne lieky na systémové použitie, tetracyklíny
ATC kód: J01AA12
Mechanizmus účinkuTigecyklín, glycylcyklínové antibiotikum, inhibuje transláciu proteínov v baktériách tak, že sa viaže
na 30S ribozomálnu subjednotku a blokuje vstup aminoacyl tRNA molekúl na A miesto ribozómu. Toto zabraňuje včleneniu sa aminokyselinových reziduí k predlžujúcim sa reťazcom peptidu.
Všeobecne sa tigecyklín považuje za bakteriostatikum. Pri 4-násobnej hodnote minimálnej inhibičnej koncentrácie (minimal inhibitory concentration, MIC) sa pri tigecyklíne pozoroval 2-log pokles počtu kolónií
Enterococcus spp.,
Staphylococcus aureus a
Escherichia coli.
Mechanizmus rezistencieTigecyklín je schopný prekonať dva významné mechanizmy rezistencie na tetracyklín, ribozómovú
ochranu a eflux. V dôsledku prítomnosti efluxných púmp spôsobujúcich multirezistenciu (multi-drug resistance, MDR) sa preukázala skrížená rezistencia voči tigecyklínu u izolátov z čeľade
Enterobacteriaceae rezistentných na minocyklín. Medzi tigecyklínom a väčšinou tried antibiotík nedochádza k skríženej rezistencii týkajúcej sa cieľového miesta.
Tigecyklín je citlivý na chromozómovo kódované efluxné pumpy spôsobujúce multirezistenciu
Proteeae a
Pseudomonas aeruginosa. Patogény z čeľade
Proteeae (
Proteus sp.,
Providencia sp. a
Morganella sp.) sú všeobecne menej citlivé na tigecyklín ako iní členovia patriaci
k
Enterobacteriaceae. Znížená citlivosť u oboch skupín sa pripisuje nadmernej expresii nešpecifickej efluxnej pumpy AcrAB spôsobujúcej multirezistenciu. Znížená citlivosť u
Acinetobacter baumannii sa
pripisuje zvýšenej expresii efluxnej pumpy AdeABC.
Hranice citlivostiHodnoty minimálnej inhibičnej konentrácie (MIC) stanovené Európskou Komisiou pre testovanie
antimikrobiálnej citlivosti (European Committee on Antimicrobial Susceptibility Testing, EUCAST)
sú nasledovné:
 Staphylococcus
Staphylococcus sp.
S £ 0,5 mg/l a R > 0,5 mg/l
Streptococcus sp. okrem
S. pneumoniae S £ 0,25 mg/l a R > 0,5 mg/l
Enterococcus sp. S £ 0,25 mg/l a R > 0,5 mg/l
Enterobacteriaceae S £ 1(^) mg/l a R > 2 mg/l
(^)Tigecyklín má zníženú aktivitu
in vitro voči
Proteus, Providencia a
Morganella sp.
Pre anaeróbne baktérie existujú klinické dôkazy účinnosti voči polymikrobiálnym intraabdominálnym infekciám, ale nie je k dispozícii korelácia medzi hodnotami MIC, farmakokinetickými/ farmakodynamickými údajmi a klinickým výsledkom. Preto sa neudávajú hranice citlivosti. Je potrebné poznamenať, že distribúcie MIC pre organizmy rodov Bacteroides a Clostridium sú široké
a môžu zahŕňať hodnoty vyššie ako 2 mg/l tigecyklínu.
K dispozícii sú limitované dôkazy o klinickej účinnosti tigecyklínu na Enterococci. Ukázalo sa však, že v klinických skúšaniach polymikrobiálne intraabdominálne infekcie reagujú na liečbu tigecyklínom.
CitlivosťPrevalencia získanej rezistencie sa môže v prípade určitých kmeňov líšiť v závislosti od geografických
podmienok a času a potrebné sú miestne informácie o rezistencii, najmä ak sa liečia závažné infekcie. Podľa potreby sa má vyhľadať odborná rada, ak je miestne rozšírenie rezistencie také, že je použitie
lieku aspoň u niektorých typov infekcií otázne.
PatogénBežne citlivé kmeneGrampozitívneaeróbyEnterococcus spp.†
Staphylococcus aureus* Staphylococcus epidermidis Staphylococcus haemolyticus Streptococcus agalactiae*Skupina
Streptococcus anginosu* (zahŕňa
S. anginosus, S. intermedius a S. constellatus)
Streptococcus pyogenes*Streptokoky zo skupiny Viridans
GramnegatívneaeróbyCitrobacter freundii* Citrobacter koseri Escherichia coli* Klebsiella oxytoca*AnaeróbyClostridium perfringens†
Peptostreptococcus sp.†
Prevotella sp.
 Patogén
Kmene, u ktorých môže byť problémom získaná rezistencia
Gramnegatívne
aeróby
Acinetobacter baumannii Burkholderia cepacia Enterobacter aerogenes Enterobacter cloacae* Klebsiella pneumoniae* Morganella morganii Proteus
Patogén
Kmene, u ktorých môže byť problémom získaná rezistencia
Gramnegatívne
aeróby
Acinetobacter baumannii Burkholderia cepacia Enterobacter aerogenes Enterobacter cloacae* Klebsiella pneumoniae* Morganella morganii Proteus sp.
Providencia sp.
Serratia marcescensStenotrophomonas maltophiliaAnaeróbyskupina Bacteroides fragilis †
Organizmy s prirodzenou rezistenciou GramnegatívneaeróbyPseudomonas aeruginosa*označuje kmene, aktivita voči ktorým sa považuje za uspokojivo dokázanú v klinických štúdiách.
† pozri časť 5.1
Hranice citlivosti, vyššie.
Elektrofyziologické vyšetreniesrdcaV randomizovanej, placebom i aktívne kontrolovanej prekríženej štúdii so 46 zdravými osobami a so
štyrmi ramenami, zameranej na QTc interval, sa nezistil žiadny významný vplyv na QTc interval po podaní jednej intravenóznej dávky tigecyklínu 50 mg alebo 200 mg
Pediatrická populáciaV otvorenej štúdii so zvyšujúcou sa viacnásobnou dávkou sa 39 deťom vo veku 8 až 11 rokov s cIAI
alebo cSSTI podával tigecyklín (0,75; 1 alebo 1,25 mg/kg). Všetci pacienti dostávali i.v. tigecyklín počas minimálne 3 po sebe nasledujúcich dní do maximálne 14 po sebe nasledujúcich dní
s možnosťou prechodu na perorálne antibiotikum v 4. dni alebo neskôr.
Klinické vyliečenie sa hodnotilo medzi 10. a 21. dňom po poslednej dávke. Súhrn výsledkov klinickej odpovede v modifikovanej populácii so zámerom liečiť sa (modified intent-to-treat, mITT) je uvedený v nasledujúcej tabuľke.
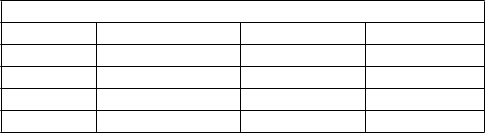 Klinické vyliečenie, populácia mITT
Klinické vyliečenie, populácia mITT0,75 mg/kg 1 mg/kg 1,25 mg/kg Indikácia n/N (%) n/N (%) n/N (%) cIAI 6/6 (100,0) 3/6 (50,0) 10/12 (83,3) cSSTI 3/4 (75,0) 5/7 (71,4) 2/4 (50,0) Celkom 9/10 (90,0) 8/13 (62,0) 12/16 (75,0)
Údaje týkajúce sa účinnosti, ktoré sú uvedené vyššie, je potrebné hodnotiť s opatrnosťou, pretože
v tejto štúdii bolo povolené súbežné podávanie aj iných antibiotík. Okrem toho je potrebné vziať do úvahy aj malý počet pacientov.
5.2 Farmakokinetické vlastnostiAbsorpciaTigecyklín sa podáva intravenózne, a preto je jeho biologická dostupnosť 100 %.
Distribúcia
Väzba tigecyklínu na proteíny plazmy in vitro bola pri koncentráciách pozorovaných v klinických
štúdiách (0,1 do 1,0 mcg/ml) v rozmedzí približne od 71 % do 89 %. Farmakokinetické štúdie na zvieratách a ľuďoch preukázali, že sa tigecyklín ľahko distribuuje do tkanív.
U potkanov, ktorý dostávali jednu alebo viacnásobné dávky 14C-tigecyklínu, bola rádioaktivita dobre distribuovaná do väčšiny tkanív. Najvyššia celková expozícia sa pozorovala v kostnej dreni, slinných žľazách, štítnej žľaze, slezine a obličkách. U ľudí bol distribučný objem tigecyklínu v rovnovážnom stave v priemere od 500 do 700 l (7 až 9 l/kg), čo naznačuje, že sa rozsiahlo distribuuje nad rámec objemu plazmy a koncentruje sa v tkanivách.'
Nie sú k dispozícii žiadne údaje o tom, či tigecyklín prechádza cez ľudskú hematoencefalickú bariéru. V klinických farmakologických štúdiách s použitím terapeutického dávkovacieho režimu 100 mg
nasledovaných 50 mg každých 12 hodín bola hodnota Cmax tigecyklínu v plazme v rovnovážnom stave
866±233 ng/ml v prípade 30 minútových infúzií a 634±97 ng/ml v prípade 60 minútových infúzií.
Hodnota AUC0-12h v ustálenom stave bola 2349±850 ng•h/ml.
Biotransformácia
V priemere sa odhaduje, že pred vylúčením sa metabolizuje menej ako 20 % tigecyklínu. U zdravých
dobrovoľníkov, mužov, bol po podaní 14C-tigecyklínu nezmenený tigecyklín primárnym 14C- značeným materiálom prítomným v moči a stolici, ale glukuronid, N-acetylovaný metabolit a epimér
tigecyklínu boli taktiež prítomné.
Štúdie in vitro s ľudskými pečeňovými mikrozómami naznačujú, že tigecyklín neinhibuje kompetitívnou inhibíciou metabolizmus sprostredkovaný niektorým z týchto 6 izoforiem cytochrómu P450 (CYP): 1A2, 2C8, 2C9, 2C19, 2D6, a 3A4. Okrem toho, tigecyklín nepreukázal závislosť od NADPH pri inhibícii CYP2C9, CYP2C19, CYP2D6 a CYP3A, čo naznačuje chýbanie inhibície založenej na mechanizme týchto CYP enzýmov.
Eliminácia
Návratnosť celkovej rádioaktivity v stolici a moči po podaní 14C-tigecyklínu naznačuje, že 59 % dávky sa eliminuje bil iárnou/fekálnou cestou a 33 % sa vylučuje v moči. Celkovo je primárnou cestou eliminácie tigecyklínu biliárna exkrécia nezmeneného tigecyklínu. Sekundárnymi cestami sú glukuronidácia a renálna exkrécia nezmeneného tigecyklínu.
Po intravenóznej infúzii je celkový klírens tigecyklínu 24 l/hod. Renálny klírens je približne 13 % celkového klírensu. Tigecyklín vykazuje polyexponenciálny priebeh eliminácie z plazmy so stredným terminálnym eliminačným polčasom 42 hodín po viacnásobných dávkach, aj keď existuje vysoká interindividuálna variabilita.
Štúdie in vitro s bunkami Caco-2 ukazujú, že tigecyklín neinhibuje tok digoxínu a nasvedčujú, že tigecyklín nie je inhibítor P-glykoproteínu (P-gp). Táto in vitro informácia je zhodná s nedostatočným účinkom tigecyklínu na klírens digoxínu zaznamenanom v in vivo štúdii na liekovú interakciu opísanej vyššie (pozri časť 4.5).
Tigecyklín je P-gp substrát na základe in vitro štúdie s bunkovou líniou exprimujúcou P-gp. Možný prínos P-gp sprostredkovaného transportu do in vivo povahy tigecyklínu nie je známy. Súčasné podávanie P-gp inhibítorov (napr. ketokonazol alebo cyklosporín) alebo P-gp induktora (napr. rifampicín) môže ovplyvniť farmakokinetiku tigecyklínu.
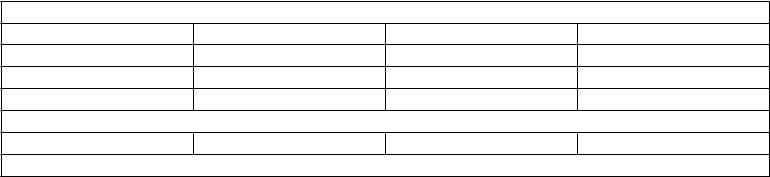 Osobitné skupinypacientovPorucha funkcie pečene
Osobitné skupinypacientovPorucha funkcie pečeneFarmakokinetická dispozícia tigecyklínu po podaní jednorazovej dávky sa nemenila u pacientov
s miernou poruchou funkcie pečene. Avšak systémový klírens tigecyklínu sa u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene (stupne B a C skóre podľa Childa a Pugha) znížil o 25
% a 55 % a biologický polčas sa predĺžil o 23 % a 43 % (pozri časť 4.2).
Porucha funkcie obličiekFarmakokinetická dispozícia tigecyklínu po podaní jednorazovej dávky sa nemenila u pacientov
s obličkovou nedostatočnosťou (klírens kreatinínu <30 ml/min, n=6). Pri závažnej poruche funkcie obličiek bol AUC parameter o 30 % vyšší ako u osôb s normálnou funkciou obličiek (pozri časť 4.2).
Staršie osobyNepozorovali sa žiadne celkové rozdiely vo farmakokinetike zdravých starších a mladších osôb (pozri časť 4.2).
Pediatrická populáciaFarmakokinetika tigecyklínu sa skúmala v dvoch štúdiách. Do prvej štúdii boli zaradené deti vo veku
8-16 rokov (n=24), ktoré dostali jednu dávku tigecyklínu ( 0,5 mg/kg až do maximálnej dávky 50 mg,
1 mg/kg až do maximálnej dávky 100 mg alebo 2 mg/kg až do maximálnej dávky 150 mg) podávanú intravenózne v 30 minútovej infúzii. Druhá štúdia sa uskutočnila u detí vo veku od 8 do 11 rokov, ktoré dostali opakované dávky tigecyklínu (0,75, 1 alebo 1,25 mg/kg až po maximálnu dávku 50 mg) podávané každých 12 hodín v 30 minútovej infúzii. V týchto štúdiách nebola podaná nasycovacia dávka. Farmakokinetické parametre sú zhrnuté v tabuľke nižšie.
Dávka normalizovaná na 1 mg/kg priemer ± SD tigecyklín Cmax a AUC u detíVek N Cmax (ng/ml) AUC (ng•h/ml)*
Jedna dávka
8 – 11 8 3 881 ± 6 637 4 034 ± 2 874
12 - 16 16 8 508 ± 11 433 7 026 ± 4 088
Opakovaná dávka
8 - 11 42 1 911 ± 3 032 2 404 ± 1 000
* jedna dávka AUC0-∞, opakovaná dávka AUC0-12h
Cieľová hladina AUC0-12h u dospelých po odporučenej dávke 100 mg nasýtenia a 50 mg každých
12 hodín bola priemerne 2500 ng•h/ml.
PK analýza populácie z obidvoch štúdií identifikovala telesnú hmotnosť ako kovariát klírensu tigecyklínu u detí vo veku 8 rokov a starších. Dávkovací režim 1,2 mg/kg každých 12 hodín (do maximálnej dávky 50 mg každých 12 hodín) u detí vo veku 8 až < 12 rokov a 50 mg každých 12 hodín u dospievajúcich vo veku 12 až < 18 rokov by pravdepodobne viedol k expozíciám, ktoré sú porovnateľné s expozíciami pozorovanými u dospelých liečených schváleným dávkovacím režimom.
V týchto štúdiách sa u niekoľkých detí pozorovali vyššie hodnoty Cmax, ako u dospelých pacientov. V dôsledku toho sa má u detí a dospievajúcich venovať pozornosť rýchlosti infúzie tigecyklínu.
PohlavieNepozorovali sa klinicky významné rozdiely v klírense tigecyklínu medzi mužmi a ženami. AUC
parameter bol odhadnutý o 20 % vyšší u žien ako u mužov.
RasaNepozorovali sa rozdiely v klírense tigecyklínu v závislosti od rasy.
Telesná hmotnosť
Klírens, klírens normalizovaný na hmotnosť a AUC neboli značne odlišné u pacientov s rozdielnou telesnou hmotnosťou, vrátane tých, ktorí vážili 125 kg. AUC parameter bol o 24 % nižší
u pacientov vážiacich 125 kg. Nie sú k dispozícii údaje od pacientov s telesnou hmotnosťou 140
kg a vyššou.
5.3 Predklinické údaje o bezpečnosti
V štúdiách toxicity opakovanej dávky u potkanov a psov sa pozorovali pri expozícii tigecyklínu
8-násobne a 10-násobne vyššej ako je ľudská denná dávka, určenej na základe AUC u potkanov
a psov, lymfoidná deplécia/atrofia lymfatických uzlín, sleziny a týmusu, znížený počet erytrocytov, retikulocytov, leukocytov a krvných doštičiek v súvislosti s hypocelularitou kostnej drene a nežiaduce účinky na obličky a gastrointestinálny trakt. Tieto zmeny sa ukázali byť reverzibilné po dvoch týždňoch dávkovania.
U potkanov sa pozorovala zmena farby kostí, ktorá po dvoch týždňoch dávkovania nebola reverzibilná.
Výsledky štúdií na zvieratách naznačujú, že tigecyklín prechádza placentou a je prítomný v tkanive plodu. V štúdiách reprodukčnej toxicity s tigecyklínom sa pozoroval pokles telesnej hmotnosti plodov potkanov a králikov (spojený s oneskorenou osifikáciou) a potraty u králikov. U potkanov a králikov nebol tigecyklín teratogénny. Tigecyklín neovplyvňoval schopnosť párenia alebo fertilitu u potkanov pri expozíciách vyšších ako 4,7-násobok dennej dávky u ľudí založenej na AUC. U samíc potkanov sa pri expozíciách vyšších ako 4,7-násobok denne dávky u ľudí založenej na AUC nezistili žiadne účinky na vaječníky alebo estrálne cykly, ktoré by súviseli so zlúčeninou.
Výsledky zo štúdií na zvieratách s použitím 14C-označeného tigecyklínu naznačujú, že sa tigecyklín rýchlo vylučuje do mlieka dojčiacich potkanov. V zhode s limitovanou perorálnou biodostupnosťou tigecyklínu sa u dojčených mláďat zaznamenala len nízka alebo žiadna systémová expozícia tigecyklínu ako následok expozície cestou materského mlieka.
Celoživotné štúdie na zvieratách za účelom hodnotenia karcinogénneho potenciálu tigecyklínu sa neuskutočnili, ale krátkodobé štúdie genotoxicity tigecyklínu boli negatívne.
V štúdiách na zvieratách bolo bolusové intravenózne podanie tigecyklínu spojené s histamínovou odpoveďou. Tieto účinky sa pozorovali pri expozíciách 14- a 3-násobne vyšších ako je ľudská denná dávka, založených na AUC potkanov a psov.
U potkanov, ktorým sa podal tigecyklín, sa nepozorovala fotosenzitivita.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát laktózy
Kyselina chlorovodíková Hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nasledujúce liečivá sa nemajú podávať súbežne s tigecyklínom cez rovnaké rameno trojcestného kohútika: amfotericín B, lipidový komplex amfotericínu B, diazepam, ezomeprazol, omeprazol
a intravenózne roztoky, ktoré môžu zvýšiť pH na viac ako 7.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
2 roky
Po rekonštitúcii a riedení vo vaku alebo inej vhodnej infúznej nádobe (napr. sklenej fľaši) sa má tigecyklín ihneď použiť.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25 °C.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Číre sklené injekčné liekovky typu I s objemom 5 ml uzavreté sivými zátkami z butylovej gumy a hliníkovými uzávermi so snímateľnou čiapočkou. Tygacil sa dodáva v baleniach obsahujúcich podložku s desiatimi injekčnými liekovkami.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prášok sa má pripraviť pomocou 5,3 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), injekčného roztoku glukózy 50 mg/ml (5 %) alebo injekčného Ringerovho roztoku s laktátom, aby sa dosiahla koncentrácia 10 mg/ml tigecyklínu. Injekčnou liekovkou sa má jemne otáčať, kým sa liek nerozpustí. Následne sa má ihneď odobrať z injekčnej liekovky 5 ml pripraveného roztoku a pridať do 100 ml vaku na intravenóznu infúziu alebo do inej vhodnej infúznej nádoby (napr. sklenej fľaše).
Na prípravu dávky 100 mg rozpustite dve injekčné liekovky do 100 ml vaku na intravenóznu infúziu alebo do inej vhodnej infúznej nádoby (napr. sklenej fľaše). Upozornenie: Injekčná liekovka obsahuje
6 % prebytok. Preto je 5 ml pripraveného roztoku zodpovedá 50 mg liečiva. Pripravený roztok má byť
žltej až oranžovej farby. Ak taký nie je, má sa zlikvidovať. Parenterálne lieky sa majú pred podaním vizuálne skontrolovať na prítomnosť častíc a zmenu farby (napr. zelená alebo čierna).
Tigecyklín sa má podávať intravenózne cez samostatnú linku alebo cez rameno trojcestného kohútika. Ak sa použije rovnaká intravenózna linka pre sekvenčnú infúziu niekoľkých liečiv, má sa pred infúziou tigecyklínu a po nej prepláchnuť buď injekčným roztokom chloridu sodného 9 mg/ml (0,9%) alebo injekčným roztokom glukózy 50 mg/ml (5 %). Injekcia sa má podať s infúznym roztokom kompatibilným s tigecyklínom a ktorýmkoľvek iným liekom podávaným cez spoločnú linku (pozri časť 6.2).
Roztok je len na jedno použitie; nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.> Kompatibilné intravenózne roztoky zahŕňajú: injekčný roztok chloridu sodného 9 mg/ml (0,9 %), injekčný roztok glukózy 50 mg/ml (5 %) a Ringerov roztok s laktátom.
Ak sa podáva cez rameno trojcestného kohútika, kompatibilita tigecyklínu zriedeného 0,9 % injekčným roztokom chloridu sodného sa preukázala s nasledujúcimi liekmi alebo roztokmi na riedenie: amikacín, dobutamín, dopamíniumchlorid, gentamicín, haloperidol, Ringerov roztok
s laktátom, lidokaíniumchlorid, metoklopramid, morfín, noradrenalín, piperacilín/tazobaktám
(s obsahom EDTA), chlorid draselný, propofol, ranitidíniumchlorid, teofylín a tobramycín.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Limited Ramsgate Road Sandwich
Kent CT13 9NJ
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/336/001
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 24. apríl 2006
Dátum posledného predĺženia registrácie: 22. február 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
http://www.ema.europa.eu.