
si vyžadujú liečbu ritonavirom potencovanými inhibítormi proteáz, je najmä v začiatočnej fáze potrebné dôsledne monitorovať pacientovu znášanlivosť voči Tracleeru, predovšetkým riziko hypotenzie a hepatálne pečeňové testy. Zvýšené dlhodobé riziko hepatálnej toxicity a hematologických nežiaducich účinkov nemožno vylúčiť, ak sa bosentan používa v kombinácii s antiretrovirálnymi liekmi. Vzhľadom na možnosť interakcií vzťahujúcich sa najmä na indukčný účinok bosentanu na CYP 450 (pozri časť 4.5), ktoré by mohli ovplyvniť účinnosť antiretrovirálnej liečby, musia byť títo pacienti tiež dôkladne monitorovaní
s ohľadom na liečbu svojej HIV infekcie.
Sekundárna pľúcna hypertenzia v súvislostischronickouobštrukčnouchoroboupľúc(CHOCHP)
Bezpečnosť a tolerancia bosentanu boli sledované v 12týždňovej výskumnej nekontrolovanej štúdii
u 11 pacientov s pľúcnou hypertenziou, ktorá vznikla sekundárne v súvislosti so závažnou CHOCHP (stupeň III podľa klasifikácie GOLD). Boli pozorované zvýšenie minútovej ventilácie a pokles saturácie kyslíkom. Najčastejším nežiaducim účinkom bolo dyspnoe (sťažené dýchanie), ktoré rozhodlo o prerušení terapie bosentanom.
Súbežné použitie s inýmiliečivami
Súbežné používanie Tracleeru s cyklosporínom A je kontraindikované (pozri časti 4.3 a 4.5).
Súbežné používanie Tracleeru s glibenklamidom, flukonazolom a rifampicínom sa neodporúča. Pre podrobnejšie informácie pozri časť 4.5.
Je nutné vyhnúť sa súbežnému podávaniu Tracleeru s inhibítormi CYP3A4 a s inhibítormi CYP2C9
(pozri časť 4.5).
4.5 Liekové a iné interakcie
Bosentan je induktor cytochrómu P 450 (CYP) izoenzýmov CYP2C9 a CYP3A4. In vitro získané dáta tiež naznačujú indukciu CYP2C19. V prípade súbežného podávania Tracleeru sa plazmatické hladiny liečiv metabolizovaných týmito izoenzýmami znížia. Je nutné zohľadniť možnosť zmeny účinnosti liečiv, ktoré sú týmito izoenzýmami metabolizované. Dávkovanie týchto liekov môže vyžadovať úpravu po začatí, zmene dávky alebo po prerušení súbežnej liečby Tracleerom.
Bosentan je metabolizovaný enzýmami CYP2C9 a CYP3A4. Inhibícia týchto izoenzýmov môže zvýšiť plazmatickú hladinu bosentanu (pozri ketokonazol). Vplyv inhibítorov CYP2C9 na hladinu bosentanu nebol študovaný. Táto kombinácia sa má používať veľmi opatrne.
Flukonazol a iné inhibítory CYP2C9 a CYP3A4: Súbežné podávanie s flukonazolom, ktorý inhibuje najmä CYP2C9 a do určitej miery aj CYP3A4, môže viesť k výraznému zvýšeniu plazmatických hladín bosentanu, a preto sa táto kombinácia neodporúča. Z toho istého dôvodu sa s Tracleerom neodporúča súbežné podávanie silného inhibítora CYP3A4 (napr. ketokonazolu, itrakonazolu alebo ritonaviru) a inhibítora CYP2C9 (napr. vorikonazolu).
Cyklosporín A: Súbežné podávanie Tracleeru a cyklosporínu A (inhibítor kalcineurínu) je kontraindikované (pozri časť 4.3). Ak sa obidva lieky podávali súbežne, bola najnižšia nameraná začiatočná hladina bosentanu približne tridsaťkrát vyššia ako hladina nameraná pri používaní iba samotného bosentanu. Pri rovnovážnom stave boli plazmatické hladiny bosentanu 3- až 4krát vyššie než v prípade monoterapie bosentanom. Mechanizmus tejto interakcie spočíva s najväčšou pravdepodobnosťou v inhibícii transportným proteínom mediovaného vychytávania bosentanu do hepatocytov cyklosporínom. Krvné hladiny cyklosporínu A (substrát CYP3A4) sa znížili približne o
50 %. Toto je pravdepodobne v dôsledku indukcie CYP3A4 bosentanom.
Takrolimus, sirolimus: Súbežné podávanie takrolimu alebo sirolimu a Tracleeru u ľudí nebolo skúmané, ale spoločné podávanie takrolimu alebo sirolimu a Tracleeru môže spôsobiť zvýšenie plazmatickej hladiny bosentanu rovnako ako súbežné podávanie s cyklosporínom A. Súbežné podávanie Tracleeru môže znížiť plazmatickú hladinu takrolimu a sirolimu. Preto sa súbežné podávanie Tracleeru a takrolimu alebo sirolimu neodporúča. Pacienti, ktorí vyžadujú podávanie spomínanej kombinácie, musia byť starostlivo sledovaní, či nedochádza k výskytu nežiaducich účinkov súvisiacich s hladinami Tracleeru a takrolimu a sirolimu v krvi.
Glibenklamid: Súbežné podávanie bosentanu 125 mg 2krát denne počas 5 dní znížilo plazmatickú hladinu glibenklamidu (substrát CYP3A4) o 40 %, s potenciálne signifikantným znížením hypoglykemického efektu. Plazmatické hladiny bosentanu tiež poklesli o 29 %. Okrem toho bol
u pacientov, ktorí sa podrobili súbežnej liečbe glibenklamidom, pozorovaný častejší vzostup hladín
aminotransferáz. Glibenklamid a bosentan inhibujú exportnú pumpu žlčových solí, čím by sa dali vysvetliť zvýšené hladiny aminotransferáz. Táto kombinácia sa nemá používať. Žiadne údaje
o liekových interakciách s ostatnými derivátmi sulfonylmočoviny nie sú k dispozícii.
Rifampicín: Súbežné podávanie bosentanu 125 mg dvakrát denne počas 7 dní s rifampicínom, potentným induktorom CYP2C9 a CYP3A4, 9 zdravým osobám znížilo plazmatické hladiny bosentanu o 58 %, pričom tento pokles dosahoval u jednotlivcov až 90 %. Ako výsledok možno preto pri súbežnom podávaní bosentanu s rifampicínom očakávať významné zníženie účinku bosentanu. Súbežné podávanie Tracleeru s rifampicínom sa preto neodporúča. Údaje o iných induktoroch CYP3A4 ako karbamazepín, fenobarbital, fenytoín a ľubovník bodkovaný nie sú dostupné, ale predpokladá sa, že ich súbežné podávanie s Tracleerom môže viesť k zníženej systémovej expozícii bosentanu. Klinicky významné zníženie účinku nemožno vylúčiť.
Lopinavir + ritonavir (a ďalšie ritonavirom potencované inhibítory proteáz): Súbežné podávanie bosentanu 125 mg dvakrát denne a lopinaviru + ritonaviru 400 + 100 mg dvakrát denne počas 9,5 dňa zdravým dobrovoľníkom viedlo k zvýšeniu plazmatickej hladiny bosentanu, ktorá bola 48-násobne vyššia ako pri podaní samotného bosentanu. Na deviaty deň bola plazmatická hladina bosentanu 5- násobne vyššia ako v prípade, keď bol bosentan podávaný samostatne. Táto interakcia je spôsobená pravdepodobne inhibíciou vychytávania bosentanu do hepatocytov, ktoré je sprostredkované transportným proteínom a inhibíciou CYP3A4 ritonavirom a redukciou klírens bosentanu. Pokiaľ sa Tracleer podáva súbežne s lopinavirom + ritonavirom alebo inými ritonavirom potencovanými inhibítormi proteáz, je potrebné sledovať znášanlivosť pacientov voči Tracleeru.
Po 9,5 dňoch súbežného podávania bosentanu, plazmatické hladiny lopinaviru a ritonaviru klesli na klinicky nevýznamné hodnoty (o približne 14 % a 17 %, v uvedenom poradí). Aj keď sa nemusí dosiahnuť celková indukcia bosentanu, nemožno vylúčiť ďalší pokles inhibítorov proteáz. Odporúča sa primeraný monitoring terapie HIV. Podobné účinky možno očakávať aj s inými ritonavirom potencovanými inhibítormi proteáz (pozri časť 4.4).
Iné antiretrovirálne lieky: Vzhľadom na nedostatok informácií nemožno formulovať ďalšie špeciálne odporúčania ohľadom užívania iných dostupných antiretrovirálnych liekov. Treba zdôrazniť, že výrazná hepatotoxicita nevirapinu môže potencovať pečeňovú toxicitu bosentanu, táto kombinácia sa neodporúča.
Hormonálna antikoncepcia: Súbežné podávanie bosentanu 125 mg dvakrát denne s jednou dávkou perorálneho kontraceptíva obsahujúceho noretisterón 1 mg + etinylestradiol 35 µg počas 7 dní znížilo AUC noretisterónu o 14 % a etinylestradiolu o 31 %. U jednotlivcov však bolo pozorované zníženie expozície až o 56 % pri noretisteróne a o 66 % pri etinylestradiole. Práve preto sa používanie len hormonálnej antikoncepcie ako jedinej metódy antikoncepcie nezávisle od spôsobu aplikácie (t.j. perorálnej, injekčnej, transdermálnej alebo implantabilnej) nepovažuje za spoľahlivú metódu antikoncepcie (pozri časti 4.4 a 4.6).
Warfarín: Súbežné podávanie bosentanu 500 mg dvakrát denne počas 6 dní znížilo plazmatické hladiny S-warfarínu (substrát CYP2C9) o 29 % a R-warfarínu (substrát CYP3A4) o 38 %. Počas klinických štúdií u pacientov s PAH, ktorým boli súbežne podávané bosentan a warfarín, neboli pozorované žiadne klinicky relevantné zmeny INR (International Normalized Ratio) ani dávky warfarínu (porovnanie vstupných hodnôt s hodnotami na konci klinických štúdií). Okrem toho, zmeny dávkovania warfarínu v priebehu testov z dôvodu zmeny INR alebo kvôli nežiaducim účinkom, boli rovnako časté u pacientov liečených bosentanom aj u pacientov s placebom. Na začiatku liečby bosentanom nie je nutné upravovať dávky warfarínu alebo podobných perorálnych antikoagulancií, ale odporúča sa intenzívnejší monitoring INR, najmä na začiatku liečby a v období titrácie vyššej dávky.
Simvastatín: Súbežné podávanie bosentanu 125 mg dvakrát denne počas 5 dní znížilo plazmatickú
hladinu simvastatínu (substrát CYP3A4) o 34 % a jeho aktívneho metabolitu β-hydroxykyseliny
o 46 %. Plazmatické hladiny bosentanu neboli súbežne podávaným simvastatínom ovplyvnené. Má sa
zvážiť sledovanie hladiny cholesterolu a prípadná úprava dávky.
Ketokonazol: Súbežné podávanie bosentanu 62,5 mg dvakrát denne počas 6 dní spolu
s ketokonazolom, potentným inhibítorom CYP3A4, zvýšilo plazmatickú hladinu bosentanu približne dvakrát. Nie je nutné upravovať dávku Tracleeru. Napriek tomu, že neboli vykonané štúdie in vivo, podobné zvýšenie plazmatických hladín bosentanu sa dá očakávať aj s inými potentnými inhibítormi CYP3A4 (napr. itrakonazol alebo ritonavir). V prípade kombinácie s inhibítorom CYP3A4
u pacientov s pomalým metabolizmom CYP2C9 avšak existuje riziko závažného zvýšenia
plazmatických hladín bosentanu, ktoré by mohlo vyvolať škodlivé nežiaduce účinky.
Epoprostenol: Obmedzené údaje získané zo štúdie (AC-052-356, [BREATHE-3]) s 10 detskými a dospievajúcimi pacientmi, ktorí dostali kombináciu bosentanu a epoprostenolu, naznačujú, že po jednotlivej aj opakovanej dávke boli hodnoty Cmax a AUC bosentanu podobné u pacientov s kontinuálnou infúziou epoprostenolu alebo bez nej (pozri časť 5.1).
Sildenafil: Súbežné podávanie bosentanu 125 mg dvakrát denne (rovnovážny stav) so sildenafilom
80 mg trikrát denne (rovnovážny stav) počas šiestich dní u zdravých dobrovoľníkov viedlo k 63 % zníženiu AUC sildenafilu a 50 % zvýšeniu AUC bosentanu. Súbežné podávanie týchto látok si vyžaduje zvýšenú pozornosť.
Digoxín: Súbežné podávanie bosentanu 500 mg dvakrát denne počas 7 dní s digoxínom znížilo AUC o 12 %, Cmax o 9 % a Cmin digoxínu o 23 %. Mechanizmom tejto interakcie môže byť indukcia P- glykoproteínu. Nie je pravdepodobné, že by táto interakcia mala klinický význam.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Štúdie na zvieratách preukázali reprodukčnú toxicitu (teratogenitu, embryotoxicitu; pozri časť 5.3).
Neexistujú žiadne spoľahlivé údaje o užívaní Tracleeru u tehotných žien. Stále nie je známe potenciálne riziko u ľudí. Tracleer je kontraindikovaný počas gravidity (pozri časť 4.3).
Ženy vo fertilnom veku
Pred začatím liečby Tracleerom u žien vo fertilnom veku je potrebné overiť, že žena nie je tehotná,
poskytnúť jej potrebné informácie o spoľahlivých metódach antikoncepcie a musí začať používať spoľahlivú antikoncepciu. Pacienti a lekári si musia byť vedomí, že v dôsledku potenciálnych farmakokinetických interakcií Tracleer môže spôsobiť neúčinnosť hormonálnych kontraceptív (pozri časť 4.5). Preto ženy vo fertilnom veku nesmú používať hormonálnu antikoncepciu (vrátane perorálnej, injekčnej, transdermálnej alebo implantabilnej formy) ako jedinú metódu antikoncepcie, ale musia
používať spoľahlivú doplnkovú alebo alternatívnu antikoncepčnú metódu. Pokiaľ sú akékoľvek
pochybnosti, ktorá antikoncepcia sa má odporučiť individuálnej pacientke, odporúča sa konzultácia s gynekológom. Vzhľadom na možné zlyhanie hormonálnej antikoncepcie počas liečby Tracleerom ako aj fakt, že počas gravidity sa závažne zhoršuje pľúcna hypertenzia, počas liečby Tracleerom sa odporúčajú testy gravidity raz za mesiac, ktoré umožnia skorú detekciu gravidity.
Dojčenie
Nie je známe, či bosentan prechádza do ľudského materského mlieka. Dojčenie sa počas liečby
Tracleerom neodporúča.
Fertilita
Štúdie na zvieratách preukázali testikulárne účinky (pozri časť 5.3). V štúdii skúmajúcej účinky
bosentanu na funkciu semenníkov u mužských pacientov s PAH sa po 3 alebo 6 mesiacoch liečby
bosentanom u 8 z 24 pacientov ukázalo zníženie koncentrácie spermy v porovnaní so začiatočnými
hodnotami najmenej o 42 %. Na základe týchto zistení a predklinických údajov nie je možné vylúčiť, že bosentan môže mať u mužov škodlivý účinok na spermatogenézu. U chlapcov po liečbe bosentanom nie je možné vylúčiť dlhodobý vplyv na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne špecifické štúdie, ktoré by priamo sledovali vplyv Tracleeru na schopnosť viesť vozidlá a obsluhovať stroje. Avšak, Tracleer môže vyvolať hypotenziu, závraty, rozmazané videnie alebo synkopy, ktoré môžu ovplyvniť schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
V 20 placebom kontrolovaných štúdiách bol bosentan podávaný vo viacerých indikáciách v dennej dávke 100-2000 mg 2486 pacientom a v skupine s placebom bolo 1838 pacientov. Priemerná dĺžka liečby bola 45 týždňov. Nežiaduce účinky boli definované ako udalosti, ktoré sa vyskytovali minimálne u 1 % pacientov liečených bosentanom, vo frekvencii aspoň o 0,5 % častejšie ako u skupiny s placebom. Najčastejšie nežiaduce reakcie sú bolesť hlavy (11,5 %), edém / retencia tekutín (13,2 %), poruchy pečeňových testov (10,9 %) a anémia / pokles hladiny hemoglobínu (9,9 %).
Liečba bosentanom sa spájala s dávkovo závislým zvýšením pečeňových aminotransferáz ako aj
s poklesom hladiny hemoglobínu (pozri časť 4.4).
Nežiaduce reakcie pozorované v 20 placebom kontrolovaných štúdiách a počas postmarketingového sledovania s bosentanom sú zoradené podľa frekvencie výskytu: veľmi časté (≥ 1/10); časté (≥ 1/100 do < 1/10); menej časté (≥ 1/1000 do < 1/100); zriedkavé (≥ 1/10000 do < 1/1000); veľmi zriedkavé (< 1/10000); neznáme (nedajú sa určiť z dostupných údajov).
V každej kategórii sú nežiaduce reakcie zoradené podľa klesajúcej závažnosti. Neboli pozorované žiadne klinicky významné rozdiely v nežiaducich reakciách medzi celým súborom a jednotlivými schválenými indikáciami.
Orgánový systém Frekvencia Nežiaduca reakcia
Poruchy krvi a lymfatického systému
Časté Anémia, pokles hemoglobínu
(pozri časť 4.4)
Neznáme Anémia alebo pokles hemoglobínu, ktorý si vyžaduje transfúziu červených krviniek1
Menej časté Trombocytopénia1
Menej časté Neutropénia, leukopénia1
Poruchy imunitného systému Časté Hypersenzitívne reakcie (zahŕňajú dermatitídu, svrbenie a vyrážku)2
Zriedkavé Anafylaxia a/alebo angioedém1
Poruchy nervového systému Veľmi časté Bolesť hlavy3
Časté Synkopy1, 4
Poruchy oka Neznáme Rozmazané videnie1
Poruchy srdca a srdcovej
činnosti
Časté Palpitácie1, 4
Cievne poruchy Časté Sčervenanie
Časté Hypotenzia1, 4

Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestiálneho traktu
Časté Upchatý nos1
Časté Refluxná choroba pažeráka, hnačka
Poruchy pečene a žlčových ciest Veľmi časté Abnormálne pečeňové testy
(pozri časť 4.4) Menej časté Zvýšenie hladiny
aminotransferáz spojené
s hepatitídou (vrátane možného zhoršenia skrytej hepatitídy) a/alebo žltačkou1 (pozri časť
4.4)
Zriedkavé Cirhóza pečene, zlyhanie pečene1
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Erytém
Veľmi časté Edém, retencia tekutín5
1Údaje boli získané z postmarketingového sledovania, frekvencie sú založené na štatistickom modelovaní v placebom kontrolovaných klinických štúdiách.
2Hypersenzitívne reakcie boli hlásené u 9,9 % pacientov liečených bosentanom a 9,1 % pacientov v skupine s placebom.
3Bolesť hlavy bola hlásená u 11,5 % pacientov liečených bosentanom a 9,8 % pacientov, ktorým bolo podávané placebo.
4 Tento typ reakcií môže tiež súvisieť s prebiehajúcim ochorením.
5 Edém alebo retencia tekutín boli hlásené u 13,2 % pacientov liečených bosentanom a 10,9 % pacientov, ktorým bolo podávané placebo.
Počas postmarketingového obdobia boli hlásené zriedkavé prípady neobjasnenej cirhózy pečene, ktoré sa vyskytovali u polymorbídnych pacientov, ktorí okrem inej terapie dlhodobo užívali Tracleer. Vyskytovali sa aj zriedkavé prípady zlyhania pečene. Tieto prípady len zvýrazňujú potrebu dodržiavať počas liečby Tracleerom pravidelný mesačný monitoring pečeňových funkcií (pozri časť 4.4).
Pediatrická populácia
Nekontrolované klinické štúdie s pediatrickými pacientmi
Bezpečnostný profil v prvej pediatrickej nekontrolovanej štúdii vykonanej s filmom obalenými tabletami (BREATHE-3: n = 19, medián veku 10 rokov [rozpätie 3 až 15 rokov], otvorená, bosentan
2 mg/kg dvakrát denne; dĺžka liečby 12 týždňov) bol podobný s bezpečnostným profilom pozorovaným v pivotných štúdiách u dospelých pacientov s PAH. V štúdii BREATHE-3 boli najčastejšími nežiaducimi reakciami sčervenanie (21 %), bolesť hlavy a abnormálne pečeňové testy (každé 16 %).
Súhrnná analýza nekontrolovaných pediatrických štúdií vykonaných u PAH s bosentanom, v dávke
32 mg vo forme dispergovateľných tabliet (FUTURE 1/2, FUTURE 3/predĺženie) zahŕňala celkom
100 detí liečených bosentanom v dávke 2 mg/kg dvakrát denne (n = 33), 2 mg/kg trikrát denne (n = 31) alebo 4 mg/kg dvakrát denne (n = 36). Pri zaradení bolo 6 pacientov vo veku medzi 3 mesiacmi až 1 rokom, 15 detí bolo vo veku medzi 1 rokom a menej ako 2 rokmi a 79 detí bolo vo veku medzi 2 rokmi až 12 rokmi. Medián času trvania liečby bol 71,8 týždňa (rozpätie 0,4 až 258 týždňov).
Bezpečnostný profil bol v tejto súhrnnej analýze nekontrolovaných pediatrických štúdií podobný
bezpečnostnému profilu pozorovanému v pivotných štúdiách u dospelých pacientov s PAH
s výnimkou infekcií, ktoré boli hlásené častejšie ako u dospelých (69,0 % vs 41,3 %). Tento rozdiel v početnosti infekcií môže byť sčasti spôsobený vyšším mediánom času expozície liečby
v pediatrickom súbore (medián 71,8 týždňov) v porovnaní so súborom dospelých pacientov (medián
17,4 týždňa). Najčastejšími nežiaducimi udalosťami boli infekcie horných dýchacích ciest (25 %), pľúcna (arteriálna) hypertenzia (20 %), nazofaryngitída (17 %), pyrexia (15 %), vracanie (13 %), bronchitída (10 %), bolesti brucha (10 %) a hnačka (10 %). V početnosti nežiaducich udalostí nebol medzi pacientami staršími a mladšími ako 2 roky žiadny relevantný rozdiel; toto zistenie je však založené len na 21 deťoch mladších ako 2 roky, vrátane 6 pacientov vo veku medzi 3 mesiacmi a 1
rokom. Nežiaduce udalosti pečeňových abnormalít sa objavili u 9 % pacientov a nežiaduce udalosti anémie/poklesu hemoglobínu u 5 % pacientov.
V randomizovanej placebom kontrolovanej štúdii (FUTURE-4), ktorá bola vykonaná u pacientov s PPHN bolo bosentanom, v dávke 2 mg/kg dvakrát denne vo forme dispergovateľných tabliet, liečených celkom 13 novorodencov (8 pacientov bolo na placebe). Medián času liečby bol 4,5 dňa pri bosentane (rozpätie 0,5 až 10,0 dní) a 4,0 dní pri placebe (rozpätie 2,5 až 6,5 dňa). Najčastejšími
nežiaducimi udalosťami u pacientov liečených bosentanom a u pacientov na placebe boli, v uvedenom poradí: anémia alebo pokles hemoglobínu (7 a 2 pacienti), generalizovaný edém (3 a 0 pacientov)
a vracanie (2 a 0 pacientov).
Laboratórne abnormality
Abnormality pečeňových testov
V priebehu klinického programu sa obvykle počas prvých 26 týždňov objavilo dávkovo závislé zvýšenie hladín pečeňových aminotransferáz, rozvinulo sa postupne a bolo prevažne asymptomatické. V post- marketingovej praxi boli hlásené zriedkavé prípady cirhózy pečene a zlyhania pečene.
Mechanizmus tohto nežiaduceho účinku nie je jasný. Hoci sa zvýšená hladina aminotransferáz môže vrátiť do normy spontánne počas pokračujúcej liečby udržiavacou dávkou Tracleeru alebo po znížení dávky, v niektorých prípadoch treba zvážiť prerušenie, prípadne ukončenie liečby (pozri časť 4.4).
V 20 placebom kontrolovaných štúdiách bolo pozorované zvýšenie pečeňových aminotransferáz
≥ 3 × HHN u 11,2 % pacientov liečených bosentanom v porovnaní s 2,4 % pacientov v skupine
s placebom. Zvýšenie na ≥ 8 × HHN bolo zaznamenané u 3,6 % bosentanom liečených pacientov a 0,4 % pacientov, ktorým bolo podávané placebo. U 0,2 % pacientov (5 pacientov) liečených bosentanom a 0,3 % pacientov (6 pacientov), ktorým bolo podávané placebo, zvýšenia hladiny aminotransferáz boli spojené so zvýšenými hladinami bilirubínu (≥ 2× HHN) bez dôkazu obštrukcie žlčových ciest.
V súhrnnej analýze vykonanej u 100 detí s PAH z nekontrolovaných pediatrických štúdií FUTURE
1/2 a FUTURE 3/predĺženie bolo pozorované zvýšenie pečeňových aminotransferáz ≥ 3 × HHN u 2 %
pacientov.
V štúdii FUTURE-4 zahŕňajúcej 13 novorodencov s PPHN liečených bosentanom, v dávke 2 mg/kg dvakrát denne v čase kratšom ako 10 dní (rozpätie 0,5 až 10,0 dní) sa počas liečby bosentanom nevyskytli žiadne prípady zvýšenia pečeňovej aminotransferázy ≥ 3 × HHN, ale 3 dni po ukončení liečby bosentanom sa vyskytol jeden prípad hepatitídy.
Hemoglobín
V placebom kontrolovaných štúdiách u dospelých bol hlásený pokles hladiny hemoglobínu zo vstupnej hodnoty na hodnotu menej ako 10 g/dl u 8,0 % pacientov liečených bosentanom a u 3,9 % pacientov vskupine s placebom (pozri časť 4.4).
V súhrnnej analýze u 100 detí s PAH z nekontrolovaných pediatrických štúdií FUTURE 1/2 a FUTURE 3/predĺženie bol u 10,0 % pacientov hlásený pokles hladiny hemoglobínu zo začiatočnej hodnoty na hodnotu menej ako 10 g/dl. K poklesu pod 8 g/dl nedošlo.
V štúdii FUTURE-4 sa počas liečby u 6 z 13 novorodencov s PPHN liečených bosentanom, vyskytol pokles hladiny hemoglobínu z referenčného rozpätia na začiatku na nižšie hodnoty ako je limit normálu.
Hlásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieBosentan bol aplikovaný ako jednotlivá dávka zdravým jedincom až do množstva 2400 mg a pacientom s iným ochorením než pľúcna hypertenzia až do množstva 2000 mg/deň počas dvoch mesiacov. Najčastejšou nežiaducou reakciou bola bolesť hlavy miernej až strednej intenzity.
Masívne predávkovanie môže mať za následok výraznú hypotenziu vyžadujúcu aktívnu kardiovaskulárnu podporu. Počas postmarketingového sledovania bol zaznamenaný jeden prípad predávkovania Tracleerom dávkou 10 000 mg, ktorú užil dospievajúci pacient mužského pohlavia. Prejavili sa u neho symptómy nauzea, vracanie, hypotenzia, závraty, potenie a rozmazané videnie. Za podpory tlaku krvi sa stav pacienta do 24 hodín navrátil do pôvodného stavu. Poznámka: bosentan sa neodstraňuje dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: iné antihypertenzíva, ATC kód: C02KX01
Mechanizmus účinkuBosentan je duálny antagonista receptora pre endotelín (ERA) s afinitou pre endotelínový receptor A a
B (ETA a ETB). Bosentan znižuje obidve, pľúcnu aj systémovú vaskulárnu rezistenciu, čo má za
následok zvýšenie minútového vývrhového objemu srdca bez zvýšenia frekvencie srdca.
Neurohormón endotelín-1 (ET-1) je jedným z najúčinnejších známych vazokonstriktorov a môže tiež podporovať fibrózu, proliferáciu buniek, srdcovú hypertrofiu a remodeláciu a pôsobí prozápalovo. Tieto účinky sú sprostredkované väzbou endotelínu na ETA a ETB receptory lokalizované v endoteli a v bunkách hladkého svalstva ciev. Hladina ET-1 v tkanivách a plazme je zvýšená pri rôznych kardiovaskulárnych poruchách a chorobách spojivových tkanív vrátane PAH, sklerodermie, akútneho a chronického zlyhania srdca, ischémie myokardu, systémovej hypertenzie a aterosklerózy, čo naznačuje patogénnu úlohu ET-1 pri týchto ochoreniach. Pri PAH a zlyhaní srdca v neprítomnosti antagonizmu endotelínového receptora, zvýšená hladina ET-1 silne koreluje so závažnosťou a prognózou ochorenia.
Bosentan kompetuje väzbu ET-1 a iných ET peptidov na obidva ETA a ETB receptory, s mierne zvýšenou afinitou k ETA receptorom (Ki = 4,1-43 nanomolov) než k ETB receptorom (Ki = 38-
730 nanomolov). Bosentan je špecifický antagonista ET receptorov, pričom sa neviaže na iné
receptory.
ÚčinnosťZvieracie modelyU zvieracích modelov pľúcnej hypertenzie opakované perorálne podávanie bosentanu znižovalo pľúcnu vaskulárnu rezistenciu a zvrátilo pľúcnu vaskulárnu rezistenciu a hypertrofiu pravej komory. U zvieracích modelov pľúcnej fibrózy bosentan znižoval ukladanie kolagénu v pľúcach.
Účinnosť u dospelých pacientov s pľúcnou artériovou hypertenziouUskutočnili sa dve randomizované, dvojito zaslepené, multicentrické, placebom kontrolované štúdie s
32 (štúdia AC-052-351) a 213 (štúdia AC-052-352, [BREATHE-1]) dospelými pacientmi s PAH III.-IV. triedy podľa funkčnej klasifikácie WHO (primárna pľúcna hypertenzia alebo pľúcna
hypertenzia sekundárna, najmä pri sklerodermii). Po 4 týždňoch liečby bosentanom v dávke 62,5 mg dvakrát denne boli udržiavacie dávky sledované v týchto štúdiách 125 mg dvakrát denne v AC-052-
351 a 125 mg dvakrát denne a 250 mg dvakrát denne v AC-052-352.
Bosentan bol pridaný k aktuálnej terapii pacientov, ktorá mohla obsahovať kombináciu antikoagulancií, vazodilatancií (napr. blokátorov kalciových kanálov), diuretík, kyslíka a digoxínu, ale nie epoprostenolu. Kontrolná skupina dostala k aktuálnej terapii placebo.
Primárnym kritériom hodnotenia každej štúdie bola zmena vzdialenosti v teste 6-minútovej chôdze
v 12. týždni v prvej štúdii a v 16. týždni v druhej štúdii. V oboch testoch malo liečenie bosentanom za následok signifikantné zvýšenie záťažovej kapacity. Placebom korigované predĺženie vzdialenosti chôdze oproti vstupným hodnotám v zmysle primárneho kritéria hodnotenia každej štúdie bolo
76 metrov (p = 0,02; t-test), resp. 44 metrov (p = 0,0002; Mann-Whitney U test). Rozdiely medzi skupinami so 125 mg dvakrát denne a s 250 mg dvakrát denne neboli štatisticky signifikantné, ale
existoval trend smerom ku zlepšeniu záťažovej kapacity v skupine liečenej dávkou 250 mg dvakrát
denne.
V dvojito slepej štúdii, ktorá bola vykonaná u časti pacientov, bolo zlepšenie v prejdenej vzdialenosti
zreteľné po 4 týždňoch liečby, výrazné po 8 týždňoch a udržalo sa až do 28. týždňa.
V retrospektívnej analýze respondérov, založenej na zmenách v zdolanej vzdialenosti testu chôdze, podľa funkčnej klasifikácie WHO a v dýchavičnosti u 95 pacientov randomizovaných na liečbu bosentanom 125 mg dvakrát denne v placebom kontrolovanom teste, bolo zistené, že v 8. týždni sa u 66 pacientov prejavilo zlepšenie, 22 bolo stabilizovaných a u 7 pacientov sa prejavilo zhoršenie. Z 22 pacientov stabilizovaných v 8. týždni sa u 6 prejavilo zlepšenie v týždňoch 12/16 a u 4 sa
v porovnaní so vstupnými hodnotami výkon zhoršil. Zo 7 pacientov, u ktorých sa prejavilo zhoršenie
8. týždeň, sa u 3 v týždňoch 12/16 výkon zlepšil a u 4 nastalo zhoršenie v porovnaní so vstupným meraním.
Invazívne hemodynamické parametre boli hodnotené iba v prvej štúdii. Liečba bosentanom viedla k signifikantnému zvýšeniu srdcového indexu spojenému so signifikantným znížením pľúcneho artériového tlaku, pľúcnej vaskulárnej rezistencie a stredného tlaku pravej predsiene.
Pri liečbe bosentanom bolo pozorované zníženie symptómov PAH. Miera dyspnoe počas testu chôdze sa u pacientov liečených bosentanom zlepšila. V štúdii AC-052-352 bolo 92 % z 213 pacientov klasifikovaných podľa vstupných hodnôt ako III. trieda podľa funkčnej klasifikácie WHO a 8 % ako trieda IV. Liečba bosentanom viedla ku zlepšeniu triedy funkčnej klasifikácie WHO u 42,4 % pacientov (placebo 30,4 %). Celková zmena triedy podľa funkčnej klasifikácie WHO v priebehu oboch štúdií bola signifikantne lepšia medzi pacientmi liečenými Tracleerom v porovnaní s pacientmi v skupine s placebom. V 28. týždni bola liečba bosentanom spojená so signifikantným znížením podielu klinického zhoršenia v porovnaní s placebom (10,7 % oproti 37,1 %; p = 0,0015).
V randomizovanej, dvojito slepej, multicentrickej, placebom kontrolovanej štúdii (AC-052-364
[EARLY]) dostávalo 185 pacientov s PAH funkčnej triedy II podľa WHO (s priemernou vzdialenosťou
v 6-minútovom teste chôdze 435 metrov) najskôr 62,5 mg bosentanu dvakrát denne počas 4 týždňov a následne počas 6 mesiacov buď 125 mg bosentanu dvakrát denne (n = 93), alebo placebo (n = 92). Zaradení boli pacienti s doposiaľ neliečenou PAH (n = 156) alebo liečení stabilnou dávkou sildenafilu (n = 29). Primárnymi cieľovými ukazovateľmi boli percentuálna zmena pľúcnej vaskulárnej rezistencie
(PVR) oproti začiatku a zmena vzdialenosti 6-minútového testu chôdze po 6 mesiacoch od začiatku
v porovnaní s placebom. Nasledujúca tabuľka uvádza výsledky analýz definovaných v protokole.
Začiatok;
PVR (dyn.sec/cm5) 6-minútový test chôdze -vzdialenosť (m)Placebo (n=88) Bosentan (n=80) Placebo (n=91) Bosentan (n=86)
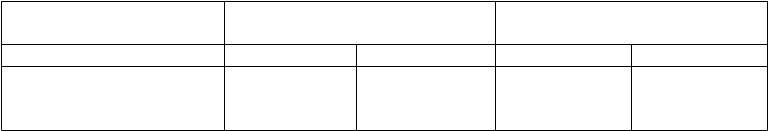
802
aritmetický priemer (SO)
(365) 851 (535)
431 (92)
443 (83)
Zmena oproti začiatku;
aritmetický priemer (SO) 128 (465)

-69 (475)
-8 (79)
11 (74)
Účinky liečby -22,6 % 19
95 % CL -34, -10 -4, 42
Hladina štatistickej významnosti – p
< 0,0001 0,0758
CL – medze intervalu spoľahlivosti; PVR – pľúcna vaskulárna rezistencia; SO – smerodajná odchýlka.
Liečba bosentanom preukázala v porovnaní s placebom redukciu početnosti klinického zhoršenia, definovaného ako kombinovaný parameter zložený zo zhoršenia symptómov, hospitalizácií v dôsledku PAH alebo úmrtia (proporcionálne zníženie rizika 77 %, 95 % interval spoľahlivosti [CI] 20-94 %,
p = 0,0114). Hlavnou zložkou preukázaného liečebného účinku bolo zníženie početnosti zhoršenia symptómov. V skupine liečenej bosentanom sa vyskytla len jedna hospitalizácia súvisiaca s PAH, kým v skupine s placebom sa vyskytli 3 hospitalizácie. Počas 6 mesiacov dvojito slepej štúdie sa
v každej zo skupín vyskytlo len jedno úmrtie. V súvislosti s ovplyvnením prežívania nemožno preto
formulovať žiadne závery.
Dlhodobé údaje boli získané od 173 pacientov, ktorí boli liečení bosentanom v kontrolovanej fáze a/alebo u ktorých bola zmenená liečba z placeba na bosentan v otvorenom predĺžení štúdie EARLY. Priemerná dĺžka užívania bosentanu bola 3,6 ± 1,8 roka (maximálne 6,1 rokov), pričom 73 % pacientov bolo liečených najmenej 3 roky a 62 % pacientov bolo liečených najmenej 4 roky. Počas predĺženia otvorenej štúdie pacienti s PAH mohli užívať doplnkovú liečbu. Väčšina pacientov bola diagnostikovaná s idiopatickou alebo dedičnou PAH (61 %). Celkovo 78 % pacientov ostalo v II. stupni PAH podľa funkčnej klasifikácie WHO. V treťom roku prežilo 90 % a v štvrtom roku 85 %
pacientov podľa Kaplan Majerovej krivky mortality. Zároveň u 88 %, resp. 79 % pacientov sa v týchto
časových intervaloch nezaznamenalo zhoršenie PAH (definované ako celková mortalita, transplantácia pľúc, atriálna septostómia alebo začiatok intravenóznej alebo subkutánnej liečby prostanoidmi). Podiel predchádzajúcej liečby placebom v dvojito zaslepenej štúdii alebo podiel ostatných liečiv v otvorenom predĺžení štúdie nie je známy.
V prospektívnej, multicentrickej, randomizovanej, dvojito slepej, placebom kontrolovanej štúdii (AC-
052-405 [BREATHE-5]) dostávali pacienti s PAH III. triedy podľa funkčnej klasifikácie WHO a Eisenmengerovým syndrómom združeným s vrodenými srdcovými chybami bosentan 62,5 mg dvakrát denne počas štyroch týždňov, potom 125 mg dvakrát denne počas ďalších 12 týždňov (n = 37, z ktorých
31 malo primárne pravoľavý, obojstranný skrat). Primárnym cieľom bolo ukázať, že bosentan nespôsobil zhoršenie hypoxémie. Po 16 týždňoch sa priemerná saturácia kyslíkom u skupiny liečenej bosentanom zvýšila o 1,0 % (95 % CI 0,7-2,8 %) v porovnaní so skupinou s placebom (n = 17), čo názorne dokazuje, že bosentan nespôsobil zhoršenie hypoxémie. Priemerná pľucna vaskulárna rezistencia sa signifikantne znížila u skupiny liečenej bosentanom (s prevažujúcim účinkom pozorovaným u pacientov
s obojsmerným vnútrosrdcovým skratom). Po 16 týždňoch bolo priemerné placebom korigované predĺženie vzdialenosti pri 6-minútovom teste chôdze 53 metrov (p = 0,0079), čo odráža zlepšenie funkčnej kapacity. Dvadsaťšesť pacientov pokračovalo v užívaní bosentanu v 24 týždňovom, otvorenom predĺžení (AC-052-409) štúdie BREATHE-5 (priemerné trvanie liečby 24,4 ± 2,0 týždňa) a účinnosť sa vo všeobecnosti udržala.
Otvorená, nekomparatívna štúdia (AC-052-362; [BREATHE-4]) bola vykonaná u 16 pacientov s PAH III. triedy podľa funkčnej klasifikácie WHO a s infekciou HIV. Pacienti boli liečení bosentanom 62,5 mg dvakrát denne počas štyroch týždňov a následne 125 mg dvakrát denne počas ďalších 12 týždňov. Po
16 týždňoch liečby nastalo oproti východiskovej hodnote významné zlepšenie v 6-minútovom teste
chôdze: pri priemernej začiatočnej hodnote 332,6 metrov bolo priemerné predĺženie vzdialenosti
91,4 metrov (p < 0,001). Nemožno vysloviť žiadny formálny záver týkajúci sa účinkov bosentanu na účinnosť antiretrovirálnych liekov (pozri tiež časť 4.4).
Nie sú známe žiadne klinické štúdie, ktoré by poukazovali na priaznivé účinky liečby Tracleerom na prežívanie. Dlhodobé prežívanie bolo ale zaznamenané u všetkých 235 pacientov, ktorí boli liečení bosentanom v 2 pivotných placebom kontrolovaných klinických štúdiách (AC-052-351 a AC-052-352)
a/alebo v ich dvoch nekontrolovaných, otvorených predĺženiach. Priemerné trvanie podávania bosentanu bolo 1,9 ± 0,7 rokov (min. 0,1 roka; max. 3,3 roka), pričom pacienti boli sledovaní priemerne 2,0 ± 0,6 roka. Vačšina z nich mala diagnostikovanú primárnu pľúcnu hypertenziu (72 %) a bola v III. triede
podľa funkčnej klasifikácie WHO (84 %). V tejto populácii bolo predpokladané prežívanie podľa
Kaplan-Meierovej metódy 93 % po 1 roku a 84 % po 2 rokoch od začiatku liečby bosentanom. Prežívanie bolo horšie v podskupine pacientov so sekundárnou pľúcnou hypertenziou pri systémovej skleróze. Hodnoty môžu byť ovplyvňované tým, že u 43 z 235 pacientov sa liečba začala epoprostenolom.
Štúdie uskutočnené u detí s pľúcnou artériovou hypertenziou
BREATHE-3 (AC-052-356)
Bosentan, filmom obalené tablety, bol hodnotený v otvorenej nekontrolovanej štúdii u 19 pediatrických pacientov s PAH vo veku 3 až 15 rokov. Táto štúdia bola v prvom rade určená ako farmakokinetická štúdia (pozri časť 5.2). Pacienti mali primárnu pľúcnu hypertenziu (10 pacientov) alebo PAH
súvisiacu s kongenitálnym ochorením srdca (9 pacientov) a na začiatku sledovania boli v II. (n = 15,
79 %) alebo III. triede (n = 4, 21 %) podľa funkčnej klasifikácie WHO. Podľa hmotnosti boli pacienti rozdelení do 3 skupín, z ktorých každá užívala počas 12 týždňov dávku bosentanu približne 2 mg/kg dvakrát denne. Polovica pacientov v každej skupine bola už liečená intravenózne podávaným epoprostenolom a dávka epoprostenolu zostala konštantná po celú dobu trvania štúdie.
Hemodynamické parametre boli merané u 17 pacientov. Srdcový index sa oproti vstupným hodnotám zvýšil priemerne o 0,5 l/min/m2, pľúcny artériovy tlak sa znížil priemerne o 8 mmHg a PVR sa znížila priemerne o 389 dyn·sec·cm-5. Tieto hemodynamické zlepšenia oproti vstupným hodnotám boli podobné pri súbežnom podávaní epoprostenolu alebo bez súbežného podávania epoprostenolu. Zmeny parametrov záťažovej kapacity v 12. týždni oproti vstupným hodnotám boli veľmi variabilné a žiadna
z nich nebola signifikantná.
FUTURE 1/2 (AC-052-365/AC-052-367)
Štúdia FUTURE 1 bola otvorená nekontrolovaná štúdia, ktorá bola vykonaná s bosentanom vo forme dispergovateľných tabliet podávaných v udržiavacej dávke 4 mg/kg dvakrát denne 36 pacientom vo veku od 2 do 11 rokov. Štúdia bola primárne navrhnutá ako farmakokinetická štúdia (pozri časť 5.2). Na začiatku mali pacienti idiopatickú (31 pacientov [86 %]) alebo familiárnu (5 pacientov [14 %]) PAH a spadali do II. (n = 23, 64 %) alebo III. triedy (n = 13, 36 %) podľa funkčnej klasifikácie WHO. V štúdii FUTURE 1 bol medián expozície hodnotenej liečby 13,1 týždňa (rozpätie 8,4 až 21,1). 33
z týchto pacientov bola poskytnutá pokračujúca liečba bosentanom vo forme dispergovateľných tabliet
v dávke 4 mg/kg dvakrát denne v nekontrolovanej predĺženej fáze štúdie FUTURE 2 po medián celkového trvania liečby 2,3 roka (v rozmedzí 0,2 až 5,0 rokov). Na začiatku štúdie FUTURE 1 užívalo 9 pacientov epoprostenol. 9 pacientom bola v priebehu štúdie novo iniciovaná liečba špecifická pre PAH. Odhad neprítomnosti zhoršenia PAH (úmrtie, transplantácia pľúc alebo hospitalizácia kvôli zhoršeniu PAH) bol po 2 rokoch podľa Kaplanovej-Meierovej metódy 78,9%. Celkový odhad prežitia podľa Kaplanovej-Meierovej metódy po 2 rokoch bol 91,2 %.
FUTURE 3 (AC-025-373)
V tejto otvorenej randomizovanej štúdii s 32 mg bosentanu vo forme dispergovateľných tabliet bolo 64 detí so stabilnou PAH vo veku od 3 mesiacov do 11 rokov randomizovaných do skupiny liečených 24 týždňov bosentanom v dávke 2 mg/kg dvakrát denne (n=33) alebo 2 mg/kg trikrát denne (n=31). 43 detí (67,2 %) bolo vo veku ≥ 2 roky až 11 rokov, 15 detí (23,4 %) bolo vo veku medzi 1 rokom a 2 rokmi
a 6 detí (9,4 %) bolo vo veku medzi 3 mesiacmi a 1 rokom. Štúdia bola primárne navrhnutá ako
farmakokinetická štúdia (pozri časť 5.2) a výsledky hodnotenia účinnosti boli len exploratórne. Etiológia PAH, podľa „Dana Point” klasifikácie, zahŕňala idiopatickú PAH (46 %), dedičnú PAH (3
%), PAH súvisiacu s korekčným chirurgickým zákrokom na srdci (38 %) a PAH súvisiacu
s kongenitálnym ochorením srdca spojenú so systémovými-pľúcnymi skratmi vrátane Eisenmengorovho syndrómu (13 %). Na začiatku liečby hodnoteným liečivom boli pacienti v I. triede (n=19 29 %), v II. triede (n = 27, 42 %) alebo III. triede (n = 18, 28 %) podľa funkčnej klasifikácie WHO. Pri vstupe do štúdie boli pacienti liečení liekmi proti PAH (najčastejšie inhibítorom fosfodiesterázy typu 5 [sildenafil] samotným [35,9 %], bosentanom samotným [10,9 %] a
kombináciou bosentanu, iloprostu a sildenafilu [10,9 %]) a v priebehu štúdie svoj liek proti PAH ďalej
užívali.
Na začiatku štúdie bola menej ako polovica zaradených pacientov liečená bosentanom samotným (45,3 % [29/64]) nekombinovaným s ďalšími liekmi proti PAH, 40,6 % (26/64) ostalo počas 24 týždňov hodnotenej liečby na monoterapii bosentanom. Analýza zahŕňajúca celkovú populáciu (64 pacientov) ukázala, že väčšina zostala prinajmenšom stabilná (t.j. bez zhoršenia) na základe nepediatrického špecifického funkčného hodnotenia podľa WHO (97 % dvakrát denne, 100 % trikrát denne) a na základe celkového klinického pocitu lekára (94 % dvakrát denne, 93 % trikrát denne) počas liečby. Odhad neprítomnosti zhoršenia PAH (úmrtie, transplantácia pľúc alebo hospitalizácia pre zhoršenie PAH) bol po 24 týždňoch podľa Kaplanovej-Meierovej metódy 96,9 % v skupine
s dávkou dvakrát denne a 96,7 % v skupine s podávanou dávkou trikrát denne.
Pri podávaní dávky 2 mg/kg trikrát denne v porovnaní s dávkou 2 mg/kg dvakrát denne nebol dokázaný žiadny klinický prínos.
Štúdie uskutočnené u novorodencov s pretrvávajúcou pľúcnou hypertenziou novorodencov (PPHN)
FUTURE 4 (AC-025-391)
Išlo o dvojito zaslepenú placebom kontrolovanú randomizovanú štúdiu u predčasne narodených alebo v termíne narodených novorodencov (gestačný vek 36 až 42 týždňov) s PPHN. Pacienti so suboptimálnou odpoveďou na inhalovaný oxid dusnatý (iNO) napriek najmenej 4 hodinám kontinuálnej liečby boli liečení bosentanom vo forme dispergovateľných tabliet v dávke 2 mg/kg dvakrát denne (N=13) alebo placebom (N=8) podávaným nazogastrickou sondou ako prídavná liečba k iNO do kompletného vysadenia iNO alebo do zlyhania liečby (definovaného ako potreba extrakorporálnej membránovej oxygenácie [ECMO] alebo nasadenia alternatívneho pľúcneho vazodilatátora) počas maximálne 14 dní.
Medián expozície hodnotenej liečby bol 4,5 (rozpätie 0,5 až 10,0) dňa v skupine liečených
bosentanom a 4,0 (rozpätie 2,5 až 6,5) dni v skupine na placebe.
Výsledky u tejto populácie nenaznačili dodatočný prínos bosentanu:
• medián času do úplného vysadenia iNO bol 3,7 dňa (95 % medze intervalu spoľahlivosti [CLs]
1,17; 6,95) pri bosentane a 2,9 dňa (95 % CLs 1,26; 4,23) pri placebe (p = 0,34).
• medián času do úplného vysadenia mechanickej ventilácie bol 10,8 dňa (95 % CLs 3,21; 12,21
dňa) pri bosentane a 8,6 dňa (95 % CLs 3,71; 9,66 dňa) pri placebe (p = 0,24).
• u jedného pacienta v skupine liečenej bosentanom došlo k zlyhaniu liečby (potreba ECMO podľa definície podľa protokolu), ktoré sa prejavilo na základe zvyšujúcich sa hodnôt oxygenačného indexu v priebehu 8 hodín po prvej dávke hodnoteného liečiva. Tento pacient sa počas 60 dní následného pozorovania vyliečil.
Kombinácia s epoprostenolom
Kombinácia bosentanu a epoprostenolu bola sledovaná v dvoch štúdiách: AC-052-355 (BREATHE-2) a AC-052-356 (BREATHE-3). AC-052-355 bola multicentrická, randomizovaná, placebom kontrolovaná, dvojito zaslepená štúdia bosentanu u 33 pacientov so závažnou PAH, ktorí dostávali súbežne epoprostenol. AC-052-356 bola otvorená, nekontrolovaná štúdia, v ktorej počas 12 týždňov 10 z 19 pediatrických pacientov dostávalo súbežne bosentan a epoprostenol. Bezpečnostný profil kombinácie sa
nelíšil od profilu očakávaného u každej zložky a kombinovaná liečba bola dobre znášaná deťmi a
dospelými. Klinický prínos kombinácie nebol preukázaný.
Systémová skleróza s vredovou chorobou prstov
Boli uskutočnené dve randomizované, dvojito zaslepené, multicentrické a placebom kontrolované štúdie u 122 (štúdia AC-052-401, [RAPIDS-1]) a 190 (štúdia AC-052-331, [RAPIDS-2]) dospelých pacientov
so systémovou sklerózou a vredovou chorobou prstov (buď pretrvávajúce vredy prstov, alebo údaj
o vredoch prstov v priebehu predchádzajúceho roka). V štúdii AC-052-331 museli mať pacienti aspoň jeden nedávno vzniknutý vred na prste a počas obidvoch štúdií muselo mať 85 % pacientov vznikajúci vred na prste na začiatku. Po 4 týždňoch liečby bosentanom v dávke 62,5 mg dvakrát denne bola
sledovaná udržiavacia dávka v obidvoch štúdiách 125 mg dvakrát denne. Dĺžka dvojito zaslepenej liečby
bola v štúdii AC-052-401 16 týždňov a v štúdii AC-052-331 24 týždňov.
Pôvodná liečba systémovej sklerózy a vredov na prstoch bola prípustná, ak ostali bez zmeny najmenej
počas jedného mesiaca predchádzajúceho začiatku liečby a v priebehu trvania dvojito zaslepenej štúdie.
Počet nových vredov prstov od začiatku do ukončenia štúdie bol primárnym kritériom hodnotenia pre obidve štúdie. V priebehu štúdie liečba bosentanom viedla v porovnaní s placebom ku zníženiu výskytu nových vredov prstov. V štúdii AC-052-401 sa v priebehu 16 týždňov dvojito zaslepenej liečby
v skupine pacientov liečených bosentanom objavilo v priemere 1,4 nových vredov na prstoch oproti 2,7 nových vredov na prstoch v skupine s placebom (p = 0,0042). V štúdii AC-052-331 v priebehu 24 týždňov dvojito zaslepenej liečby boli zhodné údaje pre nové vredy na prstoch 1,9, resp. 2,7 (p =
0,0351). V obidvoch štúdiách boli pacienti na bosentane menej náchylní k vzniku viacpočetných nových
vredov na prstoch v priebehu štúdie a dlhšie trvalo, než sa každý nasledujúci vred rozvinul, než tomu bolo u pacientov v skupine s placebom. Účinok bosentanu na zníženie počtu nových vredov na prstoch bol viac zrejmý u pacientov s viacpočetnými vredmi na prstoch.
Ani v jednej z obidvoch štúdií sa nepozoroval žiadny účinok bosentanu na rýchlosť vyhojenia vredov na
prstoch.
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti bosentanu boli dokumentované najmä u zdravých dobrovoľníkov.
Obmedzené údaje pacientov ukazujú, že vplyv bosentanu na dospelých pacientov s PAH je približne
2-krát väčší než u dospelých zdravých dobrovoľníkov.
U zdravých dobrovoľníkov bosentan vykazuje farmakokinetiku závislú od dávky a času. Klírens
a distribučný objem sa znižujú so zvýšenými intravenóznymi dávkami a stúpa s časom. Po perorálnom podaní je systémová dostupnosť úmerná dávke až po dávku 500 mg. Pri vyšších perorálnych dávkach sa zvyšuje Cmax a AUC menej než úmerne dávke.
Absorpcia
U zdravých dobrovoľníkov je absolútna biologická dostupnosť bosentanu približne 50 % a nie je
ovplyvnená potravou. Maximálna plazmatická hladina sa dosiahne v priebehu 3-5 hodín.
Distribúcia
Bosentan je výrazne viazaný (> 98 %) na plazmatické proteíny, najmä albumín. Bosentan nepreniká
do erytrocytov.
Distribučný objem (Vss) asi 18 litrov bol stanovený po intravenóznej dávke 250 mg. Biotransformácia a eliminácia
Po jednorazovej intravenóznej dávke 250 mg bol klírens 8,2 l/h. Polčas eliminácie (t1/2) je 5,4 hodiny.
Pri viacnásobnom dávkovaní sa plazmatické hladiny bosentanu postupne znižujú až na 50-65 % pôvodnej hodnoty stanovenej po podaní jednorazovej dávky. Toto zníženie je pravdepodobne dôsledkom autoindukcie metabolických pečeňových enzýmov. Rovnovážny stav bol dosiahnutý v priebehu 3-5 dní.
Bosentan je eliminovaný žlčou po metabolizácii v pečeni izoenzýmami CYP2C9 a CYP3A4
cytochrómu P450. Menej než 3 % perorálne aplikovanej dávky sa nachádzajú v moči.
Bosentan vytvára tri metabolity a iba jeden z nich je farmakologicky aktívny. Tento metabolit sa
vylučuje prevažne nezmenený žlčou. U dospelých pacientov je systémová dostupnosť aktívneho
metabolitu väčšia než u zdravých dobrovoľníkov. U pacientov so známkami cholestázy môže byť systémová dostupnosť aktívneho metabolitu zvýšená.
Bosentan je induktor CYP2C9 a CYP3A4 a možno tiež CYP2C19 a P-glykoproteínu. In vitro
bosentan inhibuje exportnú pumpu žlčových solí v kultúrach hepatocytov.
In vitro štúdie ukázali, že bosentan nemal relevantný inhibičný vplyv na testované izoenzýmy CYP (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4). Preto sa pod vplyvom bosentanu neočakáva zvyšovanie plazmatickej hladiny liečiv metabolizovaných týmito izoenzýmami.
Farmakokinetika v osobitných skupinách pacientov
Na základe skúmaného rozmedzia každého parametra sa neočakáva, že farmakokinetika bosentanu
bude v dospelej populácii relevantne ovplyvnená pohlavím, hmotnosťou, rasou alebo vekom.
Pediatrická populácia
Farmakokinetika bola u pediatrických pacientov hodnotená v 4 klinických štúdiách (BREATHE-3, FUTURE 1, FUTURE-3 a FUTURE-4; pozri časť 5.1). Kvôli obmedzeným údajom od detí mladších ako 2 roky je farmakokinetika v tejto vekovej skupine stále nedostatočne charakterizovaná.
Štúdia AC-052-356 (BREATHE-3) hodnotila farmakokinetiku jednorazových a opakovaných perorálnych dávok filmom obalených tabliet bosentanu u 19 detí vo veku od 3 rokov do 15 rokov s PAH, pričom dávkovanie záviselo od telesnej hmotnosti 2 mg/kg dvakrát denne. V tejto štúdii sa expozícia bosentanu znížila s časom spôsobom konzistentným so znalosťami o autoindukčných vlastnostiach bosentanu. Stredné hodnoty AUC (CV %) bosentanu u pediatrických pacientov liečených 31,25; 62,5 alebo 125 mg dvakrát denne boli 3496 (49), 5428 (79) a 6124 (27) ng·h/ml
v uvedenom poradí a boli nižšie než hodnota 8149 (47) ng·h/ml pozorovaná u dospelých pacientov s PAH, ktorí boli liečení dávkou 125 mg dvakrát denne. V rovnovážnom stave systémová dostupnosť u pediatrických pacientov vážiacich 10–20 kg, 20–40 kg a > 40 kg tvorila v uvedenom poradí 43 %,
67 % a 75 % systémovej dostupnosti u dospelých.
V štúdii AC-052-365 (FUTURE 1) boli podávané dispergovateľné tablety u 36 detí s PAH vo veku 2–
11 rokov. Nepozorovala sa žiadna dávková úmernosť, rovnovážne plazmatické hladiny bosentanu a AUC boli podobné pri perorálnej dávke 2 a 4 mg/kg (AUCτ bol 3577 ng·h/ml pri dávke
2 mg/kg dvakrát denne a 3371 ng·h/ml pri dávke 4 mg/kg dvakrát denne). Priemerná expozícia
bosentanu u týchto pediatrických pacientov predstavovala približne polovicu expozície u dospelých pacientov pri udržiavacej dávke 125 mg dvakrát denne, ale preukázala výraznú zhodu so závermi
u dospelých pacientov.
V štúdii AC-052-373 (FUTURE 3), používajúcej dispergovateľné tablety bola expozícia bosentanu
u pacientov liečených dávkou 2 mg/kg dvakrát denne porovnateľná s expozíciou v štúdii FUTURE 1. V celkovej populácii (n = 31) viedla dávka 2 mg/kg dvakrát denne k dennej expozícii 8535 ng·h/ml;
AUCτ bola 4268 ng·h/ml (CV: 61%). U pacientov vo veku medzi 3 mesiacmi a 2 rokmi bola denná expozícia 7879 ng·h/ml; AUCτ bola 3939 ng·h/ml (CV: 72%). U pacientov vo veku medzi 3 mesiacmi a 1 rokom (n = 2) bola AUCτ 5914 ng·h/ml (CV: 85%) a u pacientov vo veku medzi 1 rokom a 2
rokmi (n = 7) bola AUCτ 3507 ng·h/ml (CV: 70%). U pacientov starších ako 2 roky (n = 22) bola denná expozícia 8820 ng·h/ml; AUCτ bola 4410 ng·h/ml (CV: 58%). Dávkovanie bosentanu 2 mg/kg trikrát denne expozíciu nezvyšovalo; denná expozícia bola 7275 ng·h/ml, (CV: 83 %, n = 27).
Na základe záverov štúdií BREATHE-3, FUTURE 1 a FUTURE 3 sa javí, že u pediatrických pacientov expozícia bosentanu dosahuje plateau pri nižších dávkach ako u dospelých pacientov a že dávky vyššie ako 2 mg/kg dvakrát denne (4 mg/kg dvakrát denne alebo 2 mg/kg trikrát denne)
u pediatrických pacientov nedosiahnu vyššiu expozíciu bosentanu.
V štúdii AC-052-391 (FUTURE 4) vykonanej na novorodencoch sa koncentrácie bosentanu v priebehu prvého dávkovacieho intervalu pomaly a kontinuálne zvyšovali, čo viedlo k nízkej expozícii (AUC0-12 v celej krvi: 164 ng·h/ml, n = 11). V rovnovážnom stave AUC bola 6165 ng·h/ml (CV:
133%, n = 7), čo sa podobá expozícii pozorovanej u dospelých pacientov s PAH liečených 125 mg dvakrát denne, pričom sa berie do úvahy distribučný pomer krv/plazma 0,6.
Dôsledky týchto záverov, ohľadom hepatotoxicity, nie sú známe. Pohlavie a súbežné používanie intravenózne aplikovaného epoprostenolu nemajú signifikantný vplyv na farmakokinetiku bosentanu.
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre A) neboli vo farmakokinetike pozorované žiadne významné zmeny. V rovnovážnom stave bola hodnota AUC
bosentanu o 9 % vyššia a hodnota AUC aktívneho metabolitu Ro 48-5033 o 33 % vyššia u pacientov
s miernou poruchou funkcie pečene v porovnaní so zdravými dobrovoľníkmi.
Vplyv miernej poruchy funkcie pečene (Childovo-Pughovo skóre B) na farmakokinetiku bosentanu
a jeho primárneho metabolitu Ro 48-5033 bol sledovaný v štúdii, ktorá zahŕňala 5 pacientov s pľúcnou arteriálnou hypertenziou s pridruženou portálnou hypertenziou a poruchou pečeňových funkcií (Childovo-Pughovo skóre B) a 3 pacientov s PAH z iných príčin a s normálnou funkciou pečene.
U pacientov s poruchou funkcie pečene (Childovo-Pughovo skóre B) bola AUC v rovnovážnom stave (95 % CI) 360 (212-613) ng h/ml, t.j. 4,7krát vyššia a priemerná AUC (95 % CI) aktívneho metabolitu Ro 48-5033 bola 106 (58,4-192) ng h/ml t.j. 12,4krát vyššia ako u pacientov s normálnou funkciou pečene (bosentan: priemerná AUC [95 % CI]: 76,1 [9,07-638] ng h/ml; Ro 48-5033: priemerná AUC
[95 % CI] 8,57 [1,28-57,2] ng h/ml). Hoci počet zaradených pacientov bol obmedzený a vysoko variabilný, tieto dáta naznačujú výrazné zvýšenie expozícíe bosentanu a jeho primárneho metabolitu Ro 48-5033 u pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre B).
Farmakokinetika bosentanu sa neskúmala u pacientov s poruchou funkcie pečene (Childovo-Pughovo skóre C). Tracleer je kontraindikovaný u pacientov s miernou a závažnou poruchou funkcie pečene t. j. Childovo-Pughovo skóre B alebo C (pozri časť 4.3).
Porucha funkcie obličiek
U pacientov s vážnou poruchou funkcie obličiek (klírens kreatinínu 15-30 ml/min) sa plazmatická hladina bosentanu znížila približne o 10 %. Plazmatické hladiny metabolitov bosentanu sa u týchto pacientov zvýšili asi dvakrát v porovnaní s osobami s normálnou funkciou obličiek. Ak pacient trpí poruchou funkcie obličiek, úprava dávky nie je nutná. Neexistuje špecifická klinická skúsenosť
s pacientmi podstupujúcimi dialýzu. Na základe fyzikálno-chemických vlastností a vysokého stupňa väzby na plazmatické proteíny sa neočakáva, že by bosentan bol v signifikantnej miere odstránený
z cirkulácie dialýzou (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Dvojročná štúdia karcinogenity u myší ukázala zvýšený kombinovaný výskyt hepatocelulárnych adenómov a karcinómov u samcov, nie však u samíc, s plazmatickými hladinami asi dvakrát až štyrikrát vyššími než plazmatické hladiny, ktoré sa dosiahli v liečebnej dávke u ľudí. U potkanov vyvolalo perorálne podávanie bosentanu počas 2 rokov nízke, signifikantné zvýšenie kombinovaného výskytu tyroidných folikulárnych bunkových adenómov a karcinómov u samcov, nie však u samíc,
s plazmatickými hladinami asi 9- až 14krát vyššími než plazmatické hladiny, ktoré sa dosiahli pri liečebnej dávke u ľudí. Bosentan bol negatívny v testoch genotoxicity. U potkanov bola bosentanom vyvolaná mierna tyroidná hormonálna dysbalancia. Nedokázalo sa však, že by bosentan ovplyvňoval tyroidné funkcie u ľudí (tyroxín, TSH).
Vplyv bosentanu na mitochondriálne funkcie nie je známy.
Ukázalo sa, že bosentan je teratogénny u potkanov pri plazmatických hladinách 1,5krát vyšších než plazmatické hladiny, ktoré boli dosiahnuté v liečebnej dávke u ľudí. Teratogénne efekty, vrátane malformácie hlavy, faciálnych oblastí a veľkých ciev boli závislé od dávky. Podobné typy malformácií pozorované s inými antagonistami ET receptora a u myší s vyradenými ET receptormi naznačujú skupinový efekt. U žien vo fertilnom veku sa musia prijať príslušné preventívne opatrenia (pozri časti
4.3, 4.4 a 4.6).
S chronickým podávaním antagonistov endotelínového receptora hlodavcom sa spája rozvoj testikulárnej tubulárnej atrofie a zhoršenie fertility.
V štúdiách fertility samcov a samíc potkanov, nebol pozorovaný žiadny vplyv na počet spermií, motilitu a životnosť, ani na schopnosť páriť sa alebo na plodnosť, pri 21-násobných respektíve 43- násobných plazmatických hladinách než sú očakávané liečebné hladiny u ľudí. Neexistoval ani žiadny nežiaduci vplyv na vývoj pre-implantovaného embrya alebo na implantáciu.
Ľahko zvýšená incidencia testikulárnej tubulárnej atrofie bola pozorovaná u potkanov, ktorým sa podával bosentan perorálne v dávkach 125 mg/kg/deň (asi 4-násobok maximálnej odporúčanej dávky u ľudí a najnižšej testovacej dávky) počas 2 rokov, nie však v dávkach až 1500 mg/kg/deň (asi 50- násobok maximálnej odporúčanej dávky u ľudí) počas 6 mesiacov. V štúdii toxicity na juvenilných potkanoch, kde boli potkany liečené od 4. dňa po vrhu do dospelosti, bolo po odstavení pozorované zníženie absolútnej hmotnosti semenníkov a nadsemenníkov a zníženie počtu spermií
v nadsemenníkoch. Hladina pri ktorej sa nepozorujú žiadne účinky (NOAEL) bola 21-násobkom (21
dní po vrhu) a 2,3-násobok (69 dní po vrhu) ľudskej terapeutickej expozície.
21 dní po vrhu neboli po 7-násobku (samci) a 19-násobku (samice) ľudskej terapeutickej expozície zistené žiadne účinky na celkový vývoj, rast, senzorické a kognitívne funkcie a reprodukčné schopnosti. V dospelosti (69 dní po vrhu) neboli zistené žiadne účinky bosentanu pri 1,3-násobku (samci) a 2,6-násobku (samice) terapeutickej expozície u detí s PAH.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety: Kukuričný škrob Hydrolyzát škrobu
Sodná soľ karboxymetylškrobu
Povidón Glyceroldibehenát Magnéziumstearát
Obal tablety: Hypromelóza Glyceroltriacetát Mastenec
Oxid titaničitý (E171) Oxid železitý žltý (E172) Oxid železitý červený (E172) Etylcelulóza
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
V prípade bielych fliaš z polyetylénu s vysokou hustotou použite do 30 dní od prvého otvorenia.
6.4 Špeciálne upozornenia na uchovávanie
PVC/PE/PVDC/Al blistre:
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Biele fľaše z polyetylénu s vysokou hustotou:
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Tracleer 62,5 mg filmom obalené tablety
PVC/PE/PVDC/Al blistre obsahujúce 14 filmom obalených tabliet. Balenia obsahujúce 14, 56 alebo 112 filmom obalených tabliet.
Biele fľaše z polyetylénu s vysokou hustotou so silikagélovým sušidlom obsahujúce 56 filmom obalených tabliet.
Balenia obsahujúce 56 filmom obalených tabliet.
Tracleer 125 mg filmom obalené tablety
PVC/PE/PVDC/Al blistre obsahujúce 14 filmom obalených tabliet. Balenia obsahujúce 56 alebo 112 filmom obalených tabliet.
Biele fľaše z polyetylénu s vysokou hustotou so silikagélovým sušidlom obsahujúce 56 filmom obalených tabliet.
Balenia obsahujúce 56 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a zaobchádzanie s liekom
Žiadne zvláštne požiadavky.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Actelion Registration Ltd
Chiswick Tower, 13th Floor
389 Chiswick High Road
Londýn W4 4AL Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLA
Tracleer 62,5 mg filmom obalené tablety
EU/1/02/220/001
EU/1/02/220/002
EU/1/02/220/003
EU/1/02/220/007
Tracleer 125 mg filmom obalené tablety
EU/1/02/220/004
EU/1/02/220/005
EU/1/02/220/008
9
. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 15. mája 2002
Dátum predĺženia: 20. apríla 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1
. NÁZOV LIEKU
Tracleer 32 mg dispergovateľné tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá dispergovateľná tableta obsahuje 32 mg bosentanu (vo forme monohydrátu).
Pomocná látka: každá dispergovateľná tableta obsahuje 3,7 mg aspartámu (E951).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Dispergovateľná tableta:
Bledožlté až takmer biele tablety, v tvare štvorlístka, na jednej strane predelené na štyri časti a na druhej strane označené “32”. Dispergovateľná tableta sa môže rozdeliť na štyri rovnaké časti.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Liečba pľúcnej artériovej hypertenzie (PAH) na zlepšenie záťažovej kapacity a symptómov pacientov
III. triedy podľa funkčnej klasifikácie WHO. Účinnosť bola preukázaná pri:
• primárnej (idiopatickej a hereditárnej) pľúcnej artériovej hypertenzii
• pľúcnej artériovej hypertenzii sekundárnej so sklerodermiou bez signifikantného
intersticiálneho pľúcneho ochorenia
• pľúcnej artériovej hypertenzii spojenej s vrodeným ľavo-pravým skratom a Eisenmengerovým syndrómom
Isté zlepšenie bolo tiež preukázané u pacientov s pľúcnou artériovou hypertenziou II. triedy podľa funkčnej klasifikácie WHO (pozri časť 5.1).
Tracleer je indikovaný tiež na zníženie počtu nových vredov na prstoch u pacientov so systémovou sklerózou a pokračujúcou vredovou chorobou prstov (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Spôsob podávania
Tablety sa užívajú perorálne ráno a večer, s jedlom alebo bez jedla.
Pred požitím sa má pridať k dispergovateľnej tablete na lyžičke malé množstvo vody a premiešať, aby sa pred prehltnutím rozpustila. Na lyžičku sa má následne pridať malé množstvo vody, ktoré pacient tiež požije, aby sa zabezpečilo, že pacientovi bol podaný liek úplne. Pokiaľ je to možné, dávka by sa mala zapiť pohárom vody, aby sa zabezpečilo požitie celej dávky. Ak je to potrebné, dispergovateľnú tabletu možno rozdeliť pozdĺž deliacej ryhy na povrchu (pozri časť 6.6).
Dispergovateľné tablety boli testované len u pediatrických pacientov. Porovnanie biologickej dostupnosti medzi dispergovateľnými tabletami a filmom obalenými tabletami u dospelých ukázalo nižšiu dostupnosť bosentanu v dispergovateľných tabletách (pozri časť 5.2). Práve preto by malo byť ich použitie určené len pre dospelých pacientov, ktorí nemôžu užívať filmom obalené tablety.
Dávkovanie
Pľúcna
artériová
h
y
p
ertenzia
Liečbu má začať a monitorovať iba lekár so skúsenosťami s liečbou PAH.
Dospelí
U dospelých pacientov sa má liečba Tracleerom začať dávkou 62,5 mg dvakrát denne počas štyroch týždňov, a potom zvýšiť na udržiavaciu dávku 125 mg dvakrát denne. Rovnaké odporúčania platia aj
pre znovuzačatie liečby Tracleerom po prerušení terapie (pozri časť 4.4).
Pediatrická populácia
Pediatrické farmakokinetické údaje ukázali, že plazmatické koncentrácie bosentanu u detí s PAH vo veku od 1 roka do 15 rokov boli priemerne nižšie ako u dospelých pacientov a nezvyšovali sa so
zvýšením dávky Tracleeru nad 2 mg/kg telesnej hmotnosti ani zvýšením frekvencie podávania z
dvakrát denne na trikrát denne (pozri časť 5.2). Zvýšenie dávky alebo zvýšenie frekvencie podávania pravdepodobne nevedie k ďalšiemu klinickému prínosu.
Na základe týchto farmakokinetických výsledkov, ak sa používa u detí s PAH vo veku 1 rok a starších,
odporúča sa počiatočná a udržiavacia dávka 2 mg/kg ráno a večer.
Prínos bosentanu v štandardnej liečbe u novorodencov s pretrvávajúcou pľúcnou hypertenziou novorodencov (PPHN) nebol preukázaný. Nie je možné uviesť žiadne odporúčania na dávkovanie (pozri časti 5.1 a 5.2).
Postup pri klinickom zhoršení PAH
Pri klinickom zhoršení (napr. skrátenie vzdialenosti pri šesťminútovom teste chôdzou aspoň o 10 % v porovnaní s meraním pred začiatkom liečby) napriek liečbe Tracleerom počas minimálne 8 týždňov (cieľová dávka počas minimálne 4 týždňov), sa majú zvážiť alternatívne možnosti liečby. Avšak niektorí pacienti, ktorí nevykazujú žiadnu odozvu po 8 týždňoch liečby Tracleerom, môžu priaznivo reagovať po ďalších 4 až 8 týždňoch liečby.
Pri neskoršom klinickom zhoršení napriek liečbe Tracleerom (t.j. po niekoľkých mesiacoch liečby) sa má liečba opätovne prehodnotiť. Záťažová kapacita niektorých pacientov, ktorí dobre nereagujú na Tracleer v dávke 125 mg dvakrát denne, sa môže mierne zlepšiť, ak sa dávka zvýši na 250 mg dvakrát denne. Má sa starostlivo stanoviť pomer medzi prínosom a rizikom a zvážiť skutočnosť, že pečeňová toxicita je závislá od dávky (pozri časti 4.4 a 5.1).
Ukončenie liečby
S náhlym ukončením liečby Tracleerom u pacientov s PAH existujú obmedzené skúsenosti. Nebol pozorovaný žiadny dôkaz akútneho “rebound” efektu. Aby sa však zamedzilo možnému škodlivému klinickému zhoršeniu v dôsledku potenciálneho “rebound” efektu, má sa zvážiť postupné znižovanie dávky (polovica dávky počas 3 až 7 dní). V priebehu vysadzovania lieku sa odporúča intenzívnejší monitoring. Ak sa rozhodne o vysadení Tracleeru, má byť postupné, so súbežným začiatkom alternatívnej liečby.
Systémová skleróza s pokračujúcou vredovou chorobou prstov
Liečbu má začať a monitorovať iba lekár so skúsenosťami s liečbou systémovej sklerózy.
Dospelí
Liečba Tracleerom má byť zahájená dávkou 62,5 mg dvakrát denne počas štyroch týždňov, a potom zvýšená na udržiavaciu dávku 125 mg dvakrát denne. Rovnaké odporúčania platia aj pre znovuzačatie liečby Tracleerom po prerušení terapie (pozri časť 4.4).
Skúsenosti z kontrolovaných klinických štúdií sú pre túto indikáciu obmedzené na 6 mesiacov (pozri
časť 5.1).
Odozva pacientov na liečbu a potreba pokračujúcej liečby majú byť pravidelne prehodnocované. Má sa dôkladne vyhodnocovať pomer prínosu a rizika, pričom sa zohľadňuje hepatotoxicita bosentanu (pozri časti 4.4 a 4.8).
Pediatrická populácia
Nie sú žiadne údaje o bezpečnosti a účinnosti u pacientov mladších ako 18 rokov. Farmakokinetické údaje Tracleeru nie sú dostupné pre mladšie deti s touto chorobou.
Osobitné skupiny pacientov
Porucha funkcie pečene
Tracleer je kontraindikovaný u pacientov so strednou až vážnou pečeňovou dysfunkciou (pozri časti
4.3, 4.4 a 5.2). U pacientov s miernou poruchou funkcie pečene (tj. Childovo-Pughovo skóre A) (pozri
časť 5.2) nie je potrebná úprava dávky.
Porucha funkcie obličiek
U pacientov s miernou poruchou funkcie obličiek nie je potrebná úprava dávky. Dávka sa nemusí
upravovať ani u dialyzovaných pacientov (pozri časť 5.2).
Starší pacienti
Úprava dávky pre pacientov starších než 65 rokov nie je nutná.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1
• Stredná až ťažká porucha funkcie pečene, t.j. Childovo-Pughovo skóre B alebo C (pozri
časť 5.2)
• Hodnoty aminotransferáz pečene, t.j. aspartátaminotransferázy (AST) a/alebo alanínaminotransferázy (ALT) väčšie ako 3 × horná hranica normy (HHN) pred začiatkom liečby (pozri časť 4.4)
• Súbežné užívanie cyklosporínu A (pozri časť 4.5)
• Gravidita (pozri časti 4.4 a 4.6)
• Ženy vo fertilnom veku, ktoré nepoužívajú spoľahlivé metódy antikoncepcie (pozri časti 4.4, 4.5 a
4.6)
4.4 Osobitné upozornenia a opatrenia pri používaní
Účinnosť Tracleeru nebola stanovená u pacientov so závažnou PAH.
Ak sa klinický stav zhorší, má sa zvážiť prechod k terapii, ktorá sa odporúča pri závažnom stupni ochorenia (napr. epoprostenol) (pozri časť 4.2).
Vyváženosť vzťahu prínos/riziko bosentanu nebola stanovená u pacientov s PAH I. triedy podľa funkčnej
klasifikácie WHO.
Liečba Tracleerom sa má začať iba vtedy, ak je systémový systolický krvný tlak vyšší než 85 mmHg.
Tracleer nepreukázal priaznivý účinok na hojenie existujúcich vredov prstov.
 Funkcia pečene
Funkcia pečeneZvýšenie aminotransferáz pečene, t.j. aspartátaminotransferázy a alanínaminotransferázy (AST a/alebo
ALT) spojené s užívaním bosentanu závisí od dávky. Zmeny hodnôt pečeňových enzýmov sa zvyčajne objavia počas prvých 26 týždňov liečby, ale môžu sa vyskytnúť aj neskôr v priebehu liečby (pozri
časť 4.8). Toto zvýšenie môže byť čiastočne dôsledkom kompetitívnej inhibície vylučovania žlčových solí z hepatocytov, ale na výskyte dysfunkcie pečene sa pravdepodobne podieľajú aj iné mechanizmy, ktoré doposiaľ neboli celkom objasnené. Akumulácia bosentanu v hepatocytoch, ktorá vedie
k cytolýze s potenciálne závažným poškodením pečene, alebo imunologický mechanizmus, nie sú vylúčené. Riziko dysfunkcie pečene môže byť tiež zvýšené, ak sa súbežne s bosentanom podávajú
liečivá, ktoré sú inhibítormi exportnej pumpy žlčových solí, napr. rifampicín, glibenklamid a cyklosporín A (pozri časti 4.3 a 4.5). K dispozícii sú ale iba obmedzené údaje.
Hladina aminotransferáz pečene sa musí merať pred začiatkom liečby a následne v mesačných intervaloch počas liečby Tracleerom.
Okrem toho sa hladina aminotransferáz pečene musí merať 2 týždne po každom zvýšení dávky.Odporúčania prizvýšení ALT/ASTHladiny ALT/AST Odporúčania na liečbu a monitoring> 3 a ≤ 5 × HHN Výsledok má byť potvrdený druhým pečeňovým testom; ak sa výsledok potvrdí, s užívaním Tracleeru treba postupovať individuálne, možno redukovať dávkou alebo podávanie Tracleeru ukončiť (pozri časť 4.2). V sledovaní hladín aminotransferáz sa má pokračovať najmenej každé 2 týždne. Ak sa hladiny aminotransferáz vrátia k hodnotám pred začiatkom liečby, sa má zvážiť pokračovanie liečby alebo opätovné nasadenie Tracleeru podľa podmienok popísaných nižšie.
> 5 a ≤ 8 × HHN Výsledok má byť potvrdený druhým pečeňovým testom; ak sa výsledok potvrdí, liečba sa má ukončiť a najmenej každé 2 týždne sa majú monitorovať hladiny aminotransferáz Ak sa hladiny aminotransferáz vrátia k hodnotám pred začiatkom liečby, má sa zvážiť opätovné nasadenie Tracleeru podľa podmienok popísaných nižšie.
> 8 × HHN Liečba musí byť ukončená. Opätovné nasadenie Tracleeru nie je možné.
Prisprievodných klinických symptómoch poškodenia pečene, t.j. nevoľnosť, vracanie, horúčka, bolesti brucha, žltačka, neobvyklá apatia alebo únava, syndróm podobný chrípke (bolesti kĺbov, svalov, horúčka)
, musí byť liečba ukončená a opätovné nasadenie Tracleeru nie je možné.ObnovenieliečbyO obnovení liečby Tracleerom je možné uvažovať iba vtedy, ak potenciálny prínos liečby preváži možné riziká a ak sú hladiny pečeňových aminotransferáz v rozmedzí hodnôt pred začiatkom liečby. Odporúča sa konzultácia s hepatológom. Pri obnovení liečby sa musia rešpektovať pokyny rozpísané v časti 4.2.
Hladiny aminotransferáz sa musia skontrolovať do 3 dní po obnovení liečby, následne po2 týždňoch a potom podľa odporúčaní uvedených vyššie.HHN= horná hranica normy
Hladina hemoglobínuLiečba bosentanom bola spojená s dávkovo závislým znížením hladiny hemoglobínu (pozri časť 4.8).
Znížené hladiny hemoglobínu v súvislosti s užívaním bosentanu neboli v placebom kontrolovaných štúdiách progresívne a stabilizovali sa po prvých 4-12 týždňoch liečby. Odporúča sa skontrolovať hladinu hemoglobínu pred začiatkom liečby, každý mesiac v priebehu prvých 4 mesiacov liečby, a potom každý štvrťrok. Ak sa objaví klinicky relevantné zníženie hladiny hemoglobínu, výsledky sa musia vyhodnotiť a musí sa uskutočniť vyšetrenie s cieľom stanoviť príčiny a potreby špecifickej liečby. V postmarketingových sledovaniach boli zaznamenané prípady anémie, ktoré si vyžadovali transfúziu erytrocytov (pozri časť 4.8).
Ženy vo fertilnom veku
Keďže Tracleer môže zabrániť účinku hormonálnej antikoncepcie, a vzhľadom na riziko zhoršenia pľúcnej hypertenzie v tehotenstve a tiež teratogénne účinky pozorované u zvierat:
• Liečba Tracleerom sa nesmie začať u žien vo fertilnom veku, pokiaľ nepoužívajú spoľahlivú
metódu antikoncepcie a výsledok tehotenského testu pred liečbou nie je negatívny
• Hormonálna antikoncepcia nesmie byť jedinou používanou metódou antikoncepcie počas liečby
Tracleerom
• Odporúča sa vykonať tehotenský test každý mesiac, aby sa umožnila skorá detekcia tehotenstva
Pre ďalšie informácie pozri časti 4.5 a 4.6.
Pľúcna venookluzívna choroba
Pri používaní vazodilatancií (najmä prostacyklínov) u pacientov s pľúcnou venookluzívnou chorobou
boli zaznamenané prípady pľúcneho edému. Preto je potrebné zvážiť prítomnosť súčasnej venookluzívnej choroby, ak sa pri podávaní Tracleeru pacientom s PAH objavia príznaky pľúcneho edému. V postmarketingovom období sa vyskytli ojedinelé prípady pľúcneho edému u pacientov liečených Tracleerom so suspektnou diagnózou pľúcnej venookluzívnej choroby.
Pacienti s pľúcnou artériovou hypertenziou so súčasným zlyhaním ľavej komory
U pacientov s pľúcnou artériovou hypertenziou a súčasnou dysfunkciou ľavej komory nebola
uskutočnená žiadna špeciálna štúdia. Avšak 1 611 pacientov (804 liečených bosentanom a 807
v skupine s placebom) s ťažkým chronickým zlyhaním srdca (CHZS) sa počas priemerne 1,5 roka liečilo v placebom kontrolovanej štúdii (štúdia AC-052-301/302 [ENABLE 1 & 2]). V tejto štúdii sa objavil zvýšený výskyt hospitalizácií kvôli CHZS v priebehu prvých 4-8 týždňov liečby bosentanom, ktorý by mohol byť dôsledkom retencie tekutín. Retencia tekutín v tejto štúdii sa prejavila začiatočným zvýšením hmotnosti, zníženou hladinou hemoglobínu a zvýšeným výskytom edému dolných končatín. Na konci štúdie sa neprejavil rozdiel medzi skupinou pacientov liečených bosentanom a pacientov, ktorí dostávali placebo, ani v celkovom počte hospitalizácií kvôli zlyhaniu srdca, ani v mortalite. Preto sa odporúča vyšetriť pacientom príznaky retencie tekutín (napr. zvyšovanie hmotnosti), najmä ak súčasne trpia závážnou systolickou dysfunkciou. V prípade výskytu uvedených príznakov odporúčame začať liečbu diuretikami, resp. ak už sú podávané, zvýšiť ich dávku. U pacientov so známkami retencie tekutín pred začiatkom liečby Tracleerom je potrebné zvážiť liečbu diuretikami.
Pľúcna artériová hypertenzia so súčasnou infekciou HIV
Z klinických štúdií sú iba obmedzené skúsenosti s podávaním Tracleeru pacientom s PAH so
súčasnou infekciou HIV, ktorá je liečená antiretrovirálnymi liekmi (pozri časť 5.1). Štúdia liekových interakcií medzi bosentanom a lopinavirom + ritonavirom u zdravých jedincov ukázala zvýšené plazmatické hladiny bosentanu, s maximálnou hladinou počas prvých štyroch dní terapie (pozri časť
4.5). Na začiatku liečby Tracleerom u pacientov, ktorí si vyžadujú liečbu ritonavirom potencovanými inhibítormi proteáz, je najmä v začiatočnej fáze potrebné dôsledne monitorovať pacientovu znášanlivosť voči Tracleeru, predovšetkým riziko hypotenzie a hepatálne pečeňové testy. Zvýšené dlhodobé riziko hepatálnej toxicity a hematologických nežiaducich účinkov nemožno vylúčiť, ak sa bosentan používa v kombinácii s antiretrovirálnymi liekmi. Vzhľadom na možnosť interakcií vzťahujúcich sa najmä na indukčný účinok bosentanu na CYP 450 (pozri časť 4.5), ktoré by mohli ovplyvniť účinnosť antiretrovirálnej liečby, musia byť títo pacienti tiež dôkladne monitorovaní
s ohľadom na liečbu svojej HIV infekcie.
Sekundárna pľúcna hypertenzia v súvislostischronickouobštrukčnouchoroboupľúc(CHOCHP)
Bezpečnosť a tolerancia bosentanu boli sledované v 12týždňovej výskumnej nekontrolovanej štúdii
u 11 pacientov s pľúcnou hypertenziou, ktorá vznikla sekundárne v súvislosti so závažnou CHOCHP
(stupeň III podľa klasifikácie GOLD). Boli pozorované zvýšenie minútovej ventilácie a pokles
saturácie kyslíkom. Najčastejším nežiaducim účinkom bolo dyspnoe (sťažené dýchanie), ktoré
rozhodlo o prerušení terapie bosentanom.
Súbežné použitie s inýmiliečivami
Súbežné používanie Tracleeru s cyklosporínom A je kontraindikované (pozri časti 4.3 a 4.5).
Súbežné používanie Tracleeru s glibenklamidom, flukonazolom a rifampicínom sa neodporúča. Pre podrobnejšie informácie pozri časť 4.5.
Je nutné vyhnúť sa súbežnému podávaniu Tracleeru s inhibítormi CYP3A4 a s inhibítormi CYP2C9
(pozri časť 4.5).
Tracleer 32 mg dispergovateľné tablety obsahuje zdroj fenylalanínu (Aspartám – E951). Môže byť
škodlivý pre pacientov s fenylketonúriou.
4.5 Liekové a iné interakcie
Bosentan je induktor cytochrómu P 450 (CYP) izoenzýmov CYP2C9 a CYP3A4. In vitro získané dáta tiež naznačujú indukciu CYP2C19. V prípade súbežného podávania Tracleeru sa plazmatické hladiny liečiv metabolizovaných týmito izoenzýmami znížia. Je nutné zohľadniť možnosť zmeny účinnosti liečiv, ktoré sú týmito izoenzýmami metabolizované. Dávkovanie týchto liekov môže vyžadovať úpravu po začatí, zmene dávky alebo po prerušení súbežnej liečby Tracleerom.
Bosentan je metabolizovaný enzýmami CYP2C9 a CYP3A4. Inhibícia týchto izoenzýmov môže zvýšiť plazmatickú hladinu bosentanu (pozri ketokonazol). Vplyv inhibítorov CYP2C9 na hladinu bosentanu nebol študovaný. Táto kombinácia sa má používať veľmi opatrne.
Flukonazol a iné inhibítory CYP2C9 a CYP3A4: Súbežné podávanie s flukonazolom, ktorý inhibuje najmä CYP2C9 a do určitej miery aj CYP3A4, môže viesť k výraznému zvýšeniu plazmatických hladín bosentanu, a preto sa táto kombinácia neodporúča. Z toho istého dôvodu sa s Tracleerom neodporúča súbežné podávanie silného inhibítora CYP3A4 (napr. ketokonazolu, itrakonazolu alebo ritonaviru) a inhibítora CYP2C9 (napr. vorikonazolu).
Cyklosporín A: Súbežné podávanie Tracleeru a cyklosporínu A (inhibítor kalcineurínu) je kontraindikované (pozri časť 4.3). Ak sa obidva lieky podávali súbežne, bola najnižšia nameraná začiatočná hladina bosentanu približne tridsaťkrát vyššia ako hladina nameraná pri používaní iba samotného bosentanu. Pri rovnovážnom stave boli plazmatické hladiny bosentanu 3- až 4krát vyššie než v prípade monoterapie bosentanom. Mechanizmus tejto interakcie spočíva s najväčšou pravdepodobnosťou v inhibícii transportným proteínom mediovaného vychytávania bosentanu do hepatocytov cyklosporínom. Krvné hladiny cyklosporínu A (substrát CYP3A4) sa znížili približne o
50 %. Toto je pravdepodobne v dôsledku indukcie CYP3A4 bosentanom.
Takrolimus, sirolimus: Súbežné podávanie takrolimu alebo sirolimu a Tracleeru u ľudí nebolo skúmané, ale spoločné podávanie takrolimu alebo sirolimu a Tracleeru môže spôsobiť zvýšenie plazmatickej hladiny bosentanu rovnako ako súbežné podávanie s cyklosporínom A. Súbežné užívanie Tracleeru môže znížiť plazmatickú hladinu takrolimu a sirolimu. Preto sa súbežné podávanie
Tracleeru a takrolimu alebo sirolimu neodporúča. Pacienti, ktorí vyžadujú podávanie spomínanej kombinácie, musia byť starostlivo sledovaní, či nedochádza k výskytu nežiaducich účinkov súvisiacich s hladinami Tracleeru a takrolimu a sirolimu v krvi.
Glibenklamid: Súbežné podávanie bosentanu 125 mg 2krát denne počas 5 dní znížilo plazmatickú hladinu glibenklamidu (substrát CYP3A4) o 40 %, s potenciálne signifikantným znížením hypoglykemického efektu. Plazmatické hladiny bosentanu tiež poklesli o 29 %. Okrem toho bol
u pacientov, ktorí sa podrobili súbežnej liečbe glibenklamidom, pozorovaný častejší vzostup hladín aminotransferáz. Glibenklamid a bosentan inhibujú exportnú pumpu žlčových solí, čím by sa dali
vysvetliť zvýšené hladiny aminotransferáz. Táto kombinácia sa nemá používať. Žiadne údaje
o liekových interakciách s ostatnými derivátmi sulfonylmočoviny nie sú k dispozícii.
Rifampicín: Súbežné podávanie bosentanu 125 mg dvakrát denne počas 7 dní s rifampicínom, potentným induktorom CYP2C9 a CYP3A4, 9 zdravým osobám znížilo plazmatické hladiny bosentanu o 58 %, pričom tento pokles dosahoval u jednotlivcov až 90 %. Ako výsledok možno preto pri súbežnom podávaní bosentanu s rifampicínom očakávať významné zníženie účinku bosentanu. Súbežné podávanie Tracleeru s rifampicínom sa preto neodporúča. Údaje o iných induktoroch CYP3A4 ako karbamazepín, fenobarbital, fenytoín a ľubovník bodkovaný nie sú dostupné, ale predpokladá sa, že ich súbežné podávanie s Tracleerom môže viesť k zníženej systémovej expozícii bosentanu. Klinicky významné zníženie účinku nemožno vylúčiť.
Lopinavir + ritonavir (a ďalšie ritonavirom potencované inhibítory proteáz): Súbežné podávanie bosentanu 125 mg dvakrát denne a lopinaviru + ritonaviru 400 + 100 mg dvakrát denne počas 9,5 dňa zdravým dobrovoľníkom viedlo k zvýšeniu plazmatickej hladiny bosentanu, ktorá bola 48-násobne vyššia ako pri podaní samotného bosentanu. Na deviaty deň bola plazmatická hladina bosentanu 5- násobne vyššia ako v prípade, keď bol bosentan podávaný samostatne. Táto interakcia je spôsobená pravdepodobne inhibíciou vychytávania bosentanu do hepatocytov, ktoré je sprostredkované transportným proteínom a inhibíciou CYP3A4 ritonavirom a redukciou klírens bosentanu. Pokiaľ sa Tracleer podáva súbežne s lopinavirom + ritonavirom alebo inými ritonavirom potencovanými inhibítormi proteáz, je potrebné sledovať znášanlivosť pacientov voči Tracleeru.
Po 9,5 dňoch súbežného podávania bosentanu, plazmatické hladiny lopinaviru a ritonaviru klesli na klinicky nevýznamné hodnoty (o približne 14 % a 17 %, v uvedenom poradí). Aj keď sa nemusí dosiahnuť celková indukcia bosentanu, nemožno vylúčiť ďalší pokles inhibítorov proteáz. Odporúča sa primeraný monitoring terapie HIV. Podobné účinky možno očakávať aj s inými ritonavirom potencovanými inhibítormi proteáz (pozri časť 4.4).
Iné antiretrovirálne lieky: Vzhľadom na nedostatok informácií nemožno formulovať ďalšie špeciálne odporúčania ohľadom užívania iných dostupných antiretrovirálnych liekov. Treba zdôrazniť, že výrazná hepatotoxicita nevirapinu môže potencovať pečeňovú toxicitu bosentanu, táto kombinácia sa neodporúča.
Hormonálna antikoncepcia: Súbežné podávanie bosentanu 125 mg dvakrát denne s jednou dávkou perorálneho kontraceptíva obsahujúceho noretisterón 1 mg + etinylestradiol 35 µg počas 7 dní znížilo AUC noretisterónu o 14 % a etinylestradiolu o 31 %. U jednotlivcov však bolo pozorované zníženie expozície až o 56 % pri noretisteróne a o 66 % pri etinylestradiole. Práve preto sa používanie len hormonálnej antikoncepcie ako jedinej metódy antikoncepcie nezávisle od spôsobu aplikácie (t.j. perorálnej, injekčnej, transdermálnej alebo implantabilnej) nepovažuje za spoľahlivú metódu antikoncepcie (pozri časti 4.4 a 4.6).
Warfarín: Súbežné podávanie bosentanu 500 mg dvakrát denne počas 6 dní znížilo plazmatické hladiny S-warfarínu (substrát CYP2C9) o 29 % a R-warfarínu (substrát CYP3A4) o 38 %. Počas klinických štúdií u pacientov s PAH, ktorým boli podávané súbežne bosentan a warfarín, neboli pozorované žiadne klinicky relevantné zmeny INR (International Normalized Ratio) ani dávky warfarínu (porovnanie vstupných hodnôt s hodnotami na konci klinických štúdií). Okrem toho, zmeny dávkovania warfarínu v priebehu testov z dôvodu zmeny INR alebo kvôli nežiaducim účinkom, boli rovnako časté u pacientov liečených bosentanom aj u pacientov s placebom. Na začiatku liečby bosentanom nie je nutné upravovať dávky warfarínu alebo podobných perorálnych antikoagulancií, ale odporúča sa intenzívnejší monitoring INR, najmä na začiatku liečby a v období titrácie vyššej dávky.
Simvastatín: Súbežné podávanie bosentanu 125 mg dvakrát denne počas 5 dní znížilo plazmatickú
hladinu simvastatínu (substrát CYP3A4) o 34 % a jeho aktívneho metabolitu β-hydroxykyseliny
o 46 %. Plazmatické hladiny bosentanu neboli súbežne podávaným simvastatínom ovplyvnené. Má sa
zvážiť sledovanie hladiny cholesterolu a prípadná úprava dávky.
Ketokonazol: Súbežné podávanie bosentanu 62,5 mg dvakrát denne počas 6 dní spolu
s ketokonazolom, potentným inhibítorom CYP3A4, zvýšilo plazmatickú hladinu bosentanu približne dvakrát. Nie je nutné upravovať dávku Tracleeru. Napriek tomu, že neboli vykonané štúdie in vivo, podobné zvýšenie plazmatických hladín bosentanu sa dá očakávať aj s inými potentnými inhibítormi CYP3A4 (napr. itrakonazol alebo ritonavir). V prípade kombinácie s inhibítorom CYP3A4
u pacientov s pomalým metabolizmom CYP2C9 avšak existuje riziko závažného zvýšenia
plazmatických hladín bosentanu, ktoré by mohlo vyvolať škodlivé nežiaduce účinky.
Epoprostenol: Obmedzené údaje získané zo štúdie (AC-052-356, [BREATHE-3]) s 10 detskými a dospievajúcimi pacientmi, ktorí dostali kombináciu bosentanu a epoprostenolu, naznačujú, že po jednotlivej aj opakovanej dávke boli hodnoty Cmax a AUC bosentanu boli podobné u pacientov s kontinuálnou infúziou epoprostenolu alebo bez nej (pozri časť 5.1).
Sildenafil: Súbežné podávanie bosentanu 125 mg dvakrát denne (rovnovážny stav) so sildenafilom
80 mg trikrát denne (rovnovážny stav) počas šiestich dní u zdravých dobrovoľníkov viedlo k 63 % zníženiu AUC sildenafilu a 50 % zvýšeniu AUC bosentanu. Súbežné podávanie týchto látok si vyžaduje zvýšenú pozornosť.
Digoxín: Súbežné podávanie bosentanu 500 mg dvakrát denne počas 7 dní s digoxínom znížilo AUC o 12 %, Cmax o 9 % a Cmin digoxínu o 23 %. Mechanizmom tejto interakcie môže byť indukcia P- glykoproteínu. Nie je pravdepodobné, že by táto interakcia mala klinický význam.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Štúdie na zvieratách preukázali reprodukčnú toxicitu (teratogenitu, embryotoxicitu; pozri časť 5.3).
Neexistujú žiadne údaje o užívaní Tracleeru u tehotných žien. Stále nie je známe potenciálne riziko u
ľudí. Tracleer je kontraindikovaný počas gravidity (pozri časť 4.3).
Ženy vo fertilnom veku
Pred začatím liečby Tracleerom u žien vo fertilnom veku je potrebné overiť, že žena nie je tehotná,
poskytnúť jej potrebné informácie o spoľahlivých metódach antikoncepcie a musí začať používať spoľahlivú antikoncepciu. Pacienti a lekári si musia byť vedomí, že v dôsledku potenciálnych farmakokinetických interakcií Tracleer môže spôsobiť neúčinnosť hormonálnych kontraceptív (pozri časť 4.5). Preto ženy vo fertilnom veku nesmú používať hormonálnu antikoncepciu (vrátane perorálnej, injekčnej, transdermálnej alebo implantabilnej formy) ako jedinú metódu antikoncepcie, ale musia
používať spoľahlivú doplnkovú alebo alternatívnu antikoncepčnú metódu. Pokiaľ sú akékoľvek
pochybnosti, ktorá antikoncepcia sa má odporučiť individuálnej pacientke, odporúča sa konzultácia s gynekológom. Vzhľadom na možné zlyhanie hormonálnej antikoncepcie počas liečby Tracleerom ako aj fakt, že počas gravidity sa závažne zhoršuje pľúcna hypertenzia, počas liečby Tracleerom sa odporúčajú testy gravidity raz za mesiac, ktoré umožnia skorú detekciu gravidity.
Laktácia
Nie je známe, či bosentan prechádza do ľudského materského mlieka. Dojčenie sa počas liečby
Tracleerom neodporúča.
Fertilita
Štúdie na zvieratách preukázali testikulárne účinky (pozri časť 5.3). V štúdii skúmajúcej účinky
bosentanu na funkciu semenníkov u mužských pacientov s PAH sa po 3 alebo 6 mesiacoch liečby
bosentanom u 8 z 24 pacientov ukázalo zníženie koncentrácie spermy v porovnaní so začiatočnými
hodnotami najmenej o 42 %. Na základe týchto zistení a predklinických údajov nie je možné vylúčiť, že bosentan môže mať u mužov škodlivý účinok na spermatogenézu. U chlapcov po liečbe bosentanom nie je možné vylúčiť dlhodobý vplyv na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne špecifické štúdie, ktoré by priamo sledovali vplyv Tracleeru na schopnosť viesť vozidlá a obsluhovať stroje. Avšak, Tracleer môže vyvolať hypotenziu, závraty, rozmazané videnie alebo synkopy, ktoré môžu ovplyvniť schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
V 20 placebom kontrolovaných štúdiách bol bosentan podávaný vo viacerých indikáciách v dennej dávke 100-2000 mg 2486 pacientom a v skupine s placebom bolo 1838 pacientov. Priemerná dĺžka liečby bola 45 týždňov. Nežiaduce účinky boli definované ako udalosti, ktoré sa vyskytovali minimálne u 1 % pacientov liečených bosentanom, vo frekvencii aspoň o 0,5 % častejšie ako
u skupiny s placebom. Najčastejšie nežiaduce reakcie sú boli bolesť hlavy (11,5 %), edém / retencia
tekutín (13,2 %), poruchy pečeňových testov (10,9 %) a anémia / pokles hladiny hemoglobínu (9,9 %).
Liečba bosentanom sa spájala s dávkovo závislým zvýšením pečeňových aminotransferáz ako aj
s poklesom hladiny hemoglobínu (pozri časť 4.4).
Nežiaduce reakcie pozorované v 20 placebom kontrolovaných štúdiách a počas postmarketingového sledovania s bosentanom sú zoradené podľa frekvencie výskytu: veľmi časté (≥ 1/10); časté (≥ 1/100 do < 1/10); menej časté (≥ 1/1000 do < 1/100); zriedkavé (1/10000 do < 1/1000); veľmi zriedkavé
(< 1/10000), neznáme (nedajú sa určiť z dostupných údajov).
V každej kategórii sú nežiaduce reakcie zoradené podľa klesajúcej závažnosti. Neboli pozorované žiadne klinicky významné rozdiely v nežiaducich reakciách medzi celým súborom a jednotlivými schválenými indikáciami.
Orgánový systém Frekvencia Nežiaduca reakcia
Poruchy krvi a lymfatického systému
Časté Anémia, pokles hemoglobínu
(pozri časť 4.4)
Neznáme Anémia alebo pokles hemoglobínu, ktorý si vyžaduje transfúziu červených krviniek1'
Menej časté Trombocytopénia1
Menej časté Neutropénia, leukopénia1
Poruchy imunitného systému Časté Hypersenzitívne reakcie (zahŕňajú dermatitídu, svrbenie a vyrážku)2
Zriedkavé Anafylaxia a/alebo angioedém1
Poruchy nervového systému Veľmi časté Bolesť hlavy3
Časté Synkopy1, 4
Poruchy oka Neznáme Rozmazané videnie1
Poruchy srdca a srdcovej
činnosti
Časté Palpitácie1, 4
Cievne poruchy Časté Sčervenanie
Časté Hypotenzia1, 4
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Časté Upchatý nos1
Časté Refluxná choroba pažeráka,
hnačka
Poruchy pečene a žlčových ciest Veľmi časté Abnormálne pečeňové testy
(pozri časť 4.4) Menej časté Zvýšenie hladiny
aminotransferáz spojené
s hepatitídou (vrátane možného zhoršenia skrytej hepatitídy) a/alebo žltačkou1 (pozri časť 4.4)
Zriedkavé Cirhóza pečene, zlyhanie pečene1
Poruchy kože a podkožného tkaniva
Celkové poruchy a reakcie v mieste podania
Časté Erytém
Veľmi časté Edém, retencia tekutín5
1 Údaje boli získané z postmarketingového sledovania, frekvencie sú založené na štatistickom modelovaní v placebom kontrolovaných klinických štúdiách
2 Hypersenzitívne reakcie boli hlásené u 9,9 % pacientov liečených bosentanom a 9,1 % pacientov v skupine s placebom.
3 Bolesť hlavy bola hlásená u 11,5 % pacientov liečených bosentanom a 9,8 % pacientov, ktorým bolo podávané placebo.
4 Tento typ reakcií môže tiež súvisieť s prebiehajúcim ochorením.
5 Edém alebo retencia tekutín boli hlásené u 13,2 % pacientov liečených bosentanom a 10,9 % pacientov, ktorým bolo podávané placebo.
Počas postmarketingového obdobia boli hlásené zriedkavé prípady neobjasnenej cirhózy pečene, ktoré sa vyskytovali u polymorbídnych pacientov, ktorí okrem inej terapie dlhodobo užívali Tracleer. Vyskytovali sa aj zriedkavé prípady zlyhania pečene. Tieto prípady len zvýrazňujú potrebu dodržiavať počas liečby Tracleerom pravidelný mesačný monitoring pečeňových funkcií (pozri časť 4.4).
Pediatrická populáciaNekontrolované klinické štúdie s pediatrickými pacientmiBezpečnostný profil v prvej pediatrickej nekontrolovanej štúdii vykonanej s filmom obalenými tabletami (BREATHE-3: n = 19, medián veku 10 rokov [rozpätie 3 až 15 rokov], otvorená, bosentan
2 mg/kg dvakrát denne; dĺžka liečby 12 týždňov) bol podobný s bezpečnostným profilom pozorovaným v pivotných štúdiách u dospelých pacientov s PAH. V štúdii BREATHE-3 boli najčastejšími nežiaducimi reakciami sčervenanie (21 %), bolesť hlavy a abnormálne pečeňové testy (každé 16 %).
Súhrnná analýza nekontrolovaných pediatrických štúdií vykonaných u PAH s bosentanom, v dávke

32 mg vo forme dispergovateľných tabliet (FUTURE 1/2, FUTURE 3/predĺženie) zahŕňala celkom
100 detí liečených bosentanom v dávke 2 mg/kg dvakrát denne (n = 33), 2 mg/kg trikrát denne (n = 31) alebo 4 mg/kg dvakrát denne (n = 36). Pri zaradení bolo 6 pacientov vo veku medzi 3 mesiacmi až 1 rokom, 15 detí bolo vo veku medzi 1 rokom a menej ako 2 rokmi a 79 detí bolo vo veku medzi 2 rokmi až 12 rokmi. Medián času trvania liečby bol 71,8 týždňa (rozpätie 0,4 až 258 týždňov).
Bezpečnostný profil bol v tejto súhrnnej analýze nekontrolovaných pediatrických štúdií podobný
bezpečnostnému profilu pozorovanému v pivotných štúdiách u dospelých pacientov s PAH
s výnimkou infekcií, ktoré boli hlásené častejšie ako u dospelých (69,0 % vs 41,3 %). Tento rozdiel v početnosti infekcií môže byť sčasti spôsobený vyšším mediánom času expozície liečby
v pediatrickom súbore (medián 71,8 týždňov) v porovnaní so súborom dospelých pacientov (medián
17,4 týždňa). Najčastejšími nežiaducimi udalosťami boli infekcie horných dýchacích ciest (25 %), pľúcna (arteriálna) hypertenzia (20 %), nazofaryngitída (17 %), pyrexia (15 %), vracanie (13 %), bronchitída (10 %), bolesti brucha (10 %) a hnačka (10 %). V početnosti nežiaducich udalostí nebol medzi pacientami staršími a mladšími ako 2 roky žiadny relevantný rozdiel; toto zistenie je však založené len na 21 deťoch mladších ako 2 roky, vrátane 6 pacientov vo veku medzi 3 mesiacmi a 1 rokom. Nežiaduce udalosti pečeňových abnormalít sa objavili u 9 % pacientov a nežiaduce udalosti
anémie/poklesu hemoglobínu u 5 % pacientov.
V randomizovanej placebom kontrolovanej štúdii (FUTURE-4), ktorá bola vykonaná u pacientov s PPHN bolo bosentanom, v dávke 2 mg/kg dvakrát denne vo forme dispergovateľných tabliet, liečených celkom 13 novorodencov (8 pacientov bolo na placebe). Medián času liečby bol 4,5 dňa pri bosentane (rozpätie 0,5 až 10,0 dní) a 4,0 dní pri placebe (rozpätie 2,5 až 6,5 dňa). Najčastejšími
nežiaducimi udalosťami u pacientov liečených bosentanom a u pacientov na placebe boli, v uvedenom poradí, anémia, alebo pokles hemoglobínu (7 a 2 pacienti), generalizovaný edém (3 a 0 pacientov)
a vracanie (2 a 0 pacientov).
Laboratórne abnormalityAbnormality pečeňových testovV priebehu klinického programu sa obvykle počas prvých 26 týždňov objavilo dávkovo závislé zvýšenie hladín pečeňových aminotransferáz, rozvinulo sa postupne a bolo prevažne asymptomatické. V post- marketingovej praxi boli hlásené zriedkavé prípady cirhózy pečene a zlyhania pečene.
Mechanizmus tohto nežiaduceho účinku nie je jasný. Hoci sa zvýšená hladina aminotransferáz môže vrátiť do normy spontánne počas pokračujúcej liečby udržiavacou dávkou Tracleeru alebo po znížení dávky, v niektorých prípadoch treba zvážiť prerušenie, prípadne ukončenie liečby (pozri časť 4.4).
V 20 placebom kontrolovaných štúdiách bolo pozorované zvýšenie pečeňových aminotransferáz ≥ 3 × HHN u 11,2 % pacientov liečených bosentanom v porovnaní s 2,4 % pacientov v skupine s placebom. Zvýšenie na ≥ 8 × HHN bolo zaznamenané u 3,6 % bosentanom liečených pacientov a 0,4 % pacientov, ktorým bolo podávané placebo. U 0,2 % pacientov (5 pacientov) liečených bosentanom
a 0,3 % pacientov (6 pacientov), ktorým bolo podávané placebo, zvýšenia hladiny aminotransferáz
boli spojené so zvýšenými hladinami bilirubínu (≥ 2 × HHN) bez dôkazu obštrukcie žlčových ciest.
V súhrnnej analýze vykonanej u 100 detí s PAH z nekontrolovaných pediatrických štúdií FUTURE
1/2 a FUTURE 3/predĺženie bolo pozorované zvýšenie pečeňových aminotransferáz ≥ 3 × HHN u 2 %
pacientov.
V štúdii FUTURE-4 zahŕňajúcej 13 novorodencov s PPHN liečených bosentanom, v dávke 2 mg/kg dvakrát denne v čase kratšom ako 10 dní (rozpätie 0,5 až 10,0 dní) sa počas liečby bosentanom nevyskytli žiadne prípady zvýšenia pečeňovej aminotransferázy ≥ 3 × HHN, ale 3 dni po ukončení liečby bosentanom sa vyskytol jeden prípad hepatitídy.
HemoglobínV placebom kontrolovaných štúdiách u dospelých bol hlásený pokles hladiny hemoglobínu zo vstupnej hodnoty na hodnotu menej ako 10 g/dl u 8,0 % pacientov liečených bosentanom a u 3,9 % pacientov v skupine s placebom (pozri časť 4.4).
V súhrnnej analýze u 100 detí s PAH z nekontrolovaných pediatrických štúdií FUTURE 1/2 a FUTURE 3/predĺženie bol u 10,0 % pacientov hlásený pokles hladiny hemoglobínu zo začiatočnej hodnoty na hodnotu menej ako 10 g/dl. K poklesu pod 8 g/dl nedošlo.
V štúdii FUTURE-4 sa počas liečby u 6 z 13 novorodencov s PPHN liečených bosentanom, vyskytol pokles hladiny hemoglobínu z referenčného rozpätia na začiatku na nižšie hodnoty ako je limit normálu.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4
.9 Predávkovanie
Bosentan bol aplikovaný ako jednotlivá dávka zdravým jedincom až do množstva 2 400 mg a pacientom s iným ochorením než pľúcna hypertenzia až do množstva 2000 mg/deň počas dvoch mesiacov. Najčastejšou nežiaducou reakciou bola bolesť hlavy miernej až strednej intenzity.
Masívne predávkovanie môže mať za následok výraznú hypotenziu vyžadujúcu aktívnu kardiovaskulárnu podporu. Počas postmarketingového sledovania bol zaznamenaný jeden prípad predávkovania Tracleerom dávkou 10 000 mg, ktorú užil dospievajúci pacient mužského pohlavia. Prejavili sa u neho symptómy nauzea, vracanie, hypotenzia, závraty, potenie a rozmazané videnie. Za podpory tlaku krvi sa stav pacienta do 24 hodín navrátil do pôvodného stavu. Poznámka: bosentan sa neodstraňuje dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: iné antihypertenzíva, ATC kód: C02KX01
Mechanizmus účinku
Bosentan je duálny antagonista receptora pre endotelín (ERA) s afinitou pre endotelínový receptor A a
B (ETA a ETB). Bosentan znižuje obidve, pľúcnu aj systémovú vaskulárnu rezistenciu, čo má za
následok zvýšenie minútového vývrhového objemu srdca bez zvýšenia frekvencie srdca.
Neurohormón endotelín-1 (ET-1) je jedným z najúčinnejších známych vazokonstriktorov a môže tiež podporovať fibrózu, proliferáciu buniek, srdcovú hypertrofiu a remodeláciu a pôsobí prozápalovo. Tieto účinky sú sprostredkované väzbou endotelínu na ETA a ETB receptory lokalizované v endoteli a v bunkách hladkého svalstva ciev. Hladina ET-1 v tkanivách a plazme je zvýšená pri rôznych kardiovaskulárnych poruchách a chorobách spojivových tkanív vrátane PAH, sklerodermie, akútneho a chronického zlyhania srdca, ischémie myokardu, systémovej hypertenzie a aterosklerózy, čo naznačuje patogénnu úlohu ET-1 pri týchto ochoreniach. Pri PAH a zlyhaní srdca v neprítomnosti antagonizmu endotelínového receptora, zvýšená hladina ET-1 silne koreluje so závažnosťou a prognózou ochorenia.
Bosentan kompetuje väzbu ET-1 a iných ET peptidov na oba ETA a ETB receptory, s mierne zvýšenou afinitou k ETA receptorom (Ki = 4,1-43 nanomolov) než k ETB receptorom (Ki = 38-730 nanomolov). Bosentan je špecifický antagonista ET receptorov, pričom sa neviaže na iné receptory.
Účinnosť
Zvieracie modely
U zvieracích modelov pľúcnej hypertenzie opakované perorálne podávanie bosentanu znižovalo pľúcnu vaskulárnu rezistenciu a zvrátilo pľúcnu vaskulárnu rezistenciu a hypertrofiu pravej komory. U zvieracích modelov pľúcnej fibrózy bosentan znižoval ukladanie kolagénu v pľúcach.
Účinnosť u dospelých pacientov s pľúcnou artériovou hypertenziou
Uskutočnili sa dve randomizované, dvojito zaslepené, multicentrické, placebom kontrolované štúdie s
32 (štúdia AC-052-351) a 213 (štúdia AC-052-352, [BREATHE-1]) dospelými pacientmi s PAH III.-IV. triedy podľa funkčnej klasifikácie WHO (primárna pľúcna hypertenzia alebo pľúcna hypertenzia sekundárna, najmä pri sklerodermii). Po 4 týždňoch liečby bosentanom v dávke 62,5 mg dvakrát denne boli udržiavacie dávky sledované v týchto štúdiách 125 mg dvakrát denne v AC-052-
351 a 125 mg dvakrát denne a 250 mg dvakrát denne v AC-052-352.
Bosentan bol pridaný k aktuálnej terapii pacientov, ktorá mohla obsahovať kombináciu antikoagulancií, vazodilatancií (napr. blokátorov kalciových kanálov), diuretík, kyslíka a digoxínu, ale nie epoprostenolu. Kontrolná skupina dostala k aktuálnej terapii placebo.
Primárnym kritériom hodnotenia každej štúdie bola zmena vzdialenosti v teste 6-minútovej chôdze
v 12. týždni v prvej štúdii a v 16. týždni v druhej štúdii. V oboch testoch malo liečenie bosentanom za následok signifikantné zvýšenie záťažovej kapacity. Placebom korigované predĺženie vzdialenosti chôdze oproti vstupným hodnotám v zmysle primárneho kritéria hodnotenia každej štúdie bolo
76 metrov (p = 0,02; t-test), resp. 44 metrov (p = 0,0002; Mann-Whitney U test). Rozdiely medzi skupinami so 125 mg dvakrát denne a s 250 mg dvakrát denne neboli štatisticky signifikantné, ale existoval trend smerom ku zlepšeniu záťažovej kapacity v skupine liečenej dávkou 250 mg dvakrát denne.
V dvojito slepej štúdii, ktorá bola vykonaná u časti pacientov, bolo zlepšenie v prejdenej vzdialenosti
zreteľné po 4 týždňoch liečby, výrazné po 8 týždňoch a udržalo sa až do 28. týždňa.
V retrospektívnej analýze respondérov, založenej na zmenách v zdolanej vzdialenosti testu chôdze, podľa funkčnej klasifikácie WHO a v dýchavičnosti u 95 pacientov randomizovaných na liečbu bosentanom 125 mg dvakrát denne v placebom kontrolovanom teste, bolo zistené, že v 8. týždni sa u 66 pacientov prejavilo zlepšenie, 22 bolo stabilizovaných a u 7 pacientov sa prejavilo zhoršenie. Z 22 pacientov stabilizovaných v 8. týždni sa u 6 prejavilo zlepšenie v týždňoch 12/16 a u 4 sa
v porovnaní so vstupnými hodnotami výkon zhoršil. Zo 7 pacientov, u ktorých sa prejavilo zhoršenie
8. týždeň, sa u 3 v týždňoch 12/16 výkon zlepšil a u 4 nastalo zhoršenie v porovnaní so vstupným
meraním.
Invazívne hemodynamické parametre boli hodnotené iba v prvej štúdii. Liečba bosentanom viedla k signifikantnému zvýšeniu srdcového indexu spojenému so signifikantným znížením pľúcneho artériového tlaku, pľúcnej vaskulárnej rezistencie a stredného tlaku pravej predsiene.
Pri liečbe bosentanom bolo pozorované zníženie symptómov PAH. Miera dyspnoe počas testu chôdze sa u pacientov liečených bosentanom zlepšila. V štúdii AC-052-352 bolo 92 % z 213 pacientov klasifikovaných podľa vstupných hodnôt ako III. trieda podľa funkčnej klasifikácie WHO a 8 % ako trieda IV. Liečba bosentanom viedla ku zlepšeniu triedy funkčnej klasifikácie WHO u 42,4 % pacientov (placebo 30,4 %). Celková zmena triedy podľa funkčnej klasifikácie WHO v priebehu oboch štúdií bola signifikantne lepšia medzi pacientmi liečenými Tracleerom v porovnaní s pacientmi v skupine s placebom. V 28. týždni bola liečba bosentanom spojená so signifikantným znížením podielu klinického zhoršenia v porovnaní s placebom (10,7 % oproti 37,1 %; p = 0,0015).
V randomizovanej, dvojito slepej, multicentrickej, placebom kontrolovanej štúdii (AC-052-364;
[EARLY]) dostávalo 185 pacientov s PAH funkčnej triedy II podľa WHO (s priemernou vzdialenosťou
v 6-minútovom teste chôdze 435 metrov) najskôr 62,5 mg bosentanu dvakrát denne počas 4 týždňov a následne počas 6 mesiacov buď 125 mg bosentanu dvakrát denne (n = 93), alebo placebo (n = 92). Zaradení boli pacienti s doposiaľ neliečenou PAH (n = 156) alebo liečení stabilnou dávkou sildenafilu
(n = 29). Primárnymi cieľovými ukazovateľmi boli percentuálna zmena pľúcnej vaskulárnej rezistencie
(PVR) oproti začiatku a zmena vzdialenosti 6-minútového testu chôdze po 6 mesiacoch od začiatku
v porovnaní s placebom. Nasledujúca tabuľka uvádza výsledky analýz definovaných v protokole.
Začiatok;
PVR (dyn.sec/cm5) 6-minútový test chôdze -
vzdialenosť (m)
Placebo (n=88) Bosentan (n=80) Placebo (n=91) Bosentan (n=86)
aritmetický priemer (SO) 802 (365)
851 (535)
431 (92)
443 (83)
Zmena oproti začiatku;
aritmetický priemer (SO) 128 (465)

-69 (475)
-8 (79)
11 (74)
Účinky liečby -22,6 % 19
95 % CL -34, -10 -4, 42

Hladina štatistickej významnosti – p
< 0,0001 0,0758
CL – medze intervalu spoľahlivosti; PVR – pľúcna vaskulárna rezistencia; SO – smerodajná odchýlka.
Liečba bosentanom preukázala v porovnaní s placebom redukciu početnosti klinického zhoršenia, definovaného ako kombinovaný parameter zložený zo zhoršenia symptómov, hospitalizácií v dôsledku PAH alebo úmrtia (proporcionálne zníženie rizika 77 %, 95 % interval spoľahlivosti [CI] 20-94 %,
p = 0,0114). Hlavnou zložkou preukázaného liečebného účinku bolo zníženie početnosti zhoršenia symptómov. V skupine liečenej bosentanom sa vyskytla len jedna hospitalizácia súvisiaca s PAH, kým v skupine s placebom sa vyskytli 3 hospitalizácie. Počas 6 mesiacov dvojito slepej štúdie sa
v každej zo skupín vyskytlo len jedno úmrtie. V súvislosti s ovplyvnením prežívania nemožno preto
formulovať žiadne závery.
Dlhodobé údaje boli získané od 173 pacientov, ktorí boli liečení bosentanom v kontrolovanej fáze a/alebo u ktorých bola zmenená liečba z placeba na bosentan v otvorenom predĺžení štúdie EARLY. Priemerná dĺžka užívania bosentanu bola 3,6 ± 1,8 roka (maximálne 6,1 rokov), pričom 73 % pacientov bolo liečených najmenej 3 roky a 62 % pacientov bolo liečených najmenej 4 roky. Počas predĺženia otvorenej štúdie pacienti s PAH mohli užívať doplnkovú liečbu. Väčšina pacientov bola diagnostikovaná s idiopatickou alebo dedičnou PAH (61 %). Celkovo 78 % pacientov ostalo v II. stupni PAH podľa funkčnej klasifikácie WHO. V treťom roku prežilo 90 % a v štvrtom roku 85 %
pacientov podľa Kaplan Majerovej krivky mortality. Zároveň u 88 %, resp. 79 % pacientov sa v týchto
časových intervaloch nezaznamenalo zhoršenie PAH (definované ako celková mortalita, transplantácia pľúc, atriálna septostómia alebo začiatok intravenóznej alebo subkutánnej liečby prostanoidmi). Podiel predchádzajúcej liečby placebom v dvojito zaslepenej štúdii alebo podiel ostatných liečiv v otvorenom predĺžení štúdie nie je známy.
V prospektívnej, multicentrickej, randomizovanej, dvojito slepej, placebom kontrolovanej štúdii (AC-
052-405 [BREATHE-5]) dostávali pacienti s PAH III. triedy podľa funkčnej klasifikácie WHO a Eisenmengerovým syndrómom združeným s vrodenými srdcovými chybami bosentan 62,5 mg dvakrát denne počas štyroch týždňov, potom 125 mg dvakrát denne počas ďalších 12 týždňov (n = 37, z ktorých
31 malo primárne pravoľavý, obojstranný skrat). Primárnym cieľom bolo ukázať, že bosentan nespôsobil zhoršenie hypoxémie. Po 16 týždňoch sa priemerná saturácia kyslíkom u skupiny liečenej bosentanom zvýšila o 1,0 % (95 % CI -0,7-2,8 %) v porovnaní so skupinou s placebom (n = 17), čo názorne
dokazuje, že bosentan nespôsobil zhoršenie hypoxémie. Priemerná pľúcna vaskulárna rezistencia sa signifikantne znížila u skupiny liečenej bosentanom (s prevažujúcim účinkom pozorovaným u pacientov s obojsmerným vnútrosrdcovým skratom). Po 16 týždňoch bolo priemerné placebom korigované predĺženie vzdialenosti pri 6-minútovom teste chôdze 53 metrov (p = 0,0079), čo odráža zlepšenie
funkčnej kapacity. Dvadsaťšesť pacientov pokračovalo v užívaní bosentanu v 24-týždňovom, otvorenom
predĺžení (AC-052-409) štúdie BREATHE-5 (priemerné trvanie liečby 24,4 ± 2,0 týždňa) a účinnosť
sa vo všeobecnosti udržala.
Otvorená, nekomparatívna štúdia (AC-052-362; [BREATHE-4]) bola vykonaná u 16 pacientov s PAH III. triedy podľa funkčnej klasifikácie WHO a s infekciou HIV. Pacienti boli liečení bosentanom 62,5 mg dvakrát denne počas štyroch týždňov a následne 125 mg dvakrát denne počas ďalších 12 týždňov. Po
16 týždňoch liečby nastalo oproti východiskovej hodnote významné zlepšenie v 6-minútovom teste
chôdze: pri priemernej začiatočnej hodnote 332,6 metrov bolo priemerné predĺženie vzdialenosti
91,4 metrov (p < 0,001). Nemožno vysloviť žiadny formálny záver týkajúci sa účinkov bosentanu na účinnosť antiretrovirálnych liekov (pozri tiež časť 4.4).
Nie sú známe žiadne klinické štúdie, ktoré by poukazovali na priaznivé účinky liečby Tracleerom na prežívanie. Dlhodobé prežívanie bolo ale zaznamenané u všetkých 235 pacientov, ktorí boli liečení bosentanom v 2 pivotných placebom kontrolovaných klinických štúdiách (AC-052-351 a AC-052-352) a/alebo v ich dvoch nekontrolovaných, otvorených predĺženiach. Priemerné trvanie podávania bosentanu bolo 1,9 ± 0,7 rokov (min. 0,1 roka; max. 3,3 roka), pričom pacienti boli sledovaní priemerne 2,0 ± 0,6 roka. Vačšina z nich mala diagnostikovanú primárnu pľúcnu hypertenziu (72 %) a bola v III. triede
podľa funkčnej klasifikácie WHO (84 %). V tejto populácii bolo predpokladané prežívanie podľa
Kaplan-Meierovej metódy 93 % po 1 roku a 84 % po 2 rokoch od začiatku liečby bosentanom. Prežívanie bolo horšie v podskupine pacientov so sekundárnou pľúcnou hypertenziou pri systémovej skleróze. Hodnoty môžu byť ovplyvňované tým, že u 43 z 235 pacientov sa liečba začala epoprostenolom.
Štúdie uskutočnené u detí s pľúcnou artériovou hypertenziou
BREATHE-3 (AC-052-356)
Bosentan, filmom obalené tablety, bol hodnotený v otvorenej nekontrolovanej štúdii u 19 pediatrických pacientov s PAH vo veku 3 až 15 rokov.Táto štúdia bola v prvom rade určená ako farmakokinetická štúdia (pozri časť 5.2.). Pacienti mali primárnu pľúcnu hypertenziu (10 pacientov) alebo PAH
súvisiacu s kongenitálnym ochorením srdca (9 pacientov) a na začiatku sledovania boli v II. (n = 15,
79 %) alebo III. triede (n = 4, 21 %) podľa funkčnej klasifikácie WHO. Podľa hmotnosti boli pacienti rozdelení do 3 skupín, z ktorých každá užívala počas 12 týždňov dávku bosentanu približne 2 mg/kg dvakrát denne. Polovica pacientov v každej skupine bola už liečená intravenózne podávaným epoprostenolom a dávka epoprostenolu zostala konštantná po celú dobu trvania štúdie.
Hemodynamické parametre boli merané u 17 pacientov. Srdcový index sa oproti vstupným hodnotám zvýšil priemerne o 0,5 l/min/m2, pľúcny arteriovy tlak sa znížil priemerne o 8 mmHg a PVR sa znížila priemerne o 389 dyn·sec·cm-5. Tieto hemodynamické zlepšenia oproti vstupným hodnotám boli podobné pri súbežnom podávaní epoprostenolu alebo bez súbežného podávania epoprostenolu. Zmeny parametrov záťažovej kapacity v 12. týždni oproti vstupným hodnotám boli veľmi variabilné a žiadna
z nich nebola signifikantná.
FUTURE 1/2 (AC-052-365/AC-052-367)
Štúdia FUTURE 1 bola otvorená nekontrolovaná štúdia, ktorá bola vykonaná s bosentanom vo forme dispergovateľných tabliet podávaných v udržiavacej dávke 4 mg/kg dvakrát denne 36 pacientom vo veku od 2 do 11 rokov. Štúdia bola primárne navrhnutá ako farmakokinetická štúdia (pozri časť 5.2). Na začiatku mali pacienti idiopatickú (31 pacientov [86 %]) alebo familiárnu (5 pacientov [14 %]) PAH a spadali do II. (n = 23, 64 %) alebo III. triedy (n = 13, 36 %) podľa funkčnej klasifikácie WHO.
V štúdii FUTURE 1 bol medián expozície hodnotenej liečby 13,1 týždňa (rozpätie 8,4 až 21,1). 33
z týchto pacientov bola poskytnutá pokračujúca liečba bosentanom vo forme dispergovateľných tabliet v dávke 4 mg/kg dvakrát denne v nekontrolovanej predĺženej fáze štúdie FUTURE 2 po medián celkového trvania liečby 2,3 roka (v rozmedzí 0,2 až 5,0 rokov). Na začiatku štúdie FUTURE 1
užívalo 9 pacientov epoprostenol. 9 pacientom bola v priebehu štúdie novo iniciovaná liečba špecifická pre PAH. Odhad neprítomnosti zhoršenia PAH (úmrtie, transplantácia pľúc alebo hospitalizácia kvôli zhoršeniu PAH) bol po 2 rokoch podľa Kaplanovej-Meierovej metódy 78,9%. Celkový odhad prežitia podľa Kaplanovej-Meierovej metódy po 2 rokoch bol 91,2 %.
FUTURE 3 (AC-025-373)
V tejto otvorenej randomizovanej štúdii s 32 mg bosentanu vo forme dispergovateľných tabliet bolo 64 detí so stabilnou PAH vo veku od 3 mesiacov do 11 rokov randomizovaných do skupiny liečených 24 týždňov bosentanom v dávke 2 mg/kg dvakrát denne (n = 33) alebo 2 mg/kg trikrát denne (n = 31). 43 detí (67,2 %) bolo vo veku ≥ 2 roky až 11 rokov, 15 detí (23,4 %) bolo vo veku medzi 1 rokom a 2 rokmi a 6 detí (9,4 %) bolo vo veku medzi 3 mesiacmi a 1 rokom. Štúdia bola primárne navrhnutá ako farmakokinetická štúdia (pozri časť 5.2) a výsledky hodnotenia účinnosti boli len exploratórne. Etiológia PAH, podľa „Dana Point” klasifikácie, zahŕňala idiopatickú PAH (46 %), dedičnú PAH (3
%), PAH súvisiacu s korekčným chirurgickým zákrokom na srdci (38 %) a PAH súvisiacu
s kongenitálnym ochorením srdca spojenú so systémovými-pľúcnymi skratmi vrátane Eisenmengorovho syndrómu (13 %). Na začiatku liečby hodnoteným liečivom boli pacienti v I. triede (n = 19 29 %), v II. triede (n = 27, 42 %) alebo III. triede (n = 18, 28 %) podľa funkčnej klasifikácie WHO. Pri vstupe do štúdie boli pacienti liečení liekmi proti PAH (najčastejšie inhibítorom fosfodiesterázy typu 5 [sildenafil] samotným [35,9 %], bosentanom samotným [10,9 %] a
kombináciou bosentanu, iloprostu a sildenafilu [10,9 %]) a v priebehu štúdie svoj liek proti PAH ďalej
užívali.
Na začiatku štúdie bola menej ako polovica zaradených pacientov liečená bosentanom samotným (45,3 % [29/64]) nekombinovaným s ďalšími liekmi proti PAH, 40,6 % (26/64) ostalo počas 24 týždňov hodnotenej liečby na monoterapii bosentanom. Analýza zahŕňajúca celkovú populáciu (64 pacientov) ukázala, že väčšina zostala prinajmenšom stabilná (t.j. bez zhoršenia) na základe nepediatrického špecifického funkčného hodnotenia podľa WHO (97 % dvakrát denne, 100 % trikrát denne) a na základe celkového klinického pocitu lekára (94 % dvakrát denne, 93 % trikrát denne) počas liečby. Odhad neprítomnosti zhoršenia PAH (úmrtie, transplantácia pľúc alebo hospitalizácia pre zhoršenie PAH) bol po 24 týždňoch podľa Kaplanovej-Meierovej metódy 96,9 % v skupine
s dávkou dvakrát denne a 96,7 % v skupine s podávanou dávkou trikrát denne.
Pri podávaní dávky 2 mg/kg trikrát denne v porovnaní s dávkou 2 mg/kg dvakrát denne nebol dokázaný žiadny klinický prínos.
Štúdie uskutočnené u novorodencov s pretrvávajúcou pľúcnou hypertenziou novorodencov (PPHN)
FUTURE 4 (AC-025-391)
Išlo o dvojito zaslepenú placebom kontrolovanú randomizovanú štúdiu u predčasne narodených alebo v termíne narodených novorodencov (gestačný vek 36 až 42 týždňov) s PPHN. Pacienti so suboptimálnou odpoveďou na inhalovaný oxid dusnatý (iNO) napriek najmenej 4 hodinám kontinuálnej liečby boli liečení bosentanom vo forme dispergovateľných tabliet v dávke 2 mg/kg dvakrát denne (N =
13) alebo placebom (N = 8) podávaným nazogastrickou sondou ako prídavná liečba k iNO do kompletného vysadenia iNO alebo do zlyhania liečby (definovaného ako potreba extrakorporálnej membránovej xygenácie [ECMO] alebo nasadenia alternatívneho pľúcneho vazodilatátora) počas maximálne 14 dní.
Medián expozície hodnotenej liečby bol 4,5 (rozpätie 0,5 až 10,0) dňa v skupine liečených
bosentanom a 4,0 (rozpätie 2,5 až 6,5) dni v skupine na placebe.
Výsledky u tejto populácie nenaznačili dodatočný prínos bosentanu:
• medián času do úplného vysadenia iNO bol 3,7 dňa (95 % medze intervalu spoľahlivosti [CLs]
1,17; 6,95) pri bosentane a 2,9 dňa (95 % CLs 1,26; 4,23) pri placebe (p = 0,34).
• medián času do úplného vysadenia mechanickej ventilácie bol 10,8 dňa (95 % CLs 3,21; 12,21
dňa) pri bosentane a 8,6 dňa (95 % CLs 3,71; 9,66 dňa) pri placebe (p = 0,24).
• u jedného pacienta v skupine liečenej bosentanom došlo k zlyhaniu liečby (potreba ECMO podľa definície podľa protokolu), ktoré sa prejavilo na základe zvyšujúcich sa hodnôt oxygenačného indexu v priebehu 8 hodín po prvej dávke hodnoteného liečiva. Tento pacient sa počas 60 dní následného pozorovania vyliečil.
Kombinácia s epoprostenolom
Kombinácia bosentanu a epoprostenolu bola sledovaná v dvoch štúdiách: AC-052-355 (BREATHE-2) a AC-052-356 (BREATHE-3). AC-052-355 bola multicentrická, randomizovaná, placebom kontrolovaná, dvojito zaslepená štúdia bosentanu u 33 pacientov so závažnou PAH, ktorí dostávali súbežne epoprostenol. AC-052-356 bola otvorená, nekontrolovaná štúdia, v ktorej počas 12 týždňov 10 z 19 pediatrických pacientov dostávalo súbežne bosentan a epoprostenol. Bezpečnostný profil kombinácie sa
nelíšil od profilu očakávaného u každej zložky a kombinovaná liečba bola dobre znášaná deťmi a
dospelými. Klinický prínos kombinácie nebol preukázaný.
Systémová skleróza s vredovou chorobou prstov
Boli uskutočnené dve randomizované, dvojito zaslepené, multicentrické a placebom kontrolované štúdie u 122 (štúdia AC-052-401, [RAPIDS-1]) a 190 (štúdia AC-052-331, [RAPIDS-2]) dospelých pacientov
so systémovou sklerózou a vredovou chorobou prstov (buď pretrvávajúce vredy prstov alebo údaj
o vredoch prstov v priebehu predchádzajúceho roka). V štúdii AC-052-331 museli mať pacienti aspoň jeden nedávno vzniknutý vred na prste a počas obidvoch štúdií muselo mať 85 % pacientov vznikajúci vred na prste na začiatku. Po 4 týždňoch liečby bosentanom v dávke 62,5 mg dvakrát denne bola sledovaná udržiavacia dávka v obidvoch štúdiách 125 mg dvakrát denne. Dĺžka dvojito zaslepenej liečby bola v štúdii AC-052-401 16 týždňov a v štúdii AC-052-331 24 týždňov.
Pôvodná liečba systémovej sklerózy a vredov na prstoch bola prípustná, ak ostali bez zmeny najmenej
počas jedného mesiaca predchádzajúceho začiatku liečby a v priebehu trvania dvojito zaslepenej štúdie.
Počet nových vredov prstov od začiatku do ukončenia štúdie bol primárnym kritériom hodnotenia pre obidve štúdie. V priebehu štúdie liečba bosentanom viedla v porovnaní s placebom ku zníženiu výskytu nových vredov prstov. V štúdii AC-052-401 sa v priebehu 16 týždňov dvojito zaslepenej liečby, v skupine pacientov liečených bosentanom objavilo v priemere 1,4 nových vredov na prstoch oproti 2,7 nových vredov na prstoch v skupine s placebom (p = 0,0042). V štúdii AC-052-331 v priebehu 24 týždňov dvojito zaslepenej liečby boli zhodné údaje pre nové vredy na prstoch 1,9, resp. 2,7 (p =
0,0351). V obidvoch štúdiách boli pacienti na bosentane menej náchylní k vzniku viacpočetných nových vredov na prstoch v priebehu štúdie a dlhšie trvalo, než sa každý nasledujúci vred rozvinul, než tomu bolo u pacientov v skupine s placebom. Účinok bosentanu na zníženie počtu nových vredov na prstoch bol viac zrejmý u pacientov s viacpočetnými vredmi na prstoch.
Ani v jednej z obidvoch štúdií sa nepozoroval žiadny účinok bosentanu na rýchlosť vyhojenia vredov na
prstoch.
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti bosentanu boli dokumentované najmä u zdravých dobrovoľníkov. Obmedzené údaje pacientov ukazujú, že vplyv bosentanu na dospelých pacientov s PAH je približne 2 krát väčší než u dospelých zdravých dobrovoľníkov.
U zdravých dobrovoľníkov bosentan vykazuje farmakokinetiku závislú od dávky a času. Klírens
a distribučný objem sa znižujú so zvýšenými intravenóznymi dávkami a stúpa s časom. Po perorálnom podaní je systémová dostupnosť úmerná dávke až po dávku 500 mg. Pri vyšších perorálnych dávkach sa zvyšuje Cmax a AUC menej než úmerne dávke.
Absorpcia
U zdravých dobrovoľníkov je absolútna biologická dostupnosť bosentanu približne 50 % a nie je
ovplyvnená potravou. Maximálna plazmatická hladina sa dosiahne v priebehu 3-5 hodín. Distribúcia
Bosentan je výrazne viazaný (> 98 %) na plazmatické proteíny, najmä albumín. Bosentan nepreniká
do erytrocytov.
Distribučný objem (Vss) asi 18 litrov bol stanovený po intravenóznej dávke 250 mg. Biotransformácia a eliminácia
Po jednorazovej intravenóznej dávke 250 mg bol klírens 8,2 l/h. Polčas eliminácie (t1/2) je 5,4 hodiny.
Pri viacnásobnom dávkovaní sa plazmatické hladiny bosentanu postupne znižujú až na 50-65 % pôvodnej hodnoty stanovenej po podaní jednorazovej dávky. Toto zníženie je pravdepodobne dôsledkom autoindukcie metabolických pečeňových enzýmov. Rovnovážny stav bol dosiahnutý v priebehu 3-5 dní.
Bosentan je eliminovaný žlčou po metabolizácii v pečeni izoenzýmami CYP2C9 a CYP3A4
cytochrómu P450. Menej než 3 % perorálne aplikovanej dávky sa nachádzajú v moči.
Bosentan vytvára tri metabolity a iba jeden z nich je farmakologicky aktívny. Tento metabolit sa vylučuje prevažne nezmenený žlčou. U dospelých pacientov je systémová dostupnosť aktívneho metabolitu väčšia než u zdravých dobrovoľníkov. U pacientov so známkami cholestázy môže byť systémová dostupnosť aktívneho metabolitu zvýšená.
Bosentan je induktor CYP2C9 a CYP3A4 a možno tiež CYP2C19 a P-glykoproteínu. In vitro
bosentan inhibuje exportnú pumpu žlčových solí v kultúrach hepatocytov.
In vitro štúdie ukázali, že bosentan nemal relevantný inhibičný vplyv na testované izoenzýmy CYP (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4). Preto sa pod vplyvom bosentanu neočakáva zvyšovanie plazmatickej hladiny liečiv metabolizovaných týmito izoenzýmami.
Porovnanie liekových foriem
Vo farmakokinetickej štúdii (AC-052-116), dostalo 16 zdravých dobrovoľníkov 62,5 mg bosentanu
v liekovej forme filmom obalená tableta alebo 64 mg bosentanu v liekovej forme dispergovateľná tableta 32 mg. Sledovanie liečby dispergovateľnými tabletami ukázalo nižšie hladiny bosentamu ako u filmom obalených tabliet (pomer priemerov pre AUC0-∞ 0,87 [90 % CI: 0,78, 0,97]). Tmax a t1/2 bosentanu neboli signifikantne liekovou formou ovplyvnené.
Farmakokinetika v osobitných skupinách pacientov
Na základe skúmaného rozmedzia každého parametra sa neočakáva, že farmakokinetika bosentanu
bude v dospelej populácii relevantne ovplyvnená pohlavím, hmotnosťou, rasou alebo vekom.
Pediatrická populácia
Farmakokinetika bola u pediatrických pacientov hodnotená v 4 klinických štúdiách (BREATHE-3, FUTURE 1, FUTURE-3 a FUTURE-4; pozri časť 5.1). Kvôli obmedzeným údajom od detí mladších ako 2 roky je farmakokinetika v tejto vekovej skupine stále nedostatočne charakterizovaná.
Štúdia AC-052-356 (BREATHE-3) hodnotila farmakokinetiku jednorazových a opakovaných perorálnych dávok filmom obalených tabliet bosentanu u 19 detí vo veku od 3 rokov do 15 rokov s PAH, pričom dávkovanie záviselo od telesnej hmotnosti 2 mg/kg dvakrát denne. V tejto štúdii sa expozícia bosentanu znížila s časom spôsobom konzistentným so znalosťami o autoindukčných vlastnostiach bosentanu. Stredné hodnoty AUC (CV %) bosentanu u pediatrických pacientov liečených 31,25; 62,5 alebo 125 mg dvakrát denne boli 3496 (49), 5428 (79) a 6124 (27) ng·h/ml
v uvedenom poradí a boli nižšie než hodnota 8149 (47) ng·h/ml pozorovaná u dospelých pacientov s PAH, ktorí boli liečení dávkou 125 mg dvakrát denne. V rovnovážnom stave systémová dostupnosť u pediatrických pacientov vážiacich 10-20 kg, 20-40 kg a > 40 kg tvorila v uvedenom poradí 43 %, 67
% a 75 % systémovej dostupnosti u dospelých.
V štúdii AC-052-365 (FUTURE 1) boli podávané dispergovateľné tablety u 36 detí s PAH vo veku 2-
11 rokov. Nepozorovala sa žiadna dávková úmernosť, rovnovážne plazmatické hladiny bosentanu a AUC boli podobné pri perorálnej dávke 2 a 4 mg/kg (AUCτ bol 3577 ng·h/ml pri dávke
2 mg/kg dvakrát denne a 3371 ng·h/ml pri dávke 4 mg/kg dvakrát denne). Priemerná expozícia
bosentanu u týchto pediatrických pacientov predstavovala približne polovicu expozície u dospelých pacientov pri dávke 125 mg dvakrát denne, ale preukázala výraznú zhodu so závermi u dospelých pacientov.
V štúdii AC-052-373 (FUTURE 3), používajúcej dispergovateľné tablety bola expozícia bosentanu
u pacientov liečených dávkou 2 mg/kg dvakrát denne porovnateľná s expozíciou v štúdii FUTURE 1. V celkovej populácii (n = 31) viedla dávka 2 mg/kg dvakrát denne k dennej expozícii 8535 ng·h/ml; AUCτ bola 4268 ng·h/ml (CV: 61 %). U pacientov vo veku medzi 3 mesiacmi a 2 rokmi bola denná expozícia 7879 ng·h/ml; AUCτ bola 3939 ng·h/ml (CV: 72 %). U pacientov vo veku medzi 3 mesiacmi a 1 rokom (n = 2) bola AUCτ 5914 ng·h/ml (CV: 85 %) a u pacientov vo veku medzi 1 rokom a 2 rokmi (n = 7) bola AUCτ 3507 ng·h/ml (CV: 70 %). U pacientov starších ako 2 roky
(n = 22) bola denná expozícia 8820 ng·h/ml; (AUCτ bola 4410 ng·h/ml) (CV: 58 %). Dávkovanie bosentanu 2 mg/kg trikrát denne expozíciu nezvyšovalo; denná expozícia bola 7275 ng·h/ml, (CV:
83 %, n = 27).
Na základe záverov štúdií BREATHE-3, FUTURE 1 a FUTURE 3 sa javí, že u pediatrických pacientov expozícia bosentanu dosahuje plateau pri nižších dávkach ako u dospelých pacientov, a že
dávky vyššie ako 2 mg/kg dvakrát denne (4 mg/kg dvakrát denne alebo 2 mg/kg trikrát denne)
u pediatrických pacientov nedosiahnu vyššiu expozíciu bosentanu.
V štúdii AC-052-391 (FUTURE 4) vykonanej na novorodencoch sa koncentrácie bosentanu v priebehu prvého dávkovacieho intervalu pomaly a kontinuálne zvyšovali, čo viedlo k nízkej expozícii (AUC0-12 v celej krvi: 164 ng·h/ml, n = 11). V rovnovážnom stave AUC bola 6165 ng·h/ml (CV:
133%, n = 7), čo sa podobá expozícii pozorovanej u dospelých pacientov s PAH liečených 125 mg
dvakrát denne, pričom sa berie do úvahy distribučný pomer krv/plazma 0,6.
Dôsledky týchto záverov ohľadom hepatotoxicity, nie sú známe. Pohlavie a súbežné používanie intravenózne aplikovaného epoprostenolu nemajú signifikantný vplyv na farmakokinetiku bosentanu.
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre A) neboli vo farmakokinetike pozorované žiadne významné zmeny. V rovnovážnom stave bola hodnota AUC
bosentanu o 9 % vyššia a hodnota AUC aktívneho metabolitu Ro 48-5033 o 33 % vyššia u pacientov s miernou poruchou funkcie pečene v porovnaní so zdravými dobrovoľníkmi.
Vplyv miernej poruchy funkcie pečene (Childovo-Pughovo skóre B) na farmakokinetiku bosentanu
a jeho primárneho metabolitu Ro 48-5033 bol sledovaný v štúdii, ktorá zahŕňala 5 pacientov s pľúcnou arteriálnou hypertenziou s pridruženou portálnou hypertenziou a poruchou pečeňových funkcií (Childovo-Pughovo skóre B) a 3 pacientov s PAH z iných príčin a s normálnou funkciou pečene.
U pacientov s poruchou funkcie pečene (Childovo-Pughovo skóre B) bola AUC v rovnovážnom stave (95 % CI) 360 (212-613) ng h/ml, t.j. 4,7krát vyššia a priemerná AUC (95 % CI) aktívneho metabolitu Ro 48-5033 bola 106 (58,4-192) ng h/ml t.j. 12,4krát vyššia ako u pacientov s normálnou funkciou
pečene (bosentan: priemerná AUC [95 % CI]: 76,1 [9,07-638] ng h/ml; Ro 48-5033: priemerná AUC
[95 % CI] 8,57 [1,28-57,2] ng h/ml). Hoci počet zaradených pacientov bol obmedzený a vysoko variabilný, tieto dáta naznačujú výrazné zvýšenie expozícíe bosentanu a jeho primárneho metabolitu Ro 48-5033 u pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre B).
Farmakokinetika bosentanu sa neskúmala u pacientov s poruchou funkcie pečene (Childovo-Pughovo skóre C). Tracleer je kontraindikovaný u pacientov s miernou a závažnou poruchou funkcie pečene t. j. Childovo-Pughovo skóre B alebo C (pozri časť 4.3).
Porucha funkcie obličiek
U pacientov s vážnou poruchou funkcie obličiek (klírens kreatinínu 15-30 ml/min) sa plazmatická hladina bosentanu znížila približne o 10 %. Plazmatické hladiny metabolitov bosentanu sa u týchto pacientov zvýšili asi dvakrát v porovnaní s osobami s normálnou funkciou obličiek. Ak pacient trpí poruchou funkcie obličiek, úprava dávky nie je nutná. Neexistuje špecifická klinická skúsenosť
s pacientmi podstupujúcimi dialýzu. Na základe fyzikálno-chemických vlastností a vysokého stupňa väzby na plazmatické proteíny sa neočakáva, že by bosentan bol v signifikantnej miere odstránený
z cirkulácie dialýzou (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnostiDvojročná štúdia karcinogenity u myší ukázala zvýšený kombinovaný výskyt hepatocelulárnych adenómov a karcinómov u samcov, nie však u samíc, s plazmatickými hladinami asi dvakrát až štyrikrát vyššími než plazmatické hladiny, ktoré sa dosiahli v liečebnej dávke u ľudí. U potkanov vyvolalo perorálne podávanie bosentanu počas 2 rokov nízke, signifikantné zvýšenie kombinovaného výskytu tyroidných folikulárnych bunkových adenómov a karcinómov u samcov, nie však u samíc,
s plazmatickými hladinami asi 9- až 14krát vyššími než plazmatické hladiny, ktoré sa dosiahli pri liečebnej dávke u ľudí. Bosentan bol negatívny v testoch genotoxicity. U potkanov bola bosentanom vyvolaná mierna tyroidná hormonálna dysbalancia. Nedokázalo sa však, že by bosentan ovplyvňoval tyroidné funkcie u ľudí (tyroxín, TSH).
Vplyv bosentanu na mitochondriálne funkcie nie je známy.
Ukázalo sa, že bosentan je teratogénny u potkanov pri plazmatických hladinách 1,5krát vyšších než plazmatické hladiny, ktoré boli dosiahnuté v liečebnej dávke u ľudí. Teratogénne efekty, vrátane malformácie hlavy, faciálnych oblastí a veľkých ciev boli závislé od dávky. Podobné typy malformácií pozorované s inými antagonistami ET receptora a u myší s vyradenými ET receptormi naznačujú skupinový efekt. U žien vo fertilnom veku sa musia prijať príslušné preventívne opatrenia (pozri časti
4.3, 4.4 a 4.6).
S chronickým podávaním antagonistov endotelínového receptora hlodavcom sa spája rozvoj testikulárnej tubulárnej atrofie a zhoršenie fertility.
V štúdiách fertility samcov a samíc potkanov, nebol pozorovaný žiadny vplyv na počet spermií, motilitu a životnosť, ani na schopnosť páriť sa alebo na plodnosť, pri 21-násobných respektíve 43- násobných plazmatických hladinách než sú očakávané liečebné hladiny u ľudí. Neexistoval ani žiadny nežiaduci vplyv na vývoj pre-implantovaného embrya alebo na implantáciu.
Ľahko zvýšená incidencia testikulárnej tubulárnej atrofie bola pozorovaná u potkanov, ktorým sa podával bosentan perorálne v dávkach 125 mg/kg/deň (asi 4-násobok maximálnej odporúčanej dávky u ľudí a najnižšej testovacej dávky) počas 2 rokov, nie však v dávkach až 1500 mg/kg/deň (asi 50- násobok maximálnej odporúčanej dávky u ľudí) počas 6 mesiacov. V štúdii toxicity na juvenilných potkanoch, kde boli potkany liečené od 4. dňa po vrhu do dospelosti, bolo po odstavení pozorované zníženie absolútnej hmotnosti semenníkov a nadsemenníkov a zníženie počtu spermií
v nadsemenníkoch. Hladina pri ktorej sa nepozorujú žiadne účinky (NOAEL) bola 21-násobkom (21
dní po vrhu) a 2,3-násobok (69 dní po vrhu) ľudskej terapeutickej expozície.
21 dní po vrhu neboli po 7-násobku (samci) a 19-násobku (samice) ľudskej terapeutickej expozície zistené žiadne účinky na celkový vývoj, rast, senzorické a kognitívne funkcie a reprodukčné schopnosti. V dospelosti (69 dní po vrhu) neboli zistené žiadne účinky bosentanu pri 1,3-násobku (samci) a 2,6-násobku (samice) terapeutickej expozície u detí s PAH.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza
Bezvodý hydrogénfosforečnan vápenatý
Sodná soľ kroskarmelózy Koloidný oxid kremičitý bezvodý Kyselina vínna
Tutti frutti príchuť
Aspartám (E951) Acesulfám draselný Magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
5 rokov
Zostávajúce časti rozdelenej tablety možno skladovať pri izbovej teplote a treba ich spotrebovať
v priebehu nasledujúcich 7 dní.
6
.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 25 °C.
6.5 Druh obalu a obsah baleniaAlu / Alu blistre s obsahom 14 dispergovateľných tabliet. Škatuľka obsahuje 56 dispergovateľných tabliet.
6.6 Špeciálne opatrenia na likvidáciu a zaobchádzanie s liekomDispergovateľné tablety sú uložené v blistri bezpečnom pre deti.
Každá dispergovateľná tableta sa rozpúšťa vo vode, vzniká tak tekutý liek pridaním tablety
k menšiemu množstvu vody na lyžičke, použitím dostatočného množstva vody, tak aby pokrylo celú
tabletu. Po úplnom rozpustení tablety sa tekutina podá pacientovi.
Pokiaľ je to potrebné, tabletu možno deliť pozdĺž deliacich rýh na povrchu. Podržte tabletu medzi palcom a ukazovákom, deliacou ryhou smerom hore a rozlomte tabletu pozdĺž deliacej ryhy (pozri obrázok nižšie).
Zostávajúce časti rozdelenej dispergovateľnej tablety možno skladovať pri izbovej teplote a treba ich spotrebovať v priebehu nasledujúcich 7 dní.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIActelion Registration Ltd
Chiswick Tower, 13th Floor
389 Chiswick High Road
Londýn W4 4AL Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLAEU/1/02/220/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. mája 2002
Dátum predĺženia: 20. apríla 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.