
se, nezávisle od jedla. Môže sa aplikovať do oblasti brucha, stehna alebo nadlaktia. Miesto podávania injekcie a čas podávania sa môže meniť bez úpravy dávky. Avšak, je vhodnejšie, keď sa Saxenda podáva približne v rovnakom čase dňa, ktorý bol zvolený ako najvýhodnejší čas dňa.
Saxenda sa nemá miešať s inými injekčne podávanými liekmi (napr. inzulínmi).
Ak sa dávka vynechá do 12 hodín od času, keď sa obvykle používa, pacient má čo najskôr použiť vynechanú dávku. Ak zostáva menej ako 12 hodín do použitia ďalšej dávky, pacient nemá použiť vynechanú dávku a má pokračovať v režime používania jednej dávky za deň, ďalšou plánovanou dávkou. Na kompenzáciu vynechanej dávky sa nemá použiť dávka navyše, ani zvýšiť dávku, aby sa nahradila vynechaná dávka. Ďalšie pokyny týkajúce sa podávania, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liraglutid alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Liraglutid sa nesmie používať u pacientov s diabetom ako náhrada za inzulín.
Skúsenosti s použitím u pacientov s kongestívnym zlyhávaním srdca triedy I-II podľa klasifikácie
New York Heart Association (NYHA) sú obmedzené, a preto treba byť opatrný pri použití liraglutidu.
Nie sú žiadne skúsenosti s použitím u pacientov s kongestívnym zlyhávaním srdca triedy III-IV podľa
klasifikácie NYHA, a preto sa liraglutid u týchto pacientov neodporúča.
Bezpečnosť a účinnosť liraglutidu pri regulácii telesnej hmotnosti neboli sledované u pacientov:
– vo veku 75 rokov alebo starších,
– liečených inými liekmi na reguláciu telesnej hmotnosti,
– s obezitou ako druhotným príznakom endokrinologického ochorenia, alebo poruchami príjmu
potravy, alebo pri užívaní liekov, ktoré môžu spôsobiť nárast telesnej hmotnosti,
– so závažnou poruchou funkcie obličiek,
– so závažnou poruchou funkcie pečene.
Použitie u týchto pacientov sa neodporúča (pozri časť 4.2).
Kedže liraglutid v súvislosti s reguláciou telesnej hmotnosti, nebol sledovaný u pacientov s miernym
alebo stredne závažným poškodením funkcie pečene, je potrebná opatrnosť pri užívaní u týchto
pacientov (pozri časti 4.4 a 5.2).
Skúsenosti s použitím u pacientov so zápalovým ochorením čriev a diabetickou gastroparézou sú obmedzené. Požívanie liraglutidu sa u týchto pacientov neodporúča, pretože je spojené s prechodnými gastrointestinálnymi nežiaducimi reakciami, ako je nauzea, vracanie a hnačka.
Pankreatitída
Použitie agonistov GLP-1 receptora bolo spojené s rizikom vzniku akútnej pankreatitídy. V súvislosti s používaním liraglutidu bolo hlásených niekoľko prípadov akútnej pankreatitídy. Pacienti majú byť
informovaní o typických príznakoch akútnej pankreatitídy. V prípade podozrenia na pankreatitídu sa má liraglutid vysadiť; ak sa pankreatitída potvrdí, liraglutid sa nemá znovu používať. Opatrnosť je
potrebná u pacientov s anamnézou pankreatitídy.
Cholelitiáza a cholecystitída
V klinických štúdiách zameraných na reguláciu telesnej hmotnosti bola pozorovaná vyššia miera cholelitiázy a cholecystitídy u pacientov liečených liraglutidom, ako u pacientov, ktorým sa podávalo placebo. Fakt, že značný úbytok telesnej hmotnosti môže zvýšiť riziko cholelitiázy, a tým aj cholecystitídy, len čiastočne vysvetľuje vyššiu mieru ich výskytu v súvislosti s liraglutidom. Cholelitiáza a cholecystitída môžu viesť k hospitalizácii a cholecystektómii. Pacienti majú byť informovaní o typických príznakoch cholelitiázy a cholecystitídy.
Ochorenieštítnejžľazy
Počas klinických štúdií u diabetikov 2. typu boli hlásené nežiaduce účinky súvisiace so štítnou žľazou,
vrátane zvýšenia kalcitonínu v krvi, strumy a novotvaru štítnej žľazy, a to predovšetkým u pacientov s predchádzajúcim ochorením štítnej žľazy. Boli pozorované aj prípady zvýšenia kalcitonínu v krvi v
klinických štúdiách zameraných na reguláciu telesnej hmotnosti. Preto sa má liraglutid používať
opatrne u pacientov s ochorením štítnej žľazy.
Tepová frekvencia
V klinických štúdiách bolo v súvislosti s liraglutidom pozorované zvýšenie tepovej frekvencie (pozri časť 5.1). Klinicky významné zvýšenie tepovej frekvencie v súvislosti s liečbou liraglutidom je nejasné, predovšetkým u pacientov so srdcovým alebo srdcovo-cievnym ochorením z dôvodu obmedzenej expozície u týchto pacientov v klinických štúdiách. Tepová frekvencia sa má monitorovať v pravidelných intervaloch v súlade so zaužívanou klinickou praxou. Pacienti majú byť informovaní
o príznakoch zvýšenej tepovej frekvencie (búšenie srdca alebo pocit veľmi rýchleho tlkotu srdca
v pokoji). U pacientov s klinicky významným trvalým zvýšením tepovej frekvencie v pokoji, sa má
liečba liraglutidom ukončiť.
Dehydratácia
Prejavy a symptómy dehydratácie, vrátane poškodenia funkcie a akútneho zlyhania obličiek, boli
zaznamenané u pacientov liečených agonistami GLP-1 receptora. Pacienti liečení liraglutidom majú byť poučení o potenciálnom riziku dehydratácie v súvislosti s gastrointestinálnymi vedľajšími účinkami a majú vykonať preventívne opatrenia, aby zabránili strate tekutín.
Hypoglykémia u pacientov s diabetom 2. typu
U pacientov s diabetom 2. typu používajúcich liraglutid v kombinácii so sulfonylureou, môže byť zvýšené riziko hypoglykémie. Riziko hypoglykémie sa môže zmenšiť znížením dávky sulfonylurey. Pridanie lieku Saxenda pacientom, ktorí už boli liečení inzulínom nebolo hodnotené.
Pomocné látky
Saxenda obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, preto tento liek v podstate
„neobsahuje sodík“.
4.5 Liekové a iné interakcie
V testoch in vitro sa preukázal veľmi nízky potenciál liraglutidu vstupovať do farmakokinetických interakcií s inými liečivami súvisiacimi s cytochrómom P450 (CYP) a väzbou na plazmatické proteíny.
Malé spomalenie vyprázdňovania žalúdka spôsobené liraglutidom môže mať vplyv na absorpciu súčasne podávaných perorálnych liekov. Interakčné štúdie nepreukázali žiadne klinicky relevantné spomalenie absorpcie, a preto nie je potrebná úprava dávky.
Uskutočnili sa interakčné štúdie s 1,8 mg liraglutidu. Účinok na rýchlosť vyprázdňovania žalúdka bol ekvivalentný medzi 1,8 mg a 3 mg liraglutidu, (paracetamol AUC0-300 min). Niekoľko pacientov liečených liraglutidom hlásilo minimálne jeden prípad závažnej hnačky. Hnačka môže ovplyvniť absorpciu súbežne podávaných perorálnych liekov.
Warfarín a iné kumarínové deriváty
Neuskutočnili sa žiadne interakčné štúdie. Nedajú sa vylúčiť klinicky relevantné interakcie s liečivami so slabou rozpustnosťou alebo s úzkym terapeutickým indexom, ako je warfarín. Po začatí liečby
liraglutidom sa u pacientov liečených warfarínom alebo inými kumarínovými derivátmi odporúča
častejšie sledovanie INR (International Normalised Ratio).
Paracetamol (Acetaminofén)
Liraglutid nespôsobil zmenu celkovej expozície paracetamolu po podaní jednorazovej dávky 1000 mg. Hodnota Cmax paracetamolu sa znížila o 31 % a stredná hodnota tmax sa spomalila na 15 minút. Pri súbežnom podávaní paracetamolu sa nevyžaduje žiadna úprava dávky.
Atorvastatín
Liraglutid nespôsobil zmenu celkovej expozície atorvastatínu po podaní jednorazovej dávky 40 mg atorvastatínu. Preto nie je potrebná žiadna úprava dávky atorvastatínu pri jeho podávaní s liraglutidom. Pri podávaní s liraglutidom sa hodnota Cmax atorvastatínu znížila o 38 % a stredná hodnota tmax sa oneskorila z 1 na 3 hodiny.
Grizeofulvín
Liraglutid nespôsobil zmenu celkovej expozície grizeofulvínu po podaní jednorazovej dávky 500 mg grizeofulvínu. Hodnota Cmax grizeofluvínu sa zvýšila o 37 %, zatiaľ čo stredná hodnota tmax sa nezmenila. Úpravy dávok grizeofulvínu a iných zložiek s nízkou rozpustnosťou a vysokou priepustnosťou nie sú potrebné.
Digoxín
Podanie jednorazovej dávky 1 mg digoxínu spolu s liraglutidom malo za následok zníženie hodnoty
AUC digoxínu o 16 %; hodnota Cmax klesla o 31 %. Stredná hodnota digoxínu tmax sa spomalila z
1 hodiny na 1,5 hodiny. Na základe týchto výsledkov nie je potrebná žiadna úprava dávky digoxínu.
Lizinopril
Podanie jednorazovej dávky 20 mg lizinoprilu spolu s liraglutidom malo za následok zníženie AUC lizinoprilu o 15 %; hodnota Cmax klesla o 27 %. Stredná hodnota lizinoprilu tmax s liraglutidom sa spomalila zo 6 hodín na 8 hodín. Na základe týchto výsledkov nie je potrebná žiadna úprava dávky lizinoprilu.
Perorálne kontraceptíva
Po podaní jednorazovej dávky perorálneho kontraceptíva znížil liraglutid hodnotu Cmax etinylestradiolu o 12 % a levonorgestrelu o 13 %. Liraglutid spôsobil spomalenie hodnoty tmax o 1,5 hodiny pre obe zložky. Neprejavil sa žiaden klinicky relevantný účinok na celkovú expozíciu etinylestradiolu ani levonorgestrelu. Preto sa predpokladá, že antikoncepčný účinok nie je ovplyvnený spoločným podávaním s liraglutidom.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii sú len obmedzené údaje o používaní liraglutidu u gravidných žien. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí.
Liraglutid sa nemá používať počas gravidity. Ak si pacientka želá otehotnieť alebo otehotnie, liečba
liraglutidom sa má prerušiť.
Laktácia
Nie je známe, či sa liraglutid vylučuje do ľudského materského mlieka. Štúdie na zvieratách preukázali nízky prechod liraglutidu a štrukturálne blízkych metabolitov do materského mlieka. Predklinické štúdie preukázali spomalenie neonatálneho rastu u dojčených mláďat potkanov
v súvislosti s liečbou (pozri časť 5.3). Pre nedostatok skúseností sa Saxenda nemá používať počas dojčenia.
Fertilita
Okrem mierneho poklesu počtu živých implantátov, štúdie na zvieratách nepreukázali škodlivé účinky na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Saxenda nemá žiadny, alebo má len zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať
stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu:
Program klinických štúdií pre liek Saxenda pozostáva zo 6 dokončených klinických štúdií, ktorých sa zúčastnilo 5813 obéznych pacientov alebo pacientov s nadváhou minimálne s jednou komorbiditou súvisiacou s telesnou hmotnosťou. Celkovo boli najčastejšími nežiaducimi reakciami hlásenými počas liečby liekom Saxenda gastrointestinálne nežiaduce reakcie (pozri časť „Opis vybraných nežiaducich reakcií“).
Tabuľkovýzoznamnežiaducichreakcií
V Tabuľke 2 sú uvedené nežiaduce reakcie hlásené počas dlhodobých kontrolovaných klinických štúdií fázy 3. Nežiaduce reakcie sú rozdelené podľa tried orgánových systémov a frekvencie. Kategórie frekvencií sú definované takto: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10000 až < 1/1000); veľmi zriedkavé (< 1/10000). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
T
abuľka 2. Nežiaduce reakcie hlásené v kontrolovaných štúdiách fázy 3
MedDRA trieda
orgánových systémov
|
V
eľmi
časté
|
Č
asté
|
Menej časté
|
Z
riedkavé
|
Poruchy imunitného
systému
|
|
|
|
Anafylaktická
reakcia
|
Poruchy metabolizmu a
výživy
|
|
Hypoglykémia*
|
Dehydratácia
|
|
Psychické poruchy
|
|
Nespavosť**
|
|
|
Poruchy nervového
systému
|
|
Závrat
Porucha chuti
|
|
|
Poruchy srdca a srdcovej
činnosti
|
|
|
Tachykardia
|
|
Poruchy gastro-
intestinálneho traktu
|
Nauzea
Vracanie
Hnačka
Zápcha
|
Pocit suchých úst
Dyspepsia
Gastritída Gastroezofageálny reflux
Bolesť v nadbrušku
Nadúvanie
Grganie
Abdominálna distenzia
|
Pankreatitída***
|
|
Poruchy pečene a
žlčových ciest
|
|
Cholelitiáza***
|
Cholecystitída***
|
|
Poruchy kože a
podkožného tkaniva
|
|
|
Urtikária
|
|
Poruchy obličiek a
močových ciest
|
|
|
|
Akútne zlyhanie
obličiek
Porucha funkcie
obličiek
|
Celkové poruchy a
reakcie v mieste podávania injekcie
|
|
Reakcie v mieste
podávania injekcie
Asténia
Únava
|
Malátnosť
|
|
*Hypoglykémia (na základe príznakov hlásených pacientami, ktorá nebola potvrdená meraním hladiny glukózy v krvi) hlásená u pacientov bez diabetu 2. typu liečených liekom Saxenda v kombinácii
s diétou a cvičením. Ďalšie informácie nájdete v časti „Opis vybraných nežiaducich reakcií“.
**Nespavosť bola pozorovaná hlavne počas prvých 3 mesiacov liečby.
***Pozri časť 4.4.
Opis vybraných nežiaducich reakcií:Hypoglykémia u pacientov bez diabetu 2. typuV klinických štúdiách neboli u pacientov s nadváhou alebo obéznych pacientov bez diabetu 2. typu
liečených liekom Saxenda v kombinácii s diétou a cvičením hlásené žiadne prípady závažnej hypoglykémie (vyžadujúce pomoc tretej osoby). Príznaky hypoglykémie hlásilo 1,6 % pacientov liečených liekom Saxenda a 1,1 % pacientov, ktorým sa podávalo placebo; avšak tieto prípady neboli potvrdené meraním hladiny glukózy v krvi. Väčšina týchto prípadov mala mierny charakter.
Hypoglykémia u pacientov s diabetom 2. typuV klinickej štúdii bola u pacientov s nadváhou alebo obéznych pacientov s diabetom 2. typu liečených
liekom Saxenda v kombinácii s diétou a cvičením hlásená závažna hypoglykémia (vyžadujúca pomoc tretej osoby) u 0,7 % pacientov liečených liekom Saxenda a len u pacientov súbežne liečených sulfonylureou. Aj u týchto pacientov bola hlásená symptomatická hypoglykémia, a to u 43,6 % pacientov liečených liekom Saxenda a u 27,3 % pacientov, ktorým sa podávalo placebo. Spomedzi pacientov, ktorí neboli súbežne liečení sulfonylureou, 15,7 % pacientov liečených liekom Saxenda
a 7,6 % pacientov, ktorým sa podávalo placebo, hlásilo zaznamenané prípady symptomatickej hypoglykémie (definovanej ako hladina glukózy v plazme ≤ 3,9 mmol/l so sprievodnými príznakmi).
Gastrointestinálne nežiaduce reakcieVäčšina epizód gastrointestinálnych prípadov mala mierny až stredne závažný, prechodný charakter a väčšina z nich neviedla k ukončeniu liečby. Reakcie sa zvyčajne objavovali počas prvých týždňov
liečby a pominuli v priebehu niekoľkých dní alebo týždňov pokračujúcej liečby.
U pacientov vo veku ≥ 65 rokov sa môže prejaviť pri liečbe liekom Saxenda viac gastrointestinálnych účinkov.
U pacientov s miernym alebo stredným poškodením funkcie obličiek (klírens kreatínu ≥30 ml/min) sa
môže prejaviť pri liečbe liekom Saxenda viac gastrointestinálnych nežiaducich účinkov.
Akútne zlyhávanieobličiekU pacientov liečených agonistami GLP-1 receptora bolo pozorované akútne zlyhávanie funkcie obličiek. Väčšina hlásených prípadov sa vyskytla u pacientov, u ktorých sa objavila nevoľnosť, vracanie alebo hnačky, ktoré viedli k úbytku objemu mimobunkovej tekutiny (pozri časť 4.4).
Alergické reakciePri užívaní liraglutidu po jeho uvedení na trh bolo hlásených niekoľko prípadov anafylaktických reakcií ako hypotenzia, búšenie srdca, dýchavičnosť a edém. Anafylaktické reakcie môžu byť potenciálne život ohrozujúce. Ak je podozrenie na anafylaktickú reakciu, liraglutid sa má vysadiť a liečba sa nemá znovu obnoviť (pozri časť 4.3).
Reakcie v mieste podávania injekcieU pacientov liečených liekom Saxenda boli hlásené reakcie v mieste podávania injekcie. Tieto reakcie
boli zvyčajne mierne a prechodné a väčšina z nich zvyčajne počas ďalšej liečby zmizla.
TachykardiaV klinických štúdiách bola tachykardia hlásená u 0,6 % pacientov liečených liekom Saxenda a u 0,1 %
pacientov, ktorým bolo podávané placebo. Väčšina týchto prípadov mala mierny alebo stredne
závažný charakter. Tieto prípady boli ojedinelé a väčšina z nich zvyčajne počas ďalšej liečby liekom
Saxenda zmizla.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovaniePredávkovanie bolo hlásené počas klinických štúdií a po uvedení liraglutidu na trh až pri dávke 72 mg (24-násobok odporúčanej dávky na reguláciu telesnej hmotnosti). Tieto prípady zahŕňali závažnú nauzeu a závažné vracanie, čo sú tiež očakávané príznaky predávkovania liraglutidom. Žiadne
z hlásení nezahŕňalo závažnú hypoglykémiu. Všetci pacienti sa zotavili bez komplikácií.
V prípade predávkovania sa má začať vhodná podporná liečba podľa klinických prejavov a symptómov pacienta. U pacientov treba sledovať klinické prejavy dehydratácie a má sa monitorovať glykémia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnosti Farmakoterapeutická skupina: Antidiabetiká, iné antidiabetiká s výnimkou inzulínov. ATC kód: A10BX07
Mechanizmusúčinku
Liraglutid je analóg acylovaného ľudského glukagónu podobného peptidu-1 (GLP-1) s 97 %
sekvenčnou homológiou aminokyselín s endogénnym ľudským GLP-1. Liraglutid aktivuje a je viazaný na receptor GLP-1 (GLP-1R).
GLP-1 je fyziologický regulátor chuti do jedla a príjmu potravy, ale presný mechanizmus jeho účinku nie je úplne jasný. Pri štúdiách na zvieratách viedlo periférne podávanie liraglutidu k pochopeniu funkcie špecifických mozgových oblastí, ktoré sa podieľajú na regulácii chuti do jedla, kde liraglutid prostredníctvom špecifickej aktivácie GLP-1R zosilňoval kľúčové signály pocitu sýtosti a zoslaboval kľúčové signály pocitu hladu, čo v konečnom dôsledku viedlo k úbytku telesnej hmotnosti.
Farmakodynamickéúčinky
Liraglutid znižuje telesnú hmotnosť u ľudí predovšetkým prostredníctvom úbytku tukového tkaniva spolu s relatívnym úbytkom telesného tuku, ktorý bol vyšší ako úbytok podkožného tuku. Liraglutid reguluje chuť do jedla zvyšovaním pocitu sýtosti a nasýtenia a súčasným zmierňovaním pocitu hladu a chuti konzumovať ďalšiu potravu, čo v konečnom dôsledku vedie k zníženému príjmu potravy. Liraglutid nezvyšuje energetický výdaj v porovnaní s placebom.
Liraglutid stimuluje sekréciu inzulínu a znižuje nadmerne zvýšenú sekréciu glukagónu mechanizmom závislým od glukózy, výsledkom čoho je zníženie hladiny glukózy nalačno a po jedle. Účinok zníženia hladiny glukózy je výraznejší u pacientov s prediabetom alebo diabetom v porovnaní s pacientami
s normálnou glykémiou. Klinické štúdie naznačujú, že liraglutid zlepšuje a udržiava funkciu beta buniek na základe modelu homeostázy (HOMA-B) a pomer proinzulínu a inzulínu.
Klinickáúčinnosťabezpečnosť
Účinnosť a bezpečnosť liraglutidu na reguláciu telesnej hmotnosti v spojení so znížením kalorického príjmu a zvýšením fyzickej aktivity boli skúmané v rámci 4 randomizovaných, dvojito zaslepených
štúdií fázy 3 s kontrolovaným podávaním placeba, na ktorých sa zúčastnilo celkovo 5358 pacientov.
• Štúdia 1 (SCALE Obesity & Pre-Diabetes - 1839): 56-týždňová štúdia hodnotiaca úbytok telesnej hmotnosti u 3731 randomizovaných (2590 dokončilo štúdiu) obéznych pacientov
a pacientov s nadváhou s jednou s nasledujúcich komorditíd: prediabetes, hypertenzia alebo
dyslipidémia. 61 % pacientov malo na začiatku štúdie prediabetes.
• Štúdia 2 (SCALE Diabetes - 1922): 56-týždňová štúdia hodnotiaca úbytok telesnej hmotnosti
u 846 randomizovaných (628 dokončilo štúdiu) obéznych pacientov a pacientov s nadváhou s nedostatočne regulovaným diabetom typu 2 (HbA1c v rozsahu 7-10 %). Základná liečba na začiatku štúdie zahŕňala len diétu a cvičenie, metformín, sulfonylureu alebo glitazón samostatne, alebo akúkoľvek ich kombináciu.
• Štúdia 3 (SCALE Sleep apnoe - 3970): 32-týždňová štúdia hodnotiaca závažnosť spánkového
apnoe a úbytok telesnej hmotnosti u 359 randomizovaných (276 dokončilo štúdiu) obéznych pacientov so stredne závažným alebo závažným obštrukčným spánkovým apnoe.
• Štúdia 4 (SCALE Maintenance - 1923): 56-týždňová štúdia hodnotiaca udržiavanie telesnej
hmotnosti a úbytok telesnej hmotnosti u 422 randomizovaných (305 dokončilo štúdiu) obéznych pacientov a pacientov s nadváhou s hypertenziou, alebo dyslipidémiou po predchádzajúcom úbytku telesnej hmotnosti ≥ 5 % v dôsledku nízkokalorickej diéty.
Telesnáhmotnosť
U obéznych pacientov/pacientov s nadváhou vo všetkých sledovaných skupinách sa dosiahol výrazný úbytok telesnej hmotnosti s liraglutidom v porovnaní s placebom. V rámci všetkých populácií štúdie dosiahol väčší podiel pacientov úbytok telesnej hmotnosti ≥ 5 % a > 10 % s liraglutidom, ako s placebom (tabuľky 3-5). V štúdii 4 si viac pacientov udržalo úbytok telesnej hmotnosti dosiahnutý pred začatím liečby s liraglutidom ako s placebom (81,4 %, a 48,9 %). Špecifické údaje o úbytku telesnej hmotnosti, pacientoch, časovom priebehu a kumulatívnej distribúcii zmeny telesnej hmotnosti (%) pre štúdie 1-4 sú uvedené v tabuľkách 3-6 a na obrázkoch 1, 2 a 3.
O
dozva v súvislosti s úbytkom telesnej hmotnosti po 12 týždňoch
l
i
ečby
liraglutidom (3,0 mg)
Ako včasne reagujúci boli definovaní pacienti, ktorí dosiahli úbytok telesnej hmotnosti ≥ 5 % po
12 týždňoch s liečebnou dávkou liraglutidu (4 týždne so zvyšovaním dávok a 12 týždňov s liečebnou dávkou). Počas štúdie 1 dosiahlo 67,5 % pacientov úbytok telesnej hmotnosti ≥ 5 % po 12 týždňoch. Počas štúdie 2 dosiahlo 50,4 % pacientov úbytok telesnej hmotnosti ≥ 5 % po 12 týždňoch. S pokračujúcou liečbou liraglutidom sa u 86,2 % týchto včasne reagujúcich pacientov predpokladá dosiahnutie úbytku telesnej hmotnosti ≥ 5 % a u 51 % sa predpokladá dosiahnutie úbytku telesnej hmotnosti ≥ 10 % po 1 roku liečby. Predpokladaný priemerný úbytok telesnej hmotnosti u včasne reagujúcich pacientov po 1 roku liečby je 11,2 % z ich východiskovej telesnej hmotnosti (9,7 %
u mužov a 11,6 % u žien). U pacientov, ktorí dosiahli úbytok telesnej hmotnosti < 5 % po 12 týždňoch s liečebnou dávkou liraglutidu, podiel pacientov, ktorí nedosahujú úbytok telesnej hmotnosti ≥ 10 %
po 1 roku, je 93,4 %.
Glykemická kontrola
Liečba liraglutidom výrazne zlepšuje glykemické parametre v rámci jednotlivých sub-populácií s normoglykémiou, prediabetom a s diabetom 2. typu. V štúdii 1 diabetes 2. typu sa rozvinul
u menšieho počtu pacientov liečených liraglutidom v porovnaní s pacientami, ktorým bolo podávané
placebo (0,2 % oproti 1,1 %). Viac pacientov s prediabetom na začiatku štúdie sa vrátilo do normoglykemickej populácie v skupine s účinnou látkou ako s placebom (69,2 % oproti 32,7 %).
Kardiometabolické rizikové faktory
Pri liečbe s liraglutidom sa výrazne zlepšil systolický krvný tlak a znížil sa obvod pásu v porovnaní s placebom (tabuľky 3 a 4).
Index apnoe - hypopnoe (AHI)
Liečba s liraglutidom výrazne znížila závažnosť obštrukčného spánkového apnoe pri vyhodnotení
zmeny východiskovej hodnoty indexu AHI v porovnaní s placebom (tabuľka 5).
T
abuľka 3 Štúdia 1: Zmeny telesnej hmotnosti, glykémie a kardiometabolických parametrov
oproti východiskovým hodnotám v 56. týždni
Saxenda (N=2437) Placebo (N=1225) Saxenda v porovnaní s placebom
 T
elesná hmotnosť
T
elesná hmotnosť
Východisková hodnota, kg (SD) 106,3 (21,2) 106,3 (21,7) -
Priemerná zmena v 56. týždni,
% (95 % CI)
Priemerná zmena v 56. týždni, kg (95 % CI)
Podiel pacientov s úbytkom telesnej hmotnosti ≥ 5 % v 56. týždni, % (95 % CI)

Podiel pacientov s úbytkom telesnej hmotnosti ˃ 10 % v 56. týždni, % (95 % CI)
-8,0 -2,6 -5,4** (-5,8; -5,0)
-8,4 -2,8 -5,6** (-6,0; -5,1)
63,5 26,6 4,8** (4,1; 5,6)
32,8 10,1 4,3** (3,5; 5,3)
G
lykémia a kardiometabolické faktory
Východisková hodnota
Zmena Východisková hodnota
Zmena

HbA1c, % 5,6 -0,3 5,6 -0,1 -0,23** (-0,25; -0,21) FPG, mmol/l 5,3 -0,4 5,3 -0,01 -0,38** (-0,42; -0,35) Systolický krvný tlak, mmHg 123,0 -4,3 123,3 -1,5 -2,8** (-3,6; -2,1) Diastolický krvný tlak, mmHg 78,7 -2,7 78,9 -1,8 -0,9* (-1,4; -0,4) Obvod pása, cm 115,0 -8,2 114,5 -4,0 -4,2** (-4,7; -3,7)
Analýza celého súboru. Pre telesnú hmotnosť, HbA1c, FPG, krvný tlak a obvod pása sú východiskové hodnoty
priemerné, zmeny oproti východiskovým hodnotám v 56. týždni sú odhadované priemerné hodnoty (metóda
najmenších štvorcov) a liečebné kontrasty v 56. týždni sú odhadované rozdiely v liečbe. Pre podiel pacientov s úbytkom telesnej hmotnosti ≥ 5/> 10 % sú uvádzané odhadované pomery pravdepodobnosti. Chýbajúce hodnoty nasledujúce po východiskových hodnotách boli pripočítané na základe posledných vykonaných meraní.
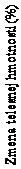
* p < 0,05. ** p < 0,0001. CI=intervaly spoľahlivosti. FPG=glukóza v plazme nalačno. SD=štandardná odchýlka.
Čas v týždňoch
Saxenda Placebo Posledné vykonané pozorovanie
Pozorované hodnoty pacientov absolvujúcich všetky naplánované návštevy
Obrázok 1 Zmena telesnej hmotnosti (%) oproti východiskovej hodnote podľa času v Štúdii 1
Zmena telesnej hmotnosti (%)
Saxenda
Placebo
Posledné vykonané pozorovanie.
Obrázok 2 Kumulatívna distribúcia zmeny telesnej hmotnosti (%) po 56 týždňoch liečby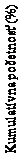 v Štúdii 1Tabuľka 4 Štúdia 2: Zmeny telesnej hmotnosti, glykémie a kardiometabolických parametrov oproti východiskovým hodnotám v 56. týždni Saxenda (N=412) Placebo (N=211) Saxenda v porovnaní s placebom
v Štúdii 1Tabuľka 4 Štúdia 2: Zmeny telesnej hmotnosti, glykémie a kardiometabolických parametrov oproti východiskovým hodnotám v 56. týždni Saxenda (N=412) Placebo (N=211) Saxenda v porovnaní s placebom
 Telesná hmotnosť
Telesná hmotnosťVýchodisková hodnota, kg (SD) 105,6 (21,9) 106,7 (21,2) -
Priemerná zmena v 56. týždni, % (95 % CI)
Priemerná zmena v 56. týždni, kg
(95 % CI)
Podiel pacientov s úbytkom telesnej hmotnosti ≥ 5 % v 56. týždni, % (95 % CI)
Podiel pacientov s úbytkom telesnej hmotnosti ˃ 10 % v 56. týždni, % (95 % CI)
-5,9 -2,0 -4,0** (-4,8; -3,1)
-6,2 -2,2 -4,1** (-5,0; -3,1)
49,8 13,5 6,4** (4,1; 10,0)
22,9 4,2 6,8** (3,4; 13,8)
G
lykémia a kardiometabolické faktory
Východisková hodnota
Zmena Východisková hodnota
Zmena

HbA1c, % 7,9 -1,3 7,9 -0,4 -0,9** (-1,1; -0,8) FPG, mmol/l 8,8 -1,9 8,6 -0,1 -1,8** (-2,1; -1,4) Systolický krvný tlak, mmHg 128,9 -3,0 129,2 -0,4 -2,6* (-4,6; -0,6) Diastolický krvný tlak, mmHg 79,0 -1,0 79,3 -0,6 -0,4 (-1,7; 1,0) Obvod pása, cm 118,1 -6,0 117,3 -2,8 -3,2** (-4,2; -2,2)'
Analýza celého súboru. Pre telesnú hmotnosť, HbA1c, FPG, krvný tlak a obvod pása sú východiskové hodnoty
priemerné, zmeny oproti východiskovým hodnotám v 56. týždni sú odhadované priemerné hodnoty (metóda
najmenších štvorcov) a liečebné kontrasty v 56. týždni sú odhadované rozdiely v liečbe. Pre podiel pacientov s úbytkom telesnej hmotnosti ≥ 5/> 10 % sú uvádzané odhadované pomery pravdepodobnosti. Chýbajúce hodnoty nasledujúce po východiskových hodnotách boli pripočítané na základe posledných vykonaných meraní.
* p < 0,05. ** p < 0,0001. CI=intervaly spoľahlivosti. FPG=glukóza v plazme nalačno. SD=štandardná odchýlka.
T
abuľka 5 Štúdia 3: Zmeny telesnej hmotnosti a indexu apnoe - hypopnoe oproti
východiskovým hodnotám v 32. týždni
Saxenda (N=180) Placebo (N=179) Saxenda v porovnaní s placebom
 T
elesná hmotnosť
T
elesná hmotnosť
Východisková hodnota, kg (SD) 116,5 (23,0) 118,7 (25,4) -
Priemerná zmena v 32. týždni, % (95 % CI)
Priemerná zmena v 32. týždni, kg
(95 % CI)
Podiel pacientov s úbytkom telesnej hmotnosti ≥ 5 % v 32. týždni, % (95 % CI)

Podiel pacientov s úbytkom telesnej hmotnosti ˃ 10 % v 32. týždni, % (95 % CI)
-5,7 -1,6 -4,2** (-5,2; -3,1)
-6,8 -1,8 -4,9** (-6,2; -3,7)
46,4 18,1 3,9** (2,4; 6,4)
22,4 1,5 19,0** (5,7; 63,1)
Východisková hodnota
Zmena Východisková hodnota
Zmena
I
ndex apnoe – hypopnoe, prípady/hodina
49,0 -12,2 49,3 -6,1 -6,1* (-11,0; -1,2)

Analýza celého súboru. Východiskové hodnoty sú priemerné, zmeny oproti východiskovým hodnotám v 32. týždni sú odhadované priemerné hodnoty (metóda najmenších štvorcov) a liečebné kontrasty v 32. týždni sú odhadované rozdiely v liečbe (95 % CI). Pre podiel pacientov s úbytkom telesnej hmotnosti ≥ 5/> 10 % sú uvádzané odhadované pomery pravdepodobnosti. Chýbajúce hodnoty nasledujúce po východiskových hodnotách boli pripočítané na základe posledných vykonaných meraní. * p < 0,05. ** p < 0,0001. CI=intervaly
spoľahlivosti. SD=štandardná odchýlka.
Tabuľka 6 Štúdia 4: Zmeny v telesnej hmotnosti oproti východiskovým hodnotám v 56. týždni
| Saxenda (N=207)
| Placebo (N=206)
| Saxenda v porovnaní s placebom
|
Východisková hodnota, kg (SD)
| 100,7 (20,8)
| 98,9 (21,2)
| -
|
Priemerná zmena v 56. týždni, % (95 % CI)
| -6,3
| -0,2
| -6,1** (-7,5; -4,6)
|
Priemerná zmena v 56. týždni, kg (95 % CI)
| -6,0
| -0,2
| -5,9** (-7,3; -4,4)
|
Podiel pacientov s úbytkom telesnej
hmotnosti ≥ 5 % v 56. týždni, % (95 % CI)
|
50,7
|
21,3
|
3,8** (2,4; 6,0)
|
Podiel pacientov s úbytkom telesnej hmotnosti ˃ 10 % v 56. týždni, % (95 % CI)
|
27,4
|
6,8
|
5,1** (2,7; 9,7)
|
Analýza celého súboru. Východiskové hodnoty sú priemerné, zmeny oproti východiskovým hodnotám v 56.
týždni sú odhadované priemerné hodnoty (metóda najmenších štvorcov) a liečebné kontrasty v 56. týždni sú odhadované rozdiely v liečbe. Pre podiel pacientov s úbytkom telesnej hmotnosti ≥ 5/> 10 % sú uvádzané odhadované pomery pravdepodobnosti. Chýbajúce hodnoty nasledujúce po východiskových hodnotách boli pripočítané na základe posledných vykonaných meraní. ** p < 0,0001. CI=intervaly spoľahlivosti. SD=štandardná odchýlka.
Čas v týždňoch
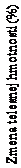
Saxenda Placebo Posledné vykonané pozorovanie
Pozorované hodnoty pacientov absolvujúcich všetky naplánované návštevy
Obrázok 3 Zmena telesnej hmotnosti (%) od randomizácie (týždeň 0) podľa času v Štúdii 4Pred týždňom 0 boli pacienti liečení len nízkokalorickou diétou a cvičením. V týždni 0 boli pacienti randomizovaní na užívanie buď lieku Saxenda, alebo placeba.
ImunogenicitaV súlade s potenciálne imunogénnymi vlastnosťami liekov s obsahom proteínov alebo peptidov sa
u pacientov po liečbe liraglutidom môžu vytvoriť protilátky proti liraglutidu. V klinických štúdiách sa u 2,5 % pacientov liečených liraglutidom vytvorili protilátky proti liraglutidu. Tvorba protilátok
nebola spojená so zníženou účinnosťou liraglutidu.
Kardiovaskulárne hodnotenieVýznamné nežiaduce kardiovaskulárne udalosti (MACE) posudzovala externá nezávislá skupina odborníkov a boli definované ako infarkt myokardu bez následku smrti, mozgová príhoda bez
následku smrti a kardiovaskulárna smrť. V dlhodobých klinických štúdiách s liekom Saxenda došlo k
6 prípadom MACE u pacientov liečených liraglutidom a 10 prípadom MACE u pacientov, ktorým
bolo podávané placebo. Pomer rizika a 95 % CI je 0,33 [0,12; 0,90] pre liraglutid v porovnaní s placebom. V klinických štúdiách fázy 3 s liraglutidom bolo pozorované priemerné zvýšenie tepovej frekvencie v porovnaní s východiskovou hodnotou o 2,5 úderu za minútu (v rámci štúdií sa táto hodnota pohybovala v rozmedzí 1,6 až 3,6 úderov za minútu). Najvyššie hodnoty tepovej frekvencie
sa dosahovali po približne 6 týždňoch. Dlhodobý klinický význam tohto priemerného zvýšenia tepovej frekvencie nebol stanovený. Táto zmena tepovej frekvencie sa po vysadení liraglutidu vrátila na
pôvodné hodnoty (pozri časť 4.4).
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre liek Saxenda
u jednej alebo vo viacerých vekových podskupinách pediatrickej populácie pri liečbe obezity a liečbe
Praderovho-Williho syndrómu (informácie o použití u pediatrickej populácie nájdete v časti 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaAbsorpcia liraglutidu po subkutánnom podaní bola pomalá, pričom maximálna koncentrácia sa dosahovala po približne 11 hodinách od podania dávky. Priemerná koncentrácia liraglutidu
v ustálenom stave (AUCτ/24) dosahovala hodnotu približne 31 nmol/l u obéznych pacientov (BMI 30 –
40 kg/m2) po podaní 3 mg liraglutidu. Expozícia liraglutidu stúpala proporcionálne s dávkou. Úplná
biologická dostupnosť liraglutidu po subkutánnom podaní je približne 55 %.
DistribúciaPriemerný evidentný distribučný objem po subkutánnom podaní je 20-25 l (u osôb s telesnou
hmotnosťou približne 100 kg). Liraglutid sa vo veľkej miere viaže na plazmatický proteín (> 98 %).
Biotransformácia
Počas 24 hodín od podania jednorazovej dávky [3H]-liraglutidu zdravým pacientom bol hlavnou zložkou v plazme nezmenený liraglutid. Boli zistené dva vedľajšie metabolity v plazme (≤ 9 % a
≤ 5 % celkovej rádioaktivity v plazme).
Eliminácia
Liraglutid je endogénne metabolizovaný podobným spôsobom ako veľké proteíny, pričom žiadny orgán nebol identifikovaný ako hlavná dráha eliminácie. Po podaní dávky [3H]-liraglutidu nebol nezmenený liraglutid zistený v moči ani v stolici. Iba malá časť podanej rádioaktivity bola vylúčená ako metabolity súvisiace s liraglutidom v moči alebo v stolici (6 %, resp. 5 %). Rádioaktivita bola
v moči a v stolici vylučovaná hlavne počas prvých 6-8 dní, pričom v oboch prípadoch išlo o tri
vedľajšie metabolity.
Priemerný klírens po subkutánnom podaní dávky liraglutidu je približne 0,9-1,4 l/hod. s polčasom
eliminácie približne 13 hodín.
Špecifické populácie
Starší pacienti
Na základe výsledkov populačnej farmakokinetickej analýzy údajov o pacientoch s nadváhou alebo obéznych pacientoch (18 až 82 rokov) nemal vek žiadny klinicky relevantný účinok na farmakokinetiku liraglutidu. Nevyžaduje sa žiadna úprava dávkovania podľa veku.
Pohlavie
Na základe výsledkov populačnej farmakokinetickej analýzy majú ženy o 24 % nižší klírens liraglutidu podľa telesnej hmotnosti v porovnaní s mužmi. Na základe údajov reakcie na expozíciu nie je potrebná žiadna úprava dávkovania podľa pohlavia.
Etnický pôvod
Podľa výsledkov populačnej farmakokinetickej analýzy, ktorá zahŕňala belošské, černošské, ázijské
a hispánske/nehispánske skupiny pacientov s nadváhou a obezitou, nemal etnický pôvod žiadny
klinicky relevantný účinok na farmakokinetiku liraglutidu.
Telesnáhmotnosť
Expozícia liraglutidu sa znižuje so zvyšujúcou sa východiskovou hodnotou telesnej hmotnosti. Denná dávka 3 mg liraglutidu poskytovala primerané systémové expozície v rozsahu telesnej hmotnosti 60-
234 kg hodnotené v rámci analýzy reakcie na expozíciu počas klinických štúdií. Expozícia liraglutidu sa neskúmala u pacientov s telesnou hmotnosťou > 234 kg.
Poruchafunkciepečene
V štúdii s jednou dávkou (0,75 mg) sa hodnotila farmakokinetika liraglutidu u pacientov s rôznymi
stupňami poškodenia funkcie pečene. Expozícia liraglutidu bola u pacientov s miernym až stredne závažným poškodením funkcie pečene znížená o 13-23 % v porovnaní so zdravými pacientami. Expozícia bola výrazne nižšia (44 %) u pacientov so závažným poškodením funkcie pečene (Child- Pughovo skóre > 9).
Poruchafunkcieobličiek
V štúdii s jednou dávkou (0,75 mg) bola expozícia liraglutidu znížená u pacientov s poruchou funkcie
obličiek v porovnaní s pacientami s normálnou funkciou obličiek. Expozícia liraglutidu bola znížená o 33 %, 14 %, 27 %, a 26 % u pacientov s miernou (klírens kreatínu CrCl 50-80 ml/min), strednou (CrCl 30-50 ml/min.) a závažnou (CrCl < 30 ml/min.) poruchou funkcie obličiek a u pacientov
v terminálnom štádiu renálneho ochorenia vyžadujúceho si dialýzu.
Pediatrická populácia
Účinky lieku Saxenda sa neskúmali u pediatrických pacientov.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po
opakovanom podávaní, alebo genotoxicity neodhalili žiadne osobitné riziko pre ľudí.
V 2-ročných štúdiách sledovania karcinogénneho potenciálu u potkanov a myší boli pozorované tumory C-buniek štítnej žľazy bez smrtiacich účinkov. U potkanov sa nesledovala hodnota NOAEL (no observed adverse affect level – úroveň, kedy nie sú pozorované žiadne nežiaduce účinky). Tieto tumory neboli pozorované u opíc liečených 20 mesiacov. Výsledky u hlodavcov sú spôsobené ne- genotoxickým mechanizmom sprostredkovaným špecifickým receptorom pre GLP-1, na ktorý sú zvlášť citlivé hlodavce. Je pravdepodobné, že význam u ľudí bude nízky, ale nedá sa úplne vylúčiť. Žiadne ďalšie tumory súvisiace s liečbou neboli zistené.
Štúdie na zvieratách nepreukázali priame škodlivé účinky na fertilitu okrem mierne zvýšenej predčasnej embryonálnej úmrtnosti pri najvyššej dávke. Podávanie liraglutidu v strednej gestačnej fáze spôsobilo pokles telesnej hmotnosti matky a spomalenie fetálneho rastu s nejednoznačnými účinkami na rebrá u potkanov a kostrovú variabilitu u králikov. U potkanov bol po expozícii liraglutidu spomalený neonatálny rast, pričom v skupine s vysokou dávkou tento stav pretrval aj v období po odstavení mláďaťa. Nie je známe, či je spomalený rast mláďat spôsobený zníženým príjmom materského mlieka v dôsledku priameho účinku GLP-1, alebo zníženou produkciou materského
mlieka v dôsledku zníženého prísunu kalórií.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát hydrogenfosforečnanu sodného
Propylénglykol
Fenol
Kyselina chlorovodíková (na úpravu pH) Hydroxid sodný (na úpravu pH)
Voda na injekciu
6.2 Inkompatibility
Látky pridané k lieku Saxenda môžu spôsobiť degradáciu liraglutidu. Nevykonali sa štúdie
kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
30 mesiacov
Po prvom použití: 1 mesiac
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke. Neuchovávajte v blízkosti mraziacej časti.
Po prvom použití: Uchovávajte pri teplote do 30 °C, alebo uchovávajte v chladničke (2°C – 8°C). Nechajte kryt na pere na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Jednorazové naplnené pero na viac dávok vyrobené z polypropylénu, polyacetálu, polykarbonátu a akrylonitrilbutadiénstyrénu obsahujúce náplň (sklo typu 1) s piestom (brómobutyl) a zátkou (brómobutyl/polyizoprén).
Každé pero obsahuje 3 ml roztoku a slúži na podávanie dávok po 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg a 3,0 mg.
Veľkosť balenia je 1, 3 alebo 5 naplnených pier.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom Roztok sa nemá používať, ak nie je číry, bezfarebný alebo takmer bezfarebný. Saxenda sa nemá používať, ak bola zmrazená.
Pero je určené na použitie s jednorazovými injekčnými ihlami NovoFine alebo NovoTwist s dĺžkou do
8 mm a s hrúbkou do 32G.
Injekčné ihly nie sú súčasťou balenia.
Pacient má byť upozornený, aby injekčnú ihlu po každom podaní injekcie zlikvidoval, a aby pero uchovával bez nasadenej injekčnej ihly. Tým sa zabráni kontaminácii, infekcii a vytekaniu. Zaistí sa tým aj presnosť dávkovania.
Akýkoľvek nepoužitý liek alebo odpadový materiál treba vrátiť do lekárne v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovo Nordisk A/S Novo Allé
DK-2880 Bagsværd
Dánsko
8. REGISTRAČNÉ ČÍSLOEU/1/15/992/001-003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.