
čas prvých mesiacov liečby vykonávať častejšie.
V klinických skúšaniach bolo pozorované kongestívne srdcové zlyhanie. V prípade významného zhoršenia kardiovaskulárneho ochorenia sa musí prehodnotiť potreba pokračovania liečby Revestivom.
M
anažment
t
ekutín
počas
liečby
Revestivom
Parenterálna podpora u pacientov užívajúcich Revestive sa musí znižovať opatrne a nesmie sa vysadiť
náhle. Po redukcii parenterálnej podpory sa musí vyhodnotiť stav hydratácie pacienta a podľa potreby sa má vykonať zodpovedajúca úprava.
Súbežne podávané lieky
Pacienti, ktorí súbežne užívajú perorálne lieky vyžadujúce titrovanie dávky alebo lieky s úzkym terapeutickým indexom, sa musia pozorne sledovať vzhľadom na možné zvýšenie absorpcie (pozri
časť 4.5).
Osobitné klinické stavy
Revestive sa neskúmal u pacientov so závažnými, klinicky nestabilnými súbežnými ochoreniami,
(napr. kardiovaskulárne, respiračné, ochorenia obličiek, infekčné, endokrinné, ochorenia pečene
alebo CNS) alebo u pacientov so zhubnými nádormi počas posledných piatich rokov (pozri časť 4.3).
Pri predpisovaní Revestivu sa musí postupovať s opatrnosťou.
Poruchafunkciepečene
Revestive sa neskúmal u pacientov so závažnou poruchou funkcie pečene. Údaje z používania u jedincov so stredne závažnou poruchou funkcie pečene nenaznačujú potrebu obmedzeného používania.
Ukončenieliečby
Pri ukončovaní liečby Revestivom sa musí postupovať s opatrnosťou kvôli riziku dehydratácie.
Pediatrická populácia
Pozrite tiež všeobecné opatrenia pre dospelých v tejto časti.
Kolorektálne polypy/neoplázie
Pred začatím liečby Revestivom sa musí vykonať u všetkých detí a dospievajúcich test na okultné krvácanie v stolici. Testovanie musí následne pokračovať každoročne po dobu užívania Revestivu.
Pred začatím liečby Revestivom musia deti a dospievajúci vo veku 12 rokov a staršie podstúpiť kolonoskopiu/sigmoidoskopiu, ak nebola vykonaná počas posledného roku. Deti do 12 rokov musia tiež podstúpiť takéto vyšetrenie, ak majú nevysvetliteľnú krv v stolici. Kolonoskopia je odporúčaná pre všetky deti a dospievajúcich po jednom roku liečby a následne aspoň raz za 5 rokov pri kontinuálnej liečbe Revestivom.
Pomocné látky
Revestive obsahuje menej ako 1 mmol sodíka (23 mg) na dávku. To znamená, že obsahuje v podstate
zanedbateľné množstvo sodíka.
Potrebná je opatrnosť pri podávaní Revestivu osobám so známou precitlivenosťou na tetracyklín
(pozri časť 4.3).
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické štúdie liekových interakcií. Štúdia in vitro naznačuje, že teduglutid neinhibuje enzýmy cytochrómu P450 zodpovedné za metabolizmus liekov. Na základe farmakodynamického účinku teduglutidu existuje možnosť zvýšenej absorpcie súbežne užívaných liekov (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Nie sú k dispozícii údaje o použití Revestivu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Revestivu počas gravidity.
Dojčenie
Nie je známe, či sa teduglutid vylučuje do ľudského mlieka. U potkanov bola priemerná koncentrácia teduglutidu v mlieku menej ako 3 % plazmatickej koncentrácie u matky po jednorazovej subkutánnej injekcii 25 mg/kg. Riziko u dojčených novorodencov/dojčiat nemôže byť vylúčené. Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Revestivu počas dojčenia.
Fertilita
Neexistujú žiadne údaje o účinkoch teduglutidu na fertilitu ľudí. Údaje u zvierat nenaznačujú žiadnu poruchu fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Revestive má malý vplyv na schopnosť viesť vozidlá, jazdiť na bicykli a obsluhovať stroje. Avšak v klinických štúdiách boli hlásené prípady výskytu synkopy (pozri časť 4.8). Takéto príhody môžu mať vplyv na schopnosť viesť vozidlá, jazdiť na bicykli alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Nežiaduce reakcie sa získali z 2 placebom kontrolovaných klinických štúdií s teduglutidom
u 109 dospelých pacientov s SBS liečených dávkami 0,05 mg/kg/deň a 0,10 mg/kg/deň až 24 týždňov.
Nežiaduce reakcie sa objavili u približne 52 % pacientov liečených teduglutidom (oproti 36 %
z pacientov, ktorým sa podávalo placebo). Najčastejšie hlásenými nežiaducimi reakciami boli bolesť
brucha a nafúknutie brucha (45 %), infekcie dýchacích ciest (28 %) (vrátane zápalu nosohltana, chrípky, infekcie horných dýchacích ciest a infekcie dolných dýchacích ciest), nauzea (26 %), reakcie v mieste podania injekcie (26 %), bolesť hlavy (16 %) a vracanie (14 %). U približne 38 % liečených pacientov so stómiou sa objavili gastrointestinálne komplikácie stómie. Väčšina týchto reakcií bola mierna až stredne závažná.
U pacientov vystavených pôsobeniu dávky teduglutidu 0,05 mg/kg/deň po dobu do 30 mesiacov sa v dlhodobej nezaslepenej predĺženej štúdii nezistili žiadne nové signály týkajúce sa bezpečnosti.
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie podľa tried orgánových systémov MedDRA a podľa frekvencie sú uvedené nižšie. Frekvencie sú definované ako veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci každej skupiny frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti. Všetky nežiaduce účinky zistené po uvedení lieku na trh sú napísané kurzívou.
F
rekvencia
Veľmi časté Časté Menej časté Neznáme
T
rieda orgánového
s
y
s
t
é
m
u
Infekcie a nákazy Infekcia dýchacích ciest*
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Chrípke podobné ochorenie
Znížená chuť do jedla
Preťaženie tekutinami
Hypersenzitivita
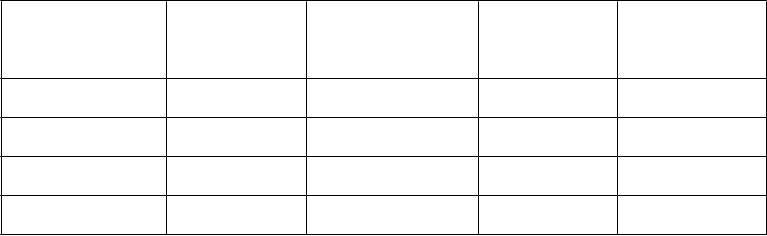
Psychické poruchy Úzkosť
Insomnia
Poruchy nervového systému
Bolesť hlavy
Poruchy srdca Kongestívne zlyhanie srdca
Poruchy ciev Synkopa
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho
traktu
Poruchy pečene
a žlčových ciest
Nafúknutie brucha
Bolesť brucha Nauzea Vracanie
Kašeľ
Dyspnoe
Kolorektálny polyp Stenóza hrubého čreva Nadúvanie Nepriechodnosť čriev Stenóza
pankreatického vývodu Pankreatitída†
Stenóza tenkého čreva
Cholecystitída
Akútna cholecystitída
Duodenálny polyp
Polyp žaludku
Celkového poruchy a reakcie v mieste podania
Úrazy, otravy a komplikácie liečebného postupu
Reakcia v mieste podania injekcie ‡
Gastrointestinálne komplikácie stómie
Periférny edém Zadržiavanie tekutín
* Zahŕňa nasledujúce preferované termíny: zápal nosohltana, chrípka, infekcia horných dýchacích ciest
a infekcia dolných dýchacích ciest.
† Zahŕňa nasledujúce preferované termíny: pankreatitída,
akútna pankreatitída a chronická pankreatitída.
‡ Zahŕňa nasledujúce preferované termíny: hematóm v mieste podania injekcie, erytém v mieste podania
injekcie, bolesť v mieste podania injekcie, opuch v mieste podania injekcie a krvácanie v mieste podania injekcie.
Popis vybraných nežiaducich reakciíImunogenicitaV súlade s prípadnými imunogénnymi vlastnosťami liekov obsahujúcich peptidy môže podanie Revestivu potenciálne vyvolať tvorbu protilátok. Na základe integrovaných dát z dvoch skúšaní u dospelých s SBS (6 mesačné randomizované, placebom kontrolované skúšanie, nasledované
24 mesačným otvoreným skúšaním) bol vývoj protilátok proti teduglutidu u jedincov, ktorí dostávali podkožným podaním 0,05 mg / kg teduglutidu raz denne 3 % (2 / 60) v 3. mesiaci, 17 % (13 / 77)
v 6. mesiaci, 24 % (16 / 67) v 12. mesiaci, 33 % (11 / 33) v 24. mesiaci a 48 % (14 / 29) v 30. mesiaci. V štúdiách fázy 3 s pacientmi s SBS, ktorí dostávali teduglutid ≥ 2 roky vyvinulo 28 % pacientov protilátky proti proteínu
E. coli (zvyškový proteín hostiteľskej bunky z výroby). Tvorba protilátok
nesúvisela s klinicky významnými bezpečnostnými zisteniami, zníženou účinnosťou alebo zmenenou farmakokinetikou Revestivu.
Reakcie v mieste podania injekcieU 26 % pacientov s SBS liečených teduglutidom sa objavili reakcie v mieste podania injekcie
v porovnaní s 5 % pacientov v skupine s placebom. Reakcie zahŕňali hematóm v mieste podania injekcie, erytém v mieste podania injekcie, bolesť v mieste podania injekcie, opuch v mieste podania injekcie a krvácanie v mieste podania injekcie (pozri tiež časť 5.3). Väčšina reakcií bola stredne závažná a žiadna z udalostí neviedla k vysadeniu lieku.
C-reaktívny proteínMierne zvýšenie C-reaktívneho proteínu približne o 25 mg/l bolo pozorované počas prvých siedmich dní liečby teduglutidom, ktorý pri podávaní injekcií denne neustále klesal. Po 24 týždňoch liečby
s teduglutidom sa u pacientov ukázal v priemere malý celkový nárast C-reaktívneho proteínu
o približne 1,5 mg/l. Tieto zmeny neboli spojené so žiadnymi inými zmenami ostatných laboratórnych parametrov a ani so žiadnymi hlásenými klinickými príznakmi. Po dlhodobej liečbe teduglutidom po
dobu do 30 mesiacov sa nevyskytli žiadne klinicky významné priemerné zvýšené hodnoty C- reaktívneho proteínu oproti východiskovej hodnote.
Pediatrická populáciaV jednej ukončenej klinickej štúdii bolo zahrnutých 37 pediatrických subjektov (vo veku
od 1 do 14 rokov), ktorých liečba teduglutidom trvala 12 týždňov. Žiadny subjekt neprerušil štúdiu
v dôsledku nežiaducej udalosti. Vo všeobecnosti bol bezpečnostný profil teduglutidu u detí
a dospievajúcich (vo veku 1-17 rokov) podobný ako u dospelých. Nasledujúce udalosti boli hlásené s vyššou frekvenciou u detských pacientov v porovnaní s dospelými: únava (veľmi časté), bolestivá
defekácia (veľmi časté) a závrat (časté). Avšak databáza údajov o bezpečnosti u detí a dospievajúcich
je obmedzená.
Údaje o dlhodobej bezpečnosti u pediatrickej populácie nie sú doteraz k dispozícii. Pre deti do 1 roku veku nie sú dostupné žiadne údaje.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieMaximálna dávka teduglutidu, ktorá sa skúmala počas klinického vývoja bola 86 mg/deň počas 8 dní.
Nepozorovali sa žiadne neočakávané systémové nežiaduce reakcie (pozri časť 4.8).
V prípade predávkovania musí pacienta pozorne sledovať zdravotnícky pracovník.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá pre tráviaci trakt a metabolizmus, rôzne liečivá tráviaceho
traktu a metabolizmu, ATC kód: A16AX08
MechanizmusúčinkuPrirodzene sa vyskytujúci ľudský peptid podobný glukagónu-2 (GLP- 2) je peptid vylučovaný L
bunkami čreva, ktorý, ako je známe, zvyšuje prietok krvi črevami a portálny prietok krvi, inhibuje sekréciu žalúdočnej kyseliny a znižuje motilitu čriev. Teduglutid je analógom GLP-2. V niekoľkých predklinických štúdiách sa preukázalo, že teduglutid udržiava integritu sliznice podporovaním obnovy a normálneho rastu čreva prostredníctvom zvýšenia výšky klkov a hĺbky črevnej žľazy.
FarmakodynamickéúčinkyPodobne ako GLP-2, teduglutid je tvorený reťazcom 33 aminokyselín, v ktorom je aminokyselina alanín nahradená glycínom v polohe 2 N-konca. Samotná substitúcia aminokyseliny v porovnaní
s prirodzeným GLP-2 vedie k rezistencii voči degradácii enzýmom dipeptidylpeptidázou-IV (DDP-IV)
in vivo, čo má za následok predĺžený polčas. Teduglutid zvyšuje výšku klkov a hĺbku črevnej žľazy
epitelu čreva.
Na základe zistení z predklinických štúdií (pozri časti 4.4 a 5.3) a navrhovaného mechanizmu účinku trofických účinkov na črevnú sliznicu sa zdá, že existuje riziko podpory neoplázie tenkého a/alebo hrubého čreva. Vykonané klinické štúdie toto zvýšené riziko nepotvrdili, ani nevyvrátili. Počas skúšaní sa vyskytlo niekoľko prípadov benígnych kolorektálnych polypov, avšak frekvencia nebola vyššia ako u pacientov, ktorí dostávali placebo. Okrem nutnosti vykonania kolonoskopie na odstránenie polypov na začiatku liečby (pozri časť 4.4) musí byť každý pacient posúdený s ohľadom na potrebu zvýšeného dozoru na základe charakteristiky pacienta (napr. vek a základné ochorenie, predchádzajúci výskyt polypov atď.).
K
l
i
n
i
cká
účinnosť
P
ediatrická populácia
Teduglutid bol skúmaný v 12-týždňovej otvorenej klinickej štúdii so 42 pediatrickými subjektmi
vo veku od 1 do 14 rokov s SBS, ktorí boli závislí na parenterálnej výžive. Cieľom tejto štúdie bolo zhodnotiť bezpečnosť, znášanlivosť a účinnosť teduglutidu v porovnaní so štandardnou
starostlivosťou. Po dobu 12 týždňov sa skúmali tri (3) dávky teduglutidu; 0,0125 mg/kg/deň (n = 8);
0,025 mg/kg/deň (n = 14) a 0,05 mg/kg/deň (n = 15). Päť (5) subjektov bolo zahrnutých do kohorty štandardnej starostlivosti.
Kompletné odstavenie
Trom subjektom (3/15, 20 %) na odporúčanej dávke teduglutidu bola do 12. týždňa vysadená
parenterálna výživa. Po 4-týždňovom vymývacom období bola u dvoch z týchto pacientov znovu
začatá parenterálna nutričná podpora.
Zníženie objemu parenterálnej výživy
V 12. týždni bola v populácii ITT, na základe údajov predpísaných lekárom, zaznamenaná priemerná zmena v objeme parenterálnej výživy −2,57 (± 3,56) l/týždeň oproti východiskovej hodnote, čo
zodpovedá −39,11 % (± 40,79) priemernému poklesu v porovnaní s 0,43 (± 0,75) l/týždeň, čo
zodpovedá 7,38 % (± 12,76) nárastu v kohorte so štandardnou starostlivosťou. V 16. týždni (4 týždne po ukončení liečby) bolo zníženie objemu parenterálnej výživy ešte viditeľné, ale menej, než aké bolo
pozorované v 12. týždni, keď sa ešte subjektom podával teduglutid (priemerný pokles o −31,80 %
(± 39,26), v porovnaní s 3,92 % (± 16,62) nárastom v skupine štandardnej starostlivosti.
Zníženie počtu kalórií v parenterálnej výžive
V 12. týždni bola v populácii ITT, na základe údajov predpísaných lekárom, zaznamenaná priemerná zmena spotreby kalórií v parenterálnej výžive −35,11 % (± 53,04) oproti východiskovej hodnote. Zodpovedajúca zmena v kohorte so štandardnou starostlivosťou bola 4,31 % (± 5,36). V 16. týždni spotreba kalórií v parenterálnej výžive naďalej klesala, a to s priemernou percentuálnou zmenou
–39,15 % (± 39,08) oproti východiskovej hodnote v porovnaní s 0,87 % (± 9,25) v kohorte štandardnej starostlivosti.
Zvýšenie objemu enterálnej výživy
V 12. týždni bola v populácii ITT na základe stanovených údajov zaznamenaná priemerná percentuálna zmena objemu enterálnej výživy oproti východiskovej hodnote 25,82 % (± 41,59)
v porovnaní s 53,65 % (± 57,01) v kohorte so štandardnou starostlivosťou. V 16. týždni kohorta
s teduglutidom ako aj kohorta so štandardnou starostlivosťou preukázali nárast objemu enterálnej výživy.
Zvýšenie počtu kalórií v enterálnej výžive
Zvýšenie objemu enterálnej výživy zodpovedalo zvýšeniu počtu enterálnych kalórií, ktoré bolo najvyššie pri podaní odporúčanej dávky. V 12. týždni bol v populácii ITT zaznamenaný percentuálny nárast 58,80 % (± 64,20) oproti východiskovému stavu predpísaných enterálnych kalórií v porovnaní s 57,02 % (± 55,25) v kohorte so štandardnou starostlivosťou. V 16. týždni pokračoval nárast príjmu
kalórií v enterálnej výžive s percentuálnym nárastom oproti východiskovej hodnote 64,57 % (± 57,53)
v porovnaní s 59,63 % (± 52,62) v kohorte so štandardnou starostlivosťou.
Skrátenie doby infúzie
V 12. týždni bol v populácii ITT na základe údajov predpísaných lekárom, zaznamenaný priemerný pokles v počte dní/týždeň na parenterálnej výžive −1,36 (± 2,37) dní/týždeň oproti východiskovej
hodnote, čo zodpovedá percentuálnemu poklesu −24,49 % (± 42,46). V kohorte so štandardnou
starostlivosťou nebola zaznamenaná žiadna zmena oproti východiskovej hodnote. Štyri subjekty
(26,7 %) na odporúčanej dávke teduglutidu dosiahli najmenej tri dni so znížením potreby parenterálnej
výživy.
V 12. týždni došlo na základe údajov z denníka subjektu k priemernému percentuálnemu poklesu
o 35,55 % (± 35,23) hodín denne v porovnaní s východiskovou hodnotou, čo zodpovedalo poklesu
−4,18 (± 4,08) v hodinách/deň pri použití parenterálnej výživy, zatiaľ čo subjekty v kohorte so štandardnou starostlivosťou vykázali minimálnu zmenu tohto parametra v rovnakom čase.
V tomto skúšaní neboli pozorované žiadne nové neočakávané signály ohľadom bezpečnosti.
Dospelí
Teduglutid sa skúmal u 17 pacientov s SBS, ktorí boli rozdelení do piatich liečebných skupín
používajúcich teduglutid v dávkach 0,03; 0,10 alebo 0,15 mg/kg raz denne alebo 0,05 alebo
0,075 mg/kg dvakrát denne v 21-dňovej otvorenej, multicentrickej štúdii na stanovenie rozmedzia dávky. Liečba viedla k zvýšenej absorpcii tekutín v gastrointestinálnom trakte
na približne750 - 1000 ml/deň so zlepšením absorpcie makro živín a elektrolytov, so znížením tekutiny v oblasti stómie alebo tekutiny v stolici a vylučovania makro živín a k zvýšeniu kľúčových štrukturálnych a funkčných adaptácií v sliznici čreva. Štrukturálne adaptácie boli prechodného
charakteru a na východiskové hladiny sa vrátili do troch týždňov po ukončení liečby.
V pivotnej dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u pacientov s SBS, ktorí vyžadovali parenterálnu výživu, bolo 43 pacientov randomizovaných na dávku teduglutidu
0,05 mg/kg/deň a 43 pacientov na placebo po dobu až 24 týždňov.
Podiel pacientov liečených teduglutidom, ktorí dosiahli 20 % až 100 % zníženie parenterálnej výživy v 20 a 24 týždni sa štatisticky významne odlišoval od placeba (27 zo 43 pacientov, 62,8 % oproti 13 zo 43 pacientov, 30,2 %, p = 0,002). Liečba teduglutidom viedla v 24 týždni k zníženiu potrieb parenterálnej výživy o 4,4 l/ týždeň (z východiskových 12,9 litrov pred liečbou) oproti 2,3 l/týždeň
(z východiskových 13,2 litra pred liečbou) pri placebe. Dvadsaťjeden (21) pacientov liečených teduglutidom (48,8 %) oproti 9 u placeba (20,9 %) dosiahlo minimálne jednodňové zníženie v podaní parenterálnej výživy (p = 0,008).
Deväťdesiatsedem percent (97 %) pacientov (37 z 39 pacientov liečených teduglutidom), ktorý ukončili placebom kontrolovanú štúdiu, bolo vybraných, aby pokračovali v dlhodobej rozšírenej štúdii, v ktorej všetci pacienti dostávali 0,05 mg/kg Revestivu denne počas ďalších maximálne
2 rokov. Tejto rozšírenej štúdie sa zúčastnilo celkovo 88 pacientov, z ktorých 39 bolo podávané placebo a 12 bolo zaradených, ale nie randomizovaných, v predchádzajúcej štúdii; 65 z 88 pacientov
absolvovalo rozšírenú štúdiu. V nej sa naďalej preukazovala zvýšená reakcia na liečbu po dobu
2,5 roka vo všetkých skupinách vystavených pôsobeniu teduglutidu, týkajúca sa zníženia objemu
parenterálnej výživy, nárastu ďalších dní bez parenterálnej výživy týždenne a dosiahnutia vysadenia parenterálnej podpory.
Tridsať (30) zo 43 pacientov liečených teduglutidom v hlavnej štúdii, ktorí vstúpili do rozšírenej štúdie, absolvovalo spolu 30 mesiacov liečby. Z nich 28 pacientov (93 %) dosiahlo najmenej 20% zníženie parenterálnej podpory. Z pacientov reagujúcich na liečbu v hlavnej štúdii, ktorí absolvovali rozšírenú štúdiu, si 21 z 22 (96 %) udržalo reakciu na teduglutid po dobu ďalších 2 rokoch nepretržitej liečby.
Priemerné zníženie parenterálnej výživy (n=30) bolo 7,55 l/týždeň (65,6% zníženie oproti východiskovej hodnote). U desiatich (10) pacientov sa vysadila parenterálna podpora počas užívania teduglutidu po dobu 30 mesiacov. Jedinci boli udržiavaní na teduglutide, aj keď sa už nevyžadovala parenterálna výživa. Týchto 10 jedincov v minulosti potrebovalo podpornú parenterálnu výživu po dobu 1,2 až 15,5 roka a pred liečbou teduglutidom potrebovali 3,5 l/týždeň až 13,4 l/týždeň podpornej parenterálnej výživy. Na konci štúdie 21 (70 %), 18 (60 %) a 18 (60 %) z 30 pacientov, ktorí absolvovali štúdiu, dosiahlo zníženie o 1, 2 alebo 3 dni parenterálnej podpory za týždeň, v príslušnom poradí.
Z 39 jedincov užívajúcich placebo 29 dokončilo 24 mesiacov liečby teduglutidom. Priemerné zníženie parenterálnej výživy bolo 3,11 l/týždeň (dodatočné 28,3% zníženie). Šestnásť (16;55,2 %) z 29 pacientov, ktorí absolvovali liečbu, dosiahlo najmenej 20% zníženie parenterálnej výživy. Na konci štúdie 14 (48,3 %), 7 (24,1 %) a 5 (17,2 %) pacientov dosiahlo zníženie parenterálnej výživy o 1, 2 alebo 3 dni týždenne, v príslušnom poradí. U dvoch (2) jedincov bola vysadená parenterálna podpora počas užívania teduglutidu.
Z 12 jedincov, ktorí neboli randomizovaní do hlavnej štúdie, 6 jedincov dokončilo 24 mesiacov liečby teduglutidom. Priemerné zníženie parenterálnej výživy bolo 4,0 l/týždeň (39,4% zníženie oproti východiskovej hodnote – začiatok rozšírenej štúdie) a 4 zo 6 jedincov, ktorí absolvovali liečbu,
(66,7 %) dosiahli najmenej 20% zníženie parenterálnej podpory. Na konci štúdie 3 (50 %), 2 (33 %) a 2 (33 %) pacienti dosiahli zníženie parenterálnej výživy o 1, 2 alebo 3 dni týždenne, v príslušnom poradí. U jedného jedinca bola vysadená parenterálna podpora počas užívania teduglutidu.
V ďalšej dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u pacientov s SBS, ktorí vyžadovali parenterálnu výživu, pacienti dostávali dávku 0,05 mg/kg/deň (n=35), dávku 0,10 mg/kg/deň (n=32) teduglutidu alebo placebo (n = 16) až 24 týždňov.
Primárna analýza výsledkov štúdie vzhľadom na účinosť nepreukázala žiadny štatisticky významný rozdiel medzi skupinou na teduglutide 0,10 mg/kg/deň a placebom, zatiaľ čo podiel jedincov, ktorí dostávali odporúčanú dávku teduglutidu 0,05 mg/kg/deň a ktorí dosiahli minimálne 20 % redukciu parenterálnej výživy v 20. a 24. týždni, bol štatisticky signifikantne odlišný oproti placebu (46 % oproti 6,3 %, p < 0,01). Liečba teduglutidom viedla k zníženiu požiadaviek parenterálnej výživy
o 2,5 l/týždeň (z východiskových 9,6 litra pred liečbou) oproti 0,9 l/týždeň (z východiskových
10,7 litra pred liečbou) pri placebe v 24. týždni.
Liečba teduglutidom indukovala expanziu absorpčného epitelu signifikantným zvýšením výšky klkov
v tenkom čreve.
Šesťdesiatpäť (65) pacientov vstúpilo do následnej SBS štúdie až na ďalších 28 týždňov liečby. Pacienti používajúci teduglutid si udržali svoje predchádzajúce zaradenie dávky počas predĺženého obdobia, zatiaľ čo pacienti, ktorí dostávali placebo, boli randomizovaní na aktívnu liečbu, buď 0,05 alebo 0,10 mg/kg/deň.
Z pacientov, ktorí dosiahli minimálne 20 % redukciu parenterálnej výživy v 20. a 24. týždni v úvodnej štúdii, si 75 % udržalo túto odpoveď na teduglutid po maximálne 1 roku nepretržitej liečby.
Priemerné zníženie týždenného objemu parenterálnej výživy bolo 4,9 l/týždeň (52% zníženie z východiskovej hodnoty) po jednom roku nepretržitej liečby teduglutidom.
Dvaja (2) pacienti na odporúčanej dávke teduglutidu boli odstavení z parenterálnej výživy do 24. týždňa. V následnej štúdii bol od parenterálnej výživy odstavený ďalší jeden pacient.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Revestivom
v jednej alebo vo viacerých vekových podskupinách pediatrickej populácie na liečbu SBS (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absorpcia teduglutidu z miesta podania subkutánnej injekcie bola rýchla s maximálnymi plazmatickými hladinami, ktoré sa objavili približne 3 – 5 hodín po podaní dávky pri všetkých hladinách dávky. Absolútna biologická dostupnosť subkutánneho teduglutidu je vysoká (88 %). Po opakovaných subkutánnych podaniach sa nepozorovala žiadna kumulácia teduglutidu.
Distribúcia
Po subkutánnom podaní je zjavný distribučný objem teduglutidu u pacientov s SBS 26 litrov.
B
i
otransformácia
Metabolizmus teduglutidu nie je celkom známy. Keďže teduglutid je peptid, je pravdepodobné, že
podlieha základnému mechanizmu metabolizmu peptidov.
Eliminácia
Terminálny polčas eliminácie teduglutidu je približne 2 hodiny. Po intravenóznom podaní teduglutidu bol plazmatický klírens približne 127 ml/hod/kg, ktorý je ekvivalentný rýchlosti glumerulárnej
filtrácie (GFR). V štúdii skúmajúcej farmakokinetiku u jedincov s poruchou funkcie obličiek sa potvrdila eliminácia obličkami. Po opakovaných subkutánnych podaniach sa nepozorovala žiadna
kumulácia teduglutidu.
Linearita dávky
Rýchlosť a rozsah absorpcie teduglutidu sú proporcionálne dávke pri jednorazových a opakovaných subkutánnych dávkach až do 20 mg.
Farmakokinetika v podskupinách pacientov
Pediatrická populácia
Na základe populačného farmakokinetického modelovania bola vo všetkých vekových skupinách po subkutánnom podaní preukázaná podobná Cmax teduglutidu. Avšak u pediatrických pacientov vo veku
1 až 17 rokov bola pozorovaná nižšia expozícia (AUC) a kratší polčas v porovnaní s dospelými.
Farmakokinetický profil Revestivu v tejto pediatrickej populácii, ako bolo hodnotené podľa klírensu
a objemu distribúcie, bol po korekcii na telesnú hmotnosť rozdielny od toho, ktorý bol pozorovaný
u dospelých. Konkrétne klírens klesá so zvyšujúcim sa vekom od 1 roku až po dospelých.
U pediatrických pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek a v terminálnom
štádiu ochorenia obličiek (end-stage renal disease, ESRD) nie sú k dispozícii žiadne údaje.
Pohlavie
V klinických štúdiách sa nepozorovali žiadne klinicky významné rozdiely týkajúce sa pohlavia.
Starší pacienti
V štúdii fázy 1 sa nezistila žiadna odlišnosť vo farmakokinetike teduglutidu medzi zdravými jedincami mladšími ako 65 rokov oproti jedincom starším ako 65 rokov. Skúsenosť u jedincov vo veku 75 rokov a starších je obmedzená.
Poruchafunkciepečene
V štúdii fázy 1 sa skúmal vplyv poruchy funkcie pečene na farmakokinetiku teduglutidu po subkutánnom podaní 20 mg teduglutidu. Maximálna expozícia a celkový rozsah expozície teduglutidu po jednorazových subkutánnych dávkach 20 mg boli nižšie (10-15 %) u jedincov so stredne závažnou poruchou funkcie pečene v porovnaní so zdravými kontrolnými jedincami.
Poruchafunkcieobličiek
V štúdii fázy 1 sa skúmal vplyv poruchy funkcie obličiek na farmakokinetiku teduglutidu po
subkutánnom podaní 10 mg teduglutidu. So zvyšujúcim sa stupňom poruchy funkcie obličiek až do (a
vrátane) terminálneho štádia ochorenia obličiek sa primárne farmakokinetické parametre teduglutidu zvyšovali až na 2,6-násobok (AUCinf) a 2,1-násobok (Cmax) v porovnaní so zdravými jedincami.
5.3 Predklinické údaje o bezpečnosti
V štúdiách subchronickej a chronickej toxikológie sa pozorovali hyperplázie žlčníka, pečeňového/žlčového vývodu a pakreatického vývodu. Tieto pozorovania pravdepodobne súviseli s očakávaným zamýšľaným farmakologickým účinkom teduglutidu a líšili sa v stupni reverzibility v rámci obdobia 8 - 13 týždňov zotavovania sa po dlhodobom podávaní.
Reakcie v mieste vpichu
V predklinických štúdiách sa zistili závažné granulomatózne zápaly spojené s miestom vpichu.
K
a
r
cinogenita/mutagenita
Počas testovania v štandardných sériách testov na genotoxicitu bol teduglutid negatívny.
V štúdii karcinogenity na potkanoch, s liečbou súvisiace benígne novotvary zahŕňali nádory epitelu žlčovodu u samcov vystavených plazmatickým hladinám teduglutidu približne 32 a 155 krát vyšším ako u pacientov, ktorým bola podávaná odporúčaná denná dávka (výskyt 1 zo 44 a 4 zo 48
v príslušnom poradí). Adenómy sliznice lačníka boli pozorované u 1 z 50 samcov a 5 z 50 samcov vystavených plazmatickým hladinám teduglutidu približne 10 a 155 krát (v príslušnom poradí) vyšším
ako u pacientov, ktorým bola podávaná odporúčaná denná dávka. Okrem toho bol pozorovaný
jejunálny adenokarcinóm u samcov potkanov pri podaní najnižšej testovanej dávky (zviera:človek
rozdiel v plazmatickej expozícii približne 10 násobok).
Reprodukčnáa vývinová toxicita
Štúdie reprodukčnej a vývinovej toxicity hodnotiace teduglutid sa vykonali na potkanoch a králikoch pri dávkach 0, 2, 10 a 50 mg/kg/deň subkutánne. Teduglutid sa nespájal s účinkami na reprodukčný
výkon, na in utero alebo vývinové parametre merané v štúdiách na zistenie fertility, embryofetálneho
vývinu a prenatálneho a postnatálneho vývinu. Farmakokinetické údaje preukázali, že expozícia teduglutidu je u plodov králika a dojčených mláďat potkana veľmi malá.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
L-histidín manitol
nátriumfosfát monohydrát
dinátriumfosfát heptahydrát
Rozpúšťadlo
voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorené injekčné liekovky
24 mesiacov
Rekonštituovaný liek
Chemická a fyzikálna stabilita sa pri použití preukázala počas 24 hodín pri teplote do 25 °C.
Z mikrobiologického hľadiska, s výnimkou použitia rekonštitučnej metódy vylučujúcej riziko mikrobiálnej kontaminácie, sa musí liek použiť okamžite.
Ak sa liek nepoužije okamžite, za čas a podmienky uchovávania po prvom otvorení zodpovedá používateľ. Za normálnych okolností to nebude dlhšie ako 24 hodín pri 2 °C – 8 °C, pokiaľ rekonštitúcia neprebehla v riadených a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok
3 ml injekčná liekovka (sklo) s gumovým uzáverom (brómbutyl) s obsahom 1,25 mg teduglutidu.
Rozpúšťadlo
Naplnená injekčná striekačka (sklo) s piestom (brómbutyl) s obsahom 0,5 ml rozpúšťadla.
Veľkosť balenia: 28 injekčných liekoviek s práškom, s 28 naplnenými injekčnými striekačkami.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Určenie počtu injekčných liekoviek potrebných na podanie jednej dávky musí byť založené na individuálnej hmotnosti pacienta a odporúčanej dávke 0,05 mg/kg/deň. Lekár musí pri každej návšteve pacienta odvážiť, stanoviť dennú dávku, ktorá sa má podať do ďalšej kontroly a podľa toho
informovať pacienta.
Tabuľka s injekčnými objemami na základe odporúčanej dávky na telesnú hmotnosť pre pediatrických
pacientov je uvedená v časti 4.2.
Naplnená injekčná striekačka musí byť opatrená ihlou na rekonštitúciu.
Prášok v injekčnej liekovke sa musí potom rozpustiť pridaním všetkého rozpúšťadla z naplnenej
injekčnej striekačky.
Injekčná liekovka sa nesmie pretrepávať, ale môže sa gúľať medzi dlaňami a raz jemne obrátiť hore dnom. Po príprave číreho bezfarebného injekčného roztoku v injekčnej liekovke sa má vzniknutý roztok odobrať do 1-ml injekčnej striekačky (alebo 0,5-ml alebo menšej injekčnej striekačky
pre pediatrické použitie) s dielikmi po 0,02 ml alebo menej (nie je súčasťou balenia).
Ak sú potrebné dve injekčné liekovky, postup sa musí zopakovať pri druhej injekčnej liekovke a ďalší roztok treba natiahnuť do injekčnej striekačky obsahujúcej roztok z prvej injekčnej liekovky. Akýkoľvek objem presahujúci predpísanú dávku v ml treba odstrániť a zlikvidovať.
Roztok sa musí aplikovať subkutánne do vyčistenej oblasti na bruchu, alebo ak to nie je možné do
stehna (pozri časť 4.2 Spôsob podávania) pomocou tenkej ihly na subkutánnu injekciu vhodnej na pediatrické použitie.
Podrobné pokyny na prípravu a injekciu Revestivu sú uvedené v písomnej informácii pre používateľa.
Roztok sa nesmie použiť ak je zakalený alebo obsahuje častice.
Len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Všetky ihly a injekčné striekačky majú byť zlikvidované v kontajneri určenom na likvidáciu ostrých predmetov.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Shire Pharmaceuticals Ireland Limited
5 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
Tel.: +800 6774 4357
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/12/787/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 30. augusta 2012
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na
nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKURevestive 5 mg prášok a rozpúšťadlo na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJedna injekčná liekovka prášku obsahuje 5 mg teduglutidu*.
Po rekonštitúcii každá injekčná liekovka obsahuje 5 mg teduglutidu v 0,5 ml roztoku, čo zodpovedá koncentrácii 10 mg/ml.
*Analóg peptidu podobného glukagónu-2 (
glucagon-like peptide-2, GLP-2) produkovaný v bunkách
Escherichia coli DNA rekombinantnou technológiou.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPrášok a rozpúšťadlo na injekčný roztok.
Prášok je biely a rozpúšťadlo je číre a bezfarebné.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieRevestive je indikovaný na liečbu pacientov vo veku 1 rok a viac so syndrómom krátkeho čreva (
ShortBowel Syndrome, SBS). Pacienti musia byť stabilizovaní po období črevnej adaptácie po operácii.
4.2 Dávkovanie a spôsob podávaniaLiečba sa má začať pod dohľadom profesionálneho lekára so skúsenosťou s liečbou SBS.
Liečba sa nesmie začať, kým sa opodstatnene nepredpokladá, že pacient je stabilizovaný po období
črevnej adaptácie. Pred začatím liečby sa musí optimalizovať a stabilizovať nutričná podpora
a intravenózny prísun tekutín.
Pri klinickom hodnotení lekárom je potrebné zvážiť jednotlivé liečebné ciele a preferencie pacienta. Pokiaľ sa nedosiahne celkové zlepšenie stavu pacienta, liečba sa musí zastaviť. Účinnosť a bezpečnosť u všetkých pacientov je potrebné priebežne starostlivo sledovať podľa klinických liečebných
postupov.
DávkovanieDospelíOdporúčaná dávka Revestivu je 0,05 mg/kg telesnej hmotnosti jedenkrát denne. Injekčný objem
k telesnej hmotnosti je uvedený nižšie v tabuľke 1. Vzhľadom k rozmanitosti populácie SBS možno
u niektorých pacientov zvážiť starostlivo sledovanú titráciu dennej dávky k optimalizácii znášanlivosti
liečby. Ak sa dávka vynechá, dávka sa musí injekovať čo najskôr v ten istý deň.
Liečebný účinok sa musí vyhodnotiť po 6 mesiacoch. U pacientov, ktorí prestali užívať parenterálnu výživu, sa odporúča pokračovanie liečby.
Tabuľka 1
Telesná hmotnosť
Sila 5 mgInjekčný objem
38-41 kg 0,20 ml
42-45 kg 0,22 ml
46-49 kg 0,24 ml
50-53 kg 0,26 ml
54-57 kg 0,28 ml
58-61 kg 0,30 ml

62-65 kg 0,32 ml
66-69 kg 0,34 ml
70-73 kg 0,36 ml
74-77 kg 0,38 ml
78-81 kg 0,40 ml
82-85 kg 0,42 ml
86-89 kg 0,44 ml
90-93 kg 0,46 ml
Pediatrická populácia (≥ 1 rok)Liečba sa má začať pod dohľadom profesionálneho lekára so skúsenosťou s liečbou pediatrických pacientov so syndrómom krátkeho čreva (SBS).
Odporúčaná dávka Revestive u detí a dospievajúcich (vo veku od 1 do 17 rokov) je rovnaká ako u dospelých (0,05 mg/kg telesnej hmotnosti raz denne). Injekčný objem k telesnej hmotnosti, pri použití injekčnej liekovky so silou 5 mg je uvedený nižšie v tabuľke 2. Injekčné liekovky so silou
1,25 mg sú tiež dostupné na pediatrické použitie (pacienti s telesnou hmotnosťou <20 kg).
V prípade vynechania dávky sa musí táto dávka injekovať čo najskôr v ten istý deň. Odporúča sa liečebné obdobie 12 týždňov, po ktorom sa musí hodnotiť účinok liečby. K dispozícii nie sú žiadne údaje u pediatrických pacientov po 12 týždňoch.
Tabuľka 2Telesná hmotnosť
Sila 5 mgInjekčný objem
10-11 kg 0,05 ml
12-13 kg 0,06 ml
14-17 kg 0,08 ml
18-21 kg 0,10 ml
22-25 kg 0,12 ml

26-29 kg 0,14 ml
30-33 kg 0,16 ml
34-37 kg 0,18 ml
38-41 kg 0,20 ml
42-45 kg 0,22 ml
46-49 kg 0,24 ml
≥50 kg Pozrite tabuľku 1 v časti „Dospelí“.
O
s
obitné populácie
Starší pacienti
Nie je potrebné upraviť dávku pacientom starším ako 65 rokov.
Porucha funkcie obličiek
Dospelým alebo pediatrickým pacientom s miernou poruchou funkcie obličiek nie je potrebné upraviť dávku. U dospelých alebo pediatrických pacientov so stredne ťažkou a ťažkou poruchou funkcie obličiek (klírens kreatinínu je nižší ako 50 ml/min) a koncovým štádiom ochorenia obličiek musí byť denná dávka znížená o 50 % (pozri časť 5.2).
Porucha funkcie pečene
Pacientom s miernou a stredne ťažkou poruchou funkcie pečene nie je potrebné upraviť dávku, na základe štúdie vykonanej u ľudí s ochorením klasifikovaným ako typ B podľa Childovho-Pughovho
skóre. Revestive nebol skúmaný u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.4 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť Revestive u detí vo veku menej ako 1 rok neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Rekonštituovaný roztok sa musí podať subkutánne injekciou jedenkrát denne, striedaním miest do jedného zo štyroch abdominálnych kvadrantov. V prípade, že sa injekcia nemôže podať do brucha kvôli bolesti, zjazveniu alebo tvrdnutiu tkaniva, môže sa tiež podať do stehna. Revestive sa nesmie podať intravenózne alebo intramuskulárne.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 alebo na stopové množstvá tetracyklínu.
Akútna alebo suspektná malignita.
Pacienti s nádorovým ochorením gastrointestinálneho traktu v anamnéze, vrátane hepatobiliárneho systému a pankreasu v priebehu posledných piatich rokov.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pri každom podaní Revestivu sa dôrazne odporúča zaznamenať názov a číslo šarže lieku, aby sa uchovala spojitosť medzi pacientom a šaržou lieku.
Dospelí
Kolorektálne polypy
Na začiatku liečby Revestivom sa má vykonať kolonoskopia s odstránením polypov. Počas prvých
2 rokov liečby Revestivom sa odporúča raz ročne vykonať kontrolné kolonoskopie (alebo alternatívne zobrazovacie vyšetrenie). Ďalšie kolonoskopie sa odporúča vykonávať v minimálne päť ročných
intervaloch. Individuálne posúdenie, či je nutná zvýšená frekvencia dohľadu nad pacientom, sa musí
vykonať na základe charakteristík pacienta (napr. vek, základné ochorenie). Pozri tiež časť 5.1. Ak sa zistí polyp, odporúča sa dodržiavať súčasné odporúčania na sledovanie polypov. V prípade zhubného
nádoru sa musí liečba Revestivom ukončiť (pozri časť 4.3).
Neoplázie gastrointestinálneho traktu vrátane hepatobiliárneho traktu
V štúdii karcinogenity na potkanoch sa v tenkom čreve a v extrahepatálnych žlčovodoch našli benígne
nádory. Tieto pozorovania neboli potvrdené klinickými štúdiami s trvaním viac ako jeden rok. Ak je
neoplázia zistená, musí sa odstrániť. V prípade zhubného nádoru sa musí liečba Revestivom ukončiť
(pozri časti 4.3 a 5.3).
Žlčníkažlčovody
V klinických štúdiách sa hlásili prípady cholecystitídy, cholangitídy a cholelitiázy. V prípade výskytu
príznakov súvisiacich so žlčníkom alebo žlčovodom sa musí prehodnotiť potreba pokračovania liečby
Revestivom.
Ochorenia pankreasu
V klinických štúdiách sa hlásili nežiaduce udalosti týkajúce sa pankreasu, napr. chronická a akútna pankreatitída, zúženie vývodu podžalúdkovej žľazy, infekcia pankreasu a zvýšené hladiny amylázy
a lipázy v krvi. V prípade výskytu nežiaducich udalostí týkajúcich sa pankreasu sa musí prehodnotiť
potreba pokračovania liečby Revestivom.
Sledovanietenkéhočreva,žlčníkaažlčovýchciestapankreasu
Pacienti s SBS majú byť pod starostlivým dohľadom podľa klinických liečebných pokynov. Zvyčajne
to zahŕňa monitorovanie funkcie tenkého čreva, žlčníka a žlčových ciest a pankreasu na prejavy a príznaky a keď je to indikované, ďalšie laboratórne vyšetrenia a zodpovedajúce zobrazovacie
techniky.
Nepriechodnosťčriev
V klinických štúdiách sa hlásili prípady nepriechodnosti čriev. V prípade opätovného výskytu
nepriechodnosti čriev sa musí prehodnotiť potreba pokračovania liečby Revestivom.
Preťaženietekutinami
V klinických skúšaniach bolo pozorované preťaženie tekutinami. Nežiaduce udalosti súvisiace
s preťažením tekutinami sa vyskytli najčastejšie počas prvých 4 týždňov liečby a časom klesali.
Vzhľadom na zvýšenú absorpciu tekutín sa musia pacienti s kardiovaskulárnym ochorením, ako napr. insuficiencia srdca a hypertenzia, sledovať s ohľadom na preťaženie tekutinami, najmä počas začiatočnej fázy liečby. Pacienti musia byť poučení, aby v prípade náhleho zvýšenia telesnej hmotnosti, opuchu členkov a/alebo dyspnoe kontaktovali svojho lekára. Preťaženiu tekutinami sa vo všeobecnosti môže predísť vhodným a včasným zhodnotením potrieb parenterálnej výživy. Toto zhodnotenie sa musí počas prvých mesiacov liečby vykonávať častejšie.
V klinických skúšaniach bolo pozorované kongestívne srdcové zlyhanie. V prípade významného zhoršenia kardiovaskulárneho ochorenia sa musí prehodnotiť potreba pokračovania liečby Revestivom.
ManažmenttekutínpočasliečbyRevestivom
Parenterálna podpora u pacientov užívajúcich Revestive sa musí znižovať opatrne a nesmie sa vysadiť náhle. Po redukcii parenterálnej podpory sa musí vyhodnotiť stav hydratácie pacienta a podľa potreby sa má vykonať zodpovedajúca úprava.
Súbežne podávané lieky
Pacienti, ktorí súbežne užívajú perorálne lieky vyžadujúce titrovanie dávky alebo lieky s úzkym terapeutickým indexom, sa musia pozorne sledovať vzhľadom na možné zvýšenie absorpcie (pozri časť 4.5).
Osobitné klinické stavy
Revestive sa neskúmal u pacientov so závažnými, klinicky nestabilnými súbežnými ochoreniami,
(napr. kardiovaskulárne, respiračné, ochorenia obličiek, infekčné, endokrinné, ochorenia pečene
alebo CNS) alebo u pacientov so zhubnými nádormi počas posledných piatich rokov (pozri časť 4.3).
Pri predpisovaní Revestivu sa musí postupovať s opatrnosťou.
Poruchafunkciepečene
Revestive sa neskúmal u pacientov so závažnou poruchou funkcie pečene. Údaje z používania
u jedincov so stredne závažnou poruchou funkcie pečene nenaznačujú potrebu obmedzeného
používania.
Ukončenieliečby
Pri ukončovaní liečby Revestivom sa musí postupovať s opatrnosťou kvôli riziku dehydratácie.
Pediatrická populácia
Pozrite tiež všeobecné opatrenia pre dospelých v tejto časti.
Kolorektálne polypy/neoplázie
Pred začatím liečby Revestivom sa musí vykonať u všetkých detí a dospievajúcich test na okultné krvácanie v stolici. Testovanie musí následne pokračovať každoročne po dobu užívania Revestivu.
Pred začatím liečby Revestivom musia deti a dospievajúci vo veku 12 rokov a staršie podstúpiť kolonoskopiu/sigmoidoskopiu, ak nebola vykonaná počas posledného roku. Deti do 12 rokov musia tiež podstúpiť takéto vyšetrenie, ak majú nevysvetliteľnú krv v stolici. Kolonoskopia je odporúčaná pre všetky deti a dospievajúcich po jednom roku liečby a následne aspoň raz za 5 rokov pri kontinuálnej liečbe Revestivom.
Pomocné látky
Revestive obsahuje menej ako 1 mmol sodíka (23 mg) na dávku. To znamená, že obsahuje v podstate
zanedbateľné množstvo sodíka.
Potrebná je opatrnosť pri podávaní Revestivu osobám so známou precitlivenosťou na tetracyklín
(pozri časť 4.3).
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické štúdie liekových interakcií. Štúdia in vitro naznačuje, že teduglutid neinhibuje enzýmy cytochrómu P450 zodpovedné za metabolizmus liekov. Na základe farmakodynamického účinku teduglutidu existuje možnosť zvýšenej absorpcie súbežne užívaných liekov (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití Revestivu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Revestivu počas gravidity.
Dojčenie
Nie je známe, či sa teduglutid vylučuje do ľudského mlieka. U potkanov bola priemerná koncentrácia teduglutidu v mlieku menej ako 3 % plazmatickej koncentrácie u matky po jednorazovej subkutánnej
injekcii 25 mg/kg. Riziko u dojčených novorodencov/dojčiat nemôže byť vylúčené. Ako preventívne
opatrenie je vhodnejšie vyhnúť sa používaniu Revestivu počas dojčenia.
Fertilita
Neexistujú žiadne údaje o účinkoch teduglutidu na fertilitu ľudí. Údaje u zvierat nenaznačujú žiadnu poruchu fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Revestive má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Avšak v klinických štúdiách boli hlásené prípady výskytu synkopy (pozri časť 4.8). Takéto príhody môžu mať vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn
bezpečnostného
profilu
Nežiaduce reakcie sa získali z 2 placebom kontrolovaných klinických štúdií s teduglutidom
u 109 pacientov s SBS liečených dávkami 0,05 mg/kg/deň a 0,10 mg/kg/deň až 24 týždňov. Nežiaduce
reakcie sa objavili u približne 52 % pacientov liečených teduglutidom (oproti 36 % z pacientov, ktorým sa podávalo placebo). Najčastejšie hlásenými nežiaducimi reakciami boli bolesť brucha
a nafúknutie brucha (45 %), infekcie dýchacích ciest (28 %) (vrátane zápalu nosohltana, chrípky,
infekcie horných dýchacích ciest a infekcie dolných dýchacích ciest), nauzea (26 %), reakcie v mieste podania injekcie (26 %), bolesť hlavy (16 %) a vracanie (14 %). U približne 38 % liečených pacientov so stómiou sa objavili gastrointestinálne komplikácie stómie. Väčšina týchto reakcií bola mierna až stredne závažná.
U pacientov vystavených pôsobeniu dávky 0,05 mg/kg/deň teduglutidu po dobu do 30 mesiacov sa v dlhodobej nezaslepenej predĺženej štúdii nezistili žiadne nové signály týkajúce sa bezpečnosti.
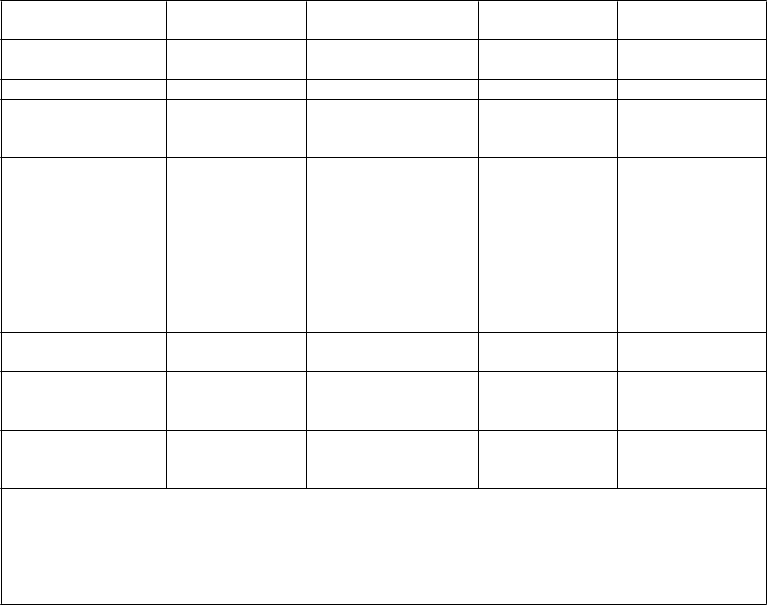
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie podľa tried orgánových systémov MedDRA a podľa frekvencie sú uvedené nižšie. Frekvencie sú definované ako veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000
až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov). V rámci každej skupiny frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti. Všetky nežiaduce účinky zistené po uvedení lieku na trh sú napísané kurzívou.
F
rekvencia
Veľmi časté Časté Menej časté Neznáme
T
rieda orgánového
s
y
s
t
é
m
u
Infekcie a nákazy Infekcia dýchacích ciest*
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Chrípke podobné ochorenie
Znížená chuť do jedla Preťaženie tekutinami
Hypersenzitivita
Psychické poruchy Úzkosť
Insomnia
Poruchy nervového systému
Bolesť hlavy
Poruchy srdca Kongestívne zlyhanie srdca
Poruchy ciev Synkopa
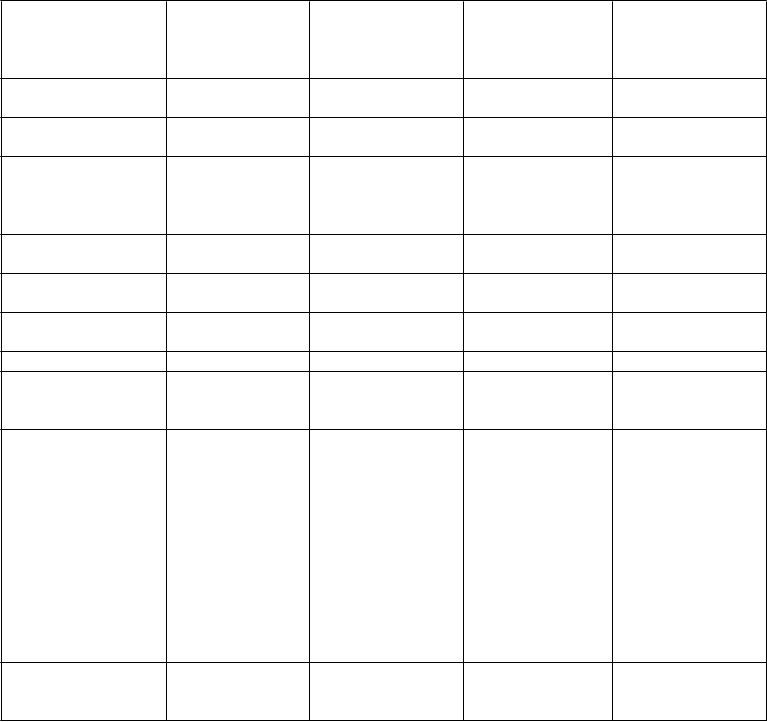
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy
gastrointestinálneho traktu
Poruchy pečene
a žlčových ciest
Nafúknutie brucha Bolesť brucha Nauzea
Vracanie
Kašeľ
Dyspnoe
Kolorektálny polyp Stenóza hrubého čreva
Nadúvanie Nepriechodnosť čriev
Stenóza pankreatického vývodu Pankreatitída† Stenóza tenkého čreva Cholecystitída Akútna cholecystitída
Duodenálny polyp
Polyp žaludku
Celkového poruchy a reakcie v mieste podania
Úrazy, otravy a komplikácie liečebného postupu
Reakcia v mieste podania injekcie ‡
Gastrointestinálne komplikácie stómie
Periférny edém Zadržiavanie tekutín

* Zahŕňa nasledujúce preferované termíny: zápal nosohltana, chrípka, infekcia horných dýchacích ciest a infekcia dolných dýchacích ciest.
† Zahŕňa nasledujúce preferované termíny: pankreatitída,
akútna pankreatitída a chronická pankreatitída.
‡ Zahŕňa nasledujúce preferované termíny: hematóm v mieste podania injekcie, erytém v mieste podania injekcie, bolesť v mieste podania injekcie, opuch v mieste podania injekcie a krvácanie v mieste podania injekcie.'
Popis vybraných nežiaducich reakciíImunogenicitaV súlade s prípadnými imunogénnymi vlastnosťami liekov obsahujúcich peptidy môže podanie
Revestivu potenciálne vyvolať tvorbu protilátok. Na základe integrovaných dát z dvoch skúšaní u dospelých s SBS (6 mesačné randomizované, placebom kontrolované skúšanie, nasledované
24 mesačným otvoreným skúšaním) bol vývoj protilátok proti teduglutidu u jedincov, ktorí dostávali podkožným podaním 0,05 mg / kg teduglutidu raz denne 3 % (2 / 60) v 3. mesiaci, 17 % (13 / 77)
v 6. mesiaci, 24 % (16 / 67) v 12. mesiaci, 33 % (11 / 33) v 24. mesiaci a 48 % (14 / 29) v 30. mesiaci.
V štúdiách fázy 3 s pacientmi s SBS, ktorí dostávali teduglutid ≥ 2 roky vyvinulo 28 % pacientov protilátky proti proteínu
E. coli (zvyškový proteín hostiteľskej bunky z výroby). Tvorba protilátok nesúvisela s klinicky významnými bezpečnostnými zisteniami, zníženou účinnosťou alebo zmenenou farmakokinetikou Revestivu.
Reakcie v mieste podania injekcieU 26 % pacientov s SBS liečených teduglutidom sa objavili reakcie v mieste podania injekcie
v porovnaní s 5 % pacientov v skupine s placebom. Reakcie zahŕňali hematóm v mieste podania injekcie, erytém v mieste podania injekcie, bolesť v mieste podania injekcie, opuch v mieste podania injekcie a krvácanie v mieste podania injekcie (pozri tiež časť 5.3). Väčšina reakcií bola stredne závažná a žiadna z udalostí neviedla k vysadeniu lieku.
C-reaktívny proteínMierne zvýšenie C-reaktívneho proteínu približne o 25 mg/l bolo pozorované počas prvých siedmich dní liečby teduglutidom, ktorý pri podávaní injekcií denne neustále klesal. Po 24 týždňoch liečby
s teduglutidom sa u pacientov ukázal v priemere malý celkový nárast C-reaktívneho proteínu približne o 1,5 mg/l. Tieto zmeny neboli spojené so žiadnymi inými zmenami ostatných laboratórnych
parametrov a ani so žiadnymi hlásenými klinickými príznakmi. Po dlhodobej liečbe teduglutidom po dobu do 30 mesiacov sa nevyskytli žiadne klinicky významné priemerné zvýšené hodnoty C- reaktívneho proteínu oproti východiskovej hodnote.
Pediatrická populáciaV jednej ukončenej klinickej štúdii bolo zahrnutých 37 pediatrických subjektov (vo veku
od 1 do 14 rokov), ktorých liečba teduglutidom trvala 12 týždňov. Žiadny subjekt neprerušil štúdiu v dôsledku nežiaducej udalosti. Vo všeobecnosti bol bezpečnostný profil teduglutidu u detí
a dospievajúcich (vo veku 1-17 rokov) podobný ako u dospelých. Nasledujúce udalosti boli hlásené s
vyššou frekvenciou u detských pacientov v porovnaní s dospelými: únava (veľmi časté), bolestivá defekácia (veľmi časté) a závrat (časté). Avšak databáza údajov o bezpečnosti u detí a dospievajúcich
je obmedzená.
Údaje o dlhodobej bezpečnosti u pediatrickej populácie nie sú doteraz k dispozícii. Pre deti do 1 roku veku nie sú dostupné žiadne údaje.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Maximálna dávka teduglutidu, ktorá sa skúmala počas klinického vývoja bola 86 mg/deň počas 8 dní.
Nepozorovali sa žiadne neočakávané systémové nežiaduce reakcie (pozri časť 4.8).
V prípade predávkovania musí pacienta pozorne sledovať zdravotnícky pracovník.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné liečivá pre tráviaci trakt a metabolizmus, rôzne liečivá tráviaceho
traktu a metabolizmu, ATC kód: A16AX08
Mechanizmus účinku
Prirodzene sa vyskytujúci ľudský peptid podobný glukagónu-2 (GLP- 2) je peptid vylučovaný L
bunkami čreva, ktorý, ako je známe, zvyšuje prietok krvi črevami a portálny prietok krvi, inhibuje sekréciu žalúdočnej kyseliny a znižuje motilitu čriev. Teduglutid je analógom GLP-2. V niekoľkých predklinických štúdiách sa preukázalo, že teduglutid udržiava integritu sliznice podporovaním obnovy a normálneho rastu čreva prostredníctvom zvýšenia výšky klkov a hĺbky črevnej žľazy.
Farmakodynamickéúčinky
Podobne ako GLP-2, teduglutid je tvorený reťazcom 33 aminokyselín, v ktorom je aminokyselina alanín nahradená glycínom v polohe 2 N-konca. Samotná substitúcia aminokyseliny v porovnaní
s prirodzeným GLP-2 vedie k rezistencii voči degradácii enzýmom dipeptidylpeptidázou-IV (DDP-IV)
in vivo, čo má za následok predĺžený polčas. Teduglutid zvyšuje výšku klkov a hĺbku črevnej žľazy epitelu čreva.
Na základe zistení z predklinických štúdií (pozri časti 4.4 a 5.3) a navrhovaného mechanizmu účinku trofických účinkov na črevnú sliznicu sa zdá, že existuje riziko podpory neoplázie tenkého a/alebo hrubého čreva. Vykonané klinické štúdie toto zvýšené riziko nepotvrdili, ani nevyvrátili. Počas skúšaní sa vyskytlo niekoľko prípadov benígnych kolorektálnych polypov, avšak frekvencia nebola vyššia ako u pacientov, ktorí dostávali placebo. Okrem nutnosti vykonania kolonoskopie na odstránenie polypov na začiatku liečby (pozri časť 4.4) musí byť každý pacient posúdený s ohľadom na potrebu zvýšeného dozoru na základe charakteristiky pacienta (napr. vek a základné ochorenie, predchádzajúci výskyt polypov atď.).
Klinickáúčinnosť
Dospelí
Teduglutid sa skúmal u 17 pacientov s SBS, ktorí boli rozdelení do piatich liečebných skupín
používajúcich teduglutid v dávkach 0,03, 0,10 alebo, 0,15 mg/kg raz denne alebo 0,05 alebo
0,075 mg/kg dvakrát denne v 21-dňovej otvorenej, multicentrickej štúdii na stanovenie rozmedzia
dávky. Liečba viedla k zvýšenej absorpcii tekutín v gastrointestinálnom trakte
na približne750 - 1000 ml/deň so zlepšením absorpcie makro živín a elektrolytov, so znížením tekutiny v oblasti stómie alebo tekutiny v stolici a vylučovania makro živín a k zvýšeniu kľúčových štrukturálnych a funkčných adaptácií v sliznici čreva. Štrukturálne adaptácie boli prechodného charakteru a na východiskové hladiny sa vrátili do troch týždňov po ukončení liečby.
V pivotnej dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u pacientov s SBS, ktorí vyžadovali parenterálnu výživu, bolo 43 pacientov randomizovaných na dávku teduglutidu
0,05 mg/kg/deň a 43 pacientov na placebo po dobu až 24 týždňov.
Podiel pacientov liečených teduglutidom, ktorí dosiahli 20 % až 100 % zníženie parenterálnej výživy v 20 a 24 týždni sa štatisticky významne odlišoval od placeba (27 zo 43 pacientov, 62,8 % oproti 13 zo 43 pacientov, 30,2 %, p = 0,002). Liečba teduglutidom viedla v 24 týždni k zníženiu potrieb parenterálnej výživy o 4,4 l/ týždeň (z východiskových 12,9 litrov pred liečbou) oproti 2,3 l/týždeň
(z východiskových 13,2 litra pred liečbou) pri placebe. Dvadsaťjeden (21) pacientov liečených
teduglutidom (48,8 %) oproti 9 u placeba (20,9 %) dosiahlo minimálne jednodňové zníženie v podaní parenterálnej výživy (p = 0,008).
Deväťdesiatsedem percent (97 %) pacientov (37 z 39 pacientov liečených teduglutidom), ktorý ukončili placebom kontrolovanú štúdiu, bolo vybraných, aby pokračovali v dlhodobej rozšírenej štúdii, v ktorej všetci pacienti dostávali 0,05 mg/kg Revestivu denne počas ďalších maximálne
2 rokov. Tejto rozšírenej štúdie sa zúčastnilo celkovo 88 pacientov, z ktorých 39 bolo podávané placebo a 12 bolo zaradených, ale nie randomizovaných, v predchádzajúcej štúdii; 65 z 88 pacientov
absolvovalo rozšírenú štúdiu. V nej sa naďalej preukazovala zvýšená reakcia na liečbu po dobu
2,5 roka vo všetkých skupinách vystavených pôsobeniu teduglutidu, týkajúca sa zníženia objemu parenterálnej výživy, nárastu ďalších dní bez parenterálnej výživy týždenne a dosiahnutia vysadenia
parenterálnej podpory.
Tridsať (30) zo 43 pacientov liečených teduglutidom v hlavnej štúdii, ktorí vstúpili do rozšírenej štúdie, absolvovalo spolu 30 mesiacov liečby. Z nich 28 pacientov (93 %) dosiahlo najmenej 20% zníženie parenterálnej podpory. Z pacientov reagujúcich na liečbu v hlavnej štúdii, ktorí absolvovali rozšírenú štúdiu si 21 z 22 (96 %) udržalo reakciu na teduglutid po dobu ďalších 2 rokoch nepretržitej liečby.
Priemerné zníženie parenterálnej výživy (n=30) bolo 7,55 l/týždeň (65,6% zníženie oproti východiskovej hodnote). U desiatich (10) pacientov sa vysadila parenterálna podpora počas užívania teduglutidu po dobu 30 mesiacov. Jedinci boli udržiavaní na teduglutidu, aj keď sa už nevyžadovala parenterálna výživa. Tých 10 jedincov v minulosti potrebovali podpornú parenterálnu výživu po dobu
1,2 až 15,5 roka a pred liečbou teduglutidom potrebovali 3,5 l//týždeň až 13,4 l/týždeň podpornej parenterálnej výživy. Na konci štúdie 21 (70 %), 18 (60 %) a 18 (60 %) z 30 pacientov, ktorí
absolvovali štúdiu, dosiahlo zníženie o 1, 2 alebo 3 dni parenterálnej podpory za týždeň, v príslušnom poradí.
Z 39 jedincov užívajúcich placebo 29 dokončilo 24 mesiacov liečby teduglutidom. Priemerné zníženie parenterálnej výživy bolo 3,11 l/týždeň (dodatočné 28,3% zníženie). Šestnásť (16, 55,2 %) z 29 pacientov, ktorí absolvovali liečbu, dosiahlo najmenej 20% zníženie parenterálnej výživy. Na konci štúdie 14 (48,3 %), 7 (24,1 %) a 5 (17,2 %) pacientov dosiahlo zníženie parenterálnej výživy o 1, 2 alebo 3 dni týždenne, v príslušnom poradí. U dvoch (2) jedincov bola vysadená parenterálna podpora počas užívania teduglutidu.
Z 12 jedincov, ktorí neboli randomizovaní do hlavnej štúdie, 6 jedincov dokončilo 24 mesiacov liečby teduglutidom. Priemerné zníženie parenterálnej výživy bolo 4,0 l/týždeň (39,4% zníženie oproti východiskovej hodnote – začiatok rozšírenej štúdie) a 4 zo 6 jedincov, ktorí absolvovali liečbu,
(66,7 %) dosiahli najmenej 20% zníženie parenterálnej podpory. Na konci štúdie 3 (50 %), 2 (33 %)
a 2 (33 %) pacienti dosiahli zníženie parenterálnej výživy o 1, 2 alebo 3 dni týždenne, v príslušnom poradí. U jedného jedinca bola vysadená parenterálna podpora počas užívania teduglutidu.
V ďalšej dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 u pacientov s SBS, ktorí vyžadovali parenterálnu výživu, pacienti dostávali dávku 0,05 mg/kg/deň (n=35), dávku 0,10 mg/kg/deň (n=32) teduglutidu alebo placebo (n = 16) až 24 týždňov.
Primárna analýza výsledkov štúdie vzhľadom na účinosť nepreukázala žiadny štatisticky významný rozdiel medzi skupinou na teduglutide 0,10 mg/kg/deň a placebom, zatiaľ čo podiel jedincov, ktorí dostávali odporúčanú dávku teduglutidu 0,05 mg/kg/deň a ktorí dosiahli minimálne 20 % redukciu parenterálnej výživy v 20. a 24. týždni, bol štatisticky signifikantne odlišný oproti placebu (46 % oproti 6,3 %, p < 0,01). Liečba teduglutidom viedla k zníženiu požiadaviek parenterálnej výživy
o 2,5 l/týždeň (z východiskových 9,6 litra pred liečbou) oproti 0,9 l/týždeň (z východiskových
10,7 litra pred liečbou) pri placebe v 24. týždni.
Liečba teduglutidom indukovala expanziu absorpčného epitelu signifikantným zvýšením výšky klkov v tenkom čreve.
Šesťdesiatpäť (65) pacientov vstúpilo do následnej SBS štúdie až na ďalších 28 týždňov liečby. Pacienti používajúci teduglutid si udržali svoje predchádzajúce zaradenie dávky počas predĺženého obdobia, zatiaľ čo pacienti, ktorí dostávali placebo, boli randomizovaní na aktívnu liečbu, buď 0,05 alebo 0,10 mg/kg/deň.
Z pacientov, ktorí dosiahli minimálne 20 % redukciu parenterálnej výživy v 20. a 24. týždni v úvodnej štúdii, si 75 % udržalo túto odpoveď na teduglutid po maximálne 1 rok nepretržitej liečby.
Priemerné zníženie týždenného objemu parenterálnej výživy bolo 4,9 l/týždeň (52% zníženie
z východiskovej hodnoty) po jednom roku nepretržitej liečby teduglutidom.
Dvaja (2) pacienti na odporúčanej dávke teduglutidu boli odstavení z parenterálnej výživy do 24. týždňa. V následnej štúdii bol od parenterálnej výživy odstavený ďalší jeden pacient.
Pediatrická populácia
Teduglutid bol skúmaný v 12-týždňovej otvorenej klinickej štúdii so 42 pediatrickými subjektmi
vo veku od 1 do 14 rokov s SBS, ktorí boli závislí na parenterálnej výžive. Cieľom tejto štúdie bolo zhodnotiť bezpečnosť, znášanlivosť a účinnosť teduglutidu v porovnaní so štandardnou starostlivosťou. Po dobu 12 týždňov sa skúmali tri (3) dávky teduglutidu; 0,0125 mg/kg/deň (n = 8);
0,025 mg/kg/deň (n = 14) a 0,05 mg/kg/deň (n = 15). Päť (5) subjektov bolo zahrnutých do kohorty štandardnej starostlivosti.
Kompletné odstavenie
Trom subjektom (3/15, 20 %) na odporúčanej dávke teduglutidu bola do 12. týždňa vysadená
parenterálna výživa. Po 4-týždňovom vymývacom období bola u dvoch z týchto pacientov znovu
začatá parenterálna nutričná podpora.
Zníženie objemu parenterálnej výživy
V 12. týždni bola v populácii ITT, na základe údajov predpísaných lekárom, zaznamenaná priemerná zmena v objeme parenterálnej výživy −2,57 (± 3,56) l/týždeň oproti východiskovej hodnote, čo
zodpovedá −39,11 % (± 40,79) priemernému poklesu v porovnaní s 0,43 (± 0,75) l/týždeň, čo
zodpovedá 7,38 % (± 12,76) nárastu v kohorte so štandardnou starostlivosťou. V 16. týždni (4 týždne po ukončení liečby) bolo zníženie objemu parenterálnej výživy ešte viditeľné, ale menej, než aké bolo pozorované v 12. týždni, keď sa ešte subjektom podával teduglutid (priemerný pokles o −31,80 %
(± 39,26), v porovnaní s 3,92 % (± 16,62) nárastom v skupine štandardnej starostlivosti.
Zníženie počtu kalórií v parenterálnej výžive
V 12. týždni bola v populácii ITT, na základe údajov predpísaných lekárom, zaznamenaná priemerná zmena spotreby kalórií v parenterálnej výžive −35,11 % (± 53,04) oproti východiskovej hodnote.
Zodpovedajúca zmena v kohorte so štandardnou starostlivosťou bola 4,31 % (± 5,36). V 16. týždni spotreba kalórií v parenterálnej výžive naďalej klesala, a to s priemernou percentuálnou zmenou
–39,15 % (± 39,08) oproti východiskovej hodnote v porovnaní s 0,87 % (± 9,25) v kohorte štandardnej starostlivosti.
Zvýšenie objemu enterálnej výživy
V 12. týždni bola v populácii ITT na základe stanovených údajov zaznamenaná priemerná percentuálna zmena objemu enterálnej výživy oproti východiskovej hodnote 25,82 % (± 41,59) v porovnaní s 53,65 % (± 57,01) v kohorte so štandardnou starostlivosťou. V 16. týždni kohorta
s teduglutidom ako aj kohorta so štandardnou starostlivosťou preukázali nárast objemu enterálnej výživy.
Zvýšenie počtu kalórií v enterálnej výžive
Zvýšenie objemu enterálnej výživy zodpovedalo zvýšeniu počtu enterálnych kalórií, ktoré bolo najvyššie pri podaní odporúčanej dávky. V 12. týždni bol v populácii ITT zaznamenaný percentuálny nárast 58,80 % (± 64,20) oproti východiskovému stavu predpísaných enterálnych kalórií v porovnaní s 57,02 % (± 55,25) v kohorte so štandardnou starostlivosťou. V 16. týždni pokračoval nárast príjmu
kalórií v enterálnej výžive s percentuálnym nárastom oproti východiskovej hodnote 64,57 % (± 57,53)
v porovnaní s 59,63 % (± 52,62) v kohorte so štandardnou starostlivosťou.
Skrátenie doby infúzie
V 12. týždni bol v populácii ITT na základe údajov predpísaných lekárom, zaznamenaný priemerný pokles v počte dní/týždeň na parenterálnej výžive −1,36 (± 2,37) dní/týždeň oproti východiskovej
hodnote, čo zodpovedá percentuálnemu poklesu −24,49 % (± 42,46). V kohorte so štandardnou
starostlivosťou nebola zaznamenaná žiadna zmena oproti východiskovej hodnote. Štyri subjekty
(26,7 %) na odporúčanej dávke teduglutidu dosiahli najmenej tri dni so znížením potreby parenterálnej výživy.
V 12. týždni došlo na základe údajov z denníka subjektu k priemernému percentuálnemu poklesu
o 35,55 % (± 35,23) hodín denne v porovnaní s východiskovou hodnotou, čo zodpovedalo poklesu
−4,18 (± 4,08) v hodinách/deň pri použití parenterálnej výživy, zatiaľ čo subjekty v kohorte so štandardnou starostlivosťou vykázali minimálnu zmenu tohto parametra v rovnakom čase.
V tomto skúšaní neboli pozorované žiadne nové neočakávané signály ohľadom bezpečnosti. Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Revestivom
v jednej alebo vo viacerých vekových podskupinách pediatrickej populácie na liečbu SBS (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absorpcia teduglutidu z miesta podania subkutánnej injekcie bola rýchla s maximálnymi plazmatickými hladinami, ktoré sa objavili približne 3 – 5 hodín po podaní dávky pri všetkých hladinách dávky. Absolútna biologická dostupnosť subkutánneho teduglutidu je vysoká (88 %). Po opakovaných subkutánnych podaniach sa nepozorovala žiadna kumulácia teduglutidu.
Distribúcia
Po subkutánnom podaní je zjavný distribučný objem teduglutidu u pacientov s SBS 26 litrov.
Biotransformácia
Metabolizmus teduglutidu nie je celkom známy. Keďže teduglutid je peptid, je pravdepodobné, že podlieha základnému mechanizmu metabolizmu peptidov.
Eliminácia
Terminálny polčas eliminácie teduglutidu je približne 2 hodiny. Po intravenóznom podaní teduglutidu bol plazmatický klírens približne 127 ml/hod/kg, ktorý je ekvivalentný rýchlosti glumerulárnej filtrácie (GFR). V štúdii skúmajúcej farmakokinetiku u jedincov s poruchou funkcie obličiek sa potvrdila eliminácia obličkami. Po opakovaných subkutánnych podaniach sa nepozorovala žiadna kumulácia teduglutidu.
L
i
nearita dávky
Rýchlosť a rozsah absorpcie teduglutidu sú proporcionálne dávke pri jednorazových a opakovaných subkutánnych dávkach až do 20 mg.
Farmakokinetika v podskupinách pacientov
Pediatrická populácia
Na základe populačného farmakokinetického modelovania bola vo všetkých vekových skupinách po subkutánnom podaní preukázaná podobná Cmax teduglutidu. Avšak u pediatrických pacientov vo veku
1 až 17 rokov bola pozorovaná nižšia expozícia (AUC) a kratší polčas v porovnaní s dospelými. Farmakokinetický profil Revestivu v tejto pediatrickej populácii, ako bolo hodnotené podľa klírensu
a objemu distribúcie, bol po korekcii na telesnú hmotnosť rozdielny od toho, ktorý bol pozorovaný u dospelých. Konkrétne klírens klesá so zvyšujúcim sa vekom od 1 roku až po dospelých.
U pediatrických pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek a v terminálnom
štádiu ochorenia obličiek (end-stage renal disease, ESRD) nie sú k dispozícii žiadne údaje.
Pohlavie
V klinických štúdiách sa nepozorovali žiadne klinicky významné rozdiely týkajúce sa pohlavia.
Starší pacienti
V štúdii fázy 1 sa nezistila žiadna odlišnosť vo farmakokinetike teduglutidu medzi zdravými jedincami
mladšími ako 65 rokov oproti jedincom starším ako 65 rokov. Skúsenosť u jedincov vo veku 75 rokov a starších je obmedzená.
Poruchafunkciepečene
V štúdii fázy 1 sa skúmal vplyv poruchy funkcie pečene na farmakokinetiku teduglutidu po
subkutánnom podaní 20 mg teduglutidu. Maximálna expozícia a celkový rozsah expozície teduglutidu po jednorazových subkutánnych dávkach 20 mg boli nižšie (10-15 %) u jedincov so stredne závažnou poruchou funkcie pečene v porovnaní so zdravými kontrolnými jedincami.
Poruchafunkcieobličiek
V štúdii fázy 1 sa skúmal vplyv poruchy funkcie obličiek na farmakokinetiku teduglutidu po subkutánnom podaní 10 mg teduglutidu. So zvyšujúcim sa stupňom poruchy funkcie obličiek až do (a vrátane) terminálneho štádia ochorenia obličiek sa primárne farmakokinetické parametre teduglutidu zvyšovali až na 2,6-násobok (AUCinf) a 2,1-násobok (Cmax) v porovnaní so zdravými jedincami.
5.3 Predklinické údaje o bezpečnosti
V štúdiách subchronickej a chronickej toxikológie sa pozorovali hyperplázie žlčníka, pečeňového/žlčového vývodu a pakreatického vývodu. Tieto pozorovania pravdepodobne súviseli s očakávaným zamýšľaným farmakologickým účinkom teduglutidu a líšili sa v stupni reverzibility v rámci obdobia 8 - 13 týždňov zotavovania sa po dlhodobom podávaní.
Reakcie v mieste vpichu
V predklinických štúdiách sa zistili závažné granulomatózne zápaly spojené s miestom vpichu.
Karcinogenita/mutagenita
Počas testovania v štandardných sériách testov na genotoxicitu bol teduglutid negatívny.
V štúdii karcinogenity na potkanoch, s liečbou súvisiace benígne novotvary zahŕňali nádory epitelu žlčovodu u samcov vystavených plazmatickým hladinám teduglutidu približne 32 a 155 krát vyšším ako u pacientov, ktorým bola podávaná odporúčaná denná dávka (výskyt 1 zo 44 a 4 zo 48
v príslušnom poradí). Adenómy sliznice lačníka boli pozorované u 1 z 50 samcov a 5 z 50 samcov vystavených plazmatickým hladinám teduglutidu približne 10 a 155 krát (v príslušnom poradí) vyšším ako u pacientov, ktorým bola podávaná odporúčaná dennádávka. Okrem toho bol pozorovaný jejunálny adenokarcinóm u samcov potkanov pri podaní najnižšej testovanej dávky (zviera:človek rozdiel v plazmatickej expozícii približne 10 násobok).
R
eprodukčná
a vývinová toxicita
Štúdie reprodukčnej a vývinovej toxicity hodnotiace teduglutid sa vykonali na potkanoch a králikoch pri dávkach 0, 2, 10 a 50 mg/kg/deň subkutánne. Teduglutid sa nespájal s účinkami na reprodukčný
výkon, na in utero alebo vývinové parametre merané v štúdiách na zistenie fertility, embryofetálneho
vývinu a prenatálneho a postnatálneho vývinu. Farmakokinetické údaje preukázali, že expozícia teduglutidu je u plodov králika a dojčených mláďat potkana veľmi malá.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
L-histidín manitol
nátriumfosfát monohydrát
dinátriumfosfát heptahydrát hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
Rozpúšťadlo
voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorené injekčné liekovky
4 roky.
Rekonštituovaný liek
Chemická a fyzikálna stabilita sa pri použití preukázala počas 3 hodín pri 25 °C.
Z mikrobiologického hľadiska, s výnimkou použitia rekonštitučnej metódy vylučujúcej riziko mikrobiálnej kontaminácie lieku, sa musí roztok použiť okamžite.
Ak sa liek nepoužije okamžite, za čas a podmienky uchovávania po prvom otvorení zodpovedá používateľ. Za normálnych okolností to nebude dlhšie ako 24 hodín pri 2 °C – 8 °C, pokiaľ rekonštitúcia neprebehla v riadených a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte pri teplote do 25 °C. Neuchovávajte v mrazničke.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok
3 ml injekčná liekovka (sklo) s gumovým uzáverom (brómbutyl) s obsahom 5 mg teduglutidu
R
ozpúšťadlo
Naplnená injekčná striekačka (sklo) s piestom (brómbutyl) s obsahom 0,5 ml rozpúšťadla.
Veľkosti balenia: 1 injekčná liekovka s práškom, s 1 naplnenou injekčnou striekačkou alebo
28 injekčných liekoviek s práškom, s 28 naplnenými injekčnými striekačkami.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Určenie počtu injekčných liekoviek potrebných na podanie jednej dávky musí byť založené na individuálnej hmotnosti pacienta a odporúčanej dávke 0,05 mg/kg/deň. Lekár musí pri každej návšteve pacienta odvážiť, stanoviťdennú dávku, ktorá sa má podať do ďalšej kontroly a podľa toho informovať pacienta.
Tabuľky s injekčnými objemami na základe odporúčanej dávky na telesnú hmotnosť pre dospelých aj
pediatrických pacientov sú uvedené v časti 4.2.
Naplnená injekčná striekačka musí byť opatrená ihlou na rekonštitúciu.
Prášok v injekčnej liekovke sa musí potom rozpustiť pridaním všetkého rozpúšťadla z naplnenej
injekčnej striekačky.
Injekčná liekovka sa nesmie pretrepávať, ale môže sa gúlať medzi dlaňami a raz jemne obrátiť hore dnom. Po príprave číreho bezfarebného injekčného roztoku v injekčnej liekovke sa má vzniknutý roztok odobrať do 1-ml injekčnej striekačky (alebo 0,5-ml alebo menšej injekčnej striekačky
pre pediatrické použitie) s dielikmi po 0,02 ml alebo menej (nie je súčasťou balenia).
Ak sú potrebné dve injekčné liekovky, postup sa musí zopakovať pri druhej injekčnej liekovke a ďalší roztok treba natiahnuť do injekčnej striekačky obsahujúcej roztok z prvej injekčnej liekovky. Akýkoľvek objem presahujúci predpísanú dávku v ml treba odstrániť a zlikvidovať.
Roztok sa musí aplikovať subkutánne do vyčistenej oblasti na bruchu, alebo ak to nie je možné do
stehna (pozri časť 4.2 Spôsob podávania) pomocou tenkej ihly na subkutánnu injekciu.
Podrobné pokyny na prípravu a injekciu Revestivu sú uvedené v písomnej informácii pre používateľa.
Roztok sa nesmie použiť ak je zakalený alebo obsahuje častice.
Len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Všetky ihly a injekčné striekačky majú byť zlikvidované v kontajneri určenom na likvidáciu ostrých
predmetov.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Shire Pharmaceuticals Ireland Limited
5 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
Tel.: +800 6774 4357
8. REGISTRAČNÉ ČÍSLO/ČÍSLA
EU/1/12/787/001
EU/1/12/787/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 30. augusta 2012
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.