
túdii simulovaného vedenia vozidla
u zdravých subjektov lasmiditan významne narúšal schopnosť viesť vozidlo (pozri časť 4.7). Je
potrebné pacientov upozorniť, aby neviedli vozidlo ani nevykonávali iné činnosti vyžadujúce zvýšenú pozornosť najmenej 8 hodín po užití každej dávky lasmiditanu, aj keď sa na to dostatočne dobre cítia.
Pacienti, ktorí nedodržiavajú tieto usmernenia, nesmú užívať lasmiditan.
Sérotonínový syndróm
Sérotonínový syndróm bol hlásený a môže sa vyskytovať pri užívaní lasmiditanu alebo ak sa podáva
s ďalšími sérotonergickými liekmi [napr. selektívnymi inhibítormi vychytávania sérotonínu (selective serotonin reuptake inhibitors, SSRI), inhibítormi spätného vychytávania sérotonínu a norepinefrínu (serotonin norepinephrine reuptake inhibitors, SNRI), tricyklickými antidepresívami (tricyclic antidepressants, TCA) a inhibítormi monoaminooxidázy (IMAO)]. Klinické skúsenosti s používaním lasmiditanu a triptánov v blízkej časovej nadväznosti sú obmedzené. Riziká vzniku sérotonínového syndrómu môžu byť aditívne. Symptómy sérotonínového syndrómu môžu zahŕňať zmeny duševného stavu (napr. nepokoj, halucinácie, kómu), autonómnu nestabilitu (napr. tachykardiu, labilný krvný tlak, hypertermiu), neuromuskulárne prejavy (napr. hyperreflexiu, poruchu koordinácie), a/alebo gastrointestinálne prejavy a príznaky (napr. nauzeu, vracanie, hnačku). Tieto reakcie môžu byť závažné. Príznaky sa obvykle začnú objavovať v priebehu minút až hodín po podaní novej alebo
vyššej dávky sérotonergických liekov. Ak je súbežná liečba inými sérotonergickými liekmi klinicky odôvodnená, odporúča sa vhodné sledovanie pacienta, najmä na začiatku liečby a pri zvyšovaní dávky. Pri podozrení na sérotonínový syndróm sa má liečba lasmiditanom ukončiť.
Látky tlmiace CNS
Vzhľadom na sedatívny potenciál lasmiditanu, ako aj jeho potenciál vyvolávať kognitívne a/alebo
neuropsychiatrické nežiaduce reakcie, v kombinácii s alkoholom alebo inými látkami tlmiacimi CNS
sa má lasmiditan používať opatrne.
Možnosti nesprávneho používania alebo zneužívanialiekov
V štúdii vykonanej u ľudí zameranej na potenciál zneužívania liekov rekreačnými užívateľmi drog sa
jednorazové 100 alebo 200 mg dávky lasmiditanu spájali s vyššou obľubou užívania ako placebo.
V inej štúdii sa nevyskytol žiadny dôkaz o fyzických abstinenčných príznakoch u zdravých jedincov po náhlom prerušení po 7 dňoch podávania. U pacientov sa má vyhodnotiť riziko zneužívania liekov a majú byť sledovaní na prejavy nesprávneho užívania alebo zneužívania lasmiditanu.
Bolesť hlavyznadmernéhoužívanialiekov(MOH- medicationoveruseheadache)
Nadmerné užívanie akéhokoľvek typu liekov na bolesti hlavy môže tieto bolesti zhoršiť. Ak takáto
situácia nastane alebo sa vyskytne podozrenie, je potrebné vyhľadať lekársku pomoc a liečba sa má ukončiť. Diagnóza MOH sa má zvážiť u pacientov, ktorí majú časté alebo každodenné bolesti hlavy napriek (alebo kvôli) pravidelnému užívaniu liekov na bolesť hlavy.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej tablete, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Lieky znižujúce srdcovú frekvenciu
Lasmiditan sa spája so znížením srdcovej frekvencie (SF). Propranolol a lasmiditan spolu znížili SF
o priemerné maximum 19,3 úderov za minútu (t.j. ďalšie zníženie o 5,1 úderov za minútu v porovnaní s propranololom samotným). Toto je potrebné vziať do úvahy u pacientov, u ktorých môže tento rozsah poklesu SF vyvolávať obavy, vrátane pacientov užívajúcich lieky znižujúce srdcovú frekvenciu.
Sérotonergické lieky
Súbežné podávanie lasmiditanu a liekov (napr. SSRI, SNRI, TCA), ktoré zvyšujú hladinu sérotonínu,
môže zvyšovať riziko sérotonínového syndrómu. Klinické skúsenosti s používaním lasmiditanu
a triptánov v blízkej časovej nadväznosti sú obmedzené. Riziká vzniku sérotonínového syndrómu môžu byť aditívne. Odporúča sa opatrnosť (pozri časť 4.4).
Možný vplyv lasmiditanunainélieky
Každodenné podávanie lasmiditanu nemenilo PK midazolamu, kofeínu ani tolbutamidu, čo sú
substráty CYP3A, CYP1A2 a CYP2C9, v uvedenom poradí. Súbežné podávanie lasmiditanu
so sumatriptánom (substrát IMAO-A a OCT1) alebo s propranololom (substrát CYP2D6) neviedlo
k žiadnym klinicky významným zmenám v expozícii týmto liekom. Po jednorazovej dávke lasmiditanu sa renálny klírens kreatinínu v priebehu 24 hodín v porovnaní s placebom mierne znížil (11 %), bezo zmien v GFR.
Možný vplyv inýchliekovnalasmiditan
Pri súbežnom podávaní lasmiditanu so sumatriptánom alebo propranololom nebola pozorovaná žiadna
zmena PK lasmiditanu. Na základe jeho dráh metabolického klírensu je nepravdepodobné, že by inhibítory alebo induktory CYP mali vplyv na expozíciu lasmiditanu. Pri súbežnom podávaní
s topiramátom (induktor CYP3A4 a inhibítor CYP2C19) nebola zaznamenaná žiadna zmena v PK
lasmiditanu.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii je len obmedzené množstvo údajov o použití lasmiditanu u gravidných žien. Štúdie na
zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Účinky lasmiditanu na vývoj ľudského plodu nie sú známe. RAYVOW sa neodporúča užívať počas gravidity.
Dojčenie
Lasmiditan a/alebo jeho metabolity sa vylučovali do mlieka laktujúcich potkanov (časť 5.3).
K dispozícii nie sú žiadne údaje o prítomnosti lasmiditanu v ľudskom materskom mlieku, o účinkoch lasmiditanu na dojčené dieťa ani o vplyve lasmiditanu na produkciu mlieka.
Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu RAYVOWOM sa má urobiť
po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu. Expozíciu novorodenca môžeme
minimalizovať zamedzením dojčenia počas 24 hodín od liečby.
Fertilita
Nie je známe, či má lasmiditan vplyv na ľudský reprodukčný potenciál. Štúdie na zvieratách
nepreukázali žiaden vplyv na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Lasmiditan má veľký vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Schopnosť viesť vozidlo sa hodnotila pomocou počítačovej simulácie jazdy. Primárnym meraným ukazovateľom bol rozdiel oproti placebu v štandardnej odchýlke laterálnej polohy (SDLP - Standard Deviation of Lateral Position), ukazovateľ schopnosti viesť vozidlo. Podanie jednej 50 mg, 100 mg alebo 200 mg dávky lasmiditanu významne narušilo schopnosť osôb viesť vozidlo 90 minút od podania dávky. V ďalšej štúdii so 100 mg alebo 200 mg lasmiditanu nedosiahla schopnosť viesť vozidlo prah poruchy vedenia vozidla po 8 hodinách alebo neskôr od podania ktorejkoľvek dávky RAYVOWU.
Pacientov je potrebné upozorniť, aby sa najmenej 8 hodín po užití každej dávky lasmiditanu nezúčastňovali aktivít vyžadujúcich zvýšenú pozornosť, ako napríklad obsluhy strojov alebo vedenia vozidla, dokonca ani vtedy, ak sa na to dostatočne dobre cítia. Pacienti, ktorí nedodržiavajú tieto usmernenia, nesmú užívať lasmiditan (pozri časť 4.4).
4.8 Nežiaduce účinky
Z
hrnutie bezpečnostného profiluNajčastejšie sa vyskytujúcimi nežiaducimi reakciami sú závrat (19,9 %), somnolencia (7,8 %), únava
(7,7 %), parestézia (6,8 %), nauzea (4,9 %), vertigo (2,6 %), hypestézia (2,5 %), a svalová slabosť
(2,3 %). Väčšina nežiaducich účinkov vykazovala závislosť na dávke.
Zoznam nežiaducich reakcií v tabuľkeV tejto tabuľke sú nežiaduce reakcie zoradené podľa triedy orgánových systémov MedDRA
a frekvencie výskytu. V rámci každej skupiny frekvencií sú nežiaduce reakcie zoradené podľa klesajúcej závažnosti. Stupne frekvencie sú nasledovné: veľmi časté (≥ 1/10), časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000).
Trieda orgánových systémov
| Veľmi časté
| Časté
| Menej časté
| Zriedkavé
|
Poruchy
imunitného systému
|
|
| precitlivenosť
|
|
Psychické poruchy
|
| spánkové abnormality
| zmätenosť, halucinácie, euforická nálada, úzkosť,
nepokoj
|
|
Poruchy nervového systému
| závrat
| porucha koordinácie,
parestézia, hypestézia, somnolencia
| letargia, porucha
pozornosti, kognitívna porucha,
duševná porucha, triaška,
rečové abnormality
| sérotonínový syndróm
|
Poruchy oka
|
| porucha zraku
|
|
|
Poruchy ucha a
labyrintu
|
| vertigo
|
|
|
Poruchy srdca
a srdcovej činnosti
|
| palpitácie
|
|
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
|
| dýchavičnosť
|
|
Poruchy gastrointestinálneho traktu
|
| vracanie, nauzea
|
|
|
Poruchy kostrovej
a svalovej sústavy
a spojivového
tkaniva
|
| svalová slabosť
| svalový kŕč, diskomfort
končatín
|
|
Celkové poruchy a reakcie v mieste
podania
|
| neobvyklý pocit, únava,
malátnosť
| diskomfort na hrudníku,
pocit horúčavy alebo pocit chladu
|
|
O
pis vybraných nežiaducichreakciíZníženie srdcovej frekvencieV klinicko-farmakologických štúdiách sa lasmiditan spájal so znížením srdcovej frekvencie o 5 až
10 úderov za minútu (beats per minute, bpm) v porovnaní so znížením o 2-5 bpm pri placebe. Výskyt bradykardie (< 50 bpm a pokles oproti vstupným hodnotám ≥ 15 bpm) pozorovaný u pacientov liečených lasmiditanom bol 7 % u 50 mg, 3 % u 100 mg, 4 % u 200 mg a 1 % u placeba.
Zvýšenie krvného tlakuPodanie jednej dávky lasmiditanu môže viesť k prechodnému zvýšeniu krvného tlaku. Jednu hodinu od podania 200 mg lasmiditanu bolo u zdravých dobrovoľníkov (nie starších) pozorované priemerné zvýšenie ambulantného systolického a diastolického tlaku oproti vstupným hodnotám o približne
2 - 3 mm Hg, v porovnaní so zvýšením o približne 1 mm Hg u placeba. U zdravých dobrovoľníkov vo veku vyššom ako 65 rokov bolo jednu hodinu od podania 200 mg lasmiditanu priemerné zvýšenie ambulantného systolického krvného tlaku oproti vstupným hodnotám 7 mm Hg, v porovnaní
s priemerným zvýšením o 4 mm Hg u placeba. Do 2 hodín po užití lasmiditanu už nebolo žiadne zvýšenie priemerného krvného tlaku v porovnaní s placebom. Klinické údaje o užívaní lasmiditanu u pacientov s ischemickou chorobou srdca sú obmedzené.
PrecitlivenosťU pacientov liečených lasmiditanom sa vyskytli prípady hypersenzitivity vrátane angioedému, vyrážky a fotosenzitívnej reakcie. V klinických štúdiách bola precitlivenosť zaznamenaná u 0,1 %
pacientov liečených lasmiditanom oproti žiadnym pacientom v skupine s placebom. Všetky prípady
boli mierne až stredne závažné a vyskytovali sa v priebehu minút až jedného dňa od podania dávky lasmiditanu. Ak sa vyskytne vážna alebo závažná reakcia z precitlivenosti, musí sa začať s vhodnou liečbou a podávanie lasmiditanu sa má ukončiť.
ZávratV klinických skúšaniach bol závrat najčastejšou nežiaducou reakciou, hlásenou u 19,9 % pacientov. Závažnosť bola spravidla mierna až stredne závažná (silné závraty 1,2 %) a sama ustupovala,
so strednou hodnotou času do nástupu 0,7 hodiny a strednou hodnotou trvania 2 hodiny. U pacientov, ktorí hlásili závrat, neboli hlásené žiadne nehody ani zranenia. Frekvencia hlásenia závratov a iných častých nežiaducich účinkov pacientmi sa obvykle pri opakovanom podávaní znižuje.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieS predávkovaním lasmiditanom sú z klinických skúšaní len obmedzené skúsenosti. V prípadoch hlásených ako predávkovanie sa vyskytli podobné nežiaduce účinky ako pri nižších dávkach, vrátane závratov, somnolencie, únavy, parestézie a hypestézie, ale nespájali sa so zvýšením závažnosti ani frekvencie výskytu. Pretože sa však v prípade predávkovania môžu vyskytovať nežiaduce reakcie, pacienti majú byť sledovaní na akékoľvek prejavy alebo príznaky nežiaducich reakcií a má sa začať
s vhodnou symptomatickou liečbou. Nie je známe žiadne antidotum pri predávkovaní lasmiditanom.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: analgetiká, antimigreniká, ATC kód: N02CC08
Mechanizmus účinku
Lasmiditan je vysoko afinitný, centrálne penetrujúci agonista receptora 5-hydroxytriptamínu 1F
(5-HT1F). Presný mechanizmus účinku nie je známy, terapeutické účinky lasmiditanu v liečbe migrény však pravdepodobne zahŕňajú agonistické účinky na receptor 5-HT1F, pokles uvoľňovania neuropeptidov a inhibíciu dráh bolesti, vrátane trojklaného nervu.
Farmakodynamické účinky
Vo väzbových štúdiách in-vitro lasmiditan vykazoval > 440-násobnú selektivitu pre receptor 5-HT1F
oproti receptorom 5-HT1B a 5-HT1D. Lasmiditan nezužuje ex-vivo ľudské koronárne artérie, ex-vivo ľudské vnútorné prsné artérie ani ex-vivo ľudské stredné meningeálne artérie, pravdepodobne kvôli svojej nízkej afinite k vazokonstrikčnému receptoru 5-HT1B.
Elektrofyziológia srdca
V dôkladnej štúdii QT sa lasmiditan spájal s poklesom srdcovej frekvencie o 6 bpm v porovnaní
s placebom a podávanie supraterapeutickej dávky 400 mg naznačovalo predĺženie QTc u žien. Analýzy podskupín naznačovali rozdiely súvisiace s pohlavím, pretože v podskupine žien bolo
pozorované významnejšie predĺženie QTc. Keďže je ale najvyššia odporúčaná dávka obmedzená
na 200 mg, neočakáva sa žiadny klinicky významný účinok.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť lasmiditanu sa skúmala v troch randomizovaných, placebom kontrolovaných,
dvojito zaslepených štúdiách fázy 3 u dospelých pacientov (N = 5910). Do štúdií boli zaradení pacienti vo veku 18 rokov a starší s 3 - 8 záchvatmi migrény za mesiac a s aspoň stredne ťažkým postihnutím vplyvom migrény (skóre hodnotenia migrénového postihnutia - Migraine Disability Assessment (MIDAS) ≥ 11).
Štúdie skúmajúce jeden záchvat
V skupine pacientov zaradenej do štúdií s jedným záchvatom (SAMURAI a SPARTAN) boli najmä ženy (84 %) v priemernom veku 42,3 rokov. Tri mesiace pred zaradením do štúdie mali pacienti
v priemere 5,2 záchvatov migrény za mesiac a priemerné celkové skóre MIDAS 31,7. Zo štúdie SAMURAI, nie však zo štúdie SPARTAN, boli vylúčení pacienti so známym ochorením koronárnych artérií, klinicky významnou arytmiou či nekontrolovanou hypertenziou. 78,3 % pacientov malo okrem migrény ešte aj ≥ 1 kardiovaskulárny rizikový faktor vrátane veku > 40 (54,2 %), nízkeho
HDL-cholesterolu (31,1 %), vysokého krvného tlaku/hypertenzie (21,3 %), aktuálneho fajčenia
(14,3 %), vysokého celkového cholesterolu (10,9 %) a anamnézy diabetu (5,9 %). 21,7 % pacientom boli predpísané preventívne lieky na migrénu a 37 % užívalo počas 3 mesiacov pred zaradením do štúdie triptán. Najnepríjemnejším príznakom (MBS - most bothersome symptom) bola fotofóbia (50,3 %), po nej nasledovala nauzea (22,2 %) a fonofóbia (20,6 %). V týchto štúdiách bola povolená druhá dávka skúšaného alebo iného lieku za 2 až 24 hodín po počiatočnej liečbe pri pretrvávajúcej alebo opakovanej migréne.
Primárnym a kľúčovým sekundárnym koncovým ukazovateľom v oboch štúdiách bolo percento pacientov bez bolesti a percento pacientov bez MBS 2 hodiny po liečbe, v porovnaní s placebom.
Obe štúdie splnili primárne aj kľúčové sekundárne ukazovatele. Všetky dávky lasmiditanu vykázali štatisticky signifikantné a klinicky významné zlepšenie v percentuálnom podiele pacientov dosahujúcich stav bez bolesti, stav bez MBS a úľavu od bolesti (definovanú ako zníženie závažnosti bolesti zo stredne závažnej alebo závažnej na začiatku liečby na miernu až žiadnu alebo z miernej na žiadnu) 2 hodiny po liečbe (pozri tabuľku č. 1). Čas nástupu stavu bez bolesti je uvedený na obrázku č. 1. Čas nástupu úľavy od bolesti mal u 50 mg a 100 mg rovnaký priebeh ako stav bez bolesti, kým odlišný priebeh v porovnaní s placebom bol u 200 mg dávky pozorovaný skôr, po 30 minútach
(17,7 % u 200 mg oproti 11,6 % u placeba, p = 0,004 v štúdii SAMURAI, 18,6 % u 200 mg oproti
14,7 % u placeba, p = 0,014 v štúdii SPARTAN).
Tabuľka č. 1. Štúdie SAMURAI a SPARTAN: Zhrnutie údajov o účinnosti
| SAMURAI
lasmiditan
| SPARTAN
lasmiditan
|
100 mg 200 mg
| Placebo
| 50 mg 100 mg 200 mg
| Placebo
|
Bez bolesti po 2 hodinách
|
N
| 503
| 518
| 524
| 556
| 532
| 528
| 540
|
Respondéri
(%)
| 28,2
| 32,2
| 15,3
| 28,6
| 31,4
| 38,8
| 21,3
|
p-hodnota
| < 0,001
| < 0,001
|
| 0,006
| < 0,001
| < 0,001
|
|
Bez MBS po 2 hodinách
|
N
| 469
| 481
| 488
| 512
| 500
| 483
| 514
|
Respondéri
(%)
| 40,9
| 40,7
| 29,5
| 40,8
| 44,2
| 48,7
| 33,5
|
p-hodnota
| < 0,001
| < 0,001
|
| 0,018
| < 0,001
| < 0,001
|
|
Úľava od bolesti po 2 hodinách
|
N
| 562
| 555
| 554
| 598
| 571
| 565
| 576
|
Respondéri
(%)
|
54,1
|
54,6
|
39,2
|
55,5
|
59,7
|
60,7
| 44,9
|
p-hodnota
| < 0,001
| < 0,001
|
| < 0,001
| < 0,001
| < 0,001
|
|
Obrázok č. 1. Percento pacientov dosahujúcich stav bez migrénovej bolesti do 2 hodín v štúdiáchSAMURAI a SPARTAN.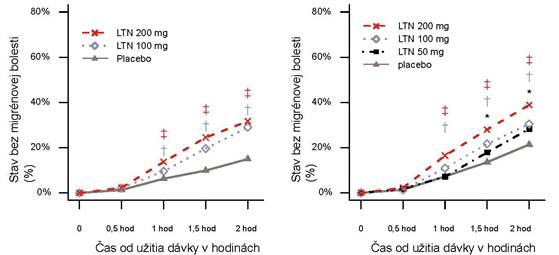
SAMURAI SPARTAN
‡ Štatistický význam u 200 mg LTN oproti placebu;
† Štatistický význam u 100 mg LTN oproti placebu:
* Štatistický význam u 50 mg LTN oproti placebu
Skratky: LTN = lasmiditan
Štúdia konzistentnosti účinkuV štúdii hodnotiacej konzistentnosť účinku boli pacienti liečení lasmiditanom 100 mg, 200 mg alebo kontrolnou liečbou počas 4 záchvatov migrény (štúdia CENTURION). V kontrolnej skupine pacienti dostávali jednu dávku lasmiditanu 50 mg na liečbu ich tretieho alebo štvrtého záchvatu a placebo na
ostatné záchvaty. V populácii zaradenej do štúdie prevažovali ženy (84 %) v priemernom veku
41,4 rokov. Tri mesiace pred zaradením do štúdie mali pacienti priemerne 4,9 záchvatov migrény za mesiac a priemerné celkové skóre MIDAS 31,9. Zo štúdie neboli vylúčení pacienti
s kardiovaskulárnymi ochoreniami a 58,5 % pacientov malo okrem migrény ≥ 1 faktor kardiovaskulárneho rizika vrátane veku > 40 rokov
(52,8 %), vysokého celkového cholesterolu
(10,8 %), vysokého krvného tlaku/hypertenzie (16,9 %) a anamnézy diabetu (3,1 %). 28,8 % pacientov
malo aktuálne predpísané preventívne lieky na migrénu a 65,0 % predtým užívalo triptán. MBS bola fotofóbia (39,7 %), po nej nasledovala nauzea (31,9 %) a fonofóbia (19,3 %).
Koprimárnymi koncovými ukazovateľmi bolo percento pacientov, ktorí boli 2 hodiny po podaní dávky bez bolesti po prvom záchvate a pacienti, ktorí boli bez bolesti aspoň pri 2 z 3 záchvatov, v porovnaní
s placebom.
Štúdia splnila svoj primárny a všetky kľúčové sekundárne koncové ukazovatele. Obe dávky, 100 mg a 200 mg lasmiditanu, preukázali štatisticky signifikantné a klinicky významné zlepšenie percenta pacientov dosahujúcich 2 hodiny od liečby stav bez bolesti, úľavu od bolesti (zníženie závažnosti bolesti zo stredne závažnej alebo závažnej na začiatku liečby na miernu až žiadnu alebo z miernej na žiadnu), stav bez MBS a udržaný stav bez bolesti po 24 hodinách (pozri tabuľku č. 2). Čas nástupu stavu bez bolesti je uvedený na obrázku č. 2. Úľava od bolesti mala u 50 mg a 100 mg rovnaký priebeh ako stav bez bolesti a u dávky 200 mg bola pozorovaná skôr, po 30 minútach (22,4 % u
200 mg oproti 14,0 % u placeba, p = 0,001).
Obe dávky preukázali konzistentnosť účinku so štatisticky signifikantným a klinicky významným zlepšením percenta pacientov dosahujúcich stav bez bolesti a úľavu od bolesti aspoň pri 2 z 3 záchvatov (pozri tabuľku č. 2).
Tabuľka č. 2. Štúdia CENTURION: Zhrnutie údajov o účinnosti
| lasmiditan
|
100 mg 200 mg
| Placebo
|
Koncové ukazovatele jedného záchvatu (ITT) N = 419 N = 434 N = 443
|
Stav bez bolesti po 2 hodinách po podaní dávky počas prvého záchvatu
|
Respondéri (%)
| 25,8
| 29,3
| 8,4
|
p-hodnota oproti placebu
| < 0,001
| < 0,001
| '
|
Úľava od bolesti po 2 hodinách po podaní dávky
počas prvého záchvatu
|
Respondéri (%)
| 65,4
| 65,2
| 41,3
|
p-hodnota oproti placebu
| < 0,001
| < 0,001
|
|
Udržaný stav bez bolesti do 24 hodín po podaní dávky počas prvého záchvatu
|
Respondéri (%)
| 13,6
| 17,3
| 4,3
|
p-hodnota oproti placebu
| < 0,001
| < 0,001
|
|
Stav bez MBS po 2 hodinách po podaní dávky počas
prvého záchvatu N = 376 N = 395 N = 396
|
Respondéri (%)
| 40,4
| 39,0
| 28,0
|
p-hodnota oproti placebu
| < 0,001
| 0,001
|
|
Koncové ukazovatele konzistentnosti (ITT
konzistentnosť)
|
Stav bez bolesti po 2 hodinách po podaní dávky N = 340 N = 336 N = 373
najmenej pri 2 z 3 záchvatov
|
Respondéri (%)
| 14,4
| 24,4
| 4,3
|
p-hodnota oproti placebu
| < 0,001
| < 0,001
|
|
Úľava od bolesti po 2 hodinách po podaní dávky N = 332 N = 333 N = 320
najmenej pri 2 z 3 záchvatov
|
|
l
asmiditan
|
100 mg 200 mg
|
Placebo
|
Respondéri (%)
|
62,3
|
66,7
|
36,9
|
p-hodnota oproti placebu
|
< 0,001
|
< 0,001
|
|
O
brázok č. 2. Percento pacientov dosahujúcich stav bez migrénovej bolesti do 2 hodín v štúdii
CENTUR
ION.

‡ Štatistický význam u 200 mg LTN oproti placebu; † Štatistický význam u 100 mg LTN oproti
placebu
Skratky: LTN = lasmiditan
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s RAYVOWOM
v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe migrénových bolestí hlavy
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo perorálnom podaní sa lasmiditan rýchlo absorbuje so strednou hodnotou tmax 1,8 hodiny.
U pacientov s migrénou nebola farmakokinetika lasmiditanu počas záchvatu migrény iná ako počas
interiktálneho obdobia. V rozmedzí klinických dávok 50 až 200 mg sa na základe výsledkov
z populačnej PK analýzy predpokladá, že absolútna biologická dostupnosť bude 50 % až 58 %. Súbežné podávanie lasmiditanu s jedlom s vysokým obsahom tuku zvýšilo priemerné hodnoty
lasmiditanu Cmax o 22 % a AUC o 19 % a oddialilo strednú hodnotu tmax o 1 hodinu. Neočakáva sa, že by tento rozdiel v expozícii bol klinicky významný. V štúdiách klinickej účinnosti bol lasmiditan
podávaný bez ohľadu na jedlo.
DistribúciaVäzba lasmiditanu na ľudské plazmatické proteíny je približne 55 % až 60 % a je nezávislá od
koncentrácie od 15 do 500 ng/ml. Odhadovaný priemerný distribučný objem bol 304 l.
B
i
otransformácia
Lasmiditan podlieha hepatálnemu a extrahepatálnemu metabolizmu primárne prostredníctvom
enzýmov iných ako CYP, pričom hlavnou dráhou je redukcia ketónov na S-M8. Na metabolizme lasmiditanu sa nezúčastňovali tieto enzýmy: MAO-A, MAO-B, flavínmonooxygenáza 3,
CYP450-reduktáza, xantínoxidáza, alkoholdehydrogenáza, aldehyddehydrogenáza a aldo-ketoreduktázy.
Lasmiditan sa tiež oxiduje v piperidínovom kruhu na M7. V porovnaní s lasmiditanom sú metabolity farmakologicky neaktívne. Lasmiditan je substrát P-gp in-vitro.
Lasmiditan a jeho hlavné metabolity sú in-vitro induktory enzýmov CYP. Lasmiditan in-vitro inhibuje CYP2D6. Lasmiditan a jeho hlavný metabolit nie sú inhibítormi MAO-A. Lasmiditan in-vitro inhibuje efluxné transportéry P-gp, BCRP a OCT1. Lasmiditan in-vitro inhibuje renálne transportéry OCT2, MATE1 a MATE2-K.
Eliminácia
Lasmiditan bol eliminovaný s geometrickou priemernou hodnotou t½ približne 5,7 hodín. Pri
každodennom podávaní nebola zaznamenaná žiadna akumulácia lasmiditanu. Odhadovaný priemerný
celkový telesný klírens bol 66,2 l/h. Lasmiditan obvykle vykazuje lineárnu PK v rozmedzí klinických dávok 50 až 200 mg. Lasmiditan sa primárne eliminuje prostredníctvom metabolizmu. Renálne vylučovanie je vedľajšou dráhou klírensu lasmiditanu s približne 3 % dávky, ktorá sa vylúči v moči ako nezmenený lasmiditan. Metabolit S-M8 predstavoval približne 66 % dávky v moči, pričom väčšina sa vylúčila do 48 hodín od podania dávky.
Osobitné skupiny pacientov
Vek, pohlavie, rasa, etnická príslušnosť a telesná hmotnosť
Vek, pohlavie, rasa, etnická príslušnosť ani telesná hmotnosť nemali významný účinok na expozíciu v populačnej farmakokinetickej analýze lasmiditanu. V jednej štúdii malo pohlavie vplyv na PK
lasmiditanu s vyšším Cmax (~ 20 - 30 %) a AUC (~ 30 %) u žien v porovnaní s mužmi, bez ohľadu na to, či bol lasmiditan podávaný s jedlom alebo nalačno. Úprava dávky podľa veku, pohlavia, rasy,
etnickej príslušnosti ani hmotnosti nie je potrebná.
Porucha funkcie obličiek
Podanie lasmiditanu osobám so závažnou poruchou funkcie obličiek (eGFR <30 ml/min/1,73 m2)
preukázalo o 18 % vyššiu expozíciu v AUC(0-∞) a o 13 % vyššiu hodnotu Cmax, oproti osobám
s normálnou funkciou obličiek. Neočakáva sa, že by tento rozdiel v expozícii bol klinicky významný.
U pacientov s miernou, stredne závažnou a závažnou poruchou funkcie obličiek nie je potrebná žiadna úprava dávky.
Porucha funkcie pečene
U osôb s miernou a stredne závažnou poruchou funkcie pečene (Child-Pughova trieda A a B,
v uvedenom poradí) bola expozícia lasmiditanu 11 % a 35 %, v uvedenom poradí, vyššia [AUC(0-∞)] ako expozícia u osôb s normálnou funkciou pečene. Cmax bola u osôb s miernou poruchou funkcie pečene vyššia o 19 % a u osôb so stredne závažnou poruchou funkciou pečene bola vyššia o 33 %. Neočakáva sa, že by tento rozdiel v expozícii bol klinicky významný. U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. Použitie lasmiditanu u osôb so závažnou poruchou funkcie pečene sa neskúmalo, a preto sa pre túto skupinu pacientov neodporúča.
5.3 Predklinické údaje o bezpečnosti
Karcinogenita bola hodnotená v dvojročnej štúdii na potkanoch a šesťmesačnej štúdii na transgénnych myšiach. U potkanov sa pozoroval zvýšený počet úmrtí súvisiacich s nádorom hypofýzy u samcov.
Význam týchto zistení z hľadiska rizika u ľudí nie je známy. U myší sa nepozorovali žiadne dôkazy karcinogenity.
Lasmiditan nebol genotoxický na základe výsledkov Amesovho testu na baktériách, štúdie chromozómových aberácií na ovariálnych bunkách čínskeho škrečka ani v mikronukleových testoch na myšiach.
Vývojová a reprodukčnátoxicita
V štúdiách na potkanoch sa neprejavili žiadne účinky na samčiu alebo samičiu fertilitu.
V štúdiách embryofetálneho vývoja na potkanoch a králikoch došlo k zníženiu telesnej hmotnosti plodu a kostrovým zmenám. U králikov došlo k miernemu zvýšeniu post-implantačných strát (embryofetálnej mortality) a bol pozorovaný nízky výskyt kardiovaskulárnych defektov plodu (malformácií). Expozícia pri dávkach bez pozorovaných nežiaducich účinkov 175 mg/kg/denne (potkany) a 75 mg/kg/denne (králiky) bola približne 37 a 1,5-násobne (v uvedenom poradí) vyššia než expozícia u ľudí pri dávke 200 mg.
V pred- a postnatálnej štúdii na potkanoch sa pri najvyššej skúšanej dávke 225 mg/kg/denne vyskytla predĺžená gestácia a pôrod, zvýšený počet mŕtvo narodených mláďat a zvýšená frekvencia postnatálneho úmrtia. Pri tejto vysokej expozícii sa pokles priemernej telesnej hmotnosti mláďat F1 pozorovaný v priebehu fázy pred odstavením u oboch pohlaví udržal aj počas fázy dozrievania F1 bez normalizácie. Odhaduje sa, že pri dávke 150 mg/kg/denne bez pozorovaných nežiaducich účinkov bola expozícia > 19-násobne vyššia ako u ľudí pri dávke 200 mg.
Všetky účinky sa vyskytli pri expozíciách toxických pre matku, ktoré prekročili expozíciu u ľudí
pri klinickej dávke 200 mg.
Štúdie na zvieratách preukázali, že lasmiditan a/alebo jeho metabolity sa vylučovali do mlieka laktujúcich potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
mikrokryštalická celulóza
sodná soľ kroskarmelózy stearát horečnatý
predželatínovaný škrob
laurylsíran sodný
Obal tablety (50 mg a 200 mg)
polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec
čierny oxid železitý (E172)
Obal tablety (100 mg)
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastenec
čierny oxid železitý (E172)
červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Polychlórtrifluóretylénové/polyvinylchloridové (PCTFE/PVC) perforované blistre s jednotlivými dávkami uzavreté viečkom z hliníkovej fólie v baleniach po 2 x 1, 4 x 1, 6 x 1, 12 x 1 a 16 x 1 filmom obalených tabletách.
Polyvinylchloridové (PVC) perforované blistre s jednotlivými dávkami uzavreté viečkom z hliníkovej fólie v baleniach po 2 x 1, 4 x 1, 6 x 1, 12 x 1 a 16 x 1 filmom obalených tabletách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
RAYVOW 50 mgfilmomobalenétablety
EU/1/21/1587/001
EU/1/21/1587/002
EU/1/21/1587/003
EU/1/21/1587/004
EU/1/21/1587/005
EU/1/21/1587/006
EU/1/21/1587/007
EU/1/21/1587/008
EU/1/21/1587/009
EU/1/21/1587/010
RAYVO
W 100 mg filmomobalenétabletyEU/1/21/1587/011
EU/1/21/1587/012
EU/1/21/1587/013
EU/1/21/1587/014
EU/1/21/1587/015
EU/1/21/1587/016
EU/1/21/1587/017
EU/1/21/1587/018
EU/1/21/1587/019
EU/1/21/1587/020
RAYVOW 200 mgfilmomobalenétabletyEU/1/21/1587/021
EU/1/21/1587/022
EU/1/21/1587/023
EU/1/21/1587/024
EU/1/21/1587/025
EU/1/21/1587/026
EU/1/21/1587/027
EU/1/21/1587/028
EU/1/21/1587/029
EU/1/21/1587/030
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu