
ktoré sú substrátmi pre transport OAT1/3 a BCRP môže zvýšiť plazmatické koncentrácie takých liekov. Lumakaftor
a ivakaftor nie sú inhibítory OATP1B1, OATP1B3 a transportéra organického katiónu (organic cation
transporter ‒ OCT) 1 a 2. Ivakaftor nie je inhibítor OAT1 a OAT3.
Overenéainépotenciálnevýznamnéliekovéinterakcie
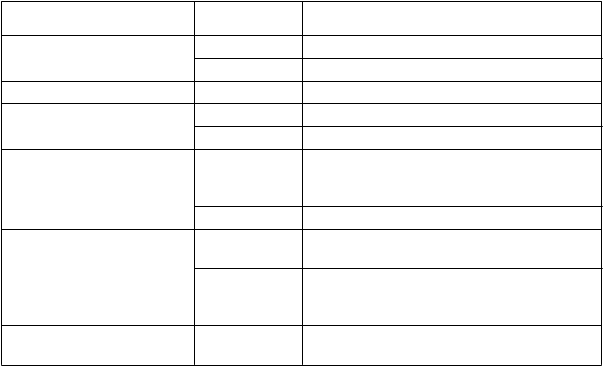
Tabuľka 3 zahŕňa overené alebo predpokladané účinky lumakaftoru/ivakaftoru na iné lieky alebo účinok iných liekov na lumakaftor/ivakaftor. Informácie uvedené v tabuľke 3 väčšinou pochádzajú
z in vitro štúdií. Odporúčania poskytnuté v rámci „Klinického komentára“ v tabuľke 3 sú založené na liekových interakčných štúdiách, klinickej závažnosti alebo predpokladaných interakciách vzhľadom
na cesty eliminácie. Liekové interakcie, ktoré sú klinicky najvýznamnejšie, sú uvedené ako prvé.
T
abuľka 3: Stanovené a ďalšie potenciálne významné liekové interakcie – odporúčané dávkovanie pre použitie lumakaftoru/ivakaftoru s ďalšími liekmi
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Súbežne podávané lieky s najväčším klinickým významom
A
ntialergiká:
montelukast ↔ LUM, IVA
↓ montelukast
kvôli indukcii CYP3A/2C8/2C9 prostredníctvom LUM
Nie je potrebná úprava dávky
montelukastu. Pri súbežnom podávaní s lumakaftorom/ivakaftorom má byť začaté vhodné klinické monitorovanie, ak je potrebné. Lumakaftor/ivakaftor môže znížiť expozíciu montelukastu, čo môže znížiť jeho účinnosť.
fexofenadín ↔ LUM, IVA
A
ntibiotiká: klaritromycín, telitromycín
↑ alebo ↓ fexofenadín

vzhľadom k potenciálnej indukcii alebo inhibícii
P-gp
↔ LUM
↑ IVA
kvôli inhibícii CYP3A prostredníctvom klaritromycínu, telitromycínu
↓ klaritromycín, telitromycín
kvôli indukcii CYP3A
prostredníctvom LUM
Úprava dávky fexofenadínu môže byť
potrebná k získaniu požadovaného klinického účinku. Lumakaftor/ivakaftor
môže zmeniť expozíciu fexofenadínu.
Pri začatí liečby klaritromycínom alebo telitromycínom u pacientov užívajúcich lumakaftor/ivakaftor nie je potrebná úprava dávky lumakaftoru/ivakaftoru.
Dávka lumakaftoru/ivakaftoru má byť
v prvom týždni liečby znížená na jednu tabletu denne pri začatí liečby
lumakaftorom/ivakaftorom u pacientov
súbežsne užívajúcich klaritromycín alebo telitromycín.
Je potrebné zvážiť alternatívu týchto antibiotík, ako je azitromycín. Lumakaftor/ivakaftor môže znížiť expozíciu klaritromycínu
a telitromycínu, čo môže znížiť ich účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
erytromycín ↔ LUM
↑ IVA
Kvôli inhibícii CYP3A
prostredníctvom erytromycínu
Neodporúča sa úprava dávky
lumakaftoru/ivakaftoru pri súbežnom podávaní s erytromycínom.
 A
ntikonvulzíva:
A
ntikonvulzíva: karbamazepín, fenobarbital, fenytoín
Antimykotiká: itrakonazol*, ketokonazol, posakonazol, vorikonazol
↓ erytromycín
kvôli indukcii CYP3A
prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A prostredníctvom týchto antikonvulzív
↓ karbamazepín, fenobarbital, fenytoín kvôli indukcii CYP3A prostredníctvom LUM
↔ LUM
↑ IVA
kvôli inhibícii CYP3A prostredníctvom týchto antimykotík
↓ itrakonazol, ketokonazol, vorikonazol kvôli indukcii CYP3A prostredníctvom LUM
↓ posakonazol
kvôli indukcii UGT
prostredníctvom LUM
Je potrebné zvážiť alternatívu erytromycínu, ako je azitromycín. Lumakaftor/ivakaftor môže znížiť expozíciu erytromycínu, čo môže znížiť jeho účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antikonvulzívami sa neodporúča. Expozície ivakaftoru a antikonvulzív môžu byť významne znížené, čo môže znížiť účinnosť obidvoch liečiv.
Neodporúča sa úprava dávky lumakaftoru/ivakaftoru na začiatku liečby týmito antimykotikami, ak pacienti súbežne užívajú lumakaftor/ivakaftor.
Dávka lumakaftoru/ivakaftoru má byť znížená na jednu tabletu denne v prvom týždni liečby lumakaftorom/ivakaftorom u pacientov, ktorí súbežne užívajú tieto antimykotiká.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antimykotikami sa neodporúča. Pacienti majú byť pozorne monitorovaní kvôli prielomovým mykotickým infekciám,
ak sú tieto lieky nevyhnutné. Lumakaftor/ivakaftor môže znížiť expozíciu týchto antimykotík, čo môže znížiť ich účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
flukonazol ↔ LUM
↑ IVA
kvôli inhibícii CYP3A
prostredníctvom flukonazolu
Neodporúča sa úprava dávky
lumakaftoru/ivakaftoru pri súbežnom podávaní s flukonazolom.
A
ntiflogistiká:
↓ flukonazol
kvôli indukcii prostredníctvom LUM;
flukonazol je vylúčený
primárne renálnou exkréciou ako nezmenené liečivo, hoci mierna redukcia expozície flukonazolu bola pozorovaná u silných induktorov
Pre žiaduci klinický efekt sú
požadované vyššie dávky flukonazolu.
Lumakaftor/ivakaftor môže znížiť expozíciu flukonazolu, čo môže znížiť jeho účinnosť.
ibuprofen ↔ LUM, IVA
 A
ntimykobakteriálne liečivá:
A
ntimykobakteriálne liečivá:
rifabutín, rifampicín*,
rifapentín
↓ ibuprofen
kvôli indukcii
CYP3A/2C8/2C9
prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A
prostredníctvom antimykobakteriálnych liečiv
↓ rifabutín
kvôli indukcii CYP3A
prostredníctvom LUM
↔ rifampicín, rifapentín
Pre žiaduci klinický efekt môžu byť
požadované vyššie dávky ibuprofenu.
Lumakaftor/ivakaftor môže znížiť expozíciu ibuprofenu, čo môže znížiť jeho účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antimykobakteriálnymi liečivami sa neodporúča. Expozícia ivakaftoru bude znížená, čo môže znížiť účinnosť lumakaftoru/ivakaftoru.
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky rifabutínu. Lumakaftor/ivakaftor môže znížiť expozíciu rifabutínu, čo môže znížiť jeho účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
B
enzodiazepíny:
midazolam, triazolam ↔ LUM, IVA
 H
ormonálna antikoncepcia:
H
ormonálna antikoncepcia: etinylestradiol, noretindron a ďalšie progestagény
Imunosupresíva: cyklosporín, everolimus, sirolimus, takrolimus (používané po orgánovej transplantácii)
Inhibítory protónovej pumpy: esomeprazol, lansoprazol, omeprazol
Rastlinné prípravky: ľubovník bodkovaný (
Hypericum perforatum)
↓ midazolam, triazolam
kvôli indukcii CYP3A
prostredníctvom LUM
↓ etinylestradiol, noretindrón a ďalšie progestagény
kvôli indukcii CYP3A/UGT prostredníctvom LUM
↔ LUM, IVA
↓ cyklosporín, everolimus, sirolimus, takrolimus
kvôli indukcii CYP3A
prostredníctvom LUM
↔ LUM, IVA
↓ esomeprazol, lansoprazol, omeprazol kvôli indukcii CYP3A/2C19 prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A prostredníctvom ľubovníka bodkovaného
Súbežné užívanie
lumakaftoru/ivakaftoru s týmito benzodiazepínmi sa neodporúča. Lumakaftor/ivakaftor znižuje expozíciu midazolamu a triazolamu, čo znižuje ich účinnosť.
Hormonálna antikoncepcia, vrátane perorálnej, injekčnej, transdermálnej a implantačnej, nemôže byť pri súbežnom podávaní
s lumakaftorom/ivakaftorom
považovaná za spoľahlivý spôsob antikoncepcie. Lumakaftor/ivakaftor
môže znížiť expozíciu hormonálnej
antikoncepcie, čo môže znížiť jej účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito imunosupresívami sa neodporúča. Lumakaftor/ivakaftor znižuje expozíciu imunosupresív, čo môže znížiť ich účinnosť. Použitie lumakaftoru/ivakaftoru u pacientov po orgánovej transplantácii nebolo študované.
Pre žiaduci klinický efekt sú požadované vyššie dávky inhibítorov protónovej pumpy. Lumakaftor/ivakaftor môže znížiť expozíciu inhibítorov protónovej pumpy, čo môže znížiť ich účinnosť.
Súbežné používanie lumakaftoru/ivakaftoru s ľubovníkom bodkovaným sa neodporúča. Expozícia ivakaftoru sa zníži, čo môže znížiť účinnosť lumakaftoru/ivakaftoru.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Ď
alšie klinicky významné súbežne podávané liečivá
A
ntiarytmiká:
digoxín ↔ LUM, IVA
A
ntikoagulanciá:
↑ alebo ↓ digoxín
kvôli potenciálnej indukcii alebo inhibícii P-gp
Sérová koncentrácia digoxínu má byť
monitorovaná a dávka má byť titrovaná podľa žiaduceho klinického účinku.
Lumakaftor/ivakaftor môže meniť
expozíciu digoxínu.
dabigatran ↔ LUM, IVA
↑ alebo ↓ dabigatran kvôli
potenciálnej indukcii alebo inhibícii P-gp
Pri súbežnom podávaní
lumakaftoru/ivakaftoru by má byť použité zodpovedajúce klinické monitorovanie. Úprava dávky dabigatranu môže byť potrebná
k získaniu požadovaného klinického účinku. Lumakaftor/ivakaftor môže
meniť expozíciu dabigatranu.
warfarín ↔ LUM, IVA
A
ntidepresíva: citalopram, escitalopram, sertralín
↑ alebo ↓ warfarín
kvôli potenciálnej indukcii alebo inhibícii CYP2C9
prostredníctvom LUM
↔ LUM, IVA
↓ citalopram, escitalopram, sertralín kvôli indukcii CYP3A/2C19 prostredníctvom LUM
Pri súbežnom podávaní
lumakaftoru/ivakaftoru s warfarínom má byť monitorovaná hladina
medzinárodneho normalizovaného
pomeru (international normalised ratio,
INR). Lumakaftor/ivakaftor môže meniť expozíciu warfarínu.
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky antidepresív. Lumakaftor/ivakaftor môže znížiť expozíciu antidepresív, čo môže znížiť ich účinnosť.
bupropión ↔ LUM, IVA

↓ bupropión
kvôli indukcii CYP2B6
prostredníctvom LUM
Pre žiaduci klinický efekt môžu byť
požadované vyššie dávky bupropiónu.
Lumakaftor/ivakaftor môže znížiť expozíciu bupropiónu, čo môže znížiť jeho účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Systémové
kortikosteroidy: metylprednizolón, prednizón
 Blokátory H2:
Blokátory H2:↔ LUM, IVA
↓ metylprednizolón, prednizónu
kvôli indukcii CYP3A
prostredníctvom LUM
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky týchto systémových kortikosteroidov. Lumakaftor/ivakaftor môže znížiť expozíciu systémových kortikosteroidov, čo môže znížiť ich účinnosť.
ranitidín ↔ LUM, IVA
P
e
rorálne antidiabetiká: repaglinid
↑ alebo ↓ ranitidín
kvôli indukcii alebo inhibícii P-gp
↔ LUM, IVA
↓ repaglinid kvôli indukcii CYP3A/2C8 prostredníctvom LUM
Pre žiaduci klinický efekt môže byť
potrebná zmena dávky ranitidínu.
Lumakaftor/ivakaftor môže meniť expozíciu ranitidínu.
Pre žiaduci klinický efekt môže byť požadovaná vyššia dávka repaglinidu. Lumakaftor/ivakaftor môže znížiť expozíciu repaglinidu, čo môže znížiť jeho účinnosť.
Vysvetlivky: ↑ = nárast, ↓ = pokles, ↔ = bez zmeny; LUM = lumakaftor; IVA = ivakaftor.
Založené na klinických liekových interakčných štúdiách. Všetky ostatné liekové interakcie sú predpokladané.
Falošne pozitívne močové testy na THC
U pacientov užívajúcich Orkambi boli hlásené falošne pozitívne skríningové testy moču na tetrahydrokanabinol (THC). Na potvrdenie výsledkov sa má zvážiť alternatívna konfirmačná metóda.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít)
o použití lumakaftoru/ivakaftoru u gravidných žien. Štúdie na zvieratách s lumakaftorom a ivakaftorom nepreukázali priame alebo nepriame účinky z hľadiska vývoja a reprodukčnej toxicity, zatiaľ čo iba s ivakaftorom boli účinky zaznamenané v dávkach toxických pre matku (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lumakaftoru/ivakaftoru počas gravidity, pokiaľ klinický stav matky nevyžaduje liečbu lumakaftorom/ivakaftorom.
Dojčenie
Nie je známe, či sa lumakaftor a/alebo ivakaftor a metabolity vylučujú do ľudského mlieka. Dostupné farmakokinetické údaje u zvierat preukázali vylučovanie lumakaftoru aj ivakaftoru do mlieka laktujúcich samíc potkanov. Ako také riziko u dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu lumakaftorom/ivakaftorom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre matku.
Fertilita
Lumakaftor nemala vplyv na plodnosť a indexy reprodukčnej výkonnosti u samcov a samíc potkanov. Ivakaftor zhoršoval plodnosť a indexy reprodukčnej výkonnosti u samcov a samíc potkanov. Pri dávke
≤ 100 mg/kg/deň nebol pozorovaný žiaden vplyv na plodnosť samcov a samíc potkanov a indexy reprodukčnej výkonnosti (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ivakaftor, ktorý je jedným z liečiv v Orkambi, má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Ivakaftor môže spôsobiť závraty (pozri časť 4.8).
Pacienti, u ktorých sa vyskytujú závraty v priebehu užívania Orkambi, majú byť poučení, aby neviedli vozidlá ani neobsluhovali stroje, kým symptómy neustúpia.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie, ktoré sa vyskytli u pacientov vo veku 12 rokov a starších, ktorí užívali
lumakaftor/ivakaftor v združených, placebom kontrolovaných štúdiách fázy 3, boli dyspnoe (14,0 % oproti 7,8 % pri placebe), hnačka (11,0 % oproti 8,4 % pri placebe) a nauzea (10,2 % oproti 7,6 % pri placebe).
Závažné nežiaduce účinky, vyskytujúce sa najmenej u 0,5 % pacientov zahŕňajú poruchy pečene a žlčových ciest, napr. zvýšené hodnoty transamináz, cholestatickú hepatitídu a hepatickú encefalopatiu.
Zoznamnežiaducichreakciívtabuľke
Nežiaduce reakcie identifikované v 24-týždňových, placebom kontrolovaných štúdiách fázy 3(skúšania 1 a 2) u pacientov vo veku 12 rokov a starších a v 24-týždňovej, placebom
kontrolovanej štúdii u pacientov vo veku 6 až 11 rokov (skúšanie 7), ktorí sú homozygotní nosiči pre
mutáciu F508del v géne CFTR sú uvedené v tabuľke 4 a sú zoradené podľa triedy orgánových systémov, frekvencie a nežiaducich účinkov. Pozorované nežiaduce reakcie ivakaftoru samotného sú tiež uvedené v tabuľke 4. Nežiaduce reakcie sú zoskupené podľa klasifikácie frekvencie MedDRA: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé
(≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Tabuľka 4: Nežiaduce reakcie u pacientov liečených lumakaftorom/ivakaftorom a ivakaftorom samotným.
T
rieda orgánových systémov
F
r
ekvencia Nežiaduce reakcie
Infekcie a nákazy veľmi časté Nasofaringitída*
časté Infekcia horných dýchacích ciest, rinitída
Poruchy ciev menej časté Hypertenzia
Poruchy nervového systému veľmi časté Bolesť hlavy, závrat*
menej časté Hepatálna encefalopatia †
Poruchy ucha a labyrintu časté Bolesť ucha*, ušné problémy*, tinnitus*, hyperémia tympanickej membrány*, vestibulárna porucha*
menej časté Kongescia ucha*
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
veľmi časté Kongescia nosa, dyspnoe, produktívny kašeľ, zvýšené vylučovanie hlienu
časté Abnormálne dýchanie, orofaryngeálna bolesť, kongescia sínusov*, rinorea, faryngeálny erytém*
veľmi časté Abdominálna bolesť*, bolesť hornej časti brucha, hnačka, nevoľnosť
T
rieda orgánových systémov
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy reprodukčného systému a prsníkov
Laboratórne a funkčné vyšetrenia
Frekvencia Nežiaduce reakciečasté Nadúvanie, vracanie
časté Zvýšené hodnoty transamináz menej časté Cholestatická hepatitída‡

časté Vyrážka
časté Nepravidelná menštruácia, dysmenorea, metrorágia, hrčka v prsníku*
menej časté Menorágia, amenorea, polymenorea, zápal prsníku*, gynekomastia*, poruchy bradavky*, bolesť bradavky*, oligomenorea
veľmi časté Baktérie v spúte
časté Zvýšená hladina kreatínfosfokinázy v krvi menej časté Zvýšený krvný tlak
* Nežiaduce reakcie a frekvencie pozorované u pacientov v klinických štúdiách v monoterapii ivakaftorom (zložka Orkambi).
† 1 pacient zo 738
‡ 2 pacienti z 738
Údaje o bezpečnosti u 1 029 pacientov vo veku 12 rokov a starších, ktorí boli homozygotní nosiči mutácie F508del v géne CFTR, liečených lumakaftorom/ivakaftorom počas ďalších až
96 týždňov v dlhodobej pokračovacej štúdii bezpečnosti a účinnosti (skúšanie 3) boli podobné 24-
týždňovej, placebom kontrolovanej štúdii (pozri časť 5.1).
Popisvybranýchnežiaducichreakcií
Poruchy pečene a žlčových ciest
Počas skúšaní 1 a 2 bola incidencia maximálnej hodnoty transaminázy (ALT alebo AST) > 8, > 5 a
> 3 x ULN 0,8 %, 2,0 % a 5,2 %; a 0,5 %, 1,9 % a 5,1 % u pacientov liečených lumakaftorom/ivakaftorom, respektíve placebom. Incidencia nežiaducich reakcií spojených so zvýšenými hodnotami transamináz bola 5,1 % a 4,6 % u pacientov liečených lumakaftorom/ivakaftorom, respektíve u pacientov s placebom. Sedem pacientov užívajúcich lumakaftor/ivakaftor malo závažné nežiaduce reakcie spojené s poruchou pečene a zvýšenými hodnotami transamináz, zahŕňajúce 3 pacientov so súbežne zvýšeným celkovým bilirubínom. Prerušenie liečby lumakaftorom/ivakaftorom u všetkých pacientov vrátilo funkčné pečeňové testy na vstupnú hodnotu alebo ich zlepšilo (pozri časť 4.4).
Medzi 7 pacientmi s pre-existujúcou cirhózou alebo portálnou hypertenziou, ktorí užívali lumakaftor/ivakaftor v placebom kontrolovanej štúdii fázy 3, bolo pozorované zhoršenie funkčných pečeňových testov so vzostupom ALT, AST, bilirubínu a hepatickej encefalopatie u jedného pacienta. Prípad sa vyskytol do 5 dní po začatí liečby a vyriešil sa prerušením liečby lumakaftorom/ivakaftorom (pozri časť 4.4).
U pacientov s CF s pre-existujúcou cirhózou pečene s portálnou hypertenziou, ktorí boli liečení lumakaftorom/ivakaftorom, boli po uvedení lieku na trh hlásené prípady dekompenzácie funkcie pečene, vrátane zlyhania pečene vedúceho k úmrtiu (pozri časť 4.4).
Respiračné príhody
Počas skúšaní 1 a 2 bola incidencia nežiaducich reakcií dýchacej sústavy (napr. hrudný dyskomfort, dyspnoe a respiračné problémy) u pacientov liečených lumakaftorom/ivakaftorom 26,3 % v porovnaní
so 17,0 % u pacientov s placebom. Incidencia týchto udalostí bola častejšia u pacientov s nižším FEV1 pred liečbou. Približne tri štvrtiny týchto udalostí začali v prvom týždni liečby a u väčšiny pacientov odozneli bez prerušenia liečby. Väčšina príhod boli mierne alebo stredne závažné, nezávažné
a neviedli k prerušeniu liečby (pozri časť 4.4).
Počas 24-týždňovej, otvorenej klinickej štúdie fázy 3b (skúšanie 5) u 46 pacientov vo veku 12 rokov a starších s pokročilým pľúcnym ochorením (ppFEV1 < 40) [priemerný východiskový ppFEV1 29,1 (rozmedzie: 18,3 až 42,0)] bola incidencia respiračných príhod 65,2 %. V podskupine 28 pacientov,
u ktorých sa liečba začala celou dávkou lumakaftoru/ivakaftoru (2 tablety každých 12 hodín) bola incidencia 71,4 % a u 18 pacientov, u ktorých sa liečba začala zníženou dávkou lumakaftoru/ivakaftoru (1 tableta každých 12 hodín počas 2 týždňov a následne zvýšenie na celú dávku) bola incidencia 55,6 %. Medzi pacientmi, u ktorých sa liečba začala celou dávkou lumakaftoru/ivakaftoru sa u jedného pacienta vyskytla závažná respiračná príhoda, u troch pacientov sa znižovala dávka a u troch pacientov sa liečba ukončila. Medzi pacientmi, u ktorých sa liečba začala polovičnou dávkou, sa nezaznamenali žiadne závažné respiračné príhody, znižovanie dávky ani ukončenie liečby (pozri časť 4.4).
Abnormality menštruačného cyklu
Počas skúšaní 1 a 2 bola incidencia kombinovaných prípadov abnormalít menštruačného cyklu
(amenorea, dysmenorea, menorágia, nepravidelná menštruácia, metrorágia, oligomenorea a
polymenorea) 9,9 % u žien liečených lumakaftorom/ivakaftorom a 1,7 % u žien s placebom. Tieto menštruačné problémy sa vyskytovali častejšie v skupine žien, ktoré užívali hormonálnu antikoncepciu (25,0 %) oproti pacientkám, ktoré hormonálnu antikoncepciu neužívali (3,5 %) (pozri časť 4.5). Väčšina týchto reakcií bola mierna alebo stredná, nezávažná. U pacientok liečených lumakaftorom/ivakaftorom približne dve tretiny týchto reakcií odozneli a medián trvania bol 10 dní.
Zvýšený krvný tlak
V priebehu skúšaní 1 a 2 boli hlásené nežiaduce reakcie spojené so zvýšením krvného tlaku (napr. hypertenzia, zvýšenie krvného tlaku) u 0,9 % (7 / 738) pacientov liečených lumakaftorom/ivakaftorom a neboli hlásené u žiadnych pacientov, ktorí dostávali placebo.
U pacientov liečených lumakaftorom/ivakaftorom (priemerný východiskový tlak 114 mmHg systolický a 69 mmHg diastolický), bol maximálny priemerný nárast oproti východiskovému systolickému a diastolickému krvnému tlaku 3,1 mmHg a 1,8 mmHg, v uvedenom poradí.
U pacientov, ktorí dostávali placebo (priemerný východiskový tlak 114 mmHg systolický a 69 mmHg diastolický), bol maximálny priemerný nárast oproti východiskovému systolickému a diastolickému krvnému tlaku 0,9 mmHg a 0,9 mmHg, v uvedenom poradí.
Podiel pacientov, ktorí mali hodnotu systolického krvného tlaku > 140 mmHg alebo diastolického krvného tlaku > 90 mmHg aspoň dvakrát, bol 3,4 % a 1,5 % u pacientov liečených lumakaftorom/ivakaftorom, v uvedenom poradí, v porovnaní s 1,6 % a 0,5 % u pacientov, ktorí dostávali placebo (pozri časť 4.4).
Pediatrickápopulácia
Bezpečnostné údaje boli hodnotené u 60 pacientov vo veku 2 až 5 rokov, 161 pacientov vo veku 6 až
11 rokov (skúšania 6, 7 a 8) a u 194 pacientov vo veku 12 až 17 rokov s CF, ktorí sú homozygotní nosiči mutácie F508del a ktorí užívali lumakaftor/ivakaftor v klinických štúdiách. Pacienti vo veku 12
až 17 rokov boli zahrnutí v skúšaniach 1 a 2.
Bezpečnostný profil u týchto pediatrických pacientov je všeobecne zhodný s tým u dospelých pacientov. Ďalšie nežiaduce reakcie zo skúšania 6 sú zahrnuté v tabuľke 4.
Popis vybraných nežiaducich reakcií u pacientov vo veku 6 až 11 rokov
Hepatobiliárne príhody
Počas 24-týždňovej, otvorenej klinickej štúdie fázy 3 u 58 pacientov vo veku 6 až 11 rokov
(skúšanie 6) bola incidencia maximálnych hodnôt transamináz (ALT alebo AST) > 8, > 5 a > 3 x ULN
5,3 %, 8,8 %, a 19,3 %. Žiaden pacient nemal hladinu celkového bilirubínu > 2 x ULN. Dávkovanie lumakaftoru/ivakaftoru bolo zachované alebo úspešne obnovené po prerušení u všetkých pacientov so zvýšenými hodnotami transamináz, okrem 1 pacienta, ktorý ukončil liečbu trvale.
Počas 24-týždňovej, placebom kontrolovanej klinickej štúdie fázy 3 u 204 pacientov vo veku 6 až
11 rokov (skúšanie 7) bola incidencia maximálnych hodnôt transamináz (ALT alebo AST) > 8, > 5,
a > 3 x ULN 1,0 %, 4,9 % a 12,6 % u pacientov s lumakaftorom/ivakaftorom a 2,0 %, 3,0 %, a 7,9 %
u pacientov liečených placebom. Žiaden pacient nemal hladinu celkového bilirubínu > 2 x ULN. Dvaja pacienti v skupine s lumakaftorom/ivakaftorom a dvaja pacienti v skupine s placebom prerušili liečbu trvale kvôli zvýšeným hodnotám transamináz.
Respiračné príhodyPočas 24-týždňovej, otvorenej klinickej štúdie fázy 3(skúšanie 6) u 58 pacientov vo veku 6 až
11 rokov (priemerná hodnota ppFEV1 bola 91,4) bola incidencia respiračných nežiaducich reakcií
6,9 % (4/58).
Počas 24-týždňovej, placebom kontrolovanej klinickej štúdie fázy 3 (skúšanie 7) u pacientov vo veku
6 až 11 rokov (priemerná hodnota ppFEV1 bola 89,8) bola incidencia respiračných nežiaducich reakcií
18,4 % u pacientov s lumakaftorom/ivakaftorom a 12,9 % u pacientov s placebom. Po začatí liečby
bol pozorovaný pokles v hodnote ppFEV1 pri meraní spirometrie po podaní. Absolútna zmena medzi hodnotou pred podaním a hodnotou 4-6 hodín po podaní bola -7,7 v 1. dni a -1,3 v 15. dni u pacientov s lumakaftorom/ivakaftorom. Pokles po podaní odoznel do 16. týždňa.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadne špecifické antidotum pre predávkovanie lumakaftorom/ivakaftorom. Liečba predávkovania pozostáva zo všeobecných podporných opatrení vrátane monitorovania životných funkcií a pozorovania klinického stavu pacienta.
Nežiaduce účinky, ktoré sa objavili vo zvýšenej incidencii ≥ 5 % v období podávania supraterapeutickej dávky v porovnaní s terapeutickou dávkou boli bolesť hlavy, generalizovaná vyrážka a zvýšené hodnoty transamináz.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá respiračného systému; ATC kód: R07AX30
MechanizmusúčinkuProteín CFTR je chloridový kanál prítomný na povrchu epitelových buniek vo viacerých orgánoch. Mutácia
F508del vplýva na proteín CFTR viacerými spôsobmi, primárne zapríčinením defektu bunkového spracovania a transportu, ktorý znižuje množstvo CFTR na bunkovom povrchu. Malé množstvo F508del-CFTR, ktoré sa dostane na bunkový povrch, má malú schopnosť otvárať kanál (porucha vrátkovania). Lumakaftor je korektor CFTR, ktorý účinkuje priamo na mutáciu
F508del-CFTR za účelom zlepšenia bunkového spracovania a transportu, čím zvyšuje množstvo funkčného CFTR na bunkovom povrchu. Ivakaftor je potenciátor CFTR, ktorý uľahčuje zvýšenie transportu chloridov potencovaním schopnosti otvárať kanál (alebo vrátkovania) proteínu CFTR na bunkovom povrchu. Kombinovaným efektom lumakaftoru a ivakaftoru je zvýšenie množstva
a funkcie F508del-CFTR na bunkovom povrchu vedúce k zvýšeniu transportu chloridov. Presný spôsob, akým lumakaftor zlepšuje bunkové spracovanie a transport F508del-CFTR, a ivakaftor
potencuje F508del-CFTR, nie je známy.
Farmakodynamické
účinky
Ú
činky na chloridy v pote:
Zmeny chloridov v pote ako odpoveď na lumakaftor samotný alebo v kombinácii s ivakaftorom boli hodnotené v dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní fázy 2 u pacientov s CF
vo veku 18 rokov a starších. V tomto skúšaní 10 pacientov (homozygotní nosiči mutácie
F508del-CFTR) dokončili užívanie lumakaftoru samotného v dávke 400 mg každých 12 hod po dobu
28 dní nasledované pridaním ivakaftoru 250 mg každých 12 hod po dobu 28 dní a 25 pacientov
(homozygotní alebo heterozygotní nosiči mutácie
F508del) dokončili užívanie placeba. Rozdiel medzi liečbou samotným lumakaftorom 400 mg každých 12 hod a placebom, hodnotený ako stredná zmena
chloridov v pote od východiskovej hodnoty po 28. deň, bol štatisticky významný −8,2 mmol/l (95 %
CI: −14, −2). Rozdiel medzi liečbou kombináciou lumakaftor 400 mg/ivakaftor 250 mg každých
12 hod a placebom, hodnotený ako stredná zmena chloridov v pote od východiskovej hodnoty po 56. deň, bol štatisticky významný −11 mmol/l (95 % CI: −18, −4).

V skúšaní 7 (pozri Klinická účinnosť a bezpečnosť) u pacientov vo veku 6 až 11 rokov, ktorí boli homozygotní nosiči mutácie
F508del génu
CFTR, bol rozdiel v liečbe (priemer stanovený metódou najmenších štvorcov) v chloridoch v pote ako priemer absolútnej zmeny v 24. týždni v porovnaní
s placebom -24,9 mmol/l (nominálna P hodnota < 0,0001). Rozdiel v liečbe (priemer stanovený metódou najmenších štvorcov) v chloridoch v pote ako priemer absolútnej zmeny v 15. dni
a v 4. týždni v porovnaní s placebom bol –20,8 mmol/l (95% CI: -23,4; -18,2; nominálna hodnota
P < 0,0001).
Zmeny v FEV1:Zmeny v ppFEV1 v odpovedi na lumakaftor samostatne alebo v kombinácii s ivakaftorom boli tiež
hodnotené v dvojito zaslepenom placebom kontrolovanom skúšaní fázy 2 u pacientov s CF vo veku
18 rokov a starších. Rozdiel medzi liečbou lumakaftorom 400 mg každých 12 hodín samotným
a placebom, hodnotený ako stredná absolútna zmena ppFEV1, bol -4,6 percentuálnych bodov (95 % CI: -9,6; 0,4) od východiskového stavu do 28. dňa, 4,2 percentuálnych bodov (95 % CI: -1,3; 9.7) od východiskového stavu do 56. dňa a 7,7 percentuálnych bodov (95 % CI: 2,6; 12,8; štatisticky významné) odo dňa 28. až 56. dňa (po pridaní ivakaftoru k lumakaftorovej monoterapii).
Zníženie tepovej frekvencieV priebehu 24 týždňov trvajúcich, placebom kontrolovaných štúdií fázy 3 bol pozorovaný maximálny pokles priemernej srdcovej frekvencie o 6 úderov za minútu (
beats per minute, bpm) od východiskovej úrovne v 1. a 15. deň, približne 4 až 6 hodín po podaní. V období po 15. dni nebol srdcový tep po podaní v týchto štúdiách sledovaný. Od 4. týždňa bola zmena priemernej tepovej frekvencie pred podaním v rozmedzí od 1 do 2 bpm pod východiskovú úroveň u pacientov liečených lumakaftorom/ivakaftorom. Percento pacientov s hodnotami srdcovej frekvencie < 50 bpm počas liečby bolo 11 % u pacientov, ktorí dostávali lumakaftor/ivakaftor, v porovnaní s 4,9 % u pacientov, ktorí dostávali placebo.
KlinickáúčinnosťabezpečnosťSkúšania u pacientov s CF vo veku 12 rokov a starších, ktorí sú homozygotní nosiči mutácie F508del
v géne CFTRÚčinnosť lumakaftoru/ivakaftoru u pacientov s CF, ktorí sú homozygotní nosiči mutácie
F508delv géne
CFTR bola hodnotená v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach s 1 108 klinicky stabilnými pacientmi s CF, z ktorých
737 pacientov bolo randomizovaných k užívaniu lumakaftoru/ivakaftoru. Pacienti v oboch skúšaniach boli randomizovaní v pomere 1:1:1 a dostávali lumakaftor 600 mg jedenkrát denne/ivakaftor 250 mg každých 12 hod, lumakaftor 400 mg každých 12 hod/ivakaftor 250 mg každých 12 hod alebo placebo.
Pacienti užívali študovaný liek s jedlom obsahujúcim tuky po dobu 24 týždňov navyše k predpísanej terapii CF (napr. bronchodilatátory, inhalačné antibiotiká, dornáza alfa a hypertonický roztok chloridu
sodného). Pacienti z týchto skúšaní boli vhodní k zaradeniu do zaslepenej predĺženej štúdie.
Skúšanie 1 hodnotilo 549 pacientov s CF vo veku 12 rokov a starších (priemerný vek 25,1 rokov) s percentom predpokladaného FEV1 (ppFEV1) pri skríningu medzi 40 - 90 (stredná hodnota na začiatku štúdie ppFEV1 60,7 [rozmedzie: od 31,1 do 94,0]). Skúšanie 2 hodnotilo 559 pacientov vo
veku 12 rokov a starších (priemerný vek 25,0 rokov) s ppFEV1 pri skríningu medzi 40 - 90 (stredný ppFEV1 na počiatku 60,5 [rozmedzie: od 31,3 do 99,8]). Pacienti s anamnézou kolonizácie organizmami ako Burkholderia cenocepacia, Burkholderia dolosa alebo Mycobacterium abscessus alebo ktorí mali 3 alebo viac abnormálnych funkčných pečeňových testov (ALT, AST, AP, GGT
≥ 3-násobok ULN alebo celkový bilirubín ≥ 2-násobok ULN) boli vyradení.
Primárnym koncovým ukazovateľom účinnosti v obidvoch štúdiách bola absolútna zmena ppFEV1 od východiskovej hodnoty po hodnotu v 24. týždni. Ďalšie premenné účinnosti, ktoré boli zahrnuté, sú relatívna zmena ppFEV1 od východiskovej hodnoty, absolútna zmena BMI od východiskovej hodnoty, absolútna zmena respiračnej domény dotazníku CFQ-R od východiskovej hodnoty, podiel pacientov, ktorí dosiahli ≥ 5 % relatívnej zmeny od východiskovej hodnoty ppFEV1 v 24. týždni a množstvo pulmonálnych exacerbácií (zahŕňajúcich tie, ktoré vyžadovali hospitalizáciu alebo intravenóznu antibiotickú terapiu) do 24. týždňa.
V obidvoch skúšanich viedla liečba lumakaftorom/ivakaftorom k štatisticky významnému zlepšeniu ppFEV1 (tabuľka 5). Stredné zlepšenie ppFEV1 bolo prudké na začiatku (15. deň) a udržalo sa po celú dobu 24-týždňovej liečby. V 15. dni bol rozdiel medzi liečbou lumakaftorom 400 mg/ivakaftorom
250 mg každých 12 hod a placebom v strednej absolútnej zmene (95 % CI) ppFEV1 od východiskovej hodnoty 2,51 percentuálnych bodov združených skúšaní 1 a 2 (P < 0,0001). Zlepšenie v ppFEV1 bolo pozorované bez ohľadu na vek, závažnosť ochorenia, pohlavie a zemepisný región. Fáza 3 skúšania lumakaftoru/ivakaftoru zahŕňala 81 pacientov s ppFEV1 < 40 na začiatku štúdie. Rozdiel v liečbe
v tejto podskupine bol porovnateľný s podskupinou pacientov s ppFEV1 ≥ 40. V 24. týždni bol rozdiel
medzi liečbou lumakaftorom 400 mg/ivakaftorom 250 mg každých 12 hod a placebom v strednej
absolútnej zmene (95 % CI) ppFEV1 od východiskovej hodnoty v združených skúšaniach 1 a 2
3,39 percentuálnych bodov (P = 0,0382) pre pacientov s ppFEV1 < 40 a 2,47 percentuálnych bodov
(P < 0,0001) pre pacientov s ppFEV1 ≥ 40.
T
abuľka 5: Súhrn primárnych a kľúčových sekundárnych výsledkov v skúšaní 1 a skúšaní 2*
S
k
ú
šanie 1 Skúšanie 2 Združené (skúšanie 1
a skúšanie 2)
Placebo
(
n = 184)
L
U
M 400 mg každých
1
2 hod/IVA 250 mg
k
až
d
ýc
h 12 hod
(
n = 182)
Placebo
(
n = 187)
L
U
M 400 mg každých
1
2 hod/IVA
2
5
0 mg každých
1
2 hod
(
n = 187)
Placebo
(
n = 371)
L
U
M 400 mg každých
1
2 hod/IVA
2
5
0 mg každých
1
2 hod
(
n = 369)
Ab
solútna zmena
v ppFEV
1
v 24. týždni
Rozdiel v liečbe
– 2,41
(P = 0,0003) †
– 2,65
(P = 0,0011) †
– 2,55
(P < 0,0001)
(p
erce
nt
u
ál
n
e
b
o
d
y
)
Rozdiel
v rámci skupiny
Rozdiel
-0,73
(P = 0,216
8)
1,68
(P = 0,0051)
4,15
-0,02
(P = 0,973
0)
2,63
(P < 0,0001)
4,69
-0,39
(P < 0,349
4)
2,16
(P < 0,0001)
4,4
R
ela
t
í
vn
a zmena
v liečbe –
(P = 0,0028)† –
(P = 0,0009)† –
(P < 0,0001)
v ppFEV
1
v 24. týždni (%)
Rozdiel v rámci skupiny
-0,85
(P = 0,393
4)
3,3
(P = 0,0011)
0,16
(P = 0,879
3)
4,85
(P < 0,0001)
-0,34
(P = 0,637
5)
4,1
(P < 0,0001)
Ab
solútna zmena
Rozdiel
– 0,13
0,36
†
– 0,24
v BMI
v 24. týždni
(
k
g
/
m
2
)
Ab
solútna zmena
v liečbe
Rozdiel v rámci skupiny
Rozdiel
0,19
(P = 0,006
5)
(P = 0,1938)
0,32
(P < 0,0001)
1,5
0,07
(P = 0,289
2)
(P < 0,0001)
0,43
(P < 0,0001)
2,9
0,13
(P = 0,006
6)
(P = 0,0004)
0,37
(P < 0,0001)
2,2
v CFQ-R
re
spiračnej
v liečbe –
(P = 0,3569) –
(P = 0,0736) –
(P = 0,0512)
d
o
m
é
n
y Skóre v 24. týždni (body)
Podiel pacientov
Rozdiel v rámci skupiny
1,1
(P = 0,342
3)
2,6
(P = 0,0295)
2,8
(P = 0,015
2)
5,7
(P < 0,0001)
1,9
(P = 0,021
3)
4,1
(P < 0,0001)
s ≥ 5 %
re
l
a
t
í
vn
y
m
% 25 % 32 % 26 % 41 % 26 % 37 %
roz
d
i
e
lo
m
v ppFEV
1
v 24. týždni
Odds ratio –
Počet príhod
1,43
(P = 0,1208) –
1,90
(P = 0,0032) –
1,66
(P = 0,0013)
Počet pulmonálnych exacerbácií počas
(pomerný výskyt za 48 týžd.)
112 (1,07) 73 (0,71) 139 (1,18) 79 (0,67) 251 (1,14) 152 (0,70)
2
4 týždňov
Relatívne
riziko –
0,66
(P = 0,0169) –
0,57
– (P = 0,0002)
0,61
(P < 0,0001)

*V každej štúdii bolo vykonané hierarchické testovanie v rámci každej aktívnej liečebnej skupiny na primárne a sekundárne koncové ukazovatele oproti placebu; pri každom kroku, P ≤ 0,0250 a všetky predchádzajúce testy tiež spĺňali túto hodnotu významnosti, ktorá bola požadovaná.
†Ukazovatele štatistickej významnosti sa potvrdili v hierarchickom testovaní.
V 24. týždni bol podiel pacientov bez pulmonálnej exacerbácie významne vyšší pre pacientov
liečených lumakaftorom/ivakaftorom v porovnaní s placebom. V združenej analýze bolo relatívne riziko exacerbácie počas 24 týždňov u jedincov liečených lumakaftorom/ivakaftorom (lumakaftor
400 mg/ivakaftor 250 mg každých 12 hod; n = 369) 0,61 (P < 0,0001), predstavujúce relatívne
zníženie o 39 % oproti placebu. Počet prípadov za rok, anualizovaného do 48 týždňov bol 0,70 v skupine s lumakaftorom/ivakaftorom a 1,14 v skupine s placebom. Liečba lumakaftorom/ivakaftorom významne znížila riziko exacerbácií vyžadujúcich hospitalizáciu oproti placebu o 61 % (relatívne riziko = 0,39; P < 0,0001; počet prípadov za 48 týždňov 0,17
pre lumakaftor/ivakaftor a 0,45 pre placebo) a znížila exacerbácie vyžadujúce liečbu intravenóznymi antibiotikami o 56 % (relatívne riziko = 0,44; P < 0,0001; počet prípadov za 48 týždňov 0,25
pre lumakaftor/ivakaftor a 0,58 pre placebo). Tieto výsledky neboli považované za štatisticky
významné v rámci hierarchických testov jednotlivých štúdií.
Dlhodobé pokračovacie skúšanie bezpečnosti a účinnostiSkúšanie 3 bola multicentrická, pokračovacia, predĺžená štúdia fázy 3 s paralelnými skupinami pre pacientov s CF, ktorá zahŕňala pacientov vo veku 12 rokov a starších zo skúšania 1 a skúšania 2. Toto predlžené skúšanie bola navrhnuté za účelom zhodnotiť bezpečnosť a účinnosť dlhodobej liečby lumakaftorom/ivakaftorom. Z 1 108 pacientov, ktorí dostali akúkoľvek liečbu v skúšaní 1 alebo 2,
dostávalo 1 029 (93 %) pacientov dávku a aktívnu liečbu (lumakaftor 600 mg raz denne/ivakaftor
250 mg každých 12 hod alebo lumakaftor 400 mg každých 12 hod/ivakaftor 250 mg každých 12 hod) v skúšaní 3 počas ďalších až 96 týždňov (t.j. celkovo až 120 týždňov). Primárna analýza účinnosti tejto predlženej štúdie zahŕňala údaje do týždňa 72 skúšania 3 a analýza senzitivity zahŕňala údaje
do týždňa 96 skúšania 3.
Pacienti liečení lumakaftorom/ivakaftorom v skúšaní 1 alebo v skúšaní 2 preukázali účinok, ktorý bol udržiavaný s ohľadom na východiskovú hodnotu po ďalších 96 týždňoch počas skúšania 3.
U pacientov, ktorí prešli z placeba na aktívnu liečbu boli pozorované podobné zmeny ako u pacientov liečených lumakaftorom/ivakaftorom v skúšaní 1 alebo skúšaní 2 (pozri Tabuľku 5). Výsledky
zo skúšania 3 sú zhrnuté na obrázku 1 a v tabuľke 6.
Obrázok 1. Absolútna zmena od východiskovej hodnoty v percentách predpokladaného FEV1pri každej návšteve†
Zač Ď Týž Týž Týž Týž Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž.
15 4 8 16 24 Ď15 týž 8 týž 16 týž 24 týž 36 týž 48 týž 60 týž 72 týž 84 týž 96
Návšteva LUM 400 mg každých 12 hod/IVA 250 mg každých 12 hod Placebo/LUM 400 mg každých12 hod/IVA 250 mg každých 12 hod Placebo
† zo skúšaní 1, 2 a 3.
Tabuľka 6: Dlhodobý účinok lumakaftoru/ivakaftoru v skúšaní 3*Placebo s prechodom na
lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele Východisková hodnota
Priemer
(SD)
štvorcov
 (95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
pp
FEV
1‡ 60,2 (14,7) 60,5 (14,1)
Absolútna zmena od východiskovej hodnoty ppFEV1 (percentuálne body)
Týždeň 72 predĺženia
(n = 134)
1,5 (0,2; 2,9)
0,0254
(n = 273)
0,5
(-0,4; 1,5)
0,2806
(n = 75)
Týždeň 96 predĺženia 0,8
(-0,8; 2,3)
(n = 147)
0,3495 0,5
(-0,7; 1,6)
0,4231
Placebo s prechodom na lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Relatívna zmena od východiskovej hodnoty ppFEV
1
(
%)
Týždeň 72 predĺženia
(n = 134)
2,6 (0,2; 5,0)
0,0332
(n = 273)
1,4
(-0,3; 3,2)
0,1074
(n = 75)
Týždeň 96 predĺženia 1,1
(-1,7; 3,9)
(n = 147)
0,4415 1,2
(-0,8; 3,3)
0,2372
V
ý
c
h
o
d
iskové BMI (kg/m
2
)‡
20,9 (2,8) 21,5 (3,0)
Absolútna zmena od východiskového BMI (kg/m
2
)
Týždeň 72 predĺženia
(n = 145)
0,62 (0,45; 0,79)
(n = 80)
< 0,0001
(n = 289)
0,69 (0,56; 0,81)
(n = 155)
< 0,0001
Týždeň 96 predĺženia 0,76 (0,56; 0,97)
< 0,0001 0,96 (0,81; 1,11)
< 0,0001
V
ý
c
h
o
d
iskové CFQ-R
s
k
ó
r
e respiračnej domény
(body)‡
70,4 (18,5) 68,3 (18,0)
Absolútna zmena CFQ-R skóre respiračnej domény (body)
Týždeň 72 predĺženia
(n = 135)
3,3 (0,7; 5,9)
0,0124
(n = 269)
5,7 (3,8; 7,5)
< 0,0001

(n = 81)
Týždeň 96 predĺženia 0,5
(-2,7; 3,6)
(n = 165)
0,7665 3,5 (1,3; 5,8)
0,0018
Placebo s prechodom na lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele
Priemer
(SD)
štvorcov
 (95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Počet pulmonálnych exacerbácií (udalosti) ** † ***
Počet udalostí na pacientorok
(95 % CI) (pomerný výskyt za 48 týždňov)
Počet udalostí vyžadujúcich hospitalizáciu na pacientorok (95 % CI) (pomerný výskyt za 48 týždňov)
Počet udalostí vyžadujúcich antibiotiká intravenózne na pacientorok (95 % CI) (pomerný výskyt za 48 týždňov)
0,69 (0,56; 0,85)
0,30 (0,22; 0,40)
0,37 (0,29; 0,49)
0,65 (0,56; 0,75)
0,24 (0,19; 0,29)
0,32 (0,26; 0,38)
*Celkovo 82 % (421 z 516 vhodných pacientov) dokončilo 72 týždňov tejto štúdie; 42 % dokončilo 96 týždňov.
U väčšiny pacientov došlo k prerušeniu liečby z iných ako bezpečnostných dôvodov.
** Celková expozícia u pacientov prevedených zo skúšaní 1 a 2 (skupina prevedená z placebo na lumakaftor/ivakaftor) bola až 96 týždňov. Podanie lumakaftoru 400 mg každých 12 hodín/ivakaftoru 250 mg každých 12 hodín v dávkovacej skupine je konzistentné s odporučeným dávkovaním.
*** Počet prípadov na pacientorok bol anualizovaný na 48 týždňov.
† Celková expozícia u pacientov prevedených zo skúšaní 1 a 2 (skupina pokračovala v používaní
kombinácie lumakaftor/ivakaftor) bola až 120 týždňov. Podanie lumakaftoru 400 mg každých 12 hodín/ivakaftoru
250 mg každých 12 hodín v dávkovacej skupine je konzistentné s odporučeným dávkovaním..
‡ Za východiskové hodnoty pre skupinu prevedenú z placeba na lumakaftor 400 mg každých 12 hodín/ivakaftoru 250 mg
každých 12 hodín sa brala východisková hodnota zo skúšania 3. Za východiskové hodnoty pre skupinu na lumakaftore
400 mg každých 12 hodín/ivakaftore 250 mg každých 12 hodín sa brala východisková hodnota zo skúšaní 1 a 2.
Skúšanie u pacientov s CF, ktorí sú heterozygotní nosiči mutácie F508del v géne CFTR
Skúšanie 4 bolo multicentrické, dvojito zaslepené, randomizované, placebom kontrolované skúšanie fázy 2 pre 125 pacientov s CF vo veku 18 rokov a starších, ktorí majú ppFEV1 od 40 - 90 vrátane,
a majú mutáciu F508del na jednej alele plus druhú alelu s mutáciou predpovedajúcou nedostatok
produkcie CFTR alebo CFTR, ktoré nereaguje na ivakaftor in vitro.
Pacienti užívali buď lumakaftor/ivakaftor (n = 62) alebo placebo (n = 63) navyše k svojej predpísanej terapii CF. Primárnym koncovým ukazovateľom bolo zlepšenie funkcie pľúc určené ako stredná hodnota absolútneho rozdielu ppFEV1 medzi východiskovou hodnotou a hodnotou v 56. deň. Liečba lumakaftorom/ivakaftorom nemala za následok významné zlepšenie ppFEV1 v porovnaní s placebom u pacientov s CF, heterozygotných nosičov mutácie F508del v géne CFTR (rozdiel v liečbe 0,60
[P = 0,5978]) a žiadne významné zlepšenie BMI alebo hmotnosti (pozri časť 4.4).
Skúšanie u pacientov s CF vo veku 6 až 11 rokov, ktorí sú homozygotní nosiči mutácie F508del v géne
CFTR
Skúšanie 7 bola 24- týždňová, placebom kontrolovaná klinická štúdia fázy 3 u 204 pacientov s CF vo veku 6 až 11 rokov (priemerný vek 8,8 rokov). Skúšanie 7 hodnotilo osoby s indexom pľúcneho klírensu (lung clearance index- LCI2,5) ≥ 7,5 pri skríningu pri prvej návšteve (priemer LCI2,5 bol 10,28 pred začiatkom liečby [rozmedzie: 6,55 až 16,38]) a ppFEV1 ≥ 70 pri skríningu (priemer ppFEV1 bol
89,8 pred začiatkom liečby [rozmedzie: 48,6 až 119,6]). Pacienti dostávali buď lumakaftor
200 mg/ivakaftor 250 mg každých 12 hodín (n = 103) alebo placebo (n = 101) okrem ich predpísanej liečby CF. Pacienti, ktorí mali 2 alebo viackrát vyššie hodnoty funkčných pečeňových testov (ALT,
AST, AP, GGT ≥ 3-krát ULN), alebo ALT alebo AST > 5-krát ULN alebo celkový bilirubín > 2-krát
ULN boli vylúčení.
Primárny koncový ukazovateľ účinnosti bola absolútna zmena v LCI2,5 od východiskovej hodnoty do
24. týždňa. Sekundárny kľúčový koncový ukazovateľ zahrnoval priemer absolútnej zmeny chloridov
v pote od východiskovej hodnoty do 15. dňa, 4. týždňa a 24. týždňa (pozri Farmakodynamické vlastnosti), absolútnu zmenu BMI od východiskovej hodnoty v 24. týždni, absolútnu zmenu v CFQ-R
respiračnej domény od východiskovej hodnoty do 24. týždňa. Tieto výsledky sú zhrnuté v tabuľke 7
nižšie:
Tabuľka 7: Súhrn primárnych a kľúčových sekundárnych výsledkov v skúšaní 7
LUM 200 mg/IVA
P
r
i
m
árny koncový ukazovateľ Absolútna zmena v indexe pľúcneho klírensu (LCI
2,5
) od východiskovej hodnoty do
24. týždňa
Placebo
(
n = 101)
Rozdiel v liečbe –
Zmena vrámci skupiny 0,08
(P = 0,5390)
250 mg každých
12 hod
(n = 103)
-1,09
(P < 0,0001)
-1,01 (P < 0,0001)
Kĺ
ú
čové sekundárne koncové body*
A
bsolútna zmena v BMI
2 Rozdiel v liečbe –
(P = 0,2522)
A
bsolútna zmena v skóre CFQ-R respiračnej domény do 24. týždňa (body)
Zmena v rámci skupiny 0,27
(P = 0,0002)
Rozdiel v liečbe –
Zmena v rámci skupiny 3,0
(P = 0,0035)
0,38
(P < 0,0001)
2,5
(P = 0,0628)
5,5
(P < 0,0001)

* Skúšanie obsahovalo kľúčový sekundárny a ďalšie sekundárne koncové ukazovateľe.
Percentuálny predpoklad FEV1 bol tiež vyhodnotený ako klinicky zmysluplný ďalší sekundárny
koncový bod. U pacientov s lumakaftorom/ivakaftorom bol rozdiel v liečbe pre absolútnu zmenu
v ppFEV1 od východiskovej hodnoty do 24. týždňa 2,4 (P
= 0,0182).
PediatrickápopuláciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Orkambi v jednej
alebo vo viacerých podskupinách pediatrickej populácie s cystickou fibrózou. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnostiExpozícia (AUC) lumakaftoru je približne 2-násobne vyššia u zdravých dospelých dobrovoľníkov v porovnaní s expozíciou u pacientov s CF. Expozícia ivakaftoru je podobná medzi zdravými dospelými dobrovoľníkmi a pacientmi s CF. Po dávkovaní dvakrát denne bol ustálený stav plazmatickej koncentrácie u zdravých jedincov dosiahnutý všeobecne približne po 7 dňoch liečby
s akumulačným pomerom približne 1,9 pre lumakaftor. Ustálený stav expozície ivakaftoru je nižší ako pri 1. dni kvôli indukčnému účinku CYP3A lumakaftorom (pozri časť 4.5).
Po perorálnom podaní lumakaftoru 400 mg každých 12 hod/ivakaftoru 250 mg každých 12 hod po jedle boli namerané hodnoty v ustálenom stave (±SD) pre AUC0-12hod a Cmax 198 (64,8) µg∙h/ml a 25,0 (7,96) µg/ml pre lumakaftor v uvedenom poradí a 3,66 (2,25) µg∙h/ml a 0,602 (0,304) µg/ml pre ivakaftor. Po perorálnom podaní ivakaftoru samotného ako 150 mg každých 12 hod boli namerané
hodnoty v rovnovážnom stave (±SD) pre AUC0-12h a Cmax pri 9,08 (3,20) µg∙h/ml a 1,12 (0,319) µg/m
v uvedenom poradí.
Absorpcia
Po opakovanom podaní perorálnej dávky lumakaftoru sa expozícia lumakaftoru zvyčajne úmerne zvýšila s dávkou od 50 mg do 1 000 mg každých 24 hodín. Expozícia lumakaftoru sa zvýšila približne
2,0-násobne pri podaní s jedlom obsahujúcim tuky v porovnaní s podaním nalačno. Medián
(rozmedzie) tmax lumakaftoru po jedle je približne 4,0 hodiny (2,0; 9,0).
Po opakovanom podaní perorálnej dávky ivakaftoru v kombinácii s lumakaftorom sa expozícia ivakaftoru zvyčajne zvýšila úmerne s dávkou od 150 mg každých 12 hodín do 250 mg každých
12 hodín. Expozícia ivakaftoru pri podaní v kombinácii s lumakaftorom sa zvýšila u zdravých dobrovoľníkov približne 3-násobne pri podávaní s jedlom obsahujúcim tuky. Preto sa má
lumakaftor/ivakaftor podávať s jedlom obsahujúcim tuky. Medián (rozmedzie) tmax po jedle je
približne 4,0 hodiny (2,0; 6,0).
Distribúcia
Približne 99 % lumakaftoru sa viaže na plazmatické proteíny, predovšetkým na albumín. Po
perorálnom podaní 400 mg lumakaftoru každých 12 hodín u pacientov s CF po jedle bol zdanlivý distribučný objem pre centrálne a periférne kompartmenty (CV) 23,5 l (48,7 %) a 33,3 l(30,5 %).
Približne 99 % ivakaftoru sa viaže na plazmatické proteíny, predovšetkým na alfa 1-kyslý glykoproteín a ambumín. Po perorálnom podaní 250 mg ivakaftoru každých12 hodín v kombinácii
s lumakaftorom boli zdanlivé distribučné objemy pre centrálne a periférne kompartmenty [variačný koeficient ako percento (CV)] (CV) 95,0 l (53,9 %), respektíve 201 l, (26,6 %).
In vitro štúdie naznačujú, že lumakaftor je substrát proteínu zodpovedného za rezistenciu pri
karcinóme prsníka (BCRP).
Biotransformácia
Lumakaftor nie je u ľudí extenzívne metabolizovaný s väčšinou lumafaktoru vylúčenou v nezmenenej forme v stolici. In vitro a in vivo údaje naznačujú, že lumakaftor je metabolizovaný predovšetkým oxidáciou a glukuronidáciou.
Ivakaftor je u ľudí extenzívne metabolizovaný. In vitro a in vivo údaje naznačujú, že ivakaftor je metabolizovaný predovšetkým prostredníctvom CYP3A. M1 a M6 sú dva hlavné metabolity ivakaftoru u ľudí. Účinnosť metabolitu M1 zodpovedá približne jednej šestine účinnosti ivakaftoru a M1 sa považuje za farmakologicky aktívny. M6 má menej ako jednu päťdesiatinu účinnosti ivakaftoru a nepovažuje sa za farmakologicky aktívny.
Eliminácia
Po perorálnom podaní lumakaftoru sa väčšina lumakaftoru (51 %) vylúči nezmenená v stolici. Zanedbateľné množstvo lumakaftoru sa vylúči ako nezmenené liečivo močom. Zdanlivý terminálny
polčas je približne 26 hodín. Zdanlivý klírens CL/F (CV) lumakaftoru pre pacientov s CF bol
2,38 l/hod (29,4 %) .
Po perorálnom podaní samotného ivakaftoru sa väčšina (87,8 %) eliminuje v stolici po metabolickej premene. Zanedbateľné množstvo ivakaftoru sa vylúči močom v nezmenenej podobe. U zdravých jedincov je polčas ivakaftoru po podaní s lumakaftorom približne 9 hodín. Typický CL/F (CV) ivakaftoru po podaní v kombinácii s lumakaftorom bol u pacientov s CF 25,1 l/h (40,5 %).
Osobitnéskupinypacientov
Poruchafunkciepečene
Po opakovanom podávaní lumakaftoru/ivakaftoru 10 dní mali jedinci so stredne závažnou poruchou funkcie pečene (Child-Pughova trieda B, skóre 7 až 9) vyššiu expozíciu (AUC0-12hod o približne 50 % a Cmax o približne 30 %) v porovnaní so zdravými demograficky zodpovedajúcimi jedincami. Vplyv miernej poruchy funkcie pečene (Child-Pughova trieda A, skóre 5 až 6) na farmakokinetiku
lumakaftoru v kombinácii s ivakaftorom sa neskúmal, ale predpokladá sa zvýšenie expozície nižšie ako 50 %.
U pacientov so závažnou poruchou funkcie pečene (Child-Pughova trieda C, skóre10 až 15) sa neuskutočnili štúdie, ale predpokladá sa, že expozícia bude vyššia ako u pacientov so stredne závažnou poruchou funkcie pečene (pozri časti 4.2, 4.4 a 4.8).
Poruchafunkcieobličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili farmakokinetické štúdie
s lumakaftorom/ivakaftorom. Vo farmakokinetickej štúdii u ľudí sa zistila minimálna eliminácia lumakaftoru a jeho metabolitov močom (iba 8,6 % z celkovej rádioaktivity sa vylúčilo močom
a 0,18 % sa vylúčilo v nezmenenej podobe). Vo farmakokinetickej štúdii u ľudí s ivakaftorom sa
zistila minimálna eliminácia ivakaftoru a jeho metabolitov močom (iba 6,6 % z celkovej dávky rádioaktivity sa vylúčilo v moči). Populačná farmakokinetická analýza klírensu v porovnaní
s klírensom kreatinínu nevykazuje žiadny trend u jedincov s miernou a stredne ťažkou poruchou
funkcie obličiek (pozri časť 4.2).
Staršie osobyí
Bezpečnosť a účinnosť lumakaftoru/ivakaftoru u pacientov vo veku 65 rokov alebo starších neboli skúmané.
Pohlavie
Vplyv pohlavia na farmakokinetiku lumakaftoru sa hodnotil pomocou údajov populačnej
farmakokinetiky z klinických štúdií lumakaftoru v kombinácii s ivakaftorom. Výsledky neukázali žiaden klinicky významný rozdiel vo farmakokinetických parametroch lumakaftoru alebo ivakaftoru medzi mužmi a ženami. Na základe pohlavia nie je potrebná úprava dávky.
Pediatrickápopulácia
Expozície u dospelých a v pediatreckej populácii sú podobné, na základe populačných analýz (PK)
znázornených v tabuľke 8 nižšie:
T
abuľka 8: Priemerná (SD) expozícia lumakaftoru a ivakaftoru podľa veku
P
r
i
e
m
erná (SD)
P
r
i
e
m
erná (SD)
V
eková skupina Dávka
AUC
ss
l
umakaftoru (μg/ml*hod)
AUC
ss
i
vakaftoru (μg/ml*hod)
Pacienti vo veku 6 až
11 rokov
Pacienti vo veku 12 až menej ako 18 rokov
lumakaftor 200 mg/ivakaftor
250 mg každých 12 hodín lumakaftor 400 mg/ivakaftor
250 mg každých 12 hodín
203 (57,4) 5,26 (3,08)
241 (61,4) 3,90 (1,56)
 5.3 Predklinické údaje o bezpečnosti
L
umakaftor
5.3 Predklinické údaje o bezpečnosti
L
umakaftor
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu a reprodukčnej toxicity a vývinu
neodhalili žiadne osobitné riziko pre ľudí. Špecifické štúdie týkajúce sa hodnotenia fototoxického
potenciálu lumakaftoru neboli vykonané, hoci na základe zhodnotenia dostupných predklinických a klinických údajov sa fototoxicita nepredpokladá.
IvakaftorÚčinky v štúdiách s opakovaným podaním sa pozorovali iba pri expozíciách považovaných za dostatočne vyššie (> 25-násobne, > 45-násobne a > 35-násobne pre myši, mačky, potkany a psov) ako
je maximálna expozícia ivakaftoru u ľudí pri podávaní Orkambi, čo poukazuje na malý význam týchto
zistení pre klinické použitie. Predklinické údaje, založené na obvyklých štúdiách genotoxicity a karcinogénneho potenciálu neodhalili žiadne zvláštne riziko pre ľudí.
B
ezpečnostná farmakológia
Ivakaftor vyvolal od koncentrácie závislý inhibičný účinok na koncové časti proteínu hERG (human ether-à-go-go related gene) s IC15 5,5 µM, v porovnaní s Cmax (1,5 µM) pre ivakaftor
pri terapeutickom dávkovaní lumakaftoru/ivakaftoru. V telemetrickej štúdii u psov s jednorazovými
dávkami až do 60 mg/kg ani pri EKG meraniach zo štúdií s opakovanou dávkou s trvaním až do
1 roka pri dávkovej hladine 60 mg/kg/deň u psov (Cmax po 365 dňoch = 36,2 až 47,6 μM) sa však nepozorovalo predĺženie QT intervalu indukované ivakaftorom. Ivakaftor vyvolal od dávky závislé, avšak prechodné zvýšenie parametrov krvného tlaku u psov v jednorazových perorálnych dávkach až do 60 mg/kg. V dôkladnej klinickej štúdii hodnotiacej QT interval pri podaní buď lumakaftor 600 mg raz denne/ivakaftor 250 mg každých 12 hodín alebo lumakaftor 1 000 mg raz denne/ivakaftor 450 mg každých 12 hodín, sa nepozorovali žiadne zmeny QTc intervalu alebo krvného tlaku, preukazujúce nemožnosť prenosu týchto predklinických výsledkov do klinickej praxe.
Gravidita a fertilita
Ivakaftor nebol teratogénny pri perorálnom podaní gravidným potkanom a králikom počas štádia organogenézy fetálneho vývoja pri dávkach približne 7-násobku (expozícia ivakaftoru a metabolitov), respektíve 46-násobku expozície ivakaftoru u človeka pri terapeutickej dávke lumakaftoru/ivakaftoru. Pri dávkach toxických pre matku spôsobil ivakaftor u potkanov zníženie telesnej hmotnosti plodu, zvýšenie incidencie variácií krčných rebier, hypoplastických rebier, zvlnenie rebier a sternálnych nepravidelností, vrátane fúzií. Význam týchto zistení pre ľudí nie je známy.
Ivakaftor znížil ukazovatele fertility a reprodukčnej výkonnosti u samcov a samíc potkanov pri dávke
200 mg/kg/deň (dosahujúc expozície 11-násobku, respektíve 7-násobku expozícií pri maximálnej odporúčanej dávke pre ľudí ivakaftorovej zložky Orkambi, založených na sumárnych AUC ivakaftoru a jeho metabolitov extrapolovaných z expozícií dosiahnutých na 90. deň pri podaní 150 mg/kg/deň
v 6-mesiacov trvajúcej štúdii toxicity s opakovaným podaním a z expozícií v 17. gestačnom dni
v pilotnej štúdii embryofetálneho vývoja u tohto druhu), keď boli samice dávkované pred a počas skorého štádia gravidity. Nebol pozorovaný žiaden vplyv na ukazovatele fertility a reprodukčnej výkonnosti u samcov a samíc pri dávke ≤ 100 mg/kg/deň (dosahujúc expozície približne 8-násobku, respektíve 5-násobku expozícií pri maximálnej odporúčanej dávke ivakaftoru, zložke Orkambi, založených na sumárnych AUC ivakaftoru a jeho metabolitov extrapolovaných z expozícií z 90. dňa pri podaní 100 mg/kg/deň v 6-mesiacov trvajúcej štúdii toxicity s opakovaným podaním
a v embryofetálnej štúdii v 17. gestačnom dni u tohto druhu). U gravidných samíc potkanov a králikov sa pozoroval transplacentárny prestup ivakaftoru.
Perinatálny a postnatálny vývoj
Ivakaftor nespôsobil vývojové defekty u potomkov gravidných potkanov, ktorí dostávali liek perorálne od gravidity po pôrod a odstavenie v dávke 100 mg/kg/deň. Dávky nad 100 mg/kg/deň viedli
k zníženiu indexov prežívania a laktácie na 92 % a 98 % kontrolných hodnôt, v uvedenom poradí,
rovnako ako k zníženiu telesnej hmotnosti mláďat.
Juvenilné zvieratá
Prípady katarakty boli pozorované u mláďat potkanov pri dávke ivakaftoru 0,32-násobku maximálnej odporúčanej dávky na základe systémovej expozície ivakaftoru a jeho metabolitov, ak je podávaný
s lumakaftorom ako Orkambi. Prípady katarakty neboli pozorované u plodov pochádzajúcich z potkaních matiek liečených počas organogenézy fetálneho vývoja, potkaních mláďat vystavených do
určitej miery požitím mlieka až do odstavenia alebo v štúdiách toxicity po opakovanom podaní ivakaftoru. Potenciálny význam týchto zistení u ľudí nie je známy.
Lumakaftoraivakaftor
Štúdie toxicity po opakovanom podaní zahŕňajúce súbežné podávanie lumakaftoru a ivakaftoru neobjavili žiadne špeciálne riziká pre ľudí z hľadiska potenciálu prídavnej alebo synergickej toxicity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
J
adro
t
a
blety Mikrokryštalická celulóza Sodná soľ kroskarmelózy Acetát sukcinát hypromelózy Povidón (K30)
Laurylsíran sodný
Stearan horečnatý
Filmovýobaltablety
Polyvinylalkohol
Oxid titaničitý (E171) Makrogol 3350
Mastenec
Karmín (E120)
Hlinitý lak žiarivá modrá FCF (E 133) Hlinitý lak indigokarmínu (E 132)
Atramentovápotlač
Šelak
Čierny oxid železitý (E172) Propylénglykol
Hydroxid amónny
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Orkambi100mg/125mgfilmomobalenétablety
3 roky
Orkambi200mg/125mgfilmomobalenétablety
4 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Pretlačovacie balenie pozostáva z polychlórtrifluóretylénu (PCTFE)/ polyvinylchloridu (PVC)
s krytom z hliníkovej fólie vystuženej papierom.
Orkambi100mg/125mgfilmomobalenétablety
Balenie obsahuje 112 filmom obalených tabliet (4 balenia, v každom 28 tabliet).
Orkambi200mg/125mgfilmomobalenétablety
Balenie obsahuje 28 filmom obalených tabliet.
Multibalenie obsahuje 56 filmom obalených tabliet (2 balenia, v každom 28 tabliet). Multibalenie obsahuje 112 filmom obalených tabliet (4 balenia, v každom 28 tabliet).
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIVertex Pharmaceuticals (Ireland) Limited
28-32 Pembroke Street Upper
Dublin 2, D02 EK84
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1059/001
EU/1/15/1059/002
EU/1/15/1059/003
EU/1/15/1059/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. novembra 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUOrkambi 100 mg/125 mg granulát vo vrecku
Orkambi 150 mg/188 mg granulát vo vrecku
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEOrkambi100mg/125 mg granulátvovreckuKaždé vrecko obsahuje 100 mg lumakaftoru (
lumacaftorum) a 125 mg ivakaftoru (
ivacaftorum).
Orkambi150mg/188 mg granulátvovreckuKaždé vrecko obsahuje 150 mg lumakaftoru (
lumacaftorum) a 188 mg ivakaftoru (
ivacaftorum).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAGranulát
Biely až takmer biely granulát.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieOrkambi granulát je indikovaný na liečbu cystickej fibrózy (CF) deťom vo veku 2 rokov a starším,
ktoré sú homozygotní nosiči mutácie
F508del v géne
CFTR (pozri časti 4.2, 4.4 a 5.1).
4.2 Dávkovanie a spôsob podávaniaOrkambi majú predpisovať len lekári so skúsenosťami s liečbou cystickej fibrózy. Ak je genotyp pacienta neznámy, má byť vykonaná genotypizácia presnou a validovanou metódou na potvrdenie prítomnosti mutácie
F508del na obidvoch alelách génu
CFTR.DávkovanieOdporúčané dávkovanie pozri v tabuľke 1.
Tabuľka 3: Odporúčané dávkovanie Orkambi u pacientov vo veku 2 rokov a staršíchVek Dávka Orkambi Celková denná dávka
2 až 5 rokov
s hmotnosťou pod
14 kg
2 až 5 rokov
s hmotnosťou
14 kg a viac
Jedno vrecko lumakaftor 100 mg/ivakaftor
125 mg každých 12 hodín
Jedno vrecko lumakaftor 150 mg/ivakaftor
188 mg každých 12 hodín
lumakaftor 200 mg/
ivakaftor 250 mg
lumakaftor 300 mg/
ivakaftor 376 mg
6 rokov a starší Viac informácií pozri v SPC Orkambi tablety
Orkambi sa má užívať s jedlom obsahujúcim tuky. Jedlo alebo občerstvenie obsahujúce tuky sa má konzumovať tesne pred alebo tesne po užití lieku (pozri časť 5.2).
Vynechaná dávka
Ak uplynulo menej ako 6 hodín od vynechanej dávky, plánovaná dávka Orkambi má byť ihneď užitá
s jedlom obsahujúcim tuky. Ak uplynulo viac ako 6 hodín, pacientovi má byť povedané, aby počkal do
nasledujúcej pravidelnej dávky. Dvojitá dávka sa nemá užívať ako náhrada za vynechanú dávku.
Súbežné používanie inhibítorov CYP3A
Nie je potrebná úprava dávky u pacientov, ktorí začínajú používať inhibítory CYP3A, ak súbežne
užívajú Orkambi. Ak ale Orkambi začínajú užívať pacienti užívajúci silné inhibítory CYP3A, dávka má byť znížená na jedno vrecko každý druhý deň (lumakaftor 100 mg/ivakaftor 125 mg u pacientov vo veku 2 až 5 rokov s hmotnosťou pod 14 kg; lumakaftor 150 mg/ivakaftor 188 mg u pacientov vo veku 2 až 5 rokov s hmotnosťou 14 kg a viac) v prvom týždni liečby, aby sa zohľadnil indukčný účinok lumakaftoru v ustálenom stave. Následne po tomto období sa má pokračovať v užívaní odporúčanej dennej dávky.
Ak sa liečba Orkambi preruší na viac ako jeden týždeň a potom obnoví, kým pacient užíva silné inhibítory CYP3A, dávka Orkambi sa má znížiť na jedno vrecko každý druhý deň (lumakaftor
100 mg/ivakaftor 125 mg u pacientov vo veku 2 až 5 rokov s hmotnosťou pod 14 kg; lumakaftor
150 mg/ivakaftor 188 mg u pacientov vo veku 2 až 5 rokov s hmotnosťou 14 kg a viac) v prvom týždni po opätovnom začatí liečby. Následne po tomto období sa má pokračovať v užívaní odporúčanej dennej dávky (pozri časť 4.5).
Osobitnéskupinypacientov
Staršie osoby
Bezpečnosť a účinnosť Orkambi u pacientov vo veku 65 rokov alebo starších neboli stanovené.
Porucha funkcie obličiek
U pacientov s miernou až stredne závažnou poruchou funkcie obličiek nie je potrebná úprava dávky. Opatrnosť sa odporúča pri používaní Orkambi u pacientov so závažnou poruchou funkcie obličiek
(klírens kreatinínu menší alebo rovný 30 ml/min) alebo s renálnym ochorením v terminálnom štádiu
(pozri časti 4.4 a 5.2).
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (Child-Pughova trieda A) nie je potrebná úprava dávky. U pacientov so stredne závažnou poruchou funkcie pečene (Child-Pughova trieda B) sa odporúča znížená dávka.
Nie sú žiadne skúsenosti s používaním Orkambi u pacientov so závažnou poruchou funkcie pečene (Child-Pughova trieda C), ale predpokladá sa vyššia expozícia ako u pacientov so stredne závažnou poruchou funkcie pečene. Po zvážení rizík a prínosov liečby sa preto má Orkambi používať
s opatrnosťou v zníženej dávke (pozri časti 4.4, 4.8 a 5.2).
Úpravu dávky u pacientov s poruchou funkcie pečene pozri v tabuľke 2.
T
abuľka 4: Úprava dávky odporúčaná u pacientov s poruchou funkcie pečene
P
orucha funkcie pečene Úprava dávky Celková denná dávka
U pacientov
vo
veku
2
až
5
rokov
s hmotnosťou
< 14
kg
lumakaftor 200 mg + ivakaftor
Mierna porucha funkcie pečene
(Child-Pughova trieda A)
Bez úpravy dávky
250 mg
U pacientovvoveku2až5rokovs hmotnosťou≥14kg
lumakaftor 300 mg + ivakaftor
376 mg
Stredne závažná porucha funkcie pečene
(Child-Pughova trieda B)
1 vrecko každé ráno a každý druhý deň 1 vrecko večer
U pacientovvoveku2až5rokov
s hmotnosťou< 14kg
Prvý deň: lumakaftor
200 mg + ivakaftor 250 mg Druhý deň: lumakaftor 100 mg + ivacaftor 125 mg
U pacientovvoveku2až5rokovs hmotnosťou≥14kg
Prvý deň: lumakaftor 300 mg +
ivacaftor 376 mg
Druhý deň: lumakaftor 150 mm +
ivakaftor 188 mg
Závažná porucha funkcie pečene
(Child-Pughova trieda C)
1 vrecko denne alebo menej často
U pacientovvoveku2až5rokov
s hmotnosťou< 14kg
lumakaftor 100 mg + ivakaftor
125 mg
U pacientovvoveku2až5rokovs hmotnosťou≥14kg
lumakaftor 150 mm + ivakaftor
188 mg
P
ediatrická populácia
Bezpečnosť a účinnosť Orkambi u detí mladších ako 2 roky neboli doteraz stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Spôsob podávaniaNa perorálne použitie.
Každé vrecko je len na jedno použitie.
Celý obsah vrecka s granulátom sa má zmiešať s jednou čajovou lyžičkou (5 ml) kašovitého jedla alebo tekutiny vhodných pre daný vek a následne sa má celý skonzumovať. Príklady kašovitého jedla sú ovocné pyré, ochutený jogurt alebo mlieko a džús. Jedlo alebo tekutina majú mať izbovú alebo nižšiu ako izbovú teplotu. Preukázalo sa, že liek je po zmiešaní stabilný jednu hodinu a preto sa má
v takomto čase skonzumovať.
4.3 Kontraindikácie
Precitlivenosť na liečivá alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
PacientisCF,ktorísúheterozygotnínosičimutácie F508del vgéneCFTR
Lumakaftor/ivakaftor nie je účinný u pacientov s CF, ktorí majú mutáciu F508del na jednej alele plus druhú alelu s mutáciou predpovedajúcou nedostatok produkcie CFTR alebo in vitro nereaguje na
ivakaftor (pozri časť 5.1).
PacientisCF,ktorímajúmutáciuvrátkovania(triedaIII)vgéneCFTR
Lumakaftor/ivakaftor nebol študovaný u pacientov s CF, ktorí majú mutáciu vrátkovania (trieda III)
v géne CFTR na jednej alele, s alebo bez mutácie F508del na druhej alele. Keďže expozícia ivakaftoru je pri podávaní v kombinácii s lumakaftorom veľmi významne znížená, nemá sa kombinácia lumakaftor/ivakaftor u týchto pacientov používať.
Respiračnépríhody
Respiračné príhody (napr. hrudný dyskomfort, dyspnoe a zmenené dýchanie) boli častejšie na začiatku liečby lumakaftorom/ivakaftorom. Závažné respiračné príhody boli častejšie pozorované u pacientov
s hodnotou úsilného expiračného objemu v prvej sekunde vyjadreného v percentách predpokladanej hodnoty (percent predicted forced expiratory volume in 1 second, ppFEV1) < 40, a môžu viesť ukončeniu podávania lieku. Klinické skúsenosti u pacientov s ppFEV1< 40 sú obmedzené a v priebehu začiatku liečby sa odporúča ďalšie sledovanie týchto pacientov (pozri časť 4.8). U niektorých pacientov bol tiež pozorovaný prechodný pokles hodnoty FEV1 po začatí liečby lumakaftorom/ivakaftorom. Nie sú žiadne skúsenosti so začatím liečby lumakaftorom/ivakaftorom u pacientov s pľúcnou exacerbáciou a preto začatie liečby u pacientov s pľúcnou exacerbáciou nie je vhodné.
Vplyvnakrvnýtlak
U niektorých pacientov liečených lumakaftorom/ivakaftorom bol pozorovaný zvýšený krvný tlak. Počas liečby by sa mal krvný tlak pravidelne sledovať u všetkých pacientov (pozri časť 4.8).
Pacientispokročilýmochorenímpečene
U pacientov s CF môžu byť prítomné abnormality pečeňovej funkcie, zahŕňajúce pokročilé ochorenie pečene. U pacientov s pokročilým ochorením pečene bolo hlásené zhoršenie funkcie pečene.
U pacientov s CF s pre-existujúcou cirhózou pečene s portálnou hypertenziou, ktorí užívajú
lumakaftor/ivakaftor, bola hlásená dekompenzácia funkcie pečene, vrátane zlyhania pečene vedúceho k úmrtiu. Lumakaftor/ivakaftor sa má používať s opatrnosťou u pacientov s pokročilým ochorením pečene a iba ak sa predpokladá, že prínosy prevážia nad rizikami. Ak je lumakaftor/ivakaftor používaný u týchto pacientov, majú byť pozorne sledovaní po začatí liečby a dávka má byť znížená (pozri časti 4.2, 4.8 a 5.2).
Hepatobiliárnepríhody
Zvýšené hodnoty transamináz boli často hlásené u pacientov s CF užívajúcich lumakaftor/ivakaftor.
V niektorých prípadoch boli tieto elevácie spojené so súčasnou eleváciou celkového bilirubínu v sére. Zvýšené hodnoty transamináz sa pozorovali častejšie u pediatrických pacientov ako u dospelých pacientov (pozri časť 4.8). V rámci pediatrických kohort rozdielnych podľa veku sa zvýšenia hodnôt transamináz pozorované u pacientov vo veku 2 až 5 rokov vyskytovali častejšie ako u pacientov vo veku 6 až 11 rokov (pozri časť 4.8).
Pretože nemôže byť vylúčená asociácia s poškodením pečene, je pred začatím liečby lumakaftorom/ivakaftorom každé 3 mesiace počas prvého roka liečby a následne raz ročne odporúčané vykonanie funkčných pečeňových testov (ALT, AST a bilirubín). U pacientov so zvýšenými
hodnotami ALT, AST alebo bilirubínu sa má zvážiť častejšie monitorovanie.
V prípade významného zvýšenia hodnôt ALT alebo AST, s alebo bez elevácie bilirubínu (ALT alebo AST > 5 x horná hranica normy [the upper limit of normal - ULN] alebo ALT alebo AST > 3 x ULN s bilirubínom > 2 x ULN), má byť podávanie lumakaftoru/ivakaftoru prerušené a majú sa
vykonávať laboratórne testy, kým sa abnormality nevyriešia. Následne po vyriešení zvýšených hodnôt transamináz majú byť zvážené prínosy a riziká podávania (pozri časti 4.2, 4.8 a 5.2).
Liekovéinterakcie
Substráty CYP3A
Lumakaftor je silný induktor CYP3A. Súbežné používanie so senzitívnymi substrátmi CYP3A alebo substrátmi CYP3A s úzkym terapeutickým indexom sa neodporúča (pozri časť 4.5).
Hormonálna antikoncepcia zahŕňajúca formu perorálnu, injekčnú, transdermálnu a implantačnú, nemá byť považovaná za spoľahlivú pri súbežnom podávaní Orkambi (pozri časť 4.5).
Silné induktory CYP3A
Ivakaftor je substrátom CYP3A4 a CYP3A5. Preto súbežné používanie so silnými induktormi CYP3A (napr. rifampicín, ľubovník bodkovaný [Hypericum perforatum]) sa neodporúča (pozri bod 4.5).
Poruchafunkcieobličiek
Opatrnosť sa odporúča pri používaní lumakaftoru/ivakaftoru u pacientov so závažnou poruchou funkcie obličiek alebo s renálnym ochorením v terminálnom štádiu (pozri časti 4.2 a 5.2).
Katarakta
U pediatrických pacientov liečených lumakaftorom/ivakaftorom a monoterapiou ivakaftorom boli hlásené prípady nekongenitálneho zakalenia šošovky bez vplyvu na zrak. Hoci v niektorých prípadoch
boli prítomné iné rizikové faktory (ako používanie kortikosteroidov, expozícia ožiarenia), možné
riziko pripísateľné ivakaftoru nemôže byť vylúčené (pozri časť 5.3). U pediatrických pacientov, ktorí začali liečbu lumakaftorom/ivakaftorom, sú odporúčané základné a následné oftalmologické vyšetrenia.
Pacientipotransplantáciiorgánov
Lumakaftor/ivakaftor sa neskúmal u pacientov s CF, ktorí podstúpili transplantáciu orgánov. Preto sa používanie u pacientov po transplantácii neodporúča. Pozri časť 4.5 pre interakcie
s imunosupresívami.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Na základe expozícií a indikovaných dávok sa profil liekových interakcií považuje za rovnaký pre všetky sily a liekové formy.
Lumakaftor je silný induktor CYP3A a ivakaftor je slabý inhibítor CYP3A, ak sú podané
v monoterapii. Pri súbežnom používaní je riziko, že iné lieky ovplyvnia lumakaftor/ivakaftor a tiež, že lumakaftor/ivakaftor ovplyvní iné lieky.
Potenciálinýchliekovovplyvniťlumakaftor/ivakaftor
Inhibítory CYP3A
Súbežné podávanie lumakaftoru/ivakaftoru s itrakonazolom, silným inhibítorom CYP3A, nemalo vplyv na expozíciu lumakaftoru, ale zvýšilo expozíciu ivakaftoru 4,3-násobne. Kvôli indukčnému
vplyvu lumakaftoru na CYP3A v ustálenom stave sa neočakáva, že by čistá expozícia ivakaftoru pri
súbežnom podávaní s inhibítorom CYP3A prekročila čistú expozíciu pri podaní bez
lumakaftoru v dávke 150 mg každých 12 hodín, čo je schválená dávka ivakaftoru pri monoterapii.
U pacientov, ktorí začali užívať inhibítory CYP3A a súbežne užívajú lumakaftor/ivakaftor nie je potrebná úprava dávky. Ak však začínajú užívať lumakaftor/ivakaftor pacienti užívajúci silné inhibítory CYP3A, dávka má byť upravená (pozri časti 4.2 a 4.4).
Pri používaní so stredne silnými alebo slabými inhibítory CYP3A sa neodporúča úprava dávky.
Induktory CYP3A
Súbežné podávanie lumakaftoru/ivakaftoru s rifampicínom, silným induktorom CYP3A, malo minimálny účinok na expozíciu lumakaftoru, ale znížilo expozíciu ivakaftoru (AUC) o 57 %. Preto sa
neodporúča súbežné podávanie lumakaftoru/ivakaftoru so silnými induktormi CYP3A (pozri
časti 4.2 a 4.4).
Pri používaní so stredne silnými alebo slabými induktormi CYP3A sa neodporúča úprava dávky.
Potenciállumakaftoru/ivakaftoruovplyvniťinélieky
Substráty CYP3A
Lumakaftor je silný induktor CYP3A. Ivakaftor je slabý inhibítor CYP3A pri podaní v monoterapii. Ako výsledný efekt terapie lumakaftorom/ivakaftorom sa predpokladá silná indukcia CYP3A. Preto
súbežné podávanie lumakaftoru/ivakaftoru so substrátmi CYP3A môže znížiť expozíciu týchto substrátov (pozri časť 4.4).
Substráty P-gp
In vitro štúdie ukazujú, že lumakaftor má potenciál inhibovať aj indukovať P-gp. Okrem toho klinická štúdia s ivakaftorom v monoterapii ukázala, že ivakaftor je slabý inhibítor P-gp. Preto môže súbežné
podávanie lumakaftoru/ivakaftoru so substrátmi P-gp (napr. digoxínom) zmeniť expozíciu týchto
substrátov.
Substráty CYP2B6 a CYP2C
Interakcie so substrátmi CYP2B6 a CYP2C sa neskúmali in vivo. Na základe in vitro štúdií sa predpokladá, že lumakaftor má potenciál indukovať CYP2B6, CYP2C8, CYP2C9 a CYP2C19; avšak
inhibícia CYP2C8 a CYP2C9 bola tiež pozorovaná in vitro. Okrem toho in vitro štúdia naznačuje, že ivakaftor môže inhibovať CYP2C9. Preto súbežné podávanie lumakaftoru/ivakaftoru môže zmeniť (tj.
buď zvýšiť, alebo znížiť) expozíciu substrátov CYP2C8 a CYP2C9, znížiť expozíciu CYP2C19
substrátov a podstatne znížiť expozíciu substrátov CYP2B6.
Potenciállumakaftoru/ivakaftoruinteragovaťstransportérmi
In vitro experimenty preukazujú, že lumakaftor je substrát pre proteín zodpovedný za rezistenciu pri
karcinóme prsníka (Breast Cancer Resistance Protein ‒ BCRP). Súbežné podanie lieku Orkambi s liekmi, ktoré inhibujú BCRP, môže zvýšiť plazmatické koncentrácie lumakaftoru. Lumakaftor inhibuje transportér organického aniónu (organic anion transporter ‒ OAT) 1 a 3. Lumakaftor
a ivakaftor sú inhibítory BCRP. Súbežné podanie lieku Orkambi s liekmi, ktoré sú substrátmi pre
transport OAT1/3 a BCRP môže zvýšiť plazmatické koncentrácie takých liekov. Lumakaftor
a ivakaftor nie sú inhibítory OATP1B1, OATP1B3 a transportéra organického katiónu (organic cation
transporter ‒ OCT) 1 a 2. Ivakaftor nie je inhibítor OAT1 a OAT3.
Overenéainépotenciálnevýznamnéliekovéinterakcie
Tabuľka 3 zahŕňa overené alebo predpokladané účinky lumakaftoru/ivakaftoru na iné lieky alebo účinok iných liekov na lumakaftor/ivakaftor. Informácie uvedené v tabuľke 3 väčšinou pochádzajú
z in vitro štúdií. Odporúčania poskytnuté v rámci „Klinického komentára“ v tabuľke 3 sú založené na
liekových interakčných štúdiách, klinickej závažnosti alebo predpokladaných interakciách vzhľadom na cesty eliminácie. Liekové interakcie, ktoré sú klinicky najvýznamnejšie, sú uvedené ako prvé.
T
abuľka 3: Stanovené a ďalšie potenciálne významné liekové interakcie – odporúčané dávkovanie pre použitie lumakaftoru/ivakaftoru s ďalšími liekmi
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Súbežne podávané lieky s najväčším klinickým významom
A
ntialergiká:
montelukast ↔ LUM, IVA
↓ montelukast
kvôli indukcii CYP3A/2C8/2C9 prostredníctvom LUM
Nie je potrebná úprava dávky
montelukastu. Pri súbežnom podávaní s lumakaftorom/ivakaftorom má byť začaté vhodné klinické monitorovanie, ak je potrebné. Lumakaftor/ivakaftor môže znížiť expozíciu montelukastu, čo môže znížiť jeho účinnosť.
fexofenadín ↔ LUM, IVA
A
ntibiotiká: klaritromycín, telitromycín
↑ alebo ↓ fexofenadín
vzhľadom k potenciálnej indukcii alebo inhibícii

P-gp
↔ LUM
↑ IVA
kvôli inhibícii CYP3A
prostredníctvom klaritromycínu, telitromycínu
↓ klaritromycín, telitromycín
kvôli indukcii CYP3A
prostredníctvom LUM
Úprava dávky fexofenadínu môže byť
potrebná k získaniu požadovaného klinického účinku. Lumakaftor/ivakaftor môže zmeniť expozíciu fexofenadínu.
Pri začatí liečby klaritromycínom alebo telitromycínom u pacientov užívajúcich lumakaftor/ivakaftor nie je potrebná úprava dávky lumakaftoru/ivakaftoru.
Dávka lumakaftoru/ivakaftoru má byť
v prvom týždni liečby znížená na jedno vrecko denne pri začatí liečby lumakaftoromu/ivakaftorom u pacientov súbežne užívajúcich klaritromycín alebo telitromycín.
Je potrebné zvážiť alternatívu týchto antibiotík, ako je azitromycín. Lumakaftor/ivakaftor môže znížiť expozíciu klaritromycínu
a telitromycínu, čo môže znížiť ich účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
erytromycín ↔ LUM
↑ IVA
Kvôli inhibícii CYP3A
prostredníctvom erytromycínu
Neodporúča sa úprava dávky
lumakaftoru/ivakaftoru pri súbežnom podávaní s erytromycínom.
A
ntikonvulzíva: karbamazepín, fenobarbital, fenytoín
Antimykotiká: itrakonazol*, ketokonazol, posakonazol, vorikonazol
↓ erytromycín
kvôli indukcii CYP3A
prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A prostredníctvom týchto antikonvulzív
↓ karbamazepín, fenobarbital, fenytoín kvôli indukcii CYP3A prostredníctvom LUM
↔ LUM
↑ IVA
kvôli inhibícii CYP3A prostredníctvom týchto antimykotík
↓ itrakonazol, ketokonazol, vorikonazol kvôli indukcii CYP3A prostredníctvom LUM
↓ posakonazol
kvôli indukcii UGT
prostredníctvom LUM
Je potrebné zvážiť alternatívu erytromycínu, ako je azitromycín. Lumakaftor/ivakaftor môže znížiť expozíciu erytromycínu, čo môže znížiť jeho účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antikonvulzívami sa neodporúča. Expozície ivakaftoru a antikonvulzív môžu byť významne znížené, čo môže znížiť účinnosť obidvoch liečiv.
Neodporúča sa úprava dávky lumakaftoru/ivakaftoru na začiatku liečby týmito antimykotikami, ak pacienti súbežne užívajú lumakaftor/ivakaftor.
Dávka lumakaftoru/ivakaftoru má byť znížená na jedno vrecko denne v prvom týždni liečby lumakaftorom/ivakaftorom u pacientov, ktorí súbežne užívajú tieto antimykotiká.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antimykotikami sa neodporúča. Pacienti majú byť pozorne monitorovaní kvôli prielomovým mykotickým infekciám,
ak sú tieto lieky nevyhnutné. Lumakaftor/ivakaftor môže znížiť expozíciu týchto antimykotík, čo môže znížiť ich účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
flukonazol ↔ LUM
↑ IVA
kvôli inhibícii CYP3A
prostredníctvom flukonazolu
Neodporúča sa úprava dávky
lumakaftoru/ivakaftoru pri súbežnom podávaní s flukonazolom.
A
ntiflogistiká:
↓ flukonazol
kvôli indukcii prostredníctvom LUM;
flukonazol je vylúčený
primárne renálnou exkréciou ako nezmenené liečivo, hoci mierna redukcia expozície flukonazolu bola pozorovaná u silných induktorov
Pre žiaduci klinický efekt sú
požadované vyššie dávky flukonazolu.
Lumakaftor/ivakaftor môže znížiť expozíciu flukonazolu, čo môže znížiť jeho účinnosť.
ibuprofen ↔ LUM, IVA
A
ntimykobakteriálne liečivá:
rifabutín, rifampicín*,
rifapentín
↓ ibuprofen
kvôli indukcii
CYP3A/2C8/2C9
prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A
prostredníctvom antimykobakteriálnych liečiv
↓ rifabutín
kvôli indukcii CYP3A
prostredníctvom LUM
↔ rifampicín, rifapentín
Pre žiaduci klinický efekt môžu byť
požadované vyššie dávky ibuprofenu.
Lumakaftor/ivakaftor môže znížiť expozíciu ibuprofenu, čo môže znížiť jeho účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito antimykobakteriálnymi liečivami sa neodporúča. Expozícia ivakaftoru bude znížená, čo môže znížiť účinnosť lumakaftoru/ivakaftoru.
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky rifabutínu. Lumakaftor/ivakaftor môže znížiť expozíciu rifabutínu, čo môže znížiť jeho účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
B
enzodiazepíny:
midazolam, triazolam ↔ LUM, IVA
H
ormonálna antikoncepcia: etinylestradiol, noretindron a ďalšie progestagény
Imunosupresíva: cyklosporín, everolimus, sirolimus, takrolimus (používané po orgánovej transplantácii)
Inhibítory protónovej pumpy: esomeprazol, lansoprazol, omeprazol
Rastlinné prípravky: ľubovník bodkovaný (
Hypericum perforatum)
↓ midazolam, triazolam
kvôli indukcii CYP3A
prostredníctvom LUM
↓ etinylestradiol, noretindrón a ďalšie progestagény
kvôli indukcii CYP3A/UGT prostredníctvom LUM
↔ LUM, IVA
↓ cyklosporín, everolimus, sirolimus, takrolimus
kvôli indukcii CYP3A
prostredníctvom LUM
↔ LUM, IVA
↓ esomeprazol, lansoprazol, omeprazol kvôli indukcii CYP3A/2C19 prostredníctvom LUM
↔ LUM
↓ IVA
kvôli indukcii CYP3A prostredníctvom ľubovníka bodkovaného
Súbežné užívanie
lumakaftoru/ivakaftoru s týmito benzodiazepínmi sa neodporúča. Lumakaftor/ivakaftor znižuje expozíciu midazolamu a triazolamu, čo znižuje ich účinnosť.
Hormonálna antikoncepcia, vrátane perorálnej, injekčnej, transdermálnej a implantačnej, nemôže byť pri súbežnom podávaní
s lumakaftorom/ivakaftorom považovaná za spoľahlivý spôsob
antikoncepcie. Lumakaftor/ivakaftor môže znížiť expozíciu hormonálnej
antikoncepcie, čo môže znížiť jej účinnosť.
Súbežné užívanie lumakaftoru/ivakaftoru s týmito imunosupresívami sa neodporúča. Lumakaftor/ivakaftor znižuje expozíciu imunosupresív, čo môže znížiť ich účinnosť. Použitie lumakaftoru/ivakaftoru u pacientov po orgánovej transplantácii nebolo študované.
Pre žiaduci klinický efekt sú požadované vyššie dávky inhibítorov protónovej pumpy. Lumakaftor/ivakaftor môže znížiť expozíciu inhibítorov protónovej pumpy, čo môže znížiť ich účinnosť.
Súbežné používanie lumakaftoru/ivakaftoru s ľubovníkom bodkovaným sa neodporúča. Expozícia ivakaftoru sa zníži, čo môže znížiť účinnosť lumakaftoru/ivakaftoru.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Ď
alšie klinicky významné súbežne podávané liečivá
A
ntiarytmiká:
digoxín ↔ LUM, IVA
A
ntikoagulanciá:
↑ alebo ↓ digoxín
kvôli potenciálnej indukcii alebo inhibícii P-gp
Sérová koncentrácia digoxínu má byť
monitorovaná a dávka má byť titrovaná podľa žiaduceho klinického účinku.
Lumakaftor/ivakaftor môže meniť
expozíciu digoxínu.
dabigatran ↔ LUM, IVA
↑ alebo ↓ dabigatran kvôli
potenciálnej indukcii alebo inhibícii P-gp
Pri súbežnom podávaní
lumakaftoru/ivakaftoru by má byť použité zodpovedajúce klinické monitorovanie. Úprava dávky dabigatranu môže byť potrebná
k získaniu požadovaného klinického účinku. Lumakaftor/ivakaftor môže
meniť expozíciu dabigatranu.
warfarín ↔ LUM, IVA
A
ntidepresíva: citalopram, escitalopram, sertralín
↑ alebo ↓ warfarín
kvôli potenciálnej indukcii alebo inhibícii CYP2C9
prostredníctvom LUM
↔ LUM, IVA
↓ citalopram, escitalopram, sertralín kvôli indukcii CYP3A/2C19 prostredníctvom LUM
Pri súbežnom podávaní
lumakaftoru/ivakaftoru s warfarínom má byť monitorovaná hladina
medzinárodneho normalizovaného
pomeru (international normalised ratio,
INR). Lumakaftor/ivakaftor môže meniť expozíciu warfarínu.
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky antidepresív. Lumakaftor/ivakaftor môže znížiť expozíciu antidepresív, čo môže znížiť ich účinnosť.
bupropión ↔ LUM, IVA
↓ bupropión
kvôli indukcii CYP2B6
prostredníctvom LUM
Pre žiaduci klinický efekt môžu byť
požadované vyššie dávky bupropiónu.
Lumakaftor/ivakaftor môže znížiť expozíciu bupropiónu, čo môže znížiť jeho účinnosť.
T
rieda súbežne
podávaného lieku:
N
ázov účinnej látky Účinok Klinický komentár
Systémové
kortikosteroidy: metylprednizolón, prednizón
Blokátory H2:↔ LUM, IVA
↓ metylprednizolón, prednizónu
kvôli indukcii CYP3A
prostredníctvom LUM
Pre žiaduci klinický efekt môžu byť požadované vyššie dávky týchto systémových kortikosteroidov. Lumakaftor/ivakaftor môže znížiť expozíciu systémových kortikosteroidov, čo môže znížiť ich účinnosť.
ranitidín ↔ LUM, IVA
P
e
rorálne antidiabetiká: repaglinid
↑ alebo ↓ ranitidín
kvôli indukcii alebo inhibícii P-gp
↔ LUM, IVA
↓ repaglinid kvôli indukcii CYP3A/2C8 prostredníctvom LUM
Pre žiaduci klinický efekt môže byť
potrebná zmena dávky ranitidínu.
Lumakaftor/ivakaftor môže meniť expozíciu ranitidínu.
Pre žiaduci klinický efekt môže byť požadovaná vyššia dávka repaglinidu. Lumakaftor/ivakaftor môže znížiť expozíciu repaglinidu, čo môže znížiť jeho účinnosť.
Vysvetlivky: ↑ = nárast, ↓ = pokles, ↔ = bez zmeny; LUM = lumakaftor; IVA = ivakaftor.
* Založené na klinických liekových interakčných štúdiách. Všetky ostatné liekové interakcie sú predpokladané.
Falošne pozitívne močové testy na THC
U pacientov užívajúcich Orkambi boli hlásené falošne pozitívne skríningové testy moču na tetrahydrokanabinol (THC). Na potvrdenie výsledkov sa má zvážiť alternatívna konfirmačná metóda.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov (menej ako 300 ukončených gravidít)
o použití lumakaftoru/ivakaftoru u gravidných žien. Štúdie na zvieratách s lumakaftorom
a ivakaftorom nepreukázali priame alebo nepriame účinky z hľadiska vývoja a reprodukčnej toxicity, zatiaľ čo iba s ivakaftorom boli účinky zaznamenané v dávkach toxických pre matku (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lumakaftoru/ivakaftoru počas gravidity,
pokiaľ klinický stav matky nevyžaduje liečbu lumakaftorom/ivakaftorom.
Dojčenie
Nie je známe, či sa lumakaftor a/alebo ivakaftor a metabolity vylučujú do ľudského mlieka. Dostupné farmakokinetické údaje u zvierat preukázali vylučovanie lumakaftoru aj ivakaftoru do mlieka laktujúcich samíc potkanov. Ako také riziko u dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu lumakaftorom/ivakaftorom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre matku.
Fertilita
Lumakaftor nemala vplyv na plodnosť a indexy reprodukčnej výkonnosti u samcov a samíc potkanov. Ivakaftor zhoršoval plodnosť a indexy reprodukčnej výkonnosti u samcov a samíc potkanov. Pri dávke
≤ 100 mg/kg/deň nebol pozorovaný žiaden vplyv na plodnosť samcov a samíc potkanov a indexy
reprodukčnej výkonnosti (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Ivakaftor, ktorý je jedným z liečiv v Orkambi, má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Ivakaftor môže spôsobiť závraty (pozri časť 4.8).
Pacienti, u ktorých sa vyskytujú závraty v priebehu užívania Orkambi, majú byť poučení, aby neviedli vozidlá ani neobsluhovali stroje, kým symptómy neustúpia.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie, ktoré sa vyskytli u pacientov vo veku 12 rokov a starších, ktorí užívali lumakaftor/ivakaftor v združených, placebom kontrolovaných štúdiách fázy 3, boli dyspnoe (14,0 %
oproti 7,8 % pri placebe), hnačka (11,0 % oproti 8,4 % pri placebe) a nauzea (10,2 % oproti 7,6 % pri
placebe).
Závažné nežiaduce účinky, vyskytujúce sa najmenej u 0,5 % pacientov zahŕňajú poruchy pečene a žlčových ciest, napr. zvýšené hodnoty transamináz, cholestatickú hepatitídu a hepatickú encefalopatiu.
Zoznamnežiaducichreakciívtabuľke
Nežiaduce reakcie identifikované v 24-týždňových, placebom kontrolovaných štúdiách fázy 3 (skúšania 1 a 2) u pacientov vo veku 12 rokov a starších a v 24-týždňovej, placebom kontrolovanej
štúdii u pacientov vo veku 6 až 11 rokov (skúšanie 7), ktorí sú homozygotní nosiči pre mutáciu
F508del v géne CFTR sú uvedené v tabuľke 4 a sú zoradené podľa triedy orgánových systémov, frekvencie a nežiaducich účinkov. Pozorované nežiaduce reakcie ivakaftoru samotného sú tiež uvedené v tabuľke 4. Nežiaduce reakcie sú zoskupené podľa klasifikácie frekvencie MedDRA: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Tabuľka 4: Nežiaduce reakcie u pacientov liečených lumakaftorom/ivakaftorom a ivakaftorom samotným.
T
rieda orgánových systémov
F
r
ekvencia Nežiaduce reakcie
Infekcie a nákazy veľmi časté Nasofaringitída*
časté Infekcia horných dýchacích ciest, rinitída
Poruchy ciev menej časté Hypertenzia
Poruchy nervového systému veľmi časté Bolesť hlavy, závrat*
menej časté Hepatálna encefalopatia †
Poruchy ucha a labyrintu časté Bolesť ucha*, ušné problémy*, tinnitus*, hyperémia tympanickej membrány*, vestibulárna porucha*
menej časté Kongescia ucha*

Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
veľmi časté Kongescia nosa, dyspnoe, produktívny kašeľ, zvýšené vylučovanie hlienu
časté Abnormálne dýchanie, orofaryngeálna bolesť, kongescia sínusov*, rinorea, faryngeálny erytém*
veľmi časté Abdominálna bolesť*, bolesť hornej časti brucha, hnačka, nevoľnosť
T
rieda orgánových systémov
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy reprodukčného systému a prsníkov
Laboratórne a funkčné vyšetrenia
Frekvencia Nežiaduce reakciečasté Nadúvanie, vracanie
časté Zvýšené hodnoty transamináz menej časté Cholestatická hepatitída‡
časté Vyrážka
časté Nepravidelná menštruácia, dysmenorea, metrorágia, hrčka v prsníku*
menej časté Menorágia, amenorea, polymenorea, zápal prsníku*, gynekomastia*, poruchy bradavky*, bolesť bradavky*, oligomenorea
veľmi časté Baktérie v spúte
časté Zvýšená hladina kreatínfosfokinázy v krvi menej časté Zvýšený krvný tlak
* Nežiaduce reakcie a frekvencie pozorované u pacientov v klinických štúdiách v monoterapii ivakaftorom (zložka Orkambi).
† 1 pacient zo 738
‡ 2 pacienti z 738
Údaje o bezpečnosti u 1 029 pacientov vo veku 12 rokov a starších, ktorí boli homozygotní nosiči mutácie F508del v géne CFTR, liečených lumakaftorom/ivakaftorom počas ďalších až
96 týždňov v dlhodobej pokračovacej štúdii bezpečnosti a účinnosti (skúšanie 3) boli podobné 24-'
týždňovej, placebom kontrolovanej štúdii (pozri časť 5.1).
Popisvybranýchnežiaducichreakcií
Poruchy pečene a žlčových ciest
Počas skúšaní 1 a 2 bola incidencia maximálnej hodnoty transaminázy (ALT alebo AST) > 8, > 5 a
> 3 x ULN 0,8 %, 2,0 % a 5,2 %; a 0,5 %, 1,9 % a 5,1 % u pacientov liečených lumakaftorom/ivakaftorom, respektíve placebom. Incidencia nežiaducich reakcií spojených so zvýšenými hodnotami transamináz bola 5,1 % a 4,6 % u pacientov liečených lumakaftorom/ivakaftorom, respektíve u pacientov s placebom. Sedem pacientov užívajúcich lumakaftor/ivakaftor malo závažné nežiaduce reakcie spojené s poruchou pečene a zvýšenými hodnotami transamináz, zahŕňajúce 3 pacientov so súbežne zvýšeným celkovým bilirubínom. Prerušenie liečby lumakaftorom/ivakaftorom u všetkých pacientov vrátilo funkčné pečeňové testy na vstupnú hodnotu alebo ich zlepšilo (pozri časť 4.4).
Medzi 7 pacientmi s pre-existujúcou cirhózou alebo portálnou hypertenziou, ktorí užívali lumakaftor/ivakaftor v placebom kontrolovanej štúdii fázy 3, bolo pozorované zhoršenie funkčných pečeňových testov so vzostupom ALT, AST, bilirubínu a hepatickej encefalopatie u jedného pacienta. Prípad sa vyskytol do 5 dní po začatí liečby a vyriešil sa prerušením liečby lumakaftorom/ivakaftorom (pozri časť 4.4).
U pacientov s CF s pre-existujúcou cirhózou pečene s portálnou hypertenziou, ktorí boli liečení lumakaftorom/ivakaftorom, boli po uvedení lieku na trh hlásené prípady dekompenzácie funkcie pečene, vrátane zlyhania pečene vedúceho k úmrtiu (pozri časť 4.4).
Respiračné príhody
Počas skúšaní 1 a 2 bola incidencia nežiaducich reakcií dýchacej sústavy (napr. hrudný dyskomfort, dyspnoe a respiračné problémy) u pacientov liečených lumakaftorom/ivakaftorom 26,3 % v porovnaní
so 17,0 % u pacientov s placebom. Incidencia týchto udalostí bola častejšia u pacientov s nižším FEV1 pred liečbou. Približne tri štvrtiny týchto udalostí začali v prvom týždni liečby a u väčšiny pacientov odozneli bez prerušenia liečby. Väčšina príhod boli mierne alebo stredne závažné, nezávažné
a neviedli k prerušeniu liečby (pozri časť 4.4).
Počas 24-týždňovej, otvorenej klinickej štúdie fázy 3b (skúšanie 5) u 46 pacientov vo veku 12 rokov a starších s pokročilým pľúcnym ochorením (ppFEV1 < 40) [priemerný východiskový ppFEV1 29,1 (rozmedzie: 18,3 až 42,0)] bola incidencia respiračných príhod 65,2 %. V podskupine 28 pacientov,
u ktorých sa liečba začala celou dávkou lumakaftoru/ivakaftoru (2 tablety každých 12 hodín) bola incidencia 71,4 % a u 18 pacientov, u ktorých sa liečba začala zníženou dávkou lumakaftoru/ivakaftoru (1 tableta každých 12 hodín počas 2 týždňov a následne zvýšenie na celú dávku) bola incidencia 55,6 %. Medzi pacientmi, u ktorých sa liečba začala celou dávkou lumakaftoru/ivakaftoru sa u jedného pacienta vyskytla závažná respiračná príhoda, u troch pacientov sa znižovala dávka a u troch pacientov sa liečba ukončila. Medzi pacientmi, u ktorých sa liečba začala polovičnou dávkou, sa nezaznamenali žiadne závažné respiračné príhody, znižovanie dávky ani ukončenie liečby (pozri časť 4.4).
Abnormality menštruačného cyklu
Počas skúšaní 1 a 2 bola incidencia kombinovaných prípadov abnormalít menštruačného cyklu
(amenorea, dysmenorea, menorágia, nepravidelná menštruácia, metrorágia, oligomenorea a polymenorea) 9,9 % u žien liečených lumakaftorom/ivakaftorom a 1,7 % u žien s placebom. Tieto
menštruačné problémy sa vyskytovali častejšie v skupine žien, ktoré užívali hormonálnu
antikoncepciu (25,0 %) oproti pacientkám, ktoré hormonálnu antikoncepciu neužívali (3,5 %) (pozri časť 4.5). Väčšina týchto reakcií bola mierna alebo stredná, nezávažná. U pacientok liečených lumakaftorom/ivakaftorom približne dve tretiny týchto reakcií odozneli a medián trvania bol 10 dní.
Zvýšený krvný tlak
V priebehu skúšaní 1 a 2 boli hlásené nežiaduce reakcie spojené so zvýšením krvného tlaku (napr. hypertenzia, zvýšenie krvného tlaku) u 0,9 % (7 / 738) pacientov liečených lumakaftorom/ivakaftorom a neboli hlásené u žiadnych pacientov, ktorí dostávali placebo.
U pacientov liečených lumakaftorom/ivakaftorom (priemerný východiskový tlak 114 mmHg systolický a 69 mmHg diastolický), bol maximálny priemerný nárast oproti východiskovému systolickému a diastolickému krvnému tlaku 3,1 mmHg a 1,8 mmHg, v uvedenom poradí.
U pacientov, ktorí dostávali placebo (priemerný východiskový tlak 114 mmHg systolický a 69 mmHg diastolický), bol maximálny priemerný nárast oproti východiskovému systolickému a diastolickému krvnému tlaku 0,9 mmHg a 0,9 mmHg, v uvedenom poradí.
Podiel pacientov, ktorí mali hodnotu systolického krvného tlaku > 140 mmHg alebo diastolického krvného tlaku > 90 mmHg aspoň dvakrát, bol 3,4 % a 1,5 % u pacientov liečených lumakaftorom/ivakaftorom, v uvedenom poradí, v porovnaní s 1,6 % a 0,5 % u pacientov, ktorí dostávali placebo (pozri časť 4.4).
Pediatrickápopulácia
Bezpečnostné údaje boli hodnotené u 60 pacientov vo veku 2 až 5 rokov, 161 pacientov vo veku 6 až
11 rokov (skúšania 6, 7 a 8) a u 194 pacientov vo veku 12 až 17 rokov s CF, ktorí sú homozygotní nosiči mutácie F508del a ktorí užívali lumakaftor/ivakaftor v klinických štúdiách. Pacienti vo veku 12
až 17 rokov boli zahrnutí v skúšaniach 1 a 2.
Bezpečnostný profil u týchto pediatrických pacientov je všeobecne zhodný s tým u dospelých pacientov. Ďalšie nežiaduce reakcie zo skúšania 6 sú zahrnuté v tabuľke 4.
Popis vybraných nežiaducich reakcií u pediatrických pacientov vo veku 2 až 11 rokov
Hepatobiliárnepríhody
Počas 24-týždňovej, otvorenej klinickej štúdie fázy 3 u 58 pacientov vo veku 6 až 11 rokov
(skúšanie 6) bola incidencia maximálnych hodnôt transamináz (ALT alebo AST) > 8, > 5 a > 3 x ULN
5,3 %, 8,8 %, a 19,3 %. Žiaden pacient nemal hladinu celkového bilirubínu > 2 x ULN. Dávkovanie lumakaftoru/ivakaftoru bolo zachované alebo úspešne obnovené po prerušení u všetkých pacientov so zvýšenými hodnotami transamináz, okrem 1 pacienta, ktorý ukončil liečbu trvale.
Počas 24-týždňovej, placebom kontrolovanej klinickej štúdie fázy 3 u 204 pacientov vo veku 6 až
11 rokov (skúšanie 7) bola incidencia maximálnych hodnôt transamináz (ALT alebo AST) > 8, > 5,
a > 3 x ULN 1,0 %, 4,9 % a 12,6 % u pacientov s lumakaftorom/ivakaftorom a 2,0 %, 3,0 %, a 7,9 %
u pacientov liečených placebom. Žiaden pacient nemal hladinu celkového bilirubínu > 2 x ULN. Dvaja pacienti v skupine s lumakaftorom/ivakaftorom a dvaja pacienti v skupine s placebom prerušili liečbu trvale kvôli zvýšeným hodnotám transamináz.
Počas 24-týždňovej, otvorenej klinickej štúdie fázy 3 u 60 pacientov vo veku 2 až 5 rokov (skúšanie 8)
bola incidencia maximálnych hodnôt transamináz (ALT alebo AST) > 8, > 5 a > 3 x ULN 8,3 % (5/60), 11,7 % (7/60) a 15,0 % (9/60). Žiadny pacient nemal hladinu celkového bilirubinu > 2 x ULN. Traja pacienti v skupine s lumakaftorom/ivakaftorom trvale ukončili liečbu kvôli zvýšeným hodnotám transamináz.
RespiračnépríhodyPočas 24-týždňovej, otvorenej klinickej štúdie fázy 3(skúšanie 6) u 58 pacientov vo veku 6 až
11 rokov (priemerná hodnota ppFEV1 bola 91,4) bola incidencia respiračných nežiaducich reakcií
6,9 % (4/58).
Počas 24-týždňovej, placebom kontrolovanej klinickej štúdie fázy 3 (skúšanie 7) u pacientov vo veku
6 až 11 rokov (priemerná hodnota ppFEV1 bola 89,8) bola incidencia respiračných nežiaducich reakcií
18,4 % u pacientov s lumakaftorom/ivakaftorom a 12,9 % u pacientov s placebom. Po začatí liečby
bol pozorovaný pokles v hodnote ppFEV1 pri meraní spirometrie po podaní. Absolútna zmena medzi hodnotou pred podaním a hodnotou 4 až 6 hodín po podaní bola -7,7 v 1. dni a -1,3 v 15. dni u pacientov s lumakaftorom/ivakaftorom. Pokles po podaní odoznel do 16. týždňa.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadne špecifické antidotum pre predávkovanie lumakaftorom/ivakaftorom. Liečba predávkovania pozostáva zo všeobecných podporných opatrení vrátane monitorovania životných funkcií a pozorovania klinického stavu pacienta.
Nežiaduce účinky, ktoré sa objavili vo zvýšenej incidencii ≥ 5 % v období podávania supraterapeutickej dávky v porovnaní s terapeutickou dávkou boli bolesť hlavy, generalizovaná vyrážka a zvýšené hodnoty transamináz.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá respiračného systému; ATC kód: R07AX30
MechanizmusúčinkuProteín CFTR je chloridový kanál prítomný na povrchu epitelových buniek vo viacerých orgánoch. Mutácia
F508del vplýva na proteín CFTR viacerými spôsobmi, primárne zapríčinením defektu
bunkového spracovania a transportu, ktorý znižuje množstvo CFTR na bunkovom povrchu. Malé množstvo F508del-CFTR, ktoré sa dostane na bunkový povrch, má malú schopnosť otvárať kanál
(porucha vrátkovania). Lumakaftor je korektor CFTR, ktorý účinkuje priamo na mutáciu
F508del-CFTR za účelom zlepšenia bunkového spracovania a transportu, čím zvyšuje množstvo funkčného CFTR na bunkovom povrchu. Ivakaftor je potenciátor CFTR, ktorý uľahčuje zvýšenie
transportu chloridov potencovaním schopnosti otvárať kanál (alebo vrátkovania) proteínu CFTR na
bunkovom povrchu. Kombinovaným efektom lumakaftoru a ivakaftoru je zvýšenie množstva
a funkcie F508del-CFTR na bunkovom povrchu vedúce k zvýšeniu transportu chloridov. Presný spôsob, akým lumakaftor zlepšuje bunkové spracovanie a transport F508del-CFTR, a ivakaftor potencuje F508del-CFTR, nie je známy.
FarmakodynamickéúčinkyÚčinky na chloridy v pote:Zmeny chloridov v pote ako odpoveď na lumakaftor samotný alebo v kombinácii s ivakaftorom boli hodnotené v dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní fázy 2 u pacientov s CF
vo veku 18 rokov a starších. V tomto skúšaní 10 pacientov (homozygotní nosiči mutácie
F508del-CFTR) dokončili užívanie lumakaftoru samotného v dávke 400 mg každých 12 hod po dobu
28 dní nasledované pridaním ivakaftoru 250 mg každých 12 hod po dobu 28 dní a 25 pacientov (homozygotní alebo heterozygotní nosiči mutácie
F508del) dokončili užívanie placeba. Rozdiel medzi liečbou samotným lumakaftorom 400 mg každých 12 hod a placebom, hodnotený ako stredná zmena chloridov v pote od východiskovej hodnoty po 28. deň, bol štatisticky významný −8,2 mmol/l (95 % CI: −14, −2). Rozdiel medzi liečbou kombináciou lumakaftor 400 mg/ivakaftor 250 mg každých
12 hod a placebom, hodnotený ako stredná zmena chloridov v pote od východiskovej hodnoty po 56. deň, bol štatisticky významný −11 mmol/l (95 % CI: −18, −4).

V skúšaní 7 (pozri Klinická účinnosť a bezpečnosť) u pacientov vo veku 6 až 11 rokov, ktorí boli homozygotní nosiči mutácie
F508del génu
CFTR, bol rozdiel v liečbe (priemer stanovený metódou najmenších štvorcov) v chloridoch v pote ako priemer absolútnej zmeny v 24. týždni v porovnaní
s placebom -24,9 mmol/l (nominálna hodnota P < 0,0001). Rozdiel v liečbe (priemer stanovený metódou najmenších štvorcov) v chloridoch v pote ako priemer absolútnej zmeny v 15. dni
a vo 4. týždni v porovnaní s placebom bol –20,8 mmol/l (95% CI: -23,4; -18,2; nominálna hodnota
P < 0,0001). V skúšaní 8 u pacientov vo veku 2 až 5 rokov, ktorí boli homozygotní nosiči mutácie
F508del-CFTR bola priemerná absolútna zmena chloridov v pote vrámci skupiny v 24. týždni,
v porovnaní s východiskovou hodnotou -31,7 mmol/l (95 % Cl: -35,7; -27,6). Navyše, priemerná absolútna zmena chloridov v pote od 24. týždňa po 26. týždeň, po ktorom nasledovalo 2-týždňové
obdobie vymývania (za účelom zhodnotenia odpovede na obdobie bez liečby), sa zvýšila
o 33,0 mmol/l (95 % Cl: 28,9; 37,1; nominálna P hodnota < 0,0001), čo po období vymývania zodpovedalo návratu k východiskovému stavu. V 24. týždni malo 16 % detí hodnoty chloridy v pote
nižšie ako 60 mmol/l a žiadne dieťa nemalo hodnoty nižšie ako 30 mmol/l.
Zmeny v FEV1:Zmeny v ppFEV1 v odpovedi na lumakaftor samostatne alebo v kombinácii s ivakaftorom boli tiež
hodnotené v dvojito zaslepenom placebom kontrolovanom skúšaní fázy 2 u pacientov s CF vo veku
18 rokov a starších. Rozdiel medzi liečbou lumakaftorom 400 mg každých 12 hodín samotným
a placebom, hodnotený ako stredná absolútna zmena ppFEV1, bol -4,6 percentuálnych bodov (95 % CI: -9,6; 0,4) od východiskového stavu do 28. dňa, 4,2 percentuálnych bodov (95 % CI: -1,3; 9.7) od východiskového stavu do 56. dňa a 7,7 percentuálnych bodov (95 % CI: 2,6; 12,8; štatisticky významné) odo dňa 28. až 56. dňa (po pridaní ivakaftoru k lumakaftorovej monoterapii).
Zníženie tepovej frekvencieV priebehu 24 týždňov trvajúcich, placebom kontrolovaných štúdií fázy 3 bol pozorovaný maximálny pokles priemernej srdcovej frekvencie o 6 úderov za minútu (
beats per minute, bpm) od
východiskovej úrovne v 1. a 15. deň, približne 4 až 6 hodín po podaní. V období po 15. dni nebol srdcový tep po podaní v týchto štúdiách sledovaný. Od 4. týždňa bola zmena priemernej tepovej
frekvencie pred podaním v rozmedzí od 1 do 2 bpm pod východiskovú úroveň u pacientov liečených lumakaftorom/ivakaftorom. Percento pacientov s hodnotami srdcovej frekvencie < 50 bpm počas liečby bolo 11 % u pacientov, ktorí dostávali lumakaftor/ivakaftor, v porovnaní s 4,9 % u pacientov,
ktorí dostávali placebo.
KlinickáúčinnosťabezpečnosťSkúšania u pacientov s CF vo veku 12 rokov a starších, ktorí sú homozygotní nosiči mutácie F508del v géne CFTRÚčinnosť lumakaftoru/ivakaftoru u pacientov s CF, ktorí sú homozygotní nosiči mutácie
F508delv géne
CFTR bola hodnotená v dvoch randomizovaných, dvojito zaslepených, placebom
kontrolovaných klinických skúšaniach s 1 108 klinicky stabilnými pacientmi s CF, z ktorých
737 pacientov bolo randomizovaných k užívaniu lumakaftoru/ivakaftoru. Pacienti v oboch skúšaniach boli randomizovaní v pomere 1:1:1 a dostávali lumakaftor 600 mg jedenkrát denne/ivakaftor 250 mg každých 12 hod, lumakaftor 400 mg každých 12 hod/ivakaftor 250 mg každých 12 hod alebo placebo. Pacienti užívali študovaný liek s jedlom obsahujúcim tuky po dobu 24 týždňov navyše k predpísanej terapii CF (napr. bronchodilatátory, inhalačné antibiotiká, dornáza alfa a hypertonický roztok chloridu sodného). Pacienti z týchto skúšaní boli vhodní k zaradeniu do zaslepenej predĺženej štúdie.
Skúšanie 1 hodnotilo 549 pacientov s CF vo veku 12 rokov a starších (priemerný vek 25,1 rokov) s percentom predpokladaného FEV1 (ppFEV1) pri skríningu medzi 40 - 90 (stredná hodnota na začiatku štúdie ppFEV1 60,7 [rozmedzie: od 31,1 do 94,0]). Skúšanie 2 hodnotilo 559 pacientov vo veku 12 rokov a starších (priemerný vek 25,0 rokov) s ppFEV1 pri skríningu medzi 40 - 90 (stredný ppFEV1 na počiatku 60,5 [rozmedzie: od 31,3 do 99,8]). Pacienti s anamnézou kolonizácie organizmami ako Burkholderia cenocepacia, Burkholderia dolosa alebo Mycobacterium abscessus alebo ktorí mali 3 alebo viac abnormálnych funkčných pečeňových testov (ALT, AST, AP, GGT
≥ 3-násobok ULN alebo celkový bilirubín ≥ 2-násobok ULN) boli vyradení.
Primárnym koncovým ukazovateľom účinnosti v obidvoch štúdiách bola absolútna zmena ppFEV1 od východiskovej hodnoty po hodnotu v 24. týždni. Ďalšie premenné účinnosti, ktoré boli zahrnuté, sú relatívna zmena ppFEV1 od východiskovej hodnoty, absolútna zmena BMI od východiskovej hodnoty, absolútna zmena respiračnej domény dotazníku CFQ-R od východiskovej hodnoty, podiel pacientov, ktorí dosiahli ≥ 5 % relatívnej zmeny od východiskovej hodnoty ppFEV1 v 24. týždni a množstvo pulmonálnych exacerbácií (zahŕňajúcich tie, ktoré vyžadovali hospitalizáciu alebo intravenóznu antibiotickú terapiu) do 24. týždňa.
V obidvoch skúšanich viedla liečba lumakaftorom/ivakaftorom k štatisticky významnému zlepšeniu ppFEV1 (tabuľka 5). Stredné zlepšenie ppFEV1 bolo prudké na začiatku (15. deň) a udržalo sa po celú dobu 24-týždňovej liečby. V 15. dni bol rozdiel medzi liečbou lumakaftorom 400 mg/ivakaftorom
250 mg každých 12 hod a placebom v strednej absolútnej zmene (95 % CI) ppFEV1 od východiskovej hodnoty 2,51 percentuálnych bodov združených skúšaní 1 a 2 (P < 0,0001). Zlepšenie v ppFEV1 bolo pozorované bez ohľadu na vek, závažnosť ochorenia, pohlavie a zemepisný región. Fáza 3 skúšania lumakaftoru/ivakaftoru zahŕňala 81 pacientov s ppFEV1 < 40 na začiatku štúdie. Rozdiel v liečbe
v tejto podskupine bol porovnateľný s podskupinou pacientov s ppFEV1 ≥ 40. V 24. týždni bol rozdiel medzi liečbou lumakaftorom 400 mg/ivakaftorom 250 mg každých 12 hod a placebom v strednej
absolútnej zmene (95 % CI) ppFEV1 od východiskovej hodnoty v združených skúšaniach 1 a 2
3,39 percentuálnych bodov (P = 0,0382) pre pacientov s ppFEV1 < 40 a 2,47 percentuálnych bodov
(P < 0,0001) pre pacientov s ppFEV1 ≥ 40.
T
abuľka 5: Súhrn primárnych a kľúčových sekundárnych výsledkov v skúšaní 1 a skúšaní 2*
S
k
ú
šanie 1 Skúšanie 2 Združené (skúšanie 1
a skúšanie 2)
Placebo
(
n = 184)
L
U
M 400 mg každých
1
2 hod/IVA 250 mg
k
až
d
ýc
h 12 hod
(n
=182)
Placebo
(
n = 187)
L
U
M 400 mg každých
1
2 hod/IVA
2
5
0 mg každých
1
2 hod
(
n = 187)
Placebo
(
n = 371)
L
U
M 400 mg každých
1
2 hod/IVA
2
5
0 mg každých
1
2 hod
(
n = 369)
Ab
solútna zmena
v ppFEV
1
v 24. týždni
Rozdiel v liečbe
– 2,41
(P = 0,0003) †
– 2,65
(P = 0,0011) †
– 2,55
(P < 0,0001)
(p
erce
nt
u
ál
n
e
b
o
d
y
)
Rozdiel
v rámci skupiny
Rozdiel
-0,73
(P = 0,216
8)
1,68
(P = 0,0051)
4,15
-0,02
(P = 0,973
0)
2,63
(P < 0,0001)
4,69
-0,39
(P < 0,349
4)
2,16
(P < 0,0001)
4,4
R
ela
t
í
vn
a zmena
v liečbe –
(P = 0,0028)† –
(P = 0,0009)† –
(P < 0,0001)
v ppFEV
1
v 24. týždni (%)
Rozdiel v rámci skupiny
-0,85
(P = 0,393
4)
3,3
(P = 0,0011)
0,16
(P = 0,879
3)
4,85
(P < 0,0001)
-0,34
(P = 0,637
5)
4,1
(P < 0,0001)
Ab
solútna zmena
Rozdiel
– 0,13
0,36
†
– 0,24
v BMI
v 24. týždni
(
k
g
/
m
2
)
Ab
solútna zmena
v liečbe
Rozdiel v rámci skupiny
Rozdiel
0,19
(P = 0,006
5)
(P = 0,1938)
0,32
(P < 0,0001)
1,5
0,07
(P = 0,289
2)
(P < 0,0001)
0,43
(P < 0,0001)
2,9
0,13
(P = 0,006
6)
(P = 0,0004)
0,37
(P < 0,0001)
2,2
v CFQ-R
re
spiračnej
v liečbe –
(P = 0,3569) –
(P = 0,0736) –
(P = 0,0512)
d
o
m
é
n
y Skóre v 24. týždni (body)
Podiel pacientov
Rozdiel v rámci skupiny
1,1
(P = 0,342
3)
2,6
(P = 0,0295)
2,8
(P = 0,015
2)
5,7
(P < 0,0001)
1,9
(P = 0,021
3)
4,1
(P < 0,0001)
s ≥ 5 %
re
l
a
t
í
vn
y
m
% 25 % 32 % 26 % 41 % 26 % 37 %
roz
d
i
e
lo
m
v ppFEV
1
v 24. týždni
Odds ratio –
Počet príhod
1,43
(P = 0,1208) –
1,90
(P = 0,0032) –
1,66
(P = 0,0013)
Počet pulmonálnych exacerbácií počas
(pomerný výskyt za 48 týžd.)
112 (1,07) 73 (0,71) 139 (1,18) 79 (0,67) 251 (1,14) 152 (0,70)
2
4 týždňov
Relatívne
riziko –
0,66
(P = 0,0169) –
0,57
– (P = 0,0002)
0,61
(P < 0,0001)

*V každej štúdii bolo vykonané hierarchické testovanie v rámci každej aktívnej liečebnej skupiny na primárne a sekundárne koncové ukazovatele oproti placebu; pri každom kroku, P ≤ 0,0250 a všetky predchádzajúce testy tiež spĺňali túto hodnotu významnosti, ktorá bola požadovaná.
†Ukazovatele štatistickej významnosti sa potvrdili v hierarchickom testovaní.
V 24. týždni bol podiel pacientov bez pulmonálnej exacerbácie významne vyšší pre pacientov
liečených lumakaftorom/ivakaftorom v porovnaní s placebom. V združenej analýze bolo relatívne riziko exacerbácie počas 24 týždňov u jedincov liečených lumakaftorom/ivakaftorom (lumakaftor
400 mg/ivakaftor 250 mg každých 12 hod; n = 369) 0,61 (P < 0,0001), predstavujúce relatívne
zníženie o 39 % oproti placebu. Počet prípadov za rok, anualizovaného do 48 týždňov bol 0,70 v skupine s lumakaftorom/ivakaftorom a 1,14 v skupine s placebom. Liečba lumakaftorom/ivakaftorom významne znížila riziko exacerbácií vyžadujúcich hospitalizáciu oproti placebu o 61 % (relatívne riziko = 0,39; P < 0,0001; počet prípadov za 48 týždňov 0,17
pre lumakaftor/ivakaftor a 0,45 pre placebo) a znížila exacerbácie vyžadujúce liečbu intravenóznymi antibiotikami o 56 % (relatívne riziko = 0,44; P < 0,0001; počet prípadov za 48 týždňov 0,25
pre lumakaftor/ivakaftor a 0,58 pre placebo). Tieto výsledky neboli považované za štatisticky
významné v rámci hierarchických testov jednotlivých štúdií.
Dlhodobé pokračovacie skúšanie bezpečnosti a účinnostiSkúšanie 3 bola multicentrická, pokračovacia, predĺžená štúdia fázy 3 s paralelnými skupinami pre pacientov s CF, ktorá zahŕňala pacientov vo veku 12 rokov a starších zo skúšania 1 a skúšania 2. Toto predlžené skúšanie bola navrhnuté za účelom zhodnotiť bezpečnosť a účinnosť dlhodobej liečby lumakaftorom/ivakaftorom. Z 1 108 pacientov, ktorí dostali akúkoľvek liečbu v skúšaní 1 alebo 2,
dostávalo 1 029 (93 %) pacientov dávku a aktívnu liečbu (lumakaftor 600 mg raz denne/ivakaftor
250 mg každých 12 hod alebo lumakaftor 400 mg každých 12 hod/ivakaftor 250 mg každých 12 hod) v skúšaní 3 počas ďalších až 96 týždňov (t.j. celkovo až 120 týždňov). Primárna analýza účinnosti tejto predlženej štúdie zahŕňala údaje do týždňa 72 skúšania 3 a analýza senzitivity zahŕňala údaje
do týždňa 96 skúšania 3.
Pacienti liečení lumakaftorom/ivakaftorom v skúšaní 1 alebo v skúšaní 2 preukázali účinok, ktorý bol udržiavaný s ohľadom na východiskovú hodnotu po ďalších 96 týždňoch počas skúšania 3.
U pacientov, ktorí prešli z placeba na aktívnu liečbu boli pozorované podobné zmeny ako u pacientov liečených lumakaftorom/ivakaftorom v skúšaní 1 alebo skúšaní 2 (pozri Tabuľku 5). Výsledky
zo skúšania 3 sú zhrnuté na obrázku 1 a v tabuľke 6.
Obrázok 1. Absolútna zmena od východiskovej hodnoty v percentách predpokladaného FEV1pri každej návšteve†
Zač Ď Týž Týž Týž Týž Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž. Predlž.
15 4 8 16 24 Ď15 týž 8 týž 16 týž 24 týž 36 týž 48 týž 60 týž 72 týž 84 týž 96
Návšteva LUM 400 mg každých 12 hod/IVA 250 mg každých 12 hod Placebo/LUM 400 mg každých12 hod/IVA 250 mg každých 12 hod Placebo
† zo skúšaní 1, 2 a 3.
Tabuľka 6: Dlhodobý účinok lumakaftoru/ivakaftoru v skúšaní 3*Placebo s prechodom na
lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele Východisková hodnota
Priemer
(SD)
štvorcov
 (95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
pp
FEV
1‡ 60,2 (14,7) 60,5 (14,1)
Absolútna zmena od východiskovej hodnoty ppFEV1 (percentuálne body)
Týždeň 72 predĺženia
(n = 134)
1,5 (0,2; 2,9)
0,0254
(n = 273)
0,5
(-0,4; 1,5)
0,2806
(n = 75)
Týždeň 96 predĺženia 0,8
(-0,8; 2,3)
(n = 147)
0,3495 0,5
(-0,7; 1,6)
0,4231
Placebo s prechodom na lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Relatívna zmena od východiskovej hodnoty ppFEV
1
(
%)
Týždeň 72 predĺženia
(n = 134)
2,6 (0,2; 5,0)
0,0332
(n = 273)
1,4
(-0,3; 3,2)
0,1074
(n = 75)
Týždeň 96 predĺženia 1,1
(-1,7; 3,9)
(n = 147)
0,4415 1,2
(-0,8; 3,3)
0,2372
V
ý
c
h
o
d
iskové BMI (kg/m
2
)‡
20,9 (2,8) 21,5 (3,0)
Absolútna zmena od východiskového BMI (kg/m
2
)
Týždeň 72 predĺženia
(n = 145)
0,62 (0,45; 0,79)
(n = 80)
< 0,0001
(n = 289)
0,69 (0,56; 0,81)
(n = 155)
< 0,0001
Týždeň 96 predĺženia 0,76 (0,56; 0,97)
< 0,0001 0,96 (0,81; 1,11)
< 0,0001
V
ý
c
h
o
d
iskové CFQ-R
s
k
ó
r
e respiračnej domény
(body)‡
70,4 (18,5) 68,3 (18,0)
Absolútna zmena CFQ-R skóre respiračnej domény (body)
Týždeň 72 predĺženia
(n = 135)
3,3 (0,7; 5,9)
0,0124
(n = 269)
5,7 (3,8; 7,5)
< 0,0001

(n = 81)
Týždeň 96 predĺženia 0,5
(-2,7; 3,6)
(n = 165)
0,7665 3,5 (1,3; 5,8)
0,0018
Placebo s prechodom na lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=176)**
Priemery stanovené metódou najmenších
Lumakaftor 400 mg
k
a
ž
d
ý
c
h 12 hod
/ ivakaftor 250 mg
k
a
ž
d
ý
c
h 12 hod
(n=369)†
Priemery stanovené metódou najmenších
V
ý
c
h
o
d
iskové a koncové ukazovatele
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Priemer
(SD)
štvorcov
(95 % CI) P hodnota
Počet pulmonálnych exacerbácií (udalosti) ** † ***
Počet udalostí na pacientorok
(95 % CI) (pomerný výskyt za 48 týždňov)
Počet udalostí vyžadujúcich hospitalizáciu na pacientorok (95 % CI) (pomerný výskyt
za 48 týždňov)
Počet udalostí vyžadujúcich antibiotiká intravenózne na pacientorok (95 % CI) (pomerný výskyt za 48 týždňov)
0,69 (0,56; 0,85)
0,30 (0,22; 0,40)
0,37 (0,29; 0,49)
0,65 (0,56; 0,75)
0,24 (0,19; 0,29)
0,32 (0,26; 0,38)
*Celkovo 82 % (421 z 516 vhodných pacientov) dokončilo 72 týždňov tejto štúdie; 42 % dokončilo 96 týždňov. U väčšiny pacientov došlo k prerušeniu liečby z iných ako bezpečnostných dôvodov.
** Celková expozícia u pacientov prevedených zo skúšaní 1 a 2 (skupina prevedená z placebo na lumakaftor/ivakaftor)
bola až 96 týždňov. Podanie lumakaftoru 400 mg každých 12 hodín/ivakaftoru 250 mg každých 12 hodín v dávkovacej skupine je konzistentné s odporučeným dávkovaním.
*** Počet prípadov na pacientorok bol anualizovaný na 48 týždňov.
† Celková expozícia u pacientov prevedených zo skúšaní 1 a 2 (skupina pokračovala v používaní
kombinácie lumakaftor/ivakaftor) bola až 120 týždňov. Podanie lumakaftoru 400 mg každých 12 hodín/ivakaftoru
250 mg každých 12 hodín v dávkovacej skupine je konzistentné s odporučeným dávkovaním..
‡ Za východiskové hodnoty pre skupinu prevedenú z placeba na lumakaftor 400 mg každých 12 hodín/ivakaftoru 250 mg
každých 12 hodín sa brala východisková hodnota zo skúšania 3. Za východiskové hodnoty pre skupinu na lumakaftore
400 mg každých 12 hodín/ivakaftore 250 mg každých 12 hodín sa brala východisková hodnota zo skúšaní 1 a 2.
Skúšanie u pacientov s CF, ktorí sú heterozygotní nosiči mutácie F508del v géne CFTR
Skúšanie 4 bolo multicentrické, dvojito zaslepené, randomizované, placebom kontrolované skúšanie fázy 2 pre 125 pacientov s CF vo veku 18 rokov a starších, ktorí majú ppFEV1 od 40 - 90 vrátane,
a majú mutáciu F508del na jednej alele plus druhú alelu s mutáciou predpovedajúcou nedostatok
produkcie CFTR alebo CFTR, ktoré nereaguje na ivakaftor in vitro.
Pacienti užívali buď lumakaftor/ivakaftor (n = 62) alebo placebo (n = 63) navyše k svojej predpísanej terapii CF. Primárnym koncovým ukazovateľom bolo zlepšenie funkcie pľúc určené ako stredná hodnota absolútneho rozdielu ppFEV1 medzi východiskovou hodnotou a hodnotou v 56. deň. Liečba lumakaftorom/ivakaftorom nemala za následok významné zlepšenie ppFEV1 v porovnaní s placebom u pacientov s CF, heterozygotných nosičov mutácie F508del v géne CFTR (rozdiel v liečbe 0,60
[P = 0,5978]) a žiadne významné zlepšenie BMI alebo hmotnosti (pozri časť 4.4).
Skúšanie u pacientov s CF vo veku 6 až 11 rokov, ktorí sú homozygotní nosiči mutácie F508del v géne
CFTR
Skúšanie 7 bola 24- týždňová, placebom kontrolovaná klinická štúdia fázy 3 u 204 pacientov s CF vo veku 6 až 11 rokov (priemerný vek 8,8 rokov). Skúšanie 7 hodnotilo osoby s indexom pľúcneho klírensu (lung clearance index- LCI2,5) ≥ 7,5 pri skríningu pri prvej návšteve (priemer LCI2,5 bol 10,28 pred začiatkom liečby [rozmedzie: 6,55 až 16,38]) a ppFEV1 ≥ 70 pri skríningu (priemer ppFEV1 bol
89,8 pred začiatkom liečby [rozmedzie: 48,6 až 119,6]). Pacienti dostávali buď lumakaftor
200 mg/ivakaftor 250 mg každých 12 hodín (n = 103) alebo placebo (n = 101) okrem ich predpísanej liečby CF. Pacienti, ktorí mali 2 alebo viackrát vyššie hodnoty funkčných pečeňových testov (ALT,
AST, AP, GGT ≥ 3-krát ULN), alebo ALT alebo AST > 5-krát ULN alebo celkový bilirubín > 2-krát
ULN boli vylúčení.
Primárny koncový ukazovateľ účinnosti bola absolútna zmena v LCI2,5 od východiskovej hodnoty do
24. týždňa. Sekundárny kľúčový koncový ukazovateľ zahrnoval priemer absolútnej zmeny chloridov
v pote od východiskovej hodnoty do 15. dňa, 4. týždňa a 24. týždňa (pozri Farmakodynamické vlastnosti), absolútnu zmenu BMI od východiskovej hodnoty v 24. týždni, absolútnu zmenu v CFQ-R
respiračnej domény od východiskovej hodnoty do 24. týždňa. Tieto výsledky sú zhrnuté v tabuľke 7
nižšie:
Tabuľka 7: Súhrn primárnych a kľúčových sekundárnych výsledkov v skúšaní 7
LUM 200 mg/IVA
P
r
i
m
árny koncový ukazovateľ Absolútna zmena v indexe pľúcneho klírensu (LCI
2,5
) od východiskovej hodnoty do
24. týždňa
Placebo
(
n = 101)
Rozdiel v liečbe – Zmena vrámci skupiny 0,08
(P = 0,5390)
250 mg každých
12 hod
(n = 103)
-1,09
(P < 0,0001)
-1,01 (P < 0,0001)
Kĺ
ú
čové sekundárne koncové body* Absolútna zmena v BMI
2 Rozdiel v liečbe –
(P = 0,2522)
A
bsolútna zmena v skóre CFQ-R respiračnej domény do 24. týždňa (body)
Zmena v rámci skupiny 0,27
(P = 0,0002)
Rozdiel v liečbe –
Zmena v rámci skupiny 3,0
(P = 0,0035)
0,38
(P < 0,0001)
2,5
(P = 0,0628)
5,5
(P < 0,0001)
* Skúšanie obsahovalo kľúčový sekundárny a ďalšie sekundárne koncové ukazovateľe.
Percentuálny predpoklad FEV1 bol tiež vyhodnotený ako klinicky zmysluplný ďalší sekundárny
koncový bod. U pacientov s lumakaftorom/ivakaftorom bol rozdiel v liečbe pre absolútnu zmenu
v ppFEV1 od východiskovej hodnoty do 24. týždňa 2,4 (P
= 0,0182).
Skúšanie 8: Štúdia týkajúca sa bezpečnosti a znášanlivosti u pediatrických pacientov s CF vo veku 2až 5 rokov, ktorí sú homozygotní nosiči mutácie F508del v géne CFTRSkúšanie 8 hodnotilo 60 pacientov vo veku 2 až 5 rokov (priemerný vek 3,7 rokov) pri skríningu.
Podľa telesnej hmotnosti pri skríningu dostávali pacienti granulát zmiešaný s jedlom každých 12 hodín v dávke lumakaftor 100 mg/ivakaftor 125 mg u pacientov s hmotnosťou pod 14 kg (n = 19) alebo lumakaftor 150 mg/ivakaftor 188 mg u pacientov s hmotnosťou 14 kg alebo viac (n = 41) po dobu
24 týždňov spolu s ostatnou predpísanou terapiou na CF. Na zhodnotenie účinkov obdobia bez liečby mali pacienti bezpečnostnú následnú návštevu po 2 týždňoch obdobia vymývania.
Sekundárne koncové ukazovatele zahŕňali absolútnu zmenu chloridov v pote v 24. týždni od východiskovej hodnoty a absolútnu zmenu chloridov v pote v 26.týždni od. 24. týždňa (pozri Farmakodynamické účinky) takisto ako koncové ukazovatele uvedené v tabuľke 8. Klinický význam rozsahu týchto zmien u detí vo veku 2 až 5 rokov s cystickou fibrózou nebol pri dlhodobej liečbe jednoznačne stanovený.
Tabuľka 8: Zhodnotenie sekundárnych výsledkov v skúšaní 8 Sekundárne koncové ukazovatele* LUM/IVA
Sekundárne koncové ukazovatele* LUM/IVAAbsolútna zmena body mass index (BMI) od n = 57
východiskovej hodnoty 0,27
95 % CI: 0,07; 0,47; P = 0,0091
Absolútna zmena z-skóre BMI vzhľadom na vek od východiskovej hodnoty
Absolútna zmena hmotnosti od východiskovej hodnoty (kg)
Absolútna zmena z-skóre hmotnosti vzhľadom na vek od východiskovej hodnoty
Absolútna zmena vzrastu od východiskovej hodnoty (v cm)
Absolútna zmena z-skóre vzrastu vzhľadom na vek od východiskovej hodnoty
Absolútna zmena hodnôt fekálnej elastázy-1
(FE-1) od východiskovej hodnoty (µg/g)**
n = 57
0,29
95 % CI: 0,14; 0,45; P = 0,0003
n = 57
1,4
95 % CI: 1,2; 1,7; P < 0,0001
n = 57
0,26
95 % CI: 0,15; 0,38; P < 0,0001
n = 57
3,6
95 % CI: 3,3; 3,9; P < 0,0001
n = 57
0,09
95 % CI: 0,02; 0,15; P = 0,0104
n = 35
52,6
95 % CI: 22,5; 82,7; P = 0,0012

LCI 2,5 n = 17
-0,58
95 % CI: -1,17; 0,02; P = 0,0559
Poznámka: P hodnoty v tabuľke sú nominálne
* Absolútnou zmenou od východiskovej hodnoty pre uvedené koncové ukazovatele je priemerná absolútna zmena od východiskovej hodnoty v 24.týždni.
** Všetci pacienti vykazovali na začiatku skúšania pankreatickú nedostatočnosť. Traja zo 48 pacientov, ktorí mali východiskové hodnoty fekálnej elastázy-1 < 100 µg/g, dosiahli v 24. týždni hodnoty ≥ 200 µg/g.
PediatrickápopuláciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Orkambi v jednej
alebo vo viacerých podskupinách pediatrickej populácie s cystickou fibrózou. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnostiExpozícia (AUC) lumakaftoru je približne 2-násobne vyššia u zdravých dospelých dobrovoľníkov v porovnaní s expozíciou u pacientov s CF. Expozícia ivakaftoru je podobná medzi zdravými dospelými dobrovoľníkmi a pacientmi s CF. Po dávkovaní dvakrát denne bol ustálený stav plazmatickej koncentrácie u zdravých jedincov dosiahnutý všeobecne približne po 7 dňoch liečby
s akumulačným pomerom približne 1,9 pre lumakaftor. Ustálený stav expozície ivakaftoru je nižší ako pri 1. dni kvôli indukčnému účinku CYP3A lumakaftorom (pozri časť 4.5).
Po perorálnom podaní lumakaftoru 400 mg každých 12 hod/ivakaftoru 250 mg každých 12 hod po jedle boli namerané hodnoty v ustálenom stave (±SD) pre AUC0-12hod a Cmax 198 (64,8) µg∙h/ml a 25,0 (7,96) µg/ml pre lumakaftor v uvedenom poradí a 3,66 (2,25) µg∙h/ml a 0,602 (0,304) µg/ml pre ivakaftor. Po perorálnom podaní ivakaftoru samotného ako 150 mg každých 12 hod boli namerané hodnoty v rovnovážnom stave (±SD) pre AUC0-12h a Cmax pri 9,08 (3,20) µg∙h/ml a 1,12 (0,319) µg/m v uvedenom poradí.
AbsorpciaPo opakovanom podaní perorálnej dávky lumakaftoru sa expozícia lumakaftoru zvyčajne úmerne zvýšila s dávkou od 50 mg do 1 000 mg každých 24 hodín. Expozícia lumakaftoru sa zvýšila približne
2,0-násobne pri podaní s jedlom obsahujúcim tuky v porovnaní s podaním nalačno. Medián
(rozmedzie) tmax lumakaftoru po jedle je približne 4,0 hodiny (2,0; 9,0).
Po opakovanom podaní perorálnej dávky ivakaftoru v kombinácii s lumakaftorom sa expozícia ivakaftoru zvyčajne zvýšila úmerne s dávkou od 150 mg každých 12 hodín do 250 mg každých
12 hodín. Expozícia ivakaftoru pri podaní v kombinácii s lumakaftorom sa zvýšila u zdravých
dobrovoľníkov približne 3-násobne pri podávaní s jedlom obsahujúcim tuky. Preto sa má lumakaftor/ivakaftor podávať s jedlom obsahujúcim tuky. Medián (rozmedzie) tmax po jedle je približne 4,0 hodiny (2,0; 6,0).
Distribúcia
Približne 99 % lumakaftoru sa viaže na plazmatické proteíny, predovšetkým na albumín. Po perorálnom podaní 400 mg lumakaftoru každých 12 hodín u pacientov s CF po jedle bol zdanlivý distribučný objem pre centrálne a periférne kompartmenty (CV) 23,5 l (48,7 %) a 33,3 l(30,5 %).
Približne 99 % ivakaftoru sa viaže na plazmatické proteíny, predovšetkým na alfa 1-kyslý glykoproteín a ambumín. Po perorálnom podaní 250 mg ivakaftoru každých12 hodín v kombinácii
s lumakaftorom boli zdanlivé distribučné objemy pre centrálne a periférne kompartmenty [variačný
koeficient ako percento (CV)] (CV) 95,0 l (53,9 %), respektíve 201 l, (26,6 %).
In vitro štúdie naznačujú, že lumakaftor je substrát proteínu zodpovedného za rezistenciu pri
karcinóme prsníka (BCRP).
Biotransformácia
Lumakaftor nie je u ľudí extenzívne metabolizovaný s väčšinou lumafaktoru vylúčenou v nezmenenej forme v stolici. In vitro a in vivo údaje naznačujú, že lumakaftor je metabolizovaný predovšetkým
oxidáciou a glukuronidáciou.
Ivakaftor je u ľudí extenzívne metabolizovaný. In vitro a in vivo údaje naznačujú, že ivakaftor je metabolizovaný predovšetkým prostredníctvom CYP3A. M1 a M6 sú dva hlavné metabolity ivakaftoru u ľudí. Účinnosť metabolitu M1 zodpovedá približne jednej šestine účinnosti ivakaftoru a M1 sa považuje za farmakologicky aktívny. M6 má menej ako jednu päťdesiatinu účinnosti ivakaftoru a nepovažuje sa za farmakologicky aktívny.
Eliminácia
Po perorálnom podaní lumakaftoru sa väčšina lumakaftoru (51 %) vylúči nezmenená v stolici. Zanedbateľné množstvo lumakaftoru sa vylúči ako nezmenené liečivo močom. Zdanlivý terminálny polčas je približne 26 hodín. Zdanlivý klírens CL/F (CV) lumakaftoru pre pacientov s CF bol
2,38 l/hod (29,4 %) .
Po perorálnom podaní samotného ivakaftoru sa väčšina (87,8 %) eliminuje v stolici po metabolickej premene. Zanedbateľné množstvo ivakaftoru sa vylúči močom v nezmenenej podobe. U zdravých jedincov je polčas ivakaftoru po podaní s lumakaftorom približne 9 hodín. Typický CL/F (CV) ivakaftoru po podaní v kombinácii s lumakaftorom bol u pacientov s CF 25,1 l/h (40,5 %).
Osobitnéskupinypacientov
Poruchafunkciepečene
Po opakovanom podávaní lumakaftoru/ivakaftoru 10 dní mali jedinci so stredne závažnou poruchou funkcie pečene (Child-Pughova trieda B, skóre 7 až 9) vyššiu expozíciu (AUC0-12hod o približne 50 % a Cmax o približne 30 %) v porovnaní so zdravými demograficky zodpovedajúcimi jedincami. Vplyv miernej poruchy funkcie pečene (Child-Pughova trieda A, skóre 5 až 6) na farmakokinetiku lumakaftoru v kombinácii s ivakaftorom sa neskúmal, ale predpokladá sa zvýšenie expozície nižšie
ako 50 %.
U pacientov so závažnou poruchou funkcie pečene (Child-Pughova trieda C, skóre10 až 15) sa neuskutočnili štúdie, ale predpokladá sa, že expozícia bude vyššia ako u pacientov so stredne závažnou poruchou funkcie pečene (pozri časti 4.2, 4.4 a 4.8).
P
orucha
f
unkcie
obličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili farmakokinetické štúdie
s lumakaftorom/ivakaftorom. Vo farmakokinetickej štúdii u ľudí sa zistila minimálna eliminácia lumakaftoru a jeho metabolitov močom (iba 8,6 % z celkovej rádioaktivity sa vylúčilo močom
a 0,18 % sa vylúčilo v nezmenenej podobe). Vo farmakokinetickej štúdii u ľudí s ivakaftorom sa
zistila minimálna eliminácia ivakaftoru a jeho metabolitov močom (iba 6,6 % z celkovej dávky rádioaktivity sa vylúčilo v moči). Populačná farmakokinetická analýza klírensu v porovnaní
s klírensom kreatinínu nevykazuje žiadny trend u jedincov s miernou a stredne ťažkou poruchou
funkcie obličiek (pozri časť 4.2).
Staršie osoby
Bezpečnosť a účinnosť lumakaftoru/ivakaftoru u pacientov vo veku 65 rokov alebo starších neboli skúmané.
Pohlavie
Vplyv pohlavia na farmakokinetiku lumakaftoru sa hodnotil pomocou údajov populačnej farmakokinetiky z klinických štúdií lumakaftoru v kombinácii s ivakaftorom. Výsledky neukázali žiaden klinicky významný rozdiel vo farmakokinetických parametroch lumakaftoru alebo ivakaftoru medzi mužmi a ženami. Na základe pohlavia nie je potrebná úprava dávky.
Pediatrickápopulácia
Expozície u dospelých a v pediatreckej populácii sú podobné, na základe populačných analýz (PK)
znázornených v tabuľke 9 nižšie:
T
abuľka 9: Priemerná (SD) expozícia lumakaftoru a ivakaftoru podľa veku
P
r
i
e
m
erná (SD)
P
r
i
e
m
erná (SD)
V
eková skupina Dávka
AUC
ss
l
umakaftoru (μg/ml*hod)
AUC
ss
i
vakaftoru (μg/ml*hod)
Pacienti vo veku 2 až
5 rokov s hmotnosťou pod
14 kg
Pacienti vo veku 2 až
5 rokov s hmotnosťou
14 kg a viac
vrecko lumakaftor
100 mg/ivakaftor 125 mg
každých 12 hodín 180 (45,5) 5,92 (4,61)
vrecko lumakaftor
150 mg/ivakaftor 188 mg
každých 12 hodín 217 (48,6) 5,90 (1,93)
Pacienti vo veku 6 až
11 rokov
Pacienti vo veku 12 až menej ako 18 rokov
lumakaftor 200 mg/ivakaftor
250 mg každých 12 hodín lumakaftor 400 mg/ivakaftor
250 mg každých 12 hodín
203 (57,4) 5,26 (3,08)
241 (61,4) 3,90 (1,56)
 5.3 Predklinické údaje o bezpečnosti
L
umakaftor
5.3 Predklinické údaje o bezpečnosti
L
umakaftor
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu a reprodukčnej toxicity a vývinu
neodhalili žiadne osobitné riziko pre ľudí. Špecifické štúdie týkajúce sa hodnotenia fototoxického
potenciálu lumakaftoru neboli vykonané, hoci na základe zhodnotenia dostupných predklinických a klinických údajov sa fototoxicita nepredpokladá.
IvakaftorÚčinky v štúdiách s opakovaným podaním sa pozorovali iba pri expozíciách považovaných za dostatočne vyššie (> 25-násobne, > 45-násobne a > 35-násobne pre myši, mačky, potkany a psov) ako
je maximálna expozícia ivakaftoru u ľudí pri podávaní Orkambi, čo poukazuje na malý význam týchto zistení pre klinické použitie. Predklinické údaje, založené na obvyklých štúdiách genotoxicity
a karcinogénneho potenciálu neodhalili žiadne zvláštne riziko pre ľudí.
B
ezpečnostná farmakológia
Ivakaftor vyvolal od koncentrácie závislý inhibičný účinok na koncové časti proteínu hERG (human ether-à-go-go related gene) s IC15 5,5 µM, v porovnaní s Cmax (1,5 µM) pre ivakaftor
pri terapeutickom dávkovaní lumakaftoru/ivakaftoru. V telemetrickej štúdii u psov s jednorazovými dávkami až do 60 mg/kg ani pri EKG meraniach zo štúdií s opakovanou dávkou s trvaním až do
1 roka pri dávkovej hladine 60 mg/kg/deň u psov (Cmax po 365 dňoch = 36,2 až 47,6 μM) sa však nepozorovalo predĺženie QT intervalu indukované ivakaftorom. Ivakaftor vyvolal od dávky závislé, avšak prechodné zvýšenie parametrov krvného tlaku u psov v jednorazových perorálnych dávkach až do 60 mg/kg. V dôkladnej klinickej štúdii hodnotiacej QT interval pri podaní buď lumakaftor 600 mg raz denne/ivakaftor 250 mg každých 12 hodín alebo lumakaftor 1 000 mg raz denne/ivakaftor 450 mg každých 12 hodín, sa nepozorovali žiadne zmeny QTc intervalu alebo krvného tlaku, preukazujúce nemožnosť prenosu týchto predklinických výsledkov do klinickej praxe.
Gravidita a fertilita
Ivakaftor nebol teratogénny pri perorálnom podaní gravidným potkanom a králikom počas štádia organogenézy fetálneho vývoja pri dávkach približne 7-násobku (expozícia ivakaftoru a metabolitov),
respektíve 46-násobku expozície ivakaftoru u človeka pri terapeutickej dávke lumakaftoru/ivakaftoru.
Pri dávkach toxických pre matku spôsobil ivakaftor u potkanov zníženie telesnej hmotnosti plodu, zvýšenie incidencie variácií krčných rebier, hypoplastických rebier, zvlnenie rebier a sternálnych
nepravidelností, vrátane fúzií. Význam týchto zistení pre ľudí nie je známy.
Ivakaftor znížil ukazovatele fertility a reprodukčnej výkonnosti u samcov a samíc potkanov pri dávke
200 mg/kg/deň (dosahujúc expozície 11-násobku, respektíve 7-násobku expozícií pri maximálnej odporúčanej dávke pre ľudí ivakaftorovej zložky Orkambi, založených na sumárnych AUC ivakaftoru
a jeho metabolitov extrapolovaných z expozícií dosiahnutých na 90. deň pri podaní 150 mg/kg/deň
v 6-mesiacov trvajúcej štúdii toxicity s opakovaným podaním a z expozícií v 17. gestačnom dni
v pilotnej štúdii embryofetálneho vývoja u tohto druhu), keď boli samice dávkované pred a počas skorého štádia gravidity. Nebol pozorovaný žiaden vplyv na ukazovatele fertility a reprodukčnej
výkonnosti u samcov a samíc pri dávke ≤ 100 mg/kg/deň (dosahujúc expozície približne 8-násobku,
respektíve 5-násobku expozícií pri maximálnej odporúčanej dávke ivakaftoru, zložke Orkambi, založených na sumárnych AUC ivakaftoru a jeho metabolitov extrapolovaných z expozícií z 90. dňa
pri podaní 100 mg/kg/deň v 6-mesiacov trvajúcej štúdii toxicity s opakovaným podaním
a v embryofetálnej štúdii v 17. gestačnom dni u tohto druhu). U gravidných samíc potkanov a králikov sa pozoroval transplacentárny prestup ivakaftoru.
Perinatálny a postnatálny vývoj
Ivakaftor nespôsobil vývojové defekty u potomkov gravidných potkanov, ktorí dostávali liek perorálne od gravidity po pôrod a odstavenie v dávke 100 mg/kg/deň. Dávky nad 100 mg/kg/deň viedli
k zníženiu indexov prežívania a laktácie na 92 % a 98 % kontrolných hodnôt, v uvedenom poradí,
rovnako ako k zníženiu telesnej hmotnosti mláďat.
Juvenilné zvieratá
Prípady katarakty boli pozorované u mláďat potkanov pri dávke ivakaftoru 0,32-násobku maximálnej odporúčanej dávky na základe systémovej expozície ivakaftoru a jeho metabolitov, ak je podávaný
s lumakaftorom ako Orkambi. Prípady katarakty neboli pozorované u plodov pochádzajúcich z
potkaních matiek liečených počas organogenézy fetálneho vývoja, potkaních mláďat vystavených do určitej miery požitím mlieka až do odstavenia alebo v štúdiách toxicity po opakovanom podaní ivakaftoru. Potenciálny význam týchto zistení u ľudí nie je známy.
Lumakaftoraivakaftor
Štúdie toxicity po opakovanom podaní zahŕňajúce súbežné podávanie lumakaftoru a ivakaftoru neobjavili žiadne špeciálne riziká pre ľudí z hľadiska potenciálu prídavnej alebo synergickej toxicity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Mikrokryštalická celulóza Sodná soľ kroskarmelózy Acetát sukcinát hypromelózy Povidón (K30)
Laurylsíran sodný
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Preukázaná stabilita zmesi po zmiešaní je jedna hodina.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Orkambi granulát je zabalený vo vrecku z laminátovej fólie [biaxiálne orientovaný polyetyléntereftalát/polyetylén/fólia/polyetylén (BOPET/PE/fólia/PE)].
Balenie obsahuje 56 vreciek (obsahuje 4 samostatné obaly so 14 vreckami v jednom obale).
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Vertex Pharmaceuticals (Ireland) Limited
28-32 Pembroke Street Upper
Dublin 2, D02 EK84
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/15/1059/006
EU/1/15/1059/007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 19. novembra 2015
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.