
i s poškodenou funkciou obličiek vyššie riziko
hypotenzie a anémie. Preto sa má zvážiť monitorovanie tlaku krvi a hemoglobínu. Nie sú žiadne klinické skúsenosti s používaním macitentanu u pacientov s PAH so závažnou poruchou obličiek.
Opatrnosť sa odporúča pri tejto skupine pacientov. Nie sú žiadne skúsenosti s používaním macitentanu u dialyzovaných pacientov, preto sa Opsumit v tejto populácii neodporúča (pozri časti 4.2 a 5.2).
Starší pacienti
S užívaním macitentanu u pacientov starších ako 75 rokov sú obmedzené skúsenosti, preto sa má
Opsumit v tejto populácii užívať s opatrnosťou (pozri časť 4.2).
Pomocné látky
Tablety Opsumitu obsahujú laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej
intolerancie, lapónskeho deficitu laktázy alebo glukózo – galaktózovej malabsorbcie nesmú užívať
tento liek.
Tablety Opsumitu obsahujú lecitín získaný zo sóje. Ak sú pacienti alergickí na sóju, Opsumit nesmú
užívať (pozri časť 4.3).
4.5 Liekové a iné interakcie
I
n vitro
š
t
údie
Izoenzýmy cytochrómu P450 CYP3A4, CYP2C8, CYP2C9 a CYP2C19 sa podieľajú na metabolizme
macitentanu a tvorbe jeho metabolitov (pozri časť 5.2). Macitentan a jeho aktívny metabolit nemajú
klinicky významný efekt na inhibičnú a indukčnú aktivitu cytochrómu P 450.
Macitentan a jeho aktívny metabolit nie sú v klinicky významnej koncentrácii inhibítormi pečeňových alebo obličkových transportérov spätného vychytávania vrátane organických anionových transportných polypeptidov (OATP1B1 a OATP1B3). Macitentan a jeho aktívne metabolity nie sú významnými substrátmi OATP1B1 a OATP1B3, avšak pasívnou difúziou prestupujú do pečene.
Macitentan a jeho aktívny metabolit nie sú v klinicky významných koncentráciách inhibítormi pečeňových a obličkových efluxných púmp vrátane multirezistentných proteínov (P-gp, MDR-1) a multirezistentných extrúznych transportérov (MATE1 a MATE2-K). Macitentan inhibuje proteín rezistencie rakoviny prsníka (BCRP) v klinicky relevantných intestinálnych koncentráciách. Macitentan nie je substrátom P-gp/MDR-1.
V klinicky významných koncentráciách macitentan a jeho aktívny metabolit neinteragujú s proteínmi aktívnymi v transporte pečeňovo žlčových solí napr. exportná pumpa pečeňových solí (BSEP)
a sodíkovo závislý taurocholát kotransportný polypeptid (NTCP).
In vivo štúdie
Interakčné štúdie boli vykonané len u dospelých.
Warfarín: Macitentan podávaný v opakovaných dávkach po 10 mg jedenkrát denne nemal žiadny vplyv na expozíciu S warfarínu (CYP2C9 substrát) alebo R warfarínu (CYP3A4 substrát) po jednorázovom podaní 25 mg warfarínu. Farmakodynamický účinok warfarínu na International Normalized Ratio - INR nebol macitentanom ovplyvnený. Farmakokinetika macitentanu a jeho aktívneho metabolitu nebola warfarínom ovplyvnená.
Sildenafil: V rovnovážnej koncentrácii sa expozícia sildenafilu 20 mg trikrát denne zvýšila o 15 % počas súčasného podávania macitentanu v dávke 10 mg jedenkrát denne. Sildenafil, substrát CYP3A4, neovplyvnil farmakokinetiku macitentanu, kým bolo zaznamenané 15% zníženie expozície aktívnemu metabolitu macitentanu. Tieto zmeny sa nepovažujú za klinicky významné. V placebom kontrolovanej klinickej štúdii u pacientov s PAH sa preukázala účinnosť a bezpečnosť macitentanu v kombinácii so sildenafilom.
Ketokonazol: V prítomnosti silného inhibítora CYP3A4 ketokonazolu 400 mg jedenkrát denne stúpla expozícia macicentanu na približne 2-násobok. Predpokladaný nárast bol približne 3 násobný v prítomnosti ketokonazolu 200 mg dvakrát denne pri použití fyziologického farmakokinetického modelovania (PBPK). Treba zohľadniť nepresnosti takéhoto modelovania. Expozícia aktívnemu metabolitu macitentanu klesla o približne 26 %. Treba postupovať opatrne, keď sa macitentan súčasne podáva so silnými inhibítormi CYP3A4 (pozri časť 4.4).
Cyclosporín A: Súčasná liečba s cyklosporínom A 100 mg dvakrát denne v kombinácii s inhibítorom CYP3A4 a OATP neovplyvnila v klinicky významnom rozsahhu rovnovážnu expozíciu macicentanu a jeho aktívnemu metabolitu.
Silné induktory CYP3A4: Súčasná liečba rifampicínom 600 mg denne, silným induktorom CYP3A4, , znížila rovnovážnu expozíciu macitentanu o 79 %, ale neovplyvnila expozíciu k aktívnemu
metabolitu. Treba zvážiť zníženú účinnosť macitentanu za prítomnosti silného induktora CYP3A4 ako
je rifampicín. Je potrebné sa vyhnúť kombinácii macitentanu so silnými induktormi CYP3A4 (pozri
časť 4.4).
H
ormonálna antikoncepcia: Napriek tomu, že neboli vykonané špecifické liekové interakčné štúdie
s hormonálnou antikoncepciou, macitentan neovplyvňoval expozíciu iným substrátom CYP3A4 ako je sildenafil. Preto sa neočakáva znížená účinnosť hormonálnej antikoncepcie.
4.6 Fertilita, gravidita a laktáciaGraviditaNie sú k dispozícii údaje o používaní macitentanu u gravidných žien. Štúdie na zvieratách preukázali
reprodukčnú toxicitu (pozri časť 5.3). Potenciálne riziko u ľudí je stále neznáme. Opsumit je
kontraindikovaný počas tehotenstva a u žien vo fertilnom veku, ktoré nepoužívajú spoľahlivú
antikoncepciu (pozri časť 4.3).
Použitie u žien vo fertilnom vekuLiečba Opsumitom u žien vo fertilnom veku sa má začať len, ak bolo potvrdené, že žena nie je

tehotná, bolo jej poskytnuté vhodné poradenstvo o antikoncepcii a používa spoľahlivú antikoncepciu
(pozri časti 4.3 a 4.4). Ženy nesmú otehotnieť 1 mesiac po ukončení liečby Opsumitom. Počas liečby
Opsumitom sa odporúča vykonávať tehotenské testy v mesačných intervaloch, aby sa zabezpečilo
skoré zistenie tehotenstva.
LaktáciaNie je známe, či sa macitentan vylučuje do ľudského materského mlieka. U potkanov sa macitentan
a jeho metabolity vylučujú do mlieka počas laktácie (pozri časť 5.3). Riziko pre dojčené dieťa nie je možné vylúčiť. Opsumit je kontraindikovaný počas dojčenia (pozri časť 4.3).
Fertilita mužov
Po liečbe macitentanom sa pozoroval rozvoj testikulárnej tubulárnej atrofie u samcov zvierat (pozri
časť 5.3). Význam tohto zistenia u ľudí nie je známy, ale nemožno vylúčiť poškodenie
spermatogenézy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeMacitentan môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri zvažovaní schopnosti viesť vozidlá a obsluhovať stroje je potrebné brať do úvahy klinický stav pacienta a profil nežiaducich účinkov macitentanu (ako je bolesť hlavy, hypotenzia).
4.8 Nežiaduce účinkyZhrnutie bezpečnostného profiluNajčastejšie hlásené nežiaduce účinky sú nazofaryngingitída (14,0 %), bolesť hlavy (13,6 %) a anémia
(13,2 %, pozri časť 4.4). Väčšina nežiaducich účinkov je miernej až strednej intenzity.
Tabuľkový zoznamnežiaducichúčinkovBezpečnosť macitentanu bola hodnotená v dlhodobých, placebom kontrolovaných štúdiách u 742
pacientov so symptomatickou PAH. Priemerná dĺžka liečby bola 103,9 týždňa v skupine s dávkou
10 mg macitentanu a 85,3 týždňa v placebovej skupine. Nežiaduce účinky súvisiace s macitentanom získané z tejto klinickej štúdie sú uvedené v tabuľke nižšie.
Frekvencia je definovaná ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000
až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000).
T
rieda orgánových systémov
|
F
r
ekvencia
|
N
ežiaduci účinok
|
Infekcie a nákazy
|
Veľmi časté
|
Nazofaryngtída
|
|
Veľmi časté
|
Bronchitída
|
|
Časté
|
Faryngitída
|
|
Časté
|
Chrípka
|
|
Časté
|
Infekcie močový ciest
|
Poruchy krvi a lymfatického systému
|
Veľmi časté
|
Anémia
|
Poruchy nervového systému
|
Veľmi časté
|
Bolesť hlavy
|
Poruchy ciev
|
Časté
|
Hypotenzia
|
Popis vybraných nežiaducich
účinkov
* Hypotenzia súvisela s používaním ERA. V dlhodobej dvojito zaslepenej štúdii u pacientov s PAH
bola hypotenzia hlásená u 7,0 % pacientov liečených 10 mg macitentanu a 4,4 % pacientov s placebom. Toto korešponduje s 3,5 udalosťami/100 pacientorokov v skupine pacientov liečených 10 mg macitentanu v porovnaní 2,7 udalosťami/100 pacientorokov v skupine s placebom.
Edém/zadržiavanie tekutín súviselo s používaním ERA a je tiež klinickou manifestáciou pravostranného zlyhávania a základného PAH ochorenia. V dlhodobej dvojito zaslepenej štúdii u pacientov s PAH bola incidencia edému v skupinách pacientov liečených macitentanom 10 mg 11 udalostí/100 pacientorokov a 12,5 udalostí/100 pacientorokov v skupine s placebom.
Laboratórne odchýlkyPečeňové aminotransferázyV dvojito zaslepenej štúdii u pacientov s PAH bola incidencia zvýšenia hladín aminotransferáz
(ALT/AST) > 3 × ULN 3,4 % v skupine s macitentanom 10 mg a 4,5 % v skupine s placebom. Zvýšenie > 5 × ULN sa vyskytlo u 2,5 % pacientov na macicentane 10 mg oproti 2 % pacientov s
skupine s placebom.
HemoglobínV dvojito zaslepenej štúdii u pacientov s PAH bol macitentan 10 mg spájaný s priemerným poklesom
hemoglobínu 1 g/dL oproti placebu. Pokles koncentrácie hemoglobínu oproti východiskovým hodnotám na hodnotu pod 10 g/dL bol hlásený u 8,7 % pacientov liečených macitentanom 10 mg a 3,4
% pacientov v skupine s placebom.
Leukocyty
V dvojito zaslepenej štúdii u pacientov s PAH sa macitentan 10 mg spájal s poklesom priemerného

počtu leukocytov 0,7 × 109/l oproti východiskovým hodnotám v porovnaní so žiadnou zmenou u pacientov s placebom.
TrombocytyV dvojito zaspelenej štúdii u pacientov s PAH sa macitentan 10 mg spájal s priemerným poklesom
počtu trombocytov 17 × 109/l v porovnaní s priemerným poklesom 11 × 109/l v skupine liečenej placebom.
Pediatrická populácia
Bezpečnosť a účinnosť macitentanu u detí neboli doteraz stanovené.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieZdravým jedincom sa macitentan podával ako jednorázová dávka až do 600 mg. Pozorovali sa nežiaduce účinky ako bolesť hlavy, nevoľnosť a vracanie. V prípade predávkovania musia byť podľa potreby vykonané štandardné podporné opatrenia. Kvôli vysokému stupňu väzby macicentanu na bielkoviny nie je pravdepodobné, že by dialýza bola účinná.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: iné antihypertenzíva, ATC kód: C02KX04
Mechanizmus účinkuEndotelín (ET)-1 a jeho receptory (ETA a ETB) sprostredkúvajú viacero účinkov, ako je
vazokonstrikcia, fibróza, proliferácia, hypertrofia a zápal. Pri chorobách ako je PAH dochádza
k zvýšeniu aktivity lokálneho ET systému a podieľa sa na cievnej hypertrofii a orgánovom poškodení.
Macitentan je perorálne aktívny silný antagonista endotelínových receptorov, aktívny na obidvoch typoch receptorov ETA a ETB a in vitro je približne 100-krát selektívnejší na ETA v porovnaní s ETB receptormi. Macitentan sa vyznačuje vysokou afinitou a dlhodobou väzbou na ET receptory v ľudských bunkách hladkých svalov pľúcnych artérií. Toto zabráni endotelínom sprostredkovanej aktivácii systému druhých poslov, ktorá vedie k vazokonstrikcii a proliferácii buniek hladkých svalov.
Klinická účinnosť abezpečnosťÚčinnosť u pacientov s pľúcnou artériovou hypertenziouMulticentrická, dvojito zaslepená, placebom kontrolovaná paralelná štúdia fázy 3 zameraná na sledovanie príhod a výsledky (AC-055-302/SERAPHIN) zameraná bola vykonaná u 742 pacientov so symptomatickou PAH, ktorí boli randomizovaní do 3 liečebných skupín (placebo [N = 250], 3 mg
[N = 250] alebo 10 mg [N = 242] macitentanu jedenkrát denne) s cieľom určiť dlhodobý účinok na
morbiditu a mortalitu.
Na začiatku bola väčšina zaradených pacientov (64 %) liečená stabilnou dávkou terapie špecifickej pre
PAH, buď perorálnymi inhibítormi fosfodiesterázy (61 %) a/alebo inhalačnými/orálnymi
prostanoidmi (6 %).
Primárnym cieľovým ukazovateľom bol čas do prvého výskytu udalosti morbidity alebo mortality až do konca dvojito zaslepenej liečby definovanej ako smrť alebo atriálna septostómia, transplantácia pľúc alebo zavedenie intravenóznej (i.v.) alebo subkutánnej (s.c.) terapie prostanoidmi alebo iné zhoršenie PAH. Iné zhoršenie PAH bolo definované ako prítomnosť všetkých troch nasledujúcich zložiek: trvalý pokles v 6 minútovom teste chôdze (6MWD) minimálne o 15 % voči východiskovej hodnote, zhoršenie symptómov PAH (zhoršenie funkčnej triedy WHO alebo pravostranné zlyhávanie
srdca) a potreba novej liečby PAH. Všetky príhody boli potvrdené nezávislou komisiou
posudzovateľov, ktorá nemala informácie o pridelenej liečbe.
Vitálny stav bol sledovaný u všetkých pacientov až do konca štúdie (end-of-study (EOS)). EOS bol určený ako dosiahnutie vopred definovaného počtu primárnych cieľových ukazovateľov. V období medzi ukončením liečby a ukončením štúdie mohli pacienti užívať 10 mg macitentanu odslepene alebo alternatívnu liečbu PAH. Celkový medián trvania dvojito zaslepenej liečby bol 115 týždňov (maximálne 188 týždňov liečby macitentanom).
Stredná hodnota veku všetkých pacientov bola 46 rokov (rozpätie 12 až 85 rokov vrátane 20 pacientov mladších ako 18 rokov, 706 pacientov vo veku od 18 až 74 rokov a 16 pacientov vo veku 75 rokov a viac) s väčšinou belochov(55 %) a belošiek (77 %). Približne 52 % bolo vo WHO funkčnej triede II,
46 % bolo vo WHO funkčnej triede III a 2 % pacientov boli vo WHO funkčnej triede IV.
Najčastejšia etiológia u hodnotenej populácie bola idiopatická lebo dedičná PAH (57 %), nasledovala PAH v súvislosti s poruchami spojivového tkaniva (31 %), PAH v súvislosti s korigovanou jednoduchou vrodenou chorobou srdca (8 %)a PAH spojená s inou etiológiou(lieky a toxíny [3 %] a HIV [1 %]).
Cieľové ukazovateleLiečba macitentanom 10 mg viedla do bodu ukončenia liečby v porovnaní s palcebom k 45 % poklesu
rizika (hazard ratio [HR] 0,55; 97,5 % Interval spoľahlivosti: 0,39 až 0,76; logrank p < 0,0001)
kombinovaných cieľových ukazovateľov morbidity a mortality [Obrázok 1 and Tabuľka 1]. Liečebný účinok bol stanovený skoro a udržal sa.
Účinnosť macitentanu 10 mg na primárny cieľový ukazovateľ bol konzistentný vo všetkých podskupinách podľa veku, pohlavia, etnickej príslušnosti, geografického pôvodu,
etiológie, monoterapie alebo v kombinovanej terapii s inými liekmi na PAH podľa WHO funkčnej
triedy (I/II a III/IV).
Obrázok 1 Kaplan-Meier odhad prvej príhody morbidity/mortality v SERAPHIN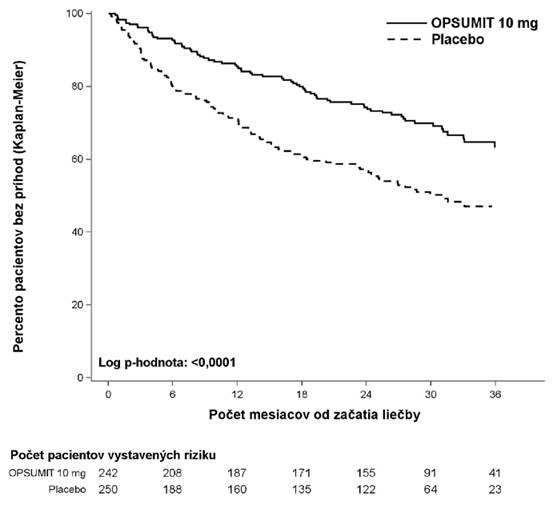
T
abuľka 1 Súhrn cieľových udalostí
C
i
eľové ukazovatele & štatistika
|
P
acienti s príhodami
|
P
orovnanie liečby:
m
acitentan 10 mg vs placebo
|
Placebo
(
N = 250)
|
Macitentan
10 mg
(
N = 242)
|
absolutné zníženie rizika
a
|
R
elatívne
z
níženie rizika (97.5% CI)
|
H
R
a
(
97,5% interval spolehlivos ti)
|
H
odnota p logrank testu
|
U
dalosť morbidita- mortalita
b
|
53%
|
37%
|
16%
|
45% (24%; 61%)
|
0,55 (0,39; 0,76)
|
< 0,0001
|
Ú
m
rtia
c
n (%)
|
19 (7,6%)
|
14 (5,8%)
|
2%
|
36%
(-42%; 71%)
|
0,64
(0,29; 1,42)
|
0,20
|
Z
horšenie
P
A
H
n (%)
|
93 (37,2%)
|
59 (24,4%)
|
13%
|
49% (27%, 65%)
|
0,51 (0,35, 0,73)
|
< 0,0001
|
i
.v./s.c.
Iniciácia Prostanoidu n (%)
|
6 (2,4%)
|
1 (0,4%)
|
2%
|
a = na základe Coxovej regresnej analýzy
b = % pacientov s príhodou do 36 mesiacov = 100 × (1 - KM odhad)
c = vsetky príčiny úmrtia až do ukončenia štúdie bez ohľadu na predchádzajúce zhoršovanie
|
Počet úmrtí zo všetkých príčin do ukončenia štúdie (EOS) v skupine liečenej macitentanom 10 mg bol
35 oproti 44 v skupine s placebom (HR 0,77; 97,5 % Interval spoľahlivosti: 0,46 až 1,28).
Riziko smrti alebo hospitalizácie v súvislosti s PAH sa do ukončenia liečby znížilo o 50 % (HR 0,50;
97,5 % Interval spoľahlivosti (CI): 0,34 až 0,75; logrank p < 0,0001) (EOT) u pacientov liečených
macitentanom 10 mg (50 udalostí) v porovaní s placebom (84 udalostí). Po 36 mesiacoch bolo hospitalizovaných v súvislosti s PAH alebo zomrelo z dôvodu súvisiaceho s PAH 44,6 % pacientov v skupine s placebom a 29,4 % pacientov v skupine s macitentanom 10 mg (zníženie absolútneho rizika 15,2 %).
Symptomatické cieľové ukazovateleNámahová kapacita bola hodnotená ako sekundárny cieľový ukazovateľ. Liečba macitentanom 10 mg
vyústila v mesiaci 6 do placebom korigovaného nárastu strednej hodnoty v 6 minútovom teste chôdze
(6MWD) o 22 metrov (97,5 % Interval spoľahlivosti (CI) 3 až 41; p = 0,0078). Vyhodnotenie 6
minútového testu chôdze podľa funkčnej triedy viedlo v 6 mesiaci k zvýšeniu placebom korigovanej strednej hodnoty predĺženia o 37 metrov (97,5 % Interval spoľahlivosti (CI): 5 až 69,) voči východiskovým hodnotám u pacientov funkčnej triedy III/IV respektíve o12 metrov (97,5% Interval spoľahlivosti (CI): 8 až 33) u pacientov funkčnej triedy I/II. Predĺženie 6MWD dosiahnuté s macitentanom sa udržalo v priebeu trvania štúdie.
Liečba macitentanom 10 mg viedla v 6. mesiaci k 74 % vyššej pravdepodobnosti zlepšenia WHO
funkčnej triedy (risk ratio (RR) 1,74; 97,5 % Interval spoľahlivosti (CI): 1,10 až 2,74; p = 0,0063). Macitentan 10 mg zlepšoval kvalitu života hodnotenú dotazníkom SF-36.
Hemodynamické výsledkyHemodynamické parametre boli hodnotené v podskupine pacientov (placebo [N = 67],
macitentan 10 mg [N = 57]) po 6 mesiacov liečby. Pacienti liečení macitentanom 10 mg dosiahli oproti placebovej skupine medián zníženia pľúcnej vaskulárnej rezistencie 36,5 % (97,5 % Interval spoľahlivosti (CI): 21,7 až 49,2 %) a zvýšenie srdcového indexu 0,58 l/min/m2 (97,5 % Interval spoľahlivosti (CI): 0,28 až 0,93 l/min/m2).
Pediatrická populácia
Európska lieková agentúra udelila odklad povinnosti predložiť výsledky štúdií s macitentanom vo
všetkých podskupinách pediatrickej populácie s PAH (pozri časť 4.2 informácie o používaní v pediatrickej populácii).
5.2 Farmakokinetické vlastnosti
Farmakokinetika macitentanu a jeho aktívneho metabolitu bola dokumentovaná predovšetkým
u zdravých jedincov. Expozícia macitentanu u pacientov s PAH bola približne 1,2-krát väčšia ako u zdravých jedincov. Expozícia pacientov aktívnemu metabolitu, ktorý je približne 5-krát menej
účinný ako macitentan, bola približne 1,3 krát vyššia ako u zdravých jedincov. Farmakokinetika
macitentanu u pacientov s PAH nebola ovplyvnená závažnosťou choroby.
Farmakokinetika macitentanu je po opakovanom podávaní závislá na dávke do 30 mg vrátane. Absorpcia
Maximálne plazmatické koncentrácie macitentanu sa dosiahnu asi za 8 hodín po podaní. Potom
plazmatické hladiny macitentanu a jeho aktívneho metabolitu pomaly klesajú so zdanlivým polčasom
eliminácie 16 respektíve 48 hodín.
U zdravých osôb sa expozícia macitentanu a jeho aktívneho metabolitu nemení za prítomnosti jedla, a preto možno macitentan užívať s jedlom aj bez jedla.
Distribúcia
Macitentan a jeho aktívny metabolit majú vysokú väzbu na plazmatické bielkoviny (> 99 %), prevažne
na albumíny a v menšom rozsahu na alfa 1 kyslý glykoproteín. Macitentan a jeho aktívny metabolit
ACT-132577 sa dobre distribuujú do tkanív ako ukazuje zdanlivý distribučný objem (Vss/F)
macitentanu približne 50 litrov a 40litrov u ACT-132577.
Biotransformácia
Macitentan má štyri primárne metabolické cesty. Oxidatívnu depropyláciu sulfamidov, ktorá vedie
k farmakologicky aktívnym metabolitom. Táto reakcia je závislá na systéme cytochrómu P450, najmä CYP3A4 (približne 99 %) s minimálnym podielom CYP2C8, CYP2C9 a CYP2C19. Aktívny metabolit cirkuluje v ľudskej plazme a môže prispievať k farmakologickému účinku. Ďalšie metabolické cesty vedú k farmkologicky inaktívnym metabolitom. Do formovania týchto metabolitov sa zapájajú viacerí členovia CYP2C rodiny, menovite CYP2C8, CYP2C9 a CYP2C19, ako aj CYP3A4.
Eliminácia
Macitentan sa vylučuje len po intenzívnom metabolizme. Dominantná cesta eliminácie je močom ,
ktorou sa vylúči približne 50 % dávky.
Špeciálne populácie
Vek, pohlavie a etnická skupina nemajú klinicky významný účinok na farmakokinetiku macitentanu
a jeho aktívneho metabolitu.
Poškodenie obličiek
U pacientov so závažným poškodením obličiek stúpla expozícia macitentanu a jeho aktívneho
metabolitu na 1,3 resp 1,6 násobok. Tento nárast sa nepovažuje za klinicky významný (pozri časti 4.2
a 4.4).
Poškodenie funkcie pečene
U jedincov s miernym, stredne závažným a závažným znížením funkcie pečene klesla hladina
macitentanu o 21 %, 34 % a 6 % a hladina aktívneho metabolitu o 20 %, 25 % a 25 %. Tento pokles sa nepovažuje za klinicky významný (pozri časti 4.2 a 4.4).
5.3 Predklinické údaje o bezpečnosti
U psov znižoval macitentan tlak krvi pri expozícii podobnej ľudskej terapeutickej dávke. Zhrubnutie intimy koronárnych artérií bolo pozorované pri 17 násobku expozícii u ľudí po 4 – 39 týždňoch liečby. S ohľadom na špecifickú druhovú senzitivitu a bezpečnostnú rezervu sa tento záver nepovažuje za významný pre ľudí.
U myší, potkanov a psov bola po liečbe macitentanom pozorovaná zvýšená hmotnosť pečene
a hepatocelulárna hypertrofia. Tieto zmeny boli väčšinou reverzibilné a nepovažovali sa za nežiaducu
adaptáciu pečene na zvýšené metabolické nároky.
V štúdii karcinogenity u myší macitentan indukoval vo všetkých dávkach minimálnu až miernu hyperpláziu mukózy a zápalovú infiltráciu submukózy nosovej dutiny. Počas 3 mesačnej štúdie toxicity u potkanov a psov neboli zaznamenané žiadne nálezy v nosovej dutine.
V štandardnej sérii in vitro a in vivo testov nebol macitentan genotoxický. Macitentan nebol fototoxický v in vivo podmienkach po jednorazovej dávke pri expozíciach až do 24 násobku ľudskej expozície. Dvojročné štúdie karcinogenity u potkanov a myší neodhalili karcinogénny potenciál pri expozíciach 20 respektíve 140 násobne vyššej oproti expozícii u ľudí.
V štúdiách dlhodobej toxicity bola u samcov potkanov a psov pozorovaná testikulárna tubulárna dilatácia s bezpečnostnou rezervou 11,6 respektíve 5,8. Tubulárna dilatácia bola úplne reverzibilná. Po dvoch rokoch liečby bola u potkanov pri 4-násobku expozície u ľudí pozorovaná testikulárna
tubulárna atrofia. Hypospermatogenéza bola pozorovaná v dlhodobých štúdiách karcinogenity
u potkanov a v štúdiách toxicity pri opakovanom podaní psom, pri expozícii s mierou bezpečnosti 9,7 u potkanov a 23 u psov. Miera bezpečnosti pre fertilitu pre samcov potkanov bola 18 a pre samice potkanov 44. Nezaznamenali sa žiadne testikulárne nálezy u myší pri liečbe do 2 rokov. Účinok macitentanu na fertilitu u mužov nie je známy (pozri časť 4.6).
Macitentan bol teratogénny u králikov a potkanov vo všetkých testovaných dávkach. U obidvoch druhov boli zaznamenané kardiovaskulárne abnormality a abnormality fúzie mandibulárneho oblúku.
Podávanie macitentanu samiciam potkanov v období od neskorej gravidity až do obdobia laktácie pri expozícii matky 5 násobne vyššej ľudskej expozícii spôsobilo znížené prežívanie mláďat a poškodenie reprodukčnej schopnosti potomkov, ktorí boli vystavení macitentanu počas neskorého intrauteriného obdobia a prostredníctvom mlieka počas sania.
Liečba juvenilných mláďat potkanov od postnatálneho dňa 4 do dňa 114 spôsobila zníženie prírastku celkovej hmotnosti vedúce k sekundárnym účinkom na vývoj (mierne oneskorenie zostupu semeníkov, reverzibilná skátenie dĺžky dlhých kostí, predĺženie estrogénového cyklu). Pri expozícii 7-násobne vyššej expozícii u ľudí sa pozoroval: mierny nárast pre a post implantačných strát, pokles priemerného počtu mláďat, zníženie hmotnosti semenníkov a nadsemenníkov. Pri 3,8 násobku expozície u ľudí bola pozorovaná testikulárna tubulárna atrofia a minimálne účinky na reprodukčné schopnosti a morfológiu spermií.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety Monohydrát laktózy Mikrokryštalická celulóza (E460i)
Sodná soľ karboxymetylškrobu (typ A)
Povidón Magnéziumstearát (E572) Polysorbát 80 (E433)
Filmový obal tablety Polyvinylalkohol (E1203) Oxid titaničitý(E171) Mastenec (E553b)
Sójový lecitín (E322) Xantánová guma (E415)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30 °C.
6.5 Druh obalu a obsah balenia
Biele, nepriehľadné PVC/PVDC/hliníkové blistre v papierových škatuľkách s obsahom 15 alebo 30
filmom obalených tabliet.
Biele fľaštičky z polyetylénu s vysokou hustotou (HDPE) so silikagélovým vysušovadlom, v
škatuľkách s obsahom 30 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Actelion Registration Ltd
Chiswick Tower 13th Floor
389 Chiswick High Road
London W4 4AL
Spojené Kráľovstvo
8. REGISTRAĆNÉ ČÍSLO
EU/1/13/893/001
EU/1/13/893/002
EU/1/13/893/003
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
P
R
ÍLOHA II
A
. VÝROBCA ZODPOVEDNÝ ZA UVOĽNENIE ŠARŽE
B
. PODMIENKY ALEBO OBMEDZENIA TÝKAJÚCE SA VÝDAJA A POUŽITIA
C
. ĎALŠIE PODMIENKY A POŽIADAVKY REGISTRÁCIE D. PODMIENKY ALEBO OBMEDZENIA TÝKAJÚCE SA
B
E
Z
P
EČNÉ
H
O A ÚČINNÉHO POUŽÍVANIA LIEKU
A
. VÝROBCA ZODPOVEDNÝ ZA UVOĽNENIE ŠARŽE
N
ázov a adresa výrobcu zodpovednéhozauvoľneniešarže
Actelion Pharmaceuticals Deutschland GmbH
Basler Strasse 63-65
79100 Freiburg
Nemecko
B. PODMIENKY ALEBO OBMEDZENIA TÝKAJÚCE SA VÝDAJA A POUŽITIA
Výdaj lieku je viazaný na lekársky predpis s obmedzením predpisovania (pozri Prílohu I: Súhrn charakteristických vlastností lieku, časť 4.2).
C. ĎALŠIE PODMIENKY A POŽIADAVKY REGISTRÁCIE
• Periodicky aktualizované správy o bezpečnosti
Držiteľ rozhodnutia o registrácii predloží prvú periodicky aktualizovanú správu o bezpečnosti tohto
lieku do 6 mesiacov po registrácii. Držiteľ rozhodnutia o registrácii následne predloží periodicky aktualizované správy o bezpečnosti tohto lieku v súlade s požiadavkami stanovenými v zozname referenčných dátumov Únie (zoznam EURD) stanovenom v ods. 7 v článku 107c smernice
2001/83/ES a uverejnenom na európskom internetovom portáli pre lieky.
D. PODMIENKY ALEBO OBMEDZENIA TÝKAJÚCE SA BEZPEČNÉHO A ÚČINNÉHO POUŽÍVANIA LIEKU
• Plán riadenia rizík (RMP)
Držiteľ rozhodnutia o registrácii vykoná požadované činnosti a zásahy v rámci dohľadu nad liekmi,
ktoré sú podrobne opísané v odsúhlasenom RMP predloženom v module 1.8.2 registračnej
dokumentácie a v rámci všetkých ďalších aktualizácii plánu riadenia rizík.
Aktualizovaný RMP je potrebné predložiť:
• na žiadosť Európskej agentúry pre lieky,
• vždy v prípade zmeny systému riadenia rizík, predovšetkým v dôsledku získania nových informácií, ktoré môžu viesť k výraznej zmene pomeru prínosu a rizika, alebo v dôsledku dosiahnutia dôležitého medzníka (v rámci dohľadu nad liekmi alebo minimalizácie rizika).
V prípade, že sa dátum predloženia periodicky aktualizovanej správy o bezpečnosti lieku (PSUR)
zhoduje s dátumom aktualizácie RMP, môžu sa predložiť súčasne.
• Dodatočnéopatrenianaminimalizáciu rizika
MAH má v spolupráci s národnou kompetentnou autoritou odsúhlasiť podrobnosti Súpravy pre
predpisujúcich lekárov a zabezpečiť kontrolovaný systém distribúcie a implementovať ich ešte pred zavedením lieku na trh. MAH má zabezpečiť, že všetci odborníci predtým ako predpíšu (a/alebo
vydajú) Opsumit budú mať Súpravu pre predpisujúcich lekárov obsahujúcu nasledovné materiály:
• Súhrn charakteristických vlastností Opsumitu
• Kontrolný zoznam pre predpisujúceho lekára.
• Brožúru pre zdravotníckych pracovníkov, obsahujúcu informácie o Opsumite
• Informačná kartička pre pacienta
Kontrolný zoznam pre predpisujúceho lekára má upovedomiť predpisujúceho lekára na kontraindikácie, upozornenia a opatrenia ako aj na kľúčové informácie aby:
• Poskytol pacientom primerané informácie ohľadom bezpečného užívania lieku;
• Zabezpečil, aby ženy v produktívnom veku neboli pred začatím liečby Opsumitom tehotné a používali spoľahlivé metódy antikoncepcie;
• Zabezpečil pacientovi Kartičku pre pacienta;
• Zdôraznil nutnosť počiatočných a mesačných tehotenských testov a monitoringu hladiny hemoglobínu a pečeňových funkcií.
Brožúra pre zdravotníckych profesionálov/lekárov musí obsahovať nasledujúce kľúčové prvky:
• Že pacienti musia byť schopní dodržiavať požiadavky na bezpečné používanie
Opsumitu;
• Riziko anémie, hepatotoxicity, teratogenity a potrebu spoľahlivej antikoncepcie;
• Nutnosť vykonávať počiatočné a:
• mesačné tehotenské testy;
• pravidelné monitorovania hladín hemoglobínu;
• pravidelné monitorovania pečeňových funkcií;
• význam okamžitého informovania lekára o možnom tehotenstve, ktoré sa vyskytne
počas liečby Opsumitom.
Informačná kartička pre pacienta, určená pre pacientov, ktorým bol predpísaný Opsumit musí
obsahovať nasledujúce kľúčové body:
• Opsumit je teratogénny pre zvieratá;
• Opsumit nesmú užívať tehotné ženy;
• Ženy vo fertilnom veku musia používať spoľahlivé metódy antikoncepcie;
• Nutnosť pravidelných mesačných tehotenských testov;
• Nutnosť pravidelných krvných testov, pretože Opsumit spôsobuje pokles hemoglobínu;
• Nutnosť pravidelného monitorovania pečeňových funkcií, pretože Opsumit má hepatotoxický potenciál.
P
R
ÍLOHA III
O
Z
NAČEN
IE OBALU A PÍSOMNÁ INFORMÁCIA PRE POUŽÍVATEĽA
A
. OZNAČENIE OBALU





 ÚDA
JE, KTORÉ MAJÚ BYŤ UVEDENÉ NA VONKAJŠOM OBALE
ŠKATUĽKA/BLISTRE
1. NÁZOV LIEKU
ÚDA
JE, KTORÉ MAJÚ BYŤ UVEDENÉ NA VONKAJŠOM OBALE
ŠKATUĽKA/BLISTRE
1. NÁZOV LIEKU
Opsumit 10 mg filmom obalené tablety
macitentan
2. LIEČIVO (LIEČIVÁ)Každá filmom obalená tableta obsahuje 10 mg macitentanu.
3. ZOZNAM POMOCNÝCH LÁTOKKaždá filmom obalená tableta obsahuje tiež laktózu a sójový lecitín (E322). Ďalšie informácie, pozri
písomnú informáciu pre používateľa.
4. LIEKOVÁ FORMA A OBSAH15 filmom obalených tabliet
30 filmom obalených tabliet
5. SPÔSOB A CESTA (CESTY) PODANIANa vnútorné použitie.
Pred použitím si prečítajte písomnú informáciu pre používateľa.
6. ŠPECIÁLNE UPOZORNENIE, ŽE LIEK SA MUSÍ UCHOVÁVAŤ MIMO DOHĽADUA DOSAHU DETÍUchovávajte mimo dohľadu a dosahu detí.
7. INÉ ŠPECIÁLNE UPOZORNENIE (UPOZORNENIA), AK JE TO POTREBNÉ8. DÁTUM EXSPIRÁCIEEXP

 9. ŠPECIÁLNE PODMIENKY NA UCHOVÁVANIE
9. ŠPECIÁLNE PODMIENKY NA UCHOVÁVANIE
Uchovávajte pri teplote do 30 °C
10. ŠPECIÁLNE UPOZORNENIA NA LIKVIDÁCIU NEPOUŽITÝCH LIEKOV ALEBO ODPADOV Z NICH VZNIKNUTÝCH, AK JE TO VHODNÉ11. NÁZOV A ADRESA DRŽITEĽA ROZHODNUTIA O REGISTRÁCIIActelion Registration Ltd
Chiswick Tower 13th Floor
389 Chiswick High Road
London W4 4AL
Spojené Kráľovstvo
12. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/893/001
EU/1/13/893/002
13. ČÍSLO VÝROBNEJ ŠARŽELot
14. ZATRIEDENIE LIEKU PODĽA SPÔSOBU VÝDAJAVýdaj lieku je viazaný na lekársky predpis.
15. POKYNY NA POUŽITIE16. INFORMÁCIE V BRAILLOVOM PÍSMEOpsumit 10 mg
 MINIMÁLNE ÚDAJE, KTORÉ MAJÚ BYŤ UVEDENÉ NA BLISTROCH
B
L
ISTRE
1. NÁZOV LIEKU
MINIMÁLNE ÚDAJE, KTORÉ MAJÚ BYŤ UVEDENÉ NA BLISTROCH
B
L
ISTRE
1. NÁZOV LIEKU
Opsumit 10 mg filmom obalené tablety
macitentan
2. NÁZOV DRŽITEĽA ROZHODNUTIA O REGISTRÁCIIActelion
3. DÁTUM EXSPIRÁCIEEXP
4. ČÍSLO VÝROBNEJ ŠARŽELot
5. INÉ
 ÚDA
JE, KTORÉ MAJÚ BYŤ UVEDENÉ NA VONKAJŠOM OBALE
ŠKATUĽKA / FĽAŠTIČKA
1. NÁZOV LIEKU
ÚDA
JE, KTORÉ MAJÚ BYŤ UVEDENÉ NA VONKAJŠOM OBALE
ŠKATUĽKA / FĽAŠTIČKA
1. NÁZOV LIEKU
Opsumit 10 mg filmom obalené tablety
macitentan
2. LIEČIVO (LIEČIVÁ)Každá filmom obalená tableta obsahuje 10 mg macitentanu.
3. ZOZNAM POMOCNÝCH LÁTOKKaždá filmom obalená tableta obsahuje tiež laktózu a sójový lecitín (E 322). Ďalšie informácie si,
pozrite v písomnej informácii pre používateľa.
4. LIEKOVÁ FORMA A OBSAH30 filmom obalených tabliet
5. SPÔSOB A CESTA PODANIANa vnútorné použitie.
Pred použitím si prečítajte písomnú informáciu pre používateľa.
6. ŠPECIÁLNE UPOZORNENIE, ŽE LIEK SA MUSÍ UCHOVÁVAŤ MIMO DOHĽADUA DOSAHU DETÍUchovávajte mimo dohľadu a dosahu detí.
7. INÉ ŠPECIÁLNE UPOZORNENIE (UPOZORNENIA), AK JE TO POTREBNÉ8. DÁTUM EXSPIRÁCIEEXP
9. ŠPECIÁLNE PODMIENKY NA UCHOVÁVANIEUchovávajte pri teplote do 30 °C.
10. ŠPECIÁLNE UPOZORNENIA NA LIKVIDÁCIU NEPOUŽITÝCH LIEKOV ALEBO ODPADOV Z NICH VZNIKNUTÝCH, AK JE TO VHODNÉ
11. NÁZOV A ADRESA DRŽITEĽA ROZHODNUTIA O REGISTRÁCII
Actelion Registration Ltd
Chiswick Tower 13th Floor
389 Chiswick High Road
London W4 4AL
Spojené Kráľovstvo
12. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/893/003
13. ČÍSLO VÝROBNEJ ŠARŽELot
14. ZATRIEDENIE LIEKU PODĽA SPÔSOBU VÝDAJAVýdaj lieku je viazaný na lekársky predpis.
15. POKYNY NA POUŽITIE16. INFORMÁCIE V BRAILOVOM PÍSMEOpsumit 10 mg
 MINIMÁLNE ÚDAJE, KTORÉ MAJÚ BYŤ UVEDENÉ NA FĽAŠTIČKÁCH
F
ĽA
ŠTIČKY
1. NÁZOV LIEKU
MINIMÁLNE ÚDAJE, KTORÉ MAJÚ BYŤ UVEDENÉ NA FĽAŠTIČKÁCH
F
ĽA
ŠTIČKY
1. NÁZOV LIEKU
Opsumit 10 mg filmom obalené tablety
macitentan
2. LIEČIVO (LIEČIVÁ)Každá filmom obalená tableta obsahuje 10 mg macitentanu
3. ZOZNAM POMOCNÝCH LÁTOKKaždá filmom obalená tableta obsahuje tiež laktózu a sójový lecitín (E 322). Ďalšie informácie si
pozrite v písomnej informácii pre používateľa.
4. LIEKOVÁ FORMA A OBSAH30 filmom obalených tabliet
5. SPÔSOB A CESTA (CESTY) PODANIANa vnútorné použitie.
6. ŠPECIÁLNE UPOZORNENIE, ŽE LIEK SA MUSÍ UCHOVÁVAŤ MIMO DOHĽADUA DOSAHU DETÍUchovávajte mimo dohľadu a dosahu detí
7. INÉ ŠPECIÁLNE UPOZORNENIE (UPOZORNENIA), AK JE TO POTREBNÉ8. DÁTUM EXSPIRÁCIEEXP
9. ŠPECIÁLNE PODMIENKY NA UCHOVÁVANIEUchovávajte pri teplote do 30 °C
 10. ŠPECIÁLNE UPOZORNENIA NA LIKVIDÁCIU NEPOUŽITÝCH LIEKOV ALEBO ODPADOV Z NICH VZNIKNUTÝCH, AK JE TO VHODNÉ
10. ŠPECIÁLNE UPOZORNENIA NA LIKVIDÁCIU NEPOUŽITÝCH LIEKOV ALEBO ODPADOV Z NICH VZNIKNUTÝCH, AK JE TO VHODNÉ
'
11. NÁZOV A ADRESA DRŽITEĽA ROZHODNUTIA O REGISTRÁCII
Actelion Registration Ltd
Chiswick Tower 13th Floor
389 Chiswick High Road
London W4 4AL
Spojené Kráľovstvo
12. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/893/003
13. ČÍSLO VÝROBNEJ ŠARŽELot
14. ZATRIEDENIE LIEKU PODĽA SPÔSOBU VÝDAJAVýdaj lieku je viazaný na lekársky predpis.
15. POKYNY NA POUŽITIE16. INFORMÁCIE V BRAILOVOM PÍSMEOpsumit 10 mg
K
artička pacienta
Strana 1 (predná) Strana 2
Na liečbu pľúcnej arteriálnej hypertenzie
Táto kartička obsahuje dôležité informácie o bezpečnosti, o ktorých musíte byť oboznámený pred liečbou Opsumitom. Noste túto kartičku vždy so sebou a ukážte ju každému ošetrujúcemu lekárovi.
Opsumit® 10 mg
macitentan filmom obalené tablety
SK
Je dôležité, aby ste svojmu ošetrujúcemu lekárovi počas liečby
Opsumitomokamžite hlásili, každé tehotenstvo alebo vedľajší účinok.
Liečebné centrum: Meno predpisujúceho lekára: Telefónne číslo predpisujúceho lekára:
Strana 3 (vnútri vľavo) Strana 4 (vnútri v strede)
T
ehotenstvo
Opsumit môže poškodiť vývoj plodu. Preto nesmiete užívať Opsumit, ak ste tehotná a tiež nesmiete otehotnieť, ak užívate Opsumit. Naviac, ak
mate pľúcnu artériovú hypertenziu, tehotenstvo môže vážne zhoršiť
príznaky vašej choroby.
Antikoncepcia
Počas liečby Opsumitom, potrebujete spoľahlivú (antikoncepciu. S
každou otázkou sa neváhajte obrátiť na svojho lekára.
Pred začiatkom liečby Opsumitom si musíte urobiť tehotenský test každý nasledujúci mesiac aj vtedy ak si myslíte že nie ste tehotná.
Podobne ako ostatné lieky z tejto skupiny Opsumit môže spôsobiť anémiu (pokles počtu červených krviniek) a môže mať vplyv na pečeň. Pred začiatkom liečby a počas liečby Opsumitom vám váš lekár vykoná krvné testy, aby zistil:
• či máte anémiu (znížený počet červených krviniek)
• či vaša pečeň pracuje správne
Strana 5 (vnútri vpravo) Strana 6 (vzadu)


Prejavy toho, že vaša pečeň nepracuje správne, môžu byť:
• nevoľnosť (nutkanie na vracanie)
• vracanie
• horúčka (vysoká telesná teplota)
• bolesti žalúdka (brucha)
• žltačka (žltnutie kože a očných bielkov)
• tmavý moč
• svrbenie kože
• letargia alebo únava (neobvyklá únava alebo vyčerpanie)
• syndróm podobný chrípke (bolesti kĺbov, svalov, horúčka)
Ak spozorujete ktorýkoľvek z týchto prejavov, okamžite informujte svojho lekára.Odporúčaná dávka Opsumitu je jedna 10 mg tableta jedenkrát denne. Tabletu prehltnite celú, zapite pohárom vody, nehryzte ani nelámte. Opsumit sa môže užívať s jedlom alebo bez jedla.
Ak zabudnete užiť Opsumit, dávku užite hneď ako si spomeniete a potom pokračujte v užívaní tabliet v obvyklý čas. Neužívajte dvojnásobnú dávku, aby ste nahradili zabudnutú tabletu.
Ďalšie informácie o Opsumite si prosím starostlivo prečítajte v písomnej informácii pre používateľa.Ak máte akékoľvek ďalšie otázky o vašej liečbe obráťte sa nasvojho lekára alebo lekárnika.©2013 Actelion Pharmaceuticals Ltd
Opsumit je ochrannou známkou firmy Actelion Pharmaceuticals Ltd
B
. PÍSOMNÁ INFORMÁCIA PRE POUŽÍVATEĽA
Písomná informácia pre používateľa
O
psumit 10 mg filmom obalené tablety
macitentan
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Môžete prispieť tým, že nahlásite akékoľvek vedľajšie účinky, ak sa u vás vyskytnú. Informácie o tom ako hlásiť vedľajšie účinky nájdete na konci časti 4.
Pozorne si prečítajte celú písomnú informáciu predtým, ako začnete užívať tento liek, pretožeobsahuje pre vás dôležité informácie.- Túto písomnú informáciu si uschovajte. Možno bude potrebné, aby ste si ju znovu prečítali.
- Ak máte akékoľvek ďalšie otázky, obráťte sa na svojho lekára alebo lekárnika.
- Tento liek bol predpísaný iba vám. Nedávajte ho nikomu inému. Môže mu uškodiť, dokonca aj
vtedy, ak má rovnaké príznaky ochorenia ako vy.
- Ak sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára alebo lekárnika.
To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej informácii
pre používateľa. Pozri časť 4.
V tejto písomnej informácii pre používateľa sa dozviete1. Čo je Opsumit a na čo sa používa
2. Čo potrebujete vedieť predtým, ako užijete Opsumit
3. Ako užívať Opsumit
4. Možné vedľajšie účinky
5. Ako uchovávať Opsumit
6. Obsah balenia a ďalšie informácie
1. Čo je Opsumit a na čo sa používaOpsumit obsahuje liečivo macitentan, ktoré patrí do skupiny liekov nazývanej „antagonisty endotelínových receptorov“.
Opsumit sa používa na liečbu pľúcnej artériovej hypertenzie (PAH) u dospelých, môže sa užívať samostatne alebo s inými liekmi na PAH. PAH je vysoký krvný tlak v cievach, ktoré transportujú krv zo srdca do pľúc (pľúcnych tepnách). U ľudí s PAH sa tieto tepny zužujú, takže srdce musí pracovať silnejšie, aby nimi prečerpalo krv. To spôsobuje u ľudí pocit únavy, závrat a dychavičnosť.
Opsumit rozširuje pľúcne tepny, uľahčuje tak srdcu čerpanie krvi pľúcnymi tepnami. Toto znižuje tlak krvi, zmierňuje príznaky a zlepšuje priebeh choroby.
2. Čo potrebujete vedieť predtým, ako užijete OpsumitNeužívajte Opsumit:• ak ste alergický na macitentan alebo na ktorúkoľvek z ďalších zložiek tohto lieku (uvedených v časti 6)
• ak ste tehotná, plánujete otehotnieť alebo môžete otehotnieť, pretože nepoužívate spoľahlivú antikoncepčnú metódu. Prečítajte si, prosím, informácie v časti „Tehotenstvo”.
• ak dojčíte. Prečítajte si, prosím, informácie v časti „Dojčenie”.
• ak máte chorobu pečene alebo ak máte veľmi vysoké hladiny pečeňových enzýmov v krvi.
Oznámte to svojmu lekárovi, ktorý rozhodne, či je tento liek pre vás vhodný. Ak sa vás týka niektorý so spomínaných bodov, oznámte to svojmu lekárovi.
U
pozornenia a opatrenia
B
uďte zvlášť opatrný pri užívaní Opsumitu:
Ak máte anémiu (znížený počet červených krviniek).
Váš lekár rozhodne, kedy si máte daťurobiťkrvné vyšetrenie:
Predtým ako začnete užívať Opsumit a v pravidelných intervaloch počas jeho užívania, vám váš lekár
urobí krvný vyšetrenie, aby zistil:
• či máte anémiu (znížené množstvo červených krviniek)
• či vaša pečeň správne funguje
Prejavy toho, že vaša pečeň nefunguje správne zahŕňajú:
• nevoľnosť (nauzea)
• vracanie
• horúčka
• bolesť žalúdka (brucha)
• žltnutie kože alebo očných bielkov (žltačka)
• tmavo sfarbený moč
• svrbenie kože
• neobvyklá únava alebo vyčerpanie (letargia alebo únava)
• príznaky podobné chrípke (bolesť kĺbov a svalov s horúčkou)
Ak spozorujete akýkoľvek z týchto prejavov, povedzte to ihneď svojmu lekárovi.
Ak máte problémy s obličkami, povedzte to svojmu lekárovi predtým ako začnete užívať Opsumit. Macitentan môže viesť u pacientov s problémami obličiek k ďalšiemu poklesu tlaku krvi a zníženiu hladiny hemoglobínu.
Deti a dospievajúci
Nedávajte tento liek deťom mladším ako 18 rokov.
Starší pacienti
Sú obmedzené skúsenosti s používaním Opsumitu u pacientov starších ako 75 rokov. Opatrnosť treba
venovať používaniu Opsumitu u tejto vekovej skupiny.
Iné lieky a Opsumit
Opsumit môže ovplyvniť iné lieky.
Ak užívate Opsumit spolu s inými liekmi, vrátane liekov uvedených nižšie, účinky Opsumitu alebo iných liekov môžu byť ovplyvnené. Oznámte prosím svojmu lekárovi alebo lekárnikovi, ak užívate niektoré z nasledujúcich liekov:
• rifampicín, klaritromycín, telitromycín (antibiotiká, používané na liečbu infekcií),
• fenytoín (liek používaný na liečbu záchvatov),
• karbamazepín ( používaný na liečbu depresie a epilepsie),
• ľubovník bodkovaný (Hypericum perforatum, rastlinný prípravok, používaný na liečbu
depresie),
• ritonavir, sachinavir (používané pri liečbu infekcie HIV),
• nefazodón (používaný na liečbu depresie),
• ketokonazol (okrem šampónu), itrakonazol, vorikonazol (lieky používané proti plesňovým
infekciám)
Ak teraz užívate, alebo ste v poslednom čase užívali, či práve budete užívať ďalšie lieky, povedzte to svojmu lekárovi alebo lekárnikovi.
T
ehotenstvo
Ak ste tehotná alebo dojčíte, ak si myslíte, že ste tehotná alebo ak plánujete otehotnieť, poraďte sa so
svojím lekárom predtým, ako začnete užívať tento liek.
Opsumit môže poškodiť plod, počatý pred, počas alebo krátko po liečbe.
• Ak je možné, že by ste mohli počas užívania Opsumitu otehotnieť, používajte spoľahlivý spôsob zabránenia počatia (antikoncepcie). Porozprávajte sa o tom so svojím lekárom.
• Neužívajte Opsumit, ak ste tehotná alebo plánujete otehotnieť.
• Ak počas užívania Opsumitu otehotniete alebo si myslíte, že by ste mohla byť tehotná, ihneď
navštívte svojho lekára.
Ak ste žena, ktorá by mohla otehotnieť, váš lekár vás požiada, aby ste si dali urobiť tehotenský
test predtým, ako začnete užívať Opsumit a v pravidelných intervaloch (raz za mesiac) počas užívania
tohto lieku.
Dojčenie
Nie je známe, či Opsumit prechádza do materského mlieka. Počas užívania Opsumitu nedojčite.
Porozprávajte sa o tom so svojím lekárom.
Vedenie vozidiel a obsluha strojov
Opsumit môže spôsobiť vedľajšie účinky ako bolesť hlavy (uvedené v časti 4) a zároveň príznaky
vášho ochorenia môžu taktiež znížiť vašu schopnosť viesť vozidlá alebo obsluhovať stroje.
Opsumit obsahuje laktózu a lecitín (sójový)
Tablety Opsumitu obsahujú malé množstvo cukru nazývaného laktóza. Ak vám váš lekár povedal, že neznášate niektoré cukry, kontaktujete svojho lekára pred prvým užitím tohto lieku.
Tablety Opsumitu obsahujú lecitín, získaný zo sóje. Neužívajte tento liek, ak ste alergický na sóju
(pozri časť 2 „Neužívajte Opsumit“).
3. Ako užívať Opsumit
Opsumit má predpisovať iba lekár, ktorý má skúsenosti s liečbou pľúcnej artériovej hypertenzie.
Vždy užívajte tento liek presne tak, ako vám povedal váš lekár. Ak si nie ste niečím istý, overte si to
u svojho lekára alebo lekárnika.
Odporúčaná dávka Opsumitu je jedna 10 mg tableta jedenkrát denne. Tabletu prehltnite
vcelku a zapite pohárom vody, nežujte a nelámte tabletu. Opsumit môžete užívať s jedlom alebo bez
jedla. Najlepšie je užívať Opsumit každý deň v rovnakom čase.
Ak užijete viac Opsumitu, ako máte
Ak užijete viac tabliet Opsumitu ako ste mali, poraďte sa so svojím lekárom.
Ak zabudnete užiť Opsumit
Ak zabudnete užiť Opsumit, užite dávku ihneď, ako si spomeniete, potom pokračujte v užívaní vašich tabliet vo zvyčajnom čase. Neužívajte dvojnásobnú dávku, aby ste nahradili vynechanú tabletu.
Ak prestanete užívať Opsumit
Opsumit je liek, ktorý budete musieť užívať pravidelne, aby ste zvládli svoju pľúcnu artériovú
hypertenziu. Neprestávajte užívať Opsumit, pokiaľ vám k tomu nedal súhlas váš lekár.
Ak máte akékoľvek ďalšie otázky týkajúce sa použitia tohto lieku, opýtajte sa svojho lekára alebo lekárnika.
4. Možné vedľajšie účinky
Tak ako všetky lieky, aj tento liek môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého.
Veľmi časté vedľajšie účinky (môžu postihovať viac ako 1 z 10 osôb)
• Málokrvnosť (znížený počet červených krviniek) alebo pokles hemoglobínu
• Bolesť hlavy
• Bronchitída (zápal dýchacích ciest)
• Nazofaryngitída (zápal sliznice nosohltana)
• Opuch/zadržiavanie tekutín (zdurenie)
Časté vedľajšie účinky (môžu postihovať až 1 z 10 osôb)
• Faryngitída (zápal hltanu)
• Chrípka
• Infekcie močového systému (infekcie močového mechúra)
• Hypotenzia (nízky tlak krvi )
Hlásenie vedľajších účinkov
Ak sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára alebo lekárnika. To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej informácii pre používateľa. Vedľajšie účinky môžete hlásiť aj priamo prostredníctvom národného systému hlásenia uvedeného v
Prílohe V. Hlásením vedľajších účinkov môžete prispieť k získaniu ďalších informácií o bezpečnosti tohto lieku.
5. Ako uchovávať OpsumitTento liek uchovávajte mimo dohľadu a dosahu detí.
Nepoužívajte tento liek po dátume exspirácie, ktorý je uvedený na škatuľke a blistri po EXP. Dátum exspirácie sa vzťahuje na posledný deň v danom mesiaci.
Uchovávajte pri teplote do 30 °C.
Nelikvidujte lieky odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Tieto opatrenia pomôžu chrániť životné prostredie.
6. Obsah balenia a ďalšie informácieČo Opsumit obsahujeLiečivo je macitentan. Každá tableta obsahuje 10 mg macitentanu.
Ďalšie zložky v tablete sú monohydrát laktózy, mikrokryštalická celulóza (E 460i), povidón, sodná soľ
karboxymetylškrobu Typ A, magnéziumstearát (E 572), polysorbát 80 (E 433), polyvinylalkohol (E
1203), oxid titaničitý (E 171), mastenec (E 553b), sójový lecitín (E 322), and xantánová guma (E 415).
Ako vyzerá Opsumit a obsah baleniaOpsumit 10 mg tablety sú biele až sivobiele dvojito vypuklé okrúhle filmom obalené tablety s “10” na
jednej strane.
Opsumit je dodávaný ako 10 mg filmom obalené tablety v blistroch po 15 alebo 30 tabletách alebo vo fľaštičkách po 30 tabletách.
Na trh nemusia byť uvedené všetky veľkosti balenia.
D
ržiteľ rozhodnutia o registrácii
Actelion Registration Ltd
Chiswick Tower 13th Floor
389 Chiswick High Road
London W4 4AL Spojené Kráľovstvo Tel: +44 20 8987 3320
Výrobca
Actelion Pharmaceuticals Deutschland GmbH Basler Strasse 65
79100 Freiburg
Nemecko
Ak potrebujete akúkoľvek informáciu o tomto lie
rozhodnutia o registrácii:
België/Belgique/Belgien
Actelion Pharmaceuticals Belgium N.V.
Tél/Tel: +32-(0)15 284 777
|
ku, kontaktujte, miestneho zástupcu držiteľa
Lietuva
Nycomed atstovybė
Tel: +370 5210 9070
|
Б
ъл
гария
Аквахим ЕООД
Teл.: +359 2 807 50 00
|
L
uxembourg/Luxemburg
Actelion Pharmaceuticals Belgium N.V. Tél/Tel: +32-(0)15 284 777
|
Č
eská republika
Actelion Pharmaceuticals CZ, s.r.o. Tel: +420 221 968 006
|
Magyarország
Actelion Pharmaceuticals Hungaria Kft. Tel: +36 1 413 3270
|
D
anmark
Actelion Danmark,
Filial af Actelion Pharmaceuticals Sverige AB, Sverige
Tlf: +45 3694 45 95
|
Malta
Actelion Pharmaceuticals Ltd
Tel: +44 845 075 0555
|
D
eutschland
Actelion Pharmaceuticals Deutschland GmbH Tel: +49 761 45 64 0
|
N
ederland
Actelion Pharmaceuticals Nederland B.V. Tel: +31 (0)348 435950
|
E
esti
Nycomed SEFA AS
Tel: +372 6112 569
|
N
orge
Actelion Pharmaceuticals Sverige AB,
Filial Norge
Tlf: +47 22480370
|
Ε
λλάδα
Actelion Pharmaceuticals Eλλάς Α.Ε.
Τηλ: +30 210 675 25 00
|
Ö
sterreich
Actelion Pharmaceuticals Austria GmbH
Tel: +43 1 505 4527
|
E
spaña
Actelion Pharmaceuticals España S.L. Tel: +34 93 366 4399
3
|
P
o
l
ska
Actelion Pharma Polska Sp. z o.o. Tel: +48 (22) 262 31 00
3
|
|
|
F
rance
Actelion Pharmaceuticals France SAS Tél: +33 (0)1 58 62 32 32
Portugal
Actelion Pharmaceuticals Portugal Lda. Tel: +351 21 358 6120
H
rvatska
Medis Adria d.o.o.
Tel: +385 (0) 1 2303 446
România
Geneva Romfarm International
Tel: + 40 (021) 231 3561
Ireland
Actelion Pharmaceuticals UK Ltd
Tel: +353 1890 771 648
Slovenija
Medis d.o.o.
Tel: +386-(0)1 589 69 00
Ísland
Actelion Pharmaceuticals Sverige AB Sími: +46 (0)8 544 982 50
Slovenská republika
Actelion Pharmaceuticals SK, s.r.o. Tel: +420 221 968 006
Italia
Actelion Pharmaceuticals Italia S.r.l.
Tel: +39 0542 64 87 40
Suomi/Finland
Actelion Pharmaceuticals Sverige AB,
Filial Finland
Puh/Tel: +358 9 2510 7720
Κύ
π
ρος
Actelion Pharmaceuticals Eλλάς Α.Ε.
Τηλ: +30 210 675 25 00
Sverige
Actelion Pharmaceuticals Sverige AB
Tel: +46 8 544 982 50
L
atvija
Nycomed Latvija
Tel: +371 784 0082
United Kingdom
Actelion Pharmaceuticals UK Ltd
Tel: +44 845 075 0555
T
áto písomná informácia pre používateľa bola naposledy aktualizovaná v
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
P
r
í
l
oha k článku 127a
P
odmienky alebo obmedzenia týkajúce sa bezpečného a účinného používania lieku, ktoré majú byť implementované členskými štátmi
P
odmienky alebo obmedzenia týkajúce sa bezpečného a účinného používania lieku, ktoré majú byť implementované členskými štátmi
Členské štáty majú zabezpečiť, implementovanie všetkých podmienok alebo obmedzení týkajúcich sa
bezpečného a účinného používania lieku, ktoré sú opísané nižšie:
1. Členský štát sa má dohodnúť s Držiteľom rozhodnutia o registrácii na podrobnostiach Súpravy pre predpisujúcich lekárov a spôsobe kontrolovanej distribúcie. Tento program musí implementovať ešte pred zavedením lieku na trh, aby zabezpečil, že:
• Všetci zdravotnícki odborníci, ktorí môžu predpisovať (alebo, a po dohode s príslušným vnútroštátnym orgánom, podávať liek), obdržia ešte pred predpísaním lieku Súpravou pre predpisujúceho lekára, ktorá bude obsahovať:
o Vzdelávací materiál pre zdravotníckych odborníkov
o Vzdelávaciu brožúru pre pacientov
o Kartičku pacienta
o Súhrn charakteristických vlastností lieku, Písomnú informáciu pre používateľa a označenie obalu