
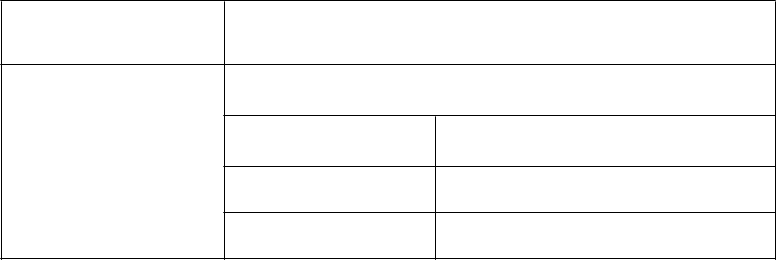
Nový liečebný cyklus sa nesmie začať, kým sa hnačka nezmierni na 1.
alebo nižší stupeň (2 – 3 stolice/deň viac ako frekvencia pred liečbou).
2. stupeň Nový liečebný cyklus sa nesmie začať, kým sa hnačka nezmierni na 1.
alebo nižší stupeň (2 – 3 stolice/deň viac ako frekvencia pred liečbou).
Stupeň (hodnota) toxicity podľa NCI CTCAE v 4.0
1
Úprava dávkovania ONIVYDE/5-FU
(u pacientov, ktorí nie sú homozygotní pre alelu UGT1A1*28)
3. alebo 4. stupeň
Prvý výskyt
Znížte dávku ONIVYDE na 60 mg/m
Znížte dávku 5-FU o 25 % (1 800 mg/m )
Znížte dávku ONIVYDE na 50 mg/m2
Druhý výskyt
Znížte dávku 5-FU o ďalších 25 % (1 350 mg/m2)
Tretí výskyt
Ukončite liečbu
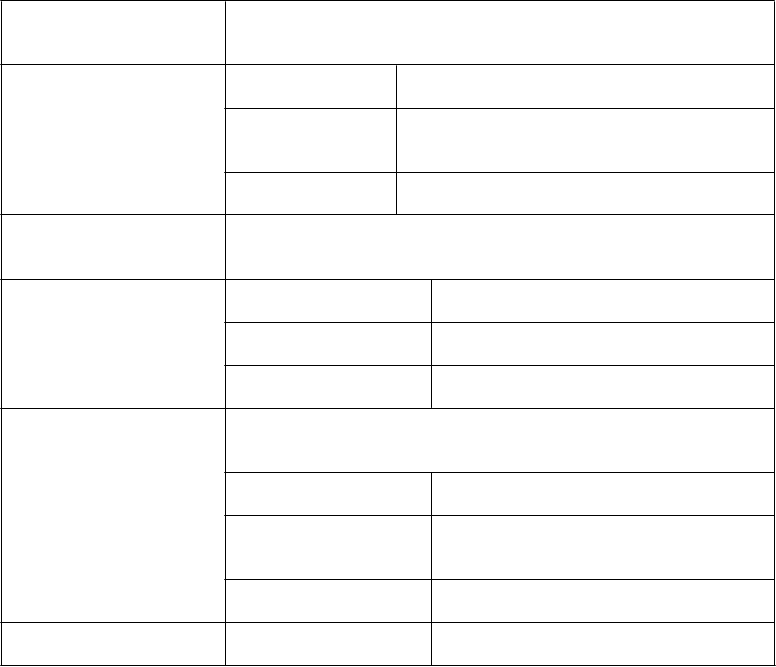
Nevoľnosť/vracanieNový liečebný cyklus sa nesmie začať, kým sa nevoľnosť/vracanie nezmierni na 1. alebo nižší stupeň alebo na východiskový stav
3. alebo 4. stupeň
(napriek
antiemetickej liečbe)
Hepatické, renálne,
respiračné alebo
iné
2
toxicity
3. alebo 4. stupeň
Prvý výskyt
Optimalizujte antiemetickú liečbu
Znížte dávku ONIVYDE na 60 mg/m
Druhý výskyt Optimalizujte antiemetickú liečbu
Znížte dávku ONIVYDE na 50 mg/m
Tretí výskyt Ukončite liečbu
Nový liečebný cyklus sa nesmie začať, kým sa nežiaduca reakcia nezmierni na 1. alebo nižší stupeň
Prvý výskyt Znížte dávku ONIVYDE na 60 mg/m
Znížte dávku 5-FU o 25 % (1 800 mg/m )
Znížte dávku ONIVYDE na 50 mg/m2
Druhý výskyt
Znížte dávku 5-FU o ďalších
25 % (1 350 mg/m2)
Tretí výskyt
Ukončite liečbu
Anafylaktická reakcia Prvý výskyt Ukončite liečbu
1 NCI CTCAE v 4.0 = Spoločné terminologické kritériá pre nežiaduce udalosti Národného onkologického ústavu (National Cancer Institute Common Terminology Criteria for Adverse Events), verzia 4.0
2 Okrem asténie a anorexie; asténia a anorexia 3. stupňa si nevyžadujú úpravu dávkovania.
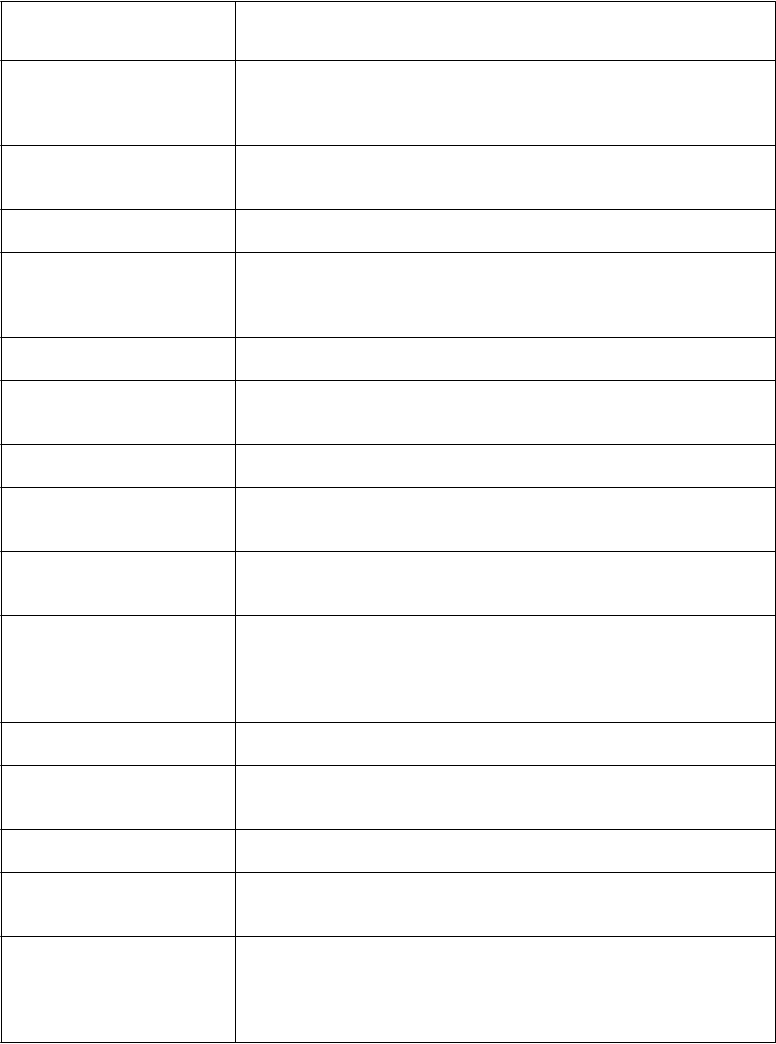
Tabuľka 2: Odporúčané úpravy dávkovania pre ONIVYDE +5-FU/LV pri toxicitách
3. a 4. stupňa u pacientov, ktorí sú homozygotní pre alelu UGT1A1*28
Stupeň (hodnota) toxicity podľa NCI CTCAE v 4.0
1
Úprava dávkovania ONIVYDE/5-FU
(u pacientov, ktorí sú homozygotní pre alelu UGT1A1*28
bez predchádzajúceho zvýšenia dávky na 80 mg/m
2
)
Nový liečebný cyklus sa nesmie začať, kým sa nežiaduca udalosť
nezmierni na 1. alebo nižší stupeň
Nežiaduce reakcie
2
3. alebo 4. stupňa
Prvý výskyt
Znížte dávku ONIVYDE na 50 mg/m
Úprava dávky 5-FU ako v tabuľke 1
Druhý výskyt Znížte dávku ONIVYDE na 40 mg/m
Úprava dávky 5-FU ako v tabuľke 1
Tretí výskyt Ukončite liečbu
1 NCI CTCAE v 4.0 = Spoločné terminologické kritériá pre nežiaduce udalosti Národného onkologického ústavu (National Cancer Institute Common Terminology Criteria for Adverse Events), verzia 4.0
2 Okrem asténie a anorexie; asténia a anorexia 3. stupňa si nevyžadujú úpravu dávkovania.
Osobitné populácie
Poškodenie funkcie pečene
Neuskutočnila sa žiadna špecifická štúdia s ONIVYDE u pacientov s poškodením funkcie pečene. ONIVYDE sa nemá používať u pacientov s hladinami bilirubínu > 2,0 mg/dl alebo aspartátaminotransferázy (AST) a alanínaminotransferázy (ALT) > 2,5-násobok hornej hranice normálnych hodnôt (upper limit of normal, ULN) alebo > 5-násobok ULN, ak sú prítomné metastázy v pečeni (pozri časť 4.4).
Poškodenie funkcie obličiek
Neuskutočnila sa žiadna špecifická štúdia s ONIVYDE u pacientov s poškodením funkcie obličiek. U pacientov s miernym až stredne závažným poškodením funkcie obličiek sa neodporúča žiadna úprava dávkovania (pozri časti 4.4 a 5.2). ONIVYDE sa neodporúča používať u pacientov so závažným poškodením funkcie obličiek (CLcr < 30 ml/min).
Starší pacienti
Štyridsaťjeden percent (41 %) pacientov liečených pomocou ONIVYDE v rámci klinického programu bolo vo veku ≥ 65 rokov. Neodporúča sa žiadna úprava dávkovania.
Pediatrická populácia
Bezpečnosť a účinnosť ONIVYDE u detí a dospievajúcich vo veku ≤ 18 rokov nebola doteraz stanovená. K dispozícii nie sú žiadne údaje.
Spôsob podávania
ONIVYDE je určený na intravenózne použitie. Koncentrát sa musí pred podaním zriediť a podáva sa
ako jednorazová intravenózna infúzia po dobu 90 minút. Ďalšie informácie, pozri časť 6.6.
Opatrenia pred zaobchádzaním alebo podaním lieku
ONIVYDE je cytotoxický liek. Pri zaobchádzaní alebo podávaní ONIVYDE sa odporúča používať
rukavice, okuliare a ochranné oblečenie. Gravidné ženy nemajú s ONIVYDE pracovať.
4.3 Kontraindikácie
Anamnéza závažnej precitlivenosti na irinotekán alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Všeobecné
ONIVYDE je lipozomálna formulácia irinotekánu s inými farmakokinetickými vlastnosťami ako
nelipozomálny irinotekán. Koncentrácia a sila dávky sú iné ako u nelipozomálnych irinotekánov. ONIVYDE nie je ekvivalentný s inými nelipozomálnymi formuláciami irinotekánu a nemá sa s nimi zamieňať.
U obmedzeného počtu pacientov s predošlou expozíciou nelipozomálnemu irinotekánu nebol preukázaný žiadny prínos ONIVYDE.
Myelosupresia/neutropénia
Počas liečby ONIVYDE sa odporúča sledovať úplný krvný obraz. Pacientov je potrebné informovať
o riziku neutropénie a závažnosti horúčky. Medián počtu dní po dolnú hodnotu (nadir) pre 3. a vyšší stupeň neutropénie je 23 (v rozsahu 8 – 104) dní po prvej dávke liečby ONIVYDE. Febrilná neutropénia (telesná teplota > 38 °C a počet neutrofilov ≤ 1 000 buniek/mm³) sa musí urýchlene liečiť
v nemocnici podaním širokospektrálnych intravenóznych antibiotík. Ak sa vyskytne neutropenická horúčka alebo ak absolútny počet neutrofilov klesne pod 1 500/mm3, liečba ONIVYDE sa má prerušiť. U pacientov s metastatickým adenokarcinómom pankreasu liečených ONIVYDE bola pozorovaná sepsa s neutropenickou horúčkou a následným septickým šokom s fatálnym koncom.
U pacientov, u ktorých sa vyskytli závažné hematologické udalosti, sa odporúča zníženie dávky alebo ukončenie liečby (pozri časť 4.2). Pacienti so závažným zlyhaním kostnej drene by nemali byť liečení ONIVYDE.
Anamnéza predchádzajúceho ožarovania orgánov v abdominálnej oblasti zvyšuje riziko závažnej neutropénie a febrilnej neutropénie po liečbe ONIVYDE. U pacientov s anamnézou ožarovania orgánov v abdominálnej oblasti sa odporúča pozorne sledovať krvný obraz a je potrebné zvážiť použitie myeloidných rastových faktorov. Pozornosť je potrebné venovať pacientom, ktorým je ONIVYDE podávaný súbežne s ožarovaním.
U pacientov s nedostatočnou glukuronidáciou bilirubínu, ako sú pacienti s Gilbertovým syndrómom, môže pri liečbe ONIVYDE existovať zvýšené riziko myelosupresie.
U ázijských pacientov existuje v porovnaní s pacientmi bielej rasy zvýšené riziko vzniku závažnej a febrilnej neutropénie po liečbe ONIVYDE+5-FU/LV (pozri časti 4.8 a 5.2).
Imunosupresívne účinky a vakcíny
Podávanie živých alebo živých atenuovaných vakcín pacientom, ktorí sú imunokompromitovaní
chemoterapeutikami vrátane ONIVYDE, môže mať za následok závažné alebo fatálne infekcie. Preto je nevyhnutné zabrániť očkovaniu živými vakcínami. Mŕtve alebo inaktivované vakcíny sa môžu podávať, odpoveď na takéto vakcíny však môže byť znížená.
Interakcie so silnými induktormi CYP3A4
ONIVYDE sa nemá podávať so silnými induktormi enzýmu CYP3A4, ako sú napríklad
antikonvulzíva (fenytoín, fenobarbital alebo karbamazepín), rifampín, rifabutín a ľubovník bodkovaný, iba v prípade, ak neexistuje žiadna alternatívna liečba. Primeraná počiatočná dávka
u pacientov užívajúcich tieto antikonvulzíva alebo iné silné induktory nebola definovaná. Najmenej
2 týždne pred začatím liečby ONIVYDE je potrebné zvážiť substitučnú liečbu liekmi, ktoré neindukujú enzýmy (pozri časť 4.5).
Interakcie so silnými inhibítormi CYP3A4 alebo silnými inhibítormi UGT1A1
ONIVYDE sa nemá podávať so silnými inhibítormi enzýmu CYP3A4 (napr. grapefruitová šťava,
klaritromycín, indinavir, itrakonazol, lopinavir, nefazodón, nelfinavir, ritonavir, saquinavir, telaprevir, vorikonazol). Liečbu silnými inhibítormi CYP3A4 je potrebné ukončiť najmenej 1 týždeň pred začatím liečby ONIVYDE.
ONIVYDE sa nemá podávať so silnými inhibítormi UGT1A (napr. atazanavir, gemfibrozil, indinavir), iba v prípade, ak neexistuje žiadna alternatívna liečba.
Hnačka
Hnačka sa môže vyskytnúť skoro (nástup v priebehu ≤ 24 hodín po začatí liečby ONIVYDE) alebo
oneskorene (> 24 hodín) (pozri časť 4.8).
U pacientov s hnačkou so skorým nástupom je potrebné zvážiť terapeutickú a profylaktickú liečbu atropínom, pokiaľ nie je kontraindikovaná. Pacientov je potrebné upozorniť na riziko oneskorenej hnačky, ktorá môže byť oslabujúca a v zriedkavých prípadoch život ohrozujúca, pretože pretrvávajúca riedka alebo vodnatá stolica môže viesť k dehydratácii, nerovnováhe elektrolytov, kolitíde, gastrointestinálnej (GI) ulcerácii, infekcii alebo sepse.
Hneď, ako sa objaví prvá tekutá stolica, má pacient začať prijímať veľké množstvá tekutín s obsahom elektrolytov. Pacienti musia mať okamžite k dispozícii loperamid (alebo ekvivalent) na začatie liečby v prípade oneskorenej hnačky. Liečba loperamidom sa má začať pri prvom výskyte mäkkej alebo
riedkej stolice alebo hneď po nástupe častejšieho vyprázdňovania ako zvyčajne. Loperamid sa má podávať, až kým pacient nie je aspoň 12 hodín bez hnačky.
Ak počas liečby hnačka pretrváva viac ako 24 hodín, aj keď pacient užíva loperamid, je potrebné zvážiť podpornú liečbu perorálnymi antibiotikami (napr. fluórchinolón počas 7 dní). Loperamid sa nemá používať dlhšie ako 48 po sebe nasledujúcich hodín kvôli riziku vzniku paralytického ileusu. Ak hnačka pretrváva viac ako 48 hodín, ukončite podávanie loperamidu, monitorujte a doplňte tekutiny
s obsahom elektrolytov a pokračujte v podpornej antibiotickej liečbe až do vymiznutia sprievodných príznakov.
Liečbu ONIVYDE je potrebné odložiť, až kým sa hnačka nezmierni na 1. alebo nižší stupeň (2 –
3 stolice/deň častejšie ako frekvencia pred liečbou). ONIVYDE sa nesmie podávať pacientom s obštrukciou čriev a chronickým zápalovým ochorením čriev, až kým nedôjde k náprave.
Pri hnačke 3. alebo 4. stupňa sa nasledujúca dávka ONIVYDE musí znížiť (pozri časť 4.2).
Cholinergné reakcie
Hnačka so skorým nástupom môže byť sprevádzaná cholinergnými príznakmi ako rinitída, nadmerné
slinenie, návaly tepla, diaforéza, bradykardia, mióza a hyperperistaltika. V prípade výskytu cholinergných príznakov sa má podať atropín.
Akútne reakcie na infúziu a reakcie súvisiace s infúziou
U pacientov liečených ONIVYDE boli hlásené reakcie na infúziu, najmä vyrážka, urtikária,
periorbitálny edém alebo pruritus. Nové udalosti (všetky 1. alebo 2. stupňa) sa vo všeobecnosti vyskytovali na začiatku liečby ONIVYDE, pričom len 2 z 10 pacientov zaznamenali udalosti po piatej dávke. Môžu sa vyskytnúť reakcie z precitlivenosti vrátane akútnej reakcie na infúziu. V prípade závažných reakcií z precitlivenosti sa má liečba ONIVYDE ukončiť.
Predchádzajúca Whippleova operácia
Pacienti s Whippleovou operáciou v anamnéze sú vystavení vyššiemu riziku závažných infekcií po
liečbe ONIVYDE v kombinácii s 5-FU a leukovorínom (pozri časť 4.8). U pacientov je potrebné sledovať prejavy infekcií.
Pľúcna toxicita
U pacientov liečených nelipozomálnym irinotekánom sa vyskytli udalosti podobné intersticiálnemu
ochoreniu pľúc (Interstitial Lung Disease, ILD)-s fatálnym koncom. V klinických štúdiách
s ONIVYDE neboli hlásené žiadne udalosti podobné ILD. Rizikové faktory zahŕňajú už existujúce ochorenie pľúc, používanie pneumotoxických liekov, faktory stimulujúce kolónie alebo predchádzajúcu rádioterapiu. U pacientov s rizikovými faktormi je potrebné pred začiatkom liečby ONIVYDE a počas nej pozorne sledovať respiračné príznaky. U malého percenta pacientov zaradených do klinickej štúdie s irinotekánom bola pozorovaná retikulonodulárna kresba pri RTG hrudníka. Ak sa objaví nové alebo progresívne dyspnoe, kašeľ a horúčka, liečba ONIVYDE sa má bezodkladne prerušiť až do vyhodnotenia diagnózy. U pacientov s potvrdenou diagnózou ILD sa má liečba ONIVYDE ukončiť.
Poškodenie funkcie pečene
Pacienti s hyperbilirubinémiou mali vyššie koncentrácie celkového SN-38 (pozri časť 5.2) a preto je
u nich zvýšené riziko neutropénie. U pacientov s celkovým bilirubínom 1,0 – 2,0 mg/dl sa musí pravidelne vyšetrovať úplný krvný obraz. Pacientom s poškodením funkcie pečene (bilirubín > 2- násobok hornej hranice normálnych hodnôt [upper limit of normal, ULN]; transaminázy > 5-násobok ULN) je potrebné venovať osobitnú pozornosť. Pri podávaní ONIVYDE v kombinácii s inými hepatotoxickými liekmi je potrebné postupovať opatrne, najmä u pacientov s už existujúcim poškodením funkcie pečene.
Poškodenie funkcie obličiek
Použitie ONIVYDE u pacientov so závažným poškodením funkcie obličiek nebolo stanovené (pozri
časť 5.2).
Pacienti s nízkou telesnou hmotnosťou (index telesnej hmotnosti < 18,5 kg/m2)
V klinickej štúdii hodnotiacej liečbu ONIVYDE+5-FU/LV sa u 5 z 8 pacientov s nízkou telesnou
hmotnosťou vyskytli nežiaduce reakcie 3. alebo 4. stupňa, najčastejšie myelosupresia, pričom
u 7 z 8 pacientov bola potrebná úprava dávkovania, a to oddialenie dávky, zníženie dávky alebo ukončenie liečby. Pri použití ONIVYDE u pacientov s indexom telesnej hmotnosti < 18,5 kg/m2 je potrebná zvýšená pozornosť.
Pomocné látky
Každý ml ONIVYDE obsahuje 0,144 mmolu (3,31 mg) sodíka. Je potrebné to vziať do úvahy
u pacientov na diéte s kontrolovaným príjmom sodíka.
4.5 Liekové a iné interakcie
Informácie o liekových interakciách s ONIVYDE sú prevzaté zo zverejnenej vedeckej literatúry, ktorá sa zaoberá nelipozomálnym irinotekánom.
Interakcie ovplyvňujúce použitie ONIVYDE
Silné induktory CYP3A4
U pacientov liečených súbežne nelipozomálnym irinotekánom a antikonvulzívami indukujúcimi enzým CYP3A4, ako fenytoín, fenobarbital alebo karbamazepín, je významne znížená expozícia irinotekánu (pokles AUC o 12 % s ľubovníkom bodkovaným a o 57 % – 79 % s fenytoínom, fenobarbitalom alebo karbamazepínom) a SN-38 (pokles AUC o 42 % s ľubovníkom bodkovaným
a o 36 % – 92 % s fenytoínom, fenobarbitalom alebo karbamazepínom). Súbežné podávanie
ONIVYDE a induktorov CYP3A4 môže preto znižovať systémovú expozíciu ONIVYDE.
Silné inhibítory CYP3A4 a inhibítory UGT1A1
U pacientov liečených súbežne nelipozomálnym irinotekánom a inhibítorom CYP3A4 a UGT1A1, ketokonazolom, je zvýšená expozícia SN-38 o 109 %. Súbežné podávanie ONIVYDE a iných
inhibítorov CYP3A4 (napr. grapefruitová šťava, klaritromycín, indinavir, itrakonazol, lopinavir,
nefazodón, nelfinavir, ritonavir, saquinavir, telaprevir, vorikonazol) môže preto zvyšovať systémovú expozíciu ONIVYDE. Na základe liekovej interakcie nelipozomálneho irinotekánu a ketokonazolu môže súbežné podávanie ONIVYDE s inými inhibítormi UGT1A1 (napr. atazanavir, gemfibrozil, indinavir) tiež zvyšovať systémovú expozíciu ONIVYDE.
Na základe analýzy populačnej farmakokinetiky nemení súbežné podávanie ONIVYDE+5-FU/LV
farmakokinetiku ONIVYDE.
Nie sú známe žiadne interakcie ONIVYDE (lipozomálneho irinotekánu) s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/antikoncepcia u mužov a žien
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby a 1 mesiac po skončení
liečby ONIVYDE. Muži musia používať kondómy počas liečby a 4 mesiace po ukončení liečby
ONIVYDE.
Gravidita
K dispozícii nie sú dostatočné údaje o použití ONIVYDE u gravidných žien. ONIVYDE môže
spôsobiť poškodenie plodu, ak sa podáva gravidným ženám, pretože u zvierat boli preukázané embryotoxické a teratogénne účinky hlavnej zložky irinotekánu (pozri časť 5.3). Na základe výsledkov štúdií na zvieratách a mechanizmu účinku irinotekánu sa preto ONIVYDE nemá používať počas gravidity, pokiaľ to nie je bezpodmienečne nutné. Ak sa ONIVYDE používa počas gravidity alebo ak pacientka počas liečby otehotnie, je potrebné ju informovať o možnom riziku pre plod.
Dojčenie
Nie je známe, či sa ONIVYDE alebo jeho metabolity vylučujú do ľudského mlieka. ONIVYDE je
kontraindikovaný počas dojčenia (pozri časť 4.3) kvôli možnému riziku závažných nežiaducich reakcií
ONIVYDE u dojčiat. Pacientky nesmú dojčiť až mesiac po podaní poslednej dávky.
Fertilita
K dispozícii nie sú žiadne údaje o vplyve ONIVYDE na ľudskú fertilitu. U zvierat bolo preukázané, že
nelipozomálny irinotekán spôsobuje atrofiu samčích a samičích reprodukčných orgánov po viacerých denných dávkach irinotekánu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
ONIVYDE má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť počas liečby opatrní pri vedení vozidiel alebo obsluhe strojov.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Nasledujúce nežiaduce reakcie, považované za možno alebo pravdepodobne súvisiace s podávaním
ONIVYDE, boli hlásené u 264 pacientov s metastatickým adenokarcinómom pankreasu, z ktorých 147
dostávalo ONIVYDE v monoterapii (120 mg/m2) a 117 dostávalo ONIVYDE (80 mg/m2)
v kombinácii s 5-FU/LV.
Najčastejšie nežiaduce reakcie (výskyt ≥ 20 %) pri liečbe ONIVYDE+5FU/LV boli: hnačka, nevoľnosť, vracanie, znížená chuť do jedla, neutropénia, únava, asténia, anémia, stomatitída a pyrexia. Najčastejšie závažné nežiaduce reakcie (≥ 2 %) pri liečbe ONIVYDE boli hnačka, vracanie, febrilná neutropénia, nevoľnosť, pyrexia, sepsa, dehydratácia, septický šok, pneumónia, akútne zlyhanie obličiek a trombocytopénia.
Výskyt nežiaducich reakcií, ktoré viedli k trvalému ukončeniu liečby, bol 11 % v skupine liečenej
ONIVYDE+5-FU/LV a 12 % v skupine liečenej monoterapiou.
Najčastejšie hlásenými nežiaducimi reakciami, ktoré viedli k ukončeniu liečby, boli infekcia a hnačka v skupine liečenej ONIVYDE+5-FU/LV a vracanie a hnačka v skupine liečenej monoterapiou.
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie, ktoré sa môžu vyskytnúť počas liečby ONIVYDE, sú zhrnuté nižšie a usporiadané
podľa triedy orgánových systémov a kategórie frekvencie (tabuľka 3). V rámci každej triedy orgánových systémov a kategórie frekvencie sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Kategórie frekvencií nežiaducich reakcií sú nasledovné: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100) a zriedkavé (≥1/10 000 až <1/1 000)**.
Tabuľka 3: Nežiaduce reakcie hlásené pri liečbe ONIVYDE v klinickej štúdii NAPOLI-1
Trieda orgánových
systémov podľa MedDRA*
Frekvencia nežiaducej reakcie**
Infekcie a nákazy Časté: Septický šok, sepsa, pneumónia, febrilná neutropénia, gastroenteritída, orálna kandidóza
Menej časté: Biliárna sepsa
Poruchy krvi a lymfatického
systému
Veľmi časté:Neutropénia, leukopénia, anémia, trombocytopénia
Časté: Lymfopénia
Poruchy imunitného systému Menej časté: Precitlivenosť
Poruchy metabolizmu
a výživy
Veľmi časté:, Hypokaliémia, hypomagneziémia, dehydratácia, znížená
chuť do jedla
Časté: Hypoglykémia, hyponatriémia, hypofosfatémia
Psychické poruchy Časté:Nespavosť
Poruchy nervového systému Veľmi časté:Závrat
Časté: Cholinergný syndróm, dysgeúzia
Poruchy srdca a srdcovej
činnosti
Časté:Hypotenzia
Poruchy ciev Časté: Pľúcna embólia, embólia, hlboká žilová trombóza
Menej časté: Trombóza
Poruchy dýchacej sústavy,
hrudníka a mediastína
Časté: Dyspnoe, dysfónia
Menej časté: Hypoxia
Poruchy gastrointestinálneho
traktu
Veľmi časté:Hnačka, vracanie, nevoľnosť, abdominálna bolesť,
stomatitída
Časté:Kolitída, hemoroidy
Menej časté: Ezofagitída, proktitída
Poruchy pečene a žlčových
ciest
Poruchy kože a podkožného tkaniva
Časté:Hypoalbuminémia
Veľmi časté:Alopécia
Menej časté: Makulopapulárna vyrážka, zmena farby nechtov
Poruchy obličiek
a močových ciest
Celkové poruchy a reakcie v mieste podania
Časté: Akútne zlyhanie obličiek
Veľmi časté:Pyrexia, periférny edém, mukózny zápal, únava, asténia
Časté: Reakcia súvisiaca s infúziou, edém
Laboratórne a funkčné
vyšetrenia
Veľmi časté: Úbytok hmotnosti
Časté: Zvýšený bilirubín, zvýšená alanínaminotransferáza, zvýšená aspartátaminotransferáza, zvýšený medzinárodný normalizovaný pomer
* MedDRA verzia 14.1
** Zriedkavý výskyt sa nedá odhadnúť zo štúdie NAPOLI-1 z dôvodu malej veľkosti vzorky
Opis vybraných nežiaducich reakciíNasledujúce nežiaduce reakcie boli pozorované v klinickej štúdii NAPOLI-1:
M
yelosupresia
Myelosupresia (neutropénia/leukopénia, trombocytopénia a anémia) bola častejšie pozorovaná v skupine liečenej ONIVYDE+5-FU/LV než v kontrolnej skupine liečenej 5-FU/LV.
Neutropénia/leukopénia
Najvýznamnejšia pozorovateľná hematologická toxicita bola neutropénia/leukopénia. Neutropénia
3. alebo vyššieho stupňa sa častejšie vyskytla u pacientov liečených ONIVYDE+5-FU/LV (27,4 %) v porovnaní s pacientmi liečenými 5-FU/LV (1,5 %). Neutropenická horúčka/sepsa sa častejšie vyskytla v skupine liečenej kombináciou ONIVYDE+5-FU/LV [u 4 pacientov (3,4 %)] v porovnaní s kontrolnou skupinou liečenou 5-FU/LV [u 1 pacienta (0,7 %)].
Trombocytopénia
Trombocytopénia 3. alebo vyššieho stupňa sa vyskytla u 2,6 % pacientov liečených
ONIVYDE+5-FU/LV a u 0 % pacientov liečených 5-FU/LV.
Anémia
Anémia 3. alebo vyššieho stupňa sa vyskytla u 10,3 % pacientov liečených ONIVYDE+5-FU/LV
a u 6,7 % pacientov liečených 5-FU/LV.
Akútne zlyhanie obličiek
Bolo zaznamenané poškodenie funkcie obličiek a akútne zlyhanie obličiek, zvyčajne u pacientov
s depléciou objemu v dôsledku nevoľnosti/vracania a/alebo hnačky. Akútne zlyhanie obličiek bolo hlásené u 6 zo 117 pacientov (5,1 %) v skupine liečenej ONIVYDE+5-FU/LV, u 10 zo 147 pacientov (6,8 %) v skupine liečenej monoterapiou ONIVYDE a u 6 zo 134 pacientov (4,5 %) v skupine liečenej
5-FU/LV.
Hnačka a súvisiace nežiaduce reakcie
Hnačka je veľmi častá nežiaduca reakcia spôsobujúca kolitídu, ileus, gastroenteritídu, únavu, dehydratáciu, úbytok hmotnosti, obličkové toxicity, hyponatriémiu a hypokaliémiu. Bolo
zaznamenané poškodenie funkcie obličiek a akútne zlyhanie obličiek, ktoré sa zvyčajne vyskytlo
u pacientov s depléciou objemu v dôsledku závažného vracania a/alebo hnačky. V klinickej štúdii sa hnačka 3. alebo 4. stupňa vyskytla u 15 zo 117 pacientov (12,8 %) liečených ONIVYDE+5-FU/LV. U pacientov s oneskorenou hnačkou bol medián času nástupu oneskorenej hnačky 8 dní po
predchádzajúcej dávke ONIVYDE. Po podaní dávky sa môže vyskytnúť hnačka so skorým nástupom, ktorá sa zvyčajne objaví ≤ 24 hodín a je zvyčajne prechodná. Hnačku so skorým nástupom môžu sprevádzať cholinergné príznaky, ktoré môžu zahŕňať rinitídu, nadmerné slinenie, návaly tepla, diaforézu, bradykardiu, miózu a hyperperistaltiku, ktorá môže spôsobiť abdominálne kŕče. V klinickej štúdii sa hnačka so skorým nástupom vyskytla u 35 pacientov (29,9 %) a cholinergné udalosti sa vyskytli u 4 pacientov (3,4 %) liečených ONIVYDE+5-FU/LV.
U hnačky 2. – 4. stupňa zastavte liečbu ONIVYDE a začnite liečbu hnačky. Pri zlepšení hnačky na
1. stupeň obnovte liečbu ONIVYDE v zníženej dávke (pozri časť 4.2).
Reakcia na infúziu
Akútne reakcie na infúziu boli hlásené u 8 zo 117 pacientov (6,8 %) v skupine liečenej ONIVYDE+5-FU/LV, u 3 zo 147 pacientov (2,0 %) v skupine liečenej monoterapiou ONIVYDE a u 8 zo 134 pacientov (6,0 %) v skupine liečenej 5-FU/LV.
Ďalšie osobitné populácie
Starší pacienti
Celkovo neboli hlásené žiadne klinicky významné rozdiely v bezpečnosti alebo účinnosti u pacientov vo veku ≥ 65 rokov a pacientov vo veku < 65 rokov, aj keď u prvej skupiny liečenej pomocou
ONIVYDE+5-FU/LV v štúdii NAPOLI-1 bola zaznamenaná vyššia frekvencia ukončenia liečby
(14,8 % verzus 7,9 %) a v niektorých prípadoch nežiaduce reakcie neustúpili. Nežiaduce reakcie
3. alebo vyššieho stupňa a závažné nežiaduce reakcie vyplývajúce z liečby boli častejšie u pacientov
vo veku < 65 rokov (84,1 % a 50,8 %) v porovnaní s pacientmi vo veku ≥ 65 rokov (68,5 % a 44,4 %).
Naopak, pri liečbe ONIVYDE+5-FU/LV v rámci štúdie adenokarcinómu pankreasu sa závažné nežiaduce reakcie, odloženie dávky, zníženie dávkovania a ukončenie liečby častejšie vyskytovalo u pacientov vo veku > 75 rokov (n = 12) v porovnaní s pacientmi vo veku ≤ 75 rokov (n = 105).
Ázijská populáciaU ázijských pacientov sa pozoroval nižší výskyt hnačky v porovnaní s pacientmi bielej rasy
[u 14 (19,2 %) zo 73 belochov sa vyskytla hnačka 3. a vyššieho stupňa a u 1 z 33 (3,3 %) Ázijcov sa vyskytla hnačka 3. a vyššieho stupňa], ale vyšší výskyt neutropénie s vyššou závažnosťou.
U pacientov liečených ONIVYDE+5-FU/LV bol výskyt neutropénie 3. a vyššieho stupňa vyšší
u ázijských pacientov [18 z 33 (55 %)] než u pacientov bielej rasy [13 zo 73 (18 %)]. Neutropenická horúčka/neutropenická sepsa boli hlásené u 6 % ázijských pacientov v porovnaní s 1 % pacientov bielej rasy. To sa zhoduje s analýzou populačnej farmakokinetiky, ktorá ukázala nižšiu expozíciu irinotekánu a vyššiu expozíciu jeho aktívnemu metabolitu SN-38 u ázijských pacientov než
u pacientov bielej rasy.
Pacienti s poškodením funkcie pečeneV klinických štúdiách s nelipozomálnym irinotekánom podávaným v rozvrhu týždenných dávok bola u pacientov s mierne zvýšenými východiskovými hladinami celkového bilirubínu v sére
(1,0 až 2,0 mg/dl) oveľa väčšia pravdepodobnosť výskytu neutropénie 3. alebo 4. stupňa v prvom cykle ako u pacientov s hladinami bilirubínu nižšími ako 1,0 mg/dl.
Pacienti s predchádzajúcou Whippleovou operáciouV klinickej štúdii hodnotiacej liečbu ONIVYDE+5-FU/LV bolo u pacientov s predchádzajúcou
Whippleovou operáciou zaznamenané vyššie riziko závažných infekcií po liečbe
ONIVYDE+5-FU/LV [9 z 29 (30 %)] v porovnaní s 11 z 88 (12,5 %) pacientov bez predchádzajúcej
Whippleovej operácie.
Pacienti s alelou UGT1A1Jedinci, ktorí sú homozygotní pre alelu UGT1A1*28 (genotyp 7/7), sú vystavení vyššiemu riziku neutropénie pri liečbe nelipozomálnym irinotekánom. V klinickej štúdii hodnotiacej
ONIVYDE+5-FU/LV bola frekvencia 3. a vyššieho stupňa neutropénie u týchto pacientov
[2 zo 7 (28,6 %)] podobná ako frekvencia u pacientov, ktorí nie sú homozygotní pre
alelu UGT1A1*28 a dostali počiatočnú dávku ONIVYDE 80 mg/m2 [30 zo 110 (27,3 %)] (pozri
časť 5.1).
Pacienti s nízkou telesnou hmotnosťou (index telesnej hmotnosti < 18,5 kg/m2)V klinickej štúdii hodnotiacej liečbu ONIVYDE+5-FU/LV sa u 5 z 8 pacientov s nízkou telesnou hmotnosťou vyskytli nežiaduce reakcie 3. alebo 4. stupňa, najčastejšie myelosupresia, pričom
u 7 z 8 pacientov bola potrebná úprava dávkovania, a to oddialenie dávky, zníženie dávky alebo ukončenie liečby (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických skúšaniach sa ONIVYDE podával v dávkach až 240 mg/m2 pacientom s rôznymi druhmi rakoviny. Nežiaduce reakcie u týchto pacientov boli podobné tým, ktoré boli hlásené pri odporúčanom režime dávok.
Boli zaznamenané prípady predávkovania nelipozomálnym irinotekánom v dávkach približne dvojnásobne vyšších než je odporúčaná liečebná dávka irinotekánu, čo môže mať fatálne následky. Najvýznamnejšími hlásenými nežiaducimi reakciami boli závažná neutropénia a závažná hnačka.
Na predávkovanie ONIVYDE nie je žiadna známa protilátka. Na prevenciu dehydratácie v dôsledku hnačky a na liečbu akýchkoľvek infekčných komplikácií je potrebné začať maximálnu podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastické látky, iné antineoplastické látky, ATC kód: L01XX19
Mechanizmus účinku
Liečivo v ONIVYDE je irinotekán (inhibítor topoizomerázy I) zapuzdrený v dvojvrstvovej lipidovej
vezikule alebo lipozóme.
Irinotekán je derivát kamptotecínu. Kamptotecíny pôsobia ako špecifické inhibítory enzýmu DNA topoizomerázy I. Irinotekán a jeho aktívny metabolit SN-38 sa reverzibilne viažu na komplex topoizomeráza I-DNA a indukujú jednovláknové lézie DNA, ktoré blokujú replikačnú vidlicu DNA
a sú zodpovedné za cytotoxicitu. Irinotekán je metabolizovaný karboxylesterázou na SN-38. SN-38 je približne 1 000-násobne účinnejší ako irinotekán v inhibícii topoizomerázy I purifikovanej
z nádorových bunkových línií ľudí a hlodavcov.
Farmakodynamické účinky
V modeloch so zvieratami sa preukázalo, že ONIVYDE zvyšuje plazmatické hladiny irinotekánu
a predlžuje expozíciu aktívnemu metabolitu SN-38 v mieste nádoru.
Klinická účinnosť a bezpečnosť
Bezpečnosť a účinnosť ONIVYDE bola skúmaná v multinárodnom, randomizovanom, otvorenom,
kontrolovanom klinickom skúšaní (NAPOLI-1), v ktorom boli testované dva liečebné režimy
u pacientov s metastatickým adenokarcinómom pankreasu, u ktorých došlo k progresii ochorenia po liečbe gemcitabínom alebo po liečbe obsahujúcej gemcitabín. Klinické skúšanie bolo zamerané na posúdenie klinickej účinnosti a bezpečnosti monoterapie ONIVYDE alebo liečby ONIVYDE+5- FU/LV v porovnaní s aktívnou kontrolnou skupinou liečenou 5-FU/LV.
Pacienti randomizovaní do skupiny liečenej ONIVYDE+5-FU/LV dostávali 80 mg/m2 ONIVYDE ako intravenóznu infúziu po dobu 90 minút, následne LV v dávke 400 mg/m2 intravenózne po dobu
30 minút a potom 5-FU v dávke 2 400 mg/m2 intravenózne po dobu 46 hodín s podávaním každé
2 týždne. Pacienti, ktorí sú homozygotní pre alelu UGT1A1*28, dostali nižšiu počiatočnú dávku
ONIVYDE (pozri časť 4.2). Pacienti randomizovaní do skupiny liečenej 5-FU/LV
dostávali 200 mg/m2 leukovorínu intravenózne po dobu 30 minút a následne 5-FU v dávke
2 000 mg/m2 intravenózne po dobu 24 hodín s podávaním v 1., 8., 15. a 22. deň 6-týždňového cyklu. Pacienti randomizovaní do skupiny liečenej monoterapiou ONIVYDE dostávali 120 mg/m2 ako intravenóznu infúziu po dobu 90 minút podávanú každé 3 týždne.'
Hlavnými kritériami vhodnosti pre zaradenie pacientov s metastatickým adenokarcinómom pankreasu do klinickej štúdie NAPOLI-1 bol stav výkonnosti podľa Karnofského (Karnofsky Performance Status, KPS) ≥ 70, normálna hladina bilirubínu, hladiny transaminázy ≤ 2,5-násobok ULN alebo ≤ 5- násobok ULN u pacientov s metastázami v pečeni a hladina albumínu ≥ 3,0 g/dl.
Celkovo 417 pacientov bolo randomizovaných do skupiny liečenej ONIVYDE+5-FU/LV (N = 117), do skupiny liečenej monoterapiou ONIVYDE (N = 151) a do skupiny liečenej 5-FU/LV (N = 149). Demografické charakteristiky a stav ochorenia pacientov pri vstupe do skúšania boli v jednotlivých skupinách skúšania v rovnováhe.
Medián veku (celej randomizovanej) populácie so zámerom liečiť bol 63 rokov (v rozpätí 31 –
87 rokov), 57 % bolo mužov, 61 % tvorili belosi a 33 % bolo Aziatov. Priemerná východisková hladina albumínu bola 3,6 g/dl a 55 %pacientov malo východiskový KPS 90 – 100. Charakteristiky ochorenia zahŕňali 68 % pacientov s metastázami v pečeni a 31 % pacientov s metastázami v pľúcach,
12 % pacientov nepodstúpilo žiadnu predchádzajúcu líniu liečby metastáz, 56 % podstúpilo
1 predchádzajúcu líniu liečby metastáz, 32 % pacientov podstúpilo 2 a viac predchádzajúcich línií liečby metastáz.
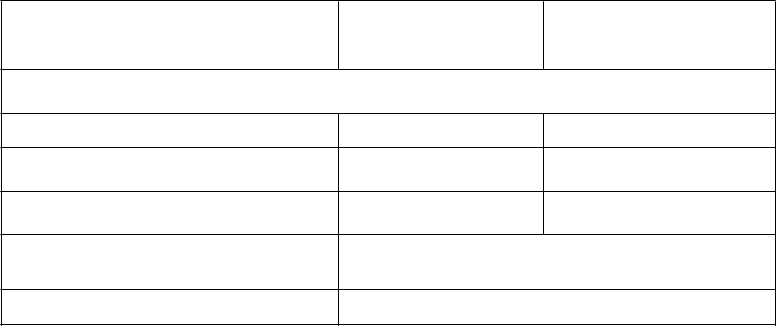
Pacienti boli liečení až do progresie ochorenia alebo neprijateľnej toxicity. Primárnym meradlom účinnosti bolo celkové prežívanie (Overall Survival, OS). Medzi ďalšie meradlá účinnosti patrilo prežívanie bez progresie ochorenia (Progression Free Survival, PFS) a miera objektívnej odpovede (Objective Response Rate, ORR). Výsledky sú znázornené v tabuľke 4. Celkové prežívanie je znázornené na obrázku 1.
Tabuľka 4 Výsledky účinnosti z klinickej štúdie NAPOLI-1
ONIVYDE+5-FU/LV
(N = 117)
5-FU/LV
(N = 119)
 Celkové prežívanie
1
Počet úmrtí, n (%) 75 (64) 80 (67)
Celkové prežívanie
1
Počet úmrtí, n (%) 75 (64) 80 (67)
Medián celkového prežívania (mesiace) 6,1 4,2
(95 % CI) (4,8; 8,9) (3,3; 5,3)
Pomer rizika (95 % CI)3 0,67 (0,49-0,92)
p-hodnota4 0,0122
Prežívanie bez progresie ochorenia1,2Smrť alebo progresia, n (%) 83 (71) 92 (77)
Medián prežívania bez progresie (mesiace) 3,1 1,5
(95 % CI) (2,7; 4,2) (1,4; 1,8)
Pomer rizika (95 % CI)3 0,56 (0,41-0,75)
p-hodnota4 0,0001
ONIVYDE+5-FU/LV (N = 117)
5-FU/LV (N = 119)
 Miera objektívnej odpovede
2
Miera objektívnej odpovede
2
N 19 1
ORR (%) 16,2 0,8
95 % CI miery5 9,6; 22,9 0,0; 2,5
Rozdiel mier (95 % CI)5 15,4 (8,5; 22,3)
p-hodnota6 < 0,0001
1 Medián je odhad mediánu doby prežívania podľa Kaplan-Meiera
2 Podľa kritérií RECIST, verzia 1.1.
3 Analýza Coxovho modelu
4 Nestratifikovaný log-rank test
5 Na základe normálnej aproximácie
6 Fisherov presný test
Skratky: 5-FU/LV=5-fluóruracil/leukovorín; CI = interval spoľahlivosti
Obrázok 1 Celkové prežívanie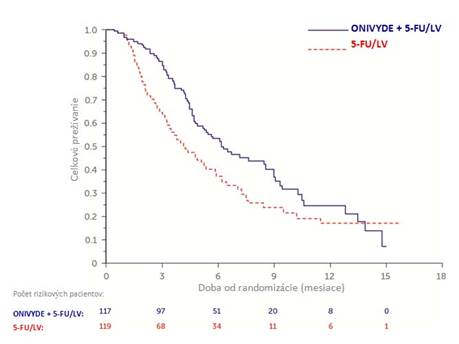
U obmedzeného počtu pacientov s predošlou expozíciou nelipozomálnemu irinotekánu nebol preukázaný žiadny prínos ONIVYDE.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s ONIVYDE vo
všetkých podskupinách pediatrickej populácie v liečbe adenokarcinómu pankreasu (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Lipozomálne puzdro irinotekánu predlžuje čas cirkulácie a obmedzuje distribúciu v porovnaní
s nelipozomálnym irinotekánom.
Farmakokinetika celkového irinotekánu a celkového SN-38 v plazme bola hodnotená u pacientov
s rakovinou liečených ONIVYDE v monoterapii alebo v rámci kombinovanej chemoterapie v dávkach medzi 60 a 180 mg/m2. Farmakokinetické parametre celkového irinotekánu a analytov SN-38 po podaní ONIVYDE v dávke 80 mg/m2 sú znázornené v tabuľke 5.
Tabuľka 5: Súhrn priemerných hodnôt (±štandardná odchýlka) celkového irinotekánu a celkového SN-38
Analyt Farmakokinetické parametre Jednotka Geometrický
priemer ONIVYDE (95 % CI)
a
80 mg/m
2
(n = 353)
b
Priemerný (SD)
nelipozomálny irinotekán
125 mg/m
2
(n = 99)
c
Celkový irinotekán
Celkovo
SN-38
AUC h ng/ml 919228 (845653-999204)
Cmax ng/ml 28353 (27761-28958)
Klírens (CL) l/h/m2 0,087 (0,080-0,094)
Objem (V) l/m2 2,6 (2,6-2,7)
t1/2 efektívny h 20,8 (19,4-22,3)
AUC h ng/ml 341 (326-358)
Cmax ng/ml 3,0 (2,9-3,1)
t1/2 efektívny h 40,9 (39,8-42,0)
10529 (3786)
1492 (452)
13,0 (5,6)
138 (60,9)
6,07 (1,19)
267 (115)
27,8 (11,6)
11,7 (4,29)
SD = štandardná odchýlka
AUC= plocha pod krivkou plazmatickej koncentrácie (extrapolovaná po nekonečno pre ONIVYDE
a AUC24h pre nelipozomálny irinotekán) Cmax = maximálna koncentrácia v plazme t1/2 efektívny = efektívny polčas
aHodnoty sú stanovené z analýzy populačnej farmakokinetiky
bN = 353 zodpovedá všetkým jedincom zahrnutým v analýze populačnej farmakokinetiky
cHodnoty boli získané zo zverejnených údajov [Schaaf LJ et al.
Clin CancerRes. 2006 Jun 15;12:3782-91]
DistribúciaPriame meranie lipozomálneho irinotekánu ukazuje, že 95 % irinotekánu ostáva počas cirkulácie
zapuzdrených v lipozóme. Ne-lipozomálny irinotekán vykazuje veľký distribučný objem (138 l/m2). Distribučný objem ONIVYDE pri dávke 80 mg/m2 bol 2,6 l/m2, čo naznačuje, že ONIVYDE sa prevažne zdržuje v priestore vaskulárnej tekutiny.
Viazanie ONIVYDE na proteíny v plazme je zanedbateľné (< 0,44 % celkového irinotekánu
v ONIVYDE). Väzba nelipozomálneho irinotekánu na proteíny v plazme je stredná (30 % až 68 %)
a SN-38 sa na proteíny v ľudskej plazme viaže veľmi pevne (približne 95 %).
Biotransformácia
Irinotekán uvoľnený z lipozomálneho puzdra má podobnú metabolickú cestu ako nelipozomálny
irinotekán.
Metabolická premena irinotekánu na aktívny metabolit SN-38 je sprostredkovaná enzýmami karboxylesterázy. Štúdie in vitro naznačujú, že irinotekán, SN-38 a ďalší metabolit, aminopentán karboxylová kyselina (APC), neinhibujú izoenzýmy cytochrómu P-450. SN-38 je následne konjugovaný prevažne enzýmom UDP-glukuronosyltransferáza 1A1 (UGT1A1) a vzniká glukuronidový metabolit. Aktivita UGT1A1 je znížená u jedincov s genetickým polymorfizmom, ktorý vedie k zníženej aktivite enzýmov, napr. polymorfizmus UGT1A1*28. V analýze populačnej farmakokinetiky u pacientov liečených ONIVYDE s použitím výsledkov z podskupiny s testovaním
genotypu UGT1A1*28, v ktorej bola analýza prispôsobená na nižšiu dávku podávanú pacientom, ktorí sú homozygotní pre alelu UGT1A1*28, mali pacienti, ktorí sú homozygotní pre túto alelu (N = 14), priemerné rovnovážne koncentrácie celkového SN-38 1,06 ng/ml a u pacientov, ktorí nie sú homozygotní pre túto alelu (N = 244), bola táto hodnota 0,95 ng/ml.
Eliminácia
Dispozícia ONIVYDE a nelipozomálneho irinotekánu nebola u ľudí plne objasnená.
Vylučovanie nelipozomálneho irinotekánu močom predstavuje 11 % až 20 %, SN-38 < 1 %
a glukuronidu SN-38 3 %. Kumulatívna exkrécia irinotekánu a jeho metabolitov (SN-38 a glukuronidu
SN-38) žlčou a močom počas 48 hodín po podaní irinotekánu sa u dvoch pacientov pohybovala v rozmedzí približne 25 % (100 mg/m2) až 50 % (300 mg/m2).
Poškodenie funkcie obličiek
Neuskutočnila sa žiadna špecifická farmakokinetická štúdia u pacientov s poškodením funkcie
obličiek. V analýze populačnej farmakokinetiky nemalo mierne až stredne závažné poškodenie funkcie obličiek žiadny účinok na expozíciu celkovému SN-38 po prispôsobení dávky podľa plochy telesného povrchu (Body Surface Area, BSA). Analýza zahŕňala 68 pacientov so stredne závažným poškodením funkcie obličiek (CLcr 30 – 59 ml/min), 147 pacientov s miernym poškodením funkcie obličiek (CLcr 60 – 89 ml/min) a 135 pacientov s normálnou funkciou obličiek (CLcr > 90 ml/min). K dispozícii nie sú dostatočné údaje u pacientov so závažným poškodením funkcie obličiek
(CLcr < 30 ml/min), ktoré by umožnili posúdiť účinok na farmakokinetiku (pozri časti 4.2 a 4.4).
Poškodenie funkcie pečene
Neuskutočnila sa žiadna špecifická farmakokinetická štúdia u pacientov s poškodením funkcie pečene.
V analýze populačnej farmakokinetiky mali pacienti s východiskovými koncentráciami celkového bilirubínu 1 – 2 mg/dl (n = 19) priemerné rovnovážne koncentrácie celkového SN-38 zvýšené
o 37 % (0,98 [95 % CI: 0,94 – 1,02] a 1,29 [95 % CI: 1,11 – 1,5] ng/ml) v porovnaní s pacientmi
s východiskovými koncentráciami bilirubínu < 1 mg/dl (n = 329). Zvýšené koncentrácie ALT/AST
však nemali žiadny vplyv na koncentrácie celkového SN-38. K dispozícii nie sú žiadne údaje u pacientov s celkovým bilirubínom vyšším ako 2-násobok ULN.
Ďalšie osobitné populácie
Vek a pohlavie
Analýza populačnej farmakokinetiky u pacientov vo veku 28 až 87 rokov, z ktorých 11 % bolo vo veku ≥ 75 rokov, naznačuje, že vek nemá žiadny klinicky významný účinok na expozíciu irinotekánu a SN-38.
Analýza populačnej farmakokinetiky u 196 pacientov mužského pohlavia a 157 pacientov ženského pohlavia naznačuje, že pohlavie nemá žiadny klinicky významný účinok na expozíciu irinotekánu
a SN-38 po prispôsobení ploche telesného povrchu (body surface area, BSA).
Etnicita
Analýza populačnej farmakokinetiky naznačuje, že Aziati majú o 56 % nižšiu priemernú rovnovážnu koncentráciu celkového irinotekánu (3,93 [95 % CI: 3,68 – 4,2] a 1,74 [95 % CI: 1,58 – 1,93] mg/l)
a o 8 % vyššiu priemernú rovnovážnu koncentráciu celkového SN-38 (0,97 [95 % CI: 0,92 – 1,03]
a 1,05 [95 % CI: 0,98 – 1,11] ng/ml) ako belosi.
Farmakokinetický/farmakodynamický vzťah
V súhrnnej analýze u 353 pacientov boli vyššie plazmatické hodnoty Cmax SN-38 spojené so zvýšenou
pravdepodobnosťou výskytu neutropénie a vyššie plazmatické hodnoty Cmax irinotekánu so zvýšenou
pravdepodobnosťou výskytu hnačky.
V klinickom skúšaní preukazujúcom účinnosť ONIVYDE boli vyššie plazmatické expozície celkovému irinotekánu a SN-38 u pacientov v liečebnej skupine s ONIVYDE+5-FU/LV spojené s dlhším celkovým prežívaním a prežívaním bez progresie, ako aj s vyššou mierou objektívnej odpovede.
5.3 Predklinické údaje o bezpečnosti
V štúdiách toxicity s jednorazovou a opakovanou dávkou u myší, potkanov a psov boli cieľovými orgánmi toxicity gastrointestinálny trakt a hematologický systém. Závažnosť účinkov súvisela s dávkou a bola reverzibilná. Hladina bez pozorovaných nežiaducich účinkov
(no-observed-adverse-effect level, NOAEL) u potkanov a psov po 90 minútovej intravenóznej infúzii
ONIVYDE podávanej každé 3 týždne počas 18 týždňov bola najmenej 180 mg/m2.
Vo farmakologických štúdiách bezpečnosti u psov sa nezistil žiadny účinok ONIVYDE na kardiovaskulárne, hemodynamické, elektrokardiografické alebo respiračné parametre pri dávkach do
21 mg/kg (420 mg/m2). V štúdiách toxicity s opakovanou dávkou u potkanov neboli pozorované žiadne zistenia poukazujúce na toxicitu súvisiacu s centrálnym nervovým systémom.
Genotoxický a karcinogénny potenciál
Neuskutočnili sa žiadne štúdie genotoxicity s ONIVYDE. Nelipozomálny irinotekán a SN-38 boli
genotoxické in vitro v teste chromozomálnych aberácii v bunkách CHO, ako aj v mikronukleárnom teste in vivo na myšiach. V iných štúdiách s irinotekánom sa však v Amesovom teste žiadny mutagénny potenciál nedokázal.
Neuskutočnili sa žiadne štúdie karcinogenity s ONIVYDE. U potkanov liečených nelipozomálnym irinotekánom jedenkrát týždenne počas 13 týždňov v maximálnej dávke 150 mg/m² nebol hlásený vznik žiadnych nádorov súvisiacich s liečbou 91 týždňov po ukončení liečby. V týchto podmienkach sa v súvislosti s dávkou pozoroval výrazný lineárny trend výskytu kombinovaných endometriálnych stromálnych polypov v rohoch maternice a endometriálnych stromálnych sarkómov. Vzhľadom na mechanizmus účinku sa má za to, že irinotekán má karcinogénny potenciál.
Reprodukčná toxicita
Neuskutočnili sa žiadne štúdie reprodukčnej a vývojovej toxicity s ONIVYDE.
Nelipozomálny irinotekán bol teratogénny u potkanov a králikov v dávkach nižších ako ľudská liečebná dávka. U mláďat potkanov, ktoré sa narodili liečeným zvieratám a mali externé abnormality, bol preukázaný pokles fertility. Tento pokles nebol pozorovaný u morfologicky normálnych mláďat. U gravidných potkanov bol pozorovaný pokles hmotnosti placenty a u potomkov zníženie životaschopnosti plodu a zvýšenie behaviorálnych abnormalít.
Nelipozomálny irinotekán spôsoboval atrofiu mužských reprodukčných orgánov u potkanov a psov po viacerých denných dávkach 20 mg/kg a 0,4 mg/kg. Tieto účinky boli reverzibilné po ukončení liečby.
6
. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Lipidy tvoriace lipozómy
1,2-distearoyl-sn-glycero-3-fosfocholín (DSPC) Cholesterol
N-(karbonyl-metoxypolyetylénglykol-2000)-1,2-distearoyl-sn-glycero-3-fosfoetanolamín
(MPEG-2000-DSPE)
Ďalšie pomocné látky
Oktasulfát sacharózy
kyselina 2- [ 4- (2-hydroxyetyl)piperazín-1-yl] etánsulfónová (tlmivý roztok HEPES) Chlorid sodný
Voda na injekcie
6.2 Inkompatibility
ONIVYDE sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
30 mesiacov.
Po riedení
Chemická a fyzikálna stabilita zriedeného infúzneho roztoku bola preukázaná po dobu do 6 hodín pri teplote 15 – 25 °C alebo po dobu najviac 24 hodín pri uchovávaní v chladničke (2 ºC – 8 ºC).
Z mikrobiologického hľadiska sa má liek okamžite použiť. Ak nie je použitý ihneď, za dĺžku a podmienky skladovania zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšej škatuli na ochranu pred svetlom. Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka zo skla typu I so sivou chlorobutylovou zátkou a hliníkovým uzáverom s odklápacím viečkom s obsahom 10 ml koncentrátu.
Každé balenie obsahuje jednu injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
ONIVYDE je cytotoxický liek a zaobchádzanie s ním si vyžaduje opatrnosť. Pri zaobchádzaní alebo podávaní ONIVYDE sa odporúča používať rukavice, okuliare a ochranné oblečenie. V prípade kontaktu roztoku s pokožkou sa má pokožka ihneď dôkladne umyť mydlom a vodou. V prípade kontaktu roztoku so sliznicami sa sliznice majú dôkladne vypláchnuť vodou. Tehotné ženy nemajú
s ONIVYDE pracovať z dôvodu cytotoxickej povahy lieku.
Príprava roztoku a podávanie
ONIVYDE sa dodáva ako sterilná lipozomálna disperzia v koncentrácii 5 mg/ml a pred podaním sa
musí zriediť. Na prípravu roztoku s príslušnou dávkou zrieďte ONIVYDE s 5 % injekčným roztokom glukózy alebo injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml (0,9 %) na konečný objem 500 ml. Zmiešajte zriedený roztok jemným otáčaním. Zriedený roztok je číry až mierne biely alebo mierne opaleskujúci bez viditeľných častíc.
ONIVYDE sa má podávať pred LV, po ktorom nasleduje 5-FU. ONIVYDE sa nesmie podávať ako bolusová injekcia ani ako nezriedený roztok.
Počas prípravy infúzie sa musia dodržiavať aseptické techniky. ONIVYDE je určený len na jednorazové použitie.
Je potrebné postupovať opatrne, aby sa zabránilo extravazácii a miesto infúzie treba sledovať pre prípad prejavov zápalu. Ak dôjde k extravazácii, odporúča sa vypláchnuť miesto injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml (0,9 %) a/alebo sterilizovanou vodou a aplikovať ľad.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBaxalta Innovations GmbH Industriestrasse 67
1221 Viedeň
Rakúsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1130/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.