
od internej
normy kalibrovanej podľa aktuálnej normy WHO pre lieky s faktorom VIII.
Jedna jednotka (U) aktivity faktora VIII zodpovedá množstvu faktora VIII v jednom ml normálnej
ľudskej plazmy.
Odporúčaná počiatočná dávka je 200 U na kilogram telesnej hmotnosti podávaná intravenóznou
injekciou (pozri časť 6.6).
Požadovaná počiatočná dávka OBIZURU u pacienta sa vypočíta podľa tohto vzorca:
Počiatočná dávka (U/kg) ÷ Sila lieku (U/injekčná liekovka) × telesná hmotnosť (kg) = Počet injekčných liekoviek
napr. pre pacienta s hmotnosťou 70 kg sa počet injekčných liekoviek na počiatočnú dávku vypočíta
nasledovne:
200 U/kg ÷ 500 U/injekčná liekovka × 70 kg = 28 injekčných liekoviek
Monitorujte aktivitu faktora VIII a klinický stav 30 minút po prvej injekcii a 3 hodiny po podaní
OBIZURU.
Monitorujte aktivitu faktora VIII bezprostredne pred a 30 minút po nasledujúcich dávkach a použite
nižšie uvedenú tabuľku pre odporúčané cieľové minimálne hladiny faktora VIII.
Na stanovenie hladiny faktora VIII sa odporúča použitie jednostupňového testu zrážania, pretože bol
použitý na stanovenie účinnosti OBIZURU a priemernej rýchlosti obnovy (pozri časti 4.4 a 5.2).
Dávka a frekvencia podávania majú byť založené na výsledkoch aktivity faktora VIII (udržiavaných v rámci odporúčaných obmedzení) a na dosiahnutej klinickej odpovedi.
Údaje o účinnosti a bezpečnosti u pacientov so získanou hemofíliou sú obmedzené (pozri časť 5.1).
Úvodná fáza
T
yp krvácania Cieľová minimálna
aktivita faktora VIII (jednotky na dl alebo % normálnej hodnoty)
Mierne až stredne závažné
krvácanie z povrchových
Počiatočná
dávka (jednotky na kg)
Nasledujúca
dávka
Titrujte nasledujúce dávky podľa klinickej
Frekvencia a trvanie
nasledujúceho dávkovania
Dávku podávajte každých 4 až 12 hodín, frekvenciu možno upraviť podľa
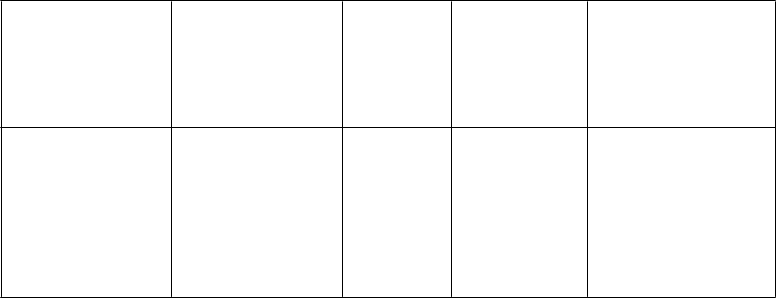
svalov/bez
neurovaskulárneho poškodenia a krvácanie do kĺbov
> 50% 200
odpovede a na
udržanie cieľovej minimálnej
klinickej odpovede a
nameranej aktivity faktora VIII
T
yp krvácania Cieľová minimálna aktivita faktora VIII (jednotky na
dl alebo %
normálnej hodnoty)
Väčšie stredne závažné až závažné intramuskulárne,
retroperitoneálne,
Počiatočná dávka (jednotky na kg)
Nasledujúca dávka
aktivity faktora
VIII
Frekvencia a trvanie nasledujúceho dávkovania
gastrointestinálne, intrakraniálne krvácanie
> 80%
 F
áza hojenia
F
áza hojenia
Keď krvácanie zareaguje na liečbu, zvyčajne v priebehu prvých 24 hodín, pokračujte v podávaní OBIZURU v dávke, ktorá udržiava minimálnu aktivitu FVIII na úrovni 30 – 40 %, až kým nebude krvácanie pod kontrolou. Maximálna aktivita FVIII v krvi nesmie presiahnuť 200 %.
Dĺžka liečby závisí od klinického posúdenia.
Pediatrická populáciaPoužitie u detí a dospievajúcich mladších ako 18 rokov s vrodenou hemofíliou alebo v zriedkavých
prípadoch získanej hemofílie nie je v súčasnosti schválené.
Spôsob podávaniaNa intravenózne použitie.
Celkový objem rekonštituovaného OBIZURU sa má podávať rýchlosťou 1 až 2 ml za minútu.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 KontraindikácieZnáme anafylaktické reakcie na liečivo, škrečí proteín alebo na ktorúkoľvek z pomocných látok
uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníPrecitlivenosťPri liečbe OBIZUROM môže dôjsť k reakciám z precitlivenosti alergického typu. Liek obsahuje
stopové množstvá škrečích proteínov.
Pacienti majú byť upozornení, že ak sa objavia príznaky precitlivenosti, musia používanie lieku okamžite prerušiť a obrátiť sa svojho lekára. Pacienti majú byť informovaní o prvotných prejavoch reakcií z precitlivenosti vrátane žihľavky, generalizovanej žihľavky, pocitu tiesne na hrudníku, sipotu, hypotenzie a anafylaxie.
V prípade šoku sa má podať štandardná lekárska liečba šoku.
T
v
orba inhibičných protilátok
Pred expozíciou a po expozícii OBIZURU boli detekované inhibičné protilátky proti prasaciemu
faktoru VIII (merané pomocou modifikovaného Nijmegenovho variantu Bethesda testu). Na začiatku liečby boli zaznamenané titre inhibítorov až do 29 jednotiek Bethesda, ale napriek tomu pacienti priaznivo reagovali na podávanie OBIZURU. Odporúča sa, aby liečba bola založená na klinickom posúdení a nie na detekcii inhibičných protilátok podľa Bethesda testu.
Nie sú k dispozícii dostatočné klinické informácie o tvorbe inhibičných protilátok proti OBIZURU po opakovanom podávaní. Preto sa má OBIZUR podávať len vtedy, ak sa to považuje za klinicky nevyhnutné. Rozsiahla kožná purpura si nevyhnutne nevyžaduje liečbu.
OBIZUR sa vyrába technológiou rekombinantnej DNA v obličkových bunkách mláďat škrečkov. Protilátky proti proteínu obličkových buniek mláďat škrečkov neboli u pacientov detekované po expozícii OBIZURU.
Vysoká a udržiavaná aktivita faktora VIII v krvi môže znamenať predispozíciu na vznik tromboembolických udalostí. Osoby s už existujúcim kardiovaskulárnym ochorením a starší ľudia sú vystavení osobitnému riziku.
Ak je potrebná venózna katetrizácia, treba vziať do úvahy komplikácie súvisiace s katétrom, napr. trombózu v mieste zavedenia katétra.
Aktivita faktora VIII stanovená chromogénnym testom je zvyčajne nižšia ako aktivita faktora VIII stanovená jednostupňovým testom zrážania. Meranie aktivity faktora VIII sa má u každého pacienta vždy vykonávať s použitím rovnakej metodiky stanovenia. Odporúča sa použitie jednostupňového testu zrážania, pretože bol použitý na stanovenie účinnosti a priemernej rýchlosti obnovy OBIZURU (pozri časti 4.2 a 5.2).
Názovačíslovýrobnejšarže
Dôrazne odporúča zaznamenať názov a číslo výrobnej šarže lieku pri každom podaní OBIZURU
pacientovi, aby sa zachovalo prepojenie medzi pacientom a výrobnou šaržou lieku.
Obsah sodíka
Každá injekčná liekovka obsahuje 4,4 mg (198 mmol) sodíka na 1 ml rekonštituovaného roztoku.
Toto musia vziať do úvahy pacienti na diéte s kontrolovaným príjmom sodíka.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne interakcie OBIZURU s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Nevykonali sa reprodukčné štúdie na zvieratách s OBIZUROM. Skúsenosti týkajúce sa použitia OBIZURU počas gravidity a laktácie nie sú k dispozícii. Preto sa má OBIZUR používať počas gravidity a laktácie, len ak je to jasne indikované.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
OBIZUR nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu:
Môže sa vyskytnúť precitlivenosť alebo alergické reakcie (ktoré môžu zahŕňať angioedém, pálenie a
pichanie v mieste vpichu, zimnicu, návaly horúčavy, generalizovanú žihľavku, bolesť hlavy, žihľavku,
hypotenziu, letargiu, nevoľnosť, nepokoj, tachykardiu, pocit tiesne na hrudníku, mravčenie, vracanie, sipot) a môžu viesť až k závažnej anafylaxii (vrátane šoku) (pozri časť 4.4).
Pacienti so získanou hemofíliou môžu vytvárať inhibičné protilátky proti prasaciemu faktoru VIII.
Tabuľkový zoznamnežiaducichreakcií:
Tabuľka uvedená nižšie je zostavená podľa klasifikácie orgánových systémov MedDRA (trieda
orgánových systémov a uprednostňovaný názov). V klinickom skúšaní OBIZURU pri získanej hemofílii sa u 29 pacientov hodnotila bezpečnosť.
Frekvencie výskytu boli vyhodnotené podľa nasledujúcej konvencie: veľmi časté (≥1/10), časté (≥1/100 až < 1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov).
T
rieda orgánových systémov
Laboratórne a funkčné
vyšetrenia
Uprednostňovaný názov MedDRA Pozitívny test na inhibičné protilátky proti prasaciemu faktoru VIII (pozri časť 4.4)
Frekvencia
Časté
 H
l
ásenie podozrení na nežiaduce reakcie
H
l
ásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieÚčinky vyšších ako odporúčaných dávok OBIZURU, neboli charakterizované.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká, krvný koagulačný faktor VIII, prasacia sekvencia. ATC kód: B02BD14
MechanizmusúčinkuOBIZUR je rekombinantný faktor VIII bez B-domény, prasacia sekvencia (susoktokog alfa). Je to glykoproteín.
Faktor VIII sa ihneď po uvoľnení do obehu pacienta naviaže na von Willebrandov faktor (vWF).
Komplex faktora VIII/von Willebrandovho faktora tvoria dve molekuly (faktor VIII a von Willebrandov faktor) s odlišnými fyziologickými funkciami. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX a urýchľuje premenu faktora X na aktivovaný faktor X, ktorý nakoniec premieňa protrombín na trombín. Trombín následne premieňa fibrinogén na fibrín a môže dôjsť k vzniku krvnej zrazeniny.
Získaná hemofília je zriedkavá porucha krvácania, pri ktorej pacienti s normálnymi génmi pre faktor VIII vytvárajú inhibičné autoprotilátky namierené proti faktoru VIII. Tieto autoprotilátky neutralizujú cirkulujúci ľudský faktor VIII a tým vytvárajú nedostatok dostupného faktora VIII. Cirkulujúce protilátky (inhibítory) zamerané proti ľudskému faktoru VIII majú minimálnu alebo nemajú žiadnu krížovú reaktivitu proti OBIZURU.
OBIZUR dočasne nahrádza inhibovaný endogénny faktor VIII, ktorý je potrebný na účinnú
hemostázu.
Klinickáúčinnosťabezpečnosť
Bezpečnosť a účinnosť OBIZURU pri liečbe závažných krvácavých epizód u pacientov so získanou
hemofíliou s autoimúnnymi inhibičnými protilátkami proti ľudskému faktoru VIII bola skúmaná v prospektívnom, nerandomizovanom, otvorenom klinickom skúšaní s 28 pacientmi
(18 kaukazského, 6 černošského a 4 ázijského pôvodu). Klinické skúšanie zahŕňalo pacientov so život
a/alebo končatiny ohrozujúcim krvácaním vyžadujúcim hospitalizáciu.
Všetky počiatočné krvácavé epizódy vykazovali podľa hodnotenia hlavného skúšajúceho pozitívnu odpoveď na liečbu do 24 hodín po počiatočnej dávke. Pozitívnou odpoveďou bolo zastavenie alebo zmiernenie krvácania s klinickým zlepšením alebo s aktivitou faktora VIII vyššou ako vopred špecifikovaný cieľ.
Pozitívna odpoveď sa pozorovala u 95 % (19/20) pacientov hodnotených po 8 hodinách a u 100 % pacientov hodnotených po 16 hodinách (18/18) po podaní dávky. Okrem odpovede na liečbu bol celkový úspech liečby stanovený skúšajúcim na základe jeho/jej schopnosti ukončiť podávanie alebo znížiť dávku a/alebo frekvenciu dávkovania OBIZURU. U celkovo 24/28 (86 %) pacientov sa dosiahla úspešná kontrola (zastavenie) počiatočnej krvácavej epizódy. U pacientov, ktorí boli liečení OBIZUROM ako liečbou prvej línie definovanou ako žiadne nedávne predchádzajúce použitie antihemoragických látok pred prvou liečbou OBIZUROM, bol u 16/17 (94 %) pacientov hlásený konečný úspech liečby. U jedenástich pacientov bolo hlásené, že pred prvou liečbou OBIZUROM dostali antihemoragiká (napr. rFVIIa, koncentrát aktivovaného protrombínového komplexu, kyselina tranexamová). Z týchto 11 pacientov bola liečba nakoniec úspešná u ôsmich pacientov (73 %).
Priemerná dávka na injekciu potrebná na úspešnú liečbu primárneho krvácania bola 133 U/kg a priemerná celková dávka bola 1 523 U/kg počas mediánu 6 dní. Medián počtu infúzií na pacienta bol 1,76 (v rozsahu: 0,2 až 5,6). Počas prvých 24 hodín bola priemerná celková dávka použitá v
klinickej štúdii 493 U/kg s mediánom 3 infúzií. Ak bola po uplynutí 24 hodín potrebná ďalšia liečba,
priemerná celková dávka použitá na kontrolu krvácavej epizódy bola 1 050 U/kg s mediánom 10,5 infúzií (priemerná dávka 100 U/kg).
Ďalšieinformácie
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s OBIZUROM vo
všetkých podskupinách pediatrickej populácie v liečbe získanej hemofílie (informácie o použití v
pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn
charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Farmakokinetické údaje namerané u 5 pacientov so získanou hemofíliou v stave bez krvácania sú
uvedené v tabuľke 1.
Tabuľka 1: Individuálne farmakokinetické údaje o aktivite faktora VIII po podaní poslednej dávky OBIZURU 5 pacientom so získanou hemofíliou. Pacienti boli v stave bez krvácania. Aktivita faktora VIII bola meraná jednostupňovým testom zrážania.
P
acient
D
ávka
(
U)
D
ávka
(
U
/
kg)
A
ktivita hFVIII (%) na začiatk u liečby
t
½ (h)
T
m
ax
(
h)
A
m
ax
(
%)
AUC
0-t
(
%
·t)
AUC
0-
∞ (%·t)
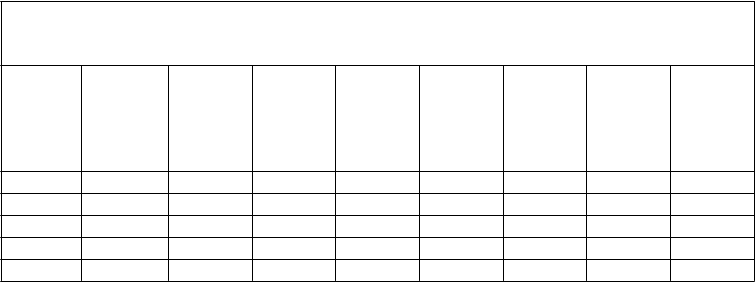
1 5000 76,7 89 17 0,42 213 3124 4988
2 2934 30,0 18 4,6 0,42 100 694 712
3 7540 144,2 3 5,3 0,45 74 473 492
4 9720 206,8 0 1,8 0,50 53 122 135
5 10000 133,3 N/A 4,2 0,75 178 1583 1686
Amax = maximálna pozorovaná aktivita v %; AUC0-t = plocha pod krivkou koncentrácia – čas od času 0 po poslednú merateľnú koncentráciu; AUC0-∞ = plocha pod krivkou koncentrácia – čas od
času 0 extrapolovaná do nekonečna; t½ = polčas rozpadu; Tmax = čas maximálnej pozorovanej aktivity
v %, N/A = nie je k dispozícii.
Priemerná rýchlosť obnovy po počiatočnej dávke 200 U/kg bola 1,06 ± 0,75 U/ml na U/kg (v rozsahu 0,10-2,61) meraná pomocou jednostupňového koagulačného testu.
Aj keď aktivita faktora VIII stanovená chromogénnym testom je zvyčajne nižšia ako aktivita faktora VIII stanovená jednostupňovým testom zrážania, po infúzii mali aktivity faktora VIII u pacientov so získanou hemofíliou v klinickej štúdii OBI-1-301 tendenciu byť vyššie, ak boli stanovené chromogénnym testom v porovnaní s jednostupňovým testom zrážania (pozri časť 4.4).
Inhibičné protilátky proti OBIZURU boli merané pomocou metódy modifikovaného Nijmegenovho variantu Bethesda testu. U troch pacientov zahrnutých do farmakokinetickej analýzy bol na začiatku liečby detekovateľný titer inhibítorov proti prasaciemu faktoru VIII (≥ 0,6 jednotiek Bethesda (BU)/ml). U troch z piatich pacientov neboli po liečbe detekovateľné žiadne titre proti prasaciemu faktoru VIII (< 0,6 BU/ml na základe posledného hláseného výsledku), u dvoch pacientov bol detekovateľný titer proti prasaciemu faktoru VIII (≥ 0,6 BU/ml).
Priemerný polčas života OBIZURU u deviatich hodnotených pacientov v stave krvácania bol
(približne) 10 hodín (v rozsahu od 2,6 do 28,6 hod.).
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti alebo toxicity po opakovanom podávaní neodhalili žiadne osobitné riziko pre ľudí. V štúdiách toxicity po opakovanom podávaní mal však výskyt a závažnosť glomerulopatie pozorovanej u opíc, ktorým bol intravenózne podávaný OBIZUR v dávkach 75, 225 a 750 U/kg/deň, tendenciu časom narastať. Nevykonali sa reprodukčné štúdie na zvieratách s OBIZUROM.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
Polysorbát 80
Chlorid sodný
Dihydrát chloridu vápenatého
Sacharóza Tris báza Tris HCl'
Dihydrát citrónanu sodného
Rozpúšťadlo
Sterilizovaná voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Rekonštituovaný roztok sa má použiť ihneď alebo najneskôr do 3 hodín po rekonštitúcii.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Podmienky na uchovávanie po rekonštitúcii, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Jedno balenie OBIZURU obsahuje 1, 5 alebo 10 kusov každej z nasledujúcich položiek
• injekčné liekovky s práškom (sklo typu I) so zátkou (butylová guma) a odtrhávacím viečkom
• naplnené injekčné striekačky (sklo typu I) so zátkou (butylová guma), gumeným krytom hrotu a adaptérom Luer Lock
• pomôcku na prenos tekutiny s integrovaným plastovým hrotom
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Po rekonštitúcii je roztok číry, bezfarebný, bez častíc a má pH 6,8 až 7,2. Osmolalita purfu sa pohybuje v rozmedzí od 59 do 65 10 % mOsm/kg H2O.
Rekonštituovaný liek sa má pred podaním vizuálne skontrolovať, či sa v ňom nenachádzajú častice
a či nezmenil farbu. Roztoky, ktoré obsahujú častice alebo zmenili farbu, sa nesmú podávať.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
Príprava
Pred začatím rekonštitúcie budete potrebovať:
• Vypočítaný počet injekčných liekoviek s práškom
• Rovnaký počet 1 ml injekčných striekačiek s rozpúšťadlom sterilných adaptérov injekčnej
liekovky
• Alkoholové tampóny
• Veľkú sterilnú injekčnú striekačku na konečný objem rekonštituovaného lieku
Postupy uvedené nižšie predstavujú všeobecné usmernenia na prípravu a rekonštitúciu OBIZURU. Nasledujúce pokyny na rekonštitúciu opakujte pri každej injekčnej liekovke s práškom určenej na rekonštitúciu.
RekonštitúciaPočas postupu rekonštitúcie používajte aseptickú techniku.
1. Zohrejte injekčnú liekovku s práškom OBIZUR a naplnenú injekčnú striekačku s rozpúšťadlom
na izbovú teplotu.
2. Odstráňte plastový uzáver z injekčnej liekovky s práškom OBIZUR (
Obrázok A).
3. Pred použitím očistite gumenú zátku alkoholovým tampónom (nie je súčasťou balenia) a nechajte uschnúť.
4. Odtrhnite kryt z balenia adaptéra injekčnej liekovky (
Obrázok B). Nedotýkajte sa hrotu luer
lock v strede adaptéra injekčnej liekovky. Nevyberajte adaptér injekčnej liekovky z balenia.
5. Umiestnite balenie s adaptérom injekčnej liekovky na čistý povrch tak, aby hrot luer lock
smeroval nahor.
6. Odstráňte bezpečnostný kryt z naplnenej injekčnej striekačky s ropúšťadlom (
Obrázok C).
7. Pevne držte balenie s adaptérom injekčnej liekovky a pripojte naplnenú injekčnú striekačku s rozpúšťadlom k adaptéru zatlačením hrotu injekčnej striekačky nadol na luer lock v strede
adaptéra a potom injekčnú striekačku otáčajte v smere hodinových ručičiek, kým bezpečne nezapadne. Spojenie príliš neuťahujte (
Obrázok D).
8. Odstráňte plastový obal (
Obrázok E).
9. Umiestnite injekčnú liekovku s práškom OBIZUR na čistý, rovný a tvrdý povrch. Umiestnite adaptér injekčnej liekovky na injekčnú liekovku s práškom OBIZUR a pevne pretlačte hrot filtra
adaptéra injekčnej liekovky cez stred gumeného krúžka injekčnej liekovky s práškom OBIZUR,
až kým priesvitný plastový uzáver nezacvakne na injekčnú liekovku (
Obrázok F).
10. Stlačte piest nadol a pomaly vstreknite celé rozpúšťadlo z injekčnej striekačky do injekčnej
liekovky s práškom OBIZUR.
11. Jemne krúžte injekčnou liekovkou s práškom OBIZUR bez toho, aby ste odstránili injekčnú striekačku, až kým sa všetok prášok úplne nerozpustí/nerekonštituuje (
Obrázok G).
Rekonštituovaný roztok sa má pred podaním vizuálne skontrolovať, či neobsahuje žiadne
častice. Nepoužívajte tento liek, ak spozorujete častice alebo zmenu farby.
12. Jednou rukou držte injekčnú liekovku a adaptér injekčnej liekovky a druhou rukou pevne uchopte telo naplnenej injekčnej striekačky s rozpúšťadlom a pohybom proti smeru hodinových
ručičiek odskrutkujte injekčnú striekačku z adaptéra injekčnej liekovky (
Obrázok H).
13. Ak sa OBIZUR uchováva pri izbovej teplote, použite ho ihneď alebo do 3 hodín po rekonštitúcii.
Obrázok A Obrázok B Obrázok C Obrázok D
O
brázok E Obrázok F Obrázok G Obrázok H
 Podávanie
L
en na intravenóznu injekciu!
Podávanie
L
en na intravenóznu injekciu!
• Pred podaním skontrolujte rekonštituovaný roztok OBIZUR, či neobsahuje častice a či nezmenil farbu. Roztok má byť číry a bezfarebný. Nepodávajte ho, ak spozorujete častice alebo zmenu farby.
• Nepodávajte OBIZUR rovnakou hadičkou alebo z rovnakej nádoby s inými injekčne
podávanými liekmi.
S použitím aseptickej techniky podajte liek nasledovným postupom:
1. Po rekonštitúcii všetkých injekčných liekoviek, pripojte veľkú injekčnú striekačku k adaptéru injekčnej liekovky jemným zatlačením hrotu injekčnej striekačky nadol na luer lock v strede
adaptéra injekčnej liekovky a potom injekčnú striekačku otáčajte v smere hodinových ručičiek,
kým bezpečne nezapadne.
2. Obráťte injekčnú liekovku dnom nahor, vtlačte vzduch z injekčnej striekačky do injekčnej
liekovky a natiahnite rekonštituovaný OBIZUR do injekčnej striekačky (
Obrázok I).
Obrázok I
3. Pohybom proti smeru hodinových ručičiek odskrutkujte veľkú injekčnú striekačku z adaptéra injekčnej liekovky a zopakujte tento postup so všetkými rekonštituovanými injekčnými liekovkami OBIZURU, až kým nezískate celkový objem určený na podanie.
4. Podajte rekonštituovaný OBIZUR intravenózne rýchlosťou 1 až 2 ml za minútu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBaxalta Innovations GmbH Industriestrasse 67
A-1221 Viedeň
Rakúsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1035/001
EU/1/15/1035/002
EU/1/15/1035/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.