
ii mohla kedykoľvek v priebehu štúdie zdvojnásobiť. Laserová
fotokoagulácia bola ako záchranná liečba povolená po 3. mesiaci v obidvoch skupinách liečby. Štúdia mala dve časti: výskumnú časť (prvých 42 pacientov analyzovaných po 6 mesiacoch) a potvrdzujúcu časť (zvyšných 109 pacientov analyzovaných po 12 mesiacoch). Stredná priemerná zmena BCVA od
1. mesiaca do 12. mesiaca v porovnaní s východiskovou hodnotou bola +7,8 (±7,72) písmen
u pacientov liečených ranibizumabom (n=102) celkove v oboch častiach klinického skúšania,
v porovnaní s -0,1 (±9,77) písmen u pacientov pri simulovanej liečbe (p<0,0001 pre rozdiel v liečbe).
V klinickom skúšaní fázy III, D2301 (RESTORE), bolo randomizovaných 345 pacientov
s poškodením zraku v dôsledku makulárneho edému, aby dostávali buď intravitreálnu injekciu 0,5 mg ranibizumabu ako monoterapiu a simulovanú laserovú fotokoaguláciu (n=116), kombináciu 0,5 mg
ranibizumabu a laserovej fotokoagulácie (n=118), alebo simulovanú injekciu a laserovú
fotokoaguláciu (n=111). Liečba ranibizumabom sa začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej zrakovej ostrosti pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Liečba sa obnovila, keď sa pozorovalo zníženie BCVA v dôsledku progresie DME. Laserová fotokoagulácia sa podala pri vstupe do štúdie v ten istý deň najmenej
30 minút pred injekciou ranibizumabu a následne podľa potreby na základe kritérií ETDRS.
240 pacientov, ktorí predtým ukončili 12-mesačné klinické skúšanie RESTORE, bolo zaradených do otvorenej, multicentrickej extenzie skúšania trvajúcej 24 mesiacov (RESTORE Extension). Pacienti dostávali 0,5 mg ranibizumabu pro re nata (PRN) do toho istého oka, ktoré bolo zvolené ako oko pre klinické skúšanie v skúšaní D2301 (RESTORE). Liečba sa znovu začala s mesačnými intervalmi až do poklesu BCVA v dôsledku DME a pokračovala až do dosiahnutia stabilnej BCVA. Okrem toho sa podávala liečba laserom, ak bola potrebná podľa úsudku skúšajúceho lekára a na základe kritérií ETDRS.
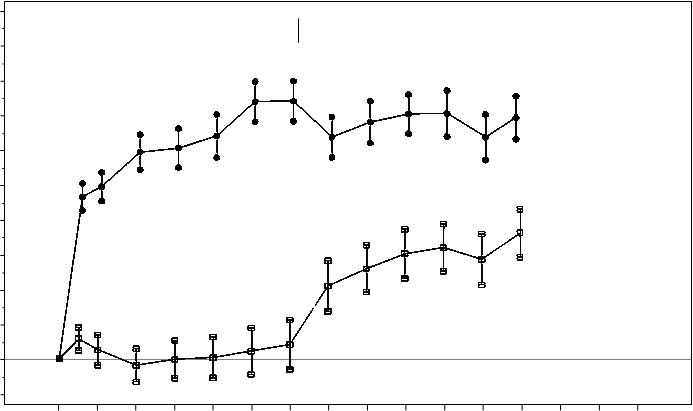
Kľúčové merané parametre sú zhrnuté v Tabuľke 2 (RESTORE a Extension) a na Obrázku 2 (RESTORE).
Obrázok 2 Stredná zmena zrakovej ostrosti v čase oproti východiskovej hodnote v štúdiiD2301 (RESTORE)
12
10
+ 6,8/+ 6,4
+ 6,2/+ 5,4*
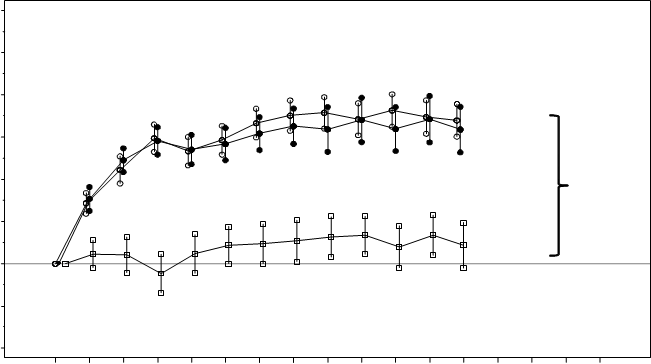
+ 0,9
Mesiac
11 12

Ranibizumab 0,5 mg (n=115)


Ranibizumab 0,5 mg+laser (n=118) Laser (n=110)
BL=východisková hodnota; SE=stredná chyba priemeru
* Rozdiel metódou najmenších štvorcov, p<0,0001/0,0004 na základe dvojstranného stratifikovaného
testu podľa Cochrana-Mantela-Haenszela
Účinok po 12 mesiacoch sa zhodoval u väčšiny podskupín. Avšak pre osoby s pomerne dobrou východiskovou hodnotou BCVA (>73 písmen) spolu s makulárnym edémom s hrúbkou centrálnej retiny <300 mm sa liečba ranibizumabom v porovnaní laserovou fotokoaguláciou nezdala prínosom.
T
abuľka 2 Výsledky po 12 mesiacoch v klinickom skúšaní D2301 (RESTORE) a po
36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension)
Meraný parameter po 12 mesiacoch v
porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
|
Ranibizumab
0,5 mg n=115
|
Ranibizumab
0,5 mg + laser n=118
|
Laser
n=110
|
Stredná priemerná zmena BCVA od 1. do
12. mesiacaa (±SD)
|
6,1 (6,4)a
|
5,9 (7,9)a
|
0,8 (8,6)
|
Stredná zmena BCVA po 12. mesiacoch
(±SD)
|
6,8 (8,3)a
|
6,4 (11,8)a
|
0,9 (11,4)
|
Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
|
37,4a
|
43,2a
|
15,5
|
Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
|
22,6
|
22,9
|
8,2
|
|
Meraný parameter po 36 mesiacoch
v skúšaní D2301-E1 (RESTORE Extension) v porovnaní s východiskovou
hodnotou v skúšaní D2301 (RESTORE)
|
Predtým
ranibizumab
0,5 mg n=83
|
Predtým
ranibizumab
0,5 mg + laser n=83
|
Predtým
laser
n=74*
|
Stredná zmena BCVA po 24 mesiacoch
(SD)
|
7,9 (9,0)
|
6,7 (7,9)
|
5,4 (9,0)
|
Stredná zmena BCVA po 36 mesiacoch
(SD)
|
8,0 (10,1)
|
6,7 (9,6)
|
6,0 (9,4)
|
Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
47,0
|
44,6
|
41,9
|
Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
27,7
|
30,1
|
21,6
|
|
|
ap<0,0001 pre porovnania skupín ranibizumabu oproti skupine laseru.
n v D2301-E1 (RESTORE Extension) je počet pacientov s východiskovou hodnotou (0. mesiac)
v D2301 (RESTORE) a tiež s hodnotou z návštevy po 36. mesiaci.
*zo 74 pacientov, ktorí predtým dostali liečbu laserom, 59 (79 %) dostalo ranibizumab v extenzii
klinického skúšania.
Zlepšenie zrakovej ostrosti pozorované pri Lucentise 0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby vzhľadom na väčšinu funkcií súvisiacich so zrakom, čo sa stanovilo prostredníctvom skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Pri iných podškálach tohto dotazníka sa nezistili rozdiely v súvislosti s liečbou. Rozdiel medzi Lucentisom
0,5 mg a kontrolnou skupinou sa stanovil s hodnotami p 0,0137 (monoterapia ranibizumabom) a
0,0041 (liečba ranibizumabom a laserom) kombinovaného skóre VFQ-25.
Stredný počet injekcií podaných v klinickom skúšani RESTORE trvajúcom 12 mesiacov bol 7,0
v skupine 0,5 mg ranibizumabu, 6,8 v skupine ranibizumabu a lasera a 7,3 simulovaných injekcií
v skupine monoterapie laserom. V priemere pripadlo na pacienta 6,4 injekcií ranibizumabu podaných za obdobie 24 mesiacov extenzie u pacientov, ktorí dostávali ranibizumab v klinickom skúšaní D2301 (RESTORE). Zo 74 pacientov v skupine liečby laserom v skúšaní D2301 (RESTORE) dostalo ranibizumab niekedy počas fázy extenzie 59 (79 %) pacientov. V priemere dostalo týchto 59 pacientov počas 24-mesačnej extenzie 8,1 injekcií ranibizumabu. Podiely pacientov, u ktorých nebola potrebná počas fázy extenzie žiadna liečba ranibizumabom, boli v skupinách predtým liečených ranibizumabom
19 %, ranibizumabom a laserom 25 % a laserom 20 %.
Profil dlhodobej bezpečnosti ranibizumabu, ktorý sa pozoroval v 24-mesačnej extenzii klinického skúšania, sa zhoduje so známym profilom bezpečnosti Lucentisu.
V klinickom skúšaní fázy IIIb, D2304 (RETAIN), bolo randomizovaných 372 pacientov s poškodením
zraku v dôsledku DME na nasledujúce podávanie intravitreálnych injekcií:
· 0,5 mg ranibizumabu súbežne s laserovou fotokoaguláciou v režime podávania a predlžovania
intervalov medzi podaniami (treat-and-extend, TE) (n=121),
· 0,5 mg ranibizumabu v monoterapii v režime TE (n=128),
· 0,5 mg ranibizumabu v monoterapii v režime PRN (n=123).
Vo všetkých skupinách sa liečba ranibizumabom začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej BCVA pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Laserová fotokoagulácia sa podala pri vstupe do skúšania v ten istý deň ako prvá
injekcia ranibizumabu a následne podľa potreby na základe kritérií ETDRS. Pri TE sa ranibizumab
potom podával podľa plánu liečby s intervalmi 2-3 mesiace. Pri PRN sa každý mesiac hodnotila
BCVA a ak to bolo potrebné, ranibizumab sa potom podal počas tej istej návštevy. Vo všetkých
skupinách sa obnovilo podávanie každý mesiac pri poklese BCVA v dôsledku progresie DME
a pokračovalo sa v ňom až do opätovného dosiahnutia stabilnej BCVA. Skúšanie trvalo 24 mesiacov.
V klinickom skúšaní RETAIN, po 3 začiatočných každomesačných návštevách s podaním liečby, počet plánovaných návštev s podaním liečby potrebný podľa režimu TE bol 13 oproti 20 mesačným návštevám, ktoré boli potrebné podľa režimu PRN. Pri oboch režimoch liečby si viac ako 70 % pacientov udržalo ich BCVA pri frekvencii návštev ≥2 mesiace. Počas 24 mesiacov bol stredný počet (medián) injekcií 12,4 (12,0) v skupine liečby ranibizumabom a laserom pri TE, 12,8 (12,0) v skupine liečby samotným ranibizumabom pri TE a 10,7 (10,0) v skupine liečby ranibizumabom pri PRN. Pridanie lasera sa nespájalo so zníženým stredným počtom injekcií ranibizumabu pri režime TE.
Kľúčové merané parametre sú zhrnuté v Tabuľke 3.
Tabuľka 3 Výsledky v klinickom skúšaní D2304 (RETAIN) Meraný parameter
v porovnaní s východiskovou hodnotou
| Ranibizumab
0,5 mg + laser pri TE
n=117
| Samotný ranibizumab
0,5 mg pri TE
n=125
| Ranibizumab
0,5 mg pri PRN
n=117
| Stredná priemerná
zmena BCVA od 1. do
12. mesiaca (SD)
|
5,9 (5,5) a
|
6,1 (5,7) a
|
6,2 (6,0)
| Stredná priemerná
zmena BCVA od 1. do
24. mesiaca (SD)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
| Stredná zmena BCVA
po 24 mesiacoch (SD)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
| Zisk ≥10 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
43,6
|
40,8
|
45,3
| Zisk ≥15 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
25,6
|
28,0
|
30,8
|
|
|
ap<0,0001 pre stanovenie neinferiority oproti PRN
V klinických skúšaniach pri DME sprevádzalo zlepšenie BCVA postupom času zníženie strednej
CSFT vo všetkých skupinách liečby.
Liečba
poškodenia
zraku
v
dôsledku makulárneho edému po RVO
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku makulárneho edému po RVO sa vyhodnotili v randomizovaných, dvojito maskovaných, kontrolovaných štúdiách BRAVO a CRUISE, do ktorých boli zaradené osoby s BRVO (n=397) a CRVO (n=392). V oboch štúdiách pacienti dostávali intravitreálne buď 0,3 mg, alebo 0,5 mg ranibizumabu, alebo simulované injekcie. Po 6 mesiacoch pacienti z kontrolnej skupiny simulovaného podania prešli na 0,5 mg ranibizumabu. V štúdii BRAVO bola vo všetkých skupinách od 3. mesiaca povolená laserová fotokoagulácia ako záchranná liečba.
Kľúčové merané parametre z BRAVO a CRUISE sú zhrnuté v Tabuľkách 4 a 5 a na Obrázkoch 3 a 4.
Tabuľka 4 Výsledky po 6 a 12 mesiacoch (BRAVO)
| Simulované podanie/ Lucentis 0,5 mg
(n=132)
| Lucentis 0,5 mg
(n=131)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 7,3 (13,0)
| 18,3 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 12,1 (14,4)
| 18,3 (14,6)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 28,8
| 61,1
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 43,9
| 60,3
| Podiel (%), ktorý dostal záchrannú liečbu
laserom počas 12 mesiacov
| 61,4
| 34,4
|
|
|
ap<0,0001
O
brázok 3 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(B
RAVO
)
+ 18,3
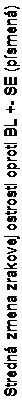
20
18
+ 18,3
16
14
12 + 12,1
10
8
6
4
2
kontrolované + 7,3kontrolná skupina simulovaného
+ 7,3kontrolná skupina simulovaného
s
im
u
l
ovaným podaním
0
podania preradená na ranibizumab
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac


Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=132) Ranibizumab 0,5 mg (n=131)
BL=východisková hodnota; SE=stredná chyba priemeru
Tabuľka 5 Výsledky po 6 a 12 mesiacoch (CRUISE)
| Simulované podanie/ Lucentis 0,5 mg (n=130)
| Lucentis 0,5 mg
(n=130)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 0,8 (16,2)
| 14,9 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 7,3 (15,9)
| 13,9 (14,2)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 16,9
| 47,7
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 33,1
| 50,8
|
|
|
ap<0,0001
O
brázok 4 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(
CRU
ISE)
20
kontrolované18
simulovaným podaním16
 + 14,9kontrolná skupina simulovaného podania preradená na ranibizumab
+ 14,9kontrolná skupina simulovaného podania preradená na ranibizumab
14
+ 13,912
10
8
+ 7,36
4
+ 0,82
0
-2
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac


Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=130) Ranibizumab 0,5 mg (n=130)
BL=východisková hodnota; SE=stredná chyba priemeru
V oboch štúdiách zlepšenie zraku sprevádzalo kontinuálne a významné zmenšovanie makulárneho
edému, merané ako hrúbka centrálnej retiny.
Pacienti s BRVO (BRAVO a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, dosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~15 písmen) ako pacienti, ktorí boli liečení ranibizumabom od začiatku (~16 písmen). Avšak počet pacientov, ktorí ukončili 2 roky liečby, bol obmedzený,
a v štúdii HORIZON boli plánované len štvrťročné kontrolné návštevy. Preto v súčasnosti nie je dosť
údajov, z ktorých by sa dali vyvodiť odporúčania, kedy sa má začať liečba ranibizumabom u pacientov s BRVO.
Pacienti s CRVO (CRUISE a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, nedosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~6 písmen) oproti pacientom, ktorí boli liečení ranibizumabom od začiatku (~12 písmen).
Zlepšenie zrakovej ostrosti pozorované pri liečbe ranibizumabom po 6 a 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby stanovený prostredníctvom podškál National Eye Institute Visual Function Questionnaire (NEI VFQ-25) vzhľadom na „aktivity vyžadujúce videnie do blízka“ a
„aktivity vyžadujúce videnie do diaľky“. Rozdiel medzi Lucentisom 0,5 mg a kontrolnou skupinou sa vyhodnotil po 6. mesiaci s hodnotami p 0,02 až 0,0002.
Liečba
poškodenia
zraku
v
dôsledku CNV pri PM
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku CNV pri PM sa stanovili na základe údajov z 12 mesiacov randomizovaného, dvojito maskovaného, účinnou liečbou kontrolovaného pivotného klinického skúšania F2301 (RADIANCE). Toto skúšanie bolo určené na vyhodnotenie dvoch rôznych režimov dávkovania 0,5 mg ranibizumabu podávaného ako intravitreálna injekcia v porovnaní s PDT verteporfínom (vPDT, fotodynamická liečba Visudynom). Do jednej
z nasledujúcich skupín bolo randomizovaných 277 pacientov:
· Skupina I (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „stability“, definovanými ako žiadna zmena BCVA v porovnaní s dvomi predchádzajúcimi mesačnými hodnoteniami).
· Skupina II (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „aktivity choroby“, definovanými ako zhoršenie zraku, ktoré možno pripísať intra- alebo subretinálnej tekutine alebo aktívnemu presakovaniu v dôsledku CNV lézie, stanovené prostredníctvom OCT a/alebo
FA).
· Skupina III (vPDT – pacienti mali od 3. mesiaca povolené dostať liečbu ranibizumabom). Počas 12 mesiacov klinického skúšania pacienti dostali v priemere 4,6 injekcií (rozmedzie 1-11) v skupine I a 3,5 injekcií (rozmedzie 1-12) v skupine II. V skupine II, ktorá zodpovedá odporúčanému
dávkovaniu (pozri časť 4.2), 50,9 % pacientov potrebovalo 1 alebo 2 injekcie, 34,5 % potrebovalo 3 až
5 injekcií a 14,7 % potrebovalo 6 až 12 injekcií počas 12 mesiacov trvania skúšania. 62,9 % pacientov v skupine II nepotrebovalo injekcie v druhom 6-mesačnom období skúšania.
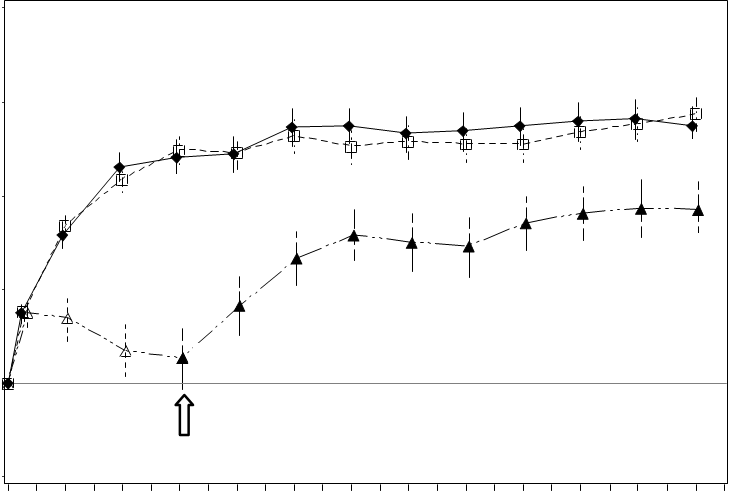
Najdôležitejšie výsledky RADIANCE sú zhrnuté v Tabuľke 6 a na Obrázku 5.
Tabuľka 6 Výsledky po 3. a 12. mesiaci (RADIANCE)
| Skupina I
ranibizumab
0,5 mg
„stabilita zraku“
(n=105)
| Skupina II
ranibizumab
0,5 mg
„aktivita choroby“
(n=116)
| Skupina III
vPDTb
(n=55)
| 3. mesiac
|
|
|
| Priemerná zmena BCVA od 1. do 3. mesiaca v porovnaní s východiskovou hodnotoua (písmená)
| +10,5
| +10,6
| +2,2
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
61,9 %
38,1 %
|
65,5 %
43,1 %
|
27,3 %
14,5 %
| 12. mesiac
|
|
|
| Počet injekcií do 12. mesiaca:
Priemer
Medián
|
4,6
4
|
3,5
2,0
|
N/A N/A
| Priemerná zmena BCVA od 1. do
12. mesiaca v porovnaní s východiskovou hodnotou (písmená)
| +12,8
| +12,5
| N/A
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
69,5 %
53,3 %
|
69,0 %
51,7 %
|
N/A N/A
|
|
|
a p<0,00001 v porovnaní s kontrolou vPDT
b Porovnávacia kontrola do 3. mesiaca. Pacienti randomizovaní do skupiny vPDT mali od 3. mesiaca
povolené dostať liečbu ranibizumabom (v skupine III dostalo ranibizumab od 3. mesiaca 38 pacientov)
O
brázok 5 Priemerná zmena oproti východiskovej hodnote BCVA do 12. mesiaca
(
RAD
IANCE)
20
15 +12,5
+14,4
10 +12,1
+13,8
+9,3

5
+1,4
0
Povolený ranibizumab
-5

|
|
| Mesiac
|
| Skupina I - ranibizumab 0,5 mg
|
|
|
| podľa stabilizácie (N=105)
|
|
|
| Skupina III - PDT verteporfínom
|
|
|
| (N=55)
|
|
|
|
|
0 1 2 3 4 5 6 7 8 9 10 11 12
Zlepšenie zraku sprevádzal pokles hrúbky centrálnej retiny.
Skupina II - ranibizumab 0,5 mg


podľa aktivity choroby (N=116)
Skupina III - ranibizumab 0,5 mg/PDT

verteporfínom od 3. mesiaca (N=55)

Prínos liečby hlásený pacientmi sa pozoroval vo väčšej miere v skupinách ranibizumabu v porovnaní
s vPDT (hodnota p<0,05) z hľadiska zlepšenia kombinovaného skóre a niekoľkých podškál NEI VFQ-
25 (celkové videnie, aktivity vyžadujúce videnie do blízka, duševné zdravie a odkázanosť na iných).
Deti a dospievajúciBezpečnosť a účinnosť ranibizumabu sa zatiaľ neskúmali u detí a dospievajúcich.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Lucentis vo všetkých vekových podskupinách detí a dospievajúcich pri neovaskulárnej VPDM, poškodení zraku v dôsledku DME, poškodení zraku v dôsledku makulárneho edému po RVO a poškodení zraku
v dôsledku CNV pri PM (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po intravitreálnom podaní Lucentisu raz mesačne pacientom s neovaskulárnou VPDM boli sérové koncentrácie ranibizumabu vo všeobecnosti nízke, s maximálnymi hladinami (Cmax) spravidla pod koncentráciou ranibizumabu potrebnou na inhibovanie biologickej aktivity VEGF o 50 %
(11-27 ng/ml, ako sa stanovilo in vitro v teste bunkovej proliferácie). Cmax bola úmerná dávke
v rozmedzí dávok 0,05 až 1,0 mg/oko. Koncentrácie v sére obmedzeného počtu pacientov s DME
naznačujú, že nemožno vylúčiť o niečo vyššiu systémovú expozíciu v porovnaní s expozíciou, aká sa pozorovala u pacientov s neovaskulárnou VPDM. Koncentrácie ranibizumabu v sére pacientov s RVO
boli podobné alebo mierne vyššie v porovnaní s koncentráciami, ktoré sa pozorovali u pacientov
s neovaskulárnou VPDM.
Na základe analýzy populačnej farmakokinetiky a vymiznutia ranibizumabu zo séra pacientov s neovaskulárnou VPDM liečených dávkou 0,5 mg, priemerný vitreálny eliminačný polčas ranibizumabu je približne 9 dní. Pri intravitreálnom podávaní Lucentisu 0,5 mg/oko raz mesačne sa predpokladá, že Cmax ranibizumabu v sére, ktorá sa dosiahne približne 1 deň po podaní, bude vo všeobecnosti v rozmedzí medzi 0,79 a 2,90 ng/ml, a Cmin sa vo všeobecnosti predpokladá v rozmedzí medzi 0,07 a 0,49 ng/ml. Predpokladaná sérová koncentrácia ranibizumabu je približne 90 000- násobne nižšia ako vitreálna koncentrácia ranibizumabu.
Pacienti s poškodením funkcie obličiek: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky Lucentisu u pacientov s poškodením funkcie obličiek. V populačnej farmakokinetickej analýze pacientov s neovaskulárnou VPDM malo 68 % (136 z 200) pacientov poškodenie funkcie obličiek (46,5 % ľahké [50-80 ml/min], 20 % stredne ťažké [30-50 ml/min] a 1,5 % ťažké [<30 ml/min]).
U pacientov s RVO malo 48,2 % (253 z 525) poškodenie funkcie obličiek (36,4 % ľahké, 9,5 %
stredne ťažké a 2,3 % ťažké). Systémový klírens bol trochu nižší, čo však nebolo klinicky významné.
Pacienti s poškodením funkcie pečene: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky
Lucentisu u pacientov s poškodením funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Bilaterálne intravitreálne podávanie ranibizumabu opiciam rodu Cynomolgus v dávkach medzi
0,25 mg/oko a 2,0 mg/oko raz za 2 týždne až do 26 týždňov malo za následok účinky na oči závislé od
dávky.
Intraokulárne sa zaznamenalo od dávky závislé zosilnenie zápalu a zvýšenie počtu buniek v prednej očnej komore s maximom 2 dni po podaní injekcie. Závažnosť zápalovej odpovede sa spravidla znížila pri podaní ďalších injekcií alebo počas zotavenia. V zadnom segmente sa pozorovala vitreálna infiltrácia buniek a zákaly sklovca, ktoré tiež mali tendenciu závisieť od dávky a spravidla pretrvávali do konca liečebného obdobia. V štúdii trvajúcej 26 týždňov sa intenzita zápalu sklovca zvyšovala
s počtom injekcií. Po zotavení sa však pozorovali dôkazy reverzibility. Povaha a načasovanie zápalu zadného segmentu poukazuje na imunitne sprostredkovanú odpoveď protilátok, čo môže byť klinicky
nevýznamné. Pri niektorých zvieratách sa pozoroval vznik katarakty po relatívne dlhom období
intenzívneho zápalu, čo naznačuje, že zmeny na šošovke sú sekundárne po ťažkom zápale. Prechodné zvýšenie vnútroočného tlaku po podaní sa pozorovalo po intravitreálnych injekciách bez ohľadu na
dávku.
Mikroskopické očné zmeny súviseli so zápalom a nepoukazovali na degeneratívne procesy. V niektorých očiach sa zaznamenali granulomatózne zápalové zmeny na papile. Tieto zmeny v zadnom segmente ustupovali a v niektorých prípadoch vymizli počas zotavovania.
Po intravitreálnom podaní sa nezistili žiadne známky systémovej toxicity. V podsúbore liečených zvierat sa našli sérové a sklovcové protilátky voči ranibizumabu.
Nie sú dostupné údaje o karcinogenite alebo mutagenite.
U gravidných opíc nespôsobilo intravitreálne podávanie ranibizumabu, ktoré malo za následok maximálne systémové expozície 0,9- až 7-násobne vyššie ako najhorší prípad klinickej expozície, vývojovú toxicitu alebo teratogenitu a nemalo žiadny vplyv na hmotnosť alebo štruktúru placenty,
hoci ranibizumab sa vzhľadom na jeho mechanizmus účinku má považovať za potenciálne teratogénny
a embryo- a fetotoxický.
Neprítomnosť účinkov na vývoj embrya a plodu sprostredkovaných ranibizumabom pravdepodobne súvisí hlavne s neschopnosťou fragmentu Fab prestupovať cez placentu. Napriek tomu bol popísaný prípad vysokých hladín ranibizumabu v sére matky a prítomnosti ranibizumabu v sére plodu, čo naznačuje, že protilátka proti ranibizumabu fungovala ako transportná bielkovina (obsahujúca segment Fc) pre ranibizumab, čím sa znižoval klírens zo séra matky a umožňoval sa prestup cez placentu. Keďže sledovania vývoja embryí a plodov sa robili u zdravých gravidných zvierat a ochorenie (napr. diabetes) môže meniť priepustnosť placenty pre fragment Fab, štúdia sa má interpretovať s opatrnosťou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát α,α-trehalózy Monohydrát histidíniumchloridu Histidín
Polysorbát 20
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorená injekčná liekovka uchovávať pri izbovej teplote (25°C) najviac
24 hodín.
6.5 Druh obalu a obsah balenia
0,23 ml sterilného roztoku v injekčnej liekovke (sklo typu I) so zátkou (chlórobutylová guma), 1 tupá ihla s filtrom (18G x 1½″, 1,2 mm x 40 mm, 5 µm), 1 injekčná ihla (30G x ½″, 0,3 mm x 13 mm) a
1 injekčná striekačka (polypropylén) (1 ml). Balenie obsahuje 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka, injekčná ihla, ihla s filtrom a injekčná striekačka sú určené len na jednorazové použitie. Opakované použitie môže mať za následok infekciu alebo iné ochorenie/poškodenie zdravia. Všetky zložky sú sterilné. Akákoľvek zložka, ktorej obal vykazuje známky poškodenia alebo nesprávnej manipulácie, sa nesmie použiť. Sterilitu nemožno zaručiť, ak uzáver obalu zložky nie je neporušený.
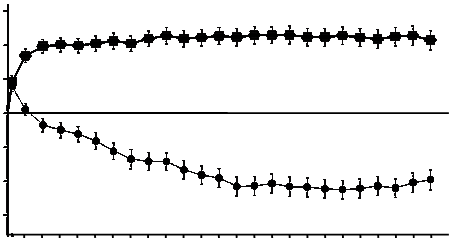
Pri príprave Lucentisu na intravitreálne podanie dodržiavajte, prosím, nasledujúce pokyny:
1. Pred odobratím obsahu sa má dezinfikovať vonkajšia časť gumenej zátky injekčnej liekovky.
2. Asepticky nasaďte ihlu s 5 µm filtrom (18G x 1½″, 1,2 mm x 40 mm, z balenia) na 1 ml injekčnú striekačku (z balenia). Vtlačte hrubú ihlu s filtrom do stredu zátky liekovky, až sa ihla dotkne dna injekčnej liekovky.
3. Odoberte všetku tekutinu z injekčnej liekovky, pri čom liekovka má byť vo zvislej polohe, mierne naklonená, aby sa uľahčilo úplné odobratie obsahu.
4. Dbajte na to, aby ste pri vyprázdnení injekčnej liekovky dostatočne potiahli piest, aby sa ihla
s filtrom úplne vyprázdnila.
5. Hrubú ihlu s filtrom nechajte v injekčnej liekovke a odpojte injekčnú striekačku od hrubej ihly s filtrom. Ihla s filtrom sa má po odobratí obsahu injekčnej liekovky zahodiť a nemá sa použiť na intravitreálnu injekciu.
6. Asepticky pevne nasaďte injekčnú ihlu (30G x ½″, 0,3 mm x 13 mm, z balenia) na injekčnú striekačku.
7. Opatrne odstráňte kryt z injekčnej ihly bez toho, aby ste odpojili injekčnú ihlu od injekčnej striekačky.
Poznámka: Pri odstránení krytu pridržte žltý násadec injekčnej ihly.
8. Opatrne vytlačte vzduch z injekčnej striekačky a upravte dávku na rysku označujúcu 0,05 ml na striekačke. Injekčná striekačka je pripravená na podanie injekcie.
Poznámka: Injekčnú ihlu neutierajte. Nepotiahnite piest injekčnej striekačky.
Po podaní injekcie nenasaďte kryt späť na injekčnú ihlu, ani ihlu neodpojte od injekčnej striekačky. Zahoďte použitú injekčnú striekačku spolu s ihlou do odpadovej nádoby na ostré predmety, alebo zlikvidujte v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLO
EU/1/06/374/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 22. január 2007
Dátum posledného predĺženia: 24. január 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Lucentis 10 mg/ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jeden ml obsahuje 10 mg ranibizumabu*. Každá injekčná liekovka obsahuje 2,3 mg ranibizumabu v 0,23 ml roztoku.
*Ranibizumab je fragment humanizovanej monoklonálnej protilátky vytvorenej v bunkách
Escherichia coli rekombinantnou DNA technológiou.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Číry, bezfarebný až svetložltý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Lucentis je indikovaný u dospelých na:
· Liečbu neovaskulárnej (vlhkej) vekom podmienenej degenerácie makuly (VPDM)
· Liečbu poškodenia zraku v dôsledku diabetického makulárneho edému (DME)
· Liečbu poškodenia zraku v dôsledku makulárneho edému po oklúzii žily sietnice (vetvovej RVO
alebo kmeňovej RVO)
· Liečbu poškodenia zraku v dôsledku neovaskularizácie chorioidey (CNV) pri patologickej myopii (PM)
4.2 Dávkovanie a spôsob podávania
Lucentis smie podávať kvalifikovaný oftalmológ so skúsenosťami s podávaním intravitreálnych
injekcií.
Odporúčaná dávka Lucentisu je 0,5 mg, podávaná ako jednorazová intravitreálna injekcia. To zodpovedá objemu injekcie 0,05 ml. Interval medzi dvoma dávkami podanými do toho istého oka má byť najmenej štyri týždne.
Liečba sa začína jednou injekciou za mesiac až do dosiahnutia maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity ochorenia, t.j. žiadna zmena zrakovej ostrosti a iných prejavov a príznakov choroby pri pokračujúcej liečbe. U pacientov s vlhkou VPDM, DME a RVO môže byť na začiatku potrebné podať po sebe tri alebo viac mesačných injekcií.
Následné sledovanie a intervaly medzi podaniami má určovať lekár a majú byť založené na aktivite choroby, stanovenej prostredníctvom zrakovej ostrosti a/alebo anatomických parametrov.
Ak zrakové a anatomické parametre podľa názoru lekára ukazujú, že pokračujúca liečba pre pacienta nie je prínosom, podávanie Lucentisu sa má ukončiť.
Sledovanie aktivity choroby môže zahŕňať klinické vyšetrenie, testovanie funkcie alebo zobrazovacie
techniky (napr. optickú koherentnú tomografiu alebo fluoresceínovú angiografiu).
Ak sa pacient lieči v režime podávania a predlžovania intervalov medzi podaniami, po dosiahnutí maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity choroby sa intervaly medzi podaniami môžu postupne predlžovať až do opätovného objavenia sa prejavov aktivity choroby alebo zhoršenia zraku. Interval medzi podaniami sa nemá naraz predĺžiť o viac ako dva týždne pri vlhkej VPDM a môže sa naraz predĺžiť až o jeden mesiac pri DME. Pri RVO sa intervaly medzi podaniami tiež môžu postupne predlžovať, avšak nie je dostatok údajov na určenie dĺžky týchto intervalov. Ak sa opäť objaví aktivita choroby, interval medzi podaniami sa má patrične skrátiť.
Pri liečbe poškodenia zraku v dôsledku CNV pri PM u mnohých pacientov môžu byť potrebné len jedna alebo dve injekcie počas prvého roka, zatiaľ čo niektorí pacienti môžu potrebovať častejšiu liečbu (pozri časť 5.1).
Lucentis a laserová fotokoagulácia pri DME a pri makulárnom edéme po BRVO
S podávaním Lucentisu súčasne s laserovou fotokoaguláciou sú určité skúsenosti (pozri časť 5.1). Pri
podávaní v ten istý deň sa Lucentis má podať minimálne 30 minút po laserovej fotokoagulácii.
Lucentis sa môže podávať pacientom, ktorí boli v minulosti liečení laserovou fotokoaguláciou.
Lucentis a fotodynamická liečba Visudynom pri CNV v dôsledku PM
Nie sú skúsenosti so súbežným podávaním Lucentisu a Visudynu.
Osobitné skupiny pacientov
Poškodenie funkcie pečene
Lucentis sa neskúmal u pacientov s poškodením funkcie pečene. U tejto populácie však nie sú potrebné zvláštne opatrenia.
Poškodenie funkcie obličiek
Úprava dávky nie je potrebná u pacientov s poškodením funkcie obličiek (pozri časť 5.2).
Starší pacienti
Úprava dávkovania u starších ľudí nie je potrebná. Skúsenosti s pacientmi s DME staršími ako
75 rokov sú obmedzené.
Deti a dospievajúci
Bezpečnosť a účinnosť Lucentisu u detí a dospievajúcich vo veku menej ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje.
Spôsob podania
Injekčná liekovka na jednorazové použitie len na intravitreálne podanie.
Lucentis sa má pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu sfarbenia.
Podanie injekcie sa má uskutočniť za aseptických podmienok, ktoré zahŕňajú chirurgickú dezinfekciu rúk, použitie sterilných rukavíc, sterilného rúška a sterilného rozvierača mihalníc (alebo náhrady)
a dostupnosť sterilnej paracentézy (pre prípad potreby). Pred vykonaním intravitreálneho zákroku sa má dôsledne preskúmať pacientova anamnéza so zreteľom na reakcie z precitlivenosti (pozri časť 4.4). Pred podaním injekcie sa má v súlade s lokálnou klinickou praxou podať náležitá anestézia
a širokospektrálny lokálny mikrobicídny prostriedok na dezinfekciu kože v okolí oka, mihalnice a povrchu oka.
Informácie o príprave Lucentisu, pozri časť 6.6.
Injekčná ihla sa zavádza 3,5-4,0 mm za limbom do dutiny sklovca, vyhýba sa horizontálnemu poludníku a smeruje do centra očnej gule. Potom sa podá objem injekcie 0,05 ml; pri následných injekciách sa má použiť iné miesto na sklére.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s aktívnymi alebo suspektnými očnými alebo periokulárnymi infekciami. Pacienti s aktívnym ťažkým vnútroočným zápalom.
4.4 Osobitné upozornenia a opatrenia pri používaní
Reakcie súvisiace s intravitreálnou injekciou
Podanie intravitreálnych injekcií vrátane injekcií Lucentisu sa spájalo s endoftalmitídou, vnútroočným zápalom, rhegmatogénnym odlúčením sietnice, trhlinou v sietnici a iatrogénnou traumatickou kataraktou (pozri časť 4.8). Pri podaní Lucentisu sa musia vždy dodržať náležité aseptické injekčné postupy. Okrem toho je potrebné pacientov sledovať počas týždňa po podaní injekcie, čo umožní včasnú liečbu v prípade infekcie. Pacienti majú byť poučení, aby bezodkladne oznámili akýkoľvek príznak, ktorý poukazuje na endoftalmitídu alebo na niektorú z vyššie uvedených príhod.
Zvýšeniavnútroočnéhotlaku
V priebehu 60 minút po podaní injekcie Lucentisu sa pozorovalo prechodné zvýšenie vnútroočného tlaku (IOP). Zistilo sa aj trvalé zvýšenie IOP (pozri časť 4.8). Vnútroočný tlak aj perfúzia hlavy zrakového nervu sa musia monitorovať a náležite liečiť.
Bilaterálnaliečba
Obmedzené údaje o bilaterálnom použití Lucentisu (vrátane podania v ten istý deň) nenaznačujú zvýšené riziko systémových nežiaducich príhod v porovnaní s unilaterálnou liečbou.
Imunogenita
Lucentis môže byť imunogénny. Keďže je možnosť zvýšenej systémovej expozície u osôb s DME, nemožno vylúčiť zvýšené riziko vzniku precitlivenosti u tejto populácie pacientov. Pacienti majú byť tiež poučení, aby hlásili akékoľvek zvýšenie závažnosti vnútroočného zápalu, ktoré môže byť klinickým príznakom zodpovedajúcim tvorbe protilátok vo vnútri oka.
Súčasnépoužitiesinýmianti-VEGF (vaskulárny endoteliálny rastový faktor)
Lucentis sa nemá podávať súčasne s inými anti-VEGF liekmi (systémovými alebo okulárnymi).
PrerušenieliečbyLucentisom
Dávka sa nemá podať a v liečbe sa nemá pokračovať skôr ako počas najbližšej plánovanej návštevy
v prípade:
· poklesu najlepšie korigovanej zrakovej ostrosti (BCVA) o ≥30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
· intraokulárneho tlaku ≥30 mmHg;
· trhliny v sietnici;
· subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je ≥50 %
celkovej plochy lézie;
· uskutočneného alebo plánovaného intraokulárneho chirurgického zákroku počas uplynulých alebo nasledujúcich 28 dní.
T
r
hlina v pigmentovom epiteli sietnice
Rizikové faktory, ktoré sa spájajú so vznikom trhliny v pigmentovom epiteli sietnice po anti-VEGF
liečbe pri vlhkej VPDM, zahŕňajú veľké a/alebo vysoko uložené odlúčenie pigmentového epitelu sietnice. Pri začatí liečby Lucentisom je potrebná opatrnosť u pacientov s týmito rizikovými faktormi pre trhliny v pigmentovom epiteli sietnice.
Regmatogénneodlúčeniesietnicealebomakulárnediery
Liečba sa má ukončiť u osôb s regmatogénnym odlúčením sietnice alebo makulárnymi dierami 3.
alebo 4. stupňa.
Skupiny pacientov, u ktorých sú skúsenosti obmedzené
S liečbou osôb s DME spôsobeným diabetom typu I sú len obmedzené skúsenosti. Lucentis sa neskúmal u pacientov, ktorí v minulosti dostali intravitreálne injekcie, u pacientov s aktívnymi
systémovými infekciami, proliferujúcou diabetickou retinopatiou, alebo u pacientov so sprievodnými
očnými ochoreniami, napr. s odlúčením sietnice alebo makulárnou dierou. Nie sú tiež žiadne
skúsenosti s liečbou Lucentisom u pacientov s diabetom s HbA1c vyšším ako 12 % a nekontrolovanou
hypertenziou. Pri liečbe takýchto pacientov má lekár vziať do úvahy tento nedostatok informácií.
Údaje o účinku Lucentisu sú obmedzené u pacientov s PM, ktorí v minulosti podstúpili neúspešnú fotodynamickú liečbu verteporfínom (vPDT). Zatiaľ čo sa pozoroval zhodný účinok u osôb so subfoveálnymi a juxtafoveálnymi léziami, nie je tiež dostatok údajov, z ktorých by bolo možné usudzovať na účinok Lucentisu u osôb s PM, ktoré majú extrafoveálne lézie.
Systémovéúčinkypointravitreálnompoužití
Po intravitreálnej injekcii inhibítorov VEGF sa zaznamenali systémové nežiaduce udalosti vrátane
krvácaní mimo oka a artériových tromboembolických príhod.
Údaje o bezpečnosti liečby u pacientov s DME, makulárnym edémom po RVO a s CNV v dôsledku PM, ktorí majú v anamnéze cievnu mozgovú príhodu alebo tranzitórne ischemické ataky, sú obmedzené. Pri liečbe takýchto pacientov je potrebné postupovať opatrne (pozri časť 4.8).
Epizódy RVO, ischemickej vetvovej RVO a kmeňovejRVOvminulosti
Skúsenosti s liečbou pacientov s epizódami RVO v minulosti a pacientov s ischemickou vetvovou
RVO (BRVO) a kmeňovou RVO (CRVO) sú obmedzené. U pacientov s RVO, u ktorých sa objavia klinické príznaky ireverzibilnej ischemickej straty zrakovej funkcie, sa liečba neodporúča.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne formálne interakčné štúdie.
Adjuvantné použitie fotodynamickej liečby (PDT) verteporfínom a Lucentisu pri vlhkej VPDM a PM,
pozri časť 5.1.
Adjuvantné použitie laserovej fotokoagulácie a Lucentisu pri DME a BRVO, pozri časti 4.2 a 5.1.
V klinických skúšaniach liečby poškodenia zraku v dôsledku DME súbežná liečba tiazolidíndiónmi neovplyvnila výsledky týkajúce sa zrakovej ostrosti alebo hrúbky čiastkového poľa centrálnej sietnice (CSTF) u pacientov liečených Lucentisom.
4.6 Fertilita, gravidita a laktácia
Ž
eny
vo
fertilnom
veku/antikoncepcia
u
žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby.
Gravidita
Nie sú dostupné klinické údaje o použití ranibizumabu v gravidite. Štúdie na makakoch krabožravých nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity alebo vývinu embrya/plodu (pozri časť 5.3). Systémová expozícia ranibizumabu po podaní do oka je nízka, ale vzhľadom na jeho mechanizmus účinku sa ranibizumab musí považovať za potenciálne teratogénny a embryo-
/fetotoxický. Preto sa ranibizumab nemá používať v gravidite, pokiaľ očakávaný prínos nie je väčší
ako možné riziko pre plod. Ženám, ktoré chcú otehotnieť a boli liečené ranibizumabom, sa odporúča počkať s počatím dieťaťa aspoň 3 mesiace od poslednej dávky ranibizumabu.
Laktácia
Nie je známe, či sa Lucentis vylučuje do ľudského mlieka. Dojčenie sa neodporúča počas používania
Lucentisu.
Fertilita
Nie sú dostupné žiadne údaje o fertilite.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liečba Lucentisom môže vyvolať dočasné poruchy videnia, čo môže ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8). Pacienti, u ktorých sa vyskytnú tieto príznaky, nesmú viesť vozidlá alebo obsluhovať stroje, kým tieto dočasné poruchy zraku neustúpia.
4.8 Nežiaduce účinky
Zhrnutieprofilubezpečnosti
Väčšina nežiaducich reakcií hlásených po podaní Lucentisu súvisí s postupom intravitreálnej injekcie.
Najčastejšie hlásené nežiaduce reakcie týkajúce sa očí po podaní injekcie Lucentisu sú: bolesť oka, hyperémia oka, zvýšený vnútroočný tlak, vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko a svrbenie oka.
Najčastejšie hlásené nežiaduce reakcie netýkajúce sa očí sú bolesť hlavy, nazofaryngitída a bolesť kĺbov.
Medzi menej často hlásené, ale závažnejšie nežiaduce reakcie patria endoftalmitída, slepota, odlúčenie sietnice, trhlina v sietnici a iatrogénna traumatická katarakta (pozri časť 4.4).
Pacientov je potrebné informovať o prejavoch týchto potenciálnych nežiaducich reakcií a poučiť ich, aby informovali svojho lekára, ak u nich vzniknú príznaky ako bolesť očí alebo zvýšenie nepríjemných pocitov, zhoršujúce sa sčervenanie očí, neostré alebo zhoršené videnie, zvýšený počet malých čiastočiek v zornom poli, alebo zvýšená citlivosť na svetlo.
Nežiaduce reakcie, ktoré sa vyskytli po podaní Lucentisu v klinických skúšaniach, sú zhrnuté
v tabuľke nižšie.
T
abuľkový
zoznam
nežiaducich
reakcií
#
Nežiaduce reakcie sú zatriedené podľa orgánových systémov a frekvencie podľa nasledujúcich konvencií: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
Veľmi časté Nazofaryngitída
Časté Infekcia močových ciest*
Poruchy krvi a lymfatického systému
Časté Anémia
Poruchy imunitného systému
Časté Precitlivenosť
Psychické poruchy
Časté Úzkosť
Poruchy nervového systému
Veľmi časté Bolesť hlavy
Poruchy oka
Veľmi časté Vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, bolesť oka, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko, hyperémia oka, svrbenie oka.
Časté Degenerácia sietnice, porucha sietnice, odlúčenie sietnice, trhlina v sietnici, odlúčenie pigmentového epitelu sietnice, trhlina v pigmentovom epiteli sietnice, znížená zraková ostrosť, krvácanie do sklovca, porucha sklovca, uveitída, iritída, iridocyklitída, katarakta, subkapsulárna katarakta, opacifikácie zadného puzdra šošovky, bodkovitá keratitída, abrázia rohovky, zápal prednej očnej komory, neostré videnie, krvácanie v mieste podania injekcie, krvácanie do oka, konjunktivitída, alergická konjunktivitída, výtok z oka, fotopsia, fotofóbia, nepríjemné pocity v oku, edém mihalnice, bolesť mihalnice, hyperémia spojoviek.
Menej časté Slepota, endoftalmitída, hypopyon, hyféma, keratopatia, adhézia dúhovky, depozity v rohovke, edém rohovky, strie rohovky,
bolesť v mieste podania injekcie, podráždenie v mieste podania
injekcie, abnormálne pocity v oku, podráždenie mihalnice.
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Kašeľ
Poruchy gastrointestinálneho traktu
Časté Nauzea
Poruchy kože a podkožného tkaniva
Časté Alergické reakcie (exantém, urtikária, pruritus, erytém)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Veľmi časté Artralgia
Laboratórne a funkčné vyšetrenia
Veľmi časté Zvýšenie vnútroočného tlaku
# Nežiaduce reakcie boli definované ako nežiaduce udalosti (u najmenej 0,5 percentuálneho bodu
pacientov), ktoré sa vyskytli častejšie (najmenej 2 percentuálne body) u pacientov liečených
Lucentisom 0,5 mg, ako u pacientov, ktorí dostávali kontrolnú liečbu (simulované podanie alebo PDT
verteporfínom).
* pozorované iba u populácie s DME
NežiaducereakciesúvisiacesoskupinouliekovV klinických skúšaniach fázy III pri vlhkej VPDM bola celková frekvencia krvácaní mimo oka, čo je nežiaduca udalosť potenciálne súvisiaca so systémovou inhibíciou VEGF (vaskulárneho endoteliálneho rastového faktora), mierne zvýšená u pacientov liečených ranibizumabom. Charakteristika rôznych krvácaní však nebola zhodná. Po intravitreálnom použití inhibítorov VEGF existuje teoretické riziko arteriálnych tromboembolických príhod vrátane cievnej mozgovej príhody
a infarktu myokardu. V klinických skúšaniach Lucentisu sa pozorovala nízka incidencia arteriálnych tromboembolických príhod u pacientov s VPDM, DME, RVO a PM a neboli významné rozdiely medzi skupinami liečenými ranibizumabom v porovnaní s kontrolnou liečbou.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieBoli hlásené prípady náhodného predávkovania v klinických skúšaniach pri vlhkej VPDM a z údajov po uvedení lieku na trh. Nežiaduce reakcie súvisiace s týmito hlásenými prípadmi boli zvýšenie vnútroočného tlaku, prechodná slepota, znížená zraková ostrosť, edém rohovky, bolesť rohovky
a bolesť oka. Ak dôjde k predávkovaniu, je potrebné sledovať a liečiť vnútroočný tlak, ak to ošetrujúci
lekár považuje za potrebné.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: oftalmologiká, látky s antineovaskularizačným účinkom, ATC kód:
S01LA04
Ranibizumab je fragment humanizovanej rekombinantnej monoklonálnej protilátky, ktorého cieľom je ľudský vaskulárny endoteliálny rastový faktor A (VEGF-A). Viaže sa s vysokou afinitou na izoformy VEGF-A (napr. VEGF110, VEGF121 a VEGF165), a tým zabraňuje vzniku väzby VEGF-A na jeho receptory VEGFR-1 a VEGFR-2. Naviazanie VEGF-A na jeho receptory má za následok proliferáciu endotelových buniek a neovaskularizáciu, ako aj únik tekutín z ciev, čo všetko sa považuje za faktory prispievajúce k progresii neovaskulárnej formy vekom podmienenej degenerácie makuly, patologickej myopie alebo poškodeniu zraku spôsobeného buď diabetickým makulárnym edémom, alebo makulárnym edémom po RVO.
Liečba
vlhkej VPDMPri vlhkej VPDM sa klinická bezpečnosť a účinnosť Lucentisu hodnotila v troch randomizovaných, dvojito maskovaných, simulovane alebo aktívne kontrolovaných klinických skúšaniach trvajúcich
24 mesiacov u pacientov s neovaskulárnou VPDM. Do týchto klinických skúšaní bolo zaradených
celkovo 1 323 pacientov (879 v aktívnej a 444 v kontrolnej skupine).
V klinickom skúšaní FVF2598g (MARINA) 716 pacientov s minimálne klasickou alebo okultnou bez klasickej chorioidálnej neovaskularizácie (CNV) dostávalo každý mesiac intravitreálne injekcie Lucentisu 0,3 mg (n=238) alebo 0,5 mg (n=240) alebo simulované injekcie (n=238).
V klinickom skúšaní FVF2587g (ANCHOR) 423 pacientov s prevažne klasickými CNV léziami dostávalo buď: 1) každý mesiac intravitreálne injekcie Lucentis 0,3 mg a simulovanú PDT (n=140);
2) každý mesiac intravitreálne injekcie Lucentisu 0,5 mg a simulovanú PDT (n=140); alebo
3) simulované intravitreálne injekcie a aktívnu PDT verteporfínom (n=143). Simulovaná alebo aktívna PDT verteporfínom sa podala s úvodnou injekciou Lucentisu a potom každé 3 mesiace, ak fluoresceínová angiografia ukázala pretrvávanie alebo recidívu úniku tekutín z ciev.
Kľúčové merané parametre sú zhrnuté v Tabuľke 1 a na Obrázku 1.
Tabuľka 1 Výsledky po 12 mesiacoch a 24 mesiacoch v klinickom skúšaní FVF2598g(MARINA) a FVF2587g (ANCHOR)
|
| FVF2598g (MARINA)
| FVF2587g (ANCHOR)
|
| Meraný
parameter
| Mesiac
| Simulované
podanie
(n=238)
| Lucentis
0,5 mg
(n=240)
| PDT
verteporfínom
(n=143)
| Lucentis
0,5 mg
(n=140)
| Strata <15 písmen
zrakovej ostrosti (%)a (zachovanie vízu, primárny ukazovateľ)
| 12. mesiac
| 62 %
| 95 %
| 64 %
| 96 %
| 24. mesiac
| 53 %
| 90 %
| 66 %
| 90 %
| Zisk ≥15 písmen
zrakovej ostrosti
(%)a
| 12. mesiac
| 5 %
| 34 %
| 6 %
| 40 %
| 24. mesiac
| 4 %
| 33 %
| 6 %
| 41 %
| Priemerná zmena
zrakovej ostrosti
(písmená) (SD)a
| 12. mesiac
| -10,5 (16,6)
| +7,2 (14,4)
| -9,5 (16,4)
| +11,3 (14,6)
| 24. mesiac
| -14,9 (18,7)
| +6,6 (16,5)
| -9,8 (17,6)
| +10,7 (16,5)
|
|
|
a p<0,01
O
brázok 1 Priemerná zmena zrakovej ostrosti od východiskovej hodnoty do 24. mesiaca v klinickom skúšaní FVF2598g (MARINA) a v klinickom skúšaní FVF2587g (ANCHOR)
15
10
5
0
-5

-10
-15
Klinické skúšanie (MARINA)

0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+6,6
-14,9
+21,5
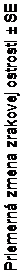
15
10

5
0
-5
-10
-15
Klinické skúšanie FVF2587g (ANCHOR)

+10,7
-9,8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+20,5
MARINA ANCHOR
Výsledky oboch klinických skúšaní ukázali, že pokračujúca liečba ranibizumabom môže predstavovať
prínos aj pre pacientov, ktorí stratili ≥15 písmen najlepšie korigovanej zrakovej ostrosti (BCVA)
v prvom roku liečby.
Klinické skúšanie FVF3192g (PIER) bolo randomizované, dvojito maskované, simulovane
kontrolované klinické skúšanie, určené na stanovenie bezpečnosti a účinnosti Lucentisu
u 184 pacientov so všetkými formami neovaskulárnej VPDM. Pacienti dostávali Lucentis 0,3 mg
(n=60) alebo 0,5 mg (n=61) intravitreálnymi injekciami alebo simulované injekcie (n=63) raz za mesiac v 3 po sebe idúcich dávkach, po ktorých nasledovalo podávanie dávky každé 3 mesiace. Od
14. mesiaca klinického skúšania sa pacientom so simulovaným podaním povolila zmena skupiny
liečby, aby dostávali ranibizumab, a od 19. mesiaca boli možné častejšie podania. Pacienti liečení
Lucentisom v PIER dostali priemerne 10 kompletných liečebných cyklov.
Primárnym cieľovým ukazovateľom účinnosti bola priemerná zmena zrakovej ostrosti po
12 mesiacoch v porovnaní s východiskovými hodnotami. Po počiatočnom zvýšení zrakovej ostrosti (po podávaní raz za mesiac) sa zraková ostrosť pacientov v priemere zhoršovala pri podávaní raz za štvrťrok a vrátila sa na východiskovú hodnotu v 12. mesiaci, pričom tento účinok sa zachoval
u väčšiny pacientov liečených ranibizumabom (82 %) do 24. mesiaca. Údaje u obmedzeného počtu osôb, ktoré zmenili skupinu liečby, aby dostávali ranibizumab po viac ako roku simulovanej liečby,
naznačili, že včasný začiatok liečby sa môže spájať s lepším zachovaním zrakovej ostrosti.
V klinických skúšaniach MARINA aj ANCHOR zlepšenie zrakovej ostrosti pozorované pri Lucentise
0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby, stanovený prostredníctvom
skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Rozdiely medzi Lucentisom
0,5 mg a dvomi kontrolnými skupinami boli vyhodnotené s hodnotami p v rozmedzí od 0,009 do
<0,0001.
Účinnosť Lucentisu v liečbe vlhkej VPDM okrem toho potvrdili štúdie VPDM ukončené po zaregistrovaní lieku. Údaje z dvoch štúdií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) nepreukázali prídavný účinok pri kombinovanom podaní verteporfínu (Visudyne PDT) a Lucentisu oproti monoterapii Lucentisom.
Liečbapoškodeniazrakuvdôsledku DME
Bezpečnosť a účinnosť Lucentisu sa vyhodnotili v dvoch randomizovaných, dvojito maskovaných
štúdiách kontrolovaných simulovaným podaním alebo účinným liekom trvajúcich 12 mesiacov
u pacientov s poškodením zraku v dôsledku diabetického makulárneho edému. Do týchto štúdií bolo
zaradených celkovo 496 pacientov (336 s aktívnou liečbou a 160 ako kontrola), z ktorých väčšina
mala diabetes typu II, pričom 28 pacientov liečených ranibizumabom malo diabetes typu I.
V štúdii fázy II, D2201 (RESOLVE), dostávalo 151 pacientov ranibizumab (6 mg/ml, n=51,
10 mg/ml, n=51) alebo simulovanú liečbu (n=49) intravitreálnymi injekciami raz za mesiac až do splnenia vopred určených kritérií pre ukončenie liečby. Začiatočná dávka ranibizumabu (0,3 mg alebo
0,5 mg) sa po prvej injekcii mohla kedykoľvek v priebehu štúdie zdvojnásobiť. Laserová
fotokoagulácia bola ako záchranná liečba povolená po 3. mesiaci v obidvoch skupinách liečby. Štúdia mala dve časti: výskumnú časť (prvých 42 pacientov analyzovaných po 6 mesiacoch) a potvrdzujúcu časť (zvyšných 109 pacientov analyzovaných po 12 mesiacoch). Stredná priemerná zmena BCVA od
1. mesiaca do 12. mesiaca v porovnaní s východiskovou hodnotou bola +7,8 (±7,72) písmen
u pacientov liečených ranibizumabom (n=102) celkove v oboch častiach klinického skúšania,
v porovnaní s -0,1 (±9,77) písmen u pacientov pri simulovanej liečbe (p<0,0001 pre rozdiel v liečbe).
V klinickom skúšaní fázy III, D2301 (RESTORE), bolo randomizovaných 345 pacientov
s poškodením zraku v dôsledku makulárneho edému, aby dostávali buď intravitreálnu injekciu 0,5 mg ranibizumabu ako monoterapiu a simulovanú laserovú fotokoaguláciu (n=116), kombináciu 0,5 mg
ranibizumabu a laserovej fotokoagulácie (n=118), alebo simulovanú injekciu a laserovú
fotokoaguláciu (n=111). Liečba ranibizumabom sa začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej zrakovej ostrosti pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Liečba sa obnovila, keď sa pozorovalo zníženie BCVA v dôsledku progresie DME. Laserová fotokoagulácia sa podala pri vstupe do štúdie v ten istý deň najmenej
30 minút pred injekciou ranibizumabu a následne podľa potreby na základe kritérií ETDRS.
240 pacientov, ktorí predtým ukončili 12-mesačné klinické skúšanie RESTORE, bolo zaradených do otvorenej, multicentrickej extenzie skúšania trvajúcej 24 mesiacov (RESTORE Extension). Pacienti dostávali 0,5 mg ranibizumabu pro re nata (PRN) do toho istého oka, ktoré bolo zvolené ako oko pre klinické skúšanie v skúšaní D2301 (RESTORE). Liečba sa znovu začala s mesačnými intervalmi až do poklesu BCVA v dôsledku DME a pokračovala až do dosiahnutia stabilnej BCVA. Okrem toho sa podávala liečba laserom, ak bola potrebná podľa úsudku skúšajúceho lekára a na základe kritérií ETDRS.
Kľúčové merané parametre sú zhrnuté v Tabuľke 2 (RESTORE a Extension) a na Obrázku 2 (RESTORE).
Obrázok 2 Stredná zmena zrakovej ostrosti v čase oproti východiskovej hodnote v štúdiiD2301 (RESTORE)12
10
+ 6,8/+ 6,4
+ 6,2/+ 5,4*
+ 0,9
Mesiac
11 12
Ranibizumab 0,5 mg (n=115)
Ranibizumab 0,5 mg+laser (n=118) Laser (n=110)
BL=východisková hodnota; SE=stredná chyba priemeru
* Rozdiel metódou najmenších štvorcov, p<0,0001/0,0004 na základe dvojstranného stratifikovaného
testu podľa Cochrana-Mantela-Haenszela
Účinok po 12 mesiacoch sa zhodoval u väčšiny podskupín. Avšak pre osoby s pomerne dobrou východiskovou hodnotou BCVA (>73 písmen) spolu s makulárnym edémom s hrúbkou centrálnej retiny <300 mm sa liečba ranibizumabom v porovnaní laserovou fotokoaguláciou nezdala prínosom.
Tabuľka 2 Výsledky po 12 mesiacoch v klinickom skúšaní D2301 (RESTORE) a po36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension) Meraný parameter po 12 mesiacoch v
porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
| Ranibizumab
0,5 mg n=115
| Ranibizumab
0,5 mg + laser n=118
| Laser
n=110
| Stredná priemerná zmena BCVA od 1. do
12. mesiacaa (±SD)
| 6,1 (6,4)a
| 5,9 (7,9)a
| 0,8 (8,6)
| Stredná zmena BCVA po 12. mesiacoch
(±SD)
| 6,8 (8,3)a
| 6,4 (11,8)a
| 0,9 (11,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 37,4a
| 43,2a
| 15,5
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 22,6
| 22,9
| 8,2
|
| Meraný parameter po 36 mesiacoch
v skúšaní D2301-E1 (RESTORE Extension) v porovnaní s východiskovou
hodnotou v skúšaní D2301 (RESTORE)
| Predtým
ranibizumab
0,5 mg n=83
| Predtým
ranibizumab
0,5 mg + laser n=83
| Predtým
laser
n=74*
| Stredná zmena BCVA po 24 mesiacoch
(SD)
|
7,9 (9,0)
|
6,7 (7,9)
|
5,4 (9,0)
| Stredná zmena BCVA po 36 mesiacoch
(SD)
|
8,0 (10,1)
|
6,7 (9,6)
|
6,0 (9,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
47,0
|
44,6
|
41,9
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
27,7
|
30,1
|
21,6
|
|
|
ap<0,0001 pre porovnania skupín ranibizumabu oproti skupine laseru.
n v D2301-E1 (RESTORE Extension) je počet pacientov s východiskovou hodnotou (0. mesiac)
v D2301 (RESTORE) a tiež s hodnotou z návštevy po 36. mesiaci.
*zo 74 pacientov, ktorí predtým dostali liečbu laserom, 59 (79 %) dostalo ranibizumab v extenzii
klinického skúšania.
Zlepšenie zrakovej ostrosti pozorované pri Lucentise 0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby vzhľadom na väčšinu funkcií súvisiacich so zrakom, čo sa stanovilo prostredníctvom skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Pri iných podškálach tohto dotazníka sa nezistili rozdiely v súvislosti s liečbou. Rozdiel medzi Lucentisom
0,5 mg a kontrolnou skupinou sa stanovil s hodnotami p 0,0137 (monoterapia ranibizumabom) a
0,0041 (liečba ranibizumabom a laserom) kombinovaného skóre VFQ-25.
Stredný počet injekcií podaných v klinickom skúšani RESTORE trvajúcom 12 mesiacov bol 7,0
v skupine 0,5 mg ranibizumabu, 6,8 v skupine ranibizumabu a lasera a 7,3 simulovaných injekcií
v skupine monoterapie laserom. V priemere pripadlo na pacienta 6,4 injekcií ranibizumabu podaných za obdobie 24 mesiacov extenzie u pacientov, ktorí dostávali ranibizumab v klinickom skúšaní D2301
(RESTORE). Zo 74 pacientov v skupine liečby laserom v skúšaní D2301 (RESTORE) dostalo ranibizumab niekedy počas fázy extenzie 59 (79 %) pacientov. V priemere dostalo týchto 59 pacientov
počas 24-mesačnej extenzie 8,1 injekcií ranibizumabu. Podiely pacientov, u ktorých nebola potrebná
počas fázy extenzie žiadna liečba ranibizumabom, boli v skupinách predtým liečených ranibizumabom
19 %, ranibizumabom a laserom 25 % a laserom 20 %.
Profil dlhodobej bezpečnosti ranibizumabu, ktorý sa pozoroval v 24-mesačnej extenzii klinického skúšania, sa zhoduje so známym profilom bezpečnosti Lucentisu.
V klinickom skúšaní fázy IIIb, D2304 (RETAIN), bolo randomizovaných 372 pacientov s poškodením
zraku v dôsledku DME na nasledujúce podávanie intravitreálnych injekcií:
· 0,5 mg ranibizumabu súbežne s laserovou fotokoaguláciou v režime podávania a predlžovania
intervalov medzi podaniami (treat-and-extend, TE) (n=121),
· 0,5 mg ranibizumabu v monoterapii v režime TE (n=128),
· 0,5 mg ranibizumabu v monoterapii v režime PRN (n=123).
Vo všetkých skupinách sa liečba ranibizumabom začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej BCVA pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Laserová fotokoagulácia sa podala pri vstupe do skúšania v ten istý deň ako prvá injekcia ranibizumabu a následne podľa potreby na základe kritérií ETDRS. Pri TE sa ranibizumab potom podával podľa plánu liečby s intervalmi 2-3 mesiace. Pri PRN sa každý mesiac hodnotila BCVA a ak to bolo potrebné, ranibizumab sa potom podal počas tej istej návštevy. Vo všetkých skupinách sa obnovilo podávanie každý mesiac pri poklese BCVA v dôsledku progresie DME
a pokračovalo sa v ňom až do opätovného dosiahnutia stabilnej BCVA. Skúšanie trvalo 24 mesiacov.
V klinickom skúšaní RETAIN, po 3 začiatočných každomesačných návštevách s podaním liečby, počet plánovaných návštev s podaním liečby potrebný podľa režimu TE bol 13 oproti 20 mesačným návštevám, ktoré boli potrebné podľa režimu PRN. Pri oboch režimoch liečby si viac ako 70 % pacientov udržalo ich BCVA pri frekvencii návštev ≥2 mesiace. Počas 24 mesiacov bol stredný počet (medián) injekcií 12,4 (12,0) v skupine liečby ranibizumabom a laserom pri TE, 12,8 (12,0) v skupine liečby samotným ranibizumabom pri TE a 10,7 (10,0) v skupine liečby ranibizumabom pri PRN. Pridanie lasera sa nespájalo so zníženým stredným počtom injekcií ranibizumabu pri režime TE.
Kľúčové merané parametre sú zhrnuté v Tabuľke 3.
Tabuľka 3 Výsledky v klinickom skúšaní D2304 (RETAIN) Meraný parameter
v porovnaní s východiskovou
hodnotou
| Ranibizumab
0,5 mg + laser pri TE
n=117
| Samotný ranibizumab
0,5 mg pri TE
n=125
| Ranibizumab
0,5 mg pri PRN
n=117
| Stredná priemerná
zmena BCVA od 1. do
12. mesiaca (SD)
|
5,9 (5,5) a
|
6,1 (5,7) a
|
6,2 (6,0)
| Stredná priemerná
zmena BCVA od 1. do
24. mesiaca (SD)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
| Stredná zmena BCVA
po 24 mesiacoch (SD)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
| Zisk ≥10 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
43,6
|
40,8
|
45,3
| Zisk ≥15 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
25,6
|
28,0
|
30,8
|
|
|
ap<0,0001 pre stanovenie neinferiority oproti PRN
V klinických skúšaniach pri DME sprevádzalo zlepšenie BCVA postupom času zníženie strednej
CSFT vo všetkých skupinách liečby.
Liečba
poškodenia
zraku
v
dôsledku makulárneho edému po RVO
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku makulárneho edému po RVO sa vyhodnotili v randomizovaných, dvojito maskovaných, kontrolovaných štúdiách BRAVO a CRUISE, do ktorých boli zaradené osoby s BRVO (n=397) a CRVO (n=392). V oboch štúdiách pacienti dostávali intravitreálne buď 0,3 mg, alebo 0,5 mg ranibizumabu, alebo simulované injekcie. Po 6 mesiacoch pacienti z kontrolnej skupiny simulovaného podania prešli na 0,5 mg ranibizumabu. V štúdii BRAVO bola vo všetkých skupinách od 3. mesiaca povolená laserová fotokoagulácia ako záchranná liečba.
Kľúčové merané parametre z BRAVO a CRUISE sú zhrnuté v Tabuľkách 4 a 5 a na Obrázkoch 3 a 4.
Tabuľka 4 Výsledky po 6 a 12 mesiacoch (BRAVO)
| Simulované podanie/ Lucentis 0,5 mg
(n=132)
| Lucentis 0,5 mg
(n=131)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 7,3 (13,0)
| 18,3 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 12,1 (14,4)
| 18,3 (14,6)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 28,8
| 61,1
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 43,9
| 60,3
| Podiel (%), ktorý dostal záchrannú liečbu
laserom počas 12 mesiacov
| 61,4
| 34,4
|
|
|
ap<0,0001
O
brázok 3 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(B
RAVO
)
+ 18,3
20
18
+ 18,3
16
14
12 + 12,1
10
8
6
4
2
kontrolované+ 7,3kontrolná skupina simulovaného
s
im
u
l
ovaným podaním
0
podania preradená na ranibizumab
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=132) Ranibizumab 0,5 mg (n=131)
BL=východisková hodnota; SE=stredná chyba priemeru
Tabuľka 5 Výsledky po 6 a 12 mesiacoch (CRUISE)
| Simulované podanie/ Lucentis 0,5 mg (n=130)
| Lucentis 0,5 mg
(n=130)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 0,8 (16,2)
| 14,9 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 7,3 (15,9)
| 13,9 (14,2)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 16,9
| 47,7
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 33,1
| 50,8
|
|
|
ap<0,0001
O
brázok 4 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(
CRU
ISE)
20
kontrolované18
simulovaným podaním16
+ 14,9kontrolná skupina simulovaného podania preradená na ranibizumab
14
+ 13,912
10
8
+ 7,36
4
+ 0,82
0
-2
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=130) Ranibizumab 0,5 mg (n=130)
BL=východisková hodnota; SE=stredná chyba priemeru
V oboch štúdiách zlepšenie zraku sprevádzalo kontinuálne a významné zmenšovanie makulárneho
edému, merané ako hrúbka centrálnej retiny.
Pacienti s BRVO (BRAVO a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, dosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~15 písmen) ako pacienti, ktorí boli liečení ranibizumabom od začiatku (~16 písmen). Avšak počet pacientov, ktorí ukončili 2 roky liečby, bol obmedzený,
a v štúdii HORIZON boli plánované len štvrťročné kontrolné návštevy. Preto v súčasnosti nie je dosť
údajov, z ktorých by sa dali vyvodiť odporúčania, kedy sa má začať liečba ranibizumabom u pacientov s BRVO.
Pacienti s CRVO (CRUISE a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, nedosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~6 písmen) oproti pacientom, ktorí boli liečení ranibizumabom od začiatku (~12 písmen).
Zlepšenie zrakovej ostrosti pozorované pri liečbe ranibizumabom po 6 a 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby stanovený prostredníctvom podškál National Eye Institute Visual Function Questionnaire (NEI VFQ-25) vzhľadom na „aktivity vyžadujúce videnie do blízka“ a
„aktivity vyžadujúce videnie do diaľky“. Rozdiel medzi Lucentisom 0,5 mg a kontrolnou skupinou sa vyhodnotil po 6. mesiaci s hodnotami p 0,02 až 0,0002.
Liečba
poškodenia
zraku
v
dôsledku CNV pri PM
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku CNV pri PM sa stanovili na základe údajov z 12 mesiacov randomizovaného, dvojito maskovaného, účinnou liečbou kontrolovaného pivotného klinického skúšania F2301 (RADIANCE). Toto skúšanie bolo určené na vyhodnotenie dvoch rôznych režimov dávkovania 0,5 mg ranibizumabu podávaného ako intravitreálna injekcia v porovnaní s PDT verteporfínom (vPDT, fotodynamická liečba Visudynom). Do jednej
z nasledujúcich skupín bolo randomizovaných 277 pacientov:
· Skupina I (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „stability“, definovanými ako žiadna zmena BCVA v porovnaní s dvomi predchádzajúcimi mesačnými hodnoteniami).
· Skupina II (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „aktivity choroby“, definovanými ako zhoršenie zraku, ktoré možno pripísať intra- alebo subretinálnej tekutine alebo aktívnemu presakovaniu v dôsledku CNV lézie, stanovené prostredníctvom OCT a/alebo
FA).
· Skupina III (vPDT – pacienti mali od 3. mesiaca povolené dostať liečbu ranibizumabom). Počas 12 mesiacov klinického skúšania pacienti dostali v priemere 4,6 injekcií (rozmedzie 1-11) v skupine I a 3,5 injekcií (rozmedzie 1-12) v skupine II. V skupine II, ktorá zodpovedá odporúčanému
dávkovaniu (pozri časť 4.2), 50,9 % pacientov potrebovalo 1 alebo 2 injekcie, 34,5 % potrebovalo 3 až
5 injekcií a 14,7 % potrebovalo 6 až 12 injekcií počas 12 mesiacov trvania skúšania. 62,9 % pacientov v skupine II nepotrebovalo injekcie v druhom 6-mesačnom období skúšania.
Najdôležitejšie výsledky RADIANCE sú zhrnuté v Tabuľke 6 a na Obrázku 5.
Tabuľka 6 Výsledky po 3. a 12. mesiaci (RADIANCE)
| Skupina I
ranibizumab
0,5 mg
„stabilita zraku“
(n=105)
| Skupina II
ranibizumab
0,5 mg
„aktivita choroby“
(n=116)
| Skupina III
vPDTb
(n=55)
| 3. mesiac
|
|
|
| Priemerná zmena BCVA od 1. do 3. mesiaca v porovnaní s východiskovou hodnotoua (písmená)
| +10,5
| +10,6
| +2,2
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
61,9 %
38,1 %
|
65,5 %
43,1 %
|
27,3 %
14,5 %
| 12. mesiac
|
|
|
| Počet injekcií do 12. mesiaca:
Priemer
Medián
|
4,6
4
|
3,5
2,0
|
N/A N/A
| Priemerná zmena BCVA od 1. do
12. mesiaca v porovnaní s východiskovou hodnotou (písmená)
| +12,8
| +12,5
| N/A
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
69,5 %
53,3 %
|
69,0 %
51,7 %
|
N/A N/A
|
|
|
a p<0,00001 v porovnaní s kontrolou vPDT
b Porovnávacia kontrola do 3. mesiaca. Pacienti randomizovaní do skupiny vPDT mali od 3. mesiaca
povolené dostať liečbu ranibizumabom (v skupine III dostalo ranibizumab od 3. mesiaca 38 pacientov)
O
brázok 5 Priemerná zmena oproti východiskovej hodnote BCVA do 12. mesiaca
(
RAD
IANCE)
20
15 +12,5
+14,4
10 +12,1
+13,8
+9,3
5
+1,4
0
Povolený ranibizumab-5
|
|
| Mesiac
|
| Skupina I - ranibizumab 0,5 mg
|
|
|
| podľa stabilizácie (N=105)
|
|
|
| Skupina III - PDT verteporfínom
|
|
|
| (N=55)
|
|
|
|
|
0 1 2 3 4 5 6 7 8 9 10 11 12
Zlepšenie zraku sprevádzal pokles hrúbky centrálnej retiny.
Skupina II - ranibizumab 0,5 mg
podľa aktivity choroby (N=116)
Skupina III - ranibizumab 0,5 mg/PDT
verteporfínom od 3. mesiaca (N=55)
Prínos liečby hlásený pacientmi sa pozoroval vo väčšej miere v skupinách ranibizumabu v porovnaní
s vPDT (hodnota p<0,05) z hľadiska zlepšenia kombinovaného skóre a niekoľkých podškál NEI VFQ-
25 (celkové videnie, aktivity vyžadujúce videnie do blízka, duševné zdravie a odkázanosť na iných).
Deti a dospievajúciBezpečnosť a účinnosť ranibizumabu sa zatiaľ neskúmali u detí a dospievajúcich.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Lucentis vo všetkých vekových podskupinách detí a dospievajúcich pri neovaskulárnej VPDM, poškodení zraku v dôsledku DME, poškodení zraku v dôsledku makulárneho edému po RVO a poškodení zraku
v dôsledku CNV pri PM (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po intravitreálnom podaní Lucentisu raz mesačne pacientom s neovaskulárnou VPDM boli sérové koncentrácie ranibizumabu vo všeobecnosti nízke, s maximálnymi hladinami (Cmax) spravidla pod koncentráciou ranibizumabu potrebnou na inhibovanie biologickej aktivity VEGF o 50 %
(11-27 ng/ml, ako sa stanovilo in vitro v teste bunkovej proliferácie). Cmax bola úmerná dávke
v rozmedzí dávok 0,05 až 1,0 mg/oko. Koncentrácie v sére obmedzeného počtu pacientov s DME
naznačujú, že nemožno vylúčiť o niečo vyššiu systémovú expozíciu v porovnaní s expozíciou, aká sa pozorovala u pacientov s neovaskulárnou VPDM. Koncentrácie ranibizumabu v sére pacientov s RVO
boli podobné alebo mierne vyššie v porovnaní s koncentráciami, ktoré sa pozorovali u pacientov
s neovaskulárnou VPDM.
Na základe analýzy populačnej farmakokinetiky a vymiznutia ranibizumabu zo séra pacientov s neovaskulárnou VPDM liečených dávkou 0,5 mg, priemerný vitreálny eliminačný polčas ranibizumabu je približne 9 dní. Pri intravitreálnom podávaní Lucentisu 0,5 mg/oko raz mesačne sa predpokladá, že Cmax ranibizumabu v sére, ktorá sa dosiahne približne 1 deň po podaní, bude vo všeobecnosti v rozmedzí medzi 0,79 a 2,90 ng/ml, a Cmin sa vo všeobecnosti predpokladá v rozmedzí medzi 0,07 a 0,49 ng/ml. Predpokladaná sérová koncentrácia ranibizumabu je približne 90 000- násobne nižšia ako vitreálna koncentrácia ranibizumabu.
Pacienti s poškodením funkcie obličiek: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky Lucentisu u pacientov s poškodením funkcie obličiek. V populačnej farmakokinetickej analýze pacientov s neovaskulárnou VPDM malo 68 % (136 z 200) pacientov poškodenie funkcie obličiek (46,5 % ľahké [50-80 ml/min], 20 % stredne ťažké [30-50 ml/min] a 1,5 % ťažké [<30 ml/min]).
U pacientov s RVO malo 48,2 % (253 z 525) poškodenie funkcie obličiek (36,4 % ľahké, 9,5 %
stredne ťažké a 2,3 % ťažké). Systémový klírens bol trochu nižší, čo však nebolo klinicky významné.
Pacienti s poškodením funkcie pečene: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky
Lucentisu u pacientov s poškodením funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Bilaterálne intravitreálne podávanie ranibizumabu opiciam rodu Cynomolgus v dávkach medzi
0,25 mg/oko a 2,0 mg/oko raz za 2 týždne až do 26 týždňov malo za následok účinky na oči závislé od
dávky.
Intraokulárne sa zaznamenalo od dávky závislé zosilnenie zápalu a zvýšenie počtu buniek v prednej očnej komore s maximom 2 dni po podaní injekcie. Závažnosť zápalovej odpovede sa spravidla znížila pri podaní ďalších injekcií alebo počas zotavenia. V zadnom segmente sa pozorovala vitreálna infiltrácia buniek a zákaly sklovca, ktoré tiež mali tendenciu závisieť od dávky a spravidla pretrvávali do konca liečebného obdobia. V štúdii trvajúcej 26 týždňov sa intenzita zápalu sklovca zvyšovala
s počtom injekcií. Po zotavení sa však pozorovali dôkazy reverzibility. Povaha a načasovanie zápalu zadného segmentu poukazuje na imunitne sprostredkovanú odpoveď protilátok, čo môže byť klinicky
nevýznamné. Pri niektorých zvieratách sa pozoroval vznik katarakty po relatívne dlhom období
intenzívneho zápalu, čo naznačuje, že zmeny na šošovke sú sekundárne po ťažkom zápale. Prechodné zvýšenie vnútroočného tlaku po podaní sa pozorovalo po intravitreálnych injekciách bez ohľadu na
dávku.
Mikroskopické očné zmeny súviseli so zápalom a nepoukazovali na degeneratívne procesy. V niektorých očiach sa zaznamenali granulomatózne zápalové zmeny na papile. Tieto zmeny v zadnom segmente ustupovali a v niektorých prípadoch vymizli počas zotavovania.
Po intravitreálnom podaní sa nezistili žiadne známky systémovej toxicity. V podsúbore liečených zvierat sa našli sérové a sklovcové protilátky voči ranibizumabu.
Nie sú dostupné údaje o karcinogenite alebo mutagenite.
U gravidných opíc nespôsobilo intravitreálne podávanie ranibizumabu, ktoré malo za následok maximálne systémové expozície 0,9- až 7-násobne vyššie ako najhorší prípad klinickej expozície, vývojovú toxicitu alebo teratogenitu a nemalo žiadny vplyv na hmotnosť alebo štruktúru placenty,
hoci ranibizumab sa vzhľadom na jeho mechanizmus účinku má považovať za potenciálne teratogénny
a embryo- a fetotoxický.
Neprítomnosť účinkov na vývoj embrya a plodu sprostredkovaných ranibizumabom pravdepodobne súvisí hlavne s neschopnosťou fragmentu Fab prestupovať cez placentu. Napriek tomu bol popísaný prípad vysokých hladín ranibizumabu v sére matky a prítomnosti ranibizumabu v sére plodu, čo naznačuje, že protilátka proti ranibizumabu fungovala ako transportná bielkovina (obsahujúca segment Fc) pre ranibizumab, čím sa znižoval klírens zo séra matky a umožňoval sa prestup cez placentu. Keďže sledovania vývoja embryí a plodov sa robili u zdravých gravidných zvierat a ochorenie (napr. diabetes) môže meniť priepustnosť placenty pre fragment Fab, štúdia sa má interpretovať s opatrnosťou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát α,α-trehalózy Monohydrát histidíniumchloridu Histidín
Polysorbát 20
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorená injekčná liekovka uchovávať pri izbovej teplote (25°C) najviac
24 hodín.
6.5 Druh obalu a obsah balenia
0,23 ml sterilného roztoku v injekčnej liekovke (sklo typu I) so zátkou (chlórobutylová guma). Balenie obsahuje 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka je určená len na jednorazové použitie. Po podaní injekcie sa všetok nespotrebovaný liek musí zlikvidovať. Liekovka, ktorá vykazuje známky poškodenia alebo nesprávnej manipulácie, sa nesmie použiť. Sterilitu nemožno zaručiť, ak uzáver obalu nie je neporušený.
Na prípravu a podanie intravitreálnej injekcie sú potrebné nasledujúce pomôcky na jednorazové
použitie:
- ihla s 5 µm filtrom (18G)
- 1 ml sterilná injekčná striekačka
- injekčná ihla (30G x ½″).
Tieto pomôcky nie sú súčasťou balenia Lucentisu.
Pri príprave Lucentisu na intravitreálne podanie dodržiavajte, prosím, nasledujúce pokyny:
1. Pred odobratím obsahu sa má dezinfikovať vonkajšia časť gumenej zátky injekčnej liekovky.
2. Asepticky nasaďte ihlu s 5 µm filtrom (18G) na 1 ml injekčnú striekačku. Vtlačte ihlu s filtrom
do stredu zátky liekovky, až sa ihla dotkne dna injekčnej liekovky.
3. Odoberte všetku tekutinu z injekčnej liekovky, pri čom liekovka má byť vo zvislej polohe,
mierne naklonená, aby sa uľahčilo úplné odobratie obsahu.
4. Dbajte na to, aby ste pri vyprázdnení injekčnej liekovky dostatočne potiahli piest, aby sa ihla
s filtrom úplne vyprázdnila.
5. Ihlu s filtrom nechajte v injekčnej liekovke a odpojte injekčnú striekačku od ihly s filtrom. Ihla s filtrom sa má po odobratí obsahu injekčnej liekovky zahodiť a nemá sa použiť na intravitreálnu injekciu.
6. Asepticky a pevne nasaďte injekčnú ihlu (30G x ½″) na injekčnú striekačku.
7. Opatrne odstráňte kryt z injekčnej ihly bez toho, aby ste odpojili injekčnú ihlu od injekčnej striekačky.
Poznámka: Pri odstránení krytu pridržte násadec injekčnej ihly.
8. Opatrne vytlačte vzduch z injekčnej striekačky a upravte dávku na rysku označujúcu 0,05 ml na
striekačke. Injekčná striekačka je pripravená na podanie injekcie.
Poznámka: Injekčnú ihlu neutierajte. Nepotiahnite piest injekčnej striekačky.
Po podaní injekcie nenasaďte kryt späť na injekčnú ihlu, ani ihlu neodpojte od injekčnej striekačky. Zahoďte použitú injekčnú striekačku spolu s ihlou do odpadovej nádoby na ostré predmety, alebo zlikvidujte v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/374/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 22. január 2007
Dátum posledného predĺženia: 24. január 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu1. NÁZOV LIEKULucentis 10 mg/ml injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEJeden ml obsahuje 10 mg ranibizumabu*. Jedna naplnená injekčná striekačka obsahuje 0,165 ml, čo zodpovedá 1,65 mg ranibizumabu. Objem, ktorý možno získať z jednej naplnenej injekčnej striekačky, je 0,1 ml. Poskytuje využiteľné množstvo na podanie jednorazovej dávky 0,05 ml, ktoré obsahujú
0,5 mg ranibizumabu.
*Ranibizumab je fragment humanizovanej monoklonálnej protilátky vytvorenej v bunkách
Escherichia coli rekombinantnou DNA technológiou.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok.
Číry, bezfarebný až svetložltý vodný roztok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLucentis je indikovaný u dospelých na:
· Liečbu neovaskulárnej (vlhkej) vekom podmienenej degenerácie makuly (VPDM)
· Liečbu poškodenia zraku v dôsledku diabetického makulárneho edému (DME)
· Liečbu poškodenia zraku v dôsledku makulárneho edému po oklúzii žily sietnice (vetvovej RVO
alebo kmeňovej RVO)
· Liečbu poškodenia zraku v dôsledku neovaskularizácie chorioidey (CNV) pri patologickej myopii (PM)
4.2 Dávkovanie a spôsob podávaniaLucentis smie podávať kvalifikovaný oftalmológ so skúsenosťami s podávaním intravitreálnych
injekcií.
Odporúčaná dávka Lucentisu je 0,5 mg, podávaná ako jednorazová intravitreálna injekcia. To zodpovedá objemu injekcie 0,05 ml. Interval medzi dvoma dávkami podanými do toho istého oka má byť najmenej štyri týždne.
Liečba sa začína jednou injekciou za mesiac až do dosiahnutia maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity ochorenia, t.j. žiadna zmena zrakovej ostrosti a iných prejavov a príznakov choroby pri pokračujúcej liečbe. U pacientov s vlhkou VPDM, DME a RVO môže byť na začiatku potrebné podať po sebe tri alebo viac mesačných injekcií.
Následné sledovanie a intervaly medzi podaniami má určovať lekár a majú byť založené na aktivite
choroby, stanovenej prostredníctvom zrakovej ostrosti a/alebo anatomických parametrov.
Ak zrakové a anatomické parametre podľa názoru lekára ukazujú, že pokračujúca liečba pre pacienta nie je prínosom, podávanie Lucentisu sa má ukončiť.
Sledovanie aktivity choroby môže zahŕňať klinické vyšetrenie, testovanie funkcie alebo zobrazovacie
techniky (napr. optickú koherentnú tomografiu alebo fluoresceínovú angiografiu).
Ak sa pacient lieči v režime podávania a predlžovania intervalov medzi podaniami, po dosiahnutí maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity choroby sa intervaly medzi podaniami môžu postupne predlžovať až do opätovného objavenia sa prejavov aktivity choroby alebo zhoršenia zraku. Interval medzi podaniami sa nemá naraz predĺžiť o viac ako dva týždne pri vlhkej VPDM a môže sa naraz predĺžiť až o jeden mesiac pri DME. Pri RVO sa intervaly medzi podaniami tiež môžu postupne predlžovať, avšak nie je dostatok údajov na určenie dĺžky týchto intervalov. Ak sa opäť objaví aktivita choroby, interval medzi podaniami sa má patrične skrátiť.
Pri liečbe poškodenia zraku v dôsledku CNV pri PM u mnohých pacientov môžu byť potrebné len jedna alebo dve injekcie počas prvého roka, zatiaľ čo niektorí pacienti môžu potrebovať častejšiu liečbu (pozri časť 5.1).
Lucentis a laserová fotokoagulácia pri DME a pri makulárnom edéme po BRVOS podávaním Lucentisu súčasne s laserovou fotokoaguláciou sú určité skúsenosti (pozri časť 5.1). Pri
podávaní v ten istý deň sa Lucentis má podať minimálne 30 minút po laserovej fotokoagulácii.
Lucentis sa môže podávať pacientom, ktorí boli v minulosti liečení laserovou fotokoaguláciou.
Lucentis a fotodynamická liečba Visudynom pri CNV v dôsledku PMNie sú skúsenosti so súbežným podávaním Lucentisu a Visudynu.
Osobitné skupiny pacientovPoškodenie funkcie pečeneLucentis sa neskúmal u pacientov s poškodením funkcie pečene. U tejto populácie však nie sú potrebné zvláštne opatrenia.
Poškodenie funkcie obličiekÚprava dávky nie je potrebná u pacientov s poškodením funkcie obličiek (pozri časť 5.2).
Starší pacientiÚprava dávkovania u starších ľudí nie je potrebná. Skúsenosti s pacientmi s DME staršími ako
75 rokov sú obmedzené.
Deti a dospievajúciBezpečnosť a účinnosť Lucentisu u detí a dospievajúcich vo veku menej ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje.
Spôsob podaniaNaplnená injekčná striekačka na jednorazové použitie len na intravitreálne podanie. Naplnená injekčná striekačka obsahuje viac lieku, ako je odporúčaná dávka 0,5 mg. Objem, ktorý možno získať z naplnenej injekčnej striekačky (0,1 ml), sa nemá celý použiť. Nadbytočné množstvo sa má vytlačiť zo striekačky pre podaním injekcie. Podanie celého objemu naplnenej injekčnej striekačky môže mať za následok predávkovanie. Vzduchovú bublinu spolu s nadbytočným liekom vytlačte pomalým posúvaním piesta, až sa hrana pod vypuklou gumenou zátkou kryje s čiernou čiarkou vyznačujúcou dávku na injekčnej striekačke (zodpovedá 0,05 ml, t.j. 0,5 mg ranibizumabu).
Lucentis sa má pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu sfarbenia.
Podanie injekcie sa má uskutočniť za aseptických podmienok, ktoré zahŕňajú chirurgickú dezinfekciu rúk, použitie sterilných rukavíc, sterilného rúška a sterilného rozvierača mihalníc (alebo náhrady)
a dostupnosť sterilnej paracentézy (pre prípad potreby). Pred vykonaním intravitreálneho zákroku sa
má dôsledne preskúmať pacientova anamnéza so zreteľom na reakcie z precitlivenosti (pozri časť 4.4).
Pred podaním injekcie sa má v súlade s lokálnou klinickou praxou podať náležitá anestézia
a širokospektrálny lokálny mikrobicídny prostriedok na dezinfekciu kože v okolí oka, mihalnice a povrchu oka.
Informácie o príprave Lucentisu, pozri časť 6.6.
Injekčná ihla sa zavádza 3,5-4,0 mm za limbom do dutiny sklovca, vyhýba sa horizontálnemu poludníku a smeruje do centra očnej gule. Potom sa podá objem injekcie 0,05 ml; pri následných injekciách sa má použiť iné miesto na sklére. Každá naplnená injekčná striekačka sa má použiť len na podanie do jedného oka.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s aktívnymi alebo suspektnými očnými alebo periokulárnymi infekciami. Pacienti s aktívnym ťažkým vnútroočným zápalom.
4.4 Osobitné upozornenia a opatrenia pri používaníReakcie súvisiace s intravitreálnou injekciouPodanie intravitreálnych injekcií vrátane injekcií Lucentisu sa spájalo s endoftalmitídou, vnútroočným zápalom, rhegmatogénnym odlúčením sietnice, trhlinou v sietnici a iatrogénnou traumatickou
kataraktou (pozri časť 4.8). Pri podaní Lucentisu sa musia vždy dodržať náležité aseptické injekčné postupy. Okrem toho je potrebné pacientov sledovať počas týždňa po podaní injekcie, čo umožní
včasnú liečbu v prípade infekcie. Pacienti majú byť poučení, aby bezodkladne oznámili akýkoľvek
príznak, ktorý poukazuje na endoftalmitídu alebo na niektorú z vyššie uvedených príhod.
ZvýšeniavnútroočnéhotlakuV priebehu 60 minút po podaní injekcie Lucentisu sa pozorovalo prechodné zvýšenie vnútroočného tlaku (IOP). Zistilo sa aj trvalé zvýšenie IOP (pozri časť 4.8). Vnútroočný tlak aj perfúzia hlavy
zrakového nervu sa musia monitorovať a náležite liečiť.
BilaterálnaliečbaObmedzené údaje o bilaterálnom použití Lucentisu (vrátane podania v ten istý deň) nenaznačujú zvýšené riziko systémových nežiaducich príhod v porovnaní s unilaterálnou liečbou.
ImunogenitaLucentis môže byť imunogénny. Keďže je možnosť zvýšenej systémovej expozície u osôb s DME,
nemožno vylúčiť zvýšené riziko vzniku precitlivenosti u tejto populácie pacientov. Pacienti majú byť tiež poučení, aby hlásili akékoľvek zvýšenie závažnosti vnútroočného zápalu, ktoré môže byť
klinickým príznakom zodpovedajúcim tvorbe protilátok vo vnútri oka.
Súčasnépoužitiesinýmianti-VEGF (vaskulárny endoteliálny rastový faktor)Lucentis sa nemá podávať súčasne s inými anti-VEGF liekmi (systémovými alebo okulárnymi).
Prerušenie
l
i
ečby
Lucentisom
Dávka sa nemá podať a v liečbe sa nemá pokračovať skôr ako počas najbližšej plánovanej návštevy
v prípade:
· poklesu najlepšie korigovanej zrakovej ostrosti (BCVA) o ≥30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
· intraokulárneho tlaku ≥30 mmHg;
· trhliny v sietnici;
· subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je ≥50 %
celkovej plochy lézie;
· uskutočneného alebo plánovaného intraokulárneho chirurgického zákroku počas uplynulých
alebo nasledujúcich 28 dní.
Trhlina v pigmentovom epiteli sietniceRizikové faktory, ktoré sa spájajú so vznikom trhliny v pigmentovom epiteli sietnice po anti-VEGF liečbe pri vlhkej VPDM, zahŕňajú veľké a/alebo vysoko uložené odlúčenie pigmentového epitelu sietnice. Pri začatí liečby Lucentisom je potrebná opatrnosť u pacientov s týmito rizikovými faktormi pre trhliny v pigmentovom epiteli sietnice.
Regmatogénneodlúčeniesietnicealebomakulárne dieryLiečba sa má ukončiť u osôb s regmatogénnym odlúčením sietnice alebo makulárnymi dierami 3.
alebo 4. stupňa.
Skupiny pacientov, u ktorých sú skúsenosti obmedzenéS liečbou osôb s DME spôsobeným diabetom typu I sú len obmedzené skúsenosti. Lucentis sa neskúmal u pacientov, ktorí v minulosti dostali intravitreálne injekcie, u pacientov s aktívnymi systémovými infekciami, proliferujúcou diabetickou retinopatiou, alebo u pacientov so sprievodnými očnými ochoreniami, napr. s odlúčením sietnice alebo makulárnou dierou. Nie sú tiež žiadne
skúsenosti s liečbou Lucentisom u pacientov s diabetom s HbA1c vyšším ako 12 % a nekontrolovanou
hypertenziou. Pri liečbe takýchto pacientov má lekár vziať do úvahy tento nedostatok informácií.
Údaje o účinku Lucentisu sú obmedzené u pacientov s PM, ktorí v minulosti podstúpili neúspešnú fotodynamickú liečbu verteporfínom (vPDT). Zatiaľ čo sa pozoroval zhodný účinok u osôb so subfoveálnymi a juxtafoveálnymi léziami, nie je tiež dostatok údajov, z ktorých by bolo možné usudzovať na účinok Lucentisu u osôb s PM, ktoré majú extrafoveálne lézie.
SystémovéúčinkypointravitreálnompoužitíPo intravitreálnej injekcii inhibítorov VEGF sa zaznamenali systémové nežiaduce udalosti vrátane
krvácaní mimo oka a artériových tromboembolických príhod.
Údaje o bezpečnosti liečby u pacientov s DME, makulárnym edémom po RVO a s CNV v dôsledku PM, ktorí majú v anamnéze cievnu mozgovú príhodu alebo tranzitórne ischemické ataky, sú obmedzené. Pri liečbe takýchto pacientov je potrebné postupovať opatrne (pozri časť 4.8).
Epizódy RVO, ischemickej vetvovej RVO a kmeňovejRVOvminulostiSkúsenosti s liečbou pacientov s epizódami RVO v minulosti a pacientov s ischemickou vetvovou
RVO (BRVO) a kmeňovou RVO (CRVO) sú obmedzené. U pacientov s RVO, u ktorých sa objavia
klinické príznaky ireverzibilnej ischemickej straty zrakovej funkcie, sa liečba neodporúča.
4.5 Liekové a iné interakcieNeuskutočnili sa žiadne formálne interakčné štúdie.
Adjuvantné použitie fotodynamickej liečby (PDT) verteporfínom a Lucentisu pri vlhkej VPDM a PM,
pozri časť 5.1.
Adjuvantné použitie laserovej fotokoagulácie a Lucentisu pri DME a BRVO, pozri časti 4.2 a 5.1.
V klinických skúšaniach liečby poškodenia zraku v dôsledku DME súbežná liečba tiazolidíndiónmi neovplyvnila výsledky týkajúce sa zrakovej ostrosti alebo hrúbky čiastkového poľa centrálnej sietnice (CSTF) u pacientov liečených Lucentisom.
4.6 Fertilita, gravidita a laktáciaŽenyvofertilnomveku/antikoncepciaužienŽeny vo fertilnom veku majú používať účinnú antikoncepciu počas liečby.
GraviditaNie sú dostupné klinické údaje o použití ranibizumabu v gravidite. Štúdie na makakoch krabožravých nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity alebo vývinu embrya/plodu (pozri časť 5.3). Systémová expozícia ranibizumabu po podaní do oka je nízka, ale vzhľadom na jeho mechanizmus účinku sa ranibizumab musí považovať za potenciálne teratogénny a embryo-
/fetotoxický. Preto sa ranibizumab nemá používať v gravidite, pokiaľ očakávaný prínos nie je väčší ako možné riziko pre plod. Ženám, ktoré chcú otehotnieť a boli liečené ranibizumabom, sa odporúča počkať s počatím dieťaťa aspoň 3 mesiace od poslednej dávky ranibizumabu.
LaktáciaNie je známe, či sa Lucentis vylučuje do ľudského mlieka. Dojčenie sa neodporúča počas používania
Lucentisu.
FertilitaNie sú dostupné žiadne údaje o fertilite.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeLiečba Lucentisom môže vyvolať dočasné poruchy videnia, čo môže ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8). Pacienti, u ktorých sa vyskytnú tieto príznaky, nesmú viesť vozidlá alebo obsluhovať stroje, kým tieto dočasné poruchy zraku neustúpia.
4.8 Nežiaduce účinkyZhrnutieprofilubezpečnostiVäčšina nežiaducich reakcií hlásených po podaní Lucentisu súvisí s postupom intravitreálnej injekcie.
Najčastejšie hlásené nežiaduce reakcie týkajúce sa očí po podaní injekcie Lucentisu sú: bolesť oka, hyperémia oka, zvýšený vnútroočný tlak, vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko a svrbenie oka.
Najčastejšie hlásené nežiaduce reakcie netýkajúce sa očí sú bolesť hlavy, nazofaryngitída a bolesť kĺbov.
Medzi menej často hlásené, ale závažnejšie nežiaduce reakcie patria endoftalmitída, slepota, odlúčenie sietnice, trhlina v sietnici a iatrogénna traumatická katarakta (pozri časť 4.4).
Pacientov je potrebné informovať o prejavoch týchto potenciálnych nežiaducich reakcií a poučiť ich, aby informovali svojho lekára, ak u nich vzniknú príznaky ako bolesť očí alebo zvýšenie nepríjemných pocitov, zhoršujúce sa sčervenanie očí, neostré alebo zhoršené videnie, zvýšený počet malých čiastočiek v zornom poli, alebo zvýšená citlivosť na svetlo.
Nežiaduce reakcie, ktoré sa vyskytli po podaní Lucentisu v klinických skúšaniach, sú zhrnuté
v tabuľke nižšie.
Tabuľkovýzoznamnežiaducichreakcií#Nežiaduce reakcie sú zatriedené podľa orgánových systémov a frekvencie podľa nasledujúcich konvencií: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé
(≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov). V rámci
jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
Veľmi časté Nazofaryngitída
Časté Infekcia močových ciest*
Poruchy krvi a lymfatického systému
Časté Anémia
Poruchy imunitného systému
Časté Precitlivenosť
Psychické poruchy
Časté Úzkosť
Poruchy nervového systému
Veľmi časté Bolesť hlavy
Poruchy oka
Veľmi časté Vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, bolesť oka, opacity v sklovci, krvácanie do spojovky, podráždenie
oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída,
suché oko, hyperémia oka, svrbenie oka.
Časté Degenerácia sietnice, porucha sietnice, odlúčenie sietnice, trhlina v sietnici, odlúčenie pigmentového epitelu sietnice, trhlina v pigmentovom epiteli sietnice, znížená zraková ostrosť, krvácanie do sklovca, porucha sklovca, uveitída, iritída, iridocyklitída, katarakta, subkapsulárna katarakta, opacifikácie zadného puzdra šošovky, bodkovitá keratitída, abrázia rohovky, zápal prednej očnej komory, neostré videnie, krvácanie v mieste podania injekcie, krvácanie do oka, konjunktivitída, alergická konjunktivitída, výtok z oka, fotopsia, fotofóbia, nepríjemné pocity v oku, edém mihalnice, bolesť mihalnice, hyperémia spojoviek.
Menej časté Slepota, endoftalmitída, hypopyon, hyféma, keratopatia, adhézia dúhovky, depozity v rohovke, edém rohovky, strie rohovky,
bolesť v mieste podania injekcie, podráždenie v mieste podania
injekcie, abnormálne pocity v oku, podráždenie mihalnice.
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Kašeľ
Poruchy gastrointestinálneho traktu
Časté Nauzea
Poruchy kože a podkožného tkaniva
Časté Alergické reakcie (exantém, urtikária, pruritus, erytém)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Veľmi časté Artralgia
Laboratórne a funkčné vyšetrenia
Veľmi časté Zvýšenie vnútroočného tlaku
# Nežiaduce reakcie boli definované ako nežiaduce udalosti (u najmenej 0,5 percentuálneho bodu
pacientov), ktoré sa vyskytli častejšie (najmenej 2 percentuálne body) u pacientov liečených Lucentisom 0,5 mg, ako u pacientov, ktorí dostávali kontrolnú liečbu (simulované podanie alebo PDT verteporfínom).
* pozorované iba u populácie s DME
NežiaducereakciesúvisiacesoskupinouliekovV klinických skúšaniach fázy III pri vlhkej VPDM bola celková frekvencia krvácaní mimo oka, čo je nežiaduca udalosť potenciálne súvisiaca so systémovou inhibíciou VEGF (vaskulárneho
endoteliálneho rastového faktora), mierne zvýšená u pacientov liečených ranibizumabom.
Charakteristika rôznych krvácaní však nebola zhodná. Po intravitreálnom použití inhibítorov VEGF
existuje teoretické riziko arteriálnych tromboembolických príhod vrátane cievnej mozgovej príhody
a infarktu myokardu. V klinických skúšaniach Lucentisu sa pozorovala nízka incidencia arteriálnych tromboembolických príhod u pacientov s VPDM, DME, RVO a PM a neboli významné rozdiely
medzi skupinami liečenými ranibizumabom v porovnaní s kontrolnou liečbou.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieBoli hlásené prípady náhodného predávkovania v klinických skúšaniach pri vlhkej VPDM a z údajov po uvedení lieku na trh. Nežiaduce reakcie súvisiace s týmito hlásenými prípadmi boli zvýšenie vnútroočného tlaku, prechodná slepota, znížená zraková ostrosť, edém rohovky, bolesť rohovky
a bolesť oka. Ak dôjde k predávkovaniu, je potrebné sledovať a liečiť vnútroočný tlak, ak to ošetrujúci lekár považuje za potrebné.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: oftalmologiká, látky s antineovaskularizačným účinkom, ATC kód:
S01LA04
Ranibizumab je fragment humanizovanej rekombinantnej monoklonálnej protilátky, ktorého cieľom je ľudský vaskulárny endoteliálny rastový faktor A (VEGF-A). Viaže sa s vysokou afinitou na izoformy VEGF-A (napr. VEGF110, VEGF121 a VEGF165), a tým zabraňuje vzniku väzby VEGF-A na jeho receptory VEGFR-1 a VEGFR-2. Naviazanie VEGF-A na jeho receptory má za následok proliferáciu endotelových buniek a neovaskularizáciu, ako aj únik tekutín z ciev, čo všetko sa považuje za faktory prispievajúce k progresii neovaskulárnej formy vekom podmienenej degenerácie makuly, patologickej myopie alebo poškodeniu zraku spôsobeného buď diabetickým makulárnym edémom, alebo makulárnym edémom po RVO.
Liečbavlhkej VPDMPri vlhkej VPDM sa klinická bezpečnosť a účinnosť Lucentisu hodnotila v troch randomizovaných,
dvojito maskovaných, simulovane alebo aktívne kontrolovaných klinických skúšaniach trvajúcich
24 mesiacov u pacientov s neovaskulárnou VPDM. Do týchto klinických skúšaní bolo zaradených
celkovo 1 323 pacientov (879 v aktívnej a 444 v kontrolnej skupine).
V klinickom skúšaní FVF2598g (MARINA) 716 pacientov s minimálne klasickou alebo okultnou bez klasickej chorioidálnej neovaskularizácie (CNV) dostávalo každý mesiac intravitreálne injekcie Lucentisu 0,3 mg (n=238) alebo 0,5 mg (n=240) alebo simulované injekcie (n=238).
V klinickom skúšaní FVF2587g (ANCHOR) 423 pacientov s prevažne klasickými CNV léziami dostávalo buď: 1) každý mesiac intravitreálne injekcie Lucentis 0,3 mg a simulovanú PDT (n=140);
2) každý mesiac intravitreálne injekcie Lucentisu 0,5 mg a simulovanú PDT (n=140); alebo
3) simulované intravitreálne injekcie a aktívnu PDT verteporfínom (n=143). Simulovaná alebo aktívna
PDT verteporfínom sa podala s úvodnou injekciou Lucentisu a potom každé 3 mesiace, ak fluoresceínová angiografia ukázala pretrvávanie alebo recidívu úniku tekutín z ciev.
Kľúčové merané parametre sú zhrnuté v Tabuľke 1 a na Obrázku 1.
Tabuľka 1 Výsledky po 12 mesiacoch a 24 mesiacoch v klinickom skúšaní FVF2598g(MARINA) a FVF2587g (ANCHOR)
|
| FVF2598g (MARINA)
| FVF2587g (ANCHOR)
|
| Meraný
parameter
| Mesiac
| Simulované
podanie
(n=238)
| Lucentis
0,5 mg
(n=240)
| PDT
verteporfínom
(n=143)
| Lucentis
0,5 mg
(n=140)
| Strata <15 písmen
zrakovej ostrosti (%)a (zachovanie vízu, primárny ukazovateľ)
| 12. mesiac
| 62 %
| 95 %
| 64 %
| 96 %
| 24. mesiac
| 53 %
| 90 %
| 66 %
| 90 %
| Zisk ≥15 písmen
zrakovej ostrosti
(%)a
| 12. mesiac
| 5 %
| 34 %
| 6 %
| 40 %
| 24. mesiac
| 4 %
| 33 %
| 6 %
| 41 %
| Priemerná zmena
zrakovej ostrosti
(písmená) (SD)a
| 12. mesiac
| -10,5 (16,6)
| +7,2 (14,4)
| -9,5 (16,4)
| +11,3 (14,6)
| 24. mesiac
| -14,9 (18,7)
| +6,6 (16,5)
| -9,8 (17,6)
| +10,7 (16,5)
|
|
|
a p<0,01
Obrázok 1 Priemerná zmena zrakovej ostrosti od východiskovej hodnoty do 24. mesiaca v klinickom skúšaní FVF2598g (MARINA) a v klinickom skúšaní FVF2587g (ANCHOR)
15
10
5
0
-5
-10
-15
Klinické skúšanie (MARINA)
0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+6,6
-14,9
+21,5
15
10
5
0
-5
-10
-15
Klinické skúšanie FVF2587g (ANCHOR)
+10,7
-9,8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+20,5
MARINA ANCHOR
Výsledky oboch klinických skúšaní ukázali, že pokračujúca liečba ranibizumabom môže predstavovať
prínos aj pre pacientov, ktorí stratili ≥15 písmen najlepšie korigovanej zrakovej ostrosti (BCVA)
v prvom roku liečby.
Klinické skúšanie FVF3192g (PIER) bolo randomizované, dvojito maskované, simulovane
kontrolované klinické skúšanie, určené na stanovenie bezpečnosti a účinnosti Lucentisu
u 184 pacientov so všetkými formami neovaskulárnej VPDM. Pacienti dostávali Lucentis 0,3 mg
(n=60) alebo 0,5 mg (n=61) intravitreálnymi injekciami alebo simulované injekcie (n=63) raz za mesiac v 3 po sebe idúcich dávkach, po ktorých nasledovalo podávanie dávky každé 3 mesiace. Od
14. mesiaca klinického skúšania sa pacientom so simulovaným podaním povolila zmena skupiny
liečby, aby dostávali ranibizumab, a od 19. mesiaca boli možné častejšie podania. Pacienti liečení
Lucentisom v PIER dostali priemerne 10 kompletných liečebných cyklov.
Primárnym cieľovým ukazovateľom účinnosti bola priemerná zmena zrakovej ostrosti po
12 mesiacoch v porovnaní s východiskovými hodnotami. Po počiatočnom zvýšení zrakovej ostrosti (po podávaní raz za mesiac) sa zraková ostrosť pacientov v priemere zhoršovala pri podávaní raz za štvrťrok a vrátila sa na východiskovú hodnotu v 12. mesiaci, pričom tento účinok sa zachoval
u väčšiny pacientov liečených ranibizumabom (82 %) do 24. mesiaca. Údaje u obmedzeného počtu osôb, ktoré zmenili skupinu liečby, aby dostávali ranibizumab po viac ako roku simulovanej liečby,
naznačili, že včasný začiatok liečby sa môže spájať s lepším zachovaním zrakovej ostrosti.
V klinických skúšaniach MARINA aj ANCHOR zlepšenie zrakovej ostrosti pozorované pri Lucentise
0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby, stanovený prostredníctvom
skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Rozdiely medzi Lucentisom
0,5 mg a dvomi kontrolnými skupinami boli vyhodnotené s hodnotami p v rozmedzí od 0,009 do
<0,0001.
Účinnosť Lucentisu v liečbe vlhkej VPDM okrem toho potvrdili štúdie VPDM ukončené po zaregistrovaní lieku. Údaje z dvoch štúdií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) nepreukázali prídavný účinok pri kombinovanom podaní verteporfínu (Visudyne PDT) a Lucentisu oproti monoterapii Lucentisom.
Liečbapoškodeniazrakuvdôsledku DMEBezpečnosť a účinnosť Lucentisu sa vyhodnotili v dvoch randomizovaných, dvojito maskovaných
štúdiách kontrolovaných simulovaným podaním alebo účinným liekom trvajúcich 12 mesiacov
u pacientov s poškodením zraku v dôsledku diabetického makulárneho edému. Do týchto štúdií bolo
zaradených celkovo 496 pacientov (336 s aktívnou liečbou a 160 ako kontrola), z ktorých väčšina
mala diabetes typu II, pričom 28 pacientov liečených ranibizumabom malo diabetes typu I.
V štúdii fázy II, D2201 (RESOLVE), dostávalo 151 pacientov ranibizumab (6 mg/ml, n=51,
10 mg/ml, n=51) alebo simulovanú liečbu (n=49) intravitreálnymi injekciami raz za mesiac až do splnenia vopred určených kritérií pre ukončenie liečby. Začiatočná dávka ranibizumabu (0,3 mg alebo
0,5 mg) sa po prvej injekcii mohla kedykoľvek v priebehu štúdie zdvojnásobiť. Laserová
fotokoagulácia bola ako záchranná liečba povolená po 3. mesiaci v obidvoch skupinách liečby. Štúdia mala dve časti: výskumnú časť (prvých 42 pacientov analyzovaných po 6 mesiacoch) a potvrdzujúcu časť (zvyšných 109 pacientov analyzovaných po 12 mesiacoch). Stredná priemerná zmena BCVA od
1. mesiaca do 12. mesiaca v porovnaní s východiskovou hodnotou bola +7,8 (±7,72) písmen
u pacientov liečených ranibizumabom (n=102) celkove v oboch častiach klinického skúšania,
v porovnaní s -0,1 (±9,77) písmen u pacientov pri simulovanej liečbe (p<0,0001 pre rozdiel v liečbe).
V klinickom skúšaní fázy III, D2301 (RESTORE), bolo randomizovaných 345 pacientov
s poškodením zraku v dôsledku makulárneho edému, aby dostávali buď intravitreálnu injekciu 0,5 mg ranibizumabu ako monoterapiu a simulovanú laserovú fotokoaguláciu (n=116), kombináciu 0,5 mg
ranibizumabu a laserovej fotokoagulácie (n=118), alebo simulovanú injekciu a laserovú
fotokoaguláciu (n=111). Liečba ranibizumabom sa začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej zrakovej ostrosti pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Liečba sa obnovila, keď sa pozorovalo zníženie BCVA v dôsledku progresie DME. Laserová fotokoagulácia sa podala pri vstupe do štúdie v ten istý deň najmenej
30 minút pred injekciou ranibizumabu a následne podľa potreby na základe kritérií ETDRS.
240 pacientov, ktorí predtým ukončili 12-mesačné klinické skúšanie RESTORE, bolo zaradených do otvorenej, multicentrickej extenzie skúšania trvajúcej 24 mesiacov (RESTORE Extension). Pacienti dostávali 0,5 mg ranibizumabu
pro re nata (PRN) do toho istého oka, ktoré bolo zvolené ako oko pre klinické skúšanie v skúšaní D2301 (RESTORE). Liečba sa znovu začala s mesačnými intervalmi až do poklesu BCVA v dôsledku DME a pokračovala až do dosiahnutia stabilnej BCVA. Okrem toho sa podávala liečba laserom, ak bola potrebná podľa úsudku skúšajúceho lekára a na základe kritérií ETDRS.
Kľúčové merané parametre sú zhrnuté v Tabuľke 2 (RESTORE a Extension) a na Obrázku 2 (RESTORE).
Obrázok 2 Stredná zmena zrakovej ostrosti v čase oproti východiskovej hodnote v štúdiiD2301 (RESTORE)12
10
+ 6,8/+ 6,4
+ 6,2/+ 5,4*
+ 0,9
Mesiac
11 12
Ranibizumab 0,5 mg (n=115)
Ranibizumab 0,5 mg+laser (n=118) Laser (n=110)
BL=východisková hodnota; SE=stredná chyba priemeru
* Rozdiel metódou najmenších štvorcov, p<0,0001/0,0004 na základe dvojstranného stratifikovaného
testu podľa Cochrana-Mantela-Haenszela
Účinok po 12 mesiacoch sa zhodoval u väčšiny podskupín. Avšak pre osoby s pomerne dobrou východiskovou hodnotou BCVA (>73 písmen) spolu s makulárnym edémom s hrúbkou centrálnej retiny <300 mm sa liečba ranibizumabom v porovnaní laserovou fotokoaguláciou nezdala prínosom.
Tabuľka 2 Výsledky po 12 mesiacoch v klinickom skúšaní D2301 (RESTORE) a po36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension) Meraný parameter po 12 mesiacoch v
porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
| Ranibizumab
0,5 mg n=115
| Ranibizumab
0,5 mg + laser n=118
| Laser
n=110
| Stredná priemerná zmena BCVA od 1. do
12. mesiacaa (±SD)
| 6,1 (6,4)a
| 5,9 (7,9)a
| 0,8 (8,6)
| Stredná zmena BCVA po 12. mesiacoch
(±SD)
| 6,8 (8,3)a
| 6,4 (11,8)a
| 0,9 (11,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 37,4a
| 43,2a
| 15,5
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 22,6
| 22,9
| 8,2
|
| Meraný parameter po 36 mesiacoch
v skúšaní D2301-E1 (RESTORE Extension) v porovnaní s východiskovou
hodnotou v skúšaní D2301 (RESTORE)
| Predtým
ranibizumab
0,5 mg n=83
| Predtým
ranibizumab
0,5 mg + laser n=83
| Predtým
laser
n=74*
| Stredná zmena BCVA po 24 mesiacoch
(SD)
|
7,9 (9,0)
|
6,7 (7,9)
|
5,4 (9,0)
| Stredná zmena BCVA po 36 mesiacoch
(SD)
|
8,0 (10,1)
|
6,7 (9,6)
|
6,0 (9,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
47,0
|
44,6
|
41,9
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
27,7
|
30,1
|
21,6
|
|
|
ap<0,0001 pre porovnania skupín ranibizumabu oproti skupine laseru.
n v D2301-E1 (RESTORE Extension) je počet pacientov s východiskovou hodnotou (0. mesiac)
v D2301 (RESTORE) a tiež s hodnotou z návštevy po 36. mesiaci.
*zo 74 pacientov, ktorí predtým dostali liečbu laserom, 59 (79 %) dostalo ranibizumab v extenzii
klinického skúšania.
Zlepšenie zrakovej ostrosti pozorované pri Lucentise 0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby vzhľadom na väčšinu funkcií súvisiacich so zrakom, čo sa stanovilo prostredníctvom skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Pri iných podškálach tohto dotazníka sa nezistili rozdiely v súvislosti s liečbou. Rozdiel medzi Lucentisom
0,5 mg a kontrolnou skupinou sa stanovil s hodnotami p 0,0137 (monoterapia ranibizumabom) a
0,0041 (liečba ranibizumabom a laserom) kombinovaného skóre VFQ-25.
Stredný počet injekcií podaných v klinickom skúšani RESTORE trvajúcom 12 mesiacov bol 7,0
v skupine 0,5 mg ranibizumabu, 6,8 v skupine ranibizumabu a lasera a 7,3 simulovaných injekcií
v skupine monoterapie laserom. V priemere pripadlo na pacienta 6,4 injekcií ranibizumabu podaných za obdobie 24 mesiacov extenzie u pacientov, ktorí dostávali ranibizumab v klinickom skúšaní D2301
(RESTORE). Zo 74 pacientov v skupine liečby laserom v skúšaní D2301 (RESTORE) dostalo ranibizumab niekedy počas fázy extenzie 59 (79 %) pacientov. V priemere dostalo týchto 59 pacientov
počas 24-mesačnej extenzie 8,1 injekcií ranibizumabu. Podiely pacientov, u ktorých nebola potrebná
počas fázy extenzie žiadna liečba ranibizumabom, boli v skupinách predtým liečených ranibizumabom
19 %, ranibizumabom a laserom 25 % a laserom 20 %.
Profil dlhodobej bezpečnosti ranibizumabu, ktorý sa pozoroval v 24-mesačnej extenzii klinického skúšania, sa zhoduje so známym profilom bezpečnosti Lucentisu.
V klinickom skúšaní fázy IIIb, D2304 (RETAIN), bolo randomizovaných 372 pacientov s poškodením
zraku v dôsledku DME na nasledujúce podávanie intravitreálnych injekcií:
· 0,5 mg ranibizumabu súbežne s laserovou fotokoaguláciou v režime podávania a predlžovania
intervalov medzi podaniami (treat-and-extend, TE) (n=121),
· 0,5 mg ranibizumabu v monoterapii v režime TE (n=128),
· 0,5 mg ranibizumabu v monoterapii v režime PRN (n=123).
Vo všetkých skupinách sa liečba ranibizumabom začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej BCVA pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Laserová fotokoagulácia sa podala pri vstupe do skúšania v ten istý deň ako prvá injekcia ranibizumabu a následne podľa potreby na základe kritérií ETDRS. Pri TE sa ranibizumab potom podával podľa plánu liečby s intervalmi 2-3 mesiace. Pri PRN sa každý mesiac hodnotila BCVA a ak to bolo potrebné, ranibizumab sa potom podal počas tej istej návštevy. Vo všetkých skupinách sa obnovilo podávanie každý mesiac pri poklese BCVA v dôsledku progresie DME
a pokračovalo sa v ňom až do opätovného dosiahnutia stabilnej BCVA. Skúšanie trvalo 24 mesiacov.
V klinickom skúšaní RETAIN, po 3 začiatočných každomesačných návštevách s podaním liečby, počet plánovaných návštev s podaním liečby potrebný podľa režimu TE bol 13 oproti 20 mesačným návštevám, ktoré boli potrebné podľa režimu PRN. Pri oboch režimoch liečby si viac ako 70 % pacientov udržalo ich BCVA pri frekvencii návštev ≥2 mesiace. Počas 24 mesiacov bol stredný počet (medián) injekcií 12,4 (12,0) v skupine liečby ranibizumabom a laserom pri TE, 12,8 (12,0) v skupine liečby samotným ranibizumabom pri TE a 10,7 (10,0) v skupine liečby ranibizumabom pri PRN. Pridanie lasera sa nespájalo so zníženým stredným počtom injekcií ranibizumabu pri režime TE.
Kľúčové merané parametre sú zhrnuté v Tabuľke 3.
Tabuľka 3 Výsledky v klinickom skúšaní D2304 (RETAIN) Meraný parameter
v porovnaní s východiskovou
hodnotou
| Ranibizumab
0,5 mg + laser pri TE
n=117
| Samotný ranibizumab
0,5 mg pri TE
n=125
| Ranibizumab
0,5 mg pri PRN
n=117
| Stredná priemerná
zmena BCVA od 1. do
12. mesiaca (SD)
|
5,9 (5,5) a
|
6,1 (5,7) a
|
6,2 (6,0)
| Stredná priemerná
zmena BCVA od 1. do
24. mesiaca (SD)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
| Stredná zmena BCVA
po 24 mesiacoch (SD)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
| Zisk ≥10 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
43,6
|
40,8
|
45,3
| Zisk ≥15 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
25,6
|
28,0
|
30,8
|
|
|
ap<0,0001 pre stanovenie neinferiority oproti PRN
V klinických skúšaniach pri DME sprevádzalo zlepšenie BCVA postupom času zníženie strednej
CSFT vo všetkých skupinách liečby.
Liečba
poškodenia
zraku
v
dôsledku makulárneho edému po RVO
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku makulárneho edému po RVO sa vyhodnotili v randomizovaných, dvojito maskovaných, kontrolovaných štúdiách BRAVO a CRUISE, do ktorých boli zaradené osoby s BRVO (n=397) a CRVO (n=392). V oboch štúdiách pacienti dostávali intravitreálne buď 0,3 mg, alebo 0,5 mg ranibizumabu, alebo simulované injekcie. Po 6 mesiacoch pacienti z kontrolnej skupiny simulovaného podania prešli na 0,5 mg ranibizumabu. V štúdii BRAVO bola vo všetkých skupinách od 3. mesiaca povolená laserová fotokoagulácia ako záchranná liečba.
Kľúčové merané parametre z BRAVO a CRUISE sú zhrnuté v Tabuľkách 4 a 5 a na Obrázkoch 3 a 4.
Tabuľka 4 Výsledky po 6 a 12 mesiacoch (BRAVO)
| Simulované podanie/ Lucentis 0,5 mg
(n=132)
| Lucentis 0,5 mg
(n=131)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 7,3 (13,0)
| 18,3 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 12,1 (14,4)
| 18,3 (14,6)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 28,8
| 61,1
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 43,9
| 60,3
| Podiel (%), ktorý dostal záchrannú liečbu
laserom počas 12 mesiacov
| 61,4
| 34,4
|
|
|
ap<0,0001
Obrázok 3 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca(BRAVO)
+ 18,3
20
18
+ 18,3
16
14
12 + 12,1
10
8
6
4
2
kontrolované+ 7,3kontrolná skupina simulovaného
s
im
u
l
ovaným podaním
0
podania preradená na ranibizumab
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=132) Ranibizumab 0,5 mg (n=131)
BL=východisková hodnota; SE=stredná chyba priemeru
Tabuľka 5 Výsledky po 6 a 12 mesiacoch (CRUISE)
| Simulované podanie/ Lucentis 0,5 mg (n=130)
| Lucentis 0,5 mg
(n=130)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 0,8 (16,2)
| 14,9 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 7,3 (15,9)
| 13,9 (14,2)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 16,9
| 47,7
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 33,1
| 50,8
|
|
|
ap<0,0001
O
brázok 4 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(
CRU
ISE)
20
kontrolované18
simulovaným podaním16
+ 14,9kontrolná skupina simulovaného podania preradená na ranibizumab
14
+ 13,912
10
8
+ 7,36
4
+ 0,82
0
-2
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=130) Ranibizumab 0,5 mg (n=130)
BL=východisková hodnota; SE=stredná chyba priemeru
V oboch štúdiách zlepšenie zraku sprevádzalo kontinuálne a významné zmenšovanie makulárneho
edému, merané ako hrúbka centrálnej retiny.
Pacienti s BRVO (BRAVO a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, dosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~15 písmen) ako pacienti, ktorí boli liečení ranibizumabom od začiatku (~16 písmen). Avšak počet pacientov, ktorí ukončili 2 roky liečby, bol obmedzený,
a v štúdii HORIZON boli plánované len štvrťročné kontrolné návštevy. Preto v súčasnosti nie je dosť
údajov, z ktorých by sa dali vyvodiť odporúčania, kedy sa má začať liečba ranibizumabom u pacientov s BRVO.
Pacienti s CRVO (CRUISE a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, nedosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~6 písmen) oproti pacientom, ktorí boli liečení ranibizumabom od začiatku (~12 písmen).
Zlepšenie zrakovej ostrosti pozorované pri liečbe ranibizumabom po 6 a 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby stanovený prostredníctvom podškál National Eye Institute Visual Function Questionnaire (NEI VFQ-25) vzhľadom na „aktivity vyžadujúce videnie do blízka“ a
„aktivity vyžadujúce videnie do diaľky“. Rozdiel medzi Lucentisom 0,5 mg a kontrolnou skupinou sa vyhodnotil po 6. mesiaci s hodnotami p 0,02 až 0,0002.
Liečba
poškodenia
zraku
v
dôsledku CNV pri PM
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku CNV pri PM sa stanovili na základe údajov z 12 mesiacov randomizovaného, dvojito maskovaného, účinnou liečbou kontrolovaného pivotného klinického skúšania F2301 (RADIANCE). Toto skúšanie bolo určené na vyhodnotenie dvoch rôznych režimov dávkovania 0,5 mg ranibizumabu podávaného ako intravitreálna injekcia v porovnaní s PDT verteporfínom (vPDT, fotodynamická liečba Visudynom). Do jednej
z nasledujúcich skupín bolo randomizovaných 277 pacientov:
· Skupina I (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „stability“, definovanými ako žiadna zmena BCVA v porovnaní s dvomi predchádzajúcimi mesačnými hodnoteniami).
· Skupina II (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „aktivity choroby“, definovanými ako zhoršenie zraku, ktoré možno pripísať intra- alebo subretinálnej tekutine alebo aktívnemu presakovaniu v dôsledku CNV lézie, stanovené prostredníctvom OCT a/alebo
FA).
· Skupina III (vPDT – pacienti mali od 3. mesiaca povolené dostať liečbu ranibizumabom). Počas 12 mesiacov klinického skúšania pacienti dostali v priemere 4,6 injekcií (rozmedzie 1-11) v skupine I a 3,5 injekcií (rozmedzie 1-12) v skupine II. V skupine II, ktorá zodpovedá odporúčanému
dávkovaniu (pozri časť 4.2), 50,9 % pacientov potrebovalo 1 alebo 2 injekcie, 34,5 % potrebovalo 3 až
5 injekcií a 14,7 % potrebovalo 6 až 12 injekcií počas 12 mesiacov trvania skúšania. 62,9 % pacientov v skupine II nepotrebovalo injekcie v druhom 6-mesačnom období skúšania.
Najdôležitejšie výsledky RADIANCE sú zhrnuté v Tabuľke 6 a na Obrázku 5.
Tabuľka 6 Výsledky po 3. a 12. mesiaci (RADIANCE)
| Skupina I
ranibizumab
0,5 mg
„stabilita zraku“
(n=105)
| Skupina II
ranibizumab
0,5 mg
„aktivita choroby“
(n=116)
| Skupina III
vPDTb
(n=55)
| 3. mesiac
|
|
|
| Priemerná zmena BCVA od 1. do 3. mesiaca v porovnaní s východiskovou hodnotoua (písmená)
| +10,5
| +10,6
| +2,2
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
61,9 %
38,1 %
|
65,5 %
43,1 %
|
27,3 %
14,5 %
| 12. mesiac
|
|
|
| Počet injekcií do 12. mesiaca:
Priemer
Medián
|
4,6
4
|
3,5
2,0
|
N/A N/A
| Priemerná zmena BCVA od 1. do
12. mesiaca v porovnaní s východiskovou hodnotou (písmená)
| +12,8
| +12,5
| N/A
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
69,5 %
53,3 %
|
69,0 %
51,7 %
|
N/A N/A
|
|
|
a p<0,00001 v porovnaní s kontrolou vPDT
b Porovnávacia kontrola do 3. mesiaca. Pacienti randomizovaní do skupiny vPDT mali od 3. mesiaca
povolené dostať liečbu ranibizumabom (v skupine III dostalo ranibizumab od 3. mesiaca 38 pacientov)
O
brázok 5 Priemerná zmena oproti východiskovej hodnote BCVA do 12. mesiaca
(
RAD
IANCE)
20
15 +12,5
+14,4
10 +12,1
+13,8
+9,3
5
+1,4
0
Povolený ranibizumab-5
|
|
| Mesiac
|
| Skupina I - ranibizumab 0,5 mg
|
|
|
| podľa stabilizácie (N=105)
|
|
|
| Skupina III - PDT verteporfínom'
|
|
|
| (N=55)
|
|
|
|
|
0 1 2 3 4 5 6 7 8 9 10 11 12
Zlepšenie zraku sprevádzal pokles hrúbky centrálnej retiny.
Skupina II - ranibizumab 0,5 mg
podľa aktivity choroby (N=116)
Skupina III - ranibizumab 0,5 mg/PDT
verteporfínom od 3. mesiaca (N=55)
Prínos liečby hlásený pacientmi sa pozoroval vo väčšej miere v skupinách ranibizumabu v porovnaní
s vPDT (hodnota p<0,05) z hľadiska zlepšenia kombinovaného skóre a niekoľkých podškál NEI VFQ-
25 (celkové videnie, aktivity vyžadujúce videnie do blízka, duševné zdravie a odkázanosť na iných).
Deti a dospievajúciBezpečnosť a účinnosť ranibizumabu sa zatiaľ neskúmali u detí a dospievajúcich.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Lucentis vo všetkých vekových podskupinách detí a dospievajúcich pri neovaskulárnej VPDM, poškodení zraku v dôsledku DME, poškodení zraku v dôsledku makulárneho edému po RVO a poškodení zraku
v dôsledku CNV pri PM (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po intravitreálnom podaní Lucentisu raz mesačne pacientom s neovaskulárnou VPDM boli sérové koncentrácie ranibizumabu vo všeobecnosti nízke, s maximálnymi hladinami (Cmax) spravidla pod koncentráciou ranibizumabu potrebnou na inhibovanie biologickej aktivity VEGF o 50 %
(11-27 ng/ml, ako sa stanovilo in vitro v teste bunkovej proliferácie). Cmax bola úmerná dávke
v rozmedzí dávok 0,05 až 1,0 mg/oko. Koncentrácie v sére obmedzeného počtu pacientov s DME
naznačujú, že nemožno vylúčiť o niečo vyššiu systémovú expozíciu v porovnaní s expozíciou, aká sa pozorovala u pacientov s neovaskulárnou VPDM. Koncentrácie ranibizumabu v sére pacientov s RVO
boli podobné alebo mierne vyššie v porovnaní s koncentráciami, ktoré sa pozorovali u pacientov
s neovaskulárnou VPDM.
Na základe analýzy populačnej farmakokinetiky a vymiznutia ranibizumabu zo séra pacientov s neovaskulárnou VPDM liečených dávkou 0,5 mg, priemerný vitreálny eliminačný polčas ranibizumabu je približne 9 dní. Pri intravitreálnom podávaní Lucentisu 0,5 mg/oko raz mesačne sa predpokladá, že Cmax ranibizumabu v sére, ktorá sa dosiahne približne 1 deň po podaní, bude vo všeobecnosti v rozmedzí medzi 0,79 a 2,90 ng/ml, a Cmin sa vo všeobecnosti predpokladá v rozmedzí medzi 0,07 a 0,49 ng/ml. Predpokladaná sérová koncentrácia ranibizumabu je približne 90 000- násobne nižšia ako vitreálna koncentrácia ranibizumabu.
Pacienti s poškodením funkcie obličiek: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky Lucentisu u pacientov s poškodením funkcie obličiek. V populačnej farmakokinetickej analýze pacientov s neovaskulárnou VPDM malo 68 % (136 z 200) pacientov poškodenie funkcie obličiek (46,5 % ľahké [50-80 ml/min], 20 % stredne ťažké [30-50 ml/min] a 1,5 % ťažké [<30 ml/min]).
U pacientov s RVO malo 48,2 % (253 z 525) poškodenie funkcie obličiek (36,4 % ľahké, 9,5 %
stredne ťažké a 2,3 % ťažké). Systémový klírens bol trochu nižší, čo však nebolo klinicky významné.
Pacienti s poškodením funkcie pečene: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky
Lucentisu u pacientov s poškodením funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Bilaterálne intravitreálne podávanie ranibizumabu opiciam rodu Cynomolgus v dávkach medzi
0,25 mg/oko a 2,0 mg/oko raz za 2 týždne až do 26 týždňov malo za následok účinky na oči závislé od
dávky.
Intraokulárne sa zaznamenalo od dávky závislé zosilnenie zápalu a zvýšenie počtu buniek v prednej očnej komore s maximom 2 dni po podaní injekcie. Závažnosť zápalovej odpovede sa spravidla znížila pri podaní ďalších injekcií alebo počas zotavenia. V zadnom segmente sa pozorovala vitreálna infiltrácia buniek a zákaly sklovca, ktoré tiež mali tendenciu závisieť od dávky a spravidla pretrvávali do konca liečebného obdobia. V štúdii trvajúcej 26 týždňov sa intenzita zápalu sklovca zvyšovala
s počtom injekcií. Po zotavení sa však pozorovali dôkazy reverzibility. Povaha a načasovanie zápalu zadného segmentu poukazuje na imunitne sprostredkovanú odpoveď protilátok, čo môže byť klinicky
nevýznamné. Pri niektorých zvieratách sa pozoroval vznik katarakty po relatívne dlhom období
intenzívneho zápalu, čo naznačuje, že zmeny na šošovke sú sekundárne po ťažkom zápale. Prechodné zvýšenie vnútroočného tlaku po podaní sa pozorovalo po intravitreálnych injekciách bez ohľadu na
dávku.
Mikroskopické očné zmeny súviseli so zápalom a nepoukazovali na degeneratívne procesy. V niektorých očiach sa zaznamenali granulomatózne zápalové zmeny na papile. Tieto zmeny v zadnom segmente ustupovali a v niektorých prípadoch vymizli počas zotavovania.
Po intravitreálnom podaní sa nezistili žiadne známky systémovej toxicity. V podsúbore liečených zvierat sa našli sérové a sklovcové protilátky voči ranibizumabu.
Nie sú dostupné údaje o karcinogenite alebo mutagenite.
U gravidných opíc nespôsobilo intravitreálne podávanie ranibizumabu, ktoré malo za následok maximálne systémové expozície 0,9- až 7-násobne vyššie ako najhorší prípad klinickej expozície, vývojovú toxicitu alebo teratogenitu a nemalo žiadny vplyv na hmotnosť alebo štruktúru placenty,
hoci ranibizumab sa vzhľadom na jeho mechanizmus účinku má považovať za potenciálne teratogénny
a embryo- a fetotoxický.
Neprítomnosť účinkov na vývoj embrya a plodu sprostredkovaných ranibizumabom pravdepodobne súvisí hlavne s neschopnosťou fragmentu Fab prestupovať cez placentu. Napriek tomu bol popísaný prípad vysokých hladín ranibizumabu v sére matky a prítomnosti ranibizumabu v sére plodu, čo naznačuje, že protilátka proti ranibizumabu fungovala ako transportná bielkovina (obsahujúca segment Fc) pre ranibizumab, čím sa znižoval klírens zo séra matky a umožňoval sa prestup cez placentu. Keďže sledovania vývoja embryí a plodov sa robili u zdravých gravidných zvierat a ochorenie (napr. diabetes) môže meniť priepustnosť placenty pre fragment Fab, štúdia sa má interpretovať s opatrnosťou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát α,α-trehalózy Monohydrát histidíniumchloridu Histidín
Polysorbát 20
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
Uchovávajte naplnenú injekčnú striekačku v zatavenej podložke v škatuli na ochranu pred svetlom.
Pred použitím sa môže neotvorená podložka uchovávať pri izbovej teplote (25°C) najviac 24 hodín.
6.5 Druh obalu a obsah balenia
0,165 ml sterilného roztoku v naplnenej injekčnej striekačke (sklo typu I) s piestom s uzatváracou zátkou z brómbutylovej gumy a s viečkom striekačky, ktoré sa skladá z bieleho, pevného tesnenia na zistenie nedovolenej manipulácie so šedým krytom špičky z brómbutylovej gumy s adaptérom Luer lock. Naplnená injekčná striekačka má piest a zarážku na uchopenie a je zabalená v zatavenej podložke.
Balenie obsahuje jednu naplnenú injekčnú striekačku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Naplnená injekčná striekačka je určená len na jednorazové použitie. Naplnená injekčná striekačka je sterilná. Sterilitu naplnenej injekčnej striekačky nemožno zaručiť, ak podložka nie je zatavená. Naplnenú injekčnú striekačku nepoužite, ak je roztok sfarbený, zakalený alebo obsahuje pevné častice.
Naplnená injekčná striekačka obsahuje viac lieku, ako je odporúčaná dávka 0,5 mg. Objem, ktorý možno získať z naplnenej injekčnej striekačky (0,1 ml), sa nemá celý použiť. Nadbytočné množstvo sa má vytlačiť zo striekačky pre podaním injekcie. Podanie celého objemu naplnenej injekčnej striekačky môže mať za následok predávkovanie. Vzduchovú bublinu spolu s nadbytočným liekom vytlačte pomalým posúvaním piesta, až sa hrana pod vypuklou gumenou zátkou kryje s čiernou čiarkou vyznačujúcou dávku na injekčnej striekačke (zodpovedá 0,05 ml, t.j. 0,5 mg ranibizumabu).
Na intravitreálnu injekciu sa má použiť sterilná injekčná ihla 30G x ½″.
Pri príprave Lucentisu na intravitreálne podanie dodržiavajte, prosím, pokyny na použitie:
Úvod
| Pozorne si prečítajte všetky pokyny na použitie predtým, ako použijete naplnenú
injekčnú striekačku.
Naplnená injekčná striekačka je určená len na jednorazové použitie. Naplnená injekčná striekačka je sterilná. Nepoužite liek, ak je balenie poškodené. Otvorenie
zatavenej podložky a všetky následné úkony sa majú vykonať za aseptických
podmienok.
Upozornenie: Dávka sa musí nastaviť na 0,05 ml.
|
Popis naplnenej injekčnej striekačky
|
viečko značka pre dávku zarážka na uchopenie striekačky 0,05 ml
Luer lock gumená zátka piest
Obrázok 1
|
Pripravte si
| 1. Overte si, že balenie obsahuje:
· sterilnú naplnenú injekčnú striekačku v zatavenej podložke.
2. Stiahnite kryt z podložky s injekčnou striekačkou a za aseptických podmienok opatrne vyberte injekčnú striekačku.
|
Skontrolujte
injekčnú striekačku
| 3. Overte si, že:
· viečko striekačky nie je odpojené od Luer
lock.
· striekačka nie je poškodená.
· roztok je číry, bezfarebný až svetložltý
a neobsahuje žiadne pevné častice.
4. Ak čokoľvek z uvedeného nie je splnené,
zlikvidujte naplnenú injekčnú striekačku
a použite novú striekačku.
|
|
O
dstráňte viečko
i
njekčnej striekačky
|
5. Odlomte (neodkrúcajte) viečko injekčnej
striekačky (pozri Obrázok 2).
6. Zahoďte viečko injekčnej striekačky
(pozri Obrázok 3).
|
O
brázok 2
O
brázok 3
|
N
asaďte ihlu
|
7. Nasaďte sterilnú injekčnú ihlu 30G x ½″
na injekčnú striekačku pevným priskrutkovaním na Luer lock (pozri
Obrázok 4).
8. Potiahnutím nahor opatrne odstráňte kryt
z injekčnej ihly (pozri Obrázok 5).
Poznámka: Injekčnú ihlu nikdy neutierajte.
|
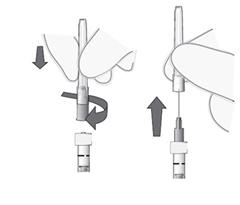
O
brázok 4 Obrázok 5
|
V
ytlačte
vzduchové bubliny
|
9. Držte injekčnú striekačku vo zvislej
polohe.
10. Ak sú v injekčnej striekačke vzduchové bubliny, opatrne na ňu klepte prstom, až kým bubliny vyplávu nahor (pozri Obrázok 6).
|
O
brázok 6
|
N
astavte dávku
|
11. Držte injekčnú striekačku vo výške očí
a opatrne stláčajte piest, až sa hrana pod vypuklou gumenou zátkou bude kryť so
značkou pre dávku (pozri Obrázok 7).
Vytlačí sa tak vzduch a nadbytočný
roztok a dávka sa nastaví na 0,05 ml.
Poznámka: Piest nie je spojený s gumenou zátkou – bráni to nasatiu vzduchu do injekčnej striekačky.
|
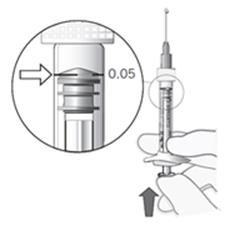
O
brázok 7
|
P
odajte injekciu
|
Podanie injekcie sa má vykonať za aseptických podmienok.
12. Injekčná ihla sa má zaviesť 3,5-4,0 mm za limbom do dutiny sklovca, vyhnúť sa horizontálnemu poludníku a smerovať do centra očnej gule.
13. Pomaly podávajte injekčný roztok, až gumená zátka dosiahne dno injekčnej striekačky a je podaný objem 0,05 ml.
14. Pri následných injekciách sa má použiť iné miesto na sklére.
15. Po podaní injekcie nenasaďte kryt späť na injekčnú ihlu, ani ihlu neodpojte od injekčnej striekačky. Zahoďte použitú injekčnú striekačku spolu s ihlou do odpadovej nádoby na ostré predmety, alebo zlikvidujte v súlade
s národnými požiadavkami.
|
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/374/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 22. január 2007
Dátum posledného predĺženia: 24. január 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
1. NÁZOV LIEKU
Lucentis 10 mg/ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jeden ml obsahuje 10 mg ranibizumabu*. Každá injekčná liekovka obsahuje 2,3 mg ranibizumabu v 0,23 ml roztoku.
*Ranibizumab je fragment humanizovanej monoklonálnej protilátky vytvorenej v bunkách
Escherichia coli rekombinantnou DNA technológiou.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok.
Číry, bezfarebný až svetložltý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Lucentis je indikovaný u dospelých na:
· Liečbu neovaskulárnej (vlhkej) vekom podmienenej degenerácie makuly (VPDM)
· Liečbu poškodenia zraku v dôsledku diabetického makulárneho edému (DME)
· Liečbu poškodenia zraku v dôsledku makulárneho edému po oklúzii žily sietnice (vetvovej RVO
alebo kmeňovej RVO)
· Liečbu poškodenia zraku v dôsledku neovaskularizácie chorioidey (CNV) pri patologickej myopii (PM)
4.2 Dávkovanie a spôsob podávania
Lucentis smie podávať kvalifikovaný oftalmológ so skúsenosťami s podávaním intravitreálnych
injekcií.
Odporúčaná dávka Lucentisu je 0,5 mg, podávaná ako jednorazová intravitreálna injekcia. To zodpovedá objemu injekcie 0,05 ml. Interval medzi dvoma dávkami podanými do toho istého oka má byť najmenej štyri týždne.
Liečba sa začína jednou injekciou za mesiac až do dosiahnutia maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity ochorenia, t.j. žiadna zmena zrakovej ostrosti a iných prejavov a príznakov choroby pri pokračujúcej liečbe. U pacientov s vlhkou VPDM, DME a RVO môže byť na začiatku potrebné podať po sebe tri alebo viac mesačných injekcií.
Následné sledovanie a intervaly medzi podaniami má určovať lekár a majú byť založené na aktivite
choroby, stanovenej prostredníctvom zrakovej ostrosti a/alebo anatomických parametrov.
Ak zrakové a anatomické parametre podľa názoru lekára ukazujú, že pokračujúca liečba pre pacienta
nie je prínosom, podávanie Lucentisu sa má ukončiť.
Sledovanie aktivity choroby môže zahŕňať klinické vyšetrenie, testovanie funkcie alebo zobrazovacie
techniky (napr. optickú koherentnú tomografiu alebo fluoresceínovú angiografiu).
Ak sa pacient lieči v režime podávania a predlžovania intervalov medzi podaniami, po dosiahnutí maximálnej zrakovej ostrosti a/alebo kým nie sú prítomné prejavy aktivity choroby sa intervaly medzi podaniami môžu postupne predlžovať až do opätovného objavenia sa prejavov aktivity choroby alebo zhoršenia zraku. Interval medzi podaniami sa nemá naraz predĺžiť o viac ako dva týždne pri vlhkej VPDM a môže sa naraz predĺžiť až o jeden mesiac pri DME. Pri RVO sa intervaly medzi podaniami tiež môžu postupne predlžovať, avšak nie je dostatok údajov na určenie dĺžky týchto intervalov. Ak sa opäť objaví aktivita choroby, interval medzi podaniami sa má patrične skrátiť.
Pri liečbe poškodenia zraku v dôsledku CNV pri PM u mnohých pacientov môžu byť potrebné len jedna alebo dve injekcie počas prvého roka, zatiaľ čo niektorí pacienti môžu potrebovať častejšiu liečbu (pozri časť 5.1).
Lucentis a laserová fotokoagulácia pri DME a pri makulárnom edéme po BRVO
S podávaním Lucentisu súčasne s laserovou fotokoaguláciou sú určité skúsenosti (pozri časť 5.1). Pri
podávaní v ten istý deň sa Lucentis má podať minimálne 30 minút po laserovej fotokoagulácii.
Lucentis sa môže podávať pacientom, ktorí boli v minulosti liečení laserovou fotokoaguláciou.
Lucentis a fotodynamická liečba Visudynom pri CNV v dôsledku PM
Nie sú skúsenosti so súbežným podávaním Lucentisu a Visudynu.
Osobitné skupiny pacientov
Poškodenie funkcie pečene
Lucentis sa neskúmal u pacientov s poškodením funkcie pečene. U tejto populácie však nie sú potrebné zvláštne opatrenia.
Poškodenie funkcie obličiek
Úprava dávky nie je potrebná u pacientov s poškodením funkcie obličiek (pozri časť 5.2).
Starší pacienti
Úprava dávkovania u starších ľudí nie je potrebná. Skúsenosti s pacientmi s DME staršími ako
75 rokov sú obmedzené.
Deti a dospievajúci
Bezpečnosť a účinnosť Lucentisu u detí a dospievajúcich vo veku menej ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje.
Spôsob podania
Injekčná liekovka na jednorazové použitie len na intravitreálne podanie.
Lucentis sa má pred podaním vizuálne skontrolovať na prítomnosť cudzorodých častíc a zmenu sfarbenia.
Podanie injekcie sa má uskutočniť za aseptických podmienok, ktoré zahŕňajú chirurgickú dezinfekciu rúk, použitie sterilných rukavíc, sterilného rúška a sterilného rozvierača mihalníc (alebo náhrady)
a dostupnosť sterilnej paracentézy (pre prípad potreby). Pred vykonaním intravitreálneho zákroku sa má dôsledne preskúmať pacientova anamnéza so zreteľom na reakcie z precitlivenosti (pozri časť 4.4).
Pred podaním injekcie sa má v súlade s lokálnou klinickou praxou podať náležitá anestézia
a širokospektrálny lokálny mikrobicídny prostriedok na dezinfekciu kože v okolí oka, mihalnice a povrchu oka.
Informácie o príprave Lucentisu, pozri časť 6.6.
Injekčná ihla sa zavádza 3,5-4,0 mm za limbom do dutiny sklovca, vyhýba sa horizontálnemu poludníku a smeruje do centra očnej gule. Potom sa podá objem injekcie 0,05 ml; pri následných injekciách sa má použiť iné miesto na sklére.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s aktívnymi alebo suspektnými očnými alebo periokulárnymi infekciami. Pacienti s aktívnym ťažkým vnútroočným zápalom.
4.4 Osobitné upozornenia a opatrenia pri používaní
Reakcie súvisiace s intravitreálnou injekciou
Podanie intravitreálnych injekcií vrátane injekcií Lucentisu sa spájalo s endoftalmitídou, vnútroočným zápalom, rhegmatogénnym odlúčením sietnice, trhlinou v sietnici a iatrogénnou traumatickou
kataraktou (pozri časť 4.8). Pri podaní Lucentisu sa musia vždy dodržať náležité aseptické injekčné
postupy. Okrem toho je potrebné pacientov sledovať počas týždňa po podaní injekcie, čo umožní včasnú liečbu v prípade infekcie. Pacienti majú byť poučení, aby bezodkladne oznámili akýkoľvek príznak, ktorý poukazuje na endoftalmitídu alebo na niektorú z vyššie uvedených príhod.
Zvýšeniavnútroočnéhotlaku
V priebehu 60 minút po podaní injekcie Lucentisu sa pozorovalo prechodné zvýšenie vnútroočného tlaku (IOP). Zistilo sa aj trvalé zvýšenie IOP (pozri časť 4.8). Vnútroočný tlak aj perfúzia hlavy zrakového nervu sa musia monitorovať a náležite liečiť.
Bilaterálnaliečba
Obmedzené údaje o bilaterálnom použití Lucentisu (vrátane podania v ten istý deň) nenaznačujú zvýšené riziko systémových nežiaducich príhod v porovnaní s unilaterálnou liečbou.
Imunogenita
Lucentis môže byť imunogénny. Keďže je možnosť zvýšenej systémovej expozície u osôb s DME,
nemožno vylúčiť zvýšené riziko vzniku precitlivenosti u tejto populácie pacientov. Pacienti majú byť tiež poučení, aby hlásili akékoľvek zvýšenie závažnosti vnútroočného zápalu, ktoré môže byť klinickým príznakom zodpovedajúcim tvorbe protilátok vo vnútri oka.
Súčasnépoužitiesinýmianti-VEGF (vaskulárny endoteliálny rastový faktor)
Lucentis sa nemá podávať súčasne s inými anti-VEGF liekmi (systémovými alebo okulárnymi).
PrerušenieliečbyLucentisom
Dávka sa nemá podať a v liečbe sa nemá pokračovať skôr ako počas najbližšej plánovanej návštevy
v prípade:
· poklesu najlepšie korigovanej zrakovej ostrosti (BCVA) o ≥30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
· intraokulárneho tlaku ≥30 mmHg;
· trhliny v sietnici;
· subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je ≥50 %
celkovej plochy lézie;
· uskutočneného alebo plánovaného intraokulárneho chirurgického zákroku počas uplynulých
alebo nasledujúcich 28 dní.
Trhlina v pigmentovom epiteli sietnice
Rizikové faktory, ktoré sa spájajú so vznikom trhliny v pigmentovom epiteli sietnice po anti-VEGF liečbe pri vlhkej VPDM, zahŕňajú veľké a/alebo vysoko uložené odlúčenie pigmentového epitelu sietnice. Pri začatí liečby Lucentisom je potrebná opatrnosť u pacientov s týmito rizikovými faktormi pre trhliny v pigmentovom epiteli sietnice.
R
egmatogénne
odlúčenie sietnice alebo makulárne diery
Liečba sa má ukončiť u osôb s regmatogénnym odlúčením sietnice alebo makulárnymi dierami 3.
alebo 4. stupňa.
Skupiny pacientov, u ktorých sú skúsenosti obmedzené
S liečbou osôb s DME spôsobeným diabetom typu I sú len obmedzené skúsenosti. Lucentis sa neskúmal u pacientov, ktorí v minulosti dostali intravitreálne injekcie, u pacientov s aktívnymi systémovými infekciami, proliferujúcou diabetickou retinopatiou, alebo u pacientov so sprievodnými očnými ochoreniami, napr. s odlúčením sietnice alebo makulárnou dierou. Nie sú tiež žiadne
skúsenosti s liečbou Lucentisom u pacientov s diabetom s HbA1c vyšším ako 12 % a nekontrolovanou
hypertenziou. Pri liečbe takýchto pacientov má lekár vziať do úvahy tento nedostatok informácií.
Údaje o účinku Lucentisu sú obmedzené u pacientov s PM, ktorí v minulosti podstúpili neúspešnú fotodynamickú liečbu verteporfínom (vPDT). Zatiaľ čo sa pozoroval zhodný účinok u osôb so subfoveálnymi a juxtafoveálnymi léziami, nie je tiež dostatok údajov, z ktorých by bolo možné usudzovať na účinok Lucentisu u osôb s PM, ktoré majú extrafoveálne lézie.
Systémovéúčinkypointravitreálnompoužití
Po intravitreálnej injekcii inhibítorov VEGF sa zaznamenali systémové nežiaduce udalosti vrátane
krvácaní mimo oka a artériových tromboembolických príhod.
Údaje o bezpečnosti liečby u pacientov s DME, makulárnym edémom po RVO a s CNV v dôsledku PM, ktorí majú v anamnéze cievnu mozgovú príhodu alebo tranzitórne ischemické ataky, sú obmedzené. Pri liečbe takýchto pacientov je potrebné postupovať opatrne (pozri časť 4.8).
Epizódy RVO, ischemickej vetvovej RVO a kmeňovejRVOvminulosti
Skúsenosti s liečbou pacientov s epizódami RVO v minulosti a pacientov s ischemickou vetvovou
RVO (BRVO) a kmeňovou RVO (CRVO) sú obmedzené. U pacientov s RVO, u ktorých sa objavia
klinické príznaky ireverzibilnej ischemickej straty zrakovej funkcie, sa liečba neodporúča.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne formálne interakčné štúdie.
Adjuvantné použitie fotodynamickej liečby (PDT) verteporfínom a Lucentisu pri vlhkej VPDM a PM,
pozri časť 5.1.
Adjuvantné použitie laserovej fotokoagulácie a Lucentisu pri DME a BRVO, pozri časti 4.2 a 5.1.
V klinických skúšaniach liečby poškodenia zraku v dôsledku DME súbežná liečba tiazolidíndiónmi neovplyvnila výsledky týkajúce sa zrakovej ostrosti alebo hrúbky čiastkového poľa centrálnej sietnice (CSTF) u pacientov liečených Lucentisom.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepciaužien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby.
Gravidita
Nie sú dostupné klinické údaje o použití ranibizumabu v gravidite. Štúdie na makakoch krabožravých nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity alebo vývinu embrya/plodu (pozri časť 5.3). Systémová expozícia ranibizumabu po podaní do oka je nízka, ale vzhľadom na jeho mechanizmus účinku sa ranibizumab musí považovať za potenciálne teratogénny a embryo-
/fetotoxický. Preto sa ranibizumab nemá používať v gravidite, pokiaľ očakávaný prínos nie je väčší ako možné riziko pre plod. Ženám, ktoré chcú otehotnieť a boli liečené ranibizumabom, sa odporúča počkať s počatím dieťaťa aspoň 3 mesiace od poslednej dávky ranibizumabu.
Laktácia
Nie je známe, či sa Lucentis vylučuje do ľudského mlieka. Dojčenie sa neodporúča počas používania
Lucentisu.
Fertilita
Nie sú dostupné žiadne údaje o fertilite.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liečba Lucentisom môže vyvolať dočasné poruchy videnia, čo môže ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje (pozri časť 4.8). Pacienti, u ktorých sa vyskytnú tieto príznaky, nesmú viesť vozidlá alebo obsluhovať stroje, kým tieto dočasné poruchy zraku neustúpia.
4.8 Nežiaduce účinky
Zhrnutieprofilubezpečnosti
Väčšina nežiaducich reakcií hlásených po podaní Lucentisu súvisí s postupom intravitreálnej injekcie.
Najčastejšie hlásené nežiaduce reakcie týkajúce sa očí po podaní injekcie Lucentisu sú: bolesť oka, hyperémia oka, zvýšený vnútroočný tlak, vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko a svrbenie oka.
Najčastejšie hlásené nežiaduce reakcie netýkajúce sa očí sú bolesť hlavy, nazofaryngitída a bolesť kĺbov.
Medzi menej často hlásené, ale závažnejšie nežiaduce reakcie patria endoftalmitída, slepota, odlúčenie sietnice, trhlina v sietnici a iatrogénna traumatická katarakta (pozri časť 4.4).
Pacientov je potrebné informovať o prejavoch týchto potenciálnych nežiaducich reakcií a poučiť ich, aby informovali svojho lekára, ak u nich vzniknú príznaky ako bolesť očí alebo zvýšenie nepríjemných pocitov, zhoršujúce sa sčervenanie očí, neostré alebo zhoršené videnie, zvýšený počet malých čiastočiek v zornom poli, alebo zvýšená citlivosť na svetlo.
Nežiaduce reakcie, ktoré sa vyskytli po podaní Lucentisu v klinických skúšaniach, sú zhrnuté
v tabuľke nižšie.
Tabuľkovýzoznamnežiaducichreakcií#
Nežiaduce reakcie sú zatriedené podľa orgánových systémov a frekvencie podľa nasledujúcich konvencií: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé
(≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov). V rámci
jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Infekcie a nákazy
Veľmi časté Nazofaryngitída
Časté Infekcia močových ciest*
Poruchy krvi a lymfatického systému
Časté Anémia
Poruchy imunitného systému
Časté Precitlivenosť
Psychické poruchy
Časté Úzkosť
Poruchy nervového systému
Veľmi časté Bolesť hlavy
Poruchy oka
Veľmi časté Vitritída, odlúčenie sklovca, retinálne krvácanie, poruchy videnia, bolesť oka, opacity v sklovci, krvácanie do spojovky, podráždenie oka, pocit cudzieho telieska v očiach, zvýšené slzenie, blefaritída, suché oko, hyperémia oka, svrbenie oka.
Časté Degenerácia sietnice, porucha sietnice, odlúčenie sietnice, trhlina
v sietnici, odlúčenie pigmentového epitelu sietnice, trhlina v pigmentovom epiteli sietnice, znížená zraková ostrosť, krvácanie do sklovca, porucha sklovca, uveitída, iritída, iridocyklitída, katarakta, subkapsulárna katarakta, opacifikácie zadného puzdra šošovky, bodkovitá keratitída, abrázia rohovky, zápal prednej očnej komory, neostré videnie, krvácanie v mieste podania injekcie, krvácanie do oka, konjunktivitída, alergická konjunktivitída, výtok z oka, fotopsia, fotofóbia, nepríjemné pocity v oku, edém mihalnice, bolesť mihalnice, hyperémia spojoviek.
Menej časté Slepota, endoftalmitída, hypopyon, hyféma, keratopatia, adhézia dúhovky, depozity v rohovke, edém rohovky, strie rohovky, bolesť v mieste podania injekcie, podráždenie v mieste podania injekcie, abnormálne pocity v oku, podráždenie mihalnice.
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté Kašeľ
Poruchy gastrointestinálneho traktu
Časté Nauzea
Poruchy kože a podkožného tkaniva
Časté Alergické reakcie (exantém, urtikária, pruritus, erytém)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Veľmi časté Artralgia
Laboratórne a funkčné vyšetrenia
Veľmi časté Zvýšenie vnútroočného tlaku
# Nežiaduce reakcie boli definované ako nežiaduce udalosti (u najmenej 0,5 percentuálneho bodu pacientov), ktoré sa vyskytli častejšie (najmenej 2 percentuálne body) u pacientov liečených
Lucentisom 0,5 mg, ako u pacientov, ktorí dostávali kontrolnú liečbu (simulované podanie alebo PDT
verteporfínom).
* pozorované iba u populácie s DME
Nežiaducereakciesúvisiace so skupinou liekov
V klinických skúšaniach fázy III pri vlhkej VPDM bola celková frekvencia krvácaní mimo oka, čo je nežiaduca udalosť potenciálne súvisiaca so systémovou inhibíciou VEGF (vaskulárneho
endoteliálneho rastového faktora), mierne zvýšená u pacientov liečených ranibizumabom.
Charakteristika rôznych krvácaní však nebola zhodná. Po intravitreálnom použití inhibítorov VEGF
existuje teoretické riziko arteriálnych tromboembolických príhod vrátane cievnej mozgovej príhody
a infarktu myokardu. V klinických skúšaniach Lucentisu sa pozorovala nízka incidencia arteriálnych
tromboembolických príhod u pacientov s VPDM, DME, RVO a PM a neboli významné rozdiely
medzi skupinami liečenými ranibizumabom v porovnaní s kontrolnou liečbou.
H
l
ásenie podozrení na
nežiaduce
r
eakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieBoli hlásené prípady náhodného predávkovania v klinických skúšaniach pri vlhkej VPDM a z údajov po uvedení lieku na trh. Nežiaduce reakcie súvisiace s týmito hlásenými prípadmi boli zvýšenie vnútroočného tlaku, prechodná slepota, znížená zraková ostrosť, edém rohovky, bolesť rohovky
a bolesť oka. Ak dôjde k predávkovaniu, je potrebné sledovať a liečiť vnútroočný tlak, ak to ošetrujúci lekár považuje za potrebné.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: oftalmologiká, látky s antineovaskularizačným účinkom, ATC kód:
S01LA04
Ranibizumab je fragment humanizovanej rekombinantnej monoklonálnej protilátky, ktorého cieľom je ľudský vaskulárny endoteliálny rastový faktor A (VEGF-A). Viaže sa s vysokou afinitou na izoformy VEGF-A (napr. VEGF110, VEGF121 a VEGF165), a tým zabraňuje vzniku väzby VEGF-A na jeho receptory VEGFR-1 a VEGFR-2. Naviazanie VEGF-A na jeho receptory má za následok proliferáciu endotelových buniek a neovaskularizáciu, ako aj únik tekutín z ciev, čo všetko sa považuje za faktory prispievajúce k progresii neovaskulárnej formy vekom podmienenej degenerácie makuly, patologickej myopie alebo poškodeniu zraku spôsobeného buď diabetickým makulárnym edémom, alebo makulárnym edémom po RVO.
Liečbavlhkej VPDMPri vlhkej VPDM sa klinická bezpečnosť a účinnosť Lucentisu hodnotila v troch randomizovaných,
dvojito maskovaných, simulovane alebo aktívne kontrolovaných klinických skúšaniach trvajúcich
24 mesiacov u pacientov s neovaskulárnou VPDM. Do týchto klinických skúšaní bolo zaradených
celkovo 1 323 pacientov (879 v aktívnej a 444 v kontrolnej skupine).
V klinickom skúšaní FVF2598g (MARINA) 716 pacientov s minimálne klasickou alebo okultnou bez klasickej chorioidálnej neovaskularizácie (CNV) dostávalo každý mesiac intravitreálne injekcie Lucentisu 0,3 mg (n=238) alebo 0,5 mg (n=240) alebo simulované injekcie (n=238).
V klinickom skúšaní FVF2587g (ANCHOR) 423 pacientov s prevažne klasickými CNV léziami dostávalo buď: 1) každý mesiac intravitreálne injekcie Lucentis 0,3 mg a simulovanú PDT (n=140);
2) každý mesiac intravitreálne injekcie Lucentisu 0,5 mg a simulovanú PDT (n=140); alebo
3) simulované intravitreálne injekcie a aktívnu PDT verteporfínom (n=143). Simulovaná alebo aktívna PDT verteporfínom sa podala s úvodnou injekciou Lucentisu a potom každé 3 mesiace, ak fluoresceínová angiografia ukázala pretrvávanie alebo recidívu úniku tekutín z ciev.
Kľúčové merané parametre sú zhrnuté v Tabuľke 1 a na Obrázku 1.
Tabuľka 1 Výsledky po 12 mesiacoch a 24 mesiacoch v klinickom skúšaní FVF2598g(MARINA) a FVF2587g (ANCHOR)
|
| FVF2598g (MARINA)
| FVF2587g (ANCHOR)
|
| Meraný
parameter
| Mesiac
| Simulované
podanie
(n=238)
| Lucentis
0,5 mg
(n=240)
| PDT
verteporfínom
(n=143)
| Lucentis
0,5 mg
(n=140)
| Strata <15 písmen
zrakovej ostrosti (%)a (zachovanie vízu, primárny ukazovateľ)
| 12. mesiac
| 62 %
| 95 %
| 64 %
| 96 %
| 24. mesiac
| 53 %
| 90 %
| 66 %
| 90 %
| Zisk ≥15 písmen
zrakovej ostrosti
(%)a
| 12. mesiac
| 5 %
| 34 %
| 6 %
| 40 %
| 24. mesiac
| 4 %
| 33 %
| 6 %
| 41 %
| Priemerná zmena
zrakovej ostrosti
(písmená) (SD)a
| 12. mesiac
| -10,5 (16,6)
| +7,2 (14,4)
| -9,5 (16,4)
| +11,3 (14,6)
| 24. mesiac
| -14,9 (18,7)
| +6,6 (16,5)
| -9,8 (17,6)
| +10,7 (16,5)
|
|
|
a p<0,01
O
brázok 1 Priemerná zmena zrakovej ostrosti od východiskovej hodnoty do 24. mesiaca v klinickom skúšaní FVF2598g (MARINA) a v klinickom skúšaní FVF2587g (ANCHOR)
15
10
5
0
-5
-10
-15
Klinické skúšanie (MARINA)
0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+6,6
-14,9
+21,5
15
10
5
0
-5
-10
-15
Klinické skúšanie FVF2587g (ANCHOR)
+10,7
-9,8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mesiac
+20,5
MARINA ANCHOR
Výsledky oboch klinických skúšaní ukázali, že pokračujúca liečba ranibizumabom môže predstavovať
prínos aj pre pacientov, ktorí stratili ≥15 písmen najlepšie korigovanej zrakovej ostrosti (BCVA)
v prvom roku liečby.
Klinické skúšanie FVF3192g (PIER) bolo randomizované, dvojito maskované, simulovane kontrolované klinické skúšanie, určené na stanovenie bezpečnosti a účinnosti Lucentisu
u 184 pacientov so všetkými formami neovaskulárnej VPDM. Pacienti dostávali Lucentis 0,3 mg
(n=60) alebo 0,5 mg (n=61) intravitreálnymi injekciami alebo simulované injekcie (n=63) raz za mesiac v 3 po sebe idúcich dávkach, po ktorých nasledovalo podávanie dávky každé 3 mesiace. Od
14. mesiaca klinického skúšania sa pacientom so simulovaným podaním povolila zmena skupiny
liečby, aby dostávali ranibizumab, a od 19. mesiaca boli možné častejšie podania. Pacienti liečení
Lucentisom v PIER dostali priemerne 10 kompletných liečebných cyklov.
Primárnym cieľovým ukazovateľom účinnosti bola priemerná zmena zrakovej ostrosti po
12 mesiacoch v porovnaní s východiskovými hodnotami. Po počiatočnom zvýšení zrakovej ostrosti (po podávaní raz za mesiac) sa zraková ostrosť pacientov v priemere zhoršovala pri podávaní raz za štvrťrok a vrátila sa na východiskovú hodnotu v 12. mesiaci, pričom tento účinok sa zachoval
u väčšiny pacientov liečených ranibizumabom (82 %) do 24. mesiaca. Údaje u obmedzeného počtu osôb, ktoré zmenili skupinu liečby, aby dostávali ranibizumab po viac ako roku simulovanej liečby,
naznačili, že včasný začiatok liečby sa môže spájať s lepším zachovaním zrakovej ostrosti.
V klinických skúšaniach MARINA aj ANCHOR zlepšenie zrakovej ostrosti pozorované pri Lucentise
0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby, stanovený prostredníctvom skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Rozdiely medzi Lucentisom
0,5 mg a dvomi kontrolnými skupinami boli vyhodnotené s hodnotami p v rozmedzí od 0,009 do
<0,0001.
Účinnosť Lucentisu v liečbe vlhkej VPDM okrem toho potvrdili štúdie VPDM ukončené po zaregistrovaní lieku. Údaje z dvoch štúdií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) nepreukázali prídavný účinok pri kombinovanom podaní verteporfínu (Visudyne PDT) a Lucentisu oproti monoterapii Lucentisom.
Liečbapoškodeniazrakuvdôsledku DME
Bezpečnosť a účinnosť Lucentisu sa vyhodnotili v dvoch randomizovaných, dvojito maskovaných
štúdiách kontrolovaných simulovaným podaním alebo účinným liekom trvajúcich 12 mesiacov
u pacientov s poškodením zraku v dôsledku diabetického makulárneho edému. Do týchto štúdií bolo
zaradených celkovo 496 pacientov (336 s aktívnou liečbou a 160 ako kontrola), z ktorých väčšina
mala diabetes typu II, pričom 28 pacientov liečených ranibizumabom malo diabetes typu I.
V štúdii fázy II, D2201 (RESOLVE), dostávalo 151 pacientov ranibizumab (6 mg/ml, n=51,
10 mg/ml, n=51) alebo simulovanú liečbu (n=49) intravitreálnymi injekciami raz za mesiac až do splnenia vopred určených kritérií pre ukončenie liečby. Začiatočná dávka ranibizumabu (0,3 mg alebo
0,5 mg) sa po prvej injekcii mohla kedykoľvek v priebehu štúdie zdvojnásobiť. Laserová
fotokoagulácia bola ako záchranná liečba povolená po 3. mesiaci v obidvoch skupinách liečby. Štúdia mala dve časti: výskumnú časť (prvých 42 pacientov analyzovaných po 6 mesiacoch) a potvrdzujúcu časť (zvyšných 109 pacientov analyzovaných po 12 mesiacoch). Stredná priemerná zmena BCVA od
1. mesiaca do 12. mesiaca v porovnaní s východiskovou hodnotou bola +7,8 (±7,72) písmen
u pacientov liečených ranibizumabom (n=102) celkove v oboch častiach klinického skúšania,
v porovnaní s -0,1 (±9,77) písmen u pacientov pri simulovanej liečbe (p<0,0001 pre rozdiel v liečbe).
V klinickom skúšaní fázy III, D2301 (RESTORE), bolo randomizovaných 345 pacientov
s poškodením zraku v dôsledku makulárneho edému, aby dostávali buď intravitreálnu injekciu 0,5 mg ranibizumabu ako monoterapiu a simulovanú laserovú fotokoaguláciu (n=116), kombináciu 0,5 mg
ranibizumabu a laserovej fotokoagulácie (n=118), alebo simulovanú injekciu a laserovú
fotokoaguláciu (n=111). Liečba ranibizumabom sa začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej zrakovej ostrosti pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Liečba sa obnovila, keď sa pozorovalo zníženie BCVA v dôsledku progresie DME. Laserová fotokoagulácia sa podala pri vstupe do štúdie v ten istý deň najmenej
30 minút pred injekciou ranibizumabu a následne podľa potreby na základe kritérií ETDRS.
240 pacientov, ktorí predtým ukončili 12-mesačné klinické skúšanie RESTORE, bolo zaradených do otvorenej, multicentrickej extenzie skúšania trvajúcej 24 mesiacov (RESTORE Extension). Pacienti dostávali 0,5 mg ranibizumabu pro re nata (PRN) do toho istého oka, ktoré bolo zvolené ako oko pre klinické skúšanie v skúšaní D2301 (RESTORE). Liečba sa znovu začala s mesačnými intervalmi až do poklesu BCVA v dôsledku DME a pokračovala až do dosiahnutia stabilnej BCVA. Okrem toho sa podávala liečba laserom, ak bola potrebná podľa úsudku skúšajúceho lekára a na základe kritérií ETDRS.
Kľúčové merané parametre sú zhrnuté v Tabuľke 2 (RESTORE a Extension) a na Obrázku 2 (RESTORE).
Obrázok 2 Stredná zmena zrakovej ostrosti v čase oproti východiskovej hodnote v štúdiiD2301 (RESTORE)12
10
+ 6,8/+ 6,4
+ 6,2/+ 5,4*
+ 0,9
Mesiac
11 12
Ranibizumab 0,5 mg (n=115)
Ranibizumab 0,5 mg+laser (n=118) Laser (n=110)
BL=východisková hodnota; SE=stredná chyba priemeru
* Rozdiel metódou najmenších štvorcov, p<0,0001/0,0004 na základe dvojstranného stratifikovaného
testu podľa Cochrana-Mantela-Haenszela
Účinok po 12 mesiacoch sa zhodoval u väčšiny podskupín. Avšak pre osoby s pomerne dobrou východiskovou hodnotou BCVA (>73 písmen) spolu s makulárnym edémom s hrúbkou centrálnej retiny <300 mm sa liečba ranibizumabom v porovnaní laserovou fotokoaguláciou nezdala prínosom.
Tabuľka 2 Výsledky po 12 mesiacoch v klinickom skúšaní D2301 (RESTORE) a po36 mesiacoch v skúšaní D2301-E1 (RESTORE Extension) Meraný parameter po 12 mesiacoch v
porovnaní s východiskovou hodnotou v skúšaní D2301 (RESTORE)
| Ranibizumab
0,5 mg n=115
| Ranibizumab
0,5 mg + laser n=118
| Laser
n=110
| Stredná priemerná zmena BCVA od 1. do
12. mesiacaa (±SD)
| 6,1 (6,4)a
| 5,9 (7,9)a
| 0,8 (8,6)
| Stredná zmena BCVA po 12. mesiacoch
(±SD)
| 6,8 (8,3)a
| 6,4 (11,8)a
| 0,9 (11,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 37,4a
| 43,2a
| 15,5
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 12 mesiacoch (%)
| 22,6
| 22,9
| 8,2
|
| Meraný parameter po 36 mesiacoch
v skúšaní D2301-E1 (RESTORE Extension) v porovnaní s východiskovou
hodnotou v skúšaní D2301 (RESTORE)
| Predtým
ranibizumab
0,5 mg n=83
| Predtým
ranibizumab
0,5 mg + laser n=83
| Predtým
laser
n=74*
| Stredná zmena BCVA po 24 mesiacoch
(SD)
|
7,9 (9,0)
|
6,7 (7,9)
|
5,4 (9,0)
| Stredná zmena BCVA po 36 mesiacoch
(SD)
|
8,0 (10,1)
|
6,7 (9,6)
|
6,0 (9,4)
| Zisk ≥10 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
47,0
|
44,6
|
41,9
| Zisk ≥15 písmen alebo BCVA ≥84 písmen
po 36 mesiacoch (%)
|
27,7
|
30,1
|
21,6
|
|
|
ap<0,0001 pre porovnania skupín ranibizumabu oproti skupine laseru.
n v D2301-E1 (RESTORE Extension) je počet pacientov s východiskovou hodnotou (0. mesiac)
v D2301 (RESTORE) a tiež s hodnotou z návštevy po 36. mesiaci.
*zo 74 pacientov, ktorí predtým dostali liečbu laserom, 59 (79 %) dostalo ranibizumab v extenzii
klinického skúšania.
Zlepšenie zrakovej ostrosti pozorované pri Lucentise 0,5 mg po 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby vzhľadom na väčšinu funkcií súvisiacich so zrakom, čo sa stanovilo prostredníctvom skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Pri iných podškálach tohto dotazníka sa nezistili rozdiely v súvislosti s liečbou. Rozdiel medzi Lucentisom
0,5 mg a kontrolnou skupinou sa stanovil s hodnotami p 0,0137 (monoterapia ranibizumabom) a
0,0041 (liečba ranibizumabom a laserom) kombinovaného skóre VFQ-25.
Stredný počet injekcií podaných v klinickom skúšani RESTORE trvajúcom 12 mesiacov bol 7,0
v skupine 0,5 mg ranibizumabu, 6,8 v skupine ranibizumabu a lasera a 7,3 simulovaných injekcií
v skupine monoterapie laserom. V priemere pripadlo na pacienta 6,4 injekcií ranibizumabu podaných za obdobie 24 mesiacov extenzie u pacientov, ktorí dostávali ranibizumab v klinickom skúšaní D2301
(RESTORE). Zo 74 pacientov v skupine liečby laserom v skúšaní D2301 (RESTORE) dostalo ranibizumab niekedy počas fázy extenzie 59 (79 %) pacientov. V priemere dostalo týchto 59 pacientov
počas 24-mesačnej extenzie 8,1 injekcií ranibizumabu. Podiely pacientov, u ktorých nebola potrebná
počas fázy extenzie žiadna liečba ranibizumabom, boli v skupinách predtým liečených ranibizumabom
19 %, ranibizumabom a laserom 25 % a laserom 20 %.
Profil dlhodobej bezpečnosti ranibizumabu, ktorý sa pozoroval v 24-mesačnej extenzii klinického skúšania, sa zhoduje so známym profilom bezpečnosti Lucentisu.
V klinickom skúšaní fázy IIIb, D2304 (RETAIN), bolo randomizovaných 372 pacientov s poškodením
zraku v dôsledku DME na nasledujúce podávanie intravitreálnych injekcií:
· 0,5 mg ranibizumabu súbežne s laserovou fotokoaguláciou v režime podávania a predlžovania
intervalov medzi podaniami (treat-and-extend, TE) (n=121),
· 0,5 mg ranibizumabu v monoterapii v režime TE (n=128),
· 0,5 mg ranibizumabu v monoterapii v režime PRN (n=123).
Vo všetkých skupinách sa liečba ranibizumabom začala intravitreálnymi injekciami raz za mesiac
a pokračovala až do dosiahnutia stabilnej BCVA pri najmenej troch po sebe nasledujúcich mesačných hodnoteniach. Laserová fotokoagulácia sa podala pri vstupe do skúšania v ten istý deň ako prvá injekcia ranibizumabu a následne podľa potreby na základe kritérií ETDRS. Pri TE sa ranibizumab potom podával podľa plánu liečby s intervalmi 2-3 mesiace. Pri PRN sa každý mesiac hodnotila BCVA a ak to bolo potrebné, ranibizumab sa potom podal počas tej istej návštevy. Vo všetkých skupinách sa obnovilo podávanie každý mesiac pri poklese BCVA v dôsledku progresie DME
a pokračovalo sa v ňom až do opätovného dosiahnutia stabilnej BCVA. Skúšanie trvalo 24 mesiacov.
V klinickom skúšaní RETAIN, po 3 začiatočných každomesačných návštevách s podaním liečby, počet plánovaných návštev s podaním liečby potrebný podľa režimu TE bol 13 oproti 20 mesačným návštevám, ktoré boli potrebné podľa režimu PRN. Pri oboch režimoch liečby si viac ako 70 % pacientov udržalo ich BCVA pri frekvencii návštev ≥2 mesiace. Počas 24 mesiacov bol stredný počet (medián) injekcií 12,4 (12,0) v skupine liečby ranibizumabom a laserom pri TE, 12,8 (12,0) v skupine liečby samotným ranibizumabom pri TE a 10,7 (10,0) v skupine liečby ranibizumabom pri PRN. Pridanie lasera sa nespájalo so zníženým stredným počtom injekcií ranibizumabu pri režime TE.
Kľúčové merané parametre sú zhrnuté v Tabuľke 3.
Tabuľka 3 Výsledky v klinickom skúšaní D2304 (RETAIN) Meraný parameter
v porovnaní s východiskovou
hodnotou
| Ranibizumab
0,5 mg + laser pri TE
n=117
| Samotný ranibizumab
0,5 mg pri TE
n=125
| Ranibizumab
0,5 mg pri PRN
n=117
| Stredná priemerná
zmena BCVA od 1. do
12. mesiaca (SD)
|
5,9 (5,5) a
|
6,1 (5,7) a
|
6,2 (6,0)
| Stredná priemerná
zmena BCVA od 1. do
24. mesiaca (SD)
|
6,8 (6,0)
|
6,6 (7,1)
|
7,0 (6,4)
| Stredná zmena BCVA
po 24 mesiacoch (SD)
|
8,3 (8,1)
|
6,5 (10,9)
|
8,1 (8,5)
| Zisk ≥10 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
43,6
|
40,8
|
45,3
| Zisk ≥15 písmen alebo
BCVA ≥84 písmen po
24 mesiacoch (%)
|
25,6
|
28,0
|
30,8
|
|
|
ap<0,0001 pre stanovenie neinferiority oproti PRN
V klinických skúšaniach pri DME sprevádzalo zlepšenie BCVA postupom času zníženie strednej
CSFT vo všetkých skupinách liečby.
Liečba
poškodenia
zraku
v
dôsledku makulárneho edému po RVO
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku makulárneho edému po RVO sa vyhodnotili v randomizovaných, dvojito maskovaných, kontrolovaných štúdiách BRAVO a CRUISE, do ktorých boli zaradené osoby s BRVO (n=397) a CRVO (n=392). V oboch štúdiách pacienti dostávali intravitreálne buď 0,3 mg, alebo 0,5 mg ranibizumabu, alebo simulované injekcie. Po 6 mesiacoch pacienti z kontrolnej skupiny simulovaného podania prešli na 0,5 mg ranibizumabu. V štúdii BRAVO bola vo všetkých skupinách od 3. mesiaca povolená laserová fotokoagulácia ako záchranná liečba.
Kľúčové merané parametre z BRAVO a CRUISE sú zhrnuté v Tabuľkách 4 a 5 a na Obrázkoch 3 a 4.
Tabuľka 4 Výsledky po 6 a 12 mesiacoch (BRAVO)
| Simulované podanie/ Lucentis 0,5 mg
(n=132)
| Lucentis 0,5 mg
(n=131)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 7,3 (13,0)
| 18,3 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 12,1 (14,4)
| 18,3 (14,6)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 28,8
| 61,1
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 43,9
| 60,3
| Podiel (%), ktorý dostal záchrannú liečbu
laserom počas 12 mesiacov
| 61,4
| 34,4
|
|
|
ap<0,0001
O
brázok 3 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(B
RAVO
)
+ 18,3
20
18
+ 18,3
16
14
12 + 12,1
10
8
6
4
2
kontrolované+ 7,3kontrolná skupina simulovaného
s
im
u
l
ovaným podaním
0
podania preradená na ranibizumab
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=132) Ranibizumab 0,5 mg (n=131)
BL=východisková hodnota; SE=stredná chyba priemeru
Tabuľka 5 Výsledky po 6 a 12 mesiacoch (CRUISE)
| Simulované podanie/ Lucentis 0,5 mg (n=130)
| Lucentis 0,5 mg
(n=130)
| Stredná zmena zrakovej ostrosti po
6 mesiacocha (písmená) (SD) (primárny
ukazovateľ)
| 0,8 (16,2)
| 14,9 (13,2)
| Stredná zmena BCVA po 12 mesiacoch
(písmená) (SD)
| 7,3 (15,9)
| 13,9 (14,2)
| Zisk ³15 písmen zrakovej ostrosti po
6 mesiacocha (%)
| 16,9
| 47,7
| Zisk ³15 písmen zrakovej ostrosti po
12 mesiacoch (%)
| 33,1
| 50,8
|
|
|
ap<0,0001
O
brázok 4 Stredná zmena BCVA oproti východiskovej hodnote v čase do 6. a 12. mesiaca
(
CRU
ISE)
20
kontrolované18
simulovaným podaním16
+ 14,9kontrolná skupina simulovaného podania preradená na ranibizumab
14
+ 13,912
10
8
+ 7,36
4
+ 0,82
0
-2
0 1 2 3 4 5 6 7 8 9 10 11 12
Mesiac
Skupina liečby Simulované podanie/Ranibizumab 0,5 mg (n=130) Ranibizumab 0,5 mg (n=130)
BL=východisková hodnota; SE=stredná chyba priemeru
V oboch štúdiách zlepšenie zraku sprevádzalo kontinuálne a významné zmenšovanie makulárneho
edému, merané ako hrúbka centrálnej retiny.
Pacienti s BRVO (BRAVO a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, dosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~15 písmen) ako pacienti, ktorí boli liečení ranibizumabom od začiatku (~16 písmen). Avšak počet pacientov, ktorí ukončili 2 roky liečby, bol obmedzený,
a v štúdii HORIZON boli plánované len štvrťročné kontrolné návštevy. Preto v súčasnosti nie je dosť
údajov, z ktorých by sa dali vyvodiť odporúčania, kedy sa má začať liečba ranibizumabom u pacientov s BRVO.
Pacienti s CRVO (CRUISE a extenzia štúdie HORIZON): Pacienti, ktorí počas prvých 6 mesiacov dostávali simulované injekcie a potom prešli na liečbu ranibizumabom, nedosiahli po 2 rokoch porovnateľné zlepšenie zrakovej ostrosti (~6 písmen) oproti pacientom, ktorí boli liečení ranibizumabom od začiatku (~12 písmen).
Zlepšenie zrakovej ostrosti pozorované pri liečbe ranibizumabom po 6 a 12 mesiacoch sprevádzal pacientmi udávaný prínos liečby stanovený prostredníctvom podškál National Eye Institute Visual Function Questionnaire (NEI VFQ-25) vzhľadom na „aktivity vyžadujúce videnie do blízka“ a
„aktivity vyžadujúce videnie do diaľky“. Rozdiel medzi Lucentisom 0,5 mg a kontrolnou skupinou sa vyhodnotil po 6. mesiaci s hodnotami p 0,02 až 0,0002.
Liečba
poškodenia
zraku
v
dôsledku CNV pri PM
Klinická bezpečnosť a účinnosť Lucentisu u pacientov s poškodením zraku v dôsledku CNV pri PM sa stanovili na základe údajov z 12 mesiacov randomizovaného, dvojito maskovaného, účinnou liečbou kontrolovaného pivotného klinického skúšania F2301 (RADIANCE). Toto skúšanie bolo určené na vyhodnotenie dvoch rôznych režimov dávkovania 0,5 mg ranibizumabu podávaného ako intravitreálna injekcia v porovnaní s PDT verteporfínom (vPDT, fotodynamická liečba Visudynom). Do jednej
z nasledujúcich skupín bolo randomizovaných 277 pacientov:
· Skupina I (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „stability“, definovanými ako žiadna zmena BCVA v porovnaní s dvomi predchádzajúcimi mesačnými hodnoteniami).
· Skupina II (ranibizumab 0,5 mg, režim dávkovania určovaný kritériami „aktivity choroby“, definovanými ako zhoršenie zraku, ktoré možno pripísať intra- alebo subretinálnej tekutine alebo aktívnemu presakovaniu v dôsledku CNV lézie, stanovené prostredníctvom OCT a/alebo
FA).
· Skupina III (vPDT – pacienti mali od 3. mesiaca povolené dostať liečbu ranibizumabom). Počas 12 mesiacov klinického skúšania pacienti dostali v priemere 4,6 injekcií (rozmedzie 1-11) v skupine I a 3,5 injekcií (rozmedzie 1-12) v skupine II. V skupine II, ktorá zodpovedá odporúčanému
dávkovaniu (pozri časť 4.2), 50,9 % pacientov potrebovalo 1 alebo 2 injekcie, 34,5 % potrebovalo 3 až
5 injekcií a 14,7 % potrebovalo 6 až 12 injekcií počas 12 mesiacov trvania skúšania. 62,9 % pacientov v skupine II nepotrebovalo injekcie v druhom 6-mesačnom období skúšania.
Najdôležitejšie výsledky RADIANCE sú zhrnuté v Tabuľke 6 a na Obrázku 5.
Tabuľka 6 Výsledky po 3. a 12. mesiaci (RADIANCE)
| Skupina I
ranibizumab
0,5 mg
„stabilita zraku“
(n=105)
| Skupina II
ranibizumab
0,5 mg
„aktivita choroby“
(n=116)
| Skupina III
vPDTb
(n=55)
| 3. mesiac
|
|
|
| Priemerná zmena BCVA od 1. do 3. mesiaca v porovnaní s východiskovou hodnotoua (písmená)
| +10,5
| +10,6
| +2,2
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
61,9 %
38,1 %
|
65,5 %
43,1 %
|
27,3 %
14,5 %
| 12. mesiac
|
|
|
| Počet injekcií do 12. mesiaca:
Priemer
Medián
|
4,6
4
|
3,5
2,0
|
N/A N/A
| Priemerná zmena BCVA od 1. do
12. mesiaca v porovnaní s východiskovou hodnotou (písmená)
| +12,8
| +12,5
| N/A
| Podiel pacientov, ktorí získali:
≥10 písmen, alebo dosiahli ≥84 písmen
BCVA
≥15 písmen, alebo dosiahli ≥84 písmen
BCVA
|
69,5 %
53,3 %
|
69,0 %
51,7 %
|
N/A N/A
|
|
|
a p<0,00001 v porovnaní s kontrolou vPDT
b Porovnávacia kontrola do 3. mesiaca. Pacienti randomizovaní do skupiny vPDT mali od 3. mesiaca
povolené dostať liečbu ranibizumabom (v skupine III dostalo ranibizumab od 3. mesiaca 38 pacientov)
O
brázok 5 Priemerná zmena oproti východiskovej hodnote BCVA do 12. mesiaca
(
RAD
IANCE)
20
15 +12,5
+14,4
10 +12,1
+13,8
+9,3
5
+1,4
0
Povolený ranibizumab-5
|
|
| Mesiac
|
| Skupina I - ranibizumab 0,5 mg
|
|
|
| podľa stabilizácie (N=105)
|
|
|
| Skupina III - PDT verteporfínom
|
|
|
| (N=55)
|
|
|
|
|
0 1 2 3 4 5 6 7 8 9 10 11 12
Zlepšenie zraku sprevádzal pokles hrúbky centrálnej retiny.
Skupina II - ranibizumab 0,5 mg
podľa aktivity choroby (N=116)
Skupina III - ranibizumab 0,5 mg/PDT
verteporfínom od 3. mesiaca (N=55)
Prínos liečby hlásený pacientmi sa pozoroval vo väčšej miere v skupinách ranibizumabu v porovnaní
s vPDT (hodnota p<0,05) z hľadiska zlepšenia kombinovaného skóre a niekoľkých podškál NEI VFQ-
25 (celkové videnie, aktivity vyžadujúce videnie do blízka, duševné zdravie a odkázanosť na iných).
Deti a dospievajúciBezpečnosť a účinnosť ranibizumabu sa zatiaľ neskúmali u detí a dospievajúcich.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií pre Lucentis vo všetkých vekových podskupinách detí a dospievajúcich pri neovaskulárnej VPDM, poškodení zraku v dôsledku DME, poškodení zraku v dôsledku makulárneho edému po RVO a poškodení zraku
v dôsledku CNV pri PM (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po intravitreálnom podaní Lucentisu raz mesačne pacientom s neovaskulárnou VPDM boli sérové koncentrácie ranibizumabu vo všeobecnosti nízke, s maximálnymi hladinami (Cmax) spravidla pod koncentráciou ranibizumabu potrebnou na inhibovanie biologickej aktivity VEGF o 50 %
(11-27 ng/ml, ako sa stanovilo in vitro v teste bunkovej proliferácie). Cmax bola úmerná dávke
v rozmedzí dávok 0,05 až 1,0 mg/oko. Koncentrácie v sére obmedzeného počtu pacientov s DME
naznačujú, že nemožno vylúčiť o niečo vyššiu systémovú expozíciu v porovnaní s expozíciou, aká sa pozorovala u pacientov s neovaskulárnou VPDM. Koncentrácie ranibizumabu v sére pacientov s RVO
boli podobné alebo mierne vyššie v porovnaní s koncentráciami, ktoré sa pozorovali u pacientov
s neovaskulárnou VPDM.
Na základe analýzy populačnej farmakokinetiky a vymiznutia ranibizumabu zo séra pacientov s neovaskulárnou VPDM liečených dávkou 0,5 mg, priemerný vitreálny eliminačný polčas ranibizumabu je približne 9 dní. Pri intravitreálnom podávaní Lucentisu 0,5 mg/oko raz mesačne sa predpokladá, že Cmax ranibizumabu v sére, ktorá sa dosiahne približne 1 deň po podaní, bude vo všeobecnosti v rozmedzí medzi 0,79 a 2,90 ng/ml, a Cmin sa vo všeobecnosti predpokladá v rozmedzí medzi 0,07 a 0,49 ng/ml. Predpokladaná sérová koncentrácia ranibizumabu je približne 90 000- násobne nižšia ako vitreálna koncentrácia ranibizumabu.
Pacienti s poškodením funkcie obličiek: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky Lucentisu u pacientov s poškodením funkcie obličiek. V populačnej farmakokinetickej analýze pacientov s neovaskulárnou VPDM malo 68 % (136 z 200) pacientov poškodenie funkcie obličiek (46,5 % ľahké [50-80 ml/min], 20 % stredne ťažké [30-50 ml/min] a 1,5 % ťažké [<30 ml/min]).
U pacientov s RVO malo 48,2 % (253 z 525) poškodenie funkcie obličiek (36,4 % ľahké, 9,5 %
stredne ťažké a 2,3 % ťažké). Systémový klírens bol trochu nižší, čo však nebolo klinicky významné.
Pacienti s poškodením funkcie pečene: Nevykonali sa formálne štúdie na sledovanie farmakokinetiky
Lucentisu u pacientov s poškodením funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Bilaterálne intravitreálne podávanie ranibizumabu opiciam rodu Cynomolgus v dávkach medzi
0,25 mg/oko a 2,0 mg/oko raz za 2 týždne až do 26 týždňov malo za následok účinky na oči závislé od
dávky.
Intraokulárne sa zaznamenalo od dávky závislé zosilnenie zápalu a zvýšenie počtu buniek v prednej očnej komore s maximom 2 dni po podaní injekcie. Závažnosť zápalovej odpovede sa spravidla znížila pri podaní ďalších injekcií alebo počas zotavenia. V zadnom segmente sa pozorovala vitreálna infiltrácia buniek a zákaly sklovca, ktoré tiež mali tendenciu závisieť od dávky a spravidla pretrvávali do konca liečebného obdobia. V štúdii trvajúcej 26 týždňov sa intenzita zápalu sklovca zvyšovala
s počtom injekcií. Po zotavení sa však pozorovali dôkazy reverzibility. Povaha a načasovanie zápalu zadného segmentu poukazuje na imunitne sprostredkovanú odpoveď protilátok, čo môže byť klinicky
nevýznamné. Pri niektorých zvieratách sa pozoroval vznik katarakty po relatívne dlhom období
intenzívneho zápalu, čo naznačuje, že zmeny na šošovke sú sekundárne po ťažkom zápale. Prechodné zvýšenie vnútroočného tlaku po podaní sa pozorovalo po intravitreálnych injekciách bez ohľadu na
dávku.
Mikroskopické očné zmeny súviseli so zápalom a nepoukazovali na degeneratívne procesy. V niektorých očiach sa zaznamenali granulomatózne zápalové zmeny na papile. Tieto zmeny v zadnom segmente ustupovali a v niektorých prípadoch vymizli počas zotavovania.
Po intravitreálnom podaní sa nezistili žiadne známky systémovej toxicity. V podsúbore liečených zvierat sa našli sérové a sklovcové protilátky voči ranibizumabu.
Nie sú dostupné údaje o karcinogenite alebo mutagenite.
U gravidných opíc nespôsobilo intravitreálne podávanie ranibizumabu, ktoré malo za následok maximálne systémové expozície 0,9- až 7-násobne vyššie ako najhorší prípad klinickej expozície, vývojovú toxicitu alebo teratogenitu a nemalo žiadny vplyv na hmotnosť alebo štruktúru placenty,
hoci ranibizumab sa vzhľadom na jeho mechanizmus účinku má považovať za potenciálne teratogénny
a embryo- a fetotoxický.
Neprítomnosť účinkov na vývoj embrya a plodu sprostredkovaných ranibizumabom pravdepodobne súvisí hlavne s neschopnosťou fragmentu Fab prestupovať cez placentu. Napriek tomu bol popísaný prípad vysokých hladín ranibizumabu v sére matky a prítomnosti ranibizumabu v sére plodu, čo naznačuje, že protilátka proti ranibizumabu fungovala ako transportná bielkovina (obsahujúca segment Fc) pre ranibizumab, čím sa znižoval klírens zo séra matky a umožňoval sa prestup cez placentu. Keďže sledovania vývoja embryí a plodov sa robili u zdravých gravidných zvierat a ochorenie (napr. diabetes) môže meniť priepustnosť placenty pre fragment Fab, štúdia sa má interpretovať s opatrnosťou.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Dihydrát α,α-trehalózy Monohydrát histidíniumchloridu Histidín
Polysorbát 20
Voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
Uchovávajte injekčnú liekovku vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorená injekčná liekovka uchovávať pri izbovej teplote (25°C) najviac
24 hodín.
6.5 Druh obalu a obsah balenia
0,23 ml sterilného roztoku v injekčnej liekovke (sklo typu I) so zátkou (chlórobutylová guma) a 1 tupá ihla s filtrom (18G x 1½″, 1,2 mm x 40 mm, 5 µm). Balenie obsahuje 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka a ihla s filtrom sú určené len na jednorazové použitie. Opakované použitie môže mať za následok infekciu alebo iné ochorenie/poškodenie zdravia. Všetky zložky sú sterilné. Akákoľvek zložka, ktorej obal vykazuje známky poškodenia alebo nesprávnej manipulácie, sa nesmie použiť. Sterilitu nemožno zaručiť, ak uzáver obalu zložky nie je neporušený.
Na prípravu a podanie intravitreálnej injekcie sú potrebné nasledujúce pomôcky na jednorazové
použitie:
- ihla s 5 µm filtrom (18G x 1½″, 1,2 mm x 40 mm, je súčasťou balenia)
- 1 ml sterilná injekčná striekačka (nie je súčasťou balenia Lucentisu)
- injekčná ihla (30G x ½″; nie je súčasťou balenia Lucentisu)
Pri príprave Lucentisu na intravitreálne podanie dodržiavajte, prosím, nasledujúce pokyny:
1. Pred odobratím obsahu sa má dezinfikovať vonkajšia časť gumenej zátky injekčnej liekovky.
2. Asepticky nasaďte ihlu s 5 µm filtrom (18G x 1½″, 1,2 mm x 40 mm, z balenia) na 1 ml injekčnú striekačku. Vtlačte hrubú ihlu s filtrom do stredu zátky liekovky, až sa ihla dotkne dna injekčnej liekovky.
3. Odoberte všetku tekutinu z injekčnej liekovky, pri čom liekovka má byť vo zvislej polohe,
mierne naklonená, aby sa uľahčilo úplné odobratie obsahu.
4. Dbajte na to, aby ste pri vyprázdnení injekčnej liekovky dostatočne potiahli piest, aby sa ihla
s filtrom úplne vyprázdnila.
5. Hrubú ihlu s filtrom nechajte v injekčnej liekovke a odpojte injekčnú striekačku od hrubej ihly s filtrom. Ihla s filtrom sa má po odobratí obsahu injekčnej liekovky zahodiť a nemá sa použiť na intravitreálnu injekciu.
6. Asepticky pevne nasaďte injekčnú ihlu (30G x ½″) na injekčnú striekačku.
7. Opatrne odstráňte kryt z injekčnej ihly bez toho, aby ste odpojili injekčnú ihlu od injekčnej striekačky.
Poznámka: Pri odstránení krytu pridržte násadec injekčnej ihly.
8. Opatrne vytlačte vzduch z injekčnej striekačky a upravte dávku na rysku označujúcu 0,05 ml na striekačke. Injekčná striekačka je pripravená na podanie injekcie.
Poznámka: Injekčnú ihlu neutierajte. Nepotiahnite piest injekčnej striekačky.
Po podaní injekcie nenasaďte kryt späť na injekčnú ihlu, ani ihlu neodpojte od injekčnej striekačky. Zahoďte použitú injekčnú striekačku spolu s ihlou do odpadovej nádoby na ostré predmety, alebo zlikvidujte v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/374/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 22. január 2007
Dátum posledného predĺženia: 24. január 2012
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu