
potrebné upraviť dávku levetiracetamu podľa funkcie obličiek, pretože klírens levetiracetamu závisí od funkcie obličiek. Toto odporúčanie je založené na štúdii s dospelými pacientmi s poruchou funkcie obličiek.
CLcr v ml/min/1,73 m2 je možné odhadnúť zo stanovenia sérového kreatinínu (mg/dl) pre mladých dospievajúcich a deti s použitím nasledujúceho vzorca (Schwartzov vzorec):
Výška (cm) x ks
CLcr (ml/min/1,73 m2) = ------------------------------------ Sérový kreatinín (mg/dl)
ks = 0,55 pre deti mladšie ako 13 rokov a dospievajúce dievčatá; ks = 0,7 pre dospievajúcich chlapcov
Úprava dávkovania pre deti a dospievajúcich pacientov s telesnou hmotnosťou menej ako 50 kg s poruchou funkcie obličiek:
Skupina Klírens kreatinínu
(ml/min/1,73 m2)
Dávka a frekvencia
Deti od 4 rokov a mladiství s hmotnosťou
do 50 kg
Normálna ≥80 10 až 30 mg/kg (0,10 až 0,30 ml/kg) dvakrát denne
Mierna 50-79 10 až 20 mg/kg (0,10 až 0,20 ml/kg) dvakrát denne
Stredne závažná 30-49 5 až 15 mg/kg (0,05 až 0,15 ml/kg) dvakrát denne
Závažná <30 5 až 10 mg/kg (0,05 až 0,10 ml/kg) dvakrát denne
Dialyzovaní pacienti v terminálnom štádiu zlyhania obličiek
-- 10 až 20 mg/kg (0,10 až 0,20 ml/kg)
jedenkrát denne (1)(2)

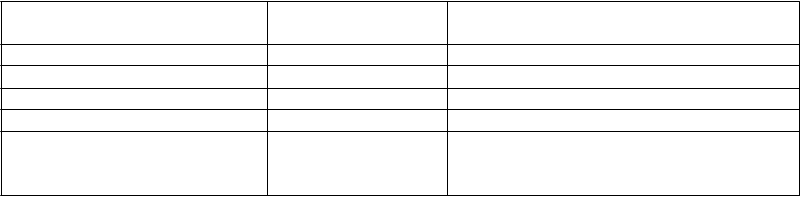
(1) V prvý deň liečby levetiracetamom sa odporúča úvodná dávka 15 mg/kg (0,15 ml/kg)
(2) Po dialýze sa odporúča dodatočná dávka 5 až 10 mg/kg (0,05 až 0,10 ml/kg)
Porucha funkcie pečeneU pacientov s miernou až stredne závažnou poruchou funkcie pečene nie je potrebná žiadna úprava dávky. U pacientov so závažnou poruchou funkcie pečene môže klírens kreatinínu podhodnocovať insuficienciu
obličiek. Preto sa pri klírense kreatinínu < 60 ml/min/1,73 m2 odporúča znížiť dennú udržiavaciu dávku o 50 %
Pediatrická populácia
Lekár má predpísať najvhodnejšiu liekovú formu, balenie a silu podľa veku, hmotnosti a dávky.
Monoterapia
Bezpečnosť a účinnosť levetiracetamu u deti a dospievajúcich vo veku do 16 rokov ako monoterapie neboli stanovené.
K dispozícii nie sú žiadne údaje.
Prídavná liečba pre deti vo veku 4 až 11 rokov a dospievajúcich (12 až 17 rokov) s hmotnosťou nižšou ako
50 kg
Začiatočná terapeutická dávka je 10 mg/kg dvakrát denne.
V závislosti od klinickej odpovede a znášanlivosti možno dávku zvýšiť až na 30 mg/kg dvakrát denne.
Zmeny dávky nemajú prekročiť zvýšenie alebo zníženie o 10 mg/kg dvakrát denne každé dva týždne.
Má sa použiť najnižšia účinná dávka.
Dávka u detí s hmotnosťou 50 kg alebo vyššou je rovnaká ako u dospelých.
Odporúčaná dávka pre deti a dospievajúcich:
Hmotnosť Začiatočná dávka:
10 mg/kg dvakrát denne
Maximálna dávka:
30 mg/kg dvakrát denne

15 kg(1) 150 mg dvakrát denne 450 mg dvakrát denne
20 kg(1) 200 mg dvakrát denne 600 mg dvakrát denne
25 kg 250 mg dvakrát denne 750 mg dvakrát denne od 50 kg(2) 500 mg dvakrát denne 1500 dvakrát denne
(1) Deti s hmotnosťou 25 kg alebo nižšou majú prednostne začať liečbu levetiracetamom 100 mg/ml perorálny roztok.
(2) Dávka u detí a dospievajúcich s hmotnosťou 50 kg alebo vyššou je rovnaká ako u dospelých.
Prídavná liečba pre dojčatá a deti mladšie ako 4 rokyBezpečnosť a účinnosť levetiracetamu infúzneho koncentrátu u dojčiat a deti vo veku menej ako 4 roky
neboli stanovené.
V súčasnosti dostupné údaje sú uvedené v častiach 4.8, 5.1 a 5.2, nie je však možné stanoviť odporúčané
dávkovanie.
Spôsob podávaniaLiečba levetiracetamom sa môže začať buď intravenóznym alebo perorálnym podaním.
Prechod na alebo z perorálneho na intravenózne podanie možno vykonať priamo bez titrácie. Má sa zachovať celková denná dávka a frekvencia podávania.
Koncentrát Levetiracetamu Hospira je určený len na intravenózne použitie a odporúčaná dávka sa má rozpustiť v najmenej 100 ml kompatibilného rozpúšťadla a podávať intravenózne ako 15-minútová
intravenózna infúzia (pozri časť 6.6).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na iné deriváty pyrolidónu alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Vysadenie
Ak sa musí levetiracetam vysadiť, podľa súčasnej klinickej praxe sa odporúča vysadzovať ho postupne
(napr. u dospelých a dospievajúcich s telesnou hmotnosťou viac ako 50 kg: znižovanie o 500 mg dvakrát
denne každé dva až štyri týždne, u detí a dospievajúcich s hmotnosťou do 50 kg: nemá zníženie dávky prekročiť pokles 10 mg/kg dvakrát denne každé dva týždne).
Renálna insuficiencia
Podávanie levetiracetamu pacientom s poruchou funkcie obličiek môže vyžadovať úpravu dávky. U
pacientov so závažnou poruchou funkcie pečene sa pred stanovením dávky odporúča posúdiť funkciu obličiek (pozri časť 4.2).
Samovražda
U pacientov liečených antiepileptikami (vrátane levetiracetamu) boli hlásené prípady samovraždy,
pokusov o samovraždu, samovražedných myšlienok a správania. Meta-analýza randomizovaných placebom kontrolovaných skúšaní s antiepileptikami preukázala malé zvýšenie rizika samovražedných
myšlienok a správania. Mechanizmus vzniku tohto rizika nie je známy.
Z tohto dôvodu sa majú u pacientov sledovať príznaky depresie a/alebo samovražedných myšlienok a správania a má sa zvážiť vhodná liečba. Pacientom (a ich opatrovateľom) sa má odporučiť, aby v prípade výskytu príznakov depresie a/alebo samovražedných myšlienok alebo správania, okamžite vyhľadali lekársku pomoc.
Pediatrická populácia
Dostupné údaje u detí nenaznačili vplyv na rast a pubertu. Avšak dlhodobé účinky u detí na schopnosť
učiť sa, inteligenciu, rast, endokrinnú funkciu, pubertu a plodnosť sú naďalej neznáme.
Pomocné látky
Tento liek obsahuje 2,5 mmol (alebo 57 mg) sodíka v maximálnej jednorazovej dávke (0,8 mmol (alebo
19 mg) v injekčnej liekovke). To je potrebné zohľadniť u pacientov s kontrolovanou sodíkovou diétou.
4.5 Liekové a iné interakcie
Antiepileptiká
Predmarketingové údaje z klinických štúdií vykonaných na dospelých ukazujú, že levetiracetam nemal
vplyv na sérové koncentrácie už podávaných antiepileptík (fenytoín, karbamazepín, kyselina valproová, fenobarbital, lamotrigín, gabapentín a primidon) a že tieto antiepileptiká neovplyvnili
farmakokinetiku levetiracetamu.
Rovnako ako u dospelých, ani u detských a dospievajúcich pacientov užívajúcich až do 60 mg/kg/deň
levetiracetamu nie je žiadny dôkaz klinicky významných liekových interakcií.
Retrospektívne hodnotenie farmakokinetických interakcií u detí a dospievajúcich s epilepsiou (4 až
17 rokov) potvrdilo, že prídavná liečba s perorálne podávaným levetiracetamom neovplyvnila sérové koncentrácie v rovnovážnom stave súbežne podávaného karbamazepínu a valproátu. Avšak údaje naznačujú o 20 % vyšší klírens levetiracetamu u detí užívajúcich enzýmy indukujúce antiepileptiká. Úprava dávky sa nevyžaduje.
Probenecid
Zistilo sa, že probenecid (500 mg štyrikrát denne), blokátor renálnej tubulárnej sekrécie, inhibuje
renálny klírens primárneho metabolitu, nie však levetiracetamu. Koncentrácia uvedeného metabolitu však
zostáva nízka. Možno očakávať, že ostatné lieky vylučované aktívnou tubulárnou sekréciou by tiež mohli znižovať renálny klírens metabolitu. Účinok levetiracetamu na probenecid sa nezisťoval a účinok levetiracetamu na ďalšie aktívne vylučované lieky, napr. NSAID, sulfónamidy a metotrexát nie je známy.
Perorálne kontraceptíva a iné farmakokinetické interakcie
Levetiracetam v dávke 1000 mg denne nemal vplyv na farmakokinetiku perorálnych kontraceptív
(etinylestradiol a levonorgestrel); endokrinné parametre (luteinizačný hormón a progesterón) sa
nezmenili. Levetiracetam v dávke 2000 mg denne nemal vplyv na farmakokinetiku digoxínu a warfarínu; protrombínové časy sa nezmenili. Súbežné podávanie s digoxínom, perorálnymi kontraceptívami a warfarínom neovplyvnilo farmakokinetiku levetiracetamu.
Alkohol
Nie sú k dispozícií žiadne údaje o interakcii levetiracetamu s alkoholom.
4.6 Fertilita, gravidita a laktácia
Gravidita
Postmarketingové údaje z niekoľkých prospektívnych registrov gravidít zdokumentovali výsledky u viac
ako 1000 žien liečených levetiracetamom v monoterapii počas prvého trimestra gravidity. Celkovo tieto údaje nenaznačujú podstatné zvýšenie rizika väčších kongenitálnych malformácií, hoci teratogénne riziko
nie je možné úplne vylúčiť. Liečba viacerými antiepileptikami je spojená s vyšším rizikom
kongenitálnych malformácií ako monoterapia a preto je potrebné zvážiť monoterapiu.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Levetiracetam sa neodporúča užívať počas gravidity a u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu, pokiaľ to nie je klinicky nevyhnutné.
Rovnako ako u iných antiepileptík, fyziologické zmeny počas gravidity môžu ovplyvniť koncentráciu levetiracetamu. Počas gravidity bol pozorovaný pokles plazmatickej koncentrácie levetiracetamu. Tento pokles je výraznejší počas tretieho trimestra (do 60 % východiskovej hodnoty koncentrácie pred graviditou). Pre gravidnú ženu liečenú levetiracetamom sa má zabezpečiť adekvátny klinický manažment. Vysadenie antiepileptickej liečby môže viesť k exacerbácii ochorenia, ktoré môže poškodiť matku a plod.
Laktácia
Levetiracetam sa vylučuje do materského mlieka. Dojčenie sa preto neodporúča. Avšak, v prípade, že je
liečba levetiracetamom potrebná počas dojčenia, pomer prínosu/rizika liečby sa má zvážiť vzhľadom k významu dojčenia.
Fertilita
V štúdiách u zvierat sa nezistil žiadny vplyv na fertilitu (pozri časť 5.3). Nie sú k dispozícii žiadne
klinické údaje, nie je známe potenciálne riziko u ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Vzhľadom na možnú rozdielnu individuálnu citlivosť niektorí pacienti môžu najmä na začiatku liečby, alebo po zvýšení dávky pociťovať ospalosť alebo iné symptómy v súvislosti s centrálnym nervovým systémom. Preto sa u týchto pacientov odporúča opatrnosť pri vykonávaní náročných aktivít, napr. pri vedení vozidiel alebo pri obsluhe strojov. Pacientom sa neodporúča viesť vozidlá ani obsluhovať stroje, kým sa nestanoví, že ich schopnosť vykonávať takéto činnosti nie je ovplyvnená.
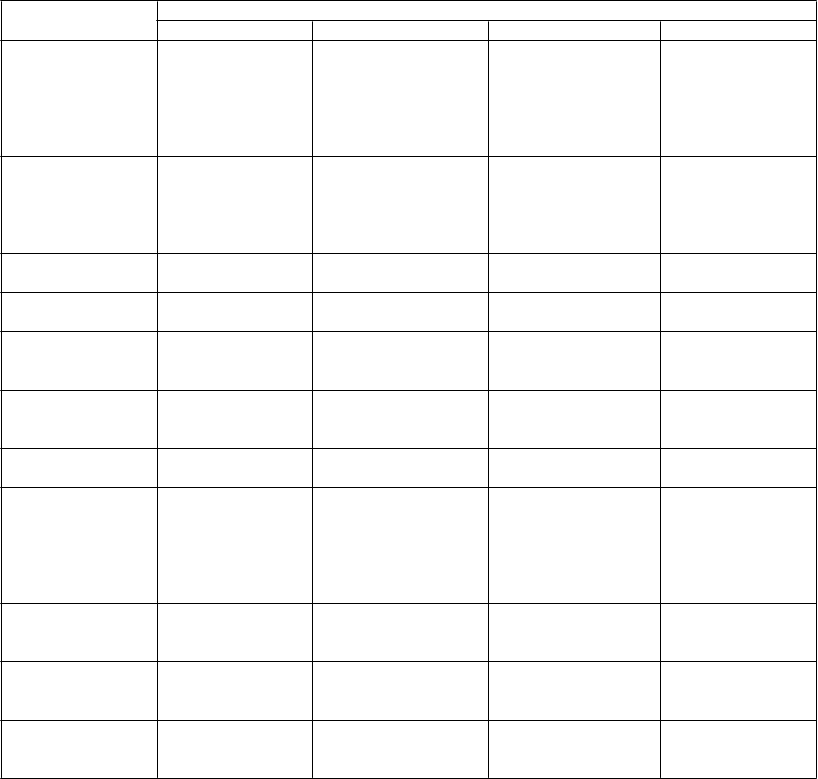
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Profil nežiaducich udalostí uvedený nižšie vychádza z analýzy združených placebom kontrolovaných
klinických skúšaní so všetkými skúmanými indikáciami s celkovým počtom 3416 pacientov liečených levetiracetamom. Tieto údaje sú doplnené o používanie levetiracetamu v zodpovedajúcom nezaslepenom predĺžení štúdií rovnako ako zo sledovania po uvedení lieku na trh. Najčastejšie hlásené nežiaduce reakcie boli nazofaryngitída, somnolencia, bolesť hlavy, únava a závrat. Profil bezpečnosti levetiracetamu je celkovo podobný vo všetkých vekových skupinách (dospelí, detskí a dospievajúci pacienti) a vo všetkých schválených epileptických indikáciách. Z dôvodu obmedzenej expozície používania intravenózneho levetiracetamu a pretože perorálna a intravenózna lieková forma sú bioekvivalentné, informácie
o bezpečnosti intravenózneho levetiracetamu sa opierajú o perorálne používanie levetiracetamu.
Zoznam nežiaducich reakcií zoradených dotabuľky
Nežiaduce reakcie hlásené v klinických štúdiách (u dospelých, dospievajúcich a detí vo veku > 1 mesiac)
a z postmarketingových skúseností sú uvedené v nasledujúcej tabuľke podľa tried orgánových systémov a podľa frekvencie. Frekvencia je definovaná nasledovne: veľmi časté (≥1/10); časté ((≥1/100 až <1/10); menej časté (≥1/1000 až <1/100); zriedkavé (≥1/10 000 až <1/1000) a veľmi zriedkavé (<1/10 000).
TOS MedDRA Kategória frekvencie
Veľmi časté Časté Menej časté Zriedkavé
Infekcie a nákazy Nazofaryngitída Infekcia
P
or
u
chy krvi a
lymfatického
systému
P
or
u
chy imunitného
s
ys
tému
P
or
u
chy
m
etabolizmu a
výživy
Trombocytopénia, leukopénia
Anorexia Zníženie hmotnosti, zvýšenie hmotnosti
Pancytopénia, neutropénia, agranulocytóza Lieková reakcia s eozinofíliou
a systémovými symptómami
(DRESS)
 P
s
y
c
h
ické poruchy
P
s
y
c
h
ické poruchy Depresia, hostilita/agresivita, anxieta, insomnia, nervozita/podráždenosť
Pokus o samovraždu, myšlienky na samovraždu, psychotická porucha,
Dokonaná samovražda, porucha osobnosti, nezvyčajné
TOS MedDRA Kategória frekvencie
Veľmi časté Časté Menej časté Zriedkavé
P
or
u
chy nervového
s
ys
tému
Somnolencia,
bolesť hlavy
Záchvat, porucha rovnováhy, závrat, letargia, tremor
abnormálne správanie, halucinácia, hnev, stav zmätenosti, panicky záchvat, citová labilita/kolísanie nálady, agitácia Amnézia, porucha pamäti, nezvyčajná koordinácia/ataxia, parestézia, porucha pozornosti
myslenie
Choreoatetóza, dyskinéza, hyperkinéza
P
or
u
chy oka Diplopia, rozmazané videnie
P
or
u
chy ucha a
labyrintu
P
or
u
chy dýchacej
sústavy, hrudníka a
mediastína
P
or
u
chy
g
astrointestinálneho
traktu
P
or
u
chy
p
ečene
a
žlčových
ciest
Vertigo
Kašeľ
Bolesť brucha, hnačka, dyspepsia, vracanie, nauzea
Abnormálne testy
pečeňovej funkcie
Pankreatitída
Zlyhanie pečene,
hepatitída
P
or
u
chy kože a
podkožného tkaniva
P
or
u
chy kostrovej a
svalovej sústavy a
spojivového tkaniva
C
elkové poruchy a
reakcie v mieste
podania
Úrazy, otravy a
komplikácie
liečebného
postupu
Vyrážka Alopécia, ekzém, pruritus
Svalová slabosť,
myalgia
Asténia/únava
Úraz
Toxická epidermálna nekrolýza, Stevensov-
Johnsonov syndróm, multiformný erytém
 Popis vybraných nežiaducich reakcií
Popis vybraných nežiaducich reakcií
Riziko anorexie je vyššie, keď sa topiramát podáva súbežne s levetiracetamom.
V niekoľkých prípadoch alopécie sa po vysadení levetiracetamu pozorovala úprava stavu. V niektorých prípadoch pancytopénie bola identifikovaná supresia kostnej drene.
Pediatrická populáciaU pacientov vo veku 1 mesiac až menej ako 4 roky bolo celkovo 190 pacientov liečených
levetiracetamom v placebom kontrolovaných a nezaslepených predĺženiach štúdií, z ktorých šesťdesiat
(60) pacientov bolo liečených levetiracetamom v placebom kontrolovaných štúdiách. U pacientov vo veku
4-16 rokov bolo celkovo 645 pacientov liečených levetiracetamom v placebom kontrolovaných štúdiách a
nezaslepenom predĺžení štúdií, z ktorých 233 pacientov bolo liečených levetiracetamom v placebom
kontrolovaných štúdiách. U oboch týchto pediatrických vekových rozmedzí boli tieto údaje doplnené o skúsenosti s používaním levetiracetamu po uvedení lieku na trh.
Profil nežiaducich udalostí levetiracetamu je celkovo podobný vo vekových skupinách a v schválených epileptických indikáciách. Výsledky bezpečnosti u detských a dospievajúcich pacientov v placebom kontrolovaných klinických štúdiách sa zhodovali s profilom bezpečnosti levetiracetamu u dospelých
s výnimkou behaviorálnych a psychiatrických nežiaducich reakcií, ktoré boli častejšie u detí ako
u dospelých. U detí a dospievajúcich vo veku 4 až 16 rokov bolo vracanie (veľmi časté, 11,2 %), agitácia
(časté, 3,4 %), kolísanie nálady (časté, 2,1 %), afektová labilita (časté, 1,7 %), agresivita (časté, 8,2 %), abnormálne správanie (časté, 5,6 %) a letargia (časté, 3,9 %) hlásené častejšie ako u iných vekových rozmedzí alebo v celkovom profile bezpečnosti. U dojčiat a detí vo veku 1 mesiac až menej ako 4 roky bolo podráždenie (veľmi časté, 11,7 %) a abnormálna koordinácia (časté, 3,3 %) hlásené častejšie ako
u iných vekových skupín alebo v celkovom profile bezpečnosti.
Dvojito zaslepená, placebom kontrolovaná pediatrická štúdia bezpečnosti s non-inferiórnym dizajnom hodnotila kognitívne a neuropsychologické účinky levetiracetamu u detí vo veku 4 až 16 rokov
s parciálnymi záchvatmi. Bolo konštatované, že levetiracetam sa neodlišoval (nebol inferiórny) od
placeba, pokiaľ ide o zmenu od východiskového stavu v skóre Leiter-R na pozornosť a pamäť, zloženom
skóre k hodnoteniu pamäti (Leiter-R Attention and Memory, Memory Screen Composite score)
u populácie splňujúcej protokol štúdie. Výsledky týkajúce sa behaviorálneho a emočného fungovania naznačovali u pacientov liečených levetiracetamom zhoršenie, pokiaľ ide o agresívne správanie, čo bolo
merané štandardizovaným a systematickým spôsobom s použitím overeného nástroja (CBCL -Achenbach
Child Behavior Checklist; Achenbachov kontrolný zoznam správania detí). Avšak, u jedincov, ktorí užívali levetiracetam v dlhodobej nezaslepenej následnej štúdii, nedošlo v priemere k zhoršeniu
behaviorálneho a emočného fungovania; obzvlášť miery agresívneho správania neboli horšie oproti
východiskovému stavu.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieSymptómyPo predávkovaní levetiracetamom sa pozorovala somnolencia, nepokoj, agresia, znížený stupeň vedomia,
depresia dýchania a kóma.
Liečba predávkovaniaNeexistuje žiadne špecifické antidotum levetiracetamu. Liečba predávkovania má byť symptomatická
a môže zahŕňať hemodialýzu. Účinnosť vylučovania levetiracetamu dialýzou je 60 % a primárneho
metabolitu 74 %.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antiepileptiká, iné antiepileptiká, ATC kód: N03AX14.
Liečivo levetiracetam je pyrolidónový derivát (S-enantiomér alfa-etyl-2-oxo-1-pyrolidín acetamidu),
chemicky nesúvisiaci s liečivami v súčasných antiepileptikách.
Mechanizmus účinku
Mechanizmus účinku levetiracetamu nebol dosiaľ celkom objasnený, zdá sa však, že je odlišný od
účinkov ostatných v súčasnosti používaných antiepileptík. Pokusy in vitro a in vivo napovedajú, že levetiracetam neovplyvňuje ani základné charakteristiky buniek ani normálny proces nervových vzruchov. In vitro štúdie ukazujú, že levetiracetam ovplyvňuje hladinu Ca2+ v neurónoch čiastočnou inhibíciou kalciových kanálov typu N a znížením uvoľňovania Ca2+ z intracelulárnych zásob v neurónoch. Levetiracetam naviac čiastočne antagonizuje zníženie aktivity GABA- a glycínových kanálov spôsobené zinkom a ß-karbolínmi. Okrem toho sa levetiracetam podľa in vitro štúdií viaže na špecifické väzbové miesto v mozgovom tkanive hlodavcov. Špecifickým väzbovým miestom je synaptický vezikulárny proteín 2A, ktorý je považovaný za súčasť procesov fúzie a exocytózy vezikúl s neurotransmitermi. Levetiracetam a jeho analógy majú rôznu afinitu k väzbe na synaptický vezikulárny proteín 2A, ktorá koreluje s ich potenciálom k zábrane vzniku záchvatov v audiogénnych modeloch u myší. Uvedený nález napovedá tomu, že interakcia medzi levetiracetamom a synaptickým vezikulárnym proteínom 2A by
mohla prispievať k antiepileptickému mechanizmu účinku lieku.
Farmakodynamické účinky
Levetiracetam poskytuje ochranu pred záchvatmi vo veľkom počte zvieracích modelov parciálnych a
primárne generalizovaných záchvatov bez toho, žeby mal pro-konvulzívny účinok. Primárny metabolit je neaktívny. Účinok pri parciálnej i generalizovanej epilepsii (epileptiformný výboj/fotoparoxyzmálna odpoveď) u človeka potvrdil široké spektrum stanoveného farmakologického profilu levetiracetamu.
Klinická účinnosť abezpečnosť
Prídavná terapia na liečbu parciálnych záchvatov so sekundárnou generalizáciou alebo bez nej u
dospelých, dospievajúcich a detí vo veku od 4 rokov s epilepsiou.
U dospelých sa účinnosť levetiracetamu dokázala v 3 dvojito zaslepených placebom kontrolovaných štúdiách pri 1000 mg, 2000 mg alebo 3000 mg/deň podávaných v 2 rozdelených dávkach s dĺžkou liečby do 18 týždňov. V sumárnej analýze bolo percento pacientov, ktorí dosiahli 50 % alebo významnejšie zníženie frekvencie parciálnych záchvatov na týždeň v porovnaní s východiskovým stavom pri stabilnej dávke (12/14 týždňov), 27,7 %, 31,6 % a 41,3 % u pacientov s 1000, 2000 alebo 3000 mg levetiracetamu a 12,6 % u pacientov s placebom.
Pediatrická populácia
U detských a dospievajúcich pacientov (vek 4 až 16 rokov) sa účinnosť levetiracetamu stanovila v dvojito- zaslepenej placebom kontrolovanej štúdii, do ktorej bolo zaradených 198 pacientov a malo dĺžku liečby 14 týždňov. V tejto štúdii pacienti užívali levetiracetam vo fixnej dávke 60 mg/kg/deň (s dávkovaním dvakrát denne).
44,6 % pacientov liečených levetiracetamom a 19,6 % pacientov s placebom malo 50 % alebo významnejšie zníženie frekvencie parciálnych záchvatov za týždeň v porovnaní s východiskovým stavom. Pri dlhodobom pokračovaní v liečbe 11,4 % pacientov nemalo záchvaty minimálne 6 mesiacov a 7,2 % nemalo záchvat minimálne 1 rok.
Monoterapia na liečbu parciálnych záchvatov so sekundárnou generalizáciou alebo bez nej u pacientov
vo veku od 16 rokov s novo diagnostikovanou epilepsiou.
Účinnosť levetiracetamu v monoterapii bola preukázaná v dvojito zaslepenej paralelnej skupine
„noninferiority“ v porovnaní s karbamazepínom s riadeným uvoľňovaním (CR) u 576 pacientov vo veku
16 rokov alebo starších s novo alebo nedávno diagnostikovanou epilepsiou. U pacientov sa mohli vyskytnúť len nevyprovokované parciálne záchvaty alebo generalizované tonicko-klonické záchvaty. Pacienti boli randomizovaní na liečbu karbamazepínom CR 400 – 1200 mg/deň alebo levetiracetamom
1000 - 3000 mg/deň, dĺžka liečby bola do 121 týždňov v závislosti od reakcie.
Šesťmesačné obdobie bez výskytu záchvatov sa dosiahlo u 73,0 % pacientov liečených levetiracetamom
a 72,8 % pacientov liečených karbamazepínom CR; upravená absolútna diferencia medzi liečbami bola
0,2 % (95 % CI: -7,8 8,2). Viac ako polovica jedincov nemala záchvaty 12 mesiacov (56,6 % jedincov s levetiracetamom a 58,5 % s karbamazepínom CR).
V štúdii odrážajúcej klinickú prax bolo možné u obmedzeného počtu pacientov, ktorí reagovali na prídavnú liečbu levetiracetamom (36 dospelých pacientov zo 69), vysadiť súbežnú antiepileptickú liečbu.
Prídavná terapia na liečbu myoklonických záchvatov u dospelých a dospievajúcich vo veku od 12 rokov s
juvenilnou myoklonickou epilepsiou.
Účinnosť levetiracetamu bola preukázaná v dvojito-zaslepenej placebom kontrolovanej štúdii s trvaním 16 týždňov u pacientov vo veku 12 rokov a starších, ktorí trpeli na idiopatickú generalizovanú epilepsiu s myoklonickými záchvatmi s rôznymi syndrómami. Väčšina pacientov mala výskyt juvenilnej myoklonickej epilepsie.
V tejto štúdii bola dávka levetiracetamu 3000 mg/deň podávaná v 2 rozdelených dávkach.
58,3 % pacientov liečených levetiracetamom a 23,3 % pacientov s placebom malo minimálne 50 % zníženie denných myoklonických záchvatov na týždeň. Pri dlhodobom pokračovaní v liečbe 28,6 % pacientov nemalo myoklonické záchvaty minimálne 6 mesiacov a 21,0 % nemalo myoklonické záchvaty minimálne 1 rok.
Prídavná terapia na liečbu primárnych generalizovaných tonicko-klonických záchvatov u dospelých a dospievajúcich vo veku od 12 rokov s idiopatickou generalizovanou epilepsiou.
Účinnosť levetiracetamu bola preukázaná v 24-týždňovej dvojito-zaslepenej placebom kontrolovanej štúdii, do ktorej boli zaradení dospelí, dospievajúci a obmedzený počet detí, ktorí trpeli na idiopatickú generalizovanú epilepsiu s primárnymi generalizovanými tonicko-klonickými (PGTC) záchvatmi
s rôznymi syndrómami (juvenilná myoklonická epilepsia, juvenilná absencia epilepsie, absencia epilepsie v detstve alebo epilepsia s Grand Mal záchvatmi pri prebudení). V tejto štúdii bola dávka'
levetiracetamu 3000 mg/deň pre dospelých a dospievajúcich alebo 60 mg/kg/deň pre deti podávaná v 2
rozdelených dávkach.
72,2 % pacientov liečených levetiracetamom a 45,2 % pacientov s placebom malo 50 % alebo významnejšie zníženie frekvencie PGTC záchvatov na týždeň. Pri dlhodobom pokračovaní v liečbe
47,4 % pacientov nemalo tonicko-klonické záchvaty minimálne 6 mesiacov a 31,5 % nemalo tonicko-
klonické záchvaty minimálne 1 rok.
5.2 Farmakokinetické vlastnosti
Farmakokinetický profil bol charakterizovaný po perorálnom podaní. Jednorazová dávka 1500 mg levetiracetamu rozpustená v 100 ml kompatibilného rozpúšťadla sa podáva intravenóznou infúziou počas 15 minút a je bioekvivalentná s 1500 mg levetiracetamu užitého perorálne, podaného v troch
500 mg tabletách.
Vyhodnotilo sa intravenózne podanie dávok až do 4000 mg rozpustených v 100 ml 0,9 % roztoku
chloridu sodného podávaných infúziou počas 15 minút a dávok až do 2500 mg rozpustených v 100 ml
0,9 % roztoku chloridu sodného podávaných infúziou počas 5 minút. Farmakokinetické a bezpečnostné profily nezistili žiadne bezpečnostné riziká.
Levetiracetam je vysoko rozpustná látka s vysokou schopnosťou prieniku. Farmakokinetický profil je lineárny pri nízkej intra- i interindividuálnej variabilite. Pri opakovanom podávaní sa nemení klírens. Časovo nezávislý farmakokinetický profil levetiracetamu sa tiež potvrdil po 1500 mg intravenóznej infúzii počas 4 dní s dávkovaním dvakrát denne.
Nie sú žiadne dôkazy o akejkoľvek príslušnej variabilite medzi pohlaviami, rasami alebo cirkadiálnej
variabilite. Farmakokinetický profil zdravých dobrovoľníkov a pacientov s epilepsiou je porovnateľný.
Dospelí a dospievajúci
Distribúcia
Maximálna plazmatická koncentrácia (Cmax) pozorovaná u 17 jedincov po jednorazovej intravenóznej
dávke 1500 mg podaných infúziou počas 15 minút bola 51 ± 19 µg/ml (aritmetický priemer ±
štandardná odchýlka).
Nie sú dostupné žiadne údaje o distribúcii v tkanivách ľudí.
Levetiracetam a ani jeho primárny metabolit sa vo významnej miere neviažu na bielkoviny v plazme
(< 10 %). Distribučný objem levetiracetamu je približne 0,5 až 0,7 l/kg, čo je hodnota blízka celkovému
objemu vody v organizme.
Biotransformácia
Levetiracetam sa u ľudí extenzívne nemetabolizuje. Hlavnou metabolickou cestou (24 % dávky) je
enzýmová hydrolýza acetamidovej skupiny. Izoenzýmy pečeňového cytochrómu P450 nepodporujú
vznik primárneho metabolitu ucb L057. Hydrolýza acetamidovej skupiny bola merateľná vo veľkom počte
tkanív vrátane krviniek. Metabolit ucb L057 j farmakologicky neúčinný.
Stanovili sa tiež dva menej významné metabolity. Jeden sa získal hydroxyláciou pyrolidónového kruhu (1,6 % dávky) a druhý otvorením pyrolidónového kruhu (0,9 % dávky). Ďalšie neidentifikované zložky predstavovali iba 0,6 % dávky.
In vivo sa nezistila žiadna enantiomerová interkonverzia pri levetiracetame ani pri jeho primárnom metabolite.
In vitro sa zistilo, že levetiracetam a jeho primárny metabolit neinhibujú hlavné izoformy ľudského pečeňového cytochrómu P450 (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 a 1A2), aktivitu glukuronylových transferáz (UGT1A1 a UGT1A6) a epoxidovej hydroxylázy. Okrem toho levetiracetam neovplyvňuje in vitro glukuronidáciu kyseliny valproovej.
V kultúrach ľudských hepatocytov mal levetiracetam minimálny alebo žiadny účinok na CYP1A2, SULT1E1 alebo UGT1A1. Levetiracetam spôsoboval miernu indukciu CYP2B6 a CYP3A4. In vitro a in vivo údaje o interakcii s perorálnymi kontraceptívami, digoxínom a warfarínom ukazujú, že in vivo sa neočakáva žiadna významná indukcia enzýmov. Preto je interakcia levetiracetamu s inými liečivami alebo naopak nepravdepodobná.
Eliminácia
Plazmatický polčas u dospelých bol 7±1 hodina a nelíšil sa ani podľa dávky, spôsobu podania ani pri
opakovanom podávaní. Priemerný celkový systémový klírens bol 0,96 ml/min/kg.
Hlavnou cestou vylučovania bol moč, ktorým sa vylučovalo priemerne 95 % dávky (približne 93 % dávky sa vylúčilo do 48 hodín). Stolicou sa vylúčilo len 0,3 % dávky. Kumulatívne vylučovanie levetiracetamu močom počas prvých 48 hodín dosiahlo 66 % dávky; v prípade jeho primárneho metabolitu 24 % dávky.
Renálny klírens levetiracetamu 0,6 ml/min/kg a pre ucb L057 je 4,2 ml/min/kg, čo ukazuje, že levetiracetam sa vylučuje glomerulárnou filtráciou s následnou tubulárnou reabsorpciou a že primárny metabolit sa okrem glomerulárnej filtrácie vylučuje aj aktívnou tubulárnou sekréciou.
Vylučovanie levetiracetamu koreluje s klírensom kreatinínu.
Starší pacienti
U starších pacientov je polčas predĺžený približne o 40 % (10 až 11 hodín). Súvisí to so znížením funkcie
obličiek u tejto populácie (pozri časť 4.2).
Porucha funkcie obličiek
Zdanlivý systémový klírens levetiracetamu a jeho primárneho metabolitu koreluje s klírensom kreatinínu.
Preto sa odporúča upraviť udržiavaciu dennú dávku levetiracetamu podľa klírensu kreatinínu u pacientov so stredne závažnou a závažnou poruchou funkcie obličiek (pozri časť 4.2).
U anurických dospelých jedincov s terminálnym štádiom zlyhania obličiek bol polčas medzi dialýzami
približne 25 hodín a počas dialýzy približne 3,1 hodiny.
Frakčné vylučovanie levetiracetamu počas typickej 4-hodinovej dialýzy tvorilo 51 %.
Porucha funkcie pečene
U osôb s miernou a stredne závažnou poruchou funkcie pečene nedochádzalo k žiadnej významnej zmene
klírensu levetiracetamu. U väčšiny osôb so závažnou poruchou funkcie pečene bol klírens levetiracetamu
znížený o vyše 50 % v dôsledku sprievodnej poruchy funkcie obličiek (pozri časť 4.2).
Pediatrická populácia
Deti (4 až 12 rokov)
Farmakokinetika sa u detských pacientov po intravenóznom podaní neskúmala. Na základe farmakokinetických vlastností levetiracetamu, farmakokinetiky u dospelých po intravenóznom podaní
a farmakokinetiky u detí po perorálnom podaní sa však očakáva podobná expozícia (AUC) levetiracetamu
u detských pacientov vo veku 4 až 12 rokov po intravenóznom a perorálnom podaní.
Po podaní jednorazovej perorálnej dávky (20 mg/kg) deťom s epilepsiou (6 až 12 rokov) bol polčas levetiracetamu 6 hodín. Zdanlivý systémový klírens bol približne o 30 % vyšší než u dospelých s epilepsiou.
Po podaní opakovaných perorálnych dávok (20 až 60 mg/kg/deň) deťom s epilepsiou (4 až 12 rokov)
sa levetiracetam rýchlo absorboval. Maximálna plazmatická koncentrácia sa pozorovala 0,5 až
1 hodinu po podaní. Pozorovalo sa lineárne a dávkovo úmerné zvýšenie maximálnych plazmatických
koncentrácií a plochy pod krivkou. Eliminačný polčas bol približne 5 hodín. Zdanlivý telesný klírens bol 1,1 ml/min/kg.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, genotoxicity, karcinogenicity neodhalili žiadne osobitné riziko pre ľudí.
Nežiaduce účinky nepozorované v klinických štúdiách, ale zistené u potkanov, a v menšom rozsahu u
myší, pri expozícii hladinám, ktoré boli podobné expozičným hladinám u človeka a s potenciálnym významom pre použitie v klinickej praxi, boli pečeňové zmeny naznačujúce adaptívnu odpoveď, ako je zvýšená hmotnosť a centrilobulárna hypertrofia, infiltrácia tuku a zvýšené pečeňové enzýmy v plazme.
U potkanov sa nepozorovali žiadne nežiaduce účinky na fertilitu alebo reprodukčnú výkonnosť samcov alebo samičiek pri dávkach až do 1800 mg/kg/deň (6-násobok maximálnej dennej dávky odporúčanej pre ľudí prepočítanej na mg/m2 alebo expozíciu) u rodičov a generácie F1.
Boli uskutočnené dve štúdie embryo-fetálneho vývoja (EFV štúdie) u potkanov s dávkami 400, 1200 a
3600 mg/kg/deň. Pri dávke 3600 mg/kg/deň došlo len v jednej z týchto dvoch EFV štúdií k nepatrnému zníženiu fetálnej hmotnosti, spojenému s hraničným nárastom počtu kostných zmien/menších anomálií.
Nedošlo k žiadnemu ovplyvneniu mortality embryí ani k zvýšeniu výskytu malformácií. NOAEL (hladina
bez pozorovaných nežiaducich účinkov) bola 3600 mg/kg/deň pre gravidné samice potkanov (12-násobok maximálnej dennej dávky odporúčanej pre ľudí prepočítanej na mg/m2 plochy povrchu tela) a 1200 mg/kg/deň pre plody.
Boli uskutočnené štyri štúdie embryo-fetálneho vývoja u králikov s dávkami 200, 600, 800, 1200 a 1800 mg/kg/deň. Dávka 1800 mg/kg/deň viedla k značnej toxicite u samíc-matiek a k zníženiu fetálnej hmotnosti, spojenému so zvýšeným výskytom plodov s kardiovaskulárnymi/kostrovými anomáliami. NOAEL bola <200 mg/kg/deň pre samice-matky a 200 mg/kg/deň pre plody (rovnajúca sa maximálnej dennej dávke odporúčanej pre ľudí prepočítanej na mg/m2 plochy povrchu tela).
Štúdia perinatálneho a postnatálneho vývoja bola realizovaná u potkanov s dávkami levetiracetamu 70,
350 a 1800 mg/kg/deň. NAOEL bola ≥1800 mg/kg/deň pre samice F0, rovnako ako pre prežitie, rast a vývoj mláďat F1 až do odstavenia (6-násobok maximálnej dennej dávky odporúčanej pre ľudí prepočítanej na mg/m2 plochy povrchu tela).
Štúdie s novorodencami a mláďatami zvierat u potkanov a psov nepreukázali žiadne nežiaduce účinky pri štandardných koncových ukazovateľoch vývoja a dozrievania v dávkach až do 1800 mg/kg/deň
(6 až 17 násobok maximálnej dennej dávky odporúčanej pre ľudí prepočítanej na mg/m2 plochy povrchu tela)
Hodnotenie environmentálneho rizika (ERA)
Levetiracetam Hospira pravdepodobne nemá nepriaznivý vplyv na životné prostredie (pozri časť 6.6), ak
sa používa podľa informácie o lieku.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Trihydrát octanu sodného Kyselina octová ľadová Chlorid sodný
Voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
2 roky
Chemická a fyzikálna stabilita zriedeného lieku v PVC vakoch bola preukázaná počas 24 hodín pri teplote
30 °C a pri teplote 2 až 8 °C. Z mikrobiologického hľadiska, pokiaľ spôsob riedenia nevylučuje riziko
mikrobiálnej kontaminácie, sa má liek použiť okamžite. Pokiaľ sa nepoužije okamžite, čas skladovania a podmienky uchovávania sú zodpovednosťou používateľa.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
Podmienky na uchovávanie nariedeného lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
5 ml sklenená injekčná liekovka (typ I) s potiahnutou bromobutylovou gumovou zátkou s hlinikovým
viečkom.
Každá škatuľa obsahuje 10 alebo 25 injekčných liekoviek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
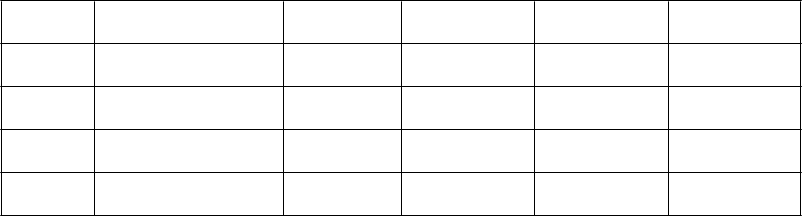
Na odporúčanú prípravu a podanie koncentrátu Levetiracetam Hospira pozri tabuľku nižšie na dosiahnutie
celkovej dennej dávky 500 mg, 1000 mg, 2000 mg a 3000 mg rozdelených do dvoch dávok.
Príprava a podanie koncentrátu Levetiracetam Hospira
D
ávka Použitý objem Objem
rozpúšťadla
Č
as infúzie Frekvencia podávania
C
elková denná dávka
250 mg 2,5 ml (polovica 5 ml
injekčnej liekovky)
500 mg 5 ml (jedna 5 ml
injekčná liekovka)
1000 mg 10 ml (dve 5 ml
injekčné liekovky)
1500 mg 15 ml (tri 5 ml
injekčné liekovky)
100 ml 15 minút Dvakrát denne 500 mg/deň
100 ml 15 minút Dvakrát denne 1000 mg/deň
100 ml 15 minút Dvakrát denne 2000 mg/deň
100 ml 15 minút Dvakrát denne 3000 mg/deň
Tento liek je iba na jednorazové použitie, všetok nepoužitý roztok má byť zlikvidovaný.
Zistilo sa, že koncentrát levetiracetamu je fyzikálne kompatibilný a chemicky stabilný pri zmiešaní s
nasledovnými rozpúšťadlami:
• Injekčný roztok chloride sodného (0.9 %)
• Ringerov roztok s laktátom
• 5 % roztok glukózy
Liek s výskytom častíc alebo zmenou farby sa nemá používať.
Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIHospira UK Limited
Queensway, Royal Leamington Spa
Warwickshire
CV31 3RW
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/13/000889/001
EU/1/13/000889/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10 DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu