
nej hmotnosti vyžadovať úpravu. Najmä v prípade veľkých chirurgických zákrokov je nevyhnutné presné monitorovanie substitučnej liečby pomocou vyšetrenia koagulácie (plazmatickej aktivity faktora VIII).
Pri použití jednostupňového testu zrážania in vitro na báze aktivovaného čiastočného tromboplastínového času (aPTT) na určenie aktivity faktora VIII v krvných vzorkách pacientov môžu byť výsledky aktivity plazmatického faktora VIII výrazne ovplyvnené typom reakčného činidla aPTT aj referenčným štandardom použitým v teste. To môže spôsobiť nadhodnotenie alebo podhodnotenie aktivity faktora VIII. Treba poznamenať, že medzi výsledkami testu získanými špecifickými reakčnými činidlami použitými v jednostupňovom teste zrážania na báze aPTT a chromogénnom teste sa môžu vyskytnúť výrazné rozdiely. To je dôležité pri monitorovaní aktivity faktora VIII lieku Jivi
a pri zmene laboratória a/alebo reakčných činidiel použitých v teste. Platí to aj pre lieky s modifikovaným dlhodobo účinkujúcim faktorom VIII.
Laboratóriá, ktoré chcú merať aktivitu lieku Jivi, by mali skontrolovať presnosť svojich postupov. Štúdia vykonaná v teréne ukázala, že aktivitu faktora VIII lieku Jivi je možné presne merať v plazme pomocou buď validovaného testu chromogénneho substrátu (ChS) alebo jednostupňového (JS) testu zrážania s použitím špecifických reakčných činidiel. Pri lieku Jivi môžu niektoré jednostupňové testy
na báze oxidu kremičitého (napr. APTT-SP, STA-PTT) podhodnocovať aktivitu faktora VIII lieku Jivi vo vzorkách plazmy. Niektoré reakčné činidlá, napr. s aktivátormi na báze kaolínu, majú potenciál
nadhodnocovať.
Klinický účinok faktora VIII je najdôležitejším prvkom pri hodnotení účinnosti liečby. Na dosiahnutie uspokojivých klinických výsledkov môže byť potrebné upraviť individuálne dávkovanie pre jednotlivých pacientov. Ak sa vypočítanou dávkou nedocielia očakávané hladiny faktora VIII alebo ak po podaní vypočítanej dávky nebude krvácanie pod kontrolou, treba u pacienta predpokladať prítomnosť cirkulujúceho inhibítora faktora VIII alebo protilátok proti PEG (pozri časť 4.4).
Dávkovanie
Dávka a trvanie substitučnej terapie závisia od závažnosti deficitu faktora VIII, miesta a rozsahu
krvácania a klinického stavu pacienta.
Počet jednotiek podaného faktora VIII sa vyjadruje v medzinárodných jednotkách (IU), ktoré sú odvodené od súčasného štandardu koncentrátu WHO pre lieky s obsahom faktora VIII. Plazmatická aktivita faktora VIII sa vyjadruje buď v percentách (vo vzťahu k normálnej ľudskej plazme), alebo prednostne v jednotkách IU (vo vzťahu k medzinárodnému štandardu pre faktor VIII v plazme).
Aktivita jednej jednotky IU faktora VIII zodpovedá množstvu faktora VIII nachádzajúcemu sa v jednom ml normálnej ľudskej plazmy.
Liečba podľa potreby
Výpočet požadovanej dávky faktora VIII sa zakladá na empirickom zistení, že 1 jednotka IU faktora VIII na kg telesnej hmotnosti zvyšuje aktivitu plazmatického faktora VIII o 1,5 – 2,5 % normálnej aktivity.
Požadovaná dávka lieku Jivi sa stanoví použitím nasledovného vzorca:
Požadované jednotky = telesná hmotnosť (kg) x požadované zvýšenie faktora VIII (% alebo IU/dl) x prevrátená hodnota pozorovaného zotavenia (t. j. 0,5 v prípade zotavenia o 2,0 %).
Množstvo, ktoré sa má podať, a frekvencia podávania majú byť vždy zamerané na klinickú účinnosť
požadovanú v individuálnych prípadoch.
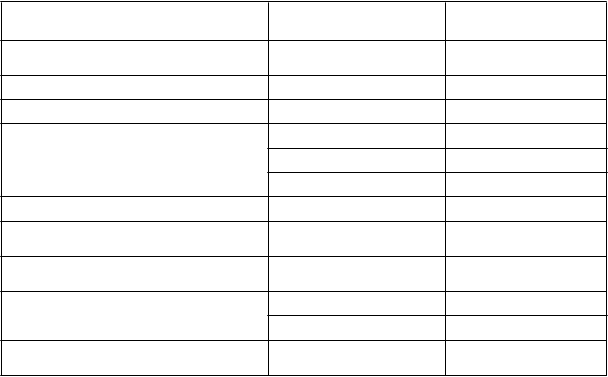
V prípade nasledujúcich hemoragických udalostí nesmie aktivita faktora VIII za príslušné obdobie klesnúť pod danú hladinu plazmatickej aktivity (v % z normálu). Nasledujúca tabuľka sa môže použiť ako návod na dávkovanie pri krvácavých epizódach a chirurgických zákrokoch:
Tabuľka 1: Návod na dávkovanie pri krvácavých epizódach a chirurgických zákrokoch u dospievajúcich a dospelých
Stupeň hemorágie/
typ chirurgického výkonu
Hemorágia
Požadovaná
hladina faktora VIII (%) (IU/dl)
Frekvencia dávok (hodiny)/
dĺžka liečby (dni)
Začínajúca hemartróza,
krvácanie do svalu alebo ústnej dutiny
20 – 40
Injekciu opakujte každých 24 – 48 hodín.
Minimálne 1 deň, kým sa krvácanie prejavované bolesťou nezastaví alebo sa nedosiahne zahojenie.
Rozsiahlejšia hemartróza,
krvácanie do svalu alebo
hematóm 30 – 60
Injekciu opakujte každých 24 – 48 hodín
počas 3 až 4 dní alebo dlhšie, kým bolesť

a akútna slabosť nevymiznú.
Ohrozujúce život
Hemorágie 60 – 100 Injekciu opakujte každých 8 až 24 hodín,
kým ohrozenie nevymizne.
Chirurgický zákrok
Malý chirurgický zákrok
vrátane extrakcie zubu
30 – 60 Každých 24 hodín, minimálne 1 deň, kým sa
nedosiahne zahojenie.
Veľký chirurgický zákrok
80 – 100 (pred operáciou a po nej)
Dávku opakujte každých 12 – 24 hodín, kým
sa rana primerane nezahojí, následne liečba minimálne ďalších 7 dní na udržanie aktivity faktora VIII na úrovni 30 % – 60 % (IU/dl).
Profylaxia
Všetky liečebné rozhodnutia na identifikovanie vhodných profylaktických liečebných režimov sa majú riadiť klinickým úsudkom na základe individuálnej charakteristiky pacienta a reakcie na liečbu.
Pri profylaxii je dávka 45 - 60 IU/kg každých 5 dní. Na základe klinických charakteristických vlastností pacienta môže byť dávka aj 60 IU/kg každých 7 dní alebo 30 - 40 IU/kg dvakrát týždenne (pozri časti 5.1 a 5.2).
Pri profylaxii nemá byť u pacientov s nadváhou maximálna dávka na injekciu vyššia ako približne
6 000 IU.
Pediatrická populácia
Jivi nie je indikovaný doposiaľ neliečeným pacientom a pacientom mladším ako 12 rokov.
Dospievajúca populácia
Dávkovanie pri individualizovanej a profylaktickej liečbe je u dospievajúcich pacientov rovnaké ako u dospelých pacientov.
Staršia populácia
Skúsenosti s pacientmi vo veku ≥ 65 rokov sú obmedzené.
Spôsob podávania
Jivi je určený na intravenózne použitie.
Jivi sa má aplikovať intravenózne počas 2 až 5 minút v závislosti od celkového objemu. Rýchlosť
podávania sa má určiť podľa pohodlia pacienta (maximálna rýchlosť injekčného podania: 2,5 ml/min). Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6 a písomnú informáciu pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Známe alergické reakcie na myšie alebo škrečie proteíny.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologických liekov, názov a číslo šarže podávaného lieku sa má
zreteľne zaznamenať.
Precitlivenosť
V súvislosti s používaním lieku Jivi sú možné reakcie z precitlivenosti alergického typu. Liek môže
obsahovať stopy myších a škrečích proteínov. Reakcie z precitlivenosti môžu tiež súvisieť
s protilátkami proti PEG (pozri odstavec „Imunitná odpoveď na polyetylénglykol (PEG)“). Ak sa vyskytnú symptómy precitlivenosti, pacientom sa má odporučiť, aby okamžite ukončili používanie tohto lieku a obrátili sa na svojho lekára. Pacienti majú byť informovaní o prvotných prejavoch reakcií z precitlivenosti vrátane žihľavky, generalizovanej žihľavky, pocitu tiesne na hrudníku, sipotu, hypotenzie a anafylaxie. Podľa potreby sa má začať so symptomatickou liečbou precitlivenosti.
V prípade anafylaxie alebo šoku treba použiť aktuálne medicínske štandardy liečby.
Inhibítory
Známou komplikáciou liečby jedincov s hemofíliou A je vznik neutralizujúcich protilátok
(inhibítorov) faktora VIII. Tieto inhibítory sú zvyčajne imunoglobulíny IgG zamerané proti prokoagulačnej aktivite faktora VIII, ktoré sú kvantifikované v jednotkách Bethesda (Bethesda Units, BU) na ml plazmy použitím modifikovaného Bethesda testu. Riziko rozvoja inhibítorov koreluje so závažnosťou ochorenia, ako aj s expozíciou faktoru VIII, pričom toto riziko býva najvyššie počas prvých 50 dní expozície (exposure days, ED), ale pokračuje počas celého života, hoci toto riziko nie je bežné. V zriedkavých prípadoch môžu inhibítory vzniknúť po prvých 50 dňoch expozície.
Klinický význam tvorby inhibítorov bude závisieť od titra inhibítora, pričom menšie riziko nedostatočnej klinickej odpovede hrozí v prípade inhibítorov nízkeho titra než v prípade vysokého titra inhibítorov.
Vo všeobecnosti sa u všetkých pacientov liečených liekmi s koagulačným faktorom VIII má pomocou náležitých klinických pozorovaní a laboratórnych vyšetrení pozorne sledovať, či nedochádza k vzniku inhibítorov.
Ak sa očakávané hladiny aktivity faktora VIII v plazme nedosiahnu, alebo ak krvácanie nie je kontrolované vhodnou dávkou, má sa vykonať testovanie prítomnosti inhibítorov faktora VIII.
U pacientov s vysokými hladinami inhibítora terapia faktorom VIII nemusí byť účinná a treba zvážiť iné možnosti liečby. Liečba takých pacientov má byť riadená lekármi, ktorí majú skúsenosti s liečbou hemofílie a s inhibítormi faktora VIII.
Imunitná odpoveď na polyetylénglykol (PEG)
Klinická imunitná odpoveď súvisiaca s protilátkami proti PEG, manifestovaná ako príznaky akútnej
precitlivenosti a/alebo straty účinku lieku, bola pozorovaná primárne v rámci prvých 4 dní expozície. Nízke hladiny faktora VIII po injekcii za neprítomnosti detegovateľných inhibítorov faktora VIII naznačujú, že strata liečebného účinku je pravdepodobne spôsobená protilátkami proti PEG. V takých prípadoch sa má Jivi vysadiť a pacienti majú prejsť na predtým účinný liek obsahujúci faktor VIII.
S pribúdajúcim vekom sa pozorovalo signifikantné zníženie rizika imunitnej odpovede na PEG. Tento efekt môže súvisieť s vývojovou zmenou imunity, a aj keď je ťažké definovať presnú vekovú hranicu zmeny rizika, tento jav sa vyskytuje vo väčšej miere u malých detí s hemofíliou.
Dôsledky akéhokoľvek možného rizika pre postihnutých pacientov s reakciou z precitlivenosti na pegylované proteíny sú neznáme. Údaje ukazujú, že u postihnutých jedincov titer IgM protilátok proti PEG po prerušení liečby liekom Jivi klesol, a po čase bol nedetegovateľný. Nepozorovala sa žiadna krížová reaktivita IgM protilátok proti PEG s ďalšími nemodifikovanými liekmi obsahujúcimi faktor VIII. Všetci pacienti mohli byť úspešne liečení svojimi predchádzajúcimi liekmi obsahujúcimi faktor VIII.
Kardiovaskulárne príhody
U pacientov s existujúcimi rizikovými faktormi kardiovaskulárnych príhod môže substitučná liečba
faktorom VIII toto riziko zvýšiť.
Komplikácie súvisiace s katétrom
Ak sa vyžaduje zariadenie na centrálny venózny vstup (central venous access devices, CVAD), má sa
zvážiť riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakteriémie a trombózy v mieste zavedenia katétra.
Pediatrická populácia
Uvedené upozornenia a opatrenia platia pre dospelých aj dospievajúcich.
Jivi nie je indikovaný pacientom vo veku < 12 rokov a predtým neliečeným pacientom.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Interakcie liekov obsahujúcich ľudský koagulačný faktor VIII (rDNA) s inými liekmi neboli hlásené.
4.6 Fertilita, gravidita a laktácia
Gravidita a dojčenie
S faktorom VIII neboli realizované reprodukčné štúdie na zvieratách. Na základe zriedkavého výskytu
hemofílie A u žien nie sú dostupné skúsenosti týkajúce sa používania faktora VIII počas gravidity
a dojčenia. Faktor VIII sa má preto používať počas gravidity a dojčenia, iba ak je to jasne indikované.
Fertilita
V štúdiách systémovej toxicity s opakovanou dávkou lieku Jivi u potkanov a králikov neboli
pozorované účinky súvisiace s liečbou na samčie pohlavné orgány (pozri časť 5.3). Účinok na fertilitu u ľudí nie je známy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Jivi nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Pozorovali sa reakcie z precitlivenosti alebo alergické reakcie (ktoré môžu zahŕňať angioedém, pálenie
a štípanie v mieste podania injekcie, triašku, začervenanie, generalizovanú urtikáriu, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, nepokoj, tachykardiu, tlak na hrudi, brnenie, vracanie, sipot), ktoré v niektorých prípadoch môžu prejsť do závažnej anafylaxie (vrátane šoku).
U pacientov s hemofíliou A, ktorí sú liečení pomocou faktora VIII vrátane lieku Jivi, sa môžu vyvinúť neutralizujúce protilátky (inhibítory) (pozri časť 5.1). Ak sa takéto inhibítory vyskytnú, stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované pracovisko zamerané na liečbu hemofílie.
Najčastejšie hlásenými nežiaducimi reakciami u predtým liečených pacientov (previously treated patients, PTP) boli v klinických skúšaniach bolesť hlavy, kašeľ a horúčka.
Tabuľkový zoznam nežiaducich účinkov
Bezpečnostnú populáciu z troch kľúčových štúdií fázy I a III [PROTECT VIII] tvorilo celkovo
221 pacientov. Medián času v štúdii pre 148 pacientov vo veku ≥12 rokov bol 713 dní. Celkový počet dní expozície (exposure days, ED) bol 18 432 s mediánom 131 ED (rozsah: 1 až 309) na pacienta; medián času v štúdii pre pediatrických pacientov vo veku < 12 rokov bol 237 dní s celkovým počtom
3 219 ED a mediánom 53 ED (rozsah: 1 - 68) na pacienta.
Tabuľka uvedená nižšie zodpovedá klasifikácii orgánových systémov MedDRA (trieda orgánových systémov a preferovaná terminologická úroveň). Frekvencie výskytu boli vyhodnotené podľa nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí s klesajúcou závažnosťou.
Tabuľka 2: Frekvencia nežiaducich liekových reakcií v klinických skúšaniach
Podľa databázy MedDRA
Trieda orgánových systémov
Nežiaduce reakcie Frekvencia
Poruchy krvi a lymfatického systému Inhibícia faktora VIII menej časté (PTP)*
Poruchy imunitného systému Precitlivenosť časté
Psychické poruchy Nespavosť časté
Poruchy nervového systému Bolesť hlavy veľmi časté Závrat časté Dysgeúzia menej časté
Poruchy ciev Začervenanie menej časté
Poruchy dýchacej sústavy, hrudníka a mediastína
Kašeľ časté
Poruchy gastrointestinálneho traktu Bolesť brucha, nauzea, vracanie
časté
Poruchy kože a podkožného tkaniva Erytém***, vyrážka**** časté
Pruritus menej časté
Celkové poruchy a reakcie v mieste podania
Reakcie v mieste podania injekcie **, horúčka
časté
*Frekvencia vychádza zo štúdií so všetkými liekmi obsahujúcimi faktor VIII, do ktorých boli zahrnutí pacienti so závažnou hemofíliou A. PTP = predtým liečení pacienti.
**Zahŕňa pruritus v mieste podania injekcie, vyrážku v mieste podania injekcie a pruritus v mieste prepichnutia cievy.
***Zahŕňa erytém a multiformný erytém.
****Zahŕňa vyrážku a papulárnu vyrážku.
Popis vybraných nežiaducich reakcií
Imunogenita
Imunogenita bola hodnotená počas klinických skúšaní s liekom Jivi u 159 (vrátane chirurgických pacientov) predtým liečených dospievajúcich (vo veku ≥ 12 rokov) a dospelých s diagnostikovanou
závažnou hemofíliou A (FVIII:C < 1 %) a s ≥ 150 dní predchádzajúcej expozície. Medián času v štúdii
bol 713 dní s mediánom 131 dní expozície (rozsah: 1 až 309 dní).
Inhibítory faktora VIII
Nevyskytli sa žiadne de novo ani potvrdené prípady inhibítora proti faktoru VIII. U jedného dospelého pacienta bol po chirurgickom zákroku hlásený jeden nepotvrdený pozitívny výsledok nízkeho titra
inhibítora faktora VIII (1,7 BU/ml).
Anti - PEG protilátky
Imunogenita proti PEG s tvorbou špecifických IgM protilátok proti PEG bola pozorovaná u jedného pacienta. Imunitná odpoveď bola po 4 injekciách lieku Jivi sprevádzaná klinickou reakciou
z precitlivenosti. Po vysadení lieku Jivi protilátky proti PEG vymizli.
Pediatrická populácia
V dokončených klinických štúdiách so 73 pediatrickými PTP vo veku < 12 rokov (44 PTP < 6 rokov,
29 PTP 6 až < 12 rokov) boli u detí mladších ako 6 rokov pozorované nežiaduce reakcie spôsobené imunitnou odpoveďou na PEG. U 10 zo 44 pacientov (23 %) vo vekovej skupine mladších ako
6 rokov bola počas prvých 4 dní expozície pozorovaná strata účinku lieku spôsobená neutralizujúcimi protilátkami proti PEG. U 3 zo 44 pacientov (7 %) bola strata účinku lieku kombinovaná s reakciami
z precitlivenosti (pozri časť 4.4). Žiadne spúšťače ani prediktory imunitnej odpovede na PEG nebolo možné identifikovať.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV klinických skúšaniach došlo k jednému prípadu predávkovania. Neboli hlásené žiadne nežiaduce účinky.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antihemoragiká: krvný koagulačný faktor VIII, ATC kód: B02BD02.
Mechanizmus účinkuKomplex faktora VIII/von Willebrandovho faktora sa skladá z dvoch molekúl (faktor VIII a von
Willebrandov faktor) s rôznymi fyziologickými funkciami. Po aplikácii pacientovi s hemofíliou sa faktor VIII viaže na von Willebrandov faktor. Aktivovaný faktor VIII pôsobí ako kofaktor pre aktivovaný faktor IX, ktorý urýchľuje konverziu faktora X na aktivovaný faktor X. Aktivovaný faktor X mení protrombín na trombín. Trombín potom mení fibrinogén na fibrín a môže dôjsť
k vytvoreniu zrazeniny. Hemofília A je pohlavne viazané dedičné ochorenie zrážavosti krvi spôsobené zníženou hladinou faktora VIII:C alebo jeho neprítomnosťou, následkom čoho dochádza ku krvácaniu do kĺbov, svalov alebo vnútorných orgánov, buď spontánnemu, alebo ako následok náhodného úrazu alebo chirurgického zákroku. Substitučnou liečbou sa hladiny faktora VIII v plazme zvýšia, čím sa umožní prechodná korekcia nedostatku faktora a korekcia tendencie ku krvácaniu.
Damoktokog alfa pegol je PEGylovanou formou rFVIII. Špecifická PEGylácia znižuje klírens faktora VIII, čo má za následok predĺžený polčas pri súčasnom zachovaní normálnych funkcií molekuly rFVIII s odstránenou B doménou (pozri časť 5.2). Damoktokog alfa pegol neobsahuje von Willebrandov faktor.
Klinická účinnosť a bezpečnosťKlinické štúdieCelkovo 232 predtým liečených pacientov so závažnou hemofíliou A bolo liečených v rámci
programu klinického skúšania, ktorý zahŕňal jednu štúdiu fázy I a dve štúdie fázy II/III. 159 pacientov bolo vo veku ≥ 12 rokov.
Fáza II/III: Farmakokinetické vlastnosti, bezpečnosť a účinnosť lieku Jivi pri individualizovanej
liečbe, pri profylaktickej liečbe s tromi režimami (dvakrát týždenne 30 - 40 IU/kg, každých
5 dní 45 - 60 IU/kg a každých 7 dní 60 IU/kg) a hemostáza počas veľkých chirurgických zákrokov sa hodnotili v medzinárodnej, otvorenej, nekontrolovanej, čiastočne
randomizovanej štúdii, ktorá bola vykonaná v súlade so schváleným plánom pediatrického výskumu. Predĺženie štúdie zahŕňalo pacientov, ktorí dokončili hlavnú štúdiu. Primárnou premennou účinnosti bola ročná miera krvácania (annualized bleed rate – ABR).
134 PTP mužského pohlavia dostalo minimálne jednu injekciu lieku Jivi (vrátane
13 pacientov vo veku 12 až 17 rokov) na profylaktickú liečbu (n = 114) alebo na individualizovanú liečbu (n = 20) počas obdobia 36 týždňov. Celkovo 121 pacientov dostávalo liečbu počas predĺženia štúdie s mediánom trvania 464 dní. V časti chirurgických zákrokov bola hodnotená hemostáza počas 20 veľkých chirurgických zákrokov
u 17 pacientov.
Fáza III (pediatrickí pacienti): Farmakokinetické vlastnosti, bezpečnosť a účinnosť lieku Jivi pri troch profylaktických režimoch (dvakrát týždenne, každých 5 a každých 7 dní) a liečba medzikrvácaní sa hodnotili v medzinárodnom, nekontrolovanom, otvorenom klinickom skúšaní u 73 pediatrických pacientov (vo veku < 12 rokov) v trvaní 50 ED a aspoň
6 mesiacov. Táto štúdia bola vykonaná v súlade so schváleným plánom pediatrického výskumu. 61 pacientov (83,6 %) dokončilo hlavnú štúdiu a 59 pacientov pokračovalo vo voliteľnej predĺženej štúdii.
Profylaktická liečba u pacientov vo veku ≥ 12 rokov
Počas obdobia hlavnej štúdie boli pacienti priradení na profylaktickú liečbu 2-krát/týždeň (n = 24) alebo randomizovaní na každých 5 dní (n = 43) alebo každých 7 dní (n = 43) alebo dostávali individualizovanú liečbu (n = 20) liekom Jivi. 99 zo 110 pacientov (90 %) zostalo v režime, ktorý im bol priradený. U 11 pacientov v skupine liečby každých 7 dní sa zvýšila frekvencia podávania. Medián dávky pre všetky profylaktické režimy bol 46,9 IU/kg/injekcia. Medián (Q1; Q3) ABR počas profylaktickej liečby bol 2,09 (0,0; 6,1) pre všetky krvácania a 0,0 (0,0; 4,2) pre spontánne krvácania
v porovnaní s 23,4 (18; 37) celkových krvácaní v skupine s individualizovanou liečbou. U 42 zo
110 pacientov v skupine s profylaktickou liečbou (38,2 %) sa nevyskytlo žiadne krvácanie.'
Počas predĺženia štúdie bolo 24 pacientov liečených 2-krát/týždeň, 37 pacientov každých 5 dní,
29 pacientov každých 7 dní a 17 pacientov zmenilo režim liečby. Medián dávky pri profylaktickej liečbe bol 47,7 IU/kg. Celkový medián (Q1; Q3) všetkých ABR bol 1,17 (0,0; 4,3) a 0,6 (0,0; 3,2) pre spontánne krvácania v kombinovaných skupinách profylaktickej liečby a celková ABR bola 33,0
v skupine s individualizovanou liečbou.
Treba poznamenať, že ABR nie je porovnateľná medzi rôznymi koncentrátmi faktorov a medzi jednotlivými klinickými štúdiami.
Liečba krvácania
Zo 702 príhod krvácania liečených liekom Jivi počas hlavnej štúdie bolo 636 (90,6 %) liečených
1 alebo 2 injekciami, z toho 81,1 % 1 injekciou. Medián (rozsah) dávky na injekciu bol 31,7 IU/kg
(14; 62). Počas predĺženia štúdie bolo 942 krvácaní liečených liekom Jivi a 92,3 % bolo kontrolovaných 1 alebo 2 injekciami, z toho 83 % 1 injekciou. Medián (rozsah) dávky bol 37,3 (18;
66) IU/kg/injekciu.
Perioperačná liečba
Celkovo sa vykonalo a vyhodnotilo 20 veľkých chirurgických zákrokov u 17 pacientov. Medián celkovej dávky pri veľkých chirurgických zákrokoch bol 219 IU/kg (rozsah: 50 - 1 500 IU/kg, vrátane pooperačného obdobia trvajúceho až 3 týždne). Perioperačná hemostatická účinnosť bola hodnotená ako dobrá alebo veľmi dobrá počas všetkých veľkých chirurgických zákrokov.
Dodatočne sa vykonalo 34 malých chirurgických zákrokov u 19 pacientov. Hemostáza bola hodnotená ako dobrá alebo veľmi dobrá vo všetkých dostupných prípadoch.
Pediatrická populácia vo veku < 12 rokov
Použitie lieku Jivi u detí mladších ako 12 rokov nie je indikované (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Celkovo 73 predtým liečených pediatrických pacientov (44 pacientov < 6 rokov a 29 pacientov vo veku 6 až < 12 rokov) dostalo profylaktickú liečbu dvakrát týždenne, každých 5 alebo každých 7 dní v štúdii fázy III. Pre 53 pacientov, ktorí dokončili hlavnú štúdiu, bol medián (Q1; Q3) ročnej miery krvácania 2,87 (1,1; 6,1) a spontánna ABR bola 0,0 (0,0; 2,6). Pri liečbe krvácaní 84,4 % krvácaní ustúpilo po podaní 1 injekcie a 91,9 % krvácaní ustúpilo po podaní 1 alebo 2 injekcií.
11 pacientov vo vekovej skupine < 6 rokov bolo vyradených z dôvodu imunitnej odpovede na PEG
súvisiacej so stratou účinnosti a/alebo reakciou z precitlivenosti počas prvých štyroch ED.
5.2 Farmakokinetické vlastnostiFarmakokinetické vlastnosti (PK) lieku Jivi sa porovnávali s PK faktora VIII v skríženej štúdii fázy I. PK sa tiež vyhodnocovali u 22 pacientov (≥ 12 rokov) a u 16 z týchto pacientov po 6 mesiacoch profylaktickej liečby v štúdii fázy II/III.
PK údaje (na základe chromogénneho testu) naznačili, že Jivi má znížený klírens (CL) s následným terminálnym polčasom, ktorý je 1,4-násobne dlhší a dávkou normalizovanou AUC, ktorá je
1,4-násobne vyššia v porovnaní s komparatívnym liekom obsahujúcim faktor VIII. Nárasty úmerné dávke boli pozorované medzi dávkami 25 a 60 IU/kg, čo svedčí o linearite dávky medzi 25 IU/kg
a 60 IU/kg.
V tabuľke 3 sú zhrnuté PK parametre po jednorazovej dávke 60 IU/kg zo štúdie fázy II/III, v ktorej sa PK vyhodnocovali u 22 pacientov. Opakované merania PK nenaznačujú žiadne významné zmeny PK charakteristík po dlhodobej liečbe.
Tabuľka 3: Farmakokinetické parametre (geometrický priemer (%CV) a aritmetický priemer(±
SD)) lieku Jivi po jednorazovej dávke 60 IU/kg na základe chromogénneho testuParametre (jednotky) JiviPacienti ≥ 12 rokovN = 22AUC (IU*h/dl) 3 710 (33,8)
3 900 ± 1 280
AUC, norm (h*kg/dl) 62,5 (33,7)
65,7 ± 21,4
Cmax (IU/dl) 163 (14,7)
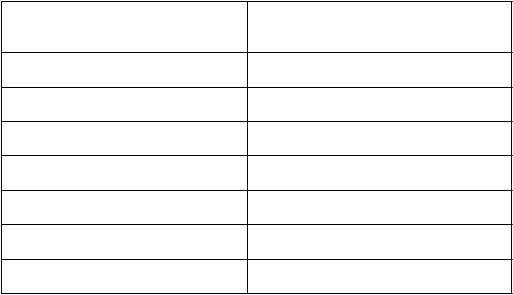
164 ± 23,8
t½ (h) 17,1 (27,1)
17,6 ± 4,26
MRTIV (h) 24,4 (27,5)
25,2 ± 6,19
Vss (dl/kg) 0,391 (16,3)
0,396 ± 0,0631
CL (dl/h/kg) 0,0160 (33,7)
0,0168 ± 0,00553
AUC: oblasť pod krivkou; AUC, norm: dávkou normalizovaná AUC; Cmax: maximálna koncentrácia lieku; t½:
terminálny polčas; MRT IV: priemerný rezidenčný čas po i.v. podaní; VSS: zrejmá objemová distribúcia v rovnovážnom stave; CL: klírens.
Narastajúce zotavenie sa stanovilo u 131 pacientov vo viacerých časových bodoch. Medián (Q1; Q3)
zotavenia bol 2,6 (2,3; 3,0) na základe chromogénneho testu.
Populačný PK model bol vyvinutý na základe všetkých dostupných meraní faktora VIII
(z intenzívneho vzorkovania PK a všetkých vzoriek počas zotavenia) v rámci 3 klinických štúdií,
umožňujúci výpočet PK parametrov pre pacientov v rôznych štúdiách. Tabuľka 4 nižšie uvádza PK
parametre na základe populačného PK modelu.
Tabuľka 4: PK parametre (geometrický priemer [%CV]) na základe populačného PK modelu s použitím chromogénneho testu
PK parameter (jednotka) 12 až <18 rokov
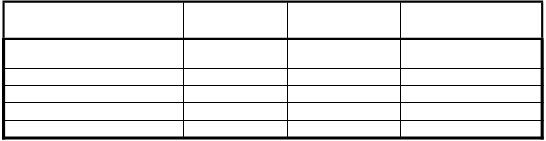 N = 12
≥ 18 rokov
N = 133
Spolu (≥ 12 rokov)
N = 145
N = 12
≥ 18 rokov
N = 133
Spolu (≥ 12 rokov)
N = 145
AUC (IU.h/dl)* 3 341 (34,2) 4 052 (31,1) 3 997 (31,6)
AUCnorm (kg.h/dl) 57,4 (32,6) 67,5 (30,6) 66,6 (31,0)
t1/2 (h) 16,8 (25,2) 17,4 (28,8) 17,4 (28,4) Vss (dl/kg) 0,423 (15,5) 0,373 (15,6) 0,376 (15,9)
CL (dl/h/kg) 0,0174 (34,2) 0,0148 (31,1) 0,0150 (31,6)
*AUC vypočítaná pre dávku 60 IU/kg.
5.3 Predklinické údaje o bezpečnosti
Jivi bol hodnotený vo farmakologických štúdiách s jednou a opakovanou dávkou, ako aj v štúdiách juvenilnej toxicity u potkanov a králikov. V dlhodobej, 6-mesačnej štúdii chronickej toxicity sa nepozorovali žiadne náznaky akumulácie PEG alebo iné účinky súvisiace s podávaním lieku Jivi. Navyše boli u dvoch druhov zvierat vykonané 4-týždňové štúdie toxicity s PEG zložkou lieku Jivi. PEG-viažúca zložka sa tiež testovala v štandardnom súbore in vivo a in vitro štúdií genotoxicity, ktoré nenaznačovali žiadny genotoxický potenciál. Tieto štúdie neodhalili žiadne bezpečnostné obavy pre ľudí.
Štúdie s jednorazovou dávkou u potkanov s rádioaktívne označenou PEG zložkou ukázali, že nič nenasvedčuje tomu, že v tele zvieraťa dochádza k hromadeniu alebo ireverzibilnej väzbe rádioaktivity. Konkrétne sa v mozgu nezistila žiadna zvyšková rádioaktivita, čo naznačuje, že rádioaktívne označená zlúčenina neprenikla cez hematoencefalickú bariéru. V štúdiách distribúcie a exkrécie u potkanov sa preukázalo, že 60 kDa PEG zložka Jivi je široko distribuovaná do orgánov a tkanív a vylučovaná
z nich močom (68,4 % až do 231. dňa po podaní) a stolicou (13,8 % až do 168. dňa po podaní).
Neboli vykonané žiadne dlhodobé štúdie na zvieratách na zhodnotenie karcinogénneho potenciálu lieku Jivi ani štúdie na zistenie účinkov lieku Jivi na reprodukciu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok sacharóza histidín glycín
chlorid sodný
dihydrát chloridu vápenatého polysorbát 80
kyselina octová, ľadová (na úpravu pH)
Rozpúšťadlo voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
Na rekonštitúciu a aplikáciu injekcie sa môžu použiť len komponenty dodávané v balení, pretože môže dôjsť ku zlyhaniu liečby v dôsledku adsorpcie faktora VIII na vnútorný povrch niektorých injekčných zariadení.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
2 roky.
Rekonštituovaný roztok
Chemická a fyzikálna stabilita po rekonštitúcii bola preukázaná počas 3 hodín pri izbovej teplote. Po rekonštitúcii neuchovávajte v chladničke.
Liek sa má z mikrobiologického hľadiska použiť ihneď po rekonštitúcii. Ak sa ihneď nepoužije, za dobu uchovávania lieku pripraveného na použitie a podmienky pred jeho použitím zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku a naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Liek uchovávaný počas jeho celkového 2-ročného času použiteľnosti (vo vonkajšom obale) sa môže uchovávať pri teplote do 25 °C obmedzený čas 6 mesiacov. Konečný dátum 6-mesačného obdobia uchovávania pri teplote do 25 °C sa má zaznamenať na vonkajší obal. Tento dátum nesmie nikdy prekročiť dátum exspirácie vytlačený na vonkajšom obale. Na konci tohto obdobia sa liek nesmie znovu uložiť do chladničky, ale má sa použiť alebo zlikvidovať.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Každé balenie lieku Jivi obsahuje:
• jednu injekčnú liekovku s práškom (10 ml injekčná liekovka typu 1 z číreho skla so sivou zátkou z brómbutylovej gumovej zmesi a s hliníkovým uzáverom),
• jednu naplnenú injekčnú striekačku s 2,5 ml rozpúšťadla (injekčná striekačka s valcom z číreho skla typu 1 so zátkou z brómbutylovej gumovej zmesi),
• jeden piest na injekčnú striekačku,
• jeden adaptér na injekčnú liekovku (s integrovaným filtrom),
• jednu súpravu na venepunkciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Podrobné pokyny na prípravu a podávanie sú obsiahnuté v písomnej informácii pre používateľa dodávanej s liekom Jivi.
Prášok lieku Jivi sa má rekonštituovať len s dodaným rozpúšťadlom (2,5 ml vody na injekciu)
v naplnenej injekčnej striekačke a adaptérom na injekčnú liekovku. Liek sa musí pripraviť na injekciu za aseptických podmienok. Ak je ktorákoľvek zložka balenia otvorená alebo poškodená, túto zložku nepoužívajte.
Po rekonštitúcii je roztok číry a bezfarebný a natiahne sa späť do injekčnej striekačky. Parenterálne lieky sa majú pred podaním vizuálne skontrolovať, či nie sú prítomné častice a nedošlo k zmene farby.
Rekonštituovaný liek sa musí pred podaním prefiltrovať, aby sa odstránili potenciálne prítomné častice v roztoku. Filtráciu možno dosiahnuť použitím adaptéra na injekčnú liekovku.
Jivi je určený len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBayer AG
51368 Leverkusen
Nemecko
8. REGISTRAČNÉ ČÍSLA
EU/1/18/1324/001 - Jivi 250 IU
EU/1/18/1324/002 - Jivi 500 IU EU/1/18/1324/003 - Jivi 1 000 IU EU/1/18/1324/004 - Jivi 2 000 IU EU/1/18/1324/005 - Jivi 3 000 IU
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: DD. mesiac RRRR
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.