
kanoch indikuje, že v prípade
súčasného podávania febuxostátu, sa má dávka merkaptopurínu/azatioprínu redukovať na 20 % alebo menej % predtým predpísanej dávky (pozri časť 4.5 a 5.3).
Liekové interakčné štúdie febuxostátu s inou cytotoxickou chemoterapiou neboli vykonané. Nie sú k dispozícii žiadne údaje týkajúce sa bezpečnosti febuxostátu počas inej cytotoxickej terapie.
Rosiglitazón/substráty CYP2C8
In vitro sa preukázalo, že febuxostát je slabým inhibítorom CYP2C8. V štúdii so zdravými
dobrovoľníkmi súčasné podanie perorálnej dávky 120 mg febuxostátu raz denne s jednou dávkou 4 mg perorálne podaného rosiglitazónu nemalo účinok na farmakokinetiku rosiglitazónu a jeho metabolit Ndemetyl rosiglitazón, febuxostát nie je inhibítorom enzýmu CYP2C8 in vivo. Súčasné podanie febuxostátu s rosiglitazónom alebo inými substrátmi CYP2C8 nevyžaduje úpravu dávky pre tieto liečivá.
Teofylín
Interakčná štúdia so zdravými dobrovoľníkmi bola vykonaná s febuxostátom na zhodnotenie, či
inhibícia xantínoxidázy (XO) môže spôsobiť zvýšenie koncentrácie teofylínu v cirkulácii, ako bolo pozorované pri použití iných inhibítorov XO. Výsledky štúdie preukázali, že súčasné podanie 80 mg febuxostátu raz denne s teofylínom 400 mg v jednej dávke nemá účinok na farmakokinetiku alebo bezpečnosť teofylínu. Osobitná opatrnosť pri súčasnom podávaní teofylínu a febuxostátu preto nie je potrebná. Nie sú dostupné údaje pre febuxostát 120 mg.
Naproxén a iné inhibítory glukuronidácie
Metabolizmus febuxostátu závisí na uridín-glukuronyl-transferáze (UGT). Lieky inhibujúce
glukuronidáciu, ako napríklad NSAID a probenecid, by teoreticky mohli ovplyvniť eliminovanie febuxostátu. U zdravých osôb bolo súčasné užívanie febuxostátu a naproxénu 250 mg dvakrát denne spojené so zvýšenou expozíciou febuxostátu (Cmax 28 %, AUC 41 % and t1/2 26 %). V klinických štúdiách nebolo užívanie naproxénu ani iných NSAID/Cox-2 inhibítorov spojené so žiadnym signifikantným zvýšením nežiaducich účinkov.
Febuxostát možno podávať spolu s naproxénom, pričom nie je potrebná žiadna úprava dávky febuxostátu alebo naproxénu.
Induktory glukuronidácie
Silné induktory enzýmu UGT by mohli viesť k zvýšenému metabolizmu a zníženej účinnosti
febuxostátu. Monitorovanie kyseliny močovej v sére sa preto odporúča 1-2 týždne po začatí liečby
silným induktorom glukuronidácie. Naopak, skončenie liečby induktorom by mohlo viesť k zvýšeným plazmatickým koncentráciám febuxostátu.
Kolchicín/indometacín/hydrochlorotiazid/warfarín
Febuxostát možno podávať súbežne s kolchicínom alebo indometacínom bez potreby upraviť dávku
febuxostátu alebo súbežne podávaného liečiva.
Pri podávaní febuxostátu s hydrochlorotiazidom nie je potrebná žiadna úprava dávky febuxostátu.
Pri podávaní febuxostátu s warfarínom nie je potrebná úprava dávky warfarínu. Podávanie febuxostátu (80 mg alebo 120 mg jedenkrát denne) s warfarínom nemá žiadny vplyv na farmakokinetiku warfarínu u zdravých osôb. INR a pôsobenie faktora VII tiež neboli ovplyvnené so súbežným podávaním febuxostátu.
Dezipramín/substráty CYP2D6
Pri pokusoch in vitro sa preukázalo, že febuxostát je slabým inhibítorom CYP2D6. V štúdii so
zdravými osobami spôsobilo 120 mg febuxostátu QD priemerne 22 % nárast AUC dezipramínu, substrátu CYP2D6, čo naznačuje možný slabý inhibičný účinok febuxostátu na enzým CYP2D6 in vivo. Preto sa nepredpokladá, že súčasné podávanie febuxostátu s inými substrátmi CYP2D6 bude vyžadovať upravenie dávky pre tieto liečivá.
Antacidá
Ukázalo sa, že pri súbežnom užití antacíd s obsahom hydroxidu horečnatého a hydroxidu hlinitého sa
oneskorí absorpcia febuxostátu (približne o 1 hodinu) a o 32 % sa zníži hodnota Cmax, ale nebola pozorovaná žiadna významná zmena AUC. Febuxostát možno preto užívať bez ohľadu na užívanie
antacíd.
4.6 Fertilita, gravidita a laktácia
Gravidita
Údaje na veľmi limitovanom počte gravidných žien užívajúcich febuxostát nepreukázali žiadne
nežiaduce účinky na graviditu alebo na zdravie plodu/novorodenca. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývin alebo pôrod (pozri časť 5.3). Nie je známe potenciálne riziko pre ľudí. Febuxostát sa nesmie používať počas gravidity.
Dojčenie
Nie je známe, či sa febuxostát vylučuje do ľudského materského mlieka. Štúdie na zvieratách
preukázali vylučovanie tohto liečiva do mlieka a zhoršený vývin dojčených mláďat. Nemožno vylúčiť riziko pre dojča. Febuxostát sa nesmie používať počas dojčenia.
Fertilita
Reprodukčné štúdie na zvieratách s dávkami do 48 mg/kg/deň nepreukázali žiadne od dávky závislé
nežiaduce účinky na fertilitu (pozri časť 5.3). Vplyv febuxostátu na ľudskú fertilitu nie je známy.
 4.
7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
4.
7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Pri užívaní febuxostátu boli hlásené ospanlivosť, závraty, parestézia a rozmazané videnie. Pacienti by mali dbať na opatrnosť pred tým, ako budú viesť vozidlá, obsluhovať stroje alebo sa podieľať na nebezpečných aktivitách, ak nie sú ubezpečení, že Febuxostat Krka nemá nežiaduci vplyv na ich výkonnosť.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofileNajčastejšie hlásené nežiaduce reakcie v klinických štúdiách (4 072 osôb liečených dávkou od
najmenej 10 mg do 300 mg) a počas obdobia po uvedení lieku na trh sú vzplanutia dny, poruchy funkcie pečene, hnačka, nauzea, bolesť hlavy, vyrážka a edém. Tieto nežiaduce účinky boli väčšinou mierne alebo stredne závažné. Zriedkavé závažné hypersenzitívne reakcie na febuxostát, z ktorých niektoré boli spojené so systémovými príznakmi a zriedkavé prípady náhleho srdcového úmrtia, sa vyskytli po uvedení lieku na trh.
Zoznam nežiaducichreakciívtabuľkeNižšie sú uvedené časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100) a zriedkavé
(≥ 1/10 000 až < 1/1 000) nežiaduce reakcie, ktoré sa vyskytovali u pacientov liečených febuxostátom.
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie v štúdiách kombinovanej fázy 3, dlhodobých rozšírených štúdiách a počas skúseností po uvedení lieku na trhPoruchy krvi a lymfatického systému
ZriedkavéPancytopénia, trombocytopénia, agranulocytóza* Poruchy imunitného systému
ZriedkavéAnafylaktická reakcia*, hypersenzitivita na liek*
Poruchy endokrinného systému
MenejčastéZvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
Poruchy oka
ZriedkavéRozmazané videnie
Poruchy metabolizmu a výživy
Časté*** Vzplanutie dny
MenejčastéDiabetes mellitus, hyperlipidémia, znížená chuť do jedla, zvýšenie hmotnosti
ZriedkavéZníženie hmotnosti, zvýšená chuť do jedla, anorexia
Psychické poruchy
MenejčastéZnížené libido, nespavosť
ZriedkavéNervozita
Poruchy nervového systému
ČastéBolesť hlavy
MenejčastéZávraty, parestézia, hemiparéza, ospanlivosť,
zmenená chuť, hypestézia, hyposmia
Poruchy ucha a labyrintu
ZriedkavéTinitus
Poruchy srdca a poruchy srdcovej činnosti
Menejčasté
Fibrilácia predsiení, palpitácie (búšenie srdca), abnormálne EKG
Zriedkavé
Náhle srdcové úmrtie*
Cievne poruchy Menejčasté
Hypertenzia, začervenenie, návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastína Menejčasté
Dyspnoe, bronchitída, infekcia horných
dýchacích ciest, kašeľ
Poruchy gastrointestinálneho traktu Časté
Hnačka**, nevoľnosť
Menejčasté
Bolesti brucha, distenzia brucha, gastroezofageálny reflux, vracanie, suchosť v ústach, dyspepsia, zápcha, častá stolica, nadúvanie, gastrointestinálne ťažkosti Zriedkavé
Pankreatitída, vredy v ústach
Poruchy pečene a žlčových ciest Časté
Abnormálna funkcia pečene** Menejčasté
Žlčové kamene
Zriedkavé
Hepatitída, žltačka*, poškodenie pečene * Poruchy kože a podkožného tkaniva Časté
Vyrážka (vrátane rôznych druhov vyrážky
hlásených s nižšou frekvenciou, pozri nižšie) Menejčasté
Dermatitída, urtikária, pruritus, poruchy
sfarbenia kože, kožné lézie, petechie, makulózna vyrážka, makulopapulózna vyrážka, papulózna vyrážka
Zriedkavé
Toxická epidermálna nekrolýza*, Stevensov- Johnsonov syndróm*, angioedém*, lieková reakcia s eozinofíliou a systémovými symptómami*, generalizovaná vyrážka (závažná)*, erytém, exfoliatívna vyrážka, folikulárna vyrážka, vezikulárna vyrážka, pustulárna vyrážka, svrbivá vyrážka*, erytematózna vyrážka, morbilliformná vyrážka, alopécia, hyperhydróza
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Menejčasté
Artralgia, artritída, myalgia, bolesti svalov a kĺbov, svalová slabosť, svalové spazmy, napätie svalov, burzitída
Zriedkavé
Rabdomyolýza*, stuhlosť kĺbov, muskuloskeletálna stuhlosť
Poruchy obličiek a močovej sústavy
MenejčastéRenálne zlyhanie, nefrolitiáza, hematúria, polakisúria, proteinúria
ZriedkavéTubulointersticiálna nefritída*, nutkanie na močenie
Poruchy reprodukčného systému a prsníkov
MenejčastéErektilná dysfunkcia
Celkové poruchy a reakcie v mieste podania
ČastéEdém
MenejčastéÚnava, bolesť na hrudi, dyskomfort na hrudi
ZriedkavéSmäd
Abnormálne laboratórne a funkčné vyšetrenia
MenejčastéZvýšenie amylázy v krvi, zníženie počtu
trombocytov, zníženie WBC, zníženie počtu lymfocytov, zvýšenie kreatínu v krvi, zvýšenie kreatinínu v krvi, pokles hemoglobínu, zvýšenie močoviny v krvi, zvýšenie triglyceridov v krvi, zvýšenie cholesterolu v krvi, zníženie hematokritu, zvýšenie laktátovaj dehydrogenázy v krvi, zvýšenie draslíka v krvi
ZriedkavéZvýšenie glukózy v krvi, predĺženie aktivovaného parciálneho tromboplastínového času, zníženie počtu červených krviniek v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie kreatínfosfokinázy v krvi*
* Nežiaduce reakcie pochádzajúce zo skúseností po uvedení lieku na trh.
** Liečba akútnej neinfekčnej hnačky a výsledky testov pečeňovej funkcie mimo normy v kombinovanej štúdii fázy 3 sú častejšie u pacientov, ktorí sú súbežne liečení kolchicínom.
*** Pozri časť 5.1 pre výskyt vzplanutia dny v randomizovaných kontrolovaných štúdiách samostatnej
fázy 3.
Opis vybranýchnežiaducichreakciíZriedkavé závažné hypersenzitívne reakcie na febuxostát, vrátane Stevensovho-Johnsonovho
syndrómu, toxickej epidermálnej nekrolýzy a anafylaktickej reakcie/šoku, sa vyskytli počas skúseností po uvedení lieku na trh. Stevensov-Johnsonov syndróm a toxická epidermálna nekrolýza sú charakterizované progresívnymi kožnými vyrážkami spojenými s pľuzgiermi alebo léziami na slizniciach a podráždením očí. Hypersenzitívne reakcie na febuxostát môžu byť spojené s nasledujúcimi symptómami: kožné reakcie charakterizované infiltrovanými makulopapulóznymi erupciami, generalizovanými alebo exfoliatívnymi vyrážkami, ale aj kožnými léziami, edémom tváre, horúčkou, hematologickými odchýlkami ako je trombocytopénia a eozinofília a zasiahnutím jedného alebo viacerých orgánov (pečene a obličky, vrátane tubulointersticiálnej nefritídy) (pozri časť 4.4).
Vzplanutia dny boli často pozorované krátko po začatí liečby a počas prvých mesiacov. Potom sa frekvencia vzplanutia dny znížila v závislosti od času. Odporúča sa profylaxia pred vzplanutím dny (pozri časti 4.2 a 4.4).
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieLiečba pacientov s prejavmi predávkovania je podporná a symptomatická.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiuratiká (liečba dny), liečivá potlačujúce tvorbu kyseliny močovej; ATC kód: M04AA03
Mechanizmus účinku
Kyselina močová je u človeka koncový produkt metabolizmu purínov a vytvára sa v stupňoch
hypoxantín → xantín → kyselina močová. Obidva stupne v uvedenom mechanizme sú katalyzované xantínoxidázou (XO). Febuxostát je derivát 2-aryltiazolu, ktorý dosahuje terapeutický účinok zníženia kyseliny močovej v sére selektívnou inhibíciou XO. Febuxostát je potentný nepurínový selektívny inhibítor XO (NP-SIXO), ktorého hodnota Ki in vitro je menej ako jeden nanomol. Ukázalo sa, že febuxostát účinne inhibuje oxidované aj redukované formy XO. V terapeutických koncentráciách febuxostát neinhibuje iné enzýmy, ktoré sa zúčastňujú na metabolizme purínu alebo pyrimidínu, konkrétne guaníndeaminázu, hypoxantínguanínfosforibozyltransferázu, orotát-fosforibozyltransferázu, orotidínmonofosfát dekarboxylázu alebo purínnukleozidfosforylázu.
Klinická účinnosť a bezpečnosť
Účinnosť febuxostátu bola preukázaná v troch pivotných štúdiách fázy 3 (dve pivotné štúdie APEX a
FACT, a v ďalšej štúdii CONFIRMS popísaných nižšie), ktoré boli uskutočnené na 4 101 pacientoch s hyperurikémiou a dnou. V každej pivotnej štúdii fázy 3 preukázal febuxostát lepšiu schopnosť znížiť a udržať sérové koncentrácie kyseliny močovej v porovnaní s alopurinolom. Primárny cieľový
parameter účinnosti v štúdiách APEX a FACT predstavoval podiel pacientov, ktorí mali sérové koncentrácie kyseliny močovej za posledné 3 mesiace < 6,0 mg/dl (357 μmol/l). V ďalšej štúdii CONFIRMS vo fáze 3, ktorej výsledky boli prvýkrát dostupné po vydaní prvého rozhodnutia o registrácii febuxostátu, primárnym cieľovým parametrom účinnosti bol podiel pacientov, ktorých sérová koncentrácia urátov bola < 6,0 mg/dl na záverečnom vyšetrení. Do týchto štúdií neboli zahrnutí pacienti s orgánovými transplantáciami (pozri časť 4.2).
Štúdia APEX: Štúdia účinnosti febuxostátu kontrolovaná alopurinolom a placebom (APEX) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 v trvaní 28 týždňov. Randomizovaných bolo tisícsedemdesiatdva pacientov (1 072): placebo (n = 134), febuxostát 80 mg raz denne (n = 267), febuxostát 120 mg raz denne (n = 269), febuxostát 240 mg raz denne (n = 134)
alebo alopurinol (300 mg raz denne [n = 258] u pacientov s východiskovou hodnotou kreatinínu v sére
≤ 1,5 mg/dl alebo 100 mg raz denne [n = 10] u pacientov s východiskovou hodnotou kreatinínu v sére
>1,5 mg/dl a ≤ 2,0 mg/dl). Dvestoštyridsať mg febuxostátu (dvojnásobne vyššia dávka ako je najvyššia odporúčaná dávka) bolo použitých ako dávka na vyhodnotenie bezpečnosti.
Štúdia APEX ukázala štatisticky významnú superioritu v obidvoch liečených skupinách s febuxostátom 80 mg raz denne a febuxostátom 120 mg raz denne oproti skupinám s konvenčne používanými dávkami alopurinolu 300 mg (n = 258)/100 mg (n = 10) v znížení kyseliny močovej pod úroveň 6 mg/dl (357 μmol/l) (pozri tabuľku 2 a obrázok 1).
Štúdia FACT: Štúdia kontrolovaná febuxostátom a alopurinolom (FACT) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 v trvaní 52 týždňov. Sedemstošesťdesiat pacientov (760) bolo randomizovaných na: febuxostát 80 mg raz denne (n = 256), febuxostát 120 mg raz denne (n = 251) alebo alopurinol 300 mg raz denne (n = 253).
Štúdia FACT ukázala štatisticky významnú superioritu v obidvoch liečených skupinách s febuxostátom 80 mg a febuxostátom 120 mg raz denne oproti liečenej skupine s konvenčne používanou dávkou alopurinolu 300 mg v znížení kyseliny močovej a udržovaní jej hodnoty pod
6 mg/dl (357 μmol/l).
V tabuľke 2 sú výsledky primárneho cieľového parametra účinnosti:
Tabuľk
a 2
Percento pacientov s koncentráciami kyseliny močovej v sére < 6,0 mg/dl (357 μmol/l) Posledné
tri mesačné návštevy
Štúdia febuxostát
80 mg QD
febuxostát
120 mg QD
alopurinol
300/
100 mg QD1
APEX
(28 týždňov) FACT
(52 týždňov)
Kombinované výsledky
48 %*
(n = 262)
53 %*
(n = 255)
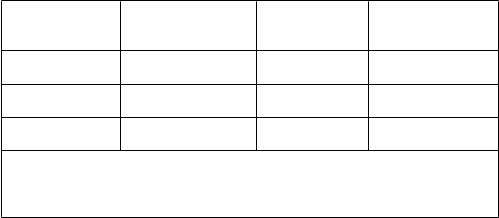
51 %*
(n = 517)
65 %*, #
(n = 269)
62 %*
(n = 250)
63 %*, #
(n = 519)
22 %
(n = 268)
21 %
(n = 251)
22 %
(n = 519)
1výsledky od subjektov, ktorým bolo podávané buď 100 mg QD (n = 10: pacienti so sérovým kreatinínom > 1,5 and ≤ 2,0 mg/dl) alebo 300 mg QD (n = 509) boli kvôli
analýzam zoskupené.
* p < 0,001 vs alopurinol, # p < 0,001 vs 80 mg
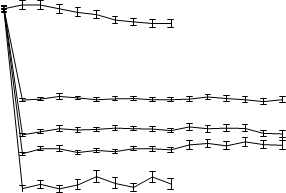
Schopnosť febuxostátu znížiť koncentrácie kyseliny močovej v sére bola rýchla a trvalá. Zníženie koncentrácie kyseliny močovej v sére na < 6,0 mg/dl (357 μmol/l) bolo zaznamenané počas návštevy v
2. týždni a udržovalo sa na rovnakej úrovni počas celej liečby. Priemerné koncentrácie kyseliny močovej v sére v čase pre každú skupinu podstupujúcu liečbu z dvoch pivotných štúdií fázy 3 sú znázornené na obrázku 1.
Obrázok 1Priemerné koncentrácie kyseliny močovej v sére v kombinácii pivotných štúdiách fázy 311
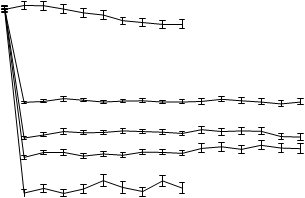
10 Placebo
9
8
7 Allopurinol
6
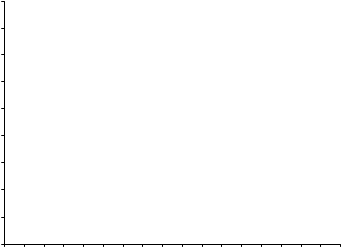
Febuxostat 80 mg
5
4 Febuxostat 120 mg
3
Febuxostat 240 mg
2
BL 2 4 6 8 12 16 20 24 28 32 36 40 44 48 52
Týždeň
BL = východisková hodnota (baseline) SEM = stredná chyba priemeru
Poznámka: 509 pacientov užívalo alopurinol v dávke 300 mg raz denne; 10 pacientov so sérovým kreatinínom > 1,5 a ≤ 2,0 mg/dl dostávalo 100 mg raz denne. (10 pacientov z 268 v štúdii APEX). Na vyhodnotenie bezpečnosti febuxostátu sa použilo 240 mg febuxostátu, čo je dvojnásobne vyššia dávka ako je najvyššia odporúčaná dávka.
Štúdia CONFIRMS: štúdia CONFIRMS bola randomizovaná kontrolovaná 26-týždňová štúdia fázy 3, ktorá hodnotila bezpečnosť a účinnosť febuxostátu 40 mg a 80 mg v porovnaní s alopurinolom 300 mg alebo 200 mg u pacientov s dnou a hyperurikémiou. Dvetisíc dvestošesťdesiatdeväť (2 269) pacientov bolo randomizovaných medzi febuxostát 40 mg raz denne (n = 757), febuxostát 80 mg raz denne (n =
756) alebo alopurinol 300/200 mg raz denne (n = 756). Najmenej 65 % pacientov malo miernu až
stredne závažnú poruchu funkcie obličiek (s klírensom kreatinínu 30 - 89 ml/min). Profylaxia pred opätovným vzplanutím dny bola povinná po dobu 26 týždňov.
Podiel pacientov, ktorí pri záverečnej návšteve mali sérové koncentrácie urátov < 6,0 mg/dl
(357 μmol/l), bol 45 % pri 40 mg febuxostátu, 67 % pri 80 mg febuxostátu a 42 % pri 300/200 mg
alopurinolu.
Primárny cieľový parameter v podskupine pacientov s poruchou funkcie obličiek
Štúdia APEX hodnotila účinnosť u 40 pacientov s poruchou funkcie obličiek (t.j. východisková koncentrácia sérového kreatinínu > 1,5 mg/dl a ≤ 2,0 mg/dl). U osôb s poruchou funkcie obličiek, ktorí boli randomizovaní na alopurinol, bola dávka ukončená na 100 mg raz denne. Febuxostát dosiahol primárny cieľový parameter účinnosti u 44 % (80 mg raz denne), 45 % (120 mg raz denne) a 60 %
(240 mg raz denne) pacientov v porovnaní s 0 % v skupine užívajúcej 100 mg alopurinolu raz denne a v skupine s placebom.
Nevyskytli sa žiadne klinicky významné rozdiely v percente zníženia koncentrácie kyseliny močovej v sére u zdravých osôb bez ohľadu na ich renálne funkcie (58 % v skupine s normálnou renálnou funkciou a 55 % v skupine so závažnou renálnou dysfunkciou).
V štúdii CONFIRMS bola prospektívne definovaná analýza pacientov s dnou a s poruchou funkcie obličiek, ktorá preukázala, že u pacientov s dnou a miernou až stredne závažnou poruchou funkcie obličiek (65 % študovaných pacientov) bol na zníženie sérových koncentrácií urátov pod 6 mg/dl významne účinnejší febuxostát ako alopurinol 300/200 mg.
Primárny cieľový parameter v podskupine pacientov s koncentráciou kyseliny močovej v sére (sUA)
≥ 10 mg/dl
Približne u 40 % pacientov (kombinácia štúdií APEX a FACT) bola východisková koncentrácia kyseliny močovej v sére (sUA) ≥ 10 mg/dl. V tejto podskupine febuxostát dosiahol primárny cieľový parameter účinnosti (sUA < 6,0 mg/dl počas posledných troch návštev) u 41 % (80 mg raz denne),
48 % (120 mg raz denne) a 66 % (240 mg raz denne) pacientov v porovnaní s 9 % v skupine užívajúcej 300 mg/100 mg alopurinolu raz denne a 0 % v skupine s placebom.
V štúdii CONFIRMS bol podiel pacientov s východiskovou sérovou koncentráciou urátov ≥ 10 mg/dl, ktorí dosiahli primárny cieľový parameter z hľadiska účinnosti (sérové koncentrácie urátov < 6,0
mg/dl pri záverečnej návšteve), 27 % (66/249) u pacientov liečených febuxostátom 40 mg podávaným
raz denne, 49 % (125/254) v prípade febuxostátu 80 mg podávaného raz denne a 31 % (72/230) pri alopurinole 300/200 mg podávaného raz denne.
Klinické výsledky: percento pacientov, ktorí vyžadovali liečbu kvôli vzplanutiu dnového záchvatu Štúdia APEX: počas 8-týždňového obdobia profylaxie väčšia časť osôb (36 %) v skupine liečenej febuxostátom 120 mg vyžadovala liečbu vzplanutia dny ako v skupine s febuxostátom 80 mg (28 %), balopurinolom 300 mg (23 %) a placebom (20 %). Výskyt záchvatov sa zvýšil po období profylaxie a postupne v priebehu času klesal. 46 - 55 % osôb dostávalo liečbu na dnové záchvaty od 8. do 28. týždňa. Dnové záchvaty sa pozorovali počas posledných 4 týždňov štúdie (24. - 28. týždeň) u 15 % (febuxostát 80 mg, 120 mg), 14 % (alopurinol 300 mg) a 20 % (placebo) osôb.
Štúdia FACT: počas 8-týždňového obdobia profylaxie väčšia časť osôb (36 %) v skupine liečenej febuxostátom 120 mg vyžadovala liečbu dnových záchvatov ako v skupine s febuxostátom 80 mg (22 %) a alopurinolom 300 mg (21 %). Po 8-týždňovom období profylaxie sa incidencia záchvatov
zvýšila a postupne v priebehu času klesla (64 % a 70 % osôb dostalo liečbu dnových záchvatov od 8. Do 52. týždňa). Záchvaty dny počas posledných 4 týždňov štúdie (49. - 52. týždeň) sa pozorovali u 6 -
8 % (febuxostát 80 mg, 120 mg) a 11 % (alopurinol 300 mg) osôb.
Percento pacientov, ktorí vyžadovali liečbu kvôli vzplanutiu dnového záchvatu (štúdia APEX a FACT) bolo číselne nižšie v skupinách, ktoré dosiahli priemerné koncentrácie urátov v sére oproti východiskovej hodnote < 6,0 mg/dl; < 5,0 mg/dl alebo < 4,0 mg/dl v porovnaní so skupinou, ktorá dosiahla priemerné koncentrácie urátov v sére oproti východiskovej hodnote ≥ 6,0 mg/dl počas posledných 32 týždňov obdobia liečby (intervaly 20. týždeň – 24. týždeň až 49. – 52. týždeň).
Počas štúdie CONFIRMS percentuálny podiel pacientov, ktorí dostali liečbu vzplanutia dny (v prvý deň až 6. mesiac) bol v skupine liečených febuxostátom 80 mg 31 % a v skupine liečenej alopurinolom 25 %. Medzi skupinami liečenými febuxostátom 80 mg a 40 mg neboli pozorované žiadne rozdiely v pomere pacientov, ktorí vyžadovali liečbu na vzplanutie dnových záchvatov.
Dlhodobé otvorené rozšírené štúdie
Štúdia EXCEL (C02-021): Štúdia EXCEL bola trojročná, otvorená, multicentrická, randomizovaná štúdia fázy 3, alopurinolom kontrolovaná bezpečnostná rozšírená štúdia u pacientov, ktorí dokončili pivotné štúdie fázy 3 (APEX a FACT). Do štúdie bolo zaradených celkom 1 086 pacientov: febuxostát
80 mg raz denne (n = 649), febuxostát 120 mg raz denne (n = 292) a alopurinol 300 mg/100 mg raz
denne (n = 145). Okolo 69 % pacientov nevyžadovalo na dosiahnutie stabilnej liečby žiadnu zmenu liečby. Pacienti, ktorí mali trikrát po sebe hladiny sUA > 6,0 mg/dl, boli vyradení.
Sérové koncentrácie urátov boli v priebehu času zachované (t.j. 91 % pacientov so začiatočnou dávkou
febuxostátu 80 mg a 93 % pacientov s dávkou febuxostátu 120 mg mali sUA < 6 mg/dl v 36. mesiaci). Údaje získané počas troch rokov ukázali, že sa znížil výskyt dnových záchvatov a menej ako 4 % pacientov vyžadovalo liečbu záchvatu (t.j. viac než 96 % pacientov nevyžadovalo liečbu záchvatu) v
16. až 24. a v 30. až 36. mesiaci.
46 % pacientom na konečnej stabilnej liečbe febuxostátom 80 mg raz denne a 38 % pacientom liečených febuxostátom 120 mg raz denne sa od pôvodného stavu až po posledné vyšetrenie úplne vyriešil primárny hmatný tofus.
Štúdia FOCUS (TMX-01-005) bola 5-ročná otvorená, multicentrická, bezpečnostná, rozšírená štúdia fázy 2 s pacientami, ktorí ukončili 4-týždne s dvojito zaslepeným podávaním febuxostátu v štúdii TMX-00-004.
Do štúdie bolo zaradených 116 pacientov, ktorí na začiatku dostávali febuxostát 80 mg raz denne.
62 % pacientov nevyžadovalo žiadnu úpravu dávkovania na udržanie sUA < 6 mg/dl a 38 % pacientov vyžadovalo úpravu až po dosiahnutí stabilnej dávky.
Pomer pacientov so sérovými koncentráciami urátov < 6,0 mg/dl (357 μmol/l) počas poslednej návštevy bol väčší ako 80 % (81 - 100 %) pre každú dávku febuxostátu.
Počas klinických štúdií fázy 3 boli pozorované mierne odchýlky v testoch funkcie pečene u pacientov liečených febuxostátom (5,0 %). Frekvencie výskytu týchto odchýlok boli podobné ako pri liečbe alopurinolom (4,2 %) (pozri časť 4.4). Zvýšené hodnoty TSH (> 5,5 μIU/ml) boli pozorované u pacientov liečených febuxostátom dlhodobo (5,5 %) a pacientov liečených alopurinolom (5,8 %) v dlhodobých otvorených rozšírených štúdiách (pozri časť 4.4).
Postmarketingové dlhodobé štúdie
Štúdia CARES bola multicentrickým, randomizovaným, dvojito zaslepeným, non-inferiority skúšaním porovnávajúcim kardiovaskulárne (KV) výstupy s febuxostátom v porovnaní s alopurinolom u pacientov s dnou a závažným kardiovaskulárnym (KV) ochorením v anamnéze, vrátane infarktu myokardu (IM), hospitalizácie pre nestabilnú angínu, koronárnej alebo cerebrálnej revaskularizácie, mozgovej príhody, prechodného ischemického záchvatu s hospitalizáciou, periférneho cievneho ochorenia alebo diabetes mellitus s evidentným mikrovaskulárnym alebo makrovaskulárnym ochorením. Pre dosiahnutie sUA menej ako 6 mg/dl bola dávka febuxostátu titrovaná od 40 mg do 80 mg (bez ohľadu na funkčnosť obličiek) a dávka alopurinolu bola titrovaná so 100 mg prírastkom od 300 do 600 mg u pacientov
s normálnou funkciou a miernym poškodením obličiek a od 200 do 400 mg u pacientov so stredne závažným poškodením obličiek.
Primárnym cieľovým ukazovateľom v štúdii CARES bol čas do prvého výskytu MACE (Major adverse cardiovascular event), kompozit nefatálneho IM, nefatálnej cievnej mozgovej príhody, KV úmrtia a nestabilnej angíny s urgentnou koronárnou revaskularizáciou.
Cieľové ukazovatele (primárne a sekundárne) sa hodnotili podľa analýzy s úmyslom liečiť (intention- to-treat - ITT) zahŕňajúcej všetkých účastníkov klinického skúšania, ktorí boli randomizovaní a dostali aspoň jednu dávku dvojito zaslepeného lieku štúdie.
Celkovo 56,6 % pacientov prerušilo liečbu v rámci skúšania predčasne a 45 % pacientov nevykonalo
všetky návštevy v skúšaní.
Celkom 6 190 pacientov bolo sledovaných po dobu s mediánom 32 mesiacov a medián trvania expozície bol 728 dní u pacientov v skupine s febuxostátom (n 3 098) a 719 dní v skupine s alopurinolom (n 3 092).
Primárny cieľový ukazovateľ MACE sa objavil v podobnej miere v skupinách liečených febuxostátom a alopurinolom (10,8 % vs. 10,4 % pacientov, v uvedenom poradí; pomer rizika [hazard ratio - HR]
1,03; obojstranný opakovaný 95 % interval spoľahlivosti [IS] 0,87 - 1,23).
Pri analýze jednotlivých zložiek MACE bola miera KV úmrtí vyššia pri febuxostáte ako pri alopurinole (4,3 % vs. 3,2 % pacientov, HR 1,34, 95 % IS 1,03 - 1,73). Miera ostatných MACE bola podobná v skupinách s febuxostátom a alopurinolom, t.j. pre nefatálny IM (3,6 % vs. 3,8 % pacientov HR 0,93, 95 % IS 0,72 až 1,21), nefatálnu mŕtvicu (2,3 % vs. 2,3 % pacientov, HR 1,01, 95 % IS 0,73
- 1,41) a urgentnú revaskularizáciu kvôli nestabilnej angíne (1,6 % vs. 1,8 % pacientov, HR 0,86,
95 % IS 0,59 - 1,26). Aj miera úmrtnosti zo všetkých príčin bola vyššia pri febuxostáte než pri alopurinole (7,8 % oproti 6,4 % pacientov, HR 1,22, 95 % IS 1,01 - 1,47), čo bolo spôsobené najmä vyššou mierou KV úmrtí v tejto skupine (pozri časť 4.4).
Miera uznanej hospitalizácie pre srdcové zlyhanie, príjmov k hospitalizácii pre arytmie bez ischémie, žilových tromboembolických udalostí a hospitalizácie pre prechodné ischemické záchvaty bola porovnateľná pre febuxostát a alopurinol.
5.2 Farmakokinetické vlastnosti
U zdravých osôb sa maximálne plazmatické koncentrácie (Cmax) a plocha pod krivkou koncentrácie a času (AUC) febuxostátu zvýšili úmerne s dávkou po jednorazových a opakovaných dávkach 10 mg až
120 mg. Pri dávkach medzi 120 mg a 300 mg bolo pozorované vyššie ako dávke úmerné zvýšenie
AUC febuxostátu. Pri podávaní dávok 10 mg až 240 mg každých 24 hodín nedochádza k žiadnej významnejšej akumulácii. Febuxostát má zdanlivý terminálny eliminačný polčas (t1/2) približne 5 až 8 hodín.
Populačné farmakokinetické/farmakodynamické analýzy boli vykonané u 211 pacientov s hyperurikémiou a dnou, ktorí boli liečení febuxostátom v dávke 40 mg - 240 mg raz denne. Vo všeobecnosti sú farmakokinetické parametre febuxostátu podľa odhadov z týchto analýz v súlade sparametrami získanými od zdravých osôb, čo znamená, že zdravé osoby sú reprezentatívnou vzorkou na hodnotenie farmakokinetických/farmakodynamických vlastností v populácii pacientov s dnou.
Absorpcia
Febuxostát sa veľmi rýchlo (tmax 1,0 - 1,5 hod.) a dobre vstrebáva (minimálne 84 %). Po jednorazovej
alebo opakovaných perorálnych dávkach 80 mg a 120 mg podávaných raz denne je Cmax približne 2,8 -
3,2 μg/ml a 5,0 - 5,3 μg/ml v uvedenom poradí. Absolútna biologická dostupnosť febuxostátu vo forme tabliet nebola stanovená.
Po opakovaných perorálnych dávkach 80 mg podávaných raz denne alebo jednorazovej dávke 120 mg s jedlom obsahujúcim vysoký podiel tukov došlo k 49 % a 38 % zníženiu Cmax a 18 % a 16 % zníženiu AUC. V testovaných prípadoch však nebola pozorovaná žiadna klinicky významná zmena v percente zníženia koncentrácie kyseliny močovej v sére (opakovaná dávka 80 mg). Febuxostát sa preto môže užívať nezávisle od jedla.
Distribúcia
Zdanlivý rovnovážny distribučný objem (Vss/F) febuxostátu je od 29 do 75 l po podaní perorálnych dávok 10 mg - 300 mg. Väzba febuxostátu na plazmatické bielkoviny je približne 99,2 %, (primárne
na albumín), a je konštantná v koncentračnom rozmedzí dosiahnutého dávkami 80 mg a 120 mg.
Väzba aktívnych metabolitov na plazmatické proteíny je približne od 82 % do 91 %.
Biotransformácia
Febuxostát sa extenzívne metabolizuje konjugáciou prostredníctvom enzýmového systému
uridíndifosfátglukuronyltransferázy (UDPGT) a oxidáciou prostredníctvom cytochrómu P450 (CYP). Boli identifikované štyri farmakologicky aktívne hydroxylové metabolity, z ktorých sa tri vyskytujú v ľudskej plazme. Štúdie in vitro s ľudskými pečeňovými mikrozómami preukázali, že tieto oxidačné metabolity boli vytvorené primárne cez CYP1A1, CYP1A2, CYP2C8 alebo CYP2C9 a febuxostát glukuronid bol vytvorený najmä v systéme UGT 1A1, 1A8 a 1A9.
Eliminácia
Febuxostát sa vylučuje pečeňou aj obličkami. Po podaní perorálnej dávky 80 mg 14C – značeného
febuxostátu sa približne 49 % dávky objavilo v moči ako nezmenený febuxostát (3 %), acylglukuronid liečiva (30 %), jeho známe oxidačné metabolity a ich konjugáty (13 %) a iné neznáme metabolity
(3 %). Okrem vylučovania obličkami sa približne 45 % dávky objavilo v stolici ako nezmenený febuxostát (12 %), acylglukuronid liečiva (1 %), jeho známe oxidačné metabolity a ich konjugáty (25 %) a iné neznáme metabolity (7 %).
Porucha funkcieobličiek
Po opakovaných dávkach 80 mg febuxostátu pacientom s miernou, stredne závažnou alebo závažnou
poruchou funkcie obličiek sa hodnota Cmax febuxostátu nezmenila v porovnaní s osobami s normálnou funkciou obličiek. Priemerná celková AUC febuxostátu sa zvýšila približne 1,8-násobne z 7,5 μg.h/ml
v skupine s normálnou funkciou obličiek na 13,2 μg.h/ml v skupine so závažnou renálnou dysfunkciou. Hodnoty Cmax a AUC aktívnych metabolitov sa zvýšili 2- a 4-násobne v uvedenom poradí. U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek však nie je potrebná žiadna úprava dávky.
Porucha funkciepečene
Po opakovaných dávkach 80 mg febuxostátu pacientom s miernou poruchou funkcie pečene
(Childovo-Pughovo skóre A) alebo so stredne závažnou poruchou funkcie pečene (Childovo-Pughovo
skóre B) sa hodnoty Cmax a AUC febuxostátu a jeho metabolitov významne nezmenili v porovnaní s osobami s normálnou funkciou pečene. Neboli uskutočnené štúdie u pacientov so závažnou poruchou
funkcie pečene (Childovo-Pughovo skóre C).
Vek
Neboli pozorované žiadne významné zmeny AUC febuxostátu alebo jeho metabolitov po
opakovaných perorálnych dávkach febuxostátu u starších ľudí v porovnaní s mladšími zdravými osobami.
Pohlavie
Po opakovaných perorálnych dávkach febuxostátu boli hodnoty Cmax o 24 % vyššie u žien ako u
mužov a hodnoty AUC boli o 12 % vyššie u žien ako u mužov. Hodnoty Cmax a AUC upravené podľa hmotnosti boli však podobné medzi oboma pohlaviami. Nie je potrebná žiadna úprava dávky podľa
pohlavia.
5.3 Predklinické údaje o bezpečnosti
Účinky v predklinických štúdiách sa všeobecne pozorovali pri expozíciách vyšších ako je maximálna expozícia u človeka.
Farmakokinetické modelovanie a simulácia údajov získaných na potkanoch naznačuje, že pri
súbežnom podávaní s febuxostátom sa klinická dávka merkaptopurínu/azatioprinu má znížiť na 20 % alebo menej % predtým predpísanej dávky, aby sa zabránilo možným hematologickým účinkom (pozri časť 4.4 a 4.5).
Karcinogenéza, mutagenéza,poruchyplodnosti
U samčích potkanov bol štatisticky významný nárast tumorov močového mechúra (papilóm z
prechodných buniek (urotel) a karcinóm) zistený iba v súvislosti s xantínovými konkrementami v skupine s vysokým dávkovaním pri približne 11-násobnej expozícii u človeka. Nebol pozorovaný signifikantný nárast žiadneho iného typu tumoru u samcov alebo samíc myší alebo potkanov. Tieto nálezy sa pokladajú za dôsledok druhovo špecifického metabolizmu purínov a zloženia moču a nemajú žiadny význam pre klinické použitie.
Štandardná séria testov genotoxicity neodhalila žiadne biologicky významné genotoxické účinky febuxostátu.
Zistilo sa, že perorálne dávky febuxostátu až 48 mg/kg/deň nemajú žiaden účinok na plodnosť a reprodukčnú schopnosť samcov a samíc potkanov.
Neboli zistené žiadne dôkazy o poruche plodnosti, teratogénnych účinkoch alebo poškodení plodu spôsobených febuxostátom. Pri vysokých dávkach sa objavila maternálna toxicita súvisiaca so znížením indexu odstavenia a znížením vývinu potomstva asi pri 4,3-násobnej expozícii u človeka. Štúdie teratogenity uskutočnené na brezivých potkanoch asi pri 4,3-násobne a na brezivých králikoch asi pri 13-násobnej expozícii u človeka, neodhalili žiadne teratogénne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Monohydrát laktózy
Mikrokryštalická celulóza
Hydroxypropylcelulóza
Sodná soľ kroskarmelózy
Koloidný hydratovaný oxid kremičitý
Stearan horečnatý
Obalová vrstva
Polyvinylalkohol
Makrogol 3350
Oxid titaničitý (E171) Mastenec
Červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaBlister (PVC/PVDC/PVC//Alu): 14, 28, 56 alebo 84 filmom obalených tabliet v škatuľke. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIKRKA, d.d., Novo mesto, Šmarješka cesta 6, 8501 Novo mesto, Slovenia
8. REGISTRAČNÉ ČÍSLA14 filmom obalených tabliet: EU/1/18/1347/001
28 filmom obalených tabliet: EU/1/18/1347/002
56 filmom obalených tabliet: EU/1/18/1347/003
84 filmom obalených tabliet: EU/1/18/1347/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. marec 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
(EMEA)
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Febuxostat Krka 120 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá filmom obalená tableta obsahuje 120 mg febuxostátu. Pomocnálátkasoznámymúčinkom
- laktóza: 109 mg
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta (tableta)
Hnedasto žlté, mierne bikonvexné filmom obalené tablety v tvare kapsuly s deliacou ryhou na obidvoch stranách. Rozmer tablety: približne 19 mm × 8 mm. Deliaca ryha slúži len na rozdelenie tablety na ľahšie prehĺtanie a nie na rozdelenie na rovnaké dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Febuxostat Krka je indikovaný na liečbu chronickej hyperurikémie pri stavoch, v ktorých už došlo k ukladaniu urátov (vrátane anamnézy alebo prítomnosti tofu a/alebo dnovej artritídy).
Febuxostat Krka je indikovaný na prevenciu a liečbu hyperurikémie u dospelých pacientov podstupujúcich chemoterapiu hematologických zhubných nádorov pri strednom až vysokom riziku syndrómu rozpadu nádoru (TLS, Tumor Lysis Syndrome).
Febuxostat Krka je určený pre dospelých.
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Dna: Odporúčaná perorálna dávka Febuxostatu Krka je 80 mg raz denne bez ohľadu na jedlo. Ak je po
2 - 4 týždňoch koncentrácia kyseliny močovej v sére > 6 mg/dl (357 μmol/l), môže sa zvážiť podávanie Febuxostatu Krka 120 mg raz denne.
Febuxostat Krka funguje dostatočne rýchlo, aby bolo možné opätovné vyšetrenie kyseliny močovej v sére po 2 týždňoch. Terapeutickým cieľom je znížiť a udržiavať kyselinu močovú v sére pod 6 mg/dl (357 μmol/l).
Odporúča sa profylaxia pred vzplanutím dnového záchvatu po dobu minimálne 6 mesiacov (pozri časť
4.4).
Syndróm rozpadu nádoru
Odporúčaná perorálna dávka Febuxostatu Krka je 120 mg raz denne bez ohľadu na jedlo. Febuxostat Krka sa má začať podávať dva dni pred začiatkom cytotoxickej liečby a pokračovať v podávaní minimálne 7 dní; liečba sa však môže predĺžiť na 9 dní podľa dĺžky trvania chemoterapie na základe klinického posúdenia.
Starší ľudia
Starším pacientom nie je potrebné upraviť dávkovanie (pozri časť 5.2).
Porucha funkcie obličiek
Účinnosť a bezpečnosť neboli úplne vyhodnotené u pacientov so závažnou poruchou funkcie obličiek
(klírens kreatinínu < 30 ml/min, pozri časť 5.2).
Úpraviť dávku nie je potrebné pacientom s miernou alebo stredne závažnou poruchou funkcie obličiek.
Porucha funkcie pečene
Účinnosť a bezpečnosť febuxostátu sa neštudovala u pacientov so závažnou poruchou funkcie pečene
(Childovo-Pughovo skóre C).
Dna: odporúčané dávkovanie pre pacientov s miernou poruchou funkcie pečene je 80 mg. Pre pacientov so stredne závažnou poruchou funkcie pečene sú k dispozícii iba obmedzené informácie.
Syndróm rozpadu nádoru: v kľúčovej štúdii tretej fázy (FLORENCE) boli z účasti zo štúdie vylúčení iba pacienti so závažnou poruchou funkcie pečene. U pacientov zaradených do štúdie nebola požadovaná úprava dávky na základe funkcie pečene.
Pediatrická populácia
Bezpečnosť a účinnosť febuxostátu u detí do 18 rokov nebola stanovená. Nie sú k dispozícii žiadne údaje.
Spôsob podávania
Perorálne použitie.
Febuxostat Krka sa má užívať ústami a môže sa užívať s jedlom alebo bez jedla.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 (pozri tiež časť 4.8).
4.4 Osobitné upozornenia a opatrenia pri používaní
Kardiovaskulárne poruchy
Liečba chronickej hyperurikémie
Liečbe febuxostatom u pacientov s už existujúcimi kardiovaskulárnymi ochoreniami (napr. infarktom myokardu, mŕtvicou alebo nestabilnou angínou pektoris) je potrebné sa vyhnúť, s jedinou výnimkou, keď nie sú vhodné žiadne iné možnosti liečby.
Číselne vyšší výskyt kardiovaskulárnych APTC príhod hlásených skúšajúcim (definovaných ako parameter z Anti-Platelet Trialists’ Collaboration (APTC) zahrňujúci kardiovaskulárnu smrť, nefatálny infarkt myokardu, nefatálnu mozgovú príhodu) bol pozorovaný v celej skupine podstupujúcej liečbu febuxostátom v porovnaní so skupinou podstupujúcou liečbu alopurinolom v štúdiách APEX a FACT (1,3 oproti 0,3 príhodám na 100 pacientorokov (PR)), ale nie v štúdii CONFIRMS (pozri časť 5.1 pre podrobné údaje o štúdiách). Výskyt skúšajúcim hlásených kardiovaskulárnych APTC príhod v kombinovaných štúdiách fázy 3 (štúdie APEX, FACT a CONFIRMS) bol 0,7 oproti 0,6 príhodám na
100 pacientorokov. V dlhodobej rozšírenej štúdii výskyt skúšajúcim hlásených APTC príhod pre
febuxostát bol 1,2 a pre alopurinol 0,6 príhod na 100 pacientorokov v uvedenom poradí. Nenašli sa žiadne štatisticky významné rozdiely a pri febuxostáte nebol potvrdený žiadny príčinný vzťah. Rizikové faktory, ktoré boli identifikované u týchto pacientov, zahŕňali anamnézu aterosklerotického ochorenia a/alebo infarktu myokardu alebo kongestívneho zlyhávania srdca.
V postregistračnom skúšaní CARES (pozri časť 5.1 pre podrobnejšiu charakteristiku štúdie) bol výskyt udalostí MACE u pacientov liečených febuxostátom podobný ako u pacientov liečených
alopurinolom (HR 1,03; 95 % IS 0,87 - 1,23), ale bola pozorovaná vyššia miera kardiovaskulárnych úmrtí (4,3 % oproti 3,2 % pacientov; HR 1,34; 95 % IS 1,03 - 1,73).
Prevencia a liečba hyperurikémie u pacientov s rizikom syndrómu rozpadu nádoru (TLS)
U pacientov, podstupujúcich chemoterapiu hematologických zhubných nádorov pri strednom až vysokom riziku syndrómu rozpadu nádoru, liečených febuxostátom, musí byť monitorovaná funkcia srdca, ako je klinicky vhodné.
Alergia nalieky/precitlivenosť
Počas skúseností po uvedení lieku na trh boli zhromaždené zriedkavé hlásenia závažných
alergických/hypersenzitívnych reakcií, vrátane život ohrozujúceho Stevensovho-Johnsonovho syndrómu, toxickej epidermálnej nekrolýzy a akútnej anafylaktickej reakcie/šoku. Vo väčšine prípadov sa tieto reakcie vyskytli počas prvého mesiaca liečby febuxostátom. Niektorí z týchto pacientov, ale nie všetci, hlásili poškodenie obličiek a/alebo predchádzajúcu hypersenzitivitu na alopurinol. Závažné hypersenzitívne reakcie vrátane liekom vyvolaných vyrážok s eozinofíliou a systémovými príznakmi (DRESS syndróm) boli v niektorých prípadoch spojené s horúčkou, hematologickými zmenami a postihnutím obličiek alebo pečene.
Pacienti majú byť poučení o prejavoch a príznakoch a majú byť dôsledne monitorovaní na výskyt symptómov alergických/hypersenzitívnych reakcií (pozri časť 4.8). Liečba febuxostátom má byť ihneď ukončená pri výskyte závažných alergických/hypersenzitívnych reakcií, vrátane Stevensovho- Johnsonovho syndrómu, pretože skoré ukončenie liečby je spojené s lepšou prognózou. Ak sa u pacienta vyvinuli alergické/hypersenzitívne reakcie, vrátane Stevensovho-Johnsonovho syndrómu a akútnej anafylaktickej reakcie/šoku, liečba týchto pacientov febuxostátom sa nesmie znovu začať.
Akútne záchvatydny(vzplanutiedny)
Liečba febuxostátom sa nesmie začať, pokiaľ úplne neodznie akútny dnový záchvat. Po začatí liečby
sa môžu vyskytnúť dnové záchvaty, a to kvôli zmene koncentrácie kyseliny močovej v sere vyplývajúcej z mobilizácie urátov uložených v tkanivách (pozri časti 4.8 a 5.1). Pri začatí liečby febuxostátom sa odporúča profylaxia záchvatu najmenej 6 mesiacov pomocou NSAID alebo kolchicínu (pozri časť 4.2).
Ak sa dnový záchvat vyskytne počas liečby febuxostátom, liečba sa nesmie prerušiť. Dnový záchvat je potrebné zvládnuť primerane podľa jednotlivého pacienta. Nepretržité podávanie febuxostátu znižuje frekvenciu a intenzitu dnových záchvatov.
Ukladanie xantínu
Pacientom, ktorí majú významne zvýšenú rýchlosť tvorby urátov (napr. malígne ochorenie a jeho
liečba, Leschov-Nyhanov syndróm), sa môže absolútna koncentrácia xantínu v moči v ojedinelých prípadoch zvýšiť natoľko, že sa bude ukladať v močovom trakte. To nebolo pozorované v kľúčovej štúdii s febuxostátom pri syndróme rozpadu nádoru. Keďže nie sú žiadne skúsenosti s užívaním febuxostátu, jeho užívanie u pacientov s Leschov-Nyhanovým syndrómom sa neodporúča.
Merkaptopurín/azatioprín
Použitie febuxostátu sa neodporúča pacientom, ktorí sú súčasne liečení merkaptopurínom alebo
azatioprínom, pretože inhibícia xantín oxidázy febuxostátom môže spôsobiť zvýšenie plazmatických hladín merkaptopurínu/azatioprínu, čo môže viesť k závažnej toxicite. U ľudí neboli uskutočnené žiadne interakčné štúdie.
Ak nie je možné sa tejto kombinácii vyhnúť, odporúča sa redukcia dávky merkaptopurínu/azatioprínu. Na základe modelových a simulačných analýz dát z preklinických štúdií na potkanoch, ktorým bol súčasne podávaný febuxostát, dávka merkaptopurínu/azatioprínu sa má redukovať na 20 % alebo menej % predtým predpísanej dávky, aby sa zabránilo možným hematologickým účinkom (pozri časť
4.5 a 5.3).
Pacienti majú byť starostlivo sledovaní a dávka merkaptopurínu/azatioprínu má byť následne upravená na základe vyhodnotenia terapeutickej odpovede a nástupu možných toxických účinkov.
Príjemcovia orgánových transplantátov
Doteraz nie sú žiadne skúsenosti s pacientmi po transplantácii orgánov, preto sa užívanie febuxostátu
týmto pacientom neodporúča (pozri časť 5.1).
Teofylín
Súčasné podanie jednej dávky 80 mg febuxostátu a jednej dávky 400 mg teofylínu zdravým
dobrovoľníkom neukázalo žiadnu farmakokinetickú interakciu (pozri časť 4.5). Febuxostát 80 mg sa môže používať u pacientov, ktorí sú súčasne liečení teofylínom bez rizika zvýšenia plazmatických koncentrácií teofylínu. Nie sú dostupné údaje pre febuxostát 120 mg.
Poruchy funkcie pečene
Počas kombinovanej tretej fázy klinických štúdií boli u pacientov liečených febuxostátom pozorované
mierne odchýlky v testoch funkcie pečene (5,0 %). Pred začatím liečby febuxostátom a potom v pravidelných intervaloch sa odporúča vyšetriť funkciu pečene na základe klinického nálezu (pozri časť
5.1).
Poruchy štítnej žľazy
V otvorených rozšírených dlhodobých štúdiách boli u pacientov počas dlhodobej liečby febuxostátom
(5,5 %) pozorované zvýšené hodnoty tyreotropného hormónu (TSH) (> 5,5 μIU/ml). Pri použití febuxostátu u pacientov so zmenenou funkciou štítnej žľazy sa vyžaduje opatrnosť (pozri časť 5.1).
Pomocné látky
Febuxostat Krka obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej
intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Merkaptopurín/azatioprín
Na základe mechanizmu účinku febuxostátu, inhibícia xantínoxidázy (XO), sa súčasné používanie
neodporúča. Inhibícia XO febuxostátom môže zvýšiť plazmatickú koncentráciu týchto liekov a viesť k ich toxicite (pozri časť 4.4). Štúdie interakcií febuxostátu s liekmi (okrem teofylínu), ktoré sú metabolizované XO neboli u ľudí uskutočnené.
Modelová a simulačná analýza dát z predklinických štúdií na potkanoch indikuje, že v prípade súčasného podávania febuxostátu, sa má dávka merkaptopurínu/azatioprínu redukovať na 20 % alebo menej % predtým predpísanej dávky (pozri časť 4.5 a 5.3).
Liekové interakčné štúdie febuxostátu s inou cytotoxickou chemoterapiou neboli vykonané. V kľúčovej štúdii bol pri syndróme rozpadu nádoru podávaný febuxostát v dávke 120 mg denne pacientom podstupujúcim rôzne chemoterapeutické režimy, vrátane monoklonálnych protilátok. Interakcie medzi liekmi neboli v priebehu tejto štúdie zaznamenané. Možné interakcie s ktorýmkoľvek súčasne podávaným cytotoxickým liekom však nie je možné vylúčiť.
Rosiglitazón/substrátyCYP2C8
In vitro sa preukázalo, že febuxostát je slabým inhibítorom CYP2C8. V štúdii so zdravými dobrovoľníkmi súčasné podanie perorálnej dávky 120 mg febuxostátu raz denne s jednou dávkou 4 mg perorálne podaného rosiglitazónu nemalo účinok na farmakokinetiku rosiglitazónu a jeho metabolit N- desmetyl rosiglitazón, febuxostát nie je inhibítorom enzýmu CYP2C8 in vivo. Súčasné podanie febuxostátu s rosiglitazónom alebo inými substrátmi CYP2C8 nevyžaduje úpravu dávky pre tieto liečivá.
Teofylín
Interakčná štúdia so zdravými dobrovoľníkmi bola vykonaná s febuxostátom na zhodnotenie, či
inhibícia xantínoxidázy (XO) môže spôsobiť zvýšenie koncentrácie teofylínu v cirkulácii ako bolo
pozorované pri použití iných inhibítorov XO. Výsledky štúdie preukázali, že súčasné podanie 80 mg febuxostátu raz denne s teofylínom 400 mg v jednej dávke nemá účinok na farmakokinetiku alebo bezpečnosť teofylínu. Osobitná opatrnosť pri súčasnom podávaní teofylínu a febuxostátu preto nie je potrebná. Nie sú dostupné údaje pre febuxostát 120 mg.
Naproxén ainéinhibítoryglukuronidácie
Metabolizmus febuxostátu závisí na uridín-glukuronyl-transferáze (UGT). Lieky inhibujúce
glukuronidáciu, ako napríklad NSAID a probenecid, by teoreticky mohli ovplyvniť eliminovanie febuxostátu. U zdravých osôb bolo súčasné užívanie febuxostátu a naproxénu 250 mg dvakrát denne spojené so zvýšenou expozíciou febuxostátu (Cmax 28 %, AUC 41 % and t1/2 26 %). V klinických štúdiách nebolo užívanie naproxénu ani iných NSAID/Cox-2 inhibítorov spojené so žiadnym signifikantným zvýšením nežiaducich účinkov.
Febuxostát možno podávať spolu s naproxénom, pričom nie je potrebná žiadna úprava dávky febuxostátu alebo naproxénu.
Induktory glukuronidácie
Silné induktory enzýmu UGT by mohli viesť k zvýšenému metabolizmu a zníženej účinnosti
febuxostátu. Monitorovanie kyseliny močovej v sére sa preto odporúča 1 - 2 týždne po začatí liečby silným induktorom glukuronidácie. Naopak, skončenie liečby induktorom by mohlo viesť k zvýšeným plazmatickým koncentráciám febuxostátu.
Kolchicín/indometacín/hydrochlorotiazid/warfarín
Febuxostát možno podávať súbežne s kolchicínom alebo indometacínom bez potreby upraviť dávku
febuxostátu alebo súbežne podávaného liečiva.
Pri podávaní febuxostátu s hydrochlorotiazidom nie je potrebná žiadna úprava dávky febuxostátu.
Pri podávaní febuxostátu s warfarínom nie je potrebná úprava dávky warfarínu. Podávanie febuxostátu (80 mg alebo 120 mg jedenkrát denne) s warfarínom nemá žiadny vplyv na farmakokinetiku warfarínu u zdravých osôb. INR a pôsobenie faktora VII tiež neboli ovplyvnené so súbežným podávaním febuxostátu.
Dezipramín/ substrátyCYP2D6
Pri pokusoch in vitro sa preukázalo, že febuxostát je slabým inhibítorom CYP2D6. V štúdii so
zdravými osobami spôsobilo 120 mg febuxostátu raz denne priemerne 22 % nárast AUC dezipramínu,
substrátu CYP2D6, čo naznačuje možný slabý inhibičný účinok febuxostátu na enzým CYP2D6 in vivo. Preto sa nepredpokladá, že súčasné podávanie febuxostátu s inými substrátmi CYP2D6 bude vyžadovať upravenie dávky pre tieto liečivá.
Antacidá

Ukázalo sa, že pri súbežnom užití antacíd s obsahom hydroxidu horečnatého a hydroxidu hlinitého sa oneskorí absorpcia febuxostátu (približne o 1 hodinu) a o 32 % sa zníži hodnota Cmax, ale nebola pozorovaná žiadna významná zmena AUC. Febuxostát možno preto užívať bez ohľadu na užívanieantacíd.
4.6 Fertility, gravidita a laktáciaGraviditaÚdaje na veľmi limitovanom počte gravidných žien užívajúcich febuxostát nepreukázali žiadne
nežiaduce účinky na graviditu alebo na zdravie plodu/novorodenca. Štúdie na zvieratách nepreukázali
priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývin alebo pôrod (pozri časť 5.3). Nie je známe potenciálne riziko pre ľudí. Febuxostát sa nesmie používať počas gravidity.
DojčenieNie je známe, či sa febuxostát vylučuje do ľudského materského mlieka. Štúdie na zvieratách
preukázali vylučovanie tohto liečiva do mlieka a zhoršený vývin dojčených mláďat. Nemožno vylúčiť
riziko pre dojča. Febuxostát sa nesmie používať počas dojčenia.
FertilitaReprodukčné štúdie na zvieratách s dávkami do 48 mg/kg/deň nepreukázali žiadne od dávky závislé
nežiaduce účinky na fertilitu (pozri časť 5.3). Vplyv Febuxostatu Krka na ľudskú fertilitu nie je
známy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojePri užívaní febuxostátu boli hlásené ospanlivosť, závraty, parestézia a rozmazané videnie. Pacienti by mali dbať na opatrnosť pred tým, ako budú viesť vozidlo, obsluhovať stroje alebo sa podieľať na nebezpečných aktivitách, ak nie sú ubezpečení, že Febuxostat Krka nemá nežiaduci vplyv na ich výkonnosť.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofileNajčastejšie hlásené nežiaduce reakcie v klinických štúdiách (4 072 osôb liečených dávkou od
najmenej 10 mg do 300 mg) a počas obdobia po uvedení na trh sú vzplanutia dny, poruchy funkcie pečene, hnačka, nauzea, bolesť hlavy, vyrážka a edém. Tieto nežiaduce účinky boli väčšinou mierne alebo stredne závažné. Zriedkavé závažné hypersenzitívne reakcie na febuxostát, z ktorých niektoré boli spojené so systémovými príznakmi a zriedkavé prípady náhleho srdcového úmrtia, sa vyskytli po uvedení .
Zoznam nežiaducichreakciívtabuľkeNižšie sú uvedené časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100) a zriedkavé (≥
1/10 000 až < 1/1 000) nežiaduce reakcie, ktoré sa vyskytovali u pacientov liečených febuxostátom.
Frekvencie výskytu sú stanovené na základe štúdií a postmarketingových skúseností u pacientov s dnou.
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie v štúdiách kombinovanej fázy 3, dlhodobých rozšírených štúdiách a počas skúseností po uvedení lieku na trhPoruchy krvi a lymfatického systému
Zriedkavé
Pancytopénia, trombocytopénia, agranulocytóza* Poruchy imunitného systému
ZriedkavéAnafylaktická reakcia*, hypersenzitivita na liek*
Poruchy endokrinného systému
MenejčastéZvýšenie hladiny hormónu stimulujúceho štítnu
žľazu v krvi
Poruchy oka
ZriedkavéRozmazané videnie
Poruchy metabolizmu a výživy
Časté*** Vzplanutie dny
MenejčastéDiabetes mellitus, hyperlipidémia, znížená chuť
do jedla, zvýšenie hmotnosti
ZriedkavéZníženie hmotnosti, zvýšená chuť do jedla, anorexia
Psychické poruchy
MenejčastéZnížené libido, nespavosť
ZriedkavéNervozita
Poruchy nervového systému
ČastéBolesť hlavy
MenejčastéZávraty, parestézia, hemiparéza, ospanlivosť, zmenená chuť, hypestézia, hyposmia
Poruchy ucha a labyrintu
ZriedkavéTinitus
Poruchy srdca a poruchy srdcovej činnosti
MenejčastéFibrilácia predsiení, palpitácie (búšenie srdca), abnormálne EKG, blok ľavého ramienka (pozri časť Syndróm rozpadu nádoru), sínusová tachykardia (pozri časť Syndróm rozpadu nádoru)
ZriedkavéNáhlesrdcovéúmrtie* Cievne poruchy
MenejčastéHypertenzia, začervenenie, návaly tepla,
hemorágia (pozri časť Syndróm rozpadu nádoru) Poruchy dýchacej sústavy, hrudníka a mediastína
MenejčastéDyspnoe, bronchitída, infekcia horných
dýchacích ciest, kašeľ
Poruchy gastrointestinálneho traktu
ČastéHnačka**, nevoľnosť
MenejčastéBolesti brucha, distenzia brucha,
gastroezofageálny reflux, vracanie, suchosť v ústach, dyspepsia, zápcha, častá stolica, nadúvanie, gastrointestinálne ťažkosti
ZriedkavéPankreatitída, vredy v ústach
Poruchy pečene a žlčových ciest
ČastéAbnormálna funkcia pečene**
Menejčasté Žlčové kamene
ZriedkavéHepatitída, žltačka*, poškodenie pečene * Poruchy kože a podkožného tkaniva
Časté
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Vyrážka (vrátane rôznych druhov vyrážky hlásených s nižšou frekvenciou, pozri nižšie) Menejčasté
Dermatitída, urtikária, pruritus, poruchy
sfarbenia kože, kožné lézie, petechie, makulózna vyrážka, makulopapulózna vyrážka, papulózna vyrážka
Zriedkavé
Toxická epidermálna nekrolýza*, Stevensov- Johnsonov syndróm*, angioedém*, lieková reakcia s eozinofíliou a systémovými symptómami*, generalizovaná vyrážka (závažná)*, erytém, exfoliatívna vyrážka, folikulárna vyrážka, vezikulárna vyrážka, pustulárna vyrážka, svrbivá vyrážka*, erytematózna vyrážka, morbilliformná vyrážka, alopécia, hyperhydróza
Menejčasté
Artralgia, artritída, myalgia, bolesti svalov a kĺbov, svalová slabosť, svalové spazmy, napätie svalov, burzitída
Zriedkavé
Rabdomyolýza*, stuhlosť kĺbov, muskuloskeletálna stuhlosť
Poruchy obličiek a močovej sústavy
MenejčastéRenálne zlyhanie, nefrolitiáza, hematúria, polakisúria, proteinúria
ZriedkavéTubulointersticiálna nefritída*, nutkanie na
močenie
Poruchy reprodukčného systému a prsníkov
MenejčastéErektilná dysfunkcia
Celkové poruchy a reakcie v mieste podania
ČastéEdém
MenejčastéÚnava, bolesť na hrudi, dyskomfort na hrudi
ZriedkavéSmäd
Abnormálne laboratórne a funkčné vyšetrenia
MenejčastéZvýšenie amylázy v krvi, zníženie počtu trombocytov, zníženie WBC, zníženie počtu lymfocytov, zvýšenie kreatínu v krvi, zvýšenie kreatinínu v krvi, pokles hemoglobínu, zvýšenie močoviny v krvi, zvýšenie triglyceridov v krvi, zvýšenie cholesterolu v krvi, zníženie hematokritu, zvýšenie laktátovaj dehydrogenázy v krvi, zvýšenie draslíka v krvi
ZriedkavéZvýšenie glukózy v krvi, predĺženie aktivovaného parciálneho tromboplastínového času, zníženie počtu červených krviniek v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie kreatínfosfokinázy v krvi*
* Nežiaduce reakcie pochádzajúce zo skúseností po uvedení lieku na trh.
** Liečba akútnej neinfekčnej hnačky a výsledky testov pečeňovej funkcie mimo normy v kombinovanej štúdii fázy 3 sú častejšie u pacientov, ktorí sú súbežne liečení kolchicínom.'
*** Pozri časť 5.1 pre výskyt vzplanutia dny v randomizovaných kontrolovanýchštúdiách samostatnej
fázy 3.
Opis vybranýchnežiaducichreakciíZriedkavé závažné hypersenzitívne reakcie na febuxostát, vrátane Stevensovho-Johnsonovho
syndrómu, toxickej epidermálnej nekrolýzy a anafylaktickej reakcie/šoku, sa vyskytli počas skúseností po uvedení lieku na trh. Stevensov-Johnsonov syndróm a toxická epidermálna nekrolýza sú charakterizované progresívnymi kožnými vyrážkami spojenými s pľuzgiermi alebo léziami na slizniciach a podráždením očí. Hypersenzitívne reakcie na febuxostát môžu byť spojené s nasledujúcimi symptómami: kožné reakcie charakterizované infiltrovanými makulopapulóznymi erupciami, generalizovanými alebo exfoliatívnymi vyrážkami, ale aj kožnými léziami, edémom tváre, horúčkou, hematologickými odchýlkami ako je trombocytopénia a eozinofília a zasiahnutím jedného alebo viacerých orgánov (pečene a obličky, vrátane tubulointersticiálnej nefritídy) (pozri časť 4.4).
Vzplanutia dny boli často pozorované krátko po začatí liečby a počas prvých mesiacov. Potom sa frekvencia vzplanutia dny znížila v závislosti od času. Odporúča sa profylaxia pred vzplanutím dny (pozri časti 4.2 a 4.4).
Syndróm rozpadunádoruZhrnutiebezpečnostnéhoprofiluV randomizovanej dvojito zaslepenej kľúčovej štúdii tretej fázy FLORENCE (FLO-01), ktorá
porovnáva febuxostát s alopurinolom (346 pacientov podstupujúcich chemoterapiu hematologických
zhubných nádorov so stredným až vysokým TLS), iba 22 (6,4 %) pacientov zaznamenalo nežiaduce účinky, konkrétne 11 (6,4 %) v každej liečenej skupine. Väčšina nežiaducich účinkov bola mierna alebo stredne silná.
Celkovo štúdia FLORENCE nepreukázala žiadne ďalšie závažné bezpečnostné skutočnosti v
porovnaní s prechádzajúcimi skúsenosťami s febuxostátom pri liečbe dny, okrem nasledujúcich troch nežiaducich účinkov (uvedených vyššie v tabuľke 1).
Poruchy srdca a srdcovej činnosti:
Menej časté: blok ľavého ramienka, sínusová tachycardia
Poruchy ciev:
Menej časté: hemorágia
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieLiečba pacientov s prejavmi predávkovania je podporná a symptomatická.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antiuratiká (liečba dny), liečivá potlačujúce tvorbu kyseliny močovej, ATC kód: M04AA03
Mechanizmusúčinku Kyselina močová je u človeka koncový produkt metabolizmu purínov a vytvára sa v stupňoch hypoxantín → xantín → kyselina močová. Obidva stupne v uvedenom mechanizme sú katalyzované xantínoxidázou (XO). Febuxostát je derivát 2-aryltiazolu, ktorý dosahuje terapeutický účinok zníženia kyseliny močovej v sére selektívnou inhibíciou XO. Febuxostát je potentný nepurínový selektívny inhibítor XO (NP-SIXO), ktorého hodnota Ki in vitro je menej ako jeden nanomol. Ukázalo sa, že febuxostát účinne inhibuje oxidované aj redukované formy XO. V terapeutických koncentráciách febuxostát neinhibuje iné enzýmy, ktoré sa zúčastňujú na metabolizme purínu alebo pyrimidínu, konkrétne guaníndeaminázu, hypoxantínguanínfosforibozyltransferázu, orotát-fosforibozyltransferázu, orotidín-monofosfát dekarboxylázu alebo purínnukleozidfosforylázu.
Klinická účinnosťabezpečnosť
Dna
Účinnosť febuxostátu bola preukázaná v troch kľúčových štúdiách fázy 3 (v dvoch kľúčových
štúdiách APEX a FACT a v pridanej CONFIRMS štúdii popísaných nižšie), ktoré boli uskutočnené na
4101 pacientoch s hyperurikémiou a dnou. V každej kľúčovej štúdii fázy 3 preukázal febuxostát vynikajúcu schopnosť znížiť a udržať koncentrácie kyseliny močovej v sére porovnateľnú s alopurinolom. Primárny parameter účinnosti v APEX a FACT štúdiách predstavoval podiel pacientov, u ktorých boli koncentrácie kyseliny močovej v sére za posledné 3 mesiace < 6,0 mg/dl (357 μmol/l). V pridanej fáze 3 štúdie CONFIRMS, ktorej výsledky boli prvýkrát dostupné po registrácii febuxostátu, primárnym parametrom účinnosti bola časť pacientov, ktorých hladina urátov v sére bola
< 6,0 mg/dl na poslednom vyšetrení. Do týchto štúdií neboli zahrnutí pacienti s orgánovými transplantáciami (pozri časť 4.2).
Štúdia APEX: Štúdia účinnosti febuxostátu kontrolovaná na základe alopurinolu a placeba (APEX) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 v trvaní 28 týždňov. Randomizovaných bolo tisícsedemdesiatdva pacientov (1072): placebo (n = 134), febuxostát 80 mg QD (n = 267), febuxostát 120 mg QD (n = 269), febuxostát 240 mg QD (n = 134) alebo alopurinol (300 mg QD [n = 258] u pacientov so základnou hodnotou kreatinínu v sére ≤ 1,5 mg/dl alebo 100 mg QD [n = 10] u pacientov so základnou hodnotou kreatinínu v sére > 1,5 mg/dl a ≤ 2,0 mg/dl). Dvestoštyridsať mg febuxostátu (dvojnásobne vyššia dávka ako je najvyššia odporúčaná dávka) bolo použitých ako dávka na vyhodnotenie bezpečnosti.
Štúdia APEX ukázala štatisticky významnú superioritu v obidvoch liečených skupinách s febuxostátom 80 mg raz denne a febuxostátom 120 mg raz denne oproti skupinám s konvenčne používanými dávkami alopurinolu 300 mg (n = 258) /100 mg (n = 10) v znižovaní kyseliny močovej pod úroveň 6 mg/dl (357 μmol/l) (pozri tabuľku 2 a obrázok 1).
Štúdia FACT: Štúdia kontrolovaná febuxostátom a alopurinolom (FACT) bola randomizovaná, dvojito zaslepená, multicentrická štúdia fázy 3 v trvaní 52 týždňov. Sedemstošesťdesiat pacientov (760) bolo randomizovaných na: febuxostát 80 mg raz denne (n = 256), febuxostát 120 mg raz denne (n = 251) alebo alopurinol 300 mg raz denne (n = 253).
Štúdia FACT ukázala štatisticky významnú superioritu v obidvoch liečených skupinách s febuxostátom 80 mg a febuxostátom 120 mg raz denne oproti liečenej skupine s konvenčne používanou dávkou alopurinolu 300 mg v znížení kyseliny močovej a udržovaní jej hodnoty pod
6 mg/dl (357 μmol/l).
V tabuľke 2 sú výsledky primárneho cieľového parametra účinnosti:
Tabuľka 2
Percento pacientov s koncentráciami kyseliny močovej v sére <6,0 mg/dl (357 μmol/l) Poslednétri mesačné návštevy
Štúdia febuxostát
80 mg QD

febuxostát
120 mg QD
alopurinol
300 /
100 mg QD1
APEX
(28 týždňov) FACT
(52 týždňov)
Kombinované výsledky
48 %*
(n = 262)
53 %*
(n = 255)
51 %*
(n = 517)
65 %*, #
(n = 269)
62 %*
(n = 250)
63 %*, #
(n = 519)
22 %
(n = 268)
21 %
(n = 251)
22 %
(n = 519)
1 výsledky od subjektov, ktorým bolo podávané buď 100 mg QD (n = 10: pacienti so sérovým kreatinínom > 1,5 a ≤ 2,0 mg/dl) alebo 300 mg QD (n = 509) boli kvôli analýzam zoskupené.
* p < 0,001 oproti allopurinolu, # p < 0,001 oproti 80 mg
Schopnosť febuxostátu znížiť koncentrácie kyseliny močovej v sére bola rýchla a trvalá. Zníženie koncentrácie kyseliny močovej v sére na < 6,0 mg/dl (357 μmol/l) bolo zaznamenané počas návštevy v
2. týždni a udržovalo sa na rovnakej úrovni počas celej liečby. Priemerné koncentrácie kyseliny močovej v sére v čase pre každú skupinu podstupujúcu liečbu z dvoch pivotných štúdií fázy 3 sú znázornené na obrázku 1.
Obrázok 1Priemerné koncentrácie kyseliny močovej v sére v kombinácii pivotných štúdiách fázy 311
10 Placebo
9
8
7 Alopurinol
6

Febuxostat 80 mg
5
4
Febuxostat 120 mg
3
Febuxostat 240 mg
2
BL 2 4 6 8 12 16 20 24 28 32 36 40 44 48 52
Týždeň
BL = východisková hodnota (baseline) SEM = stredná chyba priemeru
Poznámka: 509 pacientov užívalo alopurinol v dávke 300 mg raz denne; 10 pacientov so sérovým kreatinínom > 1,5 a ≤ 2,0 mg/dl dostávalo 100 mg raz denne. (10 pacientov z 268 v štúdii APEX). Na vyhodnotenie bezpečnosti febuxostátu sa použilo 240 mg febuxostátu, čo je dvojnásobne vyššia dávka ako je najvyššia odporúčaná dávka.
Štúdia CONFIRMS: štúdia CONFIRMS bola randomizovaná kontrolovaná 26-týždňová štúdia fázy 3, ktorá hodnotila bezpečnosť a účinnosť febuxostátu 40 mg a 80 mg v porovnaní s alopurinolom 300 mg alebo 200 mg u pacientov s dnou a hyperurikémiou. Dvetisíc dvestošesťdesiatdeväť (2 269) pacientov bolo randomizovaných medzi febuxostát 40 mg raz denne (n = 757), febuxostát 80 mg raz denne (n =
756) alebo alopurinol 300/200 mg raz denne (n = 756). Najmenej 65 % pacientov malo miernu až stredne závažnú poruchu funkcie obličiek (s klírensom kreatinínu 30 - 89 ml/min). Profylaxia pred opätovným vzplanutím dny bola povinná po dobu 26 týždňov.
Podiel pacientov, ktorí pri záverečnej návšteve mali sérové koncentrácie urátov < 6,0 mg/dl
(357 μmol/l), bol 45 % pri 40 mg febuxostátu, 67 % pri 80 mg febuxostátu a 42 % pri 300/200 mg alopurinolu.
Primárny cieľový parameter v podskupine pacientov s poruchou funkcie obličiek Štúdia APEX hodnotila účinnosť u 40 pacientov s poruchou funkcie obličiek (t.j. východisková koncentrácia sérového kreatinínu > 1,5 mg/dl a ≤ 2,0 mg/dl). U osôb s poruchou funkcie obličiek, ktorí boli randomizovaní na alopurinol, bola dávka ukončená na 100 mg raz denne. Febuxostát dosiahol primárny cieľový parameter účinnosti u 44 % (80 mg raz denne), 45 % (120 mg raz denne) a 60 %
(240 mg raz denne) pacientov v porovnaní s 0 % v skupine užívajúcej 100 mg alopurinolu raz denne a v skupine s placebom.
Nevyskytli sa žiadne klinicky významné rozdiely v percente zníženia koncentrácie kyseliny močovej v sére u zdravých osôb bez ohľadu na ich renálne funkcie (58 % v skupine s normálnou renálnou funkciou a 55 % v skupine so závažnou renálnou dysfunkciou).
V štúdii CONFIRMS bola prospektívne definovaná analýza pacientov s dnou a s poruchou funkcie obličiek, ktorá preukázala, že u pacientov s dnou a miernou až stredne závažnou poruchou funkcie obličiek (65 % študovaných pacientov) bol na zníženie sérových koncentrácií urátov pod 6 mg/dl významne účinnejší febuxostát ako alopurinol 300/200 mg.
Primárny cieľový parameter v podskupine pacientov s koncentráciou kyseliny močovej v sére (sUA) ≥
10 mg/dl
Približne u 40 % pacientov (kombinácia štúdií APEX a FACT) bola východisková koncentrácia kyseliny močovej v sére (sUA) ≥ 10 mg/dl. V tejto podskupine febuxostát dosiahol primárny cieľový parameter účinnosti (sUA < 6,0 mg/dl počas posledných troch návštev) u 41 % (80 mg raz denne),
48 % (120 mg raz denne) a 66 % (240 mg raz denne) pacientov v porovnaní s 9 % v skupine užívajúcej 300 mg/100 mg alopurinolu raz denne a 0 % v skupine s placebom.
V štúdii CONFIRMS bol podiel pacientov s východiskovou sérovou koncentráciou urátov ≥10 mg/dl, ktorí dosiahli primárny cieľový parameter z hľadiska účinnosti (sérové koncentrácie urátov <6,0 mg/dl pri záverečnej návšteve), 27 % (66/249) u pacientov liečených febuxostátom 40 mg podávaným raz denne, 49 % (125/254) v prípade febuxostátu 80 mg podávaného raz denne a 31 % (72/230) pri alopurinole 300/200 mg podávaného raz denne.
Klinické výsledky: percento pacientov, ktorí vyžadovali liečbu kvôli vzplanutiu dnového záchvatu Štúdia APEX: počas 8-týždňového obdobia profylaxie väčšia časť osôb (36 %) v skupine liečenej febuxostátom 120 mg vyžadovala liečbu vzplanutia dny ako v skupine s febuxostátom 80 mg (28 %), alopurinolom 300 mg (23 %) a placebom (20 %). Výskyt záchvatov sa zvýšil po období profylaxie a postupne v priebehu času klesal. 46 - 55 % osôb dostávalo liečbu na dnové záchvaty od 8. do 28. týždňa. Dnové záchvaty sa pozorovali počas posledných 4 týždňov štúdie (24. - 28. týždeň) u 15 % (febuxostát 80 mg, 120 mg), 14 % (alopurinol 300 mg) a 20 % (placebo) osôb.
Štúdia FACT: počas 8-týždňového obdobia profylaxie väčšia časť osôb (36 %) v skupine liečenej febuxostátom 120 mg vyžadovala liečbu dnových záchvatov ako v skupine s febuxostátom 80 mg (22 %) a alopurinolom 300 mg (21 %). Po 8-týždňovom období profylaxie sa incidencia záchvatov
zvýšila a postupne v priebehu času klesla (64 % a 70 % osôb dostalo liečbu dnových záchvatov od 8. do 52. týždňa). Záchvaty dny počas posledných 4 týždňov štúdie (49. - 52. týždeň) sa pozorovali u 6 -
8 % (febuxostát 80 mg, 120 mg) a 11 % (alopurinol 300 mg) osôb.
Percento pacientov, ktorí vyžadovali liečbu kvôli vzplanutiu dnového záchvatu (štúdia APEX a FACT) bolo číselne nižšie v skupinách, ktoré dosiahli priemerné koncentrácie urátov v sére oproti východiskovej hodnote < 6,0 mg/dl; < 5,0 mg/dl alebo < 4,0 mg/dl v porovnaní so skupinou, ktorá dosiahla priemerné koncentrácie urátov v sére oproti východiskovej hodnote ≥ 6,0 mg/dl počas posledných 32 týždňov obdobia liečby (intervaly 20. týždeň – 24. týždeň až 49. – 52. týždeň).
Počas štúdie CONFIRMS percentuálny podiel pacientov, ktorí dostali liečbu vzplanutia dny (v prvý deň až 6. mesiac) bol v skupine liečených febuxostátom 80 mg 31 % a v skupine liečenej alopurinolom 25 %. Medzi skupinami liečenými febuxostátom 80 mg a 40 mg neboli pozorované žiadne rozdiely v pomere pacientov, ktorí vyžadovali liečbu na vzplanutie dnových záchvatov.
Dlhodobé otvorené rozšírené štúdie
Štúdia EXCEL (C02-021): Štúdia EXCEL bola trojročná, otvorená, multicentrická, randomizovaná štúdia fázy 3, alopurinolom kontrolovaná bezpečnostná rozšírená štúdia u pacientov, ktorí dokončili pivotné štúdie fázy 3 (APEX a FACT). Do štúdie bolo zaradených celkom 1 086 pacientov: febuxostát
80 mg raz denne (n = 649), febuxostát 120 mg raz denne (n = 292) a alopurinol 300 mg/100 mg raz
denne (n = 145). Okolo 69 % pacientov nevyžadovalo na dosiahnutie stabilnej liečby žiadnu zmenu liečby. Pacienti, ktorí mali trikrát po sebe hladiny sUA > 6,0 mg/dl, boli vyradení.
Sérové koncentrácie urátov boli v priebehu času zachované (t.j. 91 % pacientov so začiatočnou dávkou
febuxostátu 80 mg a 93 % pacientov s dávkou febuxostátu 120 mg mali sUA < 6 mg/dl v 36. mesiaci). Údaje získané počas troch rokov ukázali, že sa znížil výskyt dnových záchvatov a menej ako 4 % pacientov vyžadovalo liečbu záchvatu (t.j. viac než 96 % pacientov nevyžadovalo liečbu záchvatu) v
16. až 24. a v 30. až 36. mesiaci.
46 % pacientom na konečnej stabilnej liečbe febuxostátom 80 mg raz denne a 38 % pacientom liečených febuxostátom 120 mg raz dennesa od pôvodného stavu až po posledné vyšetrenie úplne vyriešil primárny hmatný tofus.
Štúdia FOCUS (TMX-01-005) bola 5-ročná otvorená, multicentrická, bezpečnostná, rozšírená štúdia fázy 2 s pacientami, ktorí ukončili 4-týždne s dvojito zaslepeným podávaním febuxostátu v štúdii TMX-00-004.
Do štúdie bolo zaradených 116 pacientov, ktorí na začiatku dostávali febuxostát 80 mg raz denne.
62 % pacientov nevyžadovalo žiadnu úpravu dávkovania na udržanie sUA < 6 mg/dl a 38 % pacientov vyžadovalo úpravu až po dosiahnutí stabilnej dávky.
Pomer pacientov so sérovými koncentráciami urátov < 6,0 mg/dl (357 μmol/l) počas poslednej návštevy bol väčší ako 80 % (81 - 100 %) pre každú dávku febuxostátu.
Počas klinických štúdií fázy 3 boli pozorované mierne odchýlky v testoch funkcie pečene u pacientov liečených febuxostátom (5,0 %). Frekvencie výskytu týchto odchýlok boli podobné ako pri liečbe alopurinolom (4,2 %) (pozri časť 4.4). Zvýšené hodnoty TSH (> 5,5 μIU/ml) boli pozorované u pacientov liečených febuxostátom dlhodobo (5,5 %) a pacientov liečených alopurinolom (5,8 %) v dlhodobých otvorených rozšírených štúdiách (pozri časť 4.4).
Postmarketingové dlhodobé štúdie
Štúdia CARES bola multicentrickým, randomizovaným, dvojito zaslepeným, non-inferiority skúšaním porovnávajúcim kardiovaskulárne (KV) výstupy s febuxostátom v porovnaní s alopurinolom u pacientov s dnou a závažným kardiovaskulárnym (KV) ochorením v anamnéze, vrátane infarktu myokardu (IM), hospitalizácie pre nestabilnú angínu, koronárnej alebo cerebrálnej revaskularizácie, mozgovej príhody, prechodného ischemického záchvatu s hospitalizáciou, periférneho cievneho ochorenia alebo diabetes mellitus s evidentným mikrovaskulárnym alebo makrovaskulárnym ochorením. Pre dosiahnutie sUA menej ako 6 mg/dl bola dávka febuxostátu titrovaná od 40 mg do 80 mg (bez ohľadu na funkčnosť obličiek) a dávka alopurinolu bola titrovaná so 100 mg prírastkom od
300 do 600 mg u pacientov s normálnou funkciou a miernym poškodením obličiek a od 200 do 400
mg u pacientov so stredne závažným poškodením obličiek.
Primárnym cieľovým ukazovateľom v štúdii CARES bol čas do prvého výskytu MACE (Major
adverse cardiovascular event), kompozit nefatálneho IM, nefatálnej cievnej mozgovej príhody, KV
úmrtia a nestabilnej angíny s urgentnou koronárnou revaskularizáciou.
Cieľové ukazovatele (primárne a sekundárne) sa hodnotili podľa analýzy s úmyslom liečiť (intention-
to-treat - ITT) zahŕňajúcej všetkých účastníkov klinického skúšania, ktorí boli randomizovaní a dostali aspoň jednu dávku dvojito zaslepeného lieku štúdie.
Celkovo 56,6 % pacientov prerušilo liečbu v rámci skúšania predčasne a 45 % pacientov nevykonalo
všetky návštevy v skúšaní.
Celkom 6 190 pacientov bolo sledovaných po dobu s mediánom 32 mesiacov a medián trvania expozície bol 728 dní u pacientov v skupine s febuxostátom (n 3 098) a 719 dní v skupine s alopurinolom (n 3 092).
Primárny cieľový ukazovateľ MACE sa objavil v podobnej miere v skupinách liečených febuxostátom a alopurinolom (10,8 % vs. 10,4 % pacientov, v uvedenom poradí; pomer rizika [hazard ratio - HR]
1,03; obojstranný opakovaný 95 % interval spoľahlivosti [IS] 0,87 - 1,23).
Pri analýze jednotlivých zložiek MACE bola miera KV úmrtí vyššia pri febuxostáte ako pri alopurinole (4,3 % vs. 3,2 % pacientov, HR 1,34, 95 % IS 1,03 - 1,73). Miera ostatných MACE bola podobná v skupinách s febuxostátom a alopurinolom, t.j. pre nefatálny IM (3,6 % vs. 3,8 % pacientov HR 0,93, 95 % IS 0,72 až 1,21), nefatálnu mŕtvicu (2,3 % vs. 2,3 % pacientov, HR 1,01, 95 % IS 0,73
- 1,41) a urgentnú revaskularizáciu kvôli nestabilnej angíne (1,6 % vs. 1,8 % pacientov, HR 0,86, 95
% IS 0,59 - 1,26). Aj miera úmrtnosti zo všetkých príčin bola vyššia pri febuxostáte než pri
alopurinole (7,8 % oproti 6,4 % pacientov, HR 1,22, 95 % IS 1,01 - 1,47), čo bolo spôsobené najmä vyššou mierou KV úmrtí v tejto skupine (pozri časť 4.4).
Miera uznanej hospitalizácie pre srdcové zlyhanie, príjmov k hospitalizácii pre arytmie bez ischémie, žilových tromboembolických udalostí a hospitalizácie pre prechodné ischemické záchvaty bola porovnateľná pre febuxostát a alopurinol.
Syndróm rozpadu nádoru
Účinnosť a bezpečnosť febuxostátu v prevencii a liečbe syndrómu rozpadu nádoru skúmala štúdia
FLORENCE (FLO-01). Febuxostát C preukázal vyšší a rýchlejší účinok v znižovaní urátov v porovnaní s alopurinolom.
FLORENCE bola randomizovaná (1:1), dvojito zaslepená, pivotná štúdia fázy 3, ktorá porovnávala febuxostát v dávke 120 mg raz denne s alopurinolom v dávke 200 až 600 mg denne (priemerná denná dávka [± štandardná odchýlka]: 349,7 ± 112,90 mg) v kontrole hladiny kyseliny močovej v sére.
Vybraní pacienti boli kandidátmi na liečbu alopurinolom alebo nemali prístup k rasburikáze. Primárne cieľové parametre boli plocha pod krivkou kyseliny močovej v sére (AUC sUA1-8) a zmena hladiny sérového kreatinínu (sC), v oboch prípadoch z východiskovej hodnoty do 8. dňa.
Celkovo bolo do štúdie zaradených 346 pacientov s hematologickými malignitami podstupujúcich chemoterapiu a so stredným až vysokým rizikom syndrómu rozpadu nádoru. Priemerná AUC sUA1-8 (mgxh/dl) bola významne nižšia pri febuxostáte (514,0 ± 225,71 verzus 708,0 ± 234,42; rozdiel metódou najmenších štvorcov: -196,794 [95 % interval spoľahlivosti: -238,600; - 154,988]; p <
0,0001). Okrem toho priemerné koncentrácie kyseliny močovej v sére boli významne nižšie pri febuxostáte od prvých 24 hodín liečby aj kedykoľvek v následných časových bodoch. Medzi febuxostátom a alopurinolom sa neobjavil žiadny významný rozdiel v zmene strednej hodnoty kreatinínu v sére (%) (-0,83 ± 26, 98 vs -4,92 ± 16,70; rozdiel metódou najmenších štvorcov: 4,0970
[95 % interval spoľahlivosti: -0,6467; 8,8406]; p = 0,0903). Čo sa týka sekundárnych parametrov neboli zaznamenané významné rozdiely v incidencii laboratórneho syndrómu rozpadu nádoru (8,1 % a
9,2 % pri febuxostáte a alopurinole v uvedenom poradí; relatívne riziko: 0,875 [95 % interval spoľahlivosti: 0,4408; 1,7369]; p = 0,8488) ani klinického TLS (1,7 % a 1,2 % pri febuxostáte a alopurinole v uvedenom poradí; relatívne riziko: 0,994 [95 % interval spoľahlivosti: 0,9691; 1,0199];
p = 1,0000). Incidencia celkových liečbu vyžadujúcich prejavov a príznakov a nežiaducich reakcií
bola 67,6 % versus 64,7 % a 6,4 % versus 6,4 % pri febuxostáte a alopurinole v uvedenom poradí. V štúdii FLORENCE preukázal febuxostát vyššiu kontrolu sérových koncentrácií kyseliny močovej v porovnaní s alopurinolom u pacientov s indikáciou na liečbu alopurinolom. V súčasnosti nie sú dostupné údaje porovnávajúce febuxostát s rasburikázou. Účinnosť a bezpečnosť febuxostátu neboli stanovené u pacientov s akútnym závažným TLS, napr. u pacientov, u ktorých zlyhala iná liečba na zníženie urátov.
5.2 Farmakokinetické vlastnosti
U zdravých osôb sa maximálne plazmatické koncentrácie (Cmax) a plocha pod krivkou koncentrácie a času (AUC) febuxostátu zvýšili úmerne s dávkou po jednorazových a opakovaných dávkach 10 mg až
120 mg. Pri dávkach medzi 120 mg a 300 mg bolo pozorované vyššie ako dávke úmerné zvýšenie AUC febuxostátu. Pri podávaní dávok 10 mg až 240 mg každých 24 hodín nedochádza k žiadnej významnejšej akumulácii. Febuxostát má zdanlivý terminálny eliminačný polčas (t1/2) približne 5 až 8 hodín.
Populačné farmakokinetické/farmakodynamické analýzy boli vykonané u 211 pacientov s hyperurikémiou a dnou, ktorí boli liečení febuxostátom v dávke 40 mg - 240 mg raz denne. Vo všeobecnosti sú farmakokinetické parametre febuxostátu podľa odhadov z týchto analýz v súlade s
parametrami získanými od zdravých osôb, čo znamená, že zdravé osoby sú reprezentatívnou vzorkou na hodnotenie farmakokinetických/farmakodynamických vlastností v populácii pacientov s dnou.
Absorpcia
Febuxostát sa veľmi rýchlo (tmax 1,0 - 1,5 hod.) a dobre vstrebáva (minimálne 84 %). Po jednorazovej
alebo opakovaných perorálnych dávkach 80 mg a 120 mg podávaných raz denne je Cmax približne 2,8 -
3,2 μg/ml a 5,0 - 5,3 μg/ml v uvedenom poradí. Absolútna biologická dostupnosť febuxostátu vo forme tabliet nebola stanovená.
Po opakovaných perorálnych dávkach 80 mg podávaných raz denne alebo jednorazovej dávke 120 mg s jedlom obsahujúcim vysoký podiel tukov došlo k 49 % a 38 % zníženiu Cmax a 18 % a 16 % zníženiu AUC. V testovaných prípadoch však nebola pozorovaná žiadna klinicky významná zmena v percente zníženia koncentrácie kyseliny močovej v sére (opakovaná dávka 80 mg). Febuxostát sa preto môže užívať nezávisle od jedla.
Distribúcia
Zdanlivý rovnovážny distribučný objem (Vss/F) febuxostátu je od 29 do 75 l po podaní perorálnych dávok 10 mg - 300 mg. Väzba febuxostátu na plazmatické bielkoviny je približne 99,2 %, (primárne
na albumín), a je konštantná v koncentračnom rozmedzí dosiahnutého dávkami 80 mg a 120 mg.
Väzba aktívnych metabolitov na plazmatické proteíny je približne od 82 % do 91 %.
Biotransformácia
Febuxostát sa extenzívne metabolizuje konjugáciou prostredníctvom enzýmového systému
uridíndifosfátglukuronyltransferázy (UDPGT) a oxidáciou prostredníctvom cytochrómu P450 (CYP). Boli identifikované štyri farmakologicky aktívne hydroxylové metabolity, z ktorých sa tri vyskytujú v ľudskej plazme. Štúdie in vitro s ľudskými pečeňovými mikrozómami preukázali, že tieto oxidačné metabolity boli vytvorené primárne cez CYP1A1, CYP1A2, CYP2C8 alebo CYP2C9 a febuxostát glukuronid bol vytvorený najmä v systéme UGT 1A1, 1A8 a 1A9.
Eliminácia
Febuxostát sa vylučuje pečeňou aj obličkami. Po podaní perorálnej dávky 80 mg 14C – značeného
febuxostátu sa približne 49 % dávky objavilo v moči ako nezmenený febuxostát (3 %), acylglukuronid liečiva (30 %), jeho známe oxidačné metabolity a ich konjugáty (13 %) a iné neznáme metabolity
(3 %). Okrem vylučovania obličkami sa približne 45 % dávky objavilo v stolici ako nezmenený
febuxostát (12 %), acylglukuronid liečiva (1 %), jeho známe oxidačné metabolity a ich konjugáty
(25 %) a iné neznáme metabolity (7 %).
Porucha funkcieobličiek
Po opakovaných dávkach 80 mg febuxostátu pacientom s miernou, stredne závažnou alebo závažnou
poruchou funkcie obličiek sa hodnota Cmax febuxostátu nezmenila v porovnaní s osobami s normálnou funkciou obličiek. Priemerná celková AUC febuxostátu sa zvýšila približne 1,8-násobne z 7,5 μg.h/ml
v skupine s normálnou funkciou obličiek na 13,2 μg.h/ml v skupine so závažnou renálnou dysfunkciou. Hodnoty Cmax a AUC aktívnych metabolitov sa zvýšili 2- a 4-násobne v uvedenom poradí. U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek však nie je potrebná žiadna úprava dávky.
Porucha funkciepečene
Po opakovaných dávkach 80 mg febuxostátu pacientom s miernou poruchou funkcie pečene
(Childovo-Pughovo skóre A) alebo so stredne závažnou poruchou funkcie pečene (Childovo-Pughovo
skóre B) sa hodnoty Cmax a AUC febuxostátu a jeho metabolitov významne nezmenili v porovnaní s osobami s normálnou funkciou pečene. Neboli uskutočnené štúdie u pacientov so závažnou poruchou
funkcie pečene (Childovo-Pughovo skóre C).
Vek
Neboli pozorované žiadne významné zmeny AUC febuxostátu alebo jeho metabolitov po
opakovaných perorálnych dávkach febuxostátu u starších ľudí v porovnaní s mladšími zdravými osobami.
Pohlavie
Po opakovaných perorálnych dávkach febuxostátu boli hodnoty Cmax o 24 % vyššie u žien ako u
mužova hodnoty AUC boli o 12 % vyššie u žien ako u mužov. Hodnoty Cmax a AUC upravené podľa hmotnosti boli však podobné medzi oboma pohlaviami. Nie je potrebná žiadna úprava dávky podľa pohlavia.
5.3 Predklinické údaje o bezpečnosti
Účinky v predklinických štúdiách sa všeobecne pozorovali pri expozíciách vyšších ako je maximálna expozícia u človeka.
Farmakokinetické modelovanie a simulácia údajov získaných na potkanoch naznačuje, že pri súbežnom podávaní s febuxostátom sa klinická dávka merkaptopurínu/azatioprinu má znížiť na 20 % alebo menej % predtým predpísanej dávky, aby sa zabránilo možným hematologickým účinkom (pozri časť 4.4 a 4.5).
Karcinogenéza, mutagenéza,poruchyplodnosti
U samčích potkanov bol štatisticky významný nárast tumorov močového mechúra (papilóm z
prechodných buniek (urotel) a karcinóm) zistený iba v súvislosti s xantínovými konkrementami v skupine s vysokým dávkovaním pri približne 11-násobnej expozícii u človeka. Nebol pozorovaný signifikantný nárast žiadneho iného typu tumoru u samcov alebo samíc myší alebo potkanov. Tieto nálezy sa pokladajú za dôsledok druhovo špecifického metabolizmu purínov a zloženia moču a nemajú žiadny význam pre klinické použitie.
Štandardná séria testov genotoxicity neodhalila žiadne biologicky významné genotoxické účinky febuxostátu.
Zistilo sa, že perorálne dávky febuxostátu až 48 mg/kg/deň nemajú žiaden účinok na plodnosť a reprodukčnú schopnosť samcov a samíc potkanov.
Neboli zistené žiadne dôkazy o poruche plodnosti, teratogénnych účinkoch alebo poškodení plodu spôsobených febuxostátom. Pri vysokých dávkach sa objavila maternálna toxicita súvisiaca so znížením indexu odstavenia a znížením vývinu potomstva asi pri 4,3-násobnej expozícii u človeka. Štúdie teratogenity uskutočnené na brezivých potkanoch asi pri 4,3-násobne a na brezivých králikoch asi pri 13-násobnej expozícii u človeka, neodhalili žiadne teratogénne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Monohydrát laktózy
Mikrokryštalická celulóza
Hydroxypropylcelulóza
Sodná soľ kroskarmelózy
Koloidný hydratovaný oxid kremičitý
Stearan horečnatý
Obalová vrstvaPolyvinylalkohol
Makrogol 3350
Oxid titaničitý (E171) Mastenec
Žltý oxid železitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti2 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaBlister (PVC/PVDC/PVC//Alu): 14, 28, 56 alebo 84 filmom obalených tabliet v škatuľke. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIKRKA, d.d., Novo mesto, Šmarješka cesta 6, 8501 Novo mesto, Slovinsko
8. REGISTRAČNÉ ČÍSLA14 filmom obalených tabliet: EU/1/18/1347/005
28 filmom obalených tabliet: EU/1/18/1347/006
56 filmom obalených tabliet: EU/1/18/1347/007
84 filmom obalených tabliet: EU/1/18/1347/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. marca 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.