
i ezetimibe/simvastatíne 0,2 % a pri simvastatíne 0,1 %, pričom myopatia bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN alebo dve následné pozorovania hladiny CK ≥ 5 a < 10-násobok HHN. Incidencia rabdomyolýzy bola pri ezetimibe/simvastatíne 0,1 % a pri simvastatíne 0,2 %, pričom rabdomyolýza bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN s dôkazom poškodenia obličiek, ≥ 5-násobok HHN a < 10-násobok HHN počas dvoch následných udalostí
s dôkazom poškodenia obličiek alebo hladinou CK ≥ 10,000 IU/l bez dôkazu poškodenia obličiek
(pozri časť 4.8).
V klinickom skúšaní, v ktorom bolo viac ako 9 000 pacientov s chronickým ochorením obličiek randomizovaných na užívanie 10 mg ezetimibu v kombinácii s 20 mg simvastatínu denne (n = 4 650) alebo placeba (n = 4 620) (medián sledovaného obdobia 4,9 rokov), bol výskyt
myopatie/rabdomyolýzy 0,2 % pri ezetimibe v kombinácii so simvastatínom a 0,1 % pri placebe (pozri
časť 4.8).
Hepatálnainsuficiencia
Vzhľadom na neznáme účinky zvýšenej expozície ezetimibu sa u pacientov so stredne závažnou alebo
závažnou hepatálnou insuficienciou ezetimib neodporúča (pozri časť 5.2).
Pediatrická populácia
Účinnosť a bezpečnosť ezetimibu u pacientov vo veku 6 až 10 rokov s heterozygotnou familiárnou alebo non-familiárnou hypercholesterolémiou boli hodnotené v 12-týždňovom placebom kontrolovanom klinickom skúšaní. Účinky ezetimibu počas obdobia liečby > 12 týždňov neboli v tejto vekovej skupine skúmané (pozri časti 4.2, 4.8, 5.1 a 5.2).
Ezetimib sa neskúmal u pacientov mladších ako 6 rokov (pozri časti 4.2 a 4.8).
Účinnosť a bezpečnosť ezetimibu podávaného spolu so simvastatínom pacientom vo veku 10 až
17 rokov s heterozygotnou familiárnou hypercholesterolémiou sa hodnotila v kontrolovanom klinickom skúšaní u adolescentných chlapcov (Tannerovo štádium II alebo vyššie) a u dievčat, ktoré boli minimálne jeden rok po menarché.
V tejto limitovanej kontrolovanej štúdii nebol u adolescentných chlapcov alebo dievčat vo
všeobecnosti žiadny zistiteľný vplyv na rast alebo sexuálne dospievanie, ani akýkoľvek vplyv na dĺžku menštruačného cyklu u dievčat. Účinky ezetimibu na rast a sexuálne dospievanie počas liečebného obdobia > 33 týždňov sa však neštudovali (pozri časti 4.2 a 4.8).
Bezpečnosť a účinnosť ezetimibu podávaného spolu s dávkami simvastatínu nad 40 mg denne sa u pediatrických pacientov vo veku 10 až 17 rokov neštudovali.
Bezpečnosť a účinnosť ezetimibu podávaného spolu so simvastatínom sa u pediatrických pacientov vo
veku < 10 rokov neštudovali (pozri časti 4.2 a 4.8).
Dlhodobá účinnosť liečby ezetimibom u pacientov mladších ako 17 rokov na zníženie morbidity
a mortality v dospelosti sa neštudovala.
Fibráty
Bezpečnosť a účinnosť ezetimibu podávaného s fibrátmi neboli stanovené.
Ak je u pacienta užívajúceho ezetimib a fenofibrát podozrenie na cholelitiázu, je indikované vyšetrenie žlčníka a táto liečba sa má prerušiť (pozri časti 4.5 a 4.8).
Cyklosporín
Ak sa ezetimib začína podávať počas liečby cyklosporínom, je potrebná opatrnosť. U pacientov
užívajúcich ezetimib a cyklosporín sa majú monitorovať koncentrácie cyklosporínu (pozri časť 4.5).
Antikoagulanciá
Ak je ezetimib pridaný k warfarínu, k inému kumarínovému antikoagulantu alebo k fluindiónu, je
potrebné náležite monitorovať International Normalized Ratio (INR) (pozri časť 4.5).
Pomocnélátkysoznámymúčinkom
Tento liek obsahuje monohydrát laktózy. Pacienti so zriedkavými dedičnými problémami galaktózovej
intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať
tento liek.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
V predklinických štúdiách sa preukázalo, že ezetimib neindukuje enzýmy cytochrómu P450, ktoré metabolizujú lieky. Nepozorovali sa žiadne klinicky významné farmakokinetické interakcie medzi ezetimibom a liekmi, o ktorých je známe, že sú metabolizované cytochrómami P450 1A2, 2D6, 2C8,
2C9 a 3A4 alebo N-acetyltransferázou.
V štúdiách klinických interakcií nemal ezetimib žiadny vplyv na farmakokinetiku súbežne podávaného dapsónu, dextrometorfánu, digoxínu, perorálnych kontraceptív (etinylestradiol a levonorgestrel), glipizidu, tolbutamidu alebo midazolamu. Cimetidín podávaný súbežne
s ezetimibom nemal žiadny vplyv na biologickú dostupnosť ezetimibu.
Antacidá
Súbežné podávanie antacíd znížilo mieru absorpcie ezetimibu, ale nemalo žiadny vplyv na jeho
biologickú dostupnosť. Toto zníženie miery absorpcie sa nepovažuje za klinicky významné.
C
holestyramín
Súbežné podávanie cholestyramínu znížilo priemernú plochu pod krivkou (AUC) celkového ezetimibu
(ezetimib + ezetimib-glukuronid) približne o 55 %. Prírastok zníženia LDL cholesterolu (LDL-C)
v dôsledku pridania ezetimibu k cholestyramínu môže byť touto interakciou zmenšený (pozri časť
4.2).
Fibráty
U pacientov užívajúcich fenofibrát a ezetimib si má byť lekár vedomý možného rizika cholelitiázy
a ochorenia žlčníka (pozri časti 4.4 a 4.8).
Ak je u pacienta užívajúceho ezetimib a fenofibrát podozrenie na cholelitiázu, je indikované vyšetrenie žlčníka a táto liečba sa má prerušiť (pozri časť 4.8).
Súbežné podávanie fenofibrátu alebo gemfibrozilu mierne zvýšilo celkové koncentrácie ezetimibu
(približne 1,5 a 1,7-násobne v uvedenom poradí).
Súbežné podávanie ezetimibu s inými fibrátmi sa neštudovalo.
Fibráty môžu zvýšiť vylučovanie cholesterolu do žlče a tým viesť k cholelitiáze. V štúdiách na zvieratách ezetimib niekedy zvýšil cholesterol v žlčníkovej žlči, nie však u všetkých druhov (pozri časť 5.3). Litogénne riziko spojené s terapeutickým použitím ezetimibu nie je možné vylúčiť.
Statíny
Pri súbežnom podávaní ezetimibu s atorvastatínom, simvastatínom, pravastatínom, lovastatínom,
fluvastatínom alebo rosuvastatínom sa nepozorovali žiadne klinicky signifikantné farmakokinetické
interakcie.
Cyklosporín
V štúdii ôsmich pacientov po transplantácii obličky s klírensom kreatinínu > 50 ml/min na stabilnej
dávke cyklosporínu mala jednorazová 10-mg dávka ezetimibu za následok 3,4-násobné (rozsah 2,3 až
7,9-násobné) zvýšenie priemernej AUC celkového ezetimibu v porovnaní so zdravou kontrolnou
populáciou, ktorá dostávala samotný ezetimib v inej štúdii (n = 17). V inej štúdii mal pacient
s transplantovanou obličkou a závažnou renálnou insuficienciou, ktorý užíval cyklosporín a inú mnohopočetnú liečbu, 12-násobne vyššiu expozíciu k celkovému ezetimibu v porovnaní so súbežnými
kontrolami užívajúcimi samotný ezetimib. V dvojdobej skríženej štúdii s dvanástimi zdravými jedincami viedlo denné podávanie 20 mg ezetimibu počas 8 dní s jednorazovou 100-mg dávkou
cyklosporínu na 7. deň k priemernému 15 % nárastu AUC cyklosporínu (rozmedzie 10 % pokles až
51 % nárast) v porovnaní s jednorazovou 100-mg dávkou samotného cyklosporínu. Kontrolovaná štúdia účinku súbežného podávania ezetimibu na expozíciu cyklosporínu u pacientov
s transplantovanou obličkou sa nevykonala. Ak sa začína podávať ezetimib pri liečbe cyklosporínom, je potrebná opatrnosť. U pacientov užívajúcich ezetimib a cyklosporín sa majú monitorovať
koncentrácie cyklosporínu (pozri časť 4.4).
Antikoagulanciá
V štúdii dvanástich zdravých dospelých mužov nemalo súbežné podávanie ezetimibu (10 mg raz denne) signifikantný účinok na biologickú dostupnosť warfarínu a protrombínový čas. Po uvedení do praxe však bolo hlásené zvýšenie International Normalized Ratio (INR) u pacientov, ktorým bol ezetimib pridaný k warfarínu alebo fluindiónu. Ak je ezetimib pridaný k warfarínu, k inému kumarínovému antikoagulantu alebo fluindiónu, je potrebné náležite monitorovať INR (pozri časť
4.4).
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ezetimib súbežne podávaný so statínom je kontraindikovaný počas gravidity a dojčenia (pozri časť
4.3), oboznámte sa, prosím, s SPC príslušného statínu.
GraviditaEzetimib sa má podávať gravidným ženám iba v nevyhnutných prípadoch. Nie sú k dispozícii žiadne klinické údaje o použití ezetimibu počas gravidity. Štúdie na zvieratách s ezetimibom v monoterapii
nepreukázali žiadne priame alebo nepriame škodlivé účinky na graviditu, embryofetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
DojčenieEzetimib sa nemá užívať počas dojčenia. Štúdie na potkanoch preukázali, že ezetimib sa vylučuje do materského mlieka potkanov. Nie je známe, či sa ezetimib vylučuje do ľudského materského mlieka.
FertilitaK dispozícii nie sú žiadne údaje z klinického skúšania o účinkoch ezetimibu na fertilitu u ľudí. Ezetimib nemal žiadny účinok na fertilitu u samcov alebo samíc potkanov (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeNeuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení
vozidiel alebo obsluhovaní strojov sa však má vziať do úvahy, že bol hlásený závrat.
4.8 Nežiaduce účinkyKlinickéštúdieaskúsenostipouvedeníliekunatrh:V klinických štúdiách trvajúcich do 112 týždňov bolo denne podávané 10 mg ezetimibu samostatne
2 396 pacientom, so statínom 11 308 pacientom alebo s fenofibrátom 185 pacientom. Nežiaduce
reakcie boli zvyčajne mierne a prechodné. Celková incidencia nežiaducich účinkov bola pri ezetimibe a placebe podobná. Podobne, počet prerušení účasti v štúdii z dôvodu nežiaducich účinkov bol pre
ezetimib a placebo porovnateľný.
Ezetimib podávanýsamostatnealebospolusostatínom:U pacientov liečených ezetimibom (N = 2 396) a vo väčšej miere ako pri placebe (N = 1 159), alebo u pacientov liečených ezetimibom podávaným spolu so statínom (N = 11 308) a vo väčšej miere ako
pri podávaní samotného statínu (N = 9 361) sa pozorovali nasledujúce nežiaduce reakcie. Nežiaduce
reakcie po uvedení lieku na trh boli odvodené z hlásení pri podávaní ezetimibu samostatne alebo so
statínom.
Frekvencie sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000
až <1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< /10 000) a neznáme
(z dostupných údajov).
Monoterapia ezetimibom
| Časté
| Menej časté
|
Poruchy metabolizmu a výživy
|
| · znížená chuť do jedla
|
Poruchy ciev
|
| · nával tepla
· hypertenzia
|
Poruchy dýchacej sústavy, hrudníka
a mediastína
|
| · kašeľ
|
Poruchy gastrointestinálneho traktu
| · bolesť brucha
· hnačka
· flatulencia
| · dyspepsia
· gastroezofageálny reflux
· nauzea
|
P
oruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
|
· artralgia
· svalové spazmy
· bolesť krku
|
C
elkové poruchy a reakcie v mieste podania
|
· únava
|
· bolesť na hrudi
· bolesť
|
L
aboratórne a funkčné vyšetrenia
|
|
· zvýšená ALT a/alebo
AST
· zvýšená CPK v krvi
· zvýšená gama-glutamyl-
transferáza
· abnormálny test pečeňovej funkcie
|
Ď
alšie nežiaduce reakcie pri ezetimibe podávanom súbežne so statínom
|
Č
asté
|
Menej časté
|
P
oruchy nervového systému
|
· bolesť hlavy
|
· parestézia
|
P
oruchy gastrointestinálneho traktu
|
|
· sucho v ústach
· gastritída
|
P
oruchy kože a podkožného tkaniva
|
|
· pruritus
· vyrážka
· urtikária
|
P
oruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
· myalgia
|
· bolesť chrbta
· svalová slabosť
· bolesť v končatine
|
C
elkové poruchy a reakcie v mieste podania
|
|
· asténia
· periférny edém
|
L
aboratórne a funkčné vyšetrenia
|
· zvýšená ALT a/alebo
AST
|
|
Skúsenosti po uvedení lieku na trh (so statínom alebo bez neho)
|
N
eznáme
|
P
oruchy krvi a lymfatického systému
|
· trombocytopénia
|
P
oruchy imunitného systému
|
· precitlivenosť vrátane vyrážky, urtikárie, anafylaxie
a angioedému
|
P
sychické poruchy
|
· depresia
|
P
oruchy nervového systému
|
· závrat
· parestézia
|
P
oruchy dýchacej sústavy, hrudníka
a mediastína
|
· dyspnoe
|
P
oruchy gastrointestinálneho traktu
|
· pankreatitída
· zápcha
|
P
oruchy pečene a žlčových ciest
|
· hepatitída
· cholelitiáza
· cholecystitída
|
P
oruchy kože a podkožného tkaniva
|
· erythema multiforme
|
P
oruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
· myalgia
· myopatia/rabdomyolýza (pozri časť 4.4)
|
C
elkové poruchy a reakcie v mieste podania
|
· asténia
|
E
z
etimib podávaný súbežne s fenofibrátom
|
Č
asté
|
P
oruchy gastrointestinálneho traktu
|
· bolesť brucha
|
V multicentrickej, dvojito zaslepenej, placebom kontrolovanej klinickej štúdii u pacientov
so zmiešanou hyperlipidémiou bolo liečených 625 pacientov počas obdobia do 12 týždňov a 576 pacientov počas obdobia do 1 roka. V tejto štúdii 172 pacientov liečených ezetimibom a fenofibrátom dokončilo 12 týždňov liečby a 230 pacientov liečených ezetimibom a fenofibrátom (vrátane 109 pacientov, ktorí počas prvých 12 týždňov dostávali samotný ezetimib) dokončilo 1 rok liečby. Táto štúdia nebola usporiadaná tak, aby porovnávala liečebné skupiny podľa zriedkavých nežiaducich účinkov.
Hodnoty incidencie (95 % IS) klinicky významných zvýšení (> 3 x HHN, po sebe) sérových transamináz upravené na expozíciu liečbe boli 4,5 % (1,9; 8,8) pre monoterapiu fenofibrátom a 2,7 %
(1,2; 5,4) pre ezetimib podávaný spolu s fenofibrátom. Zodpovedajúce hodnoty incidencie pre
cholecystektómiu boli 0,6 % (0,0; 3,1) pre monoterapiu fenofibrátom a 1,7 % (0,6; 4,0) pre ezetimib
podávaný súbežne s fenofibrátom (pozri časti 4.4 a 4.5).
Pediatrickí pacienti (vo veku 6 až17rokov)V štúdii zahŕňajúcej pediatrických pacientov (vo veku 6 až 10 rokov) s heterozygotnou familiárnou
alebo non-familiárnou hypercholesterolémiou (n = 138) sa zvýšenia ALT a/alebo AST (≥ 3 x HHN, po sebe) pozorovali u 1,1 % pacientov (1 pacient) liečených ezetimibom v porovnaní s 0 % v skupine
s placebom. Zvýšenie CPK (≥ 10 x HHN) sa neobjavilo. Neboli hlásené prípady myopatie.
V separátnej štúdii zahŕňajúcej adolescentných pacientov (vo veku 10 až 17 rokov) s heterozygotnou familiárnou hypercholesterolémiou (n = 248) sa zvýšenia ALT a/alebo AST (≥ 3 x HHN, po sebe) pozorovali u 3 % pacientov (4 pacienti) liečených ezetimibom/simvastatínom v porovnaní s 2 % (2 pacienti) v skupine s monoterapiou simvastatínom. Zvýšenie CPK (≥ 10 násobok HHN) sa pozorovalo u 2 % pacientov (2 pacienti) liečených ezetimibom/simvastatínom a u 0 % v skupine s monoterapiou simvastatínom. Neboli hlásené prípady myopatie.
Tieto skúšania neboli vhodné na porovnanie zriedkavých nežiaducich liekových reakcií.
Pacienti s koronárnouchorobousrdcaapríhodouAKSvanamnézeV štúdii IMPROVE-IT (pozri časť 5.1) zahŕňajúcej 18 144 pacientov liečených ezetimibom/simvastatínom v dávke 10/40 mg (n = 9 067, z ktorých 6 % bolo vytitrovaných na
ezetimib/simvastatín 10/80 mg) alebo simvastatínom 40 mg (n = 9 077, z ktorých 27 % bolo
vytitrovaných na simvastatín 80 mg), boli bezpečnostné profily počas mediánu sledovaného obdobia
6,0 rokov podobné. Miery prerušenia liečby z dôvodu nežiaducich účinkov boli 10,6 % u pacientov liečených ezetimibom/simvastatínom a 10,1 % u pacientov liečených simvastatínom. Incidencia myopatie bola u pacientov liečených ezetimibom/simvastatínom 0,2 % a u pacientov liečených simvastatínom 0,1 %, pričom myopatia bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN alebo dve následné pozorovania hladiny CK ≥ 5
a < 10-násobok HHN. Incidencia rabdomyolýzy bola pri ezetimibe/simvastatíne 0,1 % a pri simvastatíne 0,2 %, pričom rabdomyolýza bola definovaná ako nevysvetliteľná svalová slabosť alebo bolesť s hladinou CK v sére ≥ 10-násobok HHN s dôkazom poškodenia obličiek, ≥ 5-násobok HHN
a < 10-násobok HHN počas dvoch následných udalostí s dôkazom poškodenia obličiek alebo hladinou
CK ≥ 10,000 IU/l bez dôkazu poškodenia obličiek. Incidencia následných zvýšení transamináz
(≥ 3 x HHN) bola 2,5 % pri ezetimibe/simvastatíne a 2,3 % pri simvastatíne. (Pozri časť 4.4). Nežiaduce účinky súvisiace so žlčníkom boli hlásené u 3,1 % pacientov s ezetimibom/simvastatínom oproti 3,5 % pacientov so simvastatínom. Incidencia hospitalizácií z dôvodu cholecystektómie bola
v obidvoch liečebných skupinách 1,5 %. Rakovina (definovaná ako akákoľvek nová malignita) sa počas skúšania diagnostikovala u 9,4 % pacientov s ezetimibom/simvastatínom oproti 9,5 % pacientov
so simvastatínom.
Pacienti s chronickýmochorenímobličiekV štúdii ochrany srdca a obličiek (
Study of Heart and Renal Protection, SHARP) (pozri časť 5.1), do
ktorej bolo zapojených viac ako 9 000 pacientov liečených kombináciou fixných dávok 10 mg
ezetimibu s 20 mg simvastatínu denne (n = 4 650) alebo placebom (n = 4 620), boli bezpečnostné profily počas mediánu sledovaného obdobia 4,9 rokov porovnateľné. V tomto skúšaní sa zaznamenávali len závažné nežiaduce udalosti a vysadenia lieku z dôvodu akýchkoľvek nežiaducich udalostí. Miery vysadenia lieku z dôvodu nežiaducich udalostí boli porovnateľné (10,4 % u pacientov liečených ezetimibom v kombinácii so simvastatínom, 9,8 % u pacientov liečených placebom). Výskyt myopatie/rabdomyolýzy bol 0,2 % u pacientov liečených ezetimibom v kombinácii so simvastatínom
a 0,1 % u pacientov liečených placebom. Následné zvýšenia transamináz (> 3 x HHN) sa objavili u 0,7 % pacientov liečených ezetimibom v kombinácii so simvastatínom v porovnaní s 0,6 %
pacientov liečených placebom (pozri časť 4.4). V tomto skúšaní sa neobjavili žiadne štatisticky významné zvýšenia výskytu vopred špecifikovaných nežiaducich udalostí vrátane rakoviny (9,4 % pri
ezetimibe v kombinácii so simvastatínom, 9,5 % pri placebe), hepatitídy, cholecystektómie alebo
komplikácií žlčových kameňov alebo pankreatitídy.
LaboratórnehodnotyV kontrolovaných klinických skúšaniach monoterapie bola incidencia klinicky významného zvýšenia sérových transamináz (ALT a/alebo AST ≥ 3 x HHN, po sebe) podobná pri ezetimibe (0,5 %)
a placebe (0,3 %). V skúšaniach súbežného podávania bola incidencia 1,3 % u pacientov liečených
ezetimibom v kombinácii so statínom a 0,4 % u pacientov liečených samotným statínom. Tieto zvýšenia boli vo všeobecnosti asymptomatické, neboli spojené s cholestázou a vrátili sa do
východiskových hodnôt po prerušení liečby alebo pri jej pokračovaní (pozri časť 4.4).
V klinických skúšaniach bola CPK > 10 x HHN hlásená u 4 z 1 674 (0,2 %) pacientov, ktorí dostávali ezetimib samotný oproti 1 zo 786 (0,1 %) pacientov, ktorí dostávali placebo, a u 1 z 917 (0,1 %) pacientov, ktorí dostávali súbežne ezetimib a statín oproti 4 z 929 (0,4 %) pacientov, ktorí dostávali samotný statín. V porovnaní s príslušným kontrolným ramenom (placebo alebo samotný statín) sa
v súvislosti s ezetimibom nevyskytla nadmerná myopatia alebo rabdomyolýza (pozri časť 4.4).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách bolo podávanie ezetimibu 15 zdravým jedincom v dávke 50 mg/deň až po dobu
14 dní alebo 18 pacientom s primárnou hypercholesterolémiou v dávke 40 mg/deň až po dobu 56 dní
vo všeobecnosti dobre tolerované. Po jednorazovej perorálnej dávke 5 000 mg/kg ezetimibu u potkanov a myší a 3 000 mg/kg u psov sa u týchto zvierat nepozorovala žiadna toxicita.
Bolo hlásených niekoľko prípadov predávkovania ezetimibom; väčšina nebola spojená s nežiaducimi účinkami.
Hlásené nežiaduce účinky neboli závažné. V prípade predávkovania treba použiť symptomatické
a podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: iné látky upravujúce lipidy, ATC kód: C10AX09
MechanizmusúčinkuEzetimib je nová trieda látok znižujúcich lipidy, ktoré selektívne inhibujú črevnú absorpciu cholesterolu a príbuzných rastlinných sterolov. Ezetimib je aktívny po perorálnom podaní a má mechanizmus účinku, ktorý sa líši od iných skupín látok znižujúcich cholesterol (napr. statínov, sekvestrantov žlčových kyselín [živíc], derivátov kyseliny fibrovej a rastlinných stanolov).
Molekulárnym cieľom ezetimibu je sterolový transportér Niemann-Pick C1-Like 1 (NPC1L1), ktorý je zodpovedný za intestinálne vychytávanie cholesterolu a fytosterolov.
Ezetimib sa lokalizuje na kefkovitý lem sliznice tenkého čreva a inhibuje absorpciu cholesterolu, čo
vedie k zníženej dodávke črevného cholesterolu do pečene; statíny znižujú syntézu cholesterolu
v pečeni, a spolu týmito odlišnými mechanizmami poskytujú doplňujúcu redukciu cholesterolu.
V dvojtýždňovej klinickej štúdii u 18 hypercholesterolemických pacientov inhiboval ezetimib v porovnaní s placebom absorpciu cholesterolu v čreve o 54 %.
Farmakodynamickéúčinky
Na určenie selektivity ezetimibu na inhibíciu absorpcie cholesterolu sa vykonali série predklinických štúdií. Ezetimib inhiboval absorpciu [14C]-cholesterolu, pričom nemal žiadny účinok na absorpciu triglyceridov, mastných kyselín, žlčových kyselín, progesterónu, etinylestradiolu alebo v tukoch rozpustných vitamínov A a D.
Epidemiologické štúdie preukázali, že kardiovaskulárna morbidita a mortalita sa priamoúmerne mení
s hladinou celkového-C a LDL-C a nepriamoúmerne s hladinou HDL-C.
Podávanie ezetimibu so statínom je účinné v znižovaní rizika kardiovaskulárnych príhod u pacientov s koronárnou chorobou srdca a príhodou AKS v anamnéze.
Klinickáúčinnosťabezpečnosť
V kontrolovaných klinických štúdiách ezetimib v monoterapii aj súbežne podávaný so statínom signifikantne znížil celkový cholesterol (celkový-C), cholesterol nízkodenzitných lipoproteínov
(LDL-C), apolipoproteín B (Apo B), triglyceridy (TG) a zvýšil cholesterol vysokodenzitných
lipoproteínov (HDL-C) u pacientov s hypercholesterolémiou.
Primárnahypercholesterolémia
V dvojito zaslepenej, placebom kontrolovanej, 8-týždňovej štúdii u 769 pacientov
s hypercholesterolémiou, ktorí už dostávali monoterapiu statínom a nedosiahli cieľovú hladinu LDL-C
podľa „National Cholesterol Education Program (NCEP)“ (2,6 až 4,1 mmol/l [100 až 160 mg/dl]
v závislosti od vstupných hodnôt) boli pacienti randomizovaní tak, aby dostávali k doterajšej liečbe statínom buď 10 mg ezetimibu alebo placebo.
Spomedzi pacientov liečených statínmi, ktorí pri vstupe do štúdie nespĺňali cieľovú hladinu LDL-C (~82 %), dosiahlo cieľovú hodnotu LDL-C na konci štúdie 72 % pacientov randomizovaných pre ezetimib a 19 % pacientov randomizovaných pre placebo. Korešpondujúce zníženia LDL-C boli signifikantne rozdielne (25 % pre ezetimib a 4 % pre placebo). Okrem toho, ezetimib pridaný
k prebiehajúcej liečbe statínom v porovnaní s placebom signifikantne znížil celkový-C, Apo B, TG
a zvýšil HDL-C. Ezetimib alebo placebo pridané k liečbe statínom znížili medián C-reaktívneho
proteínu o 10 % a 0 % v tomto poradí, oproti východiskovým hodnotám.
V dvoch dvojito zaslepených, randomizovaných, placebom kontrolovaných, 12-týždňových štúdiách
u 1 719 pacientov s primárnou hypercholesterolémiou ezetimibu v dávke 10 mg v porovnaní
s placebom signifikantne znížil celkový-C (13 %), LDL-C (19 %), Apo B (14 %) a TG (8 %) a zvýšil
HDL-C (3 %). Navyše ezetimib nemal žiadny účinok na plazmatické koncentrácie v tukoch rozpustných vitamínov A, D a E, nemal žiadny účinok na protrombínový čas a tak, ako ostatné látky znižujúce lipidy, neovplyvnil tvorbu adrenokortikálneho steroidného hormónu.
V multicentrickej, dvojito zaslepenej, kontrolovanej klinickej štúdii (ENHANCE) bolo 720 pacientov s heterozygotnou familiárnou hypercholesterolémiou randomizovaných na ezetimib 10 mg
v kombinácii so simvastatínom 80 mg (n = 357) alebo na simvastatín 80 mg (n = 363) počas 2 rokov.
Primárnym cieľom štúdie bolo preskúmať účinok kombinovanej liečby ezetimib/simvastatín na hrúbku vrstvy intima-média (intima-media thickness, IMT) krčnej tepny v porovnaní s monoterapiou simvastatínom. Vplyv tohto zástupného markera na kardiovaskulárnu morbiditu a mortalitu nie je stále preukázaný.
Primárny cieľový ukazovateľ, zmena priemernej hodnoty IMT všetkých šiestich segmentov krčnej tepny meraná ultrazvukom v B móde, sa medzi dvoma liečebnými skupinami signifikantne nelíšil (p = 0,29). Počas 2-ročného trvania štúdie sa hrúbka vrstvy intima-média zväčšila o 0,0111 mm pri
ezetimibe 10 mg v kombinácii so simvastatínom 80 mg (východisková priemerná hodnota karotickej IMT 0,68 mm) a o 0,0058 mm pri samotnom simvastatíne 80 mg (východisková priemerná hodnota karotickej IMT 0,69 mm).
Ezetimib 10 mg v kombinácii so simvastatínom 80 mg znížil LDL-C, celkový-C, Apo B a TG signifikantne viac ako simvastatín 80 mg. Percentuálne zvýšenie HDL-C bolo podobné pre obe liečebné skupiny. Nežiaduce reakcie hlásené pre ezetimib 10 mg v kombinácii so simvastatínom
80 mg boli zhodné s jeho známym bezpečnostným profilom.
Pediatrická populácia
V multicentrickej, dvojito zaslepenej, kontrolovanej štúdii bolo 138 pacientov (59 chlapcov
a 79 dievčat), vo veku 6 až 10 rokov (priemerný vek 8,3 rokov) s heterozygotnou familiárnou alebo
non-familiárnou hypercholesterolémiou (HeFH) s východiskovými hladinami LDL-C medzi 3,74
a 9,92 mmol/l, randomizovaných buď na ezetimib 10 mg alebo na placebo počas 12 týždňov.
V 12. týždni ezetimib v porovnaní s placebom signifikantne znížil celkový-C (-21 % oproti 0 %), LDL-C (-28 % oproti -1 %), Apo B (-22 % oproti -1 %), a non-HDL-C (-26 % oproti 0 %). Výsledky pri týchto dvoch liečebných skupinách boli podobné pre TG a HDL-C (-6 % oproti +8 % a +2 % oproti +1 % v tomto poradí).
V multicentrickej, dvojito zaslepenej, kontrolovanej štúdii bolo 142 chlapcov (Tannerovo štádium II
a vyššie) a 106 dievčat po menarché, vo veku 10 až 17 rokov (priemerný vek 14,2 rokov)
s heterozygotnou familiárnou hypercholesterolémiou (HeFH) s východiskovými hladinami LDL-C
medzi 4,1 a 10,4 mmol/l randomizovaných buď na ezetimib 10 mg podávaný spolu so simvastatínom (10, 20 alebo 40 mg) alebo na samotný simvastatín (10, 20 alebo 40 mg) počas 6 týždňov, ďalej na ezetimib podávaný spolu so simvastatínom 40 mg alebo na samotný simvastatín 40 mg počas ďalších
27 týždňov a následne na otvorené súbežné podávanie ezetimibu a simvastatínu (10, 20 alebo 40 mg)
počas ďalších 20 týždňov.
V 6. týždni ezetimib podávaný spolu so simvastatínom (všetky dávky) v porovnaní so samotným simvastatínom (všetky dávky) signifikantne znížil celkový-C (38 % oproti 26 %), LDL-C (49 % oproti
34 %), Apo B (39 % oproti 27 %), a non-HDL-C (47 % oproti 33 %). Výsledky týchto liečebných skupín boli podobné pre TG (-17 % oproti -12 %) a HDL-C (+7 % oproti +6 %). V 33. týždni boli
výsledky konzistentné s výsledkami zo 6. týždňa a signifikantne viac pacientov užívajúcich ezetimib a simvastatín 40 mg (62 %) dosiahlo ideálny cieľ podľa NCEP APP (< 2,8 mmol/l [110 mg/dl]) pre LDL-C v porovnaní s pacientmi užívajúcimi samotný simvastatín 40 mg (25 %). V 53. týždni, na
konci otvoreného predĺženia, boli účinky na lipidové parametre zachované.
Bezpečnosť a účinnosť ezetimibu podávaného spolu s dávkami simvastatínu nad 40 mg denne sa u pediatrických pacientov vo veku 10 až 17 rokov neštudovali. Bezpečnosť a účinnosť ezetimibu podávaného spolu so simvastatínom sa u pediatrických pacientov vo veku < 10 rokov neštudovali. Dlhodobá účinnosť liečby ezetimibom u pacientov mladších ako 17 rokov na zníženie morbidity
a mortality v dospelosti sa neštudovala.
Prevenciakardiovaskulárnychpríhod
Skúšanie IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) bolo
multicentrickou, randomizovanou, dvojito zaslepenou štúdiou s aktívnym komparátorom zahŕňajúcou
18 144 pacientov zaradených v priebehu 10 dní od hospitalizácie z dôvodu akútneho koronárneho syndrómu (AKS, buď akútny infarkt myokardu [IM] alebo nestabilná angína pektoris [NAP]). Pacienti
mali v čase AKS LDL-C ≤ 125 mg/dl (≤ 3,2 mmol/l), ak neužívali liečbu na zníženie tukov alebo
≤ 100 mg/dl (≤ 2,6 mmol/l), ak užívali liečbu na zníženie tukov. Všetci pacienti boli randomizovaní
v pomere 1:1 na užívanie ezetimibu/simvastatínu v dávke 10/40 mg (n = 9 067) alebo simvastatínu
40 mg (n = 9 077) a sledovaní počas mediánu 6,0 rokov.
Priemerný vek pacientov bol 63,6 rokov; 76 % bolo mužov, 84 % belochov a 27 % diabetikov. Priemerná hodnota LDL-C v čase príhody spĺňajúcej podmienky na zaradenie do štúdie bola 80 mg/dl (2,1 mmol/l) u pacientov užívajúcich liečbu na zníženie tukov (n = 6 390) a 101 mg/dl (2,6 mmol/l)
u pacientov bez predchádzajúcej liečby na zníženie tukov (n = 11 594). Pred hospitalizáciou z dôvodu príhody AKS spĺňajúcej podmienky na zaradenie do štúdie bolo 34 % pacientov na liečbe statínom.
U pacientov pokračujúcich v liečbe bola po jednom roku priemerná LDL-C 53,2 mg/dl (1,4 mmol/l)
v skupine užívajúcej ezetimib/simvastatín a 69,9 mg/dl (1,8 mmol/l) v skupine užívajúcej simvastatín
v monoterapii. Hodnoty lipidov sa vo všeobecnosti určovali pre pacientov, ktorí pokračovali
v skúšanej liečbe.
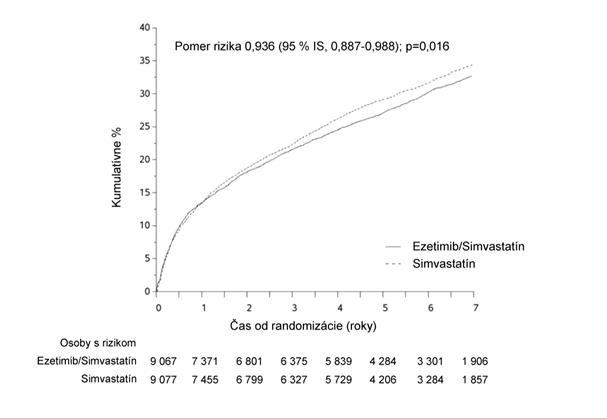
Primárny cieľový ukazovateľ bol zložený z kardiovaskulárneho úmrtia, veľkých koronárnych príhod (MCE [major coronary events], definované ako nefatálny infarkt myokardu, dokumentovaná nestabilná angína pektoris vyžadujúca hospitalizáciu alebo akýkoľvek koronárny revaskularizačný výkon, ktorý sa uskutočnil aspoň 30 dní po randomizovanom priradení liečby) a nefatálnej cievnej mozgovej príhody. Štúdia preukázala, že liečba ezetimibom súbežne so simvastatínom poskytla dodatočný prínos pri znižovaní primárneho cieľového ukazovateľa zloženého z kardiovaskulárneho úmrtia, MCE a nefatálnej cievnej mozgovej príhody v porovnaní so samotným simvastatínom (zníženie relatívneho rizika 6,4 %, p = 0,016). Primárny cieľový ukazovateľ sa vyskytol u 2 572
z 9 067 pacientov (7-ročná Kaplanova-Meierova [KM] hodnota 32,72 %) v skupine užívajúcej
ezetimib/simvastatín a u 2 742 z 9 077 pacientov (7-ročná KM hodnota 34,67 %) v skupine užívajúcej samotný simvastatín. (Pozri obrázok 1 a tabuľku 1). Očakáva sa, že tento dodatočný prínos je podobný pri súbežnom podávaní s inými statínmi, u ktorých bolo preukázané, že sú účinné v znižovaní rizika kardiovaskulárnych príhod. Celková úmrtnosť sa v tejto vysoko rizikovej skupine nezmenila (pozri tabuľku 1).
Pre všetky typy cievnej mozgovej príhody sa pozoroval celkový prínos, avšak v skupine užívajúcej ezetimib so simvastatínom sa pozoroval nesignifikantný nárast hemoragickej cievnej mozgovej príhody v porovnaní so skupinou so samotným simvastatínom (pozri tabuľku 1). Riziko hemoragickej cievnej mozgovej príhody pri podávaní ezetimibu súbežne s účinnejšími statínmi sa v dlhodobých štúdiách nehodnotilo.
Účinok liečby ezetimibom/simvastatínom bol vo všeobecnosti zhodný s celkovými výsledkami medzi mnohými podskupinami zahŕňajúcimi pohlavie, vek, rasu, diabetes mellitus v anamnéze, východiskové hladiny lipidov, predchádzajúcu liečbu statínom, predchádzajúcu cievnu mozgovú príhodu a hypertenziu.
O
brázok 1: Účinok ezetimibu/simvastatínu na primárny zložený cieľový ukazovateľ kardiovaskulárneho úmrtia, veľkej koronárnej príhody alebo nefatálnej cievnej mozgovej príhody
 T
abuľka 1: Veľké kardiovaskulárne príhody podľa liečebnej skupiny u všetkých randomizovaných pacientov v štúdii IMPROVE-IT
T
abuľka 1: Veľké kardiovaskulárne príhody podľa liečebnej skupiny u všetkých randomizovaných pacientov v štúdii IMPROVE-IT
V
ýsledok
E
z
etimib/Simvastatín
10/40 mga
(N = 9 067)
Simvastatin
40 mgb
(N = 9 077)
Pomer rizika
(95 % IS)
Hodnota
p
n K-M %c n K-M %c
 Primárny zložený cieľový ukazovateľ účinnosti
Primárny zložený cieľový ukazovateľ účinnosti(KV úmrtie, veľké koronárne

príhody a nefatálna cievna mozgová príhoda)
2 572 32,72 % 2 742 34,67 % 0,936 (0,887;
0,988)
0,016
 Sekundárny zložený cieľový ukazovateľ účinnosti
Sekundárny zložený cieľový ukazovateľ účinnosti
Úmrtie z dôvodu KCHS,
nefatálny IM, naliehavá koronárna revaskularizácia po
30 dňoch
MCE, nefatálna cievna
1 322 17,52 % 1 448 18,88 % 0,912 (0,847; 0,016
0,983)
mozgová príhoda, úmrtie
3 089 38,65 % 3 246 40,25 % 0,948 (0,903;
0,035
(
v
šetky príčiny) 0,996)
KV úmrtie, nefatálny IM,
nestabilná angína pektoris

vyžadujúca hospitalizáciu, akákoľvek revaskularizácia, nefatálna cievna mozgová príhoda
2 716 34,49 % 2 869 36,20 %
0,945 (0,897;
0,996) 0,035
V
ýsledok
E
z
etimib/Simvastatín
10/40 mga
(N = 9 067)
Simvastatin
40 mgb
(N = 9 077)
Pomer rizika
(95 % IS)
Hodnota p
n K-M %c n K-M %c
Zložky primárneho zloženého cieľového ukazovateľa a vybrané cieľové ukazovatele účinnosti(prvý výskyt špecifikovanej príhody v akomkoľvek čase)

1,000 (0,887;
1,127)
0,871 (0,798;
0,950)
1,059 (0,846;
0,997
0,002
1,326)
0,618
0,947 (0,886;
1,012) 0,107
0,802 (0,678;
0,949) 0,010
Kardiovaskulárne úmrtie
|
537
|
6,89 %
|
538
|
6,84 %
|
Veľká koronárna príhoda:
|
|
|
|
|
Nefatálny IM
|
945
|
12,77 %
|
1 083
|
14.41 %
|
Nestabilná angína pektoris
vyžadujúcahospitalizáciu
|
156
|
2,06 %
|
148
|
1,92 %
|
Koronárna revaskularizácia po
30dňoch
|
1 690
|
21.84 %
|
1 793
|
23,36 %
|
Nefatálna cievna mozgová
príhoda
|
245
|
'
3.49 %
|
305
|
4,24 %
|
IM (fatálny a nefatálny)
|
977
|
13.13 %
|
1 118
|
14,82 %
|
|
|
Cievna mozgová príhoda
0,872 (0,800;
0,950)
0,857 (0,734;
0,002
(f
a
t
á
l
naa nefatálna) 296 4.16 % 345 4,77 % 1,001) 0,052
Nehemoragická cievna
mozgová príhodad 242 3.48 % 305 4,23 %
Hemoragická cievna mozgová
príhoda 59 0.77 % 43 0,59 %
0,793 (0,670;
0,939) 0,007
1,377 (0,930;
2,040) 0,110
Úmrtie z akejkoľvek príčiny 1 215 15.36 % 1 231 15,28 % 0,989 (0,914;
1,070)
0,782
a 6 % bolo vytitrovaných na ezetimib/simvastatín 10/80 mg
b 27 % bolo vytitrovaných na simvastatín 80 mg
c Odhad v 7.roku podľa Kaplana-Meiera
d Zahŕňa ischemickú cievnu mozgovú príhodu alebo cievnu mozgovú príhodu neurčeného typu
Prevenciaveľkýchvaskulárnychpríhodprichronickomochoreníobličiek(Chronic Kidney Disease,CKD)Štúdia ochrany srdca a obličiek (
Study of Heart and Renal Protection, SHARP) bola multinárodná
randomizovaná, placebom kontrolovaná, dvojito zaslepená štúdia, ktorá sa vykonala u 9 438 pacientov s chronickým ochorením obličiek, z ktorých tretina bola na začiatku štúdie na hemodialýze. Celkovo bolo 4 650 pacientov zaradených na kombináciu fixných dávok ezetimibu 10 mg a 20 mg simvastatínu a 4 620 na placebo a pacienti boli sledovaní počas mediánu 4,9 rokov. Priemerný vek pacientov bol 62 rokov a 63 % boli muži, 72 % boli pacienti kaukazskej rasy, 23 % boli diabetici a u nehemodialyzovaných pacientov bola priemerná odhadovaná rýchlosť glomerulárnej filtrácie (eGFR)
26,5 ml/min/1,73 m2. Neexistovali žiadne kritéria na zaradenie, ktoré sa týkali lipidov. Priemerná východisková hladina LDL-C bola 108 mg/dl. Vrátane pacientov, ktorí už viac neužívali skúšaný liek,
došlo po jednom roku k zníženiu hladiny LDL-C samotným simvastatínom 20 mg o 26 %
a ezetimibom 10 mg v kombinácii s 20 mg simvastatínu o 38 % v porovnaní s placebom.
Primárnym porovnaním špecifikovaným protokolom štúdie SHARP bola analýza podľa zámeru liečiť (
intention to treat) „veľké vaskulárne príhody“ (
major vascular events MVE, definované ako nefatálny IM alebo kardiálne úmrtie, cievna mozgová príhoda alebo akákoľvek revaskularizačná procedúra) len u tých pacientov, ktorí boli na začiatku randomizovaní do skupiny s ezetimibom
v kombinácii so simvastatínom (n = 4 193) alebo do skupiny s placebom (n = 4 191). Sekundárne analýzy zahŕňali rovnaký kompozitný ukazovateľ analyzovaný v celej kohorte randomizovanej (na
začiatku štúdie alebo po 1. roku) na ezetimib v kombinácii so simvastatínom (n = 4 650) alebo na placebo (n = 4 620), ako aj zložky tohto kompozitného ukazovateľa.





Analýza primárneho cieľového ukazovateľa preukázala, že ezetimib v kombinácii so simvastatínom signifikantne znížil riziko veľkých vaskulárnych príhod (749 pacientov s príhodami v skupine
s placebom oproti 639 v skupine s ezetimibom v kombinácii so simvastatínom) s relatívnym znížením
rizika o 16 % (p = 0,001).
Tento dizajn štúdie neumožňoval zistiť separátny prínos samotného ezetimibu k účinnosti signifikantne znížiť riziko veľkých vaskulárnych príhod u pacientov s CKD.
Jednotlivé zložky MVE u všetkých randomizovaných pacientov sú uvedené v tabuľke 1. Ezetimib v kombinácii so simvastatínom signifikantne znížil riziko cievnej mozgovej príhody a akejkoľvek revaskularizácie, s nevýznamnými číselnými odlišnosťami podporujúcimi ezetimib v kombinácii so simvastatínom pri nefatálnom IM a kardiálnom úmrtí.
Tabuľka 2: Veľké vaskulárne príhody podľa liečebnej skupiny u všetkých randomizovanýchpacientov v štúdii SHARPa
Výsledok
| Ezetimib 10 mg
v kombinácii s 20 mg simvastatínu (N = 4 650)
| Placebo
(N = 4 620)
| Pomer rizika
(IS 95 %)
| Hodnota p
| Veľké vaskulárne príhody
| 701 (15,1 %)
| 814 (17,6 %)
| 0,85 (0,77-0,94)
| 0,001
| Nefatálny infarkt myokardu
| 134 (2,9 %)
| 159 (3,4 %)
| 0,84 (0,66-1,05)
| 0,12
| Kardiálne úmrtie
| 253 (5,4 %)
| 272 (5,9 %)
| 0,93 (0,78-1,10)
| 0,38
| Akákoľvek cievna mozgová
príhoda
| 171 (3,7 %)
| 210 (4,5 %)
| 0,81 (0,66-0,99)
| 0,038
| Nehemoragická cievna
mozgová príhoda
| 131 (2,8 %)
| 174 (3,8 %)
| 0,75 (0,60-0,94)
| 0,011
| Hemoragická cievna mozgová
príhoda
| 45 (1,0 %)
| 37 (0,8 %)
| 1,21 (0,78-1,86)
| 0,40
| Akákoľvek revaskularizácia
| 284 (6,1 %)
| 352 (7,6 %)
| 0,79 (0,68-0,93)
| 0,004
| Veľké aterosklerotické príhody
(Major Atherosclerotic Events, MAE)b
| 526 (11,3 %)
| 619 (13,4 %)
| 0,83 (0,74-0,94)
| 0,002
|
|
|
a Analýza podľa zámeru liečiť u všetkých pacientov v štúdii SHARP randomizovaných na ezetimib
v kombinácii so simvastatínom alebo placebo buď na začiatku alebo po 1. roku.
b MAE; definované ako súbor nefatálneho infarktu myokardu, koronárneho úmrtia, nehemoragickej cievnej mozgovej príhody alebo akejkoľvek revaskularizácie.
Absolútne zníženie LDL cholesterolu dosiahnuté ezetimibom v kombinácii so simvastatínom bolo nižšie medzi pacientmi s nižšou východiskovou hladinou LDL-C (< 2,5 mmol/l) a pacientmi na dialýze na začiatku ako u ostatných pacientov a zodpovedajúce zníženia rizika v týchto dvoch skupinách boli zoslabené.
Homozygotnáfamiliárnahypercholesterolémia(HoFH)Dvojito zaslepená, randomizovaná, 12-týždňová štúdia zahŕňala 50 pacientov s klinickou a/alebo
genotypovou diagnózou HoFH, ktorí dostávali atorvastatín alebo simvastatín (40 mg) so súbežnou LDL aferézou alebo bez nej. Ezetimib podávaný súbežne s atorvastatínom (40 alebo 80 mg) alebo simvastatínom (40 alebo 80 mg) signifikantne znížil LDL-C o 15 % v porovnaní so zvýšením dávky simvastatínu alebo atorvastatínu od 40 do 80 mg pri monoterapii.
Homozygotnásitosterolémia(fytosterolémia)V dvojito zaslepenej, placebom kontrolovanej, 8-týždňovej štúdii bolo randomizovaných 37 pacientov
s homozygotnou sitosterolémiou tak, že dostávali EZETROL 10 mg (n = 30) alebo placebo (n = 7). Niektorí pacienti dostávali inú liečbu (napr. statíny, živice). EZETROL signifikantne znížil dva hlavné rastlinné steroly, sitosterol o 21 % a kampesterol o 24 % v porovnaní s východiskovými hodnotami. Účinky zníženia sitosterolu na morbiditu a mortalitu v tejto populácii nie sú známe.
A
ortálna
stenóza
Multicentrická, dvojito zaslepená, placebom kontrolovaná štúdia „Simvastatín a Ezetimib na liečbu aortálnej stenózy“ (Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis, SEAS)
s mediánom trvania 4,4 rokov sa uskutočnila u 1 873 pacientov s asymptomatickou aortálnou stenózou
(AS) zdokumentovanou pomocou Dopplerovho merania maximálnej rýchlosti prúdenia aortou
v rozmedzí 2,5 až 4,0 m/s. Do štúdie boli zaradení len tí pacienti, u ktorých sa zvážilo, že nie je potrebná liečba statínmi za účelom zníženia rizika aterosklerotického kardiovaskulárneho ochorenia.
Pacienti boli randomizovaní v pomere 1:1 a dostávali placebo alebo im bol denne súbežne podávaný
ezetimib 10 mg a simvastatín 40 mg.
Primárnym cieľovým ukazovateľom bol súbor veľkých kardiovaskulárnych udalostí (major cardiovascular events, MCE) pozostávajúci z kardiovaskulárneho úmrtia, chirurgickej náhrady aortálnej chlopne (aortic valve replacement, AVR), kongestívneho srdcového zlyhania (congestive heart failure, CHF) v dôsledku progresie AS, nefatálneho infarktu myokardu, bypassu koronárnej artérie (coronary artery bypass grafting, CABG), perkutánnej koronárnej intervencie (percutaneous coronary intervention, PCI), hospitalizácie pre nestabilnú angínu pektoris a nehemoragickej cievnej mozgovej príhody. Sekundárnymi cieľovými ukazovateľmi boli súbory podskupín kategórií udalostí primárneho cieľového ukazovateľa.
Ezetimib/simvastatín 10/40 mg v porovnaní s placebom signifikantne neznížili riziko MCE. Primárny výsledok sa vyskytol u 333 pacientov (35,3 %) v skupine ezetimib/simvastatín
a u 355 pacientov (38,2 %) v skupine s placebom (pomer rizika v skupine ezetimib/simvastatín 0,96;
95% interval spoľahlivosti 0,83 až 1,12; p = 0,59). Náhrada aortálnej chlopne sa uskutočnila
u 267 pacientov (28,3 %) v skupine ezetimib/simvastatín a u 278 pacientov (29,9 %) v skupine s placebom (pomer rizika 1,00; 95% IS 0,84 až 1,18; p = 0,97). V skupine ezetimib/simvastatín
(n = 148) malo menej pacientov ischemické kardiovaskulárne príhody ako v skupine s placebom
(n = 187) (pomer rizika 0,78; 95% IS 0,63 až 0,97; p = 0,02) predovšetkým kvôli menšiemu počtu pacientov, ktorí podstúpili bypass koronárnej artérie.
V skupine ezetimib/simvastatín sa častejšie vyskytovala rakovina (105 oproti 70, p = 0,01). Klinický
význam tohto zistenia nie je jasný, pretože vo väčšom skúšaní SHARP sa celkový počet pacientov
s akýmkoľvek výskytom rakoviny (438 pri ezetimibe/simvastatíne oproti 439 v skupine s placebom)
nelíšil. Okrem toho, v skúšaní IMPROVE-IT sa celkový počet pacientov s akoukoľvek novou malignitou (853 v skupine užívajúcej ezetimib/simvastatín oproti 863 v skupine so simvastatínom) signifikantne nelíšil a preto zistenia zo skúšania SEAS nie je možné potvrdiť skúšaním SHARP alebo IMPROVE-IT.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po perorálnom podaní je ezetimib rýchlo absorbovaný a extenzívne konjugovaný na farmakologicky aktívny fenolový glukuronid (ezetimib-glukuronid). Priemerné maximálne plazmatické koncentrácie (Cmax) dosiahne ezetimib-glukuronid za 1 až 2 hodiny a ezetimib za 4 až 12 hodín. Absolútna biologická dostupnosť ezetimibu nemôže byť stanovená, pretože zlúčenina je prakticky nerozpustná vo vodných médiách vhodných na injekciu.
Súbežné podávanie jedla (s vysokým obsahom tuku alebo bez tuku) nemalo žiadny vplyv na perorálnu biologickú dostupnosť ezetimibu podávaného ako 10 mg tablety ezetimibu. Ezetimib sa môže podávať s jedlom alebo bez jedla.
Distribúcia
99,7 % ezetimibu a 88 až 92 % ezetimib-glukuronidu sa viaže na proteíny ľudskej plazmy.
Biotransformácia
Ezetimib je primárne metabolizovaný v tenkom čreve a v pečeni konjugáciou na glukuronid (reakcia fázy II) s následným vylučovaním do žlče. U všetkých hodnotených druhov sa pozoroval minimálny oxidatívny metabolizmus (reakcia fázy I). Ezetimib a ezetimib-glukuronid sú hlavné od liečiva
(ezetimib-glukuronid) celkového liečiva v plazme. Ezetimib aj ezetimib-glukuronid sú pomaly eliminované z plazmy s dokázaným signifikantným enterohepatálnym obehom. Polčas ezetimibu a ezetimib-glukuronidu je približne 22 hodín.
Eliminácia
Po perorálnom podaní 14C-ezetimibu (20 mg) ľudským jedincom tvoril celkový ezetimib približne
93 % celkovej rádioaktivity v plazme. Počas 10-dňového obdobia zberu sa izolovalo približne 78 %
podanej rádioaktivity v stolici a 11 % v moči. Po 48 hodinách neboli v plazme zistiteľné žiadne
úrovne rádioaktivity.
Osobitné skupiny pacientov:
Pediatrická populácia
Farmakokinetika ezetimibu je podobná medzi ≥ 6 ročnými deťmi a dospelými. Farmakokinetické
údaje pre pediatrickú populáciu < 6 rokov nie sú k dispozícii. Klinická skúsenosť u pediatrických
a adolescentných pacientov zahŕňa pacientov s HoFH alebo HeFH.
Staršíľudia
Plazmatické koncentrácie celkového ezetimibu sú asi 2-násobne vyššie u starších ľudí (≥ 65-ročných)
ako u mladých (18 až 45-ročných). Zníženie LDL-C a bezpečnostný profil sú porovnateľné u starších ľudí a mladých jedincov liečených ezetimibom. Preto u starších ľudí nie je nutná úprava dávkovania.
Poruchafunkciepečene
Po podaní jednorazovej dávky 10 mg ezetimibu sa priemerná AUC celkového ezetimibu zvýšila približne 1,7-násobne u pacientov s miernou poruchou funkcie pečene (Childovo-Pughovo skóre 5
alebo 6) v porovnaní so zdravými jedincami. V 14-dňovej štúdii opakovaných dávok (10 mg denne)
u pacientov so stredne závažnou poruchou funkcie pečene (Childovo-Pughovo skóre 7 až 9) bola
priemerná AUC celkového ezetimibu zvýšená približne 4-násobne v 1. a 14. deň v porovnaní
so zdravými jedincami. U pacientov s miernou poruchou funkcie pečene nie je potrebná úprava dávkovania. Vzhľadom na neznáme účinky zvýšenej expozície ezetimibu u pacientov so stredne
závažnou a závažnou poruchou funkcie pečene (Childovo-Pughovo skóre > 9) sa u týchto pacientov
ezetimib neodporúča (pozri časť 4.4).
Poruchafunkcieobličiek
Po podaní jednorazovej 10-mg dávky ezetimibu sa priemerná AUC celkového ezetimibu zvýšila približne 1,5-násobne u pacientov so závažnou poruchou funkcie obličiek (n = 8; priemerný klírens kreatinínu ≤ 30 ml/min/1,73 m2) v porovnaní so zdravými jedincami (n = 9). Tento výsledok nie je považovaný za klinicky významný. U pacientov s poruchou funkcie obličiek nie je potrebná úprava dávkovania.
Ďalší pacient v tejto štúdii (po transplantácii obličky, ktorý dostával mnohopočetnú liečbu vrátane cyklosporínu) mal 12-násobne vyššiu expozíciu k celkovému ezetimibu.
Pohlavie
Plazmatické koncentrácie celkového ezetimibu sú mierne vyššie (približne 20 %) u žien ako u mužov. Zníženie LDL-C a bezpečnostný profil sú porovnateľné u mužov a žien liečených ezetimibom. Preto nie je vzhľadom na pohlavie nutná úprava dávkovania.
5.3 Predklinické údaje o bezpečnosti
Štúdie toxicity po opakovanom podaní ezetimibu na zvieratách neidentifikovali žiadne cieľové orgány pre toxické účinky. U psov liečených štyri týždne ezetimibom (≥ 0,03 mg/kg/deň) sa koncentrácia cholesterolu v žlčníkovej žlči zvýšila 2,5 až 3,5-násobne. V jendoročnej štúdii na psoch, ktorým sa podávali dávky do 300 mg/kg/deň, sa však nepozorovalo zvýšenie incidencie cholelitiázy alebo iné hepatobiliárne účinky. Významnosť týchto údajov pre ľudí nie je známa. Riziko litogenity spojené
s terapeutickým užívaním ezetimibu nemožno vylúčiť.
V štúdiách súbežného podávania ezetimibu a statínov boli pozorované toxické účinky v podstate tie isté, aké sú typicky spojené s užívaním statínov. Niektoré z toxických účinkov boli výraznejšie ako pozorované pri liečbe samostatnými statínmi. Toto sa pripisuje farmakokinetickým
a farmakodynamickým interakciám kombinovanej liečby. V klinických štúdiách sa neobjavili žiadne takéto interakcie. Myopatie sa objavili u potkanov až po vystavení dávkam, ktoré boli
niekoľkonásobne vyššie ako ľudské terapeutické dávky (približne 20-násobok AUC pre statíny a 500
až 2 000-násobok AUC pre aktívne metabolity).
V sérii in vivo a in vitro hodnotení ezetimib podávaný samostatne alebo v kombinácii so statínmi neprejavil žiadny genotoxický potenciál. Dlhodobé testy karcinogenity ezetimibu boli negatívne.
Ezetimib nemal žiadny vplyv na fertilitu samcov a samíc potkanov, ani sa nezistila jeho teratogenicita u potkanov alebo u králikov a nemal ani vplyv na prenatálny alebo postnatálny vývoj. U gravidných potkanov a králikov, ktorí dostali opakované dávky ezetimibu 1 000 mg/kg/deň, prechádzal ezetimib cez placentárnu bariéru. Súbežné podávanie ezetimibu a statínov nebolo teratogénne u potkanov.
U gravidných králikov sa pozoroval malý počet deformít skeletu (spojené hrudné a kaudálne stavce, znížený počet kaudálnych stavcov). Podávanie ezetimibu v kombinácii s lovastatínom malo za
následok embryoletálne účinky.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
monohydrát laktózy laurylsíran sodný (E487) sodná soľ kroskarmelózy hypromelóza (E464) krospovidón (typ B) mikrokryštalická celulóza stearát horečnatý
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
Fľaše: použiť do 100 dní od prvého otvorenia.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Tento liek je dostupný v nasledovných druhoch balenia:
· priehľadné PVC-Aclar blistre s alumíniovou fóliou v papierovej škatuli po 14, 28, 30, 56, 84,
90, 98 a 100 tabliet, kalendárové balenie po 28 a 30 tabliet a v blistroch s perforáciou na oddelenie jednotlivých dávok po 30 x 1, 50 x 1 a 90 x 1 tabliet.
· Priehľadné PVC-Aclar odlupovacie blistre s alumíniovou fóliou v papierovej škatuli po 14, 28,
30, 56, 84, 90, 98 a 100 tabliet, kalendárové balenie po 28 a 30 tabliet a v blistroch s perforáciou na oddelenie jednotlivých dávok po 14 x 1, 28 x 1, 30 x 1, 50 x 1, 90 x 1 a 98 x 1 tabliet.
· Priehľadné PVC-PVdC blistre s alumíniovou fóliou v papierovej škatuli po 14, 28, 30, 56, 84,
90, 98 a 100 tabliet a v blistroch s perforáciou na oddelenie jednotlivých dávok po 30 x 1, 50 x
1 a 90 x 1 tabliet.
· Priehľadné PVC-PVdC odlupovacie blistre s alumíniovou fóliou v papierovej škatuli po 14, 28,
30, 56, 84, 90, 98 a 100 tabliet a v blistroch s perforáciou na oddelenie jednotlivých dávok po
14 x 1, 28 x 1, 30 x 1, 50 x 1, 90 x 1 a 98 x 1 tabliet.
· HDPE fľaša s polypropylénovým (PP) skrutkovacím viečkom a indukovanou tesniacou
výstelkou s bavlneným absorbentom po 14, 28, 50, 56, 84 a 100 tabliet.
HDPE fľaša môže byť vložená do papierovej škatule alebo môže byť bez nej, v závislosti od požiadaviek trhu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Mylan Ireland Limited Unit 35/36 Grange Parade Baldoyle Industrial Estate Dublin 13
Írsko
8. REGISTRAČNÉ ČÍSLO
31/0202/14-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 20. júna 2014
Dátum posledného predĺženia registrácie: 01. júna 2020
10. DÁTUM REVÍZIE TEXTU
10/2021