
entov s poruchou funkcie obličiek nie je potrebná úprava dávkovania (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene nie je potrebná úprava dávkovania (pozri časť 5.2). V časti 4.4
sú uvedené špeciálne opatrenia týkajúce sa pacientov so závažnou poruchou funkcie pečene.
Starší pacienti
Hoci sú skúsenosti s podávaním kaplacizumabu u starších pacientov obmedzené, nepotvrdilo sa, že je potrebná úprava dávkovania alebo osobitné opatrenia u starších pacientov (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť kaplacizumabu u pediatrických pacientov nebola doteraz stanovená. K dispozícii nie sú žiadne údaje.
Spôsobpodávania
Prvá dávka lieku Cablivi má byť podaná prostredníctvom intravenóznej injekcie. Následné dávky majú byť podané prostredníctvom subkutánnej injekcie do brucha.
Injekcie sa nemajú podávať do oblasti pupka a po sebe nasledujúce injekcie sa nemajú podávať do toho istého kvadrantu oblasti brucha.
Pacienti alebo opatrovatelia môžu podávať injekcie po absolvovaní príslušného školenia týkajúceho sa postupov podávania subkutánnych injekcií.
Pokyny na rekonštitúciu lieku Cablivi pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Krvácanie
Aktívne,klinickyvýznamnékrvácanie
V prípade aktívneho, klinicky významného krvácania sa musí prerušiť liečba liekom Cablivi.
V prípade potreby sa môže zvážiť podanie koncentrátu von Willebrandovho faktora na korekciu hemostázy. Liečba liekom Cablivi sa môže obnoviť iba na pokyn lekára so skúsenosťami s liečbou trombotických mikroangiopatií.
Zvýšené riziko krvácania
V prípade súbežného podávania perorálnych antikoagulancií alebo vysokej dávky heparínu
Vzhľadom na potenciálne zvýšené riziko krvácania sa pri začatí alebo pokračovaní liečby perorálnymi antikoagulanciami alebo vysokou dávkou heparínu musí zhodnotiť pomer prínosu a rizika liečby
a zaviesť dôkladné klinické monitorovanie.
V prípade súbežného podávania antitrombotických látok a/alebo nízkomolekulárneho heparínu (low molecular weight heparin, LMWH)
Hoci v klinických skúšaniach nebolo pozorované žiadne zvýšené riziko krvácania, v prípade súbežného podávania antitrombotickej liečby a/alebo LMWH sa musí zhodnotiť pomer prínosu
a rizika liečby a zaviesť dôkladné klinické monitorovanie.
U pacientov s koagulopatiami
Vzhľadom na potenciálne zvýšené riziko krvácania sa pri podávaní lieku Cablivi pacientom
s existujúcimi koagulopatiami (napr. hemofília, iné deficiencie koagulačného faktoru) musí zaviesť dôkladné klinické monitorovanie.
U pacientov podstupujúcich chirurgický zákrok
Ak má pacient podstúpiť elektívny chirurgický zákrok alebo dentálny zákrok, pacienta je nutné poučiť, že má informovať lekára alebo zubného lekára o tom, že užíva liek Cablivi, a najmenej 7 dní
pred plánovaným zákrokom je potrebné liečbu zastaviť. Pacient musí takisto upozorniť lekára vykonávajúceho dohľad nad liečbou liekom Cablivi na tento plánovaný zákrok.
V prípade potreby urgentného chirurgického zákroku sa môže zvážiť podanie koncentrátu von
Willebrandovho faktora na korekciu hemostázy.
Závažnáporuchafunkciepečene
Neuskutočnila sa žiadna oficiálna štúdia s kaplacizumabom u pacientov so závažnou akútnou poruchou funkcie pečene alebo s chronickou poruchou funkcie pečene a nie sú dostupné žiadne údaje
o používaní kaplacizumabu u týchto populácií. V prípade používania lieku Cablivi u tejto populácie sa
musí zhodnotiť pomer prínosu a rizika a zaviesť dôkladné klinické monitorovanie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie na vyhodnotenie používania kaplacizumabu súbežne
s perorálnymi antikoagulanciami (napr. antagonisty vitamínu K, priame perorálne antikoagulanciá
[direct oral anticoagulatns, DOAC] ako sú inhibítory trombínu alebo inhibítory faktoru Xa) alebo s vysokou dávkou heparínu (pozri časť 4.4 V prípade súbežného podávania perorálnych antikoagulancií alebo vysokej dávky heparínu).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne údaje o použití kaplacizumabu u gravidných žien. V štúdiách vykonaných na morčatách sa nepreukázal žiaden účinok kaplacizumabu na chovné samice ani na plod (pozri časť
5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lieku Cablivi počas gravidity.
Dojčenie
Nie sú k dispozícii žiadne údaje o použití kaplacizumabu u dojčiacich žien. Nie je známe, či sa kaplacizumab vylučuje do ľudského mlieka. Riziko u dieťaťa nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu sa musí urobiť po zvážení prínosu
dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinky kaplacizumabu na fertilitu u ľudí nie sú známe. V toxikologických štúdiách vykonaných na zvieratách neboli pozorované žiadne účinky kaplacizumabu na parametre fertility u samcov a samíc
(pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cablivi nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšími nežiaducimi reakciami v klinických skúšaniach boli epistaxa, bolesť hlavy a krvácanie
ďasien. Najčastejšou závažnou nežiaducou reakciou bola epistaxa.
Tabuľkovýsúhrnnežiaducichreakcií
Nežiaduce reakcie sú nižšie zoradené podľa tried orgánových systémov podľa databázy MedDRA
a podľa frekvencie. Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Trieda orgánových systémov Veľmi časté Časté
Poruchy nervového systému Bolesť hlavy Mozgový infarkt Poruchy oka Krvácanie do oka* Poruchy ciev Hematóm*
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Epistaxa* Dyspnoe, hemoptýza*
Krvácanie ďasien* Hemateméza*, hematochézia*, meléna*, krvácanie z hornej časti gastrointestinálneho traktu*, hemoroidálne krvácanie*, rektálna hemorágia*, hematóm brušnej steny*
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy obličiek a močových ciest
Poruchy reprodukčného systému a prsníkov
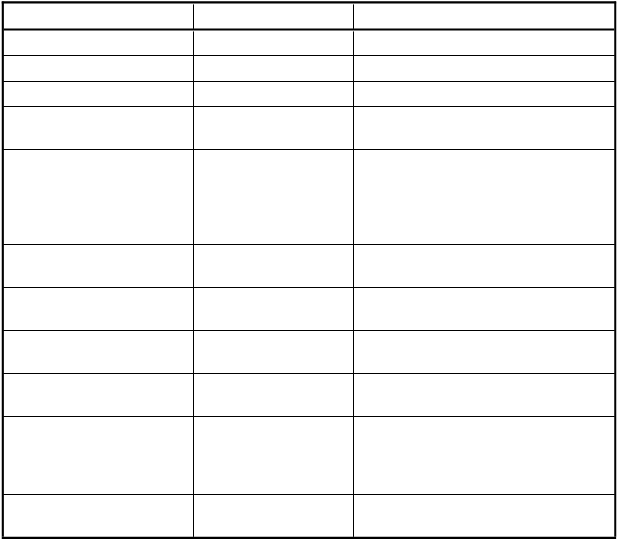
Žihľavka
Bolesť svalov
Hematúria*
Menorágia*, vaginálne krvácanie*
Celkové poruchy a reakcie v mieste podania
Úrazy, otravy a komplikácie liečebného postupu
*Krvácavé príhody: pozri nižšie
Pyrexia, únava Krvácanie v mieste podania injekcie*, pruritus v mieste podania injekcie, erytém
v mieste podania injekcie, reakcia v mieste podania injekcie
Subarachnoidálne krvácanie*
Popis vybraných nežiaducich reakciíKrvácanieV klinických štúdiách sa krvácavé príhody vyskytli v rôznych telesných systémoch nezávisle od dĺžky trvania liečby. Hoci v niektorých prípadoch boli tieto udalosti závažné a vyžadovali si lekársku starostlivosť, väčšina z nich bola prechodná a všetky ustúpili. V prípade aktívneho, klinicky významného krvácania zvážte opatrenia uvedené v časti 4.4 a 4.9.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa môže na základe farmakologického účinku kaplacizumabu zvýšiť riziko krvácania. Odporúča sa dôkladné monitorovanie prejavov a príznakov krvácania (pozri časť 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné antitrombotiká, ATC kód: B01AX07.
MechanizmusúčinkuKaplacizumab je humanizovaná bivalentná nanolátka, ktorá sa skladá z dvoch identických humanizovaných stavebných jednotiek (PMP12A2hum1) geneticky spojených tri-alanínovým
spojivom, je zameraná na A1-domému von Willebrandovho faktora a inhibuje interakciu von
Willebrandovho faktora a trombocytov. Takto kaplacizumab zabraňuje ultraveľkej adhézii trombocytov sprostredkovanej von Willebrandovým faktorom, čo je príznačné pre aTTP. Takisto
ovplyvňuje dispozíciu von Willebrandovho faktora a tým spôsobuje prechodné redukcie všetkých
hladín antigénov von Willebrandovho faktora a súbežnú redukciu hladín faktoru VIII:C počas liečby.
Farmakodynamické účinkyCieľováinhibíciaFarmakologický účinok kaplacizumabu na cieľovú inhibíciu sa hodnotil u dvoch biomarkerov aktivity von Willebrandovho faktora; zhlukovanie trombocytov vyvolané ristocetínom (ristocetin-induced platelet aggregation, RIPA) a ristocetínový kofaktor (ristocetin cofactor, RICO). To, že kaplacizumab vyvoláva úplnú inhibíciu zhlukovania trombocytov sprostredkovaného von Willebrandovým faktorom naznačuje 10 % pokles hladiny RIPA a 20 % pokles hladiny RICO. Vo všetkých klinických štúdiách kaplacizumabu sa preukázal rýchly pokles hladiny RIPA a/alebo RICO po začatí liečby a obnovenie východiskových hladín v intervale 7 dní od prerušenia liečby. Subkutánna dávka 10 mg u pacientov
s aTTP vyvolala úplnú inhibíciu zhlukovania trombocytov sprostredkovaného von Willebrandovým faktorom, čo sa potvrdilo hladinami RICO < 20 % počas celého liečebného obdobia.
CieľovádispozíciaFarmakologický účinok kaplacizumabu na cieľovú dispozíciu sa meral prostredníctvom antigénu von
Willebrandovho faktora a zrážacej aktivity faktora VIII (faktor VIII:C), ktoré boli biomarkermi.
V klinických štúdiách bol po opakovanom podávaní kaplacizumabu pozorovaný 30 – 50 % pokles hladiny antigénu von Willebrandovho faktora a dosiahnutie maxima do 1 – 2 dní od liečby. Keďže von Willebrandov faktor slúži ako nosič faktora VIII, pokles hladín antigénu von Willebrandovho faktora spôsobuje podobný pokles hladín faktora VIII:C. Pokles hladiny antigénu von Willebrandovho faktora a faktora VIII:C bol prechodný a hladiny sa vrátili na východiskové hodnoty po zastavení liečby.
Klinickáúčinnosťabezpečnosť
Účinnosť a bezpečnosť kaplacizumabu u dospelých s epizódou aTTP bola stanovená
v 2 randomizovaných, kontrolovaných štúdiách: v štúdii ALX0681-C301 v III. fáze „HERCULES“ a v štúdii ALX-0681-2.1/10 v II. fáze „TITAN“.
Účinnosť
Štúdia ALX0681-C301
V tejto dvojito zaslepenej, placebom kontrolovanej štúdii boli pacienti s epizódou aTTP boli randomizovaní v pomere 1:1 pre používanie kaplacizumabu alebo placeba v spojení sa každodennou výmenou plazmy a imunosupresiou. Pred prvou výmenou plazmy v rámci štúdie bola pacientom podaná jednorazová intravenózna bolusová injekcia 10 mg kaplacizumabu alebo placeba. Nasledovalo subkutánne podávanie 10 mg kaplacizumabu alebo placeba denne vždy po dokončení výmeny plazmy počas celého obdobia každodennej výmeny plazmy a po dobu nasledujúcich 30 dní. Ak na konci tohto liečebného obdobia bolo potvrdené pretrvávanie aktivity existujúceho ochorenia (naznačujúce bezprostredné riziko recidívy), liečba sa mohla každý týždeň predĺžiť maximálne na 4 týždne spolu
s optimalizovaním imunosupresie. Ak k recidíve došlo počas liečby skúšaným liekom, pacienti prešli
na užívanie nezaslepeného kaplacizumabu. Boli opäť liečení počas trvania každodennej výmeny plazmy a po dobu nasledujúcich 30 dní. Ak na konci tohto liečebného obdobia bolo potvrdené pretrvávanie existujúceho ochorenia, nezaslepená liečba kaplacizumabom sa mohla každý týždeň predĺžiť maximálne na 4 týždne spolu s optimalizovaním imunosupresie. Pacienti boli ďalej sledovaní po dobu 1 mesiaca od prerušenia liečby. V prípade recidívy počas obdobia ďalšieho sledovania (t. j. po zastavení celej liečby skúšaným liekom), podávanie skúšaného lieku nebolo obnovené a recidíva mala byť liečená v súlade so štandardnou starostlivosťou.
V tejto štúdii bolo randomizovaných 145 pacientov s epizódou aTTP (72 pre užívanie kaplacizumabu a 73 pre užívanie placeba). Pacienti boli vo veku od 18 do 79 rokov a priemerný vek bol 46 rokov. Polovica pacientov mala svoju prvú epizódu aTTP. Východiskové charakteristiky ochorenia boli príznačné pre aTTP.
Priemerná dĺžka liečby kaplacizumabom v dvojito zaslepenom období bola 35 dní.
Výsledkom liečby kaplacizumabom bola štatisticky významná redukcia času uplynutého do odpovede trombocytov (p < 0,01). V ktoromkoľvek časovom bode bola pravdepodobnosť dosiahnutia odpovede trombocytov u pacientov liečených kaplacizumabom 1,55-násobne vyššia než u pacientov liečených placebom.
Podľa kombinovaného výsledku z klinickej štúdie pri liečbe kaplacizumabom došlo ku 74 % redukcii percentuálneho podielu pacientov s úmrtím súvisiacim s aTTP (0/72; placebo 3/73), s exacerbáciou aTTP (3/72; placebo 28/73) alebo najmenej s jednou závažnou tromboembolickou udalosťou počas liečby skúšaným liekom (6/72; placebo 6/73) (p < 0,0001). Počas obdobia liečby skúšaným liekom nedošlo v skupine užívajúcej kaplacizumab nedošlo k žiadnemu úmrtiu a v skupine užívajúcej placebo došlo k 3 úmrtiam.
Podiel pacientov s recidívou aTTP (exacerbácia alebo relaps) za celkové obdobie štúdie (vrátane 28- dňového obdobia ďalšieho sledovania po prerušení liečby skúšaným liekom) bol o 67 % nižší
v skupine užívajúcej kaplacizumab (9/72; relaps 6/72) oproti skupine užívajúcej placebo (28/73; relaps
0/73) (p < 0,001).
Žiadni pacienti liečení kaplacizumabom (0/72) neboli refraktérny voči liečbe (definované ako neprítomnosť dvojnásobného počtu trombocytov po 4 dňoch štandardnej liečby a zvýšená hladina LDH) na rozdiel od troch pacientov liečených placebom (3/73).
Výsledkom liečby kaplacizumabom bolo zníženie priemerného počtu dní výmeny plazmy, zníženie objemu použitej plazmy, zníženie priemernej dĺžky pobytu na jednotke intenzívnej starostlivosti
a zníženie priemernej dĺžky hospitalizácie počas obdobia liečby skúšaným liekom.
Počet dní, počas ktorých dochádzalo k N
Placebo Kaplacizumab
73 71
výmene plazmy (dni)
Priemerná
(SE)
9,4 (0,81)
5,8 (0,51)
Celkový objem použitej plazmy (liter) N Priemerná (SE)
Dĺžka hospitalizácie (dni) N Priemerná (SE)
Počet dní na JIS N Priemerná (SE)
73
35,93 (4,17)
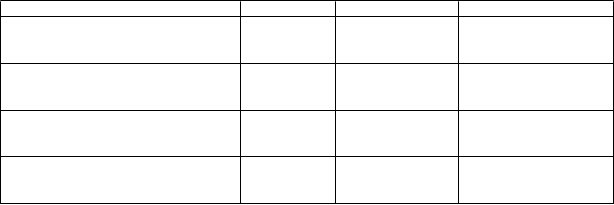
73
14,4 (1,22)
27
9,7 (2,12)
71
21,33 (1,62)
71
9,9 (0,70)
28
3,4 (0,40)
N: počet vyhodnotených pacientov; SE: Štandardná Chyba; JIS: Jednotka intenzívnej starostlivosti
Imunogenicita
V klinických štúdiách sa najviac u 9 % pacientov vyskytli protilátky proti lieku vyvolané liečbou (anti-drug antibodies, ADA). Nebol pozorovaný žiaden vplyv na klinickú účinnosť a neboli zistené žiadne závažné nežiaduce udalosti, ktoré súviseli s týmito odpoveďami ADA.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s kaplacizumabom v jednej alebo vo viacerých podskupinách pediatrickej populácie s aTTP (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika kaplacizumabu bola skúmaná u zdravých účastníkov po podaní jednorazovej intravenóznej infúzie a po podaní jednorazovej a opakovanej subkutánnej injekcie. Farmakokinetika u pacientov s aTTP bola skúmaná po podaní jednorazovej intravenóznej a opakovanej subkutánnej injekcie.'
Farmakokinetika kaplacizumabu sa preukázala ako neúmerná dávke, čo je charakterizované dispozíciou podľa cieľa. U zdravých dobrovoľníkov užívajúcich 10 mg kaplacizumabu subkutánne jedenkrát denne bola maximálna koncentrácia pozorovaná po 6 – 7 hodinách od podania dávky a ustálený stav (steady state) bol dosiahnutý po podaní prvej dávky, pričom akumulácia bola minimálna.
Absorpcia
Po subkutánnom podaní sa kaplacizumab rýchlo a takmer úplne absorbuje (odhadovaná hodnota F >
0,901) v systémovej cirkulácii.
Distribúcia
Po absorbovaní sa kaplacizumab viaže na cieľ a distribuuje sa do orgánov s dobrou perfúziou. U
pacientov s aTTP bol odhad centrálneho objemu distribúcie 6,33 l.
Biotransformácia/Eliminácia
Farmakokinetika kaplacizumabu závisí od expresie cieľového von Willebrandovho faktoru. Pri vyšších hladinách antigénu von Willebrandovho faktoru, napríklad u pacientov s aTTP, sa zvyšuje frakcia cieľového komplexu liečiva, ktorý zostáva v cirkulácii. T1/2 kaplacizumabu preto závisí od hladiny koncentrácie a cieľovej úrovne. Predpokladá sa, že kaplacizumab viazaný na cieľ sa katabolizuje v pečeni, a že neviazaný kaplacizumab sa vylučuje obličkami.
Charakteristikyvosobitnýchskupinách
Farmakokinetika kaplacizumabu bola zisťovaná na základe farmakokinetickej analýzy populácie na farmakokinetiku zo zozbieraných údajov pre farmakokinetiku. Telesná hmotnosť bola alometricky zahrnutá do modelu. Preskúmali sa rozdiely v jednotlivých subpopuláciách. V analyzovaných populáciách neovplyvňovalo pohlavie, vek, krvná skupina a rasa farmakokinetiku kaplacizumabu.
Porucha
f
unkcie
obličiek
alebo
pečene
Neboli vykonané žiadne oficiálne štúdie skúmajúce vplyv poruchy funkcie obličiek alebo pečene na farmakokinetiku kaplacizumabu. V FK/FD modeli populácie mala renálna funkcia (CRCL) štatisticky významný vplyv a spôsobovala obmedzené zvýšenie predpokladanej expozície (AUCss) pri závažnej poruche funkcie obličiek. V klinických štúdiách sa u pacientov s TTP, ktorí mali poruchu funkcie obličiek nepreukázalo dodatočné riziko vzniku nežiaducich udalostí.
5.3 Predklinické údaje o bezpečnosti
V súlade s mechanizmom účinku sa v toxikologických štúdiách kaplacizumabu preukázala vyššia tendencia krvácania u morčiat (hemoragické subkutánne tkanivo v miestach podania injekcie)
a u makakov dlhochvostých (hemoragické subkutánne tkanivo v miestach podania injekcie, krvácanie z nosa, nadmerné menštruačné krvácanie, hematóm v miestach manipulovania so zvieraťom alebo
vykonávania experimentálnych procedúr, dlhšie krvácanie v miestach podania injekcie). Okrem toho pokles hladiny antigénu von Willebrandovho faktora a následne faktora VIII:C v súvislosti
s farmakológiou bol pozorovaný u makakov dlhochvostých a v menšej miere pri faktore VIII:C u morčiat.
Vo vykonanej štúdii embryo-fetálneho vývinu u morčiat neboli hlásené žiadne prejavy toxicity.
V toxikokinetickej štúdii sa u gravidných morčiatv rámci ďalšieho sledovania hodnotila expozícia kaplacizumabu u chovných samíc a plodov. Výsledky naznačovali expozíciu kaplacizumabu
u chovných samíc a v menšej miere u plodov, pričom neboli hlásené žiadne účinky na vývin plodu.
Expozícia kaplacizumabu u plodu u primátov a u ľudí ostáva neistá, nakoľko sa predpokladá, že proteíny s chýbajúcim Fc podielom neprechádzajú voľne placentárnou bariérou.
Nevykonali sa žiadne štúdie na zhodnotenie mutagénneho potenciálu kaplacizumabu, nakoľko tieto testy nie sú relevantné pre biologiká. Na základe hodnotenia rizika karcinogenicity sa špecializované štúdie nepokladali za nevyhnutné.
Neboli vykonané špecializované štúdie na zvieratách na zhodnotenie účinku kaplacizumabu na fertilitu u samcov a samíc. V testoch zameraných na toxicitu pri opakovaných dávkach u makakov dlhochvostých nebol pozorovaný žiaden vplyv kaplacizumabu na parametre fertility u samcov (veľkosť semenníkov, fungovanie spermií, histopatologická analýza semenníka a nadsemenníka)
a u samíc (histopatologická analýza reprodukčných orgánov, periodická vaginálna cytológia).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok sacharóza
bezvodá kyselina citrónová dihydrát citronanu trisodného polysorbát 80
Rozpúšťadlo voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa Cablivi nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
4 roky.
Rekonštituovaný roztok
Bolo preukázané, že liek má počas používania chemickú a fyzikálnu stabilitu 4 hodiny pri teplote
25 ºC.
Z mikrobiologického hľadiska sa liek musí použiť ihneď, pokiaľ spôsob rekonštituovania vopred nevylučuje riziko vzniku mikrobiálnej kontaminácie.
Ak sa nepoužije ihneď, za dĺžku uchovávania počas používania a za podmienky uchovávania
zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Cablivi sa môže uchovávať pri teplote do 25 °C jednorazovo najviac po dobu 2 mesiacov, nie však po uplynutí dátumu exspirácie. Cablivi neuchovávajte znova v chladničke potom, čo bol liek uchovávaný pri izbovej teplote.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Prášok
Injekčná liekovka (sklo typu I) so zátkou (butylová guma), plombou (hliník) a uzáverom
(polypropylén) obsahujúca 10 mg kaplacizumabu.
Rozpúšťadlo
Naplnená injekčná striekačka (náplň zo skla typu I uzavretá brombutylovou gumenou zátkou)
s obsahom 1 ml vody na injekciu.
Veľkosťbalenia
· jednorazové balenie obsahujúce 1 injekčnú liekovku s práškom, 1 naplnenú injekčnú striekačku s rozpúšťadlom, 1 adaptér injekčnej liekovky, 1 hypodermickú ihlu (veľkosť 30) a 2 tampóny namočené v alkohole
· multibalenie obsahujúce 7 jednorazových balení
· viacdávkové balenie obsahujúce 7 injekčných liekoviek s práškom, 7 naplnených injekčných striekačiek s rozpúšťadlom, 7 adaptérov injekčnej liekovky, 7 hypodermických ihiel (veľkosť
30) a 14 tampónov namočených v alkohole
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Na intravenózne a subkutánne podávanie rekonštituujte prášok v injekčnej liekovke použitím adaptéra injekčnej liekovky a všetkého rozpúšťadla v naplnenej injekčnej striekačke. Rozpúšťadlo sa musí pridávať pomaly a opatrne miešať, aby sa roztok nespenil. Nechajte injekčnú liekovku s nasadenou injekčnou striekačkou stáť na rovnom povrchu po dobu 5 minút pri izbovej teplote.
Rekonštituovaný roztok je číry, bezfarebný alebo mierne žltkastý. Musí sa vizuálne skontrolovať, či sa v ňom nenachádzajú častice. Nepoužívajte roztok, v ktorom sa nachádzajú častice.
Preložte celý objem rekonštituovaného roztoku späť do sklenenej injekčnej striekačky a ihneď podajte celý objem injekčnej striekačky (pozri časť 6.3).
Cablivi je určený len na jednorazové použitie. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Ablynx NV Technologiepark 21
9052 Zwijnaarde
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1305/001
EU/1/18/1305/002
EU/1/18/1305/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 31. august 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.