
/>K dispozícii nie sú žiadne údaje.
Sledovanieliečby
Počas liečby sa odporúča stanovenie hladín faktora IX vhodným spôsobom na zistenie dávky, ktorá má byť
podaná a frekvencie opakovaných injekcií. Odpoveď jednotlivých pacientov na faktor IX sa môže líšiť, čím sa prejavujú rôzne biologické polčasy a úrovne zlepšenia. Dávka odvodená od telesnej hmotnosti môže vyžadovať úpravu u podvyživených pacientov alebo u pacientov s nadváhou. Presné sledovanie substitučnej liečby pomocou analýzy koagulácie (aktivita faktora IX v plazme) je nevyhnutné najmä
v prípade veľkých chirurgických zákrokov.
Pri použití jednostupňového testu zrážavosti na báze tromboplastínového času (aPTT) in vitro na stanovenie aktivity faktora IX vo vzorkách krvi pacientov môžu byť výsledky aktivity plazmatického faktora IX výrazne ovplyvnené typom činidla aPTT aj referenčným štandardom použitým pri teste. To je dôležité najmä pri zmene laboratória a/alebo činidiel použitých pri teste.
Výsledkom merania pomocou jednostupňového testu zrážavosti využívajúceho činidlo aPTT na báze
kaolínu bude pravdepodobne podhodnotenie úrovne aktivity.
Dávkovanie
Dávka a dĺžka substitučnej terapie závisia od závažnosti deficitu faktora IX, miesta a rozsahu krvácania a klinického stavu pacienta.
Počet jednotiek podaného rekombinantného faktora IX Fc sa vyjadruje v medzinárodných jednotkách (International Units, IU), ktoré súvisia so súčasným štandardom WHO pre lieky s obsahom faktora IX. Plazmatická aktivita faktora IX sa vyjadruje buď v percentách (v porovnaní s normálnou ľudskou
plazmou), alebo v medzinárodných jednotkách (v porovnaní s medzinárodným štandardom pre faktor IX
v plazme).
Aktivita jednej medzinárodnej jednotky (IU) rekombinantného faktora IX Fc zodpovedá množstvu faktora IX nachádzajúcom sa v jednom ml normálnej ľudskej plazmy.
Liečbapodľapotreby
Výpočet požadovanej dávky rekombinantného faktora IX Fc sa zakladá na empirickom zistení, že
1 medzinárodná jednotka (IU) faktora IX na kg telesnej hmotnosti zvyšuje aktivitu plazmatického faktora IX o 1 % normálnej aktivity (IU/dl). Požadovaná dávka sa stanoví podľa nasledujúceho vzorca:
Požadované jednotky = telesná hmotnosť (kg) x požadované zvýšenie faktora IX (%) (IU/dl) x {recipročná výťažnosť (IU/kg na IU/dl)}
Množstvo, ktoré sa má podať a frekvencia podávania majú byť vždy založené na klinickej účinnosti
v individuálnych prípadoch. Ak je na zastavenie krvácania potrebné zopakovať dávku, má sa zohľadniť predĺžený polčas ALPROLIXU (pozri časť 5.2). Nepredpokladá sa oneskorenie času do maximálnej aktivity.
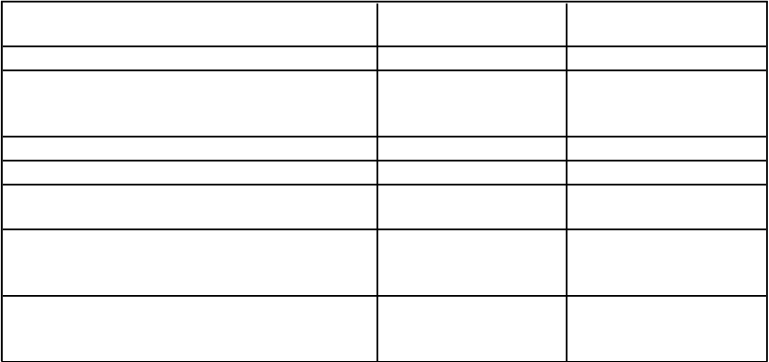
V prípade nasledujúcich krvácavých príhod nemá aktivita faktora IX počas zodpovedajúceho obdobia klesnúť pod danú úroveň aktivity v plazme (v % normálnej hodnoty alebo IU/dl). Pri epizódach krvácania a chirurgických zákrokoch možno použiť ako návod na dávkovanie tabuľku 1:
Tabuľka 1: Návod na dávkovanie ALPROLIXU na liečbu epizód krvácania a pri chirurgických zákrokoch
S
t
up
e
ň krvácania/typ
c
h
i
r
u
r
gického zákroku
K
r
v
á
c
a
n
i
e
P
o
ž
adovaná hladina
f
aktora IX (%) IU/dl
F
re
k
vencia dávkovania (hodiny)/dĺžka
l
i
e
č
b
y (dni)
Začínajúca hemartróza, krvácanie do svalu alebo do ústnej dutiny
20 – 40 Injekciu opakujte každých 48 hodín, až kým sa nezastaví krvácanie, čo sa prejaví ústupom bolesti alebo zahojením.
Rozsiahlejšia hemartróza,
krvácanie do svalu alebo
hematóm
30 – 60 Injekciu opakujte každých 24 až 48 hodín, až
kým neustúpi bolesť a akútna telesná
nespôsobilosť.
Život ohrozujúce krvácania 60 – 100 Injekciu opakujte každých 8 až 24 hodín, až
kým neustúpi stav ohrozenia života.
C
h
i
r
urgický zákrok
Menší chirurgický zákrok vrátane extrakcie zubov
30 – 60 Injekciu opakujte po 24 hodinách, podľa
potreby až do zahojenia 1.
V
e
ľ
ký
c
h
i
r
urgický
z
á
k
r
ok 80 – 100
(pred operáciou a po nej)
Injekciu podľa potreby opakujte každých 8 až
24 hodín, až kým sa dostatočne nevylieči rana, potom pokračujte v liečbe počas najmenej ďalších 7 dní na zachovanie aktivity faktora IX v rozmedzí od 30 % do 60 % (IU/dl).


1 V prípade niektorých pacientov a okolností sa interval dávkovania môže predĺžiť až na 48 hodín (pozri časť 5.2 obsahujúcu
farmakokinetické údaje).
ProfylaxiaNa dlhodobú profylaxiu krvácania sa odporúčajú nasledujúce začiatočné režimy liečby:
• 50 IU/kg jedenkrát týždenne s úpravou dávky podľa individuálnej odpovede; alebo
• 100 IU/kg jedenkrát každých 10 dní s úpravou intervalu podľa individuálnej odpovede. Najvyššia odporúčaná dávka na profylaxiu je 100 IU/kg.
StaršiapopuláciaU pacientov vo veku ≥ 65 rokov existujú iba obmedzené skúsenosti.
PediatrickápopuláciaU detí vo veku do 12 rokov sa môžu vyžadovať vyššie alebo častejšie dávky a odporúčaná začiatočná
dávka je 50 – 60 IU/kg každých 7 dní. Pre dospievajúcich vo veku od 12 rokov sa odporúčajú rovnaké
dávky ako u dospelých. Pozri časti 5.1 a 5.2.
Najvyššia odporúčaná dávka na profylaxiu je 100 IU/kg.
Spôsob podávaniaIntravenózne použitie.
Pri podávaní lieku samotným pacientom alebo ošetrovateľom sa vyžaduje vhodné zaškolenie.
ALPROLIX sa má podávať injekčne intravenóznou cestou v priebehu niekoľkých minút. Rýchlosť
podávania má zabezpečiť pohodlie pacienta a nemá prekročiť 10 ml/minútu.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo (rekombinantný ľudský koagulačný faktor IX a/alebo Fc doménu) alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Precitlivenosť
ALPROLIX môže spôsobovať reakcie z precitlivenosti alergického typu. Pacienti musia byť poučení, aby v prípade výskytu symptómov precitlivenosti, okamžite prestali užívať tento liek a kontaktovali svojho lekára. Pacienti majú byť informovaní o začiatočných prejavoch reakcií z precitlivenosti, ako sú žihľavka, generalizovaná urtikária, tlak na hrudníku, sipot, hypotenzia a anafylaxia.
V prípade anafylaktického šoku sa musí okamžite začať štandardná protišoková liečba.
Inhibítory
Po opakovanej liečbe liekmi obsahujúcimi ľudský koagulačný faktor IX sa má u pacientov monitorovať vznik neutralizačných protilátok (inhibítorov), ktoré sa majú kvantifikovať v jednotkách Bethesda (BU) použitím vhodného biologického testovania.
Správy v literatúre ukazujú koreláciu medzi výskytom inhibítora faktora IX a alergických reakcií. Preto sa má u pacientov s alergickými reakciami vyhodnotiť prítomnosť inhibítora. Treba poznamenať, že pacienti s inhibítormi faktora IX môžu byť viac ohrození anafylaxiou pri následnej liečbe faktorom IX.
Z dôvodu rizika alergických reakcií pri použití liekov obsahujúcich faktor IX sa má začiatočné podávanie faktora IX, podľa zváženia ošetrujúceho lekára, vykonávať pod lekárskym dohľadom, aby v prípade alergických reakcií bola k dispozícii adekvátna lekárska starostlivosť.
Tromboembolizmus
Pri podávaní tohto lieku pacientom s ochorením pečene, pacientom po operáciách, novorodencom alebo pacientom ohrozeným trombotickým fenoménom alebo diseminovanou intravaskulárnou koaguláciou (Disseminated Intravascular Coagulation, DIC) sa má začať klinické sledovanie začiatočných prejavov trombotickej a konzumpčnej koagulopatie s vhodným biologickým testovaním z dôvodu potenciálneho rizika trombotických komplikácií pri používaní liekov obsahujúcich faktor IX. V týchto situáciách sa má zvážiť prínos liečby ALPROLIXOM oproti rizikám týchto komplikácií.
Kardiovaskulárne udalosti
U pacientov s existujúcimi kardiovaskulárnymi rizikovými faktormi môže substitučná liečba pomocou FIX
zvyšovať kardiovaskulárne riziko.
Komplikácie súvisiace s katétrom
Ak sa vyžaduje zariadenie na centrálny venózny prístup (Central Venous Access Device, CVAD), má sa zvážiť riziko komplikácií súvisiacich s CVAD vrátane lokálnych infekcií, bakterémie a trombózy v mieste zavedenia katétra.
Z
a
z
n
a
m
e
n
a
nie
č
í
s
l
a
š
a
r
že
Pri každom podaní ALPROLIXU pacientovi sa dôrazne odporúča zaznamenať názov a číslo šarže tohto
lieku, aby sa zachovala väzba medzi pacientom a šaržou lieku.
Pediatrická populácia
Uvedené upozornenia a opatrenia platia pre dospelých aj deti.
Aspekty týkajúce sa pomocných látok
Tento liek obsahuje 0,3 mmol (alebo 6,4 mg) sodíka v injekčnej liekovke. Má sa vziať do úvahy u pacientov na diéte s kontrolovaným obsahom sodíka.
4.5 Liekové a iné interakcie
Neboli hlásené žiadne interakcie ALPROLIXU s inými liekmi. Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Gravidita a dojčenie
Žiadne štúdie reprodukcie na zvieratách s ALPROLIXOM vykonané neboli. Uskutočnila sa štúdia
prechodu placentou u myší (pozri časť 5.3). Z dôvodu zriedkavého výskytu hemofílie B u žien nie sú
k dispozícii žiadne skúsenosti týkajúce sa používania faktora IX počas gravidity a dojčenia. Preto sa má
faktor IX používať počas gravidity a dojčenia iba v prípade, ak je jednoznačne indikovaný.
Fertilita
Nie sú dostupné žiadne údaje o fertilite. Žiadne štúdie fertility na zvieratách s ALPROLIXOM vykonané neboli.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
ALPROLIX nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Zriedkavo sa pozorovali reakcie z precitlivenosti alebo alergické reakcie (ktoré môžu zahŕňať angioedém, pálenie a štípanie v mieste podania infúzie, triašku, sčervenanie, generalizovanú urtikáriu, bolesť hlavy, žihľavku, hypotenziu, letargiu, nevoľnosť, nepokoj, tachykardiu, tlak na hrudi, mravčenie, vracanie, sipot) a v niektorých prípadoch môžu prejsť až do závažnej anafylaxie (vrátane šoku). V niektorých prípadoch sa tieto reakcie rozvinuli do závažnej anafylaxie a vyskytovali sa v tesnej prechodnej súvislosti so vznikom inhibítorov faktora IX (pozri tiež časť 4.4). U pacientov s hemofíliou B s inhibítormi faktora IX
a alergickou reakciou v anamnéze sa pozoroval po pokuse o vyvolanie imunitnej znášanlivosti nefrotický syndróm.
U pacientov s hemofíliou B sa môžu vytvoriť neutralizačné protilátky (inhibítory) proti faktoru IX. Ak sa vytvoria takéto inhibítory, tento stav sa prejaví ako nedostatočná klinická odpoveď. V takýchto prípadoch sa odporúča obrátiť sa na špecializované hemofilické centrum.
Po podávaní liekov obsahujúcich faktor IX s vyšším rizikom prípravkov s nízkou čistotou existuje potenciálne riziko tromboembolických príhod. Použitie liekov obsahujúcich faktor IX s nízkou čistotou súviselo s prípadmi infarktu myokardu, diseminovanej intravaskulárnej koagulácie, venóznej trombózy
a pľúcnej embólie. Použitie faktora IX s vysokou čistotou je zriedkavo spojené s tromboembolickými komplikáciami.
Tabuľkovýzoznamnežiaducichreakcií
Frekvencie v tabuľke uvedenej nižšie sa pozorovali u spolu 153 pacientov so závažnou
hemofíliou B v klinických štúdiách fázy III a v rozširujúcej štúdii. Celkový počet dní expozície bol
17 080 s mediánom 100 (rozsah 1 – 351) dní expozície na jedného pacienta.
Tabuľka 2 uvedená nižšie zodpovedá klasifikácii orgánových systémov MedDRA (trieda orgánových systémov a preferovaný pojem miery výskytu).
Frekvencie výskytu boli hodnotené podľa nasledujúcej konvencie: veľmi časté (≥1/10), časté (≥1/100 až
<1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé
(<1/10 000), neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí s klesajúcou
závažnosťou.
Tabuľka 2: Nežiaduce reakcie hlásené pre ALPROLIX v klinických skúšaniach
T
r
i
e
d
a orgánových systémov podľa databázy
Me
d
DR
A
N
e
ž
i
aduce reakcie Kategória frekvencie
Poruchy metabolizmu a výživy znížená chuť do jedla menej časté
Poruchy nervového systému bolesť hlavy závrat dysgeúzia
časté
menej časté
menej časté
Poruchy srdca a srdcovej činnosti palpitácie menej časté
Poruchy ciev hypotenzia menej časté
Poruchy gastrointestinálneho traktu perorálna parestézia zápach z úst
Poruchy obličiek a močových ciest obštrukčná uropatia
hematúria
renálna kolika
Celkové poruchy a reakcie v mieste podania únava
bolesť v mieste podania infúzie
časté
menej časté časté
menej časté
menej časté menej časté menej časté
 P
e
d
i
a
t
r
i
c
k
á populácia
P
e
d
i
a
t
r
i
c
k
á populácia
Predpokladá sa, že frekvencia, typ a závažnosť nežiaducich reakcií u detí sú podobné ako u dospelých. Informácie o charakterizácii rozsahu a veku bezpečnostnej databázy u detí, pozri časť 5.1
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieÚčinky vyšších než odporúčaných dávok ALPROLIXU neboli popísané.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragiká, krvný koagulačný faktor IX, ATC kód: B02BD04
Mechanizmusúčinku
Faktor IX je jednoreťazcový glykoproteín s molekulárnou hmotnosťou približne 68 000 daltonov. Je to
koagulačný faktor závislý od vitamínu K. Faktor IX sa aktivuje pomocou faktora XIa vo vnútornej koagulačnej dráhe a pomocou komplexu faktora VII/tkanivového faktora vo vonkajšej dráhe. Aktivovaný faktor IX v kombinácii s aktivovaným faktorom VIII aktivuje faktor X. Aktivovaný faktor X mení protrombín na trombín. Trombín potom mení fibrinogén na fibrín a dochádza k vytvoreniu zrazeniny. Hemofília B je dedičná porucha koagulácie krvi viazaná na chromozóm X, ktorá je spôsobená zníženými hladinami faktora IX a ktorej následkom je krvácanie do kĺbov, svalov alebo vnútorných orgánov, či už spontánne alebo v dôsledku náhodného poranenia alebo chirurgického zákroku. Substitučnou liečbou sa zvýši hladina faktora IX v plazme, čím sa umožní prechodná korekcia deficitu faktora a korekcia náchylnosti na krvácanie.
ALPROLIX (eftrenonakog alfa) je dlhodobo účinkujúci, úplne rekombinantný fúzny proteín, ktorý pozostáva z ľudského koagulačného faktora IX kovalentne naviazaného na Fc doménu ľudského imunoglobulínu G1 a vyrába sa technológiou rekombinantnej DNA.
Fc doména ľudského imunoglobulínu G1 sa viaže s neonatálnym Fc receptorom. Tento receptor je exprimovaný počas celého života ako súčasť prirodzene sa vyskytujúcej dráhy, ktorá chráni imunoglobulíny pred lyzozómovou degradáciou cyklickým návratom týchto proteínov späť do krvného obehu, čo má za následok ich dlhý polčas v plazme.
Klinickáúčinnosťabezpečnosť
Bezpečnosť, účinnosť a farmakokinetické vlastnosti ALPROLIXU sa vyhodnocovali v 2 multinárodných, otvorených, pivotných štúdiách – v štúdii fázy 3 označovanej ako štúdia I a v pediatrickej štúdii
fázy 3 označovanej ako štúdia II (pozri časť „Pediatrická populácia“).
Štúdia I porovnávala účinnosť oboch profylaktických liečebných režimov (s fixným týždenným intervalom a s individualizovaným intervalom) s liečbou podľa potreby. Do tejto štúdie bolo zaradených spolu
123 predtým liečených pacientov mužského pohlavia (vo veku od 12 do 71 rokov) so závažnou
hemofíliou B (endogénna aktivita FIX ≤ 2 %). Všetci pacienti boli liečení ALPROLIXOM a sledovali sa po dobu až 77 týždňov.
Osoby v skupine s fixným týždenným intervalom dostávali ALPROLIX na bežnú profylaxiu so začiatočnou dávkou 50 IU/kg. Osoby v skupine s individualizovaným intervalom dostávali ALPROLIX na bežnú profylaxiu s fixnou dávkou 100 IU/kg so začiatočným intervalom dávkovania každých 10 dní.
Štúdia I okrem toho vyhodnocovala hemostatickú účinnosť pri liečbe prípadov krvácania a stanovovala
hemostatickú účinnosť počas perioperačnej liečby u pacientov podstupujúcich veľké chirurgické zákroky.
Profylaxiasfixnýmitýždennýmiaindividualizovanýmiintervalmi:
U hodnotených osôb zaradených do skupiny profylaxie s fixným týždenným intervalom v štúdii I sa dosahoval medián týždennej dávky na úrovni 45,17 IU/kg (medzikvartilový rozsah 38,1 - 53,7).
U hodnotených osôb zaradených do skupiny profylaxie s individualizovaným intervalom v štúdii I sa
dosahoval medián intervalu na úrovni 12,53 dňa (medzikvartilový rozsah 10,4 - 13,4).
Mediány priemernej ročnej miery výskytu krvácania (Annualised Bleeding Rates, ABR) u pacientov
s vyhodnotiteľnou účinnosťou boli na úrovni 2,95 (medzikvartilový rozsah 1,01 – 4,35) u pacientov
s profylaxiou s fixným týždenným intervalom, 1,38 (medzikvartilový rozsah 0,00 – 3,43) u pacientov
s individualizovaným intervalom a 17,69 (medzikvartilový rozsah 10,77 – 23,24) u pacientov s liečbou podľa potreby. U 42 % osôb počas individualizovanej profylaxie a u 23,0 % osôb počas týždennej profylaxie sa nevyskytovali žiadne epizódy krvácania. V skupine s profylaxiou s individualizovaným intervalom bol nižší podiel pacientov s ≥1 cieľovým kĺbom na začiatku liečby než v skupine s týždennou profylaxiou (27,6 % resp. 57,1 %, v uvedenom poradí).
Poznámka: Hodnoty ABR nie sú porovnateľné medzi rôznymi faktormi koncentrátov a medzi rôznymi
klinickými štúdiami.
Liečbakrvácania: Zo 636 prípadov krvácania pozorovaných počas štúdie I sa 90,4 % liečilo podaním
1 injekcie a spolu 97,3 % sa liečilo podaním najviac 2 injekcií. Medián priemernej dávky na injekciu na liečbu epizódy krvácania bol 46,07 (medzikvartilový rozsah 32,86 - 57,03) IU/kg. Medián celkovej dávky na liečbu epizódy krvácania bol 51,47 IU/kg (medzikvartilový rozsah 35,21 – 61,73) v skupine
s týždennou profylaxiou, 49,62 IU/kg (35,71 – 94,82) v skupine s profylaxiou s individualizovaným
intervalom a 46,58 IU/kg (33,33 – 59,41) v skupine s liečbou podľa potreby.
Pediatrická populácia
Do štúdie II bolo zaradených spolu 30 predtým liečených pediatrických pacientov mužského pohlavia so
závažnou hemofíliou B (endogénna aktivita FIX ≤ 2 %). Pacienti boli vo veku do 12 rokov (15 bolo vo
veku < 6 rokov a 15 bolo vo veku od 6 do < 12 rokov). Všetci pacienti boli liečení ALPROLIXOM
a sledovali sa po dobu najviac 52 týždňov.
Všetkých 30 pacientov bolo liečených ALPROLIXOM v režime individualizovanej profylaktickej dávky so začiatočnou dávkou 50 – 60 IU/kg podávanou každých 7 dní, ktorá sa upravovala až do maximálne
100 IU/kg, a s intervalom dávkovania upravovaným v rozsahu od minimálne jedenkrát týždenne do maximálne dvakrát týždenne.
Režimprofylaktickejindividualizovanejliečby:
Medián priemernej týždennej dávky ALPROLIXU bol na úrovni 59,40 IU/kg (medzikvartilový rozsah
52,95 až 64,78 IU/kg) pre osoby vo veku < 6 rokov a na úrovni 57,78 IU/kg (medzikvartilový rozsah
51,67 až 65,01 IU/kg) pre osoby vo veku od 6 do < 12 rokov. Celkový medián intervalu dávkovania bol na úrovni 6,99 dňa (medzikvartilový rozsah 6,94 až 7,03) bez rozdielu v mediáne intervalu dávkovania medzi vekovými kohortami. S výnimkou jedného pacienta, ktorého posledná predpísaná dávka bola 100 IU/kg každých 5 dní, boli u ostatných 29 pacientov posledné predpísané dávky na úrovni najviac 70 IU/kg každých 7 dní. U 33 % pediatrických pacientov sa nevyskytovali žiadne epizódy krvácania.
Mediány priemernej ročnej miery výskytu krvácania u pacientov vo veku <12 rokov s vyhodnotiteľnou účinnosťou boli na úrovni 1,97 (medzikvartilový rozsah 0,00 – 3,13).
Liečbaepizódkrvácania:Zo 60 prípadov krvácania pozorovaných počas štúdie II sa 75 % liečilo podaním
1 injekcie a celkovo 91,7 % epizód krvácania sa liečilo podaním najviac 2 injekcií. Medián priemernej dávky na injekciu na liečbu epizódy krvácania bol 63,51 (medzikvartilový rozsah 48,92 - 99,44) IU/kg. Medián celkovej dávky na liečbu epizódy krvácania bol 68,22 IU/kg (medzikvartilový rozsah
50,89 - 126,19).
P
e
ri
operačná
li
eč
ba
(c
hirurgická
profylaxia)
:
'
V štúdii I a v rozšírenej štúdii sa vykonalo a vyhodnotilo spolu 29 veľkých chirurgických zákrokov
u 19 osôb (17 dospelých, 1 dospievajúci a 1 pediatrický pacient vo veku < 12 rokov). Z týchto 29 veľkých
chirurgických zákrokov vyžadovalo 24 chirurgických zákrokov (82,8 %) jednu dávku pred operáciou na udržanie hemostázy počas chirurgického zákroku. Medián priemernej dávky na injekciu na udržanie hemostázy počas chirurgického zákroku bol 94,7 IU/kg (rozsah: 49 až 152 IU/kg). Celková dávka v deň chirurgického zákroku dosahovala úroveň od 51 do 318 IU/kg a celková dávka v 14-dňovom perioperačnom období dosahovala úroveň od 60 do 1 947 IU/kg.
5.2 Farmakokinetické vlastnostiVšetky farmakokinetické štúdie ALPROLIXU sa vykonali u predtým liečených pacientov so závažnou
hemofíliou B. Údaje uvedené v tejto časť boli získané pomocou jednostupňového testu zrážavosti
s činidlom aPTT na báze oxidu kremičitého kalibrovaným proti plazmatickým štandardom faktora IX.
Farmakokinetické vlastnosti sa vyhodnocovali u 22 osôb (≥ 19 rokov) dostávajúcich ALPROLIX
(rFIXFc). Po období vyplavovania lieku z tela trvajúcom najmenej 120 hodín (5 dní) dostali pacienti jednu dávku 50 IU/kg ALPROLIXU. Farmakokinetické vzorky boli odobraté pred podaním dávky, a potom následne v 11 časových bodoch až do 240 hodín (10 dní) po dávke. Farmakokinetické parametre nekompartmentálnej analýzy po dávke 50 IU/kg ALPROLIXU sú uvedené v tabuľke 3.
Tabuľka 3: Farmakokinetické parametre ALPROLIXU (dávka 50 IU/kg)
Farmakokinetické parametre1 ALPROLIXN = 22
Prírastková obnova (IU/dl na IU/kg) 0,92 (0,77 - 1,10)
AUC/dávku
(IU*h/dl na IU/kg)
31,58 (28,46 - 35,05)
Cmax (IU/dl) 46,10 (38,56 – 55,11)
Cl (ml/h/kg) 3,17 (2,85 - 3,51)
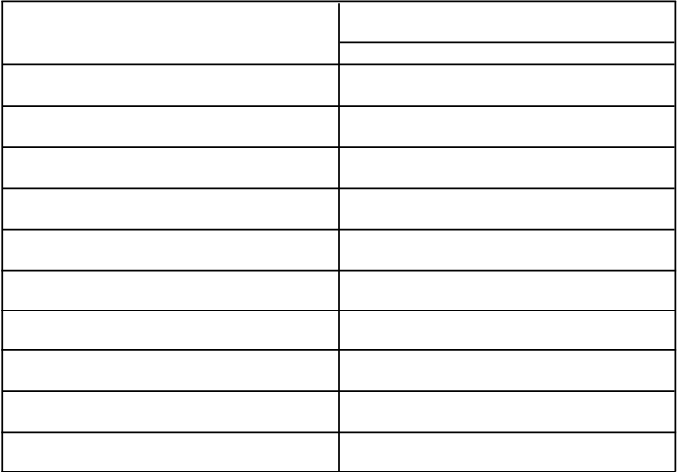
t½ (h) 77,60 (70,05 - 85,95)
t½α (h)2 5,03 (3,20 - 7,89)
t½β (h)2 82,12 (71,39 - 94,46)
MRT (h) 95,82 (88,44 – 106,21)
Vss (ml/kg) 303,4 (275,1 - 334,6)
Čas do 1 % (dni)2 11,22 (10,20 - 12,35)
1 Farmakokinetické parametre sú uvedené ako geometrická priemerná hodnota (95 % IS).
2 Tieto farmakokinetické parametre sa získali z kompartmentálnej analýzy.
Skratky: IS = interval spoľahlivosti, Cmax = čas dosiahnutia maximálnej koncentrácie, AUC = plocha pod časovou krivkou koncentrácie FIX, t
½= biologický polčas, t½α = polčas distribúcie, t½β = eliminačný polčas, Cl = klírens, Vss = distribučný objem v rovnovážnom stave, MRT = stredný pobytový čas.
Eliminačný polčas ALPROLIXU (82 hodín) je ovplyvnený oblasťou Fc, pre ktorú sa na zvieracích modeloch preukázalo sprostredkovanie prostredníctvom cyklických dráh neonatálneho receptora Fc.
Na základe údajov o aktivite FIX získaných od 161 osôb zo všetkých vekových skupín (vek od 2
do 76 rokov) s hmotnosťou od 12,5 kg do 186,7 kg v troch klinických štúdiách (12 osôb v štúdii fázy 1/2a,
123 osôb v štúdii I a 26 osôb v štúdii II) bol vyvinutý populačný farmakokinetický model. Odhad parametra Cl (klírens) ALPROLIXU pre typickú dospelú osobu s hmotnosťou 70 kg je na úrovni 2,30 dl/h a ustáleného distribučného objemu ALPROLIXU je na úrovni 194,8 dl. Pozorovaný priemerný (SD) časový profil aktivity po jednej dávke ALPROLIXU u pacientov so závažnou hemofíliou B je uvedený nižšie (pozri tabuľku 4).
Tabuľka 4: Pozorovaná priemerná (SD) aktivita FIX [IU/dl] po jednej dávke ALPROLIXU1 u pacientov
vo veku ≥ 12 rokov
Dávka
(IU/kg) 10 min 1 h 3 h 6 h 24 h 48 h 96 h 144 h 168 h 192 h 240 h 288 h
5
0 52,9 (30,6)
34,5
(7,3)
28,7
(6,7)
25,1
(5,1)
15,1
(3,9)
9,7
(3,0)
5,0
(1,6)
3,4
(1,1)
3,2
(1,9)
2,6
(1,0)
2,1
(0,9) NA
10
0 112 (24)
77,1
(12,8) NA
36,7
(8,0)
21,8
(4,8)
10,1
(2,6) NA
4,81
(1,67) NA
2,86
(0,98)
2,30
(0,94)
1 Pozri časť 4.2; NA: nie je k dispozícii.
Pediatrická populácia
Farmakokinetické parametre ALPROLIXU sa stanovili pre dospievajúcich v štúdii I (farmakokinetický odber vzoriek sa vykonal pred podaním dávky, a potom sa vykonalo vyhodnotenie vo viacerých časových bodoch až do 336 hodín (14 dní) po dávke) a pre deti v štúdii II (farmakokinetický odber vzoriek sa vykonal pred podaním dávky, a potom sa vykonalo vyhodnotenie v 7 časových bodoch až do 168 hodín
(7 dní) po dávke). V tabuľke 5 sú uvedené farmakokinetické parametre vypočítané z údajov
35 pediatrických pacientov vo veku do 18 rokov.
Tabuľka 5: Porovnanie farmakokinetických parametrov ALPROLIXU (rFIXFc) podľa vekovej kategórie
Štúdia II Štúdia I
F
K parametre
1
< 6 rokov
(2, 4)
6 až < 12 rokov
(6, 10)
12 až < 18 rokov
(12, 17)
N = 11 N = 13 N = 11
IR
(IU/dl na IU/kg) AUC/dávku (IU*h/dl na IU/kg)
0,5989 (0,5152; 0,6752)
22,71 (20,32; 25,38)
0,7170 (0,6115; 0,8407)
28,53 (24,47; 33,27)
0,8470 (0,6767; 1,0600)
29,50 (25,13; 34,63)
t
½
(h) 66,49 (55,86; 79,14)
MRT (h) 83,65 (71,76; 97,51)
Cl (ml/h/kg) 4,365 (3,901; 4,885)
Vss (ml/kg) 365,1 (316,2; 421,6)
70,34 (60,95; 81,17)
82,46 (72,65; 93,60)
3,505 (3,006; 4,087)
289,0 (236,7; 352,9)
82,22 (72,30; 93,50)
93,46 (81,77; 106,81)
3,390 (2,888; 3,979)
316,8 (267,4; 375,5)

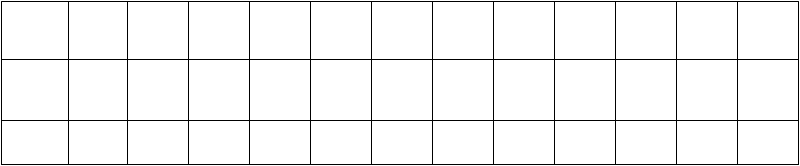
1FK parametre odvodené z nekompartmentálnej analýzy sú uvedené ako geometrická priemerná hodnota (95 % IS).
Skratky: IS = interval spoľahlivosti, IR = prírastková obnova, AUC = plocha pod časovou krivkou koncentrácieFIX, t½ = biologický
polčas, MRT = stredný pobytový čas, Cl = klírens, Vss = distribučný objem v rovnovážnom stave.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe testu trombogenicity u králikov (Wesslerov model stázy) a štúdií toxicity po opakovanom podávaní (ktoré zahŕňali vyhodnotenie lokálnej toxicity, samčích reprodukčných orgánov a elektrokardiografických parametrov) u potkanov a opíc neodhalili žiadne osobitné riziko pre ľudí. Neuskutočnili sa žiadne štúdie skúmajúce genotoxicitu, karcinogenitu, reprodukčnú toxicitu ani embryofetálny vývin. V štúdii prechodu placentou sa zistilo, že u myší prechádza ALPROLIX placentou
v malých množstvách.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok sacharóza L-histidín manitol
polysorbát 20
hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
Rozpúšťadlo chlorid sodný voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
Smú sa používať iba dodané infúzne súpravy, pretože môže dôjsť k zlyhaniu liečby v dôsledku adsorpcie
koagulačného faktora IX do vnútorných povrchov niektorých injekčných pomôcok.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
4 roky
Počas času použiteľnosti sa tento liek môže uchovávať pri izbovej teplote (do 30 °C) počas jedného obdobia nepresahujúceho 6 mesiacov. Dátum vybratia lieku z chladničky sa má zaznamenať na škatuľku. Po uchovávaní pri izbovej teplote sa liek nesmie vrátiť do chladničky. Liek sa nemá používať po dátume exspirácie vytlačenom na injekčnej liekovke alebo šesť mesiacov po vybratí škatuľky z chladničky, podľa toho, čo nastane skôr.
Po rekonštitúcii
Chemická a fyzikálna stabilita bola preukázaná počas 6 hodín pri uchovávaní pri izbovej teplote (do
30 °C). Ak sa liek nepoužije do 6 hodín, musí sa zlikvidovať. Z mikrobiologického hľadiska sa má liek
použiť okamžite po rekonštitúcii. Ak sa nepoužije ihneď, za dobu skladovania v stave pripravenom na použitie a podmienky pred použitím zodpovedá používateľ. Liek chráňte pred priamym slnečným svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Injekčnú liekovku uchovávajte vo
vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia a špeciálne zariadenie na použitie, podanie
Každé balenie obsahuje:
- prášok v sklenenej injekčnej liekovke typu 1 s chlórbutylovou gumovou zátkou,
- 5 ml rozpúšťadla v sklenenej naplnenej injekčnej striekačke typu 1 s brómbutylovou gumovou piestovou zátkou,
- plunžerový piest,
- sterilný adaptér injekčnej liekovky na rekonštitúciu,
- sterilnú infúznu súpravu,
- tampón navlhčený (tampóny navlhčené) alkoholom,
- náplasť (náplasti),
- gázový vankúšik (gázové vankúšiky).
Veľkosť balenia: 1 kus.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prášok na injekciu v každej injekčnej liekovke sa musí rekonštituovať s dodaným rozpúšťadlom (roztok chloridu sodného) z naplnenej injekčnej striekačky použitím sterilného adaptéra injekčnej liekovky na rekonštitúciu.
Injekčnou liekovkou sa má jemne krúžiť až do rozpustenia všetkého prášku.
Ďalšie informácie o rekonštitúcii a podávaní lieku si prečítajte v písomnej informácii pre používateľa. Rekonštituovaný roztok musí byť číry až mierne opalizujúci a bezfarebný. Rekonštituovaný liek sa má
pred podaním vizuálne skontrolovať, či neobsahuje pevné častice a či nedošlo ku zmene sfarbenia. Roztoky, ktoré sú zakalené alebo obsahujú usadeniny sa nemajú používať.
Tento liek je určený len na jednorazové použitie.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Biogen Idec Ltd
Innovation House
70 Norden Road
Maidenhead Berkshire SL6 4AY
Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/16/1098/001
EU/1/16/1098/002
EU/1/16/1098/003
EU/1/16/1098/004
EU/1/16/1098/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
rávne. Pomôcky nepoužívajte opakovane.