
nych zákrokov. Dentálny chirurgický zákrok môže zhoršiť stav pacientov, u ktorých počas liečby vznikne osteonekróza čeľuste. Nie sú dostupné údaje, ktoré by ukázali, či prerušenie liečby bisfosfonátom znižuje riziko osteonekrózy čeľuste
u pacientov, ktorí potrebujú dentálne zákroky. Klinický úsudok ošetrujúceho lekára založený na vyhodnotení individuálneho pomeru prospešnosti a rizika má byť určujúci pri plánovaní liečby každého pacienta.
Bolesťsvalov a kostí
Po uvedení na trh bola hlásená silná bolesť kostí, kĺbov a/alebo svalov, príležitostne znemožňujúca pohyblivosť u pacientov používajúcich bisfosfonáty. Takéto hlásenia však neboli časté. Do tejto kategórie liekov patrí Zometa (kyselina zoledrónová). Čas do nástupu symptómov sa rôznil od jedného dňa do niekoľkých mesiacov od začatia liečby. U väčšiny pacientov sa symptómy zmiernili po skončení liečby. Podsúbor pacientov mal recidívu symptómov, keď sa im znovu podal rovnaký liek alebo iný bisfosfonát.
4.5 Liekové a iné interakcie
V klinických skúšaniach sa Zometa podávala súčasne s bežne používanými protinádorovými liekmi, diuretikami, antibiotikami a analgetikami bez toho, aby sa vyskytli klinicky zjavné interakcie. Kyselina zoledrónová sa in vitro neviaže vo významnej miere na bielkoviny plazmy a neinhibuje ľudské enzýmy P450 (pozri časť 5.2), nevykonali sa však osobitné klinické štúdie interakcií. Opatrnosť sa odporúča pri súčasnom podávaní bisfosfonátov s aminoglykozidmi, pretože obidve skupiny liečiv môžu mať aditívny účinok, čo má za následok nižšiu hladinu vápnika v sére na dlhšiu dobu, ako sa požaduje. Pri použití Zomety súčasne s inými potenciálne nefrotoxickými liečivami je nutná opatrnosť. Počas liečby sa musí venovať pozornosť aj možnému vzniku hypomagneziémie.
U pacientov s mnohopočetným myelómom sa môže zvýšiť riziko poruchy funkcie obličiek pri intravenóznom podaní bisfosfonátov v kombinácii s talidomidom.
4.6 Gravidita a laktácia
Nie sú k dispozícii dostatočné údaje o použití kyseliny zoledrónovej u gravidných žien. Štúdie reprodukčnej toxicity kyseliny zoledrónovej na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Dystokia sa pozorovala pri najnižšej skúšanej dávke u potkanov (0,01 mg/kg telesnej hmotnosti). Nie je známe potenciálne riziko u ľudí. Zometa sa nemá používať počas gravidity.
Nie je známe, či sa kyselina zoledrónová vylučuje do ľudského mlieka. Zometu nemajú používať dojčiace ženy (pozri časť 4.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Frekvencia nežiaducich reakcií na Zometu 4 mg sa stanovila najmä na základe údajov získaných pri chronickej liečbe. Nežiaduce reakcie na Zometu a nežiaduce reakcie zaznamenané pri iných bisfosfonátoch sú podobné a ich výskyt možno očakávať u približne tretiny pacientov. Intravenózne podanie najčastejšie sprevádzal syndróm podobný chrípke u približne 9% pacientov, ktorý zahŕňal bolesť kostí (9,1%), horúčku (7,2%), únavu (4,1%) a zimnicu (2,9%). Príležitostne sa zaznamenali prípady bolesti kĺbov a svalov u približne 3% pacientov. Nie sú dostupné údaje o reverzibilite týchto nežiaducich účinkov.
Zníženie vylučovania vápnika obličkami často sprevádza pokles hladín fosfátu v sére (u približne 20% pacientov), ktorý je asymptomatický a nevyžaduje liečbu. Vápnik v sére môže klesnúť na asymptomatické hypokalciemické hladiny u približne 3% pacientov.
Po intravenóznej infúzii Zomety sa zaznamenali gastrointestinálne reakcie ako nauzea (5,8%) a vracanie (2,6%). Príležitostne sa v mieste podania infúzie pozorovali miestne reakcie, ako je sčervenenie alebo opuch a/alebo bolesť u menej ako 1% pacientov.
Anorexia sa zaznamenala u 1,5% pacientov liečených Zometou 4 mg.
Pozorovalo sa niekoľko prípadov exantému alebo svrbenia (menej ako 1%).
Tak ako pri iných bisfosfonátoch, prípady zápalu spojoviek sa zaznamenali u približne 1% pacientov.
Boli správy o zhoršení funkcie obličiek (2,3%), hoci v mnohých prípadoch sa na etiológii zjavne podieľalo viacero faktorov.
Podľa súhrnnej analýzy klinických skúšaní kontrolovaných placebom sa ťažká anémia (Hb < 8,0 g/dl)
zaznamenala u 5,2% pacientov, ktorí dostávali Zometu 4 mg, oproti 4,2% pacientov, ktorí dostávali placebo.
Nežiaduce reakcie na liek uvedené v Tabuľke 1 sa súhrnne zaznamenali v klinických skúšaniach po prevažne chronickej liečbe kyselinou zoledrónovou.
Tabuľka 1Nežiaduce reakcie sú zoradené podľa frekvencie, najčastejšie ako prvé, pričom frekvencia je určená nasledovne: veľmi časté (≥1/10), časté (≥1/100, <1/10), menej časté (≥1/1 000, <1/100), zriedkavé (≥1/10 000, <1/1 000), veľmi zriedkavé (<1/10 000), zahŕňajúce jednotlivé hlásenia.
Poruchy krvi a lymfatického systému
Časté: Anémia
Menej časté: Trombocytopénia, leukopénia
Zriedkavé: Pancytopénia
|
Poruchy nervového systému
Časté: Bolesť hlavy
Menej časté: Závraty, parestézie, poruchy chuti, hypestézie, hyperestézie, tremor
|
Psychické poruchy
Menej časté: Úzkosť, poruchy spánku
Zriedkavé: Zmätenosť
|
Ochorenia oka
Časté: Konjunktivitída
Menej časté: Neostré videnie
Veľmi zriedkavé: Uveitída, episkleritída
|
Gastrointestinálne poruchy
Časté: Nauzea, vracanie, anorexia
Menej časté: Hnačka, zápcha, bolesť brucha, dyspepsia, stomatitída, suchosť v ústach
|
Poruchy dýchacej sústavy, hrudníka a mediastína
Menej časté: Dyspnoe, kašeľ
|
Poruchy kože a podkožného tkaniva
Menej časté: Svrbenie, exantém (vrátane erytematózneho a makulárneho exantému), zvýšené potenie
|
Poruchy kostrového svalstva, spojivových tkanív a kostí
Časté: Bolesť kostí, bolesť svalov, bolesť kĺbov, generalizovaná bolesť
Menej časté: Svalové kŕče
|
Kardiovaskulárne poruchy
Menej časté: Hypertenzia, hypotenzia
Zriedkavé: Bradykardia
|
Poruchy obličiek a močových ciest
Časté: Zhoršenie funkcie obličiek
Menej časté: Akútne zlyhanie obličiek, hematúria, proteinúria
|
Poruchy imunitného systému
Menej časté: Reakcie z precitlivenosti
Zriedkavé: Angioneurotický edém
|
 Celkové poruchy a reakcie v mieste podania
Celkové poruchy a reakcie v mieste podania
Časté: Horúčka, syndróm podobný chrípke (zahŕňajúci únavu, zimnicu, celkový pocit nevoľnosti a návaly tepla)
Menej časté: Asténia, periférny edém, reakcie v mieste podania (zahŕňajúce bolesť, podráždenie,
opuch a induráciu), bolesť na hrudi, zvýšenie hmotnosti
Laboratórne odchýlkyVeľmi časté: Hypofosfatémia
Časté: Zvýšenie kreatinínu a močoviny v krvi, hypokalciémia
Menej časté: Hypomagneziémia, hypokaliémia
Zriedkavé: Hyperkaliémia, hypernatriémia
V jednom randomizovanom, dvojito slepom, kontrolovanom klinickom skúšaní trvajúcom 3 roky, v ktorom
sa vyhodnotila účinnosť a bezpečnosť 5 mg kyseliny zoledrónovej podávanej raz ročne v liečbe postmenopauzálnej osteoporózy (PMO) oproti placebu, celková incidencia fibrilácie predsiení bola 2,5% (96 z 3 862) u pacientok, ktoré dostávali 5 mg kyseliny zoledrónovej, a 1,9% (75 z 3 852) u pacientok, ktoré dostávali placebo. Výskyt fibrilácie predsiení ako závažnej nežiaducej udalosti bol 1,3% (51 z 3 862)
u pacientok, ktoré dostávali 5 mg kyseliny zoledrónovej, a 0,6% (22 z 3 852) u pacientok, ktoré dostávali placebo. Nerovnováha pozorovaná v tomto klinickom skúšaní sa nepozorovala v iných klinických skúšaniach s kyselinou zoledrónovou, vrátane skúšaní so 4 mg Zomety (kyselina zoledrónová) podávanými každé 3-4 týždne onkologickým pacientom. Mechanizmus, ktorý spôsobil zvýšenie incidencie fibrilácie predsiení v tomto jedinom klinickom skúšaní, nie je známy.
Skúsenosti po uvedení na trh: Prevažne u pacientov s rakovinou liečených bisfosfonátmi vrátane Zomety sa zaznamenali prípady osteonekrózy (primárne čeľuste). Mnohí z týchto pacientov mali príznaky lokálnej infekcie vrátane osteomyelitídy a väčšina hlásení sa týka pacientov s rakovinou po extrakcii zubov alebo iných dentálnych chirurgických zákrokoch. Osteonekróza čeľuste má viaceré dobre zdokumentované rizikové faktory vrátane diagnózy rakoviny, súčasne podanej liečby (napr. chemoterapie, rádioterapie, kortikosteroidov) a sprievodných ochorení (napr. anémie, koagulopatií, infekcie, ochorenia ústnej dutiny). Hoci sa nestanovila príčinná súvislosť, je vhodné vyvarovať sa dentálnych chirurgických zákrokov, pretože sa môže predĺžiť obdobie zotavenia (pozri časť 4.4). Vo veľmi zriedkavých prípadoch boli hlásené nasledujúce udalosti: hypotenzia vedúca ku synkope alebo obehovému kolapsu, najmä u pacientov so základnými rizikovými faktormi, fibrilácia predsiení, somnolencia, bronchokonstrikcia.
4.9 PredávkovanieKlinické skúsenosti s akútnym predávkovaním Zomety sú obmedzené. Pacientov, ktorí dostali vyššie ako odporúčané dávky, je potrebné starostlivo sledovať, pretože sa pozorovalo poškodenie funkcie obličiek (vrátane zlyhania obličiek) a poruchy sérových elektrolytov (vrátane vápnika, fosfátu a horčíka). Ak vznikne hypokalciémia, majú sa podať infúzie glukonanu vápenatého, ak je to klinicky indikované.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Bisfosfonáty, ATC kód: M05 BA 08
Kyselina zoledrónová patrí do skupiny bisfosfonátov a pôsobí primárne na kosti. Je to inhibítor resorpcie kostí, spôsobenej osteoklastami.
Selektívny účinok bisfosfonátov na kosti podmieňuje ich vysoká afinita k mineralizovanej kosti, ale presný mechanizmus na úrovni molekúl, ktorý vedie k inhibícii aktivity osteoklastov, zatiaľ nie je jasný. V
dlhodobých štúdiách na zvieratách kyselina zoledrónová tlmí resorpciu kosti bez nepriaznivého ovplyvnenia tvorby, mineralizácie alebo mechanických vlastností kosti.
Kyselina zoledrónová je nielen účinným inhibítorom resorpcie kostí, ale má aj viaceré protinádorové vlastnosti, ktoré by mohli prispievať k jej celkovej účinnosti pri liečbe metastatického ochorenia kostí. V predklinických skúšaniach sa ukázali nasledujúce vlastnosti:
-
In vivo: inhibícia resorpcie kostí osteoklastami, ktorá mení mikroprostredie kostnej drene a robí ho menej priaznivým pre rast nádorových buniek, antiangiogenetický účinok a analgetický účinok.
-
In vitro: inhibícia proliferácie osteoblastov, priamy cytostatický a proapoptotický účinok na nádorové bunky, synergický cytostatický účinok s inými protinádorovými liečivami, antiadhezívny a antiinvazívny účinok.
Výsledky klinických skúšaní pri prevencii príhod súvisiacich so skeletom u pacientov s postihnutím kostí pripokročilých malignitáchPrvé randomizované, dvojito slepé klinické skúšanie kontrolované placebom porovnávalo Zometu s placebom pri prevencii príhod súvisiacich so skeletom (SRE) u pacientov s rakovinou prostaty. Zometa 4 mg významne znížila podiel pacientov, u ktorých sa vyskytla aspoň jedna SRE, predĺžila medián času do prvej
SRE o > 5 mesiacov a znížila výskyt príhod pripadajúcich na pacienta za rok - mieru morbidity skeletu. Analýza viacpočetných príhod ukázala zníženie rizika vzniku SRE v skupine Zomety o 36% v porovnaní s placebom. Pacienti, ktorí dostávali Zometu, hlásili menšie stupňovanie bolesti ako pacienti, ktorí dostávali placebo, pričom rozdiel dosiahol štatistickú významnosť v 3., 9., 21., a 24. mesiaci. Menej pacientov liečených Zometou utrpelo patologické zlomeniny. Účinky liečby boli menej výrazné u pacientov s blastickými léziami. Výsledky účinnosti sú uvedené v Tabuľke 2.
V druhom klinickom skúšaní, do ktorého boli zahrnuté solídne nádory okrem rakoviny prsníka alebo prostaty, Zometa 4 mg významne znížila podiel pacientov s SRE, predĺžila medián času do prvej SRE o
> 2 mesiace a znížila mieru morbidity skeletu. Analýza viacpočetných príhod ukázala zníženie rizika vzniku
SRE v skupine Zomety o 30,7% v porovnaní s placebom. Výsledky účinnosti sú uvedené v Tabuľke 3.
Tabuľka 2: Výsledky účinnosti (pacienti s rakovinou prostaty liečení hormónmi)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
|
N
| 214
| 208
| 214
| 208
| 214
| 208
|
Podiel pacientov s
SRE (%)
| 38
| 49
| 17
| 25
| 26
| 33
|
Hodnota p
| 0,028
| 0,052
| 0,119
|
Medián času do SRE (dni)
| 488
| 321
| NR
| NR
| NR
| 640
|
Hodnota p
| 0,009
| 0,020
| 0,055
|
Miera morbidity
skeletu
| 0,77
| 1,47
| 0,20
| 0,45
| 0,42
| 0,89
|
Hodnota p
| 0,005
| 0,023
| 0,060
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
| 36
| -
| NA
| NA
| NA
| NA
|
Hodnota p
| 0,002
| NA
| NA
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
Tabuľka 3: Výsledky účinnosti (solídne nádory okrem rakoviny prsníka alebo prostaty)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
|
N
| 257
| 250
| 257
| 250
| 257
| 250
|
Podiel pacientov s
SRE (%)
| 39
| 48
| 16
| 22
| 29
| 34
|
Hodnota p
| 0,039
| 0,064
| 0,173
|
Medián času do SRE
(dni)
| 236
| 155
| NR
| NR
| 424
| 307
|
Hodnota p
| 0,009
| 0,020
| 0,079
|
Miera morbidity
skeletu
| 1,74
| 2,71
| 0,39
| 0,63
| 1,24
| 1,89
|
Hodnota p
| 0,012
| 0,066
| 0,099
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
| 30,7
| -
| NA
| NA
| NA
| NA
|
Hodnota p
| 0,003
| NA
| NA
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
V treťom randomizovanom, dvojito slepom klinickom skúšaní fázy III sa porovnávala Zometa 4 mg a pamidronát 90 mg každé 3 až 4 týždne u pacientov s mnohopočetným myelómom alebo rakovinou prsníka a aspoň jednou léziou kosti. Výsledky ukázali, že Zometa 4 mg vykazuje porovnateľnú účinnosť s 90 mg pamidronátu pri prevencii SRE. Analýza viacpočetných príhod ukázala významné zníženie rizika vzniku SRE o 16% u pacientov liečených Zometou 4 mg v porovnaní s pacientmi, ktorí dostávali pamidronát. Výsledky účinnosti sú uvedené v Tabuľke 4.
Tabuľka 4: Výsledky účinnosti (pacienti s mnohopočetným myelómom a rakovinou prsníka)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
| Zometa
4 mg
| Pam 90 mg
| Zometa
4 mg
| Pam
90 mg
| Zometa
4 mg
| Pam
90 mg
|
N
| 561
| 555
| 561
| 555
| 561
| 555
|
Podiel pacientov s
SRE (%)
| 48
| 52
| 37
| 39
| 19
| 24
|
Hodnota p
| 0,198
| 0,653
| 0,037
|
Medián času do SRE
(dni)
| 376
| 356
| NR
| 714
| NR
| NR
|
Hodnota p
| 0,151
| 0,672
| 0,026
|
Miera morbidity skeletu
| 1,04
| 1,39
| 0,53
| 0,60
| 0,47
| 0,71
|
Hodnota p
| 0,084
| 0,614
| 0,015
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
| 16
| -
| NA
| NA
| NA
| NA
|
Hodnota p
| 0,030
| NA
| NA
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
Zometa sa skúmala aj v dvojito slepom, randomizovanom klinickom skúšaní kontrolovanom placebom
u 228 pacientok s preukázanými metastázami do kostí pri rakovine prsníka, v ktorom sa vyhodnotil účinok Zomety na pomer výskytu príhod súvisiacich so skeletom (SRE), vypočítaný ako celkový počet SRE príhod (okrem hyperkalciémie a po úprave vzhľadom na predchádzajúcu zlomeninu), delený obdobím celkového rizika. Pacientky dostávali buď 4 mg Zomety, alebo placebo každé štyri týždne počas jedného roka. Pacientky boli rovnomerne rozdelené do skupín liečby Zometou a placebom.
Výskyt SRE (príhody/osoba za rok) bol 0,628 pri Zomete a 1,096 pri placebe. Podiel pacientok s aspoň jednou SRE (okrem hyperkalciémie) bol 29,8% v skupine liečenej Zometou oproti 49,6% v skupine placeba (p=0,003). Medián času do vzniku prvej SRE sa v ramene liečby Zometou na konci klinického skúšania nedosiahol a v porovnaní s placebom bol významne dlhší (p=0,007). V porovnaní s placebom Zometa znížila riziko SRE o 41% v analýze viacpočetných príhod (pomer rizika=0,59, p=0,019).
V skupine liečenej Zometou sa v porovnaní s placebom pozorovalo štatisticky významné zlepšenie pri hodnotení bolesti (pri použití Brief Pain Inventory, BPI) po 4 týždňoch a v každom neskoršom čase počas klinického skúšania (Obrázok 1). Hodnotenie bolesti pri Zomete bolo trvale pod východiskovou hodnotou a zmiernenie bolesti sprevádzala tendencia k poklesu analgetického skóre.

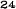
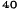
Obrázok 1. Priemerné zmeny skóre podľa BPI oproti východiskovej hodnote. Vyznačené sú štatisticky

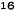

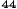
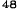
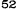
významné rozdiely (*p<0,05) pri porovnaní spôsobov liečby (Zometa oproti placebu)
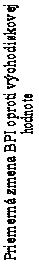
Placebo
∆

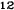
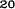
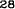

Zometa

Trvanie klinického skúšania (týždne)
Výsledky klinických skúšaní pri liečbe TIHKlinické skúšania pri hyperkalciémii vyvolanej nádorom (TIH) ukázali, že pre účinok kyseliny zoledrónovej je charakteristický pokles vápnika v sére a vylučovania vápnika močom. Vo fáze I sa v klinických skúšaniach na zistenie dávky u pacientov s miernou až stredne ťažkou hyperkalciémiou vyvolanou nádorom (TIH) skúšali účinné dávky v rozmedzí približne 1,2–2,5 mg.
Na stanovenie účinkov Zomety v porovnaní s 90 mg pamidronátu sa vo vopred plánovej analýze zlúčili výsledky dvoch pilotných multicentrických skúšaní s pacientmi s TIH. Normalizácia korigovaného vápnika v sére bola rýchlejšia na 4. deň pri Zomete 8 mg a na 7. deň pri Zomete 4 mg a 8 mg. Pozorovala sa
nasledujúca odpoveď na liečbu.
Tabuľka 5: Podiel pacientov s úplnou odpoveďou na liečbu podľa dní v zlúčených klinických skúšaniach
TIH
| 4. deň
| 7. deň
| 10. deň
|
Zometa 4 mg (N=86)
| 45,3% (p=0,104)
| 82,6% (p=0,005)*
| 88,4% (p=0,002)*
|
Zometa 8 mg (N=90)
| 55,6% (p=0,021)*
| 83,3% (p=0,010)*
| 86,7% (p=0,015)*
|
Pamidronát 90 mg (N=99)
| 33,3%
| 63,6%
| 69,7%
|
*Hodnoty p v porovnaní s pamidronátom
|
Medián času do dosiahnutia normokalciémie bol 4 dni. Medián času do relapsu (opätovné stúpnutie sérového
vápnika korigovaného podľa albumínu na ≥ 2,9 mmol/l) bol 30 až 40 dní u pacientov liečených Zometou oproti 17 dňom u pacientov liečených 90 mg pamidronátu (hodnoty p: 0,001 pre 4 mg a 0,007 pre 8 mg). Medzi oboma dávkami Zomety neboli štatisticky významné rozdiely.
V klinických skúšaniach bolo 69 pacientov s relapsom alebo bez odpovede na začiatočnú liečbu (Zometa
4 mg, 8 mg alebo pamidronát 90 mg) opäť liečených Zometou 8 mg. Podiel takýchto pacientov, u ktorých sa dosiahla odpoveď, bol asi 52%. Pretože títo pacienti boli liečení len dávkou 8 mg, nie sú údaje, ktoré by umožnili porovnanie s dávkou 4 mg.
V klinických skúšaniach s pacientmi s hyperkalciémiou vyvolanou nádorom (TIH) bol celkový profil bezpečnosti u všetkých troch skupín liečby (kyselina zoledrónová 4 mg a 8 mg a pamidronát 90 mg) podobný čo do typu a závažnosti.
5.2 Farmakokinetické vlastnostiPri jednorazových a opakovaných infúziách trvajúcich 5 a 15 minút, ktorými sa podalo 2, 4, 8 a 16 mg kyseliny zoledrónovej 64 pacientom s metastázami v kostiach, sa zistili nasledujúce farmakokinetické údaje, ktoré nezáviseli od dávky.
Po začatí infúzie kyseliny zoledrónovej sa plazmatické koncentrácie liečiva rýchlo zvýšili a dosiahli maximum na konci podania infúzie, po ktorom nasledoval rýchly pokles na < 10% maxima po 4 hodinách a
< 1% maxima po 24 hodinách, s následným dlhým obdobím veľmi nízkych koncentrácií nepresahujúcich
0,1% maxima pred druhou infúziou lieku na 28. deň.
Intravenózne podaná kyselina zoledrónová sa eliminuje trojfázovým procesom: rýchle dvojfázové vymiznutie zo systémového obehu s polčasmi t1/2α 0,24 a t1/2β 1,87 hodiny, po ktorých nasleduje dlhá eliminačná fáza s konečným polčasom eliminácie t1/2γ 146 hodín. Po opakovanom podávaní lieku každých
28 dní nedošlo k akumulácii liečiva v plazme. Kyselina zoledrónová sa nemetabolizuje a vylučuje sa
nezmenená obličkami. Počas prvých 24 hodín sa v moči nájde 39 ± 16% podanej dávky, zatiaľ čo zvyšok sa v zásade viaže na tkanivo kostí. Z kostného tkaniva sa veľmi pomaly uvoľňuje späť do systémového obehu a vylučuje sa obličkami. Celkový telesný klírens je 5,04 ± 2,5 l/hod, a to nezávisle od dávky a bez ovplyvnenia pohlavím, vekom, rasou a telesnou hmotnosťou. Predĺženie infúzie z 5 na 15 minút znížilo koncentráciu kyseliny zoledrónovej na konci infúzie o 30%, ale neovplyvnilo plochu pod krivkou koncentrácie oproti
času.
Tak ako aj pri iných bisfosfonátoch, variabilita farmakokinetických parametrov kyseliny zoledrónovej medzi pacientmi bola vysoká.
Nie sú dostupné farmakokinetické údaje o kyseline zoledrónovej u pacientov s hyperkalciémiou alebo u pacientov s insuficienciou pečene. Kyselina zoledrónová neinhibuje ľudské enzýmy P450
in vitro, nevykazuje biotransformáciu a v štúdiách na zvieratách sa v stolici našli < 3% podanej dávky, čo naznačuje, že funkcia pečene nezohráva významnú úlohu vo farmakokinetike kyseliny zoledrónovej.
Pri určení vzťahu medzi obličkovým klírensom kyseliny zoledrónovej a klírensom kreatinínu predstavoval obličkový klírens 75 ± 33% klírensu kreatinínu, ktorého priemerná hodnota bola 84 ± 29 ml/min (rozmedzie
22–143 ml/min) u 64 sledovaných pacientov s karcinómami. Analýza v tejto skupine ukázala, že u pacienta s klírensom kreatinínu 20 ml/min (ťažké poškodenie funkcie obličiek) alebo 50 ml/min (stredne ťažké poškodenie) bude zodpovedajúci predpokladaný klírens kyseliny zoledrónovej 37% alebo 72% hodnoty pacienta, ktorý má klírens kreatinínu 84 ml/min. Sú dostupné len obmedzené farmakokinetické údaje o pacientoch s ťažkou insuficienciou obličiek (klírens kreatinínu < 30 ml/min).
Kyselina zoledrónová nemá afinitu ku krvinkám a jej väzba na bielkoviny plazmy je nízka (približne 56%) a nezávisí od koncentrácie kyseliny zoledrónovej.
5.3 Predklinické údaje o bezpečnosti
Akútna toxicita
Najvyššia neletálna jednorazová intravenózna dávka bola 10 mg/kg telesnej hmotnosti u myší a 0,6 mg/kg u potkanov.
Subchronická a chronická toxicita
Kyselina zoledrónová sa dobre znášala, keď sa podávala subkutánne potkanom a intravenózne psom v dávkach do 0,02 mg/kg denne počas 4 týždňov. Podávanie 0,001 mg/kg/deň subkutánne potkanom a
0,005 mg/kg intravenózne psom raz za 2–3 dni počas až 52 týždňov sa tiež dobre znášalo.
Najčastejším nálezom v štúdiách pri opakovanom podávaní bolo zväčšenie primárnej trabekulárnej časti kosti v metafýzach dlhých kostí u rastúcich zvierat pri takmer všetkých dávkach, čo bol nález v súlade s farmakologickou antiresorpčnou účinnosťou látky.
Bezpečné rozmedzie dávok vzhľadom na účinky na obličky bolo úzke v štúdiách pri dlhodobom opakovanom parenterálnom podávaní zvieratám, ale kumulatívne hladiny bez nežiaducich príhod v štúdiách pri jednorazovom podaní (1,6 mg/kg) a pri opakovanom podávaní až do jedného mesiaca (0,06–
0,6 mg/kg/deň) nenaznačili účinky na obličky pri dávkach ekvivalentných alebo prevyšujúcich najvyššiu plánovanú terapeutickú dávku u ľudí. Dlhodobejšie opakované podávanie v dávkach, ktoré zahrnulo aj najvyššiu plánovanú terapeutickú dávku kyseliny zoledrónovej u ľudí, vyvolalo toxické účinky v iných orgánoch vrátane gastrointestinálneho traktu, pečene, sleziny a pľúc, ako aj v mieste podania intravenóznej injekcie.
Reprodukčná toxicita
Kyselina zoledrónová bola teratogénna u potkanov v subkutánnych dávkach ≥ 0,2 mg/kg. Hoci sa u králikov nepozorovala žiadna teratogenita alebo fetotoxicita, zistila sa toxicita pre matky.
Mutagenita a karcinogenita
Kyselina zoledrónová nebola mutagénna vo vykonaných testoch mutagenity a testy na karcinogenitu neposkytli žiadne dôkazy o karcinogénnom potenciále.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Injekčná liekovka s práškom: Manitol
Citrónan sodný
Ampulka s rozpúšťadlom: Voda na injekciu
6.2 Inkompatibility
Aby sa zabránilo prípadným inkompatibilitám, rekonštituovaný roztok Zomety sa má zriediť 0,9% m/V
roztokom chloridu sodného alebo 5% m/V roztokom glukózy.
Rekonštituovaný roztok Zomety sa nesmie miešať s roztokmi obsahujúcimi vápnik, napr. Ringerovým roztokom.
Skúšania so sklenenými fľašami, ako aj s niekoľkými druhmi infúznych vakov a infúznych súprav z polyvinylchloridu, polyetylénu a polypropylénu (predtým naplnenými 0,9% m/V roztokom chloridu sodného alebo 5% m/V roztokom glukózy) neukázali žiadne inkompatibility so Zometou.
6.3 Čas použiteľnosti
3 roky.
Rekonštituovaný roztok je chemicky a fyzikálne stály počas 24 hodín pri 2°C – 8°C.
6.4 Špeciálne upozornenia na uchovávanie
Žiadne zvláštne upozornenia na uchovávanie.
Po aseptickej rekonštitúcii a zriedení je najvhodnejšie okamžite použiť rekonštituovaný a zriedený liek. Ak sa nepoužije okamžite, za trvanie a podmienky uchovávania pred použitím zodpovedá používateľ. Celkový čas medzi rekonštitúciou, zriedením, uchovávaním v chladničke pri teplote 2°C – 8°C a ukončením podania nesmie byť dlhší ako 24 hodín.
6.5 Druh obalu a obsah balenia
Zometa 4 mg prášok a rozpúšťadlo na infúzny roztok sa dodáva v baleniach, ktoré obsahujú 1, 4 alebo
10 injekčných liekoviek a 1, 4 alebo 10 ampuliek s vodou na injekciu. Nie všetky veľkosti balenia musia byť
uvedené do obehu.
Injekčná liekovka s práškom: liekovka z bezfarebného skla, hydrolytická trieda I (Ph. Eur.), s objemom 6 ml. Ampulka s rozpúšťadlom: ampulka z bezfarebného skla s objemom 5 ml.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prášok sa najprv musí rekonštituovať v injekčnej liekovke, na čo sa použije 5 ml vody na injekciu z priloženej ampulky. Prášok sa musí úplne rozpustiť, kým sa roztok odoberie. Potrebné množstvo rekonštituovaného roztoku sa potom ďalej riedi 100 ml infúzneho roztoku, ktorý neobsahuje vápnik (0,9% m/V roztok chloridu sodného alebo 5% m/V roztok glukózy). Ak sa roztok uchováva v chladničke, pred podaním sa musí nechať zohriať na izbovú teplotu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB Veľká Británia
8. REGISTRAČNÉ ČÍSLO
EU/1/01/176/001-003
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 20.03.2001
Dátum prvého predĺženia registrácie: 20.03.2006
10. DÁTUM REVÍZIE TEXTU
1
. NÁZOV LIEKU
Zometa 4 mg/5 ml infúzny koncentrát
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna injekčná liekovka s 5 ml koncentrátu obsahuje 4 mg kyseliny zoledrónovej (bezvodej).
Jeden ml koncentrátu obsahuje kyselinu zoledrónovú (ako monohydrát) v množstve zodpovedajúcom 0,8 mg kyseliny zoledrónovej (bezvodej).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Infúzny koncentrát
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
− Prevencia príhod súvisiacich so skeletom (patologické zlomeniny, kompresia miechy, rádioterapia alebo chirurgický zákrok na kosti, alebo hyperkalciémia vyvolaná nádorom) u pacientov s postihnutím kostí pri pokročilých malignitách.
− Liečba hyperkalciémie vyvolanej nádorom (TIH).
4.2 Dávkovanie a spôsob podávania
Zometu smú podávať len lekári, ktorí majú skúsenosti s intravenóznym podávaním bisfosfonátov. Prevencia príhod súvisiacich so skeletom u pacientov s postihnutím kostí pri pokročilých malignitách
Dospelí a starší pacienti
Odporúčaná dávka pri prevencii príhod súvisiacich so skeletom u pacientov s postihnutím kostí pri pokročilých malignitách je 4 mg kyseliny zoledrónovej. Koncentrát sa musí ďalej zriediť 100 ml sterilného
0,9% m/V roztoku chloridu sodného alebo 5% m/V roztoku glukózy a podať ako intravenózna infúzia trvajúca nie menej ako 15 minút každé 3 až 4 týždne.
Pacientom sa má perorálne podávať aj doplnok 500 mg vápnika a 400 IU vitamínu D denne. Liečba TIH
Dospelí a starší pacienti
Odporúčaná dávka pri hyperkalciémii (vápnik v sére korigovaný podľa albumínu ≥ 12,0 mg/dl alebo
3,0 mmol/l) je 4 mg kyseliny zoledrónovej. Koncentrát sa musí ďalej zriediť 100 ml sterilného 0,9% m/V roztoku chloridu sodného alebo 5% m/V roztoku glukózy a podať ako jednorazová intravenózna infúzia trvajúca nie menej ako 15 minút. Pacienti musia byť dostatočne hydratovaní pred aj po podaní Zomety.
Poškodenie funkcie obličiek
TIH:
O liečbe Zometou u pacientov s TIH, ktorí majú aj ťažké poškodenie funkcie obličiek, sa má uvažovať len po vyhodnotení rizík a prospešnosti liečby. Z klinických skúšaní boli vylúčení pacienti s kreatinínom v sére
> 400 µmol/l alebo > 4,5 mg/dl. Nie je potrebné upraviť dávku u pacientov s TIH s kreatinínom v sére
< 400 µmol/l alebo < 4,5 mg/dl (pozri časť 4.4).
 Prevencia príhod súvisiacich so skeletom u pacientov s postihnutím kostí pri pokročilých malignitách:
Prevencia príhod súvisiacich so skeletom u pacientov s postihnutím kostí pri pokročilých malignitách:
Pri začatí liečby Zometou u pacientov s mnohopočetným myelómom alebo metastatickými léziami kostí pri solídnych nádoroch sa majú stanoviť kreatinín v sére a klírens kreatinínu (CrCl). CrCl sa vypočíta zo
sérového kreatinínu pomocou Cockcroftovho-Gaultovho vzorca. Použitie Zomety sa neodporúča
u pacientov, ktorí majú pred začatím liečby ťažké poškodenie funkcie obličiek, definované u tejto skupiny pacientov ako CrCl < 30 ml/min. Z klinických skúšaní Zomety boli vylúčení pacienti s kreatinínom v sére
> 265 µmol/l alebo > 3,0 mg/dl.
U pacientov s metastázami v kostiach, ktorí majú pred začatím liečby mierne až stredne ťažké poškodenie funkcie obličiek, definované u tejto skupiny pacientov ako CrCl 30–60 ml/min, sa odporúča nasledujúca dávka Zomety (pozri aj časť 4.4):
Východiskový klírens kreatinínu (ml/min) Odporúčaná dávka Zomety*
> 60 4,0 mg
50–60 3,5 mg*
40–49 3,3 mg*
30–39 3,0 mg*
*Dávky sa vypočítali za predpokladu cieľovej AUC 0,66 (mg•hod/l) (CrCl=75 ml/min). Očakáva sa, že zníženými dávkami sa u pacientov s poškodením funkcie obličiek dosiahne rovnaká AUC, aká sa pozoruje u pacientov s klírensom kreatinínu 75 ml/min.
Po začatí liečby sa má stanoviť kreatinín v sére pred každou dávkou Zomety a liek sa nemá podať, ak sa zhoršila funkcia obličiek. V klinických skúšaniach sa zhoršenie funkcie obličiek definovalo ako:
- Zvýšenie o 0,5 mg/dl alebo 44 µmol/l u pacientov s normálnou východiskovou hodnotou kreatinínu v sére (< 1,4 mg/dl alebo < 124 µmol/l);
- Zvýšenie o 1,0 mg/dl alebo 88 µmol/l u pacientov s abnormálnou východiskovou hodnotou kreatinínu
(> 1,4 mg/dl alebo > 124 µmol/l).
V klinických skúšaniach sa pokračovalo v liečbe Zometou, len ak sa hladina kreatinínu vrátila do rozmedzia
10% nad východiskovou hodnotou (pozri časť 4.4). V liečbe Zometou sa má pokračovať s rovnakou dávkou ako pred prerušením liečby.
Pokyny na prípravu znížených dávok ZometyPodľa potreby odoberte nasledujúci príslušný objem koncentrátu:
- 4,4 ml na dávku 3,5 mg
- 4,1 ml na dávku 3,3 mg
- 3,8 ml na dávku 3,0 mg
Odobraté množstvo koncentrátu sa musí zriediť 100 ml sterilného 0,9% m/V roztoku chloridu sodného alebo
5% m/V roztoku glukózy. Dávka sa musí podať ako jednorazová intravenózna infúzia trvajúca nie menej ako
15 minút.
Použitie Zomety u pediatrických pacientov sa nesledovalo. Zometa sa nemá používať u tejto skupiny pacientov, dokiaľ nebudú dostupné ďalšie údaje.
4.3 KontraindikácieZometa infúzny koncentrát je kontraindikovaný v gravidite, u dojčiacich žien, u pacientov s klinicky významnou precitlivenosťou na kyselinu zoledrónovú, iné bisfosfonáty alebo niektorú z pomocných látok v Zomete.
4.4 Osobitné upozornenia a opatrenia pri používaníVšeobecnéPred podaním Zomety je nutné pacientov vyšetriť, aby sa overilo, či sú dostatočne hydratovaní. U pacientov s rizikom zlyhania srdca je nutné vyvarovať sa nadbytočnej hydratácie.
Po začatí liečby Zometou sa majú starostlivo sledovať bežné metabolické parametre súvisiace s hyperkalciémiou, ako sú hladiny vápnika, fosfátu a horčíka v sére. Ak vznikne hypokalciémia, hypofosfatémia alebo hypomagneziémia, môže byť potrebná krátkodobá suplementárna liečba. Neliečení pacienti s hyperkalciémiou mávajú spravidla určitý stupeň poruchy funkcie obličiek, preto sa má zvážiť starostlivé sledovanie funkcie obličiek.
Bezpečnosť a účinnosť Zomety u pediatrických pacientov sa nestanovili. Insuficiencia obličiek
Stav pacientov s TIH, u ktorých sa preukáže zhoršenie funkcie obličiek, je nutné patrične zhodnotiť a zvážiť,
či prípadná prospešnosť liečby Zometou je väčšia ako jej možné riziko.
Pri rozhodovaní o tom, či pacientom s metastázami v kostiach podať preventívnu liečbu proti príhodám súvisiacim so skeletom, treba vziať do úvahy, že účinok liečby nastupuje za 2–3 mesiace.
Tak ako pri iných bisfosfonátoch, v súvislosti so Zometou sa zaznamenali poruchy funkcie obličiek. Medzi faktory, ktoré môžu zvýšiť možnosť zhoršenia funkcie obličiek, patrí dehydratácia, už existujúce poškodenie funkcie obličiek, viaceré liečebné cykly Zomety a iných bisfosfonátov, ako aj použitie iných nefrotoxických liekov. Hoci sa podaním Zomety v dávke 4 mg počas 15 minút toto riziko zníži, zhoršenie funkcie obličiek
sa napriek tomu môže vyskytnúť. Zhoršenie funkcie obličiek, progresia do zlyhania obličiek a dialýzy boli hlásené u pacientov po začiatočnej dávke alebo jednorazovej dávke Zomety. K zvýšeniu kreatinínu v sére dochádza aj u niektorých pacientov pri chronickom podávaní Zomety v odporúčaných dávkach na prevenciu príhod súvisiacich so skeletom, aj keď zriedkavejšie.
Hladiny kreatinínu v sére pacientov sa majú stanoviť pred každou dávkou Zomety. Pri začatí liečby pacientov s metastázami v kostiach s miernym až stredne ťažkým poškodením funkcie obličiek sa
odporúčajú nižšie dávky Zomety. Pacientom, u ktorých sa dokáže zhoršenie funkcie obličiek počas liečby, sa
Zometa nemá podať. Zometa sa má znovu podať len vtedy, keď sa ich kreatinín v sére vráti do rozmedzia
10% nad východiskovou hodnotou (pozri časť 4.2).
Vzhľadom na prípadný účinok bisfosfonátov vrátane Zomety na funkciu obličiek, nedostatok klinických údajov o bezpečnosti u pacientov s ťažkým poškodením funkcie obličiek na začiatku liečby (v klinických skúšaniach definované ako kreatinín v sére ≥ 400 µmol/l alebo ≥ 4,5 mg/dl u pacientov s TIH a ≥ 265 µmol/l alebo ≥ 3,0 mg/dl u pacientov s rakovinou a metastázami v kostiach) a len obmedzené farmakokinetické
údaje u pacientov s ťažkým poškodením funkcie obličiek na začiatku liečby (klírens kreatinínu < 30 ml/min), použitie Zomety u pacientov s ťažkým poškodením funkcie obličiek sa neodporúča.
Insuficiencia pečene
Pretože sú dostupné len obmedzené klinické údaje o pacientoch so závažnou insuficienciou pečene, nemožno dať žiadne osobitné odporúčania pre túto skupinu pacientov.
Osteonekróza čeľuste
Osteonekróza čeľuste sa zaznamenala u pacientov, prevažne u tých s rakovinou, ktorí dostávali liečbu bisfosfonátmi vrátane Zomety. Mnohí z týchto pacientov dostávali aj chemoterapiu a kortikosteroidy. Väčšina hlásených prípadov súvisela s dentálnymi zákrokmi, ako je extrakcia zuba. Mnohí mali príznaky lokálnej infekcie vrátane osteomyelitídy.
Prehliadka chrupu s náležitými preventívnymi dentálnymi zákrokmi sa má uvážiť pred liečbou bisfosfonátmi u pacientov so sprievodnými rizikovými faktormi (napr. rakovinou, chemoterapiou, kortikosteroidmi, nedostatočnou hygienou ústnej dutiny).
Ak je to možné, títo pacienti sa majú počas liečby vyvarovať invazívnych dentálnych zákrokov. Dentálny chirurgický zákrok môže zhoršiť stav pacientov, u ktorých počas liečby vznikne osteonekróza čeľuste. Nie sú dostupné údaje, ktoré by ukázali, či prerušenie liečby bisfosfonátom znižuje riziko osteonekrózy čeľuste
u pacientov, ktorí potrebujú dentálne zákroky. Klinický úsudok ošetrujúceho lekára založený na vyhodnotení individuálneho pomeru prospešnosti a rizika má byť určujúci pri plánovaní liečby každého pacienta.
Bolesť
svalov a kostí
Po uvedení na trh bola hlásená silná bolesť kostí, kĺbov a/alebo svalov, príležitostne znemožňujúca pohyblivosť u pacientov používajúcich bisfosfonáty. Takéto hlásenia však neboli časté. Do tejto kategórie liekov patrí Zometa (kyselina zoledrónová). Čas do nástupu symptómov sa rôznil od jedného dňa do niekoľkých mesiacov od začatia liečby. U väčšiny pacientov sa symptómy zmiernili po skončení liečby. Podsúbor pacientov mal recidívu symptómov, keď sa im znovu podal rovnaký liek alebo iný bisfosfonát.
4.5 Liekové a iné interakcie
V klinických skúšaniach sa Zometa podávala súčasne s bežne používanými protinádorovými liekmi, diuretikami, antibiotikami a analgetikami bez toho, aby sa vyskytli klinicky zjavné interakcie.
Kyselina zoledrónová sa in vitro neviaže vo významnej miere na bielkoviny plazmy a neinhibuje ľudské enzýmy P450 (pozri časť 5.2), nevykonali sa však osobitné klinické štúdie interakcií. Opatrnosť sa odporúča pri súčasnom podávaní bisfosfonátov s aminoglykozidmi, pretože obidve skupiny liečiv môžu mať aditívny účinok, čo má za následok nižšiu hladinu vápnika v sére na dlhšiu dobu, ako sa požaduje. Pri použití Zomety súčasne s inými potenciálne nefrotoxickými liečivami je nutná opatrnosť. Počas liečby sa musí venovať pozornosť aj možnému vzniku hypomagneziémie.
U pacientov s mnohopočetným myelómom sa môže zvýšiť riziko poruchy funkcie obličiek pri intravenóznom podaní bisfosfonátov v kombinácii s talidomidom.
4.6 Gravidita a laktácia
Nie sú k dispozícii dostatočné údaje o použití kyseliny zoledrónovej u gravidných žien. Štúdie reprodukčnej toxicity kyseliny zoledrónovej na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Dystokia sa pozorovala pri najnižšej skúšanej dávke u potkanov (0,01 mg/kg telesnej hmotnosti). Nie je známe potenciálne riziko u ľudí. Zometa sa nemá používať počas gravidity.
Nie je známe, či sa kyselina zoledrónová vylučuje do ľudského mlieka. Zometu nemajú používať dojčiace ženy (pozri časť 4.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Frekvencia nežiaducich reakcií na Zometu 4 mg sa stanovila najmä na základe údajov získaných pri chronickej liečbe. Nežiaduce reakcie na Zometu a nežiaduce reakcie zaznamenané pri iných bisfosfonátoch sú podobné a ich výskyt možno očakávať u približne tretiny pacientov. Intravenózne podanie najčastejšie sprevádzal syndróm podobný chrípke u približne 9% pacientov, ktorý zahŕňal bolesť kostí (9,1%), horúčku (7,2%), únavu (4,1%) a zimnicu (2,9%). Príležitostne sa zaznamenali prípady bolesti kĺbov a svalov u približne 3% pacientov. Nie sú dostupné údaje o reverzibilite týchto nežiaducich účinkov.
Zníženie vylučovania vápnika obličkami často sprevádza pokles hladín fosfátu v sére (u približne 20% pacientov), ktorý je asymptomatický a nevyžaduje liečbu. Vápnik v sére môže klesnúť na asymptomatické hypokalciemické hladiny u približne 3% pacientov.
Po intravenóznej infúzii Zomety sa zaznamenali gastrointestinálne reakcie ako nauzea (5,8%) a vracanie (2,6%). Príležitostne sa v mieste podania infúzie pozorovali miestne reakcie, ako je sčervenenie alebo opuch a/alebo bolesť u menej ako 1% pacientov.
Anorexia sa zaznamenala u 1,5% pacientov liečených Zometou 4 mg. Pozorovalo sa niekoľko prípadov exantému alebo svrbenia (menej ako 1%).
Tak ako pri iných bisfosfonátoch, prípady zápalu spojoviek sa zaznamenali u približne 1% pacientov.
Boli správy o zhoršení funkcie obličiek (2,3%), hoci v mnohých prípadoch sa na etiológii zjavne podieľalo viacero faktorov.
Podľa súhrnnej analýzy klinických skúšaní kontrolovaných placebom sa ťažká anémia (Hb < 8,0 g/dl)
zaznamenala u 5,2% pacientov, ktorí dostávali Zometu 4 mg, oproti 4,2% pacientov, ktorí dostávali placebo.
Nežiaduce reakcie na liek uvedené v Tabuľke 1 sa súhrnne zaznamenali v klinických skúšaniach po prevažne chronickej liečbe kyselinou zoledrónovou.
Tabuľka 1Nežiaduce reakcie sú zoradené podľa frekvencie, najčastejšie ako prvé, pričom frekvencia je určená nasledovne: veľmi časté (≥1/10), časté (≥1/100, <1/10), menej časté (≥1/1 000, <1/100), zriedkavé (≥1/10 000, <1/1 000), veľmi zriedkavé (<1/10 000), zahŕňajúce jednotlivé hlásenia.
Poruchy krvi a lymfatického systému
Časté: Anémia
Menej časté: Trombocytopénia, leukopénia
Zriedkavé: Pancytopénia
|
Poruchy nervového systému
Časté: Bolesť hlavy
Menej časté: Závraty, parestézie, poruchy chuti, hypestézie, hyperestézie, tremor
|
Psychické poruchy
Menej časté: Úzkosť, poruchy spánku
Zriedkavé: Zmätenosť
|
Ochorenia oka
Časté: Konjunktivitída
Menej časté: Neostré videnie
Veľmi zriedkavé: Uveitída, episkleritída
|
Gastrointestinálne poruchy
Časté: Nauzea, vracanie, anorexia
Menej časté: Hnačka, zápcha, bolesť brucha, dyspepsia, stomatitída, suchosť v ústach
|
Poruchy dýchacej sústavy, hrudníka a mediastína
Menej časté: Dyspnoe, kašeľ
|
Poruchy kože a podkožného tkaniva
Menej časté: Svrbenie, exantém (vrátane erytematózneho a makulárneho exantému), zvýšené potenie
|
Poruchy kostrového svalstva, spojivových tkanív a kostí
Časté: Bolesť kostí, bolesť svalov, bolesť kĺbov, generalizovaná bolesť
Menej časté: Svalové kŕče
|
Kardiovaskulárne poruchy
Menej časté: Hypertenzia, hypotenzia
Zriedkavé: Bradykardia
|
Poruchy obličiek a močových ciest
Časté: Zhoršenie funkcie obličiek
Menej časté: Akútne zlyhanie obličiek, hematúria, proteinúria
|
Poruchy imunitného systému
Menej časté: Reakcie z precitlivenosti
Zriedkavé: Angioneurotický edém
|
Celkové poruchy a reakcie v mieste podania
Časté: Horúčka, syndróm podobný chrípke
|

(zahŕňajúci únavu, zimnicu, celkový pocit nevoľnosti a návaly tepla)
Menej časté: Asténia, periférny edém, reakcie v mieste podania (zahŕňajúce bolesť, podráždenie, opuch a induráciu), bolesť na hrudi, zvýšenie hmotnosti
Laboratórne odchýlkyVeľmi časté: Hypofosfatémia
Časté: Zvýšenie kreatinínu a močoviny v krvi, hypokalciémia
Menej časté: Hypomagneziémia, hypokaliémia
Zriedkavé: Hyperkaliémia, hypernatriémia
V jednom randomizovanom, dvojito slepom, kontrolovanom klinickom skúšaní trvajúcom 3 roky, v ktorom
sa vyhodnotila účinnosť a bezpečnosť 5 mg kyseliny zoledrónovej podávanej raz ročne v liečbe postmenopauzálnej osteoporózy (PMO) oproti placebu, celková incidencia fibrilácie predsiení bola 2,5% (96 z 3 862) u pacientok, ktoré dostávali 5 mg kyseliny zoledrónovej, a 1,9% (75 z 3 852) u pacientok, ktoré dostávali placebo. Výskyt fibrilácie predsiení ako závažnej nežiaducej udalosti bol 1,3% (51 z 3 862)
u pacientok, ktoré dostávali 5 mg kyseliny zoledrónovej, a 0,6% (22 z 3 852) u pacientok, ktoré dostávali placebo. Nerovnováha pozorovaná v tomto klinickom skúšaní sa nepozorovala v iných klinických skúšaniach s kyselinou zoledrónovou, vrátane skúšaní so 4 mg Zomety (kyselina zoledrónová) podávanými každé 3-4 týždne onkologickým pacientom. Mechanizmus, ktorý spôsobil zvýšenie incidencie fibrilácie predsiení v tomto jedinom klinickom skúšaní, nie je známy.
Skúsenosti po uvedení na trh: Prevažne u pacientov s rakovinou liečených bisfosfonátmi vrátane Zomety sa zaznamenali prípady osteonekrózy (primárne čeľuste). Mnohí z týchto pacientov mali príznaky lokálnej infekcie vrátane osteomyelitídy a väčšina hlásení sa týka pacientov s rakovinou po extrakcii zubov alebo iných dentálnych chirurgických zákrokoch. Osteonekróza čeľuste má viaceré dobre zdokumentované rizikové faktory vrátane diagnózy rakoviny, súčasne podanej liečby (napr. chemoterapie, rádioterapie, kortikosteroidov) a sprievodných ochorení (napr. anémie, koagulopatií, infekcie, ochorenia ústnej dutiny). Hoci sa nestanovila príčinná súvislosť, je vhodné vyvarovať sa dentálnych chirurgických zákrokov, pretože sa môže predĺžiť obdobie zotavenia (pozri časť 4.4). Vo veľmi zriedkavých prípadoch boli hlásené nasledujúce udalosti: hypotenzia viedúca ku synkope alebo obehovému kolapsu, najmä u pacientov so základnými rizikovými faktormi, fibrilácia predsiení, somnolencia, bronchokonstrikcia.
4.9 PredávkovanieKlinické skúsenosti s akútnym predávkovaním Zomety sú obmedzené. Pacientov, ktorí dostali vyššie ako odporúčané dávky, je potrebné starostlivo sledovať, pretože sa pozorovalo poškodenie funkcie obličiek (vrátane zlyhania obličiek) a poruchy sérových elektrolytov (vrátane vápnika, fosfátu a horčíka). Ak vznikne hypokalciémia, majú sa podať infúzie glukonanu vápenatého, ak je to klinicky indikované.'
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Bisfosfonáty, ATC kód: M05 BA 08
Kyselina zoledrónová patrí do skupiny bisfosfonátov a pôsobí primárne na kosti. Je to inhibítor resorpcie kostí, spôsobenej osteoklastami.
Selektívny účinok bisfosfonátov na kosti podmieňuje ich vysoká afinita k mineralizovanej kosti, ale presný mechanizmus na úrovni molekúl, ktorý vedie k inhibícii aktivity osteoklastov, zatiaľ nie je jasný. V dlhodobých štúdiách na zvieratách kyselina zoledrónová tlmí resorpciu kosti bez nepriaznivého ovplyvnenia tvorby, mineralizácie alebo mechanických vlastností kosti.
Kyselina zoledrónová je nielen účinným inhibítorom resorpcie kostí, ale má aj viaceré protinádorové vlastnosti, ktoré by mohli prispievať k jej celkovej účinnosti pri liečbe metastatického ochorenia kostí. V predklinických skúšaniach sa ukázali nasledujúce vlastnosti:
-
In vivo: inhibícia resorpcie kostí osteoklastami, ktorá mení mikroprostredie kostnej drene a robí ho menej priaznivým pre rast nádorových buniek, antiangiogenetický účinok a analgetický účinok.
-
In vitro: inhibícia proliferácie osteoblastov, priamy cytostatický a proapoptotický účinok na nádorové bunky, synergický cytostatický účinok s inými protinádorovými liečivami, antiadhezívny a antiinvazívny účinok.
Výsledky klinických skúšaní pri prevencii príhod súvisiacich so skeletom u pacientov s postihnutím kostí pripokročilých malignitáchPrvé randomizované, dvojito slepé klinické skúšanie kontrolované placebom porovnávalo Zometu s placebom pri prevencii príhod súvisiacich so skeletom (SRE) u pacientov s rakovinou prostaty. Zometa 4 mg významne znížila podiel pacientov, u ktorých sa vyskytla aspoň jedna SRE, predĺžila medián času do prvej SRE o > 5 mesiacov a znížila výskyt príhod pripadajúcich na pacienta za rok - mieru morbidity skeletu. Analýza viacpočetných príhod ukázala zníženie rizika vzniku SRE v skupine Zomety o 36% v porovnaní s placebom. Pacienti, ktorí dostávali Zometu, hlásili menšie stupňovanie bolesti ako pacienti, ktorí dostávali placebo, pričom rozdiel dosiahol štatistickú významnosť v 3., 9., 21., a 24. mesiaci. Menej pacientov liečených Zometou utrpelo patologické zlomeniny. Účinky liečby boli menej výrazné u pacientov s blastickými léziami. Výsledky účinnosti sú uvedené v Tabuľke 2.
V druhom klinickom skúšaní, do ktorého boli zahrnuté solídne nádory okrem rakoviny prsníka alebo prostaty, Zometa 4 mg významne znížila podiel pacientov s SRE, predĺžila medián času do prvej SRE o
> 2 mesiace a znížila mieru morbidity skeletu. Analýza viacpočetných príhod ukázala zníženie rizika vzniku
SRE v skupine Zomety o 30,7% v porovnaní s placebom. Výsledky účinnosti sú uvedené v Tabuľke 3.
Tabuľka 2: Výsledky účinnosti (pacienti s rakovinou prostaty liečení hormónmi)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
| Zometa
4 mg
| Placebo
|
N
| 214
| 208
| 214
| 208
| 214
| 208
|
Podiel pacientov s
SRE (%)
| 38
| 49
| 17
| 25
| 26
| 33
|
Hodnota p
| 0,028
| 0,052
| 0,119
|
Medián času do SRE
(dni)
| 488
| 321
| NR
| NR
| NR
| 640
|
Hodnota p
| 0,009
| 0,020
| 0,055
|
Miera morbidity
skeletu
| 0,77
| 1,47
| 0,20
| 0,45
| 0,42
| 0,89
|
Hodnota p
| 0,005
| 0,023
| 0,060
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
| 36
| -
| NA
| NA
| NA
| NA
|
Hodnota p
| 0,002
| NA
| NA
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
Tabuľka 3: Výsledky účinnosti (solídne nádory okrem rakoviny prsníka alebo prostaty)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
|
Zometa
4 mg
|
Placebo
|
Zometa
4 mg
|
Placebo
|
Zometa
4 mg
|
Placebo
|
N
|
257
|
250
|
257
|
250
|
257
|
250
|
Podiel pacientov s
SRE (%)
|
39
|
48
|
16
|
22
|
29
|
34
|
Hodnota p
|
0,039
|
0,064
|
0,173
|
Medián času do SRE
(dni)
|
236
|
155
|
NR
|
NR
|
424
|
307
|
Hodnota p
|
0,009
|
0,020
|
0,079
|
Miera morbidity
skeletu
|
1,74
|
2,71
|
0,39
|
0,63
|
1,24
|
1,89
|
Hodnota p
|
0,012
|
0,066
|
0,099
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
|
30,7
|
-
|
NA
|
NA
|
NA
|
NA
|
Hodnota p
|
0,003
|
NA
|
NA
|
|
|
|
|
|
|
|
|
|
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
V treťom randomizovanom, dvojito slepom klinickom skúšaní fázy III sa porovnávala Zometa 4 mg a pamidronát 90 mg každé 3 až 4 týždne u pacientov s mnohopočetným myelómom alebo rakovinou prsníka a aspoň jednou léziou kosti. Výsledky ukázali, že Zometa 4 mg vykazuje porovnateľnú účinnosť s 90 mg pamidronátu pri prevencii SRE. Analýza viacpočetných príhod ukázala významné zníženie rizika vzniku SRE o 16% u pacientov liečených Zometou 4 mg v porovnaní s pacientmi, ktorí dostávali pamidronát. Výsledky účinnosti sú uvedené v Tabuľke 4.
Tabuľka 4: Výsledky účinnosti (pacienti s mnohopočetným myelómom a rakovinou prsníka)
| Akákoľvek SRE (+ TIH)
| Zlomeniny*
| Rádioterapia kostí
|
| Zometa
4 mg
| Pam 90 mg
| Zometa
4 mg
| Pam
90 mg
| Zometa
4 mg
| Pam
90 mg
|
N
| 561
| 555
| 561
| 555
| 561
| 555
|
Podiel pacientov s
SRE (%)
| 48
| 52
| 37
| 39
| 19
| 24
|
Hodnota p
| 0,198
| 0,653
| 0,037
|
Medián času do SRE
(dni)
| 376
| 356
| NR
| 714
| NR
| NR
|
Hodnota p
| 0,151
| 0,672
| 0,026
|
Miera morbidity
skeletu
| 1,04
| 1,39
| 0,53
| 0,60
| 0,47
| 0,71
|
Hodnota p
| 0,084
| 0,614
| 0,015
|
Zníženie rizika
utrpieť viacpočetné príhody** (%)
| 16
| -
| NA
| NA
| NA
| NA
|
Hodnota p
| 0,030
| NA
| NA
|
* Zahŕňa zlomeniny stavcov a iné zlomeniny
** Zohľadňuje všetky príhody súvisiace so skeletom, celkový počet ako aj čas do každej príhody počas klinického skúšania
NR Nedosiahol sa
NA Nemožno použiť
Zometa sa skúmala aj v dvojito slepom, randomizovanom klinickom skúšaní kontrolovanom placebom
u 228 pacientok s preukázanými metastázami do kostí pri rakovine prsníka, v ktorom sa vyhodnotil účinok Zomety na pomer výskytu príhod súvisiacich so skeletom (SRE), vypočítaný ako celkový počet SRE príhod (okrem hyperkalciémie a po úprave vzhľadom na predchádzajúcu zlomeninu), delený obdobím celkového rizika. Pacientky dostávali buď 4 mg Zomety, alebo placebo každé štyri týždne počas jedného roka. Pacientky boli rovnomerne rozdelené do skupín liečby Zometou a placebom.
Výskyt SRE (príhody/osoba za rok) bol 0,628 pri Zomete a 1,096 pri placebe. Podiel pacientok s aspoň jednou SRE (okrem hyperkalciémie) bol 29,8% v skupine liečenej Zometou oproti 49,6% v skupine placeba (p=0,003). Medián času do vzniku prvej SRE sa v ramene liečby Zometou na konci klinického skúšania nedosiahol a v porovnaní s placebom bol významne dlhší (p=0,007). V porovnaní s placebom Zometa znížila riziko SRE o 41% v analýze viacpočetných príhod (pomer rizika=0,59, p=0,019).
V skupine liečenej Zometou sa v porovnaní s placebom pozorovalo štatisticky významné zlepšenie pri hodnotení bolesti (pri použití Brief Pain Inventory, BPI) po 4 týždňoch a v každom neskoršom čase počas klinického skúšania (Obrázok 1). Hodnotenie bolesti pri Zomete bolo trvale pod východiskovou hodnotou a zmiernenie bolesti sprevádzala tendencia k poklesu analgetického skóre.

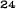
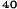
Obrázok 1. Priemerné zmeny skóre podľa BPI oproti východiskovej hodnote. Vyznačené sú štatisticky


významné rozdiely (*p<0,05) pri porovnaní spôsobov liečby (Zometa oproti placebu)
Placebo
∆
Zometa

Trvanie klinického skúšania (týždne)
Výsledky klinických skúšaní pri liečbe TIHKlinické skúšania pri hyperkalciémii vyvolanej nádorom (TIH) ukázali, že pre účinok kyseliny zoledrónovej je charakteristický pokles vápnika v sére a vylučovania vápnika močom. Vo fáze I sa v klinických skúšaniach na zistenie dávky u pacientov s miernou až stredne ťažkou hyperkalciémiou vyvolanou nádorom (TIH) skúšali účinné dávky v rozmedzí približne 1,2–2,5 mg.
Na stanovenie účinkov Zomety v porovnaní s 90 mg pamidronátu sa vo vopred plánovej analýze zlúčili výsledky dvoch pilotných multicentrických skúšaní s pacientmi s TIH. Normalizácia korigovaného vápnika v sére bola rýchlejšia na 4. deň pri Zomete 8 mg a na 7. deň pri Zomete 4 mg a 8 mg. Pozorovala sa
nasledujúca odpoveď na liečbu.
Tabuľka 5: Podiel pacientov s úplnou odpoveďou na liečbu podľa dní v zlúčených klinických skúšaniach
TIH
| 4. deň
| 7. deň
| 10. deň
|
Zometa 4 mg (N=86)
| 45,3% (p=0,104)
| 82,6% (p=0,005)*
| 88,4% (p=0,002)*
|
Zometa 8 mg (N=90)
| 55,6% (p=0,021)*
| 83,3% (p=0,010)*
| 86,7% (p=0,015)*
|
Pamidronát 90 mg (N=99)
| 33,3%
| 63,6%
| 69,7%
|
*Hodnoty p v porovnaní s pamidronátom
|
Medián času do dosiahnutia normokalciémie bol 4 dni. Medián času do relapsu (opätovné stúpnutie sérového
vápnika korigovaného podľa albumínu na ≥ 2,9 mmol/l) bol 30 až 40 dní u pacientov liečených Zometou oproti 17 dňom u pacientov liečených 90 mg pamidronátu (hodnoty p: 0,001 pre 4 mg a 0,007 pre 8 mg). Medzi oboma dávkami Zomety neboli štatisticky významné rozdiely.
V klinických skúšaniach bolo 69 pacientov s relapsom alebo bez odpovede na začiatočnú liečbu (Zometa
4 mg, 8 mg alebo pamidronát 90 mg) opäť liečených Zometou 8 mg. Podiel takýchto pacientov, u ktorých sa dosiahla odpoveď, bol asi 52%. Pretože títo pacienti boli liečení len dávkou 8 mg, nie sú údaje, ktoré by umožnili porovnanie s dávkou 4 mg.
V klinických skúšaniach s pacientmi s hyperkalciémiou vyvolanou nádorom (TIH) bol celkový profil bezpečnosti u všetkých troch skupín liečby (kyselina zoledrónová 4 mg a 8 mg a pamidronát 90 mg) podobný čo do typu a závažnosti.
5.2 Farmakokinetické vlastnostiPri jednorazových a opakovaných infúziách trvajúcich 5 a 15 minút, ktorými sa podalo 2, 4, 8 a 16 mg kyseliny zoledrónovej 64 pacientom s metastázami v kostiach, sa zistili nasledujúce farmakokinetické údaje, ktoré nezáviseli od dávky.
Po začatí infúzie kyseliny zoledrónovej sa plazmatické koncentrácie liečiva rýchlo zvýšili a dosiahli maximum na konci podania infúzie, po ktorom nasledoval rýchly pokles na < 10% maxima po 4 hodinách a
< 1% maxima po 24 hodinách, s následným dlhým obdobím veľmi nízkych koncentrácií nepresahujúcich
0,1% maxima pred druhou infúziou lieku na 28. deň.
Intravenózne podaná kyselina zoledrónová sa eliminuje trojfázovým procesom: rýchle dvojfázové vymiznutie zo systémového obehu s polčasmi t1/2α 0,24 a t1/2β 1,87 hodiny, po ktorých nasleduje dlhá eliminačná fáza s konečným polčasom eliminácie t1/2γ 146 hodín. Po opakovanom podávaní lieku každých
28 dní nedošlo k akumulácii liečiva v plazme. Kyselina zoledrónová sa nemetabolizuje a vylučuje sa
nezmenená obličkami. Počas prvých 24 hodín sa v moči nájde 39 ± 16% podanej dávky, zatiaľ čo zvyšok sa v zásade viaže na tkanivo kostí. Z kostného tkaniva sa veľmi pomaly uvoľňuje späť do systémového obehu a vylučuje sa obličkami. Celkový telesný klírens je 5,04 ± 2,5 l/hod, a to nezávisle od dávky a bez ovplyvnenia pohlavím, vekom, rasou a telesnou hmotnosťou. Predĺženie infúzie z 5 na 15 minút znížilo koncentráciu kyseliny zoledrónovej na konci infúzie o 30%, ale neovplyvnilo plochu pod krivkou koncentrácie oproti
času.
Tak ako aj pri iných bisfosfonátoch, variabilita farmakokinetických parametrov kyseliny zoledrónovej medzi pacientmi bola vysoká.
Nie sú dostupné farmakokinetické údaje o kyseline zoledrónovej u pacientov s hyperkalciémiou alebo u pacientov s insuficienciou pečene. Kyselina zoledrónová neinhibuje ľudské enzýmy P450
in vitro, nevykazuje biotransformáciu a v štúdiách na zvieratách sa v stolici našli < 3% podanej dávky, čo naznačuje, že funkcia pečene nezohráva významnú úlohu vo farmakokinetike kyseliny zoledrónovej.
Pri určení vzťahu medzi obličkovým klírensom kyseliny zoledrónovej a klírensom kreatinínu predstavoval obličkový klírens 75 ± 33% klírensu kreatinínu, ktorého priemerná hodnota bola 84 ± 29 ml/min (rozmedzie
22–143 ml/min) u 64 sledovaných pacientov s karcinómami. Analýza v tejto skupine ukázala, že u pacienta s klírensom kreatinínu 20 ml/min (ťažké poškodenie funkcie obličiek) alebo 50 ml/min (stredne ťažké poškodenie) bude zodpovedajúci predpokladaný klírens kyseliny zoledrónovej 37% alebo 72% hodnoty pacienta, ktorý má klírens kreatinínu 84 ml/min. Sú dostupné len obmedzené farmakokinetické údaje o pacientoch s ťažkou insuficienciou obličiek (klírens kreatinínu < 30 ml/min).
Kyselina zoledrónová nemá afinitu ku krvinkám a jej väzba na bielkoviny plazmy je nízka (približne 56%) a nezávisí od koncentrácie kyseliny zoledrónovej.
5.3 Predklinické údaje o bezpečnosti
Akútna toxicita
Najvyššia neletálna jednorazová intravenózna dávka bola 10 mg/kg telesnej hmotnosti u myší a 0,6 mg/kg u potkanov.
Subchronická a chronická toxicita
Kyselina zoledrónová sa dobre znášala, keď sa podávala subkutánne potkanom a intravenózne psom v dávkach do 0,02 mg/kg denne počas 4 týždňov. Podávanie 0,001 mg/kg/deň subkutánne potkanom a
0,005 mg/kg intravenózne psom raz za 2–3 dni počas až 52 týždňov sa tiež dobre znášalo.
Najčastejším nálezom v štúdiách pri opakovanom podávaní bolo zväčšenie primárnej trabekulárnej časti kosti v metafýzach dlhých kostí u rastúcich zvierat pri takmer všetkých dávkach, čo bol nález v súlade s farmakologickou antiresorpčnou účinnosťou látky.
Bezpečné rozmedzie dávok vzhľadom na účinky na obličky bolo úzke v štúdiách pri dlhodobom opakovanom parenterálnom podávaní zvieratám, ale kumulatívne hladiny bez nežiaducich príhod v štúdiách pri jednorazovom podaní (1,6 mg/kg) a pri opakovanom podávaní až do jedného mesiaca (0,06–
0,6 mg/kg/deň) nenaznačili účinky na obličky pri dávkach ekvivalentných alebo prevyšujúcich najvyššiu plánovanú terapeutickú dávku u ľudí. Dlhodobejšie opakované podávanie v dávkach, ktoré zahrnulo aj najvyššiu plánovanú terapeutickú dávku kyseliny zoledrónovej u ľudí, vyvolalo toxické účinky v iných orgánoch vrátane gastrointestinálneho traktu, pečene, sleziny a pľúc, ako aj v mieste podania intravenóznej injekcie.
Reprodukčná toxicita
Kyselina zoledrónová bola teratogénna u potkanov v subkutánnych dávkach ≥ 0,2 mg/kg. Hoci sa u králikov nepozorovala žiadna teratogenita alebo fetotoxicita, zistila sa toxicita pre matky.
Mutagenita a karcinogenita
Kyselina zoledrónová nebola mutagénna vo vykonaných testoch mutagenity a testy na karcinogenitu neposkytli žiadne dôkazy o karcinogénnom potenciále.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Manitol Citrónan sodný Voda na injekciu
6.2 Inkompatibility
Aby sa zabránilo prípadným inkompatibilitám, Zometa infúzny koncentrát sa má zriediť 0,9% m/V roztokom chloridu sodného alebo 5% m/V roztokom glukózy.
Zometa infúzny koncentrát sa nesmie miešať s roztokmi obsahujúcimi vápnik, napr. Ringerovým roztokom.
Skúšania so sklenenými fľašami, ako aj s niekoľkými druhmi infúznych vakov a infúznych súprav z polyvinylchloridu, polyetylénu a polypropylénu (predtým naplnenými 0,9% m/V roztokom chloridu sodného alebo 5% m/V roztokom glukózy) neukázali žiadne inkompatibility so Zometou.
6.3 Čas použiteľnosti
3 roky.
Roztok Zomety je stály počas 24 hodín pri teplote 2°C – 8°C po ďalšom zriedení 100 ml fyziologického roztoku chloridu sodného alebo 5% m/V roztoku glukózy
6.4 Špeciálne upozornenia na uchovávanie
Žiadne zvláštne upozornenia na uchovávanie.
Po aseptickom zriedení je najvhodnejšie okamžite použiť zriedený liek. Ak sa nepoužije okamžite, za trvanie a podmienky uchovávania pred použitím zodpovedá používateľ. Celkový čas medzi zriedením, uchovávaním v chladničke pri teplote 2°C – 8°C a ukončením podania nesmie byť dlhší ako 24 hodín.
6.5 Druh obalu a obsah balenia
Zometa infúzny koncentrát 4 mg/5 ml sa dodáva v baleniach, ktoré obsahujú 1, 4 alebo 10 injekčných liekoviek. Nie všetky veľkosti balenia musia byť uvedené do obehu.
Injekčná liekovka: plastová liekovka z číreho, bezfarebného cykloalkénového kopolyméru s objemom 5 ml, so zátkou z bromobutylovej gumy pokrytou vrstvou fluoropolyméru a hliníkovým uzáverom s plastovým strhávacím krytom.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pred podaním sa 5,0 ml koncentrátu z jednej injekčnej liekovky alebo objem koncentrátu odobratý podľa potreby musí ďalej zriediť 100 ml infúzneho roztoku, ktorý neobsahuje vápnik (0,9% m/V roztok chloridu sodného alebo 5% m/V roztok glukózy). Ak sa roztok uchováva v chladničke, pred podaním sa musí nechať zohriať na izbovú teplotu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB Veľká Británia
8. REGISTRAČNÉ ČÍSLO
EU/1/01/176/004-006
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 24.03.2003
Dátum prvého predĺženia registrácie: 20.03.2006
10. DÁTUM REVÍZIE TEXTU