
pravou alebo bez úpravy dávky Zejuly
(pozri časť 4.2). Liečba Zejulou sa má ukončiť v prípade hypertenznej krízy alebo ak sa medicínsky signifikantná hypertenzia nedá adekvátne kontrolovať antihypertenznou liečbou.
Syndrómreverzibilnejposteriórnejencefalopatie(Posterior reversible encephalopathysyndrome,PRES)
U pacientok, ktoré užívali Zejulu, boli hlásené prípady PRES (pozri časť 4.8). PRES je zriedkavá,
reverzibilná, neurologická porucha, ktorá sa môže prejavovať rýchlo sa rozvíjajúcimi príznakmi zahŕňajúcimi záchvaty kŕčov, bolesť hlavy, zmenený duševný stav, poruchu videnia alebo kortikálnu slepotu, so sprievodnou hypertenziou alebo bez nej. Na potvrdenie diagnózy PRES sa vyžaduje zobrazovacie vyšetrenie mozgu, prednostne vyšetrenie magnetickou rezonanciou (MR).
V prípade PRES sa odporúča ukončenie liečby Zejulou a liečba špecifických príznakov vrátane hypertenzie. Bezpečnosť opätovného začatia liečby Zejulou u pacientok, u ktorých sa v minulosti vyskytol PRES, nie je známa.
Tehotenstvo/antikoncepcia
Zejula sa nemá používať počas tehotenstva ani u žien vo fertilnom veku, ktoré nie sú ochotné používať
vysokoúčinnú antikoncepciu počas liečby a 6 mesiacov po užití poslednej dávky Zejuly (pozri časť 4.6). Pred začiatkom liečby majú všetky ženy vo fertilnom veku absolvovať tehotenský test.
Porucha funkcie pečene
Pacientky so závažnou poruchou funkcie pečene môžu mať zvýšenú expozíciu niraparibu,
vychádzajúc z údajov získaných u pacientok so stredne závažnou poruchou funkcie pečene, a majú byť pozorne sledované (pozri časti 4.2 a 5.2).
Laktóza
Tvrdé kapsuly Zejuly obsahujú monohydrát laktózy. Pacientky so zriedkavými dedičnými problémami
galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Tartrazín (E 102)
Tento liek obsahuje tartrazín (E 102), ktorý môže vyvolať alergické reakcie.
4.5 Liekové a iné interakcie
Farmakodynamické interakcie
Kombinácia niraparibu s vakcínami alebo imunosupresívnymi látkami nebola študovaná.
Údaje o niraparibe v kombinácii s cytotoxickými liekmi sú obmedzené. Niraparib sa má preto používať s opatrnosťou v kombinácii s vakcínami, imunosupresívnymi látkami alebo inými cytotoxickými liekmi.
Farmakokinetické interakcie
Vplyvinýchliekovnaniraparib
Niraparib ako substrát CYP (CYP1A2 a CYP3A4)
Niraparib je substrátom karboxylesteráz (carboxylesterases, CE) a UDP-glukuronozyltransferáz
(UGT) in vivo. Oxidatívny metabolizmus niraparibu je in vivo minimálny. Nie je potrebná úprava dávky Zejuly, keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú (napr. itrakonazol, ritonavir a klaritromycín) alebo indukujú CYP enzýmy (napr. rifampin, karbamazepín a fenytoín).
Niraparib ako substrát efluxných transportérov (P-gp, BCRP, BSEP, MRP2 a MATE1/2)
Niraparib je substrátom P-glykoproteínu (P-gp) a proteínu zodpovedného za rezistenciu pri rakovine prsníka (Breast Cancer Resistance Protein, BCRP). Vzhľadom na jeho vysokú permeabilitu a biologickú dostupnosť je však riziko jeho klinicky významných interakcií s liekmi, ktoré inhibujú tieto transportéry, nepravdepodobné. Nevyžaduje sa preto žiadna úprava dávky Zejuly, keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú P-gp (napr. amiodarón, verapamil) alebo BCRP (napr. osimertinib, velpatasvir a eltrombopag).
Niraparib nie je substrátom exportnej pumpy solí žlčových kyselín (bile salt export pump, BSEP) ani proteínu 2 súvisiaceho s mnohopočetnou liekovou rezistenciou (multidrug resistance-associated protein, MRP2). Hlavný primárny metabolit M1 nie je substrátom P-gp, BCRP, BSEP ani MRP2. Niraparib nie je substrátom proteínov extrúzie viacerých liekov a toxínov (multidrug and toxin extrusion proteins, MATE)-1 alebo 2, zatiaľ čo M1 je substrátom oboch.
Niraparib ako substrát transportérov hepatálneho vychytávania (OATP1B1, OATP1B3 a OCT1)
Ani niraparib, ani M1 nie je substrátom transportného polypeptidu organických aniónov 1B1 (organic anion transport polypeptide 1B1, OATP1B1), 1B3 (OATP1B3) alebo transportéra organických
katiónov 1 (organic cation transporter 1, OCT1). Nevyžaduje sa preto žiadna úprava dávky Zejuly,
keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú transportéry vychytávania OATP1B1
alebo 1B3 (napr. gemfibrozil, ritonavir) alebo OCT1 (napr. dolutegravir).
N
ir
aparib ako substrát transportérov renálneho vychytávania (OAT1, OAT3 a OCT2)
Ani niraparib, ani M1 nie je substrátom transportéra organických aniónov 1 (OAT1), 3 (OAT3)
a transportéra organických katiónov 2 (OCT2). Nevyžaduje sa preto žiadna úprava dávky Zejuly, keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú transportéry vychytávania OAT1
(napr. probenecid) alebo OAT3 (napr. probenecid, diklofenak) alebo OCT2 (napr. cimetidín, chinidín).
Vplyv niraparibu nainélieky
Inhibícia CYP (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4)
Ani niraparib, ani M1 nie je inhibítorom enzýmov CYP metabolizujúcich liečivá, konkrétne
CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4/5.
Hoci sa neočakáva inhibícia CYP3A4 v pečeni, ešte sa nestanovil potenciál relevantných koncentrácií niraparibu inhibovať CYP3A4 v tenkom čreve. Preto sa odporúča byť opatrný, keď sa niraparib
podáva v kombinácii s liečivami, ktorých metabolizmus je závislý od CYP3A4, a to najmä s liečivami,
ktoré majú úzke terapeutické rozmedzie (napr. cyklosporín, takrolimus, alfentanil, ergotamín, pimozid, kvetiapín a halofantrín).
Inhibícia UDP-glukuronozyltransferáz (UGT)
V podmienkach in vitro niraparib nevykazoval inhibičný účinok na izoformy UGT (UGT1A1,
UGT1A4, UGT1A9 a UGT2B7) až do koncentrácie 200 µmol/l. Možnosť klinicky významnej inhibície UGT niraparibom je preto minimálna.
Indukcia CYP (CYP1A2 a CYP3A4)
Ani niraparib, ani M1 nie je in vitro induktorom CYP3A4. V podmienkach in vitro niraparib podávaný raz týždenne indukuje CYP1A2 pri vysokých koncentráciách a klinický význam tohto účinku sa nedá
úplne vylúčiť. M1 nie je induktorom CYP1A2. Preto sa odporúča byť opatrný, keď sa niraparib
podáva v kombinácii s liečivami, ktorých metabolizmus je závislý od CYP1A2, a to najmä s liečivami, ktoré majú úzke terapeutické rozmedzie (napr. klozapín, teofylín a ropinirol).
Inhibícia efluxných transportérov (P-gp, BCRP, BSEP, MRP2 a MATE1/2)
Niraparib nie je inhibítorom BSEP ani MRP2. In vitro niraparib inhibuje P-gp veľmi slabo a BCRP s
IC50 = 161 µmol/l pre Pg-p a 5,8 µmol/l pre BCRP. Nedá sa preto vylúčiť klinicky významná interakcia súvisiaca s inhibíciou týchto efluxných transportérov, hoci je nepravdepodobná. Odporúča
sa preto byť opatrný, keď sa niraparib podáva v kombinácii so substrátmi BCRP (irinotekan, rosuvastatín, simvastatín, atorvastatín a metotrexát).
Niraparib je inhibítorom MATE1 s IC50 0,18 µmol/l a inhibítorom MATE2 s IC50 ≤ 0,14 µmol/l. Nedá sa vylúčiť zvýšenie plazmatických koncentrácií súbežne podávaných liekov, ktoré sú substrátmi týchto transportérov (napr. metformínu).
Hlavný primárny metabolit M1 sa nejaví ako inhibítor P-gp, BCRP, BSEP, MRP2 alebo MATE1/2.
Inhibícia transportérov hepatálneho vychytávania (OATP1B1, OATP1B3 a OCT1)
Ani niraparib, ani M1 nie je inhibítorom transportného polypeptidu organických aniónov 1B1 (OATP1B1) alebo 1B3 (OATP1B3).
In vitro niraparib podávaný raz týždenne inhibuje transportér organických katiónov 1 (OCT1)
s IC50 = 34,4 µmol/l. Odporúča sa byť opatrný, keď sa niraparib podáva v kombinácii s liečivami, ktorých vychytávanie je sprostredkované transportérom OCT1, napríklad s metformínom.
Inhibícia transportérov renálneho vychytávania (OAT1, OAT3 a OCT2)
Ani niraparib, ani M1 neinhibujú transportér organických aniónov 1 (OAT1), 3 (OAT3) a transportér organických katiónov 2 (OCT2).
Všetky klinické štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ž
eny vofertilnomveku/antikoncepciaužien
Ženy vo fertilnom veku nemajú otehotnieť počas liečby a nemajú byť tehotné na začiatku liečby. Pred
začiatkom liečby majú všetky ženy vo fertilnom veku absolvovať tehotenský test. Ženy vo fertilnom veku musia používať vysokoúčinnú antikoncepciu počas liečby a 6 mesiacov po užití poslednej dávky Zejuly.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití niraparibu u gravidných žien.
Neuskutočnili sa štúdie reprodukčnej a vývojovej toxicity na zvieratách. Na základe mechanizmu účinku však niraparib môže spôsobovať poškodenie embrya alebo plodu, vrátane embryoletálnych a
teratogénnych účinkov, pri podávaní tehotným ženám. Zejula sa nemá užívať počas gravidity.
Dojčenie
Nie je známe, či sa niraparib alebo jeho metabolity vylučujú do ľudského mlieka. Dojčenie je počas
podávania Zejuly a 1 mesiac po poslednej dávke kontraindikované (pozri časť 4.3).
Fertilita
Nie sú dostupné žiadne klinické údaje o fertilite. U potkanov a psov sa pozorovala reverzibilná
redukcia spermatogenézy (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Zejula má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientok užívajúcich
Zejulu môže dôjsť k asténii, únave, závratom alebo k ťažkostiam s koncentráciou. Pacientky,
u ktorých dôjde k týmto príznakom, majú byť opatrné pri vedení vozidiel alebo obsluhovaní strojov.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
ADR všetkých stupňov závažnosti, ktoré sa vyskytli u ≥ 10 % z 851 pacientok užívajúcich Zejulu
v monoterapii v súhrnne hodnotených klinických skúšaniach PRIMA (buď 200 mg alebo 300 mg začiatočná dávka) a NOVA, boli nauzea, anémia, trombocytopénia, únava, zápcha, vracanie, bolesť hlavy, nespavosť, znížený počet trombocytov, neutropénia, bolesti brucha, znížená chuť do jedla, hnačka, dyspnoe, hypertenzia, asténia, závraty, znížený počet neutrofilov, kašeľ, artralgia, bolesti chrbta, znížený počet leukocytov a návaly tepla.
Najčastejšími závažnými nežiaducimi reakciami, u > 1 % (frekvencie výskytu počas liečby), boli trombocytopénia a anémia.
Tabuľkový zoznamnežiaducichreakcií
Nasledujúce nežiaduce reakcie boli identifikované na základe klinických skúšaní a sledovania
po uvedení lieku na trh u pacientok užívajúcich Zejulu v monoterapii (pozri tabuľku 4). Frekvencie výskytu nežiaducich účinkov sú založené na súhrnných údajoch o nežiaducich udalostiach získaných
v štúdiách PRIMA a NOVA (fixná začiatočná dávka 300 mg/deň) u pacientok so známou expozíciou
a sú definované nasledujúcim spôsobom: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); a veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
T
abuľka 4: Tabuľkový zoznam nežiaducich reakcií
T
rieda orgánových systémov
|
F
rekvencia nežiaducich reakcií všetkých stupňov závažnosti podľa CTCAE*
|
F
rekvencia nežiaducich reakcií 3. alebo
4. stupňa závažnosti podľa CTCAE*
|
Infekcie a nákazy
|
V
eľmi časté
Infekcia močových ciest
Časté
Bronchitída, konjunktivitída
|
Menej časté
Infekcia močových ciest, bronchitída
|
Benígne a malígne nádory, vrátane nešpecifikovaných
novotvarov (cysty a polypy)
|
Č
asté
Myelodysplastický syndróm/akútna myeloidná
leukémia**
|
Č
asté Myelodysplastický syndróm/akútna
myeloidná leukémia**
|
Poruchy krvi a lymfatického systému
|
V
eľmi časté
Trombocytopénia, anémia, neutropénia, leukopénia
Menej časté
Pancytopénia, febrilná neutropénia
|
V
eľmi časté
Trombocytopénia, anémia, neutropénia
Časté
Leukopénia Menej časté Pancytopénia, febrilná neutropénia
|
Poruchy imunitného
systému
|
Č
asté
Precitlivenosť†
|
Menej časté
Precitlivenosť
|
Poruchy metabolizmu
a výživy
|
V
eľmi časté
Znížená chuť do jedla
Časté
Hypokaliémia
|
Č
asté
Hypokaliémia
Menej časté
Znížená chuť do jedla
|
Psychické poruchy
|
V
eľmi časté
Nespavosť
Časté
Úzkosť, depresia,
porucha kognitívnych funkci톆
Menej časté
Stav zmätenosti
|
Menej časté
Nespavosť, úzkosť, depresia, stav zmätenosti
|
Poruchy nervového
systému
|
V
eľmi časté
Bolesť hlavy, závraty
Časté
Dysgeúzia
Zriedkavé
Syndróm reverzibilnej posteriórnej encefalopatie (Posterior reversible
encephalopathy syndrome, PRES)**
|
Menej časté
Bolesť hlavy
|
Poruchy srdca a srdcovej činnosti
|
V
eľmi časté Palpitácie Časté Tachykardia
|
|
Poruchy ciev
|
V
eľmi časté
Hypertenzia Zriedkavé Hypertenzná kríza
|
Č
asté
Hypertenzia
|
Poruchy dýchacej
sústavy, hrudníka a mediastínia
|
V
eľmi časté
Dyspnoe, kašeľ, nazofaryngitída
Časté
Epistaxa
Menej časté
Pneumonitída
|
Menej časté
Dyspnoe, epistaxa, pneumonitída
|
T
rieda orgánových systémov
|
F
rekvencia nežiaducich reakcií všetkých stupňov závažnosti podľa CTCAE*
|
F
rekvencia nežiaducich reakcií 3. alebo
4. stupňa závažnosti
podľa CTCAE*
|
Poruchy
gastrointestinálneho
traktu
|
V
eľmi časté
Nauzea, zápcha, vracanie, bolesti brucha, hnačka, dyspepsia
Časté
Sucho v ústach, brušná distenzia, zápal sliznice, stomatitída
|
Č
asté
Nauzea, vracanie, bolesti brucha
Menej časté
Hnačka, zápcha, zápal sliznice, stomatitída,
sucho v ústach
|
Poruchy kože a
podkožného tkaniva
|
Č
asté
Fotosenzitivita, vyrážka
|
Menej časté
Fotosenzitivita, vyrážka
|
Poruchy kostrovej a
svalovej sústavy a spojivového tkaniva
|
V
eľmi časté
Bolesti chrbta, artralgia
Časté
Myalgia
|
Menej časté
Bolesti chrbta, artralgia, myalgia
|
Celkové poruchy a
reakcie v mieste podania
|
V
eľmi časté
Únava, asténia
Časté
Periférny edém
|
Č
asté
Únava, asténia
|
Laboratórne a funkčné vyšetrenia
|
Č
asté
Zvýšená hladina gamaglutamyltransferázy, zvýšená hladina AST, zvýšená hladina
kreatinínu v krvi, zvýšená hladina ALT, zvýšená
hladina alkalickej fosfatázy v krvi, znížená hmotnosť
|
Č
asté
Zvýšená hladina gamaglutamyltransferázy,
zvýšená hladina ALT
Menej časté
Zvýšená hladina AST, zvýšená hladina
alkalickej fosfatázy v krvi
|
* CTCAE = Všeobecné terminologické kritériá pre nežiaduce účinky („Common Terminology Criteria
for Adverse Events“), verzia 4.02.
** Na základe údajov z klinických skúšaní s niraparibom. Neobmedzuje sa to na pivotnú štúdiu
ENGOT-OV16 s monoterapiou.
† Zahŕňa precitlivenosť, liekovú precitlivenosť, anafylaktoidnú reakciu, liekovú erupciu, angioedém
a urtikáriu.
†† Zahŕňa poruchu pamäti, poruchu koncentrácie.
Frekvencia výskytu nežiaducich reakcií pozorovaných v skupine pacientok, ktorým bola podávaná
200 mg začiatočná dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, bola podobná alebo nižšia v porovnaní so skupinou pacientok, ktorým bola
podávaná fixná začiatočná dávka 300 mg (tabuľka 4).
Špecifické informácie týkajúce sa frekvencie výskytu trombocytopénie, anémie a neutropénie, pozri nižšie.
Opis vybraných nežiaducichreakciíHematologické nežiaduce reakcie (trombocytopénia, anémia, neutropénia), vrátane klinických diagnóz
a/alebo laboratórnych nálezov, vo všeobecnosti nastávajú v skorom štádiu liečby niraparibom, pričom ich incidencia sa postupom času znižuje.
V štúdiách NOVA a PRIMA mali pacientky vhodné na liečbu Zejulou nasledujúce východiskové hematologické parametre: absolútny počet neutrofilov (absolute neutrophil count, ANC)
≥ 1 500 buniek/µl; počet trombocytov ≥ 100 000 buniek/µl a hladina hemoglobínu ≥ 9 g/dl (NOVA)
alebo ≥ 10 g/dl (PRIMA) pred začiatkom liečby. V klinickom programe sa hematologické nežiaduce reakcie manažovali prostredníctvom laboratórneho monitoringu a úprav dávky (pozri časť 4.2).
V štúdii PRIMA bol u pacientok, ktorým bola podávaná začiatočná dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, výskyt trombocytopénie, anémie a neutropénie 3. stupňa, v uvedenom poradí, znížený zo 48 % na 21 %, z 36 % na 23 %
a z 24 % na 15 % v porovnaní so skupinou pacientok, ktorým bola podávaná fixná začiatočná dávka
300 mg. K ukončeniu liečby v dôsledku trombocytopénie, anémie a neutropénie došlo, v uvedenom poradí, u 3 %, 3 % a 2 % pacientok.
Trombocytopénia
V štúdii PRIMA sa trombocytopénia 3./4. stupňa vyskytla u 39 % pacientok liečených Zejulou
v porovnaní s 0,4 % pacientok liečených placebom, pričom medián času od podania prvej dávky
do prvého výskytu trombocytopénie bol 22 dní (rozmedzie: 15 až 335 dní) a medián jej trvania bol
6 dní (rozmedzie: 1 až 374 dní). K ukončeniu liečby v dôsledku trombocytopénie došlo
u 4 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 60 % pacientok užívajúcich Zejulu k trombocytopénii akéhokoľvek stupňa a u 34 % pacientok došlo k trombocytopénii 3. alebo 4. stupňa. U 76 % pacientok
s východiskovým počtom trombocytov nižším ako 180 × 109/l došlo k trombocytopénii akéhokoľvek stupňa a u 45 % takýchto pacientok došlo k trombocytopénii 3. alebo 4. stupňa. Medián času do
nástupu trombocytopénie akéhokoľvek stupňa bol 22 dní a medián času do nástupu trombocytopénie
3. alebo 4. stupňa bol 23 dní. Výskyt nových epizód trombocytopénie po intenzívnych modifikáciách dávky realizovaných počas prvých dvoch mesiacov liečby od 4. cyklu bol 1,2 %. Medián trvania
epizód trombocytopénie akéhokoľvek stupňa bol 23 dní a medián trvania trombocytopénie 3. alebo
4. stupňa bol 10 dní. Pacientky liečené Zejulou, u ktorých sa vyvinie trombocytopénia, môžu mať zvýšené riziko krvácania. V klinickom programe sa trombocytopénia manažovala laboratórnym
monitoringom, modifikáciou dávky a, ak to bolo potrebné, transfúziou krvných doštičiek (pozri
časť 4.2). K ukončeniu liečby v dôsledku epizód trombocytopénie (trombocytopénia a zníženie počtu trombocytov) došlo u približne 3 % pacientok.
V štúdii NOVA došlo u 48 z 367 (13 %) pacientok ku krvácaniu so súbežnou trombocytopéniou. Všetky krvácavé epizódy, ku ktorým došlo súbežne s trombocytopéniou, mali závažnosť
1. alebo 2. stupňa s výnimkou jedného prípadu petechie a hematómu 3. stupňa, ktorý bol pozorovaný súbežne so závažnou nežiaducou reakciou, pancytopéniou. K trombocytopénii dochádzalo častejšie
u pacientok, ktorých východiskový počet trombocytov bol nižší ako 180 x 109/l. U približne
76 % pacientok s nižším východiskovým počtom trombocytov (< 180 x 109/l), ktoré dostávali Zejulu, došlo k trombocytopénii akéhokoľvek stupňa a u 45 % pacientok došlo k trombocytopénii
3. alebo 4. stupňa. Pancytopénia sa pozorovala u < 1 % pacientok užívajúcich niraparib.
Anémia
V štúdii PRIMA sa anémia 3./4. stupňa vyskytla u 31 % pacientok liečených Zejulou v porovnaní
s 2 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu anémie bol 80 dní (rozmedzie: 15 až 533 dní) a medián jej trvania bol 7 dní (rozmedzie: 1 až 119 dní).
K ukončeniu liečby v dôsledku anémie došlo u 2 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 50 % pacientok k anémii akéhokoľvek stupňa a u 25 % pacientok došlo k anémii 3. alebo 4. stupňa. Medián času do nástupu anémie akéhokoľvek stupňa bol 42 dní a do anémie 3. alebo 4. stupňa to bolo 85 dní. Medián trvania anémie akéhokoľvek stupňa bol 63 dní a anémie 3. alebo 4. stupňa 8 dní. Anémia akéhokoľvek stupňa môže počas liečby Zejulou pretrvávať. V klinickom programe sa anémia manažovala prostredníctvom laboratórneho monitoringu, modifikácie dávky (pozri časť 4.2) a v prípade potreby transfúziami červených krviniek. K ukončeniu liečby v dôsledku anémie došlo u 1 % pacientok.
N
eutropénia
V štúdii PRIMA sa neutropénia 3./4. stupňa vyskytla u 21 % pacientok liečených Zejulou v porovnaní s 1 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu neutropénie bol 29 dní (rozmedzie: 15 až 421 dní) a medián jej trvania bol 8 dní (rozmedzie:
1 až 42 dní). K ukončeniu liečby v dôsledku neutropénie došlo u 2 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 30 % pacientok užívajúcich Zejulu k neutropénii akéhokoľvek stupňa a u 20 % pacientok došlo k neutropénii 3. alebo 4. stupňa. Medián času do nástupu neutropénie akéhokoľvek stupňa bol 27 dní a neutropénie 3. alebo 4. stupňa 29 dní. Medián času trvania neutropénie akéhokoľvek stupňa bol 26 dní a neutropénie 3. alebo 4. stupňa 13 dní. Okrem toho sa približne 6 % pacientok liečených niraparibom podával faktor stimulujúci kolónie granulocytov (Granulocyte Colony Stimulating Factor, G-CSF) ako súbežná liečba neutropénie. K ukončeniu liečby v dôsledku epizód neutropénie došlo u 2 % pacientok.
Myelodysplastický syndróm/akútna myeloidná leukémiaV klinických štúdiách sa MDS/AML vyskytovali u 1 % pacientok liečených Zejulou, pričom
41 % prípadov malo smrteľné následky. Výskyt bol vyšší u pacientok s recidivujúcim karcinómom vaječníka, ktoré predtým dostali 2 alebo viac línií chemoterapie na báze platiny, a s
gBRCAmut po 75-
mesačnom sledovaní prežívania. Všetky pacientky mali faktory potenciálne prispievajúce k rozvoju
MDS/AML v dôsledku predošlej chemoterapie na báze platiny. Veľa pacientok tiež dostalo iné látky poškodzujúce DNA a rádioterapiu.Väčšina hlásení bola u nositeliek
gBRCAmut. Niektoré pacientky
mali v anamnéze predchádzajúcu rakovinu alebo supresiu kostnej drene.
V štúdii PRIMA bol výskyt MDS/AML u 0,8 % pacientok, ktoré dostávali Zejulu, a u 0,4 % pacientok, ktoré dostávali placebo.
V štúdii NOVA u pacientok s recidivujúcim karcinómom vaječníka, ktoré predtým dostali aspoň dve línie chemoterapie na báze platiny, bol celkový výskyt MDS/AML 3,8 % u pacientok, ktoré dostávali Zejulu, a 1,7 % u pacientok, ktoré dostávali placebo, po 75-mesačnom sledovaní prežívania. Výskyt MDS/AML bol 7,4 % v kohorte g
BRCAmut a 1,7 % v kohorte non-g
BRCAmut u pacientok, ktoré dostávali Zejulu, a 3,1 % v kohorte g
BRCAmut a 0,9 % v kohorte non-g
BRCAmut u pacientok, ktoré dostávali placebo.
HypertenziaV štúdii PRIMA sa hypertenzia 3./4. stupňa vyskytla u 6 % pacientok liečených Zejulou v porovnaní
s 1 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu hypertenzie bol 50 dní (rozmedzie: 1 až 589 dní) a medián jej trvania bol 12 dní (rozmedzie:
1 až 61 dní). K ukončeniu liečby v dôsledku hypertenzie došlo u 0 % pacientok.
V štúdii NOVA sa hypertenzia akéhokoľvek stupňa vyskytla u 19,3 % pacientok liečených Zejulou. U 8,2 % pacientok sa vyskytla hypertenzia 3. alebo 4. stupňa. Hypertenzia sa dala jednoducho manažovať antihypertenzívami. K ukončeniu liečby v dôsledku hypertenzie došlo u < 1 % pacientok.
PediatrickápopuláciaU pediatrických pacientok sa neuskutočnili žiadne štúdie.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania Zejulou nie je k dispozícii žiadna špecifická liečba a neboli stanovené príznaky predávkovania. V prípade predávkovania majú lekári vykonať všeobecné podporné opatrenia a liečiť symptomaticky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, iné cytostatiká, ATC kód: L01XK02.
Mechanizmus účinku a farmakodynamické účinky
Niraparib je inhibítorom enzýmov nazývaných poly(ADP-ribóza) polymerázy (PARP), PARP-1
a PARP-2, ktoré hrajú rolu pri opravovaní DNA. In vitro štúdie dokázali, že niraparibom indukovaná cytotoxicita môže zahŕňať inhibíciu enzymatickej aktivity PARP a zvýšenú tvorbu komplexov
PARP-DNA, čo vedie k poškodeniu DNA, apoptóze a bunkovej smrti. Zvýšená niraparibom
indukovaná cytotoxicita sa pozorovala v nádorových bunkových líniách s deficienciou alebo bez deficiencie nádorových supresorových génov pre rakovinu prsníka (BReast CAncer, BRCA) 1 a 2. Na ortotopických xenograftových myších nádoroch (patient-derived xenograft, PDX) vzniknutých inokuláciou seróznym ľudským karcinómom vaječníka vysokého stupňa malignity sa ukázalo, že niraparib redukuje rast nádorov s mutáciami génov BRCA 1 a 2, nádorov s divokým (nezmutovaným) typom BRCA ale s deficienciou homologickej rekombinácie (HR) a nádorov s divokým typom BRCA a bez detegovateľnej deficiencie HR.
Klinická účinnosť abezpečnosť
Udržiavacialiečbaprvejlínieprikarcinómevaječníka
PRIMA bolo dvojito zaslepené, placebom kontrolované klinické skúšanie fázy 3, v ktorom boli pacientky (n = 733), u ktorých pretrvávala úplná alebo čiastočná odpoveď na prvú líniu chemoterapie
na báze platiny, randomizované 2:1 na podávanie niraparibu alebo zodpovedajúceho placeba.
V štúdii PRIMA sa liečba u 475 pacientok (317 z nich bolo randomizovaných do skupiny
s niraparibom a 158 do skupiny s placebom) začala 300 mg začiatočnou dávkou podávanou raz denne (QD) v rámci kontinuálnych 28-dňových cyklov. V štúdii PRIMA bola táto začiatočná dávka zmenená na základe 2. dodatku protokolu. Po jeho schválení bola pacientkam s východiskovou telesnou hmotnosťou ≥ 77 kg a s východiskovým počtom trombocytov ≥ 150 000/µl podávaná 300 mg dávka niraparibu (n = 34), alebo placeba raz denne (n = 21), zatiaľ čo pacientkam s východiskovou telesnou hmotnosťou < 77 kg alebo s východiskovým počtom trombocytov < 150 000/μl bola podávaná 200 mg dávka niraparibu (n = 122), alebo placeba raz denne (n = 61).
Pacientky boli randomizované po ukončení prvej línie chemoterapie na báze platiny plus/mínus chirurgický zákrok. Osoby boli randomizované do 12 týždňov od prvého dňa posledného cyklu chemoterapie. Osoby predtým dostali ≥ 6 a ≤ 9 cyklov liečby na báze platiny. Po odloženej (intervalovej) cytoredukčnej operácii dostali pacientky ≥ 2 pooperačné cykly liečby na báze platiny. Pacientky, ktoré predtým dostali bevacizumab spolu s chemoterapiou, ale nemohli dostávať bevacizumab ako udržiavaciu liečbu, neboli zo štúdie vylúčené. Pacientky nemohli predtým dostávať liečbu inhibítorom PARP (PARPi) vrátane niraparibu. Pacientky, ktoré podstúpili neoadjuvantnú chemoterapiu, po ktorej nasledovala odložená (intervalová) cytoredukčná operácia, mohli mať viditeľné reziduálne ochorenie alebo nemuseli mať žiadne reziduálne ochorenie. Pacientky
s ochorením v štádiu III, ktoré po primárnej cytoredukčnej operácii dosiahli kompletnú cytoredukciu (t. j. nemali žiadne viditeľné reziduálne ochorenie), boli zo štúdie vylúčené. Randomizácia bola stratifikovaná podľa najlepšej odpovede na liečbu dosiahnutej počas prvej línie chemoterapie na báze platiny (úplná odpoveď na liečbu vs čiastočná odpoveď na liečbu), podľa neoadjuvantnej chemoterapie (neoadjuvant chemotherapy, NACT) (áno vs nie); a podľa stavu deficiencie homologickej rekombinácie (homologous recombination deficiency, HRD) [pozitivita (deficiencia HR) vs negativita (proficiencia HR) alebo nestanovená]. Vyšetrenie na prítomnosť HRD sa vykonalo pomocou testu HRD na tumorovom tkanive získanom v čase iniciálnej diagnózy. Hladina CA-125 má byť v referenčnom rozpätí (alebo malo dôjsť k poklesu hladiny CA-125 o > 90 %) počas pacientkinej liečby prvej línie a mala byť stabilná počas aspoň 7 dní.
Pacientky začali liečbu 200 mg alebo 300 mg dávkou niraparibu alebo zodpovedajúcou dávkou
placeba v 1. cykle/1. deň (C1/D1) a užívali ich raz denne v rámci kontinuálnych 28-dňových cyklov. Pacientky sa dostavili na návštevu na klinike v každom cykle (každé 4 týždne ± 3 dni).
Primárnym koncovým ukazovateľom bolo prežívanie bez progresie ochorenia (progression-free survival, PFS) stanovené prostredníctvom zaslepeného nezávislého centrálneho hodnotenia (blinded independent central review, BICR) pomocou RECIST, verzia 1.1. Celkové prežívanie (overall survival, OS) bolo kľúčovým sekundárnym cieľom. Testovanie PFS sa vykonalo hierarchicky: najprv v populácii s deficienciou HR, potom v celkovej populácii. Medián veku bol 62 rokov a jeho rozmedzie bolo od 32 do 85 rokov u pacientok randomizovaných na niraparib a od 33 do 88 rokov
u pacientok randomizovaných na placebo. Osemdesiatdeväť percent všetkých pacientok tvorili belošky. Šesťdesiatdeväť percent pacientok randomizovaných na niraparib a 71 % pacientok
randomizovaných na placebo malo na začiatku štúdie výkonnostný stav ECOG 0. V celkovej populácii
malo 65 % pacientok ochorenie v štádiu III a 35 % pacientok malo ochorenie v štádiu IV. V celkovej populácii bol miestom primárneho nádoru u väčšiny pacientok (≥ 80 %) vaječník; väčšina pacientok
(> 90 %) mala serózny histologický podtyp nádoru. Šesťdesiatsedem percent pacientok podstúpilo
NACT. Šesťdesiatdeväť percent pacientok dosiahlo úplnú odpoveď na prvú líniu chemoterapie na báze platiny. Celkovo 6 pacientok liečených niraparibom dostalo bevacizumab ako predchádzajúcu liečbu karcinómu vaječníka.
V štúdii PRIMA sa preukázalo štatisticky významné zlepšenie PFS u pacientok randomizovaných na niraparib v porovnaní s placebom, a to v populácii s deficienciou HR a v celkovej populácii (tabuľka 5 a graf 1 a graf 2).
Sekundárne koncové ukazovatele účinnosti zahŕňali PFS po prvej nasledujúcej liečbe (PFS2) a OS (tabuľka 5).
Tabuľka 5: Výsledky účinnosti – PRIMA (stanovené prostredníctvom BICR)
| Populácia s deficienciou HR
| Celková populácia
|
niraparib
(N = 247)
| placebo
(N = 126)
| niraparib
(N = 487)
| placebo
(N = 246)
|
Medián PFS (95 % CI)
| 21,9 (19,3; NE)
| 10,4 (8,1; 12,1)
| 13,8 (11,5; 14,9)
| 8,2 (7,3; 8,5)
|
Pomer rizík (95 % CI)
| 0,43 (0,31; 0,59)
| 0,62 (0,50; 0,76)
|
p-hodnota
| < 0,0001
| < 0,0001
|
|
PFS2
Pomer rizík (95 % CI)
| 0,84 (0,485; 1,453)
| 0,81 (0,577; 1,139)
|
|
OS*
Pomer rizík (95 % CI)
| 0,61 (0,265; 1,388)
| 0,70 (0,44; 1,11)
|
PFS = prežívanie bez progresie ochorenia; CI = interval spoľahlivosti; NE = nehodnotiteľné; OS = celkové prežívanie; PFS2 = PFS po prvej nasledujúcej liečbe.
*V čase primárnej analýzy PFS bolo odhadované prežívanie po dvoch rokoch od randomizácie
84 % u pacientok užívajúcich Zejulu v porovnaní so 77 % u pacientok užívajúcich placebo v celkovej populácii.
Údaje o PFS2 a OS v súčasnosti nie sú konečné.
G
raf 1: Prežívanie bez progresie ochorenia u pacientok s nádormi s deficienciou HR – PRIMA (ITT populácia, N = 373)

Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Censored Observations: Cenzúrované pozorovania.
Graf 2: Prežívanie bez progresie ochorenia v celkovej populácii – PRIMA (ITT populácia,N = 733)
100
90
Censored Observations
Niraparib Placebo
80

HR (95% CI) 0.62 (0.502,0.755)
70
60
50
40

30
20
10
Niraparib 487
| 454
| 385
| 312
| 295
| 253
| 167
| 111
| 94
| 58
| 29
| 21
| 13
| 4
| 0
| Placebo 246
| 226
| 177
| 133
| 117
| 90
| 60
| 32
| 29
| 17
| 6
| 6
| 4
| 1
| 0
|
0
|
2
|
4
|
6
|
8
|
10
|
12
|
14
|
16
|
18
|
20
|
22
|
24
|
26
|
28
|
|
|
0
Time since Randomization (Months)
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od
randomizácie (mesiace); Censored Observations: Cenzúrované pozorovania.
A
nalýzy podskupín
V rámci populácie s deficienciou HR bol v podskupine pacientok s karcinómom vaječníka s BRCAmut
(N = 223) pozorovaný pomer rizík 0,40 (95 % CI: 0,27; 0,62). V podskupine pacientok s deficienciou
HR bez mutácie BRCA (N = 150) bol pozorovaný pomer rizík 0,50 (95 % CI: 0,31; 0,83). V populácii
s proficienciou HR (N = 249) bol pozorovaný pomer rizík 0,68 (95 % CI: 0,49; 0,94).
V exploračných analýzach podskupín bola u pacientok, ktorým bola podávaná 200 mg alebo 300 mg dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, pozorovaná porovnateľná účinnosť (PFS hodnotené skúšajúcim) s pomerom rizík 0,54 (95 % CI: 0,33;
0,91) v populácii s deficienciou HR a s pomerom rizík 0,68 (95 % CI: 0,49; 0,94) v celkovej populácii. V podskupine s proficienciou HR sa zdalo, že pri 200 mg dávke je efekt liečby nižší v porovnaní
s 300 mg dávkou.
Udržiavacialiečbarecidivujúcehokarcinómuvaječníkacitlivéhonaplatinu
Bezpečnosť a účinnosť niraparibu ako udržiavacej liečby boli skúmané v randomizovanom, dvojito zaslepenom, placebom kontrolovanom medzinárodnom klinickom skúšaní fázy 3 (NOVA) u pacientok
s recidivujúcim seróznym epitelovým karcinómom vaječníka, vajíčkovodu alebo primárnym peritoneálnym karcinómom, prevažne vysokého stupňa malignity (high-grade), ktoré boli senzitívne
na platinu, čo bolo definované úplnou odpoveďou na liečbu (complete response, CR) alebo čiastočnou
odpoveďou na liečbu (partial response, PR) pretrvávajúcou viac ako 6 mesiacov po predposlednej liečbe na báze platiny. Pacientky boli vhodné na liečbu niraparibom, ak u nich pretrvávala odpoveď na liečbu (CR alebo PR) po ukončení poslednej chemoterapie na báze platiny. Po ich poslednej liečbe na báze platiny mali mať hladiny CA-125 v referenčnom rozpätí (alebo o > 90 % nižšie v porovnaní
s východiskovou hladinou) a stabilné počas najmenej 7 dní. Pacientky nemohli dostať predchádzajúcu liečbu PARPi vrátane Zejuly. Vhodné pacientky boli zaradené do jednej z dvoch kohort na základe
výsledkov testu germinatívnych (zárodočných) mutácií génov BRCA (gBRCA). V rámci každej
kohorty boli pacientky randomizované v pomere 2:1 na podávanie niraparibu a placeba. Pacientky boli zaradené do kohorty gBRCAmut na základe vzoriek krvi na analýzu gBRCA, ktoré boli odobraté pred randomizáciou. Testovanie na tumorovú BRCA (tBRCA) mutáciu a HRD sa vykonalo pomocou testu HRD na tumorovom tkanive získanom v čase iniciálnej diagnózy alebo v čase recidívy.
Randomizácia v každej kohorte bola stratifikovaná podľa času do progresie ochorenia po predposlednej liečbe na báze platiny pred zaradením do štúdie (6 až < 12 mesiacov
a ≥ 12 mesiacov); podľa použitia alebo nepoužitia bevacizumabu v kombinácii s predposledným
alebo posledným režimom na báze platiny; a podľa najlepšej odpovede na liečbu dosiahnutej počas posledného režimu na báze platiny (úplná odpoveď a čiastočná odpoveď).
Pacientky začali liečbu niraparibom 300 mg alebo zodpovedajúcim placebom v 1. cykle/1. deň (C1/D1) a užívali ich raz denne v rámci kontinuálnych 28-dňových cyklov. Pacientky sa dostavili na návštevu na klinike v každom cykle (každé 4 týždne ± 3 dni).
V štúdii NOVA došlo u 48 % pacientok k prerušeniu podávania lieku v 1. cykle. U približne 47 %
pacientok sa liek znovu začal podávať v zníženej dávke v 2. cykle.
Najčastejšie používanou dávkou u pacientok liečených niraparibom v štúdii NOVA bolo 200 mg.
Prežívanie bez progresie ochorenia (progression-free survival, PFS) sa posudzovalo pomocou RECIST („Response Evaluation Criteria in Solid Tumors“ - Kritériá hodnotenia odpovede pri solídnych nádoroch, verzia 1.1) alebo pomocou klinických prejavov a príznakov a zvýšenej hladiny CA-125.
PFS sa hodnotilo od času randomizácie (ktorá sa realizovala do 8 týždňov od ukončenia chemoterapeutického režimu) do progresie ochorenia alebo do smrti.
Primárna analýza účinnosti z hľadiska PFS bola založená na zaslepenom centrálnom nezávislom hodnotení a bola prospektívne definovaná a vykonaná osobitne v kohorte gBRCAmut a v kohorte
non-gBRCAmut. Analýzy celkového prežívania (overall survival, OS) boli sekundárnymi výslednými ukazovateľmi.
Sekundárne koncové ukazovatele účinnosti zahŕňali interval bez chemoterapie (chemotherapy-free interval, CFI), čas do prvej nasledujúcej liečby (time to first subsequent therapy, TFST), PFS po prvej nasledujúcej liečbe (PFS2) a OS.
Demografické údaje, východiskové charakteristiky ochorenia a predchádzajúce línie liečby boli vo všeobecnosti dobre vyvážené medzi skupinou s niraparibom a skupinou s placebom v kohorte g
BRCAmut (n = 203) a kohorte nong
BRCAmut (n = 350). Medián veku bol v rozsahu od 57 do
63 rokov v liečebných skupinách a kohortách. Miestom primárneho nádoru bol u väčšiny pacientok
(> 80 %) v každej kohorte vaječník; väčšina pacientok (> 84 %) mala nádory so seróznou histológiou. Vysoké percento pacientok v oboch liečebných skupinách, v oboch kohortách, dostalo 3 alebo viacej
predchádzajúcich línií chemoterapie vrátane 49 % pacientok užívajúcich niraparib v kohorte
g
BRCAmut a 34 % pacientok užívajúcich niraparib v kohorte non-g
BRCAmut. Väčšina pacientok bola vo veku 18 až 64 rokov (78 %), kaukazskej rasy (86 %) a mala výkonnostný status ECOG rovný 0
(68 %).
V kohorte g
BRCAmut bol medián počtu cyklov liečby v skupine s niraparibom vyšší ako v skupine s placebom (14 cyklov a 7 cyklov, v uvedenom poradí). Viac pacientok v skupine s niraparibom pokračovalo v liečbe dlhšie ako 12 mesiacov než v skupine s placebom (54,4 % a 16,9 %, v uvedenom poradí). V celkovej kohorte non-g
BRCAmut bol medián počtu cyklov liečby v skupine s niraparibom vyšší ako v skupine s placebom (8 cyklov a 5 cyklov, v uvedenom poradí). Viac pacientok v skupine
s niraparibom pokračovalo v liečbe dlhšie ako 12 mesiacov než v skupine s placebom (34,2 % a
21,1 %, v uvedenom poradí).
Štúdia splnila svoj primárny cieľ, ktorým bolo štatisticky významné zlepšenie PFS pri udržiavacej liečbe niraparibom v monoterapii v porovnaní s placebom v kohorte g
BRCAmut, ako aj v celkovej kohorte non-g
BRCAmut. V tabuľke 6 a na grafoch 3 a 4 sú zobrazené výsledky týkajúce sa PFS, primárneho koncového ukazovateľa, v populácii pacientov pre primárnu analýzu účinnosti (kohorta g
BRCAmut a celková kohorta non-g
BRCAmut).
Tabuľka 6: Zhrnutie primárnych objektívnych výsledkov štúdie NOVA
| Kohorta gBRCAmut
| Kohorta non-gBRCAmut
|
niraparib
(N = 138)
| placebo
(N = 65)
| niraparib
(N = 234)
| placebo
(N = 116)
|
PFS medián (95 % CI)
| 21,0
(12,9; NE)
| 5,5
(3,8; 7,2)
| 9,3
(7,2; 11,2)
| 3,9
(3,7; 5,5)
|
p-hodnota
| < 0,0001
| < 0,0001
|
Pomer rizík
(Nir:plac) (95 % CI)
| 0,27
(0,173; 0,410)
| 0,45
(0,338; 0,607)
|
PFS = prežívanie bez progresie ochorenia; CI = interval spoľahlivosti; NE = nehodnotiteľné.
G
raf 3: Kaplanova-Meierova krivka prežívania bez progresie ochorenia v kohorte
gBRCAmut na základe hodnotenia IRC – NOVA (ITT populácia, N = 203)
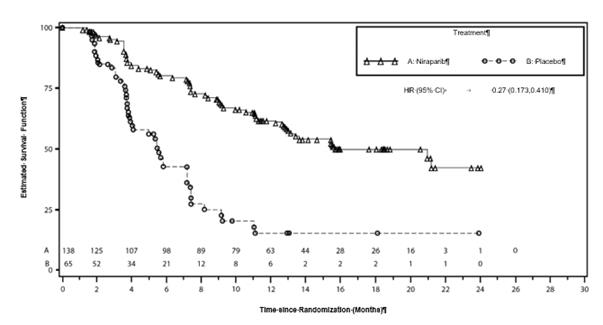
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Treatment: Liečba.
Graf 4: Kaplanova-Meierova krivka prežívania bez progresie ochorenia v celkovej kohortenon-gBRCAmut na základe hodnotenia IRC – NOVA (ITT populácia, N = 350)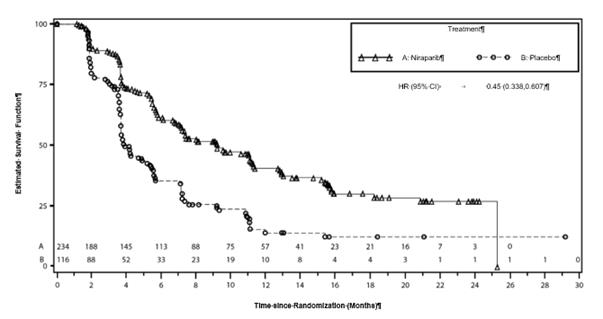
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Treatment: Liečba.
Sekundárne koncové ukazovatele účinnosti v NOVAPočas finálnej analýzy medián PFS2 v kohorte g
BRCAmut bol 29,9 mesiacov u pacientok liečených
niraparibom v porovnaní s 22,7 mesiacmi u pacientok na placebe (HR = 0,70; 95 % CI: 0,50; 0,97).
Medián PFS2 v kohorte non-g
BRCAmut bol 19,5 mesiacov u pacientok liečených niraparibom
v porovnaní so 16,1 mesiacmi u pacientok na placebe (HR = 0,80; 95 % CI: 0,63; 1,02).
Počas finálnej analýzy celkového prežívania bol medián OS v kohorte gBRCAmut (n = 203)
40,9 mesiacov u pacientok liečených niraparibom v porovnaní s 38,1 mesiacmi u pacientok na placebe
(HR = 0,85; 95 % CI: 0,61; 1,2). Zrelosť kohorty v kohorte gBRCAmut bola 76 %. Medián OS
v kohorte non-gBRCAmut (n = 350) bol 31,0 mesiacov u pacientok liečených niraparibom v porovnaní s 34,8 mesiacmi u pacientok na placebe (HR = 1,06; 95 % CI: 0,81; 1,37). Zrelosť kohorty v kohorte non-gBRCAmut bola 79 %.
Pacientkami oznamované výsledky (patient-reported outcomes, PRO) získané pomocou validovaných dotazníkov (FOSI a EQ5D) ukazujú, že u pacientok liečených niraparibom sa nezistil žiadny rozdiel oproti placebu v parametroch, ktoré sa spájajú s kvalitou života (quality of life, QoL).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Zejulou
vo všetkých podskupinách pediatrickej populácie pre karcinóm vaječníkov (s výnimkou rabdomyosarkómu a nádorov zo zárodočných buniek).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po podaní jednej 300 mg dávky niraparibu v podmienkach nalačno sa niraparib dal namerať v plazme
do 30 minút a priemerná maximálna plazmatická koncentrácia (Cmax) niraparibu sa dosiahla približne do 3 hodín [804 ng/ml (% CV: 50,2 %)]. Po viacerých perorálnych dávkach niraparibu od 30 mg
do 400 mg raz denne bola akumulácia niraparibu približne 2- až 3-násobná.
Systémová expozícia (Cmax a AUC) niraparibu sa zvyšovala úmerne s dávkou, keď sa dávka niraparibu zvyšovala z 30 mg na 400 mg. Absolútna biologická dostupnosť niraparibu je približne 73 %, z čoho vyplýva minimálny účinok prvého prechodu („first pass“ efekt). V populačnej farmakokinetickej analýze niraparibu bol pre interindividuálnu variabilitu v biologickej dostupnosti odhadnutý koeficient variácie (coefficient of variation, CV) 31 %.
Súčasná konzumácia jedla s vysokým obsahom tuku nemala žiadny signifikantný vplyv na farmakokinetiku niraparibu po podaní 300 mg niraparibu.
Bolo preukázané, že liekové formy tableta a kapsula sú bioekvivalentné. Po podaní buď jednej 300 mg tablety, alebo troch 100 mg kapsúl niraparibu nalačno u 108 pacientov so solídnymi nádormi, 90 % intervaly spoľahlivosti pomerov geometrického priemeru pre tabletu v porovnaní s kapsulami pre
Cmax, AUClast a AUC∞ spadali do limitov bioekvivalencie (0,80 a 1,25).
Distribúcia
Niraparib sa v ľudskej plazme v strednej miere viaže na proteíny (83 %), najmä na sérový albumín. V
populačnej farmakokinetickej analýze niraparibu bol zdanlivý distribučný objem (Vd/F) 1 311 l
(na základe 70 kg pacientky) u pacientok s rakovinou (CV 116 %), z čoho vyplýva rozsiahla tkanivová
distribúcia niraparibu.
Biotransformácia
Niraparib sa primárne metabolizuje prostredníctvom karboxylesteráz (CE), čím vzniká hlavný
neaktívny metabolit, M1. V štúdii hmotnostnej rovnováhy boli hlavnými cirkulujúcimi metabolitmi
M1 a M10 (následne vytvorené M1 glukuronidy).
Eliminácia
Po jednej perorálnej 300 mg dávke niraparibu bol priemerný terminálny polčas (t½) niraparibu
v rozsahu od 48 do 51 hodín (približne 2 dni). V populačnej farmakokinetickej analýze bol zdanlivý
celkový klírens (CL/F) niraparibu 16,5 l/hod. u pacientok s rakovinou (CV 23,4 %).
Niraparib sa primárne eliminuje hepatobiliárnou cestou a obličkami. Po perorálnom podaní jednej
300 mg dávky [14C]-niraparibu sa priemerne 86,2 % (rozsah 71 % až 91 %) dávky izolovalo z moču a
stolice za 21 dní. Rádioaktivita zaznamenaná v moči zodpovedala 47,5 % dávky (rozsah 33,4 % až
60,2 %) a v stolici zodpovedala 38,8 % dávky (rozsah 28,3 % až 47 %). V spojených vzorkách odoberaných 6 dní sa 40 % dávky izolovalo v moči primárne vo forme metabolitov a 31,6 % dávky sa izolovalo v stolici primárne vo forme nezmeneného niraparibu.
Osobitné populácie
Porucha funkcie obličiek
V populačnej farmakokinetickej analýze mali pacientky s miernou (klírens kreatinínu 60 - 90 ml/min)
a stredne závažnou (klírens kreatinínu 30 - 60 ml/min) poruchou funkcie obličiek mierne znížený klírens niraparibu v porovnaní s osobami s normálnou funkciou obličiek (o 7 - 17 % vyššia expozícia pri miernej poruche funkcie obličiek a o 17 - 38 % vyššia expozícia pri stredne závažnej poruche funkcie obličiek). Usudzuje sa, že tento rozdiel v expozícii nevyžaduje úpravu dávky. V klinických štúdiách sa neidentifikovala žiadna pacientka s už existujúcou závažnou poruchou funkcie obličiek alebo v konečnom štádiu ochorenia obličiek, ktorá by sa podrobovala hemodialýze (pozri časť 4.2).
Porucha funkcie pečene
V populačnej farmakokinetickej analýze údajov z klinických štúdií u pacientok sa zistilo, že už existujúca mierna porucha funkcie pečene (n = 155) nemala vplyv na klírens niraparibu. V klinickej
štúdii u pacientok s rakovinou, v ktorej sa na klasifikáciu stupňa poruchy funkcie pečene použili
kritériá NCI-ODWG, bola hodnota AUCinf niraparibu u pacientok so stredne závažnou poruchou funkcie pečene (n = 8) 1,56-násobne vyššia (90 % CI: 1,06, 2,30) ako hodnota AUCinf niraparibu
u pacientok s normálnou funkciou pečene (n = 9) po podaní jednotlivej 300 mg dávky. U pacientok
so stredne závažnou poruchou funkcie pečene sa odporúča úprava dávky niraparibu (pozri časť 4.2).
Stredne závažná porucha funkcie pečene nemala vplyv na Cmax niraparibu ani na väzbu niraparibu na proteíny. Farmakokinetika niraparibu sa nevyhodnocovala u pacientok so závažnou poruchou funkcie pečene (pozri časti 4.2 a 4.4).
Telesná hmotnosť, vek a rasa
V populačnej farmakokinetickej analýze sa zistilo, že zvyšujúca sa telesná hmotnosť zvyšuje distribučný objem niraparibu. Nezistil sa žiadny vplyv telesnej hmotnosti na klírens alebo celkovú expozíciu niraparibu. Úprava dávky podľa telesnej hmotnosti sa z farmakokinetického hľadiska nevyžaduje.
V populačnej farmakokinetickej analýze sa zistilo, že zvyšujúci sa vek znižuje klírens niraparibu. Predpokladá sa, že priemerná expozícia u 91-ročnej pacientky je o 23 % vyššia ako u 30-ročnej pacientky. Usudzuje sa, že vplyv veku nevyžaduje úpravu dávky.
K dispozícii nie sú dostatočné údaje týkajúce sa rôznych rás, aby bolo možné vyvodiť záver o vplyve
rasy na farmakokinetiku niraparibu.
Pediatrická populácia
Neuskutočnili sa žiadne štúdie na preskúmanie farmakokinetiky niraparibu u pediatrických pacientov.
5.3 Predklinické údaje o bezpečnosti
Farmakologická bezpečnosť
V podmienkach in vitro niraparib inhiboval dopamínový transportér DAT pri koncentráciách nižších,
ako sú expozície dosahované u ľudí. U myší jednorazové dávky niraparibu zvyšovali intracelulárne hladiny dopamínu a metabolitov v kortexe. Redukovaná lokomotorická aktivita sa pozorovala v jednej z dvoch štúdií s jednorazovou dávkou u myší. Klinický význam týchto zistení nie je známy. Nepozoroval sa žiadny účinok na správanie a/alebo neurologické parametre v štúdiách toxicity po opakovanom podávaní u potkanov a psov pri odhadovaných expozíciách v CNS, ktoré boli podobné alebo nižšie ako predpokladané terapeutické expozície.
Toxicita po opakovanompodávaní
U potkanov a psov bola pozorovaná znížená spermatogenéza pri expozíciách nižších, ako sú klinické
expozície a poväčšine bola reverzibilná do 4 týždňov od ukončenia dávkovania.
Genotoxicita
Niraparib nebol mutagénny v teste bakteriálnej reverznej mutácie (Amesov test), ale bol klastogénny
v in vitro teste chromozómových aberácií v cicavčích bunkách a v in vivo mikronukleovom teste
na kostnej dreni potkanov. Táto klastogenicita zodpovedá genómovej nestabilite, ktorá je výsledkom primárnej farmakológie niraparibu a svedčí o genotoxickom potenciále u ľudí.
Reprodukčná toxikológia
Štúdie reprodukčnej a vývojovej toxicity sa s niraparibom neuskutočnili.
Karcinogenita
S niraparibom sa neuskutočnili štúdie karcinogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly stearát horečnatý monohydrát laktózy
Obalkapsuly
oxid titaničitý (E 171)
želatína
briliantová modrá FCF (E 133)
erytrozín (E 127)
tartrazín (E 102)
Tlačiarenskáfarba šelak (E 904) propylénglykol (E 1520) hydroxid draselný (E 525) čierny oxid železitý (E 172) hydroxid sodný (E 524) povidón (E 1201)
oxid titaničitý (E 171)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti3 roky.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote do 30 °C.
6.5 Druh obalu a obsah baleniaAclar/PVC/hliníkové perforované blistre s jednotlivými dávkami, v škatuľkách obsahujúcich 84 × 1,
56 × 1 a 28 × 1 tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1235/001
EU/1/17/1235/002
EU/1/17/1235/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. novembra 2017
Dátum posledného predĺženia registrácie: 18. júla 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Zejula 100 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá filmom obalená tableta obsahuje monohydrát niraparib tozylátu zodpovedajúci 100 mg
niraparibu.
Pomocné látky soznámymúčinkom
Každá filmom obalená tableta obsahuje 34,7 mg monohydrátu laktózy (pozri časť 4.4).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta (tableta).
Sivá filmom obalená tableta oválneho tvaru (12 mm x 8 mm) s vyrazeným „100“ na jednej strane a
„Zejula“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Zejula je indikovaná:
• v monoterapii na udržiavaciu liečbu dospelých pacientok s pokročilým epitelovým (FIGO štádiá III a IV) karcinómom vaječníka, vajíčkovodu alebo primárnym peritoneálnym karcinómom s vysokým stupňom malignity, u ktorých pretrváva odpoveď (úplná alebo čiastočná) po ukončení prvej línie chemoterapie na báze platiny.
• v monoterapii na udržiavaciu liečbu dospelých pacientok s platina-senzitívnym, recidivujúcim, seróznym epitelovým karcinómom vaječníka, vajíčkovodu alebo primárnym peritoneálnym karcinómom s vysokým stupňom malignity, u ktorých pretrváva odpoveď (úplná alebo čiastočná) na chemoterapiu na báze platiny.
4.2 Dávkovanie a spôsob podávania
Liečbu Zejulou má začať lekár, ktorý má skúsenosti s používaním protinádorových liekov a má na ňu
aj dohliadať.
Dávkovanie
Udržiavacia liečba prvej línie pri karcinóme vaječníka
Odporúčaná začiatočná dávka Zejuly je 200 mg (dve 100 mg tablety), užívaná raz denne. Avšak
u pacientok s telesnou hmotnosťou ≥ 77 kg a s východiskovým počtom trombocytov ≥ 150 000/μl je odporúčaná začiatočná dávka Zejuly 300 mg (tri 100 mg tablety), užívaná raz denne (pozri
časti 4.4 a 4.8).
Udržiavacia liečba recidivujúceho karcinómu vaječníka
Dávkou sú tri 100 mg tablety raz denne, čo zodpovedá celkovej dennej dávke 300 mg.
Pacientky majú byť poučené, aby svoju dávku užívali každý deň približne v tom istom čase. Užívanie
pred spaním môže byť eventuálnou metódou na zvládanie nauzey.
Odporúča sa pokračovať v liečbe, kým nedôjde k progresii ochorenia alebo k vzniku toxicity.
Vynechanie dávkyAk pacientky vynechajú dávku, majú užiť nasledujúcu dávku vo svojom pravidelnom čase.
Úpravy dávky pri nežiaducich reakciáchOdporúčané úpravy dávky pri nežiaducich reakciách sú uvedené v tabuľkách 1, 2 a 3.
Vo všeobecnosti sa odporúča najprv liečbu prerušiť (ale nie na dlhšie ako na 28 za sebou nasledujúcich dní), aby sa pacientka mohla zotaviť z nežiaducej reakcie, a potom opäť začať
s rovnakou dávkou. V prípade, že dôjde k opätovnému výskytu danej nežiaducej reakcie, odporúča sa
prerušiť liečbu a potom ju znovu začať nižšou dávkou. Ak nežiaduce reakcie pretrvávajú aj po
28 dňoch od prerušenia dávkovania, odporúča sa ukončiť liečbu Zejulou. Ak sa nežiaduce reakcie nedajú zvládnuť pomocou stratégie prerušenia liečby a zníženia dávok, odporúča sa ukončiť liečbu
Zejulou.
Tabuľka 1: Odporúčané úpravy dávky pri nežiaducich reakciách
|
Veľkosť začiatočnej dávky
| 200 mg
| 300 mg
|
Prvé zníženie dávky
| 100 mg/deň
| 200 mg/deň (dve 100 mg
tablety)
|
Druhé zníženie dávky
| Ukončite liečbu Zejulou.
| 100 mg/deň* (jedna 100 mg
tablety)
|
*Ak je potrebné ďalšie zníženie dávky pod 100 mg/deň, liečbu Zejulou ukončite.
Tabuľka 2: Úpravy dávky pri nehematologických nežiaducich reakciách
|
Nehematologická nežiaduca reakcia ≥ 3. stupňa podľa
CTCAE* súvisiaca s liečbou, pri ktorej sa profylaxia nepovažuje za možnú, alebo ak nežiaduca reakcia pretrváva napriek liečbe
| Prvý výskyt:
• Prerušte liečbu Zejulou maximálne na 28 dní alebo kým nedôjde k vymiznutiu nežiaducej reakcie.
• Znovu začnite liečbu Zejulou
v zníženej dávke podľa tabuľky 1.
|
Druhý výskyt:
• Prerušte liečbu Zejulou maximálne na 28 dní alebo kým
nedôjde k vymiznutiu nežiaducej
reakcie.
• Znovu začnite liečbu Zejulou
v zníženej dávke alebo ukončite liečbu podľa tabuľky 1.
|
Nežiaduca reakcia ≥ 3. stupňa podľa CTCAE súvisiaca
s liečbou, ktorá trvá viac ako 28 dní, aj keď sa pacientke podáva Zejula v dávke 100 mg/deň
| Ukončite liečbu.
|
*CTCAE = Všeobecné terminologické kritériá pre nežiaduce účinky („Common Terminology Criteria for Adverse Events“)
 Tabuľka 3: Úpravy dávky pri hematologických nežiaducich reakciách
Tabuľka 3: Úpravy dávky pri hematologických nežiaducich reakciách
T
abuľka 3: Úpravy dávky pri hematologických nežiaducich reakciách
|
Počas liečby Zejulou sa pozorovali hematologické nežiaduce reakcie, najmä počas začiatočnej fázy
liečby. Počas prvých mesiacov liečby sa preto odporúča raz týždenne kontrolovať kompletný krvný obraz a v prípade potreby modifikovať dávku. Po prvom mesiaci sa odporúča kompletný krvný obraz kontrolovať raz za mesiac a potom v pravidelných intervaloch (pozri časť 4.4). Na základe individuálnych laboratórnych výsledkov môže byť nutné kontrolovať krvný obraz raz týždenne aj počas druhého mesiaca.
|
Hematologická nežiaduca reakcia vyžadujúca transfúziu alebo podávanie hematopoetického rastového faktora
|
• U pacientok s počtom trombocytov ≤ 10 000/μl sa má
zvážiť transfúzia trombocytov. Ak sú prítomné ďalšie rizikové faktory krvácania, ako napríklad súbežné podávanie antikoagulačných alebo antiagregačných liekov, zvážte prerušenie podávania týchto látok a/alebo transfúziu aj pri vyššom počte trombocytov.
• Znovu začnite liečbu Zejulou v zníženej dávke.
|
Počet trombocytov < 100 000/μl
|
Prvý výskyt:
• Prerušte liečbu Zejulou na maximálne 28 dní a raz týždenne kontrolujte krvný obraz, kým sa počet trombocytov nevráti na úroveň ≥ 100 000/µl.
• Znovu začnite liečbu Zejulou v rovnakej alebo zníženej dávke podľa tabuľky 1, na základe klinického posúdenia.
• Ak je počet trombocytov kedykoľvek < 75 000/μl, znovu začnite liečbu zníženou dávkou podľa tabuľky 1.
|
Druhý výskyt:
• Prerušte liečbu Zejulou na maximálne 28 dní a raz týždenne kontrolujte krvný obraz, kým sa počet
trombocytov nevráti na úroveň ≥ 100 000/µl.
• Znovu začnite liečbu Zejulou v zníženej dávke podľa tabuľky 1.
• Liečbu Zejulou ukončite, ak sa počet trombocytov nevrátil
na prijateľnú úroveň do 28 dní od prerušenia liečby alebo ak sa dávka u danej pacientky už znížila na 100 mg raz denne.
|
Počet neutrofilov < 1 000/µl alebo hemoglobín < 8 g/dl
|
• Prerušte liečbu Zejulou na maximálne 28 dní a raz týždenne kontrolujte krvný obraz, kým sa počet
neutrofilov nevráti na úroveň ≥ 1 500/µl alebo kým sa hemoglobín nevráti na úroveň ≥ 9 g/dl.
• Znovu začnite liečbu Zejulou v zníženej dávke podľa tabuľky 1.
• Liečbu Zejulou ukončite, ak sa počet neutrofilov a/alebo
hladina hemoglobínu nevrátili na prijateľnú úroveň do
28 dní od prerušenia liečby alebo ak sa dávka u danej pacientky už znížila na 100 mg denne.
|
Potvrdená diagnóza
myelodysplastického syndrómu (MDS) alebo akútnej myeloidnej leukémie (AML)
|
• Natrvalo ukončite liečbu Zejulou.
|
P
acientky s nízkou telesnou hmotnosťou pri udržiavacej liečbe recidivujúceho karcinómu vaječníka
Približne 25 % pacientok v štúdii NOVA vážilo menej ako 58 kg a približne 25 % pacientok vážilo viac ako 77 kg. Incidencia nežiaducich reakcií (adverse reactions, ADR) 3. alebo 4. stupňa bola vyššia u pacientok s nízkou telesnou hmotnosťou (78 %) ako u pacientok s vysokou telesnou
hmotnosťou (53 %). Len 13 % z pacientok s nízkou telesnou hmotnosťou pokračovalo v užívaní
300 mg dávky po 3. cykle. Pre pacientky s hmotnosťou nižšou ako 58 kg sa môže zvážiť začiatočná dávka 200 mg.
Staršie pacientkyU starších pacientok (≥ 65 rokov) nie je potrebná úprava dávky. O pacientkach vo veku 75 a viac
rokov sú k dispozícii len obmedzené klinické údaje.
Porucha funkcie obličiek
U pacientok s miernou až stredne závažnou poruchou funkcie obličiek nie je potrebná úprava dávky. Neexistujú žiadne údaje o pacientkach so závažnou poruchou obličiek alebo v konečnom štádiu ochorenia obličiek, ktoré sa podrobujú hemodialýze. U týchto pacientok buďte opatrný (pozri
časť 5.2).
Porucha funkcie pečene
U pacientok s miernou poruchou funkcie pečene (buď hladina aspartátaminotransferázy
(AST) > horná hranica referenčného rozpätia (upper limit of normal, ULN) a hladina celkového bilirubínu (total bilirubin, TB) ≤ ULN, alebo akákoľvek hladina AST
a hladina TB > 1,0- až 1,5-násobok ULN) nie je potrebná úprava dávky. U pacientok so stredne
závažnou poruchou funkcie pečene (akákoľvek hladina AST a hladina TB > 1,5- až 3-násobok ULN)
je odporúčaná začiatočná dávka Zejuly 200 mg jedenkrát denne. Neexistujú žiadne údaje o pacientkach so závažnou poruchou funkcie pečene (akákoľvek hladina AST a hladina
TB > 3-násobok ULN); u týchto pacientok buďte opatrný (pozri časti 4.4 a 5.2).
Pacientky s výkonnostným stavom ECOG 2 až 4
Nie sú k dispozícii klinické údaje o pacientkach s výkonnostným stavom ECOG 2 až 4.
Pediatrická populácia
Bezpečnosť a účinnosť niraparibu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Zejula je určená na perorálne použitie.
Zejula sa môže užívať nezávisle od jedla.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hematologické nežiaduce reakcie
U pacientok liečených Zejulou boli hlásené hematologické nežiaduce reakcie (trombocytopénia,
anémia, neutropénia) (pozri časť 4.8). Pacientky s nižšou telesnou hmotnosťou alebo s nižším východiskovým počtom trombocytov môžu mať zvýšené riziko vzniku trombocytopénie 3. a vyššieho stupňa závažnosti (pozri časť 4.2).
Odporúča sa vyšetrenie kompletného krvného obrazu raz týždenne počas prvého mesiaca, potom raz za mesiac počas nasledujúcich 10 mesiacov liečby a potom v pravidelných intervaloch, aby sa
sledovali klinicky významné zmeny v ktoromkoľvek hematologickom parametri počas liečby (pozri časť 4.2).
Ak sa u pacientky vyvinie závažná perzistentná hematologická toxicita vrátane pancytopénie, ktorá neodznie do 28 dní po prerušení liečby, má sa liečba Zejulou ukončiť.
Pre riziko trombocytopénie sa antikoagulačné látky a lieky, o ktorých je známe, že znižujú počet trombocytov, majú používať opatrne (pozri časť 4.8).
M
yelodysplastický syndróm/akútna myeloidná leukémia
U pacientok, ktoré boli liečené Zejulou v monoterapii alebo v kombinovanej liečbe v klinických
skúšaniach a v období po uvedení lieku na trh, boli hlásené prípady myelodysplastického syndrómu/akútnej myeloidnej leukémie (MDS/AML), vrátane prípadov so smrteľnými následkami (pozri časť 4.8).
V klinických skúšaniach liečba pacientok Zejulou trvala pred rozvojom MDS/AML od 0,5 mesiaca
do > 4,9 roka. Tieto prípady boli typické sekundárne MDS/AML súvisiace s liečbou rakoviny. Všetky pacientky predtým dostali chemoterapeutické režimy obsahujúce platinu a mnohé boli predtým liečené aj inými látkami poškodzujúcimi DNA a podstúpili rádioterapiu. Niektoré z pacientok mali v
anamnéze supresiu kostnej drene. V štúdii NOVA bol výskyt MDS/AML vyšší v kohorte gBRCAmut
(7,4 %) ako v kohorte non-gBRCAmut (1,7 %).
Pri podozrení na MDS/AML alebo pri protrahovaných hematologických toxicitách má byť pacientka odoslaná k hematológovi na ďalšie posúdenie. Ak sa potvrdí MDS/AML, liečba Zejulou sa má ukončiť a pacientka sa má liečiť zodpovedajúcim spôsobom.
Hypertenzia vrátane hypertenznej krízy
Počas používania Zejuly bola hlásená hypertenzia vrátane hypertenznej krízy (pozri časť 4.8).
Pred začiatkom liečby Zejulou má byť už existujúca hypertenzia adekvátne kontrolovaná. Počas liečby Zejulou sa má krvný tlak kontrolovať aspoň raz za týždeň počas prvých dvoch mesiacov, potom raz za mesiac počas prvého roka a potom v pravidelných intervaloch. U vhodných pacientok sa môže zvážiť kontrolovanie krvného tlaku v domácom prostredí a tieto pacientky majú byť poučené, aby v prípade zvýšenia krvného tlaku kontaktovali svojho lekára.
Hypertenzia má byť medikamentózne manažovaná antihypertenzívami, ako aj, v prípade potreby, úpravou dávky Zejuly (pozri časť 4.2). V klinickom programe sa krvný tak meral v 1. deň každého
28-dňového cyklu, kým pacientka užívala Zejulu. Vo väčšine prípadov bola hypertenzia adekvátne kontrolovaná štandardnou antihypertenznou liečbou spolu s úpravou alebo bez úpravy dávky Zejuly
(pozri časť 4.2). Liečba Zejulou sa má ukončiť v prípade hypertenznej krízy alebo ak sa medicínsky signifikantná hypertenzia nedá adekvátne kontrolovať antihypertenznou liečbou.
Syndrómreverzibilnejposteriórnejencefalopatie(Posterior reversible encephalopathysyndrome,PRES)
U pacientok, ktoré užívali Zejulu, boli hlásené prípady PRES (pozri časť 4.8). PRES je zriedkavá,
reverzibilná, neurologická porucha, ktorá sa môže prejavovať rýchlo sa rozvíjajúcimi príznakmi zahŕňajúcimi záchvaty kŕčov, bolesť hlavy, zmenený duševný stav, poruchu videnia alebo kortikálnu slepotu, so sprievodnou hypertenziou alebo bez nej. Na potvrdenie diagnózy PRES sa vyžaduje zobrazovacie vyšetrenie mozgu, prednostne vyšetrenie magnetickou rezonanciou (MR).
V prípade PRES sa odporúča ukončenie liečby Zejulou a liečba špecifických príznakov vrátane hypertenzie. Bezpečnosť opätovného začatia liečby Zejulou u pacientok, u ktorých sa v minulosti vyskytol PRES, nie je známa.
Tehotenstvo/antikoncepcia
Zejula sa nemá používať počas tehotenstva ani u žien vo fertilnom veku, ktoré nie sú ochotné používať
vysokoúčinnú antikoncepciu počas liečby a 6 mesiacov po užití poslednej dávky Zejuly (pozri časť 4.6). Pred začiatkom liečby majú všetky ženy vo fertilnom veku absolvovať tehotenský test.
Porucha funkcie pečene
Pacientky so závažnou poruchou funkcie pečene by mohli mať zvýšenú expozíciu niraparibu,
vychádzajúc z údajov získaných u pacientok so stredne závažnou poruchou funkcie pečene, a majú byť pozorne sledované (pozri časti 4.2 a 5.2).
Laktóza
Filmom obalené tablety Zejuly obsahujú monohydrát laktózy. Pacientky so zriedkavými dedičnými
problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Farmakodynamické interakcie
Kombinácia niraparibu s vakcínami alebo imunosupresívnymi látkami nebola študovaná.
Údaje o niraparibe v kombinácii s cytotoxickými liekmi sú obmedzené. Niraparib sa má preto používať s opatrnosťou v kombinácii s vakcínami, imunosupresívnymi látkami alebo inými cytotoxickými liekmi.
Farmakokinetické interakcie
Vplyvinýchliekovnaniraparib
Niraparib ako substrát CYP (CYP1A2 a CYP3A4)
Niraparib je substrátom karboxylesteráz (carboxylesterases, CE) a UDP-glukuronozyltransferáz (UGT) in vivo. Oxidatívny metabolizmus niraparibu je in vivo minimálny. Nie je potrebná úprava dávky Zejuly, keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú (napr. itrakonazol, ritonavir a klaritromycín) alebo indukujú CYP enzýmy (napr. rifampin, karbamazepín a fenytoín).
Niraparib ako substrát efluxných transportérov (P-gp, BCRP, BSEP, MRP2 a MATE1/2)
Niraparib je substrátom P-glykoproteínu (P-gp) a proteínu zodpovedného za rezistenciu pri rakovine prsníka (Breast Cancer Resistance Protein, BCRP). Vzhľadom na jeho vysokú permeabilitu a
biologickú dostupnosť je však riziko jeho klinicky významných interakcií s liekmi, ktoré inhibujú tieto transportéry, nepravdepodobné. Nevyžaduje sa preto žiadna úprava dávky Zejuly, keď sa podáva
súbežne s liekmi, o ktorých je známe, že inhibujú P-gp (napr. amiodarón, verapamil) alebo BCRP (napr. osimertinib, velpatasvir a eltrombopag).
Niraparib nie je substrátom exportnej pumpy solí žlčových kyselín (bile salt export pump, BSEP) ani proteínu 2 súvisiaceho s mnohopočetnou liekovou rezistenciou (multidrug resistance-associated protein, MRP2). Hlavný primárny metabolit M1 nie je substrátom P-gp, BCRP, BSEP ani MRP2. Niraparib nie je substrátom proteínov extrúzie viacerých liekov a toxínov (multidrug and toxin extrusion proteins, MATE)-1 alebo 2, zatiaľ čo M1 je substrátom oboch.
Niraparib ako substrát transportérov hepatálneho vychytávania (OATP1B1, OATP1B3 a OCT1)
Ani niraparib, ani M1 nie je substrátom transportného polypeptidu organických aniónov 1B1 (organic anion transport polypeptide 1B1, OATP1B1), 1B3 (OATP1B3) alebo transportéra organických katiónov 1 (organic cation transporter 1, OCT1). Nevyžaduje sa preto žiadna úprava dávky Zejuly,
keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú transportéry vychytávania OATP1B1
alebo 1B3 (napr. gemfibrozil, ritonavir) alebo OCT1 (napr. dolutegravir).
Niraparib ako substrát transportérov renálneho vychytávania (OAT1, OAT3 a OCT2)
Ani niraparib, ani M1 nie je substrátom transportéra organických aniónov 1 (OAT1), 3 (OAT3)
a transportéra organických katiónov 2 (OCT2). Nevyžaduje sa preto žiadna úprava dávky Zejuly, keď sa podáva súbežne s liekmi, o ktorých je známe, že inhibujú transportéry vychytávania OAT1
(napr. probenecid) alebo OAT3 (napr. probenecid, diklofenak) alebo OCT2 (napr. cimetidín, chinidín).
Vplyv niraparibu nainélieky
Inhibícia CYP (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4)
Ani niraparib, ani M1 nie je inhibítorom enzýmov CYP metabolizujúcich liečivá, konkrétne
CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP3A4/5.
Hoci sa neočakáva inhibícia CYP3A4 v pečeni, ešte sa nestanovil potenciál relevantných koncentrácií niraparibu inhibovať CYP3A4 v tenkom čreve. Preto sa odporúča byť opatrný, keď sa niraparib
podáva v kombinácii s liečivami, ktorých metabolizmus je závislý od CYP3A4, a to najmä s liečivami, ktoré majú úzke terapeutické rozmedzie (napr. cyklosporín, takrolimus, alfentanil, ergotamín, pimozid, kvetiapín a halofantrín).
Inhibícia UDP-glukuronozyltransferáz (UGT)
V podmienkach in vitro niraparib nevykazoval inhibičný účinok na izoformy UGT (UGT1A1, UGT1A4, UGT1A9 a UGT2B7) až do koncentrácie 200 µmol/l. Možnosť klinicky významnej inhibície UGT niraparibom je preto minimálna.
Indukcia CYP (CYP1A2 a CYP3A4)
Ani niraparib, ani M1 nie je in vitro induktorom CYP3A4. V podmienkach in vitro niraparib podávaný raz týždenne indukuje CYP1A2 pri vysokých koncentráciách a klinický význam tohto účinku sa nedá úplne vylúčiť. M1 nie je induktorom CYP1A2. Preto sa odporúča byť opatrným, keď sa niraparib podáva v kombinácii s liečivami, ktorých metabolizmus je závislý od CYP1A2, a to najmä s liečivami, ktoré majú úzke terapeutické rozmedzie (napr. klozapín, teofylín a ropinirol).
Inhibícia efluxných transportérov (P-gp, BCRP, BSEP, MRP2 a MATE1/2)
Niraparib nie je inhibítorom BSEP ani MRP2. In vitro niraparib inhibuje P-gp veľmi slabo a BCRP s
IC50 = 161 µmol/l pre Pg-p a 5,8 µmol/l pre BCRP. Nedá sa preto vylúčiť klinicky významná interakcia súvisiaca s inhibíciou týchto efluxných transportérov, hoci je nepravdepodobná. Odporúča sa preto byť opatrný, keď sa niraparib podáva v kombinácii so substrátmi BCRP (irinotekan, rosuvastatín, simvastatín, atorvastatín a metotrexát).
Niraparib je inhibítorom MATE1 s IC50 0,18 µmol/l a inhibítorom MATE2 s IC50 ≤ 0,14 µmol/l. Nedá sa vylúčiť zvýšenie plazmatických koncentrácií súbežne podávaných liekov, ktoré sú substrátmi týchto transportérov (napr. metformínu).
Hlavný primárny metabolit M1 sa nejaví ako inhibítor P-gp, BCRP, BSEP, MRP2 alebo MATE1/2.
Inhibícia transportérov hepatálneho vychytávania (OATP1B1, OATP1B3 a OCT1)
Ani niraparib, ani M1 nie je inhibítorom transportného polypeptidu organických aniónov 1B1 (OATP1B1) alebo 1B3 (OATP1B3).
In vitro niraparib podávaný raz týždenne inhibuje transportér organických katiónov 1 (OCT1)
s IC50 = 34,4 µmol/l. Odporúča sa byť opatrný, keď sa niraparib podáva v kombinácii s liečivami, ktorých vychytávanie je sprostredkované transportérom OCT1, napríklad s metformínom.
Inhibícia transportérov renálneho vychytávania (OAT1, OAT3 a OCT2)
Ani niraparib, ani M1 neinhibujú transportér organických aniónov 1 (OAT1), 3 (OAT3) a transportér organických katiónov 2 (OCT2).
Všetky klinické štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepciaužien
Ženy vo fertilnom veku nemajú otehotnieť počas liečby a nemajú byť tehotné na začiatku liečby. Pred začiatkom liečby majú všetky ženy vo fertilnom veku absolvovať tehotenský test. Ženy vo fertilnom veku musia používať vysokoúčinnú antikoncepciu počas liečby a 6 mesiacov po užití poslednej dávky Zejuly.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití niraparibu u gravidných žien.
Neuskutočnili sa štúdie reprodukčnej a vývojovej toxicity na zvieratách. Na základe mechanizmu účinku však niraparib môže spôsobovať poškodenie embrya alebo plodu, vrátane embryoletálnych a teratogénnych účinkov, pri podávaní tehotným ženám. Zejula sa nemá užívať počas gravidity.
Dojčenie
Nie je známe, či sa niraparib alebo jeho metabolity vylučujú do ľudského mlieka. Dojčenie je počas
podávania Zejuly a 1 mesiac po poslednej dávke kontraindikované (pozri časť 4.3).
Fertilita
Nie sú dostupné žiadne klinické údaje o fertilite. U potkanov a psov sa pozorovala reverzibilná
redukcia spermatogenézy (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Zejula má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientok užívajúcich
Zejulu môže dôjsť k asténii, únave, závratom alebo k ťažkostiam s koncentráciou. Pacientky,
u ktorých dôjde k týmto príznakom, majú byť opatrné pri vedení vozidiel alebo obsluhovaní strojov.
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
ADR všetkých stupňov závažnosti, ktoré sa vyskytli u ≥ 10 % z 851 pacientok užívajúcich Zejulu
v monoterapii v súhrnne hodnotených klinických skúšaniach PRIMA (buď 200 mg alebo 300 mg začiatočná dávka) a NOVA, boli nauzea, anémia, trombocytopénia, únava, zápcha, vracanie, bolesť hlavy, nespavosť, znížený počet trombocytov, neutropénia, bolesti brucha, znížená chuť do jedla, hnačka, dyspnoe, hypertenzia, asténia, závraty, znížený počet neutrofilov, kašeľ, artralgia, bolesti chrbta, znížený počet leukocytov a návaly tepla.
Najčastejšími závažnými nežiaducimi reakciami, u > 1 % (frekvencie výskytu počas liečby), boli trombocytopénia a anémia.
Tabuľkový zoznamnežiaducichreakcií
Nasledujúce nežiaduce reakcie boli identifikované na základe klinických skúšaní a sledovania
po uvedení lieku na trh u pacientok užívajúcich Zejulu v monoterapii (pozri tabuľku 4). Frekvencie výskytu nežiaducich účinkov sú založené na súhrnných údajoch o nežiaducich udalostiach získaných v štúdiách PRIMA a NOVA (fixná začiatočná dávka 300 mg/deň) u pacientok so známou expozíciou a sú definované nasledujúcim spôsobom: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
T
abuľka 4: Tabuľkový zoznam nežiaducich reakcií
T
rieda orgánových systémov
|
F
rekvencia nežiaducich reakcií všetkých stupňov závažnosti podľa CTCAE*
|
F
rekvencia nežiaducich reakcií 3. alebo
4. stupňa závažnosti podľa CTCAE*
|
Infekcie a nákazy
|
V
eľmi časté
Infekcia močových ciest
Časté
Bronchitída, konjunktivitída
|
Menej časté
Infekcia močových ciest, bronchitída
|
Benígne a malígne nádory, vrátane nešpecifikovaných
novotvarov (cysty a polypy)
|
Č
asté
Myelodysplastický syndróm/akútna myeloidná leukémia**
|
Č
asté Myelodysplastický syndróm/akútna
myeloidná leukémia**
|
Poruchy krvi a lymfatického systému
|
V
eľmi časté
Trombocytopénia, anémia, neutropénia, leukopénia
Menej časté
Pancytopénia, febrilná neutropénia
|
V
eľmi časté
Trombocytopénia, anémia, neutropénia
Časté
Leukopénia Menej časté Pancytopénia, febrilná neutropénia
|
Poruchy imunitného
systému
|
Č
asté
Precitlivenosť†
|
Menej časté
Precitlivenosť
|
Poruchy metabolizmu
a výživy
|
V
eľmi časté
Znížená chuť do jedla
Časté
Hypokaliémia
|
Č
asté
Hypokaliémia
Menej časté
Znížená chuť do jedla
|
Psychické poruchy
|
V
eľmi časté
Nespavosť
Časté
Úzkosť, depresia,
porucha kognitívnych funkci톆
Menej časté
Stav zmätenosti
|
Menej časté
Nespavosť, úzkosť,
depresia, stav zmätenosti
|
Poruchy nervového
systému
|
V
eľmi časté
Bolesť hlavy, závraty
Časté
Dysgeúzia
Zriedkavé
Syndróm reverzibilnej posteriórnej encefalopatie (Posterior reversible
encephalopathy syndrome, PRES)**
|
Menej časté
Bolesť hlavy'
|
Poruchy srdca a srdcovej činnosti
|
V
eľmi časté Palpitácie Časté Tachykardia
|
|
Poruchy ciev
|
V
eľmi časté
Hypertenzia Zriedkavé Hypertenzná kríza
|
Č
asté
Hypertenzia
|
Poruchy dýchacej
sústavy, hrudníka a mediastínia
|
V
eľmi časté
Dyspnoe, kašeľ, nazofaryngitída
Časté
Epistaxa
Menej časté
Pneumonitída
|
Menej časté
Dyspnoe, epistaxa, pneumonitída
|
T
rieda orgánových systémov
|
F
rekvencia nežiaducich reakcií všetkých stupňov závažnosti podľa CTCAE*
|
F
rekvencia nežiaducich reakcií 3. alebo
4. stupňa závažnosti
podľa CTCAE*
|
Poruchy
gastrointestinálneho
traktu
|
V
eľmi časté
Nauzea, zápcha, vracanie, bolesti brucha, hnačka, dyspepsia
Časté
Sucho v ústach, brušná distenzia, zápal sliznice, stomatitída
|
Č
asté
Nauzea, vracanie, bolesti brucha
Menej časté
Hnačka, zápcha, zápal sliznice, stomatitída,
sucho v ústach
|
Poruchy kože a
podkožného tkaniva
|
Č
asté
Fotosenzitivita, vyrážka
|
Menej časté
Fotosenzitivita, vyrážka
|
Poruchy kostrovej a
svalovej sústavy a spojivového tkaniva
|
V
eľmi časté
Bolesti chrbta, artralgia
Časté
Myalgia
|
Menej časté
Bolesti chrbta, artralgia, myalgia
|
Celkové poruchy a
reakcie v mieste podania
|
V
eľmi časté
Únava, asténia
Časté
Periférny edém
|
Č
asté
Únava, asténia
|
Laboratórne a funkčné vyšetrenia
|
Č
asté
Zvýšená hladina gamaglutamyltransferázy, zvýšená hladina AST, zvýšená hladina
kreatinínu v krvi, zvýšená hladina ALT, zvýšená
hladina alkalickej fosfatázy v krvi, znížená hmotnosť
|
Č
asté
Zvýšená hladina gamaglutamyltransferázy,
zvýšená hladina ALT
Menej časté
Zvýšená hladina AST, zvýšená hladina
alkalickej fosfatázy v krvi
|
* CTCAE = Všeobecné terminologické kritériá pre nežiaduce účinky („Common Terminology Criteria
for Adverse Events“), verzia 4.02.
** Na základe údajov z klinických skúšaní s niraparibom. Neobmedzuje sa to na pivotnú štúdiu
ENGOT-OV16 s monoterapiou.
† Zahŕňa precitlivenosť, liekovú precitlivenosť, anafylaktoidnú reakciu, liekovú erupciu, angioedém
a urtikáriu.
†† Zahŕňa poruchu pamäti, poruchu koncentrácie.
Frekvencia výskytu nežiaducich reakcií pozorovaných v skupine pacientok, ktorým bola podávaná
200 mg začiatočná dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, bola podobná alebo nižšia v porovnaní so skupinou pacientok, ktorým bola
podávaná fixná začiatočná dávka 300 mg (tabuľka 4).
Špecifické informácie týkajúce sa frekvencie výskytu trombocytopénie, anémie a neutropénie, pozri nižšie.
Opis vybraných nežiaducichreakciíHematologické nežiaduce reakcie (trombocytopénia, anémia, neutropénia), vrátane klinických diagnóz
a/alebo laboratórnych nálezov, vo všeobecnosti nastávajú v skorom štádiu liečby niraparibom, pričom ich incidencia sa postupom času znižuje.
V štúdiách NOVA a PRIMA mali pacientky vhodné na liečbu Zejulou nasledujúce východiskové hematologické parametre: absolútny počet neutrofilov (absolute neutrophil count,
ANC) ≥ 1 500 buniek/µl; počet trombocytov ≥ 100 000 buniek/µl a hladina hemoglobínu ≥ 9 g/dl
(NOVA) alebo ≥ 10 g/dl (PRIMA) pred začiatkom liečby. V klinickom programe sa hematologické nežiaduce reakcie manažovali prostredníctvom laboratórneho monitoringu a úprav dávky (pozri časť 4.2).
V štúdii PRIMA bol u pacientok, ktorým bola podávaná začiatočná dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, výskyt trombocytopénie, anémie a neutropénie 3. stupňa, v uvedenom poradí, znížený zo 48 % na 21 %, z 36 % na 23 %
a z 24 % na 15 % v porovnaní so skupinou pacientok, ktorým bola podávaná fixná začiatočná dávka
300 mg. K ukončeniu liečby v dôsledku trombocytopénie, anémie a neutropénie došlo, v uvedenom poradí, u 3 %, 3 % a 2 % pacientok.
Trombocytopénia
V štúdii PRIMA sa trombocytopénia 3./4. stupňa vyskytla u 39 % pacientok liečených Zejulou
v porovnaní s 0,4 % pacientok liečených placebom, pričom medián času od podania prvej dávky
do prvého výskytu trombocytopénie bol 22 dní (rozmedzie: 15 až 335 dní) a medián jej trvania bol
6 dní (rozmedzie: 1 až 374 dní). K ukončeniu liečby v dôsledku trombocytopénie došlo
u 4 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 60 % pacientok užívajúcich Zejulu k trombocytopénii akéhokoľvek stupňa a u 34 % pacientok došlo k trombocytopénii 3. alebo 4. stupňa. U 76 % pacientok
s východiskovým počtom trombocytov nižším ako 180 × 109/l došlo k trombocytopénii akéhokoľvek
stupňa a u 45 % takýchto pacientok došlo k trombocytopénii 3. alebo 4. stupňa. Medián času do nástupu trombocytopénie akéhokoľvek stupňa bol 22 dní a medián času do nástupu trombocytopénie
3. alebo 4. stupňa bol 23 dní. Výskyt nových epizód trombocytopénie po intenzívnych modifikáciách dávky realizovaných počas prvých dvoch mesiacov liečby od 4. cyklu bol 1,2 %. Medián trvania epizód trombocytopénie akéhokoľvek stupňa bol 23 dní a medián trvania trombocytopénie 3. alebo
4. stupňa bol 10 dní. Pacientky liečené Zejulou, u ktorých sa vyvinie trombocytopénia, môžu mať zvýšené riziko krvácania. V klinickom programe sa trombocytopénia manažovala laboratórnym
monitoringom, modifikáciou dávky a, ak to bolo potrebné, transfúziou krvných doštičiek (pozri
časť 4.2). K ukončeniu liečby v dôsledku epizód trombocytopénie (trombocytopénia a zníženie počtu trombocytov) došlo u približne 3 % pacientok.
V štúdii NOVA došlo u 48 z 367 (13 %) pacientok ku krvácaniu so súbežnou trombocytopéniou. Všetky krvácavé epizódy, ku ktorým došlo súbežne s trombocytopéniou, mali závažnosť
1. alebo 2. stupňa s výnimkou jedného prípadu petechie a hematómu 3. stupňa, ktorý bol pozorovaný súbežne so závažnou nežiaducou reakciou, pancytopéniou. K trombocytopénii dochádzalo častejšie
u pacientok, ktorých východiskový počet trombocytov bol nižší ako 180 x 109/l. U približne
76 % pacientok s nižším východiskovým počtom trombocytov (< 180 x 109/l), ktoré dostávali Zejulu, došlo k trombocytopénii akéhokoľvek stupňa a u 45 % pacientok došlo k trombocytopénii
3. alebo 4. stupňa. Pancytopénia sa pozorovala u < 1 % pacientok užívajúcich niraparib.
Anémia
V štúdii PRIMA sa anémia 3./4. stupňa vyskytla u 31 % pacientok liečených Zejulou v porovnaní
s 2 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu anémie bol 80 dní (rozmedzie: 15 až 533 dní) a medián jej trvania bol 7 dní (rozmedzie: 1 až 119 dní). K ukončeniu liečby v dôsledku anémie došlo u 2 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 50 % pacientok k anémii akéhokoľvek stupňa a u 25 % pacientok došlo k anémii 3. alebo 4. stupňa. Medián času do nástupu anémie akéhokoľvek stupňa bol 42 dní a do anémie 3. alebo 4. stupňa to bolo 85 dní. Medián trvania anémie akéhokoľvek stupňa bol 63 dní a anémie 3. alebo 4. stupňa 8 dní. Anémia akéhokoľvek stupňa môže počas liečby Zejulou pretrvávať. V klinickom programe sa anémia manažovala prostredníctvom laboratórneho monitoringu, modifikácie dávky (pozri časť 4.2) a v prípade potreby transfúziami červených krviniek. K ukončeniu liečby v dôsledku anémie došlo u 1 % pacientok.
N
eutropénia
V štúdii PRIMA sa neutropénia 3./4. stupňa vyskytla u 21 % pacientok liečených Zejulou v porovnaní s 1 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu neutropénie bol 29 dní (rozmedzie: 15 až 421 dní) a medián jej trvania bol 8 dní (rozmedzie:
1 až 42 dní). K ukončeniu liečby v dôsledku neutropénie došlo u 2 % pacientok užívajúcich niraparib.
V štúdii NOVA došlo u približne 30 % pacientok užívajúcich Zejulu k neutropénii akéhokoľvek stupňa a u 20 % pacientok došlo k neutropénii 3. alebo 4. stupňa. Medián času do nástupu neutropénie akéhokoľvek stupňa bol 27 dní a neutropénie 3. alebo 4. stupňa 29 dní. Medián času trvania neutropénie akéhokoľvek stupňa bol 26 dní a neutropénie 3. alebo 4. stupňa 13 dní. Okrem toho sa približne 6 % pacientok liečených niraparibom podával faktor stimulujúci kolónie granulocytov (Granulocyte Colony Stimulating Factor, G-CSF) ako súbežná liečba neutropénie. K ukončeniu liečby v dôsledku epizód neutropénie došlo u 2 % pacientok.
Myelodysplastický syndróm/akútna myeloidná leukémiaV klinických štúdiách sa MDS/AML vyskytovali u 1 % pacientok liečených Zejulou, pričom
41 % prípadov malo smrteľné následky. Výskyt bol vyšší u pacientok s recidivujúcim karcinómom vaječníka, ktoré predtým dostali 2 alebo viac línií chemoterapie na báze platiny, a s
gBRCAmut po 75-
mesačnom sledovaní prežívania. Všetky pacientky mali faktory potenciálne prispievajúce k rozvoju
MDS/AML v dôsledku predošlej chemoterapie na báze platiny. Veľa pacientok tiež dostalo iné látky poškodzujúce DNA a rádioterapiu.Väčšina hlásení bola u nositeliek
gBRCAmut. Niektoré pacientky
mali v anamnéze predchádzajúcu rakovinu alebo supresiu kostnej drene.
V štúdii PRIMA bol výskyt MDS/AML u 0,8 % pacientok, ktoré dostávali Zejulu a u 0,4 % pacientok, ktoré dostávali placebo.
V štúdii NOVA u pacientok s recidivujúcim karcinómom vaječníka, ktoré dostali aspoň dve línie chemoterapie na báze platiny, bol celkový výskyt MDS/AML 3,8 % u pacientok, ktoré dostávali Zejulu a 1,7 % u pacientok, ktoré dostávali placebo, po 75-mesačnom sledovaní prežívania. Výskyt MDS/AML bol 7,4 % v kohorte g
BRCAmut a 1,7 % v kohorte non-g
BRCAmut u pacientok, ktoré dostávali Zejulu, a 3,1 % v kohorte g
BRCAmut a 0,9 % v kohorte non-g
BRCAmut u pacientok, ktoré dostávali placebo.
HypertenziaV štúdii PRIMA sa hypertenzia 3./4. stupňa vyskytla u 6 % pacientok liečených Zejulou v porovnaní
s 1 % pacientok liečených placebom, pričom medián času od podania prvej dávky do prvého výskytu hypertenzie bol 50 dní (rozmedzie: 1 až 589 dní) a medián jej trvania bol 12 dní (rozmedzie:
1 až 61 dní). K ukončeniu liečby v dôsledku hypertenzie došlo u 0 % pacientok.
V štúdii NOVA sa hypertenzia akéhokoľvek stupňa vyskytla u 19,3 % pacientok liečených Zejulou. U 8,2 % pacientok sa vyskytla hypertenzia 3. alebo 4. stupňa. Hypertenzia sa dala jednoducho manažovať antihypertenzívami. K ukončeniu liečby v dôsledku hypertenzie došlo u < 1 % pacientok.
PediatrickápopuláciaU pediatrických pacientok sa neuskutočnili žiadne štúdie.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania Zejulou nie je k dispozícii žiadna špecifická liečba a neboli stanovené príznaky predávkovania. V prípade predávkovania majú lekári vykonať všeobecné podporné opatrenia a liečiť symptomaticky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, iné cytostatiká, ATC kód: L01XK02. Mechanizmusúčinkuafarmakodynamickéúčinky
Niraparib je inhibítorom enzýmov nazývaných poly(ADP-ribóza) polymerázy (PARP), PARP-1
a PARP-2, ktoré hrajú rolu pri opravovaní DNA. In vitro štúdie dokázali, že niraparibom indukovaná cytotoxicita môže zahŕňať inhibíciu enzymatickej aktivity PARP a zvýšenú tvorbu komplexov
PARP-DNA, čo vedie k poškodeniu DNA, apoptóze a bunkovej smrti. Zvýšená niraparibom
indukovaná cytotoxicita sa pozorovala v nádorových bunkových líniách s deficienciou alebo bez deficiencie nádorových supresorových génov pre rakovinu prsníka (BReast CAncer, BRCA) 1 a 2. Na ortotopických xenograftových myších nádoroch (patient-derived xenograft, PDX) vzniknutých inokuláciou seróznym ľudským karcinómom vaječníka vysokého stupňa malignity sa ukázalo, že niraparib redukuje rast nádorov s mutáciami génov BRCA 1 a 2, nádorov s divokým (nezmutovaným) typom BRCA, ale s deficienciou homologickej rekombinácie (HR) a nádorov s divokým typom BRCA a bez detegovateľnej deficiencie HR.
Klinická účinnosť abezpečnosť
Udržiavacialiečbaprvejlínieprikarcinómevaječníka
PRIMA bolo dvojito zaslepené, placebom kontrolované klinické skúšanie fázy 3, v ktorom boli pacientky (n = 733), u ktorých pretrvávala úplná alebo čiastočná odpoveď na prvú líniu chemoterapie
na báze platiny, randomizované 2:1 na podávanie niraparibu alebo zodpovedajúceho placeba.
V štúdii PRIMA sa liečba u 475 pacientok (317 z nich bolo randomizovaných do skupiny
s niraparibom a 158 do skupiny s placebom) začala 300 mg začiatočnou dávkou podávanou raz denne (QD) v rámci kontinuálnych 28-dňových cyklov. V štúdii PRIMA bola táto začiatočná dávka zmenená na základe 2. dodatku protokolu. Po jeho schválení bola pacientkam s východiskovou telesnou hmotnosťou ≥ 77 kg a s východiskovým počtom trombocytov ≥ 150 000/µl podávaná 300 mg dávka niraparibu (n = 34), alebo placeba raz denne (n = 21), zatiaľ čo pacientkam s východiskovou telesnou hmotnosťou < 77 kg alebo s východiskovým počtom trombocytov < 150 000/μl bola podávaná 200 mg dávka niraparibu (n = 122), alebo placeba raz denne (n = 61).
Pacientky boli randomizované po ukončení prvej línie chemoterapie na báze platiny plus/mínus chirurgický zákrok. Osoby boli randomizované do 12 týždňov od prvého dňa posledného cyklu chemoterapie. Osoby predtým dostali ≥ 6 a ≤ 9 cyklov liečby na báze platiny. Po odloženej (intervalovej) cytoredukčnej operácii dostali pacientky ≥ 2 pooperačné cykly liečby na báze platiny. Pacientky, ktoré predtým dostali bevacizumab spolu s chemoterapiou, ale nemohli dostávať bevacizumab ako udržiavaciu liečbu, neboli zo štúdie vylúčené. Pacientky nemohli predtým dostávať liečbu inhibítorom PARP (PARPi) vrátane niraparibu. Pacientky, ktoré podstúpili neoadjuvantnú chemoterapiu, po ktorej nasledovala odložená (intervalová) cytoredukčná operácia, mohli mať viditeľné reziduálne ochorenie alebo nemuseli mať žiadne reziduálne ochorenie. Pacientky
s ochorením v štádiu III, ktoré po primárnej cytoredukčnej operácii dosiahli kompletnú cytoredukciu (t. j. nemali žiadne viditeľné reziduálne ochorenie), boli zo štúdie vylúčené. Randomizácia bola stratifikovaná podľa najlepšej odpovede na liečbu dosiahnutej počas prvej línie chemoterapie na báze platiny (úplná odpoveď na liečbu vs čiastočná odpoveď na liečbu), podľa neoadjuvantnej chemoterapie (neoadjuvant chemotherapy, NACT) (áno vs nie); a podľa stavu deficiencie homologickej rekombinácie (homologous recombination deficiency, HRD) [pozitivita (deficiencia HR) vs negativita (proficiencia HR) alebo nestanovená]. Vyšetrenie na prítomnosť HRD sa vykonalo pomocou testu HRD na tumorovom tkanive získanom v čase iniciálnej diagnózy. Hladina CA-125 má byť v referenčnom rozpätí (alebo malo dôjsť k poklesu hladiny CA-125 o > 90 %) počas pacientkinej liečby prvej línie a mala byť stabilná počas aspoň 7 dní.
Pacientky začali liečbu 200 mg alebo 300 mg dávkou niraparibu alebo zodpovedajúcou dávkou
placeba v 1. cykle/1. deň (C1/D1) a užívali ich raz denne v rámci kontinuálnych 28-dňových cyklov. Pacientky sa dostavili na návštevu na klinike v každom cykle (každé 4 týždne ± 3 dni).
Primárnym koncovým ukazovateľom bolo prežívanie bez progresie ochorenia (progression-free survival, PFS) stanovené prostredníctvom zaslepeného nezávislého centrálneho hodnotenia (blinded independent central review, BICR) pomocou RECIST, verzia 1.1. Celkové prežívanie (overall survival, OS) bolo kľúčovým sekundárnym cieľom. Testovanie PFS sa vykonalo hierarchicky: najprv v populácii s deficienciou HR, potom v celkovej populácii. Medián veku bol 62 rokov a jeho rozmedzie bolo od 32 do 85 rokov u pacientok randomizovaných na niraparib a od 33 do 88 rokov
u pacientok randomizovaných na placebo. Osemdesiatdeväť percent všetkých pacientok tvorili belošky. Šesťdesiatdeväť percent pacientok randomizovaných na niraparib a 71 % pacientok
randomizovaných na placebo malo na začiatku štúdie výkonnostný stav ECOG 0. V celkovej populácii
malo 65 % pacientok ochorenie v štádiu III a 35 % pacientok malo ochorenie v štádiu IV. V celkovej populácii bol miestom primárneho nádoru u väčšiny pacientok (≥ 80 %) vaječník; väčšina pacientok
(> 90 %) mala serózny histologický podtyp nádoru. Šesťdesiatsedem percent pacientok podstúpilo
NACT. Šesťdesiatdeväť percent pacientok dosiahlo úplnú odpoveď na prvú líniu chemoterapie na báze platiny. Celkovo 6 pacientok liečených niraparibom dostalo bevacizumab ako predchádzajúcu liečbu karcinómu vaječníka.
V štúdii PRIMA sa preukázalo štatisticky významné zlepšenie PFS u pacientok randomizovaných na niraparib v porovnaní s placebom, a to v populácii s deficienciou HR a v celkovej populácii (tabuľka 5 a graf 1 a graf 2).
Sekundárne koncové ukazovatele účinnosti zahŕňali PFS po prvej nasledujúcej liečbe (PFS2) a OS (tabuľka 5).
Tabuľka 5: Výsledky účinnosti – PRIMA (stanovené prostredníctvom BICR)
| Populácia s deficienciou HR
| Celková populácia
|
niraparib
(N = 247)
| placebo
(N = 126)
| niraparib
(N = 487)
| placebo
(N = 246)
|
Medián PFS (95 % CI)
| 21,9 (19,3; NE)
| 10,4 (8,1; 12,1)
| 13,8 (11,5; 14,9)
| 8,2 (7,3; 8,5)
|
Pomer rizík (95 % CI)
| 0,43 (0,31; 0,59)
| 0,62 (0,50; 0,76)
|
p-hodnota
| < 0,0001
| < 0,0001
|
|
PFS2
Pomer rizík (95 % CI)
| 0,84 (0,485; 1,453)
| 0,81 (0,577; 1,139)
|
|
OS*
Pomer rizík (95 % CI)
| 0,61 (0,265; 1,388)
| 0,70 (0,44; 1,11)
|
PFS = prežívanie bez progresie ochorenia; CI = interval spoľahlivosti; NE = nehodnotiteľné; OS = celkové prežívanie; PFS2 = PFS po prvej nasledujúcej liečbe.
*V čase primárnej analýzy PFS bolo odhadované prežívanie po dvoch rokoch od randomizácie
84 % u pacientok užívajúcich Zejulu v porovnaní so 77 % u pacientok užívajúcich placebo v celkovej populácii.
Údaje o PFS2 a OS v súčasnosti nie sú konečné.
G
raf 1: Prežívanie bez progresie ochorenia u pacientok s nádormi s deficienciou HR – PRIMA (ITT populácia, N = 373)
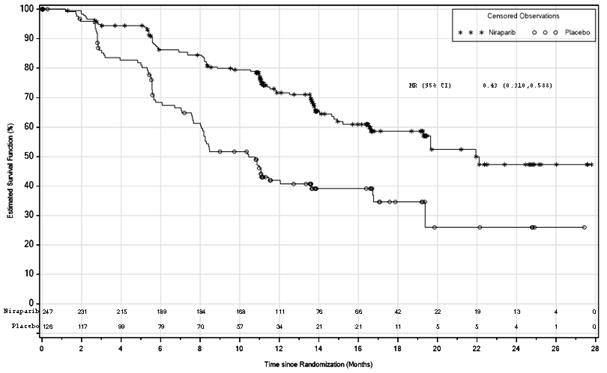
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Censored Observations: Cenzúrované pozorovania.
Graf 2: Prežívanie bez progresie ochorenia v celkovej populácii – PRIMA (ITT populácia,N = 733)
100
90
Censored Observations
Niraparib Placebo
80

HR (95% CI) 0.62 (0.502,0.755)
70
60
50
40

30
20
10
Niraparib 487
| 454
| 385
| 312
| 295
| 253
| 167
| 111
| 94
| 58
| 29
| 21
| 13
| 4
| 0
| Placebo 246
| 226
| 177
| 133
| 117
| 90
| 60
| 32
| 29
| 17
| 6
| 6
| 4
| 1
| 0
|
0
|
2
|
4
|
6
|
8
|
10
|
12
|
14
|
16
|
18
|
20
|
22
|
24
|
26
|
28
|
|
|
0
Time since Randomization (Months)
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od
randomizácie (mesiace); Censored Observations: Cenzúrované pozorovania.
A
nalýzy podskupín
V rámci populácie s deficienciou HR bol v podskupine pacientok s karcinómom vaječníka s BRCAmut
(N = 223) pozorovaný pomer rizík 0,40 (95 % CI: 0,27; 0,62). V podskupine pacientok s deficienciou
HR bez mutácie BRCA (N = 150) bol pozorovaný pomer rizík 0,50 (95 % CI: 0,31; 0,83). V populácii
s proficienciou HR (N = 249) bol pozorovaný pomer rizík 0,68 (95 % CI: 0,49; 0,94).
V exploračných analýzach podskupín bola u pacientok, ktorým bola podávaná 200 mg alebo 300 mg dávka Zejuly na základe východiskovej telesnej hmotnosti alebo východiskového počtu trombocytov, pozorovaná porovnateľná účinnosť (PFS hodnotené skúšajúcim) s pomerom rizík 0,54 (95 % CI: 0,33;
0,91) v populácii s deficienciou HR a s pomerom rizík 0,68 (95 % CI: 0,49; 0,94) v celkovej populácii. V podskupine s proficienciou HR sa zdalo, že pri 200 mg dávke je efekt liečby nižší v porovnaní
s 300 mg dávkou.
Udržiavacialiečbarecidivujúcehokarcinómuvaječníkacitlivéhonaplatinu
Bezpečnosť a účinnosť niraparibu ako udržiavacej liečby boli skúmané v randomizovanom, dvojito zaslepenom, placebom kontrolovanom medzinárodnom klinickom skúšaní fázy 3 (NOVA) u pacientok
s recidivujúcim seróznym epitelovým karcinómom vaječníka, vajíčkovodu alebo primárnym peritoneálnym karcinómom, prevažne vysokého stupňa malignity (high-grade), ktoré boli senzitívne
na platinu, čo bolo definované úplnou odpoveďou na liečbu (complete response, CR) alebo čiastočnou
odpoveďou na liečbu (partial response, PR) pretrvávajúcou viac ako 6 mesiacov po predposlednej liečbe na báze platiny. Pacientky boli vhodné na liečbu niraparibom, ak u nich pretrvávala odpoveď na liečbu (CR alebo PR) po ukončení poslednej chemoterapie na báze platiny. Po ich poslednej liečbe na báze platiny mali mať hladiny CA-125 v referenčnom rozpätí (alebo o > 90 % nižšie v porovnaní
s východiskovou hladinou) a stabilné počas najmenej 7 dní. Pacientky nemohli dostať predchádzajúcu liečbu PARPi vrátane Zejuly. Vhodné pacientky boli zaradené do jednej z dvoch kohort na základe
výsledkov testu germinatívnych (zárodočných) mutácií génov BRCA (gBRCA). V rámci každej
kohorty boli pacientky randomizované v pomere 2:1 na podávanie niraparibu a placeba. Pacientky boli zaradené do kohorty gBRCAmut na základe vzoriek krvi na analýzu gBRCA, ktoré boli odobraté pred randomizáciou. Testovanie na tumorovú BRCA (tBRCA) mutáciu a HRD sa vykonalo pomocou testu HRD na tumorovom tkanive získanom v čase iniciálnej diagnózy alebo v čase recidívy.
Randomizácia v každej kohorte bola stratifikovaná podľa času do progresie ochorenia po predposlednej liečbe na báze platiny pred zaradením do štúdie (6 až < 12 mesiacov
a ≥ 12 mesiacov); podľa použitia alebo nepoužitia bevacizumabu v kombinácii s predposledným
alebo posledným režimom na báze platiny; a podľa najlepšej odpovede na liečbu dosiahnutej počas posledného režimu na báze platiny (úplná odpoveď a čiastočná odpoveď).
Pacientky začali liečbu niraparibom 300 mg alebo zodpovedajúcim placebom v 1. cykle/1. deň (C1/D1) a užívali ich raz denne v rámci kontinuálnych 28-dňových cyklov. Pacientky sa dostavili na návštevu na klinike v každom cykle (každé 4 týždne ± 3 dni).
V štúdii NOVA došlo u 48 % pacientok k prerušeniu podávania lieku v 1. cykle. U približne 47 %
pacientok sa liek znovu začal podávať v zníženej dávke v 2. cykle.
Najčastejšie používanou dávkou u pacientok liečených niraparibom v štúdii NOVA bolo 200 mg.
Prežívanie bez progresie ochorenia (progression-free survival, PFS) sa posudzovalo pomocou RECIST („Response Evaluation Criteria in Solid Tumors“ - Kritériá hodnotenia odpovede pri solídnych nádoroch, verzia 1.1) alebo pomocou klinických prejavov a príznakov a zvýšenej hladiny CA-125.
PFS sa hodnotilo od času randomizácie (ktorá sa realizovala do 8 týždňov od ukončenia chemoterapeutického režimu) do progresie ochorenia alebo do smrti.
Primárna analýza účinnosti z hľadiska PFS bola založená na zaslepenom centrálnom nezávislom hodnotení a bola prospektívne definovaná a vykonaná osobitne v kohorte gBRCAmut a v kohorte
non-gBRCAmut. Analýzy celkového prežívania (overall survival, OS) boli sekundárnymi výslednými ukazovateľmi.
Sekundárne koncové ukazovatele účinnosti zahŕňali interval bez chemoterapie (chemotherapy-free interval, CFI), čas do prvej nasledujúcej liečby (time to first subsequent therapy, TFST), PFS po prvej nasledujúcej liečbe (PFS2) a OS.
Demografické údaje, východiskové charakteristiky ochorenia a predchádzajúce línie liečby boli vo všeobecnosti dobre vyvážené medzi skupinou s niraparibom a skupinou s placebom v kohorte g
BRCAmut (n = 203) a kohorte non-g
BRCAmut (n = 350). Medián veku bol v rozsahu od 57 do
63 rokov v liečebných skupinách a kohortách. Miestom primárneho nádoru bol u väčšiny pacientok
(> 80 %) v každej kohorte vaječník; väčšina pacientok (> 84 %) mala nádory so seróznou histológiou. Vysoké percento pacientok v oboch liečebných skupinách, v oboch kohortách, dostalo 3 alebo viacej
predchádzajúcich línií chemoterapie vrátane 49 % pacientok užívajúcich niraparib v kohorte
g
BRCAmut a 34 % pacientok užívajúcich niraparib v kohorte non-g
BRCAmut. Väčšina pacientok bola vo veku 18 až 64 rokov (78 %), kaukazskej rasy (86 %) a mala výkonnostný status ECOG rovný 0
(68 %).
V kohorte g
BRCAmut bol medián počtu cyklov liečby v skupine s niraparibom vyšší ako v skupine s placebom (14 cyklov a 7 cyklov, v uvedenom poradí). Viac pacientok v skupine s niraparibom pokračovalo v liečbe dlhšie ako 12 mesiacov než v skupine s placebom (54,4 % a 16,9 %, v uvedenom poradí).
V celkovej kohorte non-g
BRCAmut bol medián počtu cyklov liečby v skupine s niraparibom vyšší ako
v skupine s placebom (8 cyklov a 5 cyklov, v uvedenom poradí). Viac pacientok v skupine
s niraparibom pokračovalo v liečbe dlhšie ako 12 mesiacov než v skupine s placebom (34,2 % a
21,1 %, v uvedenom poradí).
Štúdia splnila svoj primárny cieľ, ktorým bolo štatisticky významné zlepšenie PFS pri udržiavacej liečbe niraparibom v monoterapii v porovnaní s placebom v kohorte g
BRCAmut, ako aj v celkovej kohorte non-g
BRCAmut. V tabuľke 6 a na grafoch 3 a 4 sú zobrazené výsledky týkajúce sa PFS, primárneho koncového ukazovateľa, v populácii pacientov pre primárnu analýzu účinnosti (kohorta g
BRCAmut a celková kohorta non-g
BRCAmut).
Tabuľka 6: Zhrnutie primárnych objektívnych výsledkov štúdie NOVA
| Kohorta gBRCAmut
| Kohorta non-gBRCAmut
|
niraparib
(N = 138)
| placebo
(N = 65)
| niraparib
(N = 234)
| placebo
(N = 116)
|
PFS medián (95 % CI)
| 21,0
(12,9; NE)
| 5,5
(3,8; 7,2)
| 9,3
(7,2; 11,2)
| 3,9
(3,7; 5,5)
|
p-hodnota
| < 0,0001
| < 0,0001
|
Pomer rizík
(Nir:plac) (95 % CI)
| 0,27
(0,173; 0,410)
| 0,45
(0,338; 0,607)
|
PFS = prežívanie bez progresie ochorenia; CI = interval spoľahlivosti; NE = nehodnotiteľné.
G
raf 3: Kaplanova-Meierova krivka prežívania bez progresie ochorenia v kohorte
gBRCAmut na základe hodnotenia IRC – NOVA (ITT populácia, N = 203)

Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Treatment: Liečba.
Graf 4: Kaplanova-Meierova krivka prežívania bez progresie ochorenia v celkovej kohortenon-gBRCAmut na základe hodnotenia IRC – NOVA (ITT populácia, N = 350)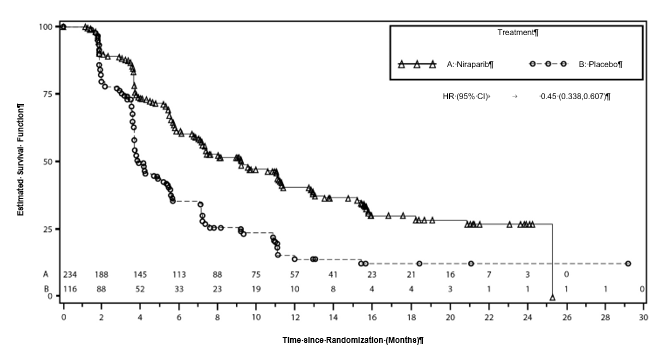
Estimated Survival Function (%): Odhad funkcie prežívania (%); Time since Randomisation (Months): Čas od randomizácie (mesiace); Treatment: Liečba..
Sekundárne koncové ukazovatele účinnosti v NOVAPočas finálnej analýzy medián PFS2 v kohorte g
BRCAmut bol 29,9 mesiacov u pacientok liečených
niraparibom v porovnaní s 22,7 mesiacmi u pacientok na placebe (HR = 0,70; 95 % CI: 0,50, 0,97).
Medián PFS2 v kohorte non-g
BRCAmut bol 19,5 mesiacov u pacientok liečených niraparibom
v porovnaní so 16,1 mesiacmi u pacientok na placebe (HR = 0,80; 95 % CI: 0,63, 1,02).
Počas finálnej analýzy celkového prežívania bol medián OS v kohorte gBRCAmut (n = 203)
40,9 mesiacov u pacientok liečených niraparibom v porovnaní s 38,1 mesiacmi u pacientok na placebe
(HR = 0,85; 95 % CI: 0,61, 1,2). Zrelosť kohorty v kohorte gBRCAmut bola 76 %. Medián OS
v kohorte non-gBRCAmut (n = 350) bol 31,0 mesiacov u pacientok liečených niraparibom v porovnaní
s 34,8 mesiacmi u pacientok na placebe (HR = 1,06; 95 % CI: 0,81, 1,37). Zrelosť kohorty v kohorte
non-gBRCAmut bola 79 %.
Pacientkami oznamované výsledky (patient-reported outcomes, PRO) získané pomocou validovaných dotazníkov (FOSI a EQ5D) ukazujú, že u pacientok liečených niraparibom sa nezistil žiadny rozdiel oproti placebu v parametroch, ktoré sa spájajú s kvalitou života (quality of life, QoL).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Zejulou
vo všetkých podskupinách pediatrickej populácie pre karcinóm vaječníkov (s výnimkou rabdomyosarkómu a nádorov zo zárodočných buniek).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po podaní jednej 300 mg dávky niraparibu v podmienkach nalačno sa niraparib dal namerať v plazme
do 30 minút a priemerná maximálna plazmatická koncentrácia (Cmax) niraparibu sa dosiahla približne do 3 hodín [804 ng/ml (% CV: 50,2 %)]. Po viacerých perorálnych dávkach niraparibu od 30 mg
do 400 mg raz denne bola akumulácia niraparibu približne 2- až 3-násobná.
Systémová expozícia (Cmax a AUC) niraparibu sa zvyšovala úmerne s dávkou, keď sa dávka niraparibu zvyšovala z 30 mg na 400 mg. Absolútna biologická dostupnosť niraparibu je približne 73 %, z čoho vyplýva minimálny účinok prvého prechodu („first pass“ efekt). V populačnej farmakokinetickej analýze niraparibu bol pre interindividuálnu variabilitu v biologickej dostupnosti odhadnutý koeficient variácie (coefficient of variation, CV) 31 %.
Súčasná konzumácia jedla s vysokým obsahom tuku nemala žiadny signifikantný vplyv na farmakokinetiku niraparibu po podaní 300 mg niraparibu.
Bolo preukázané, že liekové formy tableta a kapsula sú bioekvivalentné. Po podaní buď jednej 300 mg tablety, alebo troch 100 mg kapsúl niraparibu nalačno u 108 pacientov so solídnymi nádormi, 90 % intervaly spoľahlivosti pomerov geometrického priemeru pre tabletu v porovnaní s kapsulami pre
Cmax, AUClast a AUC∞ spadali do limitov bioekvivalencie (0,80 a 1,25).
Distribúcia
Niraparib sa v ľudskej plazme v strednej miere viaže na proteíny (83 %), najmä na sérový albumín. V
populačnej farmakokinetickej analýze niraparibu bol zdanlivý distribučný objem (Vd/F) 1 311 l
(na základe 70 kg pacientky) u pacientok s rakovinou (CV 116 %), z čoho vyplýva rozsiahla tkanivová distribúcia niraparibu.
Biotransformácia
Niraparib sa primárne metabolizuje prostredníctvom karboxylesteráz (CE), čím vzniká hlavný
neaktívny metabolit, M1. V štúdii hmotnostnej rovnováhy boli hlavnými cirkulujúcimi metabolitmi
M1 a M10 (následne vytvorené M1 glukuronidy).
Eliminácia
Po jednej perorálnej 300 mg dávke niraparibu bol priemerný terminálny polčas (t½) niraparibu
v rozsahu od 48 do 51 hodín (približne 2 dni). V populačnej farmakokinetickej analýze bol zdanlivý celkový klírens (CL/F) niraparibu 16,5 l/hod. u pacientok s rakovinou (CV 23,4 %).
Niraparib sa primárne eliminuje hepatobiliárnou cestou a obličkami. Po perorálnom podaní jednej
300 mg dávky [14C]-niraparibu sa priemerne 86,2 % (rozsah 71 % až 91 %) dávky izolovalo z moču a stolice za 21 dní. Rádioaktivita zaznamenaná v moči zodpovedala 47,5 % dávky (rozsah 33,4 % až
60,2 %) a v stolici zodpovedala 38,8 % dávky (rozsah 28,3 % až 47 %). V spojených vzorkách
odoberaných 6 dní sa 40 % dávky izolovalo v moči primárne vo forme metabolitov a 31,6 % dávky sa
izolovalo v stolici primárne vo forme nezmeneného niraparibu.
Osobitné populácie
Porucha funkcie obličiek
V populačnej farmakokinetickej analýze mali pacientky s miernou (klírens kreatinínu 60 - 90 ml/min) a stredne závažnou (klírens kreatinínu 30 - 60 ml/min) poruchou funkcie obličiek mierne znížený klírens niraparibu v porovnaní s osobami s normálnou funkciou obličiek (o 7 - 17 % vyššia expozícia pri miernej poruche funkcie obličiek a o 17 - 38 % vyššia expozícia pri stredne závažnej poruche funkcie obličiek). Usudzuje sa, že tento rozdiel v expozícii nevyžaduje úpravu dávky. V klinických štúdiách sa neidentifikovala žiadna pacientka s už existujúcou závažnou poruchou funkcie obličiek alebo v konečnom štádiu ochorenia obličiek, ktorá by sa podrobovala hemodialýze (pozri časť 4.2).
Porucha funkcie pečene
V populačnej farmakokinetickej analýze údajov z klinických štúdií u pacientok sa zistilo, že už existujúca mierna porucha funkcie pečene (n = 155) nemala vplyv na klírens niraparibu. V klinickej
štúdii u pacientok s rakovinou, v ktorej sa na klasifikáciu stupňa poruchy funkcie pečene použili
kritériá NCI-ODWG, bola hodnota AUCinf niraparibu u pacientok so stredne závažnou poruchou funkcie pečene (n = 8) 1,56-násobne vyššia (90 % CI: 1,06, 2,30) ako hodnota AUCinf niraparibu
u pacientok s normálnou funkciou pečene (n = 9) po podaní jednotlivej 300 mg dávky. U pacientok
so stredne závažnou poruchou funkcie pečene sa odporúča úprava dávky niraparibu (pozri časť 4.2). Stredne závažná porucha funkcie pečene nemala vplyv na Cmax niraparibu ani na väzbu niraparibu
na proteíny. Farmakokinetika niraparibu sa nevyhodnocovala u pacientok so závažnou poruchou funkcie pečene (pozri časti 4.2 a 4.4).
Telesná hmotnosť, vek a rasa
V populačnej farmakokinetickej analýze sa zistilo, že zvyšujúca sa telesná hmotnosť zvyšuje distribučný objem niraparibu. Nezistil sa žiadny vplyv telesnej hmotnosti na klírens alebo celkovú
expozíciu niraparibu. Úprava dávky podľa telesnej hmotnosti sa z farmakokinetického hľadiska
nevyžaduje.
V populačnej farmakokinetickej analýze sa zistilo, že zvyšujúci sa vek znižuje klírens niraparibu. Predpokladá sa, že priemerná expozícia u 91-ročnej pacientky je o 23 % vyššia ako u 30-ročnej pacientky. Usudzuje sa, že vplyv veku nevyžaduje úpravu dávky.
K dispozícii nie sú dostatočné údaje týkajúce sa rôznych rás, aby bolo možné vyvodiť záver o vplyve
rasy na farmakokinetiku niraparibu.
Pediatrická populácia
Neuskutočnili sa žiadne štúdie na preskúmanie farmakokinetiky niraparibu u pediatrických pacientov.
5.3 Predklinické údaje o bezpečnosti
Farmakologická bezpečnosť
V podmienkach in vitro niraparib inhiboval dopamínový transportér DAT pri koncentráciách nižších,
ako sú expozície dosahované u ľudí. U myší jednorazové dávky niraparibu zvyšovali intracelulárne hladiny dopamínu a metabolitov v kortexe. Redukovaná lokomotorická aktivita sa pozorovala v jednej z dvoch štúdií s jednorazovou dávkou u myší. Klinický význam týchto zistení nie je známy. Nepozoroval sa žiadny účinok na správanie a/alebo neurologické parametre v štúdiách toxicity po opakovanom podávaní u potkanov a psov pri odhadovaných expozíciách v CNS, ktoré boli podobné alebo nižšie ako predpokladané terapeutické expozície.
Toxicita po opakovanompodávaní
U potkanov a psov bola pozorovaná znížená spermatogenéza pri expozíciách nižších, ako sú klinické
expozície a poväčšine bola reverzibilná do 4 týždňov od ukončenia dávkovania.
Genotoxicita
Niraparib nebol mutagénny v teste bakteriálnej reverznej mutácie (Amesov test), ale bol klastogénny
v in vitro teste chromozómových aberácií v cicavčích bunkách a v in vivo mikronukleovom teste
na kostnej dreni potkanov. Táto klastogenicita zodpovedá genómovej nestabilite, ktorá je výsledkom primárnej farmakológie niraparibu a svedčí o genotoxickom potenciále u ľudí.
Reprodukčná toxikológia
Štúdie reprodukčnej a vývojovej toxicity sa s niraparibom neuskutočnili.
Karcinogenita
S niraparibom sa neuskutočnili štúdie karcinogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety krospovidón monohydrát laktózy stearát horečnatý
mikrokryštalická celulóza (E 460)
povidón (E 1201)
koloidný, hydratovaný oxid kremičitý
Obaltablety polyvinylalkohol (E 1203) oxid titaničitý (E 171) makrogol (E 1521) mastenec (E 533b)
čierny oxid železitý (E 172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie, uchovávajte v pôvodnom obale na ochranu tabliet pred absorpciou vody v podmienkach s vysokou vlhkosťou.
6.5 Druh obalu a obsah baleniaOPA/hliník/PVC/hliník/vinyl/akrylové blistre v škatuľkách obsahujúcich 84 × 1 a 56 × 1 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1235/004
EU/1/17/1235/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. novembra 2017
Dátum posledného predĺženia registrácie: 18. júla 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.