
duce reakcie v Triedach orgánových systémov podľa databázy MedDRA a
frekvenciami podľa štandardných konvencií: veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10) a menej časté (³ 1/1 000 až < 1/100). V rámci každej kategórie frekvencie výskytu sú nežiaduce reakcie uvedené v poradí ich klesajúcej závažnosti. Nežiaduce reakcie hlásené z klinického programu
v tabuľke uvedenej nižšie vyjadrujú frekvenciu, v ktorej sa vyskytovali v dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 (Fx-005).
Trieda orgánových systémov
| Veľmi časté
|
Infekcie a nákazy
| infekcia močových ciest
|
vaginálna infekcia
|
Poruchy gastrointestinálneho traktu
| hnačka
|
bolesť v hornej časti brucha
|
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyS predávkovaním sú minimálne klinické skúsenosti. Počas klinických skúšaní dvaja pacienti
s diagnostikovanou transtyretínovou amyloidovou kardiomyopatiou (ATTR-CM) neúmyselne užili jednu 160 mg dávku tafamidis meglumínu bez toho, aby došlo k akýmkoľvek asociovaným nežiaducim udalostiam. Najvyššia dávka tafamidis meglumínu podávaná zdravým dobrovoľníkom
v klinickom skúšaní bola 480 mg ako jednorazová dávka. Pri tejto dávke bol hlásený jeden nežiaduci účinok súvisiaci s liečbou, mierne hordeolum.
LiečbaV prípade predávkovania by sa mali podľa potreby vykonať štandardné podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá na centrálnu nervovú sústavu, ATC kód: N07XX08
Mechanizmus účinkuTafamidis je selektívny stabilizátor TTR. Tafamidis sa viaže na TTR vo väzbových miestach pre
tyroxín, pričom stabilizuje tetramér a spomaľuje jeho disociáciu na monoméry, čo je krok limitujúci rýchlosť amyloidogénneho procesu.
Farmakodynamické účinkyTranstyretínová amyloidóza je závažný invalidizujúci stav spôsobený akumuláciou rôznych
nerozpustných vláknitých proteínov, alebo amyloidu, v tkanivách v množstvách postačujúcich na poškodenie normálnej funkcie. Disociácia transtyretínového tetraméru na monoméry je krok určujúci
rýchlosť v patogenéze transtyretínovej amyloidózy. Poskladané monoméry podstúpia čiastočnú
denaturáciu a vytvoria inak poskladané monomérne amyloidogénne intermediáty. Tieto intermediáty sa potom chybne poskladajú do rozpustných oligomérov, profilamentov, filamentov a amyloidových
fibríl. Tafamidis sa s negatívnou kooperativitou viaže na dve väzobné miesta tyroxínu v prirodzenej
tetramérnej forme transtyretínu, a tak zabraňuje disociácii na monoméry. Inhibícia disociácie TTR
tetraméru je dôvodom na užívanie tafamidisu s cieľom spomaliť progresiu ochorenia u pacientov s
1.stupňom ATTR-PN.
TTR stabilizačný test sa využíval ako farmakodynamický marker a vyhodnocoval stabilitu TTR
tetraméru.
Tafamidis stabilizoval aj TTR tetramér divokého typu, aj tetraméry 14 TTR variantov, keď sa klinicky testoval po podávaní tafamidisu jedenkrát denne. Tafamidis stabilizoval aj TTR tetramér 25 variantov testovaných
ex vivo, čím sa demonštrovala stabilizácia TTR pre 40 amyloidogénnych TTR genotypov.
Klinická účinnosťabezpečnosťPivotná štúdia s tafamidis meglumínom u pacientov s 1. stupňom ATTR-PN bola 18-mesačná,
multicentrická, randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia. Táto štúdia hodnotila bezpečnosť a účinnosť tafamidis meglumínu podávaného jedenkrát denne v dávke 20 mg u 128 pacientov s ATTR-PN s mutáciou V30M a primárne s ochorením 1. stupňa. 126 zo 128 pacientov nevyžadovalo pomoc pri chôdzi. Primárnymi sledovanými ukazovateľmi boli Skóre
neuropatického postihnutia dolnej končatiny (Neuropathy Impairment Score of the Lower Limb, NIS- LL – vyhodnotenie neurologického vyšetrenia dolných končatín lekárom) a Norfolský dotazník
kvality života – diabetická neuropatia (Norfolk Quality of Life – Diabetic Neuropathy, Norfolk QOL-
DN – priamo hodnotené pacientom, skóre celkovej kvality života [total quality of life score, TQOL]). Ďalšie sledované ukazovatele zahŕňali zložené skóre funkcie veľkých nervových vlákien (vedenie
nervových vzruchov, vibračný prah a odpoveď srdcovej frekvencie na hlboké dýchanie - HRDB)
a tenkých nervových vlákien (bolesť vyvolaná teplom, prah chladu a HRDB) a výživový test
s použitím modifikovaného indexu telesnej hmotnosti (mBMI – BMI násobený albumínom v sére
v g/l). Osemdesiatšesť pacientov z 91, ktorí ukončili obdobie liečby trvajúce 18 mesiacov, bolo potom zaradených do otvorenej pokračovacej štúdie, kde počas ďalších 12 mesiacov všetci užívali tafamidis
meglumín jedenkrát denne v dávke 20 mg.
Po 18 mesiacoch liečby viac pacientov liečených tafamidis meglumínom reagovalo na liečbu podľa škály NIS-LL (zmena menej ako 2 body na stupnici NIS-LL). Výsledky vopred špecifikovaných analýz primárnych koncových ukazovateľov sú uvedené v nasledovnej tabuľke:
Vyndaqel vs placebo: NIS-LL a TQOL v 18. mesiaci (štúdia Fx-005)
|
| Placebo
| Vyndaqel
|
Vopred špecifikovaná ITT analýza
| N=61
| N=64
|
Respondenti podľa NIS-LL (% pacientov) Rozdiel (Vyndaqel mínus Placebo)
95% IS rozdielu (p-hodnota)
| 29,5 %
| 45,3 %
|
15,8 %
-0,9 %; 32,5 % (0,068)
|
TQOL zmena oproti východiskovému priemeru LS (SE)
Rozdiel v priemere LS (SE)
95% IS rozdielu (p-hodnota)
| 7,2 (2,36)
| 2,0 (2,31)
|
-5,2 (3,31)
-11,8, 1,3 (0,116)
|
Vopred špecifikovaná analýza populácie hodnotenej z hľadiska účinnosti
|
N=42
|
N=45
|
Respondenti podľa NIS-LL (% pacientov)
Rozdiel (Vyndaqel mínus placebo)
95% IS rozdielu (p-hodnota)
| 38,1 %
| 60,0 %
|
21,9 %
1,4 %; 42,4 % (0,041)
|
TQOL zmena oproti východiskovému priemeru LS (SE)
| 8,9 (3,08)
| 0,1 (2,98)
|
Rozdiel v priemere LS (SE)
95% IS rozdielu (p-hodnota)
| -8,8 (4,32)
-17,4; -0,2 (0,045)
|
| | | |
Vo vopred špecifikovanej analýze respondentov podľa ITT NIS-LL, boli tí pacienti, ktorí kvôli transplantácii pečene ukončili liečbu pred dovŕšením 18-mesačného obdobia, zatriedení ako pacienti nereagujúci na liečbu. Vopred špecifikovaná analýza populácie hodnotenej z hľadiska účinnosti použila pozorované údaje tých pacientov, ktorí túto 18-mesačnú liečbu ukončili
podľa protokolu.
Sekundárne koncové ukazovatele preukázali, že výsledkom liečby tafamidis meglumínom v porovnaní
s placebom bolo menšie zhoršenie neurologických funkcií a zlepšený nutričný stav (mBMI), ako je uvedené v nasledovnej tabuľke.
Zmeny v sekundárnych koncových ukazovateľoch podľa priemeru LS od východiskového stavu po 18. mesiac (štandardná odchýlka - SO) (populácia so zámerom liečby) (štúdia Fx-005)
|
|
Placebo
N = 61
|
Vyndaqel
N = 64
|
P
- hodnota
|
Vyndaqel % zmena vo vzťahu k placebu
|
Zmena podľa NIS-LL oproti východiskovému priemeru LS (SO)
|
5,8 (0,96)
|
2,8 (0,95)
|
0,027
|
-52 %
|
Zmena v hrubom vlákne oproti východiskovému priemeru LS (SO)
|
3,2 (0,63)
|
1,5 (0,62)
|
0,066
|
-53 %
|
Zmena v tenkom vlákne oproti východiskovému priemeru LS (SO)
|
1,6 (0,32)
|
0,3 (0,31)
|
0,005
|
-81 %
|
Zmena v mBMI oproti východiskovému priemeru LS (SO)
|
-33,8 (11,8)
|
39,3 (11,5)
|
< 0,0001
|
N/A
|
mBMI bol odvodený ako produkt sérového albumínu a Body Mass Indexu.
Založené na analýze opakovaných meraní rozdielu v liečbe pri zmene z východiskového bodu ako závislej premennej, neštruktúrovanej matici kovariancie, liečbe, mesiaci a liečbe za mesiac ako pevných efektoch a pacientovi ako náhodnom efekte modelu.
N/A = neaplikovateľné
V otvorenej pokračovacej štúdii miera zmeny podľa NIS-LL počas 12 mesiacov liečby bola podobná
miere zmeny pozorovanej u tých pacientov, ktorí boli randomizovaní a liečení tafamidisom v predchádzajúcom dvojito zaslepenom 18-mesačnom období.
Ak sa vezme do úvahy mechanizmus účinku tafamidisu a výsledky týkajúce sa stabilizácie TTR, očakáva sa, že tafamidis meglumín bude prínosným pre pacientov s ATTR-PN 1. stupňa v dôsledku mutácií iných ako V30M, hoci sú údaje obmedzené (jedna otvorená štúdia s 21 pacientmi).
Účinok tafamidisu sa hodnotil v dvojito zaslepenej, placebom kontrolovanej randomizovanej 3- ramennej štúdii u 441 pacientov s wild-type alebo hereditárnou transtyretínovou amyloidovou kardiomyopatiou (ATTR-CM). Primárna analýza tafamidis meglumínu spolu (20 mg a 80 mg) verzus placebo preukázala signifikantnú redukciu (p=0,0006) mortality zo všetkých príčin a frekvencie hospitalizácií súvisiacich s kardiologickým ochorením.
Supraterapeutická jednorazová perorálna dávka 400 mg roztoku tafamidisu u zdravých dobrovoľníkov nepreukázala žiadne predĺženie QTc intervalu.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdii s tafamidisom vo všetkých podskupinách pediatrickej populácie s transtyretínovou amyloidózou (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný za tzv. mimoriadnych okolností. To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiAbsorpciaPo perorálnom podaní mäkkej kapsuly jedenkrát denne sa maximálna vrcholová koncentrácia (Cmax)
dosiahne za strednú dobu (tmax) 4 hodiny, ak sa dávka užije nalačno. Súbežné požitie vysokokalorického jedla s vysokým obsahom tuku zmenilo rýchlosť absorpcie, ale nie rozsah
absorpcie. Tieto výsledky podporujú podávanie tafamidisu s jedlom alebo bez jedla.
DistribúciaTafamidis sa výrazne viaže na plazmatické proteíny (> 99 %). Zdanlivý distribučný objem
v ustálenom stave je 16 litrov.
Rozsah väzby tafamidisu na plazmatické proteíny sa vyhodnocoval s použitím živočíšnej a ľudskej plazmy. Afinita tafamidisu voči TTR je vyššia ako afinita pre albumín. Preto je tafamidis náchylný preferenčne sa viazať na TTR napriek signifikantne vyššej koncentrácii albumínu (600 μM)
v porovnaní s koncentráciou TTR (3,6 μM).
Biotransformáciaa eliminácia
O vylučovaní tafamidisu žlčou u ľudí neexistuje jednoznačný dôkaz. Na základe predklinických
údajov možno tvrdiť, že tafamidis sa metabolizuje glukuronidáciou a vylučuje prostredníctvom žlče. Táto cesta biotransformácie je u ľudí prijateľná, pretože približne 59 % z celkovej užitej dávky sa objaví v stolici a približne 22 % v moči. Na základe výsledkov populačnej farmakokinetiky je zdanlivý perorálny klírens tafamidis meglumínu 0,228 l/h a priemerný polčas v populácii je približne
49 hodín.
Linearita dávkyačasu
Expozícia tafamidis meglumínu pri dávkovaní jedenkrát denne sa zvyšovala so zvyšovaním dávky až
do 480 mg pri jednej dávke a pri viacerých dávkach až do 80 mg/deň. Vo všeobecnosti bolo zvyšovanie priamo úmerné alebo takmer priamo úmerné dávke a klírens tafamidisu bol v danom čase
stabilný.
Farmakokinetické parametre boli podobné po jednorazovom a opakovanom podaní 20 mg tafamidis meglumínu, čo naznačuje nedostatočnú indukciu alebo inhibíciu metabolizmu tafamidisu.
Účinky podávania tafamidis meglumínu v dávke 15 mg až 60 mg perorálneho roztoku po dobu 14 dní preukázali, že sa rovnovážny stav dosiahol na 14. deň.
Osobitné skupinypacientov
Porucha funkcie pečene
Farmakokinetické údaje svedčia o zníženej systémovej expozícii (približne 40 %) a zvýšenom celkovom klírense (0,52 l/h oproti 0,31 l/h) tafamidis meglumínu u pacientov so stredne závažnou poruchou funkcie pečene (skóre podľa Childovej-Pughovej klasifikácie v rozmedzí 7 – 9 vrátane)
v porovnaní so zdravými dobrovoľníkmi v dôsledku vyššej nenaviazanej frakcie tafamidisu. Aj keď pacienti so stredne závažnou poruchou funkcie pečene vykazujú nižšie hladiny TTR ako zdraví
jednotlivci, úprava dávky nie je potrebná, pretože stoichiometria tafamidisu s jeho cieľovým
proteínom TTR bude dostatočná na stabilizáciu TTR tetraméru. Expozícia tafamidisu u pacientov so závažnou poruchou funkcie pečene nie je známa.
Porucha funkcie obličiek
Tafamidis nebol osobitne vyhodnocovaný v štúdii zameranej na pacientov s poruchou funkcie obličiek. Vplyv klírensu kreatinínu na farmakokinetiku tafamidisu sa vyhodnocoval v populačnej farmakokinetickej analýze u pacientov s klírensom kreatinínu vyšším ako 18 ml/min. Farmakokinetické odhady neindikovali žiadny rozdiel v zdanlivom perorálnom klírense tafamidisu u pacientov s klírensom kreatinínu nižším ako 80 ml/min v porovnaní s tými, ktorých klírens kreatinínu bol vyšší alebo rovný 80 ml/min. Úprava dávky u týchto pacientov sa nepovažuje za potrebnú.
Starší
Na základe výsledkov z populačnej farmakokinetiky mali pacienti vo veku ³ 65 rokov v priemere o
15 % nižšiu odhadovanú hodnotu zdanlivého perorálneho klírensu v rovnovážnom stave v porovnaní s pacientmi mladšími ako 65 rokov. Tento rozdiel v klírense však viedol k < 20 % zvýšeniam
priemerných hodnôt Cmax a AUC v porovnaní s mladšími pacientmi a nie je klinický významný.
Farmakokinetický/farmakodynamický vzťah
Z in vitro údajov vyplýva, že tafamidis významne neinhibuje enzýmy cytochrómu P450 CYP1A2,
CYP3A4, CYP3A5, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. Neočakáva sa, že by tafamidis spôsoboval klinicky relevantné liekové interakcie v dôsledku indukcie CYP1A2, CYP2B6 alebo CYP3A4.
In vitro štúdie naznačujú, že nie je pravdepodobné, že tafamidis v klinicky relevantných koncentráciách bude spôsobovať systémové liekové interakcie so substrátmi UDP glukuronozyltransferázy (UGT). Tafamidis môže inhibovať intestinálne aktivity UGT1A1.
Tafamidis v klinicky relevantných koncentráciách preukázal nízky potenciál inhibovať proteín multiliekovej rezistencie (Multi-Drug Resistant Protein - MDR1) (známy aj ako P-glykoproteín; P-gp) systémovo a v gastointestinálnom trakte (GI), inhibovať prenášač organických katiónov 2 (OCT2), multiliekový a toxínový extrúzny prenášač 1 (multidrug and toxin extrusion transporter 1 - MATE1) a MATE2K, polypeptid prenášajúci organické anióny 1B1 (organic anion transporting polypeptide 1B1
- OATP1B1) a OATP1B3.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, reprodukčnej toxicity a vývinu, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. V štúdiách toxicity po opakovanom podávaní a štúdiách karcinogenity sa pečeň prejavila ako cieľový orgán toxicity u rozdielnych skúmaných druhov. Účinky na pečeň sa pozorovali pri expozíciách približne ³ 2,5-násobne vyšších ako je AUC u ľudí v ustálenom stave pri klinickej dávke 20 mg tafamidis meglumínu.
Vo vývinovej štúdii toxicity na zajacoch sa pozoroval mierny nárast malformácií a zmien kostry, potraty u niekoľkých samičiek, redukované embryofetálne prežívanie a zníženie fetálnej hmotnosti pri expozíciách približne ³ 7,2-násobne vyšších ako AUC v rovnovážnom stave u ľudí pri klinickej dávke
20 mg tafamidis meglumínu.
Vo vývinovej štúdii s tafamidisom zameranej na prenatálny a postnatálny vývin potkanov bolo počas gravidity a laktácie po podávaní maternálnej dávky 15 a 30 mg/kg/deň zaznamenané znížené prežívanie mláďat a pokles hmotnosti. Znížené hmotnosti mláďat samčekov sa spájali s oneskoreným pohlavným dospievaním (oddelenie predkožky) pri 15mg/kg/deň. Znížená výkonnosť v teste Morrisonovým vodným labyrintom, zameranom na učenie a pamäť, sa pozorovala pri 15 mg/kg/deň. NOAEL (hladina, pri ktorej neboli pozorované nežiaduce účinky) pre životaschopnosť a rast potomstva prvej (F1) generácie po podávaní maternálnej dávky počas gravidity a laktácie
s tafamidisom predstavovala 5 mg/kg/deň (human equivalent dose, ekvivalentná dávka pre človeka
= 0,8 mg/kg/deň), čo je približne 4,6-násobok klinickej dávky 20 mg tafamidis meglumínu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obal kapsuly
želatína (E 441)
glycerín (E 422)
žltý oxid železitý (E 172)
sorbitan
sorbitol (E 420)
manitol (E 421)
oxid titaničitý (E 171)
čistená voda
Obsah kapsuly
makrogol 400 (E 1521)
sorbitanmonooleát (E 494)
polysorbát 80 (E 433)
Potlačový atrament (purpurová Opacode)
etylalkohol
izopropylalkohol čistená voda
makrogol 400 (E 1521)
ftalát polyvinylacetátu propylénglykol (E 1520) karmín (E 120)
brilantná modrá FCF (E 133)
28 % hydroxid amónny (E 527)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25°C.
6.5 Druh obalu a obsah balenia
PVC/PA/alu/PVC-alu/PET/Papierové perforované blistre s jednotlivými dávkami.
Veľkosť balenia: balenie po 30 x 1 mäkká kapsula a multibalenie obsahujúce 90 (3 balenia po 30 x 1)
mäkkých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLA
EU/1/11/717/001
EU/1/11/717/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. november 2011
Dátum posledného predĺženia registrácie: 22. júl 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUVyndaqel 61 mg mäkké kapsuly
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Jedna mäkká kapsula obsahuje 61 mg mikronizovaného tafamidisu.
PomocnálátkasoznámymúčinkomJedna mäkká kapsula obsahuje nie viac ako 44 mg sorbitolu (E 420).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAMäkká kapsula.
Červenohnedá, nepriehľadná, podlhovastá kapsula (približne 21 mm) s vytlačeným bielym nápisom
„VYN 61“.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieVyndaqel je indikovaný na liečbu hereditárnej transtyretínovej amyloidózy alebo transtyretínovej amyloidózy divokého typu (wild type) u dospelých pacientov s kardiomyopatiou (ATTR-CM).
4.2 Dávkovanie a spôsob podávaniaLiečba pacientov sa má začať pod dohľadom odborného lekára so skúsenosťami v liečbe pacientov s amyloidózou alebo kardiomyopatiou.
V prípade podozrenia u pacientov s prítomnou špecifickou lekárskou anamnézou alebo s príznakmi srdcového zlyhávania alebo kardiomyopatie, sa má pred začiatkom podávania tafamidisu vykonať etiologická diagnostika lekárom so skúsenosťami s liečbou amyloidózy alebo kardiomyopatie, aby sa potvrdila ATTR-CM a vylúčila AL amyloidóza, a to použitím príslušných vyhodnocovacích nástrojov, ako sú: kostná scintigrafia a vyhodnotenie krvi/moču a/alebo histologické vyhodnotenie prostredníctvom biopsie a transtyretínová (TTR) genotypizácia na stanovenie wild-type alebo hereditárneho typu.
DávkovanieOdporúčaná dávka je jedna kapsula Vyndaqelu 61 mg (tafamidis) podávaná perorálne jedenkrát denne
(pozri časť 5.1).
Vyndaqel 61 mg (tafamidis) zodpovedá 80 mg tafamidis meglumínu. Tafamidis a tafamidis meglumín nie sú zameniteľné na mg báze (pozri časť 5.2).
Vyndaqel sa má začať užívať čo najskôr v priebehu ochorenia, aby klinická prospešnosť z hľadiska progresie ochorenia mohla byť evidentnejšia. Naopak, keď je kardiologické poškodenie súvisiace
s amyloidom pokročilejšie, ako pri NYHA III. triedy, rozhodnutie o tom, či začať alebo pokračovať
v liečbe je na posúdení lekára, so skúsenosťami s liečbou pacientov s amyloidózou alebo kardiomyopatiou (pozri časť 5.1). Klinické údaje o pacientoch s NYHA IV. triedy sú obmedzené.
Ak sa po užití dávky u pacienta objaví vracanie a vo zvratkoch je prítomná neporušená kapsula Vyndaqelu, potom sa má podľa možnosti užiť ďalšia dávka Vyndaqelu. Ak žiadna kapsula nie je prítomná, užitie ďalšej dávky nie je potrebné a v dávkovaní sa má pokračovať ďalší deň ako zvyčajne.
Osobitné skupinypacientovStaršíU starších pacientov (≥ 65 rokov) nie je potrebná žiadna úprava dávkovania (pozri časť 5.2).
Porucha funkcie pečene a obličiekU pacientov s poruchou funkcie obličiek alebo s miernou a stredne závažnou poruchou funkcie pečene nie je potrebná žiadna úprava dávkovania. Údaje u pacientov so závažnou poruchou funkcie obličiek (klírens kreatinínu nižší alebo rovný 30 ml/min) sú obmedzené. U pacientov so závažnou poruchou funkcie pečene sa neuskutočnili štúdie s tafamidisom a odporúča sa postupovať s opatrnosťou (pozri časť 5.2).
Pediatrická populáciaPoužitie tafamidisu sa netýka pediatrickej populácie.
Spôsob podávaniaPerorálne použitie.
Mäkké kapsuly sa majú prehltnúť celé a nemajú sa drviť ani deliť. Vyndaqel sa môže užívať s jedlom alebo bez jedla.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníŽeny vo fertilnom veku majú počas liečby tafamidisom používať primeranú antikoncepciu a pokračovať v jej používaní 1 mesiac po skončení liečby tafamidisom (pozri časť 4.6).
Tafamidis sa má pridať k štandardnej starostlivosti pri liečbe pacientov s transtyretínovou amyloidózou. V rámci tejto štandardnej starostlivosti majú lekári pacientov monitorovať a pokračovať v posudzovaní potreby inej liečby vrátane potreby orgánovej transplantácie. Keďže nie sú k dispozícii žiadne údaje týkajúce sa užívania tafamidisu po orgánovej transplantácii, liečba tafamidisom sa má ukončiť u pacientov, ktorí podstúpia orgánovú transplantáciu.
Môže sa objaviť zvýšenie funkčných testov pečene a zníženie tyroxínu (pozri časť 4.5 a 4.8). Tento liek obsahuje nie viac ako 44 mg sorbitolu v jednej kapsule.
Má sa vziať do úvahy aditívny účinok súbežne podávaných produktov obsahujúcich sorbitol (alebo fruktózu) a príjem sorbitolu (alebo fruktózy) v strave.
Obsah sorbitolu v liekoch na perorálne použitie môže ovplyvniť biologickú dostupnosť iných súbežne podávaných liekov na perorálne použitie.
4.5 Liekové a iné interakcieV klinickej štúdii u zdravých dobrovoľníkov 20 mg tafamidis meglumínu neindukovalo ani neinhibovalo enzým CYP3A4 cytochrómu P450.
Tafimidis
in vitro inhibuje eflux prenášača BCRP (proteín rezistencie rakoviny prsníka - breast cancer resistant protein) s IC50 = 1,16 µM a pri klinicky významných koncentráciách môže vyvolávať liekové interakcie so substrátmi tohto prenášača (napr. metotrexát, rosuvastatín, imatinib) po podaní
61 mg/deň dávky tafamidisu. Tafamidis takisto inhibuje vychytávanie prenášačov OAT1 s IC50 = 2,9
µM a OAT3 (prenášač organických aniónov - organic anion transporter) s IC50 = 2,36 µM
a pri klinicky významných koncentráciách môže vyvolávať liekové interakcie so substrátmi týchto prenášačov (napr. nesteroidové protizápalové lieky, bumetanid, furosemid, lamivudín, metotrexát, oseltamivir, tenofovir, ganciklovir, adefovir, cidofovir, zidovudín, zalcitabín). Na základe údajov
in vitro boli maximálne predikované zmeny v AUC pre OAT1 a OAT3 substráty stanovené menej ako
1,25 pri 61 mg dávke tafamidisu. Preto sa neočakáva, že by inhibícia prenášačov OAT1 alebo OAT3
prostredníctvom tafamidisu viedla ku klinicky signifikantným interakciám.
Neuskutočnili sa žiadne interakčné štúdie, ktoré by hodnotili účinok iných liekov na tafamidis.
AbnormalityvlaboratórnychtestochTafamidis môže znižovať sérové koncentrácie celkového tyroxínu bez sprievodnej zmeny voľného
tyroxínu (T4) alebo hormónu stimulujúceho štítnu žľazu (TSH - thyroid stimulating hormone). Toto pozorovanie týkajúce sa celkových hodnôt tyroxínu môže byť pravdepodobne spôsobené redukovaným viazaním tyroxínu na transtyretín (TTR) alebo jeho vytesňovaním z TTR v dôsledku vysokej väzobnej afinity tafamidisu k TTR receptoru pre tyroxín. Nepozorovali sa žiadne klinické nálezy, ktoré by zodpovedali dysfunkcii štítnej žľazy.
4.6 Fertilita, gravidita a laktáciaŽenyvofertilnom vekuZ dôvodu jeho predĺženého polčasu majú ženy vo fertilnom veku počas liečby tafamidisom a počas
jedného mesiaca po ukončení liečby používať antikoncepčné prostriedky.
GraviditaNie sú k dispozícii žiadne údaje o užívaní tafamidisu u gravidných žien. Štúdie na zvieratách
preukázali vývojovú toxicitu (pozri časť 5.3). Tafamidis sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
DojčenieDostupné údaje u zvierat preukázali vylučovanie tafamidisu do materského mlieka. Riziko
u novorodencov/dojčiat nemôže byť vylúčené. Tafamidis sa nemá užívať počas dojčenia.
FertilitaV predklinických štúdiách sa nepozorovala žiadna porucha fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojePodľa farmakodynamického a farmakokinetického profilu sa predpokladá, že tafamidis nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrnbezpečnostného profiluBezpečnostné údaje odrážajú expozíciu 176 pacientov s ATTR-CM 80 mg (podávanými ako
4 x 20 mg) tafamidis meglumínu podávanými denne počas 30-mesačného placebom kontrolovaného skúšania u pacientov s diagnostikovanou ATTR-CM (pozri časť 5.1).
Frekvencia nežiaducich udalostí u pacientov liečených 80 mg tafamidis meglumínu bola vo všeobecnosti podobná a porovnateľná s placebom.
Nasledovné nežiaduce udalosti boli hlásené častejšie u pacientov liečených tafamidis meglumínom 80 mg v porovnaní s placebom: flatulencia [8 pacientov (4,5%) verzus 3 pacienti (1,7%)], zvýšenie funkčných testov pečene [6 pacientov (3,4%) verzus 2 pacienti (1,1%)]. Kauzálna súvislosť nebola potvrdená.
Bezpečnostné údaje pre 61 mg tafamidisu nie sú k dispozícii, nakoľko sa táto formulácia v dvojito zaslepenej, placebom kontrolovanej, randomizovanej štúdii fázy 3 nehodnotila.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyS predávkovaním sú minimálne klinické skúsenosti. Počas klinických skúšaní dvaja pacienti
s diagnostikovanou ATTR-CM neúmyselne užili jednu 160 mg dávku tafamidis meglumínu bez toho, aby došlo k akýmkoľvek asociovaným nežiaducim udalostiam. Najvyššia dávka tafamidis meglumínu
podávaná zdravým dobrovoľníkom v klinickom skúšaní bola 480 mg ako jednorazová dávka. Pri tejto
dávke bol hlásený jeden nežiaduci účinok súvisiaci s liečbou, mierne hordeolum.
LiečbaV prípade predávkovania by sa mali podľa potreby vykonať štandardné podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné liečivá na centrálnu nervovú sústavu, ATC kód: N07XX08
Mechanizmus účinkuTafamidis je selektívny stabilizátor TTR. Tafamidis sa viaže na TTR vo väzobných miestach pre
tyroxín, pričom stabilizuje tetramér a spomaľuje jeho disociáciu na monoméry, čo je krok limitujúci rýchlosť amyloidogénneho procesu.
Farmakodynamické účinkyTranstyretínová amyloidóza je závažný invalidizujúci stav, ktorý je spôsobený akumuláciou rôznych
nerozpustných vláknitých proteínov, alebo amyloidu, v tkanivách v množstvách postačujúcich na poškodenie normálnej funkcie. Disociácia transtyretínového tetraméru na monoméry je krok určujúci rýchlosť v patogenéze transtyretínovej amyloidózy. Poskladané monoméry podstúpia čiastočnú denaturáciu a vytvoria inak poskladané monomérne amyloidogénne intermediáty. Tieto intermediáty sa potom chybne poskladajú do rozpustných oligomérov, profilamentov, filamentov
a amyloidových fibríl. Tafamidis sa s negatívnou kooperativitou viaže na dve väzobné miesta tyroxínu v prirodzenej tetramérnej forme transtyretínu, a tak zabraňuje disociácii na monoméry. Inhibícia disociácie TTR tetraméru je dôvodom na užívanie tafamidisu u pacientov s ATTR-CM.
TTR stabilizačný test sa využíval ako farmakodynamický marker a vyhodnocoval stabilitu TTR
tetraméru.
Tafamidis stabilizoval aj TTR tetramér divokého typu, aj tetraméry 14 TTR variantov, keď sa klinicky testoval po podávaní tafamidisu jedenkrát denne. Tafamidis stabilizoval aj TTR tetramér 25 variantov testovaných
ex vivo, čím sa demonštrovala stabilizácia TTR pre 40 amyloidogénnych TTR genotypov.
V multicentrickej, medzinárodnej, dvojito zaslepenej, placebom kontrolovanej, randomizovanej štúdii (pozri časť Klinická účinnosť a bezpečnosť) sa pozorovala stabilizácia TTR v 1. mesiaci a udržiavala sa až do 30. mesiaca.
Biomarkery asociované so srdcovým zlyhávaním (NT-proBNP a troponín I) favorizovali Vyndaqel oproti placebu.
Klinická účinnosťabezpečnosťÚčinnosť sa demonštrovala v multicentrickej, medzinárodnej, dvojito zaslepenej, placebom
kontrolovanej, randomizovanej 3-ramennej štúdii u 441 pacientov s wild type alebo hereditárnou
ATTR-CM.
Pacienti boli randomizovaní buď do skupiny, ktorej sa podával tafamidis meglumín 20 mg (n = 88) alebo 80 mg [podávaný ako štyri 20 mg kapsuly tafamidis meglumínu] (n = 176), alebo do zodpovedajúcej skupiny s placebom (n = 177) jedenkrát denne, navyše ku štandardnej starostlivosť (napr. diuretiká) počas 30 mesiacov. Priradenie liečby sa stratifikovalo podľa prítomnosti alebo neprítomnosti variantného TTR genotypu, ako aj podľa východiskovej závažnosti ochorenia
(triedy NYHA). Tabuľka 1 opisuje demografické údaje pacientov a ich východiskové charakteristiky.
Tabuľka 1: Demografické údaje pacientov a ich východiskové charakteristikyCharakteristika
| Tafamidis spolu
N = 264
| Placebo
N = 177
|
Vek — roky
|
Priemer (štandardná odchýlka)
| 74,5 (7,2)
| 74,1 (6,7)
|
Medián (minimum, maximum)
| 75 (46, 88)
| 74 (51, 89)
|
Pohlavie — počet (%)
|
Mužské
| 241 (91,3)
| 157 (88,7)
|
Ženské
| 23 (8,7)
| 20 (11,3)
|
TTR genotyp — počet (%)
|
ATTRm
| 63 (23,9)
| 43 (24,3)
|
ATTRwt
| 201 (76,1)
| 134 (75,7)
|
Trieda NYHA — počet (%)
|
|
|
NYHA I. triedy
| 24 (9,1)
| 13 (7,3)
|
NYHA II. triedy
| 162 (61,4)
| 101 (57,1)
|
NYHA III. triedy
| 78 (29,5)
| 63 (35,6)
|
Skratky: ATTRm = variantný transtyretínový amyloid, ATTRwt = transtyretínový amyloid divokého typu, NYHA = New
York Heart Association.
Primárna analýza využívala hierarchickú kombináciu aplikujúcu metódu podľa Finkelstein- Schoenfelda (F-S) na mortalitu zo všetkých príčin a frekvenciu hospitalizácií súvisiacich
s kardiovaskulárnym ochorením, ktorá bola definovaná ako počet hospitalizácií pacienta (tzn. prijatí
do nemocnice) z dôvodu morbidity súvisiacej s kardiovaskulárnym ochorením. Touto metódou sa porovnával každý pacient s každým iným pacientom v rámci každej vrstvy párovým spôsobom, ktorý prebiehal hierarchickým spôsobom využívajúcim mortalitu zo všetkých príčin a následne frekvenciu hospitalizácií súvisiacu s kardiovaskulárnym ochorením, keď sa pacienti nedali diferencovať na základe mortality.
Táto analýza demonštrovala signifikantnú redukciu (p = 0,0006) mortality zo všetkých príčin
a frekvencie hospitalizácií súvisiacich s kardiovaskulárnym ochorením v spojenej skupine tafamidis v dávkach 20 mg a 80 mg verzus placebo (tabuľka 2).
Tabuľka 2: Primárna analýza s použitím metódy podľa Finkelstein-Schoenfelda (F-S) pre mortalitu zo všetkých príčin a frekvencie hospitalizácií súvisiacich s kardiovaskulárnym ochorenímPrimárna analýza
| Tafamidis spolu
N = 264
| Placebo
N = 177
|
Počet (%) živých pacientov* v 30. mesiaci
| 186 (70,5)
| 101 (57,1)
|
Priemerný počet hospitalizácií súvisiacich
s kardiovaskulárnym ochorením počas 30 mesiacov (na pacienta za rok) u tých pacientov, ktorí žili v 30. mesiaci†
| 0,297
| 0,455
|
p-hodnota podľa F-S metódy
| 0,0006
|
* Transplantácia srdca a implantácia zariadenia na mechanickú podporu srdca sa považujú za indikátory blížiaceho sa terminálneho štádia. Z tohto dôvodu sa v analýze k týmto pacientom pristupovalo rovnako ako k prípadom úmrtia a títo pacienti neboli zahrnutí do „počtu živých pacientov v 30. mesiaci“, aj keď boli nažive na základe sledovacieho
vyhodnocovania životných funkcií v 30. mesiaci.
† Deskriptívny priemer tých pacientov, ktorí sú nažive po 30 mesiacoch.
Analýza individuálnych komponentov primárnej analýzy (mortalita zo všetkých príčin a hospitalizácia súvisiaca s kardiovaskulárnym ochorením) tiež demonštrovala významné redukcie pre tafamidis
verzus placebo.
Miera rizika na základe Coxovho modelu proporcionálnych rizík mortality zo všetkých príčin bola
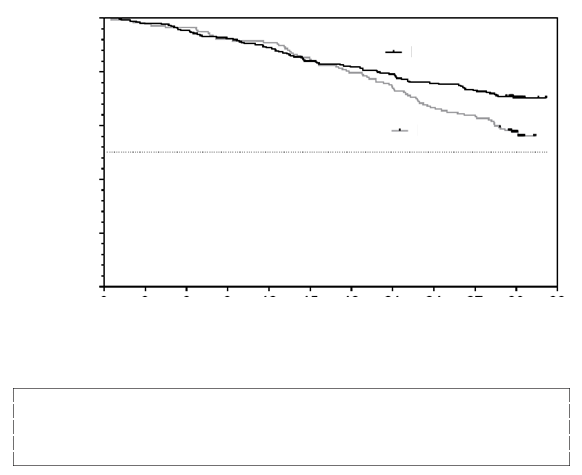
0,698 (95 % IS 0,508; 0,958), z čoho vyplýva 30,2 % redukcia rizika smrti v porovnaní s placebo skupinou (p = 0,0259). Kaplan-Meierov graf mortality zo všetkých príčin vo vzťahu k času je znázornený na obrázku 1.
Obrázok 1: Mortalita zo všetkých príčin
*
1.0
 0.8
0.6
0.4
0.8
0.6
0.4
 0.2
0.2
VYNDAQEL spolu
Placebo
0.0
0
3 6 9 12 15 18 21 24 27 30 33Čas od prvej dávky (mesiace)Naďalej ohrození pacienti
(Kumulatívne udalosti)
Spolu
| 264
| 259
| 252
| 244
| 235
| 222
| 216
| 209
| 200
| 193
| 99
| 0
|
VYNDAQEL
| 0
| 5
| 12
| 20
| 29
| 42
| 48
| 55
| 64
| 71
| 78
| 78
|
Placebo
|
177
|
173
|
171
|
163
|
161
|
150
|
141
|
131
|
118
|
113
|
51
|
0
|
| 0
| 4
| 6
| 14
| 16
| 27
| 36
| 46
| 59
| 64
| 75
| 76
|
* Transplantácia srdca a implantácia zariadenia na mechanickú podporu srdca sa považujú za indikátory blížiaceho sa
terminálneho štádia. Miera rizika na základe Coxovho modelu proporcionálnych rizík, pričom faktormi boli liečba,
TTR genotyp (variantový a štandardný) a východisková New York Heart Association (NYHA) klasifikácia (NYHA I. a II. Triedy spolu a NYHA III. triedy).
Pri podávaní tafamidisu došlo k významne nižšiemu počtu hospitalizácií súvisiacich
s kardiovaskulárnym ochorením ako pri placebe, s 32,4 % redukciou rizika (tabuľka 3).
Tabuľka 3: Frekvencia hospitalizácií súvisiacich s kardiovaskulárnym ochorením
| Tafamidis spolu
N = 264
| Placebo
N = 177
|
Celkový (%) počet pacientov s hospitalizáciami
súvisiacimi s kardiovaskulárnym ochorením
| 138 (52,3)
| 107 (60,5)
|
Hospitalizácie súvisiace s kardiovaskulárnym
ochorením za rok*
| 0,4750
| 0,7025
|
Rozdiel medzi liečbou tafamidisom a placebom
(relatívna miera rizika)*
| 0,6761
|
p-hodnota*
| < 0,0001
|
Skratka: NYHA=New York Heart Association.
* Táto analýza bola založená na Poissonovom regresnom modeli, pričom faktormi boli liečba, TTR genotyp (variantový alebo wild type), východisková New York Heart Association (NYHA) klasifikácia (spolu NYHA I. a II. triedy a NYHA III. triedy), interakcia medzi liečbou a TTR genotypom a interakcia medzi liečbou a východiskovou NYHA klasifikáciou.
Účinok liečby tafamidisom na funkčnú kapacitu a zdravotný stav sa vyhodnocoval 6-minútovým testom chôdze (6MWT) a skóre Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) (pozostávajúcom z domén pre celkové príznaky, fyzické limity, kvalitu života a sociálne limity). Významný účinok liečby favorizujúci tafamidis sa prvýkrát pozoroval v 6. mesiaci
a konzistentne pretrvával do 30. mesiaca ako pri 6MWT vzdialenosti, tak aj pri KCCQ-OS skóre
(tabuľka 4).'
Tabuľka 4: 6MWT a KCCQ-OS a zložkové doménové skóreKoncové ukazovatele
| Východiskový priemer
(SD)
| Zmena oproti východiskovej hodnote
v 30. mesiaci, LS priemer
(SE)
| Rozdiel medzi liečbou
a placebom LS priemer (95 % IS)
| p-hodnota
|
Tafamidis spolu
N = 264
| Placebo
N = 177
| Tafamidis spolu
| Placebo
|
6MWT* (metre)
| 350,55 (121,30)
| 353,26 (125,98)
| -54,87 (5,07)
| -130,55 (9,80)
| 75,68 (57,56; 93,80)
| p< 0,0001
|
KCCQ-OS*
| 67,27 (21,36)
| 65,90 (21,74)
| -7,16 (1,42)
| -20,81 (1,97)
| 13,65 (9,48; 17,83)
| p< 0,0001
|
*Vyššie hodnoty indikujú lepší zdravotný stav.
Skratky: 6MWT = 6-minútový test chôdze; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire-Overall Summary; LS = metóda najmenších štvorcov; IS = interval spoľahlivosti.
Výsledky F-S metódy prezentované prostredníctvom pomeru „výhier“ pre kombinovaný koncový ukazovateľ a jeho komponenty (mortalita zo všetkých príčin a frekvencia hospitalizácií súvisiacich s kardiovaskulárnym ochorením) konzistentne favorizovali tafamidis oproti placebu podľa dávky
a naprieč všetkými podskupinami (wild type, variantný a NYHA I. a II. triedy a III. triedy) okrem frekvencie hospitalizácií súvisiacich s kardiovaskulárnym ochorením v NYHA III. triedy (obrázok 2),
ktorých počet bol vyšší v skupine liečenej tafamidisom v porovnaní s placebom (pozri časť 4.2).
Analýzy 6MWT a KCCQ-OS tiež favorizovali tafamidis oproti placebu v každej podskupine.
Obrázok 2: Výsledky F-S metódy a komponenty podľa podskupín a dávky
F-
S Metóda *
(Pomer výhier 95 % lS)
Mortality zo všetkých príčin
 Miera rizika (95 % lS)
Frekvencia kardiovaskulárnych hospitalizácií
Pomer rizika (95 % lS)
Miera rizika (95 % lS)
Frekvencia kardiovaskulárnych hospitalizácií
Pomer rizika (95 % lS)
Celkovo – spolu
VYNDAQEL vs Placebo
|
|
|
|
|
|
|
|
|
|
|
|
|
TT
R
Genotyp
|
|
|
|
|
|
|
|
|
|
|
|
|
ATTRm (24%)
|
|
|
|
|
|
|
|
|
|
|
|
|
ATTRwt (76%)
|
|
|
|
|
|
|
|
|
|
|
|
|
NYHA východisková hodnota
|
|
|
|
|
|
|
|
|
|
|
|
|
I
. alebo II. trieda (68%)
|
|
|
|
|
|
|
|
|
|
|
|
|
III. trieda (32%)
|
|
|
|
|
|
|
|
|
|
|
|
|
Dávka
|
|
|
|
|
|
|
|
|
|
|
|
|
80 mg (40%) vs placebo (40%)
|
|
|
|
|
|
|
|
|
|
|
|
|
20 mg (20%) vs placebo (40%)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4
|
2
|
1
|
0
,
5
|
0,25
|
0,5
|
1
|
2
|
0,25
|
0.5
|
1
|
2
|
Favorizuje VYNDAQEL
Favorizuje Placebo
Favorizuje VYNDAQEL
F
a
vorizuje Placebo
Favorizuje VYNDAQEL
Favorizuje Placebo
Skratky: ATTRm = variantný transtyretínový amyloid, ATTRwt = transtyretínový amyloid divokého typu, F - S = Finkelstein-Schoenfeld, IS = interval spoľahlivosti.
* F-S výsledky prezentované použitím pomeru „výhier“(na základe mortality zo všetkých príčin a frekvencie hospitalizácií v súvislosti s kardiovaskulárnym ochorením). Pomer „výhier“ je počet párov liečených pacientov, ktorí boli úspešní,
vydelený počtom párov pacientov v placebo skupine, ktorí boli úspešní.
Transplantácia srdca a implantácia zariadenia na mechanickú podporu srdca sa považujú za rovnaké prípady ako úmrtie
Aplikovaním metódy F-S na každú dávkovú skupinu individuálne, tafamidis redukoval kombináciu mortality zo všetkých príčin a frekvenciu hospitalizácií súvisiacich s kardiologickým ochorením pre ako 80 mg tak aj 20 mg dávky v porovnaní s placebom (p=0,0030 a p=0,0048 v tomto poradí). Výsledky primárnej analýzy, 6MWT v 30. mesiaci boli štatisticky signifikantné pre dávky tafamidis meglumínu 80 mg aj 20 mg v porovnaní s placebom, s podobnými výsledkami pre obe dávky.
Dáta o účinnosti pre 61 mg tafamidisu nie sú k dispozícii, nakoľko táto formulácia sa nehodnotila v dvojito zaslepenej, placebom kontrolovanej, randomizovanej štúdii fázy 3. Relatívna biologická
dostupnosť tafamidisu 61 mg je podobná tafamidis meglumínu 80 mg v rovnovážnom stave (pozri časť 5.2).
Supraterapeutická jednorazová perorálna dávka 400 mg roztoku tafamidis meglumínu u zdravých dobrovoľníkov nepreukázala žiadne predĺženie QTc intervalu.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdii s tafamidisom vo všetkých podskupinách pediatrickej populácie s transtyretínovou amyloidózou (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po perorálnom podaní mäkkej kapsuly jedenkrát denne sa maximálna vrcholová koncentrácia (Cmax)
dosiahne za strednú dobu (tmax) 4 hodiny pre tafamidis 61 mg a 2 hodiny pre tafamidis meglumín 80 mg (4 x 20 mg), ak sa dávka užije nalačno. Súbežné požitie vysokokalorického jedla s vysokým obsahom tuku zmenilo rýchlosť absorpcie, ale nie rozsah absorpcie. Tieto výsledky podporujú
podávanie tafamidisu s jedlom alebo bez jedla.
Distribúcia
Tafamidis sa výrazne viaže na plazmatické proteíny (> 99 %). Zdanlivý distribučný objem
v ustálenom stave je 18,5 litra.
Rozsah väzby tafamidisu na plazmatické proteíny sa vyhodnocoval s použitím živočíšnej a ľudskej plazmy. Afinita tafamidisu k TTR je vyššia ako afinita pre albumín. Preto je tafamidis náchylný preferenčne sa viazať k TTR napriek signifikantne vyššej koncentrácii albumínu (600 μM)
v porovnaní s koncentráciou TTR (3,6 μM).
Biotransformácia aeliminácia
O vylučovaní tafamidisu žlčou u ľudí neexistuje jednoznačný dôkaz. Na základe predklinických
údajov možno tvrdiť, že tafamidis sa metabolizuje glukuronidáciou a vylučuje prostredníctvom žlče. Táto cesta biotransformácie je u ľudí prijateľná, pretože približne 59 % z celkovej užitej dávky sa
objaví v stolici a približne 22 % v moči. Na základe výsledkov populačnej farmakokinetiky je zdanlivý perorálny klírens tafamidis meglumínu 0,263 l/h a priemerný polčas v populácii je približne
49 hodín.
Linearitadávky ačasu
Expozícia tafamidis meglumínu pri dávkovaní jedenkrát denne sa zvyšovala so zvyšovaním dávky až
do 480 mg pri jednej dávke a pri viacerých dávkach až do 80 mg/deň. Vo všeobecnosti bolo zvyšovanie priamo úmerné alebo takmer priamo úmerné dávke a klírens tafamidisu bol v danom čase stabilný.
Relatívna biologická dostupnosť tafamidisu 61 mg je podobná relatívnej biologickej dostupnosti tafamidis meglumínu 80 mg v ustálenom stave. Tafamidis a tafamidis meglumín nie sú zameniteľné na mg báze.
Farmakokinetické parametre boli podobné po jednorazovom a opakovanom podaní 20 mg tafamidis meglumínu, čo naznačuje nedostatočnú indukciu alebo inhibíciu metabolizmu tafamidisu.
Účinky podávania tafamidis meglumínu v dávke 15 mg až 60 mg perorálneho roztoku po dobu 14 dní preukázali, že sa rovnovážny stav dosiahol na 14. deň.
Osobitnéskupiny pacientov
Porucha funkcie pečene
Farmakokinetické údaje svedčia o zníženej systémovej expozícii (približne 40 %) a zvýšenom celkovom klírense (0,52 l/h oproti 0,31 l/h) tafamidis meglumínu u pacientov so stredne závažnou poruchou funkcie pečene (skóre podľa Childovej-Pughovej klasifikácie v rozmedzí 7 – 9 vrátane)
v porovnaní so zdravými dobrovoľníkmi v dôsledku vyššej nenaviazanej frakcie tafamidisu. Aj keď pacienti so stredne závažnou poruchou funkcie pečene vykazujú nižšie hladiny TTR ako zdraví
jednotlivci, úprava dávky nie je potrebná, pretože stoichiometria tafamidisu s jeho cieľovým
proteínom TTR bude dostatočná na stabilizáciu TTR tetraméru. Expozícia tafamidisu u pacientov so závažnou poruchou funkcie pečene nie je známa.
Porucha funkcie obličiek
Tafamidis nebol osobitne vyhodnocovaný v štúdii zameranej na pacientov s poruchou funkcie obličiek. Vplyv klírensu kreatinínu na farmakokinetiku tafamidisu sa vyhodnocoval v populačnej farmakokinetickej analýze u pacientov s klírensom kreatinínu vyšším ako 18 ml/min. Farmakokinetické odhady neindikovali žiadny rozdiel v zdanlivom perorálnom klírense tafamidisu u pacientov s klírensom kreatinínu nižším ako 80 ml/min v porovnaní s tými, ktorých klírens kreatinínu bol vyšší alebo rovný 80 ml/min. Úprava dávky u týchto pacientov sa nepovažuje za potrebnú.
Starší
Na základe výsledkov z populačnej farmakokinetiky mali pacienti vo veku ³ 65 rokov v priemere
o 15 % nižšiu odhadovanú hodnotu zdanlivého perorálneho klírensu v rovnovážnom stave v porovnaní s pacientmi mladšími ako 65 rokov. Tento rozdiel v klírense však viedol k < 20 % zvýšeniam
priemerných hodnôt Cmax a AUC v porovnaní s mladšími pacientmi a nie je klinický významný.
Farmakokinetické/farmakodynamické vzťahy
Z in vitro údajov vyplýva, že tafamidis významne neinhibuje enzýmy cytochrómu P450 CYP1A2,
CYP3A4, CYP3A5, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. Neočakáva sa, že by tafamidis spôsoboval klinicky relevantné liekové interakcie v dôsledku indukcie CYP1A2, CYP2B6 alebo CYP3A4.
In vitro štúdie naznačujú, že nie je pravdepodobné, že tafamidis v klinicky relevantných koncentráciách bude spôsobovať systémové liekové interakcie so substrátmi UDP glukuronozyltransferázy (UGT). Tafamidis môže inhibovať intestinálne aktivity UGT1A1.
Tafamidis v klinicky relevantných koncentráciách preukazoval nízky potenciál inhibovať proteín multiliekovej rezistencie (Multi-Drug Resistant Protein - MDR1) (známy aj ako P-glykoproteín; P-gp) systémovo a v gastointestinálnom trakte (GI), inhibovať prenášač organických katiónov 2 (OCT2), multiliekový a toxínový extrúzny prenášač 1 (multidrug and toxin extrusion transporter 1 - MATE1) a MATE2K, polypeptid prenášajúci organické anióny 1B1 (organic anion transporting polypeptide 1B1
- OATP1B1) a OATP1B3.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, reprodukčnej toxicity a vývinu, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. V štúdiách toxicity po opakovanom podávaní a štúdiách karcinogenity sa pečeň prejavila ako cieľový orgán toxicity u rozdielnych skúmaných druhov. Účinky na pečeň sa pozorovali pri expozíciách približne rovnakých ako je AUC u ľudí v ustálenom stave pri klinickej dávke 61 mg tafamidisu.
Vo vývinovej štúdii toxicity na zajacoch sa pozoroval mierny nárast malformácií a zmien kostry, potraty u niekoľkých samičiek, redukované embryofetálne prežívanie a zníženie fetálnej hmotnosti pri expozíciách približne ³ 2,1-krát vyšších ako AUC v rovnovážnom stave u ľudí pri klinickej dávke
61 mg tafamidisu.
Vo vývinovej štúdii s tafamidisom zameranej na prenatálny a postnatálny vývin potkanov bolo počas gravidity a laktácie po podávaní maternálnej dávky 15 a 30 mg/kg/deň zaznamenané znížené prežívanie mláďat a pokles hmotnosti. Znížené hmotnosti mláďat samčekov sa spájali s oneskoreným pohlavným dospievaním (oddelenie predkožky) pri 15 mg/kg/deň. Znížená výkonnosť v teste Morrisonovým vodným labyrintom, zameranom na učenie a pamäť sa pozorovala pri 15 mg/kg/deň. NOAEL (hladina, pri ktorej neboli pozorované nežiaduce účinky) pre životaschopnosť a rast potomstva prvej (F1) generácie po podávaní maternálnej dávky počas gravidity a laktácie
s tafamidisom predstavovala 5 mg/kg/deň (human equivalent dose, ekvivalentná dávka pre človeka
= 0,8 mg/kg/deň), čo sa približne rovná klinickej dávke 61 mg tafamidisu.
6. FARMACEUTICKÉ INFORMÁCIE
6.2 Zoznam pomocných látok
Obal kapsuly
želatína (E 441)
glycerín (E 422)
červený oxid železitý (E 172)
sorbitan
sorbitol (E 420)
manitol (E 421)
čistená voda
Obsah kapsuly
makrogol 400 (E 1521)
polysorbát 20 (E 432) povidón (K-hodnota 90) butylovaný hydroxytoluén (E 321)
Potlačový atrament (biela Opacode)
etylalkohol
izopropylalkohol čistená voda
makrogol 400 (E 1521) ftalát polyvinylacetátu propylénglykol (E 1520)
oxid titaničitý (E 171)
28 % hydroxid amónny (E 527)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Žiadne.
6.5 Druh obalu a obsah baleniaPVC/PA/alu/PVC-alu/PET/Papierové perforované blistre s jednotlivými dávkami.
Veľkosť balenia: balenie po 30 x 1 mäkká kapsula a multibalenie obsahujúce 90 (3 balenia po 30 x 1)
mäkkých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLAEU/1/11/717/003
EU/1/11/717/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 16. november 2011
Dátum posledného predĺženia registrácie: 22. júl 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.