
nózne použitie. Začiatočná nasycovacia dávka sa podáva formou
intravenóznej infúzie, ktorá trvá 90 minút. Nepodávať formou intravenóznej injekcie alebo bolusu. Intravenóznu infúziu Trazimery má podávať zdravotnícky pracovník pripravený zvládnuť anafylaxiu a
má byť k dispozícii pohotovostná súprava. Pacienti sa majú sledovať najmenej šesť hodín po začatí prvej infúzie a dve hodiny po začatí nasledujúcich infúzií na príznaky ako horúčka a triaška alebo iné
príznaky súvisiace s infúziou (pozri časti 4.4 a 4.8). Prerušenie alebo spomalenie rýchlosti podávania infúzie môže pomôcť pri kontrole týchto príznakov. Po ústupe ťažkostí sa môže pokračovať v infúzii.
V prípade dobrej znášanlivosti začiatočnej nasycovacej dávky sa môžu podávať udržiavacie dávky formou 30-minútovej infúzie.
Pokyny na rekonštitúciu Trazimery na intravenózne podanie pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
• Precitlivenosť na trastuzumab, myšie bielkoviny alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• Závažná dýchavičnosť v pokoji v dôsledku komplikácií pokročilého nádorového ochorenia alebo stav vyžadujúci doplnkovú oxygenoterapiu.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovanosť
Na zlepšenie sledovanosti biologických liekov sa má zreteľne zaznamenávať obchodný názov lieku a
číslo šarže podaného lieku.
Vyšetrenie receptora HER2 sa musí vykonať v špecializovanom laboratóriu, ktoré dokáže zaručiť primeranú validáciu testovacích postupov (pozri časť 5.1).
V súčasnosti nie sú dostupné žiadne údaje z klinických skúšaní o opakovanej liečbe u pacientov s predchádzajúcou expozíciou trastuzumabu v adjuvantnom režime.
Srdcová dysfunkcia
Všeobecné faktory
Pacienti liečení Trazimerou sú vystavení zvýšenému riziku rozvoja kongestívneho srdcového zlyhania
(triedy II-IV Newyorkskej srdcovej asociácie [NYHA]) alebo asymptomatickej srdcovej dysfunkcii. Tieto udalosti sa pozorovali u pacientov liečených trastuzumabom samotným alebo v kombinácii
s paklitaxelom alebo docetaxelom, predovšetkým po chemoterapii obsahujúcej antracyklín (doxorubicín alebo epirubicín). Môžu mať stredne závažný až závažný priebeh a môžu byť spojené s úmrtím (pozri časť 4.8). Okrem toho sa odporúča opatrnosť pri liečbe pacientov so zvýšeným
rizikom srdcového poškodenia napr. hypertenzia, zdokumentovaná choroba koronárnych artérií, kongestívne srdcové zlyhanie, LVEF < 55 %, vyšší vek.
Všetci kandidáti na liečbu Trazimerou, najmä pacienti, ktorým predtým podávali antracyklíny
a cyklofosfamid (AC), sa musia zúčastniť vstupného vyšetrenia srdca, vrátane anamnézy a fyzikálneho vyšetrenia, elektrokardiogramu (EKG), echokardiogramu a/alebo scintigrafického vyšetrenia (MUGA)
alebo vyšetrenia magnetickou rezonanciou. Sledovanie môže pomôcť pri identifikácii pacientov,
u ktorých sa vyvinie srdcová dysfunkcia. Vyšetrenie srdca uskutočnené pred začiatkom liečby sa má opakovať každé 3 mesiace počas liečby a každých 6 mesiacov po vysadení liečby až do 24 mesiacov po poslednom podaní Trazimery. Pred rozhodnutím o liečbe Trazimerou sa má starostlivo zvážiť pomer rizika a prínosu tejto liečby.
Trastuzumab, ako ukazujú všetky dostupné údaje získané z populačných farmakokinetických (FK) analýz (pozri časť 5.2), môže pretrvávať v obehu až 7 mesiacov po ukončení podávania. U pacientov, ktorí dostávajú antracyklíny, po ukončení liečby trastuzumabom existuje vyššie riziko srdcovej dysfunkcie. Lekári sa majú podľa možností snažiť vyhnúť podávaniu antracyklínov najmenej
7 mesiacov po ukončení liečby trastuzumabom. Ak sa antracyklíny použijú, má sa u pacientov pozorne sledovať funkcia srdca.
Štandardizované kardiologické vyšetrenie sa má zvážiť u pacientov, u ktorých existujú obavy
z kardiovaskulárneho ochorenia po vstupnom skríningu. Počas liečby sa má u všetkých pacientov ďalej sledovať funkcia srdca (napr. každých 12 týždňov). Uvedené sledovanie môže pomôcť
identifikovať pacientov, u ktorých sa rozvíja porucha funkcie srdca. Pacienti, u ktorých sa rozvíja asymptomatická dysfunkcia srdca, môžu mať úžitok z častejšieho sledovania (napr. každých 6 až 8
týždňov). U asymptomatických pacientov s progresívnym zhoršovaním funkcie ľavej komory musí lekár zvážiť ukončenie liečby trastuzumabom, ak liečba nemá žiadny klinický prínos.
Bezpečnosť pokračujúcej alebo znovu začatej liečby trastuzumabom u pacientov, ktorí prekonali srdcovú dysfunkciu nebola prospektívne študovaná. Ak percento ejekčnej frakcie (EF) LVEF poklesne viac ako o ³ 10 bodov od bazálnej hodnoty A pod 50 %, liečba sa má zastaviť a približne do 3 týždňov sa má vykonať opakované hodnotenie LVEF. Ak sa LVEF nezlepší alebo ak sa ešte viac zhorší alebo sa vyvinie symptomatické kongestívne srdcové zlyhanie (KSZ), má sa dôkladne zvážiť ukončenie liečby trastuzumabom, ak prínos z liečby pre pacienta neprevýši jej riziká. Všetci takýto pacienti sa majú odporučiť na vyšetrenie u kardiológa a naďalej sledovať.
Ak počas liečby Trazimerou dôjde k symptomatickému srdcovému zlyhávaniu, má sa liečiť štandardnými liekmi na srdcové zlyhanie (KSZ). Väčšina pacientov, u ktorých sa vyvinulo kongestívne SZ alebo asymptomatická srdcová dysfunkcia v hlavných štúdiách, dosiahla zlepšenie štandardnou liečbou KSZ pozostávajúcou z inhibítora angiotenzín-konvertujúceho enzýmu (ACE) alebo z blokátora angiotenzínového receptora (ARB) a beta-blokátora. Väčšina pacientov
s kardiálnymi príznakmi a preukázateľným klinickým prínosom z liečby trastuzumabom naďalej pokračovala v liečbe bez toho, že by sa u nich objavili ďalšie klinicky významné srdcové udalosti.
Metastatický karcinóm prsníka
Trazimera a antracyklíny sa nemajú podávať súbežne v kombinácii v prípade MKP.
Pacienti s MKP, ktorí dostávali predtým antracyklíny, sú tiež vystavení riziku kardiálnej dysfunkcie pri liečbe Trazimerou, hoci riziko je nižšie ako pri súbežnom používaní Trazimery a antracyklínov.
Včasný karcinóm prsníka
U pacientov s VKP sa má vyšetrenie srdca, ktoré sa robilo na začiatku liečby, opakovať každé 3
mesiace počas liečby a každých 6 mesiacov po ukončení liečby až do 24 mesiacov od posledného podania Trazimery. U pacientov, ktorí dostávajú atracyklínovú chemoterapiu sa odporúča ďalšie sledovanie jedenkrát ročne počas 5 rokov od posledného podania Trazimery alebo dlhšie, ak je pozorovaný kontinuálny pokles LVEF.
U pacientov s infarktom myokardu (IM) v anamnéze, angínou pektoris vyžadujúcou liečbu,
s existujúcim kongestívnym srdcovým zlyhaním (triedy II-IV NYHA) alebo týmto ochorením
v anamnéze, LVEF < 55 %, inou kardiomyopatiou, srdcovou arytmiou vyžadujúcou liečbu, klinicky významným ochorením srdcových chlopní, slabo kontrolovanou hypertenziou (hypertenzia
kontrolovaná štandardnou liečbou) a hemodynamicky významným perikardiálnym výpotkom boli
vylúčení z adjuvantných a neoadjuvantných hlavných štúdií VKP s trastuzumabom, a preto nie je možné u týchto pacientov odporúčať liečbu.
Adjuvantná liečba
Trazimera a antracyklíny sa nemajú podávať súbežne v kombinácii v prípade adjuvantnej liečby.
U pacientov s VKP bol pozorovaný zvýšený výskyt symptomatických a asymptomatických srdcových udalostí, keď sa trastuzumab podával po chemoterapii, ktorá obsahovala antracyklíny v porovnaní
s podaním v režime docetaxel a karboplatina bez antracyklínov a bol výraznejší, keď sa trastuzumab podával súbežne s taxánmi, než keď sa podával sekvenčne. Bez ohľadu na použitý režim, väčšina
symptomatických srdcových udalostí sa vyskytla počas prvých 18 mesiacov. V jednej z 3 hlavných klinických štúdií, v ktorej bol medián sledovania 5,5 rokov (BCIRG006), bol pozorovaný kontinuálny
nárast kumulatívnej miery výskytu symptomatických srdcových alebo LVEF udalostí u pacientov, ktorým bol trastuzumab podávaný súbežne s taxánmi po liečbe antracyklínmi až 2,37 % v porovnaní s približne 1 % v dvoch porovnávacích ramenách (antracyklíny plus cyklofosfamid nasledované
taxánmi a taxány, karboplatina a trastuzumab).
Rizikové faktory pre srdcové udalosti identifikované v štyroch veľkých adjuvantných štúdiách zahŕňali vyšší vek (> 50 rokov), nízku východiskovú hodnotu LVEF (< 55 %) pred začiatkom liečby paklitaxelom alebo po jej začatí, zníženie LVEF o 10 – 15 bodov a predchádzajúce alebo súbežné používanie antihypertenzív. U pacientov liečených trastuzumabom po skončení adjuvantnej chemoterapie bolo riziko srdcovej dysfunkcie spojené s vyššou kumulatívnou dávkou antracyklínu podanou pred začatím liečby trastuzumabom a indexom telesnej hmotnosti (BMI) > 25 kg/m2.
Neoadjuvantná-adjuvantná liečba
U pacientov s VKP vhodných pre neoadjuvantnú-adjuvantnú liečbu sa Trazimera môže používať súbežne s antracyklínmi len u pacientov bez predchádzajúcej chemoterapie a len s nízko dávkovými antracyklínovými režimami napr. maximálne kumulatívne dávky doxorubicínu 180 mg/m2 alebo epirubicínu 360 mg/m2.
Ak boli pacienti liečení súbežne kompletným režimom s nízko dávkovanými antracyklínmi
a Trazimerou v neoadjuvantnej liečbe, po operácii sa nemá podávať žiadna ďalšia cytotoxická chemoterapia. V ostatných prípadoch sa rozhodnutie o potrebe ďalšej cytotoxickej chemoterapie
stanovuje na základe individuálnych faktorov.
Skúsenosti so súbežným podávaním trastuzumabu s nízkymi dávkami antracyklínov sú v súčasnosti limitované na 2 klinické skúšania (MO16432 a BO22227).
V hlavnom klinickom skúšaní MO16432 sa trastuzumab podával súbežne s neoadjuvantnou chemoterapiou, obsahujúcou tri cykly doxorubicínu (kumulatívna dávka 180 mg/m2).
Incidencia symptomatickej srdcovej dysfunkcie bola v skupinách s trastuzumabom 1,7 %.
V hlavnom klinickom skúšaní BO22227 sa trastuzumab podával súbežne s neoadjuvantnou chemoterapiou, obsahujúcou 4 cykly epirubicínu (kumulatívna dávka 300 mg/m2); pri mediáne následného sledovania (follow-up) viac ako 70 mesiacov bola incidencia srdcového zlyhávania/kongestívneho srdcového zlyhávania 0,3 % v skupine s trastuzumabom podávaným intravenózne.
Klinické skúsenosti u pacientov vo veku viac ako 65 rokov sú obmedzené. Reakciesúvisiacesinfúziou(IRR)aprecitlivenosť
Hlásené boli závažné IRR na infúziu trastuzumabu zahŕňajúce dýchavičnosť, hypotenziu, sipot,
hypertenziu, bronchospazmus, supraventrikulárnu tachyarytmiu, nižšiu saturáciu krvi kyslíkom, anafylaxiu, respiračnú tieseň, urtikáriu a angioedém (pozri časť 4.8). Na zníženie rizika výskytu udalostí súvisiacich s podaním sa môže použiť premedikácia. Väčšina z uvedených nežiaducich udalostí sa objavuje počas prvej infúzie alebo do 2,5 hodiny od začiatku prvej infúzie. Ak dôjde
k takejto infúznej reakcii, infúzia sa má ukončiť a pacienta je potrebné sledovať až do vymiznutia sledovaných príznakov (pozri časť 4.2). Tieto príznaky môžu byť liečené analgetikami/antipyretikami
napr. meperidín alebo paracetamol alebo antihistaminikami napr. difenhydramín. U väčšiny pacientov
príznaky ustúpili a následne im boli podané ďalšie infúzie trastuzumabu. Závažné reakcie sa úspešne zvládli pomocou podpornej liečby kyslíkom, beta-agonistami a kortikosteroidmi. V zriedkavých prípadoch sú tieto reakcie spojené s klinickým priebehom s fatálnymi následkami. U pacientov
s dýchavičnosťou v pokoji v dôsledku komplikácií pokročilého nádorového ochorenia a pridružených ochorení existuje zvýšené riziko fatálnej infúznej reakcie. Z toho dôvodu nesmú byť títo pacienti
liečení trastuzumabom (pozri časť 4.3).
Taktiež sa opísali prípady úvodného zlepšenia, po ktorom nasledovalo zhoršovanie klinického stavu, pričom sa tiež zaznamenali oneskorené reakcie s rýchlym zhoršovaním klinického stavu. K úmrtiam pacientov došlo v priebehu niekoľkých hodín až jedného týždňa po začatí podávania infúzie.
Vo veľmi zriedkavých prípadoch sa u pacientov objavili symptómy z podania infúzie a pľúcne symptómy viac ako šesť hodín po začatí podávania infúzie trastuzumabu. Pacienti sa majú upozorniť na možnosť vzniku neskorších ťažkostí a na nutnosť vyhľadať lekára v prípade spozorovania uvedených príznakov.
Pľúcneudalosti
Po uvedení lieku na trh sa zaznamenali prípady závažných pľúcnych udalostí v súvislosti
s používaním trastuzumabu (pozri časť 4.8). Príležitostne boli tieto udalosti fatálne. Okrem toho sa zaznamenali prípady intersticiálnej choroby pľúc, vrátane pľúcnych infiltrátov, akútneho syndrómu respiračnej tiesne, pneumónie, pneumonitídy, pleurálnych výpotkov, respiračnej tiesne, akútneho edému pľúc a respiračnej insuficiencie. Rizikové faktory spájané s intersticiálnou chorobou pľúc zahŕňajú predchádzajúcu alebo súbežnú liečbu s inými antineoplastickými terapiami ako taxány, gemcitabín, vinorelbín a radiačná liečba. Tieto udalosti môžu byť súčasťou infúznej reakcie alebo sa môžu objaviť neskôr. U pacientov s dýchavičnosťou v pokoji v dôsledku komplikácií pokročilého nádorového ochorenia a pridružených ochorení existuje zvýšené riziko pľúcnych udalostí. Preto sa nemajú títo pacienti liečiť trastuzumabom (pozri časť 4.3). Obozretnosť je potrebná pri pneumonitíde, zvlášť u pacientov, ktorí sa liečia súbežne s taxánmi.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne oficiálne interakčné štúdie. V klinických skúšaniach neboli pozorované klinicky významné interakcie medzi trastuzumabom a súbežne užívanými liekmi.
Vplyv trastuzumabunafarmakokinetikuinýchantineoplastickýchlátok
Farmakokinetické údaje zo štúdií BO15935 a M77004 u žien s HER2-pozitívnym MKP naznačili, že
expozícia paklitaxelu a doxorubicínu (a ich hlavným metabolitom 6-α hydroxyl-paklitaxelu, POH,
a doxorubicinolu, DOL) nebola zmenená za prítomnosti trastuzumabu (pri intravenóznej nasycovacej dávke 8 mg/kg s intravenóznou udržiavacou dávkou 6 mg/kg každé 3 týždne alebo pri intravenóznej nasycovacej dávke 4 mg/kg s intravenóznou udržiavacou dávkou 2 mg/kg raz týždenne). Trastuzumab však môže zvýšiť celkovú expozíciu jedného metabolitu doxorubicínu (7-deoxy-13 dihydro- doxorubicinónu, D7D). Biologická aktivita D7D a klinický vplyv zvýšenia tohto metabolitu neboli známe.
Údaje zo štúdie JP16003 s jedným ramenom s trastuzumabom (4 mg/kg intravenózna nasycovacia dávka a 2 mg/kg intravenózna dávka raz týždenne) a docetaxelom (60 mg/m2 intravenózna dávka)
u japonských žien s HER2-pozitívnym MKP naznačili, že súbežné podávanie trastuzumabu nemalo
žiadny vplyv na farmakokinetiku docetaxelu po jednorazovej dávke. Štúdia JP19959 bola podštúdia štúdie BO18255 (ToGA) u mužských a ženských japonských pacientov s pokročilým karcinómom žalúdka, v ktorej sa sledovala farmakokinetika kapecitabínu a cisplatiny používanými
s trastuzumabom alebo bez trastuzumabu. Výsledky tejto podštúdie naznačili, že expozícia biologicky aktívnym metabolitom (napr. 5-FU) kapecitabínu nebola ovplyvnená súbežným používaním cisplatiny
ani súbežným používaním cisplatiny s trastuzumabom. Samotný kapecitabín však preukázal vyššie koncentrácie a dlhší polčas pri kombinovaní s trastuzumabom. Údaje tiež naznačili, že farmakokinetika cisplatiny nebola ovplyvnená súbežným používaním kapecitabínu ani súbežným
používaním kapecitabínu s trastuzumabom.
Farmakokinetické údaje zo štúdie H4613g/GO01305 u pacientov s metastatickým alebo lokálne pokročilým neoperovateľným HER2- pozitívnym karcinómom naznačili, že trastuzumab nemal žiadny vplyv na farmakokinetiku karboplatiny.
Vplyvantineoplastických látoknafarmakokinetikutrastuzumabu
Porovnaním simulovaných sérových koncentrácií trastuzumabu po monoterapii trastuzumabom
(4 mg/kg nasycovacia dávka/2 mg/kg raz týždenne intravenózne) a pozorovaných sérových koncentrácií u japonských žien s HER2-pozitívnym MKP (štúdia JP16003) sa nezistil žiadny dôkaz o FK vplyve súbežného podávania docetaxelu na farmakokinetiku trastuzumabu.
Porovnanie FK výsledkov z dvoch štúdií fázy II (BO15935 a M77004) a jednej štúdie fázy III (H0648g), v ktorých boli pacienti liečení súbežne s trastuzumabom a paklitaxelom, a z dvoch štúdií fázy II, v ktorých bol trastuzumab podávaný v monoterapii (W016229 a MO16982), u žien s HER2- pozitívnym MKP naznačuje, že individuálne a priemerné minimálne sérové koncentrácie trastuzumabu sa odlišujú v rámci štúdií a medzi jednotlivými štúdiami, nezistil sa však žiadny jednoznačný vplyv súbežného podávania paklitaxelu na farmakokinetiku trastuzumabu. Porovnanie FK údajov o trastuzumabe zo štúdie M77004, v ktorej boli ženy s HER2-pozitívnym MKP liečené súbežne trastuzumabom, paklitaxelom a doxorubicínom, s FK údajmi trastuzumabu v štúdiách, kde bol trastuzumab podávaný ako monoterapia (H0649g) alebo v kombinácii s antracyklínom a cyklofosfamidom alebo paklitaxelom (štúdia H0648g), naznačili, že doxorubicín a paklitaxel nemali na farmakokinetiku trastuzumabu žiadny vplyv.
Farmakokinetické údaje zo štúdie H4613g/GO01305 nasvedčujú tomu, že karboplatina nemala žiadny vplyv na FK trastuzumabu.
Nezdá sa, že súbežné podávanie anastrozolu ovplyvňuje farmakokinetiku trastuzumabu.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženám vo fertilnom veku sa má odporučiť, aby používali účinnú antikoncepciu počas liečby
Trazimerou a 7 mesiacov po ukončení liečby (pozri časť 5.2).
Gravidita
Štúdie reprodukčnej toxicity sa vykonali na opiciach rodu Cynomolgus, ktoré dostávali až 25-násobok
týždennej udržiavacej dávky trastuzumabu na intravenózne použitie u ľudí 2 mg/kg. U exponovaných opíc sa nezistila žiadna porucha fertility, ani žiadny škodlivý vplyv na plod. Počas včasného (gestačné dni: 20 až 50) a neskorého (gestačné dni: 120 až 150) obdobia vývoja plodu bol pozorovaný prestup trastuzumabu cez placentu. Nie je známe, či trastuzumab ovplyvňuje schopnosť reprodukcie. Keďže na základe reprodukčných štúdií uskutočnených na zvieratách nie je vždy možné predvídať odpoveď
u ľudí, trastuzumab sa má podávať počas gravidity iba vtedy, ak jeho prínos pre matku prevýši jeho riziko pre plod.
Po uvedení lieku na trh boli u gravidných žien užívajúcich trastuzumab hlásené prípady poruchy renálneho rastu plodu a/alebo poruchy funkcie obličiek v súvislosti s oligohydramniónom, niektoré spojené s fatálnou pľúcnou hypopláziou plodu. Ženy, ktoré otehotnejú, treba informovať o možnosti poškodenia plodu. Ak je gravidná žena liečená trastuzumabom, alebo ak pacientka otehotnie počas užívania trastuzumabu alebo do 7 mesiacov po poslednej dávke trastuzumabu, je potrebná prísna kontrola multidisciplinárnym tímom.
Dojčenie
V štúdii uskutočnenej na laktujúcich opiciach rodu Cynomolgus, ktoré dostávali 25-násobok týždennej
udržiavacej dávky trastuzumabu na intravenózne použitie u ľudí 2 mg/kg sa ukázalo, že trastuzumab sa vylučuje do materského mlieka. Prítomnosť trastuzumabu v sére opičích mláďat nemala negatívny
vplyv na ich rast alebo vývoj v období medzi narodením a prvým mesiacom života. Nie je známe, či
sa trastuzumab vylučuje do materského mlieka u ľudí. Keďže ľudský imunoglobulín IgG1 sa vylučuje do materského mlieka u ľudí a možnosť negatívneho ovplyvnenia plodu nie je známa, ženy nesmú
dojčiť počas liečby Trazimerou a ešte 7 mesiacov po poslednej dávke.
Fertilita
Nie sú dostupné žiadne údaje.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Trazimera môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje (pozri časť 4.8). Pacientom, u ktorých sa objavili reakcie na podávanie infúzie (pozri časť 4.4), sa neodporúča viesť vozidlá a obsluhovať stroje až do ústupu ťažkostí.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Medzi najzávažnejšie a/alebo najčastejšie nežiaduce reakcie doteraz hlásené v súvislosti s používaním
trastuzumabu (na intravenózne a subkutánne podanie) patria srdcová dysfunkcia, reakcie súvisiace s infúziou, hematotoxicita (najmä neutropénia), infekcie a pľúcne nežiaduce reakcie.
Tabuľkový súhrnnežiaducichreakcií
V tejto časti boli použité nasledovné kategórie frekvencie výskytu: veľmi časté (> 1/10), časté
(> 1/100 až < 1/10), menej časté (> 1/1 000 až < 1/100), zriedkavé (> 1/10 000 až < 1/1 000), veľmi
zriedkavé (< 1/10 000), neznáme (nemožno odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
V tabuľke 1 sú uvedené nežiaduce reakcie, ktoré boli hlásené v súvislosti s používaním intravenózneho trastuzumabu formou monoterapie alebo v kombinácii s chemoterapiou v hlavných klinických skúšaniach a počas uvedenia lieku na trh.
Všetky reakcie sú uvedené podľa najvyššieho percenta výskytu v hlavných klinických skúšaniach. Okrem toho sú v tabuľke 1 zahrnuté aj reakcie získané po uvedení lieku na trh.
Tabuľka 1: Nežiaduce účinky hlásené v súvislosti s trastuzumabom na intravenózne použitie
v monoterapii alebo v kombinácii s chemoterapiou v hlavných klinických skúšaniach (n = 8 386)
a v sledovaní po uvedení lieku na trh
Trieda orgánových systémov
Nežiaduca reakcia Frekvencia
Infekcie a nákazy Infekcia Veľmi časté Nazofaryngitída Veľmi časté Neutropenická sepsa Časté Cystitída Časté Herpes zoster Časté Chrípka Časté Sinusitída Časté Infekcia kože Časté Rinitída Časté Infekcia horných dýchacích ciest Časté Infekcia močových ciest Časté Eryzipel Časté Celulitída Časté Faryngitída Časté
Sepsa Menej časté
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Poruchy krvi a lymfatického systému
Progresia malígneho nádoru Neznáme
Progresia nádorového ochorenia Neznáme
Febrilná neutropénia Veľmi časté Anémia Veľmi časté Neutropénia Veľmi časté Znížený počet bielych krviniek/leukopénia Veľmi časté Trombocytopénia Veľmi časté Hypoprotrombinémia Neznáme Imunitná trombocytopénia Neznáme
Poruchy imunitného systému Precitlivenosť Časté
+Anafylaktická reakcia Neznáme
+Anafylaktický šok Neznáme
Poruchy metabolizmu a výživy
Zníženie hmotnosti/úbytok hmotnosti Veľmi časté Anorexia Veľmi časté Syndróm z rozpadu nádoru Neznáme Hyperkaliémia Neznáme
Psychické poruchy Insomnia Veľmi časté Úzkosť Časté Depresia Časté Neobvyklé myslenie Časté
Trieda orgánových systémov
Nežiaduca reakcia Frekvencia
Poruchy nervového systému 1Tremor Veľmi časté Závrat Veľmi časté Bolesť hlavy Veľmi časté Parestézia Veľmi časté
Dysgeúzia Veľmi časté Periférna neuropatia Časté Hypertónia Časté Somnolencia Časté
Ataxia Časté
Paréza Zriedkavé
Edém mozgu Neznáme
Poruchy oka Konjunktivitída Veľmi časté Zvýšené slzenie Veľmi časté Suchosť oka Časté
Edém papily Neznáme
Krvácanie do sietnice Neznáme
Poruchy ucha a labyrintu Hluchota Menej časté
Poruchy srdca a srdcovej činnosti
1Znížený krvný tlak Veľmi časté
1Zvýšený krvný tlak Veľmi časté
1Nepravidelný srdcový tep Veľmi časté
1Palpitácie Veľmi časté
1Srdcový flutter Veľmi časté
Znížená ejekčná frakcia* Veľmi časté
+Kongestívne zlyhanie srdca Časté
+1Supraventrikulárna tachyarytmia Časté
Kardiomyopatia Časté Perikardiálny výpotok Menej časté Kardiogénny šok Neznáme Perikarditída Neznáme Bradykardia Neznáme Prítomnosť gallopového rytmu Neznáme
Poruchy ciev Návaly horúčavy Veľmi časté
+1Hypotenzia Časté
Vazodilatácia Časté
Poruchy dýchacej sústavy, hrudníka a mediastína
+1Sipot Veľmi časté
+Dýchavičnosť Veľmi časté
Kašeľ Veľmi časté Epistaxa Veľmi časté Rinorea Veľmi časté
+Pneumónia Časté
Astma Časté
Ochorenie pľúc Časté
+Pľúcny výpotok Časté
Pneumonitída Zriedkavé
+Pľúcna fibróza Neznáme
+Respiračná tieseň Neznáme
+Zlyhávanie dýchania Neznáme
+Infiltrácia pľúc Neznáme
+Akútny edém pľúc Neznáme
+Akútny syndróm respiračnej tiesne Neznáme
+Bronchospazmus Neznáme
+Hypoxia Neznáme
Trieda orgánových systémov
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej
Nežiaduca reakcia Frekvencia
+Znížená saturácia kyslíkom Neznáme Laryngeálny edém Neznáme Ortopnoe Neznáme Pľúcny edém Neznáme Intersticiálna choroba pľúc Neznáme Hnačka Veľmi časté Vracanie Veľmi časté Nauzea Veľmi časté
1Opuch pier Veľmi časté
Bolesť brucha Veľmi časté Dyspepsia Veľmi časté Zápcha Veľmi časté Stomatitída Veľmi časté Hemoroidy Časté
Sucho v ústach Časté Hepatocelulárne poškodenie Časté Hepatitída Časté Citlivosť pečene Časté Žltačka Zriedkavé Zlyhávanie pečene Neznáme Erytém Veľmi časté Vyrážka Veľmi časté
1Opuch tváre Veľmi časté
Alopécia Veľmi časté Ochorenie nechtov Veľmi časté Syndróm palmárno-plantárnej erytrodyzestézie Veľmi časté Akné Časté
Suchá koža Časté Ekchymóza Časté Hyperhidróza Časté Makulopapulárna vyrážka Časté Pruritus Časté Lámavosť nechtov Časté Dermatitída Časté Urtikária Menej časté Angioedém Neznáme Artralgia Veľmi časté
sústavy a spojivového tkaniva 1Svalová stuhnutosť Veľmi časté
Myalgia Veľmi časté
Artritída Časté Bolesť chrbta Časté Bolesť kostí Časté Svalové kŕče Časté Bolesť krku Časté Bolesť v končatinách Časté
Poruchy obličiek a močových Ochorenie obličiek Časté
ciest
Stavy v gravidite, v šestonedelí a perinatálnom období
Membranózna glomerulonefritída Neznáme Glomerulonefropatia Neznáme Zlyhávanie obličiek Neznáme
Oligohydramnión Neznáme Hypoplázia obličky Neznáme Hypoplázia pľúc Neznáme
Trieda orgánových systémov
Poruchy reprodukčného systému a prsníkov
Celkové poruchy a reakcie v mieste podania
Úrazy, otravy a komplikácie liečebného postupu
Nežiaduca reakcia Frekvencia
Zápal prsníka/mastitída Časté
Asténia Veľmi časté Bolesť na hrudníku Veľmi časté Triaška Veľmi časté Únava Veľmi časté Príznaky podobné chrípke Veľmi časté Reakcia súvisiaca s infúziou Veľmi časté Bolesť Veľmi časté Pyrexia Veľmi časté Zápal slizníc Veľmi časté Periférny edém Veľmi časté Nepokoj Časté
Edém Časté
Pomliaždeniny Časté
+ Označuje nežiaduce reakcie, ktoré boli hlásené v súvislosti s úmrtím.
1 Označuje nežiaduce reakcie, ktoré boli prevažne hlásené v súvislosti s reakciami na infúziu. Presné percento nie je k dispozícii.
* Pozorované pri kombinovanej liečbe po terapii antracyklínmi a v kombinácii s taxánmi.
Popisvybranýchnežiaducich reakcií
Srdcová dysfunkcia
Kongestívne zlyhanie srdca,( triedy II - IV NYHA), je častá nežiaduca reakcia súvisiaca s používaním
trastuzumabu a bola spojená s úmrtím (pozri časť 4.4). U pacientov liečených trastuzumabom sa pozorovali prejavy a symptómy srdcovej dysfunkcie, ako je dyspnoe, ortopnoe, zhoršenie kašľa, pľúcny edém, S3 galop alebo znížená ejekčná frakcia ľavej komory (pozri časť 4.4).
V 3 hlavných klinických skúšaniach adjuvantne podávaného trastuzumabu v kombinácii
s chemoterapiou bol výskyt stupňa 3/4 srdcovej dysfunkcie (hlavne symptomatického kongestívneho srdcového zlyhania) podobný u pacientov, ktorým bola podávaná samotná chemoterapia (t.j. nedostali trastuzumab) a u pacientov, ktorým bol trastuzumab podávaný sekvenčne po taxáne (0,3 – 0,4 %). Výskyt bol najvyšší u pacientov, ktorým bol trastuzumab podávaný súbežne s taxánom (2,0 %).
O súbežnom podávaní trastuzumabu s nízkou dávkou antracyklínu pri neoadjuvantnej liečbe sú obmedzené údaje (pozri časť 4.4).
Keď sa trastuzumab podával po ukončení adjuvantnej chemoterapie bolo pozorované srdcové zlyhanie triedy III-IV NYHA u 0,6 % pacientov v ramene s podávaním jeden rok pri mediáne následného sledovania 12 mesiacov. V štúdii BO16348 bola pri mediáne následného sledovania 8 rokov
incidencia závažného kongestívneho srdcového zlyhania (triedy III a IV NYHA) v ramene s 1-ročnou liečbou trastuzumabom 0,8 % a výskyt symptomatickej a asymptomatickej dysfunkcie ľavej komory miernej intenzity bol 4,6 %.
Reverzibilita závažného kongestívneho SZ (definovaná ako postupnosť aspoň dvoch po sebe idúcich hodnôt LVEF ³ 50 % po príhode) sa pozorovala u 71,4 % pacientov liečených trastuzumabom. Reverzibilita symptomatickej a asymptomatickej dysfunkcie ľavej komory miernej intenzity bola preukázaná u 79,5 % z pacientov. Približne 17 % udalostí súvisiacich so srdcovým zlyhaním sa vyskytlo po ukončení liečby trastuzumabom.
V hlavných metastatických skúšaniach s trastuzumabom na intravenózne použitie, sa výskyt srdcovej dysfunkcie pohybuje medzi 9 % a 12 %, v kombinácii s paklitaxelom v porovnaní s 1 % – 4 %
s paklitaxelom samotným. Pre monoterapiu bol výskyt 6 % – 9 %. Najvyšší výskyt srdcovej
dysfunkcie bol pozorovaný u pacientov liečených trastuzumabom súbežne s antracyklínom/cyklofosfamidom (27 %), a predstavoval signifikatne vyšší výskyt ako pri liečbe antracyklínom/cyklofosfamidom samostatným (7 % – 10 %). V následnom skúšaní, s prospektívnym sledovaním srdcovej funkcie, bola incidencia symptomatického kongestívneho SZ 2,2 % u pacientov liečených trastuzumabom a docetaxelom, v porovnaní s 0 % u pacientov, ktorým sa podával len docetaxel samotný. U väčšiny pacientov (79 %), u ktorých sa vyvinula srdcová dysfunkcia v týchto skúšaniach, došlo k zlepšeniu po podaní bežnej liečby na kongestívne SZ.
Infúzne reakcie, reakcie podobné alergickým reakciám a precitlivenosť
Predpokladá sa, že približne u 40 % pacientov liečených trastuzumabom sa vyskytne určitá forma reakcie súvisiacej s infúziou. Väčšina reakcií súvisiacich s infúziou je však miernej až stredne
závažnej intenzity (systém odstupňovania NCI-CTC) a obyčajne sa vyskytujú v počiatočnej fáze
liečby, t.j. počas podávania prvej, druhej a tretej infúzie a pri ďalších infúziách ich frekvencia klesá. Reakcie zahŕňajú triašku, horúčku, dyspnoe, hypotenziu, sipot, bronchospazmus, tachykardiu, zníženú
saturáciu kyslíkom, respiračnú tieseň, vyrážku, nauzeu,vracanie a bolesť hlavy (pozri časť 4.4).
Výskyt reakcií všetkých stupňov súvisiacich s infúziou sa líšil medzi štúdiami v závislosti od indikácie, metódy zberu údajov a podľa toho či bol trastuzumab podávaný súbežne s chemoterapiou alebo ako monoterapia.
Závažné anafylaktické reakcie vyžadujúce okamžitý dodatočný zákrok sa vyskytujú veľmi zriedkavo, a to zvyčajne počas podávania prvej alebo druhej infúzie trastuzumabu (pozri časť 4.4) a boli spojené s úmrtím.
V izolovaných prípadoch sa pozorovali anafylaktoidné reakcie.
Hematotoxicita
Febrilná neutropénia, leukopénia, anémia, trombocytopénia a neutropénia sa vyskytujú veľmi často. Frekvencia výskytu hypoprotrombinémie nie je známa. Môže byť mierne zvýšené riziko neutropénie,
keď sa podáva trastuzumab s docetaxelom po antracyklínovej liečbe.
Pľúcne udalosti
Závažné pľúcne nežiaduce reakcie sa v súvislosti s používaním trastuzumabu vyskytujú zriedkavo a boli spojené s úmrtím. Zahŕňajú nasledovné, ale neobmedzujú sa iba na ne: pľúcne infiltráty, akútny
syndróm respiračnej tiesne, pneumóniu, pneumonitídu, pleurálne výpotky, respiračnú tieseň, akútny
edém pľúc a respiračnú insuficienciu (pozri časť 4.4).
Podrobné opatrenia na minimalizáciu rizika, ktoré sú v súlade s EU plánom riadenia rizík sú uvedené v Osobitných upozorneniach a opatreniach pri používaní (časť 4.4).
Imunogenicita
V neoadjuvantnom-adjuvantnom skúšaní VKP (včasný karcinóm prsníka) (BO22227) sa pri mediáne
následného sledovania viac ako 70 mesiacov u 10,1 % (30/296) pacientov, ktorí boli liečení trastuzumabom intravenózne, vyvinuli protilátky voči trastuzumabu. U 2 z 30 pacientov v skupine s intravenózne podávaným trastuzumabom bola vo vzorkách po začatí liečby zistená prítomnosť neutralizujúcich protilátok proti trastuzumabu.
Klinický význam týchto protilátok nie je známy. Prítomnosť protilátok proti trastuzumabu nemala vplyv na farmakokinetiku, účinnosť (stanovenú patologickou kompletnou odpoveďou [pCR]
a prežívaním bez príhody [EFS]) a bezpečnosť stanovenú výskytom reakcií súvisiacich s podávaním
(ARRs) trastuzumabu intravenózne.
Nie sú k dispozícii žiadne údaje o imunogenicite týkajúce sa trastuzumabu podávaného pri karcinóme žalúdka.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
PríloheV.4.9 PredávkovanieV klinických skúšaniach u ľudí sa nezískali žiadne skúsenosti s predávkovaním. V rámci klinických skúšaní neprekročili jednotlivé dávky trastuzumabu hodnotu 10 mg/kg telesnej hmotnosti; udržiavacia dávka 10 mg/kg podaná každé 3 týždne po úvodnej nasycovacej dávke 8 mg/kg bola porozorovaná
v klinických skúšaniach u pacientov s metastatickým karcinómom žalúdka. Dávky v uvedených hodnotách boli dobre tolerované.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, monoklonálne protilátky, ATC kód: L01XC03
Trazimera je biologicky podobný liek. Podrobné informácie sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu.Trastuzumab je rekombinantná humanizovaná monoklonálna protilátka typu IgG1 proti receptoru
2 ľudského epidermálneho rastového faktora (HER2). Nadmerná expresia receptora HER2 sa pozoruje pri 20 až 30 % primárnych nádorov prsníka. V štúdiách určujúcich HER2-pozitivitu pri karcinóme
žalúdka (KŽ) s použitím imunohistochemickej metódy (IHC) a fluorescenčnej
in situ hybridizácie
(FISH) alebo chromogénnej
in situ hybridizácie (CISH) sa zistilo, že existuje široká variabilita HER2- pozitivity v rozsahu od 6,8 % do 34,0 % pri IHC a 7,1 % do 42,6 % pri FISH. V štúdiách sa zistilo, že u pacientov s karcinómom prsníka s nadmernou expresiou receptora HER2 je v porovnaní s pacientmi s tumormi bez nadmernej expresie tohto receptora skrátená doba prežívania bez prejavov ochorenia. Extracelulárna doména receptora (ECD, p105) sa môže dostať do krvi, a preto je možné ju stanoviť
vo vzorkách séra.
Mechanizmus účinkuTrastuzumab sa viaže s vysokou afinitou a špecifickosťou na subdoménu IV, juxta-membránovú
oblasť extracelulárnej domény receptora HER2. Väzba trastuzumabu na receptor HER2 inhibuje ligandovo-nezávislú signálnu dráhu receptora HER2 a bráni proteolytickému odštiepeniu jeho extracelulárnej domény, čo je mechanizmus aktivácie receptora HER2. V dôsledku toho sa v štúdiách na zvieratách aj v pokusoch
in vitro zistilo, že trastuzumab inhibuje proliferáciu ľudských nádorových buniek s nadmernou expresiou receptora HER2. Okrem toho, trastuzumab je silným mediátorom bunkami sprostredkovanej cytotoxicity závislej od protilátky (ADCC). V pokusoch
in vitro sa zistilo, že trastuzumabom sprostredkovaná ADCC je zameraná viac na nádorové bunky s nadmernou expresiou receptora HER2 ako na nádorové bunky bez nadmernej expresie receptora HER2.
Stanovenie nadmernejexpresiereceptoraHER2aleboamplifikácieHER2génuStanovenie nadmernej expresie receptora HER2 alebo amplifikácie HER2 génu pri karcinóme prsníkaTrastuzumab sa má použiť iba u pacientov, ktorých tumory vykazujú nadmernú expresiu receptora HER2 alebo amplifikáciu HER2 génu, ak je stanovené presnou a validovanou metódou. Nadmerná expresia receptora HER2 sa má stanoviť imunohistochemicky (IHC) - hodnotenie fixovaných blokov nádoru (pozri časť 4.4). Amplifikácia HER2 génu sa má stanoviť použitím fluorescenčnej
in situ hybridizácie (FISH) alebo chromogénnej
in situ hybridizácie (CISH) fixovaných blokov nádoru. Pacienti sú vhodní na liečbu Trazimerou, ak vykazujú nadmernú expresiu receptora HER2, vyjadrenú ako skóre 3+ pomocou IHC alebo majú pozitívne výsledky FISH alebo CISH.
Kvôli zaručeniu presných a opakovateľných výsledkov sa má vyšetrenie vykonať v špecializovanom laboratóriu, ktoré dokáže zabezpečiť validitu vyšetrovacích postupov.
Odporúčaný systém vyhodnocovania zafarbenia vzoriek na základe imunohistochemického vyšetrenia je v tabuľke 2:
Tabuľka 2: Odporúčaný systém vyhodnocovania imunohistochemického farbenia pri karcinóme prsníka
Skóre Spôsob sfarbenia Hodnotenie nadmernej expresie receptora HER2
0 Sfarbenie neprítomné, prípadne sfarbenie membrány je prítomné u < 10 % nádorových buniek.
1+ Slabé/ťažko pozorovateľné sfarbenie membrány je prítomné u > 10 % nádorových buniek. Farbí sa iba časť membrány nádorových buniek.
2+ Slabé až mierne sfarbenie celej membrány je prítomné u > 10 % nádorových buniek.
3+ Silné sfarbenie celej membrány je prítomné u
> 10 % nádorových buniek.
Negatívny výsledok
Negatívny výsledok
Neurčitý výsledok
Pozitívny výsledok

Všeobecne, FISH sa považuje za pozitívne, ak pomer počtu kópií HER2 génu na nádorovú bunku
k počtu kópií chromozómu 17 je väčší alebo rovný 2, alebo ak sú viac než 4 kópie HER2 génu na nádorovú bunku, pričom sa nepoužije kontrola chromozóm 17.
Všeobecne, CISH sa považuje za pozitívne, ak je viac než 5 kópií HER2 génu na jadro vo viac než
50 % nádorových buniek.
Podrobné návody na vykonanie testov a ich interpretáciu si pozrite v písomných informáciách pre stanovenie FISH a CISH. Môžu sa tiež použiť oficiálne odporúčania na testovanie HER2.
Pre ostatné metódy hodnotenia HER2 proteínu alebo génovej expresie musia byť použité analýzy vykonávané v laboratóriách, ktoré poskytujú adekvátne najmodernejšie vykonávanie validovaných metód. Tieto metódy musia byť presné a dostatočne podrobné, aby preukázali nadmernú expresiu HER2 a musia byť schopné rozoznávať medzi miernou (zodpovedá 2+) a silnou (zodpovedá 3+) nadmernou expresiou HER2.
Stanovenie nadmernej expresie receptora HER2 alebo amplifikácie HER2 génu pri karcinóme žalúdka Na stanovenie nadmernej expresie receptora HER2 alebo amplifikácie HER2 génu sa má použiť len presná a validovaná metóda. Ako prvá testovacia metóda sa odporúča IHC a v prípadoch, kde sa vyžaduje aj stav amplifikácie HER2 génu, sa musí použiť buď metóda striebrom značenej
in situ hybridizácie (SISH) alebo FISH. SISH technológia sa však odporúča preto, lebo umožňuje paralelne hodnotiť histológiu a morfológiu nádoru. Kvôli zaručeniu validácie testovacích postupov a získaniu presných a opakovateľných výsledkov musí HER2 testovanie prebiehať v laboratóriu so zaškoleným personálom. Podrobné návody na vykonanie testov a interpretáciu výsledkov sa majú prevziať
z písomnej informácie pre používateľov, kde sú opísané použité metódy na testovanie HER2.
V klinickom skúšaní ToGA (BO18255) pacienti, ktorých karcinómy boli buď IHC3+ alebo FISH
pozitívne, boli definovaní ako HER2 pozitívni, a teda boli zaradení do klinického skúšania.
Na základe výsledkov klinického skúšania boli priaznivé účinky obmedzené na pacientov s najvyššou úrovňou nadmernej expresie proteínu HER2, vyjadrenú ako skóre 3+ pomocou IHC alebo ako skóre
2+ pomocou IHC a pozitívnym výsledkom FISH.
V štúdii porovnávajúcej metódy (D008548) bol pozorovaný vysoký stupeň zhody (> 95 %) medzi
SISH a FISH technikami používanými za účelom detekcie amplifikácie HER2 génu u pacientov s karcinómom žalúdka.
Nadmerná expresia receptora HER2 sa má stanoviť imunohistochemicky (IHC) - na základe hodnotenia fixovaných blokov nádoru; amplifikácia génu HER2 sa má stanoviť použitím
in situ
hybridizácie, použitím buď SISH alebo FISH z fixovaných blokov nádoru.
Odporúčaný skórovací systém na vyhodnocovanie vzoriek označených imunohistochemicky je v tabuľke 3:
Tabuľka 3: Odporúčaný systém vyhodnocovania imunohistochemického farbenia pri karcinóme žalúdka
Skóre Chirurgické vzorky - spôsob označenia
Žiadna reaktivita alebo
Vzorky biopsie - spôsob označenia
Žiadna reaktivita alebo reaktivita
Hodnotenie nadmernej expresie receptora HER2
0 reaktivita membrány u < 10 % nádorových buniek Slabá/ťažko pozorovateľná reaktivita membrány u ³ 10 %
1+ nádorových buniek; bunky sú
reaktívne iba v časti bunkovej membrány
membrány ani v jednej nádorovej Negatívny výsledok bunke
Zhluk nádorových buniek
so slabou/ťažko pozorovateľnou
reaktivitou membrány bez ohľadu Negatívny výsledok na percento zafarbených
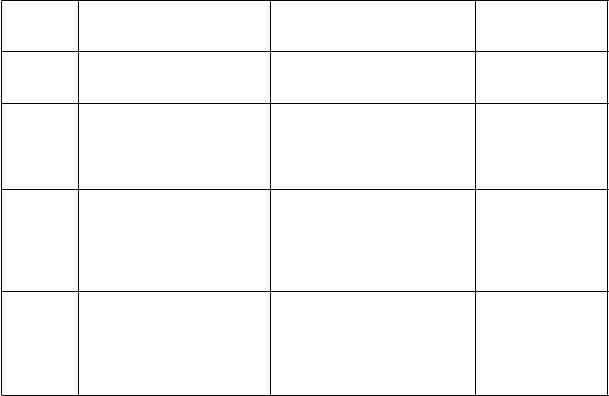
nádorových buniek
Slabá až mierna reaktivita celej, Zhluk nádorových buniek
bazolaterálnej alebo laterálnej
2+ membrány u ³ 10 %
nádorových buniek
Silná reaktivita celej, bazolaterálnej alebo laterálnej
3+ membrány u ³ 10 %
nádorových buniek
so slabou až miernou reaktivitou celej, bazolaterálnej alebo laterálnej membrány bez ohľadu na percento zafarbených nádorových buniek
Zhluk nádorových buniek so silnou reaktivitou celej, bazolaterálnej alebo laterálnej membrány bez ohľadu
na percento zafarbených nádorových buniek
Neurčitý výsledok
Pozitívny výsledok
Všeobecne, SISH alebo FISH sa považuje za pozitívnu, ak pomer počtu kópií HER2 génu na nádorovú
bunku k počtu kópií chromozómu 17 je väčší alebo rovný 2.
Klinická účinnosťabezpečnosť
Metastatický karcinóm prsníka
V klinických skúšaniach bol trastuzumab použitý v rámci monoterapie pacientov s MKP, ktorých
nádory vykazovali nadmernú expresiu receptora HER2 a u ktorých nebol úspešný jeden alebo viac režimov chemoterapie metastatického ochorenia (samotný trastuzumab).
Trastuzumab bol tiež použitý v kombinácii s paklitaxelom alebo docetaxelom na liečbu pacientov, ktorí ešte nedostali chemoterapiu kvôli metastatickému nádorovému ochoreniu. Pacienti, ktorí boli predtým liečení adjuvantnou chemoterapiou na báze antracyklínu, boli liečení paklitaxelom (175 mg/m2, podaným infúziou počas 3 hodín) v kombinácii s alebo bez trastuzumabu. V hlavnej štúdii docetaxel (100 mg/m2 podaným infúziou počas 1 hodiny) s alebo bez trastuzumabu dostávalo 60 % pacientov predtým adjuvantnú antracyklínovú chemoterapiu. Pacienti boli liečení trastuzumabom až do progresie ochorenia.
Účinnosť trastuzumabu v kombinácii s paklitaxelom nebola študovaná u pacientov, ktorí predtým nedostávali adjuvantnú antracyklínovú chemoterapiu. Avšak trastuzumab s docetaxelom bol účinný u pacientov nezávisle od toho, či dostávali alebo nedostávali antracyklíny v adjuvancii.
Na stanovenie vhodnosti pacientov pre účasť v hlavnej klinickej štúdii s trastuzumabom v monoterapii a v klinických štúdiách trastuzumabu s paklitaxelom zameraných na nadmernú expresiu receptora HER2 sa použila metóda imunohistochemického farbenia receptora HER2 vo fixovanej vzorke nádoru
prsníka pomocou myších monoklonálnych protilátok CB11 a 4D5. Tkanivá sa fixovali vo formalíne alebo v Bouinovom fixačnom roztoku. Pri hodnotení výsledkov z klinického skúšania v centrálnom laboratóriu sa používala stupnica od 0 do 3+. Pacienti s intenzitou sfarbenia 2+ alebo 3+ boli zaradení do štúdie, kým pacienti s hodnotením 0 alebo 1+ boli zo štúdie vylúčení. Viac ako 70 % z pacientov zaradených do štúdie vykazovalo nadmernú expresiu 3+. Na základe zistených údajov je možné povedať, že priaznivejšie účinky liečby sa pozorovali u pacientov s vyšším stupňom nadmernej expresie receptora HER2 (3+).
Imunohistochémia bola hlavná testovacia metóda na stanovenie HER2 pozitivity v hlavnom klinickom skúšaní docetaxel s alebo bez trastuzumabu. Menšina pacientov bola testovaná použitím
fluorescenčnej in-situ hybridizácie (FISH). V týchto skúšaniach 87 % pacientov, ktorí vstúpili
do skúšania, malo ochorenie, ktoré bolo IHC3+ a 95 % pacientov, ktorí vstúpili do skúšania, malo ochorenie IHC3+ a/alebo FISH pozitívne.
Týždenné dávkovanie pri metastatickom karcinóme prsníka
V tabuľke 4 sú zhrnuté výsledky účinnosti zo štúdií v monoterapii a v kombinovanej terapii:
Tabuľka 4: Výsledky účinnosti zo štúdií v monoterapii a kombinovanej terapii
Parameter Monoterapia Kombinovaná liečba
t
rastuzumab
1
n = 172
t
rastuzumab a
paklitaxel
2
n = 68
paklitaxel
2
n = 77
t
rastuzumab a
docetaxel
3
n = 92
docetaxel
3
n = 94
Vyhodnotenie 18 % 49 % 17 % 61 % 34 % odpovede (95 % CI) (13 – 25) (36 – 61) (9 – 27) (50 – 71) (25 – 45) Medián dĺžky
odpovede 9,1 8,3 4,6 11,7 5,7
(mesiace) (95 % CI) (5,6 – 10,3) (7,3 – 8,8) (3,7 – 7,4) (9,3 – 15,0) (4,6 – 7,6)
Medián TTP 3,2 7 1 3,0 11,7 6,1 (mesiace) (95 % CI) (2,6 – 3,5) (6,2 – 12,0) (2,0 – 4,4) (9,2 – 13,5) (5,4 – 7,2) Medián prežívania
(mesiace) (95 % CI)
16,4 24,8 17,9 31,2 22,74 (12,3 – ne) (18,6 – 33,7) (11,2 – 23,8) (27,3 – 40,8) (19,1 – 30,8)

TTP = čas trvajúci do progresie, “ne“ - nebolo možné určiť alebo ešte pokračuje. CI = interval spoľahlivosti
1. Štúdia H0649g: IHC3+ podsúbor pacientov
2. Štúdia H0648g: IHC3+ podsúbor pacientov
3. Štúdia M77001: Plne analyzovaný súbor (zameraný na liečbu), výsledky po 24 mesiacoch
Kombinovaná liečba s trastuzumabom a anastrozolomTrastuzumab sa študoval v kombinácii s anastrozolom v prvej línii liečby MKP s nadmernou expresiou HER2 a s pozitivitou hormonálneho receptora (t.j. estrogénového receptora (ER) a/alebo
progesterónového receptora (PR)) u postmenopauzálnych pacientok. Prežívanie bez progresie bolo
dvojnásobné v ramene trastuzumab plus anastrozol v porovnaní s anastrozolom samotným
(4,8 mesiacov oproti 2,4 mesiacom). Iné parametre ukazujúce zlepšenie pri kombinácii boli celková odpoveď (16,5 % oproti 6,7 %); klinický prínos (42,7 % oproti 27,9 %); čas do progresie
(4,8 mesiacov oproti 2,4 mesiacom). Nebol zaznamenaný žiadny rozdiel medzi ramenami, pokiaľ ide o odpoveď a trvanie odpovede. Medián celkového prežívania bol predĺžený na 4,6 mesiacov
u pacientov v ramene s kombinovanou liečbou. Rozdiel nebol štatisticky významný, avšak viac ako polovica pacientov v ramene so samotným anastrozolom po progresii choroby prešlo na režim
s trastuzumabom.
Trojtýždenné dávkovanie pri MKPV tabuľke 5 sú zhrnuté výsledky účinnosti z nekomparatívnych štúdií v monoterapii a v kombinovanej terapii:
Tabuľka 5: Výsledky účinnosti z nekomparatívnych štúdií v monoterapii a v kombinovanej terapii
Parameter Monoterapia Kombinovaná liečba
Vyhodnotenie odpovede (95 % CI)
Medián dĺžky odpovede (mesiace) (rozsah)
Medián TTP (mesiace)
(95 % CI)
Trastuzumab
1
n = 105
24 % (15 – 35)
10,1
(2,8 – 35,6)
3,4
(2,8 – 4,1)
Trastuzumab2
n = 72
27 % (14 – 43)
7,9
(2,1 – 18,8)
7,7
(4,2 – 8,3)
Trastuzumab a
paklitaxel3
n = 32
59 % (41 – 76)
10,5 (1,8 – 21)
12,2 (6,2 – ne)
Trastuzumab a
docetaxel4
n = 110
73 % (63 – 81)
13,4
(2,1 – 55,1)
13,6 (11 – 16)
Medián prežívania (mesiace) (95 % CI)
ne ne ne 47,3 (32 – ne)
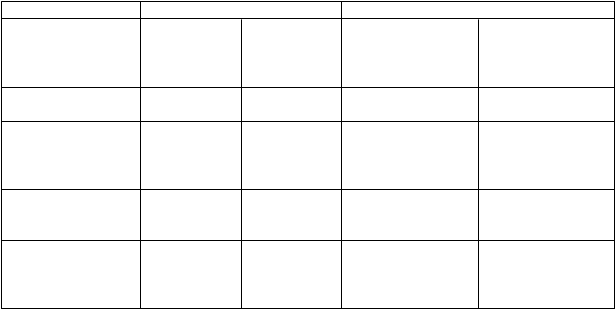
TTP = čas trvajúci do progresie; „ne“ - nebolo možné určiť alebo ešte pokračuje
CI = interval spoľahlivosti
1. Štúdia WO16229: úvodná dávka 8 mg/kg telesnej hmotnosti, pokračuje v 3-týždňových intervaloch dávkou 6
mg/kg telesnej hmotnosti
2. Štúdia MO16982: úvodná dávka 6 mg/kg telesnej hmotnosti týždenne x 3; pokračuje v 3-týždňových intervaloch dávkou 6 mg/kg telesnej hmotnosti
3. Štúdia BO15935
4. Štúdia MO16419
Miesta progresie ochoreniaU pacientov liečených trastuzumabom v kombinácii s paklitaxelom sa v porovnaní s pacientmi liečenými samotným paklitaxelom významne znížil výskyt progresie ochorenia do pečene (21,8 %
oproti 45,7 %; p = 0,004). Progresia ochorenia do centrálneho nervového systému bola častejšia u pacientov liečených trastuzumabom a paklitaxelom ako u pacientov liečených samotným
paklitaxelom (12,6 % oproti 6,5 %, p = 0,377).
Včasný karcinóm prsníka (adjuvantná liečba)Včasný karcinóm prsníka sa definuje ako nemetastatický primárny invazívny karcinóm prsníka.
Trastuzumab použitý v adjuvantnej terapii sa skúmal v 4 rozsiahlych multicentrických, randomizovaných štúdiách.
- Štúdia BO16348 bola zameraná na porovnanie jednoročnej a dvojročnej liečby trastuzumabom, ktorý sa podával každé tri týždne oproti pozorovaniu u pacientov s VKP s pozitivitou HER2 po operácii, so zavedenou chemoterapiou a rádioterapiou (ak je to
aplikovateľné). Navyše, bolo urobené porovnanie liečby trastzumabom po dobu dvoch rokov oproti liečbe trastuzumabom po dobu jedného roku. Pacienti určení na liečbu trastuzumabom
dostali úvodnú nasycovaciu dávku 8 mg/kg, po ktorej dostávali dávku 6 mg/kg každé tri týždne buď počas jedného alebo dvoch rokov.
- Štúdie NSABP B-31 a NCCTG N9831, ktoré obsahovali aj spojenú analýzu (tzv. joint analysis) boli zamerané na zhodnotenie klinického významu kombinovanej liečby
trastuzumabom s paklitaxelom po AC chemoterapii; okrem toho sa v štúdii NCCTG N9831
hodnotilo sekvenčné podanie trastuzumabu po chemoterapii AC→P u pacientov s VKP
s pozitivitou HER2 po operácii.
- Štúdia BCIRG 006 bola zameraná na preskúmanie kombinovanej liečby trastuzumabom s docetaxelom buď následne po AC chemoterapii alebo v kombinácii s docetaxelom
a karboplatinou u pacientov s VKP s pozitivitou HER2 po operácii.
V klinickom skúšaní HERA bol VKP definovaný ako operabilný, primárny, invazívny adenokarcinóm prsníka s pozitívnymi alebo negatívnymi axilárnymi uzlinami, ak bol priemer nádoru minimálne 1 cm.
V spojenej analýze štúdií NSABP B-31 a NCCTG N9831 bol včasný karcinóm prsníka definovaný na pacientky s operabilným karcinómom prsníka s vysokým rizikom, definovaným ako pozitivita HER2 a pozitívna axilárna lymfatická uzlina alebo pozitivita HER2 a lymfatická uzlina negatívna
so známkami vysokého rizika (veľkosť nádoru > 1 cm a negativita ER alebo veľkosť nádoru > 2 cm, bez ohľadu na hormonálny status).
V štúdii BCIRG 006 VKP s pozitivitou HER2 bol definovaný buď ako pozitívna lymfatická uzlina alebo negatívna uzlina s vysokým rizikom u pacientov bez postihnutia lymfatických uzlín (pN0)
a najmenej 1 z nasledujúcich faktorov: veľkosť nádoru viac než 2 cm, negativita estrogénového a progesterónového receptora, histologický stupeň a/alebo jadrový stupeň 2 – 3, alebo vek < 35 rokov).
Výsledky účinnosti zo štúdie BO16348 po mediáne následného sledovania 12 mesiacov* a 8 rokov**
sú zhrnuté v tabuľke 6:
Tabuľka 6: Výsledky účinnosti zo štúdie BO16348
Medián následného sledovania
12 mesiacov*
Medián následného sledovania
8 rokov**
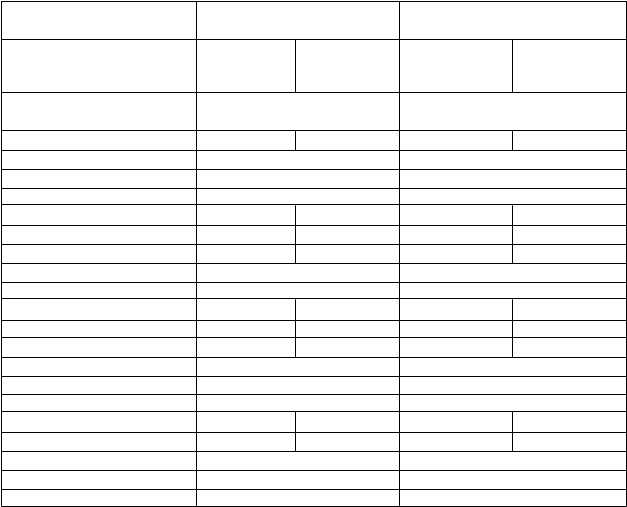
Parameter Pozorovanie Trastuzumab Pozorovanie Trastuzumab n = 1 693 1 rok n = 1 697*** 1 rok
n = 1 693 n = 1 702***
Prežívanie bez prejavov ochorenia
- Počet pacientov s príhodou 219 (12,9 %) 127 (7,5 %) 570 (33,6 %) 471 (27,7 %)
- Počet pacientov bez príhody 1 474 (87,1 %) 1 566 (92,5 %) 1 127 (66,4 %) 1 231 (72,3 %) Hodnota p oproti pozorovaniu < 0,0001 < 0,0001
Pomer rizík oproti pozorovaniu 0,54 0,76
Prežívanie bez recidívy
- Počet pacientov s príhodou 208 (12,3 %) 113 (6,7 %) 506 (29,8 %) 399 (23,4 %)
- Počet pacientov bez príhody 1 485 (87,7 %) 1 580 (93,3 %) 1 191 (70,2 %) 1 303 (76.6 %) Hodnota p oproti pozorovaniu < 0,0001 < 0,0001
Pomer rizík oproti pozorovaniu 0,51 0,73
Prežívanie bez vzdialených metastáz
- Počet pacientov s príhodou 184 (10,9 %) 99 (5,8 %) 488 (28,8 %) 399 (23,4 %)
- Počet pacientov bez príhody 1 508 (89,1 %) 1 594 (94,6 %) 1 209 (71,2 %) 1 303 (76,6 %) Hodnota p oproti pozorovaniu < 0,0001 < 0,0001
Pomer rizík oproti pozorovaniu 0,50 0,76
Celkové prežívanie (úmrtie)
- Počet pacientov s príhodou 40 (2,4 %) 31 (1,8 %) 350 (20,6 %) 278 (16,3 %)
- Počet pacientov bez príhody 1 653 (97,6 %) 1 662 (98,2 %) 1 347 (79,4 %) 1 424 (83,7 %) Hodnota p oproti pozorovaniu 0,24 0,0005
Pomer rizík oproti pozorovaniu 0,75 0,76
*Spoločný primárny koncový ukazovateľ prežívania bez prejavov ochorenia pri liečbe 1 rok oproti pozorovaniu dosiahol preddefinovanú štatistickú hranicu
**Záverečná analýza (vrátane prechodu 52 % pacientov z pozorovania do ramena liečby trastuzumabom)
*** Je nesúlad v celkovej veľkosti hodnotenej vzorky kvôli nízkemu počtu pacientov, ktorí boli randomizovaní po dátume uzávierky pre analýzu pri mediáne následného sledovania 12 mesiacov
Výsledky účinnosti z predbežnej analýzy prekročili protokolom predšpecifikovanú štatistickú hranicu pre porovnanie 1 ročnej liečby trastuzumabom oproti pozorovaniu. Pri mediáne následného sledovania
12 mesiacov bol pomer rizík (HR) 0,54 (95 % CI 0,44; 0,67) pre prežívanie bez prejavov ochorenia,
ktorý sa premieta z hľadiska 2-ročného prežívania bez prejavov ochorenia do absolútneho prínosu
7,6 percentuálnych bodov (85,8 % oproti 78,2 %) v prospech skupiny s trastuzumabom.
Finálna analýza bola vykonaná pri mediáne následného sledovania 8 rokov a ukázala, že jeden rok liečby trastuzumabom je spojený s 24 % znížením rizika v porovnaní so samotnou observáciou (HR =
0,76, 95 % CI 0,67, 0,86). Pri 8-ročnom prežívaní bez prejavov ochorenia sa to premieta do absolútneho prínosu 6,4 percentuálnych bodov v prospech 1-ročnej liečby trastuzumabom.
V tejto záverečnej analýze predĺženie liečby trastuzumabom na dva roky nepreukázalo ďalší prínos oproti jednoročnej liečbe [HR pre prežívanie bez prejavov ochorenia v ITT populácii (intent to treat)
2 roky oproti 1 roku = 0,99 (95 % CI: 0,87; 1,13), p-hodnota = 0,90 a HR pre celkové prežívanie =
0,98 (0,83; 1,15), p-hodnota = 0,78]. Výskyt asymptomatickej srdcovej dysfunkcie sa zvýšil v ramene s 2-ročnou liečbou (8,1% oproti 4,6% v ramene s 1-ročnou liečbou). Viac pacientov v ramene s 2-
ročnou liečbou malo aspoň jedenu nežiaducu príhodu stupňa 3 alebo 4 (20,4 %) v porovnaní s
ramenom s 1-ročnou liečbou (16,3 %).
V štúdiách NSABP B-31 a NCCTG N9831 sa trastuzumab podával v kombinácii s paklitaxelom po
AC chemoterapii.
Doxorubicín a cyklofosfamid boli podávané súbežne takto:
- intravenózny doxorubicín, 60 mg/m2, podávaný každé 3 týždne v 4 cykloch
- intravenózny cyklofosfamid, 600 mg/m2, viac ako 30 minút , podávaný každé 3 týždne v 4 cykloch
Paklitaxel v kombinácii s trastuzumabom bol podávaný takto:
- intravenózny paklitaxel - 80 mg/m2 v kontinuálnej intravenóznej infuzii podávanej každý týždeň po obdobie 12 týždňov
alebo
- intravenózny paklitaxel - 175 mg/m2 v kontinuálnej intravenóznej infúzii podávanej každé
3 týždne v 4 cykloch (v 1. deň každého cyklu).
Výsledky účinnosti zo spojenej analýzy klinických štúdií NSABP B-31 a NCCTG 9831 v čase
definitívnej analýzy DFS* sú zhrnuté v tabuľke 7. Medián ďalšieho následného sledovania bol
1,8 roka u pacientov v skupine AC→P a 2,0 roky u pacientov v skupine AC→PH.
Tabuľka 7: Súhrn výsledkov účinnosti zo spoločnej analýzy štúdií NSABP B-31 a NCCTG N9831
v čase definitívnej analýzy DFS*
Parameter AC→P
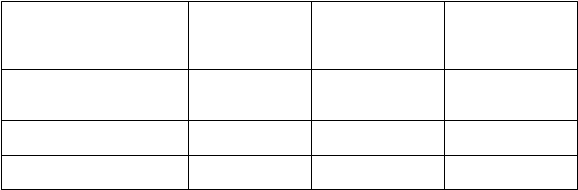
(n = 1 679)
Prežívanie bez prejavov
AC→PH (n = 1 672)
Pomer rizík oproti
AC→P (95 % CI) p-hodnota
ochorenia
Počet pacientov s príhodou
Vzdialená recidíva
261 (15,5) 133 (8,0) 0,48 (0,39, 0,59)
p < 0,0001
0,47 (0,37, 0,60)
Počet pacientov 193 (11,5) 96 (5,7)
Úmrtie (príhoda OS):
Počet pacientov s príhodou 92 (5,5) 62 (3,7)
A: doxorubicín; C: cyklofosfamid; P: paklitaxel; H: trastuzumab
p < 0,0001
0,67 (0,48, 0,92)
p = 0,014**
*pri mediáne ďalšieho následného sledovania 1,8 roka u pacientov v skupine AC→P a 2,0 roky u pacientov v skupine AC→PH
**p- hodnota pre OS neprekročila predurčenú štatistickú hranicu na porovnanie AC→PH oproti AC→P
Pre primárny koncový ukazovateľ, prežívanie bez prejavov ochorenia, pridanie trastuzumabu
ku chemoterapii paklitaxelom znížilo riziko recidívy ochorenia o 52 %. Pomer rizík sa premieta
do absolútneho prínosu 3-ročného prežívania bez prejavov ochorenia 11,8 percentuálnych bodov (87,2
% oproti 75,4 %) v prospech skupiny s AC→PH (trastuzumabom).
V čase aktualizácie údajov o bezpečnosti po mediáne 3,5 – 3,8 rokov ďalšieho následného sledovania, analýza DFS opätovne potvrdila jeho prínos preukázaný v definitívnej analýze. Napriek prechodu
na trastuzumab v kontrolnej skupine, pridanie trastuzumabu k chemoterapii paklitaxelom viedlo k 52
% zníženiu rizika návratu ochorenia. Pridanie trastuzumabu k chemoterapii paklitaxelom tiež viedlo k 37 % zníženiu rizika úmrtia.
Predbežná finálna analýza OS zo spoločnej analýzy štúdií NSABP B-31 a NCCTG N9831 bola vykonaná po 707 úmrtiach (medián následného sledovania 8,3 roka v skupine AC→P). V skupine AC→PH sa dosiahlo štatisticky významné zlepšenie OS v porovnaní s AC→P (stratifikované'
HR = 0,64; 95 % CI [0,55, 0,74]; log-rank p-hodnota < 0,0001). V 8 rokoch bolo odhadované prežívanie 86,9 % v skupine AC→PH a 79,4 % v skupine AC→P, s absolútnym prínosom 7,4 %
(95 % CI 4,9 %, 10,0 %).
Finálne výsledky zo spoločnej analýzy štúdií NSABP B-31 and NCCTG N9831 sú zhrnuté v tabuľke 8
nižšie:
Tabuľka 8: Finálna analýza celkového prežívania zo spoločnej analýzy štúdií NSABP B-31 a NCCTG N9831
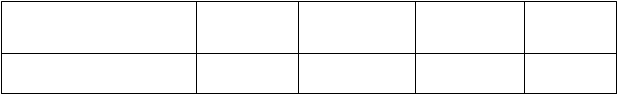
Parameter AC→P
(n = 2 032)
Úmrtie (OS udalosť):
AC→PH (n = 2 031)
p-hodnota oproti
AC→P
Pomer rizika oproti AC→P (95 % CI)
Počet pacientov s udalosťou (%) 418 (20,6 %) 289 (14,2 %) < 0,0001 0,64
A: doxorubicín; C: cyclofosfamid; P: paklitaxel; H: trastuzumab
Analýza DFS bola tiež vykonaná pri finálnej analýze OS zo spoločnej analýzy štúdií NSABP B-31
a NCCTG N9831. Aktualizované výsledky analýzy DFS (stratifikované HR = 0,61; 95 %CI [0,54,
0,69]) potvrdili podobný prínos DFS porovnateľný k definitívnej primárnej analýze DFS, napriek
24,8 % pacientom v ramene AC→P, ktorí prešli na trastuzumab. V 8 rokoch bolo odhadované prežívanie bez prejavov ochorenia 77,2 % (95 % CI: 75,4, 79,1) v skupine AC→PH, s absolútnym prínosom 11,8 % v porovnaní so skupinou AC→P.
V štúdii BCIRG 006 sa trastuzumab podával buď v kombinácii s docetaxelom po AC chemoterapii
(AC→DH), alebo v kombinácii s docetaxelom a karboplatinou (DCarbH).
Docetaxel sa podával takto:
- intravenózny docetaxel 100 mg/m2 ako 1- hodinová intravenózna infúzia podávaná každé
3 týždne v 4 cykloch (v deň 2 prvého cyklu a v deň 1 každého nasledujúceho cyklu)
alebo
- intravenózny docetaxel - 75 mg/m2 ako 1-hodinová intravenózna infúzia podávaná každé
3 týždne v 6 cykloch (v deň 2 prvého cyklu, v deň 1 každého cyklu);
po čom nasledovala:
- karboplatina - v cieľovej hodnote AUC = 6 mg/ml/min, podávaná v intravenóznej infúzii počas 30 – 60 minút, opakovane každé 3 týždne, celkovo v šiestich cykloch.
Trastuzumab sa podával týždenne s chemoterapiou a následne každé 3 týždne celkovo počas
52 týždňov.
Výsledky účinnosti zo štúdie BCIRG 006 sú zhrnuté v tabuľkách 9 a 10. Medián ďalšieho následného sledovania bol 2,9 rokov v AC→-D ramene a 3,0 roky v AC→DH a DCarbH ramenách.
Tabuľka 9: Prehľad analýz účinnosti v štúdii BCIRG 006 AC→D oproti AC→DH
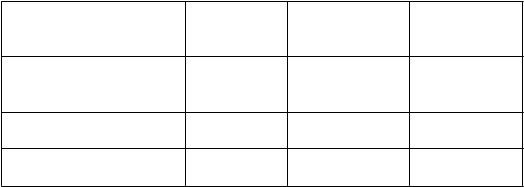
Parameter AC→D
(n = 1 073)
Prežívanie bez prejavov
AC→DI1 (n = 1 074)
Pomer rizík oproti AC→D (95 % CI) p-hodnota
ochorenia
Počet pacientov s príhodou
Vzdialená recidíva
195 134 0,61 (0,49, 0,77)
p < 0,0001
0,59 (0,46, 0,77)
Počet pacientov s príhodou 144 95
Celkové prežívanie (úmrtie)
p < 0,0001
Počet pacientov s príhodou 80 49 0,58 (0,40, 0,83)
AC→D = doxorubicín plus cyklofosfamid, po ktorých nasleduje docetaxel; AC→DH = doxorubicín plus cyklofosfamid, po ktorých nasleduje docetaxel plus trastuzumab; CI = interval spoľahlivosti
Tabuľka 10: Prehľad analýz účinnosti v štúdii BCIRG 006 AC→D oproti DCarbH
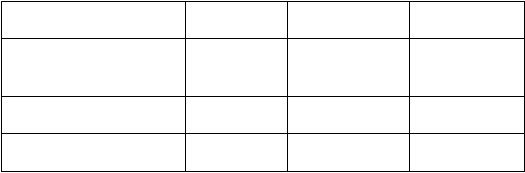
Parameter AC→D
(n = 1 073)
DCarbH (n = 1 074)
Pomer rizík oproti
AC→D (95 % CI)
Prežívanie bez prejavov ochorenia
Počet pacientov s príhodou
Vzdialená recidíva
195 145 0,67 (0,54, 0,83)
p = 0,0003
Počet pacientov s príhodou 144 103 0,65 (0,50, 0,84)
Úmrtie (príhoda OS)
Počet pacientov s príhodou 80 56
AC→D = doxorubicín plus cyklofosfamid, po ktorých nasleduje docetaxel; DCarbH = docetaxel, karboplatina a trastuzumab; CI = interval spoľahlivosti
V BCIRG 006 klinickej štúdii pre primárny koncový ukazovateľ, DFS (prežívanie bez prejavov ochorenia), pomer rizík sa premieta do absolútneho prínosu 3-ročného prežívania bez prejavov ochorenia 5,8 percentuálnych bodov (86,7 % oproti 80,9 %) v prospech skupiny s AC→DH (trastuzumabom) a 4,6 percentuálnych bodov (85,5 % oproti 80,9 %) v prospech skupiny DCarbH (trastuzumab) v porovnaní s AC→D.
V klinickej štúdii BCIRG 006, 213/1 075 pacientov v skupine DCarbH (TCH), 221/1 074 pacientov v skupine AC→DH (AC→TH) a 217/1 073 v skupine AC→D (AC→T) mali stav výkonnosti podľa Karnofského < 90 (buď 80 alebo 90). V tejto podskupine pacientov nebol zaznamenaný žiaden prínos prežívania bez prejavov ochorenia (DFS) (pomer rizík = 1,16, 95 % CI [0,73, 1,83] v DCarbH (TCH) oproti AC→D (AC→T), pomer rizík 0,97, 95 % CI [0,60, 1,55], v AC→DH (AC→TH) oproti AC→D).
V ďalšej post-hoc exploračnej analýze bola uskutočnená združená analýza súboru údajov z klinických štúdií NSABP B-31/NCCTG N9831* a BCIRG006 spájajúcich DFS príhody a symptomatické srdcové príhody, ktoré sú zhrnuté v tabuľke 11:
Tabuľka 11: Výsledky post-hoc exploračnej analýzy z klinických štúdií NSABP B-
31/NCCTGN9831* a BCIRG006 spájajúcich DFS príhody a symptomatické srdcové príhody
Primárna analýza účinnosti
DFS Pomer rizík
(95 % CI)
p-hodnota
Analýza účinnosti dlhodobého následného sledovania **
DFS Pomer rizík
(95 % CI)
p-hodnota
Post-hoc exploračná analýza s DFS a symptomatickými kardiálnymi príhodami
Dlhodobé následné sledovanie **
Pomer rizík
(95% CI)
AC → PH (oproti AC → P) (NSABP B-31 a NCCTG N9831)*
0,48 (0,39; 0,59) p < 0,0001
0,61 (0,54; 0,69) p < 0,0001
0,67 (0,60; 0,75)
AC → DH (oproti
AC → D) (BCIRG
006)
0,61 (0,49; 0,77) p < 0,0001
0,72 (0,61; 0,85) p < 0,0001
0,77 (0,66; 0,90)
DCarbH (oproti
AC → D) (BCIRG
006)
0,67 (0,54; 0,83) p = 0,0003
0,77 (0,65; 0,90) p = 0,0011
0,77 (0,66; 0,90)
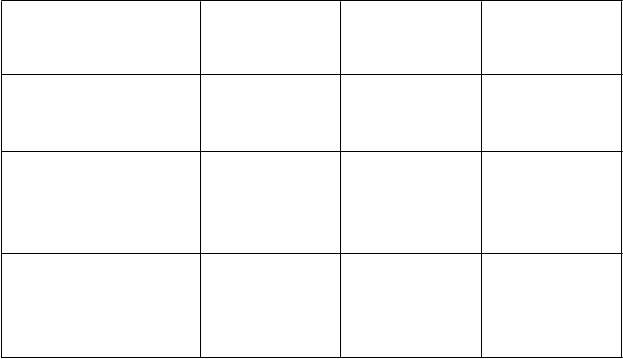
A: doxorubicín; C: cyklofosfamid; P: paklitaxel; D: docetaxel; Carb: karboplatina; H: trastuzumab CI = interval spoľahlivosti
*V čase definitívnej analýzy DFS. Medián ďalšieho následného sledovania bol 1,8 roka u pacientov v skupine
AC→P a 2,0 roky u pacientov v skupine AC→PH
**Medián trvania dlhodobého následného sledovania v spojenej analýze klinických štúdií bol u pacientov
v ramene AC→PH 8,3 rokov (rozsah 0,1 až 12,1) a 7,9 rokov (rozsah 0,0 až 12,2) v ramene AC→P; medián trvania dlhodobého následného sledovania v štúdii BCIRG006 bol 10,3 rokov v ramene AC→-D (rozsah 0,0 až
12,6) aj v ramene DCarbH (rozsah 0,0 až 13,1); a 10,4 rokov (rozsah 0,0 až 12,7) v ramene AC→DH .
Včasný karcinóm prsníka (neoadjuvantná-adjuvantná liečba)Zatiaľ nie sú dostupné výsledky porovnávajúce účinnosť trastuzumabu podávaného s chemoterapiou
v adjuvantnej liečbe s výsledkami získanými v neoadjuvantnej/adjuvantnej liečbe.
V neoadjuvantnej-adjuvantnej liečbe bola štúdia MO16432, multicentrické, randomizované klinické skúšanie, navrhnuté na skúmanie klinickej účinnosti súbežného podávania trastuzumabu s neoadjuvantnou chemoterapiou vrátane antracyklínu aj taxánu, po ktorých nasledovalo adjuvantné podávanie trastuzumabu, až do celkového trvania liečby 1 rok. Do štúdie boli zaradení pacienti
s novodiagnostikovaným lokálne pokročilým (štádium III) alebo inflamatórnym VKP. Pacienti
s HER2+ nádormi boli randomizovaní tak, aby dostávali buď neoadjuvantnú chemoterapiu súbežne
s neoadjuvantnou-adjuvantnou liečbou trastuzumabom alebo samotnú neoadjuvantnú chemoterapiu.
V štúdii MO16432 sa trastuzumab (nasycovacia dávka 8 mg/kg nasledovaná udržiavacou dávkou
6 mg/kg každé 3 týždne) podával súbežne s 10 cyklami neoadjuvantej chemoterapie nasledovne:
• Doxorubicín 60 mg/m2 a paklitaxel 150 mg/m2 podávané každé 3 týždne počas 3 cyklov, po ktorých nasledoval
• Paklitaxel 175 mg/m2 podávaný každé 3 týždne počas 4 cyklov,
po ktorých nasledovalo
• CMF v 1. a 8. deň každé 4 týždne počas 3 cyklov,
po ktorých po operácii nasledovalo
• podávanie ďalších cyklov adjuvantného trastuzumabu (na dokončenie 1 roka liečby)
Výsledky účinnosti zo štúdie MO16432 sú zhrnuté v tabuľke 12. Medián trvania následného sledovania v skupine s trastuzumabom bol 3,8 rokov.
Tabuľka 12: Výsledky účinnosti zo štúdie MO16432
Parameter Chemo + Trastuzumab

(n=115)
Prežívanie bez príhody
Len chemo
(n=116)
Pomer rizík
(95 % CI)
Počet pacientov s príhodou 46 59
0,65 (0,44; 0,96)
p = 0,0275
Celková patologická
kompletná odpoveď* (95 % CI) Celkové prežívanie
40 % (31,0; 49,6)
20,7 % (13,7; 29,2)
p=0,0014
Pomer rizík
(95 % CI)
Počet pacientov s príhodou 22 33
*definovaná ako žiadny invazívny karcinóm v prsníku a axilárnych uzlinách
0,59 (0,35; 1,02)
p = 0,0555
Absolútny prínos 13 percentuálnych bodov v prospech skupiny s trastuzumabom bol odhadovaný
z hľadiska 3-ročného prežívania bez príhody (65 % v porovnaní s 52 %).
Metastatický karcinóm žalúdka
Trastuzumab sa skúmal v randomizovanom, otvorenom klinickom skúšaní fázy III ToGA (BO18255)
v kombinácii s chemoterapiou oproti chemoterapii samotnej. Chemoterapia sa podávala takto:
- kapecitabín - 1 000 mg/m2 perorálne dvakrát denne počas 14 dní každé tri týždne, 6
cyklov (od večera 1. dňa 1 do rána 15. dňa každého cyklu)
alebo
- intravenózne 5-fluóruracil - 800 mg/m2/deň vo forme kontinuálnej intravenóznej infúzie počas 5 dní, podávanej každé 3 týždne, 6 cyklov (1. až 5. deň každého cyklu).
Každý druh chemoterapie sa podával spolu s:
- cisplatinou - 80 mg/m2 každé 3 týždne, 6 cyklov v 1. deň každého cyklu. Výsledky účinnosti z klinickej štúdie BO18225 sú zhrnuté v tabuľke 13:
Tabuľka 13: Výsledky účinnosti z klinickej štúdie BO18225
Parameter
FP
 n = 290
FP + H
n = 294
Pomer rizík (95 % CI)
p-hodnota
n = 290
FP + H
n = 294
Pomer rizík (95 % CI)
p-hodnota
Celkové prežívanie, medián
v mesiacoch 11,1
13,8 0,74 (0,60 – 0,91) 0,0046
Prežívanie bez progresie ochorenia, medián v mesiacoch
Čas do progresie ochorenia, medián v mesiacoch
5,5 6,7 0,71 (0,59 – 0,85) 0,0002
5,6 7,1 0,70 (0,58 – 0,85) 0,0003
Celkový počet odpovedí, % 34,5 % 47,3 % 1,70a (1,22, 2,38) 0,0017
Trvanie odpovede, medián v mesiacoch
4,8 6,9 0,54 (0,40 – 0,73) < 0,0001
FP + H: fluórpyrimidín/cisplatina + trastuzumab
FP: fluórpyrimidín/cisplatina
a Pomer pravdepodobnosti
Do klinického skúšania boli zaradení pacienti, ktorí neboli predtým liečení pre HER2-pozitívny inoperabilný lokálne pokročilý alebo rekurentný a/alebo metastatický adenokarcinóm žalúdka alebo gastroezofageálneho spojenia a neboli vhodní na kuratívnu liečbu. Primárnym koncovým ukazovateľom bolo celkové prežívanie, ktoré bolo definované ako čas od dátumu randomizácie do dátumu úmrtia z akejkoľvek príčiny. V čase analýzy zomrelo celkovo 349 randomizovaných pacientov: 182 pacientov (62,8 %) v kontrolnom ramene a 167 pacientov (56,8 %) v liečebnom ramene. Väčšina úmrtí bola spôsobená udalosťami spojenými so základným nádorovým ochorením.
Post-hoc analýzy podskupín naznačujú, že pozitívne liečebné účinky sú obmedzené na tumory
s vyššou hladinou HER2 proteínu (IHC 2+ /FISH+ alebo IHC 3+). Medián celkového prežívania v skupine s vysokou expresiou receptora HER2 bol 11,8 mesiaca oproti 16 mesiacom, HR 0,65
(95 % CI 0,51 - 0,83) a medián prežívania bez prejavov ochorenia bol 5,5 mesiaca oproti
7,6 mesiaca, HR 0,64 (95 % CI 0,51 – 0,79) pri FP oproti FP + H v tomto poradí. Pri celkovom prežívaní bol HR 0,75 (CI 95 % 0,51 – 1,11) v skupine IHC 2+/FISH+ a v skupine IHC
3+/FISH bol HR 0,58 (CI 95 % 0,41 – 0,81).
V exploratívnej analýze podskupiny, ktorá sa uskutočnila v klinickom skúšaní ToGA (BO18255), sa nezistil zjavný benefit na celkové prežívanie pri pridaní trastuzumabu do liečby pacientom s ECOG PS 2 pri zaradení do štúdie [HR 0,96 (95 % CI 0,51 – 1,79), s nemerateľným ochorením [HR 1,78 (95 % CI 0,87 – 3,66)] a lokálne pokročilým ochorením [HR 1,20 (95 % CI
0,29 – 4,97)].
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií
s trastuzumabom vo všetkých podskupinách pediatrickej populácie pre karcinóm prsníka a žalúdka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika trastuzumabu sa hodnotila analýzou populačného farmakokinetického modelu za použitia skupiny údajov od 1 582 jedincov, vrátane pacientov s HER2 pozitívnym MKP, VKP, MKŽ a iných typov karcinómov a zdravých dobrovoľníkov v 18 klinických skúšaniach fázy I, II a III s intravenóznym trastuzumabom. Dvojkompartmentový model s paralelnou lineárnou a nelineárnou elimináciou z centrálneho kompartmentu popísal profil trastuzumabu koncentrácia-čas. Pre nelineárnu elimináciu sa celkový klírens zvýšil pri znížených koncentráciách. Z toho dôvodu nemôže byť vyvodená hodnota konštanty biologického polčasu trastuzumabu. T1/2 sa znižuje so znižujúcimi sa koncentráciami v rámci dávkovacieho intervalu (pozri Tabuľku 16). Pacienti s MKP a VKP mali podobné farmakokinetické parametre (napr. klírens (CL), objem centrálneho kompartmentu (Vc) a predpokladané populačné expozície
v rovnovážnom stave (Cmin, Cmax a AUC) . Lineárny klírens pre MKP bol 0,136 l/deň; 0,112
l/deň pre VKP a 0,176 l/deň pre MKŽ. Nelineárne hodnoty parametrov eliminácie pre MKP,
VKP a MKŽ pacientov boli 8,81 mg/deň pre maximálnu mieru eliminácie (Vmax) a 8,92 pg/ml pre konštantu Michaelis-Menten (Km). Objem centrálnych kompartmentov bol 2,62 l
pre pacientov s MKP a VKP a 3,63 l pre pacientov s MKŽ.
Vo finálnych populačných FK modeloch boli okrem primárneho typu karcinómu navyše identifikované, ako štatisticky signifikantné kovariáty ovplyvňujúce expozíciu trastuzumabu, telesná hmotnosť, sérová aspartát-aminotransferáza a albumín. Každopádne magnitúda účinku týchto kovariátov na expozíciu trastuzumabu poukazuje na to, že je nepravdepodobné, aby tieto kovariáty mali klinicky významný účinok na koncentrácie trastuzumabu.
Predpokladané populačné FK hodnoty expozície (medián s 5. – 95. percentilami) a FK hodnoty parametrov pri klinicky relevantných koncentráciách (Cmax a Cmin) pre pacientov s MKP, VKP
a MKŽ liečených schváleným dávkovacím režimom raz týždenne a každé tri týždne sú
zobrazené v tabuľke 14 (cyklus 1), tabuľke 15 (rovnovážny stav), a tabuľke 16 (FK parametre)
nižšie.
Tabuľka 14 Predpokladané populačné FK hodnoty expozície v cykle 1 (medián s 5. – 95. percentilami) pre dávkovací režim intravenózneho trastuzumabu u pacientov s MKP, VKP a MKŽ
Režim Primárny typ n karcinómu
C
min
(pg/ml)
C
max
(pg/ml)
AUC 0 – 21 dní
(pg.deň/ml)
8 mg/kg +
MKP 805 28,7 182 1 376
(2,9 – 46,3) (134 – 280) (728 – 1 998)
30,9 176 1 390
6 mg/kg q3w
VKP 390
MKŽ 274
(18,7 – 45,5) (127 – 227) (1 039 – 1 895)
23,1 132 1 109
(6,1 – 50,3) (84,2 – 225) (588 – 1 938)
MKP 805 37,4 76,5 1073
4 mg/kg + (8,7 – 58,9) (49,4 – 114) (597 – 1 584)
2 mg/kg qw
VKP 390
38,9 76,0 1 074 (25,3 – 58,8) (54,7 – 104) (783 – 1 502)
q3w = každé 3 týždne; qw = raz týždenne
Tabuľka 15 Predpokladané populačné FK hodnoty expozície pre rovnovážny stav (medián s 5. – 95. percentilami) pre dávkovací režim intravenózneho trastuzumabu u pacientov s MKP, VKP a MKŽ
Režim Primárny typ karcinómu

Cmin,ss*
(gg/ml)
Cmax,ss**
(gg/ml)
AUCss, 0 – 21 dní
(pg.deň/ml)
Čas do
rovnováž- neho stavu (týždeň)

MKP 805 44,2 179 1 736 12 (1,8 – 85,4) (123 – 266) (618 – 2 756)
8 mg/kg + VKP 390 53,8 184 1 927 15
6 mg/kg q3w (28,7 – 85,8) (134 – 247) (1 332 – 2 771)
MKŽ 274 32,9 131 1 338 9 (6,1 – 88,9) (72,5 – 251) (557 – 2 875)
MKP 805 63,1 107 1 710 12
4 mg/kg + (11,7 – 107) (54,2 – 164) (581 – 2 715)
2 mg/kg qw 72,6 115 1 893
(46 – 109) (82,6 – 160) (1 309 – 2 734)
q3w = každé 3 týždne; qw = raz týždenne
*Cmin,ss = Cmin v rovnovážnom stave
**Cmax,ss = Cmax v rovnovážnom stave
***čas do 90 % rovnovážneho stavu
Tabuľka 16 Predpokladané populačné FK hodnoty pri rovnovážnom stave pre dávkovací režim intravenózneho trastuzumabu u pacientov s MKP, VKP a MKŽ
Režim
Primárny typ karcinómu
Celkové CL rozsah n z Cmax,ss do

Cmin,ss
(l/deň)
Rozsah t1/2 z Cmax,ss do
Cmm,ss
(deň)
8 mg/kg + 6mg/kg q3w
MKP 805 0,183 – 0,302 15,1 – 23,3
VKP 390 0,158 – 0,253 17,5 – 26,6
MKŽ 274 0,189 – 0,337 12,6 – 20,6
4 mg/kg + MKP 805 0,213 – 0,259 17,2 – 20,4
2 mg/kg qw VKP 390 0,184 – 0,221 19,7 – 23,2
q3w = každé 3 týždne; qw = raz týždenne
Vyplavenie trastuzumabu (washout)
Vyplavenie (washout) trastuzumabu sa hodnotilo po jeho intravenóznom podaní v q1w alebo q3w za použitia FK populačného modelu. Výsledky týchto simulácií naznačujú, že najmenej 95 %
pacientov dosiahne koncentrácie < 1 mikrogram/ml (približne 3 % z predpokladanej populácie Cmm,ss,
alebo približne 97 % vyplavenie) do 7 mesiacov.
HER2-ECD uvoľnený do obehu
Výskumné analýzy kovariátov s informáciou z podskupiny pacientov naznačujú, že pacienti s vyššou hladinou uvoľnenej HER2-ECD dosiahli rýchlejší nelineárny klírens (nižší Km)
(p < 0,001). Zistila sa súvislosť medzi uvoľneným antigénom a hladinou SGOT/AST, dopad uvoľneného antigénu na klírens môže byť čiastočne vysvetlený hladinami SGOT/AST.
Východiskové hladiny uvoľnenej HER2-ECD pozorované u pacientov s MKŽ (metastatický karcinóm žalúdka) boli porovnateľné s hladinami u pacientov s MKP (metastatický karcinóm prsníka) a VKP (včasný karcinóm prsníka), nebol pozorovaný žiaden evidentný dopad
na klírens trastuzumabu.
5.3 Predklinické údaje o bezpečnosti
Neexistujú žiadne dôkazy o akútnej toxicite alebo toxicite po opakovanom podávaní v štúdiách trvajúcich až 6 mesiacov, alebo reprodukčnej toxicity v teratologických štúdiách, štúdiách fertility u žien alebo v štúdiách zameraných na neskorú gestačnú toxicitu/prestup liečiva cez placenta. Trastuzumab nie je genotoxický.
Neboli vykonané žiadne dlhodobé štúdie na zvieratách na stanovene karcinogénneho potenciálu trastuzumabu, alebo kvôli zisteniu jeho vplyvu na fertilitu samcov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidíniumchlorid monohydrát
L-histidín sacharóza polysorbát 20
6.2 Inkompatibility
Tento liek sa nesmie miešať alebo riediť s inými liekmi, okrem tých, ktoré sú uvedené v časti 6.6.
Liek sa nesmie riediť roztokmi glukózy.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
4 roky
Rekonštituovaný roztok
Po rekonštitúcii sterilnou vodou na injekcie je rekonštituovaný roztok fyzikálne a chemicky stabilný
počas 48 hodín pri 2 °C – 8 °C. Nepoužitý rekonštituovaný roztok sa má zlikvidovať.
Intravenózne infúzne roztoky Trazimery v polyvinylchloridových, polyetylénových, polypropylénových alebo etylén vinylacetátových vakoch alebo sklenených intravenóznych fľašiach obsahujúcich 9 mg/ml (0,9 %) injekčného roztoku chloridu sodného sú fyzikálne
a chemicky stabilné počas 24 hodín pri teplote neprevyšujúcej 30 °C.
Z mikrobiologického hľadiska sa má rekonštituovaný roztok a infúzny roztok Trazimery použiť okamžite. Po rekonštitúcii a riedení je možné liek uchovávať iba za kontrolovaných
a validovaných aseptických podmienok. Ak sa liek nepoužije okamžite, používateľ je zodpovedný za dobu a podmienky uchovávania.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke pri teplote (2°C – 8°C). Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Neotvorené injekčné liekovky Trazimery sa môžu uchovávať pri teplote do 30 °C len počas obdobia trvajúceho maximálne 3 mesiace. Na konci tohto 3-mesačného obdobia alebo po dátume exspirácie uvedenom na injekčnej liekovke, podľa toho, čo nastane skôr, injekčnú liekovku zlikvidujte. Dátum
„zlikvidujte po“ poznačte do dátumového poľa, ktoré je poskytnuté na škatuľke. Podmienky uchovávania po rekonštitúcii lieku pozri časti 6.3 a 6.6.
6.5 Druh obalu a obsah balenia
Trazimera 150mgprášoknainfúznykoncentrát
Injekčná liekovka z číreho skla typu I s obsahom 15 ml s butylovou gumovou zátkou, potiahnutá
fluóro-živicovým filmom obsahujúca 150 mg trastuzumabu.
Každá krabička obsahuje jednu injekčnú liekovku. Trazimera420mgprášoknainfúznykoncentrát
Injekčná liekovka z číreho skla typu I s obsahom 30 ml s butylovou gumovou zátkou, potiahnutá
fluóro-živicovým filmom obsahujúca 420 mg trastuzumabu.
Každá krabička obsahuje jednu injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Počas rekonštitúcie sa má s Trazimerou manipulovať opatrne. Nadmerné spenenie počas rekonštitúcie alebo trasenie rekonštituovaným roztokom môže spôsobiť ťažkosti s natiahnutím potrebného množstva Trazimery z injekčnej liekovky.
Rekonštituovaný roztok nezmrazujte.
Trazimera 150mgprášoknainfúznykoncentrát
Za príslušných aseptických podmienok sa každá 150 mg injekčná liekovka Trazimery rekonštituuje
pomocou 7,2 ml sterilnej vody na injekcie (nie je súčasťou balenia). Na rekonštitúciu lieku sa nesmú použiť iné rozpúšťadlá.
Po rekonštitúcii získame 7,4 ml roztoku na jednorazové podanie, ktorý obsahuje približne 21 mg/ml trastuzumabu, pri pH približne 6,0. 4 % rezerva objemu umožňuje natiahnuť vyznačenú dávku 150 mg z každej injekčnej liekovky.
Trazimera 420mgprášoknainfúznykoncentrát
Za príslušných aseptických podmienok sa každá 420 mg injekčná liekovka Trazimery rekonštituuje
pomocou 20 ml sterilnej vody na injekcie (nie je súčasťou balenia). Na rekonštitúciu lieku sa nesmú použiť iné rozpúšťadlá.
Po rekonštitúcii získame 20,6 ml roztoku na jednorazové podanie, ktorý obsahuje približne 21 mg/ml trastuzumabu, pri pH približne 6,0. 5 % rezerva objemu umožňuje natiahnuť vyznačenú dávku 420 mg z každej injekčnej liekovky.
Injekčná liekovka
Trazimery
150 mg injekčná liekovka
420 mg injekčná liekovka
Objem sterilnej vody
na injekcie Finálna koncentrácia
+ 7,2 ml = 21 mg/ml
+ 20 ml = 21 mg/ml
 Návod
na
rekonštitúciu
Návod
na
rekonštitúciu
1) Pomocou sterilnej injekčnej striekačky pomaly vstreknite príslušný objem (ako je uvedené
vyššie) sterilnej vody na injekcie do injekčnej liekovky obsahujúcej lyofilizovaný prášok
Trazimery. Prúd vody na injekcie nasmerujte na lyofilizovaný koláč.
2) Pri rekonštitúcii si môžete pomôcť jemným krúžením s injekčnou liekovkou. LIEKOVKOU NETRASTE!
Po rekonštitúcii často dochádza k miernemu speneniu lieku. Injekčnú liekovku nechajte voľne stáť približne 5 minút. Rekonštituovaný roztok Trazimery má číru až svetlo hnedastožltú farbu a nesmie obsahovať žiadne viditeľné čiastočky.
Potrebný objem roztoku sa vypočíta:
• na základe nasycovacej dávky 4 mg trastuzumabu/kg telesnej hmotnosti, alebo nasledujúcej týždennej dávky 2 mg trastuzumabu/kg telesnej hmotnosti pomocou nasledujúceho vzorca:
Objem (ml) =
telesnáhmotnosť(kg)xdávka(4mg/kg -nasycovaciaalebo2mg/kg -udržiavacia)21 (mg/ml, koncentrácia rekonštituovaného roztoku)
• na základe nasycovacej dávky 8 mg trastuzumabu/kg telesnej hmotnosti, alebo nasledujúcej dávky 6 mg/kg telesnej hmotnosti podanej každé tri týždne pomocou nasledujúceho vzorca:
Objem (ml) =
telesnáhmotnosť(kg)xdávka(8mg/kg -nasycovaciaalebo6mg/kg -udržiavacia)21 (mg/ml, koncentrácia rekonštituovaného roztoku)
Príslušné množstvo roztoku sa má natiahnuť z injekčnej liekovky a pridať do infúzneho vaku alebo fľaše s obsahom 250 ml roztoku chloridu sodného 9 mg/ml (0,9 %). Nepoužívajte roztoky
s obsahom glukózy (pozri časť 6.2). Vak alebo fľašu zľahka prevracajte, aby sa roztok premiešal
a nedošlo pritom k speneniu. Po príprave sa má infúzia okamžite podať. Ak sa liek nariedi za aseptických podmienok, môže sa skladovať 24 hodín (pri teplote neprevyšujúcej 30°C).
Parenterálne lieky sa majú pred podaním skontrolovať voľným okom, či neobsahujú nejaké čiastočky a či nedošlo k zmene sfarbenia.
Medzi Trazimerou a polyvinylchloridovými, polyetylénovými, polypropylénovými alebo etylén vinylacetátovými vakmi alebo sklenenými intravenóznymi fľašami sa nepozorovali žiadne inkompatibility.
Trazimera je určená na jednorazové použitie, pretože tento liek neobsahuje žiadne konzervačné látky. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Europe MA EEIG Boulevard de la Plaine 17
1050 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/18/1295/001
EU/1/18/1295/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. júl 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu