
dimetylfumarátom a inými liekmi s obsahom fumarátov sa u pacientov s lymfopéniou (počet lymfocytov nižší ako LLN) vyskytli prípady PML. Zdá sa, že dlhotrvajúca, mierna až ťažká
lymfopénia zvyšuje riziko PML pri liečbe Tecfiderou, riziko však nemožno vylúčiť ani u pacientov
s miernou lymfopéniou.
Ďalšie faktory, ktoré by mohli prispievať k zvýšeniu rizika PML u pacientov s lymfopéniou, sú:
– dĺžka liečby Tecfiderou. Prípady PML sa vyskytli približne po 1 až 5 rokoch liečby, presná súvislosť s dĺžkou liečby však nie je známa.
– výrazné zníženie počtu CD4+ T-lymfocytov a hlavne počtu CD8+ T-lymfocytov, ktoré sú dôležité pre imunitnú obranu (pozri časť 4.8), a
– predchádzajúca imunosupresívna alebo imunomodulačná liečba (pozri nižšie).
Lekári majú zhodnotiť zdravotný stav svojich pacientov, aby určili, či príznaky
naznačujú neurologickú poruchu, a ak áno, či sú tieto príznaky typické pre SM alebo prípadne naznačujú PML.
Pri prvom prejave alebo príznaku, ktorý naznačuje PML, sa má Tecfidera vysadiť a pomocou metódy kvantitatívnej polymerázovej reťazovej reakcie (polymerase chain reaction, PCR) vrátane stanovenia DNA vírusu JCV v mozgovomiechovom moku (cerebrospinal fluid, CSF) sa musia vykonať vhodné diagnostické vyšetrenia. Príznaky PML sa môžu podobať relapsu roztrúsenej sklerózy (SM). Typické príznaky spájané s PML sú rôzne, vyvíjajú sa niekoľko dní až týždňov a zahŕňajú postupnú slabosť jednej polovice tela alebo nemotornosť končatín, poruchy videnia a zmeny v myslení, pamäti
a orientácii vedúce k zmätenosti a zmenám osobnosti. Lekári majú byť zvlášť obozretní pri príznakoch naznačujúcich PML, ktoré si pacient nemusí všimnúť. Pacientom sa má tiež odporučiť, aby o
svojej liečbe informovali svojho partnera alebo opatrovateľov, pretože si môžu všimnúť príznaky,
ktoré si pacient neuvedomuje.
PML sa môže vyskytovať iba v prípade infekcie JCV. Je potrebné vziať do úvahy, že vplyv lymfopénie na presnosť testovania sérových protilátok proti vírusu JCV sa u pacientov liečených dimetylfumarátom neskúmal. Je potrebné tiež poznamenať, že negatívny test na protilátky proti vírusu JCV (za prítomnosti normálneho počtu lymfocytov) nevylučuje možnosť následnej infekcie JCV.
Ak sa u pacienta vyvinie PML, liečba Tecfiderou sa musí natrvalo ukončiť. Predchádzajúcaliečbazahŕňajúcaimunosupresívnealeboimunomodulačnéterapie
Neboli vykonané žiadne štúdie hodnotiace účinnosť a bezpečnosť Tecfidery pri prechode pacientov
z iných liekov modifikujúcich ochorenie na Tecfideru. Podiel predchádzajúcej imunosupresívnej terapie na rozvoji PML u pacientov liečených dimetylfumarátom je možný.
Prípady PML sa vyskytujú u pacientov s rizikom PML predtým liečených natalizumabom. Lekári si majú byť vedomí, že v prípadoch PML, ktoré sa vyskytli po nedávnom ukončení liečby natalizumabom nemusí byť prítomná lymfopénia.
Okrem toho sa väčšina potvrdených prípadov PML pri liečbe Tecfiderou vyskytla u pacientov s predchádzajúcou imunomodulačnou liečbou.
Pri prechode pacientov z iných liekov modifikujúcich ochorenie na Tecfideru sa musí brať do úvahy polčas a spôsob účinku predchádzajúcej liečby, aby sa predišlo dodatočným účinkom na imunitný systém a zároveň znížilo riziko reaktivácie SM. Odporúča sa vyšetriť krvný obraz pred nasadením Tecfidery a pravidelne v priebehu liečby (pozri Krvné/laboratórne testy vyššie).
Ťažká porucha funkcie obličiek a pečene
Tecfidera nebola skúšaná u pacientov s ťažkou poruchou funkcie obličiek alebo pečene, preto je
u týchto pacientov potrebná opatrnosť (pozri časť 4.2).
Ťažké aktívne gastrointestinálne ochorenie
Tecfidera nebola skúšaná u pacientov s ťažkým aktívnym gastrointestinálnym ochorením, preto je
u týchto pacientov potrebná opatrnosť.
Sčervenanie
Počas klinických skúšaní bolo u 34 % pacientov liečených Tecfiderou zaznamenané začervenanie.
U väčšiny pacientov bolo začervenanie ľahkého až stredne ťažkého stupňa. Údaje zo štúdií so zdravými dobrovoľníkmi naznačujú, že začervenanie spájané s dimetylfumarátom je pravdepodobne
sprostredkované prostaglandínmi. U pacientov postihnutých netolerovateľným začervenaním môže
byť prospešný krátky liečebný cyklus kyselinou acetylsalicylovou bez gastrorezistentného obalu
v dávke 75 mg (pozri časť 4.5). V dvoch štúdiách so zdravými dobrovoľníkmi sa výskyt a závažnosť začervenania počas dávkovacieho obdobia znížili.
Traja pacienti z celkovo 2 560 pacientov liečených dimetylfumarátom v klinických skúšaniach mali závažné príznaky sčervenania, ktoré boli pravdepodobne hypersenzitívnymi alebo anafylaktoidnými reakciami. Tieto udalosti neboli život ohrozujúce, ale viedli k hospitalizácii. Predpisujúci lekári
i pacienti si majú byť vedomí tejto možnosti v prípade ťažkých reakcií sčervenania (pozri časti 4.2, 4.5
a 4.8).
Anafylaktické reakcie
Po podaní lieku Tecfidera boli po uvedení lieku na trh hlásené prípady anafylaxie/anafylaktoidnej
reakcie. Príznaky môžu zahŕňať dyspnoe, hypoxiu, hypotenziu, angioedém, vyrážku alebo urtikáriu. Mechanizmus anafylaxie indukovanej dimetylfumarátom nie je známy. Tieto reakcie sa väčšinou
vyskytujú po prvej dávke, ale môžu sa tiež vyskytnúť kedykoľvek v priebehu liečby a môžu byť
závažné a život ohrozujúce. Pacientov je potrebné poučiť, aby v prípade výskytu prejavov alebo príznakov anafylaxie prestali Tecfideru užívať a okamžite vyhľadali lekársku pomoc. Liečba sa nemá znovu nasadiť (pozri časť 4.8).
Infekcie
V placebom kontrolovaných skúšaniach III. fázy bol výskyt infekcií (60 % oproti 58 %) a závažných
infekcií (2 % oproti 2 %) podobný u pacientov liečených Tecfiderou ako u pacientov na placebe,
v danom poradí. Avšak, ak sa u pacienta v dôsledku imunomodulačných vlastností Tecfidery (pozri časť 5.1) rozvinie závažná infekcia, je potrebné zvážiť prerušenie liečby Tecfiderou a pred obnovením liečby sa majú prehodnotiť jej prínosy a riziká. Pacientov liečených Tecfiderou je potrebné poučiť,
aby lekárovi hlásili príznaky infekcií. U pacientov so závažnými infekciami sa liečba Tecfiderou nemá začať, kým nie je infekcia vyliečená.
Nebol pozorovaný zvýšený výskyt závažných infekcií u pacientov s počtom lymfocytov < 0,8 x 109/l alebo < 0,5 x 109/l (pozri časť 4.8). Ak liečba pokračuje aj pri stredne ťažkej až ťažkej dlhotrvajúcej lymfopénii, riziko oportúnnych infekcií, vrátane PML, nemôže byť vylúčené (pozri časť 4.4 podčasť PML).
Infekcie vyvolané vírusom herpes zoster
V súvislosti s liekom Tecfidera sa vyskytli prípady herpes zoster. Väčšina prípadov nebola závažná,
boli však hlásené aj závažné prípady zahŕňajúce diseminovaný herpes zoster, herpes zoster ophthalmicus, herpes zoster oticus, infekciu nervového systému spôsobenú vírusom herpes zoster, herpes zoster meningoencephalitis a herpes zoster meningomyelitis. Tieto udalosti sa môžu vyskytnúť kedykoľvek počas liečby. Sledujte prejavy a príznaky infekcie herpes zoster u pacientov užívajúcich Tecfideru, najmä v prípadoch, keď je hlásený súčasný výskyt lymfocytopénie. V prípade výskytu infekcie herpes zoster je potrebné podať primeranú liečbu proti infekcii herpes zoster. U pacientov so závažnými infekciami zvážte prerušenie liečby Tecfiderou do vymiznutia infekcie (pozri časť 4.8).
Začiatok liečby
Liečba Tecfiderou sa má začať postupne za účelom zníženia výskytu sčervenania
a gastrointestinálnych nežiaducich reakcií (pozri časť 4.2).
Fanconiho syndróm
V súvislosti s liekom obsahujúcim dimetylfumarát v kombinácii s inými estermi kyseliny fumarovej
boli hlásené prípady výskytu Fanconiho syndrómu. Včasná diagnóza Fanconiho syndrómu
a ukončenie liečby dimetylfumarátom sú dôležité na prevenciu vzniku poškodenia funkcie obličiek
a osteomalácie, pretože syndróm je zvyčajne reverzibilný. Najdôležitejšími prejavmi sú: proteinúria, glukozúria (s normálnymi hladinami cukru v krvi), hyperaminoacidúria a fosfatúria (možnosť
súbežného výskytu s hypofosfatémiou). Progresia môže zahŕňať príznaky ako je polyúria, polydipsia a proximálna svalová slabosť. V zriedkavých prípadoch sa môže vyskytnúť hypofosfatemická osteomalácia s nelokalizovanou bolesťou kostí, zvýšenie hladiny alkalickej fosfatázy v sére a únavové zlomeniny kostí. Dôležité je, že Fanconiho syndróm sa môže vyskytnúť bez zvýšených hladín kreatinínu alebo zníženej rýchlosti glomerulárnej filtrácie. V prípade nejasných príznakov je potrebné zvážiť, či nejde o Fanconiho syndróm a majú sa vykonať príslušné vyšetrenia.
Pediatrická populácia
Bezpečnostný profil u pediatrických pacientov je v porovnaní s dospelými kvalitatívne podobný, preto
sa aj u pediatrických pacientov uplatňujú rovnaké upozornenia a opatrenia. Kvantitatívne rozdiely v bezpečnostnom profile pozri v časti 4.8.
Dlhodobá bezpečnosť Tecfidery u pediatrickej populácie nebola doteraz stanovená.
4.5 Liekové a iné interakcie
Tecfidera nebola skúšaná v kombinácii s antineoplastickou alebo imunosupresívnou liečbou, preto je pri ich súbežnom podávaní potrebná opatrnosť. V klinických štúdiách s roztrúsenou sklerózou liečba relapsov krátkodobo súbežne podávanými intravenóznymi kortikosteroidmi nebola spojená s klinicky relevantným nárastom infekcií.
Je možné zvážiť súbežné podanie neživých očkovacích látok podľa národných očkovacích schém počas liečby Tecfiderou. V klinickej štúdii zahŕňajúcej celkom 71 pacientov s relaps-remitujúcou roztrúsenou sklerózou dosiahli pacienti, ktorí dostávali Tecfideru 240 mg dvakrát denne počas najmenej 6 mesiacov (n=38) alebo nepegylovaný interferón počas najmenej 3 mesiacov (n=33) porovnateľnú imunitnú odpoveď (definovanú ako ≥ 2-násobný vzostup titra protilátok po očkovaní oproti hodnote pred očkovaním) na podanie tetanového toxoidu (tzv. recall antigénu) a konjugovanej polysacharidovej vakcíny proti meningokoku C (neoantigén), zatiaľ čo imunitná odpoveď na rôzne sérotypy nekongujovanej 23-valentnej polysacharidovej pneumokokovej vakcíny (antigén nezávislý na T lymfocytoch) sa v oboch liečebných skupinách odlišovala. Pozitívna imunitná odpoveď, definovaná ako ≥ 4-násobný vzostup titra protilátok na tieto tri očkovacie látky, sa dosiahla
u menšieho počtu pacientov v oboch liečebných skupinách. Boli zaznamenané malé číselné rozdiely v odpovedi na tetanový toxoid a polysacharid pneumokoka sérotypu 3 v prospech nepegylovaného interferónu.
O účinnosti a bezpečnosti podania živých atenuovaných vakcín u pacientov liečených Tecfiderou nie
sú dostupné žiadne klinické údaje. U živých vakcín môže byť zvýšené riziko klinickej infekcie
a pacientom liečeným Tecfiderou nemajú byť podávané, okrem zriedkavých prípadov, keď toto potenciálne riziko pre jednotlivca je považované za menšie ako riziko vyplývajúce z nezaočkovania.
Počas liečby Tecfiderou sa nemajú súbežne podávať deriváty kyseliny fumarovej (topicky alebo systémovo).
U ľudí je dimetylfumarát intenzívne metabolizovaný esterázami ešte skôr, ako vstúpi do systémovej cirkulácie a jeho ďalšie metabolizovanie prebieha cez cyklus trikarboxylových kyselín bez účasti systému cytochrómu P450 (CYP). V in vitro štúdiách inhibície a indukcie CYP, štúdii P- glykoproteínu ani v štúdiách proteínovej väzby dimetylfumarátu a monometylfumarátu (primárny metabolit dimetylfumarátu) neboli identifikované potenciálne riziká vyplývajúce z liekovej interakcie.
Lieky bežne používané u pacientov s roztrúsenou sklerózou, ako intramuskulárny interferón beta-1a
a glatirameracetát, boli klinicky testované pre potenciálne interakcie s dimetylfumarátom a nezmenili
farmakokinetický profil dimetylfumarátu.
Dôkazy zo štúdií so zdravými dobrovoľníkmi naznačujú, že sčervenanie spájané s Tecfiderou je pravdepodobne sprostredkované prostaglandínmi. V dvoch štúdiách so zdravými dobrovoľníkmi podávanie 325 mg (alebo ekvivalentného množstva) kyseliny acetylsalicylovej bez gastrorezistentného
obalu 30 minút pred podaním Tecfidery počas 4 dní a počas 4 týždňov, v uvedenom poradí, neovplyvnilo farmakokinetický profil Tecfidery. U pacientov s relaps-remitujúcou roztrúsenou sklerózou sa musia pred súbežným podaním Tecfidery s kyselinou acetylsalicylovou zvážiť potenciálne riziká, spájané s touto liečbou. Dlhodobé (> 4 týždne) nepretržité podávanie kyseliny acetylsalicylovej sa neskúmalo (pozri časti 4.4 a 4.8).
Súbežná liečba nefrotoxickými liekmi (ako aminoglykozidy, diuretiká, nesteroidové antiflogistiká alebo lítium) môže zvýšiť potenciálne renálne nežiaduce reakcie (napr. proteinúria, pozri časť 4.8) u pacientov užívajúcich Tecfideru (pozri časť 4.4 Krvné/laboratórne testy).
Konzumovanie malých množstiev alkoholu nemalo vplyv na expozíciu dimetylfumarátu a nebolo spojené s nárastom nežiaducich reakcií. Je potrebné vyhnúť sa požívaniu veľkých množstiev silných alkoholických nápojov (viac ako 30 objemových percent alkoholu) počas jednej hodiny od užitia Tecfidery, keďže alkohol môže viesť k vyššej frekvencii gastrointestinálnych nežiaducich reakcií.
In vitro štúdie indukcie CYP nepreukázali interakciu medzi Tecfiderou a perorálnymi kontraceptívami. V in vivo štúdii súbežné podanie Tecfidery s kombinovanou perorálnou antikoncepciou (norgestimát a etinylestradiol) nevyvolalo relevantnú zmenu v expozícii perorálnej antikoncepcii. Interakčné štúdie s perorálnou antikoncepciou obsahujúcou iné progestagény sa neuskutočnili, neočakáva sa však vplyv Tecfidery na ich expozíciu.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne alebo iba obmedzené množstvo údajov o použití dimetylfumarátu
u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Tecfidera sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich vhodnú antikoncepciu (pozri časť 4.5). Tecfidera sa má užívať počas gravidity iba v prípade, ak je jednoznačne potrebná
a potenciálny prínos liečby prevyšuje potenciálne riziká pre plod.
Dojčenie
Nie je známe, či sa dimetylfumarát alebo jeho metabolity vylučujú do ľudského mlieka. Riziko
u novorodencov/dojčiat nemôže byť vylúčené. Treba urobiť rozhodnutie, či prerušiť dojčenie alebo liečbu Tecfiderou. Je potrebné zvážiť prínos dojčenia pre dieťa a prínos liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinkoch dimetylfumarátu na ľudskú fertilitu. Údaje
z predklinických štúdií nenaznačujú, že by s podávaním dimetylfumarátu vzrastalo riziko zníženia fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tecfidera nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Neuskutočnili sa žiadne štúdie účinku na schopnosť viezť vozidlá a obsluhovať stroje, avšak
v klinických štúdiách sa nepozorovali žiadne účinky súvisiace s dimetylfumarátom, ktoré by
potenciálne ovplyvňovali túto schopnosť.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofiluNajčastejšie nežiaduce reakcie (výskyt ≥ 10 %) u pacientov liečených dimetylfumarátom boli
sčervenanie a gastrointestinálne príhody (t. j. hnačka, žalúdočná nevoľnosť, bolesť brucha, bolesť
v hornej časti brucha). Sčervenanie a gastrointestinálne príhody boli u pacientov častejšie na začiatku liečby (hlavne počas prvého mesiaca) a u pacientov s výskytom sčervenania a gastrointestinálnych
príhod môže k týmto udalostiam dôjsť kedykoľvek v priebehu liečby Tecfiderou. Najčastejšie hlásené nežiaduce reakcie vedúce k ukončeniu liečby (výskyt > 1 %) u pacientov liečených Tecfiderou boli
sčervenanie (3 %) a gastrointestinálne príhody (4 %).
V placebom kontrolovaných a nekontrolovaných klinických štúdiách dostávalo Tecfideru
2 513 pacientov počas až 12 rokov s celkovou expozíciou ekvivalentnou 11 318 pacientorokom. Celkovo 1 169 pacientov bolo liečených Tecfiderou aspoň 5 rokov a 426 pacientov aspoň 10 rokov. Skúsenosti z nekontrolovaných klinických skúšaní sú konzistentné so skúsenosťami z placebom kontrolovaných klinických skúšaní.
Tabuľka s prehľadom nežiaducich reakciíNežiaduce reakcie vyplývajúce z klinických štúdií, zo štúdií bezpečnosti po uvedení lieku na trh a
spontánnych hlásení sú uvedené v tabuľke nižšie.
Nežiaduce reakcie sú uvádzané v podobe MedDRA preferovaných termínov podľa tried orgánových systémov databázy MedDRA. Výskyt nežiaducich reakcií je vyjadrený podľa nasledovných kategórií:
- Veľmi časté (≥ 1/10)
- Časté (≥ 1/100 až < 1/10)
- Menej časté (≥ 1/1 000 až < 1/100)
- Zriedkavé (≥ 1/10 000 až ˂ 1/1 000)
- Veľmi zriedkavé (< 1/10 000)
- Neznáme (z dostupných údajov)
Trieda orgánových systémov
podľa databázy MedDRA
|
Nežiaduca reakcia
|
Kategória frekvencie výskytu
|
Infekcie a nákazy
|
Gastroenteritída
|
Časté
|
Progresívna multifokálna
leukoencefalopatia (PML)
|
Neznáme
|
Herpes zoster
| Neznáme
|
Poruchy krvi a lymfatického
systému
| Lymfopénia
| Časté
|
Leukopénia
| Časté
|
Trombocytopénia
| Menej časté
|
Poruchy imunitného systému
Poruchy nervového systému
| Hypersenzitivita
| Menej časté
|
Anafylaxia
| Neznáme
|
Dyspnoe
| Neznáme
|
Hypoxia
| Neznáme
|
Hypotenzia
| Neznáme
|
Angioedém
| Neznáme
|
Pocit pálenia
| Časté
|
Poruchy ciev
| Sčervenanie
| Veľmi časté
|
Návaly tepla
| Časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Rhinorea
|
Neznáme
|
Poruchy gastrointestinálneho
traktu
|
Hnačka
|
Veľmi časté
|
Nevoľnosť
|
Veľmi časté
|
Bolesť v hornej časti brucha
|
Veľmi časté
|
Bolesť brucha
|
Veľmi časté
|
Vracanie
|
Časté
|
Dyspepsia
|
Časté
|
Gastritída
|
Časté
|
Porucha gastrointestinálneho
traktu
|
Časté
|
Poruchy pečene a žlčových ciest
|
Zvýšená hladina aspartátaminotransferáz
|
Časté
|
Zvýšená hladina alanínaminotransferáz
|
Časté
|
Poškodenie pečene indukované
liekom
|
Neznáme
|
Poruchy kože a podkožného
tkaniva
|
Pruritus
|
Časté
|
Vyrážka
|
Časté
|
Erytém
|
Časté
|
Alopécia
|
Časté
|
Poruchy obličiek a močových
ciest
|
Proteinúria
|
Časté
|
Celkové poruchy a reakcie v mieste podania
|
Pocit horúčavy
|
Časté
|
Laboratórne a funkčné vyšetrenia
|
Ketóny zistené v moči
|
Veľmi časté
|
Albumín prítomný v moči
|
Časté
|
Znížený počet bielych krviniek
|
Časté
|
Popis
v
ybraných
nežiaducich
reakcií
Sčervenanie
V placebom kontrolovaných štúdiách bol u pacientov liečených Tecfiderou oproti pacientom na placebe pozorovaný vyšší výskyt sčervenania (34 % oproti 4 %) a návalov tepla (7 % oproti 2 %),
v uvedenom poradí. Sčervenanie je obvykle popisované ako sčervenanie alebo nával tepla, ale môže
zahŕňať aj iné prejavy (napr. pocit tepla, sčervenanie, svrbenie a pocit pálenia). Sčervenanie bolo u pacientov častejšie na začiatku liečby (hlavne počas prvého mesiaca) a u pacientov s výskytom sčervenania môže k týmto udalostiam dôjsť kedykoľvek v priebehu liečby Tecfiderou. U väčšiny pacientov s výskytom sčervenania išlo o príhody ľahkého až stredne ťažkého stupňa. Celkovo 3 % pacientov liečených Tecfiderou ukončilo liečbu pre reakcie sčervenania. Výskyt závažných prípadov sčervenania, ktoré možno charakterizovať celkovým erytémom, vyrážkou a/alebo pruritom, bol pozorovaný u menej ako 1 % pacientov liečených Tecfiderou (pozri časti 4.2, 4.4 a 4.5).
GastrointestinálneVýskyt gastrointestinálnych príhod (napr. hnačka [14 % oproti 10 %], nevoľnosť [12 % oproti 9 %], bolesť v hornej časti brucha [10 % oproti 6 %], bolesť brucha [9 % oproti 4 %], vracanie [8 % oproti
5 %] a dyspepsia [5 % oproti 3 %]) bol zvýšený u pacientov liečených Tecfiderou v porovnaní
s pacientmi, ktorí dostávali placebo, v uvedenom poradí. Gastrointestinálne príhody boli u pacientov
častejšie na začiatku liečby (hlavne počas prvého mesiaca) a u pacientov s výskytom
gastrointestinálnych príhod môže k týmto príhodám dôjsť kedykoľvek v priebehu liečby Tecfiderou.
U väčšiny pacientov boli gastrointestinálne príhody ľahkého až stredne ťažkého stupňa. Štyri percentá
(4 %) pacientov liečených Tecfiderou ukončili liečbu pre gastrointestinálne príhody. Výskyt závažných gastrointestinálnych príhod, vrátane gastroenteritídy a gastritídy, bol pozorovaný u 1 % pacientov liečených Tecfiderou (pozri časť 4.2).
F
unkcia pečene
Na základe údajov z placebom kontrolovaných štúdií mala väčšina pacientov so zvýšenou koncentráciou hepatálnych transamináz koncentrácie < 3-násobok horného limitu referenčných hodnôt. Častejší výskyt zvýšenia hepatálnych transamináz u pacientov liečených Tecfiderou
v porovnaní s placebom bol pozorovaný primárne počas prvých 6 mesiacov liečby. Zvýšenie koncentrácie alanínaminotransferázy (ALT) a aspartátaminotransferázy (AST) ≥ 3-násobku horného limitu referenčných hodnôt bolo pozorované u 5 % a 2 % pacientov na placebe a u 6 % a 2 % pacientov liečených Tecfiderou. K ukončeniu liečby v dôsledku zvýšenia hepatálnych transamináz došlo v < 1 % prípadov, a podobne aj u pacientov liečených Tecfiderou alebo na placebe. V placebom kontrolovaných štúdiách nebolo pozorované zvýšenie transamináz na ≥ 3-násobok ULN pri súčasnom zvýšení celkového bilirubínu na > 2-násobok ULN nebolo v placebom kontrolovaných štúdiách pozorované.
Po uvedení lieku na trh boli po podaní Tecfidery hlásené zvýšenia hladín pečeňových enzýmov
a prípady poškodenia pečene indukované liekom (zvýšenie hladín transamináz ≥ 3-násobok ULN pri súčasnom zvýšení celkového bilirubínu > 2-násobok ULN), ktoré ustúpili po prerušení liečby.
Lymfopénia
V placebom kontrolovaných štúdiách mala väčšina (> 98 %) pacientov pred začiatkom liečby normálne hodnoty lymfocytov. Počas liečby Tecfiderou priemerné počty lymfocytov klesli v priebehu prvého roka a následne sa stabilizovali. V priemere došlo k 30 % zníženiu počtu lymfocytov oproti východiskovému stavu. Priemerná hodnota a medián počtu lymfocytov zostali v referenčnom intervale. Počty lymfocytov < 0,5 x 109/l boli pozorované u < 1 % pacientov na placebe a u 6 % pacientov liečených Tecfiderou. Počty lymfocytov < 0,2 x 109/l boli pozorované u 1 pacienta liečeného Tecfiderou a u žiadneho pacienta na placebe.
V klinických štúdiách (kontrolovaných aj nekontrolovaných) malo 41 % pacientov liečených Tecfiderou lymfopéniu (definovanú v týchto štúdiách ako < 0,91 x 109/l). Mierna lymfopénia (počet lymfocytov ≥ 0,8 x 109/l až < 0,91 x 109/l) bola pozorovaná u 28 % pacientov; stredne ťažká lymfopénia (počet lymfocytov ≥ 0,5 x 109/l až < 0,8 x 109/l) pretrvávajúca najmenej šesť mesiacov bola pozorovaná u 11 % pacientov; ťažká lymfopénia (počet lymfocytov < 0,5 x 109/l) pretrvávajúca najmenej šesť mesiacov bola pozorovaná u 2 % pacientov. V skupine s ťažkou lymfopéniou pri pokračujúcej liečbe zostal počet lymfocytov väčšinou na úrovni < 0,5 x 109/l.
Okrem toho sa v nekontrolovanej, prospektívnej štúdii po uvedení lieku na trh v 48. týždni liečby Tecfiderou (n = 185) počty CD4+ T-lymfocytov mierne (počet ≥0,2 x 109/l až <0,4 x 109/l) alebo výrazne (<0,2 x 109/l) znížili až u 37 % alebo 6 % pacientov, pričom počet CD8+ T-lymfocytov sa častejšie znížil až u 59 % pacientov s počtom < 0,2 x 109/l a 25 % pacientov s počtom <0,1 x 109/l.
V kontrolovaných a nekontrolovaných klinických štúdiách sa u pacientov, ktorí ukončili liečbu liekom Tecfidera s počtom lymfocytov pod dolnou hranicou referenčného intervalu (LLN), sledovalo obnovenie počtu lymfocytov na úroveň LLN (pozri časť 5.1).
Infekcie, vrátane PML a oportúnnych infekcií
Pri liečbe Tecfiderou boli hlásené prípady infekcií JC vírusom (JCV) spôsobujúcich progresívnu multifokálnu leukoencefalopatiu (PML) (pozri časť 4.4). PML môže byť smrteľná alebo môže spôsobiť ťažké zdravotné postihnutie. V jednom klinickom skúšaní sa u jedného pacienta, ktorý užíval Tecfideru vyvinula PML pri dlhotrvajúcej ťažkej lymfopénii (počet lymfocytov prevažne < 0,5 x 109/l po dobu 3,5 roka), prípad bol smrteľný. Po uvedení lieku na trh sa PML tiež vyskytla pri stredne
ťažkej a miernej lymfopénii (> 0,5 x 109/l až < LLN, ako je definovaný referenčný interval v miestnom laboratóriu).
V niekoľkých prípadoch PML so stanovením podskupín T-lymfocytov sa v čase diagnózy PML
zistilo, že počet CD8+ T-lymfocytov klesol na < 0,1 x 109/l, kým zníženie počtu CD4+ T-lymfocytov
bolo rôzne (od < 0,05 do 0,5 x 109/l) a viac korelovalo s celkovou závažnosťou lymfopénie
(< 0,5 x 109/l až <LLN). Následkom toho sa u týchto pacientov zvýšil pomer CD4+/CD8+.
Zdá sa, že dlhotrvajúca stredne ťažká až ťažká lymfopénia zvyšuje riziko PML pri liečbe Tecfiderou, PML sa však vyskytla aj u pacientov s miernou lymfopéniou. Okrem toho sa po uvedení lieku na trh väčšina prípadov PML vyskytla u pacientov vo veku > 50 rokov.
V súvislosti s používaním Tecfidery boli hlásené infekcie vyvolané vírusom herpes zoster.
V prebiehajúcej dlhodobej predĺženej štúdii, v ktorej sa liečilo Tecfiderou 1736 pacientov s SM, sa približne u 5 % vyskytla jedna alebo viac udalostí herpes zoster, pričom u väčšiny z nich bola miernej až strednej závažnosti. Väčšina účastníkov štúdie vrátane tých, u ktorých sa vyskytla závažná infekcia vyvolaná vírusom herpes zoster, mala počet lymfocytov nad dolnou hranicou referenčnej hodnoty.
U väčšiny osôb so súčasným počtom lymfocytov pod LLN bola lymfopénia hodnotená ako stredne ťažká alebo ťažká. Po uvedení lieku na trh nebola väčšina prípadov výskytu infekcie vyvolanej
vírusom herpes zoster závažná a infekcia po liečbe ustúpila. K dispozícii sú len obmedzené údaje
o absolútnom počte lymfocytov (absolute lymphocyte count, ALC) u pacientov s infekciou vyvolanou vírusom herpes zoster po uvedení lieku na trh. Z hlásení však vyplýva, že sa u väčšiny pacientov vyskytla stredne ťažká (≥ 0,5 x 109/l až < 0,8 x 109/l) alebo ťažká (< 0,5 x 109/l až 0,2 x 109/l) lymfopénia (pozri časť 4.4).
Laboratórne abnormality
V placebom kontrolovaných štúdiách boli koncentrácie ketónov v moči (1+ alebo vyššie) vyššie
u pacientov liečených Tecfiderou (45 %) v porovnaní s placebom (10 %). V klinických skúšaniach neboli pozorované žiadne neočakávané klinické následky.
Koncentrácie 1,25-dihydroxyvitamínu D sa znížili u pacientov liečených Tecfiderou v porovnaní s placebom (medián percentuálneho poklesu oproti východiskovej hodnote po 2 rokoch o 25 %
v porovnaní s 15 %, v uvedenom poradí), zatiaľ čo koncentrácie paratyroidného hormónu (PTH) sa
u pacientov liečených Tecfiderou zvýšili v porovnaní s placebom (medián percentuálneho nárastu
oproti východiskovej hodnote po 2 rokoch o 29 % v porovnaní s 15 %, v uvedenom poradí). Priemerné hodnoty oboch parametrov zostali v referenčnom intervale hodnôt.
Prechodné zvýšenie priemerných počtov eozinofilov bolo pozorované počas prvých dvoch mesiacoch liečby.
Pediatrická populácia
V 96-týždňovom, otvorenom, randomizovanom, aktívne kontrolovanom skúšaní u pediatrických
pacientov s RRSM vo veku od 10 rokov do menej ako 18 rokov (120 mg dvakrát denne počas 7 dní a následne 240 mg dvakrát denne počas zvyšného trvania liečby; populácia v štúdii, n = 78) sa
bezpečnostný profil u pediatrických pacientov zdal byť podobný ako u predtým
sledovaných dospelých pacientov.
Dizajn klinického skúšania u pediatrických pacientov sa líšil od dizajnu klinických skúšaní kontrolovaných placebom u dospelých. Preto nie je možné vylúčiť vplyv dizajnu klinického skúšania na numerické rozdiely v nežiaducich reakciách medzi pediatrickou a dospelou populáciou.
Nasledujúce nežiaduce udalosti boli hlásené u pediatrickej populácie častejšie (≥ 10 %) než u dospelej populácie:
• Bolesť hlavy bola hlásená u 28 % pacientov liečených Tecfiderou oproti 36 % pacientov liečených interferónom beta-1a.
• Gastrointestinálne poruchy boli hlásené u 74 % pacientov liečených Tecfiderou oproti 31 % pacientov liečených interferónom beta-1a. Spomedzi nich boli pri Tecfidere najčastejšie hlásené bolesť brucha a vracanie.
• Poruchy dýchacej sústavy, hrudníka a mediastína boli hlásené u 32 % pacientov liečených Tecfiderou oproti 11 % pacientov liečených interferónom beta-1a. Spomedzi nich boli pri Tecfidere najčastejšie hlásené orofaryngálna bolesť a kašeľ.
• Dysmenorea bola hlásená u 17 % pacientok liečených Tecfiderou oproti 7 % pacientok liečených interferónom beta-1a.
V malej, otvorenej, nekontrolovanej štúdii v trvaní 24 týždňov u pediatrických pacientov s RRSM
vo veku 13 rokov až 17 rokov (120 mg dvakrát denne počas 7 dní a následne 240 mg dvakrát denne počas zvyšného trvania liečby; populácia pre hodnotenie bezpečnosti, n = 22), po ktorej nasledovala
96-týždňová predĺžená štúdia (240 mg dvakrát denne; populácia pre hodnotenie bezpečnosti, n = 20),
sa bezpečnostný profil zdal byť podobný profilu pozorovanému u dospelých pacientov.
K dispozícii je iba obmedzené množstvo údajov u detí vo veku od 10 rokov do 12 rokov. Bezpečnosť
a účinnosť Tecfidery u detí vo veku do 10 rokov neboli doteraz stanovené.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené
v Prílohe V.4.9 PredávkovanieBoli hlásené prípady predávkovania Tecfiderou. Príznaky popisované v týchto prípadoch boli konzistentné so známym profilom nežiaducich reakcií Tecfidery. Nie sú známe žiadne terapeutické intervencie, ktoré by mohli zlepšiť elimináciu Tecfidery, a nie je známe ani antidotum. V prípade predávkovania sa odporúča na základe klinickej indikácie iniciovať symptomatickú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, iné imunosupresíva, ATC kód: L04AX07
Mechanizmus účinkuMechanizmus terapeutických účinkov dimetylfumarátu pri roztrúsenej skleróze nie je ešte úplne
známy. Predklinické štúdie naznačujú, že farmakodynamické pôsobenie dimetylfumarátu primárne sprostredkúva aktivácia transkripčnej dráhy nukleárneho faktora (erytroidný 2) typu 2 (Nrf2). Dimetylfumarát preukázateľne vyvoláva u pacientov zosilnený účinok Nrf2-dependentných antioxidačných génov (napr. NAD(P)H dehydrogenáza, chinónová oxidoreduktáza 1; [(NQO)]).
Farmakodynamické účinkyÚčinky na imunitný systémV predklinických a klinických štúdiách boli preukázané protizápalové a imunomodulačné vlastnosti
v predklinických modeloch významne redukovali zápalovými stimulmi indukovanú aktiváciu imunitných buniek a následné uvoľňovanie prozápalových cytokínov. V klinických štúdiách
u pacientov s psoriázou dimetylfumarát ovplyvňoval lymfocytárne fenotypy prostredníctvom down-
regulácie prozápalových cytokínových profilov (TH1, TH17) a pôsobil na protizápalovú produkciu
(TH2). Dimetylfumarát prejavoval terapeutickú aktivitu vo viacerých modeloch zápalového
a neurozápalového poškodenia. V štúdiách 3. fázy u pacientov s SM (DEFINE, CONFIRM
a ENDORSE) sa po nasadení Tecfidery znížili počas prvého roka priemerné počty lymfocytov
v priemere približne o 30 % oproti východiskovému stavu a potom sa ustálili. V týchto štúdiách sa u pacientov, ktorí ukončili liečbu liekom Tecfidera s počtom lymfocytov pod dolnou hranicou referenčného intervalu (LLN, 910 buniek/mm3), sledovalo obnovenie počtu lymfocytov na úroveň LLN.
Na obrázku 1 je znázornený podiel pacientov, u ktorých sa na základe Kaplanovej-Meierovej metódy odhaduje dosiahnutie LLN bez dlhotrvajúcej ťažkej lymfopénie. Východisková hodnota zotavenia (
recovery baseline, RBL) bola definovaná ako posledný ALC počas liečby pred ukončením liečby Tecfiderou. Odhadované podiely pacientov s miernou, stredne ťažkou alebo ťažkou lymfopéniou pri RBL, ktorí sa zotavili na LLN (ALC ≥ 0,9 x 109/l) v 12 a 24. týždni, sú uvedené v tabuľke 1, tabuľke 2 a tabuľke 3 s 95 % bodovými intervalmi spoľahlivosti. Štandardná chyba odhadu funkcie prežívania podľa Kaplana-Meiera je vypočítaná pomocou Greenwoodovho vzorca.
Obrázok 1: Kaplanova-Meierova metóda; podiel pacientov so zotavením na ≥ 910 buniek/mm3LLN od východiskovej hodnoty zotavenia (RBL)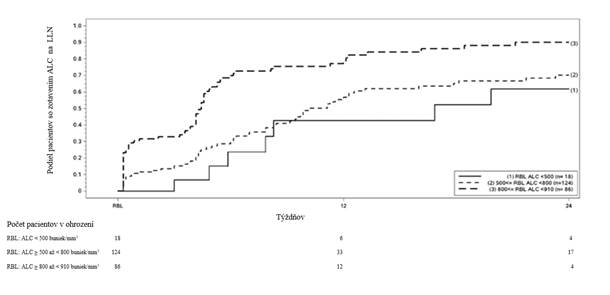 Tabuľka 1: Kaplanova-Meierova metóda; odhadovaný podiel pacientov s miernou lymfopéniou vo východiskovej hodnote zotavenia (RBL), ktorí dosiahnu LLN, po vylúčení pacientovs dlhotrvajúcou ťažkou lymfopéniou
Tabuľka 1: Kaplanova-Meierova metóda; odhadovaný podiel pacientov s miernou lymfopéniou vo východiskovej hodnote zotavenia (RBL), ktorí dosiahnu LLN, po vylúčení pacientovs dlhotrvajúcou ťažkou lymfopéniouPočet pacientov s miernou lymfopéniou a s rizikom
| Východisková hodnota
n=86
| 12. týždeň
n=12
| 24. týždeň
n=4
|
Podiel tých, ktorí dosiahli
LLN (95 % IS)
|
| 0,81
(0,71; 0,89)
| 0,90
(0,81; 0,96)
|
a Pacienti s ALC < 910 a ≥ 800 buniek/mm3 pri RBL, po vylúčení pacientov s dlhotrvajúcou ťažkou
lymfopéniou.
Tabuľka 2: Kaplanova-Meierova metóda; odhadovaný podiel pacientov so stredne ťažkou lymfopéniou vo východiskovej hodnote zotavenia (RBL), ktorí dosiahli LLN, po vylúčení pacientov s dlhotrvajúcou ťažkou lymfopéniouPočet pacientov so stredne ťažkou
lymfopénioua s rizikom
| Východisková hodnota
n=124
| 12. týždeň
n=33
| 24. týždeň
n=17
|
Podiel tých, ktorí dosiahli LLN
(95 % IS)
|
| 0,57
(0,46; 0,67)
| 0,70
(0,60; 0,80)
|
a Pacienti s ALC < 800 a ≥ 500 buniek/mm3 pri RBL, po vylúčení pacientov s dlhotrvajúcou ťažkou
lymfopéniou.
T
abuľka 3: Kaplanova-Meierova metóda; odhadovaný podiel pacientov s ťažkou lymfopéniou vo východiskovej hodnote zotavenia (RBL), ktorí dosiahli LLN, po vylúčení pacientov
s dlhotrvajúcou ťažkou lymfopéniou
P
očet pacientov s ťažkou
l
ymfopéniou
a
s rizikom
|
V
ýchodisková
hodnota n=18
|
12. týždeň
n=6
|
24. týždeň
n=4
|
Podiel tých, ktorí dosiahli LLN
(95 % IS)
|
|
0,43 (0,20; 0,75)
|
0,62 (0,35; 0,88)
|
a Pacienti s ALC < 500 buniek/mm3 pri RBL, po vylúčení pacientov s dlhotrvajúcou ťažkou lymfopéniou.
Klinická účinnosť abezpečnosťBoli vykonané dve dvojročné randomizované dvojito zaslepené placebom kontrolované štúdie
(DEFINE s 1 234 pacientmi a CONFIRM s 1 417 pacientmi) na pacientoch s relaps-remitujúcou roztrúsenou sklerózou (RRSM). Pacienti s progresívnou formou SM neboli do týchto štúdií zahrnutí.
Účinnosť (pozri tabuľku nižšie) a bezpečnosť boli preukázané na pacientoch so skóre na rozšírenej stupnici stavu invalidity (EDSS) v rozsahu od 0 do 5 vrátane, u ktorých došlo najmenej k 1 relapsu
v roku pred randomizáciou alebo do 6 týždňov pred randomizáciou im bolo urobené vyšetrenie mozgu magnetickou rezonanciou (MR), ktoré preukázalo aspoň jednu gadolíniom zvýraznenú (Gd+) léziu.
Štúdia CONFIRM mala zaslepeného hodnotiteľa (t. j. skúšajúci lekár/investigátor hodnotiaci reakcie na skúšané liečivo bol zaslepený) a ako referenčnú porovnávaciu látku glatirameracetát.
V DEFINE štúdii mali pacienti nasledujúce mediánové hodnoty vstupných parametrov: vek 39 rokov, dĺžka ochorenia 7,0 rokov, EDSS skóre 2,0. Navyše, 16 % pacientov malo EDSS skóre > 3,5; 28 % malo ≥ 2 relapsy v predchádzajúcom roku a 42 % dostávalo už predtým inú schválenú liečbu SM.
V skupine MRI 36 % pacientov zaradených do štúdie malo na začiatku Gd+ lézie (priemerný počet
Gd+ lézií 1,4).
V CONFIRM štúdii mali pacienti nasledujúce mediánové hodnoty vstupných parametrov: vek
37 rokov, dĺžka ochorenia 6,0 rokov, EDSS skóre 2,5. Navyše, 17 % pacientov malo EDSS skóre
> 3,5; 32 % malo ≥ 2 relapsy v predchádzajúcom roku a 30 % dostávalo už predtým inú schválenú liečbu SM. V skupine MR 45 % pacientov zaradených do štúdie malo na začiatku Gd+ lézie
(priemerný počet Gd+ lézií 2,4).
V porovnaní s placebom preukazovali pacienti liečení Tecfiderou klinicky a štatisticky významné zníženie primárneho ukazovateľa v štúdii DEFINE, podielu relapsujúcich pacientov po 2 rokoch,
a primárneho ukazovateľa v štúdii CONFIRM, ročného výskytu relapsov (
annualised relapse rate,
ARR) po 2 rokoch.
ARR pacientov na glatirameracetáte bol 0,286 a na placebe 0,401 v štúdii CONFIRM, čo zodpovedá
29 % zníženiu (p=0,013) a je v súlade so schválenými informáciami o lieku.
| DEFINE
| CONFIRM
|
| Placebo
| Tecfidera
240 mg dvakrát
denne
| Placebo
| Tecfidera
240 mg dvakrát
denne
| Glatiramer- acetát
|
Klinické ukazovatelea
|
Počet pacientov
| 408
| 410
| 363
| 359
| 350
|
Ročný výskyt relapsov
| 0,364
| 0,172***
| 0,401
| 0,224***
| 0,286*
|
Pomer výskytov
(95 % IS, interval spoľahlivosti)
|
| 0,47
(0,37; 0,61)
|
| 0,56
(0,42; 0,74)
| 0,71
(0,55; 0,93)
|
|
DE
FINE
|
C
O
NF
IRM
|
|
Placebo
|
T
ecfidera
240 mg dvakrát denne
|
Placebo
|
T
ecfidera
240 mg dvakrát denne
|
Gl
a
t
i
ramer-
acetát
|
Podiel relapsujúcich
|
0,461
|
0,270***
|
0,410
|
0,291**
|
0,321**
|
Pomer rizika
(95 % IS)
|
|
0,51
(0,40; 0,66)
|
|
0,66
(0,51; 0,86)
|
0,71
(0,55; 0,92)
|
Podiel s 12-týždennou
potvrdenou progresiou invalidity
|
0,271'
|
0,164**
|
0,169
|
0,128#
|
0,156#
|
Pomer rizika
(95 % IS)
|
|
0,62
(0,44; 0,87)
|
|
0,79
(0,52; 1,19)
|
0,93
(0,63; 1,37)
|
Podiel s 24-týždennou potvrdenou progresiou invalidity
|
0,169
|
0,128#
|
0,125
|
0,078#
|
0,108#
|
Pomer rizika
(95 % IS)
|
|
0,77
(0,52; 1,14)
|
|
0,62
(0,37; 1,03)
|
0,87
(0,55; 1,38)
|
MRI ukazovateleb
|
|
Počet pacientov
|
165
|
152
|
144
|
147
|
161
|
Priemerný (medián)
počet nových alebo novozväčšených T2 lézií v priebehu 2 rokov
|
16,5
(7,0)
|
3,2
(1,0)***
|
19,9
(11,0)
|
5,7
(2,0)***
|
9,6
(3,0)***
|
Priemerný pomer počtu lézií
(95 % IS)
|
|
0,15
(0,10; 0,23)
|
|
0,29
(0,21; 0,41)
|
0,46
(0,33; 0,63)
|
Priemerný (medián)
počet Gd zvýraznených lézií po 2 rokoch
|
1,8
(0)
|
0,1
(0)***
|
2,0
(0,0)
|
0,5
(0,0)***
|
0,7
(0,0)**
|
Miera
pravdepodobnosti
(95 % IS)
|
|
0,10
(0,05; 0,22)
|
|
0,26
(0,15; 0,46)
|
0,39
(0,24; 0,65)
|
Priemerný (medián)
počet nových T1
hypointenzných lézií
v priebehu 2 rokov
|
5,7 (2,0)
|
2,0 (1,0)***
|
8,1 (4,0)
|
3,8 (1,0)***
|
4,5 (2,0)**
|
Priemerný pomer počtu lézií
(95 % IS)
|
|
0,28
(0,20; 0,39)
|
|
0,43
(0,30; 0,61)
|
0,59
(0,42; 0,82)
|
aVšetky analýzy klinických ukazovateľov prebehli v rámci liečby; bMRI analýza vychádzala z MRI skupiny
pacientov.
*p-hodnota < 0,05; **p-hodnota < 0,01; ***p-hodnota < 0,0001; #štatisticky nevýznamné.
Do otvorenej nekontrolovanej 8-ročnej predĺženej štúdie (ENDORSE) bolo zaradených
1 736 vhodných pacientov s RRMS z pivotných štúdií (DEFINE a CONFIRM). Primárnym cieľom štúdie bolo posúdiť dlhodobú bezpečnosť Tecfidery u pacientov s RRMS. Z 1 736 pacientov bola približne polovica (909, 52 %) liečená 6 rokov alebo dlhšie. 501 pacientov bolo nepretržite liečených liekom Tecfidera 240 mg dvakrát denne vo všetkých 3 štúdiách a 249 pacientov, ktorí predtým dostávali placebo v štúdiách DEFINE a CONFIRM, bolo liečených dávkou 240 mg dvakrát denne
v štúdii ENDORSE. Pacienti s dávkou dvakrát denne nepretržite, boli liečení až 12 rokov.
Počas štúdie ENDORSE viac ako polovica všetkých pacientov liečených Tecfiderou 240 mg dvakrát denne nemala relaps. U pacientov liečených nepretržite dvakrát denne vo všetkých 3 štúdiách bolo upravené ARR 0,187 (95 % IS: 0,156; 0,224) v štúdiách DEFINE a CONFIRM a 0,141 (95 % IS:
0,119; 0,167) v štúdii ENDORSE. U pacientov, ktorí predtým dostávali placebo sa upravené ARR
znížilo z 0,330 (95 % IS: 0,266; 0,408) v štúdiách DEFINE a CONFIRM na 0,149 (95 % IS: 0,116;
0,190) v štúdii ENDORSE.
V štúdii ENDORSE sa u väčšiny pacientov (> 75 %) nepotvrdila progresia invalidity (meraná ako 6- mesačná trvalá progresia invalidity). Súhrnné výsledky z troch štúdií preukázali, že pacienti liečení Tecfiderou mali konzistentnú a nízku mieru potvrdenej progresie invalidity s miernym zvýšením priemerného skóre EDSS v celej štúdii ENDORSE. Hodnotenia MRI (do 6. roku, vrátane
752 pacientov, ktorí boli predtým zaradení do kohorty MRI štúdií DEFINE a CONFIRM, ukázali, že väčšina pacientov (približne 90 %) nemala žiadne gadolíniom zvýraznené lézie. Počas 6 rokov zostal
ročný upravený priemerný počet nových alebo novo zväčšených T2 lézií a nových T1 lézií nízky.
Účinnosť u pacientov s vysokou aktivitou ochorenia:
V podskupine pacientov s vysokou aktivitou ochorenia v štúdiách DEFINE a CONFIRM bol
pozorovaný konzistentný účinok liečby na relapsy, zatiaľ čo účinok na udržanie progresie invalidity do
3 mesiacov nebol jasne preukázaný. Pre potreby dizajnu týchto štúdií bolo vysokoaktívne ochorenie
definované nasledovne:
- pacienti s 2 alebo viacerými relapsmi počas jedného roka a s jednou alebo viacerými Gd- zvýraznenými léziami na MRI mozgu (n = 42 v DEFINE; n = 51 v CONFIRM) alebo
- pacienti nereagujúci na riadnu a adekvátnu liečbu (najmenej jeden rok trvajúca liečba)
interferónom beta, mali najmenej 1 relaps počas liečby v predchádzajúcom roku a najmenej 9
T2-hyperintenzívnych lézií na kraniálnom MRI alebo najmenej 1 Gd-zvýraznenú léziu, prípadne pacienti s nezmeneným alebo zvýšeným počtom relapsov v predchádzajúcom roku pri porovnaní s predchádzajúcimi 2 rokmi (n = 177 v DEFINE; n = 141 v CONFIRM).
Pediatrická populácia
Bezpečnosť a účinnosť Tecfidery u pediatrickej populácie s RRSM boli hodnotené v randomizovanej,
otvorenej, aktívne kontrolovanej (interferónom beta-1a) štúdii dvoch paralelných skupín pacientov
s RRSM vo veku od 10 rokov do 18 rokov. Randomizovaných bolo stopäťdesiat pacientov na liečbu dimetylfumarátom (240 mg perorálne dvakrát denne) alebo interferónom beta-1a (30 μg i.m. jedenkrát
za týždeň) počas 96 týždňov. Primárnym koncovým ukazovateľom bol podiel pacientov bez nových alebo novozväčšených hyperintenzívnych lézií T2 na snímkach MRI mozgu v 96. týždni. Hlavným
sekundárnym koncovým ukazovateľom bol počet nových alebo novozväčšených T2 hyperintenzívnych lézií na snímkach MRI mozgu v 96. týždni. Keďže pre primárny koncový ukazovateľ neboli vopred naplánované žiadne potvrdzujúce hypotézy, uvádzajú sa opisné štatistiky.
Podiel pacientov v populácii so zámerom liečiť (intention to treat, ITT) bez nových alebo novozväčšených T2 MRI lézií v 96. týždni v porovnaní s východiskovou hodnotou bol 12,8 %
v skupine s dimetylfumarátom oproti 2,8 % v skupine s interferónom beta-1a. Priemerný počet nových alebo novozväčšených T2 lézií v 96. týždni v porovnaní s východiskovým stavom, upravený podľa
východiskového počtu T2 lézií a veku (populácia ITT bez pacientov, ktorí nepodstúpili vyšetrenie
MRI), bol 12,4 pri dimetylfumaráte a 32,6 pri interferóne beta-1a.
Pravdepodobnosť klinického relapsu do konca obdobia 96-týždňovej otvorenej štúdie bola 34 %
v skupine s dimetylfumarátom a 48 % v skupine s interferónom beta-1a.
Bezpečnostný profil u pediatrických pacientov (vo veku od 13 rokov do menej ako 18 rokov), ktorým bola podávaná Tecfidera, bol kvalitatívne zhodný s profilom, ktorý sa predtým pozoroval u dospelých pacientov (pozri časť 4.8).
5.2 Farmakokinetické vlastnosti
Pri perorálnom podávaní dimetylfumarátu dochádza k rýchlej predsystémovej hydrolýze účinkom esteráz a k premene na primárny metabolit, monometylfumarát, ktorý je tiež aktívny. Po perorálnom podaní Tecfidery dimetylfumarát v plazme nemožno kvantifikovať. Všetky farmakokinetické analýzy pre dimetylfumarát sa preto robia na základe koncentrácií monometylfumarátu v plazme. Farmakokinetické údaje boli získané od pacientov s roztrúsenou sklerózou a od zdravých dobrovoľníkov.
Absorpcia
Tmax monometylfumarátu je 2 až 2,5 hodín. Keďže tvrdé gastrorezistentné kapsuly Tecfidery obsahujú mikrotablety, ktoré sú chránené enterosolventným obalom, k absorpcii dochádza, až keď opustia
žalúdok (obvykle za menej ako 1 hodinu). Pri podaní 240 mg dvakrát denne s jedlom bol medián
maximálnej koncentrácie (Cmax) 1,72 mg/l a celková expozícia vyjadrená ako plocha pod krivkou (AUC) 8,02 h.mg/l u pacientov s roztrúsenou sklerózou. Celkovo sa Cmax a AUC v skúšanom rozsahu dávok (120 mg až 360 mg) zvyšovali približne priamo úmerne s dávkou. Pacientom s roztrúsenou sklerózou boli dve 240 mg dávky podané s časovým odstupom 4 hodín v rámci dávkovacieho režimu trikrát denne. To vyvolalo minimálnu akumuláciu expozície so zvýšením mediánu Cmax 12 %
v porovnaní s dávkovaním dvakrát denne (1,72 mg/l pre dávkovanie dvakrát denne v porovnaní s 1,93 mg/l pri dávkovaní trikrát denne) bez akýchkoľvek bezpečnostných dôsledkov.
Jedlo nemá na expozíciu dimetylfumarátu klinicky významný účinok. Tecfidera sa však má užívať
s jedlom kvôli zlepšeniu tolerancie s ohľadom na sčervenanie a gastrointestinálne nežiaduce udalosti
(pozri časť 4.2).
Distribúcia
Zdanlivý distribučný objem po perorálnom podaní 240 mg dimetylfumarátu sa pohybuje medzi 60 l
a 90 l. Proteíny v ľudskej plazme viažu 27 % až 40 % monometylfumarátu.
Biotransformácia
U ľudí je dimetylfumarát vo veľkej miere metabolizovaný, pričom v moči sa v nezmenenej forme
dimetylfumarátu vylúči menej ako 0,1 % dávky. Skôr, ako sa dostane do systémovej cirkulácie, metabolizovanie začína účinkom esteráz, ktoré sú prítomné v gastrointestinálnom trakte, v krvi
a tkanivách. Ďalej sa metabolizuje cyklom trikarboxylových kyselín bez účasti systému cytochrómu P450 (CYP). V štúdii s jednou dávkou 240 mg 14C-dimetylfumarátu bola zistená v krvnej plazme glukóza ako prevažujúci metabolit. Iné obehové metabolity boli kyselina fumarová, kyselina citrónová a monometylfumarát. Metabolická dráha kyseliny fumarovej zahŕňa cyklus trikarboxylových kyselín, pričom ako primárna cesta eliminácie slúži vydychovanie CO2.
Eliminácia
Vydychovanie CO2 je primárnou cestou vylučovania dimetylfumarátu z tela, vylúči sa ním 60 %
dávky. Eliminácia renálnou a fekálnou cestou sú sekundárne spôsoby eliminácie, ktorými sa vylúči
15,5 % a 0,9 % dávky, v uvedenom poradí.
Terminálny polčas monometylfumarátu je krátky (asi 1 hodina) a po 24 hodinách u väčšiny jedincov už v systéme necirkuluje žiadny monometylfumarát. Nedochádza k akumulácii pôvodného liečiva alebo monometylfumarátu ani po viacerých dávkach dimetylfumarátu v terapeutickom režime.
Linearita
Expozícia dimetylfumarátu sa zväčšuje približne lineárne s dávkou ako pri jednorazovej dávke, tak
i pri viacnásobných dávkach v skúmanom rozsahu 120 mg až 360 mg.
Farmakokinetika v osobitnýchskupináchpacientov
Hoci na základe výsledkov analýzy Variance (ANOVA) je u pacientov s RRSM hlavným faktorom
expozície (Cmax a AUC) telesná hmotnosť, v klinických štúdiách nemala vplyv na hodnotené kritériá bezpečnosti a účinnosti.
Pohlavie a vek nemali významný klinický vplyv na farmakokinetiku dimetylfumarátu. Farmakokinetika nebola skúmaná u pacientov vo veku 65 rokov a viac.
Pediatrická populácia
Farmakokinetický profil dimetylfumarátu 240 mg dvakrát denne sa hodnotil v malej, nezaslepenej, nekontrolovanej štúdii u pediatrických pacientov s relaps-remitujúcou roztrúsenou sklerózou (RRSM) vo veku 13 až 17 rokov (n = 21). Farmakokinetika Tecfidery u týchto dospievajúcich pacientov bola podobná profilu predtým pozorovanému u dospelých pacientov (Cmax: 2,00±1,29 mg/l; AUC0-12hod:
3,62±1,16 h.mg/l, čo zodpovedalo celkovej dennej AUC, ktorá bola 7,24 h.mg/l).
Porucha funkcie obličiek
Pretože je renálna cesta iba sekundárnym spôsobom vylučovania dimetylfumarátu a podieľa sa na menej ako 16 % podanej dávky, hodnotenie farmakokinetiky u jednotlivcov s poruchou funkcie obličiek sa neuskutočnilo.
Porucha funkcie pečene
Keďže sú dimetylfumarát a monometylfumarát metabolizované esterázami bez účasti systému
CYP450, hodnotenie farmakokinetiky u jednotlivcov s poruchou funkcie pečene sa neuskutočnilo.
5.3 Predklinické údaje o bezpečnosti
Nežiaduce reakcie popísané v častiach Toxikológia a Reprodukčná toxicita nižšie neboli pozorované v klinických štúdiách, ale boli pozorované na zvieratách s expozičnými hladinami podobnými klinickým expozičným hladinám.
Mutagenéza
Dimetylfumarát a monometylfumarát boli negatívne v sérii in vitro analýz (Ames, chromozomálne
odchýlky v cicavčích bunkách). Dimetylfumarát bol negatívny v in vivo štúdii mikronuklea na potkanoch.
Karcinogenéza
Štúdie karcinogenity dimetylfumarátu v trvaní do 2 rokov boli uskutočnené na myšiach a potkanoch.
Dimetylfumarát bol podávaný perorálne v dávkach 25, 75, 200 a 400 mg/kg/deň u myší a v dávkach
25, 50, 100 a 150 mg/kg/deň u potkanov.
U myší sa zvýšil výskyt renálneho tubulárneho karcinómu pri dávke 75 mg/kg/deň, pri expozícii (AUC) ekvivalentnej odporúčanej dávke u ľudí. U potkanov sa zvýšil výskyt renálneho tubulárneho karcinómu a adenómu z Leydigových buniek semenníkov pri dávke 100 mg/kg/deň, čo je približne dvakrát vyššia expozícia ako odporúčaná dávka u ľudí. Význam týchto zistení pokiaľ ide o riziko
u ľudí nie je známy.
Výskyt skvamocelulárneho papilómu a karcinómu bezžľazovej časti žalúdka (predžalúdka) sa zvýšil u myší pri expozícii ekvivalentnej odporúčanej dávke u ľudí a u potkanov pri expozícii nižšej ako je odporúčaná dávka u ľudí (na základe AUC). Predžalúdok hlodavcov nemá náprotivok u ľudí.
Toxikológia
Predklinické štúdie na hlodavcoch, králikoch a opiciach sa uskutočnili so suspenziou dimetylfumarátu
(dimetylfumarát v 0,8 % hydroxypropylmetylcelulóze) podávanej perorálnou sondou do žalúdka. Uskutočnila sa chronická štúdia na psoch, počas ktorej bola perorálne podávaná kapsula dimetylfumarátu.
Po opakovanom perorálnom podávaní dimetylfumarátu myšiam, potkanom, psom a opiciam boli pozorované zmeny na obličkách. U všetkých druhov bola pozorovaná regenerácia tubulárneho epitelu obličiek, naznačujúca možnosť poškodenia. U potkanov celoživotné dávkovanie viedlo k hyperplázii renálnych tubúl (2-ročná štúdia). U psov, ktorí dostávali denné perorálne dávky dimetylfumarátu počas 11 mesiacov sa vypočítaná hladina dávky, pri ktorej sa pozorovala kortikálna atrofia, rovnala trojnásobku odporúčanej dávky na základe AUC. U opíc, ktoré dostávali denné perorálne dávky dimetylfumarátu počas 12 mesiacov, sa pozorovala nekróza jednotlivých buniek pri dávke rovnajúcej sa dvojnásobku odporúčanej dávky na základe AUC. Intersticiálna fibróza a kortikálna atrofia sa
pozorovali pri dávke šesťkrát vyššej ako je odporúčaná dávka na základe AUC. Význam týchto zistení pre človeka nie je známy.
V semenníkoch psov a potkanov bola pozorovaná degenerácia semenotvorného epitelu. Tieto zistenia boli pozorované pri približne odporúčanej dávke u potkanov a pri 3-násobku odporúčanej dávky
u psov (na základe AUC). Význam týchto zistení pre človeka nie je známy.
V predžalúdku myší a potkanov boli v štúdiách trvajúcich 3 mesiace alebo dlhšie pozorované hyperplázia skvamózneho epitelu a hyperkeratóza, zápal, skvamocelulárny papilóm a karcinóm. Predžalúdok myší a potkanov nemá náprotivok u ľudí.
Reprodukčná toxicita
Perorálne podávanie dimetylfumarátu potkaním samcom pri dávke 75, 250 a 375 mg/kg/deň pred
párením a počas párenia nemalo žiadny vplyv na samčiu plodnosť ani pri najvyššej testovanej dávke
(najmenej 2-násobok dávky odporúčanej na základe AUC). Perorálne podávanie dimetylfumarátu potkaním samiciam pri dávkach 25, 100 a 250 mg/kg/deň pred párením a počas párenia a pokračujúce po 7. deň gravidity vyvolalo zníženie počtu štádií ruje za 14 dní a zvýšenie počtu zvierat s predĺženou rujou pri najvyššej testovanej dávke (11-násobok dávky odporúčanej na základe AUC). Tieto zmeny však neovplyvnili plodnosť ani počet produkovaných životaschopných plodov.
Ukázalo sa, že dimetylfumarát prestupuje placentárnou membránou do krvi plodu u potkanov
a králikov, s pomermi plodovej koncentrácie ku koncentrácii v materskej plazme 0,48 až 0,64 k 0,1, v uvedenom poradí. U potkanov a králikov neboli malformácie pozorované pri žiadnej dávke. Podávanie dimetylfumarátu v perorálnych dávkach 25, 100 a 250 mg/kg/deň gravidným potkaním samiciam v období organogenézy vyvolalo u samíc nežiaduce účinky pri 4-násobku dávky odporúčanej na základe AUC a nízku hmotnosť plodu a oneskorenú osifikáciu (členkových článkov a prstových článkov zadných nôh) pri 11-násobku odporúčanej dávky na základe AUC. Nižšia
hmotnosť plodu a oneskorená osifikácia boli považované za následok toxicity u samíc (znížená telesná hmotnosť a spotreba potravy).
Perorálne podávanie dimetylfumarátu pri dávkach 25, 75 a 150 mg/kg/deň gravidným samiciam králika počas organogenézy nemalo žiadny vplyv na vývoj embrya a plodu a pri 7-násobku odporúčanej dávky spôsobilo zníženie telesnej hmotnosti matiek a vyšší výskyt potratov pri 16- násobku odporúčanej dávky na základe AUC.
Perorálne podávanie dimetylfumarátu pri dávkach 25, 100 a 250 mg/kg/deň potkanom počas gravidity a laktácie spôsobilo zníženie telesnej hmotnosti F1 potomstva a oneskorenie sexuálnej zrelosti u F1 samcov pri 11-násobku dávky odporúčanej na základe AUC. U F1 potomstva nebol pozorovaný žiadny vplyv na plodnosť. Zníženie telesnej hmotnosti potomstva bolo považované za následok toxicity u samíc.
Dve štúdie toxicity na mladých potkanoch s denným perorálnym podávaním dimetylfumarátu
od 28. postnatálneho dňa (postnatal day, PND) do 90. - 93. PND (čo zodpovedá približne veku
3 rokov a viac u ľudí) odhalili podobné toxicity na cieľové orgány, obličky a predžalúdok, aké sa
pozorovali u dospelých zvierat. V prvej štúdii dimetylfumarát neovplyvnil vývoj, neurobehaviorálne príznaky, ani samčiu a samičiu plodnosť pri najvyššej dávke až 140 mg/kg/deň (približne 4,6-násobok odporúčanej dávky u ľudí podľa obmedzených údajov o AUC u pediatrických pacientov). Podobne sa v druhej štúdii nepozorovali u samcov mladých potkanov žiadne účinky na samčie reprodukčné
a prídavné orgány pri najvyššej dávke dimetylfumarátu až 375 mg/kg/deň (približne 15-násobok predpokladanej AUC pri odporúčanej dávke u pediatrických pacientov). U samcov mladých potkanov
sa však prejavil znížený obsah kostných minerálov a znížená kostná denzita v stehennej kosti
a bedrových stavcoch. Zmeny v hustote kostí sa tiež pozorovali u mladých potkanov po perorálnom podaní diroximel fumarátu, ďalšieho esteru kyseliny fumarovej, ktorý sa in vivo metabolizuje na rovnaký aktívny metabolit monometyl fumarát. Hladina bez pozorovaného nežiadúceho účinku (no observable adverse effect level, NOAEL) denzitometrických zmien u mladých potkanov odpovedá približne 1,5-násobku predpokladanej AUC pri odporúčanej dávke u pediatrických pacientov. Súvislosť účinkov na kosti a nižšej telesnej hmotnosti je možná, ale priamy účinok sa nedá vylúčiť. Zistenia týkajúce sa kostí majú pre dospelých pacientov obmedzený význam. Význam pre pediatrických pacientov nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly(mikrotabletysenterosolventnýmobalom)
mikrokryštalická celulóza
kroskarmelóza, sodná soľ
mastenec
oxid kremičitý, koloidný bezvodý stearát horečnatý
trietylcitrát
kopolymér kyseliny metakrylovej s metylmetakrylátom (1:1)
kopolymér kyseliny metakrylovej s etylakrylátom (1:1), 30 % disperzia simetikón
laurylsíran sodný polysorbát 80
Obal kapsuly
želatína
oxid titaničitý (E171)
briliantová modrá FCF (E133)
žltý oxid železitý (E172)
Potlač kapsuly (čiernyatrament)
šelak
hydroxid draselný
čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote do 30 °C.
Uchovávajte blistre vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia120 mg kapsuly: 14 kapsúl v PVC/PE/PVDC-PVC hliníkových blistroch.
240 mg kapsuly: 56 alebo 168 kapsúl v PVC/PE/PVDC-PVC hliníkových blistroch.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBiogen Netherlands B.V. Prins Mauritslaan 13
1171 LP Badhoevedorp
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/13/837/001
EU/1/13/837/002
EU/1/13/837/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 30. január 2014
Dátum posledného predĺženia registrácie: 20. september 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.