
týždne pred liečbou Strimvelisom. Záložná vzorka kmeňových buniek sa odoberá na použitie ako záchranná liečba pre prípad, že by došlo k zlyhaniu v priebehu procesu výroby lieku, k zlyhaniu transplantátu alebo k dlhotrvajúcej aplázii kostnej drene po liečbe Strimvelisom.
Pacient musí byť schopný darovať dostatočný počet CD34+ buniek, aby sa dodali minimálne 4 milióny purifikovaných CD34+ buniek/kg, ktoré sú potrebné na výrobu Strimvelisu.
Strimvelis je určený len na autológne použitie (pozri časť 4.4).
Pred podaním infúzie sa musí potvrdiť, že identita pacienta sa zhoduje so základnými jedinečnými informáciami o pacientovi uvedenými na infúznom(-ych) vaku(-och) a/alebo na obale Strimvelisu (pozri časti 4.4 a 6.6).
Prípravný režim pred liečbou
Odporúča sa podávať busulfán v dávke 0,5 mg/kg intravenózne každých 6 hodín počas dvoch po sebe idúcich dní, pričom sa má začať podávať tri dni pred podaním Strimvelisu. Celková dávka busulfánu
je 4 mg/kg a podáva sa rozdelená do 8 čiastkových dávok po 0,5 mg/kg. Po podaní prvej dávky sa majú každý deň stanoviť plazmatické hladiny busulfánu pomocou vhodnej metódy analýzy opakovane
odobratých vzoriek krvi. Ak hodnota AUC busulfánu prekročí 4 000 nanogramov/ml*h
(974 µmol/l.minúta), dávka sa má podľa hodnoty AUC náležite znížiť.
Premedikácia
Odporúča sa intravenózne podať antihistaminikum 15 - 30 minút pred infúziou Strimvelisu.
Dávkovanie
Rozmedzie odporúčanej dávky Strimvelisu je medzi 2 a 20 miliónmi CD34+ buniek/kg.
Ak liek obsahuje menej ako 2 milióny CD34+ buniek/kg, ošetrujúci lekár má na základe individuálneho hodnotenia prínosu a rizika rozhodnúť, či sa prikročí k jeho podaniu. U pacientov, ktorí boli v klinických skúšaniach liečení < 2 miliónmi CD34+buniek/kg, sa pozorovalo zlyhanie liečby.
Strimvelis sa má podať iba jedenkrát. Osobitné skupiny pacientov
Staršie osoby
Strimvelis nie je určený na použitie u pacientov vo veku > 65 rokov a v tejto vekovej skupine nebol skúmaný.
Porucha funkcie obličiek
Strimvelis nebol skúmaný u pacientov s poruchou funkcie obličiek. Predpokladá sa, že u nich nebude potrebná žiadna úprava dávky.
Porucha funkcie pečene
Strimvelis nebol skúmaný u pacientov s poruchou funkcie pečene. Predpokladá sa, že u nich nebude potrebná žiadna úprava dávky.
Pediatrická populácia
Bezpečnosť a účinnosť Strimvelisu u detí mladších ako šesť mesiacov alebo starších ako 6 rokov a 1 mesiac neboli stanovené (pozri časť 4.4). K dispozícii nie sú žiadne údaje.
Spôsob podávania
Strimvelis je určený na intravenóznu infúziu.
Má sa použiť transfúzna súprava s filtrom. Majú sa používať iba filtre určené na použitie
s transfúznymi súpravami, aby sa predišlo neúmyselnému odstráneniu buniek z lieku.
Rýchlosť podávania infúzie nemá prekročiť 5 ml/kg/h. Doba podávania je približne 20 minút (pozri časť 6.6). Po podaní sa má na prepláchnutie vaku použiť 50 ml injekčná striekačka naplnená fyziologickým roztokom.
Opatrenia pred zaobchádzaním alebo podaním lieku
Tento liek obsahuje geneticky modifikované bunky. Majú sa dodržiavať národné smernice
o biologickej bezpečnosti, ktoré sa vzťahujú na takéto lieky (pozri časť 6.6).
Strimvelis nie je testovaný na prítomnosť pôvodcov prenosných infekčných ochorení. Zdravotnícki pracovníci, ktorí zaobchádzajú so Strimvelisom, preto majú prijať náležité opatrenia, aby sa zabránilo možnému prenosu infekčných ochorení.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Leukémia alebo myelodysplázia v súčasnej alebo minulej anamnéze.
Pozitívny výsledok vyšetrenia na prítomnosť vírusu ľudskej imunodeficiencie (HIV) alebo
akéhokoľvek pôvodcu ochorenia uvedeného v aktuálnej smernici EÚ o bunkách a tkanivách.
Predchádzajúca génová terapia v anamnéze.
4.4 Osobitné upozornenia a opatrenia pri používaní
Strimvelis je určený výlučne na autológne použitie a nikdy sa nesmie podať pacientovi inému ako je
pôvodný darca CD34+ buniek.
V niektorých prípadoch pacient nebude môcť dostať Strimvelis kvôli problémom súvisiacim s jeho výrobou. Po tom, ako to bude ošetrujúcemu lekárovi oznámené, môže byť nutné, aby náležite upravil liečebný program pacienta (t. j. ukončenie prípravného režimu s busulfánom a/alebo podanie lieku vyrobeného zo záložnej vzorky kmeňových buniek, ak je to vhodné).
Výsledky kontroly kvality druhého štádia budú k dispozícii až po podaní infúzie lieku. Ak sa
po podaní infúzie Strimvelisu zistia klinicky významné problémy súvisiace s jeho kvalitou, napríklad výsledky mimo špecifikácií, ošetrujúci lekár o tom bude informovaný. Lekár má pacienta vhodným spôsobom sledovať a/alebo liečiť.
Strimvelis sa má používať obozretne u pacientov starších ako 6 rokov a 1 mesiac a mladších ako
6 mesiacov, pretože k dispozícii nie sú údaje z klinických skúšaní získané v tomto vekovom rozmedzí. Starší pacienti majú zvyčajne nižšiu schopnosť darovať vysoký počet CD34+ buniek, čo môže znamenať, že starších pacientov nie je možné liečiť. Je tiež pravdepodobné, že úspešná tvorba
T-lymfocytov po podaní Strimvelisu je ovplyvnená funkciou reziduálneho tkaniva týmusu, ktorá môže byť u starších detí znížená. Použitie Strimvelisu u pacientov, ktorí sú starší ako pacienti v minulosti zaradení v štúdiách, sa má dôkladne zvážiť a vyhradiť iba pre prípady, v ktorých boli vyčerpané
všetky iné racionálne možnosti liečby.
Strimvelis sa má používať obozretne u pacientov s precitlivenosťou na aminoglykozidy alebo hovädzí sérový albumín.
Po liečbe Strimvelisom neboli hlásené žiadne prípady leukémie ani myelodysplázie. V porovnateľných
klinických skúšaniach s génovou terapiou používanou pri Wiskottovom-Aldrichovom syndróme,
SCID viazanej na chromozóm X a chronickej granulomatóze sa však v minulosti zaznamenali prípady leukémie súvisiacej s inzerciou vektorov do oblastí chromozómov. Zistilo sa, že retrovírusové
inzerčné miesta (retroviral insertion sites, RIS) sa nachádzajú v tesnej blízkosti alebo v rámci lokusov CCND2 a LMO2 a že existuje potenciálne riziko leukemickej transformácie po liečbe Strimvelisom. Odporúča sa, aby boli pacienti dlhodobo sledovaní a aby absolvovali kontrolnú prehliadku aspoň
raz ročne počas prvých jedenástich rokov a potom po 13 a 15 rokoch po liečbe Strimvelisom. Kontrolná prehliadka má zahŕňať vyšetrenie kompletného krvného obrazu s diferenciálom,
biochemické vyšetrenia a vyšetrenie hladiny hormónu stimulujúceho štítnu žľazu.
Dlhodobé účinky a trvácnosť odpovede na liečbu Strimvelisom pri ADA-SCID nie sú známe (pozri
časť 5.1).
Pacientov treba pozorne sledovať so zameraním sa na výskyt závažných a oportúnnych infekcií, parametre rekonštitúcie imunitného systému a potrebu substitučnej liečby intravenóznymi imunoglobulínmi (IVIG); v prípade nedostatočnej odpovede na liečbu sa odporúča zaviesť iné liečby ADA-SCID pod dohľadom lekára.
Vyskytli sa prípady, keď liečba Strimvelisom nebola úspešná. Niektorí pacienti museli znovu začať dlhodobú enzýmovú substitučnú liečbu a/alebo podstúpiť transplantáciu kmeňových buniek (pozri časť 5.1).
Pri prejavoch ADA-SCID neimunologického pôvodu sa nemusí dosiahnuť odpoveď na liečbu
Strimvelisom.
Neuskutočnilo sa žiadne testovanie imunogenity Strimvelisu.
U pacientov sa môže rozvinúť autoimunita. 67 % (12 z 18) pacientov liečených Strimvelisom malo autoimunitné protilátky alebo iné prejavy autoimunity (napr. autoimunitnú trombocytopéniu, autoimunitnú aplastickú anémiu, autoimunitnú hepatitídu a Guillainov-Barrého syndróm) (pozri časť 4.8).
Pacienti liečení Strimvelisom nesmú darovať krv, orgány, tkanivá a bunky na transplantáciu v nijakom
čase v budúcnosti. Táto informácia je uvedená na pohotovostnej karte pacienta.
Po liečbe Strimvelisom došlo k zvýšeniu počtu T-lymfocytov (CD3+) a NK (CD56+) buniek. Medián hodnôt po 3 rokoch od génovej terapie bol pod hodnotou referenčného rozpätia. Odporúča sa nepretržité sledovanie. Hlásené boli prípady kožných papilómov, abnormálnych výsledkov elektroforézy bielkovín v sére a jeden prípad lipofibrómu, jeden prípad pľúcnej masy a jeden prípad zmenšeného repertoáru V-beta génových rodín T-bunkového receptora (TCR). Príčinná súvislosť
s liekom sa nepreukázala.
Hlásené boli nežiaduce udalosti súvisiace s použitím centrálnych venóznych katétrov (CVK) (napr. závažné infekcie súvisiace so zavedením CVK a trombóza súvisiaca s katétrom). Pacientov treba pozorne sledovať kvôli možným nežiaducim udalostiam súvisiacim s katétrom.
Obsah sodíka
Tento liek obsahuje 0,15 mmol sodíka na ml. U pacientov na diéte s kontrolovaným obsahom sodíka to má byť brané do úvahy.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Neočakáva sa, že by Strimvelis interagoval s enzýmami z rodiny pečeňového cytochrómu P-450 ani s transportérmi liekov.
4.6 Fertilita, gravidita a laktácia
Ž
eny vo fertilnom veku
Keďže Strimvelis sa podáva po prípravnom režime s busulfánom, pacientky v plodnom veku musia
používať spoľahlivú bariérovú antikoncepciu počas podania Strimvelisu a aspoň 6 mesiacov po ňom.
Gravidita
K dispozícii nie sú žiadne klinické údaje o expozícii v období gravidity.
Štúdie reprodukčnej a vývojovej toxicity sa neuskutočnili. Strimvelis sa nemá používať počas gravidity.
Dojčenie
Nie je známe, či sa Strimvelis vylučuje do ľudského mlieka. Účinok na dojčené deti po podaní
Strimvelisu ich matkám sa neskúmal.
Strimvelis sa nemá podávať ženám, ktoré dojčia. Fertilita
K dispozícii nie sú žiadne údaje o účinkoch Strimvelisu na fertilitu ľudí. V štúdiách na zvieratách sa
nehodnotili účinky na samčiu a samičiu fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Strimvelis nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnosť Strimvelisu sa hodnotila u 18 osôb, pričom medián trvania sledovania bol 7 rokov.
Vzhľadom na malú populáciu pacientov a veľkosť kohort, nežiaduce reakcie v tabuľke neposkytujú
celistvý pohľad na charakter a frekvenciu týchto udalostí. Závažné nežiaduce reakcie zahŕňajú
autoimunitu (napr. autoimunitnú hemolytickú anémiu, autoimunitnú aplastickú anémiu, autoimunitnú hepatitídu, autoimunitnú trombocytopéniu a Guillainov-Barrého syndróm). Najčastejšie hlásenou nežiaducou reakciou bola pyrexia.
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie sú nižšie uvedené podľa triedy orgánových systémov MedDRA a frekvencie
výskytu. Použité kategórie frekvencie sú:
Veľmi časté > /10
Časté ≥ 1/100 až < 1/10
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
T
rieda orgánových systémov Veľmi časté Časté
P
oruchy krvi a lymfatického systému
Anémiaa, neutropéniaa Autoimunitná hemolytická anémia,
autoimunitná aplastická
anémia, autoimunitná trombocytopénia
P
oruchy endokrinného systému Hypotyreóza Autoimunitná tyreoiditída
Poruchy nervového systému Guillainov-Barrého
syndróm
Poruchy ciev Hypertenziaa
P
oruchy dýchacej sústavy,
hrudníka a mediastína
Astma, alergická rinitída
P
oruchy pečene a žlčových ciest Autoimunitná hepatitída
P
oruchy kože a podkožného
t
kaniva
Atopická dermatitída,
ekzém
C
elkové poruchy a reakcie
v mieste podania
Pyrexia
L
aboratórne a funkčné
vyšetrenia
Zvýšené hladiny
pečeňových enzýmova, pozitívne antinukleárne
protilátky (ANA)
Pozitívne antineutrofilové
cytoplazmatické protilátky, pozitívne
protilátky proti hladkému svalstvu
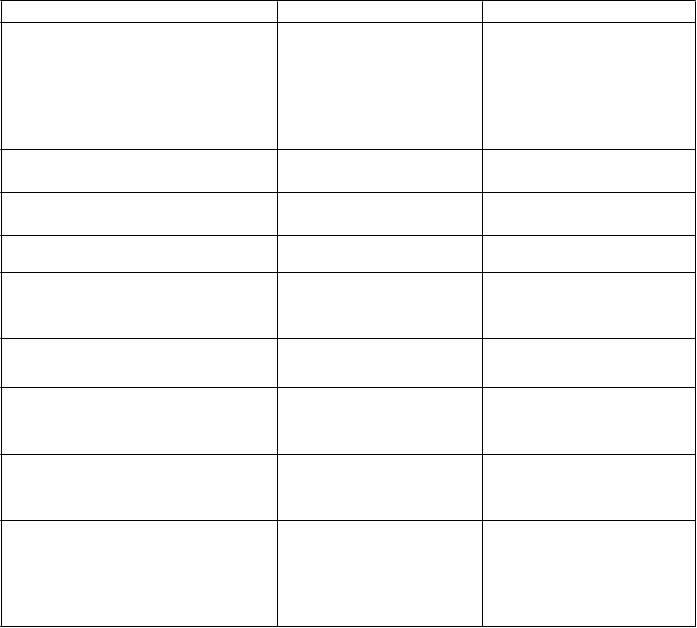
aNežiaduce reakcie považované za potenciálne súvisiace s prípravným režimom s busulfánom
Opis vybraných nežiaducich reakciíRekonštitúcia imunitného systémuVšetky zistené nežiaduce reakcie uvedené v tabuľke (okrem tých, ktoré potenciálne súviseli
s busulfánom) sa považujú za súvisiace s rekonštitúciou imunitného systému, vzhľadom na ich
charakter a čas vzniku. Tieto autoimunitné nežiaduce reakcie boli hlásené u osôb po génovej terapii. Väčšina z nich bola hlásená počas 3-mesačného až 3-ročného obdobia sledovania a väčšina z nich vymizla, s výnimkou hypotyreózy a pozitívnych výsledkov vyšetrení na prítomnosť ANA. Okrem toho, nežiaduce reakcie alergického pôvodu uvedené v tabuľke boli hlásené prevažne počas
3-mesačného až 3-ročného obdobia sledovania.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieK dispozícii nie sú žiadne údaje z klinických štúdií týkajúce sa predávkovania Strimvelisom.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunostimulanciá, iné imunostimulanciá, ATC kód: zatiaľ nepridelený
Mechanizmus účinku
Po podaní infúzie sa CD34+ bunky uchytia v kostnej dreni, kde repopulujú hematopoetický systém podielom buniek, ktoré exprimujú farmakologicky aktívne hladiny enzýmu ADA.
Predpokladá sa, že po tom, ako u pacienta dôjde k úspešnému uchyteniu CD34+ buniek, budú účinky
lieku celoživotné.
Farmakodynamickéúčinky
Medián percentuálneho počtu geneticky modifikovaných buniek v periférnej krvi po jednom a troch
rokoch po liečbe bol v uvedenom poradí 28 % (rozmedzie 6 % - 92 %) a 30 % (rozmedzie
8 % - 101 %) pokiaľ ide o CD19+ bunky a 73 % (rozmedzie 20 % - 100 %) a 67 % (rozmedzie
39 % - 82 %) pokiaľ ide o CD3+ bunky.
Prítomnosť transgénu vedie k zvýšenej expresii ADA. Jeden rok po liečbe bol medián aktivity ADA (adenozíndeaminázy v mononukleárnych bunkách) v lymfocytoch periférnej krvi
181,2 (rozmedzie 42,1 - 1 678,2) nmol/h/mg bielkoviny, v porovnaní s východiskovým mediánom
(rozmedzie) 80,6 (30,5 - 92,3) nmol/h/mg bielkoviny. Aktivita ADA zostala zvýšená počas celého trvania 3-ročného sledovania.
Klinickáúčinnosťabezpečnosť
Celkovo 18 pacientov s ADA-SCID podstúpilo liečbu Strimvelisom v jednom otvorenom pivotnom klinickom skúšaní (AD1115611; N = 12), v dvoch skôr vykonaných otvorených pilotných štúdiách (AD1117054/AD1117056; N = 3) a v rámci „compassionate use“ (program na použitie neregistrovaného lieku v nevyhnutých prípadoch) (AD1117064; N = 3). Štúdie hodnotili použitie Strimvelisu s rozmedzím 0,9 milióna - 18,2 milióna CD34+ buniek/kg. Všetci pacienti absolvovali
pred génovou terapiou prípravný režim s busulfánom, pričom väčšine z nich sa podal v celkovej dávke
4 mg/kg aplikovanej intravenózne počas 2 po sebe idúcich dní pred podaním infúzie CD34+. Štyri osoby predtým podstúpili neúspešnú transplantáciu kmeňových buniek od haploidentického darcu
a 15 z 18 osôb predtým podstúpilo enzýmovú substitučnú liečbu hovädzou adenozíndeaminázou
modifikovanou polyetylénglykolom (polyethylene-glycol-modified bovine adenosine deaminase, PEG-ADA). U pacientov, ktorí boli predtým liečení PEG-ADA, sa jej podávanie ukončilo
10 až 22 dní pred liečbou Strimvelisom. Medián veku populácie v programe klinického vývoja bol
1,7 roka (rozmedzie 0,5 až 6,1) a 61 % tvorili osoby mužského pohlavia. Osemdesiattri percent tvorili belosi (56 % kaukazského/európskeho pôvodu a 28 % arabského/severoafrického pôvodu),
11 % tvorili Afroameričania/Afričania a 6 % tvorili Ázijci.
Pacienti liečení v pivotnej štúdii
Účinnosť Strimvelisu sa hodnotila v 3-ročnej otvorenej, prospektívnej štúdii u detí, ktoré nemali HLA-zhodného súrodeneckého darcu kmeňových buniek a ktoré nedosiahli dostatočnú odpoveď na liečbu PEG-ADA, ktoré takúto liečbu netolerovali, alebo pre ktoré nebola dostupná.
Výsledky po 3 rokoch získané u pacientov liečených v pivotnej štúdii sú uvedené v tabuľke 1. Liečba Strimvelisom viedla k 100 % miere prežívania po 3 rokoch po liečbe, k zníženiu výskytu závažných infekcií, k zvýšeniu počtu T-lymfocytov (CD3+) a všetky osoby mali po liečbe hladiny deoxyadenozínového nukleotidu v erytrocytoch venóznej krvi (red blood cell deoxyadenosine nucleotide, RBC dAXP) nižšie ako patologické hladiny (> 100 nmol/ml).
T
abuľka 1. Výsledky po 3 rokoch získané v ITT populácii v pivotnej štúdii*
C
i
eľový ukazovateľ Východiskový stav/Pred liečbou
a
3. rok/3 roky po liečbe
b
'
Prežívanie
n
%
Závažné infekcie
Neaplikovateľné 12
100 %
n
Výskyt závažných infekcií na osoborok pozorovania (95 % interval
spoľahlivosti)
Počet T-lymfocytov (x 106/l)
n
medián (rozmedzie)
% osôb s hladinou RBC dAXP vo venóznej krvi < 100 nmol/ml po podaní Strimvelisud
12
1,10 (0,74 - 1,58)
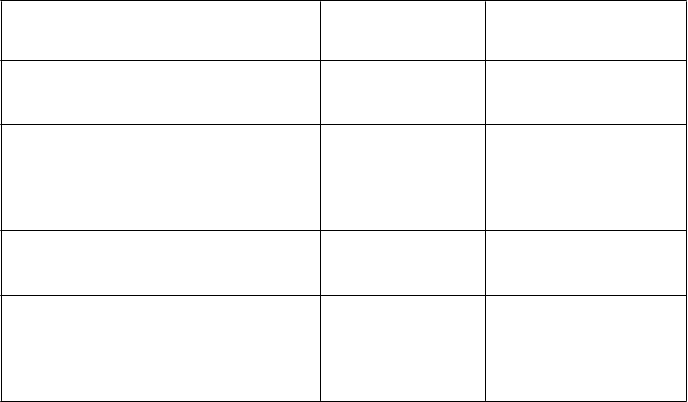
11
88,0 (19 - 2 718)
12
0,429c (0,24 - 0,72)
11
828,0 (309 - 2 458)
n Neaplikovateľnée 11
% 100 %
*Vrátane údajov získaných u jedného pacienta po intervencii s PEG-ADA [≥ 3-mesačná liečba] alebo
transplantácii hematopoetických kmeňových buniek.
a Na základe celého predliečebného obdobia pokiaľ ide o závažné infekcie (údaje získané retrospektívne)
a na základe údajov získaných na vstupnej (baseline) návšteve pokiaľ ide o počet T-lymfocytov. U pacienta
č. 10 nebol k dispozícii údaj o východiskovom počte T-lymfocytov.
b Na základe 3-ročného poliečebného obdobia pokiaľ ide o prežívanie a závažné infekcie a na základe údajov získaných na kontrolnej návšteve v 3. roku pokiaľ ide o počet T-lymfocytov a hladinu dAXP. Pacient č. 8 ukončil účasť v štúdii pred absolvovaním kontrolnej návštevy v 3. roku, a preto sa u neho nezískali žiadne údaje o počte T-lymfocytov a hladine dXAP.
c Závažné infekcie sú infekcie, ktoré vyžadujú hospitalizáciu alebo predĺženie hospitalizácie. Z výpočtu bolo
vylúčené obdobie 3-mesačnej hospitalizácie nasledujúcej bezprostredne po génovej terapii.
d dAXP=dAMP+dADP+dATP. Výsledky hladiny dAXP sú založené na analýze respondérov (t. j. pacientov odpovedajúcich na liečbu) týkajúcej sa percenta pacientov po génovej terapii, ktorí splnili definíciu adekvátnej metabolickej detoxikácie, preto je východisková hodnota neaplikovateľná.
e 9 z 11 (82 %) pacientov malo východiskovú hladinu dAXP < 100 nmol/ml. Všetci títo pacienti boli predtým
liečení PEG-ADA.
Funkcia T-lymfocytov: U pacientov liečených v pivotnej štúdii sa preukázala proliferácia
T-lymfocytov v odpovedi na stimuláciu protilátkami proti CD3 (medián 62 629 cpm, rozmedzie
4 531 až 252 173) a fytohemaglutinínom (medián 140 642 cpm, rozmedzie 11 119 až 505 607)
po 1 roku po génovej terapii a odpoveď na uvedenú stimuláciu sa udržala až do 3. roku. Zistenia, že hodnota TREC (T cell receptor excision circles; t. j. excízne kruhy preusporiadania T-bunkového receptora) v lymfocytoch periférnej krvi sa v porovnaní s jej východiskovou hodnotou zvýšila (medián
141, rozmedzie 56 až 1 542 kópií/100 ng DNA) po 1 roku a udržala sa do 3. roku po liečbe
a že u všetkých osôb sa preukázali polyklonálne V-beta reťazce v jednom alebo vo viacerých hodnotených obdobiach po génovej terapii, poskytujú ďalšie podporné dôkazy o vývoji funkčných
T-lymfocytov.
Funkcia B-lymfocytov: Všetkých 12 osôb liečených v pivotnej štúdii dostávalo liečbu IVIG v čase
skríningu a u 7 osôb (58 %) bolo používanie IVIG ukončené počas 0- až 3-ročného sledovania
po génovej terapii.
D
l
hodobé sledovanie
U všetkých 12 osôb liečených v pivotnej štúdii a tiež u 18 osôb zahrnutých do integrovanej analýzy sa pozorovala 100 % miera prežívania, pričom medián trvania sledovania bol približne 7 rokov.
V populácii z pivotnej štúdie bolo prežívanie bez potreby intervencie (definované ako prežívanie bez
potreby znovuzavedenia dlhodobej (≥ 3-mesačnej) liečby PEG-ADA, alebo bez potreby transplantácie kmeňových buniek) 92 % (11/12 osôb) (82 % (14/17 ôsob) v integrovanej populácii). U jednej osoby liečenej v pilotnej štúdii neboli k dispozícii údaje o znovuzavedení liečby PEG-ADA, a preto bola vylúčená z analýzy prežívania bez potreby intervencie v integrovanej populácii. Dlhodobá liečba
PEG-ADA (trvajúca nepretržite dlhšie ako 3 mesiace) sa zaviedla u troch osôb; dve z týchto osôb
následne podstúpili transplantáciu kmeňových buniek od zhodného súrodeneckého darcu a jedna osoba zotrvala na dlhodobej liečbe PEG-ADA. U ďalšej osoby bolo potrebné dočasne trvajúce podávanie PEG-ADA kvôli autoimunitnej nežiaducej udalosti (pozri časť 4.4).
U pacientov, ktorí boli liečení v pivotnej štúdii, výskyt závažných infekcií klesal počas celého obdobia
sledovania (tabuľka 2).
Tabuľka 2. Výskyt závažných infekcií na osoborok expozície (populácia z pivotnej štúdie)*
Pred
liečbou
Po liečbe
Časové obdobie (roky)
n/a 0,33 - 1 > 1 - 2 > 2 - 3 > 3 - 4 > 4 - 5 > 5 - 6 > 6 - 7 > 7 - 8 Celkovo
Počet osôb 12 12 11 11 11 11 9 7 3 12
Počet závažných infekcií Počet závažných infekcií na osoborok
29 6 3 0 2 0 1 0 0 12
1,10 0,63 0,27 0,00 0,18 0,00 0,12 0,00 0,00 0,17
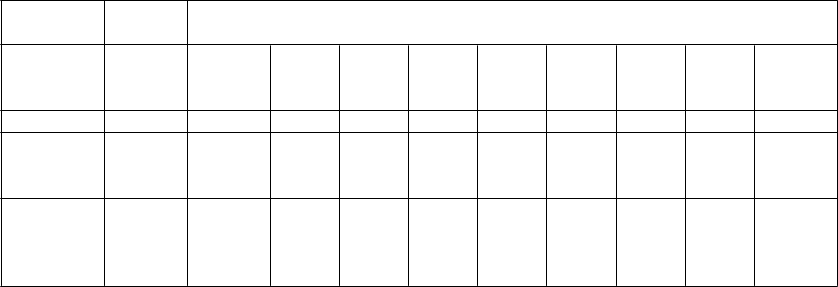
* S vylúčením údajov získaných u jedného pacienta v čase intervencie s PEG-ADA (≥ 3-mesačná liečba) alebo
transplantácie hematopoetických kmeňových buniek. n/a - neaplikovateľné.
5.2 Farmakokinetické vlastnostiStrimvelis je autológna bunková terapia. Povaha Strimvelisu je taká, že konvenčné štúdie zamerané
na farmakokinetiku, absorpciu, distribúciu, metabolizmus a elimináciu nie sú aplikovateľné.
5.3 Predklinické údaje o bezpečnostiReprodukčné a vývojové štúdie sa neuskutočnili.
Vykonala sa 4-mesačná biodistribučná štúdia na myšiach. CD34+ bunky získané z pupočníkovej krvi zdravých ľudí, ktoré boli transdukované vektorom použitým na výrobu Strimvelisu, boli intravenózne podané myšiam po prípravnom režime s busulfánom. U väčšiny myší sa do konca štúdie preukázala rekonštitúcia hematopoetického systému. Nízke hladiny ľudských buniek a sekvencie vektora sa zistili aj v nehematopoetických orgánoch, čo je v zhode s prítomnosťou krvi obsahujúcej transdukované ľudské bunky. Nevyskytli sa žiadne nežiaduce účinky na prežívanie, hematologické parametre alebo
na histopatológiu hlavných orgánov, okrem úbytku telesnej hmotnosti a atrofie semenníkov a vaječníkov, ktoré súviseli s podávaním busulfánu.
Štúdie karcinogenity sa neuskutočnili, pretože nebol k dispozícii žiadny adekvátny zvierací model na hodnotenie tumorogénneho potenciálu Strimvelisu z dôvodu nemožnosti dosiahnuť dlhodobé uchytenie transdukovaných buniek u myší.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
6 hodín.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri 15 - 30 °C.
6.5 Druh obalu a obsah balenia
50 ml etylénvinylacetátový (EVA) infúzny vak s adaptérom s hrotom a konektorom typu luer uzatvoreným uzáverom so závitom typu luer lock, zabalený v opakovane použiteľnom vonkajšom obale.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Strimvelis sa dopraví priamo do zdravotníckeho zariadenia, kde sa bude podávať infúzia. Infúzny(-e)
vak(-y) sa nachádza(-jú) vo vnútri uzatvoreného vonkajšieho obalu. Vaky sa musia uchovávať
vo vonkajšom obale, až kým nenastane čas na ich použitie.
Strimvelis je určený výlučne na autológne použitie. Pred podaním infúzie sa musí identita pacienta zhodovať so základnými jedinečnými informáciami o pacientovi uvedenými na vnútornom a/alebo vonkajšom obale.
Jemne pretrepte obsah infúzneho vaku, aby sa znovu rozptýlili akékoľvek zhluky buniek, podajte pomocou transfúznej súpravy s filtrom, aby sa odstránili akékoľvek zostávajúce zhluky buniek.
Tento liek obsahuje geneticky modifikované bunky. Majú sa dodržiavať príslušné národné smernice o biologickej bezpečnosti (pozri časť 4.2).
Strimvelis nie je testovaný na prítomnosť pôvodcov prenosných infekčných ochorení. Zdravotnícki pracovníci, ktorí zaobchádzajú so Strimvelisom, preto majú prijať náležité opatrenia, aby sa zabránilo možnému prenosu infekčných ochorení.
Pracovné povrchy a materiály, ktoré mohli prísť do styku so Strimvelisom, sa musia dekontaminovať vhodným dezinfekčným prostriedkom.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami na biologickú bezpečnosť.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
GlaxoSmithKline Trading Services Limited
Currabinny Carrigaline County Cork Írsko
8. REGISTRAČNÉ ČÍSLOEU/1/16/1097/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.