
ch
prípadov. Prípady boli hlásené v klinických skúšaniach s ertugliflozínom. V mnohých prípadoch bol
prejav tohto stavu atypický s len mierne zvýšenými hodnotami glukózy v krvi, do 14 mmol/l
(250 mg/dl). Nie je známe, či sa DKA vyskytuje s vyššou pravdepodobnosťou pri vyšších dávkach ertugliflozínu.
Riziko výskytu diabetickej ketoacidózy sa musí zvážiť v prípade nešpecifických príznakov ako sú nauzea, vracanie, anorexia, bolesť brucha, nadmerný smäd, ťažkosti s dýchaním, zmätenosť, neobvyklá únava alebo ospalosť. Ak sa vyskytnú tieto príznaky, pacientov treba okamžite vyšetriť na ketoacidózu, bez ohľadu na hladinu glukózy v krvi.
U pacientov so suspektnou alebo diagnostikovanou DKA sa má liečba Steglujanom okamžite ukončiť. Liečba sa má prerušiť u pacientov hospitalizovaných kvôli veľkým chirurgickým zákrokom alebo
akútnym závažným ochoreniam. V obidvoch prípadoch je možné liečbu Steglujanom obnoviť po stabilizovaní stavu pacienta.
Pred začatím liečby Steglujanom sa majú zvážiť faktory v anamnéze pacienta, ktoré ho môžu predisponovať ku ketoacidóze.
Pacienti, u ktorých môže byť vyššie riziko DKA, zahŕňajú pacientov s nízkou funkčnou rezervou betabuniek (napr. pacienti s diabetom 2. typu s nízkou hladinou C-peptidu alebo s latentným
autoimunitným diabetom u dospelých (latent autoimmune diabetes in adults, LADA) alebo pacienti s pankreatitídou v anamnéze), pacientov s ochoreniami vedúcimi k obmedzenému príjmu potravy alebo závažnej dehydratácii, pacientov, u ktorých sú dávky inzulínu znížené a pacientov so zvýšenou potrebou inzulínu z dôvodu akútneho ochorenia, chirurgického zákroku alebo nadmerného požívania alkoholu. U týchto pacientov sa majú používať inhibítory SGLT2 s opatrnosťou.
Obnovenie liečby inhibítormi SGLT2 u pacientov s DKA počas liečby inhibítormi SGLT2
v anamnéze sa neodporúča, pokiaľ nebol identifikovaný a vyriešený iný jednoznačný spúšťací faktor
DKA.
Bezpečnosť a účinnosť Steglujanu u pacientov s diabetom 1. typu sa nestanovili a Steglujan sa nemá používať na liečbu pacientov s diabetom 1. typu. Obmedzené údaje z klinických skúšaní naznačujú, že DKA sa vyskytuje často u pacientov s diabetom 1. typu liečených inhibítormi SGLT2.
Amputáciedolnýchkončatín
V dlhodobých klinických štúdiách s iným inhibítorom SGLT2 sa pozoroval zvýšený počet prípadov amputácie dolnej končatiny (hlavne prstov). Nie je známe, či ide o skupinový účinok. Rovnako ako u všetkých diabetických pacientov je dôležité odporučiť pacientom pravidelnú preventívnu starostlivosť o chodidlá.
Poruchafunkcieobličiek
Účinnosť ertugliflozínu závisí od funkcie obličiek a účinnosť je znížená u pacientov so stredne závažnou poruchou funkcie obličiek a pravdepodobne chýba u pacientov so závažnou poruchou
funkcie obličiek (pozri časť 4.2).
Liečba Steglujanom sa nemá začínať u pacientov s hodnotou eGFR nižšou ako 60 ml/min/1,73 m2 alebo CrCl nižšou ako 60 ml/min. Liečba Steglujanom sa má ukončiť, ak eGFR pretrváva na hodnote nižšej ako 45 ml/min/1,73 m2 alebo CrCl pretrváva na hodnote nižšej ako 45 ml/min z dôvodu zníženia účinnosti.
Sledovanie funkcie obličiek sa odporúča nasledovne:
- pred začatím liečby Steglujanom a pravidelne počas liečby (pozri časť 4.2),
- častejšie u pacientov s hodnotou eGFR nižšou ako 60 ml/min/1,73 m2 alebo CrCl nižšou ako
60 ml/min.
Hypoglykémiaprisúbežnompoužívaníinzulínualiečivstimulujúcichsekréciuinzulínu Ertugliflozín môže zvýšiť riziko hypoglykémie, keď sa používa v kombinácii s inzulínom a/alebo liečivom stimulujúcim sekréciu inzulínu, o ktorých je známe, že spôsobujú hypoglykémiu (pozri časť
4.8). Pri použití sitagliptínu v kombinácii s inzulínom alebo sulfonylureou sa pozorovala hypoglykémia. Preto na minimalizáciu rizika vzniku hypoglykémie môže byť potrebná nižšia dávka
inzulínu alebo liečiva stimulujúceho sekréciu inzulínu, ak sa používajú v kombinácii so Steglujanom
(pozri časti 4.2 a 4.5).
Mykotickéinfekciepohlavnýchorgánov
Ertugliflozín zvyšuje riziko mykotických infekcií pohlavných orgánov. V skúšaniach s inhibítormi
SGLT2 sa mykotické infekcie pohlavných orgánov objavili s väčšou pravdepodobnosťou u pacientov s mykotickými infekciami pohlavných orgánov v anamnéze a u mužov bez obriezky (pozri časť 4.8). Pacientov je potrebné sledovať a vhodne liečiť.
Infekciemočovýchciest
Vylučovanie glukózy v moči môže byť spojené so zvýšeným rizikom infekcií močových ciest. Výskyt infekcií močových ciest sa v skupinách s ertugliflozínom 5 mg a 15 mg (4,0 % a 4,1 %) a v skupine
s placebom (3,9 %) výrazne nelíšil. Väčšina udalostí bola mierna alebo stredne závažná a nebol hlásený žiadny vážny prípad. Počas liečby pyelonefritídy alebo urosepsy sa má zvážiť dočasné
prerušenie liečby ertugliflozínom.
Reakcie
z
precitlivenosti
U pacientov liečených sitagliptínom boli po uvedení lieku na trh hlásené závažné reakcie
z precitlivenosti (pozri časť 4.8). Tieto reakcie zahŕňajú anafylaxiu, angioedém a exfoliatívne kožné choroby vrátane Stevensovho-Johnsonovho syndrómu. Nástup týchto reakcií sa objavil v priebehu
prvých 3 mesiacov po nasadení liečby, pričom niektoré hlásenia sa vyskytli po prvej dávke. Ak je
podozrenie na reakciu z precitlivenosti, liečba Steglujanom sa má prerušiť. Majú sa vyhodnotiť iné možné príčiny udalosti a má sa začať náhradná liečba diabetu.
Bulóznypemfigoid
U pacientov užívajúcich inhibítory DPP-4 vrátane sitagliptínu, boli po uvedení lieku na trh hlásené prípady bulózneho pemfigoidu. Ak je podozrenie na bulózny pemfigoid, liečba Stelujanom sa má
ukončiť.
Starší pacienti
Starší pacienti môžu mať zvýšené riziko vzniku deplécie objemu. U pacientov vo veku 65 rokov a starších liečených ertugliflozínom bol vyšší výskyt nežiaducich reakcií súvisiacich s depléciou
objemu v porovnaní s mladšími pacientmi. Predpokladá sa, že Steglujan bude mať zníženú účinnosť
u starších pacientov s poruchou funkcie obličiek (pozri časti 4.2 a 4.8).
Zlyhávaniesrdca
Skúsenosť s použitím u triedy I-II podľa klasifikácie New York Heart Association (NYHA) je obmedzená a nie je žiadna skúsenosť z klinických štúdií so Steglujanom u triedy III-IV podľa klasifikácie NYHA.
Laboratórnevyšetreniamoču
Vzhľadom na mechanizmus účinku ertugliflozínu, bude výsledok vyšetrenia glukózy v moči
u pacientov užívajúcich Steglujan pozitívny. Na sledovanie kontroly glykémie sa majú použiť náhradné metódy.
Interferenciastestomna1,5-anhydroglucitol(1,5-AG)
Sledovanie kontroly glykémie pomocou testu na 1,5-AG sa neodporúča, pretože stanovenia 1,5-AG na hodnotenie kontroly glykémie u pacientov užívajúcich inhibítory SGLT2 nie sú spoľahlivé. Na sledovanie kontroly glykémie sa majú použiť náhradné metódy.
4.5 Liekové a iné interakcie
Farmakokinetické liekové interakčné štúdie so Steglujanom sa neuskutočnili; takéto štúdie však boli vykonané s ertugliflozínom a sitagliptínom, jednotlivými liečivami Steglujanu.
Ertugliflozín
Farmakodynamickéinterakcie
Diuretiká
Ertugliflozín môže zvyšovať diuretický účinok diuretík a môže zvýšiť riziko dehydratácie a hypotenzie (pozri časť 4.4).
Inzulín a liečivá stimulujúce sekréciu inzulínu
Inzulín a liečivá stimulujúce sekréciu inzulínu, ako sú deriváty sulfonylurey, spôsobujú hypoglykémiu. Ertugliflozín môže zvýšiť riziko vzniku hypoglykémie, ak sa používa v kombinácii s inzulínom
a/alebo liečivom stimulujúcim sekréciu inzulínu. Preto na zníženie rizika hypoglykémie môže byť potrebná nižšia dávka inzulínu alebo liečiva stimulujúceho sekréciu inzulínu, ak sa používajú
v kombinácii so Steglujanom (pozri časti 4.2, 4.4 a 4.8).
Farmakokinetické
interakcie
Účinky
iných
liekov
na
farmakokinetiku
ertugliflozínu
Primárnym mechanizmom odbúravania ertugliflozínu je metabolizmus sprostredkovaný UGT1A9
a UGT2B7.
Interakčné štúdie vykonané u zdravých osôb s použitím režimu s jednorazovou dávkou naznačujú, že sitagliptín, metformín, glimepirid ani simvastatín nespôsobujú zmenu farmakokinetiky ertugliflozínu.
Podávanie viacnásobných dávok rifampicínu (induktor UGT a CYP) znižuje AUC ertugliflozínu
o 39 % a Cmax ertugliflozínu o 15 %. Tento pokles expozície sa nepovažuje za klinicky významný
a preto sa neodporúča žiadna úprava dávky. Klinicky významný účinok s ostatnými induktormi (napr.
karbamazepín, fenytoín, fenobarbital) sa neočakáva.
Vplyv inhibítorov UGT na farmakokinetiku ertugliflozínu sa klinicky neskúmal, ale potenciálne zvýšenie expozície ertugliflozínu v dôsledku inhibície UGT sa nepovažuje za klinicky významné.
Účinkyertugliflozínunafarmakokinetikuinýchliekov
Interakčné štúdie vykonané u zdravých dobrovoľníkov naznačujú, že ertugliflozín nemal žiadny klinicky významný účinok na farmakokinetiku sitagliptínu, metformínu a glimepiridu.
Súbežné podávanie simvastatínu s ertugliflozínom viedlo k 24 % zvýšeniu AUC a 19 % zvýšeniu Cmax simvastatínu a k 30 % zvýšeniu AUC a 16 % zvýšeniu Cmax kyseliny simvastatínovej. Mechanizmus malých zvýšení simvastatínu a kyseliny simvastatínovej nie je známy a neprebieha prostredníctvom inhibície OATP ertugliflozínom. Tieto zvýšenia sa nepovažujú za klinicky významné.
Sitagliptín
Farmakokinetické interakcie
Účinkyinýchliekovnasitagliptín
Sitagliptín sa primárne eliminuje nezmenený v moči a metabolizmus zohráva iba malú úlohu. Štúdie in vitro ukázali, že primárnym enzýmom zodpovedným za limitovaný metabolizmus sitagliptínu je CYP3A4 s prispením CYP2C8.
Metabolizmus môže zohrať významnejšiu úlohu v eliminácii sitagliptínu v podmienkach ťažkej poruchy funkcie obličiek alebo terminálneho štádia ochorenia obličiek (ESRD). Z tohto dôvodu je možné, že silné inhibítory CYP3A4 (ako sú ketokonazol, itrakonazol, ritonavir, klaritromycín) môžu zmeniť farmakokinetiku sitagliptínu u pacientov s ťažkou poruchou funkcie obličiek alebo ESRD. Interakčné štúdie uskutočnené u pacientov s diabetom 2. typu alebo u zdravých dobrovoľníkov naznačujú, že metformín a cyklosporín nemali žiadny klinicky významný účinok na farmakokinetiku sitagliptínu.
Účinkysitagliptínunainélieky
V štúdiách liekových interakcií nemal sitagliptín klinicky významné účinky na farmakokinetiku nasledujúcich liečiv: metformín, rosiglitazón, glyburid, simvastatín, warfarín a perorálne
kontraceptíva.
Digoxín: Sitagliptín mal malý účinok na plazmatické koncentrácie digoxínu. Po podávaní 0,25 mg digoxínu súbežne so 100 mg sitagliptínu denne po dobu 10 dní sa plazmatická AUC digoxínu zvýšila priemerne o 11 % a plazmatická Cmax priemerne o 18 %. Neodporúča sa žiadna úprava dávky digoxínu. Pacienti s rizikom digoxínovej toxicity však majú byť na ňu sledovaní, ak sa sitagliptín
a digoxín podávajú súbežne.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje o použití Steglujanu u gravidných žien. K dispozícii je iba obmedzené množstvo údajov o použití ertugliflozínu u gravidných žien. Na základe výsledkov zo štúdií na zvieratách, môže ertugliflozín ovplyvniť vývin a dozrievanie obličiek (pozri časť 5.3). Preto sa Steglujan nemá používať počas gravidity.
Dojčenie
K dispozícii nie sú žiadne informácie týkajúce sa prítomnosti Steglujanu alebo jeho jednotlivých zložiek v ľudskom mlieku, účinkov na dojčené dieťa alebo účinkov na tvorbu mlieka. Štúdie na
dojčiacich zvieratách sa s kombináciou zložiek Steglujanu neuskutočnili. Ertugliflozín a sitagliptín sú
prítomné v mlieku dojčiacich potkanov. Ertugliflozín spôsobil účinky u mláďat dojčiacich potkanov.
Farmakologicky sprostredkované účinky sa pozorovali u mláďat potkanov liečených ertugliflozínom (pozri časť 5.3). Keďže k dozrievaniu obličiek u ľudí dochádza in utero a počas prvých 2 rokov života, kedy môže dôjsť k expozícii počas dojčenia, riziko u novorodencov/dojčiat sa nedá vylúčiť. Steglujan sa nemá používať počas dojčenia.
Fertilita
Účinok Steglujanu na fertilitu u ľudí sa neskúmal. V štúdiách na zvieratách sa nepozorovali žiadne účinky ertugliflozínu ani sitagliptínu na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Steglujan nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidla alebo obsluhovaní strojov však treba zobrať do úvahy, že pri používaní sitagliptínu boli hlásené závrat a somnolencia. Pacientov je navyše potrebné upozorniť na riziko hypoglykémie, keď sa Steglujan používa v kombinácii s inzulínom alebo liečivami stimulujúcimi sekréciu inzulínu
a na zvýšené riziko nežiaducich reakcií súvisiacich s depléciou objemu ako je posturálny závrat (pozri časti 4.2, 4.4 a 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Ertugliflozínasitagliptín
Bezpečnosť súbežne podávaného ertugliflozínu a sitagliptínu sa hodnotila u 990 pacientov s diabetes mellitus 2. typu liečených 26 týždňov v troch štúdiách: vo faktoriálnej štúdii ertugliflozínu 5 mg alebo
15 mg v kombinácii so sitagliptínom 100 mg jedenkrát denne v porovnaní s jednotlivými zložkami,
v placebom kontrolovanej štúdii s ertugliflozínom 5 mg alebo 15 mg ako prídavnej liečby
k sitagliptínu 100 mg a metformínu jedenkrát denne a v placebom kontrolovanej štúdii so začiatočnou liečbou ertugliflozínom 5 mg alebo 15 mg jedenkrát denne v kombinácii so sitagliptínom 100 mg
jedenkrát denne (pozri časť 5.1). Výskyt a druhy nežiaducich reakcií v týchto troch štúdiách boli
podobné nežiaducim reakciám pozorovaným pri ertugliflozíne a sú popísané v tabuľke 1. V týchto troch skúšaniach, ktoré zahŕňali sitagliptín, neboli zistené žiadne ďalšie nežiaduce reakcie v porovnaní
s tromi placebom kontrolovanými štúdiami s ertugliflozínom (pozri nižšie).
Ertugliflozín
Združený súbor placebom kontrolovaných skúšaní
Primárne hodnotenie bezpečnosti bolo vykonané v združenom súbore troch 26-týždňových placebom kontrolovaných skúšaní. Ertugliflozín sa používal vo forme monoterapie v jednom skúšaní a ako
prídavná liečba v dvoch skúšaniach (pozri časť 5.1). Tieto údaje odrážajú expozíciu ertugliflozínu
u 1 029 pacientov s priemernou dĺžkou trvania expozície približne 25 týždňov. Pacienti dostávali ertugliflozín 5 mg (N = 519), ertugliflozín 15 mg (N = 510) alebo placebo (N = 515) jedenkrát denne.
Najčastejšie hlásenými nežiaducimi reakciami v rámci klinického programu boli vulvovaginálna mykotická infekcia a iné mykotické infekcie ženských pohlavných orgánov. Zriedkavo sa objavila závažná diabetická ketoacidóza. Frekvencie výskytu pozri v časti „Popis vybraných nežiaducich reakcií“ a pozri časť 4.4.
Sitagliptín
Boli hlásené závažné nežiaduce reakcie zahŕňajúce pankreatitídu a reakcie z precitlivenosti. Pri kombinácii so sulfonylureou (4,7 %-13,8 %) a inzulínom (9,6 %) sa hlásila hypoglykémia (pozri časť
4.4).
Tabuľkovýsúhrnnežiaducichreakcií
Nežiaduce reakcie uvedené nižšie sú klasifikované podľa frekvencie a triedy orgánových systémov
(system organ class, SOC). Kategórie frekvencií sú definované podľa nasledujúceho pravidla: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka 1: Nežiaduce reakcie
Trieda orgánových systémov
Frekvencia
Infekcie a nákazy
Veľmi časté
Časté
Poruchy krvi a lymfatického systému
Nežiaduca reakcia
vulvovaginálna mykotická infekcia a iné mykotické infekcie ženských pohlavných orgánov*,†,1
kandidová balanitída a iné mykotické infekcie mužských pohlavných orgánov*,†,1
Zriedkavé trombocytopénia2
Poruchy imunitného systému
Neznáme reakcie z precitlivenosti vrátane anafylaktických odpovedí*,a,2
Poruchy metabolizmu a výživy
Časté
Zriedkavé
Poruchy nervového systému
Časté
Menej časté
Poruchy dýchacej sústavy, hrudníka a mediastína
hypoglykémia*,†,1,2
diabetická ketoacidóza*,†,1
bolesť hlavy2
závrat2
Neznáme intersticiálne ochorenie pľúca,2
Poruchy gastrointestinálneho traktu
Menej časté Neznáme Neznáme Neznáme
Poruchy kože a podkožného tkaniva
Menej časté Neznáme Neznáme Neznáme Neznáme Neznáme
Neznáme
zápcha2
vracaniea,2
akútna pankreatitídaa,*,b,2
hemoragická a nekrotizujúca pankreatitída s fatálnymi následkami alebo bez nich*,a,2
pruritusa,2 angioedéma,*,2 vyrážkaa,*,2 urtikáriaa,*,2
kožná vaskulitídaa,*,2
exfoliatívne kožné ochorenia vrátane Stevensovho- Johnsonovho syndrómua,*,2
bulózny pemfigoida,*,2
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Neznáme Neznáme Neznáme Neznáme Poruchy ciev
artralgiaa,2 myalgiaa,2 bolesť chrbtaa,2 artropatiaa,2
Časté deplécia objemu*,†,1
Poruchy obličiek a močových ciest
Časté
Menej časté
Neznáme
Neznáme
Poruchy reprodukčného systému a prsníkov
zvýšené vylučovanie moču‡,1
dyzúria1, zvýšená hladina kreatinínu v krvi/znížená rýchlosť glomerulárnej filtrácie†,1
porucha funkcie obličieka,2
akútne zlyhanie obličieka,2
Časté vulvovaginálny pruritus1
Celkové poruchy a reakcie v mieste podania
Časté smäd§,1
Laboratórne a funkčné vyšetreniaČasté zmenená hladina tukov v sére¶,1, zvýšená hladina hemoglobínu**1, zvýšená hladina BUN¶¶1
1 Nežiaduce reakcie pri ertugliflozíne.
2 Nežiaduce reakcie pri sitagliptíne.
* Pozri časť 4.4.
† Ďalšie informácie pozri v podčastiach nižšie.
‡ Zahŕňa: polakizúriu, nutkanie na močenie, polyúriu, zvýšené vylučovanie moču a noktúriu.
§ Zahŕňa: smäd a polydipsiu.
¶ Priemerné percentuálne zmeny oproti východiskovej hodnote boli pre LDL-C 5,8 % pri ertugliflozíne 5 mg a 8,4 % pri ertugliflozíne 15 mg oproti 3,2 % pri placebe; pre celkový cholesterol 2,8 % pri ertugliflozíne 5 mg a 5,7 % pri
ertugliflozíne 15 mg oproti 1,1 % pri placebe; avšak pre HDL-C 6,2 % pri ertugliflozíne 5 mg a 7,6 % pri ertugliflozíne
15 mg oproti 1,9 % pri placebe. Medián percentuálnych zmien oproti východiskovej hodnote bol pre triglyceridy -
3,9 % pri ertugliflozíne 5 mg a -1,7 % pri ertugliflozíne 15 mg oproti 4,5 % pri placebe.
** Podiel osôb s minimálne jedným zvýšením hemoglobínu > 2,0 g/dl bol vyšší v skupinách s ertugliflozínom 5 mg a 15 mg
(4,7 % a 4,1 %, v uvedenom poradí) v porovnaní s placebom (0,6 %).
¶¶ Podiel osôb, ktoré mali akýkoľvek výskyt zvýšenia hodnôt BUN ≥ 50 % a hodnota > HHN bola číselne vyššia v skupine s ertugliflozínom 5 mg a vyššia v skupine s ertugliflozínom 15 mg (7,9 % a 9,8 %, v uvedenom poradí) v porovnaní so skupinou s placebom (5.1 %).
a Nežiaduce reakcie boli identifikované počas sledovania po uvedení lieku na trh.
b Pozri nižšie
štúdiu kardiovaskulárnej bezpečnosti TECOS.
Popis vybranýchnežiaducichreakciíDepléciaobjemu(ertugliflozín)Ertugliflozín spôsobuje osmotickú diurézu, ktorá môže viesť k zníženiu intravaskulárneho objemu a nežiaducim reakciám súvisiacim s depléciou objemu. V združenom súbore placebom kontrolovaných štúdií bol výskyt nežiaducich udalostí súvisiacich s depléciou objemu (dehydratácia,
posturálny závrat, presynkopa, synkopa, hypotenzia a ortostatická hypotenzia) nízky (< 2 %) a v rámci skupín s ertugliflozínom a placebom nebol výrazne odlišný. V analýzach podskupín v širšom združenom súbore štúdií fázy 3, bol u osôb s hodnotou eGFR < 60 ml/min/1,73 m2, osôb vo veku ≥ 65 rokov a osôb užívajúcich diuretiká výskyt deplécie objemu vyšší v skupinách s ertugliflozínom
v porovnaní so skupinou s komparátorom (pozri časti 4.2 a 4.4). U osôb s hodnotou eGFR
< 60 ml/min/1,73 m2 bol výskyt 5,1 % pri ertugliflozíne 5 mg, 2,6 % pri ertugliflozíne 15 mg a 0,5 %
v skupine s komparátorom a u osôb s hodnotou eGFR 45 až < 60 ml/min/1,73 m2 bol výskyt 6,4 % pri ertugliflozíne 5 mg, 3,7 % pri ertugliflozíne 15 mg a 0 % v skupine s komparátorom.
Hypoglykémia(ertugliflozín)V združenom súbore placebom kontrolovaných štúdií, bol výskyt zdokumentovanej hypoglykémie zvýšený pre ertugliflozín 5 mg a 15 mg (5,0 % a 4,5 %) v porovnaní s placebom (2,9 %). V tejto
populácii bol výskyt závažnej hypoglykémie 0,4 % v každej skupine. Keď sa ertugliflozín používal vo
forme monoterapie, výskyt hypoglykemických udalostí v oboch skupinách s ertugliflozínom bol 2,6 % a v skupine s placebom 0,7 %. Keď sa používal ako prídavná liečba k metformínu, výskyt hypoglykemických udalostí v skupine s ertugliflozínom 5 mg bol 7,2 %, v skupine s ertugliflozínom
15 mg 7,8 % a v skupine s placebom 4,3 %.
Ak sa ertugliflozín pridal k metformínu a porovnal so sulfonylureou, výskyt hypoglykémie bol vyšší pri sulfonylurei (27 %) v porovnaní s ertugliflozínom (5,6 % pri ertugliflozíne 5 mg a 8,2 % pri ertugliflozíne 15 mg).
U pacientov so stredne závažnou poruchou funkcie obličiek používajúcich inzulín, SU alebo meglitinidy ako základnú liečbu, bola hypoglykémia zdokumentovaná u 36 % pri ertugliflozíne 5 mg,
27 % pri ertugliflozíne 15 mg a 36 % pri placebe (pozri časti 4.2, 4.4 a 4.5).
Diabetickáketoacidóza(ertugliflozín)V rámci klinického programu s ertugliflozínom sa ketoacidóza identifikovala u 3 z 3 409 (0,1 %)
pacientov liečených ertugliflozínom a u 0,0 % pacientov liečených komparátorom (pozri časť 4.4).
Zvýšená
hladina
kreatinínu
v
krvi/pokles
rýchlosti
glomerulárnej
filtrácie
a
udalosti
súvisiace
sobličkami (ertugliflozín)
Úvodné zvýšenia priemernej hladiny kreatinínu a poklesy priemernej hodnoty eGFR u pacientov
liečených ertugliflozínom boli počas prebiehajúcej liečby vo všeobecnosti prechodné. U pacientov so stredne závažnou poruchou funkcie obličiek na začiatku sa objavili väčšie priemerné zmeny, ktoré sa v 26. týždni neupravili späť na východiskový stav; tieto zmeny sa upravili po ukončení liečby.
Nežiaduce reakcie súvisiace s obličkami (napr. akútne poškodenie obličiek, porucha funkcie obličiek, akútne prerenálne zlyhanie) sa môžu objaviť u pacientov liečených ertugliflozínom, najmä u pacientov so stredne závažnou poruchou funkcie obličiek, kedy výskyt nežiaducich reakcií súvisiacich
s obličkami bol 2,5 % u pacientov liečených ertugliflozínom 5 mg, 1,3 % u pacientov liečených ertugliflozínom 15 mg a 0,6 % u pacientov užívajúcich placebo.
Mykotickéinfekciepohlavnýchorgánov(ertugliflozín)
V združenom súbore troch placebom kontrolovaných klinických skúšaní sa mykotické infekcie ženských pohlavných orgánov (napr. kandidózy pohlavných orgánov, mykotická infekcia pohlavných
orgánov, vaginálna infekcia, vulvitída, vulvovaginálne kandidózy, vulvovaginálna mykotická infekcia,
vulvovaginitída) objavili u 9,1 % žien liečených ertugliflozínom 5 mg, 12 % žien liečených ertugliflozínom 15 mg a 3,0 % žien užívajúcich placebo. U žien došlo k ukončeniu liečby z dôvodu
mykotických infekcií pohlavných orgánov u 0,6 % pacientok liečených ertugliflozínom a u 0 %
pacientok užívajúcich placebo (pozri časť 4.4).
V rovnakom združenom súbore sa mykotické infekcie mužských pohlavných orgánov (napr. kandidová balanitída, balanopostitída, infekcia pohlavných orgánov, mykotická infekcia pohlavných orgánov) objavili u 3,7 % mužov liečených ertugliflozínom 5 mg, u 4,2 % mužov liečených ertugliflozínom 15 mg a 0,4 % mužov užívajúcich placebo. Mykotické infekcie mužských pohlavných orgánov sa častejšie objavovali u mužov bez obriezky. U mužov došlo k ukončeniu liečby z dôvodu mykotických infekcií pohlavných orgánov u 0,2 % pacientov liečených ertugliflozínom a u 0 % pacientov užívajúcich placebo. V zriedkavých prípadoch sa hlásila fimóza a niekedy došlo
k vykonaniu obriezky (pozri časť 4.4).
Sitagliptín
Okrem nežiaducich reakcií popísaných vyššie v tabuľke, nežiaduce účinky, hlásené bez ohľadu na kauzálnu súvislosť s liečbou a vyskytujúce sa aspoň v 5 % a častejšie u pacientov liečených sitagliptínom, zahŕňali infekciu horných dýchacích ciest a nazofaryngitídu. Ďalšie nežiaduce účinky, hlásené bez ohľadu na kauzálnu súvislosť s liečbou, ktoré sa vyskytli častejšie u pacientov liečených sitagliptínom (nedosahujúce hodnotu 5 %, ale vyskytujúce sa s incidenciou > 0,5 % vyššou v skupine so sitagliptínom ako v kontrolnej skupine), zahŕňali osteoartrózu a bolesť v končatine.
Niektoré nežiaduce reakcie sa pozorovali častejšie v štúdiách kombinovaného používania sitagliptínu s inými antidiabetickými liekmi, v porovnaní so štúdiami sitagliptínu v monoterapii. Tieto reakcie zahŕňali hypoglykémiu (frekvencia veľmi časté v kombinácii so sulfonylureou a metformínom), chrípku (časté s inzulínom (s metformínom alebo bez neho)), nauzeu a vracanie (časté
s metformínom), plynatosť (časté s metformínom alebo pioglitazónom), zápchu (časté v kombinácii so sulfonylureou a metformínom), periférny edém (časté s pioglitazónom alebo v kombinácii
s pioglitazónom a metformínom), somnolenciu a hnačku (menej časté s metformínom) a sucho v ústach (menej časté s inzulínom (s metformínom alebo bez neho)).
TECOS (Skúšanie hodnotiace kardiovaskulárne výsledky pri sitagliptíne; The Trial Evaluating
Cardiovascular Outcomes with Sitagliptin)
Štúdia kardiovaskulárnej bezpečnosti so sitagliptínom (TECOS) zahŕňala 7 332 pacientov liečených sitagliptínom, 100 mg denne (alebo 50 mg denne ak východisková eGFR bola ≥ 30
a < 50 ml/min/1,73 m2) a 7 339 pacientov, ktorí užívali placebo v populácii podľa liečebného zámeru
(intention-to-treat). Obidve liečby sa pridali k bežnej starostlivosti zameranej na regionálne štandardy pre HbA1c a KV rizikové faktory. Celkový výskyt závažných nežiaducich udalostí u pacientov užívajúcich sitagliptín bol podobný ako u pacientov užívajúcich placebo.
V populácii podľa liečebného zámeru (intention-to-treat) bol medzi pacientmi, ktorí na začiatku používali inzulín a/alebo sulfonylureu, výskyt ťažkej hypoglykémie u pacientov užívajúcich sitagliptín
2,7 % a u pacientov užívajúcich placebo 2,5 %; medzi pacientmi, ktorí na začiatku nepoužívali inzulín
a/alebo sulfonylureu bol výskyt ťažkej hypoglykémie u pacientov užívajúcich sitagliptín 1,0 %
a u pacientov užívajúcich placebo 0,7 %. Výskyt potvrdených udalostí pankreatitídy bol u pacientov užívajúcich sitagliptín 0,3 % a u pacientov užívajúcich placebo 0,2 %.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa Steglujanom použite zvyčajné podporné opatrenia (napr. odstráňte nevstrebaný liek z gastrointestinálneho traktu, použite klinické sledovanie vrátane elektrokardiogramu a nasaďte podpornú liečbu) na základe klinického stavu pacienta.
ErtugliflozínPri ertugliflozíne sa nepreukázala žiadna toxicita u zdravých osôb pri jednorazových perorálnych dávkach až do 300 mg a viacnásobných dávkach až do 100 mg denne počas 2 týždňov.
Neidentifikovali sa žiadne možné akútne príznaky a prejavy predávkovania. Odstránenie ertugliflozínu
pomocou hemodialýzy sa neskúmalo.
SitagliptínPočas kontrolovaných klinických skúšaní u zdravých osôb sa podávali jednorazové dávky sitagliptínu až do 800 mg. V jednej štúdii pri dávke 800 mg sitagliptínu sa pozorovalo minimálne predĺženie QTc, ktoré sa nepovažovalo za klinicky významné. V klinických štúdiách nie sú žiadne skúsenosti
s dávkami vyššími ako 800 mg. V štúdiách I. fázy s opakovanými dávkami sa pri sitagliptíne
v dávkach do 600 mg denne podávaných po dobu do 10 dní, ani v dávkach 400 mg denne podávaných po dobu do 28 dní nepozorovali žiadne s dávkou súvisiace klinické nežiaduce reakcie.
Sitagliptín je mierne dialyzovateľný. V klinických štúdiách bolo približne 13,5 % dávky odstránenej po 3- až 4-hodinovej hemodialýze. Ak je to klinicky vhodné, môže sa zvážiť predĺženie hemodialýzy. Nie je známe, či je sitagliptín dialyzovateľný peritoneálnou dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antidiabetiká, kombinácie perorálnych liečiv znižujúcich hladiny glukózy v krvi, ATC kód: A10BD24
MechanizmusúčinkuSteglujan predstavuje kombináciu dvoch antihyperglykemických látok s komplementárnymi mechanizmami účinku na zlepšenie kontroly glykémie u pacientov s diabetom 2. typu: inhibítor SGLT2 ertugliflozín a inhibítor DPP-4 sitagliptíniumfosfát.
ErtugliflozínSGLT2 je hlavným transportérom zodpovedným za spätné vstrebávanie glukózy z glomerulárneho filtrátu späť do krvného obehu. Ertugliflozín je účinný, selektívny a reverzibilný inhibítor SGLT2. Inhibíciou SGLT2 ertugliflozín znižuje spätné vstrebávanie prefiltrovanej glukózy v obličkách
a znižuje prahovú hranicu glukózy v obličkách, čím zvyšuje vylučovanie glukózy močom.
Sitagliptín
Sitagliptín je člen skupiny perorálnych antihyperglykemických látok nazývaných inhibítory dipeptidyl peptidázy 4 (DPP-4). Zlepšenie kontroly glykémie pozorované pri tomto lieku môže byť sprostredkované zvýšením hladín aktívnych inkretínových hormónov. Inkretínové hormóny vrátane glukagónu podobného peptidu-1 (GLP-1) a glukózo-dependentného inzulínotropného polypeptidu (GIP) sa uvoľňujú črevom počas dňa a hladiny sa zvyšujú v odpovedi na jedlo. Inkretíny sú súčasťou endogénneho systému zapojeného do fyziologickej regulácie homeostázy glukózy. Keď sú koncentrácie glukózy v krvi normálne alebo zvýšené, GLP-1 a GIP zvyšujú syntézu a uvoľňovanie inzulínu z pankreatických beta buniek intracelulárnymi signálnymi dráhami zahŕňajúcimi cyklický AMP. Liečba s GLP-1 alebo s inhibítormi DPP-4 na zvieracích modeloch diabetu 2. typu preukázala zlepšenie odpovede beta buniek na glukózu a stimuláciu biosyntézy a uvoľňovania inzulínu. Pri vyšších hladinách inzulínu sa zvyšuje vychytávanie glukózy tkanivami. GLP-1 navyše znižuje
sekréciu glukagónu z pankreatických alfa buniek. Znížené koncentrácie glukagónu spolu s vyššími hladinami inzulínu vedú k zníženiu tvorby hepatálnej glukózy, čo vedie k zníženiu hladín glukózy
v krvi. Účinky GLP-1 a GIP sú závislé od glukózy, teda keď je koncentrácia glukózy v krvi nízka,
stimulácia uvoľňovania inzulínu a potláčanie sekrécie glukagónu prostredníctvom GLP-1 sa nepozoruje. Pri GLP-1 aj GIP je stimulácia sekrécie inzulínu zvýšená, keď glukóza stúpne nad normálne koncentrácie. Ďalej, GLP-1 neoslabuje normálnu glukagónovú odpoveď na hypoglykémiu. Aktivita GLP-1 a GIP je obmedzená enzýmom DPP-4, ktorý rýchlo hydrolyzuje inkretínové hormóny na inaktívne látky. Sitagliptín zabraňuje hydrolýze inkretínových hormónov enzýmom DPP-4, dôsledkom čoho zvyšuje plazmatické koncentrácie aktívnych foriem GLP-1 a GIP. Zvyšovaním hladiny aktívnych inkretínov sitagliptín zvyšuje uvoľňovanie inzulínu a znižuje hladiny glukagónu
v závislosti od glukózy. U pacientov s diabetom 2. typu s hyperglykémiou viedli tieto zmeny
v hladinách inzulínu a glukagónu k zníženiu hemoglobínu A1c (HbA1c) a zníženiu koncentrácií glukózy nalačno a po jedle. Od glukózy závislý mechanizmus sitagliptínu sa líši od mechanizmu sulfonylureí, ktoré zvyšujú sekréciu inzulínu, aj keď sú hladiny glukózy nízke, a môžu u pacientov
s diabetom 2. typu a u zdravých osôb viesť k hypoglykémii. Sitagliptín je silný a vysoko selektívny inhibítor enzýmu DPP-4 a pri terapeutických koncentráciách neinhibuje blízko príbuzné enzýmy DPP-8 alebo DPP-9.
V dvojdňovej štúdii u zdravých osôb zvýšil samotný sitagliptín koncentrácie aktívneho GLP-1, kým samotný metformín zvýšil koncentrácie aktívneho a celkového GLP-1 v podobnom rozsahu. Súbežné podanie sitagliptínu a metformínu malo aditívny účinok na koncentrácie aktívneho GLP-1. Sitagliptín, ale nie metformín, zvýšil koncentrácie aktívneho GIP.
Farmakodynamické účinky
Ertugliflozín
Vylučovanie glukózy močom a objem moču
U zdravých osôb a u pacientov s diabetes mellitus 2. typu sa po podaní jednorazovej dávky
a viacnásobnej dávky ertugliflozínu pozorovali zvýšenia množstva glukózy vylúčenej do moču závislé od dávky. Modelovanie odpovede na dávku naznačuje, že ertugliflozín 5 mg a 15 mg vedie k takmer
maximálnemu vylučovaniu glukózy močom (urinary glucose excretion, UGE) u pacientov s diabetes
mellitus 2. typu, čo predstavuje maximálnu inhibíciu 87 % pri ertugliflozíne 5 mg a 96 % pri ertugliflozíne 15 mg.
Klinická účinnosťabezpečnosť
Ertugliflozín v kombinácii so sitagliptínom
Účinnosť a bezpečnosť ertugliflozínu v kombinácii so sitagliptínom sa skúmali v 3 multicentrických, randomizovaných, dvojito zaslepených, placebom alebo aktívnym komparátorom kontrolovaných klinických štúdiách fázy 3 zahŕňajúcich 1 985 pacientov s diabetom 2. typu. Rasové rozdelenie pacientov v rámci týchto troch štúdií bolo 72,9 % až 90,4 % belochov, 0,0 % až 20,3 % aziatov, 1,9 % až 4,5 % černochov a 4,8 % až 5,4 % ostatných. Hispánski a latinskoamerickí pacienti tvorili 15,6 %
až 36,1 % populácie. Priemerný vek pacientov v týchto 3 štúdiách sa pohyboval od 55,1 do 59,1 rokov
(rozmedzie 21 rokov až 85 rokov). V rámci týchto troch štúdií bolo 16,2 % až 29,9 % pacientov vo veku ≥ 65 rokov a 2,3 % až 2,8 % vo veku ≥ 75 rokov.
Faktoriálna štúdia ertugliflozínu a sitagliptínu ako prídavná kombinovaná liečba s metformínom Celkovo 1 233 pacientov s diabetom 2. typu sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 26-týždňovej, aktívne kontrolovanej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu 5 mg alebo 15 mg v kombinácii so sitagliptínom 100 mg v porovnaní s jednotlivými liečivami. Pacienti s diabetom 2. typu nedostatočne kontrolovaní metformínom v monoterapii
(≥ 1 500 mg/deň) boli randomizovaní do jednej z piatich skupín aktívnej liečby: ertugliflozín 5 mg alebo 15 mg, sitagliptín 100 mg alebo sitagliptín 100 mg v kombinácii s 5 mg alebo 15 mg ertugliflozínu s podávaním jedenkrát denne ako doplnok k prebiehajúcej základnej liečbe metformínom (pozri tabuľku 2).
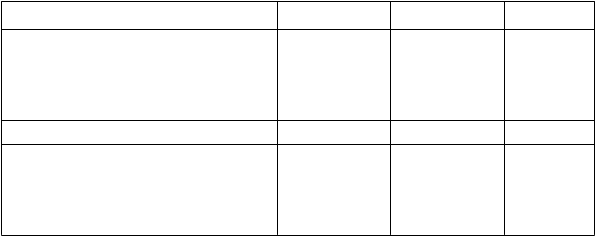
Tabuľka 2: Výsledky v 26. týždni z faktoriálnej štúdie ertugliflozínu a sitagliptínu ako prídavnej kombinovanej liečby s metfornínom v porovnaní so samostatne podávanými jednotlivými liečivami*
Ertugliflozín
5 mg
Ertugliflozín
1
5 mg
Sitagliptín
10
0 mg
Ertugliflozín 5 mg +
sitagliptín 100 mg
Ertugliflozín 15 mg
+ sitagliptín 100 mg
HbA1c (%) N = 250 N = 248 N = 247 N = 243 N = 244
Východisková hodnota (priemerná) 8,6 8,6 8,5 8,6 8,6
Zmena oproti východiskovej hodnote (priemer LS†) Rozdiel od
-1,0 -1,1 -1,1 -1,5 -1,5
sitagliptínu ertugliflozínu 5 mg ertugliflozínu 15 mg
(priemer LS†, 95% IS)
-0,4‡ (-0,6, -0,3)
-0,5‡ (-0,6, -0,3)
-0,5‡ (-0,6, -0,3)
-0,4‡ (-0,6, -0,3)
Pacienti [N (%)] s HbA1c < 7 % 66 (26,4) 79 (31,9) 81 (32,8) 127§ (52,3) 120§ (49,2)
Telesná hmotnosť (kg) N = 250 N = 248 N = 247 N = 243 N = 244
Východisková hodnota (priemerná) 88,6 88,0 89,8 89,5 87,5
Zmena oproti východiskovej
hodnote (priemer LS†)
Rozdiel od sitagliptínu
(priemer LS†, 95% IS)
-2,7 -3,7 -0,7 -2,5 -2,9
-1,8‡ (-2,5, -1,2) -2,3‡ (-2,9, -1,6)

* N zahŕňa všetkých randomizovaných liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, východiskovú hodnotu eGFR a interakciu času podľa liečby.
‡ p < 0,001 v porovnaní s kontrolnou skupinou.
§ p < 0,001 v porovnaní s príslušnou dávkou ertugliflozínu alebo sitagliptínu (na základe porovnaní upravených pomerov pravdepodobnosti z modelu logistickej regresie s použitím viacnásobného pripočítania hodnôt chýbajúcich údajov).
Ertugliflozín ako prídavná kombinovaná liečba s metformínom a sitagliptínomCelkovo 463 pacientov s diabetom 2. typu nedostatočne kontrolovaných metformínom
(≥ 1 500 mg/deň) a sitagliptínom 100 mg jedenkrát denne sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 26-týždňovej, placebom kontrolovanej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu. Pacienti boli randomizovaní na podávanie ertugliflozínu 5 mg, ertugliflozínu 15 mg alebo placeba jedenkrát denne ako doplnok k pokračujúcej základnej liečbe metformínom a sitagliptínom (pozri tabuľku 3).
Tabuľka 3: Výsledky v 26. týždni zo štúdie ertugliflozínu ako prídavnej liečby v kombinácii s metformínom a sitagliptínom*
Ertugliflozín 5 mg Ertugliflozín 15 mg Placebo
HbA1c (%) N = 156 N = 153 N = 153
Východisková hodnota (priemerná) 8,1 8,0 8,0
Zmena oproti východiskovej hodnote (priemer LS†) -0,8 -0,9 -0,1
Rozdiel od placeba (priemer†, 95% IS) -0,7‡ (-0.9; -0,5) -0,8‡ (-0,9; -0,6)
 Pacienti [N (%)] s HbA1c < 7 %
Pacienti [N (%)] s HbA1c < 7 % 50 (32,1)‡ 61 (39,9)‡ 26 (17,0)
Telesná hmotnosť (kg) N = 156 N = 153 N = 153Východisková hodnota (priemerná) 87,6 86,6 86,5
Zmena oproti východiskovej hodnote (priemer LS†) -3,3 -3,0 -1,3
Rozdiel od placeba (priemer†, 95% IS) -2,0‡ (-2,6; -1,4) -1,7‡ (-2,3; -1,1)
* N zahŕňa všetkých randomizovaných liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, predchádzajúci antihyperglykemický liek.
‡ p < 0,001 v porovnaní s placebom.
Kombinovaná liečba ertugliflozínom a sitagliptínomCelkovo 291 pacientov s diabetom 2. typu nedostatočne kontrolovaných diétou a cvičením sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, placebom kontrolovanej, 26-týždňovej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu v kombinácii so sitagliptínom. Títo pacienti, ktorí nedostávali žiadnu základnú antihyperglykemickú liečbu, boli randomizovaní na podávanie ertugliflozínu 5 mg alebo ertugliflozínu 15 mg v kombinácii so sitagliptínom (100 mg) alebo placeba jedenkrát denne (pozri tabuľku 4).
Tabuľka 4: Výsledky v 26. týždni zo štúdie kombinovanej liečby ertugliflozínu a sitagliptínu*
Ertugliflozín 5 mg +
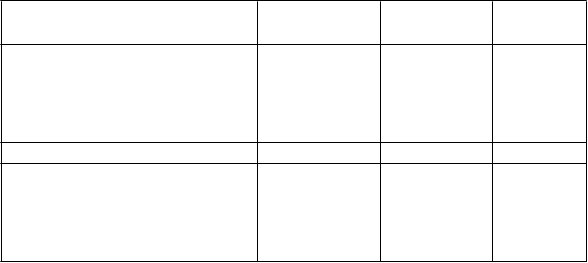 sitagliptín
Ertugliflozín
1
5 mg + sitagliptín
Placebo
sitagliptín
Ertugliflozín
1
5 mg + sitagliptín
Placebo
HbA1c (%) N = 98 N = 96 N = 96
Východisková hodnota (priemerná) 8,9 9,0 9,0
Zmena oproti východiskovej hodnote (priemer
LS†)
-1,6 -1,7 -0,4
Rozdiel od placeba (priemer LS†, 95% IS) -1,2‡ (-1,5; -0,8) -1,2‡ (-1,6; -0,9)
Pacienti [N (%)] s HbA1c < 7 % 35§ (35,7) 30§ (31,3) 8 (8,3)
Telesná hmotnosť (kg) N = 98 N = 96 N = 97
Východisková hodnota (priemerná) 90,8 91,3 95,0
Zmena oproti východiskovej hodnote (priemer
LS†)
-2,9 -3,0 -0,9
Rozdiel od placeba (priemer LS†, 95% IS) -2,0‡ (-3,0; -1,0) -2,1‡ (-3,1; -1,1)
* N zahŕňa všetkých pacientov, ktorí dostali minimálne jednu dávku skúšaného lieku a mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na základe longitudinálneho modelu zahŕňajúceho podmienky pre liečbu, čas
a interakciu času podľa liečby.
‡ p < 0,001 v porovnaní s placebom.
§ p < 0,001 v porovnaní s placebom (na základe porovnaní upravených pomerov pravdepodobnosti z modelu logistickej regresie s použitím viacnásobného pripočítania hodnôt chýbajúcich údajov).
Plazmatická hladina glukózy nalačno
V troch placebom kontrolovaných štúdiách viedol ertugliflozín k štatisticky významným zníženiam plazmatických hladín glukózy nalačno (fasting plasma glucose, FPG). Zníženia FPG upravené
s ohľadom na placebo boli 1,92 mmol/l pre ertugliflozín 5 mg a 2,44 mmol/l pre ertugliflozín 15 mg
vo forme monoterapie, 1,48 mmol/l pre ertugliflozín 5 mg a 2,12 mmol/l pre ertugliflozín 15 mg ako prídavnej liečby k metformínu a 1,40 mmol/l pre ertugliflozín 5 mg a 1,74 mmol/l pre ertugliflozín
15 mg ako prídavnej liečby k metformínu a sitagliptínu.
Kombinácia ertugliflozínu a sitagliptínu viedla k významne väčším zníženiam FPG v porovnaní so samotným sitagliptínom alebo ertugliflozínom alebo v porovnaní s placebom. Kombinácia ertugliflozínu 5 mg alebo 15 mg a sitagliptínu viedla k prírastkovým zníženiam FPG 0,46 až
0,65 mmol/l v porovnaní so samotným ertugliflozínom alebo 1,02 až 1,28 mmol/l v porovnaní so samotným sitagliptínom. Zníženia pri ertugliflozíne 5 mg alebo 15 mg upravené s ohľadom na placebo
v kombinácii so sitagliptínom boli 2,16 mmol/l a 2,56 mmol/l.
Účinnosť u pacientov s východiskovou hodnotou HbA1c ≥ 10 %
V štúdii pacientov nedostatočne kontrolovaných metformínom s východiskovou hodnotou HbA1c od
7,5-11,0 %, v podskupine pacientov s východiskovou hodnotou HbA1c ≥ 10 % viedla kombinácia ertugliflozínu 5 mg so sitagliptínom k zníženiu HbA1c o 2,35 % a kombinácia ertugliflozínu 15 mg so sitagliptínom k zníženiu HbA1c o 2,66 %, v porovnaní so znížením o 2,10 % v skupine so samotným ertugliflozínom 5 mg, 1,30 % v skupine so samotným ertugliflozínom 15 mg a 1,82 % v skupine so samotným sitagliptínom.
Hladina glukózy po jedle
Pri použití v monoterapii viedol ertugliflozín 5 mg a 15 mg k štatisticky významným zníženiam hladiny glukózy po jedle (post-prandial glucose, PPG) po 2 hodinách upraveným s ohľadom na placebo v hodnote 3,83 mmol/l a 3,74 mmol/l.
Kombinácia ertugliflozínu 5 mg so sitagliptínom a kombinácia ertugliflozínu 15 mg so sitagliptínom viedla k štatisticky významným zníženiam PPG po 2 hodinách upraveným s ohľadom na placebo
v hodnote 3,46 mmol/l a 3,87 mmol/l.
Tlak krvi
Po 26 týždňoch liečby viedla kombinácia ertugliflozínu 5 mg alebo 15 mg a sitagliptínu 100 mg
k štatisticky významným zníženiam systolického tlaku krvi (systolic blood pressure, SBP) v porovnaní so samotným sitagliptínom (-2,8 mmHg pre E5/S100 a -3,0 mmHg pre E15/S100) alebo placebom (-
4,4 mmHg pre E5/S100 a -6,4 mmHg pre E15/S100). Okrem toho, pri pridaní k základnej liečbe metformínom a sitagliptínom viedol ertugliflozín 5 mg k štatisticky významnému zníženiu SBP upravenému s ohľadom na placebo o 2,9 mmHg a ertugliflozín 15 mg k štatisticky významnému
zníženiu SBP upravenému s ohľadom na placebo o 3,9 mmHg.
Analýza podskupín
U pacientov s diabetom 2. typu liečených ertugliflozínom v kombinácii so sitagliptínom bolo zlepšenie
HbA1c podobné medzi skupinami definovanými na základe veku, pohlavia, rasy a dĺžky trvania diabetes mellitus 2. typu.
TECOS štúdia kardiovaskulárnej bezpečnosti
TECOS bola randomizovaná štúdia u 14 671 pacientov v populácii podľa liečebného zámeru (intention-to-treat) s HbA1c ≥ 6,5 až 8,0 % so stanoveným KV ochorením, ktorí dostávali sitagliptín (7 332) 100 mg denne (alebo 50 mg denne ak východisková eGFR bola ≥ 30 a < 50 ml/min/1,73 m2) alebo placebo (7 339) pridané k bežnej starostlivosti zameranej na regionálne štandardy pre HbA1c
a KV rizikové faktory. Do štúdie neboli zaradení pacienti s eGFR < 30 ml/min/1,73 m2. Populácia
štúdie zahŕňala 2 004 pacientov vo veku ≥ 75 rokov a 3 324 pacientov s poruchou funkcie obličiek
(eGFR < 60 ml/min/1,73 m2).
Počas trvania štúdie bol celkový odhadovaný priemerný (SD) rozdiel v HbA1c medzi skupinou užívajúcou sitagliptín a skupinou užívajúcou placebo 0,29 % (0,01), 95% IS (−0,32; −0,27); p < 0,001. Primárny kardiovaskulárny cieľový ukazovateľ bol zložený z prvého výskytu kardiovaskulárneho úmrtia, nefatálneho infarktu myokardu, nefatálnej cievnej mozgovej príhody alebo hospitalizácie
z dôvodu nestabilnej angíny pectoris. Sekundárne kardiovaskulárne cieľové ukazovatele zahŕňali prvý výskyt kardiovaskulárneho úmrtia, nefatálneho infarktu myokardu alebo nefatálnej cievnej mozgovej
príhody; prvý výskyt jednotlivých zložiek primárnej kombinácie; úmrtnosť z akejkoľvek príčiny;
a hospitalizáciu z dôvodu kongestívneho zlyhania srdca.
Po mediáne sledovania 3 roky, sitagliptín pridaný k bežnej starostlivosti, nezvýšil riziko hlavných kardiovaskulárnych nežiaducich udalostí alebo riziko hospitalizácie z dôvodu zlyhania srdca
v porovnaní s bežnou starostlivosťou bez sitagliptínu u pacientov s diabetom 2. typu (pozri tabuľku 5).
Tabuľka 5: Hodnoty zložených kardiovaskulárnych výsledkov a kľúčových sekundárnych výsledkov
Sitagliptín 100 mg Placebo
N (%)
Výskyt na 100 paciento-
rokov* N (%)
Výskyt na
100 paciento- rokov*
Pomer rizika
(95% IS)
Hodnota p†
Analýza v populácii podľa liečebného zámeru (intention-to-treat)
Počet pacientov
7 332 7 339
Primárny zložený cieľový ukazovateľ
(Kardiovaskulárne úmrtie,
nefatálny infarkt myokardu, nefatálna cievna mozgová
príhoda alebo hospitalizácia z dôvodu nestabilnej angíny
pectoris) 839 (11,4) 4,1 851 (11,6) 4,2
Sekundárny zložený cieľový ukazovateľ
(Kardiovaskulárne úmrtie,
nefatálny infarkt myokardu, nefatálna cievna mozgová'
0,98 (0,89–1,08) < 0,001
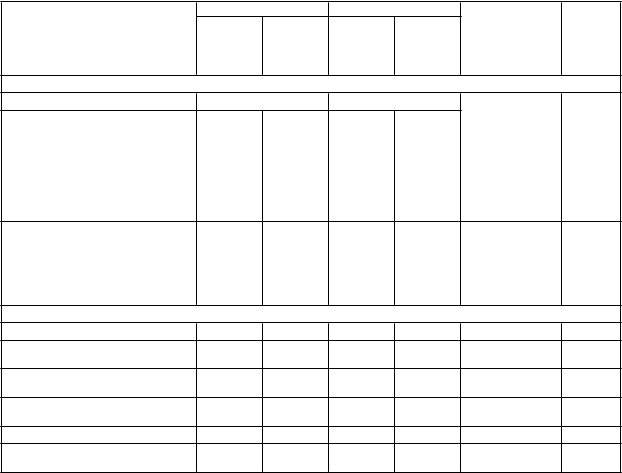
príhoda) 745 (10,2) 3,6 746 (10,2) 3,6 0,99 (0,89–1,10) < 0,001
Sekundárny výsledokKardiovaskulárne úmrtie 380 (5,2) 1,7 366 (5,0) 1,7 1,03 (0,89-1,19) 0,711
Infarkt myokardu (fatálny
a nefatálny) 300 (4,1) 1,4 316 (4,3) 1,5 0,95 (0,81–1,11) 0,487
Cievna mozgová príhoda
(fatálna a nefatálna) 178 (2,4) 0,8 183 (2,5) 0,9 0,97 (0,79–1,19) 0,760
Hospitalizácia z dôvodu
nestabilnej angíny pectoris 116 (1,6) 0,5 129 (1,8) 0,6 0,90 (0,70–1,16) 0,419
Úmrtie z akejkoľvek príčiny 547 (7,5) 2,5 537 (7,3) 2,5 1,01 (0,90–1,14) 0,875
Hospitalizácia z dôvodu
zlyhania srdca‡ 228 (3,1) 1,1 229 (3,1) 1,1 1,00 (0,83–1,20) 0,983
*Výskyt na 100 pacientorokov je vypočítaný ako 100 x (celkový počet pacientov s ≥ 1 udalosťou počas hodnoteného obdobia expozície na celkový počet pacientorokov počas obdobia sledovania).
†Na základe Coxovho modelu stratifikovaného podľa regiónu. Pre zložené cieľové ukazovatele hodnoty p zodpovedajú testu
neinferiority so snahou dokázať, že pomer rizika je menší ako 1,3. Pre všetky ostatné cieľové ukazovatele hodnoty p zodpovedajú testu rozdielov v pomere rizika.
‡Analýza hospitalizácie z dôvodu zlyhania srdca bola upravená na základe východiskovej anamnézy zlyhania srdca.
PediatrickápopuláciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií Steglujanu vo všetkých podskupinách pediatrickej populácie v liečbe diabetes mellitus 2. typu (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiSteglujanPreukázalo sa, že Steglujan je bioekvivalentný súbežnému podaniu zodpovedajúcich dávok tabliet ertugliflozínu a sitagliptínu.
Pri podaní Steglujanu sú účinky jedla s vysokým obsahom tuku na farmakokinetiku ertugliflozínu
a sitagliptínu porovnateľné s účinkami pozorovanými pri podaní oddelene v jednotlivých tabletách.
Podanie Steglujanu s jedlom znížilo Cmax ertugliflozínu o 29 % a nemalo významný vplyv na AUCinf
ertugliflozínu alebo na AUCinf a Cmax sitagliptínu.
Ertugliflozín
Všeobecnýúvod
U zdravých osôb a pacientov s diabetom 2. typu je farmakokinetika ertugliflozínu podobná. Priemerná plazmatická AUC v rovnovážnom stave bola 398 ng∙hod/ml a Cmax v rovnovážnom stave 81 ng/ml pri liečbe ertugliflozínom 5 mg jedenkrát denne a 1,193 ng∙hod/ml a 268 ng/ml pri liečbe ertugliflozínom
15 mg jedenkrát denne. Rovnovážny stav sa dosiahol po 4 až 6 dňoch podávania ertugliflozínu jedenkrát denne. Ertugliflozín nevykazoval časovo závislú farmakokinetiku a pri viacnásobnom dávkovaní sa hromadil v plazme až do 10–40 %.
Absorpcia
Po perorálnom podaní jednorazovej dávky ertugliflozínu 5 mg a 15 mg, sa maximálna plazmatická koncentrácia (medián Tmax) ertugliflozínu objavila 1 hodinu po podaní dávky nalačno. Plazmatická Cmax a AUC ertugliflozínu sa zvyšuje dávkovo úmerným spôsobom po jednorazových dávkach od
0,5 mg do 300 mg a po viacnásobných dávkach od 1 mg do 100 mg. Absolútna perorálna biologická
dostupnosť ertugliflozínu po podaní 15 mg dávky je približne 100 %.
Podanie ertugliflozínu s jedlom s vysokým obsahom tukov a kalórií znižuje Cmax ertugliflozínu o 29 % a predlžuje Tmax o 1 hodinu, ale nespôsobuje zmenu AUC v porovnaní so stavom nalačno. Pozorovaný vplyv jedla na farmakokinetiku ertugliflozínu sa nepovažuje za klinicky významný a ertugliflozín sa môže podávať s jedlom alebo bez jedla. V klinických skúšaniach fázy 3 sa ertugliflozín podával bez ohľadu na jedlo.
Ertugliflozín je substrátom transportérov P-glykoproteínu (P-gp) a proteínu rezistencie rakoviny prsníka (breast cancer resistance protein, BCRP).
Distribúcia
Priemerný distribučný objem ertugliflozínu v rovnovážnom stave je po intravenóznej dávke 86 l. Väzba ertugliflozínu na plazmatické bielkoviny je 93,6 % a nie je závislá od plazmatických koncentrácií ertugliflozínu. Väzba na plazmatické bielkoviny nie je významne zmenená u pacientov
s poruchou funkcie obličiek alebo pečene. Pomer koncentrácie ertugliflozínu v krvi a plazme je 0,66.
Ertugliflozín nie je substrátom transportérov organických aniónov (OAT1, OAT3), organických katiónov (OCT1, OCT2) ani transportných polypeptidov pre organické anióny (OATP1B1, OATP1B3) in vitro.
Biotransformácia
Metabolizmus je primárnym mechanizmom odbúravania ertugliflozínu. Hlavnou metabolickou cestou pre ertugliflozín je O-glukuronidácia sprostredkovaná UGT1A9 a UGT2B7 na dva glukuronidy, ktoré sú pri klinicky významných koncentráciách farmakologicky neúčinné. Metabolizmus ertugliflozínu sprostredkovaný CYP (oxidatívny) je minimálny (12 %).
Eliminácia
Priemerný systémový plazmatický klírens po intravenóznej dávke 100 µg bol 11 l/hod. Priemerný polčas eliminácie u pacientov s diabetom 2. typu s normálnou funkciou obličiek bol odhadnutý na 17
hodín na základe analýzy populačnej farmakokinetiky. Po podaní perorálneho roztoku
[14C]-ertugliflozínu zdravým osobám sa približne 41 % rádioaktivity súvisiacej s liekom vylúčilo stolicou a 50 % močom. Len 1,5 % podanej dávky sa vylúčilo vo forme nezmeneného ertugliflozínu
močom a 34 % vo forme nezmeneného ertugliflozínu stolicou, čo je pravdepodobne v dôsledku
vylučovania glukuronidových metabolitov žlčou a následnej hydrolýzy na materskú zlúčeninu.
Osobitné skupinypacientov
Porucha funkcie obličiek
V klinickej farmakologickej štúdii fázy 1 u pacientov s diabetom 2. typu a miernou, stredne závažnou alebo závažnou poruchou funkcie obličiek (na základe stanovenia eGFR) boli po podaní jednorazovej dávky 15 mg ertugliflozínu priemerné zvýšenia AUC ertugliflozínu ≤ 1,7-násobné v porovnaní
s osobami s normálnou funkciou obličiek. Tieto zvýšenia AUC ertugliflozínu sa nepovažujú za klinicky významné. Medzi rôznymi skupinami funkcie obličiek neboli žiadne klinicky významné
rozdiely v hodnotách Cmax ertugliflozínu. 24-hodinové vylučovanie glukózy močom klesalo so zvyšujúcou sa závažnosťou poruchy funkcie obličiek (pozri časť 4.4). Väzba ertugliflozínu na plazmatické bielkoviny nebola ovplyvnená u pacientov s poruchou funkcie obličiek.
Porucha funkcie pečene
Stredne závažná porucha funkcie pečene (na základe klasifikácie podľa Childa-Pugha) neviedla
k zvýšeniu expozície ertugliflozínu. AUC ertugliflozínu sa znížila približne o 13 % a Cmax sa znížila približne o 21 % v porovnaní s osobami s normálnou funkciou pečene. Tento pokles v expozícii ertugliflozínu sa nepovažuje za klinicky významný. Neexistuje žiadna klinická skúsenosť u pacientov s poruchou funkcie pečene triedy C (závažná) podľa Childa-Pugha. Väzba ertugliflozínu na
plazmatické bielkoviny nebola ovplyvnená u pacientov so stredne závažnou poruchou funkcie pečene.
Pediatrická populácia
U pediatrických pacientov sa nevykonali žiadne štúdie s ertugliflozínom.
Vplyvy veku, telesnej hmotnosti, pohlavia a rasy
Na základe analýzy populačnej farmakokinetiky nemá vek, telesná hmotnosť, pohlavie ani rasa klinicky významný účinok na farmakokinetiku ertugliflozínu.
Sitagliptín
Absorpcia
Po perorálnom podaní 100-mg dávky zdravým osobám sa sitagliptín rýchlo absorboval, pričom
k maximálnym plazmatickým koncentráciám (medián Tmax) došlo 1 až 4 hodiny po podaní dávky. Priemerná plazmatická AUC sitagliptínu bola 8,52 mM•h, Cmax bola 950 nM. Absolútna biologická dostupnosť sitagliptínu je približne 87 %. Vzhľadom na to, že súbežné podanie sitagliptínu s jedlom obsahujúcim vysoké množstvo tukov nemalo žiadny vplyv na farmakokinetiku, môže sa Steglujan podávať s jedlom alebo bez jedla.
Plazmatická AUC sitagliptínu stúpala úmerne dávke. Dávková proporcionalita sa nestanovila pre Cmax
a C24h (Cmax sa zvýšila viac než úmerne dávke a C24h sa zvýšila menej než úmerne dávke).
Distribúcia
Priemerný distribučný objem v rovnovážnom stave po podaní jednorazovej 100-mg intravenóznej dávky sitagliptínu zdravým osobám je približne 198 litrov. Frakcia sitagliptínu reverzibilne viazaná
na plazmatické bielkoviny je nízka (38 %).
Biotransformácia
Sitagliptín sa primárne eliminuje nezmenený v moči a metabolizmus je menej dôležitá cesta. Približne
79 % sitagliptínu sa vylúči nezmenených v moči.
Po perorálnej dávke [14C] sitagliptínu sa približne 16 % rádioaktivity vylúčilo vo forme metabolitov sitagliptínu. Šesť metabolitov bolo zistených v stopových množstvách a nepredpokladá sa, že by prispievali k plazmatickej DPP-4 inhibičnej aktivite sitagliptínu. Štúdie in vitro ukazujú, že hlavný enzým zodpovedný za limitovaný metabolizmus sitagliptínu bol CYP3A4, s prispením CYP2C8.
Údaje in vitro preukázali, že sitagliptín nie je inhibítorom CYP izoenzýmov CYP3A4, 2C8, 2C9, 2D6,
1A2, 2C19 alebo 2B6 a nie je induktorom CYP3A4 a CYP1A2.
Eliminácia
Po podaní perorálnej dávky [14C]-sitagliptínu zdravým osobám sa približne 100 % podanej rádioaktivity eliminovalo v stolici (13 %) alebo v moči (87 %) počas jedného týždňa po podaní dávky.
Zjavný terminálny t1/2 po podaní 100-mg perorálnej dávky sitagliptínu bol približne 12,4 hodín. Sitagliptín sa iba minimálne kumuluje pri viacnásobných dávkach. Renálny klírens bol približne
350 ml/min.
Eliminácia sitagliptínu sa uskutočňuje primárne prostredníctvom renálneho vylučovania a zahŕňa aktívnu tubulárnu sekréciu. Sitagliptín je substrátom pre ľudský organický aniónový transportér-3 (hOAT-3), ktorý sa môže zúčastňovať na renálnej eliminácii sitagliptínu. Klinický význam hOAT-3 pri transporte sitagliptínu nebol stanovený. Sitagliptín je tiež substrátom P-gp, ktorý sa tiež môže zúčastňovať na sprostredkovaní renálnej eliminácie sitagliptínu. Cyklosporín, inhibítor P-gp, však neznižuje renálny klírens sitagliptínu. Sitagliptín nie je substrátom transportérov OCT2, OAT1 alebo PEPT1/2. In vitro sitagliptín v terapeuticky relevantných plazmatických koncentráciách neinhiboval transport sprostredkovaný OAT3 (IC50=160 μM) alebo P-gp (až do 250 μM). V klinickej štúdii mal sitagliptín malý účinok na plazmatické koncentrácie digoxínu, čo naznačuje, že sitagliptín môže byť miernym inhibítorom P-gp.
Liekovéinterakcie
So Steglujanom a inými liekmi sa nevykonali žiadne štúdie liekových interakcií, takéto štúdie sa však uskutočnili s jednotlivými liečivami.
Hodnotenieertugliflozínuinvitro
V štúdiách in vitro ertugliflozín a glukuronidy ertugliflozínu neinhibovali ani neinaktivovali CYP
1A2, 2C9, 2C19, 2C8, 2B6, 2D6 alebo 3A4 a neindukovali CYP 1A2, 2B6 alebo 3A4. Ertugliflozín a glukuronidy ertugliflozínu neinhibovali aktivitu UGT 1A6, 1A9 ani 2B7 in vitro. Ertugliflozín bol
vo vyšších koncentráciách, ktoré neboli klinicky významné, slabým inhibítorom UGT 1A1 a 1A4 in
vitro. Glukuronidy ertugliflozínu nemali na tieto izoformy žiadny účinok. Vo všeobecnosti je nepravdepodobné, že ertugliflozín ovplyvní farmakokinetiku súbežne podávaných liečiv eliminovaných týmito enzýmami.
Ertugliflozín ani glukuronidy ertugliflozínu významne neinhibujú transportéry P-gp, OCT2, OAT1 alebo OAT3 ani transportné polypeptidy OATP1B1 a OATP1B3 pri klinicky významných koncentráciách in vitro. Vo všeobecnosti je nepravdepodobné, že ertugliflozín ovplyvní farmakokinetiku súbežne podávaných liečiv, ktoré sú substrátmi týchto transportérov.
Hodnoteniesitagliptínuinvitro
Údaje in vitro naznačujú, že sitagliptín neinhibuje ani neindukuje izoenzýmy CYP450. V klinických štúdiách sitagliptín významne nezmenil farmakokinetiku metformínu, glyburidu, simvastatínu, rosiglitazónu, warfarínu alebo perorálnych kontraceptív a poskytol in vivo dôkaz o slabej tendencii
k spôsobeniu interakcií so substrátmi CYP3A4, CYP2C8, CYP2C9 a OCT. Sitagliptín môže byť miernym inhibítorom P-gp in vivo.
Transportné štúdie in vitro preukázali, že sitagliptín je substrátom pre P-gp a OAT3. Transport sitagliptínu sprostredkovaný OAT3 bol in vitro inhibovaný probenecidom, hoci riziko klinicky významných interakcií sa považuje za nízke. Súbežné podanie inhibítorov OAT3 sa in vivo nehodnotilo.
Charakteristikaupacientov
Farmakokinetika sitagliptínu bola vo všeobecnosti podobná u zdravých osôb a u pacientov s diabetom
2. typu.
Porucha funkcie obličiek
U pacientov s normálnou funkciou obličiek má metabolizmus, vrátane metabolizmu cez CYP3A4, iba malú úlohu pri klírense sitagliptínu. Metabolizmus môže zohrávať významnejšiu úlohu pri eliminácii
sitagliptínu pri ťažkej poruche funkcie obličiek alebo pri terminálnom štádiu ochorenia obličiek
(ESRD).
V porovnaní s bežnými zdravými kontrolnými osobami bola plazmatická AUC sitagliptínu zvýšená iba mierne u pacientov s GFR ≥ 45 až < 90 ml/min. Vzhľadom na to, že zvýšenia tohto rozsahu nie sú klinicky významné, nie je u týchto pacientov potrebná úprava dávkovania.
Porucha funkcie pečene
U pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre ≤ 9)
nie je potrebná úprava dávky. U pacientov s ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre > 9) nie je žiadna klinická skúsenosť. Vzhľadom na to, že sa sitagliptín vylučuje primárne
obličkami, nepredpokladá sa, že by ťažká porucha funkcie pečene mala vplyv na farmakokinetiku
sitagliptínu.
Starší pacienti
Nie je potrebná úprava dávky v závislosti od veku. Vychádzajúc z farmakokinetickej analýzy údajov populácie 1. a 2. fázy vek nemal klinicky významný vplyv na farmakokinetiku sitagliptínu. Staršie osoby (65 až 80 rokov) mali približne o 19 % vyššie plazmatické koncentrácie sitagliptínu v porovnaní s mladšími osobami.
Pediatrická populácia
Neuskutočnili sa žiadne štúdie so sitagliptínom u pediatrických pacientov.
Charakteristika iných pacientov
Nie je potrebná úprava dávky v závislosti od pohlavia, rasy alebo indexu telesnej hmotnosti (body mass index, BMI). Tieto charakteristiky nemali klinicky významný vplyv na farmakokinetiku
sitagliptínu vychádzajúc z kompozitnej analýzy farmakokinetických údajov 1. fázy a z analýzy
farmakokinetických údajov populácie 1. a 2. fázy.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, akútnej toxicity, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
Ertugliflozín
Všeobecnátoxicita
Štúdie toxicity po opakovanom perorálnom podávaní sa vykonali u myší až do 13 týždňov, u potkanov až do 26 týždňov a u psov až do 39 týždňov. Prejavy toxicity, ktoré sa považovali za nežiaduce boli vo všeobecnosti pozorované pri expozíciách vyšších alebo rovných 77-násobku neviazanej expozície (AUC) u ľudí pri maximálnej odporúčanej dávke u ľudí (maximum recommended human dose, MRHD) 15 mg/deň. Väčšina toxicity bola konzistentná s farmakológiou súvisiacou s vylučovaním glukózy močom a zahŕňala zníženie telesnej hmotnosti a telesného tuku, zvýšenie konzumácie jedla, hnačku, dehydratáciu, zníženie hladiny glukózy v sére a zvýšenie hladín ostatných parametrov v sére odzrkadľujúcich zvýšený metabolizmus bielkovín, glukoneogenézu a nerovnováhu elektrolytov
a zmeny moču ako je napr. polyúria, glukozúria a kalciúria. Mikroskopické zmeny súvisiace
s glukozúriou a/alebo kalciúriou pozorované len u hlodavcov zahŕňali dilatáciu renálnych tubulov, hypertrofiu zona glomerulosa v nadobličkách (potkany) a zvýšenie trabekulárnej kosti (potkany).
U psov sa okrem vracania neobjavili žiadne nálezy nežiaducej toxicity pri 379-násobku neviazanej
expozície (AUC) u ľudí pri MRHD 15 mg/deň.
Karcinogenéza
V 2-ročnej štúdii karcinogenity u myší sa ertugliflozín podával perorálnou sondou do žalúdka
v dávkach 5, 15 a 40 mg/kg/deň. Neobjavili sa žiadne neoplastické nálezy súvisiace s ertugliflozínom pri dávkach až do 40 mg/kg/deň (približne 41-násobok neviazanej expozície u ľudí pri MRHD
15 mg/deň na základe AUC). V 2-ročnej štúdii karcinogenity u potkanov sa ertugliflozín podával
perorálnou sondou do žalúdka v dávkach 1,5, 5 a 15 mg/kg/deň. Neoplastické nálezy súvisiace
s ertugliflozínom zahŕňali zvýšený výskyt benígneho adrenálneho medulárneho feochromocytómu
u samcov potkanov pri 15 mg/kg/deň. Tento nález sa pripisoval malabsorpcii uhľohydrátov vedúcej k zmenenej homeostáze vápnika a nepovažoval sa za významný pre riziko u ľudí. Hladina bez pozorovaného účinku (no-observed-effect level, NOEL) pre neopláziu bola 5 mg/kg/deň (približne
16-násobok neviazanej expozície u ľudí pri MRHD 15 mg/deň).
Mutagenéza
Ertugliflozín nebol mutagénny ani klastogénny s metabolickou aktiváciou alebo bez nej v teste mikrobiálnej reverznej mutácie, cytogenetických testoch in vitro (ľudské lymfocyty) a testoch
mikrojadier in vivo u potkanov.
Reprodukčnátoxikológia
V štúdiách fertility a embryonálneho vývinu u potkanov sa samcom a samiciam potkanov podával ertugliflozín v dávke 5, 25 a 250 mg/kg/deň. Neobjavili sa žiadne účinky na fertilitu pri dávke
250 mg/kg/deň (približne 386-násobok neviazanej expozície u ľudí pri MRHD 15 mg/deň na základe
porovnaní AUC). Ertugliflozín nemal nežiaduci vplyv na výsledky vývinu u potkanov pri expozíciách u matiek zodpovedajúcich 239-násobku expozície u ľudí pri maximálnej klinickej dávke 15 mg/deň
a u králikov pri expozíciách u matiek zodpovedajúcich 1 069-násobku expozície u ľudí pri maximálnej
klinickej dávke 15 mg/deň, na základe AUC. Pri dávke toxickej pre matky u potkanov
(250 mg/kg/deň) sa pozorovala nižšia životaschopnosť plodu a vyšší výskyt viscerálnej malformácie pri expozícii u matky, ktorá bola 510-násobkom maximálnej klinickej dávky 15 mg/deň.
V štúdii pre- a postnatálneho vývinu sa pozoroval znížený postnatálny rast a vývin u potkanov, ktorým bol ertugliflozín podávaný od 6. dňa gravidity po 21. deň laktácie v dávke ≥ 100 mg/kg/deň (odhadovaný 239-násobok expozície u ľudí pri maximálnej klinickej dávke 15 mg/deň na základe AUC). Pohlavné dozrievanie bolo oneskorené u obidvoch pohlaví pri dávke 250 mg/kg/deň (odhadovaný 620-násobok MRHD pri dávke 15 mg/deň na základe AUC).
Ak sa ertugliflozín podával mláďatám potkana od 21. postnatálneho dňa (postnatal day, PND) do 90. PND, počas obdobia renálneho vývinu zodpovedajúceho neskorému druhému a tretiemu trimestru ľudskej gravidity, pozorovalo sa zvýšenie hmotnosti obličiek, dilatácia renálnej panvičky a tubulov
a mineralizácia renálnych tubulov pri expozícii 13-násobku maximálnej klinickej dávky 15 mg/deň na základe AUC. Účinky na kosť (kratšia dĺžka stehennej kosti, nárast trabekulárnej kosti v stehennej
kosti) ako aj účinky oneskoreného dospievania sa pozorovali pri expozícii 817-násobku MRHD
15 mg/deň na základe AUC. Účinky na obličky a kosť sa úplne nezvrátili po 1-mesačnom období zotavenia.
Sitagliptín
U hlodavcov sa pozorovala renálna a hepatálna toxicita pri systémových expozičných hodnotách
58-krát vyšších, ako je expozičná hladina u ľudí, pričom najvyššia neúčinná hladina bola zistená pri
19-násobku expozičnej hladiny u ľudí. Pri expozíciách 67-krát vyšších ako sú klinické expozičné hladiny, sa u potkanov pozorovali abnormality rezákov, pričom najvyššia neúčinná hladina pre tento nález bola 58-krát vyššia vychádzajúc zo 14-dňovej štúdie u potkanov. Význam týchto zistení pre ľudí nie je známy. Prechodné fyzické príznaky súvisiace s liečbou, z ktorých niektoré poukazovali na nervovú toxicitu, napr. dýchanie s otvorenými ústami, slinenie, vracanie bielej peny, ataxia, triaška, znížená aktivita a/alebo zhrbený postoj, sa pozorovali u psov pri expozičných hladinách približne
23-násobne vyšších, ako je klinická expozícia. Okrem toho sa histologicky zistila veľmi mierna až mierna degenerácia kostrových svalov pri dávkach vedúcich k systémovým expozíciám približne
23-násobne vyšším, ako je expozičná hladina u ľudí. Zistilo sa, že najvyššia neúčinná hladina pre tieto
nálezy je 6-násobok klinickej expozičnej hladiny.
V predklinických štúdiách sa nepreukázala genotoxicita sitagliptínu. Sitagliptín nebol karcinogénny
u myší. U potkanov došlo k zvýšenej incidencii hepatálnych adenómov a karcinómov pri systémových expozičných hladinách 58-násobne vyšších, ako je expozičná hladina u ľudí. Keďže sa zistilo, že hepatotoxicita koreluje s indukciou hepatálnej neoplázie u potkanov, táto zvýšená incidencia hepatálnych tumorov u potkanov bola pravdepodobne následkom chronickej hepatálnej toxicity pri
tejto vysokej dávke. Vzhľadom na vysoké bezpečnostné rozpätie (19-násobok pri najvyššej neúčinnej hladine) sa tieto neoplastické zmeny nepovažujú za významné v prípade ľudí.
Nepozorovali sa žiadne nežiaduce účinky na fertilitu samcov a samíc potkanov, ktorým bol sitagliptín podaný pred a počas párenia.
V pre-/postnatálnej vývojovej štúdii uskutočnenej na potkanoch sa nezistili žiadne nežiaduce účinky sitagliptínu.
Štúdie reprodukčnej toxicity preukázali v súvislosti s liečbou mierne zvýšenie výskytu fetálnych malformácií rebier (chýbajúce, hypoplastické a zvlnené rebrá) u potomkov potkanov vystavených systémovým expozičným hladinám vyšším, ako je 29-násobok expozičných hladín u ľudí. Tehotenská toxicita sa pozorovala u králikov pri viac ako 29-násobku expozičných hladín u ľudí. Vzhľadom na vysoké bezpečnostné rozpätie tieto zistenia nepoukazujú na významné riziko pre ľudskú reprodukciu. Sitagliptín sa do značnej miery vylučuje do mlieka dojčiacich potkanov (pomer mlieko/plazma: 4:1).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety
mikrokryštalická celulóza (E460)
bezvodý hydrogenfosforečnan vápenatý sodná soľ kroskarmelózy
stearylfumaran sodný (E487)
stearan horečnatý(E470b)
Obal tablety
hypromelóza (E464) hydroxypropylcelulóza (E463) oxid titaničitý (E171)
červený oxid železitý (E172) žltý oxid železitý (E172) čierny oxid železitý (E172) karnaubský vosk (E903)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Al/PVC/PA/Al blistre
Balenia po 14, 28, 30, 84, 90 a 98 filmom obalených tabliet v neperforovaných blistroch. Balenia po 30 x 1 filmom obalená tableta v perforovaných blistroch umožňujúcich oddelenie jednotlivej dávky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMerck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLASteglujan5mg/100 mgfilmomobalenétabletyEU/1/18/1266/001
EU/1/18/1266/002
EU/1/18/1266/003
EU/1/18/1266/004
EU/1/18/1266/005
EU/1/18/1266/006
EU/1/18/1266/013
Steglujan15mg/100 mgfilmomobalenétabletyEU/1/18/1266/007
EU/1/18/1266/008
EU/1/18/1266/009
EU/1/18/1266/010
EU/1/18/1266/011
EU/1/18/1266/012
EU/1/18/1266/014
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. marca 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.