
u QTc. Toto zahŕňa pacientov s hypokaliémiou alebo hypomagneziémiou, pacientov s vrodeným syndrómom dlhého QT, pacientov užívajúcich antiarytmiká alebo iné lieky, ktoré vedú k predĺženiu QT a pacientov liečených vysokými kumulatívnymi dávkami antracyklínu. Hypokaliémia alebo hypomagneziémia sa majú pred podaním dasatinibu upraviť.
Nežiaduce reakcie na srdce
Dasatinib sa skúmal v randomizovanej klinickej štúdií s 519 pacientmi s novo diagnostikovanou CML v chronickej fáze, ktorá zahŕňala pacientov s predošlými srdcovým ochoreniami. Nežiaduce reakcie na srdce z kongestívneho srdcového zlyhania/srdcovej dysfunkcie, perikardiálneho výpotku, arytmií, palpitácií, predĺženie QT intervalu a infarkt myokardu (vrátane úmrtia) sa hlásili u pacientov užívajúcich dasatinib. Nežiaduce srdcové udalosti boli častejšie u pacientov s rizikovými faktormi alebo so srdcovým ochorením v anamnéze. Pacienti s rizikovými faktormi (napr. hypertenzia, hyperlipidémia, diabetes) alebo srdcovým ochorením v anamnéze (napr. predošlá perkutánna koronárna intervencia, dokázané ochorenie koronárnych artérií) majú byť starostlivo sledovaní pre klinické prejavy alebo príznaky zhodné so srdcovou dysfunkciou, ako je bolesť na hrudníku, skrátené dýchanie a potenie.
Ak sa tieto klinické prejavy alebo príznaky vyvinú, lekárom sa odporúča prerušiť podávanie dasatinibu a zvážiť potrebu alternatívnej liečby špecifickej pre CML. Po vyhodnotení, má byť pred obnovením liečby dasatinibom vykonané funkčné posúdenie. Dasatinib môže byť znovu nasadený v pôvodnej dávke pri miernych/stredne závažných nežiaducich reakciách (≤ 2. stupeň) a môže pokračovať na úrovni redukovanej dávky pri závažných nežiaducich reakciách (≥ 3. stupeň) (pozri časť 4.2). Pacienti pokračujúci v liečbe, majú byť pravidelne monitorovaní.
Pacienti s nekontrolovaným alebo závažným kardiovaskulárnym ochorením neboli zaradení do klinických štúdií.
Reaktivácia hepatitídy B
Reaktivácia hepatitídy B u pacientov, ktorí sú chronickými prenášačmi tohto vírusu, sa vyskytla v prípade, že títo pacienti užívali inhibítory BCR-ABL-tyrozínkinázy. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie.
Pacienti majú byť vyšetrení na HBV infekciu pred začatím liečby liekom SPRYCEL. Pred začatím liečby u pacientov s pozitívnym sérologickým testom na hepatitídu B (vrátane pacientov s aktívnym ochorením) a u pacientov s pozitívnym testom na HBV infekciu počas liečby je potrebné konzultovať s odborníkmi na ochorenia pečene a liečbu hepatitídy B. Prenášači vírusu HBV, ktorí potrebujú liečbu liekom SPRYCEL, majú byť pozorne sledovaní na prejavy a príznaky aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby (pozri časť 4.8).
Účinky na rast a vývoj u pediatrických pacientov
V pediatrických klinických skúšaniach so SPRYCELOM s pediatrickými pacientmi rezistentnými/intolerantnými na imatinib a s predtým neliečenými pediatrickými pacientmi po minimálne 2 rokoch liečby sa hlásili nežiaduce udalosti súvisiace s liečbou spojené s rastom kostí a vývojom u 6 (4,6 %) pacientov, jedna z nich bola závažnej intenzity (spomalenie rastu 3. stupňa). Týchto 6 hlásení zahŕňalo hlásenia oneskorenej fúzie epifýz, osteopéniu, spomalenie rastu a gynekomastiu (pozri časť 5.1). Tieto výsledky je ťažké interpretovať v kontexte chronických ochorení, ako je CML a je potrebné dlhodobé následné sledovanie.
Laktóza
Tento liek obsahuje laktózu, monohydrát Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Liečivá,ktorémôžuzvýšiťplazmatickékoncentráciedasatinibu
Štúdie in vitro poukazujú na to, že dasatinib je substrátom CYP3A4. Súbežné užívanie dasatinibu a liekov alebo látok, ktoré silne inhibujú CYP3A4 (napr. ketokonazol, itrakonazol, erytromycín, klaritromycín, ritonavir, telitromycín, grapefruitový džús) môže zvýšiť expozíciu dasatinibu. Preto sa u pacientov liečených dasatinibom neodporúča systémové podávanie silného inhibítora CYP3A4.
V klinicky relevantných koncentráciách sa v priemere 96 % dasatinibu viaže na plazmatické proteíny na základe in vitro experimentov. Neboli vykonané štúdie hodnotiace interakcie dasatinibu s inými liekmi viažucimi sa na proteíny. Potenciál pre zámenu a jej klinická dôležitosť nie je známa.
Liečivá,ktorémôžuznížiťplazmatickékoncentráciedasatinibu
Keď sa dasatinib podával počas 8 dní večer so 600 mg rifampicínu, silným induktorom CYP3A4, hodnota AUC dasatinibu sa znížila o 82 %. Iné lieky, ktoré indukujú aktivitu CYP3A4 (napr. dexametazón, fenytoín, karbamazepín, fenobarbital alebo rastlinné lieky obsahujúce Hypericum perforatum, známy aj ako Ľubovník bodkovaný), môžu tiež zvýšiť metabolizmus a znížiť plazmatické koncentrácie dasatinibu. Z tohto dôvodu sa neodporúča súbežné užívanie silných induktorov CYP3A4 s dasatinibom. U pacientov, u ktorých je indikovaný rifampicín alebo iné induktory CYP3A4, sa majú použiť alternatívne lieky s menším potenciálom pre enzýmovú indukciu.
Antagonisty H2-histamínového receptora a inhibítory protónovej pumpy
Dlhodobá supresia sekrécie žalúdočnej kyseliny spôsobená H2-antagonistami alebo inhibítormi protónovej pumpy (napr. famotidínom a omeprazolom) pravdepodobne zníži expozíciu dasatinibu. V štúdii jednorazovej dávky u zdravých jedincov podanie famotidínu 10 hodín pred jednorazovou
dávkou SPRYCELU znížilo expozíciu dasatinibu o 61 %. V štúdii so14 zdravými dobrovoľníkmi pri podaní jednorazovej 100 mg dávky SPRYCELU 22 hodín počas štvrtého dňa, 40 mg dávka omeprazolu znížila v rovnovážnom stave hodnotu AUC dasatinibu o 43 % a hodnotu Cmax dasatinibu o
42 %. U pacientov liečených SPRYCELOM sa má namiesto H2-antagonistov alebo inhibítorov protónovej pumpy zvážiť použitie antacíd (pozri časť 4.4).
Antacidá
Predklinické údaje potvrdzujú, že rozpustnosť dasatinibu závisí od pH. U zdravých jedincov súbežné užívanie antacíd obsahujúcich hydroxid hlinitý/hydroxid horečnatý so SPRYCELOM znížilo hodnotu AUC jednorazovej dávky SPRYCELU o 55 % a Cmax o 58 %. Keď však boli antacidá podané 2 hodiny pred jednorazovou dávkou SPRYCELU, nepozorovali sa žiadne významné zmeny v koncentrácii ani expozícii dasatinibu. Z tohto dôvodu sa antacidá môžu podať 2 hodín pred alebo 2 hodiny po podaní SPRYCELU (pozri časť 4.4).
Liečivá,ktorýchplazmatickékoncentráciemôžedasatinibovplyvniť
Súbežné užívanie dasatinibu a substrátu CYP3A4 môže zvýšiť expozíciu substrátu CYP3A4. V štúdii so zdravými jedincami jednorazová 100 mg dávka dasatinibu zvýšila hodnotu AUC simvastatínu, známeho substrátu CYP3A4, o 20 % a hodnotu Cmax simvastatínu o 37 %. Nie je možné vylúčiť, že účinok je väčší po viacnásobných dávkach dasatinibu. Z tohto dôvodu sa substráty CYP3A4, o ktorých je známe, že majú úzku terapeutickú šírku (napr. astemizol, terfenadín, cisaprid, pimozid, chinidín, bepridil alebo námeľové alkaloidy [ergotamín, dihydroergotamín]), majú podávať opatrne u pacientov liečených dasatinibom (pozri časť 4.4).
In vitro údaje indikujú potenciálne riziko interakcie s CYP2C8 substrátmi ako sú glitazóny.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepciaumužovažien
Sexuálne aktívni muži aj ženymajú počas liečby používať účinnú metódu antikoncepcie.
Gravidita
Na základe skúseností u ľudí sa predpokladá, že dasatinib spôsobuje vrodené malformácie vrátane defektov neurálnej trubice a má škodlivé farmakologické účinky na plod, ak sa podáva počas gravidity. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
SPRYCEL sa nemá používať počas gravidity, ak si klinický stav ženy nevyžaduje liečbu dasatinibom.
Ak sa SPRYCEL použije počas gravidity, pacientka musí byť informovaná o možnom riziku pre plod.
Dojčenie
Existujú nedostatočné/obmedzené informácie o vylučovaní dasatinibu do materského mlieka u ľudí alebo zvierat. Fyzikálno-chemické a dostupné farmakodynamické/toxikologické údaje o dasatinibe poukazujú na vylučovanie do materského mlieka a nie je možné vylúčiť riziko pre dojčené dieťa. Počas liečby SPRYCELOM sa musí dojčenie prerušiť.
Fertilita
V štúdiách na zvieratách nebola fertilita samcov a samíc potkanov ovplyvnená liečbou dasatinibom (pozri časť 5.3). Lekári a iní zdravotnícki pracovníci majú prekonzultovať s mužmi primeraného veku možné účinky SPRYCELU na fertilitu a táto konzultácia môže zahŕňať zváženie konzervácie spermií.
 4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
SPRYCEL má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť informovaní, že počas liečby dasatinibom môžu mať nežiaduce reakcie, ako je závrat alebo rozmazané videnie. Z tohto dôvodu sa im pri vedení vozidla alebo obsluhe strojov má odporučiť opatrnosť.
4.8 Nežiaduce účinkyPrehľadbezpečnostnéhoprofiluÚdaje opísané nižšie vyjadrujú expozíciu SPRYCELU pri všetkých dávkach testovaných v klinických štúdiách (N=2 900) zahŕňajúcich 324 dospelých pacientov s novo diagnostikovanou chronickou fázou CML, 2 388 dospelých pacientov rezistentných alebo intolerantných na imatinib s chronickou alebo s pokročilou fázou CML alebo Ph+ ALL a 188 pediatrických pacientov.
U 2 712 dospelých pacientov buď s chronickou fázou CML, pokročilou fázou CML alebo Ph+ ALL bol medián trvania liečby 19,2 mesiaca (rozsah 0 až 93,2 mesiaca). V randomizovanom klinickom skúšaní s pacientmi s novo diagnostikovanou chronickou fázou CML bol medián trvania liečby približne 60 mesiacov. Medián trvania liečby u 1 618 dospelých pacientov s chronickou fázou CML bol 20 mesiacov (rozsah 0 až 92,9 mesiaca). Medián trvania liečby u 1 094 dospelých pacientov
s pokročilou fázou CML alebo Ph+ ALL bol 6,2 mesiaca (rozsah 0,1 až 99,6 mesiaca). U
188 pacientov v pediatrických štúdiách bol medián trvania liečby 26,3 mesiaca (rozsah 0 až
99,6 mesiaca). V podskupine 130 pediatrických pacientov s chronickou fázou CML liečených
SPRYCELOM bol medián trvania liečby 42,3 mesiaca (rozsah 0,1 až 99,6 mesiaca).
Väčšina pacientov liečených SPRYCELOM mala v určitom čase nežiaduce reakcie. V celkovej populácii 2 712 dospelých jedincov liečených SPRYCELOM malo 520 (19 %) nežiaduce reakcie, ktoré mali za následok prerušenie liečby.
Celkový bezpečnostný profil SPRYCELU v pediatrickej populácii bol podobný tomu, ktorý je v dospelej populácii bez ohľadu na liekovú formu s výnimkou toho, že v pediatrickej populácii sa nehlásil perikardiány výpotok, pleurálny výpotok, pľúcny edém alebo pľúcna hypertenzia. Zo
130 pediatrických jedincov s CML-CP liečených SPRYCELOM sa u 2 (1,5 %) vyskytli nežiaduce reakcie, ktoré mali za následok ukončenie liečby.
TabuľkovýzoznamnežiaducichreakciíNasledovné nežiaduce reakcie, s výnimkou laboratórnych anomálií, sa hlásili u pacientov v klinických štúdiách so SPRYCELOM a po uvedení lieku na trh (Tabuľka 5). Tieto reakcie sú vymenované podľa tried orgánových systémov a podľa frekvencie. Frekvencie sú definované nasledovne: veľmi časté
(≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); neznáme (nie je možné stanoviť z dostupných údajov po uvedení lieku na trh).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 5: Súhrn nežiaducich reakcií zostavených do tabuľkyInfekcie a nákazyVeľmi časté infekcia (vrátane bakteriálnej, vírusovej, plesňovej, nešpecifickej)
Časté pneumónia (vrátane bakteriálnej, vírusovej a plesňovej), infekcia/zápal horných dýchacích ciest, herpetická vírusová infekcia, enterokolitída, sepsa (vrátane menej častých prípadov s fatálnymi následkami)
Neznáme reaktivácia hepatitídy B
Poruchy krvi a lymfatického systémuVeľmi časté myelosupresia (vrátane anémie, neutropénie, trombocytopénie)
Časté febrilná neutropénia
Menej časté lymfadenopatia, lymfopénia
Zriedkavé aplázia červených krviniek
 Poruchy imunitného systému
M
enej časté
Poruchy imunitného systému
M
enej časté hypersenzitivita (vrátane erythema nodosum) Zriedkavé anafylaktický šok
Poruchy endokrinného systémuMenej časté hypotyreoidizmus
Zriedkavé hypertyreoidizmus, tyreoiditída
Poruchy metabolizmu a výživyČasté poruchy chuti do jedlaa, hyperurikémia
Menej časté syndróm z rozpadu nádoru, dehydratácia, hypoalbuminémia, hypercholesterolémia
Zriedkavé diabetes mellitus
Psychické poruchyČasté depresia, insomnia
Menej časté úzkosť, stav zmätenosti, ovplyvnenie lability, zníženie libida
Poruchy nervového systémuVeľmi časté bolesť hlavy
Časté neuropatia (vrátane periférnej neuropatie), závrat, porucha chuti, spavosť
Menej časté krvácanie do CNS*b, synkopa, tremor, amnézia, porucha rovnováhy
Zriedkavé cerebrovaskulárna príhoda, prechodný ischemický záchvat, kŕč, očná neuritída, paralýza VII. nervu, demencia, ataxia
Poruchy okaČasté poruchy videnia (vrátane zrakovej poruchy, rozmazaného videnia a zníženej zrakovej ostrosti), suchosť očí
Menej časté porucha zraku, konjunktivitída, fotofóbia, zvýšené slzenie
Poruchy ucha a labyrintuČasté tinnitus
Menej časté strata sluchu, vertigo
Poruchy srdca a srdcovej činnostiČasté kongestívne srdcové zlyhanie/srdcová dysfunkcia*c, perikardiálny výpotok*, arytmia
(vrátane tachykardie), palpitácie
Menej časté infarkt myokardu (vrátane fatálneho následku)*, QT predĺženie na elektrokardiograme*, perikarditída, ventrikulárna arytmia (vrátane ventrikulárnej tachykardie), angina pectoris, kardiomegália, nezvyčajná T vlna na elektrokardiograme, zvýšený troponín
Zriedkavé cor pulmonale, myokarditída, akútny koronárny syndróm, zastavenie srdca, predĺženie PR na elektrokardiograme, koronárne arteriálne ochorenie, pleuroperikarditída
Neznáme fibrilácia predsiení/ flutter predsiení
Poruchy cievVeľmi časté hemorágia*d
Časté hypertenzia, začervenanie
Menej časté hypotenzia, tromboflebitída, trombóza
Zriedkavé hlboká žilová trombóza, embólia, livedo reticularis
Poruchy dýchacej sústavy, hrudníka a mediastínaVeľmi časté pleurálny výpotok*, dyspnoe
Časté pľúcny edém*, pľúcna hypertenzia*, pľúcna infiltrácia, pneumonitída, kašeľ
Menej časté pľúcna arteriálna hypertenzia, bronchospazmus, astma
Zriedkavé pľúcna embólia, syndróm akútnej respiračnej tiesne
Neznáme intersticiálne ochorenie pľúc
Poruchy gastrointestinálneho traktuVeľmi časté hnačka, vracanie, nauzea, abdominálna bolesť
Časté gastrointestinálne krvácanie*, kolitída (vrátane neutropenickej kolitídy), gastritída, zápal sliznice (vrátane mukozitídy/stomatitídy), dyspepsia, abdominálna distenzia, zápcha, porucha mäkkého tkaniva v ústach
Menej časté pankreatitída (vrátane akútnej pankreatitídy), horný gastrointestinálny vred, ezofagitída, ascites*, análna fisúra, dysfágia, gastroezofageálne refluxné ochorenie
 Zriedkavé
Zriedkavé gastroenteropatia zo straty proteínov, ileus, análna fistula
Neznáme fatálna gastrointestinálna hemorágia*
Poruchy pečene a žlčových ciestMenej časté hepatitída, cholecystitída, cholestáza
Poruchy kože a podkožného tkanivaVeľmi časté kožná vyrážkae
Časté alopécia, dermatitída (vrátane ekzému), pruritus, akné, suchá koža, urtikária, hyperhidróza
Menej časté neutrofilná dermatóza, fotosenzitivita, porucha pigmentácie, panikulitída, kožný vred, bulózne ochorenia, porucha nechtov, syndróm palmárno-plantárnej erytrodyzestézie, ochorenie vlasov
Zriedkavé leukocytoklastická vaskulitída, fibróza kože
Neznáme Stevensov-Johnsonov syndrómf
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaVeľmi časté muskuloskeletálna bolesť
Časté artralgia, myalgia, svalová slabosť, muskuloskeletálna stuhnutosť, svalový kŕč
Menej časté rabdomyolýza, osteonekróza, svalový zápal, tendonitída, artritída
Zriedkavé oneskorená fúzia epifýz,g spomalenie rastug
Poruchy obličiek a močových ciestMenej časté poruchy funkcie obličiek (vrátane renálneho zlyhania), časté močenie, proteinúria
Neznáme nefrotický syndróm
Stavy v gravidite, v šestonedelí a perinatálnom obdobíZriedkavé potrat
Poruchy reprodukčného systému a prsníkovMenej časté gynekomastia, porucha menštruácie
Celkové poruchy a reakcie v mieste podaniaVeľmi časté periférny edémh, únava, pyrexia, edém tvárei
Časté asténia, bolesť, bolesť na hrudi, generalizovaný edém*j, zimnica
Menej časté pocit ťažoby, iný povrchový edém*k
Zriedkavé porucha chôdze
Laboratórne a funkčné vyšetreniaČasté zníženie telesnej hmotnosti, zvýšenie telesnej hmotnosti
Menej časté zvýšenie kreatínfosfokinázy v krvi, zvýšenie gama-glutamyltransferázy
Úrazy, otravy a komplikácie liečebného postupuČasté kontúzia
a Zahŕňa zníženú chuť do jedla, predčasnú sýtosť, zvýšenú chuť do jedla.
b Zahŕňa krvácanie do centrálneho nervového systému, cerebrálny hematóm, cerebrálne krvácanie, extradurálny hematóm, intrakraniálne krvácanie, hemoragickú cievnu mozgovú príhodu, subarachnoidálne krvácanie, subdurálny hematóm a
subdurálne krvácanie.
c Zahŕňa zvýšený mozgový natriuretický peptid, ventrikulárnu dysfunkciu, dysfunkciu ľavej komory, dysfunkciu pravej komory, srdcové zlyhanie, akútne srdcové zlyhanie, chronické srdcové zlyhanie, kongestívne srdcové zlyhanie, kardiomyopatiu, kongestívnu kardiomyopatiu, diastolickú dysfunkciu, zníženú ejekčnú frakciu a ventrikulárne zlyhanie,
zlyhanie ľavej komory, zlyhanie pravej komory a ventrikulárnu hypokinézu.
d Okrem gastrointestinálneho krvácania a krvácania do CNS; tieto nežiaduce reakcie sú hlásené pod triedou orgánového systému poruchy gastrointestinálneho traktu alebo pod triedou orgánového systému poruchy nervového systému.
e Zahŕňa liekovú erupciu, erytém, multiformný erytém, erytrózu, exfoliatívnu vyrážku, generalizovaný erytém, genitálnu
vyrážku, potničky, milie, miliariu, pustulárnu psoriázu, vyrážku, erytematóznu vyrážku, folikulárnu vyrážku, generalizovanú vyrážku, makulárnu vyrážku, makulo-papulárnu vyrážku, papulárnu vyrážku, pruritickú vyrážku, pustulárnu vyrážku, vezikulárnu vyrážku, kožnú exfoliáciu, kožné podráždenie, toxickú kožnú erupciu, vezikulóznu urtikáriu a vaskulitickú vyrážku.
f Po uvedení lieku na trh sa hlásili jednotlivé prípady Stevensovho-Johnsonovho syndrómu. Nebolo možné určiť, či tieto mukokutánne nežiaduce reakcie priamo súviseli so SPRYCELOM alebo so súbežne podávaným liekom.
g Frekvencie hlásené ako časté v pediatrických štúdiách.
h Gravitačný edém, lokalizovaný edém, periférny edém.
i Konjunktiválny edém, edém oka, opuch oka, edém očného viečka, edém tváre, edém pier, makulárny edém, edém úst, orbitálny edém, periorbitálny edém, opuch tváre.
j Preťaženie tekutinou, retencia tekutín, opuch gastrointestinálneho traktu, generalizovaný edém, periférny edém, edém, edém z dôvodu ochorenia srdca, perinefrický výpotok, edém po chirurgickom výkone, viscerálny edém.
k Opuch genitálií, edém v mieste vpichu, genitálny edém, edém pohlavného údu, opuch pohlavného údu, skrotálny edém, opuch kože, opuch semenníkov, opuchy vagíny a pošvy.
* Ďalšie podrobnosti, pozri časť "Opis vybraných nežiaducich reakcií"
Opisvybranýchnežiaducichreakcií
Myelosupresia
Liečba SPRYCELOM sa spája s anémiou, neutropéniou a trombocytopéniou. Ich výskyt je skorší a častejší u pacientov v pokročilej fáze CML alebo s Ph+ ALL ako u pacientov v chronickej fáze CML (pozri časť 4.4).
Krvácanie
U pacientov užívajúcich SPRYCEL sa hlásili nežiaduce reakcie krvácaní súvisiacich s liekom
v rozsahu od petechie a epistaxy po gastrointestinálne krvácanie 3. alebo 4. stupňa a krvácanie do CNS (pozri časť 4.4).
Retencia tekutín
Rôzne nežiaduce reakcie, ako je pleurálny výpotok, ascites, pľúcny edém a perikardiálny výpotok
s povrchovým edémom alebo bez neho, možno súhrnne opísať ako “retenciu tekutiny”. V štúdii s novo diagnostikovanou chronickou fázou CML po minimálne 60 mesiacoch následného sledovania zahŕňali nežiaduce reakcie retencie tekutiny súvisiacej s dasatinibom pleurálny výpotok (28 %). povrchový edém (14 %), pľúcnu hypertenziu (5 %) generalizovaný edém (4 %) a perikardiálny výpotok (4 %). Kongestívne srdcové zlyhanie/dysfunkcia srdca a pľúcny edém sa hlásili u < 2 % pacientov.
V priebehu času bol kumulatívny pomer pleurálneho výpotku súvisiaceho s dasatinibom (všetky stupne) 10 % po 12 mesiacoch, 14 % po 24 mesiacoch, 19 % po 36 mesiacoch, 24 % po 48 mesiacoch a 28 % po 60 mesiacoch. Celkovo 46 pacientov liečených dasatinibom malo rekurentný pleurálny výpotok. Sedemnásť pacientov malo 2 samostatné nežiaduce reakcie, 6 malo 3 nežiaduce reakcie,
18 malo 4 až 8 nežiaducich reakcií a 5 malo > 8 epizód pleurálneho výpotku.
Medián času do prvého pleurálneho výpotku súvisiaceho s dasatinibom 1. alebo 2. stupňa bol
114 týždňov (rozsah: 4 až299 týždňov). Menej než 10 % pacientov s pleurálnym výpotkom malo závažný (3. alebo 4. stupeň) pleurálnych výpotkov súvisiacich s dasatinibom. Medián času do prvého výskytu pleurálneho výpotku súvisiaceho s dasatinibom ≥ 3. stupňa bol 175 týždňov (rozsah: 114 až
274 týždňov). Medián trvania pleurálneho výpotku súvisiaceho s dasatinibom (všetky stupne) bol
283 dní (~40 týždňov).
Pleurálny výpotok bol zvyčajne vratný a bol zvládnutý prerušením liečby SPRYCELOM a použitím diuretík alebo iných vhodných podporných ošetrovacích opatrení (pozri časti 4.2 a 4.4). Medzi pacientmi liečenými dasatinibom s pleurálnym výpotkom súvisiacim s liekom (n=73) malo 45 (62 %) prerušenie liečby a 30 (41 %) malo zníženie dávky. Okrem toho 34 (47 %) dostalo diuretiká,
23 (32 %) dostalo kortikosteroidy a 20 (27 %) dostalo kortikosteroidy aj diuretiká. Deväť (12 %)
pacientov podstúpilo terapeutickú torakocentézu.
Šesť percent pacientov liečených dasatinibom prerušilo liečbu z dôvodu pleurálneho výpotku súvisiaceho s liekom.
Pleurálny výpotok nezhoršil schopnosť pacienta dosiahnuť odpoveď. Medzi pacientmi s pleurálnym výpotkom liečenými dasatinibom dosiahlo 96 % cCCyR, 82 % dosiahlo MMR a 50 % dosiahlo MR4.5 i napriek prerušeniam liečby alebo úprave dávky.
Pozri časť 4.4 ďalšie informácie o pacientoch s chronickou fázou CML a s pokročilou fázou CML
alebo Ph+ ALL.
Pľúcna arteriálna hypertenzia (PAH)
PAH (prekapilárna pľúcna arteriálna hypertenzia potvrdená pravostrannou katétrizáciou srdca) sa hlásila v súvislosti s expozíciou dasatinibom. V týchto prípadoch sa hlásila PAH po začatí liečby dasatinibom, a to po viac ako jednom roku liečby. Pacienti s PAH hlásili počas liečby dasatinibom časté užívanie súbežných liekov alebo mali komorbidity, okrem základného nádorového ochorenia. Zlepšenie hemodynamických a klinických parametrov sa pozorovalo u pacientov liečených dasatinibom s PAH po ukončení liečby dasatinibom.
Predĺženie QT intervalu
V štúdii fázy III s pacientmi s novo diagnostikovanou chronickou fázou CML, jeden pacient (< 1 %) z pacientov liečených SPRYCELOM mal QTcF > 500 ms po minimálne 12 mesiacoch následného
sledovania (pozri časť 4.4). Po minimálne 60 mesiacoch následného sledovania sa u žiadnych ďalších pacientov nehlásilo, že mali QTcF > 500 ms.
V 5 klinických štúdiách fázy II s pacientmi s rezistenciou alebo intoleranciou na predchádzajúcu
liečbu imatinibom, sa vykonalo opakované základné EKG a aj počas liečby vo vopred určených časových intervaloch a snímané centrálne pre 865 pacientov liečených SPRYCELOM 70 mg dvakrát denne. Interval QT bol korigovaný podľa srdcovej frekvencie pomocou Fridericiovej metódy. Vo všetkých časových intervaloch po dávke na 8. deň, boli priemerné zmeny oproti východiskovej hodnote intervalu QTcF 4 – 6 ms, s pridruženým horným 95 % intervalom spoľahlivosti < 7 ms. Z
2 182 pacientov s rezistenciou alebo intoleranciou na predchádzajúcu liečbu imatinibom, ktorí v
klinických štúdiách užívali SPRYCEL, 15 (1 %) pacientov hlásilo predĺženie QTc, ako nežiaducu reakciu. Dvadsaťjeden pacientov (1 %) malo QTcF > 500 ms (pozri časť 4.4).
Nežiaduce reakcie na srdce
Pacienti s rizikovými faktormi alebo srdcovým ochorením v anamnéze, majú byť starostlivo sledovaní pre prejavy alebo príznaky zhodné so srdcovou dysfunkciou a majú byť posúdení a primerane liečení (pozri časť 4.4).
Reaktivácia hepatitídy B
V súvislosti s inhibítormi BCR-ABL-tyrozínkinázy bola hlásená reaktivácia hepatitídy B. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie (pozri časť 4.4).
V klinickej štúdii dávkovej optimalizácie fázy III s pacientmi s chronickou fázou CML s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom (medián trvania liečby 30 mesiacov) bol výskyt pleurálneho výpotku a kongestívneho srdcového zlyhania/srdcovej dysfunkcie nižší u pacientov liečených SPRYCELOM dávkou 100 mg jedenkrát denne ako u pacientov liečených SPRYCELOM dávkou 70 mg dvakrát denne. Myelosupresia sa vyskytla tiež menej často v skupine liečenej dávkou
100 mg jedenkrát denne (pozri Abnormality laboratórnych testov nižšie). Medián trvania liečby
v skupine so 100 mg jedenkrát denne bol 37 mesiacov (rozmedzie 1 – 91 mesiacov). Kumulatívny výskyt vybraných nežiaducich reakcií, ktoré sa hlásili pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne sú uvedené v tabuľke6a.
Tabuľka 6a: Vybrané nežiaduce reakcie hlásené v 3. fáze štúdie optimalizácie dávky
(Intolerancia alebo rezistencia na imatinib chronická fáza CML)a
Minimálne 2-ročné následné sledovanie
Minimálne 5-ročné následné sledovanie
Minimálne 7-ročné následné sledovanie
 Všetky stupne
3
.
/4. stupeň
V
š
e
tky stupne
3./4. stupeň
Všetky stupne
3./4. stupeň
Všetky stupne
3
.
/4. stupeň
V
š
e
tky stupne
3./4. stupeň
Všetky stupne
3./4. stupeň
Preferovaný výraz Percento (%) pacientov
Hnačka 27 2 28 2 28 2
Retencia tekutín 34 4 42 6 48 7
Povrchový edém 18 0 21 0 22 0
Pleurálny výpotok 18 2 24 4 28 5
Generalizovaný edém 3 0 4 0 4 0
Perikardiálny
výpotok 2 1 2 1 3 1
Pľúcna hypertenzia 0 0 0 0 2 1
Krvácanie 11 1 11 1 12 1
Gastrointestinálne
krvácanie 2 1 2 1 2 1
a Výsledky štúdie 3. fázy optimalizácie dávky hlásené v populácii pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne (n=165)
V štúdii fázy III optimalizácie dávky s pacientmi s pokročilou fázou CML a Ph+ ALL bol medián trvania liečby 14 mesiacov pre akcelerovanú fázu CML, 3 mesiace pre myeloidnú blastovú CML,
4 mesiace pre lymfoidnú blastovú CML a 3 mesiace pre Ph+ ALL. Vybrané nežiaduce reakcie, ktoré
sa hlásili pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne sú uvedené v tabuľke 6b. Schéma
dávkovania 70 mg dvakrát denne sa tiež skúmala. Schéma dávkovania 140 mg jedenkrát denne preukázala porovnateľný profil účinnosti voči schéme dávkovania 70 mg dvakrát denne, no s priaznivejším profilom bezpečnosti.
Tabuľka 6b: Vybrané nežiaduce reakcie hlásené zo štúdie fázy III optimalizácie dávky:
 PokročiláfázaCMLaPh+ALLa 140 mg jedenkrát denne n = 304Všetky stupne 3./4. stupeň
PokročiláfázaCMLaPh+ALLa 140 mg jedenkrát denne n = 304Všetky stupne 3./4. stupeň Preferovaný výraz Percento (%) pacientovHnačka
Preferovaný výraz Percento (%) pacientovHnačka 28 3
Retencia tekutín 33 7
Povrchový edém 15 < 1
Pleurálny výpotok 20 6
Generalizovaný edém 2 0
Kongestívne srdcové 1 0
zlyhanie / srdcová dysfunkciab
Perikardiálny výpotok 2 1
Pľúcny edém 1 1
Krvácanie 23 8
Gastrointestinálnekrvácanie 8 6 a Výsledky štúdie 3. fázy optimalizácie dávky hlásené v populácii s odporúčanou začiatočnou dávkou 140 mg jedenkrát denne (n=304) pri 2-ročnej finálnej štúdii následného sledovania.
b Vrátane komorovej dysfunkcie, srdcového zlyhania, kongestívneho srdcového zlyhania, kardiomyopatie, kongestívnej kardiomyopatie, diastolickej disfunkcie, zníženie ejekčnej frakcie a komorového zlyhania.
Abnormality laboratórnych testovHematológiaV štúdii fázy III sa u pacientov s novo diagnostikovanou chronickou fázou CML užívajúcich SPRYCEL hlásili nasledovné laboratórne abnormality 3. a 4. stupňa po minimálne 12 mesiacoch následného sledovania: neutropénia (21 %), trombocytopénia (19 %) a anémia (10 %). Po minimálne
60 mesiacoch následného sledovania boli kumulatívne výsledky neutropénie, thrombocytopénie a anémie 29 %, 22 % a 13 %, v uvedenom poradí.
U pacientov s novo diagnostikovanou chronickou fázou CML liečených SPRYCELOM , u ktorých sa vyskytla myelosupresia 3. alebo 4. stupňa, zvyčajne došlo po krátkom prerušení a/alebo znížení dávky a trvalom prerušení liečby k zotaveniu, ktoré sa vyskytlo u 1,6 % pacientov po minimálne
12 mesiacoch následného sledovania. Po minimálne 60 mesiacoch následného sledovania bol kumulatívny pomer trvalého vysadenia liečby z dôvodu myelosupresie 3. alebo 4. stupňa 2,3 %.
U pacientov s CML s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom boli cytopénie (trombocytopénia, neutropénia a anémia) trvalý nález. Výskyt cytopénií bol však tiež závislý od stupňa ochorenia. Výskyt hematologických abnormalít 3. a 4. stupňa je uvedený v tabuľke 7.
Tabuľka 7: CTC hematologických laboratórnych abnormalít 3./4. stupňa v klinických štúdiách
s
pacientmi
s
rezistenciou
alebo
intoleranciou
na
p
redošlú
liečbu
imatinibom
a
Chronická fáza
Akcelerovaná
Myeloidná
blastová
Lymfoidná blastová
fáza a Ph+ ALL
(n= 165)
b
fáza
(
n=
1
5
7
)
c
fáza
(n=
74)
c
(n=
1
6
8)
c
Percento
(%)
pacientov
Hematologické
par
a
me
tre
Neutropénia 36 58 77 76
Trombocytopénia 23 63 78 74
Anémia 13 47 74 44
a Výsledky štúdie 3. fázy optimalizácie dávky hlásené po 2-ročnej štúdii následného sledovania.
b Štúdia CA180-034 výsledky pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
c Štúdia CA180-035 výsledky pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne.
CTC stupne: neutropénia (3. stupňa ≥ 0,5– < 1,0 × 109/l, 4. stupňa < 0.5 × 109/l); trombocytopénia (3. stupňa ≥ 25–
< 50 × 109/l,4. stupňa < 25 × 109/l); anémia (hemoglobín 3. stupňa ≥ 65– < 80 g/l,4. stupňa < 65 g/l).
Kumulatívne cytopénie 3. alebo 4. stupňa u pacientov liečených dávkou 100 mg jedenkrát denne boli podobné v 2. a 5. roku, zahŕňajúce: neutropénie (35 % oproti 36 %), trombocytopénie (23 % oproti
24 %) a anémie (13 % oproti 13 %).
U pacientov, ktorí mali myelosupresiu 3. alebo 4. stupňa, zvyčajne došlo k zotaveniu po krátkom prerušení liečby a/alebo znížení dávky a trvalé ukončenie liečby bolo potrebné u 5 % pacientov. Väčšina pacientov pokračovala v liečbe bez ďalších dôkazov o myelosupresii.
Biochemické vyšetrenia
V štúdii s novo diagnostikovanou chronickou fázou CML sa hlásila hypofosfatémia 3. a 4. stupňa
u 4 % pacientov liečených SPRYCELOM a vzostupy transamináz, kreatinínu a bilirubínu sa hlásili u
≤ 1 % pacientov po minimálne 12 mesiacoch následného sledovania. Po minimálne 60 mesiacoch následného sledovania bol kumulatívny pomer hypofosfatémie 3. alebo 4. stupňa 7 %, vzostupu kreatinínu a bilirubínu 3. alebo 4. stupňa 1 % a vzostupu transamináz 3. alebo 4. stupňa zostal 1 %. Nezistili sa žiadne prerušenia liečby SPRYCELOM v dôsledku týchto biochemických laboratórnych parametrov.
2-ročné následné sledovanie
Vzostupy transamináz alebo bilirubínu 3. alebo 4. stupňa sa hlásili u 1 % pacientov v chronickej fáze CML (rezistentných alebo intolerantných na imatinib), ale u pacientov v pokročilej fáze CML a u pacientov s Ph+ ALL sa vzostupy hlásili so zvýšenou 1 až 7 % frekvenciou. Zvyčajne sa zvládli znížením dávky alebo prerušením liečby. V štúdii dávkovej optimalizácie s chronickou fázou CML fázy III sa vyskytlo zvýšenie transamináz alebo bilirubínu 3. alebo 4. stupňa u ≤ 1 % pacientov s podobným nízkym výskytom vo všetkých štyroch skupinách. V štúdii dávkovej optimalizácie fázy III s pokročilou fázou CML a Ph+ALL sa vyskytlo zvýšenie transamináz alebo bilirubínu 3. alebo
4. stupňa u 1 % až 45 % pacientov v jednotlivých liečených skupinách.
Približne u 5 % pacientov liečených SPRYCELOM, ktorí mali normálne východiskové hladiny vápnika 3. alebo 4. stupňa, vznikla v určitom čase počas štúdie prechodná hypokalciémia. Obvykle nebola žiadna súvislosť medzi zníženými hladinami vápnika a klinickými symptómami. Pacienti, u ktorých vznikla hypokalciémia 3. alebo 4. stupňa sa často zotavili po perorálnej suplementácii vápnika. Hypokalciémia 3. alebo 4. stupňa, hypokaliémia a hypofosfatémia sa vyskytli u pacientov vo všetkých fázach CML, ale so zvýšenou frekvenciou u pacientov s myeloidnou alebo lymfoidnou blastovou fázou CML a Ph+ ALL. Zvýšenie kreatinínu 3. alebo 4. stupňa sa vyskytlo u < 1 % pacientov s chronickou fázou CML a hlásil sa zvýšený výskyt od 1 do 4 % u pacientov s pokročilou fázou CML.
Pediatrickápopulácia
Bezpečnostný profil u pediatrických pacientov bol porovnateľný s bezpečnostným profilom u dospelých. Frekvencia, typ a závažnosť nežiaducich reakcií u detí sa očakávajú rovnaké ako u dospelých.
V pediatrických štúdiách s CML bola miera laboratórnych anomálií zhodná so známym profilom laboratórnych parametrov u dospelých.
Osobitnépopulácie
Zatiaľ čo bezpečnostný profil SPRYCELU u starších ľudí bol podobný profilu v mladšej populácii, u pacientov vo veku 65 rokov a starších je pravdepodobnejší výskyt často hlásených nežiaducich reakcií, ako je únava, pleurálny výpotok, dyspnoe, kašeľ, krvácanie do dolného gastrointestinálneho traktu
a porucha chuti a pravdepodobnejší výskyt menej často hlásených nežiaducich reakcií, ako sú
distenzia brucha, závrat, perikardiálny výpotok, kongestívne srdcové zlyhanie a pokles telesnej hmotnosti a majú sa starostlivo sledovať (pozri časť 4.4).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
Skúsenosti s predávkovaním SPRYCELOM v klinických štúdiách sú limitované z dôvodu ojedinelých prípadov. Najväčšie predávkovanie 280 mg denne počas jedného týždňa sa vyskytlo u dvoch
pacientov a u oboch sa vyvinulo signifikantné zníženie počtu trombocytov. Pretože sa dasatinib spája s
myelosupresiou 3. alebo 4. stupňa (pozri časť 4.4), pacienti, ktorí prijali vyššiu ako odporúčanú dávku, majú byť dôkladne sledovaní na myelosupresiu a má im byť poskytnutá podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE06
Farmakodynamickéúčinky
Dasatinib inhibuje aktivitu BCR-ABL-kinázy a kináz rodiny SRC, ako aj niekoľkých ďalších vybraných onkogénnych kináz zahŕňajúcich kinázy c-KIT receptora, efrínového (EPH) receptora a PDGFβ receptora. Dasatinib je silným, subnanomolárnym inhibítorom BCR-ABL-kinázy
s účinnosťou pri koncentrácii 0,6-0,8 nM. Viaže sa na inaktívne aj aktívne konformácie enzýmu BCR-
ABL.
Mechanizmusúčinku
V podmienkach in vitro je dasatinib aktívny v leukemických bunkových líniách predstavujúcich varianty ochorenia citlivého a rezistentného na imatinib. Tieto predklinické štúdie ukazujú, že dasatinib môže prekonať rezistenciu na imatinib, ktorá je dôsledkom nadmernej expresie BCR-ABL, mutácií BCR-ABL-kinázovej domény, aktivácie alternatívnych signálnych dráh zahŕňajúcich kinázy
rodiny SRC (LYN, HCK) a nadmernej expresie génu pre rezistenciu na viaceré lieky. Dasatinib okrem toho inhibuje kinázy rodiny SCR pri subnanomolárnej koncentrácii.
V samostatných experimentoch in vivo využívajúcich myšacie modely CML dasatinib zabránil progresii chronickej fázy CML do blastickej fázy a predĺžil prežívanie myší nesúcich ľudské bunkové línie CML kultivované na rôznych miestach, vrátane centrálneho nervového systému.
Klinickáúčinnosťabezpečnosť
V štúdii fázy I. bola hematologická a cytogenetická odpoveď pozorovaná vo všetkých fázach CML a u Ph+ ALL u prvých 84 pacientov liečených a následne sledovaných počas doby 27 mesiacov. Odpovede mali trvalý charakter vo všetkých fázach CML a u Ph+ ALL.
Vykonali sa štyri jednoramenné, nekontrolované, otvorené klinické štúdie fazy II hodnotiace bezpečnosť a účinnosť dasatinibu u pacientov s CML v chronickej, akcelerovanej alebo myeloidnej
blastickej fáze, ktorí boli buď rezistentní, alebo intolerantní na imatinib. Jedna randomizovaná, nekomparatívna štúdia sa vykonala u pacientov v chronickej fáze, u ktorých zlyhala úvodná liečba so
400 alebo 600 mg imatinibu. Počiatočná dávka dasatinibu bola 70 mg dvakrát denne. Úpravy dávky
boli povolené na zlepšenie účinnosti alebo zvládnutie toxicity (pozri časť 4.2).
Dve randomizované, otvorené klinické štúdie fázy III sa vykonali s cieľom zhodnotiť účinnosť dasatinibu podávaného jedenkrát denne v porovnaní s dasatinibom podávaným dvakrát denne. Okrem toho, jedna otvorená, randomizovaná, porovnávacia štúdia fázy III bola vykonaná u dospelých pacientov s novo diagnostikovanou chronickou fázou CML.
Účinnosť dasatinibu sa zakladá na miere hematologickej a cytogenetickej odpovede.
Stálosť odpovede a odhadovaná miera prežitia poukazuje na ďalší klinický úžitok dasatinibu.
V klinických štúdiách sa hodnotil celkový počet 2 712 pacientov, z ktorých 23 % bolo vo veku
≥ 65 rokov a 5 % bolo vo veku ≥ 75 rokov.
Chronická fáza CML – novo diagnostikovaná
Medzinárodná otvorená, multicentrická, randomizovaná, porovnávacia štúdia fázy III sa vykonala s dospelými pacientmi s novo diagnostikovanou chronickou fázou CML. Pacienti boli randomizovaní do skupiny užívajúcich buď SPRYCEL 100 mg jedenkrát denne, alebo imatinib 400 mg jedenkrát
denne. Primárnym ukazovateľom bola miera potvrdenej kompletnej cytogenetickej odpovede (cCCyR)
počas 12 mesiacov. Sekundárne ukazovatele zahŕňali čas v cCCyR (miera trvania odpovede), čas do cCCyR, mieru veľkej molekulárnej odpovede (MMR), čas do MMR, prežívanie bez progresie (PFS) a celkové prežívanie (OS). Ostatné relevantné výsledky účinnosti zahŕňali CCyR a mieru kompletnej molekulárnej odpovede (CMR). Štúdia prebieha.
Celkovo 519 pacientov bolo randomizovaných do skupín: 259 do skupiny SPRYCELU a 260 k imatinibu. Základné charakteristiky boli dobre vyvážené medzi oboma liečebnými skupinami s ohľadom na vek (medián veku bol 46 rokov pre SPRYCELovú skupinu a 49 rokov pre imatinibovú skupinu s 10 % pacientov v SPRYCELOVEJ skupine a 11 % pacientov v imatinibovej skupine vo veku 65 rokov alebo starších), pohlavie (ženy 44 % v SPRYCELOVEJ skupine a 37 % v imatinibovej skupine), a rasu (kaukazská 51 % SPRYCELOVEJ skupine v a 55 % v imatinibovej skupine, ázijská
42 % v SPRYCELOVEJ skupine a 37 % imatinibovej skupine). Na začiatku, bola distribúcia Hasford skóre podobná v SPRYCELOM a imatinibom liečenej skupine (nízke riziko: 33 % a 34 %, stredné riziko 48 % a 47 %, vysoké riziko: 19 % a 19 % v SPRYCELOVEJ skupine a imatinibovej skupine v uvedenom poradí).
V minimálne 12 mesačnom následnom sledovaní, 85 % pacientov randomizovaných do skupiny so
SPRYCELOM a 81 % pacientov randomizovaných do skupiny s imatinibom stále dostávalo najprv prvolíniovú liečbu. Prerušenie počas 12 mesiacov z dôvodu progresie ochorenia sa vyskytlo u 3 % pacientov liečených SPRYCELOM a u 5 % pacientov liečených imatinibom.
V minimálne 60 mesačnom následnom sledovaní, 60 % pacientov randomizovaných do skupiny so SPRYCELOM a 63 % pacientov randomizovaných do skupiny s imatinibom stále dostávalo prvolíniovú liečbu. Prerušenie liečby počas 60 mesiacov z dôvodu progresie ochorenia sa vyskytlo u
11 % pacientov liečených SPRYCELOM a u 14 % pacientov liečených imatinibom.
Výsledky účinnosti sú uvedené v tabuľke 8. Štatisticky významne väčší podiel pacientov v SPRYCELOVEJ skupine dosiahol cCCyR v priebehu prvých 12 mesiacov liečby v porovnaní s pacientmi v imatinibovej skupine. Účinnosť SPRYCELU bola dôsledne preukázaná v rôznych podskupinách, zahŕňajúc vek, pohlavie a východiskové Hasford skóre.
Tabuľka 8: Výsledky účinnosti z 3. fázy štúdie s novo diagnostikovanými pacientmi s
chronickou
fázou
C
ML
SPRYCEL
imatinib
p-hodnota
n= 259 n= 260
Miera
o
dpovede
(95
%
CI)
Cytogenetická odpoveď
v priebehu 12 mesiacov
cCCyRa 76,8 % (71,2–81,8) 66,2 % (60,1-71,9) p< 0,007*
CCyRb 85,3 % (80,4 – 89,4) 73,5 % (67,7 – 78,7) ⎯
v priebehu 24 mesiacovcCCyRa 80,3 % 74,2 % ⎯
CCyRb 87,3 % 82,3 % ⎯
v priebehu 36 mesiacovcCCyRa 82,6 % 77,3 % ⎯
CCyRb 88,0 % 83,5 % ⎯
v priebehu 48 mesiacovcCCyRa 82,6 % 78,5 % ⎯
CCyRb 87,6 % 83,8 % ⎯
v priebehu 60 mesiacovcCCyRa 83,0 % 78,5 % ⎯
CCyRb 88 % 83,8 % ⎯
Veľká molekulárna odpoveďc
12 mesiacov 52,1 % (45,9-58,3) 33,8 % (28,1-39,9) p< 0,00003*
24 mesiacov 64,5 % (58,3 – 70,3) 50 % (43,8 – 56,2) ⎯
36 mesiacov 69,1 % (63,1 – 74,7) 56,2 % (49,9 – 62,3) ⎯
48 mesiacov 75,7 % (70,0 – 80,8) 62,7 % (56,5 – 68,6) ⎯
60mesiacov 76,4%(70,8–81,5) 64,2%(58,1–70,1) p=0,0021 Pomer rizika (HR)počas 12 mesiacov (99,99 % CI)Čas do cCCyR 1,55 (1,0 – 2,3) p< 0,0001* Čas do MMR 2,01 (1,2 – 3,4) p< 0,0001* Trvania cCCyR 0,7 (0,4 – 1,4) P< 0,035
počas 24 mesiacov (95% CI)Čas do cCCyR 1,49 (1,22 – 1,82) ⎯ Čas do MMR 1,69 (1,34 – 2,12) ⎯ Trvania cCCyR 0,77 (0,55 – 1,10) ⎯
počas 36 mesiacov (95% CI)Čas do cCCyR 1,48 (1,22 – 1,80) ⎯
Čas do MMR 1,59 (1,28 – 1,99) ⎯
Trvania cCCyR 0,77 (0,53 – 1,11) ⎯
počas 48 mesiacov (95% CI)Čas do cCCyR 1,45 (1,20 – 1,77) ⎯ Čas do MMR 1,55 (1,26 – 1,91) ⎯ Trvania cCCyR 0,81 (0,56 – 1,17) ⎯
počas 60 mesiacov (95% CI)Čas do cCCyR 1,46 (1,20 – 1,77) p=0,0001
Čas do MMR 1,54 (1,25 – 1,89) p<0,0001
TrvaniacCCyR 0,79(0,55–1,13) p=0,1983 a Potvrdená kompletná cytogenetická odpoveď (cCCyR) je definovaná ako odpoveď známa z dvoch po sebe idúcich udalostí
(najmenej 28 dní odstup).
b Kompletná cytogenetická odpoveď (CCyR) je založená na jednom cytogenetickom zhodnotení kostnej drene.
c Veľká molekulová odpoveď (v akomkoľvek čase) bola definovaná ako BCR-ABL podiely ≤ 0,1 % podľa RQ-PCR vo vzorkách periférnej krvi štandardizovaných na Medzinárodnej stupnici. Ide o kumulatívne miery predstavujúce minimálne
sledovanie pre stanovený časový rámec.
*Upravené na Hasford skóre a uvedená štatistická významnosť na preddefinovanú nominálnu hodnotu významnosti. CI = interval spoľahlivosti
Po 60 mesačnom následnom sledovaní bol medián času do cCCyR 3,1 mesiaca v SPRYCELOVEJ skupine a 5,8 mesiaca v imatinibovej skupine u pacientov s potvrdenou CCyR. Medián času do MMR bol po 60 mesačnom následnom sledovaní 9,3 mesiaca v SPRYCELOVEJ skupine a 15,0 mesiacov v imatinibovej skupine pacientov s MMR. Tieto výsledky sú zhodné s tými, ktoré sa zistili po 12, 24
a 36 mesiacoch.
Čas do MMR je zobrazený graficky na Grafe 1. Čas do MMR bol rovnomerne kratší u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
Graf 1: Odhad času veľkej molekulárnej odpovede (MMR) podľa Kaplan-Meiera
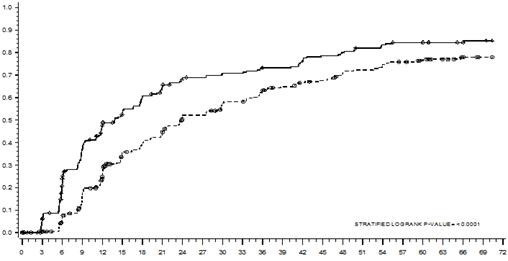 MESIACE
MESIACE Dasatinib ------ Imatinib

Cenzurovaní

Cenzurovaní
SKUPINA # RESPONDÉRY / #RANDOMIZOVANÍ POMERRIZIKA(95%CI)Dasatinib 198/259
Imatinib 167/260
Dasatinib pred imatinibom 1,54 ( 1,25 - 1,89)
Miery cCCyR v SPRYCELOM a imatinibom liečenej skupine, v uvedenom poradí, počas 3 mesiacov
(54 % a 30 %), počas 6 mesiacov (70 % a 56 %), a počas 9 mesiacov (75 % a 63 %), počas
24 mesiacov (80 % a 74 %), počas 36 mesiacov (83 % a 77 %), počas 48 mesiacov (83 % a 79 %)
a počas 60 mesiacov (83 % a 79 %) boli zhodné s primárnym koncovým ukazovateľom. Miery MMR
v SPRYCELOM a imatinibom liečenej skupine, v uvedenom poradí počas 3 mesiacov (8 % a 0,4 %),
6 mesiacov (27 % a 8 %), 9 mesiacov (39 % a 18 %), 12 mesiacov (46 % a 28 %), 24 mesiacov (64 % a 46 %), 36 mesiacov (67 % a 55 %), 48 mesiacov (73 % a 60 %) a počas 60 mesiacov (76 % a 64 %) boli tiež v zhode s primárnym koncovým ukazovateľom.
Pomery MMR pri špecifických časových bodoch sú zobrazené graficky na Grafe 2. Pomery MMR boli rovnomerne vyššie u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
 Graf 2: Hodnoty MMR v priebehu času – všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML)
Graf 2: Hodnoty MMR v priebehu času – všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML)
po
2
.
r
o
koch
64%, p<,0001
po3.rokoch
67%, p<,0055
po4.rokoch
73%, p<,0021
po5.rokoch
76%, p<,0022
po
1
.
r
o
ku

46%, p<,0001
N
Dasatinib 100 mg jedenkrát denne 259
--------- Imatinib 400 mg jedenkrát denne 260
Mesiace od randomizácie
Podiel pacientov, ktorí dosiahli pomer BCR-ABL ≤0,01 % (4-log zníženie) v akomkoľvek čase bol
vyšší v SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (54,1 % proti
45 %). Podiel pacientov, ktorí dosiahli pomer BCR-ABL ≤0,032% (4,5-log zníženie) v akomkoľvek čase bol vyšší v SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (44 % proti 34 %).
Pomery MR4.5 v priebehu času sú zobrazené graficky na Grafe 3. Pomery MR4.5 boli v priebehu času rovnomerne vyššie u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
 Graf 3: Hodnoty MR4.5 v priebehu času – všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML
Graf 3: Hodnoty MR4.5 v priebehu času – všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML
po
1
.
r
oku
5%, p<,2394
po2.rokoch19%, p<,0008
 po3.rokoch
po3.rokoch24%, p<,0013
po4.rokoch34%, p<,0055
po5.rokoch42 %, p<,0251
N
Dasatinib 100 mg jedenkrát denne 259
--------- Imatinib 400 mg jedenkrát denne 260
Mesiace od randomizácie
Miera MMR v akomkoľvek čase v každej rizikovej skupine určená Hasfordovým skóre bola vyššia v
SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (nízke riziko: 90 % a
69 %; stredné riziko: 71 % a 65 %; vysoké riziko: 67 % a 54 %) v uvedenom poradí.
V ďalšej analýze dosiahlo viac pacientov liečených dasatinibom (84 %) skorú molekulárnu odpoveď (definovanú ako hladiny BCR-ABL 10 % po 3 mesiacoch) v porovnaní s pacientmi liečenými imatinibom (64 %). Pacienti s dosiahnutou skorou molekulárnou odpoveďou mali nižšie riziko transformácie, vyšší pomer prežívania bez progresie (PFS, progression-free survival) a vyšší pomer celkového prežívania (OS, overall survival), ako je uvedené v tabuľke 9.
Tabuľka 9: Pacienti s dasatinibom s BCR-ABL 10 % a >10%po3mesiacoch
Pacienti s BCR-ABL 10 %
Pacienti s BCR-ABL
Dasatinib
N
=
235
po
3
mesiacoch
>
10
%
po
3
mesiacoch
Počet pacientov (%) 198 (84,3) 37 (15,7)
Transformácia po 60 mesiacoch, n/N (%)
Pomer PFS po 60 mesiacoch
(95 % CI)
6/198 (3,0) 5/37 (13,5)
92,0 % (89,6; 95,2) 73,8 % (52,0; 86,8)
Pomer OS po 60 mesiacoch (95 % CI) 93,8 % (89,3; 96,4) 80,6 % (63,5; 90,2)
Hodnoty OS pri špecifických časových bodoch sú zobrazené na Grafe 4. Hodnota OS bola
rovnomerne vyššia u pacientov liečených dasatinibom, ktorí dosiahli hladinu BCR-ABL ≤ 10 % po
3 mesiacoch než u tých, ktorí ju nedosiahli.
Graf 4: Grafické znázornenie orientačných bodov celkového prežívania po dasatinibe pomocou hladiny BCR-ABL ( 10 % alebo > 10 %) po 3 mesiacoch v 3. fáze štúdie s novo diagnostikovanými pacientmi s chronickou fázou CML


Rizikoví pacienti
M
ESIACE
<=10% 198 198 197 196 195 193 193 191 191 190 188 187 187 184 182 181 180 179 179 177 171 96 54 29 3 0
>10% 37 37 37 35 34 34 34 33 33 31 30 29 29 29 28 28 28 27 27 27 26 15 10 6 0 0
≤10 % ------ >10 %
Cenzurovaní

Cenzurovaní
SKUPINA # ÚMRTIA / # Pacient, ktorý ukončil štúdiu MEDIÁN (95 % CI) POMER RIZIKA (95 % CI)
≤10 % 14/198 .(. - .)
>10 % 8/37 .(. - .)
0,29 (0,12 – 0,69)
Progresia ochorenia bola definovaná ako zvýšenie bielych krviniek napriek zodpovedajúcej liečbe, strata CHR, čiastočná CyR alebo CCyR, progresia do akcelerovanej fázy alebo blastovej fázy alebo smrť. Odhadovaná miera 60-mesačného PFS bola 88,9 % (CI: 84 % – 92,4 %) v dasatinibom a imatinibom liečenej skupine. Zmena na akcelerovanú alebo blastovú fázu po 60 mesiacoch sa vyskytla u niekoľkých dasatinibom liečených pacientov (n=8; 3 %) v porovnaní s pacientmi liečenými imatinibom (n = 15; 5,8 %). Odhadovaná miera 60-mesačného prežitia pre dasatinibom a imatinibom liečených pacientov bola 90,9 % (CI: 86,6 % – 93,8 %) a 89,6 % (CI: 85,2 % – 92,8 %), v uvedenom poradí. Medzi dasatinibom a imatinibom nebol rozdiel v OS (HR 1,01; 95 % CI: 0,58 – 1,73; p=
0,9800) a PFS (HR 1,00, 95 % CI: 0,58 – 1,72, p = 0,9998).
U pacientov, u ktorých sa zaznamenala progresia ochorenia alebo ukončenie liečby dasatinibom alebo imatinibom, sa vykonalo sekvenovanie BCR-ABL na krvných vzorkách od pacientov, u ktorých boli dostupné. V obidvoch liečených ramenách sa pozoroval podobný pomer výskytu mutácií. Mutácie detekované u pacientov liečených dasatinibom boli T315I, F317I/L a V299L. V ramene liečenom imatinibom sa detekovalo iné spektrum mutácií. Na základe
in vitro údajov sa dasatinib neprejavuje ako účinný voči mutácii T315I.
Chronická fáza CML – Rezistencia alebo intolerancia na predošlú liečbu imatinibomVykonali sa dve klinické štúdie s pacientmi rezistentnými alebo intolerantnými na imatinib; koncovým ukazovateľom primárnej účinnosti v týchto štúdiách bola veľká cytogenetická odpoveď (MCyR).
1. štúdiaOtvorená, randomizovaná, nekomparatívna, multicentrická štúdia sa vykonala s pacientmi, u ktorých zlyhala úvodná liečba so 400 alebo 600 mg imatinibu. Boli randomizovaní (2:1) buď do skupiny s dasatinibom (70 mg dvakrát denne) alebo s imatinibom (400 mg dvakrát denne). Prechod do alternatívnej liečebnej skupiny bol povolený vtedy, ak pacienti vykazovali známky progresie ochorenia alebo intoleranciu, ktorú nebolo možné zvládnuť úpravou dávky. Primárny koncový ukazovateľ bol MCyR po 12 týždňoch. Sú dostupné výsledky u 150 pacientov: 101 pacientov bolo randomizovaných do skupiny s dasatinibom a 49 pacientov do skupiny s imatinibom (všetci pacienti
boli rezistentní na imatinib). Medián času od stanovenia diagnózy po randomizáciu bol 64 mesiacov v skupine s dasatinibom a 52 mesiacov v skupine s imatinibom. Všetci pacienti boli po predošlej intenzívnej liečbe. Predošlá úplná hematologická odpoveď (CHR) na imatinib sa dosiahla u 93 % z celkovej populácie pacientov. Predošlá MCyR na imatinib sa dosiahla u 28 % pacientov v skupine s dasatinibom a u 29 % pacientov v skupine s imatinibom.
Medián trvania liečby bol 23 mesiacov pre skupinu s dasatinibom (pričom dosiaľ bolo 44 % pacientov liečených počas > 24 mesiacov) a 3 mesiace pre skupinu s imatinibom (pričom dosiaľ bolo 10 % pacientov liečených počas > 24 mesiacov). Pred prechodom do druhej skupiny liečby dosiahlo CHR v skupine s dasatinibom deväťdesiattri percent pacientov a imatinibom 82 % pacientov.
Počas 3 mesiacov sledovania došlo k MCyR častejšie v skupine s dasatinibom (36 %) ako v skupine s imatinibom (29 %). Pozoruhodné je, že v skupine s dasatinibom hlásilo úplnú cytogenetickú odpoveď (CCyR) 22 % pacientov, pričom v skupine s imatinibom dosiahlo CcyR len 8 %. Pri dlhšej liečbe a sledovaní (medián 24 mesiacov) sa MCyR dosiahla u 53 % pacientov liečených dasatinibom (CCyR u
44 %) a u 33 % pacientov liečených imatinibom (CCyR u 18 %) pred prechodom do druhej skupiny. Medzi pacientmi, ktorí užívali imatinib v dávke 400 mg pred vstupom do štúdie, MCyR dosiahlo 61 % pacientov v ramene s dasatinibom a 50 % v ramene s imatinibom.
Na základe Kaplan-Meierových odhadov pomer pacientov, ktorí si udržali MCyR počas 1 roka bol
92 % (95 % CI: [85 % – 100 %]) pre dasatinib (CCyR 97 %, 95 % CI: [92 % – 100 %]) a 74 % (95 % CI: [49 % – 100 %]) pre imatinib (CCyR 100 %). Pomer pacientov, ktorí si udržali MCyR počas
18 mesiacov bol 90 % (95 % CI: [82% – 98%]) pre dasatinib (CCyR 94%, 95 % CI: [87% – 100 %]) a
74 % (95 % CI: [49 % – 100 %]) pre imatinib (CCyR 100 %).
Na základe Kaplan-Meierových odhadov pomer pacientov, ktorí mali progresiu prežívania (PFS) počas 1 roka bol 91 % (95 % CI: [85 % – 97 %]) pre dasatinib a 73 % (95 % CI: [54 % – 91 %]) pre imatinib. Pomer pacientov, ktorí mali PFS počas 2 rokov bol 86 % (95 % CI: [78% – 93%]) pre dasatinib a 65% (95 % CI: [43% – 87%]) pre imatinib.
Z celkového počtu 43 % pacientov v skupine s dasatinibom a 82 % pacientov v skupine s imatinibom došlo k zlyhaniu liečby, ktoré bolo definované ako progresia ochorenia alebo prechod do druhej skupiny liečby (nedostatočná odpoveď, intolerancia skúšaného lieku, atď.).
Výskyt veľkej cytogenetickej odpovede (definovaný ako BCR-ABL/kontrola transkriptov ≤ 0,1 % s RQ-PCR vo vzorke periférnej krvi) pred prechodom do druhej skupiny bol 29 % pre dasatinib a 12 % pre imatinib.
2. štúdia
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi rezistentnými alebo intolerantnými na imatinib (napr. pacienti, ktorí majú skúsenosť so signifikantnou toxicitou počas liečby imatinibom, ktorý vylučuje ďalšiu liečbu).
Dasatinib 70 mg dvakrát denne užívalo celkovo 387 pacientov (288 rezistentní a 99 intolerantní na
imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 61 mesiacov. Väčšina pacientov (53 %) dostala predošlú liečbu imatinibom trvajúcu viac ako 3 roky. Väčšina rezistentných pacientov (72 %) užívala > 600 mg imatinibu. Okrem imatinibu dostalo 35 % pacientov aj predošlú cytotoxickú chemoterapiu, 65 % dostalo predošlú liečbu interferónom a 10 % bolo po predošlej transplantácii kmeňových buniek. Tridsaťosem percent pacientov malo na začiatku liečby mutácie, o ktorých je známe, že vyvolávajú rezistenciu na imatinib. Medián trvania liečby dasatinibom bol 24 mesiacov, pričom dosiaľ bolo 51 % pacientov liečených počas > 24 mesiacov. Výsledky účinnosti sú uvedené v tabuľke 10. MCyR sa dosiahla u 55 % pacientov rezistentných na imatinib a 82 % pacientov intolerantných na imatinib. Pri minimálne 24-mesačnom sledovaní malo len 21 z 240 pacientov, ktorí dosiahli McyR, progresiu ochorenia a nedosiahli medián trvania MCyR.
Na základe Kaplan-Meierových odhadov si 95 % (95 % CI: [92 % – 98 %]) pacientov udržalo MCyR počas 1 roka a 88 % (95 % CI: [83 % – 93 %]) si udržalo MCyR počas 2 rokov. Pomer pacientov, ktorí si udržali CCyR počas roka bol 97 % (95 % CI: [94 % – 99 %]) a počas 2 rokov bol 90 % (95 % CI: [86 % – 95 %]). Štyridsaťdva percent pacientov rezistentných na imatinib bez predchádzajúcej MCyR na imatinib (n= 188) dosiahlo MCyR s dasatinibom.
Vyskytlo sa 45 odlišných BCR-ABL mutácií u 38 % pacientov zúčastnených v tejto štúdii. Úplná hematologická odpoveď alebo MCyR bola dosiahnutá u pacientov majúcich množstvo BCR-ABL mutácií súvisiacich s rezistenciou na imatinib okrem T315I. Podiel MCyR v 2. roku bol podobný či pacienti mali nejakú základnú BCR-ABL mutáciu, P-loop mutáciu alebo nemali mutáciu (63 %, 61 % a 62 %).
Medzi pacientmi rezistentnými na imatinib bol odhadovaný pomer PFS 88 % (95 % CI: [84 % –
92 %]) v 1. roku a 75 % (95 % CI: [69 % – 81 %]) v 2 roku. Medzi pacientmi intolerantnými na imatinib bol odhadovaný pomer PFS 98 % (95 % CI: [95% – 100%]) v 1. roku a 94% (95 % CI: [88%
– 99%]) v 2 roku.
Pomer veľkej molekulárnej odpovede po 24 mesiacoch bol 45 % (35 % pre imatinib rezistentných pacientov a 74 % pre imatinib intolerantných pacientov).
Akcelerovaná fáza CML
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi intolerantnými alebo rezistentnými na imatinib. Dasatinib 70 mg dvakrát denne užívalo celkovo 174 pacientov
(161 rezistentní a 13 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 82 mesiacov. Medián trvania liečby dasatinibom bol 14 mesiacov, pričom dosiaľ bolo 31% pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (hodnotený u
41 pacientov s CCyR) po 24 mesiacoch bol 46 %. Ďalšie výsledky účinnosti sú uvedené v tabuľke 10.
Myeloidná blastická fáza CML
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi intolerantnými alebo rezistentnými na imatinib. Dasatinib 70 mg dvakrát denne užívalo celkovo 109 pacientov
(99 rezistentní a 10 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 48 mesiacov. Medián trvania liečby dasatinibom bol 3,5 mesiacov, pričom dosiaľ bolo 12 % pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (hodnotený u
19 pacientov s CCyR) po 24 mesiacoch bol 68 %. Ďalšie výsledky účinnosti sú uvedené v tabuľke 10.
Lymfoidná blastická fáza CML a Ph+ ALL
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi v lymfoidnej blastickej fáze CML alebo s Ph+ ALL, ktorí boli rezistentní alebo intolerantní na predošlú liečbu imatinibom. Dasatinib 70 mg dvakrát denne užívalo celkovo 48 pacientov v lymfoidnej blastickej fáze CML
(42 rezistentní a 6 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 28 mesiacov. Medián trvania liečby dasatinibom bol 3 mesiace s 2 % pacientov liečených počas
> 24 mesiacov. Pomer veľkej molekulárnej odpovede (všetkých 22 liečených pacientov s CCyR) po
24 mesiacoch bol 50 %. Okrem toho dasatinib 70 mg dvakrát denne užívalo 46 pacientov s Ph+ ALL (44 rezistentní a 2 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 18 mesiacov. Medián trvania liečby dasatinibom bol 3 mesiace, pričom dosiaľ bolo 7 % pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (všetkých 25 liečených pacientov s CCyR) po 24 mesiacoch bol 52 %. Ďalšie výsledky účinnosti sú uvedené v tabuľke 10. Za zmienku stojí, že veľká hematologická odpoveď (MaHR) bola dosiahnutá rýchlo (väčšinou do 35 dní od prvého podania dasatinibu u pacientov v lymfoidnej blastickej fáze CML, a do 55 dní u pacientov
s Ph+ ALL).
Tabuľka
10:
Účinnosť
SPRYCELU
vo
fáze
II
k
linických
štúdií
s
jednou
skupinou
a
Myeloidná
Chronická fáza
Akcelerovaná
bl
a
stová fáza
Lymfoidná blastová
Ph+ ALL
 (n= 387) fáza
(n= 174) (n= 109) fáza
(n= 48) (n= 46)
Pomer hematologickej odpovede
b
(
%)
(n= 387) fáza
(n= 174) (n= 109) fáza
(n= 48) (n= 46)
Pomer hematologickej odpovede
b
(
%)
MaHR (95% CI) n/a 64% (57 –
72)
33% (24 –
43)
35% (22 –
51)
41% (27 –
57)
CHR (95% CI) 91% (88 –
94)
50% (42 – 58) 26% (18 –
35)
29% (17 –
44)
35% (21 –
50)
NEL (95% CI) n/a 14% (10 – 21) 7% (3 – 14) 6% (1 – 17) 7% (1 – 18) Trvanie MaHR (%; Kaplan-Meierove odhady)
1. rok n/a 79% (71 – 87) 71% (55 –
87)
2. rok n/a 60% (50 – 70) 41% (21 –
29% (3 – 56) 32% (8 – 56)
10% (0 – 28) 24% (2 – 47)
60)
Cytogenetická odpoveď
c
(%)
MCyR (95% CI) 62% (57 –
67)
CCyR (95% CI) 54% (48 –
40% (33 – 48) 34% (25 –
44)
33% (26 – 41) 27% (19 –
52% (37 –
67)
46% (31 –
57% (41 –
71)
54% (39 –
59) 36) 61) 69)
Prežívanie
(%;
Kaplan-Meierove
odhady)
Progresia – voľná
91% (88 –
94) 64% (57 – 72)
80% (75 –
84) 46% (38 – 54)
Celková
35% (25 –
45) 14% (3 – 25) 21% (9 – 34)
20% (11 –
29) 5% (0 – 13) 12% (2 – 23)
97% (95 –
99) 83% (77 – 89)
2. rok 94% (91 –
48% (38 –
59)
38% (27 –
30% (14 –
47)
26% (10 –
35% (20 –
51)
31% (16 –
97)
72% (64 – 79) 50) 42) 47)
Údaje uvedené v tabuľke sú zo štúdií s užívanou úvodnou dávkou 70 mg dvakrát denne. Pozri časť 4.2 pre odporučenú úvodnú dávku.
a Čísla v bold fonte sú výsledky primárneho koncového ukazovateľa.
b Kritéria hematologickej odpovede (všetky odpovede boli potvrdené po 4 týždňoch): Veľká hematologická odpoveď: (MaHR) = úplná hematologická odpoveď (CHR) + žiadna prítomnosť leukémie (NEL).
CHR (chronická CML): WBC ≤ zdravotníckeho zariadenia ULN, trombocyty < 450 000/mm3, bez blastov alebo promyelocytov v periférnej krvi, < 5% myelocytov plus metamyelocyty v periférnej krvi, bazofily v periférnej krvi
< 20% a žiadne extramedulárne postihnutie.
CHR (pokročilá CML/Ph+ ALL): WBC ≤ zdravotníckeho zariadenia ULN, ANC ≥ 1 000/mm3, trombocyty
≥ 100 000/mm3, bez blastov alebo promyelocytov v periférnej krvi, blasty kostnej drene ≤ 5%, < 5% myelocyty plus metamyelocyty v periférnej krvi, bazofily v periférnej krvi < 20% a žiadne extramedulárne postihnutie.
NEL: rovnaké kritéria ako pre CHR ale ANC ≥ 500/mm3 a < 1 000/mm3, alebo trombocyty ≥ 20 000/mm3 a
≤ 100 000/mm3.
c Kritéria cytogenetickej odpovede: úplná (0 % Ph+ metafáza) alebo čiastočná (> 0 % – 35 %). MCyR (0 % – 35 %)
kombinovaná úpná a čiastočná odpoveď.
n/a = neaplikovateľný; CI = interval spoľahlivosti; ULN = horný limit normálneho rozpätia.
Výsledok u pacientov s tranplantáciou kostnej drene po liečbe dasatinibom nie je úplne zhodnotený.
Klinické štúdie fázy III u pacientov s CML v chronickej, akcelerovanej alebo myeloidnej blastovej fázea Ph+ ALL, ktorí boli rezistentní alebo netolerovali imatinib
Dve randomizované, otvorené štúdie sa vykonali na zhodnotenie účinnosti dasatinibu podávaného jedenkrát denne v porovnaní s dasatinibom podávaným dvakrát denne. Výsledky uvedené nižšie sú založené na minimálne 2-ročnom a 7-ročnom následnom sledovaní po začatí liečby dasatinibom.
1. štúdia
V štúdii s chronickou fázou CML bola MCyR primárnym koncovým ukazovateľom u pacientov rezistentných na imatinib. MCyR bola hlavným sekundárnym koncovým ukazovateľom pri celkovej dennej dávke u pacientov rezistentných na imatinib. Ďalšie sekundárne koncové ukazovatele zahŕňali trvania MCyR, PFS a celkového prežitia. Z celkového počtu 670 pacientov, z ktorých bolo
497 rezistentných na imatinib, boli pacienti v skupinách randomizovaní na dasatinib v dávke 100 mg jedenkrát denne, 140 mg jedenkrát denne, 50 mg dvakrát denne alebo 70 mg dvakrát denne. Medián trvania liečby všetkých pacientov stále liečených s minimálne 5-ročným následným sledovaním (n=205) bol 59 mesiacov (rozpätie 28 – 66 mesiacov). Medián trvania liečby všetkých pacientov po 7- ročnom následnom sledovaní bol 29,8 mesiaca (rozpätie < 1 – 92,9 mesiaca).
Účinnosť sa dosiahla vo všetkých skupinách liečených dasatinibom v dávkovacej schéme jedenkrát denne dokazujúc porovnateľnú účinnosť (nie horšiu) dávkovacej schémy dvakrát denne primárneho koncového ukazovateľa účinnosti (rozdiel v MCyR 1,9 %, 95 % interval spoľahlivosti [-6,8 % –
10,6 %]); schéma dávkovania 100 mg jedenkrát denne však potvrdila zlepšenú bezpečnosť a znášanlivosť. Výsledky účinnosti sú uvedené v tabuľkách 11 a 12.

 Tabuľka 11: Účinnosť SPRYCELU v štúdii fázy III optimalizácie dávky: rezistencia alebo intolerancia na imatinib chronická fáza CML (2-ročné výsledky)aVšetci pacienti n=167Pacienti rezistentní na imatinib n=124 Pomerhematologickej odpovedeb (%) (95 % CI)
Tabuľka 11: Účinnosť SPRYCELU v štúdii fázy III optimalizácie dávky: rezistencia alebo intolerancia na imatinib chronická fáza CML (2-ročné výsledky)aVšetci pacienti n=167Pacienti rezistentní na imatinib n=124 Pomerhematologickej odpovedeb (%) (95 % CI)
CHR
92 % (86 – 95) Cytogenetická odpoveďc (%) (95 % CI)MCyR
Všetci pacienti
63 % (56 – 71)Pacienti rezistentní na
imatinib
59 % (50 – 68)CCyR
Všetci pacienti
50 % (42 – 58)Pacienti rezistentní na
imatinib 44 % (35 – 53)Veľká molekulárna odpoveď u pacientov, ktorí dosiahli CCyRd (%) (95 % CI)Všetci pacienti
69 % (58 – 79) Pacientirezistentnínaimatinib 72%(58–83) a Výsledky hlásené pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
b Kritéria hematologickej odpovede (všetky odpovede potvrdené po 4 týždňoch): Kompletná hematologická odpoveď (CHR) (chronická CML): WBC ≤ ULN zdravotníckeho zariadenia, trombocyty < 450,000/mm3TP, žiadne blasty alebo promyelocyty v periférnej krvi, <5 % myelocytov plus metamyelocytov v periférnej krvi, bazofily v periférnej krvi
< 20 % a žiadne extramedulárne postihnutia.
c Kritéria cytogenetickej odpovede: úplná (0 % Ph+ metafáza) alebo čiastočná (> 0 %–35 %). MCyR (0 %–35 %)
kombinácie s úplnými a čiastočnými odpoveďami.
d Kritéria veľkej molekulárnej odpovede: Definované ako BCR-ABL/kontrola transkriptorov ≤ 0,1 % pomocou RQ-PCR
vo vzorkách periférnej krvi.
Tabuľka 12: Dlhodobá účinnosť SPRYCELU v 3. fáze štúdie optimalizácie dávky: rezistencia alebo intolerancia na imatinib pacienti s chronickou fázou CML
a
Minimálne
obdobie
následného
sledovania
1.
rok 2.
roky 5.
rokov 7.
rokov
Veľká molekulárna odpoveď
Všetci pacienti NA 37 % (57/154) 44 % (71/160) 46 % (73/160)
Pacienti rezistentní na imatinib
Pacienti intolerantní na imatinib
Prežívanie bez progresie ochoreniab
NA 35 % (41/117) 42 % (50/120) 43 % (51/120) NA 43 % (16/37) 53 % (21/40) 55 % (22/40)
Všetci pacienti 90 % (86-95) 80 % (73; 87) 51 % (41; 60) 42 % (33; 51)
Pacienti rezistentní na imatinib
Pacienti intolerantní na imatinib
Celkové prežívanie
88 % (82; 94) 77 % (68; 85) 49 % (39; 59) 39 % (29; 49)
97 % (92; 100) 87 % (76; 99) 56 % (37; 76) 51 % (32; 67)
Všetci pacienti 96 % (93; 99) 91 % (86; 96) 78 % (72; 85) 65 % (56; 72)
Pacienti rezistentní na imatinib
94 % (90; 98) 89 % (84; 95) 77 % (69; 85) 63 % (53; 71)
Pacienti intolerantní na
100 % (100; 100) 95 % (88;
82 % (70; 94) 70 % (52; 82)
imatinib 100)
a Výsledky hlásené pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
b Progresia bola definovaná ako zvýšenie počtu WBC, strata CHR alebo MCyR, ≥ 30 % zvýšenie v Ph+ metafáze, potvrdená AP/BP ochorenia alebo smrť. PFS bolo analyzované na princípe úmyslu liečiť (intent-to-treat) a pacienti boli
následne sledovaní na udalosti vrátane dodatočnej liečby.
Na základe Kaplan-Meierových odhadov bol pomer pacientov liečených dasatinibom v dávke 100 mg jedenkrát denne, ktorí si udržali MCyR počas 18 mesiacov 93 % (95 % CI: [88 % – 98 %]).
Účinnosť bola tiež hodnotená u pacientov, ktorí boli intolerantní na imatinib. V tejto populácii pacientov, ktorí užívali 100 mg jedenkrát denne bola MCyR dosiahnutá u 77 % a CCyR u 67 %.
2. štúdia
V štúdii s pokročilou fázou CML a Ph+ ALL bola MaHR primárnym koncovým ukazovateľom. Z celkových 611 pacientov, boli pacienti v skupinách randomizovaní dasatinibom buď v dávke 140 mg jedenkrát denne alebo 70 mg dvakrát denne. Medián trvania liečby bol priemerne 6 mesiacov (rozpätie
0,03 – 31 mesiacov).
Dávkovacia schéma jedenkrát denne poukázala porovnateľnú účinnosť (nie horšiu) s dávkovacou schémou dvakrát denne primárneho koncového ukazovateľa účinnosti (rozdiel v MaHR 0,8 %, 95 % interval spoľahlivosti [-7,1 % – 8,7 %]); dávkovacia schéma 140 mg jedenkrát denne však potvrdila zlepšenie bezpečnosti a znášanlivosti.
Pomery odpovede sú uvedené v tabuľke 13.
Tabuľka 13: Účinnosť SPRYCELU v štúdii fázy III optimalizácie dávky: Pokročilá fáza
CML
a
Ph+
ALL
(2-ročné
výsledky)
a
L
y
mfo
idná
Akcelerovaná fáza
Myeloidná blastová
blastová fáza
Ph+ALL
(n= 158) fáza
(n=
75)
(n= 33) (n= 40)
M
a
H
R
b (95 % CI) CHRb
(95 % CI) NELb
66 % (59 – 74)
47 % (40 – 56)
19 %
28 % (18 – 40)
17 % (10 – 28)
11 %
42 % (26 – 61)
21 % (9 – 39)
21 %
38 % (23 – 54)
33 % (19 – 49)
5 %
(95 % CI) (13 – 26) (5 – 20) (9 –39) (1–17)
M
C
y
R
c (95 % CI) CCyR
39% (31 – 47)
32%
28% (18 – 40)
17%
52% (34 – 69)
39%
70% (54 – 83)
50%
(95
%
CI)
(25
–
40)
(10
–
2
8
)
(23
–
58)
(34
–
6
6
)
a Výsledky hlásené pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne (pozri časť 4.2).
b Kritéria hematologickej odpovede (všetky kritéria boli potvrdené po 4 týždňoch): Veľká hematologická odpoveď: (MaHR) = úplná hematologická odpoveď (CHR) + bez prítomnosti leukémie (NEL).
CHR: WBC ≤ ULN zdravotníckeho zariadenia, ANC ≥ 1 000/mm3, trombocyty < 100 000/mm3, žiadne blasty
alebopromyelocyty v periférnej krvi, blasty v krvi a v kostnej dreni ≤ 5 %, < 5 % myelocyty plus metamyelocyty v periférnejkrvi, bazofily v periférnej krvi < 20 % a žiadne extramedulárne postihnutia.
NEL: rovnaké kritériá ako pre CHR ale ANC ≥ 500/mm3 a < 1 000/mm3, alebo trombocyty ≥ 20 000/mm3 a
≤ 100 000/mm3.
c MCyR kombinácie obidvoch úplnej (0 % Ph+ metafáza) a čiastočnej (> 0 % – 35 %) odpovede. CI = interval spoľahlivosti ULN = horný limit normálneho rozpätia.
U pacientov s akcelerovanou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne sa nedosiahol medián trvania MaHR a medián celkového prežívania a medián PFS bol 25 mesiacov.
U pacientov s myeloidnou blastovou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne bol medián trvania MaHR 8 mesiacov, medián PFS bol 4 mesiace a medián celkového prežívania bol 8 mesiacov. U pacientov s lymfoidnou blastovou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne bol medián trvania MaHR 5 mesiacov, medián PFS bol 5 mesiacov a medián celkového prežívania bol 11 mesiacov.
Medián trvania MaHR u pacientov s Ph+ALL liečených s dávkovacím režimom 140 mg jedenkrát denne bol 5 mesiacov, medián PFS bol 4 mesiace a medián celkového prežívania bol 7 mesiacov.
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so SPRYCELOM v jednej alebo vo viacerých podskupinách pediatrickej populácie s pozitívnym chromozómom Philadelphia (BCR-ABL translokácia) s akútnou lymfoblastovou leukémiou (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
Pediatrickí pacienti s CML
Zo 130 pacientov s chronickou fázou CML (CML-CP) liečených v dvoch pediatrických štúdiách, otvorené, nerandomizované klinické skúšanie s nastavovaním dávky I. fáza a otvorené, nerandomizované klinické skúšanie II. fáza, bolo 84 pacientov (výhradne z klinického skúšania
II. fázy) novo diagnostikovaných s CML-CP a 46 pacientov (17 z klinického skúšania I. fázy a 29 z klinického skúšania II. fázy), bolo rezistentných alebo intolerantných na predchádzajúcu liečbu imatinibom. Deväťdesiat sedem zo 130 pediatrických pacientov s CML-CP sa liečilo SPRYCELOM tabletami v dávke 60 mg/m2 jedenkrát denne (maximálna dávka 100 mg jedenkrát denne u pacientov s vyšším BSA). Pacienti sa liečili až do progresie ochorenia alebo do neakcetovateľnej toxicity.
Kľúčové koncové ukazovatele účinnosti boli: kompletná cytogenetická odpoveď (CCyR), veľká cytogenetická odpoveď (MCyR) a veľká molekulárna odpoveď (MMR). Výsledky sú uvedené v tabuľke 14.
Tabuľka 14: Účinnosť SPRYCELU u pediatrických pacientov s CML-CP
Kumulatívna
odpoveď
v
priebehu
času
s
obdobím
minimálneho
následného
sledovania
3
mesiace 6
mesiacov 12
mesiacov 24
mesiacov
CCyR
(95 % CI)
Novo diagnostikovaní (N = 51)a
43,1 % (29,3; 57,8)
66,7 % (52,1; 79,2)
96,1 % (86,5; 99,5)
96,1 % (86,5; 99,5)
Predtým imatinib
(N = 46)b
45,7 %
(30,9; 61,0)
71,7 %
(56,5; 84,0)
78,3 %
(63,6; 89,1)
82,6 %
(68,6; 92,2)
MCyR
(95 % CI) Novo diagnostikovaní (N = 51)a
60,8 % (46,1; 74,2)
90,2 % (78,6; 96,7)
98,0 % (89,6; 100)
98,0 % (89,6; 100)
Predtým imatinib
(N = 46)b
60,9 %
(45,4; 74,9)
82,6 %
(68,6; 92,2)
89,1 %
(76,4; 96,4)
89,1 %
(76,4; 96,4)
MMR
(95 % CI) Novo diagnostikovaní (N = 51)a
7,8 % (2,2; 18,9)
31,4 % (19,1; 45,9)
56,9 % (42,2; 70,7)
74,5 % (60,4; 85,7)
Predtým imatinib

(N = 46)b
15,2 %
(6,3; 28,9)
26,1 %
(14,3; 41,1)
39,1 %
(25,1; 54,6)
52,2 %
(36,9; 67,1)
aPacienti z pediatrickej štúdie II. fázy s novo diagnostikovanou CML-CP, ktorí užívali perorálnu tabletovú formu
bPacienti z pediatrickej štúdie I. fázy a II. fázy s CML-CP s rezistenciou alebo intoleranciou na imatinib, ktorí užívali
perorálnu tabletovú formu
V pediatrickej štúdii I. fázy po minimálne 7 rokoch následného sledovania bol u 17 pacientov s CML- CP s rezistenciou alebo intoleranciou na imatinib medián trvania PFS 53,6 mesiaca a miera OS bola
82,4 %.
V pediatrickej štúdii II. fázy bola u pacientov, ktorí užívali tabletovú formu odhadnutá 24-mesačná miera PFS u 51 pacientov s novo diagnostikovanou CML-CP 94,0 % (82,6; 98,0) a 81,7 % (61,4;
92,0) u 29 pacientov s CML-CP s rezistenciou/intoleranciou na imatinib. Po 24 mesiacoch následného sledovania bolo OS u novo diagnostikovaných pacientov 100 % a 96,6 % u pacientov rezistentných alebo intolerantných na imatinib.
V pediatrickej štúdii II. fázy progredoval do blastovej fázy CML 1 novo diagnostikovaný pacient a
2 pacienti rezistentní alebo intolerantní na imatinib.
33 novo diagnostikovaných pediatrických pacientov s CML-CP dostalo SPRYCEL prášok na perorálnu suspenziu v dávke 72 mg/m2. Táto dávka predstavuje o 30 % nižšiu expozícii v porovnaní s odporúčanou dávkou (pozri časť 5.2. súhrn charakteristických vlastností lieku SPRYCEL prášok na perorálnu suspenziu). U týchto pacientov bolo CCyR a MMR po 12 mesiacoch CCyR: 87,9 % [95 % CI: (71,8-96,6)] a MMR: 45,5 % [95 % CI: (28,1-63,6)].
U pediatrických pacientov s CML-CP liečených dasatinibom predtým exponovaných imatinibu boli na konci liečby detekované mutácie: T315A, E255K a F317L. E255K a F317L však boli detekované aj pred liečbou. Na konci liečby neboli detekované žiadne mutácie u novo diagnostikovaných pacientov s CML-CP.
5.2 Farmakokinetické vlastnosti
Farmakokinetika dasatinibu sa hodnotila u 229 dospelých zdravých jedincov a u 84 pacientov. Absorpcia
Dasatinib sa u pacientov po perorálnom podaní rýchlo vstrebáva, pričom maximálne koncentrácie sa
dosiahnu medzi 0,5 – 3 hodinami. Po perorálnom podaní je vzostup priemernej expozície (AUCτ) približne úmerný prírastku dávky v celom rozmedzí dávok od 25 mg do 120 mg dvakrát denne. U pacientov je celkový priemerný konečný polčas dasatinibu približne 5 – 6 hodín.
Údaje od zdravých jedincov, ktorým sa podala jednorazová 100 mg dávka dasatinibu po 30 minútach po jedle s vysokým obsahom tuku, poukázali na 14 % zvýšenie priemernej hodnoty AUC dasatinibu. Požitie jedla s nízkym obsahom tuku 30 minút pred užitím dasatinibu viedlo k 21 % zvýšeniu priemernej hodnoty AUC dasatinibu. Pozorované vplyvy jedla nepredstavujú klinicky významné zmeny v expozícii.
Distribúcia
U pacientov má dasatinib veľký zdanlivý distribučný objem (2 505 l), čo svedčí o tom, že liek sa v rozsiahlej miere distribuuje v extravaskulárnom priestore. Pri klinicky významných koncentráciách dasatinibu bola väzba na plazmatické bielkoviny približne 96 % na základe in vitro experimentov.
Biotransformácia
Dasatinib sa u ľudí v rozsiahlej miere metabolizuje, pričom do tvorby metabolitov sú zapojené viaceré enzýmy. U zdravých jedincov, ktorým bolo podaných 100 mg dasatinibu označeného [14C], predstavoval nezmenený dasatinib 29 % cirkulujúcej rádioaktivity v plazme. Plazmatická koncentrácia a in vitro meraná aktivita svedčia o tom, že nie je pravdepodobné, že metabolity dasatinibu majú významnú úlohu v pozorovanej farmakológii lieku. CYP3A4 je hlavným enzýmom zodpovedným za metabolizmus dasatinibu.
Eliminácia
Prevláda vylučovanie stolicou, prevažne vo forme metabolitov. Po jednorazovej perorálnej dávke dasatinibu označeného [14C] sa približne 89 % dávky vylúčilo do 10 dní, pričom 4 % rádioaktivity sa zistili v moči a 85 % rádioaktivity sa zistilo v stolici. Nezmenený dasatinib predstavoval 0,1 % dávky v moči a 19 % dávky v stolici, pričom zvyšok dávky bol vo forme metabolitov.
Poruchafunkciepečeneaobličiek
Vplyv poruchy funkcie pečene na farmakokinetiku jednorazovej dávky dasatinibu bol hodnotený u
8 jedincov so stredne ťažkou poruchou funkcie pečene, ktorí užívali dávku 50 mg a u 5 jedincov s ťažkou poruchou funkcie pečene, ktorí užívali dávku 20 mg v porovnaní so zodpovedajúcimi zdravými jedincami, ktorí užívali dávku 70 mg dasatinibu. Priemerné Cmax dasatinibu upravené na dávku 70 mg sa znížili o 47 % a AUC o 8 % u jedincov so stredne ťažkou poruchou funkcie pečene v porovnaní s osobami s normálnou funkciou pečene. U jedincov s ťažkou poruchou funkcie pečene sa priemerné Cmax upravené na dávku 70 mg znížili o 43 % a AUC o 28 % v porovnaní s osobami s normálnou funkciou pečene (pozri časti 4.2 a 4.4).
Dasatinib a jeho metabolity sú v minimálnej miere vylučované obličkami. Pediatrickápopulácia
Farmakokinetika dasatinibu sa hodnotila u 104 pediatrických pacientov s leukémiou alebo solídnymi
nádormi (72 dostalo tabletovú formu a 32 dostalo prášok na perorálnu suspenziu).
Farmakokinetika tabletovej formy dasatinibu sa hodnotila u 72 pediatrických pacientov s recidívou leukémie alebo s refraktérnou leukémiou alebo so solídnymi nádormi s perorálnou dávkou v rozsahu od 60 do 120 mg/m2 jedenkrát denne a 50 až 110 mg/m2 dvakrát denne. Údaje dvoch štúdii boli spojené a potvrdili, že sa dasatinib rýchlo absorboval. Pri všetkých hladinách dávok a vekových skupinách sa pozoroval priemerný Tmax medzi 0,5 a 6 hodinami a priemerný polčas v rozsahu
od 2 do 5 hodín. PK dasatinibu potvrdila proporcionalitu dávky so zvýšením expozície v súvislosti s
dávkou, čo sa pozorovalo u pediatrických pacientov. Medzi deťmi a dospievajúcimi sa nepozorovali žiadne signifikantné rozdiely PK dasatinibu. Pri rôznych hladinách dávok sa geometrické priemery normalizovanej dávky dasatinibu Cmax, AUC (0-T) a AUC (INF) ukázali medzi deťmi a dospievajúcimi podobné. PPK simulácia na základe modelu predpovedala, že telesná hmotnosť odstupňuje odporúčanie na dávkovanie opísané pre tablety v časti 4.2, očakáva sa, že poskytne podobnú expozíciu pre tabletu v dávke 60 mg/m2. Tieto údaje sa majú zohľadniť, ak sa pacienti prestavujú z tabliet na prášok na perorálnu suspenziu alebo naopak.
5.3 Predklinické údaje o bezpečnosti
Profil predklinickej bezpečnosti dasatinibu sa hodnotil v sérii štúdií in vitro a in vivo na myšiach, potkanoch, opiciach a králikoch.
Primárna toxicita sa vyskytla v gastrointestinálnom, hematopoetickom a lymfoidnom systéme. Gastrointestinálna toxicita bola limitovaná dávku u potkanov a opíc, keďže črevo bolo hlavným cieľovým orgánom. U potkanov boli minimálne až mierne poklesy v parametroch erytrocytov sprevádzané zmenami kostnej drene; u opíc sa podobné zmeny vyskytli s nižšou incidenciou. Lymfoidná toxicita u potkanov pozostávala z lymfoidnej deplécie lymfatických uzlín, sleziny a
týmusu a zníženej hmotnosti lymfoidných orgánov. Zmeny v gastrointestinálnom, hematopoetickom a
lymfoidnom systéme boli reverzibilné po ukončení liečby.
Obličkové zmeny u opíc liečených počas 9 mesiacov sa obmedzovali na zvýšenie prirodzenej mineralizácie v obličkách. Kožné krvácanie bolo pozorované v štúdii akútnej jednorazovej perorálnej dávky u opíc, ale nebolo pozorované v štúdiách opakovanej dávky ani u opíc ani u potkanov.
U potkanov dasatinib inhiboval agregáciu trombocytov in vitro a predĺžil čas krvácania z kutikuly
in vivo, ale nevyvolal spontánne krvácanie.
Aktivita dasatinibu in vitro v teste hERG a teste na Purkyňových vláknach svedčí o možnom predĺžení repolarizácie srdcových komôr (QT interval). V štúdii s jednorazovou dávkou in vivo na opiciach,
ktoré boli pri vedomí a sledované telemetricky, však neboli žiadne zmeny v QT intervale, ani v tvare
vlny na EKG.
Dasatinib nebol mutagénny v testoch in vitro na bakteriálnych bunkách (Amesov test) a nebol genotoxický v teste in vivo na potkaních mikronukleoch. Dasatinib mal klastogénny účinok in vitro na deliace sa ovariálne bunky škrečka (CHO).
Dasatinib neovplyvnil obvyklú samčiu alebo samičiu fertilitu potkanov a včasnú štúdiu embryofetálneho vývoja, ale indukoval embryoletalitu v dávkach zodpovedajúcich klinickej expozícii u ľudí. V štúdiách embryofetálneho vývoja podobne spôsobil dasatinib embryoletalitu, ktorá bola spojená so zníženou veľkosťou vrhu u potkanov ako aj zmeny skeletu plodu u potkanov aj králikov. Tieto účinky sa vyskytovali pri dávkach, ktoré nespôsobovali toxické prejavy u matky naznačujúc, že dasatinib je selektívna toxická látka na reprodukciu od implementácie až počas celej organogenézy.
U myší vyvolal dasatinib imunosupresiu, ktorá súvisela s dávkou a bola účinne zvládnutá znížením dávky a/alebo zmenami dávkovacej schémy. Dasatinib mal fototoxický potenciál v teste fototoxicity in vitro zameranom na prenikanie neutrálnej červene do myších fibroblastov. Dasatinib sa považoval za nefototoxický in vivo po jednorazovom perorálnom podaní samičej bezchlpej myši v expozíciách zodpovedajúcich nanajvýš 3-násobnej expozícii u ľudí po podaní odporúčanej terapeutickej dávky (vychádzajúcej z AUC).
V dvojročnej štúdii karcinogenity sa potkanom podávali perorálne dávky dasatinibu 0,3; 1 a
3 mg/kg/deň. Najvyššia dávka viedla k plazmatickej hladine expozície (AUC) vo všeobecnosti ekvivalentnej expozícii u ľudí pri odporúčanom rozsahu úvodných dávok od 100 mg do 140 mg denne. Zistilo sa štatisticky významné zvýšenie celkovej incidencie karcinómov skvamóznych buniek a papilómov v maternici a krčku maternice u samičiek s vysokou dávkou a adenóm prostaty u samcov s nízkou dávkou. Významnosť nálezov zo štúdie karcinogenity na potkanoch pre ľudí nie je známa.
6
. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
tablety monohydrát laktózy mikrokryštalická celulóza sodná soľ kroskarmelózy hydroxypropylcelulóza magnéziumstearát
Filmovýobal hypromelóza
oxid titaničitý (E171)
makrogol 400
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
SPRYCEL20mg,SPRYCEL50mgaSPRYCEL70mgfilmomobalenétablety
Alu/Alu blistre (kalendárne blistre alebo perforované blistre umožňujúce oddelenie jednotlivej dávky). Fľaša HDPE s polypropylénovým detským bezpečnostným uzáverom.
Škatuľa obsahujúca 56 filmom obalených tabliet v 4 kalendárnych blistroch obsahujúcich 14 filmom obalených tabliet.
Škatuľa obsahujúca 60 x 1 filmom obalených tabliet v perforovaných blistroch umožňujúcich oddelenie jednotlivej dávky.
Škatuľa obsahujúca jednu fľašu so 60 filmom obalenými tabletami.
SPRYCEL80mg,SPRYCEL100mgaSPRYCEL140mgfilmomobalenétablety
Alu/Alu blistre (perforované blistre umožňujúce oddelenie jednotlivej dávky). Fľaša HDPE s polypropylénovým detským bezpečnostným uzáverom.
Škatuľa obsahujúca 30 x 1 filmom obalených tabliet v perforovaných blistroch umožňujúcich oddelenie jednotlivej dávky.
Škatuľa obsahujúca jednu fľašu s 30 filmom obalenými tabletami. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Filmom obalené tablety pozostávajú z jadra tablety, ktoré je obklopené filmovým obalom, aby sa zabránilo vystaveniu zdravotníckych pracovníkov liečivu. Pri manipulácii s tabletami, ktoré sú neúmyselne rozdrvené alebo zlomené sa na náležitú likvidáciu odporúča použiť latexové alebo nitrilové rukavice, aby sa minimalizovalo riziko kožnej expozície.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
BRISTOL-MYERS SQUIBB PHARMA EEIG Uxbridge Business Park
Sanderson Road Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLO
SPRYCEL20mgfilmomobalenétablety
EU/1/06/363/004
EU/1/06/363/007
EU/1/06/363/001
SPRYCEL50mgfilmomobalenétablety
EU/1/06/363/005
EU/1/06/363/008
EU/1/06/363/002
SPRYCEL70mgfilmomobalenétablety
EU/1/06/363/006
EU/1/06/363/009
EU/1/06/363/003
SPRYCEL80mgfilmomobalenétablety
EU/1/06/363/013
EU/1/06/363/012
SPRYCEL100mgfilmomobalenétablety
EU/1/06/363/011
EU/1/06/363/010
SPRYCEL140mgfilmomobalenétablety
EU/1/06/363/015
EU/1/06/363/014
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 20. novembra 2006
Dátum posledného predĺženia registrácie: 15. júla 2016
10
. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúre pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
SPRYCEL 10 mg/ml prášok na perorálnu suspenziu
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna fľaška prášku na perorálnu suspenziu obsahuje 990 mg dasatinibu (vo forme monohydrátu). Po príprave obsahuje jedna fľaška 99 ml perorálnej suspenzie. Každý ml perorálnej suspenzie obsahuje 10 mg dasatinibu (vo forme monohydrátu).
Pomocná látka so známym účinkom
Každý ml perorálnej suspenzie obsahuje približne 291 mg sacharózy, 2.1 mg sodíka, 0,25 mg
benzoanu sodného, 0,25 mg kyseliny benzoovej, 0,017 mg benzylalkoholu a < 10 ppm oxidu siričitého
(E220).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok na perorálnu suspenziu. Biely až takmer biely prášok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
SPRYCEL je indikovaný na liečbu pediatrických pacientov s:
§ novo diagnostikovaným pozitívnym chromozómom Philadelphia chronickej myeloidnej leukémie v chronickej fáze (Ph+ CML-CP) alebo Ph+ CML-CP s rezistenciou alebo intoleranciou na
predošlú liečbu zahŕňajúcu imatinib.
4.2 Dávkovanie a spôsob podávania
Liečbu má začať lekár, ktorý má skúsenosti v diagnostikovaní a liečbe pacientov s leukémiou. Dávkovanie
Dávkovanie je na základe telesnej hmotnosti (pozri tabuľku 1). Dasatinib sa podáva perorálne
jedenkrát denne buď vo forme SPRYCELU prášku na perorálnu suspenziu alebo ako filmom obalené tablety (pozri súhrn charakteristických vlastností lieku SPRYCEL filmom obalené tablety). Dávka sa má na základe zmien v telesnej hmotnosti prepočítať každé 3 mesiace alebo, ak je to potrebné, častejšie. Tableta sa neodporúča pre pacientov vážiacich menej ako 10 kg; u týchto pacientov sa má použiť prášok na perorálnu suspenziu. Zvýšenie alebo zníženie dávky sa odporúča na základe individuálnej odpovede pacienta a znášanlivosti. U detí vo veku do 1 roka nie sú žiadne skúsenosti s liečbou SPRYCELOM.
SPRYCEL filmom obalené tablety a SPRYCEL prášok na perorálnu suspenziu nie sú bioekvivalentné. Pacienti, ktorí sú schopní tablety prehĺtať a želajú si prestavenie liečby zo SPRYCELU prášku na perorálnu suspenziu na SPRYCEL tablety alebo pacienti, ktorí nie sú schopní tablety prehĺtať a želajú si prestavenie liečby z tabliet na perorálnu suspenziu, tak môžu spraviť pod podmienkou, že sa dodržia odporúčania správneho dávkovania pre liekovú formu.
Odporúčané začiatočné denné dávkovanie SPRYCELU prášku na perorálnu suspenziu pre pediatrických pacientov a dospelých pacientov, ktorí nemôžu prehĺtať tablety je uvedené v tabuľke 1.
Tabuľka 1: Dávkovanie SPRYCELU prášku na perorálnu suspenziu pre pacientov s
Ph+ CML-CP
(10
mg/ml
p
o
príprave
suspenzie)
Telesná
hmotnosť
(kg)
Denná
d
ávka,
ml
(mg)
5 až menej ako 10 kg 4 ml (40 mg)
10 až menej ako 20 kg 6 ml (60 mg)
20 až menej ako 30 kg 9 ml (90 mg)
30 až menej ako 45 kg 10,5 ml (105 mg)
minimálne 45 kg 12 ml (120 mg)
Dávka na použitie prášku na perorálnu suspenziu u dospelých pacientov s akcelerovanou, myeloidnou
alebo lymfoidnou blastovou fázou (pokročilá fáza) CML alebo Ph+ ALL nie je stanovená.
Trvanie liečbyV klinických štúdiách s dospelými a pediatrickými pacientmi sa v liečbe SPRYCELOM pokračovalo až do progresie ochorenia alebo až dovtedy, kým u pacienta nevznikla intolerancia. Účinok ukončenia liečby na dlhodobý výsledok ochorenia po dosiahnutí cytogenetickej alebo molekulárnej odpovede
vrátane kompletnej cytogenetickej odpovede (complete cytogenetic response, CCyR), veľkej
molekulárnej odpovede (major molecular response, MMR) a MR4.5 sa neskúmal.
Na dosiahnutie odporúčanej dávky je SPRYCEL dostupný ako 20 mg, 50 mg, 70 mg, 80 mg, 100 mg a
140 mg filmom obalené tablety a ako prášok na perorálnu suspenziu (10 mg/ml po príprave suspenzie). Zvýšenie alebo zníženie dávky sa odporúča na základe pacientovej odpovede a znášanlivosti.
Zvyšovanie dávkyU pediatrických pacientov, ktorí nedosiahli hematologickú, cytogenetickú a molekulárnu odpoveď pri odporúčaných časových intervaloch sa podľa súčasných liečebných postupov odporúčajú nasledovné zvyšovania dávky uvedené v tabuľke 2.
Tabuľka2: ZvyšovaniedávkyupacientovsPh+CML-CP Dávka(maximálnadávkanadeň) Začiatočnádávka Zvýšenie Prášok na perorálnu suspenziu 4 ml (40 mg) 5 ml (50 mg)
6 ml (60 mg) 8 ml (80 mg)
9 ml (90 mg) 12 ml (120 mg)
10,5 ml (105 mg) 14 ml (140 mg)
12 ml (120 mg) 16 ml (160 mg) Úprava dávky kvôli nežiaducim reakciámMyelosupresiaV klinických štúdiách sa myelosupresia liečila prerušením dávky, znížením dávky alebo ukončením skúšanej liečby. Transfúzia trombocytov a transfúzia erytrocytov sa použili v prípade potreby.
U pacientov s rezistentnou myelosupresiou sa použil hematopoetický rastový faktor. Odporúčania úpravy dávok sú zhrnuté v tabuľke 3.
 Tabuľka 3: Úprava dávkovania počas neutropénie a trombocytopénie u pacientov s Ph+ CML- CP
Tabuľka 3: Úprava dávkovania počas neutropénie a trombocytopénie u pacientov s Ph+ CML- CP
1. Ak cytopénia pretrváva dlhšie
Dávka (maximálna dávka na deň)
ako 3 týždne, preverte, či
Pôvodná začiatočná dávka
Zníženie dávky o jednu úroveň
Zníženie dávky o dve úrovne
cytopénia súvisí s
leukémiou (punkcia alebo biopsia kostnej drene).
2. Ak cytopénia nesúvisí s leukémiou, prerušte liečbu až dovtedy, kým nebude ANC
≥ 1,0 x 109/l a
počet trombocytov
≥ 75 x 109/l a
pokračujte s pôvodnou začiatočnou dávkou alebo so zníženou dávkou.
Prášok na perorálnu suspenziu
4 ml (40 mg) 3 ml (30 mg) 2 ml (20 mg)
6 ml (60 mg) 5 ml (50 mg) 4 ml (40 mg)
9 ml (90 mg) 7 ml (70 mg) 6 ml (60 mg)
10,5 ml (105 mg) 9 ml (90 mg) 7 ml (70 mg)
12 ml (120 mg) 10 ml (100 mg) 8 ml (80 mg)
3. Ak dôjde k
opakovanej cytopénii, zopakujte punkciu/biopsiu kostnej drene a pokračujte v
liečbe so zníženou dávkou.
ANC: absolútny počet neutrofilov
Ak sa u pediatrických pacientov s Ph+ CML-CP opakovane vyskytne neutropénia alebo trombocytopénia ≥ 3. stupňa počas kompletnej hematologickej odpovede (complete hematologic response, CHR), liečba SPRYCELOM sa má prerušiť a potom sa môže v liečbe pokračovať so zníženou dávkou. Dočasné zníženia dávok na stredne závažné stupne cytopénie a odpoveď ochorenia sa majú vykonať podľa potreby.
Nehematologické nežiaduce reakcie
Ak počas liečby dasatinibom vznikne stredne závažná nehematologická nežiaduca reakcia 2. stupňa, liečba sa má prerušiť až do vymiznutia tejto nežiaducej reakcie alebo návratu k pôvodným hodnotám. Má sa pokračovať s rovnakou dávkou, ak ide o prvý výskyt a dávka sa má znížiť, ak ide o opakujúcu sa nežiaducu reakciu. Ak počas liečby dasatinibom vznikne závažná nehematologická nežiaduca reakcia 3. alebo 4. stupňa, liečba sa musí prerušiť až do vymiznutia tejto nežiaducej reakcie. Potom sa môže v liečbe pokračovať s použitím zníženej dávky v závislosti od začiatočnej závažnosti nežiaducej reakcie. U CML-CP pediatrických pacientov s nehematologickými nežiaducimi reakciami sa má postupovať podľa odporúčaní na zníženie dávky pre hematologické nežiaduce reakcie, ktoré sú opísané vyššie.
Pleurálny výpotok
Ak je diagnostikovaný pleurálny výpotok, liečba dasatinibom sa má prerušiť až kým bude pacient vyšetrený, asymptomatický alebo sa vráti k pôvodným hodnotám. Ak sa epizóda nezlepší približne počas jedného týždňa, má sa zvážiť postup s použitím diuretík alebo kortikosteroidov alebo oboch súčasne (pozri časti 4.4 a 4.8). Po vyriešení prvej epizódy, sa má zvážiť opätovná liečba dasatinibom v dávke na rovnakej úrovni. Po vyriešení nasledujúcich epizód, sa má opätovne začať liečba
dasatinibom v zníženej dávke o jednu úroveň. Po vyriešení závažnej epizódy (3. alebo 4. stupeň) môže liečba, ak je to vhodné, pokračovať so zníženou dávkou v závislosti od začiatočnej závažnosti nežiaducej reakcie.
Špeciálnepopulácie
Pediatrická populácia
Bezpečnosť a účinnosť SPRYCELU u detí mladších ako 1 rok neboli doteraz stanovené. K dispozícii nie sú žiadne údaje (pozri časť 5.1).
Starší ľudia
U týchto pacientov sa nepozorovali žiadne klinicky významné farmakokinetické rozdiely súvisiace s vekom. U starších ľudí nie je potrebné osobitné odporúčanie dávkovania.
Porucha funkcie pečene
Pacienti s miernou, stredne závažnou alebo závažnou poruchou funkcie pečene môžu dostávať odporúčanú začiatočnú dávku. SPRYCEL sa však musí používať opatrne u pacientov s poruchou funkcie pečene (pozri časť 5.2).
Porucha funkcie obličiek
Neuskutočnili sa žiadne klinické štúdie so SPRYCELOM s pacientmi so zníženou funkciou obličiek (zo štúdie pacientov s novo diagnostikovanou chronickou fázou CML boli vylúčení pacienti s koncentráciou kreatinínu v sére > 3-násobok hornej hranice normálneho rozpätia a zo štúdií pacientov s chronickou fázou CML s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom boli vylúčení pacienti s koncentráciou kreatinínu v sére > 1,5-násobok hornej hranice normálneho rozpätia). Vzhľadom na to, že renálny klírens dasatinibu a jeho metabolitov je < 4 %, u pacientov s renálnou insuficienciou sa neočakáva pokles celkového telesného klírensu.
Spôsobpodávania
SPRYCEL sa musí podávať perorálne. Môže sa užívať s jedlom alebo bez jedla a má sa užívať dôsledne buď ráno alebo večer. Perorálne suspenzia sa nesmie užívať s grapefruitom alebo grapefruitovým džúsom (pozri časť 4.5). Pripravená perorálne suspenzia sa môže ďalej miešať s mliekom, jogurtom, jablkových džúsom alebo jablkovým kompótom.
Detaily o príprave a podávaní tohto lieku a pokyny na používanie, pozri časť 6.6.
Pacienti alebo opatrovatelia majú byť poučení, aby postupovali podľa „Pokynov na prípravu prášku na perorálnu suspenziu“ a „Pokynov na podávanie pacientovi“ na konci písomnej informácie.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Klinickyvýznamnéinterakcie
Dasatinib je substrátom a inhibítorom cytochrómu P450 (CYP) 3A4. Z tohto dôvodu existuje možnosť interakcie s inými súbežne podávanými liekmi, ktoré sú metabolizované prevažne prostredníctvom CYP3A4 alebo ktoré modulujú aktivitu CYP3A4 (pozri časť 4.5).
Súbežné užívanie dasatinibu a liekov alebo látok, ktoré silne inhibujú CYP3A4 (napr. ketokonazol, itrakonazol, erytromycín, klaritromycín, ritonavir, telitromycín, grapefruitový džús) môže zvýšiť
expozíciu dasatinibu. Z tohto dôvodu sa u pacientov liečených dasatinibom neodporúča súbežné podávanie silného inhibítora CYP3A4 (pozri časť 4.5).
Súbežné užívanie dasatinibu a liekov, ktoré indukujú CYP3A4 (napr. dexametazón, fenytoín, karbamazepín, rifampicín, fenobarbital alebo rastlinné lieky obsahujúce Hypericum perforatum, známy aj ako Ľubovník bodkovaný), môže značne znížiť expozíciu dasatinibu, a tým prípadne zvýšiť riziko zlyhania liečby. Preto sa u pacientov liečených dasatinibom má zvoliť súbežné podávanie alternatívnych liekov s menším potenciálom pre indukciu CYP3A4 (pozri časť 4.5).
Súbežné užívanie dasatinibu a substrátu CYP3A4 môže zvýšiť expozíciu substrátu CYP3A4. Z tohto dôvodu je potrebná opatrnosť, ak sa dasatinib podáva súbežne so substrátmi CYP3A4 s úzkou terapeutickou šírkou, ako sú astemizol, terfenadín, cisaprid, pimozid, chinidín, bepridil alebo námeľové alkaloidy (ergotamín, dihydroergotamín) (pozri časť 4.5).
Súbežné užívanie dasatinibu a histamínového-2 (H2) antagonistu (napr. famotidínu), inhibítora protónovej pumpy (napr. omeprazolu) alebo hydroxidu hlinitého/hydroxidu horečnatého môže znížiť expozíciu dasatinibu. Preto sa H2-antagonisty a inhibítory protónovej pumpy neodporúčajú a prípravky s obsahom hydroxidu hlinitého/hydroxidu horečnatého sa majú podať 2 hodiny pred alebo 2 hodiny po podaní dasatinibu (pozri časť 4.5).
Špeciálnepopulácie
Na základe záverov farmakokinetickej štúdie s jednorazovou dávkou, pacienti s ľahkou, stredne ťažkou alebo ťažkou poruchou funkcie pečene môžu dostávať odporúčanú začiatočnú dávku (pozri časť 5.2). Z dôvodu obmedzení klinickej štúdie sa odporúča opatrnosť pri podávaní dasatinibu pacientom s poruchou funkcie pečene.
Významnénežiaducereakcie
Myelosupresia
Liečba dasatinibom sa spája s anémiou, neutropéniou a trombocytopéniou. Ich výskyt je skorší a častejší u pacientov v pokročilej fáze CML alebo s Ph+ ALL ako u pacientov v chronickej fáze CML. U pacientov s pokročilou fázou CML alebo s Ph+ ALL sa kontrola kompletného krvného obrazu musí vykonávať raz za týždeň počas prvých 2 mesiacov a potom raz za mesiac alebo ak je to klinicky indikované. U pacientov s chronickou fázou CML sa kontrola kompletného krvného obrazu musí vykonávať raz za 2 týždne počas 12 týždňov, potom raz za 3 mesiace alebo ak je to klinicky indikované. Myelosupresia je obvykle reverzibilná a zvyčajne bola zvládnutá dočasným vysadením dasatinibu alebo redukciou dávky (pozri časti 4.2 a 4.8).
Krvácanie
U pacientov s chronickou fázou CML (n=548) malo 5 pacientov (1 %) užívajúcich dasatinib krvácanie
3. alebo 4. stupňa. V klinických štúdiách s pacientmi s pokročilou fázou CML, ktorí užívali odporúčanú dávku SPRYCELU (n=304) sa u 1 % pacientov vyskytlo ťažké krvácanie do centrálneho nervového systému (CNS). Jeden prípad bol smrteľný a spájal sa s trombocytopéniou 4. stupňa podľa Všeobecných kritérií toxicity (Common Toxicity Criteria, CTC). Gastrointestinálne krvácanie 3. alebo
4. stupňa sa vyskytlo u 46 % pacientov s pokročilou fázou CML a obvykle si vyžadovalo prerušenie liečby a podanie transfúzií. Krvácanie iného druhu 3. alebo 4. stupňa sa vyskytlo u 2 % pacientov
s pokročilou fázou CML. Väčšina nežiaducich reakcií spojených s krvácaním u týchto pacientov zvyčajne súvisela s trombocytopéniou 3. alebo 4. stupňa (pozri časť 4.8). Okrem toho hodnotenia krvných doštičiek in vitro a in vivo poukázali na to, že liečba SPRYCELOM reverzibilne ovplyvňuje aktiváciu krvných doštičiek.
Opatrnosť je potrebná, ak sa u pacientov vyžaduje užívanie liekov, ktoré potláčajú funkciu krvných doštičiek alebo antikoagulancií.
Retencia tekutín
Používanie dasatinibu sa spája s retenciou tekutín. V klinickej štúdii fázy III u pacientov s novo diagnostikovanou chronickou fázou CML, sa hlásila retencia tekutín 3. alebo 4. stupňa u 13 pacientov (5 %) v skupine liečenej dasatinibom a u 2 pacientov (1 %) v skupine liečenej imatinibom po
minimálne 60 mesiacoch následného sledovania (pozri časť 4.8). Zo všetkých pacientov s chronickou fázou CML liečených SPRYCELOM sa u 32 pacientov (6 %), ktorí dostávali SPRYCEL
v odporúčanej dávke (n=548) vyskytla závažná retencia tekutín. V klinických štúdiách s pacientmi s
pokročilou fázou CML, ktorí dostávali SPRYCEL v odporúčanej dávke (n=304) sa hlásila retencia tekutín 3. alebo 4. stupňa u 8 % pacientov, zahŕňajúca pleurálny výpotok 3. alebo 4. stupňa hlásený u
7 % pacientov a perikardiálny výpotok 3. alebo 4. stupňa hlásený u 1 % pacientov. U týchto pacientov sa hlásil pľúcny edém 3. alebo 4. stupňa a pľúcna hypertenzia 3. alebo 4. stupňa u 1 % pacientov.
Pacienti, u ktorých vzniknú príznaky svedčiace o pleurálnom výpotku, ako je dyspnoe alebo suchý kašeľ, musia byť vyšetrení pomocou röntgenu hrudníka. Pleurálny výpotok 3. alebo 4. stupňa si môže vyžadovať torakocentézu a oxygenoterapiu. Nežiaduce reakcie retencie tekutín boli zvyčajne zvládnuté pomocou podporných opatrení zahŕňajúcich podávanie diuretík a krátkodobé podávanie steroidov (pozri časti 4.2 a 4.8). U pacientov vo veku 65 rokov a starších je viac pravdepodobný výskyt pleurálneho výpotku, dyspnoe, kašľa, perikardiálneho výpotku a kongestívneho zlyhania srdca než u mladších pacientov, a preto majú byť starostlivo sledovaní.
Pľúcna arteriálna hypertenzia (PAH)
PAH (prekapilárna pľúcna hypertenzia potvrdená pravostrannou katetrizáciou srdca) sa hlásila v súvislosti s liečbou dasatinibom (pozri časť 4.8). V týchto prípadoch sa hlásila PAH po začatí liečby dasatinibom, a to po viac ako jednom roku liečby.
Pacienti majú byť vyšetrení na prejavy a príznaky základného kardiopulmonálneho ochorenia pred začatím liečby dasatinibom. Na začiatku liečby sa má vykonať echokardiografia u každého pacienta s prítomnými príznakmi srdcového ochorenia a má sa zvážiť u pacientov s rizikovými faktormi srdcového alebo pľúcneho ochorenia. Pacienti u ktorých sa po začatí liečby vyvinie dyspnoe a únava, majú byť vyšetrení na vylúčenie bežných etiológií zahŕňajúce pleurálny výpotok, pľúcny edém, anémiu alebo pľúcnu infiltráciu. V súlade s odporúčaniami manažmentu nehematologických nežiaducich reakcií (pozri časť 4.2) sa má dávka dasatinibu znížiť alebo sa má liečba prerušiť počas tohto vyšetrenia. Ak sa nenájde vysvetlenie alebo ak po znížení dávky alebo prerušení liečby nedôjde k žiadnemu zlepšeniu, má sa uvažovať o diagnóze PAH. Diagnostický prístup sa má riadiť štandardnými postupmi. Ak sa potvrdí PAH, liečba dasatinibom sa má trvale ukončiť. Monitorovanie
sa má vykonávať podľa štandardných postupov. Zlepšenie hemodynamických a klinických parametrov sa pozorovalo u pacientov liečených dasatinibom s PAH po ukončení liečby dasatinibom.
Predĺženie QT intervalu
Údaje in vitro svedčia o tom, že dasatinib môže predĺžiť repolarizáciu srdcových komôr (QT interval) (pozri časť 5.3). Z 258 pacientov liečených dasatinibom a z 258 pacientov liečených imatinibom s minimálne 60 mesačným následným sledovaním v štúdii fázy III s novo diagnostikovanou chronickou fázou CML, 1 pacient (<1 %) v každej skupine mal predĺžený QTc ako nežiaducu reakciu. Medián zmien vo QTcF podľa východiskových hodnôt bol 3,0 ms u pacientov liečených dasatinibom v porovnaní s 8,2 ms u pacientov liečených imatinibom. Jeden pacient (< 1 %), v každej skupine mal QTcF > 500 ms. U 865 pacientov s leukémiou liečených dasatinibom v klinických štúdiách fázy II
boli priemerné zmeny od východiskových hodnôt QTc intervalu s korekciou podľa metódy Fridericia
(QTcF) 4 - 6 ms; horné 95 % intervaly spoľahlivosti pre všetky priemerné zmeny od východiskových hodnôt boli < 7 ms (pozri časť 4.8).
Z 2 182 pacientov s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom, ktorí užívali dasatinib v klinických štúdiách malo 15 (1 %) predĺženie QTc hlásené ako nežiaduci účinok. Dvadsaťjeden z týchto pacientov (< 1 %) malo QTcF > 500 ms.
Dasatinib sa musí podávať opatrne pacientom, u ktorých došlo alebo u ktorých môže dôjsť k predĺženiu QTc. Toto zahŕňa pacientov s hypokaliémiou alebo hypomagneziémiou, pacientov s vrodeným syndrómom dlhého QT, pacientov užívajúcich antiarytmiká alebo iné lieky, ktoré vedú k predĺženiu QT a pacientov liečených vysokými kumulatívnymi dávkami antracyklínu. Hypokaliémia alebo hypomagneziémia sa majú pred podaním dasatinibu upraviť.
Nežiaduce reakcie na srdce
Dasatinib sa skúmal v randomizovanej klinickej štúdií s 519 pacientmi s novo diagnostikovanou CML v chronickej fáze, ktorá zahŕňala pacientov s predošlými srdcovým ochoreniami. Nežiaduce reakcie na srdce z kongestívneho srdcového zlyhania/srdcovej dysfunkcie, perikardiálneho výpotku, arytmií, palpitácií, predĺženie QT intervalu a infarkt myokardu (vrátane úmrtia) sa hlásili u pacientov užívajúcich dasatinib. Nežiaduce srdcové udalosti boli častejšie u pacientov s rizikovými faktormi alebo so srdcovým ochorením v anamnéze. Pacienti s rizikovými faktormi (napr. hypertenzia, hyperlipidémia, diabetes) alebo srdcovým ochorením v anamnéze (napr. predošlá perkutánna koronárna intervencia, dokázané ochorenie koronárnych artérií) majú byť starostlivo sledovaní pre klinické prejavy alebo príznaky zhodné so srdcovou dysfunkciou, ako je bolesť na hrudníku, skrátené dýchanie a potenie.
Ak sa tieto klinické prejavy alebo príznaky vyvinú, lekárom sa odporúča prerušiť podávanie dasatinibu a zvážiť potrebu alternatívnej liečby špecifickej pre CML. Po vyriešení sa má pred obnovením liečby dasatinibom vykonať funkčné posúdenie. Dasatinib sa môže znovu nasadiť v pôvodnej dávke pri miernych/stredne závažných nežiaducich reakciách (≤ 2. stupeň) a môže pokračovať na úrovni zníženej dávky pri závažných nežiaducich reakciách (≥ 3. stupeň) (pozri časť 4.2). Pacienti pokračujúci v liečbe, majú byť pravidelne monitorovaní.
Pacienti s nekontrolovaným alebo závažným kardiovaskulárnym ochorením neboli zaradení do klinických štúdií.
Reaktivácia hepatitídy B
Reaktivácia hepatitídy B u pacientov, ktorí sú chronickými prenášačmi tohto vírusu, sa vyskytla v prípade, že títo pacienti užívali inhibítory BCR-ABL-tyrozínkinázy. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie.
Pacienti majú byť vyšetrení na HBV infekciu pred začatím liečby liekom SPRYCEL. Pred začatím liečby u pacientov s pozitívnym sérologickým testom na hepatitídu B (vrátane pacientov s aktívnym ochorením) a u pacientov s pozitívnym testom na HBV infekciu počas liečby je potrebné konzultovať s odborníkmi na ochorenia pečene a liečbu hepatitídy B. Prenášači vírusu HBV, ktorí potrebujú liečbu liekom SPRYCEL, majú byť pozorne sledovaní na prejavy a príznaky aktívnej HBV infekcie počas celej liečby a niekoľko mesiacov po ukončení liečby (pozri časť 4.8).
Účinkynarastavývojupediatrickýchpacientov
V pediatrických klinických skúšaniach so SPRYCELOM s pediatrickými pacientmi rezistentnými/intolerantnými na imatinib a s predtým neliečenými pediatrickými pacientmi po minimálne 2 rokoch liečby sa hlásili nežiaduce udalosti súvisiace s liečbou spojené s rastom kostí a vývojom u 6 (4,6 %) pacientov, jedna z nich bola závažnej intenzity (spomalenie rastu 3. stupňa). Týchto 6 hlásení zahŕňalo hlásenia oneskorenej fúzie epifýz, osteopéniu, spomalenie rastu a gynekomastiu (pozri časť 5.1). Tieto výsledky je ťažké interpretovať v kontexte chronických ochorení, ako je CML a je potrebné dlhodobé následné sledovanie.
Pomocnélátky
Sodík
Tento liek obsahuje 2,1 mg sodíka na ml SPRYCELU perorálnej suspenzie, čo zodpovedá 2,1 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Sacharóza
Po príprave s vodou obsahuje SPRYCEL prášok na perorálnu suspenziu približne 0,29 g/ml sacharózy. Pri odporúčanom pediatrickom dávkovaní obsahuje SPRYCEL perorálna suspenzia
1,17 gramov sacharózy v 40 mg dasatinibu a 4,37 gramov sacharózy v 150 mg dasatinibu. Musí sa to vziať do úvahy u pacientov s diabetes mellitus.
Pacienti so zriedkavými dedičnými problémami intolerancie fruktózy, glukózogalaktózovej malabsorpcie alebo deficitu sacharázy a izomaltázy nesmú užívať tento liek.
Môže škodiť zubom.
Kyselina benzoová a benzoany
SPRYCEL obsahuje 0,25 mg kyseliny benzoovej v každom ml perorálnej suspenzie a 0,25 mg benzoanu sodného v každom ml perorálnej suspenzie.
Kyselina benzoová/soľ benzoanu môže zhoršiť novorodeneckú žltačku (žltnutie kože a očí) (až do veku 4 týždňov).
Benzylalkohol
SPRYCEL obsahuje 0,017 mg benzylalkoholu v každom ml perorálnej suspenzie. Benzylalkohol môže spôsobiť alergické reakcie.
Z dôvodu respiračných príznakov monitorujte pacientov mladších ako 3 roky.
SPRYCEL sa nemá používať počas gravidity, ak si klinický stav ženy nevyžaduje liečbu dasatinibom (pozri časť 4.6). Upozornite pacientky, ktoré sú tehotné alebo ktoré môžu otehotnieť, o možnom riziku pre plod, pretože dasatinib a pomocná látka benzylalkohol sa môžu v priebehu času kumulovať a spôsobiť metabolickú acidózu.
Používajte s opatrnosťou u pacientov s poruchou funkcie pečene alebo s poruchou funkcie obličiek, pretože benzylalkohol sa môže v priebehu času kumulovať a spôsobiť metabolickú acidózu.
Oxid siričitý (E220)
Zriedkavo môže vyvolať závažné reakcie z precitlivenosti a bronchospazmus.
4.5 Liekové a iné interakcie
Liečivá,ktorémôžuzvýšiťplazmatickékoncentráciedasatinibu
Štúdie in vitro poukazujú na to, že dasatinib je substrátom CYP3A4. Súbežné užívanie dasatinibu a liekov alebo látok, ktoré silne inhibujú CYP3A4 (napr. ketokonazol, itrakonazol, erytromycín, klaritromycín, ritonavir, telitromycín, grapefruitový džús) môže zvýšiť expozíciu dasatinibu. Preto sa u pacientov liečených dasatinibom neodporúča systémové podávanie silného inhibítora CYP3A4.
V klinicky relevantných koncentráciách sa v priemere 96 % dasatinibu viaže na plazmatické proteíny na základe in vitro experimentov. Neboli vykonané štúdie hodnotiace interakcie dasatinibu s inými liekmi viažucimi sa na proteíny. Potenciál pre zámenu a jej klinická dôležitosť nie je známa.
Liečivá,ktorémôžuznížiťplazmatickékoncentráciedasatinibu
Keď sa dasatinib podával počas 8 dní večer so 600 mg rifampicínu, silným induktorom CYP3A4, hodnota AUC dasatinibu sa znížila o 82 %. Iné lieky, ktoré indukujú aktivitu CYP3A4 (napr. dexametazón, fenytoín, karbamazepín, fenobarbital alebo rastlinné lieky obsahujúce Hypericum perforatum, známy aj ako Ľubovník bodkovaný), môžu tiež zvýšiť metabolizmus a znížiť plazmatické koncentrácie dasatinibu. Z tohto dôvodu sa neodporúča súbežné užívanie silných induktorov CYP3A4 s dasatinibom. U pacientov, u ktorých je indikovaný rifampicín alebo iné induktory CYP3A4, sa majú použiť alternatívne lieky s menším potenciálom pre enzýmovú indukciu.
Antagonisty H2-histamínového receptora a inhibítory protónovej pumpy
Dlhodobá supresia sekrécie žalúdočnej kyseliny spôsobená H2-antagonistami alebo inhibítormi protónovej pumpy (napr. famotidínom a omeprazolom) pravdepodobne zníži expozíciu dasatinibu. V štúdii jednorazovej dávky u zdravých jedincov podanie famotidínu 10 hodín pred jednorazovou
dávkou SPRYCELU znížilo expozíciu dasatinibu o 61 %. V štúdii so14 zdravými dobrovoľníkmi pri podaní jednorazovej 100 mg dávky SPRYCELU 22 hodín počas štvrtého dňa, 40 mg dávka omeprazolu znížila v rovnovážnom stave hodnotu AUC dasatinibu o 43 % a hodnotu Cmax dasatinibu o
42 %. U pacientov liečených SPRYCELOM sa má namiesto H2-antagonistov alebo inhibítorov protónovej pumpy zvážiť použitie antacíd (pozri časť 4.4).
Antacidá
Predklinické údaje potvrdzujú, že rozpustnosť dasatinibu závisí od pH. U zdravých jedincov súbežné užívanie antacíd obsahujúcich hydroxid hlinitý/hydroxid horečnatý so SPRYCELOM znížilo hodnotu
AUC jednorazovej dávky SPRYCELU o 55 % a Cmax o 58 %. Keď však boli antacidá podané 2 hodiny pred jednorazovou dávkou SPRYCELU, nepozorovali sa žiadne významné zmeny v koncentrácii ani expozícii dasatinibu. Z tohto dôvodu sa antacidá môžu podať 2 hodín pred alebo 2 hodiny po podaní SPRYCELU (pozri časť 4.4).
Liečivá,ktorýchplazmatickékoncentráciemôžedasatinibovplyvniť
Súbežné užívanie dasatinibu a substrátu CYP3A4 môže zvýšiť expozíciu substrátu CYP3A4. V štúdii so zdravými jedincami jednorazová 100 mg dávka dasatinibu zvýšila hodnotu AUC simvastatínu, známeho substrátu CYP3A4, o 20 % a hodnotu Cmax simvastatínu o 37 %. Nie je možné vylúčiť, že účinok je väčší po viacnásobných dávkach dasatinibu. Z tohto dôvodu sa substráty CYP3A4, o ktorých je známe, že majú úzku terapeutickú šírku (napr. astemizol, terfenadín, cisaprid, pimozid, chinidín, bepridil alebo námeľové alkaloidy [ergotamín, dihydroergotamín]), majú podávať opatrne u pacientov liečených dasatinibom (pozri časť 4.4).
In vitro údaje indikujú potenciálne riziko interakcie s CYP2C8 substrátmi ako sú glitazóny.
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/antikoncepciaumužovažien
Sexuálne aktívni muži aj ženymajú počas liečby používať účinnú metódu antikoncepcie.
Gravidita
Na základe skúseností u ľudí sa predpokladá, že dasatinib spôsobuje vrodené malformácie vrátane defektov neurálnej trubice a má škodlivé farmakologické účinky na plod, ak sa podáva počas gravidity. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
SPRYCEL sa nemá používať počas gravidity, ak si klinický stav ženy nevyžaduje liečbu dasatinibom.
Ak sa SPRYCEL použije počas gravidity, pacientka musí byť informovaná o možnom riziku pre plod.
Dojčenie
Existujú nedostatočné/obmedzené informácie o vylučovaní dasatinibu do materského mlieka u ľudí alebo zvierat. Fyzikálno-chemické a dostupné farmakodynamické/toxikologické údaje o dasatinibe poukazujú na vylučovanie do materského mlieka a nie je možné vylúčiť riziko pre dojčené dieťa. Počas liečby SPRYCELOM sa musí dojčenie prerušiť.
Tehotné alebo dojčiace ženy sa majú vyhýbať expozícii SPRYCELU prášku na perorálnu suspenziu. Fertilita
V štúdiách na zvieratách nebola fertilita samcov a samíc potkanov ovplyvnená liečbou dasatinibom
(pozri časť 5.3). Lekári a iní zdravotnícki pracovníci majú prekonzultovať s mužmi primeraného veku možné účinky SPRYCELU na fertilitu a táto konzultácia môže zahŕňať zváženie konzervácie spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
SPRYCEL má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti musia byť informovaní, že počas liečby dasatinibom môžu mať nežiaduce reakcie, ako je závrat alebo rozmazané videnie. Z tohto dôvodu sa im pri vedení vozidla alebo obsluhe strojov má odporučiť opatrnosť.
4.8 Nežiaduce účinky
Prehľadbezpečnostnéhoprofilu
Údaje opísané nižšie vyjadrujú expozíciu SPRYCELU pri všetkých dávkach testovaných v klinických štúdiách (N=2 900) zahŕňajúcich 324 dospelých pacientov s novo diagnostikovanou chronickou fázou CML, 2 388 dospelých pacientov rezistentných alebo intolerantných na imatinib s chronickou alebo pokročilou fázou CML alebo Ph+ ALL a 188 pediatrických pacientov.

U 2 712 dospelých pacientov buď s chronickou fázou CML, pokročilou fázou CML alebo Ph+ ALL bol medián trvania liečby 19,2 mesiaca (rozsah 0 až 93,2 mesiaca). V randomizovanom klinickom skúšaní s pacientmi s novo diagnostikovanou chronickou fázou CML bol medián trvania liečby približne 60 mesiacov. Medián trvania liečby u 1 618 dospelých pacientov s chronickou fázou CML bol 20 mesiacov (rozsah 0 až 92,9 mesiaca). Medián trvania liečby u 1 094 dospelých pacientov s pokročilou fázou CML alebo Ph+ ALL bol 6,2 mesiaca (rozsah 0,1 až 99,6 mesiaca). U 188 pacientov v pediatrických štúdiách bol medián trvania liečby 26,3 mesiaca (rozsah 0 až 99,6 mesiaca). V podskupine 130 pediatrických pacientov s chronickou fázou CML liečených SPRYCELOM bol medián trvania liečby 42,3 mesiaca (rozsah 0,1 až 99,6 mesiaca).
Väčšina pacientov liečených SPRYCELOM mala v určitom čase nežiaduce reakcie. V celkovej populácii 2 712 dospelých jedincov liečených SPRYCELOM malo 520 (19 %) nežiaduce reakcie, ktoré mali za následok prerušenie liečby.
Celkový bezpečnostný profil SPRYCELU v pediatrickej populácii bol podobný tomu, ktorý je v dospelej populácii bez ohľadu na liekovú formu s výnimkou toho, že v pediatrickej populácii sa nehlásil perikardiány výpotok, pleurálny výpotok, pľúcny edém alebo pľúcna hypertenzia. Zo
130 pediatrických jedincov s CML-CP liečených SPRYCELOM sa u 2 (1,5 %) vyskytli nežiaduce reakcie, ktoré mali za následok ukončenie liečby.
TabuľkovýzoznamnežiaducichreakciíNasledovné nežiaduce reakcie, s výnimkou laboratórnych anomálií, sa hlásili u pacientov v klinických štúdiách so SPRYCELOM a po uvedení lieku na trh (Tabuľka 4). Tieto reakcie sú vymenované podľa tried orgánových systémov a podľa frekvencie. Frekvencie sú definované nasledovne: veľmi časté
(≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); neznáme (nie je možné stanoviť z dostupných údajov po uvedení lieku na trh).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 4: Súhrn nežiaducich reakcií zostavených do tabuľkyInfekcie a nákazyVeľmi časté infekcia (vrátane bakteriálnej, vírusovej, plesňovej, nešpecifickej)
Časté pneumónia (vrátane bakteriálnej, vírusovej a plesňovej), infekcia/zápal horných dýchacích ciest, herpetická vírusová infekcia, enterokolitída, sepsa (vrátane menej častých prípadov s fatálnymi následkami)
Neznáme reaktivácia hepatitídy B
Poruchy krvi a lymfatického systémuVeľmi časté myelosupresia (vrátane anémie, neutropénie, trombocytopénie)
Časté febrilná neutropénia
Menej časté lymfadenopatia, lymfopénia
Zriedkavé aplázia červených krviniek
Poruchy imunitného systémuMenej časté hypersenzitivita (vrátane erythema nodosum)
Zriedkavé anafylaktický šok
Poruchy endokrinného systému Menej časté hypotyreoidizmus
Zriedkavé hypertyreoidizmus, tyreoiditída
Poruchy metabolizmu a výživyČasté poruchy chuti do jedlaa, hyperurikémia
Menej časté syndróm z rozpadu nádoru, dehydratácia, hypoalbuminémia, hypercholesterolémia
Zriedkavé diabetes mellitus
Psychické poruchyČasté depresia, insomnia
Menej časté úzkosť, stav zmätenosti, ovplyvnenie lability, zníženie libida
Poruchy nervového systému
Veľmi časté bolesť hlavy
Časté neuropatia (vrátane periférnej neuropatie), závrat, porucha chuti, spavosť
Menej časté krvácanie do CNS*b, synkopa, tremor, amnézia, porucha rovnováhy
Zriedkavé cerebrovaskulárna príhoda, prechodný ischemický záchvat, kŕč, očná neuritída, paralýza VII. nervu, demencia, ataxia
Poruchy okaČasté poruchy videnia (vrátane zrakovej poruchy, rozmazaného videnia a zníženej zrakovej ostrosti), suchosť očí
Menej časté porucha zraku, konjunktivitída, fotofóbia, zvýšené slzenie
Poruchy ucha a labyrintuČasté tinnitus
Menej časté strata sluchu, vertigo
Poruchy srdca a srdcovej činnostiČasté kongestívne srdcové zlyhanie/srdcová dysfunkcia*c, perikardiálny výpotok*, arytmia
(vrátane tachykardie), palpitácie
Menej časté infarkt myokardu (vrátane fatálneho následku)*, QT predĺženie na elektrokardiograme*, perikarditída, ventrikulárna arytmia (vrátane ventrikulárnej tachykardie), angina pectoris, kardiomegália, nezvyčajná T vlna na elektrokardiograme, zvýšený troponín
Zriedkavé cor pulmonale, myokarditída, akútny koronárny syndróm, zastavenie srdca, predĺženie PR na elektrokardiograme, koronárne arteriálne ochorenie, pleuroperikarditída
Neznáme fibrilácia predsiení/ flutter predsiení
Poruchy cievVeľmi časté hemorágia*d
Časté hypertenzia, sčervenenie
Menej časté hypotenzia, tromboflebitída, trombóza
Zriedkavé hlboká žilová trombóza, embólia, livedo reticularis
Poruchy dýchacej sústavy, hrudníka a mediastínaVeľmi časté pleurálny výpotok*, dyspnoe
Časté pľúcny edém*, pľúcna hypertenzia*, pľúcna infiltrácia, pneumonitída, kašeľ
Menej časté pľúcna arteriálna hypertenzia, bronchospazmus, astma
Zriedkavé pľúcna embólia, syndróm akútnej respiračnej tiesne
Neznáme intersticiálne ochorenie pľúc
Poruchy gastrointestinálneho traktuVeľmi časté hnačka, vracanie, nauzea, abdominálna bolesť
Časté gastrointestinálne krvácanie*, kolitída (vrátane neutropenickej kolitídy), gastritída, zápal sliznice (vrátane mukozitídy/stomatitídy), dyspepsia, abdominálna distenzia, zápcha, porucha mäkkého tkaniva v ústach
Menej časté pankreatitída (vrátane akútnej pankreatitídy), horný gastrointestinálny vred, ezofagitída, ascites*, análna fisúra, dysfágia, gastroezofageálne refluxné ochorenie
Zriedkavé gastroenteropatia zo straty proteínov, ileus, análna fistula
Neznáme fatálna gastrointestinálna hemorágia*
Poruchy pečene a žlčových ciestMenej časté hepatitída, cholecystitída, cholestáza
Poruchy kože a podkožného tkanivaVeľmi časté kožná vyrážkae
Časté alopécia, dermatitída (vrátane ekzému), pruritus, akné, suchá koža, urtikária, hyperhidróza
Menej časté neutrofilná dermatóza, fotosenzitivita, porucha pigmentácie, panikulitída, kožný vred, bulózne ochorenia, porucha nechtov, syndróm palmárno-plantárnej erytrodyzestézie, ochorenie vlasov
Zriedkavé leukocytoklastická vaskulitída, fibróza kože
Neznáme Stevensov-Johnsonov syndrómf
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Veľmi časté muskuloskeletálna bolesť
Časté artralgia, myalgia, svalová slabosť, muskuloskeletálna stuhnutosť, svalový kŕč
Menej časté rabdomyolýza, osteonekróza, svalový zápal, tendonitída, artritída
Zriedkavé oneskorená fúzia epifýz,g spomalenie rastug
Poruchy obličiek a močových ciestMenej časté poruchy funkcie obličiek (vrátane renálneho zlyhania), časté močenie, proteinúria
Neznáme nefrotický syndróm
Stavy v gravidite, v šestonedelí a perinatálnom obdobíZriedkavé potrat
Poruchy reprodukčného systému a prsníkovMenej časté gynekomastia, porucha menštruácie
Celkové poruchy a reakcie v mieste podaniaVeľmi časté periférny edémh, únava, pyrexia, edém tvárei
Časté asténia, bolesť, bolesť na hrudi, generalizovaný edém*j, zimnica
Menej časté pocit ťažoby, iný povrchový edémk
Zriedkavé porucha chôdze
Laboratórne a funkčné vyšetreniaČasté zníženie telesnej hmotnosti, zvýšenie telesnej hmotnosti
Menej časté zvýšenie kreatínfosfokinázy v krvi, zvýšenie gama-glutamyltransferázy
Úrazy, otravy a komplikácie liečebného postupuČasté kontúzia
a Zahŕňa zníženú chuť do jedla, predčasnú sýtosť, zvýšenú chuť do jedla.
b Zahŕňa krvácanie do centrálneho nervového systému, cerebrálny hematóm, cerebrálne krvácanie, extradurálny hematóm, intrakraniálne krvácanie, hemoragickú cievnu mozgovú príhodu, subarachnoidálne krvácanie, subdurálny hematóm a
subdurálne krvácanie.
c Zahŕňa zvýšený mozgový natriuretický peptid, ventrikulárnu dysfunkciu, dysfunkciu ľavej komory, dysfunkciu pravej komory, srdcové zlyhanie, akútne srdcové zlyhanie, chronické srdcové zlyhanie, kongestívne srdcové zlyhanie,
kardiomyopatiu, kongestívnu kardiomyopatiu, diastolickú dysfunkciu, zníženú ejekčnú frakciu a ventrikulárne zlyhanie, zlyhanie ľavej komory, zlyhanie pravej komory a ventrikulárnu hypokinézu.
d Okrem gastrointestinálneho krvácania a krvácania do CNS; tieto nežiaduce reakcie sú hlásené pod triedou orgánového systému poruchy gastrointestinálneho traktu alebo pod triedou orgánového systému poruchy nervového systému.
e Zahŕňa liekovú erupciu, erytém, multiformný erytém, erytrózu, exfoliatívnu vyrážku, generalizovaný erytém, genitálnu
vyrážku, potničky, milie, miliariu, pustulárnu psoriázu, vyrážku, erytematóznu vyrážku, folikulárnu vyrážku, generalizovanú vyrážku, makulárnu vyrážku, makulo-papulárnu vyrážku, papulárnu vyrážku, pruritickú vyrážku, pustulárnu vyrážku, vezikulárnu vyrážku, kožnú exfoliáciu, kožné podráždenie, toxickú kožnú erupciu, vezikulóznu urtikáriu a vaskulitickú vyrážku.
f Po uvedení lieku na trh sa hlásili jednotlivé prípady Stevensovho-Johnsonovho syndrómu. Nebolo možné určiť, či tieto mukokutánne nežiaduce reakcie priamo súviseli so SPRYCELOM alebo so súbežne podávaným liekom.
g Frekvencie hlásené ako časté v pediatrických štúdiách.
h Gravitačný edém, lokalizovaný edém, periférny edém.
i Konjunktiválny edém, edém oka, opuch oka, edém očného viečka, edém tváre, edém pier, makulárny edém, edém úst, orbitálny edém, periorbitálny edém, opuch tváre.
j Preťaženie tekutinou, retencia tekutín, opuch gastrointestinálneho traktu, generalizovaný edém, edém, edém z dôvodu ochorenia srdca, perinefrický výpotok, edém po chirurgickom výkone, viscerálny edém.
k Opuch genitálií, edém v mieste vpichu, genitálny edém, edém pohlavného údu, opuch pohlavného údu, skrotálny edém, opuch kože, opuch semenníkov, opuchy vagíny a pošvy.
* Ďalšie podrobnosti, pozri časť "Opis vybraných nežiaducich reakcií"
OpisvybranýchnežiaducichreakciíMyelosupresiaLiečba SPRYCELOM sa spája s anémiou, neutropéniou a trombocytopéniou. Ich výskyt je skorší a častejší u pacientov v pokročilej fáze CML alebo s Ph+ ALL ako u pacientov v chronickej fáze CML (pozri časť 4.4).
KrvácanieU pacientov užívajúcich SPRYCEL sa hlásili nežiaduce reakcie krvácaní súvisiacich s liekom
v rozsahu od petechie a epistaxy po gastrointestinálne krvácanie 3. alebo 4. stupňa a krvácanie do CNS (pozri časť 4.4).
Retencia tekutín
Rôzne nežiaduce reakcie, ako je pleurálny výpotok, ascites, pľúcny edém a perikardiálny výpotok
s povrchovým edémom alebo bez neho, možno súhrnne opísať ako “retenciu tekutiny”. V štúdii s novo diagnostikovanou chronickou fázou CML po minimálne 60 mesiacoch následného sledovania zahŕňali nežiaduce reakcie retencie tekutiny súvisiacej s dasatinibom pleurálny výpotok (28 %). povrchový edém (14 %), pľúcnu hypertenziu (5 %) generalizovaný edém (4 %) a perikardiálny výpotok (4 %). Kongestívne srdcové zlyhanie/dysfunkcia srdca a pľúcny edém sa hlásili u < 2 % pacientov.
V priebehu času bol kumulatívny pomer pleurálneho výpotku súvisiaceho s dasatinibom (všetky stupne) 10 % po 12 mesiacoch, 14 % po 24 mesiacoch, 19 % po 36 mesiacoch, 24 % po 48 mesiacoch a 28 % po 60 mesiacoch. Celkovo 46 pacientov liečených dasatinibom malo rekurentný pleurálny výpotok. Sedemnásť pacientov malo 2 samostatné nežiaduce reakcie, 6 malo 3 nežiaduce reakcie,
18 malo 4 až 8 nežiaducich reakcií a 5 malo > 8 epizód pleurálneho výpotku.
Medián času do prvého pleurálneho výpotku súvisiaceho s dasatinibom 1. alebo 2. stupňa bol
114 týždňov (rozmedzie: 4 až 299 týždňov). Menej než 10 % pacientov s pleurálnym výpotkom malo závažný (3. alebo 4. stupeň) pleurálnych výpotkov súvisiacich s dasatinibom. Medián času do prvého výskytu pleurálneho výpotku súvisiaceho s dasatinibom ≥ 3. stupňa bol 175 týždňov (rozmedzie: 114 až 274 týždňov). Medián trvania pleurálneho výpotku súvisiaceho s dasatinibom (všetky stupne) bol
283 dní (~40 týždňov).
Pleurálny výpotok bol zvyčajne vratný a bol zvládnutý prerušením liečby SPRYCELOM a použitím diuretík alebo iných vhodných podporných ošetrovacích opatrení (pozri časti 4.2 a 4.4). Medzi pacientmi liečenými dasatinibom s pleurálnym výpotkom súvisiacim s liekom (n=73) malo 45 (62 %) prerušenie liečby a 30 (41 %) malo zníženie dávky. Okrem toho 34 (47 %) dostalo diuretiká, 23
(32 %) dostalo kortikosteroidy a 20 (27 %) dostalo kortikosteroidy aj diuretiká. Deväť (12 %)
pacientov podstúpilo terapeutickú torakocentézu.
Šesť percent pacientov liečených dasatinibom prerušilo liečbu z dôvodu pleurálneho výpotku súvisiaceho s liekom.
Pleurálny výpotok nezhoršil schopnosť pacienta dosiahnuť odpoveď. Medzi pacientmi s pleurálnym
výpotkom liečenými dasatinibom dosiahlo 96 % cCCyR, 82 % dosiahlo MMR a 50 % dosiahlo MR4.5
i napriek prerušeniam liečby alebo úprave dávky.
Pozri časť 4.4 ďalšie informácie o pacientoch s chronickou fázou CML a s pokročilou fázou CML
alebo Ph+ ALL.
Pľúcna arteriálna hypertenzia (PAH)
PAH (prekapilárna pľúcna arteriálna hypertenzia potvrdená pravostrannou katétrizáciou srdca) sa hlásila v súvislosti s expozíciou dasatinibom. V týchto prípadoch sa hlásila PAH po začatí liečby dasatinibom, a to po viac ako jednom roku liečby. Pacienti s PAH hlásili počas liečby dasatinibom časté užívanie súbežných liekov alebo mali komorbidity, okrem základného nádorového ochorenia. Zlepšenie hemodynamických a klinických parametrov sa pozorovalo u pacientov liečených dasatinibom s PAH po ukončení liečby dasatinibom.
Predĺženie QT intervalu
V štúdii fázy III s pacientmi s novo diagnostikovanou chronickou fázou CML, jeden pacient (< 1 %) z pacientov liečených SPRYCELOM mal QTcF > 500 ms po minimálne 12 mesiacoch následného sledovania (pozri časť 4.4). Po minimálne 60 mesiacoch následného sledovania sa u žiadnych ďalších pacientov nehlásilo, že mali QTcF > 500 ms.
V 5 klinických štúdiách fázy II s pacientmi s rezistenciou alebo intoleranciou na predchádzajúcu
liečbu imatinibom, sa vykonalo opakované základné EKG a aj počas liečby vo vopred určených časových intervaloch a snímané centrálne pre 865 pacientov liečených SPRYCELOM 70 mg dvakrát denne. Interval QT bol korigovaný podľa srdcovej frekvencie pomocou Fridericiovej metódy. Vo všetkých časových intervaloch po dávke na 8. deň, boli priemerné zmeny oproti východiskovej hodnote intervalu QTcF 4 – 6 ms, s pridruženým horným 95 % intervalom spoľahlivosti < 7 ms. Z
2 182 pacientov s rezistenciou alebo intoleranciou na predchádzajúcu liečbu imatinibom, ktorí v klinických štúdiách užívali SPRYCEL, 15 (1 %) pacientov hlásilo predĺženie QTc, ako nežiaducu reakciu. Dvadsaťjeden pacientov (1 %) malo QTcF > 500 ms (pozri časť 4.4).
Nežiaduce reakcie na srdce
Pacienti s rizikovými faktormi alebo srdcovým ochorením v anamnéze, majú byť starostlivo sledovaní pre prejavy alebo príznaky zhodné so srdcovou dysfunkciou a majú byť posúdení a primerane liečení (pozri časť 4.4).
Reaktivácia hepatitídy B
V súvislosti s inhibítormi BCR-ABL-tyrozínkinázy bola hlásená reaktivácia hepatitídy B. Niektoré prípady viedli k akútnemu zlyhaniu pečene alebo k fulminantnej hepatitíde, ktorých výsledkom bola transplantácia pečene alebo úmrtie (pozri časť 4.4).
V klinickej štúdii dávkovej optimalizácie fázy III s pacientmi s chronickou fázou CML s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom (medián trvania liečby 30 mesiacov) bol výskyt pleurálneho výpotku a kongestívneho srdcového zlyhania/srdcovej dysfunkcie nižší u pacientov liečených SPRYCELOM dávkou 100 mg jedenkrát denne ako u pacientov liečených SPRYCELOM dávkou 70 mg dvakrát denne. Myelosupresia sa vyskytla tiež menej často v skupine liečenej dávkou
100 mg jedenkrát denne (pozri Abnormality laboratórnych testov nižšie). Medián trvania liečby
v skupine so 100 mg jedenkrát denne bol 37 mesiacov (rozsah 1 – 91 mesiacov). Kumulatívny výskyt vybraných nežiaducich reakcií, ktoré sa hlásili pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne sú uvedené v tabuľke 5a.
Tabuľka 5a: Vybrané nežiaduce reakcie hlásené v 3. fáze štúdie optimalizácie dávky
(Intolerancia alebo rezistencia na imatinib chronická fáza CML)a
M
inimálne 2-ročné následné sledovanie
Minimálne 5-ročné následné sledovanie
Minimálne 7-ročné následné sledovanie
Všetky stupne
3./4. stup eň
V
š
e
tky stupne
3
./4. stup eň
Všetky stupne
3
.
/4. stup eň
 Preferovaný výraz
Preferovaný výraz Percento (%) pacientov
Hnačka 27 2 28 2 28 2
Retencia tekutín 34 4 42 6 48 7
Povrchový edém 18 0 21 0 22 0
Pleurálny výpotok 18 2 24 4 28 5
Generalizovaný edém 3 0 4 0 4 0
Perikardiálny
výpotok 2 1 2 1 3 1
Pľúcna hypertenzia 0 0 0 0 2 1
Krvácanie 11 1 11 1 12 1
Gastrointestinálne
krvácanie 2 1 2 1 2 1
a Výsledky štúdie 3. fázy optimalizácie dávky hlásené v populácii pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne (n=165)
V štúdii fázy III optimalizácie dávky s pacientmi s pokročilou fázou CML a Ph+ ALL bol medián trvania liečby 14 mesiacov pre akcelerovanú fázu CML, 3 mesiace pre myeloidnú blastovú CML,
4 mesiace pre lymfoidnú blastovú CML a 3 mesiace pre Ph+ ALL. Vybrané nežiaduce reakcie, ktoré
sa hlásili pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne sú uvedené v tabuľke 5b. Schéma dávkovania 70 mg dvakrát denne sa tiež skúmala. Schéma dávkovania 140 mg jedenkrát denne preukázala porovnateľný profil účinnosti voči schéme dávkovania 70 mg dvakrát denne, no s priaznivejším profilom bezpečnosti.
Tabuľka 5b: Vybrané nežiaduce reakcie hlásené v štúdii III. fázy optimalizácie dávky:
Pokročilá
fáza
CML
a
Ph+
ALL
a
140 mg jedenkrát denne n = 304
Všetky stupne 3./4. stupeň
 Preferovaný výraz
P
ercento (%) pacientov
Hnačka
Preferovaný výraz
P
ercento (%) pacientov
Hnačka 28 3
Retencia tekutín 33 7
Povrchový edém 15 < 1
Pleurálny výpotok 20 6
Generalizovaný edém 2 0
Kongestívne srdcové 1 0
zlyhanie / srdcová dysfunkciab
Perikardiálny výpotok 2 1
Pľúcny edém 1 1
Krvácanie 23 8
Gastrointestinálnekrvácanie 8 6 a Výsledky štúdie 3. fázy optimalizácie dávky hlásené v populácii s odporúčanou začiatočnou dávkou 140 mg jedenkrát denne (n=304) pri 2-ročnej finálnej štúdii následného sledovania.
b Vrátane komorovej dysfunkcie, srdcového zlyhania, kongestívneho srdcového zlyhania, kardiomyopatie, kongestívnej kardiomyopatie, diastolickej disfunkcie, zníženie ejekčnej frakcie a komorového zlyhania.
Abnormality laboratórnych testovHematológiaV štúdii III. fázy sa u pacientov s novo diagnostikovanou chronickou fázou CML užívajúcich SPRYCEL hlásili nasledovné laboratórne abnormality 3. a 4. stupňa po minimálne 12 mesiacoch následného sledovania: neutropénia (21 %), trombocytopénia (19 %) a anémia (10 %). Po minimálne
60 mesiacoch následného sledovania boli kumulatívne výsledky neutropénie, thrombocytopénie a'
anémie 29 %, 22 % a 13 %, v uvedenom poradí.
U pacientov s novo diagnostikovanou chronickou fázou CML liečených SPRYCELOM , u ktorých sa vyskytla myelosupresia 3. alebo 4. stupňa, zvyčajne došlo po krátkom prerušení a/alebo znížení dávky a trvalom prerušení liečby k zotaveniu, ktoré sa vyskytlo u 1,6 % pacientov po minimálne
12 mesiacoch následného sledovania. Po minimálne 60 mesiacoch následného sledovania bol
kumulatívny pomer trvalého vysadenia liečby z dôvodu myelosupresie 3. alebo 4. stupňa 2,3 %.
U pacientov s CML s rezistenciou alebo intoleranciou na predošlú liečbu imatinibom boli cytopénie (trombocytopénia, neutropénia a anémia) trvalý nález. Výskyt cytopénií bol však tiež závislý od stupňa ochorenia. Výskyt hematologických abnormalít 3. a 4. stupňa je uvedený v tabuľke 6.
Tabuľka 6: CTC hematologických laboratórnych abnormalít 3./4. stupňa v klinických štúdiách s pacientmi s rezistenciou alebo intoleranciounapredošlúliečbuimatiniboma
Chronická fáza
Akcelerovaná fáza
Myeloidná blastová fáza
Lymfoidná blastová fáza a
 Ph+ ALL
Ph+ ALL
Hematologické parametre
(n= 165)
b
(n=
157)
c
(n=
7
4)
c
(n=
1
6
8)
c
Percento (%) pacientov
Neutropénia 36 58 77 76
Trombocytopénia 23 63 78 74
Anémia 13 47 74 44

a Výsledky štúdie 3. fázy optimalizácie dávky hlásené po 2-ročnej štúdii následného sledovania.
b Štúdia CA180-034 výsledky pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
c Štúdia CA180-035 výsledky pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne.
CTC stupne: neutropénia (3. stupňa ≥ 0,5– < 1,0 × 109/l, 4. stupňa < 0.5 × 109/l); trombocytopénia (3. stupňa ≥ 25– < 50 ×
109/l, 4. stupňa < 25 × 109//l); anémia (hemoglobín 3. stupňa ≥ 65– < 80 g/l, 4. stupňa < 65 g/l).
Kumulatívne cytopénie 3. alebo 4. stupňa u pacientov liečených dávkou 100 mg jedenkrát denne boli podobné v 2. a 5. roku, zahŕňajúce: neutropénie (35 % oproti 36 %), trombocytopénie (23 % oproti
24 %) a anémie (13 % oproti 13 %).
U pacientov, ktorí mali myelosupresiu 3. alebo 4. stupňa, zvyčajne došlo k zotaveniu po krátkom prerušení liečby a/alebo znížení dávky a trvalé ukončenie liečby bolo potrebné u 5 % pacientov. Väčšina pacientov pokračovala v liečbe bez ďalších dôkazov o myelosupresii.
Biochemické vyšetrenia
V štúdii s novo diagnostikovanou chronickou fázou CML sa hlásila hypofosfatémia 3. a 4. stupňa
u 4 % pacientov liečených SPRYCELOM a vzostupy transamináz, kreatinínu a bilirubínu sa hlásili u
≤ 1 % pacientov po minimálne 12 mesiacoch následného sledovania. Po minimálne 60 mesiacoch následného sledovania bol kumulatívny pomer hypofosfatémie 3. alebo 4. stupňa 7 %, vzostupu kreatinínu a bilirubínu 3. alebo 4. stupňa 1 % a vzostupu transamináz 3. alebo 4. stupňa zostal 1 %. Nezistili sa žiadne prerušenia liečby SPRYCELOM v dôsledku týchto biochemických laboratórnych parametrov.
2-ročné následné sledovanie
Vzostupy transamináz alebo bilirubínu 3. alebo 4. stupňa sa hlásili u 1 % pacientov v chronickej fáze CML (rezistentných alebo intolerantných na imatinib), ale u pacientov v pokročilej fáze CML a u pacientov s Ph+ ALL sa vzostupy hlásili so zvýšenou 1 až 7 % frekvenciou. Zvyčajne sa zvládli znížením dávky alebo prerušením liečby. V štúdii dávkovej optimalizácie s chronickou fázou CML fázy III sa vyskytlo zvýšenie transamináz alebo bilirubínu 3. alebo 4. stupňa u ≤ 1 % pacientov s podobným nízkym výskytom vo všetkých štyroch skupinách. V štúdii dávkovej optimalizácie fázy III s pokročilou fázou CML a Ph+ALL sa vyskytlo zvýšenie transamináz alebo bilirubínu 3. alebo
4. stupňa u 1 % až 45 % pacientov v jednotlivých liečených skupinách.
Približne u 5 % pacientov liečených SPRYCELOM, ktorí mali normálne východiskové hladiny vápnika 3. alebo 4. stupňa, vznikla v určitom čase počas štúdie prechodná hypokalciémia. Obvykle nebola žiadna súvislosť medzi zníženými hladinami vápnika a klinickými symptómami. Pacienti, u ktorých vznikla hypokalciémia 3. alebo 4. stupňa sa často zotavili po perorálnej suplementácii vápnika. Hypokalciémia 3. alebo 4. stupňa, hypokaliémia a hypofosfatémia sa vyskytli u pacientov vo všetkých fázach CML, ale so zvýšenou frekvenciou u pacientov s myeloidnou alebo lymfoidnou blastovou fázou CML a Ph+ ALL. Zvýšenie kreatinínu 3. alebo 4. stupňa sa vyskytlo u < 1 % pacientov s chronickou fázou CML a hlásil sa zvýšený výskyt od 1 do 4 % u pacientov s pokročilou fázou CML.
Pediatrickápopulácia
Bezpečnostný profil u pediatrických pacientov bol porovnateľný s bezpečnostným profilom u dospelých. Frekvencia, typ a závažnosť nežiaducich reakcií u detí sa očakávajú rovnaké ako u dospelých.
V pediatrických štúdiách s CML bola miera laboratórnych anomálií zhodná so známym profilom laboratórnych parametrov u dospelých.
Špeciálnepopulácie
Zatiaľ čo bezpečnostný profil SPRYCELU u starších ľudí bol podobný profilu v mladšej populácii, u pacientov vo veku 65 rokov a starších je pravdepodobnejší výskyt často hlásených nežiaducich reakcií, ako je únava, pleurálny výpotok, dyspnoe, kašeľ, krvácanie do dolného gastrointestinálneho traktu
a porucha chuti a pravdepodobnejší výskyt menej často hlásených nežiaducich reakcií, ako sú distenzia brucha, závrat, perikardiálny výpotok, kongestívne srdcové zlyhanie a pokles telesnej hmotnosti a majú sa starostlivo sledovať (pozri časť 4.4).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
Skúsenosti s predávkovaním SPRYCELOM v klinických štúdiách sú limitované z dôvodu ojedinelých prípadov. Najväčšie predávkovanie 280 mg denne počas jedného týždňa sa vyskytlo u dvoch
pacientov a u oboch sa vyvinulo signifikantné zníženie počtu trombocytov. Pretože sa dasatinib spája s
myelosupresiou 3. alebo 4. stupňa (pozri časť 4.4), pacienti, ktorí prijali vyššiu ako odporúčanú dávku, majú byť dôkladne sledovaní na myelosupresiu a má im byť poskytnutá podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE06
Farmakodynamickéúčinky
Dasatinib inhibuje aktivitu BCR-ABL-kinázy a kináz rodiny SRC, ako aj niekoľkých ďalších vybraných onkogénnych kináz zahŕňajúcich kinázy c-KIT receptora, efrínového (EPH) receptora a PDGFβ receptora. Dasatinib je silným, subnanomolárnym inhibítorom BCR-ABL-kinázy
s účinnosťou pri koncentrácii 0,6-0,8 nM. Viaže sa na inaktívne aj aktívne konformácie enzýmu BCR-
ABL.
Mechanizmusúčinku
V podmienkach in vitro je dasatinib aktívny v leukemických bunkových líniách predstavujúcich varianty ochorenia citlivého a rezistentného na imatinib. Tieto predklinické štúdie ukazujú, že dasatinib môže prekonať rezistenciu na imatinib, ktorá je dôsledkom nadmernej expresie BCR-ABL, mutácií BCR-ABL-kinázovej domény, aktivácie alternatívnych signálnych dráh zahŕňajúcich kinázy
rodiny SRC (LYN, HCK) a nadmernej expresie génu pre rezistenciu na viaceré lieky. Dasatinib okrem toho inhibuje kinázy rodiny SCR pri subnanomolárnej koncentrácii.
V samostatných experimentoch in vivo využívajúcich myšacie modely CML dasatinib zabránil progresii chronickej fázy CML do blastickej fázy a predĺžil prežívanie myší nesúcich ľudské bunkové línie CML kultivované na rôznych miestach, vrátane centrálneho nervového systému.
Klinickáúčinnosťabezpečnosť
V štúdii fázy I. bola hematologická a cytogenetická odpoveď pozorovaná vo všetkých fázach CML a u Ph+ ALL u prvých 84 pacientov liečených a následne sledovaných počas doby 27 mesiacov. Odpovede mali trvalý charakter vo všetkých fázach CML a u Ph+ ALL.
Vykonali sa štyri jednoramenné, nekontrolované, otvorené klinické štúdie fazy II hodnotiace bezpečnosť a účinnosť dasatinibu u pacientov s CML v chronickej, akcelerovanej alebo myeloidnej blastickej fáze, ktorí boli buď rezistentní, alebo intolerantní na imatinib. Jedna randomizovaná, nekomparatívna štúdia sa vykonala u pacientov v chronickej fáze, u ktorých zlyhala úvodná liečba so
400 alebo 600 mg imatinibu. Počiatočná dávka dasatinibu bola 70 mg dvakrát denne. Úpravy dávky boli povolené na zlepšenie účinnosti alebo zvládnutie toxicity (pozri časť 4.2).
Dve randomizované, otvorené klinické štúdie fázy III sa vykonali s cieľom zhodnotiť účinnosť dasatinibu podávaného jedenkrát denne v porovnaní s dasatinibom podávaným dvakrát denne. Okrem toho, jedna otvorená, randomizovaná, porovnávacia štúdia fázy III bola vykonaná u dospelých pacientov s novo diagnostikovanou chronickou fázou CML.
Účinnosť dasatinibu sa zakladá na miere hematologickej a cytogenetickej odpovede.
Stálosť odpovede a odhadovaná miera prežitia poukazuje na ďalší klinický úžitok dasatinibu.
V klinických štúdiách sa hodnotil celkový počet 2 712 pacientov, z ktorých 23 % bolo vo veku
≥ 65 rokov a 5 % bolo vo veku ≥ 75 rokov.
Chronická fáza CML - novo diagnostikovaná
Medzinárodná otvorená, multicentrická, randomizovaná, porovnávacia štúdia fázy III sa vykonala s dospelými pacientmi s novo diagnostikovanou chronickou fázou CML. Pacienti boli randomizovaní do skupiny užívajúcich buď SPRYCEL 100 mg jedenkrát denne, alebo imatinib 400 mg jedenkrát
denne. Primárnym ukazovateľom bola miera potvrdenej kompletnej cytogenetickej odpovede (cCCyR) počas 12 mesiacov. Sekundárne ukazovatele zahŕňali čas v cCCyR (miera trvania odpovede), čas do cCCyR, mieru veľkej molekulárnej odpovede (MMR), čas do MMR, prežívanie bez progresie (PFS) a celkové prežívanie (OS). Ostatné relevantné výsledky účinnosti zahŕňali CCyR a mieru kompletnej molekulárnej odpovede (CMR). Štúdia prebieha.
Celkovo 519 pacientov bolo randomizovaných do skupín: 259 do skupiny SPRYCELU a 260 k imatinibu. Základné charakteristiky boli dobre vyvážené medzi oboma liečebnými skupinami s ohľadom na vek (medián veku bol 46 rokov pre SPRYCELOVÚ skupinu a 49 rokov pre imatinibovú skupinu s 10 % pacientov v SPRYCELOVEJ skupine a 11 % pacientov v imatinibovej skupine vo veku 65 rokov alebo starších), pohlavie (ženy 44 % v SPRYCELOVEJ skupine a 37 % v imatinibovej skupine), a rasu (kaukazská 51 % SPRYCELOVEJ skupine v a 55 % v imatinibovej skupine, ázijská
42 % v SPRYCELOVEJ skupine a 37 % imatinibovej skupine). Na začiatku, bola distribúcia Hasford skóre podobná v SPRYCELOM a imatinibom liečenej skupine (nízke riziko: 33 % a 34 %, stredné riziko 48 % a 47 %, vysoké riziko: 19 % a 19 % v SPRYCELOVEJ skupine a imatinibovej skupine v uvedenom poradí).
V minimálne 12 mesačnom následnom sledovaní, 85 % pacientov randomizovaných do skupiny so SPRYCELOM a 81 % pacientov randomizovaných do skupiny s imatinibom stále dostávalo prvú líniu liečby. Prerušenie počas 12 mesiacov z dôvodu progresie ochorenia sa vyskytlo u 3 % pacientov liečených SPRYCELOM a u 5 % pacientov liečených imatinibom.
V minimálne 60 mesačnom následnom sledovaní, 60 % pacientov randomizovaných do skupiny so SPRYCELOM a 63 % pacientov randomizovaných do skupiny s imatinibom stále dostávalo prvú líniu liečby. Prerušenie počas 60 mesiacov z dôvodu progresie ochorenia sa vyskytlo u 11 % pacientov liečených SPRYCELOM a u 14 % pacientov liečených imatinibom.
Výsledky účinnosti sú uvedené v tabuľke 7. Štatisticky významne väčší podiel pacientov v SPRYCELOVEJ skupine dosiahol cCCyR v priebehu prvých 12 mesiacov liečby v porovnaní s pacientmi v imatinibovej skupine. Účinnosť SPRYCELU bola dôsledne preukázaná v rôznych podskupinách, zahŕňajúc vek, pohlavie a východiskové Hasford skóre.
Tabuľka 7: Výsledky účinnosti z 3. fázy štúdie s novo diagnostikovanými pacientmi s
chronickou
f
ázou
C
M
L
SPRYCEL
imatinib
p-hodnota
n= 259 n= 260
Miera
o
dpovede
(95
%
CI)
Cytogenetická odpoveď
v priebehu 12 mesiacov
cCCyRa 76,8 % (71,2–81,8) 66,2 % (60,1-71,9) p< 0,007* CCyRb 85,3 % (80,4-89,4) 73,5% (67,7-78,7) ⎯
v priebehu 24 mesiacovcCCyRa 80,3 % 74,2 % ⎯
CCyRb 87,3 % 82,3 % ⎯
v priebehu 36 mesiacovcCCyRa 82,6 % 77,3 % ⎯
CCyRb 88,0 % 83,5 % ⎯
v priebehu 48 mesiacovcCCyRa 82,6 % 78,5 % ⎯
CCyRb 87,6 % 83,8 % ⎯
v priebehu 60 mesiacovcCCyRa 83,0 % 78,5 % ⎯
CCyRb 88,0 % 83,8 % ⎯
Veľká molekulárna odpoveďc
12 mesiacov 52,1 % (45,9-58,3) 33,8 % (28,1-39,9) p< 0,00003*
24 mesiacov 64,5 % (58,3-70,3) 50 % (43,8-56,2) ⎯
36 mesiacov 69,1 % (63,1-74,7) 56,2 % (49,9-62,3) ⎯
48 mesiacov 75,7 % (70,0-80,8) 62,7 % (56,5-68,6) ⎯
60mesiacov 76,4%(70,8-81,5) 64,2%(58,1-70,1) p=0,0021 Pomer rizika (HR)počas 12 mesiacov (99,99 % CI)Čas do cCCyR 1,55 (1,0-2,3) p< 0,0001* Čas do MMR 2,01 (1,2-3,4) p< 0,0001* Trvania cCCyR 0,7 (0,4-1,4) p< 0,035
počas 24 mesiacov (95 % CI)Čas do cCCyR 1,49 (1,22 – 1,82) ⎯ Čas do MMR 1,69 (1,34-2,12) ⎯ Trvania cCCyR 0,77 (0,55-1,10) ⎯
počas 36 mesiacov (95 % CI)Čas do cCCyR 1,48 (1,22-1,80) ⎯ Čas do MMR 1,59 (1,28-1,99) ⎯ Trvania cCCyR 0,77 (0,53-1,11) ⎯
počas 48 mesiacov (95 % CI)Čas do cCCyR 1,45 (1,20-1,77) ⎯ Čas do MMR 1,55 (1,26-1,91) ⎯ Trvania cCCyR 0,81 (0,56-1,17) ⎯
počas 60 mesiacov (95 % CI)Čas do cCCyR 1,46 (1,20 – 1,77) p=0,0001
Čas do MMR 1,54 (1,25-1,89) p<0,0001
TrvaniacCCyR 0,79(0,55-1,13) p=0,1983 a Potvrdená kompletná cytogenetická odpoveď (cCCyR) je definovaná ako odpoveď známa z dvoch po sebe idúcich udalostí
(najmenej 28 dní odstup).
b Kompletná cytogenetická odpoveď (CCyR) je založená na jednom cytogenetickom zhodnotení kostnej drene.
c Veľká molekulová odpoveď (v akomkoľvek čase) bola definovaná ako BCR-ABL podiely ≤ 0,1% podľa RQ-PCR vo vzorkách periférnej krvi štandardizovaných na Medzinárodnej stupnici. Ide o kumulatívne miery predstavujúce minimálne
sledovanie pre stanovený časový rámec.
*Upravené na Hasford skóre a uvedená štatistická významnosť na preddefinovanú nominálnu hodnotu významnosti. CI = interval spoľahlivosti
Po 60 mesačnom následnom sledovaní bol medián času do cCCyR 3,1 mesiaca v SPRYCELOVEJ skupine a 5,8 mesiaca v imatinibovej skupine u pacientov s potvrdenou CCyR. Medián času do MMR bol po 60 mesačnom následnom sledovaní 9,3 mesiaca v SPRYCELOVEJ skupine a 15,0 mesiacov v imatinibovej skupine pacientov s MMR. Tieto výsledky sú zhodné s tými, ktoré sa zistili po 12, 24
a 36 mesiacoch.
Čas do MMR je zobrazený graficky na grafe 1. Čas do MMR bol rovnomerne kratší u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
Graf 1:
Odhad času veľkej molekulárnej odpovede (MMR) podľa Kaplan-Meiera
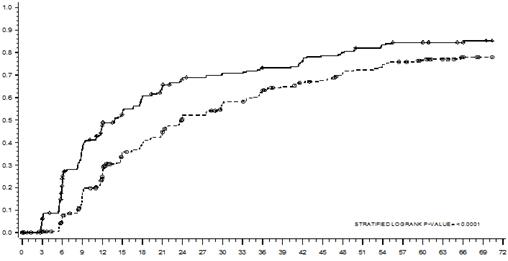
Dasatinib ------ Imatinib

Cenzurovaní

Cenzurovaní
MESIACE
SKUPINA #
RESPONDÉRY
/
#
RANDOMIZOVANÍ POMER
RIZIKA
(95
%
CI)
Dasatinib 198/259
Imatinib 167/260
Dasatinib ku imatinibu 1,54 (1,25 - 1,89)
Miery cCCyR v SPRYCELOM a imatinibom liečenej skupine, v uvedenom poradí, počas 3 mesiacov
(54 % a 30 %), počas 6 mesiacov (70 % a 56 %), a počas 9 mesiacov (75 % a 63 %), počas
24 mesiacov (80 % a 74 %), počas 36 mesiacov (83 % a 77 %), počas 48 mesiacov (83 % a 79 %)
a počas 60 mesiacov (83 % a 79 %) boli zhodné s primárnym koncovým ukazovateľom. Miery MMR
v SPRYCELOM a imatinibom liečenej skupine, v uvedenom poradí počas 3 mesiacov (8 % a 0,4 %),
6 mesiacov (27 % a 8 %), 9 mesiacov (39 % a 18 %), 12 mesiacov (46 % a 28 %), 24 mesiacov (64 % a 46 %), 36 mesiacov (67 % a 55 %), 48 mesiacov (73 % a 60 %) a počas 60 mesiacov (76 % a 64 %) boli tiež v zhode s primárnym koncovým ukazovateľom.
Pomery MMR pri špecifických časových bodoch sú zobrazené graficky na Grafe 2. Pomery MMR boli rovnomerne vyššie u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
 Graf 2: Hodnoty MMR v priebehu času - všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML)
Graf 2: Hodnoty MMR v priebehu času - všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML)
p
o
2
rokoch
64 %, p<,0001
po3rokoch
67 %, p<,0055
po4rokoch
73 %, p<,0021
po5rokoch
76 %, p<,0022
p
o
1
roku
46 %, p<,0001
N
Dasatinib 100 mg jedenkrát denne 259
--------- Imatinib 400 mg jedenkrát denne 260
Mesiace od randomizácie
Podiel pacientov, ktorí dosiahli pomer BCR-ABL ≤0,01 % (4-log zníženie) v akomkoľvek čase bol
vyšší v SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (54,1 % proti
45 %). Podiel pacientov, ktorí dosiahli pomer BCR-ABL ≤0,0032% (4,5-log zníženie) v akomkoľvek čase bol vyšší v SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (44% proti
34%).
Pomery MR4.5 v priebehu času sú zobrazené graficky na grafe 3. Pomery MR4.5 boli v priebehu času rovnomerne vyššie u pacientov liečených dasatinibom v porovnaní s pacientmi liečenými imatinibom.
Graf 3: Hodnoty MR4.5 v priebehu času - všetci randomizovaní pacienti v 3. fáze štúdie s novo diagnostikovanými pacientmi v chronickej fáze CML
po
1
r
oku
5 %, p<,2394
po2rokoch19 %, p<,0008
 po3rokoch
po3rokoch24 %, p<,0013
po4rokoch34 %, p<,0055
po5rokoch42 %, p<,0251
N
Dasatinib 100 mg jedenkrát denne 259
--------- Imatinib 400 mg jedenkrát denne 260
Mesiace od randomizácie
Miera MMR v akomkoľvek čase v každej rizikovej skupine určená Hasfordovým skóre bola vyššia v
SPRYCELOM liečenej skupine v porovnaní s imatinibom liečenej skupine (nízke riziko: 90 % a
69 %; stredné riziko: 71 % a 65 %; vysoké riziko: 67 % a 54 %) v uvedenom poradí.
V ďalšej analýze dosiahlo viac pacientov liečených dasatinibom (84 %) skorú molekulárnu odpoveď (definovanú ako hladiny BCR-ABL 10 % po 3 mesiacoch) v porovnaní s pacientmi liečenými imatinibom (64 %). Pacienti s dosiahnutou skorou molekulárnou odpoveďou mali nižšie riziko transformácie, vyšší pomer prežívania bez progresie (PFS, progression-free survival) a vyšší pomer celkového prežívania (OS, overall survival), ako je uvedené v tabuľke 8.
Tabuľka 8: Pacienti s dasatinibom s BCR-ABL 10 % a >10%po3mesiacoch
Pacienti s BCR-ABL 10 %
Pacienti s BCR-ABL
Dasatinib
N
=
235
po
3
mesiacoch
>
10
%
po
3
mesiacoch
Počet pacientov (%) 198 (84,3) 37 (15,7)
Transformácia po 60 mesiacoch, n/N (%)
Pomer PFS po 60 mesiacoch
(95 % CI)
6/198 (3,0) 5/37 (13,5)
92,0 % (89,6; 95,2) 73,8 % (52,0; 86,8)
Pomer OS po 60 mesiacoch (95 % CI) 93,8 % (89,3; 96,4) 80,6 % (63,5; 90,2)
Hodnoty OS pri špecifických časových bodoch sú zobrazené na Grafe 4. Hodnota OS bola
rovnomerne vyššia u pacientov liečených dasatinibom, ktorí dosiahli hladinu BCR-ABL ≤ 10 % po
3 mesiacoch než u tých, ktorí ju nedosiahli.
Graf 4: Grafické znázornenie orientačných bodov celkového prežívania po dasatinibe pomocou hladiny BCR-ABL ( 10 % alebo > 10 %) po 3 mesiacoch v 3. fáze štúdie s novo diagnostikovanými pacientmi s chronickou fázou CML
Rizikoví pacienti
MESIACE
<=10% 198 198 197 196 195 193 193 191 191 190 188 187 187 184 182 181 180 179 179 177 171 96 54 29 3 0
>10% 37 37 37 35 34 34 34 33 33 31 30 29 29 29 28 28 28 27 27 27 26 15 10 6 0 0
≤10 % ------ >10 %
Cenzurovaní

Cenzurovaní
SKUPINA # ÚMRTIA / #Pacient, ktorý ukončil štúdiu MEDIÁN (95 % CI) POMER RIZIKA (95 % CI)
≤10 % 14/198 .(. - .)
>10 % 8/37 .(. - .)
0,29 (0,12 - 0,69)
Progresia ochorenia bola definovaná ako zvýšenie bielych krviniek napriek zodpovedajúcej liečbe, strata CHR, čiastočná CyR alebo CCyR, progresia do akcelerovanej fázy alebo blastovej fázy alebo smrť. Odhadovaná miera 60-mesačného PFS bola 88,9 % (CI: 84 %-92,4 %) v dasatinibom a imatinibom liečenej skupine. Zmena na akcelerovanú alebo blastovú fázu po 60 mesiacoch sa vyskytla u niekoľkých dasatinibom liečených pacientov (n=8; 3 %) v porovnaní s pacientmi liečenými imatinibom (n=15; 5,8 %). Odhadovaná miera 60-mesačného prežitia pre dasatinibom a imatinibom liečených pacientov bola 90,9 % (CI: 86,6 % – 93,8 %) a 89,6 % (CI: 85,2 % – 92,8 %), v uvedenom poradí. Medzi dasatinibom a imatinibom nebol rozdiel v OS (HR 1,01; 95 % CI: 0,58-1,73; p= 0,9800) a PFS (HR 1,00, 95 % CI: 0,58-1,72, p = 0,9998).
U pacientov, u ktorých sa zaznamenala progresia ochorenia alebo ukončenie liečby dasatinibom alebo imatinibom, sa vykonalo sekvenovanie BCR-ABL na krvných vzorkách od pacientov, u ktorých boli dostupné. V obidvoch liečených ramenách sa pozoroval podobný pomer výskytu mutácií. Mutácie detekované u pacientov liečených dasatinibom boli T315I, F317I/L a V299L. V ramene liečenom imatinibom sa detekovalo iné spektrum mutácií. Na základe
in vitro údajov sa dasatinib neprejavuje ako účinný voči mutácii T315I.
Chronická fáza CML - Rezistencia alebo intolerancia na predošlú liečbu imatinibomVykonali sa dve klinické štúdie s pacientmi rezistentnými alebo intolerantnými na imatinib; koncovým ukazovateľom primárnej účinnosti v týchto štúdiách bola veľká cytogenetická odpoveď (MCyR).
1. štúdiaOtvorená, randomizovaná, nekomparatívna, multicentrická štúdia sa vykonala s pacientmi, u ktorých zlyhala úvodná liečba so 400 alebo 600 mg imatinibu. Boli randomizovaní (2:1) buď do skupiny s dasatinibom (70 mg dvakrát denne) alebo s imatinibom (400 mg dvakrát denne). Prechod do alternatívnej liečebnej skupiny bol povolený vtedy, ak pacienti vykazovali známky progresie ochorenia alebo intoleranciu, ktorú nebolo možné zvládnuť úpravou dávky. Primárny koncový ukazovateľ bol MCyR po 12 týždňoch. Sú dostupné výsledky u 150 pacientov: 101 pacientov bolo randomizovaných do skupiny s dasatinibom a 49 pacientov do skupiny s imatinibom (všetci pacienti
boli rezistentní na imatinib). Medián času od stanovenia diagnózy po randomizáciu bol 64 mesiacov v skupine s dasatinibom a 52 mesiacov v skupine s imatinibom. Všetci pacienti boli po predošlej intenzívnej liečbe. Predošlá úplná hematologická odpoveď (CHR) na imatinib sa dosiahla u 93 % z celkovej populácie pacientov. Predošlá MCyR na imatinib sa dosiahla u 28 % pacientov v skupine s dasatinibom a u 29 % pacientov v skupine s imatinibom.
Medián trvania liečby bol 23 mesiacov pre skupinu s dasatinibom (pričom dosiaľ bolo 44 % pacientov liečených počas > 24 mesiacov) a 3 mesiace pre skupinu s imatinibom (pričom dosiaľ bolo 10 % pacientov liečených počas > 24 mesiacov). Pred prechodom do druhej skupiny liečby dosiahlo CHR v skupine s dasatinibom deväťdesiattri percent pacientov a imatinibom 82 % pacientov.
Počas 3 mesiacov sledovania došlo k MCyR častejšie v skupine s dasatinibom (36 %) ako v skupine s imatinibom (29 %). Pozoruhodné je, že v skupine s dasatinibom hlásilo úplnú cytogenetickú odpoveď (CCyR) 22 % pacientov, pričom v skupine s imatinibom dosiahlo CcyR len 8 %. Pri dlhšej liečbe a sledovaní (medián 24 mesiacov) sa MCyR dosiahla u 53 % pacientov liečených dasatinibom (CCyR u
44 %) a u 33 % pacientov liečených imatinibom (CCyR u 18 %) pred prechodom do druhej skupiny. Medzi pacientmi, ktorí užívali imatinib v dávke 400 mg pred vstupom do štúdie, MCyR dosiahlo 61 % pacientov v ramene s dasatinibom a 50 % v ramene s imatinibom.
Na základe Kaplan-Meierových odhadov pomer pacientov, ktorí si udržali MCyR počas 1 roka bol
92 % (95 % CI: [85 %-100 %]) pre dasatinib (CCyR 97 %, 95 % CI: [92 %-100 %]) a 74 % (95 % CI:
[49 %-100 %]) pre imatinib (CCyR 100 %). Pomer pacientov, ktorí si udržali MCyR počas
18 mesiacov bol 90 % (95 % CI: [82%-98%]) pre dasatinib (CCyR 94 %, 95 % CI: [87 %-100 %]) a
74 % (95 % CI: [49 %-100 %]) pre imatinib (CCyR 100 %).
Na základe Kaplan-Meierových odhadov pomer pacientov, ktorí mali progresiu prežívania (PFS) počas 1 roka bol 91 % (95 % CI: [85 %-97 %]) pre dasatinib a 73 % (95 % CI: [54 %-91 %]) pre imatinib. Pomer pacientov, ktorí mali PFS počas 2 rokov bol 86 % (95 % CI: [78 %-93 %]) pre dasatinib a 65 % (95 % CI: [43 %-87 %]) pre imatinib.
Z celkového počtu 43 % pacientov v skupine s dasatinibom a 82 % pacientov v skupine s imatinibom došlo k zlyhaniu liečby, ktoré bolo definované ako progresia ochorenia alebo prechod do druhej skupiny liečby (nedostatočná odpoveď, intolerancia skúšaného lieku, atď.).
Výskyt veľkej cytogenetickej odpovede (definovaný ako BCR-ABL/kontrola transkriptov ≤ 0,1 % s RQ-PCR vo vzorke periférnej krvi) pred prechodom do druhej skupiny bol 29 % pre dasatinib a 12 % pre imatinib.
2. štúdia
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi rezistentnými alebo intolerantnými na imatinib (napr. pacienti, ktorí majú skúsenosť so signifikantnou toxicitou počas liečby imatinibom, ktorý vylučuje ďalšiu liečbu).
Dasatinib 70 mg dvakrát denne užívalo celkovo 387 pacientov (288 rezistentní a 99 intolerantní na
imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 61 mesiacov. Väčšina pacientov (53 %) dostala predošlú liečbu imatinibom trvajúcu viac ako 3 roky. Väčšina rezistentných pacientov (72 %) užívala > 600 mg imatinibu. Okrem imatinibu dostalo 35 % pacientov aj predošlú cytotoxickú chemoterapiu, 65 % dostalo predošlú liečbu interferónom a 10 % bolo po predošlej transplantácii kmeňových buniek. Tridsaťosem percent pacientov malo na začiatku liečby mutácie, o ktorých je známe, že vyvolávajú rezistenciu na imatinib. Medián trvania liečby dasatinibom bol 24 mesiacov, pričom dovtedy bolo 51 % pacientov liečených počas > 24 mesiacov. Výsledky účinnosti sú uvedené v tabuľke 9. MCyR sa dosiahla u 55 % pacientov rezistentných na imatinib a 82 % pacientov intolerantných na imatinib. Pri minimálne 24-mesačnom sledovaní malo len 21 z 240 pacientov, ktorí dosiahli McyR, progresiu ochorenia a nedosiahli medián trvania MCyR.
Na základe Kaplan-Meierových odhadov si 95 % (95 % CI: [92 %-98 %]) pacientov udržalo MCyR počas 1 roka a 88 % (95 % CI: [83 %-93 %]) si udržalo MCyR počas 2 rokov. Pomer pacientov, ktorí si udržali CCyR počas roka bol 97 % (95 % CI: [94 %-99 %]) a počas 2 rokov bol 90 % (95 % CI:
[86 %-95 %]). Štyridsaťdva percent pacientov rezistentných na imatinib bez predchádzajúcej MCyR
na imatinib (n= 188) dosiahlo MCyR s dasatinibom.
Vyskytlo sa 45 odlišných BCR-ABL mutácií u 38 % pacientov zúčastnených v tejto štúdii. Úplná hematologická odpoveď alebo MCyR bola dosiahnutá u pacientov majúcich množstvo BCR-ABL mutácií súvisiacich s rezistenciou na imatinib okrem T315I. Podiel MCyR v 2. roku bol podobný či pacienti mali nejakú základnú BCR-ABL mutáciu, P-loop mutáciu alebo nemali mutáciu (63 %, 61 % a 62 %).
Medzi pacientmi rezistentnými na imatinib bol odhadovaný pomer PFS 88 % (95 % CI: [84 %-92 %]) v 1. roku a 75 % (95 % CI: [69 %-81 %]) v 2 roku. Medzi pacientmi intolerantnými na imatinib bol odhadovaný pomer PFS 98 % (95 % CI: [95 %-100 %]) v 1. roku a 94 % (95 % CI: [88%-99 %]) v
2 roku.
Pomer veľkej molekulárnej odpovede po 24 mesiacoch bol 45 % (35 % pre imatinib rezistentných pacientov a 74 % pre imatinib intolerantných pacientov).
Akcelerovaná fáza CML
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi intolerantnými alebo rezistentnými na imatinib. Dasatinib 70 mg dvakrát denne užívalo celkovo 174 pacientov
(161 rezistentní a 13 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 82 mesiacov. Medián trvania liečby dasatinibom bol 14 mesiacov, pričom dosiaľ bolo 31 % pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (hodnotený u
41 pacientov s CCyR) po 24 mesiacoch bol 46 %. Ďalšie výsledky účinnosti sú uvedené v tabuľke 9.
Myeloidná blastická fáza CML
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi intolerantnými alebo rezistentnými na imatinib. Dasatinib 70 mg dvakrát denne užívalo celkovo 109 pacientov
(99 rezistentní a 10 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 48 mesiacov. Medián trvania liečby dasatinibom bol 3,5 mesiacov, pričom dosiaľ bolo 12 % pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (hodnotený u
19 pacientov s CCyR) po 24 mesiacoch bol 68 %. Ďalšie výsledky účinnosti sú uvedené v tabuľke 9.
Lymfoidná blastická fáza CML a Ph+ ALL
Otvorená, multicentrická štúdia s jednou skupinou sa vykonala s pacientmi v lymfoidnej blastickej fáze CML alebo s Ph+ ALL, ktorí boli rezistentní alebo intolerantní na predošlú liečbu imatinibom. Dasatinib 70 mg dvakrát denne užívalo celkovo 48 pacientov v lymfoidnej blastickej fáze CML
(42 rezistentní a 6 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 28 mesiacov. Medián trvania liečby dasatinibom bol 3 mesiace s 2 % pacientov liečených počas
> 24 mesiacov. Pomer veľkej molekulárnej odpovede (všetkých 22 liečených pacientov s CCyR) po
24 mesiacoch bol 50 %. Okrem toho dasatinib 70 mg dvakrát denne užívalo 46 pacientov s Ph+ ALL (44 rezistentní a 2 intolerantní na imatinib). Medián času od stanovenia diagnózy po začiatok liečby bol 18 mesiacov. Medián trvania liečby dasatinibom bol 3 mesiacov, pričom dosiaľ bolo 7 % pacientov liečených počas > 24 mesiacov. Pomer veľkej molekulárnej odpovede (všetkých
25 liečených pacientov s CCyR) po 24 mesiacoch bol 52 %. Ďalšie výsledky účinnosti sú uvedené v
tabuľke 9. Za zmienku stojí, že veľká hematologická odpoveď (MaHR) bola dosiahnutá rýchlo
(väčšinou do 35 dní od prvého podania dasatinibu u pacientov v lymfoidnej blastickej fáze CML, a do
55 dní u pacientov s Ph+ ALL).
Tabuľka
9
:
Účinnosť
SPRYCELU
vo
fáze
I
I
k
linických
štúdií
s
jednou
skupinou
a
C
h
r
o
ni
cká fáza
Akcelerovan á fáza
Myeloidná blastová fáza
Lymfoidná blastová fáza
Ph+ ALL
(n= 387) (n= 174) (n= 109) (n= 48) (n= 46)
Pomer
hematologickej
odpovede
b
(%)
MaHR (95 % CI) n/a
64 % (57-72) 33 % (24-43) 35 % (22-51) 41 % (27-57) CHR (95 % CI)
91 % (88-94) 50 % (42-58) 26 % (18-35) 29 % (17-44) 35 % (21-50) NEL (95 % CI) n/a 14 % (10-21) 7 % (3-14) 6 % (1-17) 7 % (1-18)
Trvanie MaHR (%; Kaplan-Meierove odhady)
1 rok n/a 79 % (71-87) 71 % (55-87) 29 % (3-56) 32 % (8-56)
2roky n/a 60%(50-70) 41%(21-60) 10%(0-28) 24%(2-47) Cytogenetickáodpoveďc (%) MCyR (95 % CI)
62 % (57-67) 40 % (33-48) 34 % (25-44) 52 % (37-67) 57 % (41-71) CCyR (95 % CI) 54 % (48-59) 33 % (26-41) 27 % (19-36) 46 % (31-61) 54 % (39-69)
Prežívanie (%; Kaplan-Meierove odhady)Bez progresie
1 rok 91 % (88-94) 64 % (57-72) 35 % (25-45) 14 % (3-25) 21 % (9-34)
2roky 80%(75-84) 46%(38-54) 20%(11-29) 5%(0-13) 12%(2-23) Celkovo
1 rok 97 % (95-99) 83 % (77-89) 48 % (38-59) 30 % (14-47) 35 % (20-51)
2roky 94%(91-97) 72%(64-79) 38%(27-50) 26%(10-42) 31%(16-47) Údaje uvedené v tabuľke sú zo štúdií s užívanou úvodnou dávkou 70 mg dvakrát denne. Pozri časť 4.2 pre odporučenú úvodnú dávku.
a Čísla v bold fonte sú výsledky primárneho koncového ukazovateľa.
b Kritéria hematologickej odpovede (všetky odpovede potvrdené po 4 týždňoch): Veľká hematologická odpoveď: (MaHR)
= úplná hematologická odpoveď (CHR) + žiadna prítomnosť leukémie (NEL).
CHR (chronická CML): WBC ≤ zdravotníckeho zariadenia ULN, trombocyty < 450 000/mm3, bez blastov alebo promyelocytov v periférnej krvi, < 5% myelocytov plus metamyelocyty v periférnej krvi, bazofily v periférnej krvi
< 20% a žiadne extramedulárne postihnutie.
CHR (pokročilá CML/Ph+ ALL): WBC ≤ zdravotníckeho zariadenia ULN, ANC ≥ 1 000/mm3, trombocyty
≥ 100 000/mm3, bez blastov alebo promyelocytov v periférnej krvi, blasty kostnej drene ≤ 5%, < 5% myelocyty plus metamyelocyty v periférnej krvi, bazofily v periférnej krvi < 20% a žiadne extramedulárne postihnutie.
NEL: rovnaké kritériá ako pre CHR ale ANC ≥ 500/mm3 a < 1 000/mm3, alebo trombocyty
≥ 20 000/mm3 a ≤ 100 000/mm3.
c Kritéria cytogenetickej odpovede: úplná (0 % Ph+ metafáza) alebo čiastočná (> 0 %-35 %). MCyR (0 %-35 %)
kombinovaná úpná a čiastočná odpoveď.
n/a = neaplikovateľný; CI = interval spoľahlivosti; ULN = horný limit normálneho rozpätia.
Výsledok u pacientov s tranplantáciou kostnej drene po liečbe dasatinibom nie je úplne zhodnotený. Klinické štúdie fázy III u pacientov s CML v chronickej, akcelerovanej alebo myeloidnej blastovej
fáze a Ph+ ALL, ktorí boli rezistentní alebo netolerovali imatinib
Dve randomizované, otvorené štúdie sa vykonali na zhodnotenie účinnosti dasatinibu podávaného jedenkrát denne v porovnaní s dasatinibom podávaným dvakrát denne. Výsledky uvedené nižšie sú založené na minimálne 2-ročnom a 7-ročnom následnom sledovaní po začatí liečby dasatinibom.
1. štúdiaV štúdii s chronickou fázou CML bola MCyR primárnym koncovým ukazovateľom u pacientov rezistentných na imatinib. MCyR bola hlavným sekundárnym koncovým ukazovateľom pri celkovej dennej dávke u pacientov rezistentných na imatinib. Ďalšie sekundárne koncové ukazovatele zahŕňali trvania MCyR, PFS a celkového prežitia. Z celkového počtu 670 pacientov, z ktorých bolo
497 rezistentných na imatinib, boli pacienti v skupinách randomizovaní na dasatinib v dávke 100 mg
jedenkrát denne, 140 mg jedenkrát denne, 50 mg dvakrát denne alebo 70 mg dvakrát denne. Medián trvania liečby všetkých pacientov stále liečených s minimálne 5-ročným následným sledovaním (n=205) bol 59 mesiacov (rozpätie 28-66 mesiacov). Medián trvania liečby všetkých pacientov po 7- ročnom následnom sledovaní bol 29,8 mesiaca (rozpätie < 1-92,9 mesiaca).
Účinnosť sa dosiahla vo všetkých skupinách liečených dasatinibom v dávkovacej schéme jedenkrát denne dokazujúc porovnateľnú účinnosť (nie horšiu) dávkovacej schémy dvakrát denne primárneho
koncového ukazovateľa účinnosti (rozdiel v MCyR 1,9 %, 95 % interval spoľahlivosti [-6,8 % -
10,6 %]); schéma dávkovania 100 mg jedenkrát denne však potvrdila zlepšenú bezpečnosť a znášanlivosť. Výsledky účinnosti sú uvedené v tabuľkách 10 a 11.
Tabuľka 10: Účinnosť SPRYCELU v štúdii fázy III optimalizácie dávky: rezistencia alebo
intolerancianaimatinibchronickáfázaCML(2-ročnévýsledky)a
Všetci pacienti n=167 Pacienti rezistentní na
imatinib n=124
Pomerhematologickejodpovedeb (%)(95%CI)
CHR 92%(86–95)
Cytogenetickáodpoveďc (%)(95%CI)
MCyR
Všetci pacienti 63 % (56-71)
Pacienti rezistentní na
imatinib 59 % (50-68)
CCyR
Všetci pacienti 50 % (42-58)
Pacienti rezistentní na
imatinib 44 % (35-53)
Veľkámolekulárnaodpoveďupacientov,ktorídosiahliCCyRd (%)(95%CI)
Všetci pacienti 69 % (58-79)
Pacientirezistentnínaimatinib 72%(58-83)
a Výsledky hlásené pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
b Kritéria hematologickej odpovede (všetky odpovede potvrdené po 4 týždňoch): Kompletná hematologická odpoveď (CHR) (chronická CML): WBC ≤ ULN zdravotníckeho zariadenia, trombocyty < 450 000/mm3, žiadne blasty alebo promyelocyty v periférnej krvi, <5 % myelocytov plus metamyelocytov v periférnej krvi, bazofily v periférnej krvi
< 20 % a žiadne extramedulárne postihnutia.
c Kritéria cytogenetickej odpovede: úplná (0 % Ph+ metafáza) alebo čiastočná (> 0 %–35 %). MCyR (0 %–35 %)
kombinácie s úplnými a čiastočnými odpoveďami.
d Kritéria veľkej molekulárnej odpovede: Definované ako BCR-ABL/kontrola transkriptorov ≤ 0,1 % pomocou RQ-PCR
vo vzorkách periférnej krvi.
Tabuľka 11: Dlhodobá účinnosť SPRYCELU v 3. fáze štúdie optimalizácie dávky: rezistencia alebo intolerancia na imatinib pacienti s chronickou fázou CML
a
Minimálne
obdobie
následného
sledovania
1
r
ok 2
r
o
k
y 5
r
okov 7
r
o
k
ov
Veľká molekulárna odpoveď
Všetci pacienti NA 37 % (57/154) 44 % (71/160) 46 % (73/160)
Pacienti rezistentní na imatinib
Pacienti intolerantní na
imatinib
Prežívanie bez progresie ochoreniab
NA 35 % (41/117) 42 % (50/120) 43 % (51/120) NA 43 % (16/37) 53 % (21/40) 55 % (22/40)
Všetci pacienti 90 % (86; 95) 80 % (73; 87) 51 % (41; 60) 42 % (33; 51)
Pacienti rezistentní na imatinib
Pacienti intolerantní na
imatinib
Celkové prežívanie
88 % (82; 94) 77 % (68; 85) 49 % (39; 59) 39 % (29; 49)
97 % (92; 100) 87 % (76; 99) 56 % (37; 76) 51 % (32; 67)
Všetci pacienti 96 % (93; 99) 91 % (86; 96) 78 % (72; 85) 65 % (56; 72)
Pacienti rezistentní na imatinib
94 % (90; 98) 89 % (84; 95) 77 % (69; 85) 63 % (53; 71)
Pacienti intolerantní na
100 % (100; 100) 95 % (88;
82 % (70; 94) 70 % (52; 82)
imatinib 100)
a Výsledky hlásené pri odporúčanej začiatočnej dávke 100 mg jedenkrát denne.
b Progresia bola definovaná ako zvýšenie počtu WBC, strata CHR alebo MCyR, ≥ 30 % zvýšenie v Ph+ metafáze, potvrdená AP/BP ochorenia alebo smrť. PFS bolo analyzované na princípe úmyslu liečiť (intent-to-treat) a pacienti boli
následne sledovaní na udalosti vrátane dodatočnej liečby.
Na základe Kaplan-Meierových odhadov bol pomer pacientov liečených dasatinibom v dávke 100 mg jedenkrát denne, ktorí si udržali MCyR počas 18 mesiacov 93 % (95 % CI: [88%-98%]).
Účinnosť bola tiež hodnotená u pacientov, ktorí boli intolerantní na imatinib. V tejto populácii pacientov, ktorí užívali 100 mg jedenkrát denne bola MCyR dosiahnutá u 77 % a CCyR u 67 %.
2. štúdia
V štúdii s pokročilou fázou CML a Ph+ ALL bola MaHR primárnym koncovým ukazovateľom. Z celkových 611 pacientov, boli pacienti v skupinách randomizovaní dasatinibom buď v dávke 140 mg jedenkrát denne alebo 70 mg dvakrát denne. Medián trvania liečby bol priemerne 6 mesiacov (rozpätie
0,03 – 31 mesiacov).
Dávkovacia schéma jedenkrát denne poukázala porovnateľnú účinnosť (nie horšiu) s dávkovacou schémou dvakrát denne primárneho koncového ukazovateľa účinnosti (rozdiel v MaHR 0,8 %, 95 % interval spoľahlivosti [-7,1 % - 8,7 %]); dávkovacia schéma 140 mg jedenkrát denne však potvrdila zlepšenie bezpečnosti a znášanlivosti.
Pomery odpovede sú uvedené v tabuľke 12.
Tabuľka 12: Účinnosť SPRYCELU v štúdii fázy III optimalizácie dávky: Pokročilá fáza
CML
a
P
h
+
ALL
(2-ročné
v
ý
sledky)
a
Akcelerovaná fáza
M
yeloidná blastová fáza
Lymfoidná blastová fáza
Ph+ALL
(n= 158) (n= 75) (n= 33) (n= 40)
M
a
H
R
b (95 % CI) CHRb
(95 % CI) NELb
66 % (59-74)
47 % (40-56)
19 %
28 % (18-40)
17 % (10-28)
11 %
42 % (26-61)
21 % (9-39)
21 %
38 % (23-54)
33 % (19 – 49)
5 %
(95 % CI) (13-26) (5-20) (9-39) (1-17)
M
C
y
R
c (95 % CI) CCyR
39 % (31-47)
32 %
28 % (18-40)
17 %
52 % (34-69)
39 %
70 % (54-83)
50 %
(95
%
CI)
(25-40) (10-28) (23-58) (34-66)
a Výsledky hlásené pri odporúčanej začiatočnej dávke 140 mg jedenkrát denne (pozri časť 4.2).
b Kritéria hematologickej odpovede (všetky kritéria boli potvrdené po 4 týždňoch): Veľká hematologická odpoveď: (MaHR) = úplná hematologická odpoveď (CHR) + žiadna prítomnosť leukémie (NEL).
CHR: WBC ≤ ULN zdravotníckeho zariadenia, ANC ≥ 1 000/mm3, trombocyty < 100 000/mm3, žiadne blasty
alebopromyelocyty v periférnej krvi, blasty v krvi a v kostnej dreni ≤ 5 %, < 5 % myelocyty plus metamyelocyty v periférnejkrvi, bazofily v periférnej krvi < 20 % a žiadne extramedulárne postihnutia.
NEL: rovnaké kritériá ako pre CHR ale ANC ≥ 500/mm3 a < 1 000/mm3, alebo trombocyty ≥ 20 000/mm3 a
≤ 100 000/mm3.
c MCyR kombinácie obidvoch úplnej (0 % Ph+ metafáza) a čiastočnej (> 0 %-35 %) odpovede. CI = interval spoľahlivosti ULN = horný limit normálneho rozpätia.
U pacientov s akcelerovanou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne sa nedosiahol medián trvania MaHR a medián celkového prežívania a medián PFS bol 25 mesiacov.
U pacientov s myeloidnou blastovou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne bol medián trvania MaHR 8 mesiacov, medián PFS bol 4 mesiace a medián celkového prežívania bol 8 mesiacov. U pacientov s lymfoidnou blastovou fázou CML liečených s dávkovacím režimom 140 mg jedenkrát denne bol medián trvania MaHR 5 mesiacov, medián PFS bol 5 mesiacov a medián celkového prežívania bol 11 mesiacov.
Medián trvania MaHR u pacientov s Ph+ALL liečených s dávkovacím režimom 140 mg jedenkrát denne bol 5 mesiacov, medián PFS bol 4 mesiace a medián celkového prežívania bol 7 mesiacov.
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so SPRYCELOM v jednej alebo vo viacerých podskupinách pediatrickej populácie s pozitívnym chromozómom Philadelphia (BCR-ABL translokácia) s akútnou lymfoblastovou leukémiou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Pediatrickí pacienti s CML
Zo 130 pacientov s chronickou fázou CML (CML‑CP) liečených v dvoch pediatrických štúdiách, otvorené, nerandomizované klinické skúšanie s nastavovaním dávky I. fáza a otvorené, nerandomizované klinické skúšanie II. fáza, bolo 84 pacientov (výhradne z klinického skúšania
II. fázy) novo diagnostikovaných s CML‑CP a 46 pacientov (17 z klinického skúšania I. fázy a 29 z klinického skúšania II. fázy) bolo rezistentných alebo intolerantných na predchádzajúcu liečbu imatinibom. Deväťdesiat sedem zo 130 pediatrických pacientov s CML-CP sa liečilo SPRYCELOM tabletami v dávke 60 mg/m2 jedenkrát denne (maximálna dávka 100 mg jedenkrát denne u pacientov s vysokým BSA). Pacienti sa liečili až do progresie ochorenia alebo do neakcetovateľnej toxicity.
Kľúčové koncové ukazovatele účinnosti boli: kompletná cytogenetická odpoveď (CCyR), veľká cytogenetická odpoveď (MCyR) a veľká molekulárna odpoveď (MMR). Výsledky sú uvedené v tabuľke 13.
Tabuľka 13: Účinnosť SPRYCELU u pediatrických pacientov s CML-CP
Kumulatívna
odpoveď
v
priebehu
času
s
obdobím
minimálneho
následného
sledovania
3
mesiace 6
mesiacov 12
mesiacov 24
mesiacov
CCyR
(95 % CI)
Novo diagnostikovaní (N = 51)a
43,1 % (29,3; 57,8)
66,7 % (52,1; 79,2)
96,1 % (86,5; 99,5)
96,1 % (86,5; 99,5)
Predtým imatinib
(N = 46)b
45,7 %
(30,9; 61,0)
71,7 %
(56,5; 84,0)
78,3 %
(63,6; 89,1)
82,6 %
(68,6; 92,2)
MCyR
(95 % CI) Novo diagnostikovaní (N = 51)a
60,8 % (46,1; 74,2)
90,2 % (78,6; 96,7)
98,0 % (89,6; 100)
98,0 % (89,6; 100)
Predtým imatinib
(N = 46)b
60,9 %
(45,4; 74,9)
82,6 %
(68,6; 92,2)
89,1 %
(76,4; 96,4)
89,1 %
(76,4; 96,4)
MMR
(95 % CI) Novo diagnostikovaní (N = 51)a
7,8 % (2,2; 18,9)
31,4 % (19,1; 45,9)
56,9 % (42,2; 70,7)
74,5 % (60,4; 85,7)
Predtým imatinib
(N = 46)b
15,2 %
(6,3; 28,9)
26,1 %
(14,3; 41,1)
39,1 %
(25,1; 54,6)
52,2 %
(36,9; 67,1)
a Pacienti z pediatrickej štúdie II. fázy s novo diagnostikovanou CML-CP, ktorí užívali perorálnu tabletovú formu
b Pacienti z pediatrických štúdií I. fázy a II. fázy rezistentní alebo intolerantní na imatinib s CML-CP, ktorí užívali perorálnu tabletovú formu
V pediatrickej štúdii I. fázy po minimálne 7 rokoch následného sledovania bol u 17 pacientov rezistentných alebo intolerantných na imatinib s CML-CP medián trvania PFS 53,6 mesiaca a miera OS bola 82,4 %.
V pediatrickej štúdii II. fázy bola u pacientov, ktorí užívali tabletovú formu odhadnutá 24-mesačná miera PFS u 51 pacientov s novo diagnostikovanou CML-CP 94,0 % (82,6; 98,0) a 81,7 % (61,4;
92,0) u 29 pacientov s CML-CP s rezistenciou/intoleranciou na imatinib. Po 24 mesiacoch následného
sledovania bolo OS u novo diagnostikovaných pacientov 100 % a 96,6 % u pacientov rezistentných alebo intolerantných na imatinib.
V pediatrickej štúdii II. fázy progredoval do blastovej fázy CML 1 novo diagnostikovaný pacient a
2 pacienti rezistentní alebo intolerantní na imatinib.
33 novo diagnostikovaných pediatrických pacientov s CML-CP dostalo SPRYCEL prášok na perorálnu suspenziu v dávke 72 mg/m2. Táto dávka predstavuje o 30 % nižšiu expozíciu v porovnaní s odporúčanou dávkou (pozri časť 5.2). U týchto pacientov po 12 mesiacoch CCyR a MMR bolo CCyR:
87,9 % [95 % CI: (71,8-96,6)] a MMR: 45,5 % [95 % CI: (28,1-63,6)].
U pediatrických pacientov s CML-CP, liečených dasatinibom, predtým exponovaných imatinibu, boli na konci liečby detekované mutácie: T315A, E255K a F317L. E255K a F317L však boli detegované aj pred liečbou. Na konci liečby neboli detegované žiadne mutácie u novo diagnostikovaných pacientov s CML-CP.
5.2 Farmakokinetické vlastnosti
Farmakokinetika dasatinibu sa hodnotila u 229 dospelých zdravých jedincov a u 84 pacientov. Absorpcia
Dasatinib sa u pacientov po perorálnom podaní rýchlo vstrebáva, pričom maximálne koncentrácie sa
dosiahnu medzi 0,5-3 hodinami. Po perorálnom podaní je vzostup priemernej expozície (AUCτ) približne úmerný prírastku dávky v celom rozmedzí dávok od 25 mg do 120 mg dvakrát denne. U pacientov je celkový priemerný konečný polčas dasatinibu približne 5 – 6 hodín.
Údaje od zdravých jedincov, ktorým sa podala jednorazová 100 mg dávka dasatinibu po 30 minútach po jedle s vysokým obsahom tuku, poukázali na 14 % zvýšenie priemernej hodnoty AUC dasatinibu. Požitie jedla s nízkym obsahom tuku 30 minút pred užitím dasatinibu viedlo k 21 % zvýšeniu priemernej hodnoty AUC dasatinibu. Pozorované vplyvy jedla nepredstavujú klinicky významné zmeny v expozícii.
Distribúcia
U pacientov má dasatinib veľký zdanlivý distribučný objem (2 505 l), čo svedčí o tom, že liek sa v rozsiahlej miere distribuuje v extravaskulárnom priestore. Pri klinicky významných koncentráciách dasatinibu bola väzba na plazmatické bielkoviny približne 96 % na základe in vitro experimentov.
Biotransformácia
Dasatinib sa u ľudí v rozsiahlej miere metabolizuje, pričom do tvorby metabolitov sú zapojené viaceré enzýmy. U zdravých jedincov, ktorým bolo podaných 100 mg dasatinibu označeného [14C], predstavoval nezmenený dasatinib 29 % cirkulujúcej rádioaktivity v plazme. Plazmatická koncentrácia a in vitro meraná aktivita svedčia o tom, že nie je pravdepodobné, že metabolity dasatinibu majú významnú úlohu v pozorovanej farmakológii lieku. CYP3A4 je hlavným enzýmom zodpovedným za metabolizmus dasatinibu.
Eliminácia
Prevláda vylučovanie stolicou, prevažne vo forme metabolitov. Po jednorazovej perorálnej dávke dasatinibu označeného [14C] sa približne 89 % dávky vylúčilo do 10 dní, pričom 4 % rádioaktivity sa zistili v moči a 85 % rádioaktivity sa zistilo v stolici. Nezmenený dasatinib predstavoval 0,1 % dávky v moči a 19 % dávky v stolici, pričom zvyšok dávky bol vo forme metabolitov.
Poruchafunkciepečeneaobličiek
Vplyv poruchy funkcie pečene na farmakokinetiku jednorazovej dávky dasatinibu bol hodnotený u
8 jedincov so stredne závažnou poruchou funkcie pečene, ktorí užívali dávku 50 mg a u 5 jedincov so závažnou poruchou funkcie pečene, ktorí užívali dávku 20 mg v porovnaní so zodpovedajúcimi zdravými jedincami, ktorí užívali dávku 70 mg dasatinibu. Priemerné Cmax dasatinibu upravené na dávku 70 mg sa znížili o 47 % a AUC o 8 % u jedincov so stredne závažnou poruchou funkcie pečene v porovnaní s osobami s normálnou funkciou pečene. U jedincov so závažnou poruchou funkcie pečene sa priemerné Cmax upravené na dávku 70 mg znížili o 43 % a AUC o 28 % v porovnaní s osobami s normálnou funkciou pečene (pozri časti 4.2 a 4.4).
Dasatinib a jeho metabolity sú v minimálnej miere vylučované obličkami. Pediatrickápopulácia
Farmakokinetika dasatinibu sa hodnotila u 104 pediatrických pacientov s leukémiou alebo solídnymi
nádormi (72 dostalo tabletovú formu a 32 dostalo prášok na perorálnu suspenziu).
Bioekvivalentná štúdia hodnotiaca prášok na perorálnu suspenziu a referenčnú tabletovú formu u
77 dospelých pacientov preukázala, že expozícia prášku na perorálnu suspenziu bola o 19 % nižšia ako expozícia u referenčných tabliet. Údaje koncentrácií u 32 pediatrických pacientov liečených práškom na perorálnu suspenziu v dávke 72 mg/m2 boli skumulované s údajmi z tabliet na populačnú farmakokinetickú (PPK) analýzu, ktorá preukázala, že expozícia prášku na perorálnu suspenziu (podľa merania priemernej koncentrácie v rovnovážnom stave podľa času [Cavgss]) pri dávke 72 mg/m2 bola
približne o 30 % nižšia ako expozícia pri tablete s dávkou 60 mg/m2. PPK simulácia na základe
modelu predpokladala, že telesná hmotnosť odstupňuje odporúčanie na dávkovanie opísané pre prášok na perorálnu suspenziu v časti 4.2 súhrnu charakteristických vlastností pre prášok na perorálnu suspenziu, očakáva sa, že poskytne podobnú expozíciu ako tableta v dávke 60 mg/m2. Tieto údaje sa majú zohľadniť, ak sa pacienti prestavujú z prášku na perorálnu suspenziu na tablety alebo opačne.
5.3 Predklinické údaje o bezpečnosti
Profil predklinickej bezpečnosti dasatinibu sa hodnotil v sérii štúdií in vitro a in vivo na myšiach, potkanoch, opiciach a králikoch.
Primárna toxicita sa vyskytla v gastrointestinálnom, hematopoetickom a lymfoidnom systéme. Gastrointestinálna toxicita bola limitovaná dávku u potkanov a opíc, keďže črevo bolo hlavným cieľovým orgánom. U potkanov boli minimálne až mierne poklesy v parametroch erytrocytov sprevádzané zmenami kostnej drene; u opíc sa podobné zmeny vyskytli s nižšou incidenciou. Lymfoidná toxicita u potkanov pozostávala z lymfoidnej deplécie lymfatických uzlín, sleziny a
týmusu a zníženej hmotnosti lymfoidných orgánov. Zmeny v gastrointestinálnom, hematopoetickom a lymfoidnom systéme boli reverzibilné po ukončení liečby.
Obličkové zmeny u opíc liečených počas 9 mesiacov sa obmedzovali na zvýšenie prirodzenej mineralizácie v obličkách. Kožné krvácanie bolo pozorované v štúdii akútnej jednorazovej perorálnej dávky u opíc, ale nebolo pozorované v štúdiách opakovanej dávky ani u opíc ani u potkanov.
U potkanov dasatinib inhiboval agregáciu trombocytov in vitro a predĺžil čas krvácania z kutikuly
in vivo, ale nevyvolal spontánne krvácanie.
Aktivita dasatinibu in vitro v teste hERG a teste na Purkyňových vláknach svedčí o možnom predĺžení repolarizácie srdcových komôr (QT interval). V štúdii s jednorazovou dávkou in vivo na opiciach,
ktoré boli pri vedomí a sledované telemetricky, však neboli žiadne zmeny v QT intervale, ani v tvare
vlny na EKG.
Dasatinib nebol mutagénny v testoch in vitro na bakteriálnych bunkách (Amesov test) a nebol genotoxický v teste in vivo na potkaních mikronukleoch. Dasatinib mal klastogénny účinok in vitro na deliace sa ovariálne bunky škrečka (CHO).
Dasatinib neovplyvnil obvyklú samčiu alebo samičiu fertilitu potkanov a včasnú štúdiu embryofetálneho vývoja, ale indukoval embryoletalitu v dávkach zodpovedajúcich klinickej expozícii u ľudí. V štúdiách embryofetálneho vývoja podobne spôsobil dasatinib embryoletalitu, ktorá bola spojená so zníženou veľkosťou vrhu u potkanov ako aj zmeny skeletu plodu u potkanov aj králikov. Tieto účinky sa vyskytovali pri dávkach, ktoré nespôsobovali toxické prejavy u matky naznačujúc, že dasatinib je selektívna toxická látka na reprodukciu od implementácie až počas celej organogenézy.
U myší vyvolal dasatinib imunosupresiu, ktorá súvisela s dávkou a bola účinne zvládnutá znížením dávky a/alebo zmenami dávkovacej schémy. Dasatinib mal fototoxický potenciál v teste fototoxicity in vitro zameranom na prenikanie neutrálnej červene do myších fibroblastov. Dasatinib sa považoval za nefototoxický in vivo po jednorazovom perorálnom podaní samičej bezchlpej myši v expozíciách zodpovedajúcich nanajvýš 3-násobnej expozícii u ľudí po podaní odporúčanej terapeutickej dávky (vychádzajúcej z AUC).
V dvojročnej štúdii karcinogenity sa potkanom podávali perorálne dávky dasatinibu 0,3; 1 a
3 mg/kg/deň. Najvyššia dávka viedla k plazmatickej hladine expozície (AUC) vo všeobecnosti ekvivalentnej expozícii u ľudí pri odporúčanom rozsahu úvodných dávok od 100 mg do 140 mg denne. Zistilo sa štatisticky významné zvýšenie celkovej incidencie karcinómov skvamóznych buniek a papilómov v maternici a krčku maternice u samičiek s vysokou dávkou a adenóm prostaty u samcov s nízkou dávkou. Významnosť nálezov zo štúdie karcinogenity na potkanoch pre ľudí nie je známa.
6
. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
sacharóza
karmelóza, sodná soľ emulzia simetikónu zložená z:
simetikón
makrogolsorbitantristearát makrogolstearát
glyceroly, metylcelulóza xantánová guma kyselina benzoová kyselina sorbová kyselina sírová
kyselina vínna
citronan sodný, bezvodý benzoan sodný (E211)
oxid kremičitý, koloidný hydrofóbny
zmes bobulovej arómy [obsahujúca benzylalkohol, oxid siričitý (E220)]
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Zatvorenáfľaška
2 roky.
Popríprave
Perorálna suspenzia je stabilná počas 60 dní. Uchovávajte v chladničke pri teplote (2°C - 8°C). Neuchovávajte v mrazničke.
Pripravená perorálna suspenzia zmiešaná s mliekom, jogurtom, jablkovým džúsom alebo jablkovým kompótom sa môže uchovávať pri alebo do 25°C až do 1 hodiny.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25C.
Podmienky na uchovávanie po príprave lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
120-ml fľaška z polyetylénu s vysokou hustotou s polypropylénovým detským bezpečnostným uzáverom s obsahom 33 g prášku na perorálnu suspenziu.
Veľkosť balenia: 1 fľaška
Každé balenie obsahuje aj adaptér na fľašku na vloženie dávkovacej striekačky (PIBA) z polyetylénu s nízkou hustotou a 12-ml perorálnu dávkovaciu striekačku (telo striekačky z polypropylénu s piestom striekačky z polyetylénu s vysokou hustotou) v zatavenom plastovom vrecku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Odporúča sa, aby lekárnik alebo kvalifikovaný zdravotnícky pracovník pripravil SPRYCEL prášok na perorálnu suspenziu pred jeho vydaním pacientovi. Prášok na perorálnu suspenziu obsahuje práškovú zmes s liečivom a pomocné látky, obsiahnuté vo fľaške na prípravu. Po príprave fľaška obsahuje
99 ml perorálnej suspenzie, z nich 90 ml je určených na dávkovanie a podávanie.
Pri manipulácii s práškom, ktorý sa neúmyselne vysypal z fľašky, sa na správnu likvidáciu odporúča použiť latexové alebo nitrilové rukavice, aby sa minimalizovalo riziko kožnej expozície.
Pokyny na prípravu prášku na perorálnu suspenziuSPRYCEL prášok na perorálnu suspenziu sa má pripraviť nasledovne:
Poznámka: Ak si musíte pripraviť viac ako jednu fľašku, najprv dokončite prípravu jednej fľašky. Pred začatím prípravy si umyte ruky. Tento postup sa má vykonať na čistom povrchu.
1. krok:Jemne poklepte dno každej fľašky (obsahujúcej 33 g SPRYCELU prášku na perorálnu suspenziu), aby sa prášok uvoľnil. Odstráňte detský bezpečnostný uzáver a fóliové tesnenie. Jednorazovo doplňte do fľašky 77,0 ml čistenej vody a uzáverom ju pevne zatvorte.
2. krok:Okamžite dobre premiešajte, nie kratšie ako 60 sekúnd, aby ste získali homogénnu suspenziu.
Ak sú ešte vždy viditeľné zhluky, pokračujte v miešaní kým sa nerozpustia. Príprava týmto spôsobom vytvorí 90 ml (objemu na podávanie) SPRYCELU perorálnej suspenzie s koncentráciou 10 mg/ml.
3. krok:Odstráňte uzáver, do hrdla fľašky vložte adaptér na fľašku na vloženie dávkovacej striekačky
(PIBA) a fľašku pevne zatvorte detským bezpečnostným uzáverom.
4. krok:Na štítok fľašky napíšte dátum exspirácie pripravenej perorálnej suspenzie (dátum exspirácie pripravenej perorálnej suspenzie je 60 dní od dátumu prípravy). Všetka nepoužitá perorálna suspenzia sa má po tomto dátume zlikvidovať alebo vrátiť lekárnikovi.
5. krok: Fľašku s vloženým PIBA, písomnou informáciou pre používateľa a perorálnou dávkovacoustriedačkou v pôvodnom balení vydajte pacientovi alebo opatrovateľovi.Pacienta alebo opatrovateľa upozornite, aby pred každým použitím fľašku intenzívne premiešali.
Pokynynapodávanieprepacienta• SPRYCEL perorálnu suspenziu užívajte nalačno alebo po jedle.
• Pred každým použitím a po každom použití sí umyte ruky.
• Pripravenú perorálnu suspenziu uchovávajte v chladničke (2°C - 8°C). Neuchovávajte v mrazničke.
• Skontrolujte celkovú predpísanú dávku a zistite počet mililitrov (ml), ktoré budete potrebovať.
• Ak je potrebné množstvo väčšie ako 11 ml, musí sa rozdeliť do 2 dávok podľa označenia v tabuľke 14.
Tabuľka 14: Ako rozdeliť dávku perorálnej suspenzie, ktorá je väčšia ako 11 mlCelková predpísaná dávka (ml) Prvá dávka (ml) Druhá dávka (ml)12 6 6

13 7 6
14 7 7
15 8 7
16 8 8
Pred prípravou dávky SPRYCELU perorálnej suspenzie na podávanie pacientovi, majte pripravené
nasledovné doplnky:
• Papierovú utierku
• 1 fľašku
SPRYCELU
perorálnej suspenzie s obsahom bielej až žltej nepriezračnej suspenzie.
• 12-ml perorálnu striekačku dodávanú s
fľaškou.
• Malú nádobu naplnenú vodou,
ktorá sa použije na vypláchnutie striekačky.
perorálna striekačka fľaška
fľaška
Pozorne si privravte SPRYCEL perorálnu suspenziu na podávanie, odmerajte si dávku a týmto
spôsobom striekačku naplňte:
1. V zatvorenej fľaške
premiešajte SPRYCEL perorálnu suspenziu pretrepávaním počas
30 sekúnd.
• Pred každým použitím ju dobre pretrepte.
Fľašku opakovan e pretrepáv ajte počas30 sekúnd.
2. Z fľašky odstráňte uzáver.
Uistite sa, že poskytnutý adaptér na fľaške na vloženie striekačky je pevne vo fľaške vsunutý.
Uistite sa, že je adaptér na fľaške pevne vsunutý.
3. Predtým ako začnete, si pozrite mierky na časti striekačky, tak budete vidieť pokiaľ ju máte naplniť. Všimnite si, že značky na striekačke sú v ml. Nájdite označenie, ktoré zodpovedá dávke, ktorú predpísal lekár. Pred každým použitím sa uistite, že piest striekačky je zatlačený na dno tela striekačky.
4. Do fľašky vo zvislej polohe vložte pevne hrot striekačky do adaptéra na fľašku.
5. Pevne držte hrot striekačky vo fľaške, fľašku so striekačkou pretočte hore dnom.
6. Pomaly natiahnite predpísané množstvo SPRYCELU perorálnej suspenzie vyťahovaním piestu striekačky, pokiaľ nedosiahnete označenie pre predpísanú dávku.
• Piest držte, aby sa zabránilo jeho pohybu.
Môže sa vytvoriť vákuum, ktoré vtiahne piest späť do valca.
• Ak nie je možné získať dávku z jednej fľašky, na
doplnenie celej predpísanej dávky použite druhú fľašku. Uistite sa, že bola druhá fľaška pred použitím pretrepaná.
7. Pevne držte hrot striekačky vo fľaške a fľašku so striekačkou pretočte späť do zvislej polohy.
8. Odstráňte striekačku z fľašky, buďte opatrný, aby ste nestlačili piest.
9. Pacientovi vo vzpriamenej polohe vložte do úst medzi vnútornú stranu úst a jazyk hrot striekačky. Pomaly stláčajte piest, až kým sa nepodá celá dávka.
• Skontrolujte, aby ste sa uistili, či pacient užil celú
dávku.
• Ak je potrebná druhá dávka na doplnenie
celkovej predpísanej dávky, zopakujte 3. až
10. krok.
• Uzáver vložte späť na fľašku a pevne zatvorte.
Uchovávajte vo zvislej polohe.
10. Po každom
použití umyte striekačku z
vonkajšej a vnútornej strany vodou a nechajte ju usušiť na vzduchu, aby sa mohla opäť použiť nasledujúci deň.
•
Neumývajte v umývačke riadu.•
Striekačku neoddeľujte,aby sa zabránilo jej poškodeniu.11. Pokyny na likvidáciu nepoužitého lieku, striekačky a fľašky nájdete na štítku fľašky.
 NEPOUŽÍVAJTE UMÝVAČKU RIADUPO KAŽDOM POUŽITÍ NECHAJTE USUŠIŤ NA VZDUCHUNEODDEĽUJTE STRIEKAČKU
NEPOUŽÍVAJTE UMÝVAČKU RIADUPO KAŽDOM POUŽITÍ NECHAJTE USUŠIŤ NA VZDUCHUNEODDEĽUJTE STRIEKAČKU
Po príprave sa má perorálna suspenzia podávať iba pomocou perorálnej dávkovacej striekačky
dodávanej v každom balení. Detailnejšie pokyny na používanie si pozrite v písomnej informácii.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
BRISTOL-MYERS SQUIBB PHARMA EEIG Uxbridge Business Park
Sanderson Road
Uxbridge UB8 1DH Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/06/363/016
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 20. novembra 2006
Dátum posledného predĺženia registrácie: 15. júla 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúre pre lieky
http://www.ema.europa.eu.
kom alebo zdravotnou sestrou.