
v>
Špeciálne
skupiny
pacientov
Starší pacienti (65 rokov a starší)
U starších pacientov je počiatočná dávka Spravata 28 mg esketamínu (deň 1, počiatočná dávka, pozri tabuľku 2 vyššie). Následné dávky sa majú zvyšovať na základe účinnosti a znášanlivosti
v prírastkoch po 28 mg až na 56 mg alebo 84 mg.
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene (trieda A podľa Childa-Pugha) alebo so stredne ťažkou poruchou funkcie pečene (trieda B podľa Childa-Pugha) nie je potrebná úprava dávky.
Maximálna dávka 84 mg sa však má používať opatrne u pacientov so stredne ťažkou poruchou funkcie
pečene.
Spravato nebol skúmaný u pacientov s ťažkou poruchou funkcie pečene (trieda C podľa Childa- Pugha). Použitie v tejto populácii sa neodporúča (pozri časť 4.4 a 5.2).
Porucha funkcie obličiek
Nie je nutná úprava dávky u pacientov s miernou až ťažkou poruchou funkcie obličiek. Použitie u pacientov na dialýze sa neskúmalo.
Rasa
Pre pacientov s japonským pôvodom je počiatočná dávka Spravata 28 mg esketamínu (1. deň, počiatočná dávka, pozri tabuľku 3 vyššie). Následné dávky sa majú zvyšovať na základe účinnosti a znášanlivosti v prírastkoch po 28 mg až na 56 mg alebo 84 mg.
Tabuľka 3: Odporúčané dávkovanie Spravata u dospelých pacientov s japonským pôvodom
Indukčná fáza Udržiavacia fáza
1.
až
4.
týždeň
: Počiatočná dávka
v 1. deň: 28 mg
Ďalšie dávky: 28 mg, 56 mg alebo 84 mg dvakrát týždenne, všetky zmeny dávky majú byť
v prírastkoch po 28 mg
Dôkaz o terapeutickom prínose sa má vyhodnotiť na konci indukčnej fázy, aby sa stanovilo, či je
potrebné pokračovať v liečbe.
5.až8.týždeň:
28 mg, 56 mg alebo 84 mg jedenkrát týždenne, všetky zmeny dávky majú byť
v prírastkoch po 28 mg
Od9.týždňa:
28 mg, 56 mg alebo 84 mg každé 2 týždne
alebo jedenkrát týždenne, všetky zmeny dávky majú byť v prírastkoch po 28 mg
Potreba pokračovania v liečbe sa má pravidelne prehodnocovať.
 Pediatrická populácia
Pediatrická populácia
Bezpečnosť a účinnosť Spravata u pediatrických pacientov vo veku 17 rokov a mladších nebola stanovená. K dispozícii nie sú žiadne údaje. Spravato sa u detí mladších ako 7 rokov v indikácii na liečbu depresie rezistentnej voči liečbe nepoužíva.
Spôsob podávaniaSpravato je určený len na nazálne použitie. Aplikátor s nosovou aerodisperziou je jednorazový
aplikátor, ktorý dodáva celkovo 28 mg esketamínu v dvoch vstrekoch (jeden vstrek do každej nosovej dierky). Aby sa zabránilo strate lieku, prípravok sa pred použitím nesmie pumpovať. Je určený na
podávanie samotným pacientom pod dohľadom zdravotníckeho pracovníka s použitím 1 aplikátora (na
podanie dávky 28 mg), 2 aplikátorov (na podanie dávky 56 mg) alebo 3 aplikátorov (na podanie dávky
84 mg) s 5-minútovou prestávkou medzi použitím každého ďalšieho aplikátora.
Kýchanie po podaníAk dôjde ku kýchnutiu ihneď po podaní, nemá sa používať ďalší aplikátor.
Použitie tej istej nosovej dierky na 2 po sebe idúce vstreky
Ak dôjde k podaniu do tej istej nosovej dierky, nemá sa používať ďalší aplikátor.
Ukončenie liečby Spravatom nevyžaduje postupné znižovanie dávky; na základe údajov z klinických skúšaní je riziko abstinenčných príznakov nízke.
4.3 Kontraindikácie
● Precitlivenosť na liečivo, ketamín, alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1
● Pacienti, u ktorých zvýšenie krvného tlaku alebo intrakraniálneho tlaku predstavuje vážne riziko
(pozri časť 4.8):
- Pacienti s aneuryzmatickým cievnym ochorením (vrátane intrakraniálnej, hrudnej alebo abdominálnej aorty alebo periférnych arteriálnych ciev).
- Pacienti s anamnézou intracerebrálneho krvácania.
- Nedávny výskyt (do 6 týždňov) kardiovaskulárnej príhody, vrátane infarktu myokardu
(IM).
4.4 Osobitné upozornenia a opatrenia pri používaní
Neuropsychiatrické a motorické poruchy
Bolo hlásené, že Spravato spôsoboval v priebehu klinických štúdií somnolenciu, sedáciu, disociačné
príznaky, poruchy vnímania, závraty, vertigo a úzkosť (pozri časť 4.8). Tieto účinky môžu zhoršiť pozornosť, úsudok, myslenie, rýchlosť reakcie a motorické zručnosti. Pri každom liečebnom sedení majú byť pacienti pod dohľadom zdravotníckeho pracovníka monitorovaní, aby sa vyhodnotilo, kedy bude pacient na základe klinického posúdenia považovaný za stabilizovaného (pozri časť 4.7).
Útlm dýchania
Po podaní vysokých dávok po rýchlej intravenóznej injekcii esketamínu alebo ketamínu používaného
na anestéziu sa môže vyskytnúť útlm dýchania. V klinických skúšaniach s nosovou aerodisperziou esketamínu (Spravato) nebol pozorovaný žiadny prípad útlmu dýchania; boli hlásené zriedkavé
prípady hlbokej sedácie. Súbežné použitie Spravata s látkami tlmiacimi CNS môže zvýšiť riziko
sedácie (pozri časť 4.5). U pacienta je nevyhnutné pozorné sledovanie sedácie a útlmu dýchania.
Účinok na krvný tlak
Spravato môže spôsobiť prechodné zvýšenia systolického a/alebo diastolického krvného tlaku, ktoré
dosahuje maximum približne 40 minút po podaní lieku a trvá približne 1 – 2 hodiny (pozri časť 4.8). Po ktoromkoľvek ošetrení môže dôjsť k značnému zvýšeniu krvného tlaku. Spravato je
kontraindikovaný u pacientov, u ktorých zvýšenie krvného tlaku alebo intrakraniálneho tlaku
predstavuje závažné riziko (pozri časť 4.3). Pred predpísaním Spravata majú byť pacienti s inými kardiovaskulárnymi a cerebrovaskulárnymi ochoreniami starostlivo vyšetrení s cieľom určiť, či potenciálne prínosy Spravata prevažujú nad jeho rizikami.
U pacientov, ktorých krvný tlak pred podaním dávky sa považuje za zvýšený (všeobecné pravidlo:
> 140/90 mmHg u pacientov vo veku < 65 rokov a > 150/90 mmHg u pacientov vo veku ≥ 65 rokov), je pred začatím liečby Spravatom vhodné upraviť životný štýl a/alebo antihypertenzívne farmakoterapie. Ak je pred podaním Spravata zvýšený krvný tlak, pri rozhodovaní o odložení liečby Spravatom sa má zvážiť pomer prínosu a rizika pre každého jednotlivého pacienta.
Po podaní dávky sa má monitorovať krvný tlak. Krvný tlak sa má merať približne 40 minút po podaní dávky a následne podľa klinického posúdenia, až kým jeho hodnoty neklesnú. Ak je krvný tlak dlhodobo zvýšený, je potrebné okamžite vyhľadať pomoc odborníkov, ktorí majú skúsenosti s liečbou krvného tlaku. Pacienti, u ktorých sa vyskytnú príznaky hypertenznej krízy, majú dostať okamžite akútnu zdravotnú starostlivosť.
Pacienti s klinicky významnými alebo nestabilnými kardiovaskulárnymi alebo respiračnými
ochoreniami
U pacientov s klinicky významnými alebo nestabilnými kardiovaskulárnymi alebo respiračnými
ochoreniami začnite liečbu Spravatom iba v prípade, že prínos prevažuje nad rizikom. Týmto pacientom sa má Spravato podávať v podmienkach, kde je k dispozícii vhodné vybavenie na resuscitáciu a zdravotnícki pracovníci vyškolení v oblasti kardiopulmonálnej resuscitácie. Medzi ochorenia, ktoré je potrebné zvážiť, okrem iných patrí:
● Závažná pľúcna insuficiencia, vrátane CHOCHP;
● Spánkové apnoe s morbídnou obezitou (BMI ≥ 35);
● Pacienti s nekontrolovanou bradyarytmiou alebo tachyarytmiou vedúcou k hemodynamickej nestabilite;
● Pacienti s anamnézou IM. Títo pacienti majú byť pred podaním klinicky stabilní a bez srdcových príznakov;
● Hemodynamicky významné ochorenie srdcových chlopní alebo srdcové zlyhávanie (NYHA
trieda III – IV).
Samovražda/samovražedné myšlienky alebo zhoršenie klinického stavu
Depresia je spojená so zvýšeným rizikom samovražedných myšlienok, sebapoškodzovania
a samovraždy (udalosti súvisiace so samovraždou). Toto riziko pretrváva, až kým nenastane významná remisia, preto je potrebné pacientov starostlivo sledovať. Je všeobecnou klinickou skúsenosťou, že
riziko samovraždy sa môže v skorých štádiách zotavovania zvýšiť.
Pacienti, ktorí majú v anamnéze udalosti súvisiace so samovraždou, a pacienti, u ktorých sa pred začatím liečby prejavuje vyššia miera samovražedných predstáv, sú vystavení väčšiemu riziku samovražedných myšlienok alebo pokusov o samovraždu, a preto majú byť počas liečby starostlivo sledovaní.
Liečba má byť sprevádzaná starostlivým dohľadom nad pacientmi, osobitne u pacientov s vysokým rizikom, najmä na začiatku liečby a po zmenách dávky. Pacientov (a ich opatrovateľov) je potrebné upozorniť na potrebu sledovať výskyt akéhokoľvek klinického zhoršenia, samovražedného správania alebo myšlienok a neobvyklých zmien v správaní a na potrebu vyhľadať lekársku pomoc ihneď, ako sa takéto príznaky objavia.
Zneužívanie, závislosť, vysadenie
Osoby s anamnézou zneužívania návykových látok alebo závislosti môžu byť vystavené väčšiemu
riziku zneužívania a nesprávneho používania Spravata. Pred predpísaním Spravata sa má vyhodnotiť riziko každého pacienta z hľadiska zneužívania a nesprávneho používania a pacienti, ktorí dostávajú
Spravato, majú byť počas liečby monitorovaní z hľadiska vývoja správania alebo stavov zneužívania a
nesprávneho používania, vrátane správania, ktoré je prejavom vyhľadávania drogy.
Pri dlhodobom používaní ketamínu bola hlásená závislosť a tolerancia. U osôb, ktoré boli závislé od ketamínu, boli pri vysadení liečby ketamínom hlásené abstinenčné príznaky vo forme silnej túžby po lieku, úzkosti, trasenia, potenia a búšenia srdca.
Ketamín, racemická zmes arketamínu a esketamínu, je liek, ktorý bol podľa údajov zneužívaný. Možnosť zneužívania a nesprávneho používania Spravata je minimalizovaná tým, že tento liek sa podáva pod priamym dohľadom zdravotníckeho pracovníka. Spravato obsahuje esketamín a môže byť predmetom zneužívania alebo nevhodného používania.
Iné populácie so zvýšeným rizikom
Spravato sa má používať opatrne u pacientov s nasledujúcimi ochoreniami. Týchto pacientov je nutné
skôr, ako sa im predpíše Spravato, starostlivo posúdiť a liečba sa má začať iba v prípade, že prínos prevažuje nad rizikom.
● Prítomnosť alebo anamnéza psychózy;
● Prítomnosť alebo anamnéza mánie alebo bipolárnej poruchy;
● Hypertyreóza, ktorá nebola dostatočne liečená;
● Anamnéza poranenia mozgu, hypertenzná encefalopatia, intratekálna terapia s ventrikulárnymi shuntmi alebo akékoľvek iné ochorenie spojené so zvýšeným intrakraniálnym tlakom.
Starší pacienti (65 rokov a starší)
U starších pacientov liečených Spravatom môže byť väčšie riziko pádu, a preto majú byť títo pacienti
starostlivo sledovaní.
Ťažká porucha funkcie pečene
Z dôvodu predpokladaného zvýšenia expozície a nedostatku klinických skúseností sa Spravato
neodporúča u pacientov s poruchou funkcie pečene triedy C podľa Childa-Pugha (ťažká).
Pri chronickom užívaní ketamínu bola hlásená hepatotoxicita, preto nie je možné vylúčiť možnosť takéhoto účinku v dôsledku dlhodobého používania Spravata.
Príznaky ochorenia močových ciest
Pri užívaní Spravata boli hlásené príznaky ochorenia močových ciest a močového mechúra (pozri časť
4.8). Počas liečby sa odporúča monitorovať ochorenia močových ciest a močového mechúra, a ak príznaky pretrvávajú, je potrebné vyhľadať príslušného odborníka.
4.5 Liekové a iné interakcie
Súbežné používanie Spravata s látkami tlmiacimi CNS (ako sú benzodiazepíny, opioidy, alkohol)
môže zvýšiť sedáciu, a preto sa má pozorne sledovať.
Krvný tlak sa má starostlivo monitorovať, keď sa Spravato používa súbežne s psychostimulanciami (napr. amfetamíny, metylfenidát, modafinil, armodafinil) alebo inými liekmi, ktoré môžu zvyšovať krvný tlak (napr. deriváty xantínu, ergometrín, hormóny štítnej žľazy, vazopresín alebo IMAO, ako sú tranylcypromín, selegilín, fenelzín).
4.6 Fertilita, gravidita a laktácia
Ženy v plodnom veku
Spravato sa neodporúča počas gravidity a u žien v plodnom veku, ktoré nepoužívajú antikoncepciu.
Gravidita
K dispozícii nie sú žiadne alebo sú len obmedzené údaje o používaní esketamínu tehotnými ženami.
Štúdie na zvieratách ukázali, že ketamín, racemická zmes arketamínu a esketamínu, vyvoláva neurotoxicitu u vyvíjajúcich sa plodov (pozri časť 5.3). Podobné riziko s esketamínom nemožno
vylúčiť.
Ak žena počas liečby Spravatom otehotnie, liečba sa má prerušiť a pacientka má byť čo najskôr informovaná o možnom riziku pre plod a klinických/terapeutických možnostiach.
Dojčenie
Nie je známe, či sa esketamín vylučuje do ľudského materského mlieka. Údaje u zvierat preukázali
vylučovanie esketamínu do mlieka. Riziko u dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu Spravatom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na zvieratách ukázali, že plodnosť a reprodukčné schopnosti neboli esketamínom nepriaznivo
ovplyvnené.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Spravato má veľký vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Bolo hlásené, že Spravato spôsobovalo v klinických štúdiách somnolenciu, sedáciu, disociačné príznaky, poruchy vnímania, závraty, vertigo a úzkosť (pozri časť 4.8). Pred podaním Spravata majú byť pacienti poučení, aby sa nezúčastňovali potenciálne nebezpečných činností vyžadujúcich úplnú duševnú bdelosť a motorickú koordináciu, ako je vedenie vozidla alebo obsluha strojov, až do nasledujúceho dňa po pokojnom spánku (pozri časť 4.4).
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Najčastejšie pozorovanými nežiaducimi reakciami u pacientov s depresiou rezistentnou voči liečbe
liečených Spravatom boli závraty (30 %), nauzea (27 %), disociácia (26 %), bolesť hlavy (24 %), somnolencia (18 %), vertigo (18 %), dysgeúzia (17 %), hypoestézia (11 %) a vracanie (10 %).
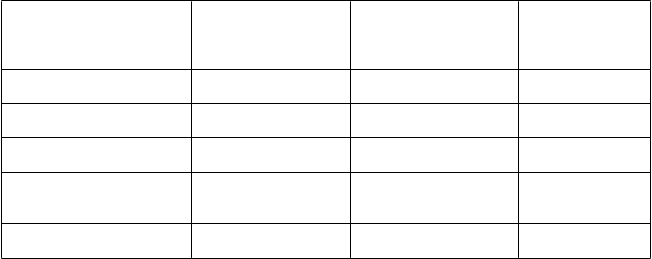
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie hlásené pri používaní esketamínu sú uvedené v tabuľke nižšie. V rámci určených
tried orgánových systémov sú nežiaduce reakcie uvedené pod nadpismi frekvencií s použitím nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných
údajov).
Trieda orgánových
systémov
Nežiaduca reakcia na liek
Frekvencia
Veľmi časté Časté Menej časté
Psychické poruchy disociácia euforická nálada, nepokoj, úzkosť, ilúzie, podráždenosť, záchvaty paniky, zmenené vnímanie času, halucinácie vrátane vizuálnych halucinácií, derealizácia
Poruchy nervového systému
závraty, bolesti hlavy, dysgeúzia, somnolencia, hypoestézia
mentálne postihnutie, tremor, letargia, dysartria, parestézia, sedácia
Poruchy oka rozmazané videnie
Poruchy ucha a labyrintu vertigo hyperakúzia, tinitus
Poruchy srdca a srdcovej činnosti
tachykardia
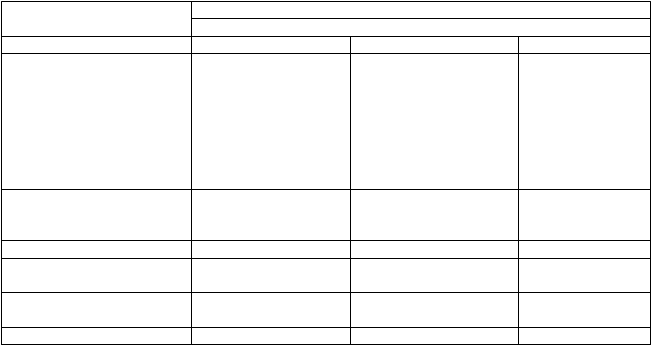 Poruchy ciev
Poruchy ciev hypertenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu Poruchy kože a
podkožného tkaniva
Poruchy obličiek a močových ciest
Celkové poruchy a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
nepríjemný pocit v nose, sucho v nose vrátane tvorby krúst, svrbenie
v nose
nevoľnosť, vracanie sucho v ústach, orálna hypestézia hyperhidróza
polakizúria, dyzúria, naliehavosť močenia neprirodzený pocit, pocit opitosti, pocit zmeny telesnej teploty
zvýšený krvný tlak
hypersekrécia slín
 Opis
vybraných nežiaducich reakcií
Disociácia
Opis
vybraných nežiaducich reakcií
Disociácia
Disociácia (26 %) bola jedným z najčastejších psychologických účinkov esketamínu. Ďalšie súvisiace pojmy zahŕňajú derealizáciu (1,9 %), depersonalizáciu (1,7 %), ilúzie (1,5 %) a skreslené vnímanie
času (1,2 %). Tieto nežiaduce reakcie boli hlásené ako prechodné reakcie, ktoré spontánne vymizli, a
vyskytli sa v deň dávkovania. Intenzita disociácie bola v rámci štúdií hlásená ako závažná pri incidencii menej ako 4 %. Príznaky disociácie typicky ustúpili po 1,5 hodine od podania dávky a ich závažnosť mala tendenciu časom sa znižovať s opakovanými liečebnými sedeniami.
Sedácia/somnolenciaNežiaduce reakcie v podobe sedácie (9,1 %) a somnolencie (18,0 %) boli primárne mierne alebo stredne závažné, vyskytli sa v deň podania dávky a spontánne vymizli v ten istý deň. Sedatívne účinky zvyčajne ustúpili do 1,5 hodiny od podania dávky. Miera somnolencie bola v priebehu dlhodobej liečby relatívne stabilná. V prípadoch sedácie neboli pozorované žiadne príznaky respiračnej tiesne
a hemodynamické parametre (vrátane vitálnych funkcií a saturácie kyslíkom) zostali v normálnom rozsahu.
Zmeny krvného tlakuV klinických štúdiách bolo zvýšenie systolického a diastolického krvného tlaku (STK a DTK)
v priebehu času približne 7 až 9 mmHg pre STK a 4 až 6 mmHg pre DTK v čase 40 minút po dávke a
2 až 5 mmHg pre STK a 1 až 3 mmHg pre DTK v čase 1,5 hodiny po dávke u pacientov používajúcich Spravato plus perorálne antidepresíva (pozri časť 4.4). Frekvencia výrazne abnormálneho zvýšenia krvného tlaku STK (zvýšenie ≥ 40 mmHg) sa pohybovala od 8 % (< 65 rokov) do 17 % (≥ 65 rokov) a DTK (zvýšenie ≥ 25 mmHg) sa pohybovala od 13 % (< 65 rokov) do 14 % (≥ 65 rokov) u pacientov, ktorí dostávajú esketamín plus perorálne antidepresívum. Incidencia zvýšenia STK (≥ 180 mmHg)
bola 3 % a DTK (≥ 110 mmHg) bola 4 %.
Kognitívne poruchy a poruchy pamätePri dlhodobom používaní ketamínu alebo jeho zneužívaní boli hlásené kognitívne poruchy a poruchy pamäte. Tieto účinky sa časom nezvyšovali a po vysadení ketamínu boli reverzibilné. V dlhodobých
klinických štúdiách sa hodnotil účinok nosovej aerodisperzie esketamínu na kognitívne funkcie v priebehu času a výkon zostával stabilný.
Príznaky týkajúce sa močových ciestPri dennom a dlhodobom používaní ketamínu vo vysokých dávkach boli hlásené prípady intersticiálnej cystitídy. V klinických štúdiách s esketamínom sa nevyskytli žiadne prípady
intersticiálnej cystitídy, u pacientov liečených esketamínom sa však pozoroval vyšší výskyt príznakov
dolných močových ciest (polakizúria, dyzúria, urgentnosť močenia, noktúria a cystitída) ako u pacientov, ktorí dostávali placebo.
Hlásenia podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieMožnosť predávkovania Spravatom zo strany pacienta je minimalizovaná formou lieku a podávaním pod dohľadom zdravotníckeho pracovníka (pozri časť 4.2).
PríznakyMaximálna testovaná jednorazová dávka nosovej aerodisperzie esketamínu u zdravých dobrovoľníkov
bola 112 mg; táto dávka nepreukázala žiadne známky toxicity a/alebo nežiaducich klinických následkov. V porovnaní s odporúčaným rozsahom dávky však bola dávka 112 mg nosovej aerodisperzie esketamínu spojená s vyšším výskytom nežiaducich reakcií vrátane závratu, hyperhidrózy, somnolencie, hypoestézie, abnormálneho pocitu, nevoľnosti a vracania.
Život ohrozujúce príznaky sa predpokladajú na základe skúseností s ketamínom podávaným
v množstve rovnajúcom sa 25-násobku obvyklej anestetickej dávky. Klinické príznaky sú opísané ako kŕče, srdcové arytmie a zastavenie dýchania. Možnosť podania porovnateľnej supraterapeutickej
dávky esketamínu intranazálnou cestou je nepravdepodobná.
LiečbaNeexistuje žiadne špecifické antidotum na predávkovanie esketamínom. V prípade predávkovania sa
má zvážiť možnosť požitia viacerých liekov. Liečba predávkovania Spravatom má pozostávať z liečby klinických príznakov a príslušného monitorovania. Starostlivý dohľad a monitorovanie má
pokračovať, kým sa pacient nezotaví.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Psychoanaleptiká; Iné antidepresíva, ATC kód: N06AX27.
Mechanizmus účinkuEsketamín je S-enantiomér racemického ketamínu. Je to neselektívny, nekompetitívny antagonista
receptora
N-metyl-
D-aspartátu (NMDA), čo je ionotropný glutamátový receptor. Prostredníctvom antagonizmu receptora NMDA spôsobuje esketamín prechodné zvýšenie uvoľňovania glutamátu, čo vedie k zvýšeniu stimulácie receptora α-amino-3-hydroxy-5-metyl-4-izoxazol propiónovej kyseliny (AMPAR) a následne k zvýšeniu neurotrofickej signalizácie, čo môže prispieť k obnoveniu synaptických funkcií v týchto oblastiach mozgu, ktoré sú spojené s reguláciou nálady a emocionálneho správania. K rýchlej reakcii môže prispieť obnovenie dopaminergnej neurotransmisie v oblastiach mozgu zapojených do odmeňovania a motivácie a znížená stimulácia mozgových oblastí zapojených
do anhedónie.
Farmakodynamické účinkyPotenciálzneužívaniaV štúdii zameranej na potenciál zneužívania u používateľov viacerých rekreačných drog (n = 41) mali jednorazové dávky nosovej aerodisperzie esketamínu (84 mg a 112 mg) a intravenózne podávaný ketamín (0,5 mg/kg podávaný infúziou počas 40 minút) ako pozitívna kontrola za následok významne vyššie skóre v porovnaní s placebom pri subjektívnom hodnotení toho, aký má pacient „pôžitok
z lieku“, a pri iných mierach subjektívnych účinkov lieku.
Klinická účinnosť a bezpečnosť
Účinnosť a bezpečnosť nosovej aerodisperzie Spravato bola skúmaná v piatich klinických štúdiách
fázy 3 u dospelých pacientov (18 až 86 rokov) s depresiou rezistentnou voči liečbe (TRD, z angl. treatment-resistant depression), ktorí splnili kritériá DSM-5 pre veľkú depresívnu poruchu a
neodpovedali na najmenej dve terapie perorálnymi antidepresívami (AD) v primeranej dávke a trvaní
v súčasnej epizóde veľkej depresívnej poruchy. Do štúdie bolo zaradených 1833 dospelých pacientov, z ktorých 1601 bolo vystavených Spravatu.
Depresia rezistentná voči liečbe – krátkodobé štúdie
Spravato bolo hodnotené v troch krátkodobých (4-týždňových) randomizovaných, dvojito zaslepených
štúdiách fázy 3 s aktívnou kontrolou u pacientov s TRD. Štúdie TRANSFORM-1 (TRD3001) a TRANSFORM-2 (TRD3002) sa uskutočnili u dospelých pacientov (18 až < 65 rokov) a štúdia TRANSFORM-3 (TRD3005) sa uskutočnila u dospelých pacientov vo veku ≥ 65 rokov. Pacienti v štúdiách TRD3001 a TRD3002 začali liečbu Spravatom v dávke 56 mg plus novo iniciované
perorálne AD podávané denne alebo novo iniciovaným perorálnym AD podávaným denne plus nosová aerodisperzia s placebom v 1. deň. Dávkovanie Spravata sa potom udržiavalo na 56 mg alebo sa
titrovalo na 84 mg alebo bola pacientom podávaná zodpovedajúca nosová aerodisperzia s placebom
dvakrát týždenne počas 4-týždňovej dvojito zaslepenej indukčnej fázy. Dávky Spravata 56 mg alebo
84 mg boli v štúdii TRD3001 fixné a v štúdii TRD3002 flexibilné. V štúdii TRD3005 začali pacienti
(≥ 65 rokov) liečbu Spravatom v dávke 28 mg plus novo iniciované perorálne AD podávané denne alebo novo iniciovaným perorálnym AD podávaným denne plus nosová aerodisperzia s placebom (1.
deň). Dávkovanie Spravata sa potom titrovalo na 56 mg alebo na 84 mg alebo bola pacientom
podávaná zodpovedajúca nosová aerodisperzia s placebom dvakrát týždenne počas 4-týždňovej
dvojito zaslepenej indukčnej fázy. V štúdiách TRD3002 a TRD3005 s flexibilnou dávkou bola titrácia dávky Spravata smerom nahor založená na klinickom posúdení a dávka sa mohla podľa znášanlivosti
titrovať smerom nadol. Novo iniciované otvorene podávané perorálne AD (SNRI: duloxetín,
venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) sa vo všetkých štúdiách začalo podávať v 1. deň. O výbere novo iniciovaného perorálneho AD rozhodol skúšajúci na základe
predchádzajúcej liečby pacienta. Vo všetkých krátkodobých štúdiách bola primárnym koncovým
ukazovateľom účinnosti zmena celkového skóre MADRS oproti východiskovej hodnote do 28. dňa.
Východiskové demografické charakteristiky a charakteristiky týkajúce sa zdravotného stavu pacienta v štúdiách TRD3002, TRD3001 a TRD3005 sú uvedené v tabuľke 4.
Tabuľka 4: Východiskové demografické charakteristiky v štúdiách TRD3002, TRD3001
a TRD3005 (kompletné súbory analýz)
Vek, roky
Štúdia TRD3002 (N = 223)
Štúdia TRD3001 (N = 342)
Štúdia TRD3005 (N = 137)
Medián (rozsah) 47,0 (19; 64) 47,0 (18; 64) 69,0 (65; 86) Pohlavie, n (%)
Muži 85 (38,1 %) 101 (29,5 %) 52 (38,0 %)
Ženy 138 (61,9 %) 241 (70,5 %) 85 (62,0 %) Rasa, n (%)
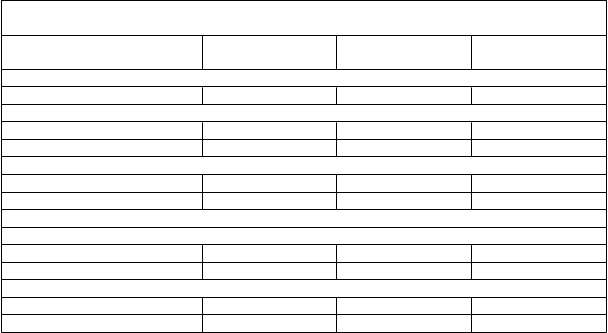
Biela 208 (93,3 %) 262 (76,6 %) 130 (94,9 %)
Čierna alebo afroamerická 11 (4,9 %) 19 (5,6 %) -- Predchádzajúce perorálne antidepresíva bez odpovede (t. j. antidepresíva, ktoré zlyhali)
Počet špecifických antidepresív, n (%)
2 136 (61,0 %) 167 (48,8 %) 68 (49,6 %)
3 alebo viac 82 (36,8 %) 167 (48,8 %) 58 (42,3 %) Novo iniciovaný perorálny antidepresívny liek, ktorý sa začal podávať pri randomizácii, n (%)
SNRI 152 (68,2 %) 196 (57,3 %) 61 (44,5 %)
SSRI 71 (31,8 %) 146 (42,7 %) 76 (55,5 %)
Vyradení zo štúdie
(z akéhokoľvek dôvodu), n/N (%)

30/227 (13,2 %) 31/346 (9,0 %) 16/138 (11,6 %)
V štúdii TRD3002 s flexibilnou dávkou dostávalo na 28. deň 67 % pacientov randomizovaných na
Spravato dávku 84 mg. V štúdii TRD3002 preukázal esketamín plus novo iniciované perorálne AD klinicky významnú a štatistickú nadradenosť v porovnaní s novo iniciovaným perorálnym AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia s placebom (tabuľka 5) a zmiernenie príznakov sa pozorovalo už 24 hodín po podaní dávky.
V štúdii TRD3001 sa pozoroval klinicky významný liečebný účinok, ktorý sa prejavil v zmene celkového skóre MADRS oproti východiskovej hodnote na konci štvortýždňovej indukčnej fázy, v prospech Spravata podávaného spolu s novo iniciovaným perorálnym AD v porovnaní s novo iniciovaným perorálnym AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI:
escitalopram, sertralín) plus nosová aerodisperzia s placebom (tabuľka 5). V štúdii TRD3001 nebol účinok liečby pre skupinu so Spravatom 84 mg plus perorálne AD v porovnaní s perorálnym AD plus
placebo štatisticky významný.
V štúdii TRD3005 dostávalo na 28. deň 64 % pacientov randomizovaných do skupiny liečenej Spravatom na dávku 84 mg, 25 % dostávalo dávku 56 mg a 10 % dostávalo dávku 28 mg. V štúdii TRD3005 sa pozoroval klinicky významný, ale nie štatisticky významný liečebný účinok, ktorý sa prejavil v zmene celkového skóre MADRS oproti východiskovej hodnote na konci štvortýždňovej indukčnej fázy, v prospech Spravata podávaného spolu s novo iniciovaným perorálnym AD
v porovnaní s novo iniciovaným perorálnym AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia s placebom (tabuľka 5). Analýzy podskupín naznačujú obmedzenú účinnosť v populácii staršej ako 75 rokov.
Tabuľka 5: Primárne výsledky účinnosti na zmenu celkového skóre MADRS pre 4-týždňové klinické štúdie (ANCOVA BOCF)
Štúdia č. Liečebná skupina
§
Počet
pacientov
Spravato 56 mg +
 Priemerné východiskové skóre (SD)Priemerná zmena LS od východiskového stavu do konca týždňa 4 (SE)Priemerný rozdiel LS (95 % CI)†
Priemerné východiskové skóre (SD)Priemerná zmena LS od východiskového stavu do konca týždňa 4 (SE)Priemerný rozdiel LS (95 % CI)†-4,3
TRD3001
perorálne AD 115 37,4 (4,8) -18,9 (1,3)
Spravato 84 mg +
perorálne AD 114 37,8 (5,6) -16,2 (1,3)
Perorálne AD + nosová
(-7,8; -0,8)#
-1,2
(-4,7; 2,3)#
aerodisperzia s placebom
Spravato (56 mg alebo
113 37,5 (6,2) -14,7 (1,3)
-3,5
TRD3002
84 mg) + perorálne AD 114 37,0 (5,7) -17,7 (1,3)
Perorálne AD + nosová
(-6,7; -0,3)‡
TRD3005 (≥ 65 rokov)
aerodisperzia s placebom
Spravato (28 mg, 56 mg alebo 84 mg) + perorálne AD
Perorálne AD + nosová aerodisperzia
s placebom
109 37,3 (5,7) -14,3 (1,3)
72 35,5 (5,9) -10,1 (1,7)
65 34,8 (6,4) -6,8 (1,7)
-2,9
(-6,5; 0,6)#

SD = štandardná odchýlka; SE = štandardná chyba; priemer LS = priemer najmenších štvorcov; CI = interval spoľahlivosti; AD = antidepresívum
§ Nazálne podávaný esketamín alebo placebo; perorálne AD = novo iniciované AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín)
† Rozdiel (Spravato + perorálne AD mínus perorálne AD + nosová aerodisperzia s placebom) v zmene priemeru najmenších štvorcov oproti východiskovej hodnote
‡ Liečebná skupina, ktorá bola štatisticky významne nadradená v porovnaní s perorálnym AD + nosová aerodisperzia s placebom
# Medián objektívneho odhadu (t. j. vážená kombinácia priemerov LS rozdielu oproti perorálnemu AD + nosová aerodisperzia s placebom) a 95 % flexibilný interval spoľahlivosti
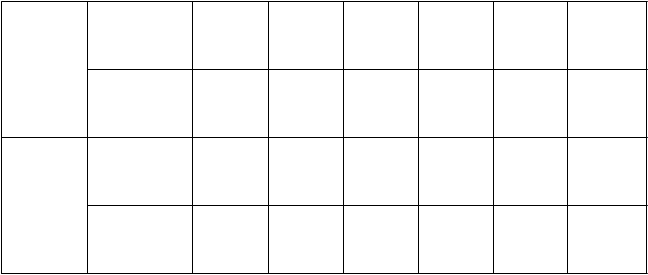
Miera odpovede a remisieOdpoveď bola definovaná ako ≥ 50 % zníženie celkového skóre MADRS od východiskovej hodnoty
v indukčnej fáze. Na základe zníženia celkového skóre MADRS oproti východiskovej hodnote bol podiel pacientov v štúdiách TRD3001, TRD3002 a TRD3005, ktorí preukázali odpoveď na liečbu
Spravatom plus perorálnym AD, väčší ako pri perorálnom AD a nosovej aerodisperzii s placebom
v priebehu 4-týždňovej dvojito zaslepenej indukčnej fázy (tabuľka 6).
Remisia bola definovaná ako celkové skóre MADRS ≤ 12. Vo všetkých troch štúdiách bol vyšší podiel pacientov liečených Spravatom plus perorálnym AD v remisii na konci 4-týždňovej dvojito zaslepenej indukčnej fázy ako pri perorálnom AD plus nosová aerodisperzia s placebom (tabuľka 6).
Tabuľka 6: Miera odpovede a remisie v 4-týždňových klinických štúdiách na základe údajov BOCF Počet pacientov (%)Miera odpovede†
Miera
Štúdia č.
Liečebná skupina§
remisie‡
24 hodín Týždeň 1 Týždeň 2 Týždeň 3 Týždeň 4 Týždeň 4

TRD3001
Spravato
56 mg +
perorálne AD
Spravato
84 mg +
perorálne AD Perorálne AD
+ nosová
aerodisperzia s placebom
20 (17,4 %)
17 (14,9 %)#
8 (7,1 %)
21 (18,3 %)
16 (14,0 %)
5 (4,4 %)
29 (25,2 %)
25 (21,9 %)
15 (13,3 %)
52 (45,2 %)
33 (28,9 %)
25 (22,1 %)
61 (53,0 %)
52 (45,6 %)
42 (37,2 %)
40 (34,8 %)
38 (33,3 %)
33 (29,2 %)
TRD3002
TRD3005
Spravato
56 mg alebo
84 mg +
perorálne AD Perorálne AD
+ nosová
aerodisperzia s placebom
Spravato
18 (15,8 %)
11 (10,1 %)
15 (13,2 %)
13 (11,9 %)
29 (25,4 %)
23 (21,1 %)
54 (47,4 %)
35 (32,1 %)
70 (61,4 %)
52 (47,7 %)
53 (46,5 %)
31 (28,4 %)
(≥ 65
rokov)
28 mg, 56 mg alebo 84 mg + perorálne AD Perorálne AD
+ nosová
aerodisperzia s placebom
NA 4 (5,6 %)
NA 3 (4,6 %)
4 (5,6 %)
8 (12,3 %)
9 (12,5 %)
8 (12,3 %)
17 (23,6 %)
8 (12,3 %)
11 (15,3 %)
4 (6,2 %)
AD = antidepresívum; NA = nie je k dispozícii
§ Nazálne podávaný Spravato alebo placebo; perorálne AD = novo iniciované AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín)
† Odpoveď bola definovaná ako ≥ 50 % zníženie celkového skóre MADRS od východiskovej hodnoty
‡ Remisia bola definovaná ako celkové skóre MADRS ≤ 12
# Prvá dávka bola Spravato 56 mg + perorálne AD
Depresia rezistentná voči liečbe – dlhodobé štúdieŠtúdiazameranánaprevenciurecidívyUdržanie antidepresívnej účinnosti bolo preukázané v štúdii zameranej na prevenciu recidívy. Štúdia
SUSTAIN-1 (TRD3003) bola dlhodobá, randomizovaná, dvojito zaslepená, aktívne kontrolovaná, multicentrická štúdia s paralelnými skupinami zameraná na prevenciu recidívy. Primárna miera
výsledku na hodnotenie prevencie recidívy depresie bola vyjadrená ako čas do recidívy. Celkovo bolo
do štúdie zaradených 705 pacientov; 437 bolo zaradených priamo; 150 bolo prevedených zo štúdie TRD3001 a 118 bolo prevedených zo štúdie TRD3002. Pacientom, ktorí boli priamo zaradení, sa podávalo Spravato (56 mg alebo 84 mg dvakrát týždenne) plus perorálne AD v 4-týždňovej indukčnej fáze otvorenej liečby. Na konci otvorenej indukčnej fázy bolo 52 % pacientov v remisii (celkové skóre MADRS ≤ 12) a 66 % pacientov dosiahlo odpoveď na liečbu (≥ 50 % zlepšenie celkového skóre MADRS). Pacienti, ktorí odpovedali na liečbu (455), pokračovali v liečbe Spravatom plus perorálnym AD v 12-týždňovej optimalizačnej fáze. Po indukčnej fáze dostávali pacienti Spravato raz týždenne počas 4 týždňov a od týždňa 8 sa na stanovenie frekvencie dávkovania použil algoritmus (založený na MADRS); pacienti v remisii (t. j. ich celkové skóre MADRS bolo ≤ 12) boli zaradení na dávkovanie každý druhý týždeň, avšak ak sa celkové skóre MADRS zvýšilo na > 12, potom sa frekvencia zvýšila na dávkovanie raz týždenne počas nasledujúcich 4 týždňov; s cieľom udržať pacienta na najnižšej frekvencii dávkovania, aby sa zachovala odpoveď/remisia. Na konci 16 týždňov liečby boli pacienti so stabilnou remisiou (n = 176) alebo stabilnou odpoveďou (n = 121) randomizovaní, aby pokračovali
v liečbe Spravatom alebo prestali používať Spravato a prešli na nosová aerodisperzia s placebom. Stabilná remisia bola definovaná ako celkové skóre MADRS ≤ 12 v najmenej 3 z posledných 4'
týždňov optimalizačnej fázy a stabilná odpoveď bola definovaná ako ≥ 50 % zníženie celkového skóre
MADRS oproti východiskovej hodnote za posledné 2 týždne optimalizačnej fázy, ale nie v stabilnej remisii.
Stabilná remisiaPacienti v stabilnej remisii, ktorí pokračovali v liečbe Spravatom plus perorálnym AD, zaznamenali štatisticky významne dlhší čas do recidívy depresívnych príznakov ako pacienti na novo iniciovanom perorálnom AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia s placebom (obrázok 1). Recidíva bola definovaná ako celkové skóre MADRS ≥ 22 počas 2 po sebe nasledujúcich týždňov alebo hospitalizácia pre zhoršenie depresie
alebo akákoľvek iná klinicky významná udalosť indikujúca recidívu. Medián času do recidívy v skupine, ktorá dostávala novo iniciované perorálne AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia s placebom, bol 273 dní, zatiaľ čo medián nebolo možné stanoviť pre Spravato plus perorálne AD, pretože táto skupina nikdy nedosiahla 50 % mieru recidívy.
Obrázok 1: Čas do recidívy u pacientov so stabilnou remisiou v štúdii TRD3003 (kompletný súbor analýz)
U pacientov so stabilnou remisiou bola miera recidív na základe odhadov podľa Kaplanovej- Meierovej metódy počas 12- a 24-týždňového dvojito zaslepeného sledovania 13 % resp. 32 % pre Spravato a 37 % resp. 46 % pre placebo.
Stabilná odpoveďVýsledky účinnosti boli takisto konzistentné u pacientov so stabilnou odpoveďou, ktorí pokračovali v liečbe Spravatom plus perorálnym AD; pacienti zaznamenali štatisticky významne dlhší čas do recidívy depresívnych príznakov ako pacienti na novo iniciovanom perorálnom AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia
s placebom (obrázok 2). Medián času do recidívy v skupine, ktorá dostávala novo iniciované perorálne AD (SNRI: duloxetín, venlafaxín s predĺženým uvoľňovaním; SSRI: escitalopram, sertralín) plus nosová aerodisperzia s placebom (88 dní), bol kratší ako v skupine, ktorá dostávala Spravato plus perorálne AD (635 dní).
Obrázok 2: Čas do recidívy u pacientov so stabilnou odpoveďou v štúdii TRD3003 (kompletný súbor analýz)

U pacientov so stabilnou odpoveďou bola miera recidív na základe odhadov podľa Kaplanovej-
Meierovej metódy počas 12- a 24-týždňového dvojito zaslepeného sledovania 21 % resp. 21 % pre
Spravato a 47 % resp. 56 % pre nosovú aerodisperziu s placebom.
Zápis do štúdie TRD3003 bol rozložený na približne 2 roky. Udržiavacia fáza mala premenlivú dobu trvania a pokračovala, až kým individuálny pacient nemal recidívu depresívnych príznakov alebo neukončil liečbu z akéhokoľvek iného dôvodu, alebo kým sa štúdia neskončila, pretože sa vyskytol požadovaný počet prípadov recidív. Čísla vyjadrujúce expozíciu boli ovplyvnené tým, že štúdia bola ukončená po vopred určenom počte recidív na základe priebežnej analýzy. Po počiatočnej 16- týždňovej liečbe kombináciou Spravato plus perorálne AD bol medián trvania expozície Spravatu
v udržiavacej fáze 4,2 mesiaca (rozsah: 1 deň až 21,2 mesiaca) u pacientov liečených Spravatom
(stabilná remisia a stabilná odpoveď). V tejto štúdii dostávalo 31,6 % pacientov Spravato dlhšie ako 6
mesiacov a 7,9 % pacientov dostalo Spravato dlhšie ako 1 rok v rámci udržiavacej fázy.
Frekvencia dávkovaniaFrekvencia dávkovania použitá väčšinu času počas udržiavacej fázy je uvedená v tabuľke 7.
Z pacientov randomizovaných na Spravato 60 % dostávalo dávku 84 mg a 40 % dostávalo dávku
56 mg.
Tabuľka 7: Frekvencia dávkovania používaná väčšinu času; udržiavacia fáza (štúdia
TRD3003)
Stabilná remisia Stabilní respondéri
Najčastejšia frekvencia dávkovania
Spravato + perorálne AD (N = 90)
Perorálne AD
+ nosová aerodisperzia s placebom
(N = 86)
Spravato + perorálne AD (N = 62)
Perorálne AD
+ nosová aerodisperzia s placebom
(N = 59)

Raz týždenne 21 (23,3 %) 27 (31,4 %) 34 (54,8 %) 36 (61,0 %) Každý druhý týždeň 62 (68,9 %) 48 (55,8 %) 21 (33,9 %) 19 (32,2 %)
Raz týždenne alebo každý
druhý týždeň 7 (7,8 %) 11 (12,8 %) 7 (11,3 %) 4 (6,8 %)
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so Spravatom
týkajúcich sa liečby veľkej depresívnej poruchy v jednej alebo vo viacerých podskupinách pediatrickej populácie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPriemerná absolútna biologická dostupnosť 84 mg esketamínu podaného vo forme nosovej
aerodisperzie je približne 48 %.
Esketamín sa po nazálnom podaní rýchlo absorbuje nosovou sliznicou a v plazme je možné ho namerať už po 7 minútach po podaní 28 mg dávky. Čas do dosiahnutia maximálnej plazmatickej koncentrácie (tmax) je typicky 20 až 40 minút po poslednom vstreku nosovej aerodisperzie počas liečebného sedenia (pozri časť 4.2).
Zvýšenia maximálnej plazmatickej koncentrácie (Cmax) a plochy pod krivkou vyjadrujúcej závislosť plazmatickej koncentrácie od času (AUC∞) v závislosti od dávky nosovej aerodisperzie esketamínu boli dosiahnuté dávkami 28 mg, 56 mg a 84 mg.
Farmakokinetický profil esketamínu je podobný po podaní jednorazovej dávky a opakovanej dávky bez akumulácie v plazme, keď sa esketamín podáva dvakrát týždenne.
DistribúciaPriemerný distribučný objem esketamínu v rovnovážnom stave podávaného intravenóznou cestou je
709 litrov.
Podiel celkovej koncentrácie esketamínu viazaného na proteíny v ľudskej plazme je v priemere 43 až
45 %. Stupeň viazania esketamínu na plazmatické proteíny nie je závislý od funkcie pečene alebo obličiek.
Esketamín nie je substrátom transportérov P-glykoproteínu (P-gp; proteín multiliekovej rezistencie 1), proteínu rezistencie rakoviny prsníka (BCRP) ani substrátom pre organický aniónový transportér (OATP) 1B1 alebo OATP1B3. Esketamín neinhibuje tieto transportéry ani proteíny extrudujúce mnoholiekové a toxické zlúčeniny 1 (MATE1) a MATE2-K, alebo organický katiónový transportér 2 (OCT2), OAT1 alebo OAT3.
Biotransformácia
Esketamín sa extenzívne metabolizuje v pečeni. Primárnou metabolickou dráhou esketamínu
v ľudských pečeňových mikrozómoch je N-demetylácia za tvorby noresketamínu. Hlavnými enzýmami cytochrómu P450 (CYP) zodpovednými za N-demetyláciu esketamínu sú CYP2B6 a CYP3A4. Ďalšie enzýmy CYP vrátane CYP2C19 a CYP2C9 prispievajú v oveľa menšej miere. Noresketamín sa následne metabolizuje dráhami závislými od CYP na iné metabolity, z ktorých niektoré podliehajú glukuronidácii.
Eliminácia
Priemerný klírens esketamínu podávaného intravenóznou cestou bol približne 89 l/hod. Po dosiahnutí
Cmax po nazálnom podaní bol pokles koncentrácií esketamínu v plazme počas prvých niekoľkých hodín rýchly a potom postupnejší. Priemerný terminálny polčas eliminácie po podaní vo forme nosovej aerodisperzie sa všeobecne pohyboval od 7 do 12 hodín.
Po intravenóznom podaní rádioaktívne označeného esketamínu sa približne 78 % podanej rádioaktivity zistilo v moči a 2 % podanej rádioaktivity sa zistilo v stolici. Po perorálnom podaní rádioaktívne označeného esketamínu sa približne 86 % podanej rádioaktivity zistilo v moči a 2 % podanej rádioaktivity sa zistilo v stolici. Získané rádioaktívne látky pozostávali predovšetkým
z metabolitov esketamínu. Pri intravenóznom a perorálnom spôsobe podania sa < 1 % dávky vylúčilo v moči ako nezmenený liek.
Linearita/nelinearita
Expozícia esketamínu sa zvyšuje s dávkou od 28 mg do 84 mg. Zvýšenie hodnôt Cmax a AUC bolo
menšie ako dávkovo proporcionálne medzi dávkou 28 mg a 56 mg alebo 84 mg, ale bolo takmer
dávkovo proporcionálne medzi dávkou 56 mg a 84 mg.
Interakcie
Účinokiných liekovnaesketamín
Inhibítory pečeňových enzýmov
Predchádzajúca liečba zdravých jedincov perorálnym tiklopidínom, inhibítorom hepatálnej aktivity
CYP2B6 (250 mg dvakrát denne počas 9 dní pred dňom podania esketamínu a v deň jeho podania), nemala žiadny vplyv na Cmax esketamínu podaného ako nosová aerodisperzia. Hodnota AUC∞ esketamínu sa zvýšila približne o 29 %. Terminálny polčas eliminácie esketamínu nebol ovplyvnený predchádzajúcou liečbou tiklopidínom.
Predchádzajúca liečba perorálnym klaritromycínom, inhibítorom aktivity pečeňového enzýmu CYP3A4 (500 mg dvakrát denne počas 3 dní pred podaním esketamínu a v deň podania), zvyšuje priemernú hodnotu Cmax resp. AUC∞ nazálne podaného esketamínu približne o 11 % resp. 4 %. Terminálny polčas eliminácie esketamínu nebol ovplyvnený predchádzajúcou liečbou klaritromycínom.
Induktory pečeňových enzýmov
Predchádzajúca liečba perorálnym rifampicínom, silným induktorom aktivity viacerých pečeňových enzýmov CYP, ako sú CYP3A4 a CYP2B6, (600 mg denne počas 5 dní pred podaním esketamínu) znížila priemerné hodnoty Cmax a AUC∞ esketamínu podávaného ako nosová aerodisperzia približne o 17 % resp. 28 %.
Iné lieky podávané vo forme nosovej aerodisperzie
Predchádzajúca liečba pacientov s anamnézou alergickej rinitídy a s predchádzajúcou expozíciou trávovému peľu s použitím oxymetazolínu podávaného vo forme nosovej aerodisperzie (2 vstreky
0,05 % roztoku podané 1 hodinu pred nazálnym podaním esketamínu) mala len menší účinok na
farmakokinetiku esketamínu.
Predchádzajúce nazálne podanie mometazónfuroátu (200 mikrogramov denne počas 2 týždňov
s poslednou dávkou mometazónfuroátu podanou 1 hodinu pred nazálnym podaním esketamínu) malo u zdravých jedincov menší účinok na farmakokinetiku esketamínu.
Účinok esketamínu nainélieky
Nazálne podávanie 84 mg esketamínu dvakrát týždenne počas 2 týždňov znížilo priemernú
plazmatickú AUC∞ perorálne podávaného midazolamu (jedna 6 mg dávka), ktorý je substrátom pečeňového CYP3A4, približne o 16 %.
Nazálne podávanie 84 mg esketamínu dvakrát týždenne počas 2 týždňov neovplyvnilo priemernú plazmatickú AUC perorálne podávaného bupropiónu (jedna 150 mg dávka), ktorý je substrátom pečeňového CYP2B6.
Špeciálne skupiny pacientov
Staršípacienti(65rokovastarší)
Farmakokinetika esketamínu podávaného vo forme nosovej aerodisperzie bola porovnávaná medzi staršími, ale inak zdravými jedincami a mladšími zdravými dospelými. Priemerné hodnoty esketamínu Cmax a AUC∞, ktoré boli dosiahnuté dávkou 28 mg, boli o 21 % a 18 % vyššie u starších jedincov (vekové rozpätie 65 až 81 rokov) v porovnaní s mladšími dospelými jedincami (vekové rozpätie 22 až
50 rokov). Priemerné hodnoty esketamínu Cmax a AUC∞, ktoré boli dosiahnuté dávkou 84 mg, boli o
67 % a 38 % vyššie u starších jedincov (vekové rozpätie 75 až 85 rokov) v porovnaní s mladšími
dospelými jedincami (vekové rozpätie 24 až 54 rokov). Terminálny polčas eliminácie esketamínu bol u starších a mladších dospelých jedincov podobný (pozri časť 4.2).
Poruchafunkcieobličiek
V porovnaní s jedincami s normálnou funkciou obličiek (klírens kreatinínu [CLCR], 88 až 140 ml/min) bol Cmax esketamínu v priemere o 20 až 26 % vyšší u jedincov s miernou (CLCR, 58 až 77 ml/min), stredne ťažkou (CLCR, 30 až 47 ml/min) alebo ťažkou (CLCR, 5 až 28 ml/min, nie na dialýze) poruchou funkcie obličiek po podaní 28 mg dávky nosovej aerodisperzie esketamínu. Hodnota AUC∞ bola o 13 až 36 % vyššia u jedincov s miernou až ťažkou poruchou funkcie obličiek.
Nie sú žiadne klinické skúsenosti s podávaním esketamínu vo forme nosovej aerodisperzie u pacientov na dialýze.
Poruchafunkciepečene
Hodnoty Cmax a AUC∞ esketamínu produkované dávkami 28 mg boli u jedincov s poruchou funkcie pečene triedy A podľa Childa-Pugha (mierna) podobné ako u zdravých jedincov. Hodnoty Cmax resp. AUC∞ esketamínu boli o 8 % vyššie resp. o 103 % vyššie u jedincov s poruchou funkcie pečene triedy B podľa Childa-Pugha (stredne ťažká) v porovnaní so zdravými jedincami.
Nie sú žiadne klinické skúsenosti s podávaním esketamínu vo forme nosovej aerodisperzie
u pacientov s poruchou funkcie pečene triedy C podľa Childa-Pugha (ťažká) (pozri časť 4.2 a 4.4).
Rasa
Farmakokinetické vlastnosti nosovej aerodisperzie esketamínu boli porovnávané medzi zdravými ázijskými jedincami a belošskými jedincami. Priemerné hodnoty Cmax a AUC∞ pre esketamín v plazme produkované jednorazovou dávkou esketamínu v dávke 56 mg boli približne o 14 % a 33 % vyššie
u čínskych jedincov v porovnaní s belošskými jedincami. Obidva parametre boli približne o 40 % vyššie u japonských jedincov v porovnaní s belošskými jedincami. Hodnota Cmax pre esketamín bola v priemere o 10 % nižšia a hodnota AUC∞ bola o 17 % vyššia u kórejských jedincov v porovnaní
s belošskými jedincami. Priemerný terminálny polčas eliminácie esketamínu v plazme ázijských
pacientov sa pohyboval od 7,1 do 8,9 hodiny a u belošských jedincov bol 6,8 hodín.
Pohlavie
a
telesná
hmotnosť
Na základe populačnej farmakokinetickej analýzy neboli pozorované z hľadiska pohlavia a celkovej telesnej hmotnosti (> 39 až 170 kg) žiadne významné rozdiely vo farmakokinetických vlastnostiach nosovej aerodisperzie esketamínu.
Alergickárinitída
Farmakokinetické vlastnosti jednorazovej dávky 56 mg esketamínu podávaného vo forme nosovej aerodisperzie boli podobné u pacientov s alergickou rinitídou, ktorí boli vystavení peľu trávy,
v porovnaní so zdravými jedincami.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní, genotoxicity, neurotoxicity, reprodukčnej toxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. Štúdie na zvieratách s ketamínom preukázali vývinovú neurotoxicitu. Možnosť, že esketamín má neurotoxické účinky na vývin plodov, nemožno vylúčiť (pozri časť 4.6).
Genotoxicita
Esketamín nebol v Amesovom teste s metabolickou aktiváciou alebo bez nej mutagénny. Genotoxické
účinky esketamínu boli pozorované pri skríningovom mikronukleovom teste in vitro v prítomnosti metabolickej aktivácie. Intravenózne podávaný esketamín však nemal genotoxické vlastnosti
v mikronukleovom teste kostnej drene in vivo u potkanov a v kométovom teste na pečeňových
bunkách potkanov in vivo.
Reprodukčná toxicita
V štúdii zameranej na embryofetálnu vývinovú toxicitu nazálne podávaného ketamínu u potkanov
nebolo potomstvo nepriaznivo ovplyvnené za prítomnosti toxicity pre matku pri dávkach, ktorých výsledkom bola expozícia až 6-násobne vyššia ako expozícia u ľudí na základe hodnôt AUC. V štúdii
zameranej na embryofetálnu vývinovú toxicitu nazálne podávaného ketamínu u králikov sa pozorovali malformácie skeletu a znížená telesná hmotnosť plodu pri dávkach toxických pre matku. Expozícia
u králikov bola na základe hodnôt AUC v oblasti hodnôt expozície ľudí.
Publikované štúdie na zvieratách (vrátane primátov) pri dávkach, ktoré viedli k ľahkej až strednej anestézii, ukazujú, že použitie anestetík počas obdobia rýchleho rastu mozgu alebo synaptogenézy vedie k úbytku buniek vo vyvíjajúcom sa mozgu, čo môže byť spojené s dlhodobými kognitívnymi deficienciami. Klinicky význam týchto predklinických nálezov nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
monohydrát kyseliny citrónovej edetát disodný
hydroxid sodný (na úpravu pH)
voda na injekcie
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaSklenená liekovka typu I so zátkou z chlórbutylovej gumy. Naplnená a uzavretá liekovka sa zostaví do manuálne aktivovaného aplikátora s nosovou aerodisperziou. Aplikátor umožňuje podanie dvoch vstrekov.
V každom balení je každý aplikátor jednotlivo zabalený v uzavretom blistri. Veľkosti balenia obsahujúce 1, 2, 3 alebo 6 aplikátorov s nosovou aerodisperziou. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen-Cilag International NV Turnhoutseweg 30
B-2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/19/1410/001 (balenie s 1 nosovou aerodisperziou)

EU/1/19/1410/002 (balenie s 2 nosovými aerodisperziami) EU/1/19/1410/003 (balenie s 3 nosovými aerodisperziami)
EU/1/19/1410/004 (balenie so 6 nosovými aerodisperziami)
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.