
y sa pred podaním Spinrazy odobral taký objem cerebrospinálneho moku (cerebral spinal fluid, CSF), ktorý je ekvivalentný objemu Spinrazy určenému na podanie injekcie.
Pri podaní Spinrazy môže byť potrebná sedácia, a to v závislosti od klinického stavu pacienta.
Pre vykonanie intratekálneho podania Spinrazy sa môže zvážiť použitie ultrasonografie (alebo inej zobrazovacej techniky), najmä u mladších pacientov a u pacientov so skoliózou. Pri príprave a podaní Spinrazy sa musí použiť aseptická technika; pozri pokyny na použitie v časti 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Procedúra lumbálnej punkcie
V súvislosti s procedúrou lumbálnej punkcie existuje riziko nežiaducich reakcií (napr. bolesť hlavy,
bolesť chrbta, vracanie; pozri časť 4.8). Možné obtiaže s touto cestou podania môžu byť u veľmi
mladých pacientov a u pacientov so skoliózou. Použitie ultrasonografie alebo inej zobrazovacej techniky sa môže zvážiť pre uľahčenie intratekálneho podania Spinrazy podľa uváženia lekára.
Trombocytopénia a koagulačné abnormality
Po subkutánnom alebo intravenóznom podaní iných antisense oligonukleotidov sa pozorovala
trombocytopénia a koagulačné abnormality, vrátane akútnej závažnej trombocytopénie. Ak je to klinicky indikované, pred podaním Spinrazy sa odporúča laboratórne vyšetrenie hodnôt krvných doštičiek a koagulácie.
Renálna toxicita
Po subkutánnom alebo intravenóznom podaní iných antisense oligonukleotidov sa pozorovala renálna
toxicita. Ak je to klinicky indikované, odporúča sa vyšetrenie bielkovín v moči (prednostne z prvej
rannej vzorky moču). V prípade pretrvávajúcej zvýšenej hladiny bielkovín v moči sa musí zvážiť ďalšie vyšetrenie.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. In vitro štúdie naznačili, že nusinersen nie je induktor ani inhibítor metabolizmu sprostredkovaného CYP450. In vitro štúdie naznačujú, že pravdepodobnosť interakcií s nusinersenom kvôli kompetícii o väzbu na plazmatické proteíny alebo kompetícii
s prenášačmi, alebo ich inhibícii je nízka.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití nusinersenu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity
(pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu Spinrazy počas
gravidity.
Dojčenie
Nie je známe, či sa nusinersen/metabolity vylučujú do ľudského mlieka.
Riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Spinrazou sa musí urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
V štúdiách toxicity na zvieratách sa nepozorovali žiadne účinky na fertilitu samcov alebo samíc (pozri
časť 5.3). K dispozícii nie sú žiadne údaje o možných účinkoch na fertilitu ľudí.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Spinraza nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Posúdenie bezpečnosti Spinrazy bolo založené na dvoch klinických štúdiách fázy 3 u dojčiat (CS3B)
a detí (CS4) s SMA, spolu s otvorenými štúdiami, ktoré zahŕňali dojčatá s presymptomaticky geneticky diagnostikovanou SMA a dojčatá a deti s SMA. Z 260 pacientov, ktorí dostávali Spinrazu
počas maximálne 4 rokov, dostávalo liečbu 154 pacientov aspoň 1 rok.
Tabuľkový zoznamnežiaducich reakcií
Posúdenie nežiaducich účinkov je založené na nasledujúcich údajoch frekvencie:
Veľmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
T
abuľka
1:
N
ežiaduce
reakcie
súvisiace
s procedúrou lumbálnej punkcie hlásené v štúdii CS4(neskorší nástup SMA) s výskytom najmenej o 5 % vyšším u pacientovliečenýchSpinrazouakou pacientov s predstieranouliečbou
Trieda orgánových systémov
podľa MedDRA
Preferovaný termín podľa
MedDRA
Kategória frekvencie Spinrazy,
n=84
Poruchy nervového systému Bolesť hlavy* Veľmi časté
Poruchy gastrointestinálneho traktu
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Vracanie* Časté
Bolesť chrbta* Veľmi časté

*Nežiaduce udalosti považované za súvisiace s procedúrou lumbálnej punkcie. Tieto udalosti možno považovať za prejavy postpunkčného syndrómu.
Popis vybraných nežiaducich reakciíPozorovali sa nežiaduce reakcie spojené s podaním Spinrazy lumbálnou punkciou. Väčšina týchto
reakcií bola hlásená v priebehu 72 hodín po procedúre. Výskyt a závažnosť týchto udalostí boli konzistentné s udalosťami očakávanými v súvislosti s lumbálnou punkciou. Žiadne závažné komplikácie lumbálnej punkcie, ako sú závažné infekcie, sa v klinickom skúšaní Spinrazy nepozorovali.
Niektoré nežiaduce udalosti bežne asociované s lumbálnou punkciou (napr. bolesť hlavy a bolesť chrbta) sa nemohli hodnotiť v populácii dojčiat vystavenej Spinraze kvôli obmedzenej komunikácii odpovedajúcej tejto vekovej skupine.
ImunogenicitaImunogénna odpoveď na nusinersen sa stanovila u 148 pacientov zo vzoriek plazmy pri
východiskovom stave a v priebehu liečby vyhodnotením hladín protilátok proti lieku (
anti-drug antibodies, ADA). Celkovo bol výskyt ADA nízky, s tvorbou ADA vznikajúcich liečbou
u 7 (5 %) pacientov, z toho bol výskyt u 2 pacientov prechodný, u 2 sa považoval za trvalý a u 3 nebol
potvrdený. Nebol zrejmý účinok tvorby ADA na klinickú odpoveď, nežiaduce udalosti ani na farmakokinetický profil nusinersenu.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách neboli hlásené žiadne prípady predávkovania spojené s nežiaducimi reakciami. V prípade predávkovania sa musí poskytnúť podporná zdravotná starostlivosť vrátane konzultácie so
zdravotníckym pracovníkom a dôsledného sledovania klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: {skupina}, ATC kód: zatiaľ nepridelený
Mechanizmus účinku
Nusinersen je antisense oligonukleotid (antisense oligonucleotide, ASO), ktorý zvyšuje podiel
zaradenia exónu 7 v transkriptoch mediátorovej ribonukleovej kyseliny (messenger ribonucleic acid, mRNA) pre SMN2 (survival motor neuron 2, SMN2) väzbou na intronic splice silencing site (ISS-N1) nachádzajúce sa v intróne 7 SMN2 premediátorovej ribonukleovej kyseliny (pre-messenger
ribonucleic acid, pre-mRNA) pre SMN2. ASO touto väzbou vytesňuje zostrihové faktory, ktoré za normálnych okolností potláčajú zostrih. Vytesnenie týchto faktorov vedie k retencii exónu 7 v SMN2
mRNA, a preto keď sa SMN2 mRNA vytvorí, môže sa prepísať do funkčného proteínu SMN v plnej
dĺžke.
SMA je progresívne neuromuskulárne ochorenie v dôsledku mutácií na chromozóme 5q v géne SMN1. Druhý gén SMN2, ktorý sa nachádza blízko SMN1, je zodpovedný za produkciu malého množstva proteínu SMN. SMA je klinické spektrum ochorenia, pričom závažnosť ochorenia je spojená s menším počtom kópií génu SMN2 a mladším vekom pri nástupe symptómov.
Klinická účinnosť a bezpečnosť
Symptomatickí pacienti
Nástup ochorenia u detí
Štúdia CS3B (ENDEAR) bola randomizovaná, dvojito zaslepená, štúdia fázy 3 kontrolovaná
s predstieranou liečbou, ktorá sa vykonala u 121 symptomatických detí vo veku ≤ 7 mesiacov
s diagnostikovanou SMA (nástup symptómov vo veku menej ako 6 mesiacov). CS3B bola navrhnutá tak, aby sa posúdil účinok Spinrazy na motorické funkcie a prežívanie. Pacienti boli randomizovaní
2:1 buď na Spinrazu (podľa schváleného dávkovacieho režimu) alebo do kontrolnej skupiny
s predstieranou liečbou, s dĺžkou liečby v rozsahu 6 až 442 dní.
Medián veku nástupu klinických prejavov a symptómov SMA bol 6,5 týždňa u pacientov liečených
Spinrazou oproti 8 týždňom v kontrolnej skupine s predstieranou liečbou, pričom 99 % pacientov
malo 2 kópie génu SMN2, a preto bol u nich vývoj SMA typu I považovaný za najviac pravdepodobný
. Medián veku, kedy bola pacientom podaná prvá dávka, bol 164,5 dňa u liečených pacientov
a 205 dní v kontrolnej skupine s predstieranou liečbou. Charakteristické znaky ochorenia na začiatku štúdie boli zväčša podobné u pacientov liečených Spinrazou a pacientov v kontrolnej skupine
s predstieranou liečbou s výnimkou toho, že pacienti liečení Spinrazou na začiatku štúdie mali
v porovnaní s pacientami v kontrolnej skupine s predstieranou liečbou vyššie percento paradoxného dýchania (89 % oproti 66 %), pneumónie alebo respiračných symptómov (35 % oproti 22 %),
problémov s prehĺtaním alebo kŕmením (51 % oproti 29 %) a potreby podpory dýchania (26 % oproti
15 %).
Pri záverečnej analýze štatisticky signifikantne vyššie percento pacientov dosiahlo definíciu respondera pre motorický míľnik v skupine so Spinrazou (51 %) v porovnaní s kontrolnou skupinou s predstieranou liečbou (0 %) (p<0,0001). Ako primárny koncový ukazovateľ sa hodnotila doba do úmrtia alebo do trvalej ventilácie (≥ 16 hodín ventilácie/deň nepretržite po dobu > 21 dní bez prítomnosti akútnej reverzibilnej udalosti alebo tracheostómie). Štatisticky signifikantné účinky na prežívanie bez udalosti, celkové prežívanie, podiel pacientov dosahujúcich definíciu respondera pre
motorický míľnik a percento pacientov so zlepšením najmenej o 4 body oproti východiskovej hodnote v detskom teste pre neuromuskulárne ochorenia Detskej nemocnice vo Philadelphii (Children’s
Hospital of Philadelphia Infant Test for Neuromuscular Disease, CHOP INTEND) sa pozorovali u pacientov v skupine so Spinrazou v porovnaní s pacientmi v kontrolnej skupine s predstieranou
liečbou (tabuľka 2).
V súbore na stanovenie účinnosti 18 pacientov (25 %) v skupine so Spinrazou a 12 pacientov (32 %)
v kontrolnej skupine s predstieranou liečbou vyžadovalo trvalú ventiláciu. Z týchto pacientov splnilo
6 pacientov (33 %) v skupine so Spinrazou a 0 pacientov (0 %) v kontrolnej skupine s predstieranou
liečbou protokolom definované kritériá respondera pre motorický míľnik.
T
abuľka 2: Primárne a sekundárne koncové ukazovatele pri záverečnej analýze – štúdia CS3B
P
arameter účinnosti Pacienti liečení Spinrazou Pacienti s predstieranou
li
e
čbou v kontrolnej skupine
P
r
ežívanie
P
r
ežívanie bez udalosti
2
Počet pacientov, ktorí zomreli alebo potrebovali trvalú ventiláciu
31 (39 %) 28 (68 %)
Pomer rizika (95% IS) 0,53 (0,32 – 0,89)
p-hodnota p = 0,0046
Celkové prežívanie2
Počet pacientov, ktorí zomreli 13 (16 %) 16 (39 %)
Pomer rizika (95% IS) 0,37 (0,18 – 0,77)
p-hodnota p = 0,0041
Motorické funkcie
Motorické míľniky3
Podiel dosiahnutých preddefinovaných kritérií respondera pre motorický míľnik (sekcia HINE 2)4,5
37 (51 %)1
p<0,0001
0 (0 %)
Podiel v 183. deň 41 % 5 % Podiel v 302. deň 45 % 0 % Podiel v 394. deň 54 % 0 %
Podiel zlepšení v celkovom skóre motorických míľnikov
Podiel zhoršení v celkovom skóre
motorických míľnikov
CHOP INTEND3
49 (67 %) 5 (14 %)
1 (1 %) 8 (22 %)
Podiel dosiahnutých zlepšení o 4 body 52 (71 %)
p<0,0001
1 (3 %)
Podiel dosiahnutých zhoršení o 4 body 2 (3 %) 17 (46 %) Podiel s akýmkoľvek zlepšením 53 (73 %) 1 (3 %) Podiel s akýmkoľvek zhoršením 5 (7 %) 18 (49 %)
1CS3B bola ukončená po pozitívnej štatistickej analýze primárneho koncového ukazovateľa v predbežnej analýze (štatisticky
signifikantne väčšie percento pacientov, ktorí dosiahli definíciu respondera pre motorický míľnik v skupine so Spinrazou
(41 %) v porovnaní s kontrolnou skupinou s predstieranou liečbou (0 %), p<0,0001).
2Pri záverečnej analýze sa hodnotili prežívanie bez udalosti a celkové prežívanie pomocou populácie s úmyslom liečiť (
Intent to Treat, ITT), (ITT Spinraza n=80; kontrolná skupina s predstieranou liečbou n=41).
3Pri záverečnej analýze sa uskutočnili analýzy CHOP INTEND a motorického míľnika s použitím súboru na stanovenie
účinnosti (Spinraza n=73; kontrolná skupina s predstieranou liečbou n=37).
4Hodnotené na poslednej návšteve v rámci štúdie v 183. deň , 302. deň a 394. deň.
5Podľa Hammersmithovho neurologického vyšetrenia detí (
Hammersmith Infant Neurological Examination, HINE) časť 2:
zvýšenie o ≥2 body [alebo najvyššie skóre] v schopnosti kopnúť, ALEBO zvýšenie o ≥1 bod v motorických míľnikoch držania hlavy, prevaľovania sa, sedenia, lezenia, stoja alebo chôdze, A zlepšenie vo viacerých kategóriách motorických míľnikov ako zhoršenia, definované ako responder pre túto primárnu analýzu.
Rozsah zlepšenia v CHOP INTEND je zobrazený na obrázku 1(zmena oproti východiskovému skóre u každého pacienta).
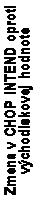 O
brázok 1: Zmena v CHOP INTEND od východiskovej hodnoty po poslednú návštevu v rámci štúdie
v 183. deň, 302. deň
a 394. deň
– štúdia Endear /CS3B (súbor pre stanovenie účinnosti,
ES) (Efficacy
Set
, ES)
O
brázok 1: Zmena v CHOP INTEND od východiskovej hodnoty po poslednú návštevu v rámci štúdie
v 183. deň, 302. deň
a 394. deň
– štúdia Endear /CS3B (súbor pre stanovenie účinnosti,
ES) (Efficacy
Set
, ES)
N = 78
Liečba
Spinraza kontrolná skupina
Poznámka 1: Najkratší stĺpec na čiare v 0 určuje hodnotu 0.
Poznámka 2: V súbore pre stanovenie účinnosti zomrelo 29 zo 110 pacientov (13 (18 %) v skupine so
Spinrazou a 16 (43 %) v kontrolnej skupine) a 3 ukončili štúdiu z iného dôvodu ako úmrtie (2 (3 %)
v skupine so Spinrazou a 1 (3 %) v kontrolnej skupine), a preto neboli zaradení do tejto analýzy ES.
Tieto výsledky sú podporené otvorenou štúdiou fázy 2 u symptomatických pacientov
s diagnostikovanou SMA (CS3A). Medián veku nástupu klinických prejavov a symptómov bol 56 dní a u pacientov boli prítomné buď 2 kópie génu SMN2 (n=17) alebo 3 kópie génu SMN2 (n=2)
(u 1 pacienta bol počet kopií génu SMN2 neznámy). U pacientov v tejto štúdii sa považoval za najviac
pravdepodobný vývoj SMA typu I. Medián veku v čase prvej dávky bol 162 dní.
V čase plánovanej predbežnej analýzy bol medián doby pacientov v štúdii 670 dní. Primárnym koncovým ukazovateľom bol podiel pacientov, ktorí sa zlepšili v jednej alebo viacerých kategóriách motorických míľnikov (podľa HINE časť 2: zvýšenie o ≥2 body [alebo najvyššie skóre] v schopnosti kopnúť alebo vedomom uchopení ALEBO zvýšenie o ≥1 bod v motorických míľnikoch držania hlavy, prevaľovania sa, sedenia, lezenia, stoja alebo chôdze). V tomto čase 13 z 20 pacientov (65 %) dosiahlo postupom času primárny koncový ukazovateľ s trvalým zlepšením v priemernom dosiahnutí motorického míľnika. Od východiskového stavu do 694. dňa sa pozorovalo trvalé zlepšenie
v priemernom skóre CHOP INTEND (priemerná zmena 16,90). Celkovo 11 z 20 pacientov (55 %)
dosiahlo koncový ukazovateľ zvýšenia v celkovom skóre CHOP INTEND o ≥4 body v čase ich
poslednej návštevy v rámci štúdie pred ukončením zberu údajov.
Neskorší nástup ochorenia
Štúdia CS4 (CHERISH) je randomizovaná, dvojito zaslepená, štúdia fázy 3 kontrolovaná
s predstieranou liečbou, ktorá sa uskutočnila u 126 symptomatických pacientov s neskorším nástupom SMA (nástup symptómov po 6 mesiacoch veku). Pacienti boli randomizovaní 2:1 buď na Spinrazu (ktorým boli podané 3 nasycovacie dávky a udržiavacie dávky každých 6 mesiacov) alebo do kontrolnej skupiny s predstieranou liečbou a dĺžka liečby bola v rozsahu 170 až 470 dní. Medián veku pri skríningu bol 3 roky a medián veku nástupu klinických prejavov a symptómov SMA bol
11 mesiacov. Väčšina pacientov (88 %) mala 3 kópie génu SMN2 (8 % malo 2 kópie, 2 % mali
4 kópie a 2 % malo počet kopií neznámy). U pacientov v tejto štúdii bol považovaný za najpravdepodobnejší vývoj SMA typu II alebo III. Charakteristické znaky ochorenia vo
východiskovom stave boli celkovo podobné s výnimkou nevyváženosti v podiele pacientov, ktorí
kedykoľvek dosiahli schopnosť stáť bez podpory (13 % pacientov v skupine so Spinrazou
a 29 % v kontrolnej skupine s predstieranou liečbou) alebo chôdze s podporou (24 % pacientov v skupine so Spinrazou a 33 % v kontrolnej skupine s predstieranou liečbou).
Predbežná analýza sa uskutočnila, keď všetci pacienti ukončili hodnotenie v 6. mesiaci a najmenej
39 pacientov ukončilo hodnotenie v 15. mesiaci, pozri tabuľku 3. Primárnym koncovým ukazovateľom, ktorý sa hodnotil v čase predbežnej analýzy, bola zmena východiskového skóre v 15. mesiaci podľa rozšírenej Hammersmithovej funkčnej motorickej škály (Hammersmith Functional Motor Scale Expanded, HFMSE). Primárna analýza sa uskutočnila v populácii IIT
(Spinraza: n=84; kontrolná skupina s predstieranou liečbou: n=42) a údaje podľa HFMSE získané
v priebehu štúdie od pacientov bez návštevy v 15. mesiaci sa hodnotili použitím metódy viacnásobnej imputácie. U pacientov liečených Spinrazou sa v porovnaní s pacientami v kontrolnej skupine
s predstieranou liečbou pozorovalo štatisticky signifikantné zlepšenie východiskového skóre HFMSE. Analýza podskupiny pacientov v ITT populácii, u ktorých sa pozorovali hodnoty v 15. mesiaci,
preukázala konzistentné, štatisticky signifikantné výsledky. Deskriptívne výsledky z doplňujúcich funkčných meraní vrátane revidovaného modulového testu hornej končatiny a dosiahnutia motorického WHO míľnika sú uvedené v tabuľke 3.
Skoršie začatie liečby po nástupe symptómov viedlo k skoršiemu a výraznejšiemu zlepšeniu motorických funkcií ako v prípade oneskoreného začatia liečby; avšak obe skupiny zaznamenali prínos liečby v porovnaní s kontrolnou skupinou s predstieranou liečbou.
Tabuľka 3: Primárne a sekundárne koncové ukazovatele v predbežnej analýze – štúdia CS41
Pacienti liečení Spinrazou Pacienti s predstieranou
liečbou v kontrolnej skupine
H
F
MSE skóre
Zmena v celkovom skóre HFMSE
v 15. mesiaci1,2 oproti východiskovej hodnote
4,0 (95% IS: 2,9; 5,1)
p=0,0000002
-1,9 (95% IS: -3,8; 0,0)
Podiel pacientov, ktorí dosiahli
zlepšenie aspoň 3 body oproti východiskovej hodnote1,3
RULM5
Priemerná zmena celkového skóre RULM1,2,3 v 15. mesiaci oproti východiskovej hodnote
Motorické WHO míľniky Podiel pacientov, ktorí dosiahli akýkoľvek nový motorický míľnik v 15. mesiaci 3,4
57,3 % 20,5 %
3,7 0,3
17,1 10,5
1CS4 bola ukončená po pozitívnej štatistickej analýze primárneho koncového ukazovateľa.
2Metóda najmenších štvorcov
3Nebolo štatisticky testované v predbežnej analýze.
4Dosiahnutie WHO míľnika sa hodnotilo použitím predbežného súboru populácie pre stanovenie účinnosti (
Interim Efficacy Set,IES) (IES, Spinraza n=35; kontrolná skupina s predstieranou liečbou n=19); v prípade chýbajúcich údajov sú analýzy založené na imputovaných údajoch.
5RULM (revidovaný modulový test hornej končatiny) (
Revised upper limb module, RULM))
Tieto výsledky sú podporené 2 otvorenými štúdiami (štúdia CS2 a štúdia CS12). Do analýzy bolo zahrnutých 28 pacientov, ktorí dostali prvú dávku v štúdii CS2 a potom pokračovali v predĺženej fáze štúdie CS12. Do týchto štúdií boli zaradení pacienti vo veku 2 až 15 rokov v čase prvej dávky.
3 z 28 pacientov mali najmenej 18 rokov v čase poslednej návštevy v rámci štúdie. 1 z 28 pacientov mal 2 kópie génu SMN2, 21 pacientov malo 3 kópie a 6 pacientov malo 4 kópie.
Hodnotenie pacientov prebiehalo počas liečebného obdobia 3 rokov. Trvalé zlepšenie sa pozorovalo u pacientov s typom II, s priemerným zlepšením východiskového skóre HFSME 12,3 (SD 5,46; n=6) s priemerným celkovým skóre 35,3 (SD 12,58) po 1 050 dňoch liečby. Žiaden ustálený stav sa nepozoroval. U pacientov s SMA typu III sa preukázalo priemerné zlepšenie východiskového skóre HFSME 1,6 (SD 3,91; n=7) s priemerným celkovým skóre 53,0 (SD 9,22) po 1 050 dňoch.
6-minútový test chôdze (six-minute walk test, 6MWT) sa uskutočnil len u ambulantných pacientov.
U týchto pacientov sa po 1 050 dňoch pozorovalo priemerné zlepšenie o 96,7 metrov (SD 42,36; n=6)
s priemernou vzdialenosťou 6MWT 278,2 metrov (SD 157,58). Samostatnú chôdzu dosiahli dvaja ambulantní pacienti, ktorí neboli predtým samostatní (typ III), a jeden neambulantný pacient (typ II).
Presymptomatické deti'
Štúdia CS5 (NURTURE) je otvorená štúdia u presymptomatických detí s geneticky diagnostikovanou
SMA, ktoré boli zaradené do štúdie vo veku 6 týždňov alebo mladšie. U pacientov v tejto štúdii bol považovaný za najpravdepodobnejší vývoj SMA typu I alebo II. Medián veku v čase prvej dávky bol
19 dní.
V predbežnej analýze 18 z 20 pacientov absolvovalo návštevu v 64. deň a tak vytvorili súbor pre stanovenie účinnosti (2 kópie génu SMN2, n=13; 3 kópie génu SMN2, n=5). Medián doby v štúdii bol
317,5 dní. Primárnym koncovým ukazovateľom hodnoteným v čase predbežnej analýzy bola doba do úmrtia alebo respiračnej intervencie (definovanej ako invazívna alebo neinvazívna ventilácia počas
≥ 6 hodín/deň nepretržite počas ≥7 nasledujúcich dní ALEBO tracheostómia). V plánovanej
predbežnej analýze žiaden pacient nedosiahol primárny koncový ukazovateľ úmrtia alebo respiračnej intervencie.
Pacienti dosiahli míľniky, ktoré sa neočakávali pri SMA typu I alebo II a konzistentnejšie
s normálnym vývinom. V porovnaní s východiskovými hodnotami dosiahlo zlepšenie v motorických
míľnikoch HINE v predbežnej analýze 16 pacientov (89 %) zo súboru pre stanovenie účinnosti. Dvanásť pacientov samostatne sedelo, 9 stálo s alebo bez neja 6 chodilo s podporou alebo bez nej. U šestnástich pacientov (89 %) sa preukázalo zlepšenie v celkovom skóre CHOP INTEND
o ≥ 4 body; 7 z nich dosiahlo maximálne celkové skóre CHOP INTEND 64. U jedného pacienta (6 %)
došlo k poklesu celkového skóre CHOP INTEND o ≥ 4 body.
Podiel pacientov s rozvíjajúcou sa klinicky manifestovanou SMA sa hodnotil u pacientov, ktorí v predbežnej analýze podstúpili návštevu v 365. deň (n=9). Kritériá definované protokolom pre klinicky manifestovanú SMA zahŕňali hmotnosť upravenú vzhľadom na vek pod piaty percentil WHO, pokles o 2 alebo viac percentilov hlavnej krivky nárastu hmotnosti, zavedenie perkutánnej gastrickej sondy a/alebo neschopnosť dosiahnuť očakávané vekovo-primerané WHO míľniky
(samostatné sedenie, státie s pomocou a lozenie po kolenách a rukách). Päť (56 %) pacientov priberalo na hmotnosti a dosiahlo WHO míľniky konzistentné s normálnym vývinom. Hoci 4 pacienti (44 %)
(každý s 2 kópiami génu SMN2) splnili kritériá definované protokolom, títo pacienti priberali na
hmotnosti a dosahovali WHO míľniky, vrátane samostatného sedenia, nekonzistentné s SMA typu I.
Porovnanie dosiahnutia motorického míľnika u pacientov so symptomatickým skorým nástupom SMA
a presymptomatickou SMA je znázornené na obrázku 2.
 O
brázok 2: Zmena v motorických
m
í
ľ
nikoch
HINE oproti
dňom
štúdie
pre štúdie CS3B (liečba
a predstieraná liečba), CS3A a CS5
O
brázok 2: Zmena v motorických
m
í
ľ
nikoch
HINE oproti
dňom
štúdie
pre štúdie CS3B (liečba
a predstieraná liečba), CS3A a CS5

= CS5 (N=18) < CS3A (N=20) 5 CS3B-liečivo (N=73) u CS3B-predstieraná liečba (N=37)
CS5
CS3A CS3B-liečivo CS3B-predstieraná
liečba
Plánovaný deň návštevy
Populácie použité na obrázku: Nurture (CS5) - súbor pre predbežné stanovenie účinnosti, CS3A – všetci pacienti s dávkou, CS3B – súbor pre stanovenie účinnosti.
Pre každú štúdiu, návštevy s n<5 nie sú plánované.
5.2 Farmakokinetické vlastnosti
U pediatrických pacientov s diagnózou SMA bola stanovená farmakokinetika (pharmacokinetics, PK)
jednotlivej a viacnásobnej dávky nusinersenu podaného intratekálnou injekciou.
Absorpcia
Intratekálna injekcia nusinersenu do CSF umožňuje, aby sa nusinersen plne distribuoval z CSF do
cieľových tkanív centrálneho nervového systému (CNS). Priemerné minimálne koncentrácie nusinersenu v CSF sa po viacnásobných nasycovacích a udržiavacích dávkach zvýšili približne 1,4- až
3-násobne a ustálený stav sa dosiahol približne v priebehu 24 mesiacov. Po intratekálnom podaní boli
minimálne plazmatické koncentrácie nusinersenu relatívne nízke v porovnaní s minimálnymi koncentráciami v CSF. Medián plazmatických hodnôt Tmax bol v rozmedzí 1,7 až 6,0 hodín. Priemerné plazmatické hodnoty Cmax a AUC sa zvyšovali približne proporcionálne k dávke naprieč hodnoteným rozmedzím dávky. Po viacnásobných dávkach nedochádzalo k žiadnemu zvýšeniu plazmatických parametrov expozície (Cmax a AUC).
Distribúcia
Údaje z pitiev pacientov (n = 3) ukazujú, že intratekálne podaný nusinersen sa do značnej miery
distribuuje v CNS a dosahuje terapeutické hladiny v cieľových tkanivách miechy. Prítomnosť
nusinersenu sa preukázala aj v neurónoch a iných typoch buniek miechy a mozgu a taktiež v periférnych tkanivách, ako sú kostrové svaly, pečeň a obličky.
Biotransformácia
Nusinersen sa metabolizuje pomaly a to najmä hydrolýzou, ktorá je spostredkovaná exonukleázou
(3’- a 5’). Nie je substrát, alebo inhibítor, alebo induktor enzýmov CYP450.
Eliminácia
Priemerný terminálny polčas eliminácie z CSF sa odhaduje na 135 až 177 dní. Ako primárna cesta
eliminácie nusinersenu a jeho metabolitov sa predpokladá vylučovanie močom.
Interakcie
In vitro štúdie ukázali, že nusinersen nie je induktor alebo inhibítor oxidatívneho metabolizmu
sprostredkovaného CYP450, a preto by nemal interferovať s inými liekmi o tieto metabolické dráhy. Nusinersen nie je substrát alebo inhibítor ľudských prenášačov BCRP, P-gp, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3 alebo BSEP.
Charakteristiky u osobitných populácií pacientov
Porucha funkcie obličiek a pečene
Farmakokinetika nusinersenu u pacientov s poruchou funkcie obličiek alebo pečene sa neskúmala. Vplyv pečeňovej alebo obličkovej insuficiencie ako premenných sa nemohol dôkladne hodnotiť
v populačnom PK modeli vzhľadom na vzácnosť pacientov s nálezom klinicky relevantnej pečeňovej
alebo obličkovej insuficiencie. Populačné PK analýzy neodhalili žiadnu zjavnú koreláciu medzi klinickými biochemickými pečeňovými a obličkovými markermi a variabilitou medzi jednotlivými pacientmi.
Rasa
Väčšina sledovaných pacientov boli belosi. Populačná PK analýza naznačuje, že je nepravdepodobné ovplyvenie PK nusinersenu rasou.
5.3 Predklinické údaje o bezpečnosti
Karcinogenita
Dlhodobé štúdie na zvieratách, ktoré by hodnotili karcinogénny potenciál nusinersenu, sa
neuskutočnili.
Mutagenita
U nusinersenu sa nepreukázali žiadne známky genotoxicity.
Reprodukčná toxicita
Štúdie reprodukčnej toxicity sa vykonali použitím subkutánneho podania nusinersenu myšiam
a králikom. Nepozoroval sa žiadny vplyv na fertilitu samcov alebo samíc, embryofetálny vývin alebo prenatálny/postnatálny vývin.
Toxikológia
V štúdiách toxicity po opakovanom (14 týždňov a 53 týždňov) intratekálnom podávaní mladým
makakom jávskym bol nusinersen dobre tolerovaný. Výnimkou bol akútny, prechodný deficit spinálnych reflexov v dolnej časti miechy, ktorý sa vyskytol pri najvyšších hladinách dávok v každej
štúdii (3 alebo 4 mg na dávku; čo je ekvivalentné 30 alebo 40 mg na intratekálnu dávku u pacientov).
Tieto účinky sa pozorovali počas niekoľkých hodín po podaní dávky a vo všeobecnosti vymizli do
48 hodín.
V 53-týždňovej štúdii s intratekálnym podávaním makakom jávskym sa nepozorovali žiadne toxické
účinky pri hladinách až 14-násobne prevyšujúcich odporúčanú ročnú klinickú udržiavaciu dávku.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát dihydrogenfosforečnanu sodného
hydrogénfosforečnan sodný
chlorid sodný chlorid draselný
dihydrát chloridu vápenatého hexahydrát chloridu horečnatého hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte v škatuli na ochranu pred svetlom.
Ak nie je k dispozícii chladnička, Spinraza sa môže uchovávať v pôvodnej škatuli na ochranu pred svetlom pri teplote do 30 °C alebo nižšej po dobu maximálne 14 dní.
Pred podaním sa v prípade potreby môžu neotvorené injekčné liekovky Spinrazy vybrať z chladničky alebo do nej vrátiť. Ak sa vyberú z pôvodnej škatule, celkový čas uchovávania po vybratí z chladničky nesmie prekročiť 30 hodín pri teplote neprevyšujúcej 25 °C.
6.5 Druh obalu a obsah balenia
5 ml v sklenenej injekčnej liekovke typu I s brómbutylovou gumovou zátkou a hliníkovým uzáverom a plastovým viečkom.
Veľkosť balenia je jedna injekčná liekovka v škatuli.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Len na jednorazové použitie.
Pokyny na prípravu lieku pred podaním
1. Injekčná liekovka Spinrazy sa musí pred podaním skontrolovať na prítomnosť častíc. Ak sa
spozorujú častice a/alebo tekutina v injekčnej liekovke nie je číra a bezfarebná, injekčná liekovka sa nesmie použiť.
2. Pri príprave roztoku Spinrazy na intratekálne podanie sa musí použiť aseptická technika.
3. Injekčná liekovka sa musí pred podaním vybrať z chladničky a nechať ohriať na izbovú teplotu
(25 °C) bez použitia vonkajších zdrojov tepla.
4. Ak sa injekčná liekovka neotvorí a roztok sa nepoužije, musí sa vrátiť späť do chladničky (pozri časť 6.4).
5. Tesne pred podaním odstráňte plastové viečko a vsuňte ihlu injekčnej striekačky do injekčnej
liekovky cez stred uzáveru, aby sa natiahol príslušný objem. Spinraza sa nesmie riediť. Nie je potrebné
používať externé filtre.
6. Roztok, ktorý bol natiahnutý do injekčnej striekačky a nepoužije sa v priebehu 6 hodín, sa musí
zlikvidovať.
7. Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBiogen Idec Ltd
Innovation House
70 Norden Road Maidenhead Berkshire
SL6 4AY
Veľká Británia
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/17/1188/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.