časť 4.4 a 4.8). V prípade potreby sa po 4 týždňoch podávania môže dávka upraviť na ďalšiu dávkovú úroveň (pozri časť 5.1). Vzhľadom na zvýšenú frekvenciu hlásení nežiaducich účinkov pri užívaní dávky 40 mg v porovnaní s nižšími dávkami lieku (pozri časť 4.8), finálna titrácia na dávky 30mg alebo 40 mg sa má zvážiť až po 4-týždňovom období liečby, a to len u pacientov s ťažkou hypercholesterolémiou a s vysokým kardiovaskulárnym rizikom (predovšetkým u pacientov s familiárnou hypercholesterolémiou), u ktorých sa pri užívaní dávky 20 mg
nedosiahol liečebný cieľ a ktorí budú pod stálym lekárskym dohľadom (pozri časť 4.4). Odporúča sa, aby pacient, ktorý začína užívať dávky 40 mg alebo 30 mg, bol pod dohľadom odborníka.
Prevencia kardiovaskulárnych príhodV štúdii zameranej na redukciu rizika kardiovaskulárnych príhod sa podávala dávka 20 mg denne (pozri časť 5.1).
Deti a dospievajúciPoužitie u detí majú určovať špecialisti.
Deti a dospievajúci vo veku 10 - 17 rokov (chlapci v Tannerovom štádiu II a vyššom a dievčatá, ktoré majú menštruáciu najmenej 1 rok).
U detí a dospievajúcich s heterozygotnou familiárnou hypercholesterolémiou je zvyčajná začiatočná dávka 5 mg denne. Zvyčajný rozsah dávky je 5 - 20 mg perorálne raz denne. Titrácia sa má u detských pacientov vykonať podľa individuálnej odpovede a znášanlivosti, ako je uvedené v odporúčaniach pre liečbu detí a dospievajúcich (pozri časť 4.4). Pred začatím liečby rosuvastatínom majú mať deti a dospievajúci štandardnú diétu na zníženie cholesterolu; táto diéta má pokračovať počas liečby rosuvastatínom. Bezpečnosť a účinnosť dávok vyšších ako 20 mg sa u tejto populácie neskúmala.
U detí sa nemajú používať 30 mg a 40 mg tablety.
Deti mladšie ako 10 rokov
Skúsenosti u detí mladších ako 10 rokov sú obmedzené na malý počet detí (vo veku medzi 8 a 10 rokov) s homozygotnou familiárnou hypercholesterolémiou. Preto sa Sorvasta neodporúča u detí mladších ako 10 rokov.
Podávanie starším osobám
Odporúčaná začiatočná dávka u pacientov starších ako 70 rokov je 5 mg (pozri časť 4.4). Nie je potrebná žiadna ďalšia úprava dávkovania pokiaľ ide o vek.
Podávanie pacientom s poškodením funkcie obličiek
U pacientov s miernym až stredne ťažkým poškodením funkcie obličiek nie je potrebné upravovať dávku. U pacientov so stredne ťažým poškodením funkcie obličiek (klírens kreatinínu < 60 ml/min) je odporúčaná začiatočná dávka 5 mg. Dávky 40 mg alebo 30 mg sú kontraindikované u pacientov so stredne ťažkým poškodením funkcie obličiek. Podávanie Sorvasty pacientom s ťažkým poškodením funkcie obličiekje kontraindikované v akejkoľvek dávke (pozri časti 4.3 a 5.2).
Podávanie pacientom s poškodením funkcie pečene
U pacientov s Childovým-Pughovým skóre 7 a nižším sa nezaznamenalo žiadne zvýšenie systémovej expozície rosuvastatínu. Zvýšenie systémovej expozície sa však pozorovalo u pacientov s Childovým-Pughovým skóre 8 a 9 (pozri časť 5.2). U týchto pacientov je potrebné zhodnotiť funkciu pečene (pozri časť 4.4). Nie sú žiadne skúsenosti s podávaním rosuvastatínu pacientom s Childovým-Pughovým skóre nad 9. Sorvasta je kontraindikovaná u pacientov s aktívnym ochorením pečene (pozri časť 4.3).
Rasa U pacientov ázijského pôvodu sa pozorovala zvýšená systémová expozícia (pozri časti 4.4 a 5.2). Odporúčaná začiatočná dávka pre pacientov ázijského pôvodu je 5 mg. Dávky 40 mg alebo 30 mg sú kontraindikované u pacientov ázijského pôvodu.
Podávanie pacientom s predispozíciou pre vznik myopatieOdporúčaná začiatočná dávka je 5 mg u pacientov s predispozičnými faktormi pre vznik myopatie (pozri časť 4.4).
Dávky 40 mg alebo 30 mg sú u týchto pacientov kontraindikované (pozri časť 4.3).
4.3 KontraindikácieSorvasta je kontraindikovaná:
- u pacientov s precitlivenosťou na rosuvastatín, alebo na ktorúkoľvek z pomocných látok
- u pacientov s aktívnym ochorením pečene, vrátane nevysvetleného pretrvávajúceho zvýšenia sérových transamináz a akéhokoľvek zvýšenia sérových transamináz nad trojnásobok hornej hranice normálnych hodnôt (ULN)
- u pacientov s ťažkým poškodením funkcie obličiek (klírens kreatinínu < 30 ml/min)
- u pacientov s myopatiou
- u pacientov, ktorí súbežne užívajú cyklosporín
- v gravidite a počas laktácie a u žien vo fertilnom veku, ktoré nepoužívajú primeranú antikoncepciu.
Dávky 40 mg a 30 mg sú kontraindikované u pacientov s nasledovnými predispozičnými faktormi pre vznik myopatie/rabdomyolýzy:
- stredne ťažké poškodenie funkcie obličiek (klírens kreatinínu < 60 ml/min)
- hypotyreoidizmus
- osobná alebo rodinná anamnéza dedičných muskulárnych porúch
- predchádzajúca anamnéza muskulárnej toxicity po podaní iných inhibítorov HMG-CoA reduktázy alebo fibrátov
- nadmerné požívanie alkoholu
- okolnosti, pri ktorých môže dôjsť k zvýšeniu plazmatických hladín
- pacienti ázijského pôvodu
- súbežné užívanie fibrátov
(pozri časti 4.4, 4.5 a 5.2).
4.4Osobitné upozornenia a opatrenia pri používaníÚčinky na obličkyU pacientov, ktorí dostávali vyššie dávky rosuvastatínu najmä 40 mg, sa pri vyšetrení moču, vykonanom pomocou diagnostických prúžkov, zistila proteinúria, ktorá bola väčšinou tubulárneho pôvodu a mala prechodný alebo intermitentný charakter. Proteinúria nepredznamenávala akútne alebo progresívne ochorenie obličiek (pozri časť 4.8). Výskyt hlásení závažných renálnych nežiaducich účinkov je pri postmarketingovom užívaní vyšší pri dávke 40 mg. U pacientov liečenými dávkami 30 alebo 40 mg sa má zvážiť zaradenie sledovania obličkových funkcií (najmenej raz za 3 mesiace) do rutinných kontrol.
Účinky na kostrový svalU pacientov užívajúcich rosuvastatín v akýchkoľvek dávkach, najmä v dávkach vyšších ako 20 mg, boli hlásené účinky na kostrový sval, akými sú napr. myalgia, myopatia a v zriedkavých prípadoch rabdomyolýza. Pri užívaní ezetimibu v kombinácii s inhibítormi HMG-CoA reduktázy bol vo veľmi zriedkavých prípadoch hlásený výskyt rabdomyolýzy. Nedajú sa celkom vylúčiť farmakodynamické interakcie (pozri časť 4.5), je preto potrebná zvýšená opatrnosť pri súbežnom užívaní týchto liekov.
Rovnako ako u iných inhibítorov HMG-CoA reduktázy je výskyt prípadov rabdomyolýzy spojených s užívaním rosuvastatínu v postmarketingovom sledovaní vyšší pri dávke 40 mg.
Stanovovanie kreatínkinázyKreatínkináza (CK) sa nemá stanovovať po fyzickej námahe, alebo ak jestvuje iná možná príčina zvýšenia hodnoty CK, ktorá môže skresliť výsledok. Ak sú východiskové hodnoty CK významne zvýšené (> 5-krát ULN), je treba vykonať potvrdzujúci test v priebehu 5-7 dní. Ak opakovaný test potvrdí CK > 5-krát ULN, liečba sa nemá začať.
Pred liečbouSorvastu podobne ako iné inhibítory reduktázy HMG-CoA, je potrebné predpisovať s opatrnosťou pacientom s nasledovnými predispozičnými faktormi pre vznik myopatie/rabdomyolýzy:
- poškodenie funkcie obličiek
- hypotyreoidizmus
- osobná alebo rodinná anamnéza dedičných muskulárnych porúch
- predchádzajúca anamnéza muskulárnej toxicity po podaní iných inhibítorov HMG-CoA reduktázy alebo fibrátov
- nadmerné požívanie alkoholu
- vek nad 70 rokov
- okolnosti, pri ktorých môže dôjsť k zvýšeniu plazmatických hladín (pozri časť 5.2)
- súbežné užívanie fibrátov.
U týchto pacientov sa má zvážiť riziko liečby v porovnaní s možným prínosom liečby a odporúča sa ich klinické monitorovanie. Ak sú východiskové hodnoty CK významne zvýšené (> 5-krát ULN), liečba sa nemá začať.
Počas liečbyPacientov je treba požiadať, aby okamžite hlásili nevysvetliteľné bolesti svalov, slabosť alebo kŕče, najmä ak sú spojené s nevoľnosťou alebo horúčkou. U týchto pacientov je potrebné stanoviť hladinu kreatínkinázy. Ak dôjde k výraznému vzostupu hladiny kreatínkinázy (> 5-krát ULN), alebo ak sú muskulárne symptómy závažné a spôsobujú ťažkosti počas dňa (aj keď sú hodnoty CK
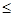
5-krát ULN), liečba sa musí vysadiť. Po úprave symptómov a hodnôt CK sa má zvážiť opätovné podávanie Sorvasty alebo alternatívneho inhibítora HMG-CoA reduktázy v najnižšej dávke a pacienta je treba starostlivo sledovať. Pravidelné sledovanie hodnôt CK u asymptomatických pacientov nie je potrebné.
V klinických skúškach s rosuvastatínom sa u malého počtu pacientov užívajúcich rosuvastatín súbežne s inou terapiou nepreukázalo zosilnenie účinkov na kostrový sval. Lenže u pacientov, ktorí užívali iné inhibítory HMG-CoA reduktázy spolu s derivátmi kyseliny fibrovej vrátane gemfibrozilu, s cyklosporínom, kyselinou nikotínovou, azolovými antimykotikami, inhibítormi proteáz a makrolidovými antibiotikami sa pozoroval zvýšený výskyt myozitídy a myopatie.
Gemfibrozil zvyšuje riziko myopatie, ak sa podáva súbežne s niektorými inhibítormi HMG-CoA reduktázy. Preto sa kombinácia Sorvasty a gemfibrozilu neodporúča. Prínos ďalšej úpravy hladín lipidov súbežným podávaním Sorvasty s fibrátmi alebo niacínom má prevýšiť potenciálne riziká takýchto kombinácií. Užívanie Sorvasty v dávkach 40 mg a 30 mg súbežne s fibrátmi je kontraindikované (pozri časti 4.5 a 4.8).
Sorvasta sa nemá podávať pacientom s akútnym závažným ochorením naznačujúcim myopatiu alebo s predispozíciou navznik obličkovej nedostatočnosti v dôsledku rabdomyolýzy (napr. sepsa, hypotenzia, veľké chirurgické zákroky, trauma, závažné metabolické, endokrinné a elektrolytové poruchy alebo nekontrolované kŕče).
Účinky na pečeň
Podobne ako pri iných inhibítoroch HMG-CoA reduktázy, aj pri podávaní Sorvasty je potrebné venovať zvýšenú pozornosť pacientom, ktorí konzumujú nadmerné množstvá alkoholu a/alebo majú v anamnéze ochorenie pečene. Pred začiatkom a 3 mesiace po nasadení liečby sa odporúča vykonať funkčné testy pečene. Liečba Sorvastou sa má prerušiť alebo dávkovanie znížiť, ak hladina sérových transamináz je 3-krát vyššia ako horná hranica normálnych hodnôt. Výskyt hlásení závažných hepatálnych nežiaducich účinkov (najmä zvýšenie hepatálnych transamináz) je pri postmarketingovom užívaní vyšší pri dávke 40 mg.
U pacientov so sekundárnou hypercholesterolémiou spôsobenou hypotyreoidizmom alebo nefrotickým syndrómom je potrebné vyliečiť základné ochorenie pred začatím terapie Sorvastou.
RasaFarmakokinetické štúdie preukázali zvýšenú expozíciu u pacientov ázijského pôvodu v porovnaní s belochmi, resp. kaukazskou rasou (pozri časti 4.2 a 5.2).
Inhibítory proteázySúbežné užívanie s inhibítormi proteázy sa neodporúča (pozri časť 4.5).
Intersticiálne ochorenie pľúcPri užívaní statínov, hlavne pri dlhodobej liečbe, boli hlásené výnimočné prípady výskytu intersticiálneho ochorenia pľúc (pozri časť 4.8). Medzi prejavy patrí dyspnoe, suchý (neproduktívny) kašeľ a celkové zhoršenie zdravotného stavu (únava, chudnutie, horúčka). Ak je podozrenie, že sa u pacienta prejavilo intersticiálne ochorenie pľúc, je nutné prerušiť terapiu statínmi.
Diabetes mellitus
U pacientov s glykémiou nalačno 5,6 - 6,9 mmol/l, liečba rosuvastatínom bola spojená so zvýšeným rizikom diabetu mellitus (pozri časť 4.8).
Deti a dospievajúci
Hodnotenie lineárneho rastu (výška), hmotnosti, BMI (body mass index) a sekundárnych znakov pohlavného dozrievania podľa Tannera je u detských pacientov vo veku 10 až 17 rokov užívajúcich rosuvastatín obmedzené na jednoročné obdobie. Po 52 týždňoch liečby v rámci štúdie sa nezistil žiadny vplyv na rast, hmotnosť, BMI alebo sexuálne dozrievanie (pozri časť 5.1). Skúsenosti z klinickej štúdie u detí a dospievajúcich sú obmedzené a dlhodobý účinok rosuvastatínu (> 1 rok) na pubertu nie je známy.
V klinických štúdiách u detí a dospievajúcich užívajúcich rosuvastatín počas 52 týždňov sa pozorovali zvýšené CK > 10-krát ULN a svalové symptómy po cvičení alebo zvýšenej fyzickej aktivite častejšie v porovnaní s pozorovaním v klinických štúdiách u dospelých (pozri časť 4.8).
Pomocné látky
Liek obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, (laponského) deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Cyklosporín: Počas súbežného podávania rosuvastatínu a cyklosporínu sa pozorovalo, že hodnoty AUC rosuvastatínu boli v priemere 7-násobne vyššie v porovnaní s hladinami pozorovanými u zdravých dobrovoľníkov (pozri časť 4.3).
Súbežné podávanie s cyklosporínom nemalo vplyv na koncentrácie cyklosporínu v plazme.
Antagonisty vitamínu K: Podobne ako pri iných inhibítoroch HMG-CoA reduktázy, môže nasadenie terapie alebo zvyšovanie dávky Sorvasty u pacientov súbežne liečených antagonistami vitamínu K (napr. warfarín alebo iné kumarínové antikoagulans) viesť k zvýšeniu INR (International Normalised Ratio). Prerušenie podávania alebo zníženie dávky Sorvasty môže viesť k zníženiu INR. Za takýchto okolností je vhodné kontrolovať INR.
Gemfibrozil a iné hypolipidemiká:Súbežné podávanie rosuvastatínu a gemfibrozilu viedlo k dvojnásobnému vzostupu C
max a AUC rosuvastatínu (pozri časť 4.4). Na základe údajov zo špecifických interakčných štúdií sa nepredpokladajú žiadne farmakokineticky relevantné interakcie s fenofibrátom, farmakodynamické interakcie sa však vyskytnúť môžu. Gemfibrozil, fenofibrát, iné fibráty a niacín (kyselina nikotínová) v dávkach znižujúcich lipidy (≥ 1 g/deň) zvyšujú riziko myopatie, ak sa podávajú súbežne s inhibítormi HMG-CoA reduktázy -pravdepodobne preto, že môžu spôsobiť myopatiu aj pri samostatnom podaní. Súbežné podávanie dávky 40 mg a 30 mg s fibrátmi je kontraindikované (pozri časti 4.3 a 4.4). U týchto pacientov sa má liečba začať tiež dávkou 5 mg.
Ezetimib: Súbežné užívanie rosuvastatínu a ezetimibu nemalo ani u jedného z nich za následok zmeny AUC alebo C
max. Farmakodynamické interakcie medzi Sorvastou a ezetimibom, čo sa týka nežiaducich účinkov, sa však nedajú vylúčiť (pozri časť 4.4).
Inhibítory proteázy:Hoci presný mechanizmus interakcie nie je známy, súbežné užívanie inhibítorov proteázy môže výrazne zvýšiť expozíciu rosuvastatínu. Súbežné podávanie 20 mg rosuvastatínu a kombinovaného lieku pozostávajúceho z dvoch inhibítorov proteázy (400 mg lopinaviru/100 mg ritonaviru) zdravým dobrovoľníkom v rámci farmakokinetického skúšania bolo spojené s približne dvojnásobným zvýšením rovnovážneho AUC
(0-24) a päťnásobným zvýšením C
max rosuvastatínu. Súbežné užívanie rosuvastatínu a inhibítorov proteázy u HIV-pozitívnych pacientov sa preto neodporúča (pozri tiež časť 4.4).
Antacidá: Súbežné podávanie rosuvastatínu a suspenzie antacíd obsahujúcej hydroxid hlinitý a horečnatý viedlo k poklesu plazmatických koncentrácií rosuvastatínu približne o 50 %. Tento účinok sa však zmiernil, ak sa antacidum podalo 2 hodiny po podaní rosuvastatínu. Klinický význam tejto interakcie sa neskúmal.
Erytromycín: Súbežné podávanie rosuvastatínu a erytromycínu viedlo k 20 % poklesu AUC (0-t) a k 30 % zníženiu hodnoty C
max rosuvastatínu. Príčinou tejto interakcie môže byť zvýšenie motility čreva vyvolané erytromycínom
Perorálne kontraceptíva/substitučná hormonálna liečba (HRT):Súbežné podávanie rosuvastatínu a perorálnych kontraceptív viedlo k vzostupu AUC etinylestradiolu o 26 % a norgestrelu o 34 %. Takéto zvýšenie hladín v plazme je treba vziať do úvahy pri určení dávok perorálneho kontraceptíva. U pacientok užívajúcich súbežne rosuvastatín a substitučnú hormonálnu liečbu nie sú dostupné farmakokinetické údaje, a preto sa nedá vylúčiť, že môže dôjsť k podobnému efektu. Takáto kombinácia sa však podávala veľkému počtu žien v klinických štúdiách a bola dobre tolerovaná.
Iné lieky:Na základe údajov zo špecifických interakčných štúdií sa neočakávajú žiadne klinicky relevantné interakcie s digoxínom.
Izoenzýmy cytochrómu P450: Výsledky skúšok
in vitro a in vivo ukázali, že rosuvastatín nie je inhibítorom ani induktorom izoenzýmov cytochrómu P450. Navyše, rosuvastatín je slabým substrátom pre tieto izoenzýmy. Medzi rosuvastatínom a flukonazolom (inhibítor CYP2C9 a CYP3A4), alebo ketokonazolom (inhibítor CYP2A6 a CYP3A4) sa nepozorovali žiadne klinicky relevantné interakcie. Súbežné podávanie itrakonazolu (inhibítor CYP3A4) a rosuvastatínu viedlo k 28 % zvýšeniu AUC rosuvastatínu. Takéto malé zvýšenie sa nepovažuje za klinicky významné. Liekové interakcie v súvislosti s metabolizmom sprostredkovaným cytochrómom P450 sa preto neočakávajú.
4.6Fertilita, gravidita a laktáciaSorvasta je kontraindikovaná (pozri časť 4.3) počas gravidity a laktácie.
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby.
Keďže cholesterol a iné produkty jeho biosyntézy sú pre vývoj plodu nenahraditeľné, potenciálne riziká vyplývajúce z inhibície HMG-CoA reduktázy prevažujú nad prínosom liečby počas gravidity. Štúdie na zvieratách poskytli obmedzené dôkazy reprodukčnej toxicity (pozri časť 5.3). Ak počas užívania tohto lieku pacientka otehotnie, liek je potrebné okamžite vysadiť.
U potkanov sa rosuvastatín vylučuje do mlieka. O vylučovaní do materského mlieka u ľudí nie sú žiadne údaje (pozri časť 4.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje.
Na základe farmakodynamických vlastností rosuvastatínu nie je pravdepodobné, že bude ovplyvňovať schopnosť viesť vozidlá a obsluhovať stroje. Pri vedení vozidiel alebo obsluhe strojov je potrebné vziať do úvahy, že počas liečby sa môže objaviť závrat.
4.8 Nežiaduce účinkyNežiaduce účinky pozorované v súvislosti s podávaním rosuvastatínu sú spravidla mierne a prechodné. V kontrolovaných klinických skúškach menej ako 4 % pacientov liečených rosuvastatínom ukončilo účasť v štúdii kvôli nežiaducim účinkom.
Frekvencia výskytu nežiaducich účinkov je usporiadaná nasledovne:
časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000), s neznámou frekvenciou (z dostupných údajov sa nedá určiť).
Poruchy imunitného systémuZriedkavé: hypersenzitívne reakcie vrátane angioedému.
Poruchy endokrinného systému
Časté: diabetes mellitus.
Poruchy nervového systémuČasté: bolesť hlavy, závrat.
Poruchy gastrointestinálneho traktu Časté: zápcha, nauzea, bolesť brucha.
Zriedkavé: pankreatitída.
Poruchy kože a podkožného tkaniva Menej časté: pruritus, vyrážka a žihľavka.
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaČasté: myalgia.
Zriedkavé: myopatia a rabdomyolýza.
Celkové poruchy a reakcie v mieste podaniaČasté: asténia.
Podobne ako pri iných inhibítoroch HMG CoA reduktázy, výskyt nežiaducich účinkov lieku má stúpajúcu tendenciu so zvyšujúcou sa dávkou.
Účinky na obličky: U pacientov užívajúcich rosuvastatín sa pri vyšetrení moču, vykonanom pomocou diagnostických prúžkov, zistila proteinúria väčšinou tubulárneho pôvodu. Zmena z negatívneho alebo stopového nálezu bielkoviny v moči na ++ alebo viac v určitom časovom úseku liečby sa pozorovala pri podávaní 10 mg a 20 mg rosuvastatínu u menej ako 1 % prípadov, pri podávaní 40 mg rosuvastatínu približne u 3 %. Pri podávaní 20 mg rosuvastatínu sa zistilo malé zvýšenie proteinúrie (z negatívneho alebo stopového nálezu na +). Počas ďalšej liečby došlo vo väčšine prípadov k spontánnemu zníženiu, resp. k vymiznutiu proteinúrie. Zhodnotenie údajov z klinických skúšaní a postmarketingového užívania doteraz neidentifikovalo príčinnú súvislosť medzi proteinúriou a akútnym alebo progresívnym ochorením obličiek.
U pacientov liečených rosuvastatínom sa pozoroval výskyt hematúrie a údaje z klinických skúšaní preukázali, že jej výskyt je nízky.
Účinky na kostrový sval: U pacientov užívajúcich rosuvastatín v akýchkoľvek dávkach, najmä v dávkach vyšších ako 20 mg, boli hlásené účinky na kostrový sval, akými sú napr. myalgia, myopatia (vrátane myozitídy) a v zriedkavých prípadoch rabdomyolýza.
U pacientov užívajúcich rosuvastatín sa pozoroval dávkovo závislý vzostup koncentrácie kreatínkinázy; vo väčšine prípadov bolo toto zvýšenie mierne, asymptomatické a prechodné. Ak sa hladiny CK zvýšia (> 5 krát ULN), liečba sa má prerušiť (pozri časť 4.4).
Účinky na pečeň: Podobne ako pri iných inhibítoroch HMG-CoA reduktázy u malého počtu pacientov užívajúcich rosuvastatín sa pozoroval dávkovo závislý vzostup transamináz; vo väčšine prípadov bolo toto zvýšenie mierne, asymptomatické a prechodné.
Postmarketingové skúsenosti:Počas postmarketingových skúseností s rosuvastatínom boli okrem vyššie uvedených hlásené nasledujúce nežiaduce účinky:
Poruchy nervového systému:
Veľmi zriedkavé: polyneuropatia, strata pamäti.
Poruchy dýchacej sústavy, hrudníka a mediastína:
S neznámou frekvenciou: kašeľ, dýchavičnosť.
Poruchy gastrointestinálneho traktu:
S neznámou frekvenciou: hnačka.
Poruchy pečene a žlčových ciest
Veľmi zriedkavé:žltačka, hepatitída.
Zriedkavé:zvýšenie hepatálnych transamináz.
Poruchy kože a podkožného tkaniva: S neznámou frekvenciou: Stevensov-Johnsonov syndróm.
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaZriedkavé:artralgia.
Poruchy obličiek a močovej sústavy
Veľmi zriedkavé
: hematúria.
Celkové poruchy a reakcie v mieste podania:
S neznámou frekvenciou: edém.
Po podaní niektorých statínov sa vyskytli nasledujúce nežiaduce účinky:
- depresia,
- poruchy spánku, vrátane nespavosti a nočných môr,
- sexuálna dysfunkcia,
- výnimočné prípady intersticiálneho ochorenia pľúc, a to najmä počas dlhodobej liečby (pozri časť 4.4).
Výskyt prípadov rabdomyolýzy, závažných renálnych nežiaducich účinkov a závažných hepatálnych nežiaducich účinkov (najmä zvýšenie hodnôt hepatálnych transamináz) je vyšší pri dávke 40 mg.
Deti a dospievajúciZvýšenie kreatínkinázy > 10-krát ULN a svalové symptómy sa pozorovali po cvičení alebo zvýšenej fyzickej aktivite v 52-týždňovej klinickej štúdii u detí a dospievajúcich častejšie v porovnaní s dospelými (pozri časť 4.4). V ostatných ohľadoch, bezpečnostný profil rosuvastatínu bol u detí a dospievajúcich podobný v porovnaní s dospelými
4.9 Predávkovanie
Neexistuje žiadna špecifická liečba predávkovania. Ak dôjde k predávkovaniu, liečba je symptomatická a podľa potreby sa majú vykonať podporné opatrenia. Je potrebné sledovať funkcie pečene a hladiny kreatínkinázy. Hemodialýza pravdepodobne nemáprospešný účinok.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: inhibítory HMG-CoA reduktázy, ATC kód:C10AA07
Spôsob účinku Rosuvastatín je selektívny a kompetitívny inhibítor HMG-CoA reduktázy, rýchlosť limitujúceho enzýmu, ktorý konvertuje 3-hydroxy-3-metylglutarylkoenzým A namevalonát, prekurzor cholesterolu. Primárnym miestom účinku rosuvastatínu je pečeň, cieľový orgán pre znižovanie hladiny cholesterolu.
Rosuvastatín zvyšuje počet LDL receptorov v pečeni na povrchu buniek, čím sa potencuje vychytávanie a katabolizmus LDL a inhibuje sasyntéza VLDL v pečeni, následkom čoho sa znižuje celkový počet častíc VLDL a LDL.
Farmakodynamické účinkyRosuvastatín znižuje zvýšenú koncentráciu LDL-cholesterolu, koncentráciu celkového cholesterolu, triglyceridov a zvyšuje hladinu HDL-cholesterolu. Znižuje tiež hladiny ApoB, nonHDL-C, VLDL-C, VLDL-TG a zvyšuje hladinu ApoA-I (pozri Tabuľku 1). Rosuvastatín tiež znižuje vzájomný pomer LDL-C/HDL-C, celkového C/HDL-C, nonHDL-C/HDL-C a pomer ApoB/ApoA-I.
Tabuľka 1 Dávkovo závislá odpoveď u pacientov s primárnou hypercholesterolémiou (typ IIa a IIb) (upravené priemerné percento zmien v porovnaní s východiskovými hodnotami)Dávka
| N
| LDL-C
| Celkový-C
| HDL-C
| TG
| nonHDL-C
| ApoB
| ApoA-I
|
Placebo
| 13
| -7
| -5
| 3
| -3
| -7
| -3
| 0
|
5
| 17
| -45
| -33
| 13
| -3
| -44
| -38
| 4
|
10
| 17
| -52
| -36
| 14
| -10
| -48
| -42
| 4
|
20
| 17
| -55
| -40
| 8
| -23
| -51
| -46
| 5
|
40
| 18
| -63
| -46
| 10
| -28
| -60
| -54
| 0
|
Terapeutický účinoksa prejaví v priebehu 1 týždňa od začiatku liečby a 90 % maximálnej odpovede sa dosiahne spravidla do 2 týždňov. Maximálna odpoveď sa obyčajne dosiahne do 4 týždňov a potom sa udržuje.'
Klinická účinnosťRosuvastatín je účinný u dospelých pacientov s hypercholesterolémiou, s hypertriglyceridémiou i bez nej, bez ohľadu na rasu, pohlavie či vek; je účinný u špeciálnych skupín pacientov, napríklad u diabetikov a u pacientov s familiárnou hypercholesterolémiou.
Kumulované údaje z fázy III klinického skúšania ukázali, že rosuvastatín bol účinný v liečbe väčšiny pacientov s hypercholesterolémiou typu IIa a IIb (priemerné východiskové hodnoty LDL - C okolo 4,8 mmol/l) podľa prijatých smerníc Európskej spoločnosti pre aterosklerózu (EAS;1998). Približne 80 % pacientov užívajúcich 10 mg rosuvastatínu dosiahlo cieľové hodnoty LDL - C (< 3 mmol/l), odporúčané smernicami EAS.
V rozsiahlej štúdii s pacientmi s heterozygótnou formou familiárnej hypercholesterolémie sa rosuvastatín podával 435 osobám v dávkach od 20 mg do 80 mg v rámci titrovania vhodnej sily lieku. Vo všetkých dávkach vykazovala Sorvasta priaznivý účinok na parametre lipidov a dosahovanie cieľov terapie. Po titrácii na dennú dávku 40 mg (12 týždňov liečby) sa hladina LDL - C znížila o 53 %. 33 % pacientov dosiahlo cieľové hodnoty pre hladinu LDL - C (< 3mmol/l) stanovené v smerniciach EAS.
V rámci titrovania vhodnej sily lieku v otvorenej štúdii sa hodnotila odpoveď 42 pacientov s homozygótnou formou familiárnej hypercholesterolémie na rosuvastatín 20 – 40 mg. V celkovej populácii sa dosiahlo priemerné zníženie hladín LDL - C o 22 %.
V klinických skúškach, do ktorých bol zaradený obmedzený počet pacientov, sa preukázalo, že rosuvastatín má v kombinácii s fenofibrátom aditívny účinok na znižovanie hladiny triglyceridov a v kombinácii s niacínom na zvyšovanie hladiny HDL - C. (pozri časť 4.4)
V multicentrickom, dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní (METEOR) bolo 984 pacientov vo veku od 45 do 70 rokov s nízkym rizikom ICHS (definovanom ako Framinghamské riziko < 10 % počas viac ako 10 rokov), s priemernou hodnotou LDL-cholesterolu 4,0 mmol/l, ale so subklinickou aterosklerózou (zistenou pomocou CIMT - Carotid Intima Media Thickness) randomizovaných na liečbu buď rosuvastatínom 40 mg jedenkrát denne alebo placebom počas 2 rokov. Rosuvastatín v porovnaní s placebom signifikantne spomalil progresiu aterosklerotického procesu zisteného pomocou CIMT na 12 miestach karotídy, a to o 0,0145 mm/rok [95 % CI - 0,0196 až - 0,0093; p < 0,0001]. Zmena z východiskovej hodnoty u skupiny pacientov liečených rosuvastatínom bola - 0,0014 mm/rok [- 0,12 %/rok (nesignifikantné)] v porovnaní s progresiou [+ 0,0131 mm/rok (1,12 %/rok (p < 0,0001] u pacientov užívajúcich placebo. Zatiaľ sa nepreukázala priama súvislosť medzi znížením CIMT a redukciou rizika výskytu kardiovaskulárnych udalostí. Populácia pacientov, ktorá sa zúčastnila klinického skúšania METEOR, je z hľadiska koronárneho ochorenia srdca nízko riziková a nepredstavuje cieľovú populáciu pre liečbu Sorvastou v dávke 40 mg. Sorvasta v dávke 40 mg sa má podávať len pacientom s ťažkou hypercholesterolémiou s vysokým kardiovaskulárnym rizikom (pozri časť 4.2).
Zdôvodnenie použitia statínov v primárnej prevencii: Intervenčná štúdia s rosuvastatínom (JUPITER štúdia), posudzovala vplyv rosuvastatínu na výskyt závažných aterosklerotických kardiovaskulárnych ochorení u 17 802 mužov (≥ 50 rokov) a žien (≥ 60 rokov).
Účastníci štúdie boli náhodne rozdelení do skupiny s placebom (n = 8901) alebo rosuvastatínom 20 mg jedenkrát denne (n = 8901) a boli sledovaní počas jej trvania priemerne 2 roky.
Koncentrácia LDL-cholesterolu sa znížila o 45 % (p < 0,001) v rosuvastatínovej skupine v porovnaní so skupinou s placebom.
V post-hoc analýze vysoko rizikovej podskupiny subjektov s východiskovým Framinghamským rizikovým skóre > 20 % (1558 osôb) došlo k významnému zníženiu kombinovaného koncového bodu kardiovaskulárnej smrti, mŕtvice a infarktu myokardu (p = 0,028) pri liečbe rosuvastatínom oproti placebu. Absolútne zníženie rizika bolo v pomere 8,8 prípadov na 1000 pacientorokov. Celková mortalita sa v tejto vysoko rizikovej skupine nezmenila (p = 0,193). V post-hoc analýze vysoko rizikovej podskupiny subjektov (celkovo 9302 osôb) s východiskovým SCORE rizikom ≥ 5 % (extrapolovaným, aby boli zahrnutí pacienti nad 65 rokov) došlo k významnému zníženiu kombinovaného koncového bodu kardiovaskulárnej smrti, mŕtvice a infarktu myokardu (p = 0,0003) pri liečbe rosuvastatínom oproti placebu. Absolútne zníženie rizika bolo v pomere 5,1 prípadov na 1000 pacientorokov. Celková mortalita v tejto vysoko rizikovej skupine sa nezmenila (p = 0,076).
V štúdii JUPITER prerušilo z dôvodu nežiaducich účinkov liečbu 6,6 % subjektov liečených rosuvastatínom a 6,2 % subjektov liečených placebom. Najčastejšie nežiaduce účinky, ktoré viedli k prerušeniu liečby boli: myalgia (0,3 % rosuvastatín, 0,2 % placebo), abdominálna bolesť (0,03 % rosuvastatín, 0,02 % placebo) a vyrážka (0,02 % rosuvastatín, 0,03 % placebo). Najčastejšie nežiaduce účinky, ktoré sa vyskytli s vyššou alebo rovnakou frekvenciou ako po podaní placeba, boli uroinfekcie (8,7 % rosuvastatín, 8,6 % placebo), nazofaryngitída (7,6 % rosuvastatín, 7,2 % placebo), bolesť chrbta (7,6 % rosuvastatín, 6,9 % placebo) a myalgia (7,6 % rosuvastatín, 6,6 % placebo).
Deti a dospievajúci
V multicentrickej dvojito zaslepenej, randomizovanej, placebom kontrolovanej, 12-týždňovej štúdii (n = 176, z toho 97 mužov a 79 žien), nasledovanej 40-týždňovou otvorenou titračnou fázou rosuvastatínu (n = 173, z toho 96 mužov a 77 žien), užívali pacienti vo veku 10 - 17 rokov (Tannerovo štádium II-V, dievčatá, ktoré majú menštruáciu najmenej 1 rok) s heterozygotnou familiárnou hypercholesterolémiou, rosuvastatín 5, 10 alebo 20 mg alebo placebo denne počas 12 týždňov a potom užívali všetci rosuvastatín denne počas 40 týždňov. Pri vstupe do štúdie bolo približne 30 % pacientov vo veku 10 - 13 rokov a približne 17 % bolo v Tannerovom štádiu II, 18 % v Tannerovom štádiu III, 40 % v Tannerovom štádiu IV a 25 % bolo v Tannerovom štádiu V. Po 12-týždňovej štúdii sa LDL - C znížil o 38,3 % s rosuvastatínom 5 mg, o 44,6 % s rosuvastatínom 10 mg a o 50,0 % s rosuvastatínom 20 mg, v porovnaní s 0,7 % znížením u placeba.
Na konci 40-týždňovej otvorenej fázy titrovaním do cieľovej hodnoty a zvyšovaním dávky do maximálne 20 mg jedenkrát denne dosiahlo cieľovú hodnotu LDL - C menej ako 2,8 mmol/l 70 zo 173 pacientov (40,5 %).
Po 52 týždňoch liečby v rámci štúdie sa nezistil vplyv na rast, hmotnosť, BMI alebo sexuálne dozrievanie (pozri časť 4.4). Skúsenosti z klinických štúdií u detí a dospievajúcich pacientov sú obmedzené a dlhodobý účinok rosuvastatínu (> 1 rok) na pubertu nie je známy. Táto štúdia (n = 176) nebola vhodná pre porovnanie zriedkavých nežiaducich účinkov.
5.2 Farmakokinetické vlastnostiAbsorpcia: Maximálne koncentrácie rosuvastatínu v plazme sa dosahujú približne 5 hodín po perorálnom podaní. Absolútna biologická dostupnosť je približne 20 %.
Distribúcia: Rosuvastatín sa extenzívne vychytáva v pečeni, ktorá je primárnym miestom syntézy cholesterolu a klírensu LDL - C. Distribučný objem rosuvastatínu je približne 134 l. Približne 90 % rosuvastatínu sa viaže na bielkoviny plazmy, hlavne na albumín.
Metabolizmus: Rosuvastatín sa čiastočne metabolizuje (približne 10 %). Metabolické štúdie
in vitro s použitím ľudských hepatocytov indikujú, že rosuvastatín je slabým substrátom pre metabolizmus sprostredkovaný cytochrómom P450. CYP2C9 bol hlavným izoenzýmom,v menšej miere 2C19, 3A4 a 2D6. Hlavnými identifikovanými metabolitmi sú N -desmetyl metabolit a laktónový metabolit. N-desmetyl metabolit je približne o 50 % menej účinný ako rosuvastatín, kým laktónová forma sa považuje za klinicky neúčinnú. Rosuvastatín je zodpovedný za viac ako 90 % inhibície aktivity HMG - CoA reduktázy v cirkulácii.
Vylučovanie: Približne 90 % rosuvastatínu sa vylúči v nezmenenej forme stolicou (vo forme absorbovaného a neabsorbovaného liečiva) a zvyšok močom. Približne 5 % sa vylučuje v nezmenenej forme močom. Polčas eliminácie z plazmy je približne 20 hodín. Polčas eliminácie sa so zvyšujúcou dávkou nezvyšuje. Geometrický priemer hodnoty plazmatického klírensu je približne 50 l/hod (koeficient zmeny 21,7 %). Podobne ako u iných inhibítorov HMG-CoA reduktázy, do hepatálnej absorpcie rosuvastatínu je zapojený membránový prenášač OATP - C. Tento prenášač je dôležitý pre elimináciu rosuvastatínu pečeňou.
Linearita: Systémová expozícia rosuvastatínu sa zvyšuje proporcionálne v závislosti od dávky.
Po podaní viacnásobných denných dávok nie sú zmeny vo farmakokinetických parametroch.
Špeciálne populácie
Vek a pohlavie: Farmakokinetika rosuvastatínunie je klinicky relevantne ovplyvnená vekom ani pohlavím.
Farmakokinetika rosuvastatínu u detí a dospievajúcich s heterozygotnou familiárnou hypercholesterolémiou bola podobná ako u dospelých dobrovoľníkov (pozri "Deti a dospievajúci" nižšie).
Rasa: Farmakokinetické štúdie preukázali približne dvojnásobné zvýšenie strednej hodnoty AUC a C
max u pacientov ázijského pôvodu (Japonci, Číňania, Filipínci, Vietnamci a Kórejci) v porovnaní s kaukazskou rasou (belochmi). U Indov sa vyskytla približne 1,3-násobná elevácia strednej hodnoty AUC a C
max. Populačná farmakokinetická analýza neodhalila klinicky relevantné rozdiely vo farmakokinetike medzi belochmi (kaukazská rasa) a černochmi.
Porucha funkcie obličiek: V klinickej skúške s pacientmi s rôznym stupňom poruchy obličkových funkcií nemali poruchy mierneho až stredného stupňa vplyv na koncentrácie rosuvastatínu alebo N‑desmetylmetabolitu v plazme. U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) sa však zistil 3 - násobný vzostup koncentrácií v plazme a 9 - násobný vzostup koncentrácie N‑desmetylmetabolitu v porovnaní so zdravými dobrovoľníkmi. Rovnovážne stavy plazmatických koncentrácií rosuvastatínu u jedincov vystavených hemodialýze boli približne o 50 % vyššie v porovnaní so zdravými dobrovoľníkmi.
Porucha funkcie pečene: V štúdii, ktorej sa zúčastnili pacienti s rôznym stupňom poškodenia funkcie pečene sa nedokázala zvýšená expozícia rosuvastatínu u pacientov s Childovým-Pughovým skóre 7 alebo nižším. U dvoch pacientov s Childovým-Pughovým skóre 8 a 9 sa však zistilo najmenej dvojnásobné zvýšenie systémovej expozície v porovnaní s pacientmi s nižším Childovým-Pughovým skóre. S pacientami s Childovým-Pughovým skóre vyšším ako 9 nie sú žiadne skúsenosti.
Deti a dospievajúciFarmakokinetické parametre u detí a dospievajúcich s heterozygotnou familiárnou hypercholesterolémiou vo veku od 10 do 17 rokov neboli úplne charakterizované. Malá farmakokinetická štúdia s rosuvastatínom (podávaným vo forme tabliet) u 18 detských pacientov ukázala, že expozícia u detských pacientov sa zdá porovnateľná s expozíciou u dospelých pacientov. Okrem toho výsledky ukazujú, že veľká odchýlka od proporcionality dávky sa neočakáva.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje na základe obvyklých štúdií farmakologickej bezpečnosti, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. Špecifické testy na účinky hERG neboli skúmané. Nežiaduce reakcie sa nepozorovali v klinických skúšaniach ale zistili sa u zvierat pri hladinách expozície podobnej klinickej expozícii nasledovne: v skúšaniach toxicity po opakovanom podávaní sa pozorovali histopatologické zmeny pečene pravdepodobne vzhľadom na farmakologický účinok rosuvastatínu u myší, potkanov a s nižším rozsahom účinkov na močový mechúr u psov, avšak nie u opíc. Okrem toho sa pozorovala testikulárna toxicita u opíc a psov pri vyšších dávkach. Reprodukčná toxicita evidentná u potkanov, zníženie počtu vrhov, zníženie hmotnosti mláďat vo vrhu a ich prežitia sa pozorovala pri dávkach toxických pre matku, kde systémová expozícia bola niekoľkokrát vyššia ako hodnota terapeutickej expozície.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety Anhydrid laktózy
Mikrokryštalická celulóza
Krospovidón
Magnéziumstearát
Koloidný bezvodý oxid kremičitý
Obal tabletyMonohydrát laktózy
Oxid titaničitý (E171)
Makrogol 6000
Bázický butylovaný metakrylátový kopolymér
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek si nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Obal: blister OPA/ALU/PVC/ALU
Veľkosť balenia: 10, 14, 20, 28, 30, 56, 60, 84, 90, 98 a 100 filmom obalených tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
TRANSMEDIC Slovakia s.r.o., Lazovná 68, 974 01 Banská Bystrica, Slovensko
8. REGISTRAČNÉ ČÍSLO
Sorvasta 5 mg: 31/0698/10-S
Sorvasta 10 mg: 31/0699/10-S
Sorvasta 15 mg: 31/0700/10-S
Sorvasta 20 mg: 31/0701/10-S
Sorvasta 30 mg: 31/0702/10-S
Sorvasta 40 mg: 31/0703/10-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
10. DÁTUM POSLEDNEJ REVÍZIE
September 2010