
adruje v mg somatropínu vzhľadom na obsah podielu somatropínu a bez zahrnutia linkera mPEG, aby sa predišlo chybám v medikácii, keď pacienti prechádzajú z dennej somatropínovej liečby.
Dávkovanie
Dávkovanie a podávanie má byť individualizované pre každého pacienta.
Počiatočná dávkaOdporúčaná počiatočná dávka Lonapegsomatropin Ascendis Pharma je 0,24 mg somatropínu/kg
telesnej hmotnosti, podávaná raz týždenne. Odporúčanú silu počiatočnej dávky pre takúto dávku podľa
hmotnostného rozsahu môžete nájsť v tabuľke 1.
Tabuľka 1 Odporúčaná dávka pre pacientov podľa hmotnosti pri predpísanýchdávkach 0,24 mg somatropínu/kg/týždeňHmotnosť (kg)
| Sila dávky somatropínu
|
11,5 – 13,9
| 3 mg
|
14 – 16,4
| 3,6 mg
|
16,5 – 19,9
| 4,3 mg
|
20 – 23,9
| 5,2 mg
|
24 – 28,9
| 6,3 mg
|
29 – 34,9
| 7,6 mg
|
35 – 41,9
| 9,1 mg
|
42 – 50,9
| 11 mg
|
51 – 60,4
| 13,3 mg
|
60,5 – 69,9
| 15,2 mg (s použitím dvoch dvojkomorových náplní po 7,6 mg)
|
70 – 84,9
| 18,2 mg (s použitím dvoch dvojkomorových náplní po 9,1 mg)
|
85 – 100
| 22 mg (s použitím dvoch dvojkomorových náplní po 11 mg)
|
Ak predpisujete dávku inú než 0,24 mg somatropínu/kg/týždeň, vypočítajte celkovú týždennú dávku
(v mg somatropínu) a vyberte príslušnú silu dávky nasledujúcim spôsobom:
• Celková týždenná dávka (mg somatropínu) = predpísaná dávka (mg somatropínu/kg) x telesná hmotnosť pacienta (kg)
• Celkovú týždennú dávku (mg somatropínu) zaokrúhlite na najbližšiu silu dávky, pričom tiež zvážte liečebné ciele a klinickú odpoveď.
Počiatočná dávka pre pacientov prechádzajúcich z denných somatropínových liekovAk meníte liečbu z denného somatropínu na lonapegsomatropín raz týždenne, medzi poslednou
dávkou somatropínu raz denne a prvou dávkou lonapegsomatropínu musí byť najmenej 8 hodín.
U detí prechádzajúcich z denného somatropínu môžu lekári upraviť počiatočnú dávku vzhľadom na
aktuálnu dávku somatropínu, individuálnu klinickú odpoveď a klinické úvahy špecifické pre pacienta.
U detí prechádzajúcich z denných somatropínových liekov v týždennej dávke rovnej alebo väčšej ako
0,24 mg somatropínu/kg telesnej hmotnosti je odporúčaná počiatočná dávka lonapegsomatropínu
0,24 mg somatropínu/kg telesnej hmotnosti (pozri Tabuľka 1).
U detí prechádzajúcich z denných somatropínových liekov v týždennej dávke menšej než 0,24 mg somatropínu/kg telesnej hmotnosti použite predtým predpísanú týždennú dávku ako odporúčanú počiatočnú dávku lonapegsomatropínu (pozri rovnicu vyššie).
Titrácia dávkyDávku lonapegsomatropínu je potrebné individuálne upraviť pre každého pacienta na základe klinickej
odpovede, nežiaducich reakcií a/alebo sérových koncentrácií inzulínu podobného rastového faktora 1 (IGF-1) mimo cieľového rozsahu. Dostupné sily dávok somatropínu nájdete v časti 1.
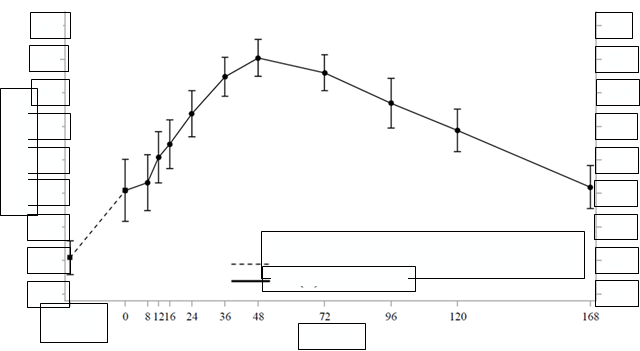
Pri titrácii dávky možno ako usmernenie použiť priemerné hladiny skóre štandardnej odchýlky (SDS) IGF-1 (odobrané 4 – 5 dní po dávkovaní) (pozri Tabuľka 2). Pred hodnotením výsledných hladín SDS IGF-1 je potrebné počkať minimálne 2 týždne po nasadení lonapegsomatropínu alebo po akejkoľvek zmene dávky. Úpravy dávkovania majú byť zamerané na dosiahnutie priemerných hladín SDS IGF-1 na hranici normálneho rozsahu, t. j. medzi -2 a +2 (pokiaľ možno blízko 0 SDS).
Hladiny SDS IGF-1 sa môžu v priebehu času líšiť, a preto sa odporúča rutinné monitorovanie
sérových hladín SDS IGF-1 v priebehu liečby, najmäpočas puberty.
Tabuľka 2 Odporúčaná zmena sily dávky somatropínu pre priemerné kategórie SDS IGF-1Priemerný rozsah SDS IGF-1 (odobraný 4. – 5. deň po dávkovaní)
| Odporúčaná zmena sily dávky somatropínu
|
> +4
| Znížiť o 3 dávkové sily
|
+3 až +4
| Znížiť o 2 dávkové sily
|
+2 až +3
| Znížiť o 1 dávkovú silu
|
-2 až +2
| Bez zmeny
|
< -2
| Zvýšiť o 1 dávkovú silu
|
HodnotenieliečbyHodnotenie účinnosti a bezpečnosti sa má zvážiť v približne 6- až 12-mesačných intervaloch a môže
byť hodnotené posúdením auxologických parametrov, biochémie (IGF-1, hormónov, glukózy a hladín
lipidov) a pubertálneho stavu. Počas puberty treba zvážiť častejšie hodnotenia.
Liečba sa má vysadiť u pacientov s ročnou rýchlosťou rastu do výšky < 2 cm/rok, po dosiahnutí
konečnej výšky, s SDS rýchlosti rastu do výšky < + 1 po prvom roku liečby alebo ak je vek kostí
> 14 rokov (dievčatá) alebo > 16 rokov (chlapci), čo korešponduje s uzavretím epifýzových rastových
platničiek.
Keď sú epifýzy zrastené, pacienti musia byť klinicky prehodnotení z hľadiska potreby liečby rastovým
hormónom.
Perorálna estrogénová liečbaU žien užívajúcich perorálnu estrogénovú liečbu sa môže vyžadovať vyššia dávka rastového hormónu
na dosiahnutie liečebného cieľa (pozri časť 4.4).
Vynechaná dávkaAk sa dávka vynechá, musí sa podať čo najskôr a nie neskôr než 2 dni po vynechanej dávke. Ak už
uplynuli viac než 2 dni, vynechaná dávka sa musí preskočiť a ďalšia dávka sa má podať v pravidelne
plánovaný deň. V každom prípade potom pacienti môžu obnoviť svoj pravidelný rozpis dávkovania
raz za týždeň.
Zmena dávkovacieho dňaDeň týždenného podania injekcie sa môže zmeniť na iný deň v týždni. Lonapegsomatropín sa môže
podať 2 dni pred alebo 2 dni po plánovanom dávkovacom dni. Musí sa zaistiť, aby medzi poslednou
dávkou a novozavedeným pravidelným dávkovacím dňom raz týždenne uplynulo aspoň 5 dní.
Osobitné populáciePoruchafunkcieobličiekNie sú k dispozícii žiadne informácie o pacientoch s poruchou funkcie obličiek a nie je možné
poskytnúť odporúčania na dávkovanie.
Porucha
funkcie
p
ečene
Nie sú k dispozícii žiadne informácie o pacientoch s poruchou funkcie pečene a nie je možné
poskytnúť odporúčania na dávkovanie.
Pediatrická populácia
Bezpečnosť a účinnosť lonapegsomatropínu u detí vo veku do 3 rokov neboli stanovené. V súčasnosti
dostupné údaje sú opísané v časti 5.1, ale neumožňujú uviesť odporúčania na dávkovanie.
Spôsob podávania
Každá injekcia sa má podávať subkutánne raz týždenne do brucha, zadku alebo stehna. Miesto
podania sa má striedať, aby nevznikla lipoatrofia.
Lonapegsomatropín je určený na podávanie po rekonštitúcii prášku na injekčný roztok s dodaným rozpúšťadlom. Lonapegsomatropín sa má podávať pomocou autoinjektora GH Auto-Injector. Pacient a opatrovateľ musia byť zaškolení, aby sa zaistilo pochopenie postupu podávania pomocou tejto pomôcky, aby sa umožnilo (samostatné) injekčné podávanie lonapegsomatropínu.
Rekonštituovaný roztok má byť bezfarebný a číry až opalizujúci a musí byť bez alebo prakticky bez viditeľných častíc (pozri časť 6.6).
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6 a pokyny uvedené na konci písomnej
informácie pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Somatropín sa nesmie použiť, ak existuje akýkoľvek dôkaz nádorovej aktivity (pozri časť 4.4).
Intrakraniálne nádory musia byť neaktívne a protinádorová liečba musí byť dokončená pred začatím
liečby rastovým hormónom. Liečba sa musí vysadiť, ak existuje dôkaz nádorového rastu.
Pacienti s akútnymi kritickými ochoreniami, ktorí utrpia komplikácie po otvorenej operácii srdca, brušnej operácii, viacnásobnej traume pri nehode, akútnom respiračnom zlyhaní alebo s podobnými stavmi nesmú byť liečení lonapegsomatropínom (pre pacientov podstupujúcich substitučnú liečbu, pozri časť 4.4).
Lonapegsomatropín sa nesmie použiť na podporu rastu u detí so zatvorenými epifýzami.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Akútne kritické ochorenie
U kriticky chorých dospelých pacientov trpiacich na komplikácie po otvorenej operácii srdca, brušnej
operácii, viacnásobnej traume pri nehode alebo akútnom respiračnom zlyhaní bola úmrtnosť vyššia
u pacientov liečených 5,3 mg alebo 8 mg somatropínu denne (t. j. 37,1 – 56 mg/týždeň) v porovnaní
s pacientmi dostávajúcimi placebo, 42 % oproti 19 %. Pretože nie sú k dispozícii žiadne informácie
o substitučnej liečbe rastovým hormónom u akútne kriticky chorých pacientov, prínosy pokračovania liečby lonapegsomatropínom v tejto situácii sa musia zvážiť oproti potenciálnym rizikám. U všetkých
pacientov, u ktorých sa vyvinie iné alebo podobné akútne kritické ochorenie, sa musí zvážiť možný prínos liečby lonapegsomatropínom v porovnaní s potenciálnym súvisiacim rizikom.
Neoplazmus
U pacientov s predchádzajúcim malígnym ochorením sa musí venovať osobitná pozornosť prejavom
a príznakom relapsu.
Pacienti s existujúcimi nádormi alebo GHD sekundárnou pri intrakraniálnej lézii sa musia pravidelne
vyšetrovať, či nedochádza k progresii alebo recidíve základného chorobného procesu.
U ľudí, ktorí v detstve prežili rakovinu, bolo hlásené riziko druhého neoplazmu u pacientov liečených
rastovým hormónom po prvom neoplazme. Intrakraniálne nádory, najmä meningiómy, boli najčastejšou formou druhého neoplazmu hláseného u pacientov liečených ožarovaním hlavy na prvý neoplazmus.
Benígna intrakraniálna hypertenzia
V prípade závažnej alebo opakovanej ataxie, bolesti hlavy, problémov s videním, nevoľnosti a/alebo
vracania sa odporúča funduskopia na papiloedém. Ak sa potvrdí papiloedém, musí sa zvážiť diagnóza
benígnej intrakraniálnej hypertenzie a ak je to vhodné, liečba rastovým hormónom sa má vysadiť.
V súčasnosti neexistujú dostatočné dôkazy na vydanie špecifického poradenstva ohľadom
pokračovania liečby rastovým hormónom u pacientov s vyliečenou intrakraniálnou hypertenziou. Ak
sa znovu začne liečba rastovým hormónom, je potrebné pozorné sledovanie, či sa nevyskytnú príznaky
intrakraniálnej hypertenzie. Funduskopické vyšetrenie sa odporúča pri nasadení a pravidelne
v priebehu liečby.
Citlivosť na inzulín
Rastový hormón môže znížiť citlivosť na inzulín. U pacientov s diabetes mellitus si dávka inzulínu
môže vyžadovať úpravu po začatí liečby lonapegsomatropínom. Pacientov s diabetes mellitus,
netoleranciou glukózy alebo ďalšími rizikovými faktormi pre diabetes mellitus je potrebné pozorne
sledovať počas liečby lonapegsomatropínom (pozri časť 4.5).
Hypoadrenalizmus
Zavedenie liečby rastovým hormónom môže mať za následok inhibíciu 11β-hydroxysteroid
dehydrogenázy typu 1 (11βHSD-1) a znížené sérové koncentrácie kortizolu. V dôsledku toho môže
byť odhalený predtým nediagnostikovaný centrálny (sekundárny) hypoadrenalizmus a môže byť
potrebná substitúcia glukokortikoidov. Okrem toho pacienti liečení substitučnou glukokortikoidovou
liečbou pre predtým diagnostikovaný hypoadrenalizmus si môžu vyžadovať zvýšenie udržiavacích
alebo nárazových dávok po začatí liečby lonapegsomatropínom (pozri časť 4.5).
Funkcia štítnej žľazy
Rastový hormón zvyšuje extratyroidálnu konverziu T4 na T3, čo môže mať za následok zníženie
sérových koncentrácií T4 a zvýšenie sérových koncentrácií T3. U všetkých pacientov sa má preto sledovať funkcia štítnej žľazy. U pacientov s hypopituitarizmom na štandardnej substitučnej liečbe sa musí pozorne sledovať potenciálny účinok liečby lonapegsomatropínom na funkciu štítnej žľazy (pozri časť 4.5 a 4.8).
Epifyzeolýza hlavice stehennej kosti
U pacientov s endokrinnými poruchami vrátane GHD môže dôjsť častejšie k epifyzeolýze hlavice
stehennej kosti než vo všeobecnej populácii. Deti s pretrvávajúcou bolesťou bedra/kolena a/alebo
s krívaním počas liečby lonapegsomatropínom sa musia klinicky vyšetriť.
Skolióza
U ktoréhokoľvek dieťaťa počas rýchleho rastu môže dôjsť k progresii skoliózy. Pretože liečba
rastovým hormónom zvyšuje rýchlosť rastu, počas liečby je potrebné sledovať prejavy a progresiu
skoliózy. Liečba rastovým hormónom však nepreukázala zvýšenie výskytu alebo závažnosti skoliózy
(pozri časť 4.8).
Pankreatitída
Hoci je to zriedkavé, ak sa u detí liečených rastovým hormónom objaví nevysvetlená bolesť brucha, je
potrebné zvážiť pankreatitídu.
Prader-Williho syndróm
Lonapegsomatropín nebol študovaný u pacientov s Prader-Williho syndrómom. Lonapegsomatropín
nie je indikovaný na dlhodobú liečbu pediatrických pacientov, ktorí majú zlyhanie rastu kvôli geneticky potvrdenému Prader-Williho syndrómu, ak nemajú zároveň diagnózu GHD. Boli hlásené prípady náhleho úmrtia po začatí liečby rastovým hormónom u pacientov s Prader-Williho
syndrómom, ktorí mali jeden alebo viac z nasledujúcich rizikových faktorov: závažná obezita,
anamnéza obštrukcie horných dýchacích ciest alebo spánkového apnoe alebo neidentifikovaná infekcia dýchacích ciest.
Leukémia
U malého počtu pacientov s GHD, z ktorých niektorí boli liečení somatropínom, bola hlásená
leukémia. Neexistujú však dôkazy, že výskyt leukémie je zvýšený u príjemcov rastového hormónu,
pokiaľ nemajú predispozičné faktory.
Použitie pri perorálnej estrogénovej liečbe
Perorálny estrogén ovplyvňuje odpoveď IGF-1 na rastový hormón. Ak pacientka, ktorá užíva
lonapegsomatropín, začne s perorálnou estrogénovou liečbou, môže byť potrebné zvýšiť dávku
lonapegsomatropínu, aby sa hladiny sérového IGF-1 udržali v normálnom rozsahu primeranom veku (pozri časť 4.2). Naopak, ak pacientka používajúca lonapegsomatropín vysadí perorálnu estrogénovú liečbu, môže byť potrebné znížiť dávku lonapegsomatropínu, aby sa predišlo nadbytku rastového hormónu a/alebo nežiaducim reakciám (pozri časť 4.5).
Protilátky
U niektorých pacientov boli sledované protilátky na lonapegsomatropín. Žiadne z týchto protilátok
neboli neutralizačné a nebol pozorovaný žiadny zjavný klinický dopad. Testovanie na prítomnosť
protilátok sa má však zvážiť u pacientov, ktorí neodpovedajú na liečbu.
4.5 Liekové a iné interakcie
Glukokortikoidová liečba
Súbežná liečba glukokortikoidmi inhibuje účinky lonapegsomatropínu podporujúce rast. Pacienti
s deficienciou adrenokortikotropného hormónu (ACTH) musia mať pozorne upravenú substitučnú
liečbu glukokortikoidmi, aby sa zabránilo akémukoľvek inhibičnému účinku na rast, a pacienti liečení
glukokortikoidmi musia mať pozorne sledovaný rast na vyhodnotenie potenciálneho dopadu liečby
glukokortikoidmi na rast.
Rastový hormón znižuje konverziu kortizónu na kortizol a môže odhaliť predtým neobjavený centrálny hypoadrenalizmus alebo spôsobiť neúčinnosť nízkych dávok glukokortikoidovej substitúcie (pozri časť 4.4).
Produkty metabolizované cytochrómom P450
Štúdie liekových interakcií neboli vykonané s lonapegsomatropínom. Údaje z interakčných štúdií so
somatropínom vykonaných u detí a dospelých s deficienciou rastového hormónu a u zdravých starších mužov naznačujú, že podávanie somatropínu môže zvýšiť klírens zložiek, o ktorých sa vie, že sú metabolizované izoenzýmami cytochrómu P450, najmä CYP3A a CYP1A2. Klírens zložiek metabolizovaných prostredníctvom CYP3A4 (napríklad pohlavné steroidy, kortikosteroidy, antikonvulzíva a cyklosporín) a CYP1A2 (napríklad teofylín) môže byť zvýšený a mať za následok zníženú expozíciu týchto zložiek. Klinická závažnosť týchto zistení nie je známa.
Inzulín a/alebo iné hypoglykemické látky
U pacientov s diabetes mellitus vyžadujúcich farmakologickú liečbu (napr. antihyperglykemické
lieky) si dávka inzulínu a/alebo perorálneho hypoglykemického lieku môže vyžadovať úpravu po
začatí liečby lonapegsomatropínom (pozri časť 4.4).
Hormóny štítnej žľazy
Pretože rastový hormón zvyšuje extratyroidálnu konverziu T4 na T3, môže byť potrebná úprava
substitučnej liečby hormónmi štítnej žľazy (pozri časť 4.4).
Perorálna estrogénová liečba
U pacientok na perorálnej estrogénovej liečbe sa môže vyžadovať vyššia dávka rastového hormónu na
dosiahnutie liečebného cieľa (pozri časť 4.2 a 4.4).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití lonapegsomatropínu
u gravidných žien. Publikované štúdie s použitím krátkodobo účinkujúceho somatropínu u gravidných žien v priebehu niekoľkých desaťročí neidentifikovali žiadne riziká závažných vrodených chýb, potratov alebo nežiaducich výsledkov pre matku alebo plod v spojitosti s liekom.
Štúdie na zvieratách sú nedostatočné z hľadiska reprodukčnej toxicity (pozri časť 5.3). Lonapegsomatropin Ascendis Pharma sa neodporúča podávať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Dojčenie
Nie sú k dispozícii žiadne údaje o prítomnosti lonapegsomatropínu v ľudskom mlieku alebo účinku na
dojčených novorodencov/deti. Pretože lonapegsomatropín sa nevstrebáva perorálne, je
nepravdepodobné, že by negatívne ovplyvnil dojčených novorodencov/deti.
Lonapegsomatropin Ascendis Pharma sa môže používať počas laktácie len na základe prísnej indikácie.
Fertilita
O účinkoch lonapegsomatropínu na fertilitu neexistujú žiadne klinické údaje. Štúdie na zvieratách sú
nedostatočné z hľadiska fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Lonapegsomatropín nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá
a obsluhovať stroje.
4
.8 Nežiaduce účinky
Z
h
r
nu
t
i
e bezpečnostného profiluNajčastejšie hlásené nežiaduce reakcie v klinických skúšaniach s lonapegsomatropínom boli bolesť
hlavy (11,1 %), artralgia (4,6 %), sekundárna hypotyreóza (2,6 %) a reakcie v mieste podania injekcie
(1,6 %). Tieto reakcie boli vo všeobecnosti prechodné a závažnosť bola mierna až stredná.
Tabuľkový zoznam nežiaducich reakciíTabuľka 3 nižšie zobrazuje nežiaduce reakcie, ktoré sa vyskytli počas liečby lonapegsomatropínom.
Nežiaduce reakcie sú zoradené podľa názvov tried orgánových systémov MedDRA a frekvencie
pomocou nasledujúcej terminológie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme
(z dostupných údajov).
Tabuľka 3 Frekvencia nežiaducich reakcií v klinických skúšaniachTrieda systémových orgánov
| Veľmi časté
| Časté
| Menej časté
|
Poruchy endokrinného systému
|
| Sekundárna hypotyreóza
| Sekundárna adrenokortikálna insuficiencia
|
Poruchy nervového systému
| Bolesť hlavy
|
|
|
Poruchy kostrovej
a svalovej sústavy a spojivového tkaniva
|
| Artralgia
| Skolióza
Artritída
Rastové bolesti
|
Poruchy reprodukčného
systému a prsníkov
|
|
| Gynekomastia
|
Celkové poruchy a reakcie v mieste podania
|
| Reakcie v mieste podania injekciea
|
|
a Reakcie v mieste podania injekcie zahŕňajú hyperémiu, atrofiu miesta podania injekcie, bolesť
v mieste podania injekcie, urtikáriu v mieste podania injekcie a miestny opuch. Reakcie v mieste
podania injekcie pozorované s lonapegsomatropínom boli všeobecne mierne a prechodné.
Popis vybraných nežiaducich reakciíImunogenitaPacienti si môžu vytvárať protilátky na lonapegsomatropín. Podiel pacientov, ktorí mali pozitívny test
na detegovateľné viažuce protilátky kedykoľvek počas liečby, bol nízky (6,3 %) a žiadni pacienti
nemali neutralizačné protilátky. Nebola pozorovaná žiadna zjavná korelácia protilátok viažucich
anti-lonapegsomatropín s nežiaducimi udalosťami alebo stratou účinnosti. V prípade inak
nevysvetlenej nedostatočnej odpovede na liečbu lonapegsomatropínom je potrebné zvážiť testovanie
na protilátky na lonapegsomatropín (pozri časť 4.4).
Nežiaduce reakcie spojené s farmakologickou triedou rastového hormónuOkrem vyššie spomínaných nežiaducich liekových reakcií boli hlásené aj reakcie uvedené nižšie
s inými liekmi obsahujúcimi rastové hormóny. Frekvencie týchto nežiaducich udalostí nemožno
odhadnúť z dostupných údajov (ak nie je uvedené inak).
• Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy): leukémia
(pozri časť 4.4).
• Poruchy metabolizmu a výživy: diabetes mellitus 2. typu (pozri časť 4.4).
• Poruchy nervového systému: benígna intrakraniálna hypertenzia (pozri časť 4.4), parestézia.
• Poruchy kostrovej a svalovej sústavy a spojivového tkaniva: myalgia.
• Poruchy reprodukčného systému a prsníkov: gynekomastia (frekvencia: menej časté).
• Poruchy kože a podkožného tkaniva: kožná vyrážka, urtikária a pruritus.
• Celkové poruchy a reakcie v mieste podania: periférny edém, edém tváre.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyAkútne predávkovanie môže spočiatku viesť k hypoglykémii a následne k hyperglykémii. Dlhodobé
predávkovanie môže mať za následok prejavy a príznaky gigantizmu.
ManažmentLiečba je symptomatická a podporná. Na predávkovanie somatropínom nie je žiadne antidotum.
Po predávkovaní sa odporúča sledovať funkciu štítnej žľazy.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Hormóny a analógy hypotalamu a hypofýzy, somatropín a agonisty somatropínu, ATC kód: zatiaľ neurčený.
Mechanizmus účinkuLonapegsomatropín je „prekurzor“ somatropínu s dlhodobým pôsobením. Lonapegsomatropín
obsahuje pôvodné liečivo somatropín, ktorý je prechodne konjugovaný na metoxypolyetylénglykolový nosič (4 x 10 kDa mPEG) pomocou patentového linkera TransCon. Nosič má tieniaci účinok, ktorý minimalizuje vylučovanie obličkami a receptorom mediovaný klírens lonapegsomatropínu. Po subkutánnom podaní lonapegsomatropín uvoľňuje plne aktívny somatropín pomocou autoštiepenia linkera TransCon. Somatropín (191 aminokyselín) má taký istý mechanizmus účinku a distribúciu ako
denný somatropín, no v subkutánnej injekcii raz týždenne.
Somatropín sa viaže k dimérickému receptoru hGH v bunkovej membráne cieľových buniek, čo má za následok intracelulárnu transdukciu signálu a viacero farmakodynamických účinkov. Somatropín má priame účinky na tkanivo a metabolizmus a nepriame účinky mediované IGF-1 vrátane stimulácie diferenciácie a proliferácie chondrocytov, stimulácie tvorby pečeňovej glukózy, syntézy a lipolýzy proteínu. Somatropín stimuluje rast kostí u pediatrických pacientov s GHD v dôsledku účinkov na rastové platničky (epifýzy) kostí.
Farmakodynamické účinkySomatropín uvoľňovaný z lonapegsomatropínu produkuje lineárnu odpoveď IGF-1 na dávku, so
zmenou dávky 0,02 mg somatropínu/kg, čo má za následok približnú zmenu v priemernom týždennom
skóre štandardnej odchýlky (SDS) IGF-1 s hodnotou 0,17.
V rovnovážnom stave hladiny SDS IGF-1 dosiahli najvyššie hodnoty približne 2 dni po dávke
s priemerným týždenným SDS IGF-1 dosiahnutým približne 4,5 dní po dávke (Obrázok 1). Hladiny SDS IGF-1 boli v normálnom rozsahu pre pacientov s GHD väčšinu týždňa, podobne ako pri dennom somatropíne.
Obrázok 1 Stredné (±SE) SDS IGF-1 v rovnovážnom stave u detí s GHD po podaní lonapegsomatropínu raz týždenne 0,24 mg somatropínu/kg/týždeň
1,0 1,0
0,5
0,5
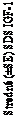
0,0
0,0
-0,5
-0,5
-1,0
-1,0
-1,5
-1,5
-2,0
-2,5
-3,0
Základná hodnota
pred začiatkom liečby (základná hodnota) do 13. dávky
(čas 0)
čas (h) po 13. dávke
Čas (h)
-2,0
-2,5
-3,0
 Kl
i
n
i
ck
á
ú
činnosť
a
b
ezpečnosť
Kl
i
n
i
ck
á
ú
činnosť
a
b
ezpečnosť
Účinnosť a bezpečnosť lonapegsomatropínu raz týždenne boli hodnotené v klinických skúšaniach
3. fázy, ktoré zahŕňali 306 pediatrických pacientov s GHD.
V 52-týždňovom multicentrickom randomizovanom, nezaslepenom, aktívne kontrolovanom klinickom skúšaní 3. fázy s paralelnými skupinami bolo randomizovaných 161 neliečených, predpubertálnych pacientov s GHD na lonapegsomatropín raz týždenne (N = 105) alebo denný somatropín (N = 56), obidve skupiny na celkovú týždennú dávku 0,24 mg somatropínu/kg. Pacienti boli vo veku od 3,2 do
13,1 roka so stredným vekom 8,5 roka. Väčšina subjektov (N = 132 (82 %)) boli chlapci. Pacienti mali
SDS strednej základnej výšky -2,93. Primárnym koncovým bodom účinnosti bola ročná rýchlosť rastu do výšky (AHV) v 52. týždni. Liečba lonapegsomatropínom raz týždenne po dobu 52 týždňov mala za následok, že hodnota AHV nebola horšia v porovnaní s denným somatropínom (tabuľka 4). Zmeny hodnôt skóre štandardnej odchýlky (SDS) výšky (zmena od základnej hodnoty) mali tiež tendenciu sa zvyšovať pre denný lonapegsomatropín pri porovnaní s denným somatropínom (tabuľka 4). Zmeny AHV a SDS výšky mali tendenciu sa zvyšovať pre lonapegsomatropín v porovnaní s hodnotami pri somatropíne od 26. týždňa do konca skúšania v 52. týždni.
Stredný pomer (SD) veku kosti k chronologickému veku postupoval podobne v oboch ramenách od základnej hodnoty do 52. týždňa: 0,69 (0,16) až 0,75 (0,15) s lonapegsomatropínom raz týždenne
a 0,70 (0,14) až 0,76 (0,14) s denným somatropínom.
Tabuľka 4 Rast a odpoveď IGF-1 v 52. týždni u pediatrických neliečených pacientov s GHD(analýza zamýšľanej liečby)
| Lonapegsomatropín raz týždenne (N = 105) (0,24 mg
somatropínu/kg/týždeň)
| Denný somatropín
(N = 56) (0,24 mg somatropínu/kg/týždeň)
| Odhad rozdielu v liečbe (lonapegsomatropín
mínus somatropín)
|
AHV (cm/rok)a, stredný LS (95 %
CI)
| 11,2 (10,7 – 11,6)
| 10,3
(9,7 – 10,9)
| 0,9b
(0,2 – 1,5)
|
SDS výšky, zmena od základnej hodnotyc, stredný LS (95 % CI)
| 1,10 (1,02 – 1,18)
| 0,96 (0,85 – 1,06)
| 0,14d
(0,03 – 0,26)
|
Kategória SDS IGF-1e, %
< 0
0 až +2
+2 až +3
> +3
|
23,1 %
69,2 %
7,7 %
0
|
40,7 %
57,4 %
1,9 %
0
|
Neanalyzované
|
a AHV: Odhady stredného LS a 95 % CI sú z modelu ANCOVA, ktorý zahŕňal základný vek, vrcholové hladiny rastového hormónu (logaritimicky transformované) pri stimulačnom teste, základné SDS výšky – priemerné SDS rodičovskej výšky ako kovariáty a liečbu a pohlavie ako faktory. Chýbajúce údaje sú imputované viacerými imputačnými metódami.
b p = 0,0088 (2-stranný) pre nadradenosť
c SDS výšky, zmena od základnej hodnoty: Odhady stredného LS a 95 % CI sú z modelu ANCOVA, ktorý zahŕňal základný vek, vrcholové hladiny rastového hormónu (logaritimicky transformované) pri stimulačnom teste, základné SDS výšky ako kovariáty, a liečbu a pohlavie ako faktory.
d p = 0,0149 (2-stranný)
e Priemerná hladina v 52. týždni
V nezaslepenom predĺženom období pacienti, ktorí pokračovali v liečbe lonapegsomatropínom, mali zvýšenie SDS výšky o 1,61 od základnej hodnoty do 104. týždňa. Pacienti, ktorí prešli z denného somatropínu na lonapegsomatropín v 52. týždni, mali zvýšenie SDS výšky o 1,49 od základnej hodnoty do 104. týždňa.
Podporné dôkazyDôkazy z ďalších klinických skúšaní s lonapegsomatropínom podporujú dlhodobú klinickú účinnosť
liečby lonapegsomatropínom.
V 26-týždňovom nezaslepenom klinickom skúšaní s jedným ramenom hodnotiacom lonapegsomatropín 0,24 mg somatropínu/kg/týždeň u 146 pediatrických pacientov s GHD vo veku od
1 do 17 rokov, z ktorých 143 dostávalo predchádzajúcu liečbu denným somatropínom v priemere (SD)
1,1 (0,7) roka, stredná (SD) ročná rýchlosť rastu do výšky bola 9 (2,7) cm/rok a stredná (SD) zmena
od základnej hodnoty SDS výšky v skúšaní bola 0,28 (0,25). Preferencia pacienta a opatrovateľa boli
vyhodnotené v 13. týždni. 84 % pacientov a 90 % opatrovateľov uprednostňovalo lonapegsomatropín
raz týždenne oproti predchádzajúcemu dennému somatropínu.
Priemerná kategória SDS IGF-1
|
Základná hodnota
(N = 143)
n (%)
|
26
. týždeň (N = 139) n (%)
|
< 0'
|
37 (25,9)
|
13 (9,4)
|
0 až +2
|
74 (51,7)
|
71 (51,1)
|
+2 až +3
|
27 (18,9)
|
33 (23,7)
|
> +3
|
5 (3,5)
|
22 (15,8)
|
|
|
Tabuľka 5 Priemerné hladiny SDS IGF-1 pri základnej hodnote a v 26. týždni u pediatrických pacientov s GHD s predchádzajúcou liečbou (analýza zamýšľanej liečby)
5
.2 Farmakokinetické vlastnosti
Farmakokinetika po podaní lonapegsomatropínu bola hodnotená po jednej dávke u celkom
73 zdravých dospelých v 2 skúšaniach. Okrem toho farmakokinetika u pediatrických pacientov s GHD bola hodnotená na základe intenzívneho odberu vzoriek v 13. týždni u 11 účastníkov a rozptýleného odberu vzoriek u 109 účastníkov v 2 skúšaniach. Demografické informácie sú uvedené v tabuľke 6 pre subjekty zahrnuté vo farmakokinetickom hodnotení lonapegsomatropínu.
Tabuľka 6 Demografické údaje účastníkov vo farmakokinetickom hodnotení Kategória
| Zdraví dospelí
| Deti s GHD
| N
| 73
| 109
| Muž/žena
| 55/19
| 87/22
| Americký Indián alebo pôvodný obyvateľ
Aljašky
| 0
| 0
| Ázijec
| 10
| 1
| Černoch alebo Afroameričan
| 13
| 2
| Pôvodný obyvateľ Havaja alebo iného
tichomorského ostrova
|
0
|
0
| Beloch
| 49
| 104 (11 s intenzívnym
odberom FK vzoriek)
| Iná rasa/viaceré
| 1
| 2
| Hispánec alebo Latinoameričan
| 23
| 5
| Nie hispánskeho alebo latinskoamerického
pôvodu
| 50
| 104
|
|
|
lonapegsomatropínu AbsorpciaPo subkutánnom podaní dávky lonapegsomatropín uvoľňuje somatropín kontrolovaným spôsobom,
ktorý nasleduje kinetiku prvého radu.
U pediatrických pacientov s GHD po subkutánnom podaní dávky lonapegsomatropínu 0,24 mg
somatropínu/kg/týždeň bola pozorovaná stredná (CV %) vrcholová sérová koncentrácia
v rovnovážnom stave (Cmax) lonapegsomatropínu 1 230 (86,3) ng somatropínu/ml pri strednom čase
Tmax 25 hodín a pre uvoľnený somatropín bola hodnota Cmax 15,2 (83,4) ng/ml pri strednom čase do
dosiahnutia Cmax 12 hodín. Stredná (CV %) expozícia somatropínu počas jednotýždňového intervalu
dávky (plocha pod krivkou) bola 500 (83,8) h*ng/ml. Akumulácia lonapegsomatropínu alebo
somatropínu po opakovanom podaní dávok sa nepozorovala.
U pediatrických pacientov s GHD sa injekcie striedali medzi bruchom, zadkom a stehnom.
Nepozorovala sa žiadna zjavná spojitosť medzi miestom podania a expozíciou somatropínu.
Absolútna biologická dostupnosť lonapegsomatropínu po subkutánnom podaní dávky nebola
skúmaná.
Distribúcia
U pediatrických pacientov s GHD bol stredný (CV %) zdanlivý distribučný objem
lonapegsomatropínu v rovnovážnom stave po subkutánnom podaní 0,24 mg somatropínu/kg/týždeň
0,13 (109) l/kg. Očakáva sa, že somatropín uvoľnený z lonapegsomatropínu má podobný distribučný
objem ako endogénny rastový hormón.
Eliminácia
Metabolizmus
Metabolická premena somatropínu zahŕňa katabolizmus proteínov v pečeni aj obličkách.
Vylučovanie
U pediatrických pacientov s GHD bol stredný (CV %) zdanlivý klírens lonapegsomatropínu
v rovnovážnom stave po subkutánnom podaní 0,24 mg somatropínu/kg/týždeň 3,2 (67) ml/h/kg so stredným (±SD) pozorovaným polčasom rozpadu 30,7 (±12,7) hodiny. Zdanlivý polčas rozpadu somatropínu uvoľneného z lonapegsomatropínu bol približne 25 hodín.
Osobitné populácie
S lonapegsomatropínom sa nevykonali žiadne farmakokinetické štúdie špecifické pre pohlavie.
Dostupná literatúra naznačuje, že farmakokinetika somatropínu je podobná u mužov aj žien.
Na základe farmakokinetickej analýzy populácie, vek, pohlavie, rasa/etnická príslušnosť a telesná
hmotnosť nemajú klinicky významný účinok na farmakokinetiku.
S lonapegsomatropínom sa nevykonali žiadne štúdie u pacientov s poruchou funkcie obličiek alebo pečene (pozri časť 4.2). Znížený klírens somatropínu po podaní denného somatropínu sa pozoroval u pacientov so závažnou dysfunkciou pečene a obličiek. Klinická závažnosť tohto zníženia nie je
známa. Očakáva sa, že farmakokinetika nosiča mPEG lonapegsomatropínu bude závisieť od funkcie
obličiek, no nebola hodnotená u pacientov s poruchou funkcie obličiek.
Lonapegsomatropín nebol študovaný u pacientov mladších než 6 mesiacov (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe farmakologických štúdií bezpečnosti, toxicity po opakovanom
podávaní, genotoxicity a karcinogenicity neodhalili žiadne osobitné riziko pre ľudí.
Reprodukčné toxikologické štúdie vykonané u potkanov a histopatologické hodnotenie reprodukčných orgánov u opíc, ktorým bol subkutánne podaný lonapegsomatropín v dávkach až 20-násobne vyšších než klinická dávka 0,24 mg somatropínu/kg/týždeň, nevyvolali nežiaduce účinky na samčiu a samičiu fertilitu ani na reprodukčné orgány. Z dôvodu tvorby protilátok narúšajúcich expozíciu u potkanov sa nemohol vytvoriť pevný záver ohľadom relevantnosti pre ľudskú fertilitu.
U potkanov sa po podaní subkutánneho lonapegsomatropínu v dávkach až 13-násobne vyšších než klinická dávka 0,24 mg somatropínu/kg/týždeň nevyskytli žiadne embryofetálne vývojové toxicity. Z dôvodu prerušovanej expozície sa nemohol vytvoriť pevný záver ohľadom embryofetálnej vývojovej štúdie u potkanov.
Embryofetálna vývojová štúdia toxicity u králikov preukázala abnormality plodu a embryofetálnu úmrtnosť pri 1,5-násobku a 6-násobku klinickej dávky 0,24 mg somatropínu/kg/týždeň, čo mohlo byť spôsobené materskou toxicitou. Klinická závažnosť týchto zistení je neistá.
V prenatálnych a postnatálnych vývojových štúdiách u potkanov neboli žiadne nežiaduce účinky na gravidné/dojčiace samice ani na vývoj zárodku a potomstva po expozícii samice od implantácie po odstavenie subkutánnym dávkam štrukturálne príbuzného prechodne pegylovaného prekurzora somatropínu až 13-násobne vyšším dávkam než klinická dávka 0,24 mg somatropínu/kg/týždeň.
Expozícia mPEG
Po roku expozície pri výške približne 10-násobku expozície ľudí komponentu mPEG
lonapegsomatropínu sa objavuje vakuolizácia v epiteliálnych bunkách choroid plexus (CP) makakov dlhochvostých. Približne pri výške 34-násobku expozície ľudí komponentu mPEG bol pozorovaný mierny nárast počtu zvierat s vakuolami v epiteliálnych bunkách CP opíc. Vakuolizácia buniek sa považuje za adaptačnú reakciu. Nepovažuje sa preto za možný nepriaznivý účinok u ľudí pri liečebnej dávke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
Kyselina jantárová
Dihydrát trehalózy
Trometamol
Rozpúšťadlo
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorené
3 roky pri uchovávaní v chladničke (2 °C – 8 °C).
Prípadne možno Lonapegsomatropin Ascendis Pharma uchovávať pri teplotách ≤ 30 °C maximálne
6 mesiacov. Do 6 mesiacov možno liek vrátiť do chladničky (2 °C – 8 °C).
Zaznamenajte dátum na škatuľke, kedy bol liek po prvýkrát vybratý z chladničky. Liek zlikvidujte po
uplynutí 6 mesiacov.
Po rekonštitúcii
Chemická a fyzikálna stabilita pri použití bola preukázaná pre rekonštituovaný liek uchovávaný
4 hodiny pri teplotách ≤ 30 °C.
Z mikrobiologického hľadiska by sa mal liek použiť ihneď po rekonštitúcii. Ak sa nepoužije ihneď, používateľ preberá zodpovednosť za skladovacie časy a podmienky pri použití, ktoré nesmú prekročiť
4 hodiny pri teplotách ≤ 30 °C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Pre alternatívne podmienky na uchovávanie pri teplotách ≤ 30 °C, pozri časť 6.3.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Sklená náplň (sklo typu 1) s dvomi komorami oddelenými gumovou zátkou (brómobutyl). Náplň je uzavretá gumovou zátkou (brómobutyl) na jednom konci a gumovým uzatváracím diskom (brómobutyl) na druhom konci. Náplň je nasadená na plastový adaptér ihly.
Každé balenie obsahuje 4 dvojkomorové náplne na jedno použitie zabalené v individuálnych blistroch a 6 jednorazových injekčných ihiel 0,25 mm x 4 mm (31 G x 5/32 palcov). Každá dvojkomorová
náplň má špecifické označenie s priradenými dvojfarebne kódovanými prúžkami, ktoré používa len
autoinjektor na výber správnych nastavení rekonštitúcie. Farby sily sú vyznačené na škatuľke
a blistrovej fólii a majú sa používať na odlíšenie jednotlivých síl.
Lonapegsomatropin Ascendis Pharma 3 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 3 mg somatropínu vo forme prášku v prvej komore a 0,279 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je žlté/zelené. Farba sily
na škatuľke a blistri je svetlooranžová.
Lonapegsomatropin Ascendis Pharma 3,6 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 3,6 mg somatropínu vo forme prášku v prvej komore a 0,329 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je žlté/azúrovo modré.
Farba sily na škatuľke a blistri je azúrovo modrá.
Lonapegsomatropin Ascendis Pharma 4,3 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 4,3 mg somatropínu vo forme prášku v prvej komore a 0,388 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je žlté/ružové. Farba sily
na škatuľke a blistri je tmavosivá.
Lonapegsomatropin Ascendis Pharma 5,2 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 5,2 mg somatropínu vo forme prášku v prvej komore a 0,464 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je zelené/ružové. Farba
sily na škatuľke a blistri je žltá.
Lonapegsomatropin Ascendis Pharma 6,3 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 6,3 mg somatropínu vo forme prášku v prvej komore a 0,285 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je azúrovo modré/žlté.
Farba sily na škatuľke a blistri je oranžová.
Lonapegsomatropin Ascendis Pharma 7,6 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 7,6 mg somatropínu vo forme prášku v prvej komore a 0,338 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je azúrovo modré/ružové.
Farba sily na škatuľke a blistri je tmavofialová.
Lonapegsomatropin Ascendis Pharma 9,1 mg prášok a
rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 9,1 mg somatropínu vo forme prášku v prvej komore a 0,4 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je ružové/žlté. Farba sily
na škatuľke a blistri je zlatohnedá.
Lonapegsomatropin Ascendis Pharma 11 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 11 mg somatropínu vo forme prášku v prvej komore a 0,479 ml
rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je ružové/zelené. Farba
sily na škatuľke a blistri je tmavomodrá.
Lonapegsomatropin Ascendis Pharma 13,3 mg prášok a rozpúšťadlo na injekčný roztok v náplni
Každá dvojkomorová náplň obsahuje 13,3 mg somatropínu vo forme prášku v prvej komore
a 0,574 ml rozpúšťadla v druhej komore. Dvojfarebné označenie náplne (spodok/vrch) je
ružové/azúrovo modré. Farba sily na škatuľke a blistri je tmavočervená.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Manipulácia
Ak sa uchováva v chladničke, nechajte vytemperovať pri izbovej teplote 15 minút pred použitím.
Každá dvojkomorová náplň Lonapegsomatropin Ascendis Pharma obsahujúca prášok a rozpúšťadlo na injekčný roztok je určená len na jedno použitie a musí sa použiť len s dodanými injekčnými ihlami
a autoinjektorom GH Auto-Injector. Autoinjektor GH Auto-Injector nie je zahrnutý v tomto balení. Prášok na injekčný roztok sa musí rekonštituovať s dodaným rozpúšťadlom pomocou autoinjektora GH Auto-Injector po pripevnení ihly k dvojkomorovej náplni.
Rekonštituovaný roztok má byť bezfarebný a číry až opalizujúci a musí byť bez alebo prakticky bez viditeľných častíc. Roztok môže občas obsahovať vzduchové bubliny. Ak roztok obsahuje častice, nesmie sa injekčne podať.
Po rekonštitúcii sa Lonapegsomatropin Ascendis Pharma podáva subkutánne (s automatickým dávkovaním) pomocou autoinjektora GH Auto-Injector.
Lonapegsomatropin Ascendis Pharma sa dávkuje ako plná jedna dávka (celkové použitie).
Pred použitím si prečítajte pokyny na použitie na prípravu Lonapegsomatropin Ascendis Pharma uvedené na konci písomnej informácie pre používateľa a pokyny na použitie dodané s autoinjektorom GH Auto-Injector.
Likvidácia
Pacientovi musí byť nariadené, aby zlikvidoval náplň aj injekčnú ihlu po každej injekcii. Všetok
nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Ascendis Pharma Endocrinology Division A/S Tuborg Boulevard 12
DK-2900 Hellerup
Dánsko
8
. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/21/1607/001
EU/1/21/1607/002
EU/1/21/1607/003
EU/1/21/1607/004
EU/1/21/1607/005
EU/1/21/1607/006
EU/1/21/1607/007
EU/1/21/1607/008
EU/1/21/1607/009
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU