
>
U starších pacientov je viac pravdepodobné, že majú zníženú funkciu obličiek. Vzhľadom na to, že sa abnormality funkcie obličiek môžu vyskytnúť po začatí liečby ertugliflozínom a o metformíne je
známe, že sa vylučuje obličkami, Segluromet sa má u starších pacientov používať s opatrnosťou.
Pravidelné monitorovanie funkcie obličiek je potrebné na to, aby napomohlo zabrániť laktátovej acidóze spojenej s metformínom, najmä u starších pacientov (pozri časť 4.4). Treba vziať do úvahy funkciu obličiek a riziko deplécie objemu (pozri časti 4.4 a 4.8).
Skúsenosť so Seglurometom u pacientov vo veku ≥ 75 rokov je obmedzená.
Pediatrická populácia
Bezpečnosť a účinnosť Seglurometu u detí vo veku do 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Segluromet sa má užívať perorálne dvakrát denne s jedlom, aby sa obmedzili gastrointestinálne nežiaduce reakcie spojené s metformínom. V prípade ťažkostí s prehĺtaním je možné tabletu prelomiť alebo podrviť, pretože ide o formu s okamžitým uvoľňovaním dávky.
4.3 Kontraindikácie
- precitlivenosť na liečivá alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1;
- akýkoľvek typ akútnej metabolickej acidózy (ako je laktátová acidóza, diabetická ketoacidóza
[DKA]);
- diabetická prekóma;
- závažné zlyhávanie obličiek (GFR menej ako 30 ml/min), terminálne štádium ochorenia obličiek (end-stage renal disease, ESRD) alebo u dialyzovaných pacientov (pozri časť 4.4);
- akútny stav, ktorý môže zmeniť funkciu obličiek, ako je:
- dehydratácia,
- závažná infekcia,
- šok;
- akútne alebo chronické ochorenie, ktoré môže spôsobiť hypoxiu tkaniva, ako je:
- zlyhanie srdca alebo dýchania,
- nedávny infarkt myokardu,
- šok;
- porucha funkcie pečene;
- akútna otrava alkoholom, alkoholizmus.
4.4 Osobitné upozornenia a opatrenia pri používaní
Všeobecné
Segluromet sa nemá používať u pacientov s diabetes mellitus 1. typu.
Laktátováacidóza
Laktátová acidóza, veľmi zriedkavá, ale závažná metabolická komplikácia, sa najčastejšie vyskytuje pri akútnom zhoršení funkcie obličiek alebo pri kardiorespiračnom ochorení, či sepse. Akumulácia
metformínu sa objavuje pri akútnom zhoršení funkcie obličiek a zvyšuje riziko laktátovej acidózy.
V prípade dehydratácie (závažné vracanie, hnačka, horúčka alebo znížený príjem tekutín) je potrebné dočasne prerušiť podávanie metformínu a odporúča sa kontaktovať zdravotníckeho pracovníka.
Lieky, ktoré môžu akútne narušiť funkciu obličiek (ako napríklad antihypertenzíva, diuretiká
a nesteroidové protizápalové lieky [non-steroidal anti-inflammatory drugs, NSAIDs]) sa majú u pacientov liečených metformínom začať podávať s opatrnosťou. Ďalšie rizikové faktory pre laktátovú acidózu sú nadmerné požívanie alkoholu, hepatálna insuficiencia, nedostatočne kontrolovaný diabetes, ketóza, dlhotrvajúce hladovanie a akékoľvek stavy spojené s hypoxiou, ako aj súbežné užívanie liekov, ktoré môžu spôsobiť laktátovú acidózu (pozri časti 4.3 a 4.5).
Pacienti a/alebo opatrovatelia musia byť informovaní o riziku laktátovej acidózy. Laktátová acidóza je charakterizovaná acidotickou dyspnoe, bolesťou brucha, svalovými kŕčmi, asténiou a hypotermiou, po ktorých nasleduje kóma. V prípade podozrenia na výskyt príznakov musí pacient prestať užívať metformín a vyhľadať okamžitú lekársku pomoc. Diagnostickými laboratórnymi nálezmi sú znížená hodnota pH krvi (< 7,35), zvýšené plazmatické hladiny laktátu (> 5 mmol/l) a zvýšená aniónová medzera a pomer laktátu/pyruvátu.
Podávaniejódovanýchkontrastných látok
Intravaskulárne podávanie jódových kontrastných látok môže viesť k nefropatii vyvolanej kontrastnou látkou, čo spôsobuje akumuláciu metformínu a zvýšené riziko laktátovej acidózy. Pred zobrazovacím
vyšetrením alebo v čase zobrazovacieho vyšetrenia je potrebné prerušiť podávanie lieku Segluromet
a v podávaní sa nesmie pokračovať skôr, ako po uplynutí minimálne 48 hodín, za predpokladu, že funkcia obličiek bola opätovne posúdená a považovaná za stabilnú (pozri časti 4.2 a 4.5).
Funkciaobličiek
Účinnosť ertugliflozínu závisí od funkcie obličiek a účinnosť je znížená u pacientov so stredne závažnou poruchou funkcie obličiek a pravdepodobne chýba u pacientov so závažnou poruchou
funkcie obličiek (pozri časť 4.2).
Liečba Seglurometom sa nemá začínať u pacientov s hodnotou GFR nižšou ako 60 ml/min. Segluromet sa má vysadiť kvôli zníženej účinnosti, ak GFR pretrváva na hodnote nižšej ako
45 ml/min.
GFR sa má posúdiť pred začiatkom liečby Seglurometom, a potom pravidelne počas liečby (pozri časť
4.2). Častejšie sledovanie funkcie obličiek sa odporúča u pacientov s hodnotou GFR nižšou ako
60 ml/min. Metformín je kontraindikovaný u pacientov s hodnotou GFR < 30 ml/min a má sa dočasne vysadiť pri výskyte stavov, ktoré menia funkciu obličiek (pozri časť 4.3).
Chirurgickýzákrok
Segluromet sa musí vysadiť v čase chirurgického zákroku s celkovou, spinálnou alebo epidurálnou anestéziou. Liečba sa nesmie opätovne nasadiť skôr ako 48 hodín po chirurgickom zákroku alebo po
obnovení perorálnej výživy a za predpokladu, že došlo k opätovnému vyhodnoteniu funkcie obličiek a po potvrdení, že je stabilná.
Hypotenzia/depléciaobjemu
Ertugliflozín spôsobuje osmotickú diurézu, ktorá môže viesť k zníženiu intravaskulárneho objemu.
Z tohto dôvodu sa po začatí liečby Seglurometom môže vyskytnúť symptomatická hypotenzia (pozri časť 4.8), najmä u pacientov s poruchou funkcie obličiek (eGFR nižšia ako 60 ml/min/1,73 m2 alebo CrCl nižšia ako 60 ml/min), u starších pacientov (≥ 65 rokov), u pacientov užívajúcich diuretiká alebo u pacientov podstupujúcich liečbu hypertenzie s hypotenziou v anamnéze. Pred začatím liečby Seglurometom sa má vyhodnotiť stav objemu a v prípade indikácie sa má upraviť. Po začatí liečby je potrebné sledovať prejavy a príznaky.
Na základe svojho mechanizmu účinku, ertugliflozín indukuje osmotickú diurézu, zvyšuje hladinu kreatinínu v sére a znižuje eGFR. Zvýšenia hladiny kreatinínu v sére a poklesy eGFR boli väčšie
u pacientov so stredne závažnou poruchou funkcie obličiek (pozri časť 4.8).
V prípade stavov, ktoré môžu viesť k strate tekutín (napr. gastrointestinálne ochorenie), sa u pacientov liečených ertugliflozínom odporúča dôkladné sledovanie stavu objemu (napr. fyzikálne vyšetrenie, merania krvného tlaku, laboratórne vyšetrenia vrátane hematokritu) a elektrolytov. Má sa zvážiť dočasné prerušenie liečby Seglurometom dovtedy, kým nedôjde k úprave straty tekutín.
Diabetickáketoacidóza
U pacientov liečených inhibítormi sodík-glukózového kotransportéra 2 (sodium glucose
co-transporter-2, SGLT2) boli v klinických skúšaniach a po uvedení lieku na trh hlásené zriedkavé prípady diabetickej ketoacidózy (diabetic ketoacidosis, DKA), vrátane život ohrozujúcich a fatálnych
prípadov. Prípady boli hlásené v klinických skúšaniach s ertugliflozínom. V mnohých prípadoch bol
prejav tohto stavu atypický, s len mierne zvýšenými hodnotami glukózy v krvi, do 14 mmol/l
(250 mg/dl). Nie je známe, či sa DKA vyskytuje s vyššou pravdepodobnosťou pri vyšších dávkach ertugliflozínu.
Riziko výskytu diabetickej ketoacidózy sa musí zvážiť v prípade nešpecifických príznakov, ako sú nauzea, vracanie, anorexia, bolesť brucha, nadmerný smäd, ťažkosti s dýchaním, zmätenosť, neobvyklá únava alebo ospalosť. Ak sa vyskytnú tieto príznaky, pacientov treba okamžite vyšetriť na ketoacidózu, bez ohľadu na hladinu glukózy v krvi.
U pacientov so suspektnou alebo diagnostikovanou DKA sa má liečba Seglurometom okamžite ukončiť.
Liečba sa má prerušiť u pacientov hospitalizovaných kvôli veľkým chirurgickým zákrokom alebo akútnym závažným ochoreniam. V obidvoch prípadoch je možné liečbu Seglurometom obnoviť po stabilizovaní stavu pacienta.
Pred začatím liečby Seglurometom sa majú zvážiť faktory v anamnéze pacienta, ktoré ho môžu predisponovať ku ketoacidóze.
Pacienti, u ktorých môže byť vyššie riziko DKA, zahŕňajú pacientov s nízkou funkčnou rezervou betabuniek (napr. pacienti s diabetom 2. typu s nízkou hladinou C-peptidu alebo s latentným autoimunitným diabetom u dospelých (latent autoimmune diabetes in adults, LADA) alebo pacienti s pankreatitídou v anamnéze), pacientov s ochoreniami vedúcimi k obmedzenému príjmu potravy
alebo závažnej dehydratácii, pacientov, u ktorých sú dávky inzulínu znížené a pacientov so zvýšenou potrebou inzulínu z dôvodu akútneho ochorenia, chirurgického zákroku alebo nadmerného požívania alkoholu. U týchto pacientov sa majú používať inhibítory SGLT2 s opatrnosťou.
Obnovenie liečby inhibítormi SGLT2 u pacientov s DKA počas liečby inhibítormi SGLT2
v anamnéze sa neodporúča, pokiaľ nebol identifikovaný a vyriešený iný jednoznačný spúšťací faktor
DKA.
Bezpečnosť a účinnosť Seglurometu u pacientov s diabetom 1. typu sa nestanovili a Segluromet sa nemá používať na liečbu pacientov s diabetom 1. typu. Obmedzené údaje z klinických skúšaní naznačujú, že DKA sa vyskytuje často u pacientov s diabetom 1. typu liečených inhibítormi SGLT2.
Amputáciedolnýchkončatín
V dlhodobých klinických štúdiách s iným inhibítorom SGLT2 sa pozoroval zvýšený počet prípadov amputácie dolnej končatiny (hlavne prstov). Nie je známe, či ide o skupinový účinok. Rovnako ako u všetkých diabetických pacientov je dôležité odporučiť pacientom pravidelnú preventívnu starostlivosť o chodidlá.
Hypoglykémiaprisúbežnompoužívaníinzulínualiečivstimulujúcichsekréciuinzulínu Ertugliflozín môže zvýšiť riziko hypoglykémie, keď sa používa v kombinácii s inzulínom a/alebo liečivom stimulujúcim sekréciu inzulínu, o ktorých je známe, že spôsobujú hypoglykémiu (pozri časť
4.8). Preto na minimalizáciu rizika vzniku hypoglykémie môže byť potrebná nižšia dávka inzulínu alebo liečiva stimulujúceho sekréciu inzulínu, ak sa používajú v kombinácii so Seglurometom (pozri
časti 4.2 a 4.5).
Mykotickéinfekciepohlavnýchorgánov
Ertugliflozín zvyšuje riziko mykotických infekcií pohlavných orgánov. V skúšaniach s inhibítormi
SGLT2 sa mykotické infekcie pohlavných orgánov objavili s väčšou pravdepodobnosťou u pacientov s mykotickými infekciami pohlavných orgánov v anamnéze a u mužov bez obriezky (pozri časť 4.8). Pacientov je potrebné sledovať a vhodne liečiť.
Infekciemočovýchciest
Vylučovanie glukózy v moči môže byť spojené so zvýšeným rizikom infekcií močových ciest. Výskyt infekcií močových ciest sa v skupinách s ertugliflozínom 5 mg a 15 mg (4,0 % a 4,1 %) a v skupine
s placebom (3,9 %) výrazne nelíšil. Väčšina udalostí bola mierna alebo stredne závažná a nebol
hlásený žiadny vážny prípad. Počas liečby pyelonefritídy alebo urosepsy sa má zvážiť dočasné prerušenie liečby ertugliflozínom.
Starší pacienti
Starší pacienti môžu mať zvýšené riziko vzniku deplécie objemu. U pacientov vo veku 65 rokov
a starších, liečených ertugliflozínom, bol vyšší výskyt nežiaducich reakcií súvisiacich s depléciou objemu v porovnaní s mladšími pacientmi. Riziko vzniku laktátovej acidózy súvisiacej s metformínom sa zvyšuje s vekom pacienta, pretože starší pacienti majú vyššiu pravdepodobnosť porúch pečene, obličiek alebo srdca než mladší pacienti. Predpokladá sa, že Segluromet bude mať zníženú účinnosť
u starších pacientov s poruchou funkcie obličiek (pozri časti 4.2 a 4.8). U starších pacientov kontrolujte funkciu obličiek častejšie.
Zlyhávaniesrdca
Skúsenosť s použitím u triedy I-II podľa klasifikácie New York Heart Association (NYHA) je obmedzená a nie je žiadna skúsenosť z klinických štúdií s ertugliflozínom u triedy III-IV podľa
klasifikácie NYHA.
Laboratórnevyšetreniamoču
Vzhľadom na mechanizmus účinku ertugliflozínu, bude výsledok vyšetrenia glukózy v moči
u pacientov užívajúcich Segluromet pozitívny. Na sledovanie kontroly glykémie sa majú použiť náhradné metódy.
Inte
r
ferencias
testom
na
1,5-anhydroglucitol
(1,5-AG)
Sledovanie kontroly glykémie pomocou testu na 1,5-AG sa neodporúča, pretože stanovenia 1,5-AG na hodnotenie kontroly glykémie u pacientov užívajúcich inhibítory SGLT2 nie sú spoľahlivé. Na sledovanie kontroly glykémie sa majú použiť náhradné metódy.
4.5 Liekové a iné interakcie
Farmakokinetické liekové interakčné štúdie so Seglurometom sa neuskutočnili; takéto štúdie však boli vykonané s ertugliflozínom a metformínom, jednotlivými liečivami Seglurometu.
Ertugliflozín
Farmakodynamickéinterakcie
Diuretiká
Ertugliflozín môže zvyšovať diuretický účinok diuretík a môže zvýšiť riziko dehydratácie a hypotenzie (pozri časť 4.4).
Inzulín a liečivá stimulujúce sekréciu inzulínu
Inzulín a liečivá stimulujúce sekréciu inzulínu, ako sú deriváty sulfonylurey, spôsobujú hypoglykémiu. Ertugliflozín môže zvýšiť riziko vzniku hypoglykémie, ak sa používa v kombinácii s inzulínom
a/alebo liečivom stimulujúcim sekréciu inzulínu. Preto na zníženie rizika hypoglykémie môže byť
potrebná nižšia dávka inzulínu alebo liečiva stimulujúceho sekréciu inzulínu, ak sa používajú v kombinácii so Seglurometom (pozri časti 4.2, 4.4 a 4.8).
Farmakokinetické interakcie
Účinkyinýchliekovnafarmakokinetikuertugliflozínu
Primárnym mechanizmom odbúravania ertugliflozínu je metabolizmus sprostredkovaný UGT1A9
a UGT2B7.
Interakčné štúdie vykonané u zdravých osôb, s použitím režimu s jednorazovou dávkou naznačujú, že sitagliptín, metformín, glimepirid ani simvastatín nespôsobujú zmenu farmakokinetiky ertugliflozínu.
Podávanie viacnásobných dávok rifampicínu (induktor UGT a CYP) znižuje AUC ertugliflozínu
o 39 % a Cmax ertugliflozínu o 15 %. Tento pokles expozície sa nepovažuje za klinicky významný,
a preto sa neodporúča žiadna úprava dávky. Klinicky významný účinok s ostatnými induktormi (napr.
karbamazepín, fenytoín, fenobarbital) sa neočakáva.
Vplyv inhibítorov UGT na farmakokinetiku ertugliflozínu sa klinicky neskúmal, ale potenciálne zvýšenie expozície ertugliflozínu v dôsledku inhibície UGT sa nepovažuje za klinicky významné.
Účinkyertugliflozínunafarmakokinetikuinýchliekov
Interakčné štúdie vykonané u zdravých dobrovoľníkov naznačujú, že ertugliflozín nemal žiadny klinicky významný účinok na farmakokinetiku sitagliptínu, metformínu a glimepiridu.
Súbežné podávanie simvastatínu s ertugliflozínom viedlo k 24 % zvýšeniu AUC a 19 % zvýšeniu Cmax simvastatínu a k 30 % zvýšeniu AUC a 16 % zvýšeniu Cmax kyseliny simvastatínovej. Mechanizmus malých zvýšení simvastatínu a kyseliny simvastatínovej nie je známy a neprebieha prostredníctvom inhibície OATP ertugliflozínom. Tieto zvýšenia sa nepovažujú za klinicky významné.
M
etformín
Súbežné
používanie,
ktoré
sa
neodporúča
Alkohol
Intoxikácia alkoholom je spojená so zvýšeným rizikom laktátovej acidózy, najmä v prípadoch hladovania, nedostatočnej výživy alebo poškodenia funkcie pečene.
Jódované kontrastné látky
Pred zobrazovacím vyšetrením alebo v čase zobrazovacieho vyšetrenia sa musí liek Segluromet vysadiť a v podávaní sa nesmie pokračovať skôr, kým neuplynie minimálne 48 hodín, za predpokladu,
že došlo k opätovnému vyhodnoteniu funkcie obličiek a zistilo sa, že je stabilná (pozri časti 4.2 a 4.4).
Kombinácievyžadujúceopatrnosťpripoužívaní
Niektoré lieky môžu nežiaduco ovplyvňovať funkciu obličiek, čo môže zvýšiť riziko laktátovej acidózy, napr. NSAIDs vrátane selektívnych inhibítorov cyklooxygenázy II (COX), inhibítorov
enzýmu konvertujúceho angiotenzín (ACE), antagonistov receptora angiotenzínu II a diuretík, najmä
kľučkových (loop) diuretík. Ak sa začína používať alebo sa používa takýto liek v kombinácii s metformínom, je potrebné dôkladné monitorovanie funkcie obličiek.
Transportéry organických katiónov (OCT)
Metformín je substrátom oboch transportérov OCT1 a OCT2.
Súbežné podávanie metformínu s
· inhibítormi OCT1 (ako je verapamil) môže znížiť účinnosť metformínu,
· induktormi OCT1 (ako je rifampicín) môže zvýšiť gastrointestinálnu absorpciu a účinnosť metformínu,
· inhibítormi OCT2 (ako sú cimetidín, dolutegravir, ranolazín, trimetoprim, vandetanib, isavukonazol) môže znížiť renálnu elimináciu metformínu a tým viesť k zvýšeniu plazmatickej
koncentrácie metformínu,
· inhibítormi oboch OCT1 a OCT2 (ako sú krizotinib, olaparib) môže zmeniť účinnosť a renálnu elimináciu metformínu.
Preto sa najmä u pacientov s poruchou funkcie obličiek odporúča opatrnosť, keď sa tieto lieky podávajú súbežne s metformínom, pretože sa môže zvýšiť plazmatická koncentrácia metformínu. Ak je to potrebné, môže sa zvážiť úprava dávky metformínu, pretože inhibítory/induktory OCT môžu zmeniť účinnosť metformínu.
Glukokortikoidy (podávané systémovo a lokálne), beta-2 agonisty a diuretiká majú vnútornú hyperglykemickú aktivitu. Pacient má byť informovaný a má sa u neho častejšie sledovať glukóza v krvi, najmä na začiatku liečby takýmito liekmi. V prípade potreby sa má dávka antihyperglykemického lieku upraviť počas liečby iným liekom a po jeho vysadení.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje o použití Seglurometu u gravidných žien.
Obmedzené množstvo údajov naznačuje, že použitie metformínu u gravidných žien nie je spojené so zvýšeným rizikom kongenitálnych malformácií. Štúdie s metformínom na zvieratách nenaznačujú škodlivé účinky pokiaľ ide o graviditu, embryonálny alebo fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
K dispozícii je iba obmedzené množstvo údajov o použití ertugliflozínu u gravidných žien. Na základe výsledkov zo štúdií na zvieratách, ertugliflozín môže ovplyvniť vývin a dozrievanie obličiek (pozri časť 5.3). Preto sa Segluromet nemá používať počas gravidity.
Dojčenie
K dispozícii nie sú žiadne informácie týkajúce sa prítomnosti ertugliflozínu v ľudskom mlieku, účinkov na dojčené dieťa alebo účinkov na tvorbu mlieka. Metformín je prítomný v ľudskom materskom mlieku. Ertugliflozín a metformín sú prítomné v mlieku dojčiacich potkanov. Ertugliflozín spôsobil účinky u mláďat dojčiacich potkanov.
Farmakologicky sprostredkované účinky sa pozorovali u mláďat potkanov liečených ertugliflozínom (pozri časť 5.3). Keďže k dozrievaniu obličiek u ľudí dochádza in utero a počas prvých 2 rokov života, kedy môže dôjsť k expozícii počas dojčenia, riziko u novorodencov/dojčiat sa nedá vylúčiť.
Segluromet sa nemá používať počas dojčenia.
Fertilita
Účinok Seglurometu na fertilitu u ľudí sa neskúmal. V štúdiách na zvieratách sa nepozorovali žiadne účinky ertugliflozínu alebo metformínu na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Segluromet nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov je potrebné upozorniť na riziko hypoglykémie, ak sa Segluromet používa
v kombinácii s inzulínom alebo liečivami stimulujúcimi sekréciu inzulínu a na zvýšené riziko
nežiaducich reakcií súvisiacich s depléciou objemu, ako je posturálny závrat (pozri časti 4.2, 4.4
a 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Ertugliflozínametformín
Bezpečnosť súbežne podávaného ertugliflozínu a metformínu sa hodnotila u 1 083 pacientov s diabetes mellitus 2. typu, liečených počas 26 týždňov v združenom súbore dvoch placebom
kontrolovaných skúšaní: ako prídavná liečba ertugliflozínom ku liečbe metformínom a ako prídavná liečba ertugliflozínom ku liečbe sitagliptínom a metformínom (pozri časť 5.1). Výskyt a typ
nežiaducich reakcií v týchto dvoch klinických skúšaniach sa podobal nežiaducim reakciám pozorovaným pri ertugliflozíne. Neboli zistené žiadne ďalšie nežiaduce reakcie pri združovaní týchto dvoch placebom kontrolovaných skúšaní, zahŕňajúcich metformín v porovnaní s tromi placebom
kontrolovanými skúšaniami s ertugliflozínom (pozri ďalej).
Ertugliflozín
Združený súbor placebom kontrolovaných skúšaní
Primárne hodnotenie bezpečnosti bolo vykonané v združenom súbore troch 26-týždňových placebom kontrolovaných skúšaní. Ertugliflozín sa používal vo forme monoterapie v jednom skúšaní a ako prídavná liečba v dvoch skúšaniach (pozri časť 5.1). Tieto údaje odrážajú expozíciu ertugliflozínu
u 1 029 pacientov s priemernou dĺžkou trvania expozície približne 25 týždňov. Pacienti dostávali ertugliflozín 5 mg (N = 519), ertugliflozín 15 mg (N = 510) alebo placebo (N = 515) jedenkrát denne.
Najčastejšie hlásenými nežiaducimi reakciami v rámci klinického programu boli vulvovaginálna mykotická infekcia a iné mykotické infekcie ženských pohlavných orgánov. Zriedkavo sa objavila závažná diabetická ketoacidóza. Frekvencie výskytu pozri v časti „Popis vybraných nežiaducich reakcií“ a pozri časť 4.4.
Tabuľkovýsúhrnnežiaducichreakcií
Nežiaduce reakcie uvedené nižšie sú klasifikované podľa frekvencie a triedy orgánových systémov (system organ class, SOC). Kategórie frekvencií sú definované podľa nasledujúceho pravidla: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka 1: Nežiaduce reakcie
Trieda orgánových systémov
Frekvencia
Infekcie a nákazy
Veľmi časté
Časté
Nežiaduca reakcia
vulvovaginálna mykotická infekcia a iné mykotické infekcie ženských pohlavných orgánov*,†,1
kandidová balanitída a iné mykotické infekcie mužských pohlavných orgánov*,†,1
Poruchy metabolizmu a výživy
Časté
Zriedkavé
Veľmi zriedkavé
hypoglykémia*,†,1
diabetická ketoacidóza*,†,1
laktátová acidóza*,2, nedostatok vitamínu B12‡,2
Poruchy nervového systému
Časté porucha chuti2
Poruchy cievČasté deplécia objemu*,†,1
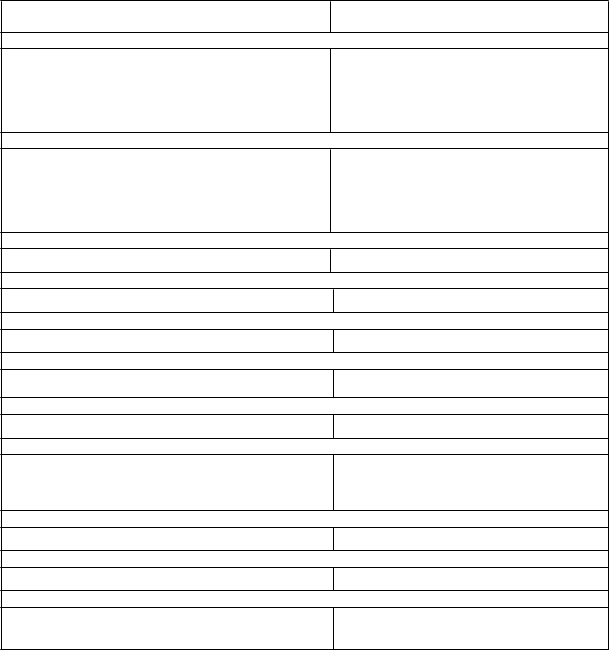 Poruchy gastrointestinálneho traktu
Poruchy gastrointestinálneho traktuVeľmi časté gastrointestinálne symptómy§,2
Poruchy pečene a žlčových ciestVeľmi zriedkavé abnormálne výsledky funkčných vyšetrení pečene2, hepatitída2
Poruchy kože a podkožného tkanivaVeľmi zriedkavé erytém2, pruritus2, utrikária2
Poruchy obličiek a močových ciest
Časté
Menej časté
Poruchy reprodukčného systému a prsníkov
zvýšené vylučovanie moču¶,1
dyzúria1, zvýšená hladina kreatinínu v krvi/znížená rýchlosť glomerulárnej filtrácie†,1
Časté vulvovaginálny pruritus1
Celkové poruchy a reakcie v mieste podania
Časté smäd#,1
Laboratórne a funkčné vyšetrenia
Časté zmenená hladina tukov v séreÞ,1, zvýšená hladina hemoglobínuß,1, zvýšená hladina BUNà,1
1 Nežiaduce reakcie pri ertugliflozíne.
2 Nežiaduce reakcie pri metformíne.
* Pozri časť 4.4.
† Ďalšie informácie pozri v podčastiach nižšie.
‡ Dlhodobá liečba metformínom bola spojená s poklesom absorpcie vitamínu B12, čo môže veľmi zriedkavo viesť ku klinicky významnému nedostatku vitamínu B12 (napr. megaloblastická anémia).
§ Gastrointestinálne symptómy ako je nauzea, vracanie, hnačka, bolesť brucha a strata chuti sa najčastejšie vyskytujú na začiatku liečby a vo väčšine prípadov sa spontánne vyriešia.
¶ Zahŕňa: polakizúriu, nutkanie na močenie, polyúriu, zvýšené vylučovanie moču a noktúriu.
# Zahŕňa: smäd a polydipsiu.
Þ Priemerné percentuálne zmeny oproti východiskovej hodnote boli pre LDL-C 5,8 % pri ertugliflozíne 5 mg a 8,4 % pri ertugliflozíne 15 mg oproti 3,2 % pri placebe; pre celkový cholesterol 2,8 % pri ertugliflozíne 5 mg a 5,7 % pri ertugliflozíne 15 mg oproti 1,1 % pri placebe, avšak pre HDL-C 6,2 % pri ertugliflozíne 5 mg a 7,6 % pri ertugliflozíne
15 mg oproti 1,9 % pri placebe. Medián percentuálnych zmien oproti východiskovej hodnote bol pre triglyceridy -3,9 % pri ertugliflozíne 5 mg a -1,7 % pri ertugliflozíne 15 mg oproti 4,5 % pri placebe.
ß Podiel osôb s minimálne 1 zvýšením hemoglobínu > 2,0 g/dl bol vyšší v skupinách s ertugliflozínom 5 mg a 15 mg (4,7 %
a 4,1 %, v uvedenom poradí) v porovnaní so skupinou s placebom (0,6 %).
à Podiel osôb, ktoré mali akýkoľvek výskyt zvýšenia hodnôt BUN ≥ 50 % a hodnota > HHN bola číselne vyššia v skupine s ertugliflozínom 5 mg a vyššia v skupine s ertugliflozínom 15 mg (7,9 % a 9,8 %, v uvedenom poradí) v porovnaní so skupinou s placebom (5,1 %).
Popis vybranýchnežiaducichreakcií
Depléciaobjemu(ertugliflozín)
Ertugliflozín spôsobuje osmotickú diurézu, ktorá môže viesť k zníženiu intravaskulárneho objemu a nežiaducim reakciám súvisiacim s depléciou objemu. V združenom súbore placebom kontrolovaných štúdií bol výskyt nežiaducich udalostí súvisiacich s depléciou objemu (dehydratácia,
posturálny závrat, presynkopa, synkopa, hypotenzia a ortostatická hypotenzia) nízky (< 2 %) a v rámci
skupín s ertugliflozínom a placebom nebol výrazne odlišný. V analýzach podskupín v širšom združenom súbore štúdií fázy 3, bol u osôb s hodnotou eGFR <60 ml/min/1,73 m2, osôb vo veku
≥65 rokov a osôb užívajúcich diuretiká výskyt deplécie objemu vyšší v skupinách s ertugliflozínom v porovnaní so skupinou s komparátorom (pozri časti 4.2 a 4.4). U osôb s hodnotou eGFR
<60 ml/min/1,73 m2 bol výskyt 5,1 % pri ertugliflozíne 5 mg, 2,6 % pri ertugliflozíne 15 mg a 0,5 %
v skupine s komparátorom a u osôb s hodnotou eGFR 45 až <60 ml/min/1,73 m2 bol výskyt 6,4 % pri ertugliflozíne 5 mg, 3,7 % pri ertugliflozíne 15 mg a 0 % v skupine s komparátorom.
Hypoglykémia(ertugliflozín)
V združenom súbore placebom kontrolovaných štúdií bol výskyt zdokumentovanej hypoglykémie zvýšený pri ertugliflozíne 5 mg a 15 mg (5,0 % a 4,5 %) v porovnaní s placebom (2,9 %). V tejto populácii bol výskyt závažnej hypoglykémie 0,4 % v každej skupine. Keď sa ertugliflozín používal vo forme monoterapie, výskyt hypoglykemických udalostí v oboch skupinách s ertugliflozínom bol 2,6 % a v skupine s placebom 0,7 %. Keď sa používal ako prídavná liečba k metformínu, výskyt hypoglykemických udalostí v skupine s ertugliflozínom 5 mg bol 7,2 %, v skupine s ertugliflozínom
15 mg 7,8 % a v skupine s placebom 4,3 %.
Ak sa ertugliflozín pridal k metformínu a porovnal so sulfonylureou, výskyt hypoglykémie bol vyšší pri sulfonylurei (27 %) v porovnaní s ertugliflozínom (5,6 % pri ertugliflozíne 5 mg a 8,2 % pri ertugliflozíne 15 mg).
U pacientov so stredne závažnou poruchou funkcie obličiek používajúcich inzulín, SU alebo meglitinidy ako základnú liečbu, bola hypoglykémia zdokumentovaná u 36 % pri ertugliflozíne 5 mg,
27 % pri ertugliflozíne 15 mg a 36 % pri placebe (pozri časti 4.2, 4.4 a 4.5).
Diabetickáketoacidóza(ertugliflozín)
V rámci klinického programu s ertugliflozínom sa ketoacidóza identifikovala u 3 z 3 409 (0,1 %)
pacientov liečených ertugliflozínom a u 0,0 % pacientov liečených komparátorom (pozri časť 4.4).
Zvýšenáhladinakreatinínuvkrvi/poklesrýchlostiglomerulárnejfiltrácieaudalostisúvisiacesobličkami(ertugliflozín)
Úvodné zvýšenia priemernej hladiny kreatinínu a poklesy priemernej hodnoty eGFR u pacientov liečených ertugliflozínom boli počas prebiehajúcej liečby vo všeobecnosti prechodné. U pacientov so stredne závažnou poruchou funkcie obličiek na začiatku sa objavili väčšie priemerné zmeny, ktoré sa
v 26. týždni neupravili späť na východiskový stav; tieto zmeny sa upravili po ukončení liečby.
Nežiaduce reakcie súvisiace s obličkami (napr. akútne poškodenie obličiek, porucha funkcie obličiek, akútne prerenálne zlyhanie) sa môžu objaviť u pacientov liečených ertugliflozínom, najmä u pacientov so stredne závažnou poruchou funkcie obličiek, kedy výskyt nežiaducich reakcií súvisiacich
s obličkami bol 2,5 % u pacientov liečených ertugliflozínom 5 mg, 1,3 % u pacientov liečených ertugliflozínom 15 mg a 0,6 % u pacientov užívajúcich placebo.
Mykotickéinfekciepohlavnýchorgánov(ertugliflozín)
V združenom súbore troch placebom kontrolovaných klinických skúšaní sa mykotické infekcie ženských pohlavných orgánov (napr. kandidózy pohlavných orgánov, mykotická infekcia pohlavných
orgánov, vaginálna infekcia, vulvitída, vulvovaginálne kandidózy, vulvovaginálna mykotická infekcia,
vulvovaginitída) objavili u 9,1 % žien liečených ertugliflozínom 5 mg, 12 % žien liečených ertugliflozínom 15 mg a 3,0 % žien užívajúcich placebo. U žien došlo k ukončeniu liečby z dôvodu
mykotických infekcií pohlavných orgánov u 0,6 % pacientok liečených ertugliflozínom a u 0 %
pacientok užívajúcich placebo (pozri časť 4.4).
V rovnakom združenom súbore sa mykotické infekcie mužských pohlavných orgánov (napr. kandidová balanitída, balanopostitída, infekcia pohlavných orgánov, mykotická infekcia pohlavných orgánov) objavili u 3,7 % mužov liečených ertugliflozínom 5 mg, u 4,2 % mužov liečených ertugliflozínom 15 mg a 0,4 % mužov užívajúcich placebo. Mykotické infekcie mužských pohlavných orgánov sa častejšie objavovali u mužov bez obriezky. U mužov došlo k ukončeniu liečby z dôvodu mykotických infekcií pohlavných orgánov u 0,2 % pacientov liečených ertugliflozínom a u 0 % pacientov užívajúcich placebo. V zriedkavých prípadoch sa hlásila fimóza a niekedy došlo
k vykonaniu obriezky (pozri časť 4.4).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania so Seglurometom použite bežné podporné opatrenia (napr. odstráňte nevstrebaný liek z gastrointestinálneho traktu, použite klinické sledovanie a nasaďte podpornú liečbu) na základe klinického stavu pacienta.
ErtugliflozínPri ertugliflozíne sa nepreukázala žiadna toxicita u zdravých osôb pri jednorazových perorálnych dávkach až do 300 mg a viacnásobných dávkach až do 100 mg denne počas 2 týždňov.
Neidentifikovali sa žiadne možné akútne príznaky a prejavy predávkovania. Odstránenie ertugliflozínu
pomocou hemodialýzy sa neskúmalo.
MetformínPredávkovanie metformíniumchloridom sa vyskytlo, vrátane požitia množstiev vyšších ako 50 g. Hypoglykémia bola hlásená u približne 10 % prípadov, ale kauzálna súvislosť s metformínium- chloridom sa nestanovila. Laktátová acidóza bola hlásená u približne 32 % prípadov predávkovania metformínom (pozri časť 4.4). Laktátová acidóza je medicínsky naliehavý stav a musí sa liečiť
v nemocnici. Metformín je dialyzovateľný s klírensom až do 170 ml/min za dobrých hemodyna- mických podmienok. Preto môže byť hemodialýza najúčinnejšia na odstránenie nahromadeného
liečiva u pacientov s podozrením na predávkovanie metformínom.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antidiabetiká, kombinácie perorálnych liečiv znižujúcich hladiny glukózy v krvi, ATC kód: A10BD23.
MechanizmusúčinkuSegluromet je kombináciou dvoch antihyperglykemických látok s komplementárnymi mechanizmami účinku na zlepšenie kontroly glykémie u pacientov s diabetom 2. typu: ertugliflozín, inhibítor SGLT2 a metformíniumchlorid, člen skupiny biguanidov.
ErtugliflozínSGLT2 je hlavným transportérom zodpovedným za spätné vstrebávanie glukózy z glomerulárneho filtrátu späť do krvného obehu. Ertugliflozín je účinný, selektívny a reverzibilný inhibítor SGLT2. Inhibíciou SGLT2 ertugliflozín znižuje spätné vstrebávanie prefiltrovanej glukózy v obličkách
a znižuje prahovú hranicu glukózy v obličkách, čím zvyšuje vylučovanie glukózy močom.
M
etformín
Metformín je antihyperglykemická látka, ktorá zlepšuje glukózovú toleranciu u pacientov s diabetom
2. typu, čím sa znižuje bazálna a postprandiálna plazmatická glukóza. Jeho farmakologické mechanizmy účinku sa líšia od iných skupín perorálnych antihyperglykemík. Metformín znižuje
tvorbu glukózy v pečeni, znižuje absorpciu glukózy v čreve a zvyšovaním periférnej absorpcie
a využitia glukózy zvyšuje inzulínovú senzitivitu. Na rozdiel od sulfonylurey, metformín nevyvoláva hypoglykémiu ani u pacientov s diabetom 2. typu ani u normálnych osôb, s výnimkou osobitných okolností (pozri časť 4.5) a nespôsobuje hyperinzulinémiu. Pri liečbe metformínom zostáva sekrécia inzulínu nezmenená, zatiaľ čo hladiny inzulínu nalačno a celodenná plazmatická odpoveď inzulínu sa môžu skutočne znížiť.
Farmakodynamické účinky
Ertugliflozín
Vylučovanie glukózy močom a objem moču
U zdravých osôb a u pacientov s diabetes mellitus 2. typu sa po podaní jednorazovej dávky
a viacnásobnej dávky ertugliflozínu pozorovali zvýšenia množstva glukózy vylúčenej do moču závislé od dávky. Modelovanie odpovede na dávku naznačuje, že ertugliflozín 5 mg a 15 mg vedie k takmer
maximálnemu vylučovaniu glukózy močom (urinary glucose excretion, UGE) u pacientov s diabetes
mellitus 2. typu, čo predstavuje maximálnu inhibíciu 87 % pri ertugliflozíne 5 mg a 96 % pri ertugliflozíne 15 mg.
Klinická účinnosťabezpečnosť
Ertugliflozín v kombinácii s metformínom
Účinnosť a bezpečnosť ertugliflozínu v kombinácii s metformínom sa skúmali v 4 multicentrických, randomizovaných, dvojito zaslepených, placebom a aktívnym komparátorom kontrolovaných
klinických štúdiách fázy 3 zahŕňajúcich 3 643 pacientov s diabetom 2. typu. Rasové rozdelenie
pacientov v rámci týchto štyroch štúdií bolo 66,2 % až 80,3 % belochov; 10,6 % až 20,3 % aziatov,
1,9 % až 10,3 % černochov a 4,5 % až 7,4 % ostatných. Hispánski alebo latinskoamerickí pacienti tvorili 15,6 % až 34,5 % populácie. Priemerný vek pacientov v týchto štyroch štúdiách sa pohyboval od 55,1 do 59,1 rokov (v rozmedzí od 21 rokov do 86 rokov); 15,6 % až 29,9 % pacientov vo veku
≥65 rokov a 0,6 % až 3,8 % vo veku ≥ 75 rokov.
Ertugliflozín ako prídavná kombinovaná liečba s metformínom
Celkovo 621 pacientov s diabetom 2. typu nedostatočne kontrolovaných metformínom v monoterapii
(≥ 1 500 mg/deň) sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 26-týždňovej, placebom kontrolovanej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu v kombinácii
s metformínom. Pacienti boli randomizovaní do skupín s ertugliflozínom 5 mg, ertugliflozínom 15 mg
alebo placebom podávaným jedenkrát denne ako doplnok k prebiehajúcej základnej liečbe metformínom (pozri tabuľku 2).
Tabuľka 2: Výsledky v 26. týždni z placebom kontrolovanej štúdie s ertugliflozínom používaným v kombinácii s metformínom*
Ertugliflozín 5 mg Ertugliflozín 15 mg Placebo
HbA1c (%) N = 207 N = 205 N = 209
Východisková hodnota (priemerná) 8,1 8,1 8,2
Zmena oproti východiskovej hodnote (priemer
LS†)
-0,7 -0,9 -0,0
Rozdiel od placeba (priemer LS†, 95% IS) -0,7‡ (-0,9; -0,5) -0,9‡ (-1,1; -0,7)
Pacienti [N (%)] s HbA1c < 7 % 73 (35,3)§ 82 (40,0)§ 33 (15,8)
Telesná hmotnosť (kg) N = 207 N = 205 N = 209
Východisková hodnota (priemerná) 84,9 85,3 84,5
Zmena oproti východiskovej hodnote (priemer
LS†))
-3,0 -2,9 -1,3
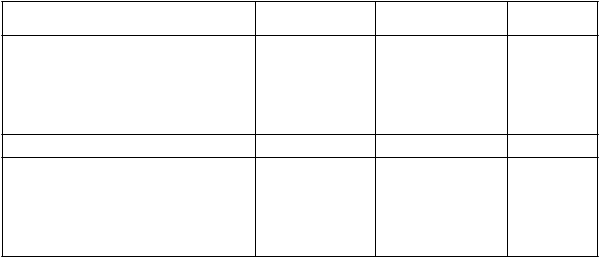
Rozdiel od placeba (priemer LS†, 95% IS) -1,7‡ (-2,2; -1,1) -1,6‡ (-2,2; -1,0)
* N zahŕňa všetkých randomizovaných liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, predchádzajúci antihyperglykemický liek (metformín v monoterapii alebo metformín + iná AHA), východisková hodnota eGFR (kontinuálna), menopauzálny stav
randomizovanej skupiny (muži, premenopauzálne ženy, perimenopauzálne ženy alebo < 3 roky po menopauze, ženy ³ 3
roky po menopauze) a interakcia času podľa liečby.
‡ p £ 0,001 v porovnaní s placebom.
§ p < 0,001 v porovnaní s placebom (na základe porovnaní upravených pomerov pravdepodobnosti z modelu logistickej regresie s použitím viacnásobného pripočítania hodnôt chýbajúcich údajov).
Faktoriálna štúdia ertugliflozínu a sitagliptínu ako prídavná kombinovaná liečba s metformínom Celkovo 1 233 pacientov s diabetom 2. typu sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 26-týždňovej, aktívne kontrolovanej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu 5 mg alebo 15 mg v kombinácii so sitagliptínom 100 mg v porovnaní s jednotlivými liečivami. Pacienti s diabetom 2. typu nedostatočne kontrolovaní metformínom v monoterapii
(≥ 1 500 mg/deň) boli randomizovaní do jednej z piatich skupín aktívnej liečby: ertugliflozín 5 mg alebo 15 mg, sitagliptín 100 mg alebo sitagliptín 100 mg v kombinácii s 5 mg alebo 15 mg
ertugliflozínu s podávaním jedenkrát denne ako doplnok k prebiehajúcej základnej liečbe
metformínom (pozri tabuľku 3).
Tabuľka 3: Výsledky v 26. týždni z faktoriálnej štúdie s ertugliflozínom a sitagliptínom ako prídavnej kombinovanej liečby s metformínom v porovnaní so samostatne podávanými jednotlivými liečivami*
Ertugliflozín
5 mg
Ertugliflozín
1
5 mg
Sitagliptín
10
0 mg
Ertugliflozín 5 mg +
S
itagliptín 100 mg
Ertugliflozín 15 mg
+ Sitagliptín 100 mg
HbA1c (%) N = 250 N = 248 N = 247 N = 243 N = 244
Východisková hodnota (priemerná) 8,6 8,6 8,5 8,6 8,6
Zmena oproti východiskovej hodnote (priemer LS†) Rozdiel od
-1,0 -1,1 -1,1 -1,5 -1,5
sitagliptínu ertugliflozínu 5 mg ertugliflozínu 15 mg
(priemer LS†, 95% IS)
-0,4‡ (-0,6; -0,3)
-0,5‡ (-0,6; -0,3)
-0,5‡ (-0,6; -0,3)
-0,4‡ (-0,6; -0,3)
Pacienti [N (%)] s HbA1c < 7 % 66 (26,4) 79 (31,9) 81 (32,8) 127§ (52,3) 120§ (49,2)
Telesná hmotnosť (kg) N = 250 N = 248 N = 247 N = 243 N = 244
Východisková hodnota (priemerná) 88,6 88,0 89,8 89,5 87,5
Zmena oproti východiskovej
hodnote (priemer LS†)
Rozdiel od sitagliptínu
(priemer LS†, 95% IS)
-2,7 -3,7 -0,7 -2,5 -2,9
-1,8‡ (-2,5; -1,2) -2,3‡ (-2,9; -1,6)
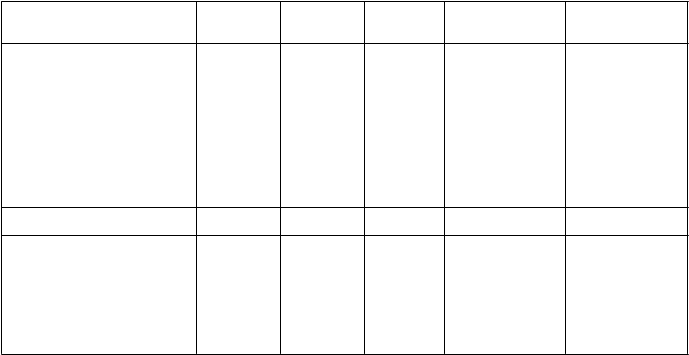
* N zahŕňa všetkých randomizovaných, liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, východiskovú hodnotu eGFR a interakciu času podľa liečby.
‡ p < 0,001 v porovnaní s kontrolnou skupinou.
§ p < 0,001 v porovnaní s príslušnou dávkou ertugliflozínu alebo sitagliptínu (na základe porovnaní upravených pomerov pravdepodobnosti z modelu logistickej regresie s použitím viacnásobného pripočítania hodnôt chýbajúcich údajov).
Ertugliflozín ako prídavná kombinovaná liečba s metformínom a sitagliptínomCelkovo 463 pacientov s diabetom 2. typu nedostatočne kontrolovaných metformínom
(≥ 1 500 mg/deň) a sitagliptínom 100 mg jedenkrát denne sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 26-týždňovej, placebom kontrolovanej štúdie na vyhodnotenie účinnosti
a bezpečnosti ertugliflozínu. Pacienti boli randomizovaní na podávanie ertugliflozínu 5 mg,
ertugliflozínu 15 mg alebo placeba jedenkrát denne ako doplnok k pokračujúcej základnej liečbe metformínom a sitagliptínom (pozri tabuľku 4).
Tabuľka 4: Výsledky v 26. týždni zo štúdie ertugliflozínu ako prídavnej liečby v kombinácii s metformínom a sitagliptínom*Ertugliflozín 5 mg Ertugliflozín 15 mg PlaceboHbA1c (%) N = 156 N = 153 N = 153Východisková hodnota (priemerná) 8,1 8,0 8,0
Zmena oproti východiskovej hodnote (priemer LS†) -0,8 -0,9 -0,1
Rozdiel od placeba (priemer LS†, 95% IS) -0,7‡ (-0,9; -0,5) -0,8‡ (-0,9; -0,6)
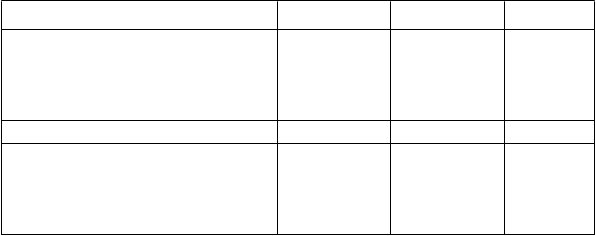 Pacienti [N (%)] s HbA1c < 7 %
Pacienti [N (%)] s HbA1c < 7 % 50 (32,1)‡ 61 (39,9)‡ 26 (17,0)
Telesná hmotnosť (kg) N = 156 N = 153 N = 153Východisková hodnota (priemerná) 87,6 86,6 86,5
Zmena oproti východiskovej hodnote (priemer LS†) -3,3 -3,0 -1,3
Rozdiel od placeba (priemer LS†, 95% IS) -2,0‡ (-2,6; -1,4) -1,7‡ (-2,3; -1,1)
* N zahŕňa všetkých randomizovaných, liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, predchádzajúci antihyperglykemický liek.
‡ p £ 0,001 v porovnaní s placebom.
Aktívne kontrolovaná štúdia ertugliflozínu oproti glimepiridu ako prídavná kombinovaná liečba s metformínom
Celkovo 1 326 pacientov s diabetom 2. typu nedostatočne kontrolovaných metformínom
v monoterapii sa zúčastnilo randomizovanej, dvojito zaslepenej, multicentrickej, 52-týždňovej, aktívnym komparátorom kontrolovanej štúdie na vyhodnotenie účinnosti a bezpečnosti ertugliflozínu
v kombinácii s metformínom. Títo pacienti, ktorí dostávali metformín v monoterapii
(≥ 1 500 mg/deň), boli randomizovaní na podávanie ertugliflozínu 5 mg, ertugliflozínu 15 mg alebo glimepiridu jedenkrát denne ako doplnok k pokračujúcej základnej liečbe metformínom. Glimepirid sa
začal podávať v dávke 1 mg/deň a dávka sa vytitrovala až na maximálnu dávku 6 alebo 8 mg/deň
(v závislosti od maximálnej schválenej dávky v každej krajine) alebo na maximálnu znášanú dávku alebo sa dávka titrovala smerom nadol na predchádzanie alebo zvládnutie hypoglykémie. Priemerná denná dávka glimepiridu bola 3,0 mg (pozri tabuľku 5).
Tabuľka 5: Výsledky v 52. týždni z aktívne kontrolovanej štúdie porovnávajúcej ertugliflozín s glimepiridom ako prídavnej liečby u pacientov nedostatočne kontrolovaných metformínom*
Ertugliflozín 5 mg Ertugliflozín 15 mg Glimepirid
HbA1c (%) N = 448 N = 440 N = 437
Východisková hodnota (priemerná) 7,8 7,8 7,8
Zmena oproti východiskovej hodnote (priemer
LS†)
-0,6 -0,6 -0,7
Rozdiel od glimepiridu (priemer LS†, 95% IS) 0,2 (0,1; 0,3) 0,1‡ (-0,0; 0,2)
 Pacienti [N (%)] s HbA1c < 7 %
Pacienti [N (%)] s HbA1c < 7 % 154 (34,4) 167 (38,0) 190 (43,5)
Telesná hmotnosť (kg) N = 448 N = 440 N = 437Východisková hodnota (priemerná) 87,9 85,6 86,8
Zmena oproti východiskovej hodnote (priemer
LS†)
-3,0 -3,4 0,9
Rozdiel od glimepiridu (priemer LS†, 95% IS) -3,9 (-4,4; -3,4) -4,3§ (-4,8; -3,8)
* N zahŕňa všetkých randomizovaných, liečených pacientov, ktorí mali aspoň jedno stanovenie výsledného parametra.
† Priemery najmenších štvorcov upravené na liečbu, čas, predchádzajúci antihyperglykemický liek (monoterapia alebo duálna liečba), východisková hodnota eGFR (prebiehajúca) a interakcia času podľa liečby. Čas sa považoval za
kategorizačný parameter.
‡ Neinferiorita sa vyhlasuje v prípade, ak je horná hranica dvojstranného 95% intervalu spoľahlivosti (IS) pre priemerný rozdiel menej ako 0,3%.
¶ p < 0,001 v porovnaní s glimepiridom.
Plazmatická hladina glukózy nalačno
V troch placebom kontrolovaných štúdiách viedol ertugliflozín k štatisticky významným zníženiam plazmatických hladín glukózy nalačno (fasting plasma glucose, FPG). Zníženia FPG upravené
s ohľadom na placebo boli 1,92 mmol/l pre ertugliflozín 5 mg a 2,44 mmol/l pre ertugliflozín 15 mg
vo forme monoterapie; 1,48 mmol/l pre ertugliflozín 5 mg a 2,12 mmol/l pre ertugliflozín 15 mg ako prídavnej liečby k metformínu a 1,40 mmol/l pre ertugliflozín 5 mg a 1,74 mmol/l pre ertugliflozín'
15 mg ako prídavnej liečby k metformínu a sitagliptínu.
Kombinácia ertugliflozínu a sitagliptínu k základnej liečbe metformínom mala za následok významne väčšie zníženia FPG v porovnaní so samotným sitagliptínom alebo ertugliflozínom. Kombinácia ertugliflozínu 5 mg alebo 15 mg a sitagliptínu viedla k prírastkovým zníženiam FPG 0,46 mmol/l
a 0,65 mmol/l v porovnaní so samotným ertugliflozínom alebo 1,02 mmol/l a 1,28 mmol/l v porovnaní so samotným sitagliptínom.
Účinnosť u pacientov s východiskovou hodnotou HbA1c ≥ 9 %
V štúdii ertugliflozínu pridaného k metformínu u pacientov s východiskovou hodnotou HbA1c od
7,0-10,5 %, boli zníženia HbA1c pre podskupinu pacientov v štúdii s východiskovou hodnotou HbA1c
≥ 9% upravené s ohľadom na placebo 1,31% pre ertugliflozín 5 mg a 1,43% pre ertugliflozín 15 mg.
V štúdii pacientov nedostatočne kontrolovaných metformínom s východiskovou hodnotou HbA1c od
7,5-11,0 % v podskupine pacientov s východiskovou hodnotou HbA1c ≥ 10 %, viedla kombinácia ertugliflozínu 5 mg so sitagliptínom k zníženiu HbA1c o 2,35 % a kombinácia ertugliflozínu 15 mg so sitagliptínom k zníženiu HbA1c o 2,66 % v porovnaní so znížením o 2,10 % v skupine so samotným ertugliflozínom 5 mg, 1,30 % v skupine so samotným ertugliflozínom 15 mg, a 1,82 % v skupine so samotným sitagliptínom.
Tlak krvi
Prídatná liečba ertugliflozínom 5 mg a ertugliflozínom 15 mg k základnej liečbe metformínom viedla k štatisticky významnému na placebo korigovanému zníženiu SBP o 3,7 mmHg a 4,5 mmHg. Prídatná liečba ertugliflozínom 5 mg a ertugliflozínom 15 mg k základnej liečbe metformínom
a sitagliptínom viedla k štatisticky významnému na placebo korigovanému zníženiu SBP o 2,9 mmHg a 3,9 mmHg.
V 52-týždňovej aktívne kontrolovanej štúdii oproti glimepiridu boli zníženia SBP oproti východiskovej hodnote 2,2 mmHg pri ertugliflozíne 5 mg a 3,8 mmHg pri ertugliflozíne 15 mg, zatiaľ čo u osôb liečených glimepiridom došlo k zvýšeniu SBP oproti východiskovej hodnote o 1,0 mmHg.
Analýza podskupín
U pacientov s diabetom 2. typu liečených ertugliflozínom v kombinácii s metformínom sa pozorovali klinicky významné zníženia HbA1c v podskupinách definovaných na základe veku, pohlavia, rasy,
etnickej príslušnosti, geografickej oblasti, východiskového BMI, východiskovej hodnoty HbA1c
a dĺžky trvania diabetes mellitus 2. typu.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so Seglurometom vo všetkých podskupinách pediatrickej populácie pri liečbe diabetu 2. typu (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Segluromet
Preukázalo sa, že Segluromet je bioekvivalentný súbežnému podaniu zodpovedajúcich dávok tabliet ertugliflozínu a metformínu.
Ertugliflozín
Všeobecnýúvod
Farmakokinetika ertugliflozínu u zdravých osôb a pacientov s diabetom 2. typu je podobná. Pri dávke
5 mg ertugliflozínu jedenkrát denne bola priemerná plazmatická AUC 398 ng∙h/ml a Cmax 81 ng/ml a pri dávke 15 mg ertugliflozínu jedenkrát denne bola priemerná plazmatická AUC 1 193 ng∙h/ml
a Cmax 268 ng/ml. Rovnovážny stav sa dosiahol po 4 až 6 dňoch dávkovania ertugliflozínu jedenkrát denne. Ertugliflozín nevykazuje časovo závislú farmakokinetiku a v plazme sa akumuluje až na 10-
40 % po opakovanom podaní.
Absorpcia
Po perorálnom podaní jednorazovej dávky 5 mg a 15 mg ertugliflozínu, sa maximálna plazmatická koncentrácia (medián Tmax) ertugliflozínu objavila 1 hodinu po podaní dávky nalačno. Plazmatická Cmax a AUC ertugliflozínu sa zvyšuje dávkovo úmerným spôsobom po jednorazových dávkach od
0,5 mg do 300 mg a po viacnásobných dávkach od 1 mg do 100 mg. Absolútna perorálna biologická
dostupnosť ertugliflozínu po podaní 15 mg dávky je približne 100%.
Podanie ertugliflozínu s jedlom s vysokým obsahom tukov a kalórií znižuje Cmax ertugliflozínu o 29% a predlžuje Tmax o 1 hodinu, ale nespôsobuje zmenu AUC v porovnaní so stavom nalačno. Pozorovaný vplyv jedla na farmakokinetiku ertugliflozínu sa nepovažuje za klinicky významný a ertugliflozín sa môže podávať s jedlom alebo bez jedla. V klinických skúšaniach fázy 3 sa ertugliflozín podával bez ohľadu na jedlo.
Účinky jedla s vysokým obsahom tuku na farmakokinetiku ertugliflozínu a metformínu podávaných vo forme tabliet Segluromet, sú porovnateľné s účinkami hlásenými pre jednotlivé tablety. Jedlo nemalo významný vplyv na AUCinf ertugliflozínu ani metformínu, ale znížilo priemernú hodnotu Cmax ertugliflozínu o približne 41 % a metformínu o približne 29 % v porovnaní so stavom nalačno.
Ertugliflozín je substrátom transportérov P-glykoproteínu (P-gp) a proteínu rezistencie rakoviny prsníka (breast cancer resistance protein, BCRP).
Distribúcia
Priemerný distribučný objem ertugliflozínu v rovnovážnom stave je po intravenóznej dávke 86 l. Väzba ertugliflozínu na plazmatické bielkoviny je 93,6 % a nie je závislá od plazmatických koncentrácií ertugliflozínu. Väzba na plazmatické bielkoviny nie je významne zmenená u pacientov
s poruchou funkcie obličiek alebo pečene. Pomer koncentrácie ertugliflozínu v krvi a plazme je 0,66.
Ertugliflozín nie je substrátom transportérov organických aniónov (OAT1, OAT3), organických katiónov (OCT1, OCT2) ani transportných polypeptidov pre organické anióny (OATP1B1, OATP1B3) in vitro.
Biotransformácia
Metabolizmus je primárnym mechanizmom odbúravania ertugliflozínu. Hlavnou metabolickou cestou pre ertugliflozín je O-glukuronidácia sprostredkovaná UGT1A9 a UGT2B7 na dva glukuronidy, ktoré sú pri klinicky významných koncentráciách farmakologicky neúčinné. Metabolizmus ertugliflozínu sprostredkovaný CYP (oxidatívny) je minimálny (12 %).
Eliminácia
Priemerný systémový plazmatický klírens po intravenóznej dávke 100 µg bol 11 l/hod. Priemerný polčas eliminácie u pacientov s diabetom 2. typu s normálnou funkciou obličiek bol odhadnutý na 17
hodín na základe analýzy populačnej farmakokinetiky. Po podaní perorálneho roztoku
[14C]-ertugliflozínu zdravým osobám sa približne 41 % rádioaktivity súvisiacej s liekom vylúčilo stolicou a 50 % močom. Len 1,5 % podanej dávky bolo vylúčených vo forme nezmeneného
ertugliflozínu močom a 34 % vo forme nezmeneného ertugliflozínu stolicou, čo je pravdepodobne
v dôsledku vylučovania glukuronidových metabolitov žlčou a následnej hydrolýzy na materskú zlúčeninu.
Osobitné skupinypacientov
Porucha funkcie obličiek
V klinickej farmakologickej štúdii fázy 1 u pacientov s diabetom 2. typu a miernou, stredne závažnou alebo závažnou poruchou funkcie obličiek (na základe stanovenia eGFR) boli po podaní jednorazovej
dávky ertugliflozínu 15 mg priemerné zvýšenia AUC ertugliflozínu ≤ 1,7-násobné v porovnaní s osobami s normálnou funkciou obličiek. Tieto zvýšenia AUC ertugliflozínu sa nepovažujú za
klinicky významné. Medzi rôznymi skupinami funkcie obličiek neboli žiadne klinicky významné rozdiely v hodnotách Cmax ertugliflozínu. 24-hodinové vylučovanie glukózy močom klesalo so zvyšujúcou sa závažnosťou poruchy funkcie obličiek (pozri časť 4.4). Väzba ertugliflozínu na plazmatické bielkoviny nebola ovplyvnená u pacientov s poruchou funkcie obličiek.
Porucha funkcie pečene
Stredne závažná porucha funkcie pečene (na základe klasifikácie podľa Childa-Pugha) neviedla
k zvýšeniu expozície ertugliflozínu. AUC ertugliflozínu sa znížila približne o 13 % a Cmax sa znížila približne o 21 % v porovnaní s osobami s normálnou funkciou pečene. Tento pokles v expozícii ertugliflozínu sa nepovažuje za klinicky významný. Neexistuje žiadna klinická skúsenosť u pacientov s poruchou funkcie pečene triedy C (závažná) podľa Childa-Pugha. Väzba ertugliflozínu na
plazmatické bielkoviny nebola ovplyvnená u pacientov so stredne závažnou poruchou funkcie pečene.
Pediatrická populácia
U pediatrických pacientov sa nevykonali žiadne štúdie s ertugliflozínom.
Vplyvy veku, telesnej hmotnosti, pohlavia a rasy
Na základe analýzy populačnej farmakokinetiky nemá vek, telesná hmotnosť, pohlavie ani rasa klinicky významný účinok na farmakokinetiku ertugliflozínu.
Liekové interakcie
Hodnotenie ertugliflozínu in vitro
V štúdiách in vitro ertugliflozín a glukuronidy ertugliflozínu neinhibovali ani neinaktivovali CYP
1A2, 2C9, 2C19, 2C8, 2B6, 2D6 alebo 3A4 a neindukovali CYP 1A2, 2B6 alebo 3A4. Ertugliflozín a glukuronidy ertugliflozínu neinhibovali aktivitu UGT 1A6, 1A9 ani 2B7 in vitro. Ertugliflozín bol
vo vyšších koncentráciách, ktoré neboli klinicky významné, slabým inhibítorom UGT 1A1 a 1A4 in
vitro. Glukuronidy ertugliflozínu nemali na tieto izoformy žiadny účinok. Vo všeobecnosti je nepravdepodobné, že ertugliflozín ovplyvní farmakokinetiku súbežne podávaných liečiv
eliminovaných týmito enzýmami.
Ertugliflozín ani glukuronidy ertugliflozínu významne neinhibujú transportéry P-gp, OCT2, OAT1 alebo OAT3 ani transportné polypeptidy OATP1B1 a OATP1B3 pri klinicky významných koncentráciách in vitro. Vo všeobecnosti je nepravdepodobné, že ertugliflozín ovplyvní farmakokinetiku súbežne podávaných liečiv, ktoré sú substrátmi týchto transportérov.
Metformín
Absorpcia
Absolútna biologická dostupnosť 500 mg tablety metformíniumchloridu podávanej nalačno je približne 50-60 %. Skúšania používajúce jednorazové perorálne dávky tabliet metformíniumchloridu
500 mg až 1 500 mg a 850 mg až 2 550 mg naznačujú, že pri zvyšujúcich sa dávkach neexistuje
proporcionalita dávky, čo je spôsobené skôr zníženou absorpciou ako zmenou eliminácie. Pri obvyklých klinických dávkach a dávkovacích schémach tabliet metformíniumchloridu sa rovnovážne plazmatické koncentrácie metformínu dosiahnu za 24-48 hodín a sú zvyčajne < 1 µg/ml.
V kontrolovaných klinických skúšaniach metformínu maximálne plazmatické hladiny metformínu neprekročili 5 µg/ml, a to ani pri maximálnych dávkach.
Potrava znižuje rozsah a mierne spomaľuje absorpciu metformínu, čo dokazuje približne o 40 % nižšia priemerná maximálna plazmatická koncentrácia Cmax, o 25 % zníženie plochy pod časovou krivkou koncentrácie (AUC) a predĺženie času dosiahnutia maximálnej plazmatickej koncentrácie (Tmax) o 35 minút po podaní jednej tablety metformínu 850 mg s jedlom v porovnaní s tabletou rovnakej sily podávanou nalačno. Klinický význam tohto poklesu nie je známy.
Distribúcia
Zdanlivý distribučný objem (V/F) metformínu je po jednorazových perorálnych dávkach tabliet metformíniumchloridu 850 mg v priemere 654 ± 358 l. Väzba metformínu na plazmatické proteíny je zanedbateľná. Metformín preniká do erytrocytov.
Biotransformácia
Metformín sa vylučuje nezmenený močom. U ľudí neboli identifikované žiadne metabolity.
Eliminácia
Renálny klírens je približne 3,5-krát vyšší ako klírens kreatinínu, čo naznačuje, že tubulárna sekrécia je hlavnou cestou eliminácie metformínu. Po perorálnom podaní sa približne 90% absorbovaného
liečiva eliminuje renálnou cestou počas prvých 24 hodín s polčasom plazmatickej eliminácie približne
6,2 hodiny.
Osobitné skupinypacientov
Porucha funkcie obličiek
U pacientov so zníženou funkciou obličiek sa polčas rozpadu metformínu v plazme a krvi predlžuje a renálny klírens sa znižuje v pomere k zníženiu eGFR (pozri časti 4.3 a 4.4).
Porucha funkcie pečene
Neboli vykonané žiadne farmakokinetické štúdie s metformínom u pacientov s hepatálnou insuficienciou.
Vplyvy veku, telesnej hmotnosti, pohlavia a rasy
Obmedzené údaje z kontrolovaných skúšaní metformínu u zdravých starších osôb naznačujú, že celkový plazmatický klírens metformínu sa znižuje, polčas sa predlžuje a Cmax sa zvyšuje v porovnaní so zdravými mladými osobami. Z týchto údajov sa zdá, že zmena farmakokinetiky metformínu
s pribúdajúcim vekom je primárne dôsledkom zmeny renálnej funkcie.
Farmakokinetické parametre metformínu sa významne nelíšili medzi normálnymi osobami a pacientmi s diabetom 2. typu, keď sa analyzovali podľa pohlavia. Podobne, v kontrolovaných klinických skúšaniach u pacientov s diabetom 2. typu bol antihyperglykemický účinok metformínu porovnateľný
u mužov a žien.
Neuskutočnili sa žiadne štúdie farmakokinetických parametrov metformínu podľa rasy.
V kontrolovaných klinických štúdiách s metformínom u pacientov s diabetom 2. typu bol antihyperglykemický účinok porovnateľný u belochov (n=249), černochov (n=51) a hispáncov (n=24).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, akútnej toxicity, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
Všeobecná toxicita
Ertugliflozín
Štúdie toxicity po opakovanom perorálnom podávaní sa vykonali u myší až do 13 týždňov, u potkanov až do 26 týždňov a u psov až do 39 týždňov. Prejavy toxicity, ktoré sa považovali za nežiaduce boli vo
všeobecnosti pozorované pri expozíciách vyšších alebo rovných 77-násobku neviazanej expozície
(AUC) u ľudí pri maximálnej odporúčanej dávke u ľudí (maximum recommended human dose, MRHD) 15 mg/deň. Väčšina toxicity bola konzistentná s farmakológiou súvisiacou s vylučovaním glukózy močom a zahŕňala zníženie telesnej hmotnosti a telesného tuku, zvýšenie konzumácie jedla, hnačku, dehydratáciu, zníženie hladiny glukózy v sére a zvýšenie hladín ostatných parametrov v sére odzrkadľujúcich zvýšený metabolizmus bielkovín, glukoneogenézu a nerovnováhu elektrolytov
a zmeny moču ako je napr. polyúria, glukozúria a kalciúria. Mikroskopické zmeny súvisiace
s glukozúriou a/alebo kalciúriou pozorované len u hlodavcov zahŕňali dilatáciu renálnych tubulov, hypertrofiu zona glomerulosa v nadobličkách (potkany) a zvýšenie trabekulárnej kosti (potkany).
U psov sa okrem vracania neobjavili žiadne nálezy nežiaducej toxicity pri 379-násobku neviazanej expozície (AUC) u ľudí pri MRHD 15 mg/deň.
Karcinogenéza
Ertugliflozín
V 2-ročnej štúdii karcinogenity u myší sa ertugliflozín podával perorálnou sondou do žalúdka
v dávkach 5, 15 a 40 mg/kg/deň. Neobjavili sa žiadne neoplastické nálezy súvisiace s ertugliflozínom pri dávkach až do 40 mg/kg/deň (približne 41-násobok neviazanej expozície u ľudí pri MRHD
15 mg/deň na základe AUC). V 2-ročnej štúdii karcinogenity u potkanov sa ertugliflozín podával perorálnou sondou do žalúdka v dávkach 1,5; 5 a 15 mg/kg/deň. Neoplastické nálezy súvisiace
s ertugliflozínom zahŕňali zvýšený výskyt benígneho, adrenálneho medulárneho feochromocytómu
u samcov potkanov pri 15 mg/kg/deň. Tento nález sa pripisoval malabsorpcii uhľohydrátov vedúcej k zmenenej homeostáze vápnika a nepovažoval sa za významný pre riziko u ľudí. Hladina bez pozorovaného účinku (no-observed-effect level, NOEL) pre neopláziu bola 5 mg/kg/deň (približne 16- násobok neviazanej expozície u ľudí pri MRHD 15 mg/deň).
Metformín
Dlhodobé skúšania karcinogenity sa vykonali na potkanoch (dávkovanie v trvaní 104 týždňov)
v dávkach do 900 mg/kg/deň a vrátane 900 mg/kg/deň a na myšiach (dávkovanie v trvaní 91 týždňov)
v dávkach 1 500 mg/kg/deň a vrátane 1 500 mg/kg/deň. Tieto dávky predstavujú približne štvornásobok maximálnej odporúčanej dennej dávky 2 000 mg pre človeka vychádzajúc z porovnaní povrchovej plochy tela. Nebol zistený žiadny dôkaz karcinogenity pri metformíne u samcov, ani
u samíc myší. Podobne, sa na samcoch potkanov nepozoroval žiadny tumorogénny potenciál súvisiaci s metformínom. Bola však zvýšený výskyt benígnych stromálnych maternicových polypov u samíc
potkanov pri dávke 900 mg/kg/deň.
Mutagenéza
Ertugliflozín
Ertugliflozín nebol mutagénny ani klastogénny s metabolickou aktiváciou alebo bez nej v teste mikrobiálnej reverznej mutácie, cytogenetických testoch in vitro (ľudské lymfocyty) a testoch mikrojadier in vivo u potkanov.
Metformín
Neexistujú žiadne dôkazy o mutagénnom potenciáli metformínu v nasledujúcich testoch in vitro: Amesov test (S. typhimurium), test génovej mutácie (bunky myšacieho lymfómu) alebo test chromozomálnych aberácií (ľudské lymfocyty). Výsledky testu mikrojadier na myšiach in vivo boli tiež negatívne.
Reprodukčná toxikológia
Ertugliflozín
V štúdiách fertility a embryonálneho vývinu na potkanoch sa samcom a samiciam potkanov podával ertugliflozín v dávke 5, 25 a 250 mg/kg/deň. Neobjavili sa žiadne účinky na fertilitu pri dávke
250 mg/kg/deň (približne 386-násobok neviazanej expozície u ľudí pri MRHD 15 mg/deň na základe
porovnaní AUC). Ertugliflozín nemal nežiaduci vplyv na výsledky vývinu u potkanov pri expozíciách u matiek zodpovedajúcich 239-násobku expozície u ľudí pri maximálnej klinickej dávke 15 mg/deň
a u králikov pri expozíciách u matiek zodpovedajúcich 1 069-násobku expozície u ľudí pri maximálnej
klinickej dávke 15 mg/deň, na základe AUC. Pri dávke toxickej pre matky u potkanov
(250 mg/kg/deň) sa pozorovala nižšia životaschopnosť plodu a vyšší výskyt viscerálnej malformácie pri expozícii u matky, ktorá bola 510-násobkom maximálnej klinickej dávky 15 mg/deň.
V štúdii pre- a postnatálneho vývinu sa pozoroval znížený postnatálny rast a vývin u potkanov, ktorým bol ertugliflozín podávaný od 6. dňa gravidity po 21. deň laktácie v dávke ≥ 100 mg/kg/deň (odhadovaný 239-násobok expozície u ľudí pri maximálnej klinickej dávke 15 mg/deň na základe AUC). Pohlavné dozrievanie bolo oneskorené u obidvoch pohlaví pri dávke 250 mg/kg/deň (odhadovaný 620-násobok MRHD pri dávke 15 mg/deň na základe AUC).
Ak sa ertugliflozín podával mláďatám potkana od 21. postnatálneho dňa (postnatal day, PND) do 90. PND, počas obdobia renálneho vývinu zodpovedajúceho neskorému druhému a tretiemu trimestru ľudskej gravidity, pozorovalo sa zvýšenie hmotnosti obličiek, dilatácia renálnej panvičky a tubulov
a mineralizácia renálnych tubulov pri expozícii 13-násobku maximálnej klinickej dávky 15 mg/deň na základe AUC. Účinky na kosť (kratšia dĺžka stehennej kosti, nárast trabekulárnej kosti v stehennej
kosti) ako aj účinky oneskoreného dospievania sa pozorovali pri expozícii 817-násobku MRHD
15 mg/deň na základe AUC. Účinky na obličky a kosť sa úplne nezvrátili po 1-mesačnom období zotavenia.
M
etformín
Fertilita samcov a samíc potkanov nebola ovplyvnená metformínom podávaným v dávkach až
600 mg/kg/deň, čo je približne trojnásobok maximálnej odporúčanej dennej dávky pre ľudí vychádzajúc z porovnaní povrchovej plochy tela. Metformín nemal nepriaznivý vplyv na výsledky
vývoja pri podávaní v dávkach až do 600 mg/kg/deň u potkanov a králikov. To predstavuje expozíciu
rovnú približne 2-násobku expozície pri maximálnej odporúčanej dávke pre ľudí 2 000 mg vychádzajúc z porovnaní povrchovej plochy tela u potkanov a až 6-násobku expozície pri maximálnej odporúčanej dávke pre ľudí 2 000 mg vychádzajúc z porovnaní povrchovej plochy tela u králikov. Stanovenie fetálnych koncentrácií dokázalo čiastočnú placentárnu bariéru pre metformín.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety
povidón K29-32 (E1201)
mikrokryštalická celulóza (E460)
krospovidón (E1202) laurylsíran sodný (E487) stearan horečnatý (E470b)
Filmovýobal
Segluromet2,5mg/850mgfilmomobalenétabletyaSegluromet7,5mg/850mgfilmomobalenétablety
hypromelóza (E464)
hydroxypropylcelulóza (E463)
oxid titaničitý (E171) červený oxid železitý (E172) žltý oxid železitý (E172) čierny oxid železitý (E172) karnaubský vosk (E903)
Segluromet2,5mg/1000mgfilmomobalenétabletyaSegluromet7,5mg/1000mgfilmomobalenétablety
hypromelóza (E464)
hydroxypropylcelulóza (E463)
oxid titaničitý (E171) červený oxid železitý (E172) karnaubský vosk (E903)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Al/PVC/PA/Al blistre.
Balenia po 14, 28, 56, 60, 168 a 180 filmom obalených tabliet v neperforovaných blistroch
a viacnásobné balenie obsahujúce 196 (4 balenia po 49) filmom obalených tabliet v neperforovaných blistroch.
Balenia po 30 x 1 filmom obalená tableta v perforovaných blistroch umožňujúcich oddelenie jednotlivej dávky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
Segluromet2,5mg/850 mgfilmomobalenétablety
EU/1/18/1265/001
EU/1/18/1265/002
EU/1/18/1265/003
EU/1/18/1265/004
EU/1/18/1265/005
EU/1/18/1265/006
EU/1/18/1265/007
EU/1/18/1265/029
Segluromet2,5mg/1 000mgfilmomobalenétablety
EU/1/18/1265/008
EU/1/18/1265/009
EU/1/18/1265/010
EU/1/18/1265/011
EU/1/18/1265/012
EU/1/18/1265/013
EU/1/18/1265/014
EU/1/18/1265/030
Segluromet7,5mg/850 mgfilmomobalenétablety
EU/1/18/1265/015
EU/1/18/1265/016
EU/1/18/1265/017
EU/1/18/1265/018
EU/1/18/1265/019
EU/1/18/1265/020
EU/1/18/1265/021
EU/1/18/1265/031
Segluromet
7,5
mg/1 000mg
filmom
obalené
tablety
EU/1/18/1265/022
EU/1/18/1265/023
EU/1/18/1265/024
EU/1/18/1265/025
EU/1/18/1265/026
EU/1/18/1265/027
EU/1/18/1265/028
EU/1/18/1265/032
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 23. marca 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.