
globín pretrváva po dobu >21 dní a je podozrenie, že
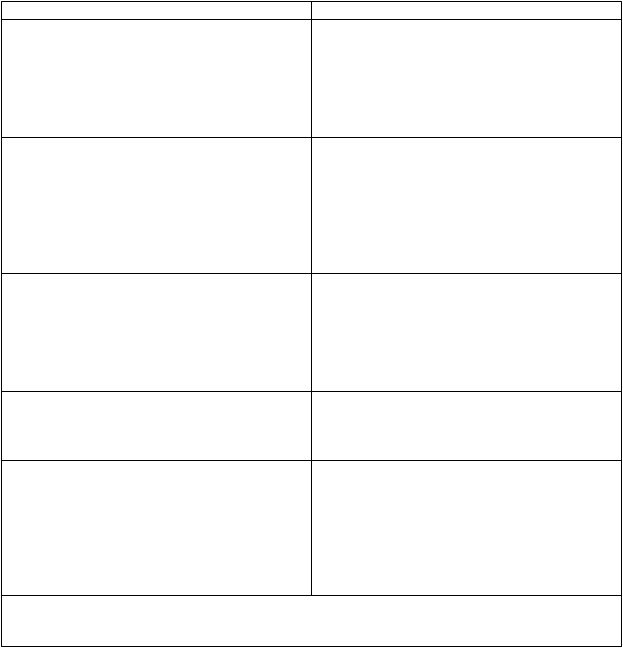
súvisí s Rydaptom.
Vysaďte liečbu Rydaptom na 3 dni (6 dávok), potom opakujte dávku 50 mg dvakrát denne a ak je tolerovaná, postupne ju zvýšte na 100 mg dvakrát denne.
Iné nehematologické toxicity 3./4. stupňa Vysaďte liečbu Rydaptom, kým sa udalosť zmení na ≤2. stupeň, potom opakujte dávku Rydaptu
50 mg dvakrát denne a ak je tolerovaná, zvýšte ju na 100 mg dvakrát denne.
Vysaďte liečbu Rydaptom, ak sa toxické účinky nezmenia na ≤2. stupeň počas 21 dní alebo sa znovu objaví závažná toxicita pri zníženej dávke
Rydaptu.
ANC: Absolútny počet neutrofilov
Závažnosť CTCAE: 1. stupeň=mierne symptómy; 2. stupeň=stredne závažné symptómy;
3. stupeň=závažné symptómy; 4. stupeň=život ohrozujúce symptómy.
Vynechaná dávka
V prípade vynechania dávky má pacient užiť ďalšiu dávku v obvyklom čase.
Ak dôjde k vracaniu, pacient nemá užiť ďalšiu dávku Rydaptu, ale má užiť ďalšiu plánovanú dávku.
Osobitné skupiny pacientov
Starší pacienti (≥65 rokov)
U pacientov starších ako 65 rokov nie je potrebná úprava dávkovania (pozri časť 5.2). U pacientov s AML vo veku 60-70 rokov sú obmedzené skúsenosti s midostaurínom a u pacientov s AML nad
70 rokov nie sú žiadne skúsenosti. U pacientov vo veku ≥60 rokov sa má Rydapt používať len
u pacientov, ktorí sú vhodní na intenzívnu indukčnú chemoterapiu s adekvátnym výkonnostným
stupňom a bez signifikantných komorbidít.
Porucha funkcie obličiek
U pacientov s mierne alebo stredne ťažkou poruchou funkcie obličiek nie je potrebná úprava
dávkovania. Klinické skúsenosti u pacientov s ťažkou poruchou funkcie obličiek sú obmedzené
a k dispozícii nie sú žiadne údaje u pacientov v terminálnom štádiu ochorenia obličiek (pozri časti 4.4
a 5.2).
Porucha funkcie pečene
U pacientov s mierne alebo stredne ťažkou poruchou funkcie pečene (trieda A alebo B podľa Childa-Pugha) nie je potrebná úprava dávkovania (pozri časť 5.2). Neuskutočnila sa žiadna štúdia u pacientov s ťažkou (trieda C podľa Childa-Pugha) poruchou funkcie pečene (pozri časť 4.4).
Akútna promyelocytová leukémia
Rydapt sa neskúmal u pacientov s akútnou promyelocytovou leukémiou, a preto sa jeho použitie
u tejto populácie pacientov neodporúča.
Pediatrická populácia
Bezpečnosť a účinnosť Rydaptu u detí a dospievajúcich mladších ako 18 rokov neboli stanovené
(pozri časť 5.1). V súčasnosti dostupné údaje sú opísané v časti 5.2, ale neumožňujú uviesť odporúčania na dávkovanie.
Spôsob podávania
Rydapt je určený na perorálne použitie.
Kapsuly sa majú prehltnúť celé a zapiť pohárom vody. Nemajú sa otvárať, drviť alebo žuvať, aby sa
zabezpečilo správne dávkovanie a zabránilo sa nepríjemnej chuti obsahu kapsuly.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Súbežné podanie silných induktorov CYP3A4, napr. rifampicín, ľubovník bodkovaný (Hypericum perforatum), karbamazepín, enzalutamid, fenytoín (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
N
eutropénia a infekcie
Neutropénia sa objavila u pacientov, ktorí dostávali Rydapt ako monoterapiu a v kombinácii
s chemoterapiou (pozri časť 4.8). Ťažká neutropénia (ANC <0,5 x 109/l) bola spravidla reverzibilná po vysadení Rydaptu až do uzdravenia a prerušenia liečby v štúdiách ASM, SM-AHN a MCL. Počty bielych krviniek (WBC, White blood cell counts,) sa majú pravidelne sledovať, najmä na začiatku
liečby.
U pacientov, u ktorých sa vyvinie nevysvetliteľná ťažká neutropénia, sa má liečba Rydaptom prerušiť, až kým je hodnota ANC ≥1,0 x 109/l, ako sa odporúča v tabuľkách 1 a 2. Liečba Rydaptom sa má prerušiť u pacientov, u ktorých sa objaví recidivujúca alebo dlhotrvajúca ťažká neutropénia, u ktorej je podozrenie, že súvisí s Rydaptom (pozri časť 4.2).
Akákoľvek aktívna závažná infekcia má byť zvládnutá pred začatím liečby Rydaptom v monoterapii. Pacienti musia byť sledovaní kvôli prejavom a príznakom infekcie, vrátane akýchkoľvek infekcií súvisiacich s použitím zdravotníckej pomôcky, a ak sa určí diagnóza infekcie, okamžite sa podľa potreby musí začať vhodná liečba, vrátane prerušenia liečby Rydaptom.
Porucha funkcie srdca
Pacienti so symptomatickým kongestívnym zlyhaním srdca boli vylúčení z klinických štúdií.
V štúdiách ASM, SM-AHN a MCL sa vyskytla porucha funkcie srdca ako kongestívne zlyhávanie srdca (SZ) (vrátanie niekoľkých úmrtí) a prechodné zníženie ejekčnej frakcie ľavej komory (LVEF, Left Ventricular Ejection Fraction). V randomizovanej štúdii AML sa medzi ramenami Rydapt + chemoterapia a placebo + chemoterapia nepozoroval žiadny rozdiel v SZ. U rizikových pacientov sa má Rydapt používať opatrne a pacient sa má starostlivo sledovať meraním LVEF, ak je klinicky indikované (na začiatku a počas liečby).
U pacientov liečených midostaurínom sa zaznamenala zvýšená frekvencia predĺženia QTc (pozri časť 4.8), avšak mechanizmus vzniku tohto javu sa nezistil. Opatrnosť je na mieste u pacientov
s rizikom predĺženia QTc (napr. z dôvodu súbežného užívania liekov a/alebo porúch elektrolytov). Stanovenie intervalu QT prostredníctvom EKG sa má zvážiť, ak sa Rydapt užíva súbežne s liekmi, ktoré môžu predĺžiť interval QT.
Pľúcna toxicita
Intersticiálna choroba pľúc (ILD, Interstitial Lung Disease) a pneumonitída, v niektorých prípadoch
fatálne, sa vyskytli u pacientov liečených Rydaptom v monoterapii alebo v kombinácii
s chemoterapiou. Pacienti sa majú sledovať kvôli príznakom svedčiacim o ILD alebo pneumonitíde a liečba Rydaptom sa má prerušiť u pacientov, u ktorých sa vyskytnú pľúcne príznaky svedčiace
o ILD alebo pneumonitíde stupňa ≥3 (NCI CTCAE).
Embryofetálna toxicita a dojčenie
Gravidné ženy musia byť informované o možnom riziku pre plod; ženy v reprodukčnom veku musia
byť poučené o vykonaní tehotenského testu počas 7 dní pred začatím liečby Rydaptom a použití účinnej antikoncepcie počas liečby Rydaptom a najmenej po dobu 4 mesiacov po ukončení liečby. Ženy, ktoré používajú hormonálnu antikoncepciu, majú navyše používať bariérovú metódu antikoncepcie.
Vzhľadom na možné závažné nežiaduce reakcie u dojčených detí spôsobené Rydaptom musia ženy prerušeniť dojčenie počas liečby Rydaptom a najmenej 4 mesiace po ukončení liečby (pozri časť 4.6).
Ť
ažká poruchafunkciepečene
Opatrnosť je na mieste pri zvažovaní podania midostaurínu u pacientov s ťažkou poruchou funkcie
pečene a pacienti majú byť starostlivo sledovaní pre toxicitu (pozri časť 5.2).
Ťažká poruchafunkcieobličiek
Opatrnosť je na mieste pri zvažovaní podania midostaurínu u pacientov s ťažkou poruchou funkcie
obličiek alebo v terminálnom štádiu ochorenia obličiek a pacienti majú byť starostlivo sledovaní pre
toxicitu (pozri časť 5.2).
Interakcie
Opatrnosť je potrebná pri predpisovaní midostaurínu súbežne s liekmi, ktoré sú silnými inhibítormi
CYP3A4 ako napríklad, ale nie výhradne, antimykotiká (napr. ketokonazol), niektoré antivirotiká (napr. ritonavir), makrolidové antibiotiká (napr. klaritromycín) a nefazodón, pretože môžu zvyšovať plazmatické koncentrácie midostaurínu, najmä pri (opätovnom) začatí liečby midostaurínom (pozri časť 4.5). Je potrebné zvážiť alternatívne lieky, ktoré nie sú silnými inhibítormi aktivity CYP3A4.
V situáciách, kde neexistujú uspokojivé terapeutické alternatívy, sa majú pacienti starostlivo sledovať
kvôli toxicite súvisiacej s midostaurínom.
Pomocné látky
Rydapt obsahuje hydroxystearoylmakrogol-glycerol, ktorý môže spôsobiť žalúdočné ťažkosti
a hnačku.
100 mg dávka Rydaptu obsahuje približne 14 objemových % bezvodého etanolu, čo zodpovedá
333 mg alkoholu. Zodpovedá to 8,4 ml piva alebo 3,5 ml vína. Alkohol môže byť škodlivý u pacientov s problémami súvisiacimi s alkoholom, epilepsiou alebo problémami s pečeňou, alebo počas gravidity
alebo dojčenia.
4.5 Liekové a iné interakcie
Midostaurín sa rozsiahle metabolizuje v pečeni hlavne prostredníctvom enzýmov CYP3A4, ktoré sú
buď indukované alebo inhibované radom súbežne podávaných liekov.
Účinok iných liekovna Rydapt
Lieky alebo liečivá, o ktorých je známe, že ovplyvňujú aktivitu CYP3A4, môžu ovplyvniť
plazmatické koncentrácie midostaurínu a teda bezpečnosť a/alebo účinnosť Rydaptu.
Silné induktory CYP3A4
Súbežné použitie Rydaptu so silnými induktormi CYP3A4 (napr. karbamazepín, rifampicín, enzalutamid, fenytoín, ľubovník bodkovaný [Hypericum perforatum]) je kontraindikované (pozri časť 4.3). Silné induktory CYP3A4 znižujú expozíciu midostaurínu a jeho aktívnych metabolitov
(CGP52421 a CGP62221). V štúdii u zdravých jedincov znížilo súbežné podávanie silného induktora
CYP3A4 rifampicínu (600 mg denne) v rovnovážnom stave s jednorazovou dávkou midostaurínu
50 mg Cmax midostaurínu v priemere o 73 % a AUCinf o 96 %, v uvedenom poradí. CGP62221
vykazoval podobnú štruktúru. Priemerná AUClast CGP52421 sa znížila o 60 %.
Silné inhibítory CYP3A4
Silné inhibítory CYP3A4 môžu zvýšiť koncentrácie midostaurínu v krvi. V štúdii u 36 zdravých jedincov viedlo súbežné podávanie silného inhibítora CYP3A4 ketokonazolu v rovnovážnom stave s jednorazovou dávkou midostaurínu 50 mg k signifikantnému zvýšeniu expozície midostaurínu (1,8-násobné zvýšenie Cmax a 10-násobné zvýšenie AUCinf) a 3,5-násobnému zvýšeniu AUCinf CGP62221, zatiaľ čo Cmax aktívnych metabolitov (CGP62221 a CGP52421) sa znížili o polovicu
(pozri časť 5.2). Midostaurín v rovnovážnom stave (50 mg dvakrát denne po dobu 21 dní) so silným
inhibítorom CYP3A4 itrakonazolom v rovnovážnom stave v podskupine pacientov (N=7) zvýšil
expozíciu midostaurínu v rovnovážnom stave (Cmin) 2,09-násobne. Cmin CGP52421 sa zvýšila
1,3-násobne, zatiaľ čo sa nepozoroval žiadny signifikantný účinok v expozícii CGP62221 (pozri
časť 4.4).
Účinok Rydaptu na iné lieky
Midostaurín nie je inhibítorom CYP3A4 in vivo. Farmakokinetika midazolamu (skúmaná citlivosť
CYP3A4) nebola ovplyvnená po trojdňovom podávaní midostaurínu zdravým jedincom.
Na základe údajov in vitro midostaurín a/alebo jeho metabolity majú potenciál inhibovať enzýmy
CYP1A2, CYP2D6, CYP2C8, CYP2C9, CYP2E1 a CYP3A4/5.
Na základe údajov in vitro midostaurín a/alebo jeho metabolity majú potenciál indukovať enzýmy CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP3A4. In vitro midostaurín inhiboval OATP1B1, BCRP a P-glykoproteín (P-gp) (pozri časť 5.2). Kombinácia údajov o autoindukcii midostaurínu in vivo po opakovanom podávaní a o zvýšení koncentrácií 4β-OH-cholesterolu v plazme naznačuje, že midostaurín môže byť in vivo aspoň stredne silný induktor CYP3A4.
Nevykonali sa štúdie in vivo pre skúmanie indukcie a inhibície enzýmov a transportérov midostaurínom a aktívnymi metabolitmi. Lieky s úzkym terapeutickým rozsahom, ktoré sú substrátmi CYP1A2 (napr. tizanidín), CYP2D6 (napr. kodeín), CYP2C8 (napr. paklitaxel), CYP2C9 (napr. warfarín), CYP2C19 (napr. omeprazol), CYP2E1 (napr. chlórzoxazón), CYP3A4/5 (napr. takrolimus), CYP2B6 (napr. efavirenz), P-gp (napr. paklitaxel), BCRP (napr. atorvastatín) alebo OATP1B1 (napr. digoxín), sa majú používať opatrne, keď sa podávajú súbežne s midostaurínom, a môže byť potrebná úprava dávkovania pre udržanie optimálnej expozície (pozri časť 5.2).
V súčasnosti nie je známe, či midostaurín môže znížiť účinnosť hormonálnej antikoncepcie, a preto ženy používajúce hormonálnu antikoncepciu majú pridať bariérovú metódu antikoncepcie (pozri časť 4.6).
Interakcie s jedlom
U zdravých jedincov sa absorpcia midostaurínu (AUC) zvýšila v priemere o 22 %, keď sa Rydapt
podával súbežne so štandardným jedlom, a v priemere o 59 %, keď sa podával súbežne s jedlom s vysokým obsahom tuku. Maximálna koncentrácia midostaurínu (Cmax) sa znížila o 20 % pri štandardnom jedle a o 27 % pri jedle s vysokým obsahom tuku oproti podaniu na lačný žalúdok (pozri časť 5.2).
Rydapt sa odporúča podávať s jedlom.
4.6 Fertilita, gravidita a laktácia
Ž
eny vofertilnomveku
Ženy vo ferilnom veku musia byť informované, že štúdie na zvieratách ukazujú, že midostaurín je
škodlivý pre vyvýjajúci sa plod. Sexuálne aktívne ženy vo fertilnom veku sú poučené o vykonaní
tehotenského testu pred začatím liečby Rydaptom a použití účinnej antikoncepcie (metódy, v dôsledku ktorých je výskyt gravidity u menej ako 1 %) pri používaní Rydaptu a najmenej po dobu 4 mesiacov
po ukončení liečby Rydaptom. V súčasnosti nie je známe, či midostaurín môže znížiť účinnosť hormonálnej antikoncepcie, a preto ženy používajúce hormonálnu antikoncepciu majú pridať bariérovú metódu antikoncepcie.
Gravidita
Midostaurín môže pri podaní gravidnej žene spôsobiť poškodenie plodu. Neexistujú žiadne adekvátne
a dobre kontrolované štúdie u gravidných žien. Reprodukčné štúdie na potkanoch a králikoch
preukázali, že midostaurín indukuje fetotoxicitu (pozri časť 5.3). Rydapt sa neodporúča užívať počas gravidity alebo u žien vo fertilnom veku nepoužívajúcich antikoncepciu. Gravidné ženy musia byť poučené o možnom riziku pre plod.
Dojčenie
Nie je známe, či sa midostaurín alebo jeho aktívne metabolity vylučujú do materského mlieka.
Dostupné údaje o zvieratách ukázali, že midostaurín a jeho aktívne metabolity prechádzajú do mlieka
dojčiacich potkanov. Dojčenie sa má prerušiť počas liečby Rydaptom a po dobu najmenej 4 mesiacov
po ukončení liečby.
Fertilita
K dispozícii nie sú žiadne údaje o účinku Rydaptu na fertilitu u ľudí. Štúdie na zvieratách
s midostaurínom preukázali poškodenie fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Rydapt má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientov užívajúcich Rydapt boli hlásené závraty a vertigo a má sa to zvážiť pri posudzovaní pacientovej schopnosti viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
AML
Vyhodnotenie bezpečnosti Rydaptu (50 mg dvakrát denne) u pacientov s novodiagnostikovanou, FLT3 mutovanou AML je založené na randomizovanej, dvojito zaslepenej, placebom kontrolovanej
štúdii fázy III u 717 pacientov. Celkový medián doby trvania expozície bol 42 dní (rozpätie 2 až
576 dní) pre pacientov v ramene s Rydaptom a štandardnou chemoterapiou oproti 34 dňom (rozpätie 1
až 465 dní) pre pacientov v ramene s placebom a štandardnou chemoterapiou. U 205 pacientov (120
v ramene s Rydaptom a 85 v ramene s placebom), ktorí sa zúčastnili udržiavacej fázy, bol medián doby trvania expozície pri udržiavaní 11 mesiacov pre obe ramená (16 až 520 dní pre pacientov
v ramene s Rydaptom a 22 až 381 dní v ramene s placebom).
Najčastejšími nežiaducimi reakciami na liek (ADR, Adverse Drug Reactions) v ramene s Rydaptom boli febrilná neutropénia (83,4 %), nauzea (83,4 %), exfoliatívna dermatitída (61,6 %), vracanie (60,7 %), bolesť hlavy (45,9 %), petechie (35,8 %) a pyrexia (34,5 %). Najčastejšími ADR
3./4. stupňa boli febrilná neutropénia (83,5 %), lymfopénia (20,0 %), infekcia súvisiaca s použitím
zdravotníckej pomôcky (15,7 %), exfoliatívna dermatitída (13,6 %), hyperglykémia (7,0 %) a nauzea (5,8 %). Najčastejšími abnormálnymi laboratórnymi hodnotami boli znížený hemoglobín (97,3 %), znížený ANC (86,7 %), zvýšená ALT (84,2 %), zvýšená AST (73,9 %) a hypokaliémia (61,7 %). Najčastejšími abnormálnymi laboratórnymi hodnotami 3./4. stupňa boli znížený ANC (85,8 %), znížený hemoglobín (78,5 %), zvýšená ALT (19,4 %) a hypokaliémia (13,9 %).
Závažné ADR sa vyskytli s podobným výskytom u pacientov s Rydaptom oproti ramenu s placebom.
Najčastejším závažným ADR v oboch ramenách bola febrilná neutropénia (16 %).
Prerušenie liečby v dôsledku akejkoľvek nežiaducej reakcie sa vyskytlo u 3,1 % pacientov v ramene s Rydaptom oproti 1,3 % v ramene s placebom. Najčastejšia nežiaduca reakcia 3./4. stupňa vedúca
k prerušeniu liečby v ramene s Rydaptom bola exfoliatívna dermatitída (1,2 %).
Bezpečnostný profil počas udržiavacej fázy
Kým v tabuľke 3 je uvedený výskyt ADR počas celej doby trvania štúdie, ak sa udržiavacia fáza
(Rydapt v monoterapii alebo placebo) hodnotila samostatne, pozoroval sa rozdiel v type a závažnosti
ADR. Celkový výskyt ADR počas udržiavacej fázy bol celkovo nižší ako počas indukčnej
a konsolidačnej fázy. Výskyt ADR počas udržiavacej fázy bol však vyšší v ramene s Rydaptom ako
v ramene s placebom. ADR vyskytujúce sa častejšie v ramene s midostaurínom oproti placebu počas udržiavacej fázy zahŕňali: nauzeu (46,4 % oproti 17,9 %), hyperglykémiu (20,2 % oproti 12,5 %), vracanie (19 % oproti 5,4 %) a predĺženie QT (11,9 % oproti 5,4 %).
Väčšina hlásených hematologických abnormalít sa vyskytla v priebehu indukčnej a konsolidačnej fázy, keď pacienti dostávali Rydapt alebo placebo v kombinácii s chemoterapiou. Najčastejšími hematologickými abnormalitami 3./4. stupňa hlásenými u pacientov počas udržiavacej fázy
s Rydaptom boli zníženie ANC (20,8 % oproti 18,8 %) a leukopénia (7,5 % oproti 5,9 %).
ADR hlásené počas udržiavacej fázy viedli k prerušeniu liečby u 1,2 % pacientov v ramene s Rydaptom a u žiadnych v ramene s placebom.
ASM, SM-AHN a MCL
Bezpečnosť Rydaptu (100 mg dvakrát denne) v monoterapii u pacientov s ASM, SM-AHN a MCL sa hodnotila u 142 pacientov v dvoch otvorených, multicentrických štúdiách s jedným ramenom. Medián doby trvania expozície Rydaptu bol 11,4 mesiacov (rozpätie: 0 až 81 mesiacov).
Najčastejšími ADR boli nauzea (82 %), vracanie (68 %), hnačka (51 %), periférny edém (35 %)
a únava (31 %). Najčastejšími ADR 3./4. stupňa boli únava (8,5 %), sepsa (7,7 %), pneumónia (7 %), febrilná neutropénia (7 %) a hnačka (6,3 %). Najčastejšími nehematologickými laboratórnymi
abnormalitami boli hyperglykémia (93,7 %), zvýšená celková hladina bilirubínu (40,1 %), zvýšená
hladina lipázy (39,4 %), zvýšená hladina aspartátaminotransferázy (AST) (33,8 %)
a alanínaminotransferázy (ALT) (33,1 %), zatiaľ čo najčastejšími hematologickými laboratórnymi abnormalitami boli zníženie absolútneho počtu lymfocytov (73,2 %) a ANC (58,5 %). Najčastejšími laboratórnymi abnormalitami 3./4. stupňa boli zníženie absolútneho počtu lymfocytov (45,8 %), zníženie ANC (26,8 %), hyperglykémia (19 %) a zvýšená hladina lipázy (17,6 %).
Modifikácie dávky (prerušenie alebo úprava) kvôli ADR sa vyskytli u 31 % pacientov. Najčastejšími
ADR, ktoré viedli k modifikácii dávky (výskyt ≥5 %), boli nauzea a vracanie.
ADR, ktoré viedli k prerušeniu liečby, sa vyskytli u 9,2 % pacientov. Najčastejšími (výskyt ≥1 %) boli febrilná neutropénia, nevoľnosť, vracanie a pleurálny výpotok.
T
abuľkový zoznamnežiaducichreakcií na liek
ADR sú uvedené podľa MedDRA tried orgánových systémov. V rámci každej triedy orgánových
systémov sú ADR zoradené podľa frekvencie, začínajúc s najčastejšími reakciami, pomocou
nasledujúcej konvencie (CIOMS III): veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000); neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
AML
V tabuľke 3 sú uvedené kategórie frekvencie ADR hlásených v štúdii fázy III u pacientov s novodiagnostikovanou AML s FLT3 mutáciou.
Tabuľka 3 Nežiaduce reakcie na liek pozorované v klinickej štúdii AML
Všetky
stupne 3./4. stupeň
N
ežiaduca reakcia na liek
Infekcie a nákazy
R
ydapt + chemo n=229
1
%
R
ydapt + chemo n=345
1
%
K
ategória frekvencie
Infekcia súvisiaca s použitím
zdravotníckej pomôcky
24 15,7 Veľmi časté
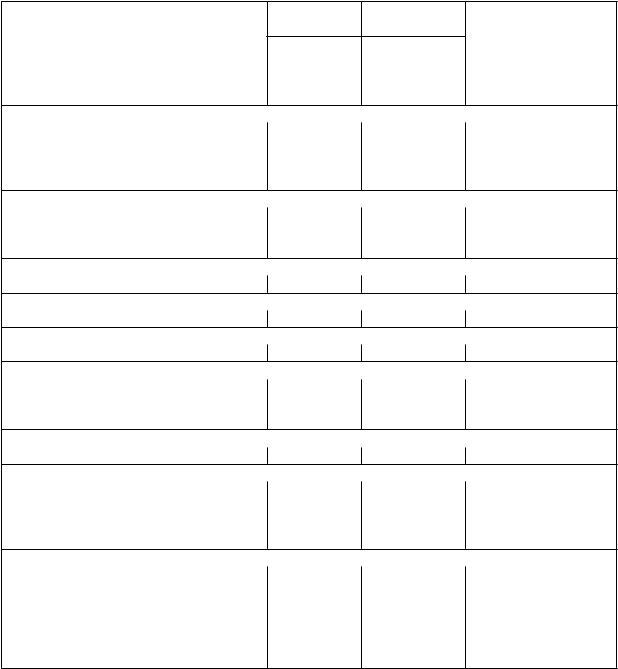
Infekcia horných dýchacích ciest 5,2 0,6 Časté Neutropenická sepsa 0,9 3,5 Menej časté
Poruchy krvi a lymfatického systémuFebrilná neutropénia 83,4 83,5 Veľmi časté
Petechie 35,8 1,2 Veľmi časté Lymfopénia 16,6 20 Veľmi časté
Poruchy imunitného systémuPrecitlivenosť 15,7 0,6 Veľmi časté
Poruchy metabolizmu a výživyHyperurikémia 8,3 0,6 Časté
Psychické poruchyInsomnia 12,2 0 Veľmi časté
Poruchy nervového systémuBolesť hlavy 45,9 2,6 Veľmi časté
Synkopa 5,2 4,6 Časté
Tremor 3,9 0 Časté
Poruchy okaOpuch očného viečka 3,1 0 Časté
Poruchy srdca a srdcovej činnostiHypotenzia 14,4 5,5 Veľmi časté
Sínusová tachykardia 9,6 1,2 Časté
Hypertenzia 7,9 2,3 Časté Perikardiálna efúzia 3,5 0,6 Časté
Poruchy dýchacej sústavy, hrudníka a mediastínaEpistaxa 27,5 2,6 Veľmi časté
Bolesť hrtana 11,8 0,6 Veľmi časté
Dyspnoe 10,9 5,5 Veľmi časté Pleurálny výpotok 5,7 0,9 Časté Nazofaryngitída 8,7 0 Časté Syndróm akútnej dychovej tiesne 2,2 2,3 Časté
P
oruchy gastrointestinálneho traktu
Nauzea 83,4 5,8 Veľmi časté
Vracanie 60,7 2,9 Veľmi časté Stomatitída 21,8 3,5 Veľmi časté Bolesť v hornej časti brucha 16,6 0 Veľmi časté Hemoroidy 15,3 1,4 Veľmi časté Anorektálny diskomfort 7 0,9 Časté Abdominálny diskomfort 3,5 0 Časté Poruchy kože a podkožného tkaniva
Exfoliatívna dermatitída 61,6 13,6 Veľmi časté
Hyperhidróza 14,4 0 Veľmi časté
Suchá koža 7 0 Časté Keratitída 6,6 0,3 Časté Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Bolesť chrbta 21,8 1,4 Veľmi časté
Artralgia 14 0,3 Veľmi časté
Bolesti kostí 9,6 1,4 Časté Bolesť v končatinách 9,6 1,4 Časté Bolesť krku 7,9 0,6 Časté Celkové poruchy a reakcie v mieste podania
Pyrexia 34,5 3,2 Veľmi časté
Trombóza súvisiaca s katétrom 3,5 2 Časté
Laboratórne a funkčné vyšetrenia
Znížený hemoglobín* 97,3 78,5 Veľmi časté
Znížený ANC* 86,7 85,8 Veľmi časté
Zvýšená ALT* 84,2 19,4 Veľmi časté Zvýšená AST* 73,9 6,4 Veľmi časté Hypokaliémia* 61,7 13,9 Veľmi časté Hyperglykémia 20,1 7 Veľmi časté Hypernatriémia* 20 1,2 Veľmi časté
Predĺžený aktivovaný parciálny tromboplastínový čas
12,7 2,6 Veľmi časté
Hyperkalciémia* 6,7 0,6 Časté
Zvýšená telesná hmotnosť 6,6 0,6 Časté
1V centrách klinických skúšaní v Severnej Amerike sa zaznamenávali všetky stupne 13 vopred
určených nežiaducich udalostí. Pri všetkých ostatných nežiaducich udalostiach sa zaznamenávali len
udalosti 3. a 4. stupňa. Preto sú nežiaduce udalosti všetkých stupňov zhrnuté len u pacientov
v centrách klinických skúšaní mimo Severnej Ameriky, zatiaľ čo nežiaduce udalosti 3. a 4. stupňa sú
zhrnuté u pacientov vo všetkých centrách klinických skúšaní.
* Frekvencia je založená na laboratórnych hodnotách.
ASM, SM-AHN a MCL
V tabuľke 4 sú uvedené kategórie frekvencie ADR na základe údajov zhromaždených z dvoch štúdií
u pacientov s ASM, SM-AHN a MCL.
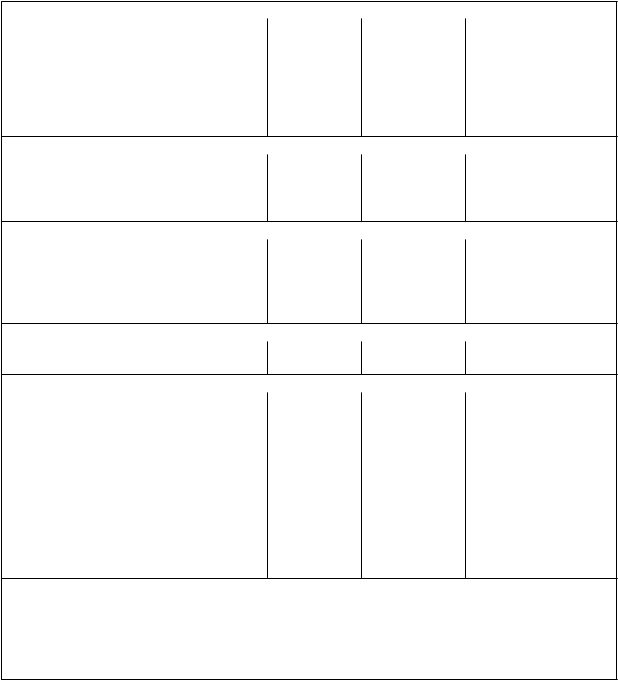
Tabuľka 4 Nežiaduce reakcie na liek pozorované v klinických štúdiách ASM, SM-AHN a MCL
N
ežiaduce reakcie na liek Rydapt (100 mg dvakrát
denne) N=142
K
ategória frekvencie
Infekcie a nákazy
V
šetky stupne
%
stupne 3./4.
%
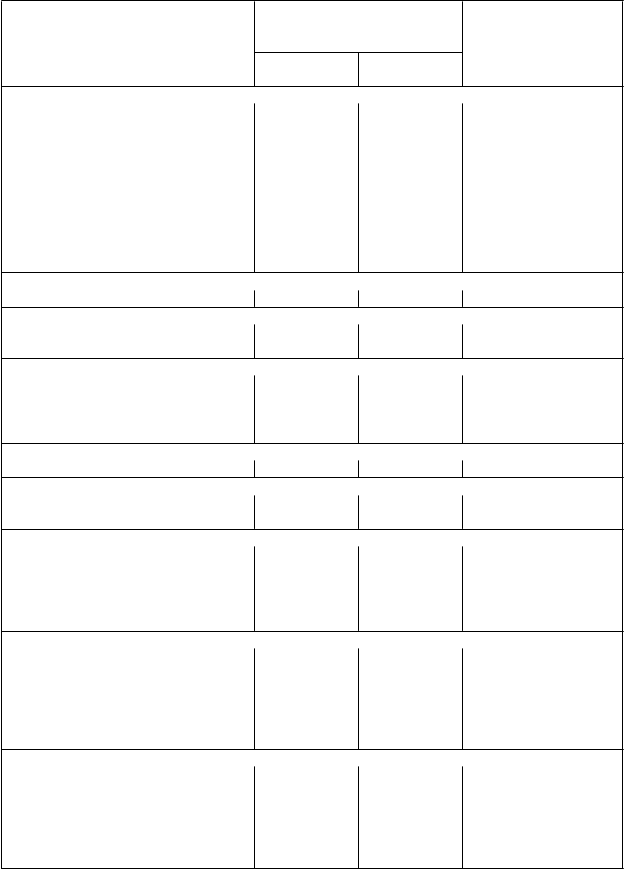
Infekcia močových ciest 13 2,8 Veľmi časté
Infekcia horných dýchacích ciest 11 1,4 Veľmi časté
Pneumónia 8,5 7,0 Časté Sepsa 7,7 7,7 Časté Bronchitída 5,6 0 Časté Orálny herpes 4,9 0 Časté Cystitída 4,2 0 Časté Sinusitída 4,2 0,7 Časté Eryzipel 3,5 1,4 Časté Herpes zoster 3,5 0,7 Časté
Poruchy krvi a lymfatického systémuFebrilná neutropénia 7,7 7,0 Časté
Poruchy imunitného systémuPrecitlivenosť 2,1 0 Časté
Anafylaktický šok 0,7 0,7 Menej časté
Poruchy nervového systémuBolesť hlavy 26 1,4 Veľmi časté
Závraty 13 0 Veľmi časté
Porucha pozornosti 7 0 Časté Tremor 6,3 0 Časté
Poruchy ucha a labyrintuVertigo 4,9 0 Časté
Poruchy cievHypotenzia 9,2 2,1 Časté
Hematóm 6,3 0,7 Časté
Poruchy dýchacej sústavy, hrudníka a mediastínaDyspnoe 18 5,6 Veľmi časté Kašeľ 16 0,7 Veľmi časté Pleurálny výpotok 13 4,2 Veľmi časté
Epistaxa 12 2,8 Veľmi časté
Orofaryngálna bolesť 4,2 0 Časté
Poruchy gastrointestinálneho traktuNauzea 82 5,6 Veľmi časté
Vracanie 68 5,6 Veľmi časté Hnačka 51 6,3 Veľmi časté Zápcha 29 0,7 Veľmi časté Dyspepsia 5,6 0 Časté Gastrointestinálne krvácanie 4,2 3,5 Časté
Celkové poruchy a reakcie v mieste podaniaPeriférny edém 35 3,5 Veľmi časté
Únava 31 8,5 Veľmi časté Pyrexia 27 4,2 Veľmi časté Asténia 4,9 0,7 Časté Zimnica 4,9 0 Časté
Edém 4,2 0,7 Časté
 L
aboratórne a funkčné vyšetrenia
L
aboratórne a funkčné vyšetrenia
Hyperglykémia (nie nalačno)* 93,7 19,0 Veľmi časté
Znížený absolútny počet lymfocytov* 73,2 45,8 Veľmi časté
Znížený ANC* 58,5 26,8 Veľmi časté Zvýšený celkový bilirubín* 40,1 4,9 Veľmi časté Zvýšená lipáza* 39,4 17,6 Veľmi časté Zvýšená AST* 33,8 2,8 Veľmi časté Zvýšená ALT* 33,1 3,5 Veľmi časté Zvýšená amyláza* 20,4 7,0 Veľmi časté Zvýšená telesná hmotnosť 5,6 2,8 Časté
Úrazy, otravy a komplikácie liečebného postupuKontúzia 6,3 0 Časté
Pád 4,2 0,7 Časté
* Frekvencia je založená na laboratórnych hodnotách.
Popisvybranýchnežiaducichreakcií na liekPoruchy gastrointestinálneho traktuU pacientov s AML, ASM, SM-AHN a MCL sa pozorovala nauzea, vracanie a hnačka. U pacientov s ASM, SM-AHN a MCL viedli tieto udalosti k úprave dávky alebo prerušeniu liečby u 26 %
a k ukončeniu liečby u 4,2 % pacientov. Väčšina udalostí sa vyskytla počas prvých 6 mesiacov liečby
a boli liečiteľné podpornými profylaktickými liekmi.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené
Prílohe V.4.9 PredávkovanieHlásené skúsenosti s predávkovaním u ľudí sú veľmi obmedzené. Jednotlivé dávky až do 600 mg sa podávali s akceptovateľnou akútnou tolerabilitou. Pozorovanými nežiaducimi reakciami boli hnačka, bolesť brucha a vracanie.
Nie je známe žiadne špecifické antidotum midostaurínu. V prípade predávkovania sa pacienti musia starostlivo sledovať kvôli prejavom alebo príznakom nežiaducich reakcií, a musí sa začať vhodná symptomatická a podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastiká, inhibítory proteínkinázy, ATC kód: L01XE39
Mechanizmus účinkuMidostaurín inhibuje viaceré receptory tyrozínkinázy, vrátane FLT3 a KIT kinázy. Midostaurín
inhibuje signalizáciu receptorov FLT3 a indukuje zastavenie bunkového cyklu a apoptózu
v leukemických bunkách, ktoré exprimujú mutantné receptory FLT3 ITD alebo TKD alebo zvýšenou
expresiou receptorov FLT3 nezmutovaného - divokého typu. Údaje
in vitro naznačujú, že midostaurín inhibuje D816V mutantné KIT receptory pri expozíciách, ktoré sa dosahujú u pacientov (priemerná dosiahnutá expozícia vyššia ako IC50). Údaje
in vitro naznačujú, že KIT receptory divokého typu sú inhibované v oveľa menšej miere pri týchto koncentráciách (priemerná dosiahnutá expozícia nižšia
ako IC50). Midostaurín interferuje s aberantnou KIT D816V-sprostredkovanou signalizáciou a inhibuje proliferáciu a prežívanie mastocytov a uvoľňovanie histamínu.
Okrem toho midostaurín inhibuje niekoľko ďalších receptorov tyrozínkinázy, ako je PDGFR (receptor pre rastový faktor odvodený od trombocytov) alebo VEGFR2 (receptor 2 pre vaskulárny endoteliálny rastový faktor), ako aj členov skupiny serín/treonínkináz PKC (proteínkinázy C). Midostaurín sa viaže na katalytické domény týchto kináz a inhibuje mitogénnu signalizáciu príslušných rastových faktorov
v bunkách, čo vedie k zastaveniu rastu.
Midostaurín v kombinácii s chemoterapeutikami (cytarabín, doxorubicín, idarubicín a daunorubicín)
viedol k synergickej inhibícii rastu bunkových línií AML s expresiou FLT3-ITD.
Farmakodynamické účinky
Dva hlavné metabolity boli identifikované na myšacích modeloch a u ľudí, t.j. CGP62221
a CGP52421. V proliferačných testoch s bunkami s expresiou FLT3-ITD, CGP62221 preukázal podobnú účinnosť v porovnaní s materskou zlúčeninou, avšak CGP52421 bol približne 10-násobne menej účinný.
Elektrofyziológia srdca
Štúdia zameraná na QT u 192 zdravých jedincov s dávkou 75 mg dvakrát denne neukázala klinicky signifikantné predĺženie QT spôsobené midostaurínom a CGP62221, ale trvanie štúdie nebolo dostatočne dlhé, aby sa odhadli účinky dlhodobo pôsobiaceho metabolitu CGP52421 na predĺženie QTc. Zmena oproti východiskovému QTcF s koncentráciou midostaurínu a oboma metabolitmi bola preto ďalej skúmaná v štúdii fázy II u 116 pacientov s ASM, SM-AHN alebo MCL. Midostaurín, CGP62221 ani CGP52421 s mediánom maximálnych koncentrácií Cmin dosiahnutým pri dávke 100 mg dvakrát denne nepreukázali schopnosť spôsobiť klinicky signifikantné predĺženie QTcF, pretože horné hranice predpokladanej zmeny v týchto hladinách koncentrácií boli menej ako 10 ms (5,8; 2,4
a 4,0 ms, v uvedenom poradí). V populácii s ASM, SM-AHN a MCL malo 25,4 % pacientov najmenej jedno EKG meranie s QTcF väčším ako 450 ms a 4,7 % väčším ako 480 ms.
Klinická účinnosť
AML
Účinnosť a bezpečnosť midostaurínu v kombinácii so štandardnou chemoterapiou v porovnaní s placebom so štandardnou chemoterapiou a v monoterapii ako udržiavacia liečba sa hodnotila
u 717 pacientov (vo veku 18 až 60 rokov) v randomizovanej, dvojito zaslepenej štúdii fázy III.
Pacienti s novodiagnostikovanou AML s mutáciou FLT3, stanovenou testom v klinickej štúdii, boli randomizovaní (1:1) na liečbu midostaurínom 50 mg dvakrát denne (n=360) alebo placebom (n=357) následne v kombinácii so štandardnou indukciou daunorubicínom (60 mg/m2 denne 1.-3. deň) / cytarabínom (200 mg/m2 denne 1.-7. deň) a konsolidáciou vysokou dávkou cytarabínu (3 g/m2 každých 12 hodín 1., 3., 5. deň), nasledované pokračujúcou liečbou midostaurínom alebo placebom podľa počiatočného zadania až do 12 ďalších cyklov (28 dní/cyklus). Kým do štúdie boli zaradení pacienti s rôznymi cytogenetickými abnormalitami súvisiacimi s AML,, pacienti s akútnou promyelocytovou leukémiou (M3) alebo AML súvisiacou s liečbou boli zo štúdie vylúčení. Pacienti boli rozdelení podľa stavu mutácie FLT3: TKD, ITD s alelickým pomerom <0,7 a ITD s alelickým pomerom ≥0,7.
Tieto dve liečebné skupiny boli všeobecne vyvážené vzhľadom na východiskové, demografické charakteristiky ochorenia. Medián veku pacientov bol 47 rokov (rozmedzie: 18 až 60 rokov), väčšina pacientov mala výkonnostný stupeň ECOG 0 alebo 1 (88,3 %) a väčšina pacientov mala de novo AML (95 %). Z pacientov, u ktorých bola zaznamenaná rasa, 88,1 % boli belosi. Väčšina pacientov (77,4 %) mala mutácie FLT3-ITD, z nich najviac (47,6 %) malo nízky alelický pomer (<0,7), a 22,6 %
pacientov malo mutácie FLT3-TKD. Mužov bolo 48 % v ramene s midostaurínom a 41 % v ramene s placebom.
Pacienti, ktorí pokračovali transplantáciou hematopoetických kmeňových buniek (SCT, Stem Cell Transplant), ukončili liečbu v rámci štúdie pred začatím skúšobného režimu SCT. Celková miera SCT bola 59,4 % (214/360) pacientov v ramene s midostaurínom a štandardnou chemoterapiou oproti
55,2 % (197/357) v ramene s placebom a štandardnou chemoterapiou. Všetci pacienti boli sledovaní pre určenie doby prežívania.
Primárnym ukazovateľom v tejto štúdii bolo celkové prežívanie (OS, Overall Survival), zistené od dátumu randomizácie až do úmrtia z akejkoľvek príčiny. Primárna analýza sa vykonala minimálne po približne 3,5 rokoch od randomizácie posledného pacienta. Štúdia preukázala štatisticky signifikantné zlepšenie OS s 23 % znížením rizika úmrtia pre midostaurín so štandardnou chemoterapiou
v porovnaní s placebom so štandardnou chemoterapiou (pozri tabuľku 6 a obrázok 1).
Obrázok 1 Kaplanova-Meierova krivka celkového prežívania, necenzurovaná pre SCT
100

80
60
Midostaurín (n=360) Medián: 74,7 mesiacov
Placebo (n=357) Medián: 25,6 mesiacov
HR: 0,774 (95 % IS, 0,629-0,953)
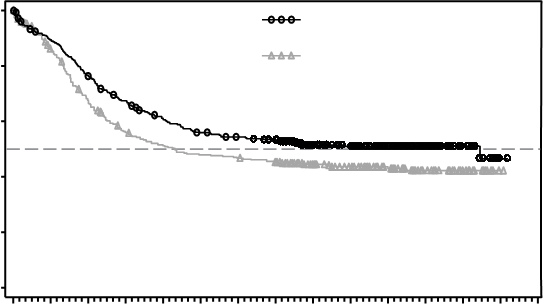 P
P = 0,0078
40
20
0
0 6 12 18 24 30 36 42 48 54 60 66 72 78 84
Mesiace
Pacienti s rizikom
M
e
s
i
ac
e Midostaurín Placebo
0 6 12 18 24 30 36 42 48 54 60 66 72
360 314 269 234 208 189 181 174 133 120 77 50 22
357 284 221 179 163 152 148 141 110 95 71 45 20
78 84
1 0
1 0
Hlavným sekundárnym ukazovateľom bolo prežívanie bez udalosti (EFS; Event-Free survival, EFS udalosť je definovaná ako nedosiahnutie úplnej remisie (CR, Complete Remission) v priebehu 60 dní od začatia liečby podľa protokolu, alebo relaps, alebo úmrtie z akejkoľvek príčiny). EFS ukázalo štatisticky signifikantné zlepšenie pri midostauríne so štandardnou chemoterapiou oproti placebu
so štandardnou chemoterapiou (HR: 0,78 [95 % IS, 0,66 až 0,93] p = 0,0024) a medián EFS
8,2 mesiacov a 3,0 mesiace, v uvedenom poradí; pozri tabuľku 5.
Tabuľka 5 Účinnosť midostaurínu pri AML
P
arameter účinnosti Midostaurín n=360
Placebo n=357
H
R
*
(
95 % IS)
P
- hodnota
¥
C
elkové prežívanie (OS)
1
Medián OS v mesiacoch (95 % IS) 74,7 (31,5; NE) 25,6 (18,6; 42,9) 0,77 (0,63; 0,95) 0,0078
Kaplanove-Meierove odhady po
5 rokoch (95 % IS)
Prežívanie bez udalosti (EFS)2
Medián EFS v mesiacoch
s ohľadom na CR v priebehu
60 dní od začatia liečby (95 % IS) Medián EFS v mesiacoch
s ohľadom na CR kedykoľvek
počas indukcie (95 % IS) Prežívanie bez ochorenia (disease free survival, DFS)
Medián DFS v mesiacoch (95 % IS)
Úplná remisia (CR)
v priebehu 60 dní od začatia liečby
(%)
0,51 (0,45; 0,56) 0,43 (0,38; 0,49)
8,2 (5,4; 10,7) 3,0 (1,9; 5,9) 0,78 (0,66; 0,93) 0,0024
10,2 (8,1; 13,9) 5,6 (2,9; 6,7) 0,73 (0,61; 0,87) 0,0001
26,7 (19,4; NE) 15,5 (11,3; 23,5) 0,71 (0,55; 0,92) 0,0051
212 (58,9) 191 (53,5) NE 0,073§
kedykoľvek počas indukcie (%) 234 (65,0) 207 (58,0) NE 0,027§
Kumulatívny výskyt relapsu (cumulative incidence of relapse, CIR)Medián (95 % IS) NE (25,7; NE) 17,6 (12,7; 46,3) 0,68 (0,52; 0,89) 0,0023
1primárny ukazovateľ; 2hlavný sekundárny ukazovateľ; NE (NE, Not Estimated): nestanovené
*Pomer rizík (HR, Hazard Ratio) odhadnutý pomocou Coxovho regresného modelu stratifikovaného podľa
randomizácie faktora mutácie FLT3.
¥1-stranná p-hodnota vypočítaná pomocou log-rank testu stratifikovaného podľa randomizácie faktora
mutácie FLT3.
§Nesignifikantné
Bol zaznamenaný trend zvýhodňujúci midostaurín vo výskyte CR do 60. dňa v ramene
s midostaurínom (58,9 % oproti 53,5 %; p = 0,073), ktorý pokračoval pri zvážení všetkých CR počas
indukcie (65,0 % oproti 58,0 %; p = 0,027). Okrem toho u pacientov, ktorí dosiahli úplnú remisiu počas indukcie, bol kumulatívny výskyt relapsu po 12 mesiacoch 26 % v ramene s midostaurínom oproti 41 % v ramene s placebom.
Analýza citlivosti OS a EFS pri cenzurovaní v čase SCT tiež podporila klinický prínos midostaurínu a štandardnej chemoterapie oproti placebu.
Výsledky OS podľa stavu SCT sú zobrazené na obrázku 2. Pre EFS, zohľadňujúc úplnú remisiu do
60. dňa od začiatku liečby v štúdii, boli HR 0,602 (95 % IS: 0,372; 0,974) u pacientov s SCT a 0,827 (95 % IS: 0,689; 0,993) u pacientov bez SCT priaznivejšie pri midostauríne.
Obrázok 2 Kaplanova-Meierova krivka celkového prežívania podľa stavu SCT v AML
100%
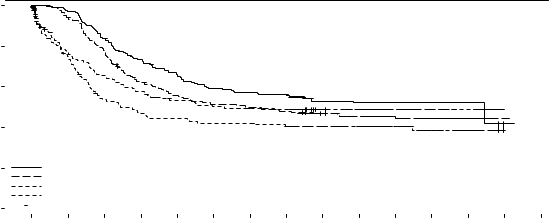
MIDOSTAURÍN – SCT
Medián
prežívania
Jedinci Udalosť (mesiace) 95 % IS
80%
PLACEBO – SCT MIDOSTAURÍN – žiadna SCT PLACEBO – žiadna SCT
214
197
146
160
100
105
71
81
74,7 37,3
35,9 22,6
31,7 16,9
14,7 10,0
N.E.
N.E. N.E.
36,9
60%
40%
20%
0%
1: MIDOSTAURÍN – SCT
2: PLACEBO – SCT
3: MIDOSTAURÍN – žiadna SCT
4: PLACEBO – žiadna SCT
Cenzurované
HR (95% IS) – SCT 0,780 (0,593; 1,026)
HR (95% IS) – žiadna SCT 0,798 (0,580; 1,098)
0 6 12 18 24 30 36 42 48 54 60 66 72 78 84
Celkové prežívanie (mesiace)
Počet pacientov stále s rizikom
1 214 207 178 154 137 122 117 112 84 76 50 33 12 1 0
2 197 184 151 118 105 97 93 90 67 58 42 28 12 1 0
3 146 107 91 80 71 67 64 62 49 44 27 17 10 0
4 160 100 70 61 58 55 55 51 43 37 29 17 8 0
V analýze podskupín sa nepozoroval žiadny zjavný prínos pre OS u žien, avšak prínos liečby sa
pozoroval u žien pri všetkých sekundárnych ukazovateľoch účinnosti (pozri tabuľku 6).
Tabuľka 6 Prehľad OS, EFS, CR, DFS a CIR podľa pohlavia pri AML
U
kazovateľ Celkove
95 % IS
OS (HR) 0,774 (0,629; 0,953)
Muži
95 % IS
0,533 (0,392; 0,725)
Ženy
95 % IS
1,007 (0,757; 1,338)
EFS (indukcia CR) (HR)
0,728 (0,613; 0,866)
0,660 (0,506; 0,861)
0,825 (0,656; 1,037)
Indukcia CR (OR) 0,743* (0,550; 1,005)
0,675* (0,425; 1,072)
0,824* (0,552; 1,230)
DFS (indukcia CR) (HR)
CIR (indukcia CR) (HR)
0,663 (0,516; 0,853)
0,676 (0,515; 0,888)
0,594 (0,408; 0,865)
0,662 (0,436; 1,006)
0,778 (0,554; 1,093)
0,742 (0,516; 1,069)
*Pomer šancí vypočítaný ako (neúplná remisia po liečbe/úplná remisia poliečbe) / (neúplná remisia pri placebe/úplná remisia pri placebe)
HR=pomer rizík (Hazard ratio); OR=pomer šancí (odds ratio)
Účinnosť a bezpečnosť sa hodnotila u pacientov vo veku 60-70 rokov v investigátorom iniciovanej štúdii fázy II s jedným ramenom s midostaurínom v kombinácii s intenzívnou indukciou, konsolidáciou vrátane alogénnej transplantácie kmeňových buniek (SCT, stem cell transplantation) a liečiva v monoterapii na udržiavanie u pacientov s FLT3-ITD mutovanou AML. Na základe predbežnej analýzy bola u pacientov starších ako 60 rokov (46 zo 145 pacientov) miera EFS po
2 rokoch (primárny ukazovateľ) 27,1 % (95 % IS: 16,6; 44,1) a medián OS 15,5 mesiacov.
ASM, SM-AHN a MCL
Účinnosť midostaurínu u pacientov s ASM, SM-AHN a MCL, súhrnne označované ako pokročilá systémová mastocytóza (SM), sa hodnotila v dvoch otvorených („open-label“), multicentrických štúdiách s jedným ramenom (celkovo 142 pacientov).
Pivotná štúdia bola multicentrická štúdia fázy II s jedným ramenom u 116 pacientov s pokročilou SM (štúdia CPKC412D2201). Midostaurín sa podával perorálne v dávke 100 mg dvakrát denne až do progresie ochorenia alebo netolerovateľnej toxicity. Zo 116 pacientov zaradených do štúdie,
89 pacientov bolo vhodných na hodnotenie odpovede na liečbu a tvorili populáciu primárnej účinnosti. Z nich 73 pacientov malo ASM (57 s AHN) a 16 pacientov malo MCL (6 s AHN). Medián veku
v populácii primárnej účinnosti bol 64 rokov približne s polovicou pacientov vo veku ≥65 rokov.
Približne jedna tretina (36 %) dostávala pred tým antineoplastickú liečbu ASM, SM-AHN alebo MCL. Na začiatku štúdie malo v populácii primárnej účinnosti 65 % pacientov >1 merateľný C nález (trombocytopénia, hypoalbuminémia, anémia, vysoká celková hladina bilirubínu, anémia závislá od transfúzií, pokles hmotnosti, neutropénia, vysoká hladina ALT alebo vysoká hladina AST). KIT D816V mutácia bola zistená u 82 % pacientov.
Primárnym ukazovateľom bola celková miera odpovede (ORR, Overall Response Rate). Miera odpovede bola hodnotená na základe modifikovaných Valentových a Chesonových kritérií a odpovede boli posudzované kontrolnou komisiou štúdie. Sekundárne ukazovatele zahŕňali trvanie odpovede, čas do odpovede a celkové prežívanie. Odpovede na midostaurín sú uvedené v tabuľke 7. Aktivita sa pozorovala bez ohľadu na počet predchádzajúcich terapií a prítomnosť alebo neprítomnosť AHN. Potvrdené odpovede sa pozorovali u pacientov s pozitívnou mutáciou KIT D816V (ORR=63 %), ako
aj u pacientov s divokým typom alebo neznámou KIT D816V (ORR=43,8 %). Avšak medián
prežívania u pacientov s pozitívnou KIT D816V bol dlhší, t.j. 33,9 mesiacov (95 % IS: 20,7; 42) ako u pacientov s divokým typom alebo neznámou KIT D816V bol 10 mesiacov (95 % IS: 6,9; 17,4).
Štyridsaťšesť percent pacientov malo pokles infiltrácie do kostnej drene, ktorý prekračoval 50 %,
a 58 % malo pokles sérových hladín tryptázy, ktorý prekračoval 50 %. Objem sleziny sa znížil
o ≥10 % u 68,9 % pacientov s aspoň 1 posúdením od začiatku (26,7 % pacientov malo zníženie
o ≥35 %, čo koreluje s 50 % poklesom pri palpácii).
Medián času do odpovede bol 0,3 mesiaca (rozpätie: 0,1 až 3,7 mesiacov). Medián trvania sledovania bol 43 mesiacov.
Tabuľka 7 Účinnosť midostaurínu pri ASM, SM-AHN a MCL: populácia primárnej účinnosti
P
r
i
m
árny ukazovateľ Celková odpoveď, n (%)'
Všetci ASM SM-AHN MCL
N=89 N=16 N=57 N=16
53 (59,6) 12 (75,0) 33 (57,9) 8 (50,0)
(95% IS) (48,6; 69,8) (47,6; 92,7) (44,1; 70,9) (24,7; 75,3)
Hlavná odpoveď, n
(%)
Čiastočná odpoveď, n (%)
Stabilizované ochorenie, n (%)
Progresívne ochorenie,
n (%) Sekundárne ukazovatele Medián trvania odpovede, mesiace (95 % IS)
Medián celkového prežívania, mesiace (95 % IS)
Kaplanove-Meierove odhady po 5 rokoch (95 % IS)
40 (44,9) 10 (62,5) 23 (40,4) 7 (43,8)
13 (14,6) 2 (12,5) 10 (17,5) 1 (6,3)
11 (12,4) 1 (6,3) 7 (12,3) 3 (18,8)
10 (11,2) 1 (6,3) 6 (10,5) 3 (18,8)
18,6 (9,9; 34,7) 36,8 (5,5; NE) 10,7 (7,4; 22,8) NR (3,6; NE)
26,8 (17,6; 34,7) 51,1 (28,7; NE) 20,7 (16,3; 33,9) 9,4 (7,5; NE)
26,1 (14,6; 39,2) 34,8 (1,7; 76,2) 19,9 (8,6; 34,5) 33,7 (12,3; 56,8)



NE: neodhadnuté, NR (NR, Not Reached): nedosiahnuté
Pacienti, ktorí dostávali inú antineoplastickú liečbu, ako bola v štúdii, boli považovaní za pacientov
s progresiou v čase novej liečby.
Hoci štúdia bola navrhnutá tak, aby bola hodnotená pomocou Valentových a Chesonových modifikovaných kritérií, ako post-hoc exploračná analýza, účinnosť sa hodnotila tiež podľa Medzinárodnej pracovnej skupiny pre výskum myeloproliferatívnych novotvarov za rok 2013 a kritérií zhody Európskej kompetenčnej siete pre liečbu mastocytózy (IWG-MRT-ECNM,
International Working Group - Myeloproliferative Neoplasms Research and Treatment - European
Competence Network on Mastocytosis). Odpoveď na Rydapt sa stanovila použitím výpočtového algoritmu aplikovaného bez akéhokoľvek posudzovania. Zo 116 pacientov malo 113 C nález definovaný podľa kritérií IWG odpovede (okrem ascitu ako C nálezu). Všetky odpovede boli posúdené a vyžadovali 12-týždňové potvrdenie (pozri tabuľku 8).
Tabuľka 8 Účinnosť midostaurínu pri ASM, SM-AHN a MCL podľa kritérií zhody
IWG-MRT-ECNM použitím algoritmického prístupu
V
šetci
hodnotení
A
SM SM-AHN MCL Neznámy
 podtyp
podtyp
pacienti
N=113 N=15 N=72 N=21 N=5
Celková miera odpovede, n

(%)
32 (28,3) 9 (60,0) 15 (20,8) 7 (33,3) 1 (20,0)
(95 % IS) (20,2; 37,6) (32,3; 83,7) (12,2; 32,0) (14,6; 57,0) (0,5; 71,6)
Najlepšia celková odpoveď, n (%)
Úplná remisia 1 (0,9) 0 0 1 ( 4,8) 0
Čiastočná remisia 17 (15,0) 5 (33,3) 8 (11,1) 3 (14,3) 1 (20,0) Klinické zlepšenie 14 (12,4) 4 (26,7) 7 (9,7) 3 (14,3) 0
Trvanie odpovede*
n/N (%) 11/32 (34,4) 4/9 (44,4) 4/15 (26,7) 3/7 (42,9) 0/1 (0,0)
medián (95 % IS) NE (27,0; NE)
36,8 (10,3; 36,8)
NE (17,3; NE)
NE NE (4,1; NE)
Celkové prežívanie
n/N (%) 65/113 (57,5)
4/15 (26,7) 49/72 (68,1)
12/21 (57,1)
0/5 (0,0)0
medián (95 % IS) 29,9 (20,3; 42,0)
*Doba pre potvrdenie odpovedí: 12 týždňov
Analýza vylučuje ascites ako C nález.
51,1 (28,7; NE)
22,1 (16,8; 32,2)
22,6 NE (8,3; NE)
Pacienti, ktorí dostávali inú antineoplastickú liečbu, ako bola v štúdii, boli považovaní za pacientov s

progresiou v čase novej liečby.
Podporná štúdia bola multicentrická, otvorená („open-label“) štúdia fázy II s jedným ramenom
u 26 pacientov s ASM, SM-AHN a MCL (CPKC412A2213). Midostaurín sa podával perorálne
v dávke 100 mg dvakrát denne v cykloch pozostávajúcich z 28 dní. Nedostatok hlavnej odpovede (MR, MajorResponse) alebo čiastočnej odpovede (PR, Partial Response) na konci druhého cyklu vyžadoval
prerušenie liečby v štúdii. Dvadsať (76,9 %) pacientov malo ASM (17 [85 %] s AHN) a 6 pacientov
(23,1 %) malo MCL (2 [33,3 %] s AHN). Medián veku bol 64,5 rokov s polovicou pacientov vo veku
≥65 rokov). Na začiatku štúdie malo 88,5 % >1 C nález a 69,2 % dostávalo predtým aspoň jednu antineoplastickú liečbu.
Primárnym ukazovateľom bola ORR hodnotená pomocou Valentových kritérií počas prvých dvoch cyklov liečby. Devätnásť pacientov (73,1 %; 95 % IS=[52,2; 88,4]) dosiahlo odpoveď v priebehu prvých dvoch cyklov liečby (13 MR; 6 PR). Medián trvania sledovania bol 73 mesiacov a medián trvania odpovede sa nedosiahol. Medián celkového prežívania bol 40,0 mesiacov (prežívanie sa sledovalo u pacientov počas jedného roka po ukončení liečby).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Rydaptom vo
všetkých podskupinách pediatrickej populácie pre liečbu malígnej mastocytózy a mastocytovej leukémie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Rydaptom v jednej alebo vo viacerých podskupinách pediatrickej populácie pre liečbu akútnej myeloidnej leukémie (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Midostaurín je zlúčenina s dobrou absorpciou a zlou rozpustnosťou. Dva z jeho metabolitov preukázali farmakologické aktivity (CGP52421 a CGP62221). Po viacnásobných dávkach boli farmakokinetika midostaurínu a CGP62221 časovo závislé, s počiatočným zvýšením pozorovaným
v prvom týždni, po ktorom nasledoval pokles koncentrácií až do dosiahnutia rovnovážneho stavu na
28 deň. Nezdá sa, že koncentrácie CGP52421 klesajú tak výrazne ako pri midostauríne a CGP62221.
Absorpcia
Absolútna biologická dostupnosť midostaurínu po perorálnom podaní nie je známa.
U ľudí bola absorpcia midostaurínu rýchla po perorálnom podaní, Tmax celkovej rádioaktivity sa pozorovala 1-3 hodiny po podaní dávky. Analýza farmakokinetiky populácie ukázala, že absorpcia u pacientov bola nižšia ako úmerná dávke pri dávkach >50 mg dvakrát denne.
U zdravých jedincov, po podaní jednorazovej dávky 50 mg midostaurínu s jedlom, AUC midostaurínu
sa zvýšila na 20 800 ng*h/ml a Cmax sa znížila na 963 ng/ml (pozri časť 4.5). Podobne sa pri
CGP52421 a CGP62221 AUC zvýšila na 19000 a 29200 ng*h/ml a Cmax sa znížila na 172 a 455 ng/ml,
v uvedenom poradí. Čas do dosiahnutia maximálnej koncentrácie sa tiež predĺžil v prítomnosti jedla
s vysokým obsahom tuku. Tmax sa predĺžil pri všetkých látkach, medián Tmax midostaurínu bol 3 h a pri
CGP52421 a CGP62221 sa Tmax predĺžil na 6 a 7 hodín, v uvedenom poradí.
V klinických štúdiách sa účinnosť a bezpečnosť Rydaptu skúmala po podaní s ľahkým jedlom. Po perorálnom podaní jednorazovej dávky 100 mg midostaurínu po jedle pacientom s ASM, SM-AHN a MCL boli pri midostauríne hodnoty AUCinf 49600 ng*h/ml, Cmax 2940 ng/ml a Tmax 3 h. Pri
CGP52421 boli hodnoty AUC0-12h 2770 ng*h/ml a Cmax 299 ng/ml. Pri CGP62221 boli hodnoty AUC0-
12h 8700 ng*h/ml a Cmax 931 ng/ml. Po opakovanom podávaní perorálnych dávok 100 mg midostaurínu
dvakrát denne boli hodnoty Cmin,ss midostaurínu v plazme u pacientov s AML a ASM, SM-AHN, MCL
919 a 1060 ng/ml, v uvedenom poradí. Pri CGP62221 boli Cmin, ss u populácií s AML a ASM, SM-
AHN, MCL 1610 ng/ml a 2020 ng/ml, v uvedenom poradí. Pri CGP52421 boli Cmin,ss u populácií s
AML a ASM, SM-AHN, MCL 8630 ng/ml a 2860 ng/ml, v uvedenom poradí.
Distribúcia
Midostaurín má distribúciu v tkanivách s geometrickým priemerom 95,2 l (Vz/F). Midostaurín a jeho
metabolity sú distribuované najmä v plazme skôr ako v červených krvinkách. Údaje in vitro
preukázali, že viac ako 98 % midostaurínu sa viaže na proteíny v plazme, ako je albumín, α1-kyslý glykoproteín (AGP, Acid Glycoprotein) a lipoproteín.
Biotransformácia
Midostaurín sa metabolizuje prostredníctvom CYP3A4 prevažne oxidáciou. Hlavné zložky v plazme
zahŕňajú midostaurín a dva hlavné aktívne metabolity, CGP62221 (prostredníctvom O-demetylácie)
a CGP52421 (prostredníctvom hydroxylácie), predstavujúce 27,7±2,7 % a 38,0±6,6 %, v uvedenom poradí, celkovej plazmatickej expozície pri 96 hodinách po jednorazovej dávke 50 mg midostaurínu.
Eliminácia
Medián terminálnych polčasov midostaurínu, CGP62221 a CGP52421 v plazme je približne 20,5; 32,3
a 482 hodín. Priemerný zdanlivý plazmatický klírens (CL/F) bol u zdravých jedincov 2,4-3,1 l/h.
U pacientov s AML a ASM, SM-AHN a MCL boli populačné farmakokinetické odhady pre klírens
midostaurínu v rovnovážnom stave 5,9 l/h a 4,4 l/h, v uvedenom poradí. Výsledky zo štúdie hmotnostnej rovnováhy u ľudí indikujú, že vylučovanie stolicou je hlavnou cestou vylučovania (78 % dávky), a väčšinou vo forme metabolitov (73 % dávky), zatiaľ čo nezmenený midostaurín predstavuje
3 % dávky. Len 4 % dávky sa vylučujú močom.
Linearita/nelinearita
Midostaurín a jeho metabolity všeobecne nepreukázali žiadnu významnú odchýlku od proporcionality
dávky po jednorazovej dávke v rozsahu 25 mg až 100 mg. Avšak po viacnásobných dávkach v rozsahu dávok 50 mg až 225 mg denne bolo zvýšenie expozície nižšie ako dávke úmerné.
Po viacnásobných perorálnych dávkach ukázal midostaurín časovo závislú farmakokinetiku
s počiatočným zvýšením plazmatických koncentrácií počas prvého týždňa (maximálna Cmin)
s následným poklesom času do rovnovážneho stavu po približne 28 dňoch (2,5-násobný pokles). Aj
keď presný mechanizmus klesajúcej koncentrácie midostaurínu nie je jasný, je pravdepodobné, že môže byť dôsledkom autoindukčných vlastností midostaurínu a jeho dvoch aktívnych metabolitov
CGP52421 a CGP62221 na CYP3A4. Farmakokinetika metabolitu CGP62221 preukázala podobný
model. Koncentrácia CGP52421 sa však zvýšila až 2,5-násobne pri ASM, SM-AHN a MCL a až
9-násobne pri AML, v porovnaní s midostaurínom po jednom mesiaci liečby.
In vitro hodnotenie potenciálu liekových interakcií
Enzýmové liekové interakcie
Inhibícia cytochrómu P450
Na základe údajov in vitro sa midostaurín a jeho aktívne metabolity, CGP52421 a CGP62221,
považujú za inhibítory a môžu potenciálne spôsobiť zvýšenie expozície súbežne podaných liekov primárne vylučovaných CYP1A2, CYP2D6, CYP2C8, CYP2C9, CYP2E1 a CYP3A4/5. Okrem toho sa pozorovala in vitro tiež časovo závislá inhibícia CYP3A4/5 midostaurínom, CGP52421
a CGP62221.
Indukcia cytochrómu P450
Na základe údajov in vitro sa midostaurín a jeho aktívne metabolity, CGP52421 a CGP62221, tiež považujú za induktory a môžu spôsobiť zníženie expozície súbežne podaných liekov primárne
vylučovaných CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP3A4 (pozri časť 4.5).
Liekové interakcie s transportérmi
In vitro experimenty preukázali, že midostaurín, CGP52421 a CPG62221 môžu potenciálne inhibovať
P-gp, BCRP a OATP1B1.
Osobitné skupiny pacientov
St arší pacienti
Na základe populačnej farmakokinetickej analýzy sa nezistil žiadny signifikantný vplyv veku na farmakokinetiku midostaurínu a jeho dvoch aktívnych metabolitov u pacientov vo veku medzi 65
a 85 rokov. U dospelých pacientov s ASM, SM-AHN a MCL alebo AML sa nevyžaduje na základe
veku žiadna úprava dávky midostaurínu.
Pediatrickí pacienti
Rydapt sa neodporúča používať u detí a dospievajúcich (pozri časť 4.2). Farmakokinetika midostaurínu u pediatrických pacientov bola preskúmaná v štúdii fázy I s eskaláciou dávky
v monoterapii u 22 pacientov (12 vo veku 0-2 rokov a 10 vo veku 10-17 rokov) s AML alebo MLL opätovne usporiadanou ALL použitím prístupu populačnej farmakokinetiky. Farmakokinetika midostaurínu bola menšia ako dávka úmerná s dávkami 30 mg/m2 a 60 mg/m2 po jednorazovej dávke a viacnásobných dávkach. Vzhľadom na obmedzené fakrmakokinetické údaje u pediatrických pacientov nie je možné urobiť žiadne porovnanie s farmakokinetikou midostaurínu u dospelých.
Pohlavie
Na základe populačnej farmakokinetickej modelovej analýzy vplyvu pohlavia na klírens midostaurínu
a jeho aktívnych metabolitov nebolo žiadne štatisticky signifikantné zistenie a očakávané zmeny
v expozícii (<20 %) sa nepovažovali za klinicky relevantné. Na základe pohlavia sa nevyžaduje žiadna
úprava dávky midostaurínu.
Rasa/et ni cká prí slušnosť
Neexistujú žiadne rozdiely vo farmakokinetickom profile medzi belochmi a černochmi. Na základe
štúdie fázy I u zdravých japonských dobrovoľníkov sú farmakokinetické profily midostaurínu a jeho metabolitov (CGP62221 a CGP52421) podobné v porovnaní s tými, ktoré sa pozorovali v iných
farmakokinetických štúdiách vykonaných u belochov a černochov. Na základe etnickej príslušnosti sa
nevyžaduje žiadna úprava dávky midostaurínu.
Porucha f unkc ie peče ne
Štúdia zameraná na poruchu funkcie pečene hodnotila systémovú expozíciu midostaurínu po perorálnom podaní 50 mg dvakrát denne po dobu 6 dní u jedincov s miernou alebo stredne ťažkou poruchou funkcie pečene na začiatku štúdie (trieda A alebo B podľa Childa-Pugha, v uvedenom poradí) a kontrolných jedincov s normálnou funkciou pečene. Maximálna koncentrácia sa dosiahla medzi 2 a 3 hodinami po podaní jednorazovej dávky alebo opakovaných dávok vo všetkých
skupinách. V 1. deň boli AUC0-12 a Cmax 8 130 ng*h/ml a 1 206 ng/ml, v uvedenom poradí, u zdravých jedincov. AUC0-12 sa znížila o 39 % a 36 % u jedincov s miernou a stredne ťažkou poruchou funkcie pečene, v uvedenom poradí. Na 7. deň bola AUCCthrough (expozícia pod krivkou Cthrough od 1. dňa do
7. dňa) 5 410 ng*h/ml u zdravých jedincov a bola znížená o 35 % a 20 % u jedincov s miernou
a stredne ťažkou poruchou funkcie pečene, v uvedenom poradí. AUCtau sa znížila o 28 % a 20 % na
7. deň, v uvedenom poradí. Napokon boli analyzované dlhodobé údaje od pacientov použitím metódy
populačnej farmakokinetiky. Žiadny vplyv poruchy funkcie pečene nie je možné identifikovať
u pacientov s miernou a stredne ťažkou poruchou funkcie pečene v populáciách s ASM, SM-AHN, MCL a AML.
U jedincov s miernou alebo stredne ťažkou poruchou funkcie pečene nedošlo v porovnaní s jedincami s normálnou funkciou pečene celkovo k žiadnemu klinicky relevantnému zvýšeniu expozície midostaurínu (AUC) v plazme. U pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene na začiatku štúdie nie je potrebná žiadna úprava dávkovania. Farmakokinetika midostaurínu sa nehodnotila u pacientov s ťažkou poruchou funkcie pečene na začiatku štúdie (trieda C podľa
Childa-Pugha) (pozri čast 4.4).
Porucha f unkc ie obl i či ek
Renálna eliminácia je menej významným spôsobom eliminácie midostaurínu. Nevykonala sa žiadna štúdia s midostaurínom zameraná na poruchu funkcie obličiek. Uskutočnili sa populačné
farmakokinetické analýzy použitím údajov z klinických štúdií u pacientov s AML (n=180) a ASM,
SM-AHN a MCL (n=141). Z 321 pacientov sa u 177 pacientov preukázala už existujúca mierna
(n=113), stredne ťažká (n=60) alebo ťažká (n=4) porucha funkcie obličiek (15 ml/min ≤ klírens kreatinínu [CrCL] <90 ml/min). U 144 pacientov sa preukázala normálna funkcia obličiek (CrCL
>90 ml/min) na začiatku štúdie. Na základe populačnej farmakokinetickej analýzy nebol klírens midostaurínu signifikantne ovplyvnený poruchou funkcie obličiek, a preto nie je potrebná žiadna
úprava dávkovania u pacientov s miernou alebo stredne ťažkou poruchou funkcie obličiek.
5.3 Predklinické údaje o bezpečnosti
Vzhľadom na dávku limitujúcu toxicitu sa nemohli dosiahnuť hladiny klinickej terapeutickej expozície na zvieratách. Všetky zistenia na zvieratách popísané nižšie sa pozorovali pri expozícii midostaurínu signifikantne nižšej ako terapeutické hladiny.
Farmakologická bezpečnosť a toxicita po jednorazovom/opakovanom podávaní
Farmakologické štúdie bezpečnosti indikujú, že midostaurín pravdepodobne neinterferuje s vitálnymi
funkciami centrálneho nervového systému. In vitro midostaurín neinhibuje aktivitu kanálov hERG až
po limit rozpustnosti 12 µM. Dva hlavné metabolity u ľudí GGP52421 a CGP62221 (tiež testované pri limite rozpustnosti) inhibovali kanál hERG v mierne bezpečnom rozmedzí. V štúdiách po
opakovanom podávaní psom sa pozoroval pokles srdcovej frekvencie, predĺženie P-Q intervalu
a sporadicky vyskytujúce sa atrioventrikulárne bloky u jednotlivých zvierat.
V štúdiách po opakovanom podávaní boli cieľovými orgánmi pre toxicitu gastrointestinálny trakt (eméza u psov a opíc, hnačka a zmeny sliznice), semenníky (znížená spermatogenéza), kostná dreň (hypocelularita) a lymfatické orgány (deplécia/atrofia). Účinok na kostnú dreň a lymfatické orgány bol sprevádzaný hematologickými zmenami, zníženým počtom bielych krviniek, lymfocytov a parametrov červených krviniek. Zvýšenie hladiny pečeňových enzýmov (ALT a AST) sa pozorovalo trvale
u potkanov, u psov a opíc v dlhodobých štúdiách s trvaním ≥3 mesiace, bez histopatologických korelácií.
Reprodukčná toxicita
V štúdiách fertility na potkanoch sa midostaurín spájal so zníženou fertilitou, testikulárnou
degeneráciou a atrofiou, zníženou pohyblivosťou spermií, oligo- a aspermiou, zvýšenou resorpciou,
zníženým počtom gravidít, počtom zahniezdených a živých embryí.
V štúdiách embryofetálneho vývoja u potkanov a králikov boli zaznamenané nárast počtu neskorých resorpcií, znížená hmotnosť plodu a znížená skeletálna osifikácia.
V štúdii prenatálneho a postnatálneho vývinu sa zaznamenali dystokia u matky a znížená veľkosť vrhu, nižšia telesná hmotnosť mláďat, predčasné úplné otvorenie očí a oneskorená ontogenéza sluchového úľakového reflexu.
Štúdie najuvenilnýchzvieratách
V štúdii toxicity na mláďatách potkanov sa midostaurín podával od 7. až do 70. dňa po pôrode. Boli
zaznamenané zníženie telesnej hmotnosti, krvácanie a zmiešané bunkové infiltrácie do pľúc
a erytrocytóza/erytrofagocytóza v mezenterických lymfatických uzlinách. Nezistili sa žiadne účinky na telesný vývoj, zmyslové funkcie alebo behaviorálne funkcie. Index párenia, index fertility a miera počatia boli znížené pri 0, 5 a 15 mg/kg/deň, ale nie pri 2 mg/kg/deň.
Genotoxicita
In vitro a in vivo štúdie genotoxicity zahŕňajúce relevantné ukazovatele genotoxicity nepreukázali
žiadne známky mutagénneho alebo klastogénneho vplyvu. Neuskutočnili sa žiadne štúdie
karcinogenity.
Hodnotenie environmentálneho rizika (ERA)
Štúdie hodnotiace environmentálne riziko preukázali, že midostaurín má potenciál byť perzistentný,
bioakumulatívny a toxický pre životné prostredie.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
O
bsah kapsuly
Hydroxystearoylmakrogol-glycerol
makrogol bezvodý etanol
mono-di-triglyceridy z kukuričného oleja
D-alfa-tokoferolacetát
Obal kapsuly
želatína
glycerol
oxid titaničitý (E171)
žltý oxid železitý (E172)
červený oxid železitý (E172)
čistená voda
Potlač
karmín (E120)
hypromelóza propylénglykol
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
PA/Al/PVC-Al blistre. Jeden blister obsahuje 4 mäkké kapsuly.
Balenie obsahujúce 56 (2 balenia po 28) alebo 112 (4 balenia po 28) mäkkých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR
Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/17/1218/001-002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE18. september 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu