
/>
Nasledujúce pokyny týkajúce sa 30-dávkového inhalátora platia aj pre 7-dávkový inhalátor. Inhalátor Ellipta obsahuje jednotlivé dávky a je pripravený na použitie.
Inhalátor je zabalený vo vaničke obsahujúcej vrecko s vysúšadlom na zníženie vlhkosti. Vrecko s vysúšadlom sa má zlikvidovať a nesmie sa jesť ani inhalovať.
Pacienta treba upozorniť, aby neotváral vaničku, kým nebude pripravený inhalovať dávku.
Inhalátor bude v polohe „zatvorený“, keď sa po prvýkrát vyberie z uzatvorenej vaničky. Je potrebné
dopísať dátum na vyhradené miesto na štítku inhalátora vedľa označenia „Zlikvidujte do“. Dátum
„Zlikvidujte do“ je 6 týždňov od dátumu prvého otvorenia vaničky. Po tomto dátume sa inhalátor už
viac nemá používať. Vanička sa po prvom otvorení môže zlikvidovať.
Ak sa kryt inhalátora otvorí a zatvorí bez inhalovania lieku, dávka sa vyplytvá. Vyplytvaná dávka sa
bezpečne zadrží vo vnútri inhalátora a viac ju už nebude možné inhalovať.
Počas jednej inhalácie nie je možné náhodne užiť vyššiu alebo dvojnásobnú dávku lieku.
a) Pripravte dávku
Keď budete pripravený užiť dávku, otvorte kryt. Inhalátorom sa nemá triasť.
Posúvajte kryt smerom nadol, až kým nezačujete „kliknutie“. Teraz je liek pripravený na inhaláciu.
Počítadlo dávok to potvrdí odpočítaním 1 dávky. Ak počítadlo dávok neodpočíta dávku, keď je počuť
„kliknutie“, inhalátor neuvoľní dávku a treba ho vziať späť k lekárnikovi a poradiť sa s ním.
b) Ako inhalovať liek
Inhalátor sa má držať mimo úst a treba vydýchnúť čo najviac, ako je to možné bez námahy.
Ale nesmie sa vydýchnuť do inhalátora.
Náustok treba vložiť medzi pery a potom ho pevne obomknúť perami. Vetracie otvory sa počas
používania nesmú zakrývať prstami.
• Inhalujte jedným dlhým, plynulým a hlbokým vdýchnutím. Potom treba zadržať dych tak
dlho, ako je to možné (aspoň 3 - 4 sekundy).
• Vyberte si inhalátor z úst.
• Pomaly a jemne vydýchnite.
Ani pri správnom použití inhalátora nemusíte pocítiť chuť či prítomnosť lieku.
c) Zatvorte inhalátor
Pred zatvorením krytu sa môže náustok inhalátora očistiť pomocou suchej papierovej vreckovky. Posuňte kryt smerom nahor až na doraz, aby ste zakryli náustok.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Astma
Umeklidíniumbromid sa nemá používať u pacientov s astmou, keďže sa u tejto skupiny pacientov nesledoval.
Paradoxný bronchospazmus
Podávanie umeklidíniumbromidu môže vyvolať paradoxný bronchospazmus, ktorý môže ohrozovať život. Ak sa vyskytne paradoxný bronchospazmus, liečba sa má ihneď ukončiť a v prípade potreby sa má začať alternatívna liečba.
Zhoršenie ochorenia
Umeklidíniumbromid je určený na udržiavaciu liečbu CHOCHP. Nemá sa používať na úľavu
od akútnych príznakov, t.j. ako záchranný liek na liečbu akútnych epizód bronchospazmu. Akútne
príznaky sa majú liečiť inhalačným krátkodobo pôsobiacim bronchodilatanciom. Zvýšené užívanie krátkodobo pôsobiacich bronchodilatancií na zmiernenie príznakov svedčí o zhoršení kontroly
ochorenia. V prípade zhoršenia CHOCHP počas liečby umeklidíniumbromidom sa má prehodnotiť
stav pacienta a liečebný režim CHOCHP.
Kardiovaskulárneúčinky
Po podávaní antagonistov muskarínových receptorov vrátane umeklidíniumbromidu sa môžu objaviť
kardiovaskulárne účinky, akými sú srdcové arytmie, napr. atriálna fibrilácia a tachykardia. Pacienti s klinicky významným nekontrolovaným kardiovaskulárnych ochorením boli z klinických štúdií
vylúčení. Preto sa má umeklidíniumbromid u pacientov so závažnými kardiovaskulárnymi poruchami,
najmä so srdcovými arytmiami, používať obozretne.
Antimuskarínovýúčinok
Vzhľadom na antimuskarínový účinok sa má umeklidíniumbromid používať obozretne u pacientov s retenciou moču alebo s glaukómom s úzkym uhlom.
Pomocné látky
Tento liek obsahuje laktózu. Pacienti so zriedkavými dedičným problémami galaktózovej intolerancie,
lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Klinicky významné interakcie sprostredkované umeklidíniumbromidom podávaným v klinických dávkach sa považujú za nepravdepodobné kvôli nízkym plazmatickým koncentráciám dosahovaným po inhalácii dávky.
Iné antimuskariniká
Súbežné podávanie umeklidíniumbromidu s inými dlhodobo pôsobiacimi antagonistami muskarínových receptorov alebo s liekmi obsahujúcimi toto liečivo sa nesledovalo a neodporúča sa,
pretože môže potencovať známe nežiaduce reakcie pri inhalačných antagonistoch muskarínových
receptorov.
I
nterakcie na úrovni metabolizmu a transportných systémov
Umeklidíniumbromid je substrát cytochrómu P450 2D6 (CYP2D6). Farmakokinetika umeklidíniumbromidu v rovnovážnom stave sa hodnotila u zdravých dobrovoľníkov s nedostatočnou aktivitou CYP2D6 (pomalí metabolizátori). Pri 4-násobne vyššej dávke ako je terapeutická dávka sa nepozoroval žiaden vplyv na hodnotu AUC alebo Cmax umeklidínia. Pri 8-násobne vyššej dávke sa pozorovalo približne 1,3-násobné zvýšenie hodnoty AUC umeklidíniumbromidu bez vplyvu na Cmax umeklidíniumbromidu. Na základe rozsahu týchto zmien sa neočakáva žiadna klinicky významná lieková interakcia, keď sa umeklidínium podáva súbežne s inhibítormi CYP2D6 alebo keď sa podáva osobám s geneticky nedostatočnou aktivitou CYP2D6 (pomalí metabolizátori).
Umeklidíniumbromid je subtrát transportného P-glykoproteínu (P-gp). Vplyv stredne silného inhibítora P-gp verapamilu (240 mg jedenkrát denne) na farmakokinetiku umeklidíniumbromidu
v rovnovážnom stave sa hodnotil u zdravých dobrovoľníkov. Nepozoroval sa žiaden vplyv verapamilu
na Cmax umeklidíniumbromidu. Pozorovalo sa približne 1,4-násobné zvýšenie hodnoty AUC umeklidíniumbromidu. Na základe rozsahu týchto zmien sa neočakáva žiadna klinicky významná interakcia, keď sa umeklidíniumbromid podáva súbežne s inhibítormi P-gp.
InéliekynaliečbuCHOCHP
Hoci sa neuskutočnili žiadne formálne štúdie interakcií v in vivo podmienkach, inhalačný umeklidíniumbromid sa používal súbežne s inými liekmi na liečbu CHOCHP vrátane krátkodobo a dlhodobo pôsobiacich bronchodilatancií so sympatomimetickým účinkom a inhalačných kortikosteroidov bez klinických dôkazov o interakciách.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje o použití umeklidíniumbromidu u gravidných žien. Štúdie na
zvieratách nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Umeklidíniumbromid sa má používať počas gravidity, len ak je očakávaný prínos pre matku podstatný v porovnaní s možným rizikom pre plod.
Dojčenie
Nie je známe, či sa umeklidíniumbromid vylučuje do ľudského mlieka. Riziko u dojčených
novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu s Roluftou, sa má urobiť po zvážení prínosu
dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch umeklidíniumbromidu na fertilitu ľudí. Štúdie
na zvieratách nepreukázali žiadne účinky umeklidíniumbromidu na fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Umeklidíniumbromid nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá
a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn
bezpečnostného
profilu
Najčastejšie hlásené nežiaduce reakcie pri podávaní Rolufty boli nazofaryngitída a infekcia horných dýchacích ciest.
Tabuľkovýsúhrnnežiaducichreakcií
Bezpečnostný profil umeklidíniumbromidu sa hodnotil u 1 663 pacientov s CHOCHP, ktorí užívali
55 mikrogramov alebo vyššiu dávku počas jedného roka. Zahŕňa to aj 576 pacientov, ktorí užívali
odporúčanú dávku 55 mikrogramov jedenkrát denne.
Frekvencie nežiaducich reakcií uvedené v nasledujúcej tabuľke zahŕňajú hrubú mieru výskytu pozorovanú v štyroch štúdiách účinnosti a v štúdii dlhodobej bezpečnosti (ktoré zahŕňali
1 412 pacientov, ktorí užívali umeklidíniumbromid).
Frekvencia nežiaducich reakcií je definovaná pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10,000) a neznáme (z dostupných údajov).
T
rieda orgánových
systémov
N
ežiaduca reakcia Frekvencia
Infekcie a nákazy Nazofaryngitída
Infekcia horných dýchacích ciest Infekcia močových ciest Sinusitída
Faryngitída
Poruchy nervového systému Bolesť hlavy
Dysgeuzia
Poruchy oka Glaukóm
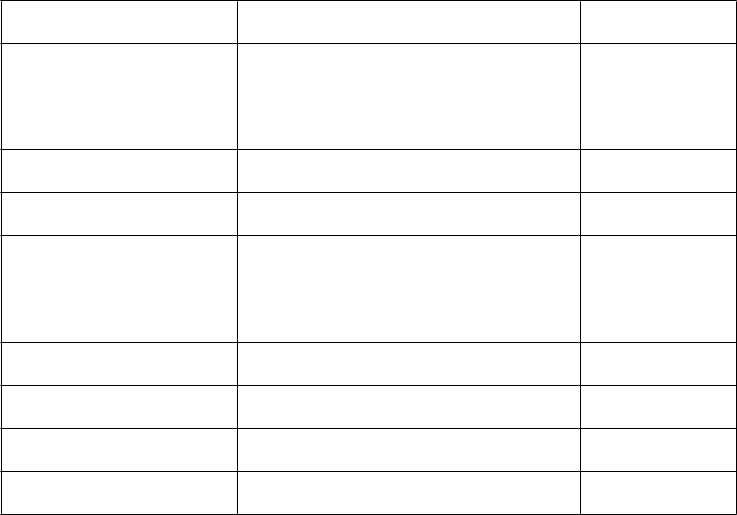
Rozmazané videnie
Časté Časté Časté Časté
Menej časté
Časté
Menej časté
Neznáme
Neznáme
Poruchy srdca a srdcovej
činnosti
Atriálna fibrilácia Idioventrikulárny rytmus Supraventrikulárna tachykardia Supraventrikulárne extrasystoly Tachykardia
Menej časté Menej časté Menej časté Menej časté Časté
Poruchy dýchacej sústavy, hrudníka a mediastína
Kašeľ Časté
Poruchy gastrointestinálneho traktu
Zápcha
Suchosť v ústach
Menej časté
Menej časté
Poruchy kože a podkožného tkaniva
Vyrážka Menej časté
Poruchy obličiek
a močových ciest
Retencia moču
Dyzúria
Neznáme
Neznáme
H
l
ásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePredávkovanie umeklidíniumbromidom pravdepodobne vyvolá prejavy a príznaky zhodné so známymi nežiaducimi účinkami inhalačných antagonistov muskarínových receptorov (napr. suchosť v ústach, poruchy zrakovej akomodácie a tachykardia).
Ak dôjde k predávkovaniu, pacient má podľa potreby dostať podpornú liečbu s náležitým sledovaním.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Liečivá na obštrukčné ochorenia dýchacích ciest, anticholinergiká, ATC kód: R03BB07
Mechanizmusúčinku
Umeklidíniumbromid je dlhodo pôsobiaci antagonista muskarínových receptorov (označovaný aj ako
anticholinergikum). Je to derivát chinuklidínu, ktorý je antagonistom muskarínových receptorov s aktivitou naprieč viacerými podtypmi cholinergných muskarínových receptorov. Umeklidíniumbromid vykazuje bronchodilatačný účinok kompetitívnou inhibíciou väzby acetylcholínu s cholinergnými muskarínovými receptormi hladkého svalstva dýchacích ciest. Vykazoval pomalú reverzibilitu na ľudskom podtype muskarínového receptora M3 v in vitro podmienkach a dlhotrvajúci účinok v in vivo podmienkach, keď sa podával priamo do pľúc
v predklinických modeloch.
Farmakodynamické účinky
V 6-mesačnej štúdii fázy III (DB2113373) sa pri Rolufte v porovnaní s placebom dosiahlo klinicky
významné zlepšenie pľúcnych funkcií (hodnotených pomocou objemu úsilného výdychu za 1 sekundu
(forced expiratory volume in 1 second) [FEV1]) počas 24 hodín po podávaní jednej dennej dávky, ktoré bolo evidentné 30 minút po podaní prvej dávky (zlepšenie v porovnaní s placebom o 102 ml, p < 0,001*). V 24. týždni bolo priemerné maximálne zlepšenie hodnoty FEV1 v priebehu prvých
6 hodín po podaní dávky v porovnaní s placebom 130 ml (p < 0,001*). Pri účinku Rolufty sa
v priebehu času nepreukázal rozvoj tachyfylaxie.
Elektrofyziológia srdca
Vplyv umeklidínia 500 mikrogramov (jednotková dávka) na QT interval sa hodnotil v placebom a moxifloxacínom kontrolovanom skúšaní zameranom na QT interval so 103 zdravými
dobrovoľníkmi. Po opakovaných dávkach umeklidínia 500 mikrogramov jedenkrát denne počas 10 dní sa nepozoroval žiaden klinicky významný vplyv na predĺženie QT intervalu (korigovaného podľa metódy Fridericia) ani vplyv na srdcovú frekvenciu.
Klinická účinnosť
Klinická účinnosť Rolufty podávanej jedenkrát denne sa hodnotila u 904 dospelých pacientov
s klinickou diagnózou CHOCHP, ktorí užívali umeklidíniumbromid alebo placebo v dvoch pivotných klinických štúdiách fázy III; v 12-týždňovej štúdii (AC4115408) a v 24-týždňovej štúdii
(DB2113373).
Pivotné štúdie účinnosti:
Vplyv na pľúcne funkcie
V oboch pivotných štúdiách, v 12-týždňovej aj v 24-týždňovej, sa pri podávaní Rolufty preukázalo štatisticky signifikantné a klinicky významné zlepšenie pľúcnych funkcií (definované zmenou trough (minimálnej hodnoty) FEV1 v porovnaní s východiskovou hodnotou v 12. týždni a v 24. týždni,
v uvedenom poradí, ktorá bola v oboch štúdiách primárnym cieľom účinnosti) v porovnaní s placebom
(pozri tabuľku 1). V oboch štúdiách bol bronchodilatačný účinok pri Rolufte v porovnaní s placebom evidentný po prvom dni liečby a zachoval sa počas 12-týždňového a 24-týždňového obdobia liečby.
V priebehu času sa nezistilo oslabenie bronchodilatačného účinku.
Tabuľka 1: Trough FEV1 (ml) v 12. týždni a v 24. týždni (primárny cieľ)
L
i
ečba s Roluftou
55 µg
12-týždňová štúdia
R
ozdiel medzi liečbami
1
24-týždňová štúdia
R
ozdiel medzi liečbami
1
v porovnaní s placebom
µg = mikrogramy
95 % interval spoľahlivostip-hodnota127 (52, 202)

< 0,001
95 % interval spoľahlivostip-hodnota115 (76, 155)
< 0,001
1 priemerná hodnota určená metódou najmenších štvorcov (95 % interval spoľahlivosti)
V 12-týždňovej pivotnej štúdii sa pri podávaní Rolufty v porovnaní s placebom preukázalo štatisticky signifikantné zvýšenie váženého priemeru hodnoty FEV1 počas 0 - 6 hodín po dávke, v porovnaní
s východiskovou hodnotou, v 12. týždni (166 ml, p < 0,001). V 24-týždňovej pivotnej štúdii sa
pri podávaní Rolufty v porovnaní s placebom preukázalo zvýšenie váženého priemeru hodnoty FEV1
počas 0 - 6 hodín po dávke, v porovnaní s východiskovou hodnotou, v 24. týždni (150 ml; p < 0,001*).
Vplyv na symptómy
Dýchavičnosť:
V 12-týždňovej štúdii sa pri podávaní Rolufty v porovnaní s placebom nepreukázalo štatisticky signifikantné zlepšenie fokálneho (t.j. súhrnného) skóre TDI v 12. týždni (o 1,0 bod; p = 0,05). V 24-týždňovej štúdii sa pri podávaní Rolufty v porovnaní s placebom preukázalo štatisticky signifikantné zlepšenie fokálneho skóre TDI v 24. týždni (o 1,0 bod, p < 0,001).
V 12-týždňovej štúdii bolo percento pacientov, ktorí v 12. týždni odpovedali na liečbu
aspoň minimálnym klinicky významným rozdielom (minimum clinically important difference, MCID)
vo fokálnom skóre TDI o 1 bod, väčšie pri podávaní Rolufty (38 %) v porovnaní s placebom (15 %). Podobne aj v 24-týždňovej štúdii dosiahlo väčšie percento pacientov rozdiel vo fokálnom skóre TDI
o ≥ 1 bod v 24. týždni pri podávaní Rolufty (53 %) v porovnaní s placebom (41 %).
Kvalita života súvisiaca so zdravotným stavom:
V 12-týždňovej štúdii sa pri podávaní Rolufty tiež preukázalo štatisticky signifikantné zlepšenie kvality života súvisiacej so zdravotným stavom hodnotenej pomocou SGRQ dotazníka (St. George’s
Respiratory Questionnaire), o čom svedčilo zníženie celkového skóre SGRQ v 12. týždni v porovnaní
s placebom (-7,90 bodu, p < 0,001). V 24-týždňovej štúdii sa pri podávaní Rolufty preukázalo väčšie
zlepšenie v zmysle zmeny celkového skóre SGRQ v porovnaní s východiskovým skóre v 24. týždni v porovnaní s placebom (-4,69 bodu, p < 0,001*).
V 12-týždňovej štúdii bolo percento pacientov, ktorí v 12. týždni odpovedali na liečbu aspoň
na úrovni MCID v skóre SGRQ (definovaným ako zníženie o 4 body v porovnaní s východiskovým skóre), väčšie pri podávaní Rolufty 55 mikrogramov (44 %) v porovnaní s placebom (26 %). Podobne aj v 24-týždňovej štúdii dosiahlo väčšie percento pacientov aspoň MCID v 24. týždni pri podávaní Rolufty (44 %) v porovnaní s placebom (34 %).
E
xacerbácie CHOCHP
V 24-týždňovej štúdii liečba s Roluftou znížila riziko exacerbácie CHOCHP v porovnaní s placebom (analýza času do prvej exacerbácie; Hazard Ratio 0,6, p = 0,035*). V 24. týždni bola pravdepodobnosť výskytu exacerbácie u pacientov liečených s Roluftou 8,9 % v porovnaní s 13,7 % pri placebe. Tieto štúdie neboli špecificky usporiadané tak, aby hodnotili vplyv liečby na exacerbácie CHOCHP
a pacienti boli zo štúdie vylúčení, ak sa u nich vyskytla exacerbácia.
Použitie záchrannej lečby
V 12-týždňovej štúdii sa pri podávaní Rolufty v porovnaní s placebom znížilo používanie záchrannej liečby s obsahom salbutamolu (zníženie v priemere o 0,7 vstreku denne počas 1. - 12. týždňa,
p = 0,025) a dosiahlo sa vyššie percento dní bez potreby záchrannej liečby (v priemere 46,3 %)'
v porovnaní s placebom (v priemere 35,2 %; nevykonala sa žiadna formálna štatistická analýza tohto
cieľového ukazovateľa). V 24-týždňovej štúdii s Roluftou bola priemerná (SD) zmena v počte
vstrekov záchrannej liečby s obsahom salbutamolu v porovnaní s východiskovým stavom počas
24-týždňového obdobia liečby -1,4 (0,20) pri placebe a -1,7 (0,16) pri Rolufte (rozdiel = -0,3;
95 % IS: -0,8; 0,2; p = 0,276). U pacientov liečených liekom Rolufta sa zaznamenalo vyššie percento dní bez potreby záchrannej liečby (v priemere 31,1 %) v porovnaní s placebom (v priemere 21,7%). Nevykonalo sa žiadne formálne štatistické testovanie tohto cieľového ukazovateľa.
Podporné štúdie účinnosti
V dvoch 12-týždňových, placebom kontrolovaných štúdiách (200109 a 200110) viedlo pridanie Rolufty k flutikazónfuroátu/vilanterolu (FF/VI) (99/22 mikrogramov) jedenkrát denne u dospelých pacientov s klinickou diagnózou CHOCHP k štatisticky signifikantným a klinicky významným zlepšeniam primárneho cieľového ukazovateľa, ktorý bol trough FEV1 na 85. deň v porovnaní
s placebom plus FF/VI (124 ml (95 % IS: 93, 154, p < 0,001) a 122 ml (95 % IS: 91, 152, p < 0,001)).
O zlepšeniach pľúcnych funkcií svedčilo znížené užívanie salbutamolu počas 1. - 12. týždňa
(-0,4 vstreku denne (95 % IS: -0,7; -0,2, p < 0,001) a -0,3 vstreku denne (95 % IS: -0,5; -0,1,
p = 0,003)) v porovnaní s placebom plus FF/VI, ale zlepšenia skóre SGRQ v 12. týždni neboli štatisticky signifikantné (200109) ani klinicky významné (200109 a 200110). Krátka dĺžka trvania
štúdií a obmedzený počet prípadov exacerbácií znemožňujú vyvodiť záver týkajúci sa dodatočného účinku Rolufty na výskyt exacerbácií CHOCHP.
V týchto štúdiách sa po pridaní Rolufty k FF/VI nezistili žiadne nové nežiaduce reakcie na liek. Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Roluftou
vo všetkých podskupinách pediatrickej populácie s CHOCHP (informácie o použití v pediatrickej
populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Po inhalačnom podaní umeklidíniumbromidu zdravým dobrovoľníkom sa Cmax dosiahla
do 5 až 15 minút. Absolútna biologická dostupnosť inhalačného umeklidíniumbromidu bola
v priemere 13 % dávky, so zanedbateľným prispením perorálnej absorpcie. Po opakovanom podávaní
inhalačného umeklidíniumbromidu sa rovnovážny stav dosiahol do 7 až 10 dní s 1,5- až 1,8-násobnou kumuláciou.
Distribúcia
Po intravenóznom podaní zdravým osobám bol priemerný distribučný objem 86 litrov. V in vitro
podmienkach bola väzba na plazmatické bielkoviny v ľudskej plazme v priemere 89 %.
B
i
otransformácia
I
n vitro štúdie ukázali, že umeklidíniumbromid sa metabolizuje hlavne prostredníctvom
cytochrómu P450 2D6 (CYP2D6) a je substrátom transportného P-glykoproteínu (P-gp). Primárne metabolické cesty umeklidíniumbromidu sú oxidatívne (hydroxylácia, O-dealkylácia) s následnou konjugáciou (glukuronidácia, atď.), ktorých výsledkom sú rôzne metabolity buď so zníženým farmakologickým účinkom, alebo s nestanoveným farmakologickým účinkom. Systémová expozícia metabolitom je nízka.
Eliminácia
Plazmatický klírens po intravenóznom podaní bol 151 litrov/hodinu. Po intravenóznom podaní sa
približne 58 % podanej rádioaktívne značenej dávky (alebo 73 % zachytenej izotopom značenej látky)
vylúčilo stolicou do 192 hodín po podaní dávky. Močom sa vylúčilo 22 % podanej rádioaktívne značenej dávky do 168 hodín (27 % zachytenej izotopom značenej látky). Vylučovanie látok súvisiacich s liečivom stolicou po intravenóznom podaní svedčilo o vylučovaní žlčou. Po perorálnom podaní zdravým mužom sa celková izotopom značená látka primárne vylúčila stolicou (92 % podanej rádioaktívne značenej dávky alebo 99 % zachytenej izotopom značenej látky) do 168 hodín po podaní dávky. Menej ako 1 % perorálne podanej dávky (1 % zachytenej izotopom značenej látky) sa vylúčilo močom, čo poukazuje na zanedbateľnú absorpciu po perorálnom podaní. Plazmatický eliminačný polčas umeklidíniumbromidu po inhalačnom podávaní počas 10 dní bol v priemere 19 hodín, s 3 %
až 4 % liečiva vylúčenými v nezmenenej forme močom v rovnovážnom stave.
Charakteristické vlastnosti v špecifických skupinách osôb alebo pacientov
Staršie osoby
Populačná farmakokinetická analýza ukázala, že farmakokinetika umeklidíniumbromidu je podobná medzi pacientmi s CHOCHP vo veku 65 a viac rokov a pacientmi mladšími ako 65 rokov.
Porucha funkcie obličiek
U osôb s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) sa nepreukázalo zvýšenie systémovej expozície umeklidíniumbromidu (Cmax a AUC) a medzi osobami s ťažkou poruchou funkcie obličiek a zdravými dobrovoľníkmi sa nepreukázala zmenená väzba na plazmatické bielkoviny.
Porucha funkcie pečene
U osôb so stredne ťažkou poruchou funkcie pečene (stupeň B Childovej-Pughovej klasifikácie) sa nepreukázalo zvýšenie systémovej expozície umeklidíniumbromidu (Cmax a AUC) a medzi osobami so stredne ťažkou poruchou funkcie pečene a zdravými dobrovoľníkmi sa nepreukázala zmenená väzba na plazmatické bielkoviny. U osôb s ťažkou poruchou funkcie pečene sa umeklidíniumbromid nehodnotil.
Ďalšie osobitné skupiny pacientov
Populačná farmakokinetická analýza ukázala, že nie je potrebná žiadna úprava dávkovania
umeklidíniumbromidu na základe veku, rasy, pohlavia, užívania inhalačných kortikosteroidov
alebo telesnej hmotnosti. Štúdia s pomalými metabolizátormi CYP2D6 nepreukázala klinicky významný vplyv genetického polymorfizmu CYP2D6 na systémovú expozíciu umeklidíniumbromidu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. V predklinických štúdiách s umeklidíniumbromidom sa zistili nálezy typicky súvisiace
s primárnym farmakologickým účinkom antagonistov muskarínových receptorov a/alebo lokálne
podráždenie.
R
eprodukčná toxicita
Umeklidíniumbromid nebol teratogénny u potkanov ani u králikov. V štúdii prenatálneho
a postnatálneho vývoja viedlo subkutánne podávanie umeklidíniumbromidu potkanom k nižšiemu prírastku telesnej hmotnosti a k nižšiemu príjmu potravy u potkaních matiek a k mierne zníženej telesnej hmotnosti mláďat pred odstavením u samíc, ktorým bola podávaná dávka
180 mikrogramov/kg/deň (približne 80-násobok klinickej expozície dosahovanej u ľudí po podaní
55 mikrogramov umeklidínia, na základe AUC).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát laktózy, magnéziumstearát.
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po otvorení vaničky: 6 týždňov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30°C. Ak sa inhalátor uchováva v chladničke, treba ho z nej vybrať aspoň
hodinu pred použitím, aby dosiahol izbovú teplotu.
Inhalátor uchovávajte vo vnútri uzatvorenej vaničky na ochranu pred vlhkosťou a vyberte ho z nej až tesne pred prvým použitím.
Inhalátor sa má používať v priebehu 6 týždňov od prvého otvorenia vaničky.
Napíšte dátum, kedy sa má inhalátor zlikvidovať, na vyhradené miesto na štítku inhalátora. Tento dátum treba doplniť hneď, ako sa inhalátor vyberie z vaničky.
6.5 Druh obalu a obsah balenia
Inhalátor Ellipta pozostáva zo šedého korpusu, svetlozeleného krytu náustka a počítadla dávok a je zabalený vo vaničke z laminátovej fólie obsahujúcej vrecko s vysúšadlom. Vanička je uzatvorená odnímateľnou fóliou.
Inhalátor obsahuje jeden blister z laminátovej hliníkovej fólie so 7 alebo 30 dávkami. Inhalátor je viaczložková pomôcka zložená z polypropylénu, polyetylénu s vysokou hustotou,
polyoxymetylénu, polybutyléntereftalátu, akrylonitrilbutadiénstyrénu, polykarbonátu a nehrdzavejúcej
ocele.
Veľkosti balenia obsahujú 7-dávkový a 30-dávkový inhalátor. Multibalenia obsahujú 3 x 30-dávkové inhalátory.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Pokyny na zaobchádzanie s liekom, pozri časť 4.2.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxoSmithKline Trading Services Limited
Currabinny
Carrigaline
County Cork
Írsko
8. REGISTRAČNÉ ČÍSLAEU/1/17/1174/001
EU/1/17/1174/002
EU/1/17/1174/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.