
e účinky
Pri užívaní liekov so sympatomimetickým účinkom vrátane Revinty Ellipta sa môžu objaviť
kardiovaskulárne účinky, akými sú srdcové arytmie, napr. supraventrikulárna tachykardia
a extrasystoly. Preto sa má flutikazónfuroát/vilanterol používať obozretne u pacientov so závažným kardiovaskulárnym ochorením.
Pacienti s poruchou funkcie pečene
U pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene sa má použiť dávka
92/22 mikrogramov a pacientov treba sledovať kvôli systémovým nežiaducim reakciám súvisiacim s kortikosteroidmi (pozri časť 5.2).
Systémové účinky kortikosteroidov
Systémové účinky sa môžu prejaviť pri akomkoľvek inhalačnom kortikosteroide, hlavne
pri dlhodobom užívaní vysokých dávok. Výskyt týchto účinkov je oveľa menej pravdepodobný ako pri perorálnych kortikosteroidoch. Možné systémové účinky zahŕňajú Cushingov syndróm, cushingoidné prejavy, útlm funkcie nadobličiek, zníženie kostnej denzity, spomalenie rastu u detí
a dospievajúcich, kataraktu a glaukóm a zriedkavejšie aj rôzne účinky na psychiku alebo správanie zahŕňajúce psychomotorickú hyperaktivitu, poruchy spánku, úzkosť, depresiu alebo agresivitu (hlavne u detí).
Flutikazónfuroát/vilanterol sa má podávať obozretne pacientom s pľúcnou tuberkulózou alebo pacientom s chronickými alebo neliečenými infekciami.
Hyperglykémia
U diabetikov bolo hlásené zvýšenie hladín glukózy v krvi a treba to vziať do úvahy pri predpisovaní
tohto lieku pacientom s diabetes mellitus v anamnéze.
Pneumónia u pacientov s CHOCHP
U pacientov s CHOCHP liečených flutikazónfuroátom/vilanterolom sa pozoroval zvýšený výskyt
pneumónie. Zaznamenal sa aj zvýšený výskyt pneumónií, ktoré mali za následok hospitalizáciu. V niekoľkých prípadoch bola pneumónia, ako nežiaduca udalosť, smrteľná (pozri časť 4.8).
U pacientov s CHOCHP musia lekári zostať ostražití kvôli možnému vzniku pneumónie, pretože klinické prejavy takýchto infekcií sa prekrývajú s príznakmi exacerbácií CHOCHP. Rizikové faktory vzniku pneumónie u pacientov s CHOCHP liečených flutikazónfuroátom/vilanterolom zahŕňajú súčasné fajčenie, pneumóniu v anamnéze, BMI < 25 kg/m2 a hodnotu FEV1 < 50 % referenčnej hodnoty. Tieto faktory treba vziať do úvahy pri predpisovaní flutikazónfuroátu/vilanterolu a v prípade výskytu pneumónie sa má liečba prehodnotiť.
Revinty Ellipta 184/22 mikrogramov nie je určený pre pacientov s CHOCHP. Dávka
184/22 mikrogramov v porovnaní s dávkou 92/22 mikrogramov nemá väčší prínos a pri jej podávaní existuje potenciálne zvýšené riziko vzniku systémových nežiaducich reakcií súvisiacich
s kortikosteroidmi (pozri časť 4.8).
Výskyt pneumónií bol u pacientov s astmou častý pri podávaní vyššej dávky. U astmatikov užívajúcich flutikazónfuroát/vilanterol 184/22 mikrogramov bol výskyt pneumónií číselne vyšší ako
u pacientov užívajúcich flutikazónfuroát/vilanterol 92/22 mikrogramov alebo placebo (pozri časť 4.8). Identifikované neboli žiadne rizikové faktory.
Pomocné látky
Pacienti so zriedkavými dedičným problémami galaktózovej intolerancie, lapónskeho deficitu laktázy
alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Klinicky významné liekové interakcie sprostredkované flutikazónfuroátom/vilanterolom v klinických dávkach sa považujú za nepravdepodobné vzhľadom na nízke plazmatické koncentrácie dosahované po inhalácii dávky.
Interakcie s betablokátormi
Blokátory beta2-adrenergných receptorov môžu oslabiť alebo antagonizovať účinok agonistov
beta2-adrenergných receptorov. Súbežnému použitiu neselektívnych aj selektívnych blokátorov
beta2-adrenergných receptorov sa treba vyhnúť, pokiaľ neexistujú závažné dôvody na ich použitie.
Interakcie s inhibítormi CYP3A4
Flutikazónfuroát aj vilanterol sú pri prvom prechode pečeňou rýchlo odstraňované vďaka rozsiahlemu
metabolizmu sprostredkovanému pečeňovým enzýmom CYP3A4.
Pri súbežnom podávaní so silnými inhibítormi CYP3A4 (napr. s ketokonazolom, ritonavirom) sa odporúča sa obozretnosť, keďže môže dôjsť k zvýšenej systémovej expozície flutikazónfuroátu aj vilanterolu. Preto je potrebné vyhnúť sa súbežnému užívaniu týchto liekov. U zdravých osôb sa uskutočnila štúdia zameraná na liekové interakcie sprostredkované CYP3A4 s opakovaným podávaním kombinácie flutikazónfuroát/vilanterol (184/22 mikrogramov) a silného inhibítora CYP3A4 ketokonazolu (400 mg). Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-24) flutikazónfuroátu o 36 % a jeho Cmax o 33 %. Zvýšenie expozície flutikazónfuroátu sa spájalo s 27 % znížením váženej priemernej hladiny kortizolu v sére nameranej počas 0 - 24 hodín. Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-t) vilanterolu o 65 % a jeho Cmax o 22 %. Zvýšenie
expozície vilanterolu nebolo spojené so zvýšením systémových účinkov súvisiacich s beta2-agonistami
na srdcovú frekvenciu, hladinu draslíka v krvi alebo QTcF interval.
Interakcie s inhibítormi P-glykoproteínu
Flutikazónfuroát aj vilanterol sú substrátmi P-glykoproteínu (P-gp). Klinická farmakologická štúdia
so zdravými osobami, ktorým bol súbežne podávaný vilanterol a verapamil, ktorý je silným inhibítorom P-gp a stredne silným inhibítorom CYP3A4, nepreukázala významný vplyv
na farmakokinetiku vilanterolu. Klinické farmakologické štúdie so špecifickým inhibítorom P-gp a flutikazónfuroátom sa neuskutočnili.
Lieky so sympatomimetickým účinkom
Súbežné podávanie iných liekov so sympatomimetickým účinkom (v monoterapii alebo ako súčasť
kombinovanej liečby) môže potencovať nežiaduce reakcie pri flutikazónfuroáte/vilanterole.
Revinty Ellipta sa nemá používať spolu s inými dlhodobo pôsobiacimi agonistami beta2-adrenergných receptorov ani s inými liekmi obsahujúcimi dlhodobo pôsobiace agonisty beta2-adrenergných receptorov.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Štúdie na zvieratách preukázali reprodukčnú toxicitu pri expozíciách, ktoré nie sú klinicky významné
(pozri časť 5.3). Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití flutikazónfuroátu a vilanteroltrifenatátu u gravidných žien.
O podávaní flutikazónfuroátu/vilanterolu gravidným ženám sa má uvažovať len vtedy, ak je očakávaný prínos pre matku väčší ako možné riziko pre plod.
Laktácia
Nie sú dostatočné informácie o vylučovaní flutikazónfuroátu alebo vilanteroltrifenatátu a/alebo
ich metabolitov do ľudského mlieka. Avšak iné kortikosteroidy a beta2-agonisty boli v ľudskom mlieku detegované (pozri časť 5.3). Riziko u dojčených novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu flutikazónfuroátom/vilanterolom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú údaje o fertilite získané u ľudí. Štúdie na zvieratách nepreukázali žiaden vplyv
flutikazónfuroátu/vilanterolu na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Flutikazónfuroát alebo vilanterol nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť
vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Na určenie frekvencie výskytu nežiaducich reakcií súvisiacich s flutikazónfuroátom/vilanterolom sa
použili údaje z rozsiahlych klinických skúšaní zameraných na astmu a CHOCHP. V programe klinického vývoja zameraného na liečbu astmy bolo do integrovaného hodnotenia nežiaducich reakcií zaradených celkovo 7 034 pacientov. V programe klinického vývoja zameraného na liečbu CHOCHP bolo do integrovaného hodnotenia nežiaducich reakcií celkovo zaradených 6 237 osôb.
Najčastejšie hlásené nežiaduce reakcie pri podávaní flutikazónfuroátu a vilanterolu boli bolesť hlavy
a nazofaryngitída. S výnimkou pneumónie a zlomenín bol bezpečnostný profil u pacientov s astmou a u pacientov s CHOCHP podobný. Počas klinických štúdií sa pneumónia a zlomeniny častejšie pozorovali u pacientov s CHOCHP.
Tabuľkový zoznam nežiaducich reakciíNežiaduce reakcie sú uvedené podľa triedy orgánových systémov a frekvencie výskytu.
Na klasifikáciu frekvencií sa použila nasledujúca konvencia: veľmi časté (≥ 1/10); časté
(≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Nežiaduca reakcia (reakcie)
| Frekvencia
|
Infekcie a nákazy
| Pneumónia*
Infekcia horných dýchacích ciest
Bronchitída
Chrípka
Kandidóza ústnej dutiny a hrdla
| Časté
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté
|
Poruchy srdca a srdcovej
činnosti
| Extrasystoly
| Menej časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
| Nazofaryngitída
Orofaryngálna bolesť
Sinusitída Faryngitída Rinitída Kašeľ Dysfónia
| Veľmi časté
Časté
|
Poruchy gastrointestinálneho traktu
| Bolesť brucha
| Časté
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Artralgia
Bolesť chrbta
Zlomeniny**
| Časté
|
Celkové poruchy a reakcie
v mieste podania
| Pyrexia
| Časté
|
*, ** Pozri „Popis vybraných nežiaducich reakcií“ uvedený nižšie
Popis vybraných nežiaducich reakcií
Pneumónia
V integrovanej analýze dvoch opakovane vykonaných jednoročných štúdií s pacientmi s CHOCHP,
ktorí prekonali exacerbáciu v predchádzajúcom roku (n = 3 255), bol počet prípadov pneumónií na 1 000 pacientorokov 97,9 pri FF/VI 184/22, 85,7 pri FF/VI 92/22 a 42,3 v skupine s VI 22.
Pri ťažkých pneumóniách sa zodpovedajúci počet prípadov na 1 000 pacientorokov rovnal 33,6; 35,5
a 7,6, v uvedenom poradí, kým pri závažných pneumóniách sa zodpovedajúci počet prípadov
na 1 000 pacientorokov rovnal 35,1 pri FF/VI 184/22, 42,9 pri FF/VI 92/22 a 12,1 pri VI 22. Napokon, počet smrteľných prípadov pneumónií upravený vzhľadom na expozíciu bol 8,8 pri FF/VI 184/22
v porovnaní s 1,5 pri FF/VI 92/22 a 0 pri VI 22FF/VI.
V integrovanej analýze 11 štúdií s astmatikmi (7 034 pacientov) bol výskyt pneumónií
na 1 000 pacientorokov 18,4 pri FF/VI 184/22 v porovnaní s 9,6 pri FF/VI 92/22 a 8,0 v skupine s placebom.
ZlomeninyV dvoch opakovane vykonaných 12-mesačných štúdiách s celkovo 3 255 pacientami s CHOCHP bol
výskyt zlomenín kostí celkovo nízky vo všetkých liečených skupinách, s vyšším výskytom
vo všetkých skupinách s Revinty Ellipta (2 %) v porovnaní so skupinou s vilanterolom v dávke
22 mikrogramov (< 1%). Hoci v skupinách s Revinty Ellipta bolo viac zlomenín v porovnaní so skupinou s vilanterolom v dávke 22 mikrogramov, zlomeniny typicky súvisiace s použitím kortikosteroidov (napr. kompresívne zlomeniny stavcov chrbtice/zlomeniny torakolumbálnych stavcov, zlomeniny bedra a acetabula) sa vyskytli u < 1 % pacientov v skupinách liečených
s Revinty Ellipta a v skupine liečenej vilanterolom.
V integrovanej analýze 11 štúdií zameraných na astmu (7 034 pacientov) bol výskyt zlomenín < 1 %
a zvyčajne súviseli s úrazom.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovaniePríznaky a prejavyPredávkovanie flutikazónfuroátom/vilanterolom môže vyvolať prejavy a príznaky spôsobené účinkom
jednotlivých zložiek vrátane tých, ktoré sú pozorované pri predávkovaní inými beta2-agonistami a zhodujú sa so známymi skupinovými účinkami inhalačných kortikosteroidov (pozri časť 4.4).
LiečbaŠpecifická liečba predávkovania flutikazónfuroátom/vilanterolom nie je k dispozícii. Ak dôjde
k predávkovaniu, pacient má podľa potreby dostať podpornú liečbu s náležitým sledovaním.
O kardioselektívnej betablokáde sa má uvažovať len v prípade závažných prejavov spôsobených predávkovaním vilanterolom, ktoré sú klinicky významné a neodpovedajú na podporné opatrenia. Kardioselektívne betablokátory sa majú použiť obozretne u pacientov s bronchospazmom v anamnéze.
Ďalšia liečba sa má riadiť klinickým stavom pacienta alebo odporúčaniami poskytnutými národným toxikologickým informačným centrom, pokiaľ sú k dispozícii.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Liečivá na obštrukčné ochorenia dýchacích ciest, adrenergiká a iné liečivá na obštrukčné ochorenia dýchacích ciest, ATC kód: R03AK10.
Mechanizmus účinku
Flutikazónfuroát a vilanterol sú zástupcami dvoch liekových skupín (syntetický kortikosteroid a
selektívny, dlhodobo pôsobiaci agonista beta2-receptorov).
Farmakodynamické účinky
Flutikazónfuroát
Flutikazónfuroát je syntetický trifluórovaný kortikosteroid so silným protizápalovým účinkom. Presný
mechanizmus, prostredníctvom ktorého flutikazónfuroát ovplyvňuje príznaky astmy a CHOCHP, nie je známy. Preukázalo sa, že kortikosteroidy majú širokú škálu účinkov na viaceré typy buniek
(napr. eozinofily, makrofágy, lymfocyty) a na mediátory (napr. cytokíny a chemokíny, ktoré sa zúčastňujú na zápale).
Vilanteroltrifenatát
Vilanteroltrifenatát je selektívny, dlhodobo pôsobiaci agonista beta2-adrenergných receptorov
(long-acting, beta2-adrenergic agonist, LABA).
Farmakologické účinky agonistov beta2-adrenergných receptorov, vrátane vilanteroltrifenatátu, možno
aspoň sčasti pripísať stimulácii vnútrobunkovej adenylátcyklázy, enzýmu, ktorý katalyzuje premenu
adenozíntrifosfátu (ATP) na cyklický-3’,5’-adenozínmonofosfát (cyklický AMP). Zvýšené hladiny cyklického AMP spôsobujú uvoľnenie hladkého svalstva priedušiek a inhibujú uvoľňovanie mediátorov okamžitej precitlivenosti z buniek, najmä z mastocytov.
Medzi kortikosteroidmi a LABA dochádza k molekulárnym interakciám, pomocou ktorých steroidy aktivujú gén pre beta2-adrenergné receptory, zvyšujú počet a citlivosť receptorov a LABA pripravujú glukokortikoidové receptory na aktiváciu závislú od steroidov a zvyšujú translokáciu do bunkového jadra. Tieto synergické interakcie sa odzrkadľujú vo zvýšenom protizápalovom účinku, ktorý sa preukázal in vitro a in vivo v rôznych zápalových bunkách, ktoré sa podieľajú na patofyziológii astmy aj CHOCHP. Štúdie s flutikazónfuroátom a vilanterolom, v ktorých sa vyšetrovali bioptické vzorky tkaniva dýchacích ciest, tiež preukázali synergické pôsobenie medzi kortikosteroidmi a LABA pri klinických dávkach liečiv podávaných pacientom s CHOCHP.
Klinická účinnosť a bezpečnosť
Astma
Tri randomizované, dvojito zaslepené štúdie fázy III (HZA106827, HZA106829 and HZA106837)
s rôznou dĺžkou trvania hodnotili bezpečnosť a účinnosť flutikazónfuroátu/vilanterolu u dospelých a dospievajúcich pacientov s perzistentnou astmou. Všetky osoby užívali ICS (inhalačný kortikosteroid) s LABA, alebo bez LABA, aspoň 12 týždňov pred 1. návštevou. V štúdii HZA106837 prekonali všetci pacienti aspoň jednu exacerbáciu, ktorá si vyžadovala liečbu perorálnymi kortikosteroidmi, v roku predchádzajúcom 1. návšteve. Štúdia HZA106827 trvala 12 týždňov
a hodnotila účinnosť flutikazónfuroátu/vilanterolu 92/22 mikrogramov [n = 201] a FF
92 mikrogramov [n = 205]) v porovnaní s placebom [n = 203], všetky lieky boli podávané jedenkrát
denne. Štúdia HZA106829 trvala 24 týždňov a hodnotila účinnosť flutikazónfuroátu/vilanterolu
184/22 mikrogramov [n = 197] a FF 184 mikrogramov [n = 194]), oba lieky boli podávané jedenkrát denne, v porovnaní s FP 500 mikrogramov dvakrát denne [n = 195].
V štúdiách HZA106827/HZA106829 boli kombinovanými primárnymi cieľmi účinnosti zmena trough
FEV1 (minimálna hodnota FEV1 pred podaním bronchodilatancia a pred podaním dávky)
na kontrolnej návšteve na konci obdobia liečby v porovnaní s východiskovou hodnotou u všetkých
osôb a vážený priemer opakovane meranej hodnoty FEV1 počas 0 - 24 hodín po podaní dávky vypočítaný v podskupine osôb na konci obdobia liečby. Sekundárnym cieľom, na hodnotenie ktorého mala štúdia dostatočnú štatistickú silu, bola zmena v percentuálnom počte dní bez potreby záchranného lieku počas liečby v porovnaní s východiskovým stavom. Výsledky primárnych
a kľúčových sekundárnych cieľov v týchto štúdiách sú uvedené v tabuľke 1.
Tabuľka 1 - Výsledky primárnych a kľúčových sekundárnych cieľov v štúdiách HZA106827 aHZA106829Číslo štúdie
| HZA106829
| HZA106827
|
Liečebná dávka
FF/VI*(mikrogramy)
| FF/VI 184/22
jedenkrát denne vs. FF 184 jedenkrát denne
| FF/VI 184/22
jedenkrát denne vs. FP 500
dvakrát denne
| FF/VI 92/22
jedenkrát denne vs. FF 92 jedenkrát denne
| FF/VI/92/22
jedenkrát denne vs. placebo jedenkrát denne
|
Zmena trough FEV1 určeného na základe posledného preneseného pozorovania (Last
Observation Carried Forward, LOCF) v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
| 193 ml
p<0,001 (108, 277)
| 210 ml
p<0,001 (127, 294)
| 36 ml
p=0,405
(-48, 120)
| 172 ml
p<0,001 (87, 258)
|
Vážený priemer opakovane meranej hodnoty FEV1 počas 0 - 24 hodín po podaní dávky
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
| 136 ml
p=0,048 (1, 270)
| 206 ml
p=0,003 (73, 339)
| 116 ml
p=0,06
(-5, 236)
| 302 ml
p<0,001 (178, 426)
|
Zmena v percentuálnom počte dní bez potreby záchranného lieku v porovnaní s východiskovým stavom
|
Rozdiel medzi liečbami p-hodnota
(95 % IS)
| 11,7 %
p<0,001 (4,9; 18,4)
| 6,3 %
p=0,067
(-0,4; 13,1)
| 10,6 %
p<0,001 (4,3; 16,8)
| 19,3 %
p<0,001 (13,0; 25,6)
|
Zmena v percentuálnom počte dní bez príznakov v porovnaní s východiskovým stavom
|
Rozdiel medzi liečbami p-hodnota
(95 % IS)
| 8,4 %
p=0,010 (2,0; 14,8)
| 4,9 %
p=0,137
(-1,6; 11,3)
| 12,1 %
p<0,001 (6,2; 18,1)
| 18,0 %
p<0,001 (12,0; 23,9)
|
Zmena v dopoludňajšej hodnote maximálneho výdychového prietoku (PEF)
v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
| 33,5 l/min
p<0,001 (22,3; 41,7)
| 32,9 l/min
p<0,001 (24,8; 41,1)
| 14,6 l/min
p<0,001 (7,9; 21,3)
| 33,3 l/min
p<0,001 (26,5; 40,0)
|
Zmena v popoludňajšej hodnote maximálneho výdychového prietoku (PEF)
v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
| 30,7 l/min
p<0,001 (22,5; 38,9)
| 26,2 l/min
p<0,001 (18,0; 34,3)
| 12,3 l/min
p<0,001 (5,8; 18,8)
| 28,2 l/min
p<0,001 (21,7; 34,8)
|
*FF/VI = flutikazónfuroát/vilanterolŠtúdia HZA106837 mala rôznu dĺžku trvania (od minimálne 24 týždňov po maximálne 76 týždňov, pričom väčšina pacientov bola liečená aspoň 52 týždňov). V štúdii HZA106837 bola pacientom náhodne pridelená buď liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov [n = 1 009], alebo liečba FF 92 mikrogramov [n = 1 010], oba lieky boli podávané jedenkrát denne. V štúdii HZA106837 bol primárnym cieľom čas do objavenia sa prvej závažnej exacerbácie astmy. Závažná exacerbácia
astmy bola definovaná ako zhoršenie astmy vyžadujúce si užívanie systémových kortikosteroidov počas aspoň 3 dní alebo hospitalizácia pacienta v nemocnici alebo návšteva lekárskej pohotovosti kvôli astme, ktorá si vyžadovala liečbu systémovými kortikosteroidmi. Upravená priemerná zmena trough FEV1 v porovnaní s východiskovou hodnotou sa taktiež hodnotila ako sekundárny cieľ.
V štúdii HZA106837 sa riziko výskytu závažnej exacerbácie astmy u pacientov liečených flutikazónfuroátom/vilanterolom 92/22 mikrogramov znížilo o 20 % v porovnaní s pacientmi liečenými samotným FF 92 mikrogramov (hazard ratio 0,795, p = 0,036, 95 % IS: 0,642; 0,985). Výskyt závažných exacerbácií astmy na pacientoroky bol 0,19 v skupine s FF 92 mikrogramov (približne 1 prípad na každých 5 rokov) a 0,14 v skupine s flutikazónfuroátom/vilanterolom
92/22 mikrogramov (približne 1 prípad na každých 7 rokov). Pomer výskytu exacerbácií pri liečbe flutikazónfuroátom/vilanterolom 92/22 mikrogramov v porovnaní s liečbou FF 92 mikrogramov bol
0,755 (95 % IS: 0,603; 0,945). To predstavuje 25 % zníženie výskytu závažných exacerbácií astmy u osôb liečených flutikazónfuroátom/vilanterolom 92/22 mikrogramov v porovnaní s osobami liečenými FF 92 mikrogramov (p = 0,014). Dvadsaťštyrihodinový bronchodilatačný účinok flutikazónfuroátu/vilanterolu sa zachoval počas celého jednoročného obdobia liečby bez toho, že by sa preukázala strata účinnosti (bez výskytu tachyfylaxie). Pri liečbe flutikazónfuroátom/vilanterolom
92/22 mikrogramov sa zhodne preukázalo zlepšenie trough FEV1 o 83 ml až 95 ml
v 12., 36. a 52. týždni a pri poslednom zaznamenanom meraní (Endpoint) na konci liečby v porovnaní
s liečbou FF 92 mikrogramov (p < 0,001, 95 % IS: 52, 126 ml pri poslednom zaznamenanom meraní
(Endpoint)). Na konci liečby 44 % pacientov v skupine s flutikazónfuroátom/vilanterolom
92/22 mikrogramov dosiahlo dobrú kontrolu ochorenia (skóre dotazníka na hodnotenie kontroly astmy - ACQ7 ≤ 0,75) v porovnaní s 36 % osôb v skupine s FF 92 mikrogramov (p < 0,001, 95 % IS:
1,23; 1,82).
Štúdie zamerané na porovnanie s kombináciou salmeterol/flutikazónpropionát
V 24-týždňovej štúdii (HZA113091) s dospelými a dospievajúcimi pacientmi s perzistentnou astmou sa tak pri liečbe flutikazónfuroátom/vilanterolom 92/22 mikrogramov podávaným jedenkrát denne večer, ako aj pri liečbe kombináciou salmeterol/FP 50/250 mikrogramov podávanou dvakrát denne preukázalo zlepšenie pľúcnych funkcií v porovnaní s východiskovým stavom. Upravené priemerné zvýšenie váženého priemeru hodnoty FEV1 meranej počas 0 - 24 hodín, ku ktorému došlo počas liečby, v porovnaní s východiskovou hodnotou, o 341 ml (pri liečbe kombináciou flutikazónfuroát/vilanterol) a o 377 ml (pri liečbe kombináciou salmeterol/FP) svedčilo o celkovom zlepšení pľúcnych funkcií počas 24 hodín pri oboch liečbach. Upravený priemerný rozdiel medzi liečbami rovnajúci sa 37 ml medzi skupinami nebol štatisticky významný (p = 0,162). Pokiaľ ide
o trough FEV1, osoby v skupine s flutikazófuroátom/vilanterolom dosiahli priemernú zmenu vypočítanú s použitím metódy najmenších štvorcov (least squares, LS) rovnajúcu sa 281 ml
v porovnaní s východiskovou hodnotou a osoby v skupine so salmeterolom/FP dosiahli zmenu rovnajúcu sa 300 ml; (rozdiel v upravenom priemere rovnajúci sa 19 ml (95 % IS: -0,073; 0,034) nebol štatisticky významný (p = 0,485)).
Neuskutočnili sa žiadne porovnávacie štúdie s kombináciou salmeterol/FP alebo s inými kombináciami ICS/LABA, ktoré by náležite porovnali vplyv na exacerbácie astmy.
Flutikazónfuroát v monoterapii
Dvadsaťštyritýždňová randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia (FFA112059)
hodnotila bezpečnosť a účinnosť FF 92 mikrogramov jedenkrát denne [n = 114]
a FP 250 mikrogramov dvakrát denne [n = 114] v porovnaní s placebom [n = 115] u dospelých
a dospievajúcich pacientov s perzistentnou astmou. Všetky osoby museli užívať stabilnú dávku ICS
aspoň 4 týždne pred 1. návštevou (skríningovou návštevou) a použitie LABA nebolo povolené počas
4 týždňov od 1. návštevy. Primárnym cieľom účinnosti bola zmena trough FEV1 (pred podaním bronchodilatancia a pred podaním dávky) na kontrolnej návšteve na konci obdobia liečby v porovnaní s východiskovou hodnotou. Sekundárnym cieľom, na hodnotenie ktorého mala štúdia dostatočnú štatistickú silu, bola zmena v percentuálnom počte dní bez potreby záchranného lieku počas
24-týždňového obdobia liečby v porovnaní s východiskovým stavom. V 24. týždni sa trough FEV1
pri podávaní FF zvýšil o 146 ml (95 % IS: 36, 257 ml, p = 0,009) a pri podávaní FP sa zvýšil o 145 ml (95 % IS: 33, 257 ml, p = 0,011) v porovnaní s placebom. Percentuálny počet dní bez potreby záchranného lieku sa pri podávaní FF zvýšil o 14,8 % (95 % IS: 6,9; 22,7, p < 0,001) a pri podávaní
FP sa zvýšil o 17,9 % (95 % IS: 10,0; 25,7, p < 0,001) v porovnaní s placebom.
Štúdia využívajúca provokáciu alergénom
Bronchoprotektívny účinok flutikazónfuroátu/vilanterolu 92/22 mikrogramov na včasnú a neskorú astmatickú odpoveď na inhalačný alergén sa hodnotil v placebom kontrolovanej štúdii s pacientmi s ľahkou astmou, so štyrmi spôsobmi skríženia liečby (four-way crossover) a s opakovaným podávaním lieku (HZA113126). Pacientom bola náhodne pridelená liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov, FF 92 mikrogramov, vilanterolom
22 mikrogramov alebo placebom, všetky lieky boli podávané jedenkrát denne počas 21 dní, po ktorej nasledovala provokácia alergénom 1 hodinu po poslednej dávke. Alergénom boli roztoče bytového prachu, mačacie lupiny alebo brezový peľ; výber bol založený na individuálnych skríningových testoch. Opakované merania hodnoty FEV1 sa porovnali s hodnotami pred provokáciou alergénom
po inhalácii fyziologického roztoku (východiskové hodnoty). Celkovo možno konštatovať, že najväčší
účinok na včasnú astmatickú odpoveď sa pozoroval pri liečbe flutikazónfuroátom/vilanterolom
92/22 mikrogramov v porovnaní s liečbou samotným FF 92 mikrogramov alebo samotným vilanterolom 22 mikrogramov. Tak liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov, ako aj liečba FF 92 mikrogramov prakticky odstránila neskorú astmatickú odpoveď v porovnaní s liečbou samotným vilanterolom. Liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov poskytla významne väčšiu ochranu pred hyperreaktivitou priedušiek vyvolanou alergénom v porovnaní s FF
a vilanterolom podávanými v monoterapii, čo sa hodnotilo na 22. deň prostredníctvom provokácie metacholínom.
Chronická obštrukčná choroba pľúc
Program klinického vývoja zameraný na liečbu CHOCHP zahŕňal 12-týždňovú (HZC113107), dve
6-mesačné (HZC112206, HZC112207) a dve jednoročné (HZC102970, HZC102871) randomizované kontrolné štúdie s pacientmi s klinickou diagnózou CHOCHP. Tieto štúdie zahŕňali hodnotenia pľúcnych funkcií, dyspnoe a stredne ťažkých až ťažkých exacerbácií.
Šesťmesačné štúdie
Štúdie HZC112206 a HZC112207 boli 24-týždňové randomizované, dvojito zaslepené, placebom kontrolované štúdie s paralelným usporiadaním skupín, porovnávajúce účinok kombinácie
so samostatne podávaným vilanterolom a FF a s placebom. Štúdia HZC112206 hodnotila účinnosť
flutikazónfuroátu/vilanterolu 46/22 mikrogramov [n = 206] a flutikazónfuroátu/vilanterolu
92/22 mikrogramov [n = 206] v porovnaní s FF 92 mikrogramov [n = 206], vilanterolom
22 mikrogramov [n = 205] a s placebom [n = 207], všetky lieky boli podávané jedenkrát denne. Štúdia
HZC112207 hodnotila účinnosť flutikazónfuroátu/vilanterolu 92/22 mikrogramov [n = 204]
a flutikazónfuroátu/vilanterolu 184/22 mikrogramov [n = 205] v porovnaní s FF 92 mikrogramov
[n = 204], FF 184 mikrogramov [n = 203] a s vilanterolom 22 mikrogramov [n = 203] a s placebom
[n = 205], všetky lieky boli podávané jedenkrát denne.
Všetci pacienti museli mať anamnézu fajčenia aspoň 10 balíčkorokov; pomer FEV1/FVC po podaní salbutamolu menší alebo rovný 0,70; hodnotu FEV1 po podaní salbutamolu menšiu alebo rovnú 70 % referenčnej hodnoty a modifikované skóre dyspnoe podľa Rady pre lekársky výskym (Modified Medical Research Council, mMRC) ≥ 2 (stupnica 0 - 4) pri skríningu. V štúdii HZC112206 bola
pri skríningu priemerná hodnota FEV1 pred podaním bronchodilatancia 42,6 % referenčnej hodnoty a priemerná reverzibilita bola 15,9 % a v štúdii HZC112207 bola pri skríningu priemerná hodnota FEV1 pred podaním bronchodilatancia 43,6 % hodnoty a priemerná reverzibilita bola 12,0 %. Kombinovanými primárnymi cieľmi v oboch štúdiách boli vážený priemer hodnoty FEV1 od nuly po 4 hodiny po podaní dávky na 168. deň a zmena trough FEV1 pred podaním dávky na 169. deň
v porovnaní s východiskovou hodnotou.
V integrovanej analýze oboch štúdií sa pri liečbe flutikazónfuroátom/vilanterolom 92/22 mikrogramov preukázalo klinický významné zlepšenie pľúcnych funkcií. Na 169. deň sa upravená priemerná
hodnota trough FEV1 pri podávaní flutikazónfuroátu/vilanterolu 92/22 mikrogramov zvýšila o 129 ml
(95 % IS: 91, 167 ml, p < 0,001) a pri podávaní vilanterolu sa zvýšila o 83 ml (95 % IS: 46, 121 ml,
p < 0,001) v porovnaní s placebom. Trough FEV1 sa pri podávaní flutikazónfuroátu/vilanterolu
92/22 mikrogramov zvýšil o 46 ml v porovnaní s vilanterolom (95 % IS: 8, 83 ml, p = 0,017).
Na 168. deň sa upravený priemerný vážený priemer hodnoty FEV1 počas 0 - 4 hodín pri podávaní flutikazónfuroátu/vilanterolu 92/22 mikrogramov zvýšil o 193 ml (95 % IS: 156, 230 ml, p < 0,001) a pri podávaní vilanterolu sa zvýšil o 145 ml (95 % IS: 108, 181 ml, p < 0,001) v porovnaní s placebom. Upravený priemerný vážený priemer hodnoty FEV1 počas 0 - 4 hodín sa pri podávaní flutikazónfuroátu/vilanterolu 92/22 mikrogramov zvýšil o 148 ml v porovnaní so samotným FF (95 % IS: 112, 184 ml, p < 0,001).
12-mesačné štúdie
Štúdie HZC102970 a HZC102871 boli 52-týždňové randomizované, dvojito zaslepené štúdie
s paralelným usporiadaním skupín porovnávajúce flutikazónfuroát/vilanterol 184/22 mikrogramov, flutikazónfuroát/vilanterol 92/22 mikrogramov, flutikazónfuroát/vilanterol 46/22 mikrogramov a vilanterol 22 mikrogramov, všetky lieky boli podávané jedenkrát denne. Sledoval sa vplyv na ročný výskyt stredne ťažkých/ťažkých exacerbácií u osôb s CHOCHP s anamnézou fajčenia aspoň
10 balíčkorokov a s pomerom FEV1/FVC po podaní salbutamolu menším alebo rovným 0,70
a s hodnotou FEV1 po podaní salbutamolu menšou alebo rovnou 70 % referenčnej hodnoty a ktoré
mali v anamnéze zdokumentovaný výskyt ≥ 1 exacerbácie CHOCHP, ktorá si vyžadovala antibiotiká
a/alebo perorálne kortikosteroidy alebo hospitalizáciu v priebehu 12 mesiacov pred 1. návštevou. Primárnym cieľom bol ročný výskyt stredne ťažkých a ťažkých exacerbácií. Stredne ťažké/ťažké exacerbácie boli definované ako zhoršujúce sa príznaky, ktoré si vyžadovali liečbu perorálnymi kortikosteroidmi a/alebo antibiotikami alebo hospitalizáciu pacienta v nemocnici. Obe štúdie mali
4-týždňovú úvodnú fázu, v rámci ktorej dostávali všetky osoby otvorenú liečbu salmeterolom/FP
50/250 dvakrát denne s cieľom štandardizovať farmakoterapiu CHOCHP a stabilizovať ochorenie pred náhodným pridelením zaslepenej liečby skúšaným liekom trvajúcej 52 týždňov. Pred začiatkom úvodnej fázy osoby prestali užívať predchádzajúce lieky na CHOCHP okrem krátkodobo pôsobiacich bronchodilatancií. Počas obdobia liečby nebolo povolené súbežné užívanie inhalačných dlhodobo pôsobiacich bronchodilatancií (beta2-agonistov a anticholinergík), liekov obsahujúcich kombináciu ipratrópium/salbutamol, perorálnych beta2-agonistov a liekov s obsahom teofylínu. Povolené boli perorálne kortikosteroidy a antibiotiká na akútnu liečbu exacerbácií CHOCHP, ktoré sa užívali podľa špecifických pokynov. Osoby užívali salbutamol podľa potreby počas celého trvania štúdií.
Výsledky oboch štúdií ukázali, že liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov jedenkrát denne viedla k nižšiemu ročnému výskytu stredne ťažkých/ťažkých exacerbácií CHOCHP v porovnaní s vilanterolom (tabuľka 2).
Tabuľka 2: Analýza výskytu exacerbácií po 12 mesiacoch liečby
Cieľ
|
HZC102970
|
HZC102871
|
Integrované výsledky HZC102970 a HZC102871
|
Vilanterol
(n=409)
|
flutikazón-
furoát/
vilanterol
92/22 (n=403)
|
Vilanterol
(n=409)
|
Flutikazón-
furoát/
vilanterol
92/22 (n=403)
|
Vilanterol
(n=818)
|
Flutikazón-
furoát/
vilanterol
92/22 (n=806)
|
Stredne ťažké a ťažké exacerbácie
|
Upravený
priemerný ročný výskyt
|
1,14
|
0,90
|
1,05
|
0,70
|
1,11
|
0,81
|
Výskyt
v porovnaní s VI
95 % IS
p-hodnota
% zníženie
(95 % IS)
|
|
0,79 (0,64;0,97)
0,024
21 (3, 36)
|
|
0,66 (0,54; 0,81)
<0,001
34 (19,46)
|
|
0,73 (0,63; 0,84)
<0,001
27 (16, 37)
|
Absolútny
rozdiel v počte exacerbácií
na rok
v porovnaní s vilanterolom (95 % IS)
|
|
0,24 (0,03; 0,41)
|
|
0,36 (0,20; 0,48)
|
|
0,30 (0,18; 0,41)
|
Čas do
objavenia sa prvej exacerbácie: Hazard ratio (95 % IS)
% zníženie rizika
p-hodnota
|
|
0,80 (0,66; 0,99)
20
0,036
|
|
0,72 (0,59; 0,89)
28
0,002
|
|
0,76 (0,66; 0,88)
24
p<0,001
|
V integrovanej analýze štúdií HZC102970 a HZC102871 sa v 52. týždni pozorovalo zlepšenie
upravenej priemernej hodnoty trough FEV1 (o 42 ml, 95 % IS: 19, 64 ml, p < 0,001) pri porovnaní liečby flutikazónfuroátom/vilanterolom 92/22 mikrogramov a liečby vilanterolom v dávke
22 mikrogramov. Dvadsaťštyrihodinový bronchodilatačný účinok flutikazónfuroátu/vilanterolu sa zachoval od podania prvej dávky po celé jednoročné obdobie liečby bez toho, že by sa preukázala strata účinnosti (bez výskytu taxyfylaxie).
Pri kombinovanom posudzovaní týchto dvoch štúdií malo celkovo 2 009 (62 %) pacientov kardiovaskulárne ochorenie v anamnéze/kardiovaskulárne rizikové faktory pri skríningu. Výskyt kardiovaskulárnych ochorení v anamnéze/kardiovaskulárnych rizikových faktorov bol medzi liečebnými skupinami podobný, pričom pacienti najčastejšie trpeli hypertenziou (46 %), po nej nasledovala hypercholesterolémia (29 %) a diabetes mellitus (12 %). V tejto podskupine pacientov sa v porovnaní s celkovou populáciou pozorovali podobné vplyv na zníženie výskytu stredne ťažkých a ťažkých exacerbácií. U pacientov s kardiovaskulárnymi ochoreniami v anamnéze/kardiovaskulárnymi rizikovými faktormi viedla liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov k významne
nižšiemu ročnému výskytu stredne ťažkých/ťažkých exacerbácií CHOCHP v porovnaní s vilanterolom
(upravený priemerný ročný výskyt 0,83 a 1,18, v uvedenom poradí, 30 % zníženie výskytu (95 % IS:
16, 42 %, p < 0,001)). V tejto podskupine sa v 52. týždni pozorovalo aj zlepšenie upravenej priemernej
hodnoty trough FEV1 (44 ml, 95 % IS: 15, 73 ml, (p = 0,003)) pri porovnaní liečby flutikazónfuroátom/vilanterolom 92/22 mikrogramov s liečbou vilanterolom v dávke 22 mikrogramov.
Štúdie zamerané na porovnanie s kombináciou salmeterol/flutikazónpropionát
V 12-týždňovej štúdii (HZC113107) s pacientmi s CHOCHP sa pri liečbe kombináciou flutikazónfuroát/vilanterol 92/22 mikrogramov podávanou jedenkrát denne ráno, ako aj pri liečbe kombináciou salmeterol/FP 50/500 mikrogramov podávanou dvakrát denne preukázalo zlepšenie pľúcnych funkcií v porovnaní s východiskovým stavom. Upravené priemerné zvýšenie váženého priemeru hodnoty FEV1 meranej počas 0 - 24 hodín, ku ktorému došlo počas liečby, v porovnaní
s východiskovou hodnotou, o 130 ml (pri liečbe kombináciou flutikazónfuroát/vilanterol) a o 108 ml
(pri liečbe kombináciou salmeterol/FP) svedčilo o celkovom zlepšení pľúcnych funkcií počas 24 hodín pri oboch liečbach. Upravený priemerný rozdiel rovnajúci sa 22 ml (95 % IS: -18, 63 ml) medzi skupinami nebol štatisticky významný (p = 0,282). Upravená priemerná zmena trough FEV1
na 85. deň v porovnaní s východiskovou hodnotou bola 111 ml v skupine
s flutikazófuroátom/vilanterolom a 88 ml v skupine so salmeterolom/FP; rozdiel medzi liečebnými skupinami rovnajúci sa 23 ml (95 % IS: -20, 66) nebol klinicky či štatisticky významný (p = 0,294). Neuskutočnili sa žiadne porovnávacie štúdie s kombináciou salmeterol/FP alebo s inými bežne podávanými bronchodilatanciami, ktoré by náležite porovnali vplyv na exacerbácie CHOCHP.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky s Revinty Ellipta
vo všetkých podskupinách pediatrickej populácie s CHOCHP (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Revinty Ellipta v jednej alebo vo viacerých podskupinách pediatrickej populácie s astmou (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absolútna biologická dostupnosť flutikazónfuroátu a vilanterolu podávaných inhalačne vo forme
kombinácie flutikazónfuroát/vilanterol bola v priemere 15,2 % a 27,3 %, v uvedenom poradí. Perorálna biologická dostupnosť flutikazónfuroátu aj vilanterolu bola nízka, v priemere 1,26 %
a < 2 %, v uvedenom poradí. Vzhľadom na nízku perorálnu biologickú dostupnosť je systémová expozícia flutikazónfuroátu a vilanterolu po inhalačnom podaní predovšetkým dôsledkom absorpcie inhalovanej dávky dodanej do pľúc.
Distribúcia
Po intravenóznom podaní sa flutikazónfuroát aj vilanterol v rozsiahlej miere distribuujú s priemerným
distribučným objemom v rovnovážnom stave 661 l a 165 l, v uvedenom poradí. Flutikazónfuroát aj vilanterol sa slabo viažu na erytrocyty. V in vitro podmienkach bola väzba flutikazónfuroátu a vilanterolu na plazmatické bielkoviny v ľudskej plazme vysoká, v priemere
> 99,6 % a 93,9 %, v uvedenom poradí. U osôb s poruchou funkcie obličiek alebo pečene sa nezistilo zníženie rozsahu väzby na plazmatické bielkoviny in vitro.
Flutikazónfuroát a vilanterol sú substrátmi P-glykoproteínu (P-gp), predpokladá sa však, že súbežné podávanie flutikazónfuroátu/vilanterolu s inhibítormi P-gp pravdepodobne nezmení systémovú expozíciu flutikazónfuroátu ani vilanterolu, keďže oba sú dobre absorbovateľnými molekulami.
Biotranformácia
Na základe in vitro údajov sú hlavné metabolické cesty flutikazónfuroátu aj vilanterolu
sprostredkované predovšetkým enzýmom CYP3A4.
Flutikazónfuroát sa primárne metabolizuje hydrolýzou S-fluórmetyl-karbotioátu na metabolity s významne zníženým kortikosteroidovým účinkom. Vilanterol sa primárne metabolizuje
O-dealkyláciou na rôzne metabolity s významne zníženým β1- a β2-agonistickým účinkom.
Eliminácia
Po perorálnom podaní sa flutikazónfuroát u ľudí eliminoval predovšetkým metabolizmom, pričom
jeho metabolity sa vylučovali takmer výhradne stolicou a < 1 % izotopom značenej dávky sa vylúčilo močom.
Po perorálnom podaní sa vilanterol eliminoval hlavne metabolizmom, s následným vylučovaním metabolitov, pričom v štúdii s izotopom značenou látkou perorálne podávanou ľuďom sa 70 % izotopom značenej dávky vylúčilo močom a 30 % sa vylúčilo stolicou. Zdanlivý plazmatický eliminačný polčas vilanterolu po jednorazovom inhalačnom podaní flutikazónfuroátu/vilanterolu bol v priemere 2,5 hodiny. Efektívny polčas kumulácie vilanterolu, stanovený na základe inhalačného
podávania opakovaných 25 mg dávok vilanterolu, je 16,0 hodín u osôb s astmou a 21,3 hodiny u osôb s CHOCHP.
Pediatrická populácia
U dospievajúcich (vo veku 12 rokov a starších) sa neodporúčajú žiadne úpravy dávky.
Farmakokinetika flutikazónfuroátu/vilanterolu u pacientov mladších ako 12 rokov sa nesledovala. Bezpečnosť a účinnosť flutikazónfuroátu/vilanterolu u detí mladších ako 12 rokov neboli doteraz stanovené.
Osobitné skupiny pacientov
Starší pacienti (> 65 rokov)
Vplyv veku na farmakokinetiku flutikazónfuroátu a vilanterolu sa stanovil v štúdiách fázy III zameraných na astmu a CHOCHP. U osôb s astmou sa nepreukázalo, že by vek (12 - 84 rokov) ovplyvňoval farmakokinetiku flutikazónfuroátu a vilanterolu.
U osôb s CHOCHP sa nepreukázalo, že by vek ovplyvňoval farmakokinetiku flutikazónfuroátu, zatiaľ čo vo vekovej skupine 41- až 84-ročných sa pozorovalo zvýšenie (37 %) hodnoty AUC(0-24) vilanterolu. Predpokladá sa, že u staršej osoby (vo veku 84 rokov) s nízkou telesnou hmotnosťou
(35 kg) bude hodnota AUC(0-24) vilanterolu o 35 % vyššia ako odhadovaná hodnota v tejto populácii (u osoby s CHOCHP vo veku 60 rokov a s telesnou hmotnosťou 70 kg), zatiaľ čo hodnota Cmax zostane nezmenená. Tieto rozdiely pravdepodobne nie sú klinicky významné.
Pre osoby s astmou a osoby s CHOCHP sa neodporúčajú žiadne úpravy dávky.
Porucha funkcie obličiek
Klinická farmakologická štúdia s flutikazónfuroátom/vilanterolom ukázala, že ťažká porucha funkcie obličiek (klírens kreatinínu < 30 ml/min) nemala za následok významne vyššiu expozíciu flutikazónfuroátu alebo vilanterolu ani výraznejšie systémové účinky spojené s kortikosteroidmi alebo beta2-agonistami v porovnaní so zdravými osobami.
U pacientov s poruchou funkcie obličiek nie je potrebná úprava dávky. Vplyv hemodialýzy sa nesledoval.
Porucha funkcie pečene
Po opakovanom podávaní flutikazónfuroátu/vilanterolu počas 7 dní došlo u osôb s poruchou funkcie pečene (stupňa A, B alebo C Childovej-Pughovej klasifikácie) v porovnaní so zdravými osobami
k zvýšeniu systémovej expozície flutikazónfuroátu (až na trojnásobok, keď sa merala na základe AUC(0–24)). Zvýšenie systémovej expozície flutikazónfuroátu u osôb so stredne ťažkou poruchou funkcie pečene (stupňa B Childovej-Pughovej klasifikácie; pri podávaní flutikazónfuroátu/vilanterolu
184/22 mikrogramov) sa v porovnaní so zdravými osobami spájalo s priemerným 34 % znížením hladiny koncentrácie kortizolu v sére. Systémová expozícia flutikazónfuroátu normalizovaná na dávku
bola u osôb so stredne ťažkou a ťažkou poruchou funkcie pečene (stupňa B alebo C
Childovej-Pughovej klasifikácie) podobná.
Po opakovanom podávaní flutikazónfuroátu/vilanterolu počas 7 dní nedošlo u osôb s miernou, stredne ťažkou alebo ťažkou poruchou funkcie pečene (stupňa A, B alebo C Childovej-Pughovej klasifikácie) k významnému zvýšeniu systémovej expozície vilanterolu (Cmax a AUC).
U osôb s miernou alebo stredne ťažkou poruchou funkcie pečene (vilanterol, 22 mikrogramov) alebo s ťažkou poruchou funkcie pečene (vilanterol, 12,5 mikrogramu) nemala kombinácia flutikazónfuroát/vilanterol klinicky významný vplyv na beta-adrenergné systémové účinky (srdcová frekvencia alebo hladina draslíka v sére) v porovnaní so zdravými osobami.
Ďalšie osobitné skupiny pacientov
U osôb s astmou bola odhadovaná hodnota AUC(0-24) flutikazónfuroátu u osôb pochádzajúcich
z východnej Ázie, Japonska a juhovýchodnej Ázie (12 - 13 % osôb) v priemere o 33 % až 53 % vyššia
v porovnaní s inými rasovými skupinami. Nepreukázalo sa však, že by sa vyššia systémová expozícia v tejto populácii spájala s vyšším účinkom na 24-hodinovú exkréciu kortizolu do moču. Predpokladá sa, že hodnota Cmax vilanterolu bude v priemere o 220 až 287 % vyššia a hodnota AUC(0-24) bude porovnateľná u osôb ázijského pôvodu v porovnaní s osobami iných rasových skupín. Nepreukázalo sa však, že by táto vyššia hodnota Cmax vilanterolu mala za následok klinicky významný vplyv
na srdcovú frekvenciu.
U osôb s CHOCHP bola odhadovaná hodnota AUC(0-24) flutikazónfuroátu u osôb pochádzajúcich
z východnej Ázie, Japonska a juhovýchodnej Ázie (13 - 14 % osôb) v priemere o 23 % až 30 % vyššia
v porovnaní s osobami kaukazskej rasy. Nepreukázalo sa však, že by sa vyššia systémová expozícia v tejto populácii spájala s vyšším účinkom na 24-hodinovú exkréciu kortizolu do moču. U osôb
s CHOCHP sa nezistil žiaden vplyv rasy na odhadované hodnoty farmakokinetických parametrov vilanterolu.
Pohlavie, telesná hmotnosť a BMI
Na základe populačnej farmakokinetickej analýzy údajov zo štúdie fázy III získaných od 1 213 osôb s astmou (712 žien) a 1 225 osôb s CHOCHP (392 žien) sa nepreukázalo, že by pohlavie, telesná hmotnosť alebo BMI (index telesnej hmotnosti) ovplyvňovali farmakokinetiku flutikazónfuroátu.
Na základe populačnej farmakokinetickej analýzy u 856 osôb s astmou (500 žien) a 1 091 osôb
s CHOCHP (340 žien) sa nepreukázalo, že by pohlavie, telesná hmotnosť alebo BMI ovplyvňovali farmakokinetiku vilanterolu.
Nie je potrebná žiadna úprava dávkovania v závislosti od pohlavia, telesnej hmotnosti alebo BMI.
5.3 Predklinické údaje o bezpečnosti
Farmakologické a toxikologické účinky pozorované pri podávaní flutikazónfuroátu alebo vilanterolu v predklinických štúdiách boli rovnaké ako tie, ktoré sa typicky spájajú buď s glukokortikosteroidmi, alebo s beta2-agonistami. Podávanie flutikazónfuroátu v kombinácii s vilanterolom neviedlo
k významnej novej toxicite.
Genotoxicita a karcinogenita
Flutikazónfuroát
Flutikazónfuroát nebol genotoxický v štandardnom súbore štúdií a nebol karcinogénny v štúdiách s jeho celoživotným inhalačným podávaním potkanom a myšiam pri expozíciách podobných tým, ktoré sa dosahujú po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Vilanteroltrifenatát
V štúdiách genetickej toxicity neboli vilanterol (vo forme alfa-fenylcinamátu) a kyselina trifenyloctová genotoxické, čo poukazuje na to, že vilanterol (vo forme trifenatátu) nepredstavuje genotoxické riziko pre ľudí.
Zhodne so zisteniami získanými pri iných beta2-agonistoch sa v štúdiách s celoživotným inhalačným podávaním zistilo, že vilanteroltrifenatát mal proliferatívne účinky na reprodukčný systém samíc potkanov a myší a na hypofýzu potkanov. U potkanov sa pri expozícii 2-násobne vyššej a u myší
pri expozícii 30-násobne vyššej ako je expozícia dosahovaná po maximálnej odporúčanej dávke pre
ľudí, na základe AUC, nezistilo zvýšenie výskytu tumorov.
Reprodukčná toxicita
Flutikazónfuroát
Účinky pozorované po inhalačnom podávaní flutikazónfuroátu v kombinácii s vilanterolom u potkanov boli podobné ako účinky pozorované po podávaní samotného flutikazónfuroátu. Flutikazónfuroát nebol teratogénny u potkanov ani u králikov, ale spomalil vývoj u potkanov a spôsobil potraty u králikov, keď bol podávaný v dávkach toxických pre gravidné samice. Nezistili sa žiadne účinky na vývoj u potkanov pri expozíciách, ktoré boli približne 3-násobne vyššie ako expozície dosahované po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Vilanteroltrifenatát
Vilanteroltrifenatát nebol teratogénny u potkanov. V štúdiách s inhalačným podávaním králikom spôsobil vilanteroltrifenatát účinky podobné tým, ktoré sú pozorované pri podávaní iných
beta2-agonistov (rázštep podnebia, predčasné otvorenie očných viečok, zrastenie sternebra
a ohnutie/malrotácia končatín). Pri subkutánnom podávaní sa nezistili žiadne účinky pri expozíciách
84-násobne vyšších ako sú expozície dosahované po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Ani flutikazónfuroát, ani vilanteroltrifenatát nemali nežiaduce účinky na fertilitu alebo prenatálny a postnatálny vývoj u potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát laktózy
Magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom použití: 6 týždňov.
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote do 25°C. Ak sa inhalátor uchováva v chladničke, treba ho z nej vybrať aspoň
hodinu pred použitím, aby dosiahol izbovú teplotu. Uchovávajte v pôvodnom balení na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah baleniaInhalátor pozostáva zo svetlošedého korpusu, svetlomodrého krytu náustka a počítadla dávok a je zabalený vo vaničke z laminátovej fólie obsahujúcej vrecko s vysúšadlom. Vanička je uzatvorená odnímateľnou fóliou.
Inhalátor obsahuje dva stripy z laminátovej hliníkovej fólie so 14 alebo 30 dávkami.
Inhalátor je viaczložková pomôcka zložená z polypropylénu, polyetylénu s vysokou hustotou, polyoxymetylénu, polybutyléntereftalátu, akrylonitrilbutadiénstyrénu, polykarbonátu a nehrdzavejúcej ocele.
Veľkosti balenia obsahujú 14-dávkový alebo 30-dávkový inhalátor. Multibalenia obsahujú
3 x 30-dávkové inhalátory.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne. Pokyny na použitie, pozri časť 4.2.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxo Group Limited.
980 Great West Road, Brentford, Middlesex TW8 9GS, Spojené kráľovstvo.
8. REGISTRAČNÉ ČÍSLAEU/1/14/929/001
EU/1/14/929/002
EU/1/14/929/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKURevinty Ellipta 184 mikrogramov/22 mikrogramov dávkovaný inhalačný prášok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá jednotlivá inhalácia poskytuje inhalovanú dávku (dávku, ktorá vyjde z náustka)
184 mikrogramov flutikazónfuroátu a 22 mikrogramov vilanterolu (vo forme trifenatátu).
To zodpovedá jednotkovej dávke 200 mikrogramov flutikazónfuroátu a 25 mikrogramov vilanterolu
(vo forme trifenatátu).
Pomocná látka so známym účinkom:
Každá inhalovaná dávka obsahuje približne 25 mg laktózy (vo forme monohydrátu). Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMADávkovaný inhalačný prášok
(Inhalačný prášok).
Biely prášok v svetlošedom inhalátore so svetlomodrým krytom náustka a počítadlom dávok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieAstmaRevinty Ellipta je indikovaný na pravidelnú liečbu astmy u dospelých a dospievajúcich
vo veku 12 rokov a starších v prípadoch, keď je vhodné použitie kombinovaného lieku (s dlhodobo pôsobiacim beta2-agonistom a inhalačným kortikosteroidom):
• u pacientov, ktorých stav nie je dostatočne kontrolovaný inhalačnými kortikosteroidmi a inhalačnými krátkodobo pôsobiacimi beta2-agonistami používanými podľa potreby.
4.2 Dávkovanie a spôsob podávaniaDávkovanieAstmaDospelí a dospievajúci vo veku 12 rokov a staršíJedna inhalácia Revinty Ellipta 184/22 mikrogramov jedenkrát denne.
Pacienti zvyčajne pocítia zlepšenie pľúcnych funkcií do 15 minút po inhalácii Revinty Ellipta. Pacientov však treba informovať, že je nutné liek užívať pravidelne každý deň, aby sa udržala kontrola príznakov astmy a že v jeho užívaní sa má pokračovať aj v asymptomatickom období.
Ak sa v čase medzi užitím jednotlivých dávok príznaky zhoršia, má sa užiť inhalačný krátkodobo pôsobiaci beta2-agonista na dosiahnutie okamžitej úľavy.
Liečba počiatočnou dávkou Revinty Ellipta 92/22 mikrogramov sa má zvážiť u dospelých
a dospievajúcich vo veku 12 rokov a starších, ktorí potrebujú nízku až stredne veľkú dávku inhalačného kortikosteroidu v kombinácii s dlhodobo pôsobiacim beta2-agonistom. Ak pacienti nie sú dostatočne kontrolovaní s Revinty Ellipta 92/22 mikrogramov, dávku možno zvýšiť
na 184/22 mikrogramov, ktorá môže ďalej zlepšiť kontrolu astmy.
Pacienti majú byť pravidelne kontrolovaní lekárom tak, aby sila flutikazónfuroátu/vilanterolu, ktorú užívajú, bola vždy optimálna a bola menená len na odporúčanie lekára. Dávka má byť titrovaná
na najnižšiu dávku, pri ktorej sa udržuje efektívna kontrola príznakov.
Použitie Revinty Ellipta 184/22 mikrogramov sa má zvážiť u dospelých a dospievajúcich
vo veku 12 rokov a starších, ktorí potrebujú vyššiu dávku inhalačného kortikosteroidu v kombinácii s dlhodobo pôsobiacim beta2-agonistom.
Maximálna odporúčaná dávka je Revinty Ellipta 184/22 mikrogramov jedenkrát denne. Pacientom s astmou sa má podávať sila Revinty Ellipta, ktorá obsahuje vhodnú dávku
flutikazónfuroátu (FF) vzhľadom na závažnosť ich ochorenia. Lekári majú vziať do úvahy, že
u pacientov s astmou je dávka 100 mikrogramov flutikazónfuroátu (FF) jedenkrát denne približne ekvivalentná s dávkou 250 mikrogramov flutikazónpropionátu (FP) dvakrát denne,
kým FF 200 mikrogramov jedenkrát denne je približne ekvivalentný s FP 500 mikrogramov dvakrát denne.
Deti mladšie ako 12 rokov
Bezpečnosť a účinnosť Revinty Ellipta u detí mladších ako 12 rokov neboli doteraz stanovené v indikácii astmy.
K dispozícii nie sú žiadne údaje.
Osobitné skupiny pacientov
Starší pacienti (> 65 rokov)
V tejto skupine pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
V tejto skupine pacientov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie pečene
Štúdie s osobami s miernou, stredne ťažkou a ťažkou poruchou funkcie pečene ukázali zvýšenú systémovú expozíciu flutikazónfuroátu (Cmax aj AUC) (pozri časť 5.2).
Pri podávaní lieku pacientom s poruchou funkcie pečene je potrebná obozretnosť, pretože môžu byť
vystavení zvýšenému riziku vzniku systémových nežiaducich reakcií súvisiacich s kortikosteroidmi. Maximálna dávka pre pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene je
92/22 mikrogramov (pozri časť 4.4).
Spôsob podávania
Revinty Ellipta je určený len na inhalačné použitie.
Má sa podávať každý deň v rovnakom čase.
Konečné rozhodnutie o tom, či sa má dávka užívať ráno alebo večer, sa má ponechať na lekára.
Ak sa dávka vynechá, ďalšia dávka sa má užiť nasledujúci deň vo zvyčajnom čase.
Ak sa inhalátor uchováva v chladničke, treba ho z nej vybrať aspoň hodinu pred použitím, aby dosiahol izbovú teplotu.
Po inhalácii si pacienti majú vypláchnuť ústa vodou a vodu vypľuť.
Pri prvom použití inhalátora nie je potrebné skontrolovať, či správne funguje a špeciálnym spôsobom ho pripraviť na použitie. Treba postupovať podľa podrobných pokynov na použitie.
Inhalátor Ellipta je zabalený vo vaničke obsahujúcej vrecko s vysúšadlom na zníženie vlhkosti. Po otvorení vaničky sa má vrecko s vysúšadlom zlikvidovať.
Keď sa inhalátor vyberie z vaničky, bude v polohe „zatvorený“. Nesmie sa otvoriť, kým pacient nebude pripravený inhalovať dávku lieku.
Nižšie uvedené podrobné pokyny na použitie 30-dávkového inhalátora Ellipta platia aj pre 14-dávkový inhalátor Ellipta.
Pokyny na použitie
1. Prečítajte si nasledujúce informácie predtým, ako inhalátor začnete používať
Ak sa kryt inhalátora otvorí a zatvorí bez inhalovania lieku, dávka sa vyplytvá. Vyplytvaná dávka sa bezpečne zadrží vo vnútri inhalátora, ale už viac nebude k dispozícii na inhaláciu.
V jednej inhalácii nie je možné náhodne užiť liek navyše alebo dvojnásobnú dávku.
Kryt
Počítadlo dávok
Vždy, keď ho
otvoríte, pripravíte jednu dávku lieku.
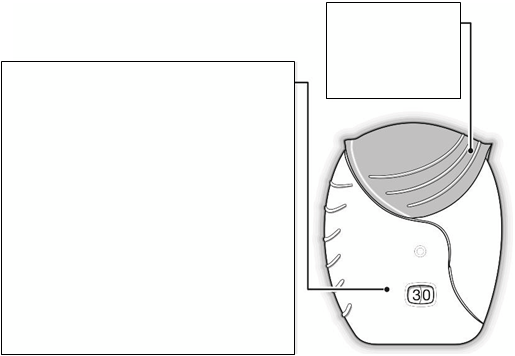
Ukazuje, koľko dávok lieku zostáva
v inhalátore.
Pred prvým použitím inhalátora ukazuje presne 30 dávok.Odpočíta
1 dávku vždy, keď otvoríte kryt.
Keď zostane menej ako 10 dávok, polovica počítadla dávok bude červená.Po použití poslednej dávky
bude polovica počítadla dávok červená a ukáže sa číslica 0. Váš inhalátor je teraz prázdny.
Ak potom otvoríte kryt, počítadlo dávok sa zmení z napoly červeného na úplne červený.
2. Ako pripraviť dávkuKeď budete pripravený užiť dávku, otvorte kryt.
Inhalátorom netraste.Posúvajte kryt smerom nadol, až kým nebudete počuť „
kliknutie“.
Teraz je liek pripravený na inhaláciu. Počítadlo dávok to potvrdí odpočítaním 1 dávky.
Ak počítadlo dávok neodpočíta dávku, keď začujete „
kliknutie“, inhalátor neuvoľní dávku. Vezmite ho späť k lekárnikovi a poraďte sa s ním.
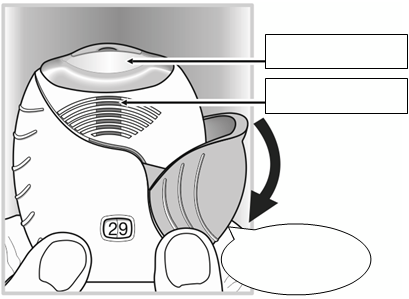 Náustok
Vetrací otvor
„
Kliknutie“
3. Ako inhalovať liek
Náustok
Vetrací otvor
„
Kliknutie“
3. Ako inhalovať liek
Držte inhalátor mimo úst a vydýchnite čo najviac, ako je to možné bez námahy. Nevydychujte do inhalátora.
Vložte si náustok medzi pery a pevne ho obomknite perami. Nezakrývajte vetracie otvory prstami.
Jedenkrát dlho, plynule a hlboko vdýchnite. Zadržte dych tak dlho, ako je to možné (aspoň
3 - 4 sekundy).
• Vyberte si inhalátor z úst.
• Pomaly a jemne vydýchnite.
Vaše pery priliehajúk vytvarovanému náustku na inhalovanie.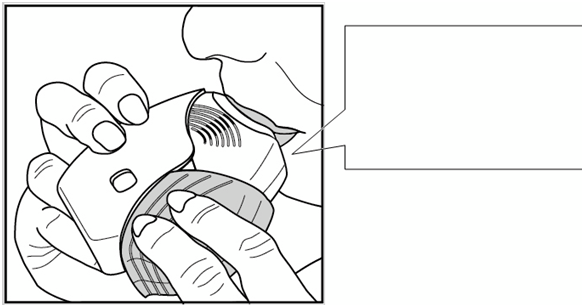 Nezakryte vetracie otvory prstami.
Nezakryte vetracie otvory prstami.Nie je možné pocítiť chuť ani prítomnosť lieku, dokonca ani pri správnom použití inhalátora.
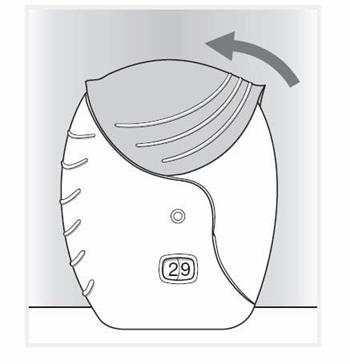
4. Zatvorte inhalátor a vypláchnite si ústaAk chcete očistiť náustok, použite
suchú papierovú vreckovku predtým, ako kryt zatvoríte. Posúvajte kryt smerom nahor kým to pôjde, aby ste zakryli náustok.
Po použití inhalátora si ústa vypláchnite vodou.
Zníži sa tým pravdepodobnosť vzniku kandidózy (kvasinkovej infekcie) ústnej dutiny a hrdla ako vedľajších účinkov.
 4.3 Kontraindikácie
4.3 KontraindikáciePrecitlivenosť na liečivá alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníZhoršenie ochoreniaFlutikazónfuroát/vilanterol sa nemá používať na liečbu akútnych príznakov astmy, ktoré si vyžadujú
krátkodobo pôsobiace bronchodilatancium. Zvýšené užívanie krátkodobo pôsobiacich bronchodilatancií na zmiernenie príznakov svedčí o zhoršení kontroly astmy a takýchto pacientov má vyšetriť lekár.
Pacienti s astmou nesmú ukončiť liečbu flutikazónfuroátom/vilanterolom bez dohľadu lekára, keďže sa im príznaky po jej ukončení môžu vrátiť.
Počas liečby flutikazónfuroátom/vilanterolom sa môžu vyskytnúť nežiaduce udalosti spojené s astmou a exacerbácie astmy. Pacientov treba upozorniť na to, aby pokračovali v liečbe s Revinty Ellipta aj
v prípade, ak po jej začatí zostanú príznaky astmy naďalej nekontrolované alebo ak sa zhoršia, ale aby zároveň vyhľadali pomoc lekára.
Paradoxný bronchospazmusPo podaní dávky môže dôjsť k paradoxnému bronchospazmu s okamžitým zhoršením piskotov. Tento
stav je nutné okamžite liečiť podaním krátkodobo pôsobiaceho inhalačného bronchodilatancia. Používanie Revinty Ellipta treba ihneď ukončiť, pacienta vyšetriť a v prípade potreby treba začať alternatívnu liečbu.
Kardiovaskulárne účinkyPri užívaní liekov so sympatomimetickým účinkom vrátane Revinty Ellipta sa môžu objaviť
kardiovaskulárne účinky, akými sú srdcové arytmie, napr. supraventrikulárna tachykardia
a extrasystoly. Preto sa má flutikazónfuroát/vilanterol používať obozretne u pacientov so závažným kardiovaskulárnym ochorením.
Pacienti s poruchou funkcie pečene
U pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene sa má použiť dávka
92/22 mikrogramov a pacientov treba sledovať kvôli systémovým nežiaducim reakciám súvisiacim s kortikosteroidmi (pozri časť 5.2).
Systémové účinky kortikosteroidov
Systémové účinky sa môžu prejaviť pri akomkoľvek inhalačnom kortikosteroide, hlavne
pri dlhodobom užívaní vysokých dávok. Výskyt týchto účinkov je oveľa menej pravdepodobný ako pri perorálnych kortikosteroidoch. Možné systémové účinky zahŕňajú Cushingov syndróm, cushingoidné prejavy, útlm funkcie nadobličiek, zníženie kostnej denzity, spomalenie rastu u detí
a dospievajúcich, kataraktu a glaukóm a zriedkavejšie aj rôzne účinky na psychiku alebo správanie zahŕňajúce psychomotorickú hyperaktivitu, poruchy spánku, úzkosť, depresiu alebo agresivitu (hlavne
u detí).
Flutikazónfuroát/vilanterol sa má podávať obozretne pacientom s pľúcnou tuberkulózou alebo pacientom s chronickými alebo neliečenými infekciami.
Hyperglykémia
U diabetikov bolo hlásené zvýšenie hladín glukózy v krvi a treba to vziať do úvahy pri predpisovaní
tohto lieku pacientom s diabetes mellitus v anamnéze.
Pneumónia u pacientov s CHOCHP
U pacientov s CHOCHP liečených flutikazónfuroátom/vilanterolom sa pozoroval zvýšený výskyt
pneumónie. Zaznamenal sa aj zvýšený výskyt pneumónií, ktoré mali za následok hospitalizáciu. V niekoľkých prípadoch bola pneumónia, ako nežiaduca udalosť, smrteľná (pozri časť 4.8).
U pacientov s CHOCHP musia lekári zostať ostražití kvôli možnému vzniku pneumónie, pretože klinické prejavy takýchto infekcií sa prekrývajú s príznakmi exacerbácií CHOCHP. Rizikové faktory vzniku pneumónie u pacientov s CHOCHP liečených flutikazónfuroátom/vilanterolom zahŕňajú súčasné fajčenie, pneumóniu v anamnéze, BMI < 25 kg/m2 a hodnotu FEV1 < 50 % referenčnej hodnoty. Tieto faktory treba vziať do úvahy pri predpisovaní flutikazónfuroátu/vilanterolu a v prípade výskytu pneumónie sa má liečba prehodnotiť.
Revinty Ellipta 184/22 mikrogramov nie je určený pre pacientov s CHOCHP. Dávka
184/22 mikrogramov v porovnaní s dávkou 92/22 mikrogramov nemá väčší prínos a pri jej podávaní existuje potenciálne zvýšené riziko vzniku systémových nežiaducich reakcií súvisiacich
s kortikosteroidmi (pozri časť 4.8).
Výskyt pneumónií bol u pacientov s astmou častý pri podávaní vyššej dávky. U astmatikov užívajúcich flutikazónfuroát/vilanterol 184/22 mikrogramov bol výskyt pneumónií číselne vyšší ako
u pacientov užívajúcich flutikazónfuroát/vilanterol 92/22 mikrogramov alebo placebo (pozri časť 4.8). Identifikované neboli žiadne rizikové faktory.
Pomocné látky
Pacienti so zriedkavými dedičným problémami galaktózovej intolerancie, lapónskeho deficitu laktázy
alebo glukózo-galaktózovej malabsorpcie nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Klinicky významné liekové interakcie sprostredkované flutikazónfuroátom/vilanterolom v klinických dávkach sa považujú za nepravdepodobné vzhľadom na nízke plazmatické koncentrácie dosahované po inhalácii dávky.
Interakcie s betablokátormi
Blokátory beta2-adrenergných receptorov môžu oslabiť alebo antagonizovať účinok agonistov
beta2-adrenergných receptorov. Súbežnému použitiu neselektívnych aj selektívnych blokátorov
beta2-adrenergných receptorov sa treba vyhnúť, pokiaľ neexistujú závažné dôvody na ich použitie.
Interakcie s inhibítormi CYP3A4
Flutikazónfuroát aj vilanterol sú pri prvom prechode pečeňou rýchlo odstraňované vďaka rozsiahlemu
metabolizmu sprostredkovanému pečeňovým enzýmom CYP3A4.
Pri súbežnom podávaní so silnými inhibítormi CYP3A4 (napr. s ketokonazolom, ritonavirom) sa odporúča sa obozretnosť, keďže môže dôjsť k zvýšenej systémovej expozície flutikazónfuroátu aj vilanterolu. Preto je potrebné vyhnúť sa súbežnému užívaniu týchto liekov. U zdravých osôb sa uskutočnila štúdia zameraná na liekové interakcie sprostredkované CYP3A4 s opakovaným podávaním kombinácie flutikazónfuroát/vilanterol (184/22 mikrogramov) a silného inhibítora CYP3A4 ketokonazolu (400 mg). Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-24) flutikazónfuroátu o 36 % a jeho Cmax o 33 %. Zvýšenie expozície flutikazónfuroátu sa spájalo s 27 % znížením váženej priemernej hladiny kortizolu v sére nameranej počas 0 - 24 hodín. Súbežné podávanie zvýšilo priemernú hodnotu AUC(0-t) vilanterolu o 65 % a jeho Cmax o 22 %. Zvýšenie
expozície vilanterolu nebolo spojené so zvýšením systémových účinkov súvisiacich s beta2-agonistami
na srdcovú frekvenciu, hladinu draslíka v krvi alebo QTcF interval.
Interakcie s inhibítormi P-glykoproteínu
Flutikazónfuroát aj vilanterol sú substrátmi P-glykoproteínu (P-gp). Klinická farmakologická štúdia
so zdravými osobami, ktorým bol súbežne podávaný vilanterol a verapamil, ktorý je silným inhibítorom P-gp a stredne silným inhibítorom CYP3A4, nepreukázala významný vplyv
na farmakokinetiku vilanterolu. Klinické farmakologické štúdie so špecifickým inhibítorom P-gp a flutikazónfuroátom sa neuskutočnili.
Lieky so sympatomimetickým účinkom
Súbežné podávanie iných liekov so sympatomimetickým účinkom (v monoterapii alebo ako súčasť
kombinovanej liečby) môže potencovať nežiaduce reakcie pri flutikazónfuroáte/vilanterole.
Revinty Ellipta sa nemá používať spolu s inými dlhodobo pôsobiacimi agonistami beta2-adrenergných receptorov ani s inými liekmi obsahujúcimi dlhodobo pôsobiace agonisty beta2-adrenergných receptorov.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Gravidita
Štúdie na zvieratách preukázali reprodukčnú toxicitu pri expozíciách, ktoré nie sú klinicky významné'
(pozri časť 5.3). Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití flutikazónfuroátu a vilanteroltrifenatátu u gravidných žien.
O podávaní flutikazónfuroátu/vilanterolu gravidným ženám sa má uvažovať len vtedy, ak je očakávaný prínos pre matku väčší ako možné riziko pre plod.
Laktácia
Nie sú dostatočné informácie o vylučovaní flutikazónfuroátu alebo vilanteroltrifenatátu a/alebo
ich metabolitov do ľudského mlieka. Avšak iné kortikosteroidy a beta2-agonisty boli v ľudskom mlieku detegované (pozri časť 5.3). Riziko u dojčených novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu flutikazónfuroátom/vilanterolom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú údaje o fertilite získané u ľudí. Štúdie na zvieratách nepreukázali žiaden vplyv
flutikazónfuroátu/vilanterolu na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Flutikazónfuroát alebo vilanterol nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť
vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Na určenie frekvencie výskytu nežiaducich reakcií súvisiacich s flutikazónfuroátom/vilanterolom sa
použili údaje z rozsiahlych klinických skúšaní zameraných na astmu a CHOCHP. V programe klinického vývoja zameraného na liečbu astmy bolo do integrovaného hodnotenia nežiaducich reakcií zaradených celkovo 7 034 pacientov. V programe klinického vývoja zameraného na liečbu CHOCHP bolo do integrovaného hodnotenia nežiaducich reakcií celkovo zaradených 6 237 osôb.
Najčastejšie hlásené nežiaduce reakcie pri podávaní flutikazónfuroátu a vilanterolu boli bolesť hlavy
a nazofaryngitída. S výnimkou pneumónie a zlomenín bol bezpečnostný profil u pacientov s astmou a u pacientov s CHOCHP podobný. Počas klinických štúdií sa pneumónia a zlomeniny častejšie pozorovali u pacientov s CHOCHP.
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a frekvencie výskytu.
Na klasifikáciu frekvencií sa použila nasledujúca konvencia: veľmi časté (≥ 1/10); časté
(≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000).
V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
|
Nežiaduca reakcia (reakcie)
|
Frekvencia
|
Infekcie a nákazy
|
Pneumónia*
Infekcia horných dýchacích ciest
Bronchitída
Chrípka
Kandidóza ústnej dutiny a hrdla
|
Časté
|
Poruchy nervového systému
|
Bolesť hlavy
|
Veľmi časté
|
Poruchy srdca a srdcovej
činnosti
|
Extrasystoly
|
Menej časté
|
Poruchy dýchacej sústavy,
hrudníka a mediastína
|
Nazofaryngitída
Orofaryngálna bolesť
Sinusitída Faryngitída Rinitída Kašeľ Dysfónia
|
Veľmi časté
Časté
|
Poruchy gastrointestinálneho
traktu
|
Bolesť brucha
|
Časté
|
Poruchy kostrovej a svalovej
sústavy a spojivového tkaniva
|
Artralgia
Bolesť chrbta
Zlomeniny**
|
Časté
|
Celkové poruchy a reakcie v mieste podania
|
Pyrexia
|
Časté
|
*, ** Pozri „Popis vybraných nežiaducich reakcií“ uvedený nižšie
Popis vybraných nežiaducich reakcií
Pneumónia
V integrovanej analýze dvoch opakovane vykonaných jednoročných štúdií s pacientmi s CHOCHP,
ktorí prekonali exacerbáciu v predchádzajúcom roku (n = 3 255), bol počet prípadov pneumónií na 1 000 pacientorokov 97,9 pri FF/VI 184/22, 85,7 pri FF/VI 92/22 a 42,3 v skupine s VI 22.
Pri ťažkých pneumóniách sa zodpovedajúci počet prípadov na 1 000 pacientorokov rovnal 33,6; 35,5
a 7,6, v uvedenom poradí, kým pri závažných pneumóniách sa zodpovedajúci počet prípadov
na 1 000 pacientorokov rovnal 35,1 pri FF/VI 184/22, 42,9 pri FF/VI 92/22 a 12,1 pri VI 22. Napokon, počet smrteľných prípadov pneumónií upravený vzhľadom na expozíciu bol 8,8 pri FF/VI 184/22
v porovnaní s 1,5 pri FF/VI 92/22 a 0 pri VI 22FF/VI.
V integrovanej analýze 11 štúdií s astmatikmi (7 034 pacientov) bol výskyt pneumónií
na 1 000 pacientorokov 18,4 pri FF/VI 184/22 v porovnaní s 9,6 pri FF/VI 92/22 a 8,0 v skupine s placebom.
ZlomeninyV dvoch opakovane vykonaných 12-mesačných štúdiách s celkovo 3 255 pacientami s CHOCHP bol
výskyt zlomenín kostí celkovo nízky vo všetkých liečených skupinách, s vyšším výskytom
vo všetkých skupinách s Revinty Ellipta (2 %) v porovnaní so skupinou s vilanterolom v dávke
22 mikrogramov (< 1%). Hoci v skupinách s Revinty Ellipta bolo viac zlomenín v porovnaní so skupinou s vilanterolom v dávke 22 mikrogramov, zlomeniny typicky súvisiace s použitím kortikosteroidov (napr. kompresívne zlomeniny stavcov chrbtice/zlomeniny torakolumbálnych stavcov, zlomeniny bedra a acetabula) sa vyskytli u < 1 % pacientov v skupinách liečených
s Revinty Ellipta a v skupine liečenej vilanterolom.
V integrovanej analýze 11 štúdií zameraných na astmu (7 034 pacientov) bol výskyt zlomenín < 1 %
a zvyčajne súviseli s úrazom.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovaniePríznaky a prejavyPredávkovanie flutikazónfuroátom/vilanterolom môže vyvolať prejavy a príznaky spôsobené účinkom
jednotlivých zložiek vrátane tých, ktoré sú pozorované pri predávkovaní inými beta2-agonistami a zhodujú sa so známymi skupinovými účinkami inhalačných kortikosteroidov (pozri časť 4.4).
LiečbaŠpecifická liečba predávkovania flutikazónfuroátom/vilanterolom nie je k dispozícii. Ak dôjde
k predávkovaniu, pacient má podľa potreby dostať podpornú liečbu s náležitým sledovaním.
O kardioselektívnej betablokáde sa má uvažovať len v prípade závažných prejavov spôsobených predávkovaním vilanterolom, ktoré sú klinicky významné a neodpovedajú na podporné opatrenia. Kardioselektívne betablokátory sa majú použiť obozretne u pacientov s bronchospazmom v anamnéze.
Ďalšia liečba sa má riadiť klinickým stavom pacienta alebo odporúčaniami poskytnutými národným toxikologickým informačným centrom, pokiaľ sú k dispozícii.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Liečivá na obštrukčné ochorenia dýchacích ciest, adrenergiká a iné liečivá na obštrukčné ochorenia dýchacích ciest, ATC kód: R03AK10.
Mechanizmus účinkuFlutikazónfuroát a vilanterol sú zástupcami dvoch liekových skupín (syntetický kortikosteroid a
selektívny, dlhodobo pôsobiaci agonista beta2-receptorov).
Farmakodynamické účinky
Flutikazónfuroát
Flutikazónfuroát je syntetický trifluórovaný kortikosteroid so silným protizápalovým účinkom. Presný
mechanizmus, prostredníctvom ktorého flutikazónfuroát ovplyvňuje príznaky astmy a CHOCHP, nie je známy. Preukázalo sa, že kortikosteroidy majú širokú škálu účinkov na viaceré typy buniek
(napr. eozinofily, makrofágy, lymfocyty) a na mediátory (napr. cytokíny a chemokíny, ktoré sa zúčastňujú na zápale).
Vilanteroltrifenatát
Vilanteroltrifenatát je selektívny, dlhodobo pôsobiaci agonista beta2-adrenergných receptorov
(long-acting, beta2-adrenergic agonist, LABA).
Farmakologické účinky agonistov beta2-adrenergných receptorov, vrátane vilanteroltrifenatátu, možno
aspoň sčasti pripísať stimulácii vnútrobunkovej adenylátcyklázy, enzýmu, ktorý katalyzuje premenu
adenozíntrifosfátu (ATP) na cyklický-3’,5’-adenozínmonofosfát (cyklický AMP). Zvýšené hladiny cyklického AMP spôsobujú uvoľnenie hladkého svalstva priedušiek a inhibujú uvoľňovanie mediátorov okamžitej precitlivenosti z buniek, najmä z mastocytov.
Medzi kortikosteroidmi a LABA dochádza k molekulárnym interakciám, pomocou ktorých steroidy aktivujú gén pre beta2-adrenergné receptory, zvyšujú počet a citlivosť receptorov a LABA pripravujú glukokortikoidové receptory na aktiváciu závislú od steroidov a zvyšujú translokáciu do bunkového jadra. Tieto synergické interakcie sa odzrkadľujú vo zvýšenom protizápalovom účinku, ktorý sa preukázal in vitro a in vivo v rôznych zápalových bunkách, ktoré sa podieľajú na patofyziológii astmy aj CHOCHP. Štúdie s flutikazónfuroátom a vilanterolom, v ktorých sa vyšetrovali bioptické vzorky tkaniva dýchacích ciest, tiež preukázali synergické pôsobenie medzi kortikosteroidmi a LABA pri klinických dávkach liečiv podávaných pacientom s CHOCHP.
Klinická účinnosť a bezpečnosť
Astma
Tri randomizované, dvojito zaslepené štúdie fázy III (HZA106827, HZA106829 and HZA106837)
s rôznou dĺžkou trvania hodnotili bezpečnosť a účinnosť flutikazónfuroátu/vilanterolu u dospelých a dospievajúcich pacientov s perzistentnou astmou. Všetky osoby užívali ICS (inhalačný kortikosteroid) s LABA, alebo bez LABA, aspoň 12 týždňov pred 1. návštevou. V štúdii HZA106837 prekonali všetci pacienti aspoň jednu exacerbáciu, ktorá si vyžadovala liečbu perorálnymi kortikosteroidmi, v roku predchádzajúcom 1. návšteve. Štúdia HZA106827 trvala 12 týždňov
a hodnotila účinnosť flutikazónfuroátu/vilanterolu 92/22 mikrogramov [n = 201] a FF
92 mikrogramov [n = 205]) v porovnaní s placebom [n = 203], všetky lieky boli podávané jedenkrát denne. Štúdia HZA106829 trvala 24 týždňov a hodnotila účinnosť flutikazónfuroátu/vilanterolu
184/22 mikrogramov [n = 197] a FF 184 mikrogramov [n = 194]), oba lieky boli podávané jedenkrát denne, v porovnaní s FP 500 mikrogramov dvakrát denne [n = 195].
V štúdiách HZA106827/HZA106829 boli kombinovanými primárnymi cieľmi účinnosti zmena trough
FEV1 (minimálna hodnota FEV1 pred podaním bronchodilatancia a pred podaním dávky)
na kontrolnej návšteve na konci obdobia liečby v porovnaní s východiskovou hodnotou u všetkých
osôb a vážený priemer opakovane meranej hodnoty FEV1 počas 0 - 24 hodín po podaní dávky vypočítaný v podskupine osôb na konci obdobia liečby. Sekundárnym cieľom, na hodnotenie ktorého mala štúdia dostatočnú štatistickú silu, bola zmena v percentuálnom počte dní bez potreby záchranného lieku počas liečby v porovnaní s východiskovým stavom. Výsledky primárnych
a kľúčových sekundárnych cieľov v týchto štúdiách sú uvedené v tabuľke 1.
Tabuľka 1 - Výsledky primárnych a kľúčových sekundárnych cieľov v štúdiách HZA106827 a
H
Z
A106829
Číslo štúdie
|
HZA106829
|
HZA106827
|
Liečebná dávka
FF/VI*(mikrogramy)
|
FF/VI 184/22
jedenkrát denne vs. FF 184 jedenkrát denne
|
FF/VI 184/22
jedenkrát denne vs. FP 500
dvakrát denne
|
FF/VI 92/22
jedenkrát denne vs. FF 92 jedenkrát denne
|
FF/VI/92/22
jedenkrát denne vs. placebo jedenkrát denne
|
Z
m
ena trough FEV
1
určeného na základe posledného preneseného pozorovania (Last
Observation Carried Forward
, LOCF) v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
|
193 ml
p<0,001 (108, 277)
|
210 ml
p<0,001 (127, 294)
|
36 ml
p=0,405
(-48, 120)
|
172 ml
p<0,001 (87, 258)
|
Vážený priemer opakovane meranej hodnoty FEV
1
počas 0 - 24 hodín po podaní dávky
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
|
136 ml
p=0,048 (1, 270)
|
206 ml
p=0,003 (73, 339)
|
116 ml
p=0,06
(-5, 236)
|
302 ml
p<0,001 (178, 426)
|
Z
m
ena v percentuálnom počte dní bez potreby záchranného lieku v porovnaní s východiskovým stavom
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
|
11,7 %
p<0,001 (4,9; 18,4)
|
6,3 %
p=0,067
(-0,4; 13,1)
|
10,6 %
p<0,001 (4,3; 16,8)
|
19,3 %
p<0,001 (13,0; 25,6)
|
Z
m
ena v percentuálnom počte dní bez príznakov v porovnaní s východiskovým stavom
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
|
8,4 %
p=0,010 (2,0; 14,8)
|
4,9 %
p=0,137
(-1,6; 11,3)
|
12,1 %
p<0,001 (6,2; 18,1)
|
18,0 %
p<0,001 (12,0; 23,9)
|
Z
m
ena v dopoludňajšej hodnote maximálneho výdychového prietoku (PEF)
v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami p-hodnota
(95 % IS)
|
33,5 l/min p<0,001
(22,3; 41,7)
|
32,9 l/min p<0,001
(24,8; 41,1)
|
14,6 l/min p<0,001
(7,9; 21,3)
|
33,3 l/min p<0,001
(26,5; 40,0)
|
Z
m
ena v popoludňajšej hodnote maximálneho výdychového prietoku (PEF)
v porovnaní s východiskovou hodnotou
|
Rozdiel medzi liečbami
p-hodnota
(95 % IS)
|
30,7 l/min
p<0,001 (22,5; 38,9)
|
26,2 l/min
p<0,001 (18,0; 34,3)
|
12,3 l/min
p<0,001 (5,8; 18,8)
|
28,2 l/min
p<0,001 (21,7; 34,8)
|
*FF/VI = flutikazónfuroát/vilanterol
Štúdia HZA106837 mala rôznu dĺžku trvania (od minimálne 24 týždňov po maximálne 76 týždňov, pričom väčšina pacientov bola liečená aspoň 52 týždňov). V štúdii HZA106837 bola pacientom náhodne pridelená buď liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov [n = 1 009], alebo liečba FF 92 mikrogramov [n = 1 010], oba lieky boli podávané jedenkrát denne. V štúdii HZA106837 bol primárnym cieľom čas do objavenia sa prvej závažnej exacerbácie astmy. Závažná exacerbácia astmy bola definovaná ako zhoršenie astmy vyžadujúce si užívanie systémových kortikosteroidov počas aspoň 3 dní alebo hospitalizácia pacienta v nemocnici alebo návšteva lekárskej pohotovosti
kvôli astme, ktorá si vyžadovala liečbu systémovými kortikosteroidmi. Upravená priemerná zmena trough FEV1 v porovnaní s východiskovou hodnotou sa taktiež hodnotila ako sekundárny cieľ.
V štúdii HZA106837 sa riziko výskytu závažnej exacerbácie astmy u pacientov liečených flutikazónfuroátom/vilanterolom 92/22 mikrogramov znížilo o 20 % v porovnaní s pacientmi liečenými samotným FF 92 mikrogramov (hazard ratio 0,795, p = 0,036, 95 % IS: 0,642; 0,985). Výskyt závažných exacerbácií astmy na pacientoroky bol 0,19 v skupine s FF 92 mikrogramov (približne 1 prípad na každých 5 rokov) a 0,14 v skupine s flutikazónfuroátom/vilanterolom
92/22 mikrogramov (približne 1 prípad na každých 7 rokov). Pomer výskytu exacerbácií pri liečbe flutikazónfuroátom/vilanterolom 92/22 mikrogramov v porovnaní s liečbou FF 92 mikrogramov bol
0,755 (95 % IS: 0,603; 0,945). To predstavuje 25 % zníženie výskytu závažných exacerbácií astmy u osôb liečených flutikazónfuroátom/vilanterolom 92/22 mikrogramov v porovnaní s osobami liečenými FF 92 mikrogramov (p = 0,014). Dvadsaťštyrihodinový bronchodilatačný účinok flutikazónfuroátu/vilanterolu sa zachoval počas celého jednoročného obdobia liečby bez toho, že by sa preukázala strata účinnosti (bez výskytu tachyfylaxie). Pri liečbe flutikazónfuroátom/vilanterolom
92/22 mikrogramov sa zhodne preukázalo zlepšenie trough FEV1 o 83 ml až 95 ml
v 12., 36. a 52. týždni a pri poslednom zaznamenanom meraní (Endpoint) na konci liečby v porovnaní
s liečbou FF 92 mikrogramov (p < 0,001, 95 % IS: 52, 126 ml pri poslednom zaznamenanom meraní
(Endpoint)). Na konci liečby 44 % pacientov v skupine s flutikazónfuroátom/vilanterolom
92/22 mikrogramov dosiahlo dobrú kontrolu ochorenia (skóre dotazníka na hodnotenie kontroly astmy - ACQ7 ≤ 0,75) v porovnaní s 36 % osôb v skupine s FF 92 mikrogramov (p < 0,001, 95 % IS:
1,23; 1,82).
Štúdie zamerané na porovnanie s kombináciou salmeterol/flutikazónpropionát
V 24-týždňovej štúdii (HZA113091) s dospelými a dospievajúcimi pacientmi s perzistentnou astmou sa tak pri liečbe flutikazónfuroátom/vilanterolom 92/22 mikrogramov podávaným jedenkrát denne večer, ako aj pri liečbe kombináciou salmeterol/FP 50/250 mikrogramov podávanou dvakrát denne preukázalo zlepšenie pľúcnych funkcií v porovnaní s východiskovým stavom. Upravené priemerné zvýšenie váženého priemeru hodnoty FEV1 meranej počas 0 - 24 hodín, ku ktorému došlo počas liečby, v porovnaní s východiskovou hodnotou, o 341 ml (pri liečbe kombináciou flutikazónfuroát/vilanterol) a o 377 ml (pri liečbe kombináciou salmeterol/FP) svedčilo o celkovom zlepšení pľúcnych funkcií počas 24 hodín pri oboch liečbach. Upravený priemerný rozdiel medzi liečbami rovnajúci sa 37 ml medzi skupinami nebol štatisticky významný (p = 0,162). Pokiaľ ide
o trough FEV1, osoby v skupine s flutikazófuroátom/vilanterolom dosiahli priemernú zmenu vypočítanú s použitím metódy najmenších štvorcov (least squares, LS) rovnajúcu sa 281 ml
v porovnaní s východiskovou hodnotou a osoby v skupine so salmeterolom/FP dosiahli zmenu rovnajúcu sa 300 ml; (rozdiel v upravenom priemere rovnajúci sa 19 ml (95 % IS: -0,073; 0,034) nebol štatisticky významný (p = 0,485)).
Neuskutočnili sa žiadne porovnávacie štúdie s kombináciou salmeterol/FP alebo s inými kombináciami ICS/LABA, ktoré by náležite porovnali vplyv na exacerbácie astmy.
Flutikazónfuroát v monoterapii
Dvadsaťštyritýždňová randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia (FFA112059)
hodnotila bezpečnosť a účinnosť FF 92 mikrogramov jedenkrát denne [n = 114]
a FP 250 mikrogramov dvakrát denne [n = 114] v porovnaní s placebom [n = 115] u dospelých
a dospievajúcich pacientov s perzistentnou astmou. Všetky osoby museli užívať stabilnú dávku ICS
aspoň 4 týždne pred 1. návštevou (skríningovou návštevou) a použitie LABA nebolo povolené počas
4 týždňov od 1. návštevy. Primárnym cieľom účinnosti bola zmena trough FEV1 (pred podaním bronchodilatancia a pred podaním dávky) na kontrolnej návšteve na konci obdobia liečby v porovnaní s východiskovou hodnotou. Sekundárnym cieľom, na hodnotenie ktorého mala štúdia dostatočnú štatistickú silu, bola zmena v percentuálnom počte dní bez potreby záchranného lieku počas
24-týždňového obdobia liečby v porovnaní s východiskovým stavom. V 24. týždni sa trough FEV1
pri podávaní FF zvýšil o 146 ml (95 % IS: 36, 257 ml, p = 0,009) a pri podávaní FP sa zvýšil o 145 ml
(95 % IS: 33, 257 ml, p = 0,011) v porovnaní s placebom. Percentuálny počet dní bez potreby záchranného lieku sa pri podávaní FF zvýšil o 14,8 % (95 % IS: 6,9; 22,7, p < 0,001) a pri podávaní FP sa zvýšil o 17,9 % (95 % IS: 10,0; 25,7, p < 0,001) v porovnaní s placebom.
Štúdia využívajúca provokáciu alergénom
Bronchoprotektívny účinok flutikazónfuroátu/vilanterolu 92/22 mikrogramov na včasnú a neskorú astmatickú odpoveď na inhalačný alergén sa hodnotil v placebom kontrolovanej štúdii s pacientmi s ľahkou astmou, so štyrmi spôsobmi skríženia liečby (four-way crossover) a s opakovaným podávaním lieku (HZA113126). Pacientom bola náhodne pridelená liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov, FF 92 mikrogramov, vilanterolom
22 mikrogramov alebo placebom, všetky lieky boli podávané jedenkrát denne počas 21 dní, po ktorej nasledovala provokácia alergénom 1 hodinu po poslednej dávke. Alergénom boli roztoče bytového prachu, mačacie lupiny alebo brezový peľ; výber bol založený na individuálnych skríningových testoch. Opakované merania hodnoty FEV1 sa porovnali s hodnotami pred provokáciou alergénom
po inhalácii fyziologického roztoku (východiskové hodnoty). Celkovo možno konštatovať, že najväčší
účinok na včasnú astmatickú odpoveď sa pozoroval pri liečbe flutikazónfuroátom/vilanterolom
92/22 mikrogramov v porovnaní s liečbou samotným FF 92 mikrogramov alebo samotným vilanterolom 22 mikrogramov. Tak liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov, ako aj liečba FF 92 mikrogramov prakticky odstránila neskorú astmatickú odpoveď v porovnaní s liečbou samotným vilanterolom. Liečba flutikazónfuroátom/vilanterolom 92/22 mikrogramov poskytla významne väčšiu ochranu pred hyperreaktivitou priedušiek vyvolanou alergénom v porovnaní s FF
a vilanterolom podávanými v monoterapii, čo sa hodnotilo na 22. deň prostredníctvom provokácie metacholínom.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Revinty Ellipta
v jednej alebo vo viacerých podskupinách pediatrickej populácie s astmou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Absolútna biologická dostupnosť flutikazónfuroátu a vilanterolu podávaných inhalačne vo forme
kombinácie flutikazónfuroát/vilanterol bola v priemere 15,2 % a 27,3 %, v uvedenom poradí. Perorálna biologická dostupnosť flutikazónfuroátu aj vilanterolu bola nízka, v priemere 1,26 %
a < 2 %, v uvedenom poradí. Vzhľadom na nízku perorálnu biologickú dostupnosť je systémová expozícia flutikazónfuroátu a vilanterolu po inhalačnom podaní predovšetkým dôsledkom absorpcie inhalovanej dávky dodanej do pľúc.
Distribúcia
Po intravenóznom podaní sa flutikazónfuroát aj vilanterol v rozsiahlej miere distribuujú s priemerným
distribučným objemom v rovnovážnom stave 661 l a 165 l, v uvedenom poradí. Flutikazónfuroát aj vilanterol sa slabo viažu na erytrocyty. V in vitro podmienkach bola väzba flutikazónfuroátu a vilanterolu na plazmatické bielkoviny v ľudskej plazme vysoká, v priemere
> 99,6 % a 93,9 %, v uvedenom poradí. U osôb s poruchou funkcie obličiek alebo pečene sa nezistilo zníženie rozsahu väzby na plazmatické bielkoviny in vitro.
Flutikazónfuroát a vilanterol sú substrátmi P-glykoproteínu (P-gp), predpokladá sa však, že súbežné podávanie flutikazónfuroátu/vilanterolu s inhibítormi P-gp pravdepodobne nezmení systémovú
expozíciu flutikazónfuroátu ani vilanterolu, keďže oba sú dobre absorbovateľnými molekulami.
Biotranformácia
Na základe in vitro údajov sú hlavné metabolické cesty flutikazónfuroátu aj vilanterolu
sprostredkované predovšetkým enzýmom CYP3A4.
Flutikazónfuroát sa primárne metabolizuje hydrolýzou S-fluórmetyl-karbotioátu na metabolity s významne zníženým kortikosteroidovým účinkom. Vilanterol sa primárne metabolizuje
O-dealkyláciou na rôzne metabolity s významne zníženým β1- a β2-agonistickým účinkom.
Eliminácia
Po perorálnom podaní sa flutikazónfuroát u ľudí eliminoval predovšetkým metabolizmom, pričom
jeho metabolity sa vylučovali takmer výhradne stolicou a < 1 % izotopom značenej dávky sa vylúčilo močom.
Po perorálnom podaní sa vilanterol eliminoval hlavne metabolizmom, s následným vylučovaním metabolitov, pričom v štúdii s izotopom značenou látkou perorálne podávanou ľuďom sa 70 % izotopom značenej dávky vylúčilo močom a 30 % sa vylúčilo stolicou. Zdanlivý plazmatický eliminačný polčas vilanterolu po jednorazovom inhalačnom podaní flutikazónfuroátu/vilanterolu bol v priemere 2,5 hodiny. Efektívny polčas kumulácie vilanterolu, stanovený na základe inhalačného
podávania opakovaných 25 mg dávok vilanterolu, je 16,0 hodín u osôb s astmou a 21,3 hodiny u osôb s CHOCHP.
Pediatrická populácia
U dospievajúcich (vo veku 12 rokov a starších) sa neodporúčajú žiadne úpravy dávky.
Farmakokinetika flutikazónfuroátu/vilanterolu u pacientov mladších ako 12 rokov sa nesledovala. Bezpečnosť a účinnosť flutikazónfuroátu/vilanterolu u detí mladších ako 12 rokov neboli doteraz stanovené.
Osobitné skupiny pacientov
Starší pacienti (> 65 rokov)
Vplyv veku na farmakokinetiku flutikazónfuroátu a vilanterolu sa stanovil v štúdiách fázy III zameraných na astmu a CHOCHP. U osôb s astmou sa nepreukázalo, že by vek (12 - 84 rokov) ovplyvňoval farmakokinetiku flutikazónfuroátu a vilanterolu.
Pre osoby s astmou a osoby s CHOCHP sa neodporúčajú žiadne úpravy dávky.
Porucha funkcie obličiek
Klinická farmakologická štúdia s flutikazónfuroátom/vilanterolom ukázala, že ťažká porucha funkcie obličiek (klírens kreatinínu < 30 ml/min) nemala za následok významne vyššiu expozíciu flutikazónfuroátu alebo vilanterolu ani výraznejšie systémové účinky spojené s kortikosteroidmi alebo beta2-agonistami v porovnaní so zdravými osobami.
U pacientov s poruchou funkcie obličiek nie je potrebná úprava dávky. Vplyv hemodialýzy sa nesledoval.
Porucha funkcie pečene
Po opakovanom podávaní flutikazónfuroátu/vilanterolu počas 7 dní došlo u osôb s poruchou funkcie pečene (stupňa A, B alebo C Childovej-Pughovej klasifikácie) v porovnaní so zdravými osobami
k zvýšeniu systémovej expozície flutikazónfuroátu (až na trojnásobok, keď sa merala na základe AUC(0–24)). Zvýšenie systémovej expozície flutikazónfuroátu u osôb so stredne ťažkou poruchou funkcie pečene (stupňa B Childovej-Pughovej klasifikácie; pri podávaní flutikazónfuroátu/vilanterolu
184/22 mikrogramov) sa v porovnaní so zdravými osobami spájalo s priemerným 34 % znížením hladiny koncentrácie kortizolu v sére. Systémová expozícia flutikazónfuroátu normalizovaná na dávku bola u osôb so stredne ťažkou a ťažkou poruchou funkcie pečene (stupňa B alebo C
Childovej-Pughovej klasifikácie) podobná.
Po opakovanom podávaní flutikazónfuroátu/vilanterolu počas 7 dní nedošlo u osôb s miernou, stredne ťažkou alebo ťažkou poruchou funkcie pečene (stupňa A, B alebo C Childovej-Pughovej klasifikácie) k významnému zvýšeniu systémovej expozície vilanterolu (Cmax a AUC).
U osôb s miernou alebo stredne ťažkou poruchou funkcie pečene (vilanterol, 22 mikrogramov) alebo s ťažkou poruchou funkcie pečene (vilanterol, 12,5 mikrogramu) nemala kombinácia flutikazónfuroát/vilanterol klinicky významný vplyv na beta-adrenergné systémové účinky (srdcová frekvencia alebo hladina draslíka v sére) v porovnaní so zdravými osobami.
Ďalšie osobitné skupiny pacientov
U osôb s astmou bola odhadovaná hodnota AUC(0-24) flutikazónfuroátu u osôb pochádzajúcich
z východnej Ázie, Japonska a juhovýchodnej Ázie (12 - 13 % osôb) v priemere o 33 % až 53 % vyššia
v porovnaní s inými rasovými skupinami. Nepreukázalo sa však, že by sa vyššia systémová expozícia v tejto populácii spájala s vyšším účinkom na 24-hodinovú exkréciu kortizolu do moču. Predpokladá sa, že hodnota Cmax vilanterolu bude v priemere o 220 až 287 % vyššia a hodnota AUC(0-24) bude porovnateľná u osôb ázijského pôvodu v porovnaní s osobami iných rasových skupín. Nepreukázalo sa však, že by táto vyššia hodnota Cmax vilanterolu mala za následok klinicky významný vplyv
na srdcovú frekvenciu.
Pohlavie, telesná hmotnosť a BMI
Na základe populačnej farmakokinetickej analýzy údajov zo štúdie fázy III získaných od 1 213 osôb s astmou (712 žien) sa nepreukázalo, že by pohlavie, telesná hmotnosť alebo BMI (index telesnej hmotnosti) ovplyvňovali farmakokinetiku flutikazónfuroátu.
Na základe populačnej farmakokinetickej analýzy u 856 osôb s astmou (500 žien) sa nepreukázalo, že by pohlavie, telesná hmotnosť alebo BMI ovplyvňovali farmakokinetiku vilanterolu.
Nie je potrebná žiadna úprava dávkovania v závislosti od pohlavia, telesnej hmotnosti alebo BMI.
5.3 Predklinické údaje o bezpečnosti
Farmakologické a toxikologické účinky pozorované pri podávaní flutikazónfuroátu alebo vilanterolu v predklinických štúdiách boli rovnaké ako tie, ktoré sa typicky spájajú buď s glukokortikosteroidmi, alebo s beta2-agonistami. Podávanie flutikazónfuroátu v kombinácii s vilanterolom neviedlo
k významnej novej toxicite.
Genotoxicita a karcinogenita
Flutikazónfuroát
Flutikazónfuroát nebol genotoxický v štandardnom súbore štúdií a nebol karcinogénny v štúdiách s jeho celoživotným inhalačným podávaním potkanom a myšiam pri expozíciách podobných tým, ktoré sa dosahujú po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Vilanteroltrifenatát
V štúdiách genetickej toxicity neboli vilanterol (vo forme alfa-fenylcinamátu) a kyselina trifenyloctová genotoxické, čo poukazuje na to, že vilanterol (vo forme trifenatátu) nepredstavuje genotoxické riziko pre ľudí.
Zhodne so zisteniami získanými pri iných beta2-agonistoch sa v štúdiách s celoživotným inhalačným podávaním zistilo, že vilanteroltrifenatát mal proliferatívne účinky na reprodukčný systém samíc potkanov a myší a na hypofýzu potkanov. U potkanov sa pri expozícii 2-násobne vyššej a u myší
pri expozícii 30-násobne vyššej ako je expozícia dosahovaná po maximálnej odporúčanej dávke pre
ľudí, na základe AUC, nezistilo zvýšenie výskytu tumorov.
Reprodukčná toxicita
Flutikazónfuroát
Účinky pozorované po inhalačnom podávaní flutikazónfuroátu v kombinácii s vilanterolom u potkanov boli podobné ako účinky pozorované po podávaní samotného flutikazónfuroátu. Flutikazónfuroát nebol teratogénny u potkanov ani u králikov, ale spomalil vývoj u potkanov a spôsobil potraty u králikov, keď bol podávaný v dávkach toxických pre gravidné samice. Nezistili sa žiadne účinky na vývoj u potkanov pri expozíciách, ktoré boli približne 3-násobne vyššie ako expozície dosahované po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Vilanteroltrifenatát
Vilanteroltrifenatát nebol teratogénny u potkanov. V štúdiách s inhalačným podávaním králikom spôsobil vilanteroltrifenatát účinky podobné tým, ktoré sú pozorované pri podávaní iných
beta2-agonistov (rázštep podnebia, predčasné otvorenie očných viečok, zrastenie sternebra
a ohnutie/malrotácia končatín). Pri subkutánnom podávaní sa nezistili žiadne účinky pri expozíciách
84-násobne vyšších ako sú expozície dosahované po maximálnej odporúčanej dávke pre ľudí, na základe AUC.
Ani flutikazónfuroát, ani vilanteroltrifenatát nemali nežiaduce účinky na fertilitu alebo prenatálny a postnatálny vývoj u potkanov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Monohydrát laktózy
Magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom použití: 6 týždňov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 25°C. Ak sa inhalátor uchováva v chladničke, treba ho z nej vybrať aspoň
hodinu pred použitím, aby dosiahol izbovú teplotu. Uchovávajte v pôvodnom balení na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
Inhalátor pozostáva zo svetlošedého korpusu, svetlomodrého krytu náustka a počítadla dávok a je zabalený vo vaničke z laminátovej fólie obsahujúcej vrecko s vysúšadlom. Vanička je uzatvorená odnímateľnou fóliou.
Inhalátor obsahuje dva stripy z laminátovej hliníkovej fólie so 14 alebo 30 dávkami.
Inhalátor je viaczložková pomôcka zložená z polypropylénu, polyetylénu s vysokou hustotou, polyoxymetylénu, polybutyléntereftalátu, akrylonitrilbutadiénstyrénu, polykarbonátu a nehrdzavejúcej ocele.
Veľkosti balenia obsahujú 14-dávkový alebo 30-dávkový inhalátor. Multibalenia obsahujú
3 x 30-dávkové inhalátory.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom Nepoužitý liek alebo odpad vzniknutý z lieku treba vrátiť do lekárne. Pokyny na použitie, pozri časť 4.2.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlaxo Group Limited.
980 Great West Road, Brentford, Middlesex TW8 9GS, Spojené kráľovstvo.
8. REGISTRAČNÉ ČÍSLAEU/1/14/929/004
EU/1/14/929/005
EU/1/14/929/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.