
o
Nezaznamenali sa žiadne signifikantné interakcie sildenafilu (50 mg) s tolbutamidom (250 mg) alebo s warfarínom (40 mg), liekmi, ktoré sú metabolizované prostredníctvom CYP2C9.
Sildenafil nemal významný vplyv na expozíciu atorvastatínu (AUC zvýšené o 11 %), čo naznačuje, že sildenafil nemá klinicky významný účinok na CYP3A4.
Medzi sildenafilom (v jednotlivej dávke 100 mg) a acenokumarolom sa nepozorovali žiadne interakcie.
Sildenafil (50 mg) nepotencioval predĺženie času krvácania zapríčineného kyselinou acetylsalicylovou
(150 mg).
Sildenafil (50 mg) nepotencioval hypotenzívny účinok alkoholu u zdravých dobrovoľníkov, ktorí mali priemernú maximálnu koncentráciu alkoholu v krvi 80 mg/dl.
V štúdii so zdravými dobrovoľníkmi vyvolal sildenafil v rovnovážnom stave (80 mg trikrát denne)
zvýšenie AUC bosentanu o 50 % (125 mg dvakrát denne).
Populačná farmakokinetická analýza údajov zo štúdie s dospelými pacientmi s PAH na základnej liečbe bosentanom (62,5 mg – 125 mg dvakrát denne) ukázala zvýšenie (20 % (95 % CI: 9,8 – 30,8) AUC bosentanu pri súčasnom podávaní sildenafilu v rovnovážnom stave (20 mg trikrát denne)
v menšom rozsahu ako to, ktoré sa pozorovalo u zdravých dobrovoľníkov, ktorým bol podávaný bosentan súčasne so sildenafilom v dávke 80 mg trikrát denne (pozri časti 4.4 a 5.1).
V špecifickej interakčnej štúdii u pacientov s hypertenziou, ktorí súčasne užívali amplodipín so sildenafilom (100 mg), sa zaznamenalo ďalšie zníženie systolického tlaku krvi v ľahu o 8 mmHg. Zodpovedajúce ďalšie zníženie diastolického tlaku krvi v ľahu bolo o 7 mmHg. Toto ďalšie zníženie tlaku krvi malo podobný rozsah, ako keď sa sildenafil podával zdravým dobrovoľníkom samostatne.
V troch špecifických liekových interakčných štúdiách sa pacientom s benígnou hyperpláziou prostaty
(BPH) stabilizovanou na liečbe doxazosínom súbežne podával alfablokátor doxazosín (4 mg, resp.
8 mg) a sildenafil (25 mg, 50 mg alebo 100 mg). V týchto štúdiách sa u sledovanej populácie pozoroval dodatočný priemerný pokles systolického a diastolického tlaku krvi v ľahu o 7/7 mmHg,
9/5 mmHg, resp. 8/4 mmHg, ako aj dodatočný priemerný pokles tlaku krvi v stoji o 6/6 mmHg,
11/4 mmHg, resp. 4/5 mmHg. Keď sa sildenafil a doxazosín súbežne podávali pacientom stabilizovaným na liečbe doxazosínom, hlásenia o výskyte symptomatickej posturálnej hypotenzie
u pacientov boli ojedinelé. Tieto zahŕňali závraty a stratu rovnováhy, ale nie synkopu. Súčasné podávanie sildenafilu pacientom užívajúcim alfablokátory môže viesť u vnímavých jedincov
k symptomatickej hypotenzii (pozri časť 4.4).
Sildenafil (100 mg v jednorazovej dávke) neovplyvnil farmakokinetiku inhibítora HIV proteáz v rovnovážnom stave, sachinaviru, ktorý je substrátom/inhibítorom CYP3A4.
V súlade so známym účinkom sildenafilu na metabolickú cestu oxid dusnatý/cGMP (pozri časť 5.1) sa preukázalo, že sildenafil potencuje hypotenzívny účinok nitrátov, a preto je jeho súčasné podanie
s donormi oxidu dusnatého alebo nitrátmi v akejkoľvek forme kontraindikované (pozri časť 4.3).
Riociguát
Predklinické štúdie ukázali aditívny systémový účinok znižujúci krvný tlak, keď sa inhibítory PDE5 podávali súčasne s riociguátom. Klinické štúdie preukázali, že riociguát zosilňuje hypotenzívne účinky inhibítorov PDE5. V skúšanej populácii nebol nájdený žiadny dôkaz o priaznivom klinickom účinku spomínanej kombinácie. Súčasné užívanie riociguátu s PDE5 inhibítormi, vrátane sildenafilu, je kontraindikované (pozri časť 4.3).
Sildenafil nemal klinicky významný vplyv na plazmatické hladiny perorálnych kontraceptív
(etinylestradiol 30 μg a levonorgestrel 150 μg).
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku a antikoncepcia u mužov a žien
Vzhľadom na nedostatok údajov o účinnosti Revatia u gravidných žien sa Revatio neodporúča užívať u žien v reprodukčnom veku, pokiaľ nepoužívajú súčasne vhodnú antikoncepciu.
Gravidita
Nie sú k dispozícii žiadne údaje o použití sildenafilu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu a embryonálny/fetálny vývoj. Štúdie
na zvieratách preukázali toxicitu na postnatálny vývoj (pozri časť 5.3).
Vzhľadom na nedostatok údajov sa má Revatio používať u gravidných žien iba v nevyhnutných prípadoch.
Dojčenie
Nie sú k dispozícii žiadne adekvátne a dobre kontrolované štúdie u dojčiacich žien. Údaje od jednej dojčiacej ženy naznačujú, že sildenafil a jeho aktívny metabolit, N-demetylsildenafil, sa vo veľmi malých množstvách vylučujú do materského mlieka. Nie sú dostupné žiadne klinické údaje týkajúce sa nežiaducich účinkov u dojčených detí, ale nepredpokladá sa , že by požité množstvá spôsobovali
nejaké nežiaduce účinky. Predpisujúci lekári majú starostlivo zvážiť klinickú potrebu matky užívať sildenafil a akékoľvek potenciálne nežiaduce účinky u dojčeného dieťaťa.
Fertilita
Predklinické údaje získané na základe obvyklých farmakologických štúdií fertility neodhalili žiadne osobitné riziko pre ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Revatio má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Keďže závrat a zmenené videnie boli hlásené v klinických štúdiách so sildenafilom, pacienti predtým, ako budú viesť vozidlá a obsluhovať stroje, majú poznať, ako reagujú na podanie Revatia.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V pivotnej, placebom kontrolovanej štúdii s Revatiom pri pľúcnej artériovej hypertenzii bolo
207 pacientov randomizovaných a liečených dávkami 20 mg, 40 mg alebo 80 mg Revatia TID a
70 pacientov bolo randomizovaných na placebo. Liečba trvala 12 týždňov. Celková frekvencia prerušenia liečby u pacientov liečených sildenafilom v dávkach 20 mg, 40 mg a 80 mg TID bola
2,9 %, 3,0 % a 8,5 % v uvedenom poradí, v porovnaní s 2,9 % u placeba. Z 277 pacientov liečených v pivotnej štúdii prešlo do dlhodobej pokračovacej štúdie 259 pacientov. V nej sa podávali dávky do
80 mg trikrát denne (4-krát vyššie ako odporúčaná dávka 20 mg trikrát denne) a po 3 rokoch 87 % zo
183 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V placebom kontrolovanej štúdii s Revatiom ako doplnkom k intravenóznemu epoprostenolu u pľúcnej artériovej hypertenzie bolo celkovo 134 pacientov liečených Revatiom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg a potom na 80 mg trikrát denne podľa znášanlivosti) a epoprostenolom a 131 pacientov bolo liečených placebom a epoprostenolom. Dĺžka liečby bola 16 týždňov. Celková frekvencia prerušenia liečby z dôvodu nežiaducich účinkov
u pacientov liečených sildenafilom/epoprostenolom bola 5,2 % v porovnaní s 10,7 % u pacientov liečených placebom/epoprostenolom. Nedávno hlásenými nežiaducimi reakciami, ktoré sa vyskytli oveľa častejšie v skupine so sildenafilom/epoprostenolom, bola okulárna hyperémia, rozmazané videnie, upchatý nos, nočné potenie, bolesť chrbta a sucho v ústach. Známe nežiaduce reakcie bolesť hlavy, návaly tepla, bolesti v končatinách a edém boli zaznamenané s vyššou frekvenciou u pacientov liečených sildenafilom/epoprostenolom v porovnaní s pacientmi liečenými placebom/epoprostenolom. Z pacientov, ktorí ukončili úvodnú liečbu, prešlo do dlhodobej pokračovacej štúdie 242 pacientov.

V nej sa podávali dávky do 80 mg TID a po 3 rokoch 68 % zo 133 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V dvoch placebom kontrolovaných štúdiách boli nežiaduce účinky obyčajne mierne až stredne závažné. Najčastejšie hlásenými nežiaducimi účinkami, ktoré sa vyskytli (³ 10 %) u Revatia
v porovnaní s placebom boli bolesti hlavy, návaly tepla, dyspepsia, hnačka a bolesť v končatinách.
Zoznam nežiaducich reakcií usporiadaný do tabuľkyNežiaduce reakcie, ktoré sa vyskytli u > 1 % pacientov liečených Revatiom a boli častejšie (> 1 % rozdiel) u Revatia v pivotnej štúdii alebo v spoločnom súbore údajov z oboch placebom kontrolovaných štúdií pri liečbe pľúcnej artériovej hypertenzie Revatiom v dávkach
20, 40 alebo 80 mg TID, sú uvedené v tabuľke nižšie podľa triedy a skupín frekvencií (veľmi časté
(³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100) a neznáme (z dostupných údajov)). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Hlásenia po uvedení lieku na trh sú uvedené kurzívou.
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakciaInfekcie a nákazyČasté celulitída, chrípka, zápal priedušiek, zápal prinosových dutín, rinitída,
gastroenteritída
Poruchy krvi a lymfatického systémuČasté anémia
Poruchy metabolizmu a výživyČasté retencia tekutín
Psychické poruchyČasté nespavosť, úzkosť
Poruchy nervového systémuVeľmi časté bolesť hlavy
Časté migréna, tremor, parestézie, pocit pálenia, hypestézia
Poruchy okaČasté krvácanie do sietnice, poškodenie zraku, rozmazané videnie, fotofóbia, chromatopsia, cyanopsia, podráždenie
očí, okulárna hyperémia
Menej časté zníženie zrakovej ostrosti, diplopia, neprirodzené pocity v oku
Neznáme nearteritická predná ischemická neuropatia zrakového nervu (NAION)
*, oklúzia ciev sietnice
*, poruchy
v zornom poli
*Poruchy ucha a labyrintuČasté závrat
Neznáme
náhla strata sluchuPoruchy cievVeľmi časté návaly tepla
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakcia
Neznáme hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté epistaxa, kašeľ, upchatý nos
Poruchy gastrointestinálneho traktu
Veľmi časté hnačka, dyspepsia
Časté gastritída, gastroezofageálna refluxná choroba, hemoroidy, nafúknutie
brucha, sucho v ústach
Poruchy kože a podkožného tkaniva
Časté
Neznáme
alopécia, erytém, nočné potenie
vyrážka
 Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Veľmi časté bolesť v končatinách
Časté myalgia, bolesť chrbta
Poruchy obličiek a močových ciestMenej časté hematúria
Poruchy reprodukčného systému a prsníkovMenej časté penilné krvácanie, hematospermia, gynekomastia
Neznáme
priapizmus, zvýšená erekciaCelkové poruchy a reakcie v mieste podaniaČasté horúčka
* Tieto nežiaduce účinky/reakcie boli hlásené s neznámou frekvenciou u pacientov užívajúcich sildenafil na liečbu erektilnej dysfunkcie u mužov (MED).
Pediatrická populáciaV placebom kontrolovanej štúdii s Revatiom, do ktorej boli zaradení pacienti s pľúcnou artériovou hypertenziou vo veku 1 až 17 rokov, bolo liečených celkovo 174 pacientov trikrát denne v dávkovacej schéme s Revatiom buď s nízkou (10 mg u pacientov s hmotnosťou > 20 kg; žiadni pacienti
s hmotnosťou ≤ 20 kg nedostávali nízku dávku), strednou (10 mg u pacientov s hmotnosťou ≥
8 – 20 kg; 20 mg u pacientov s hmotnosťou ≥ 20 – 45 kg; 40 mg u pacientov s hmotnosťou > 45 kg)
alebo vysokou dávkou (20 mg u pacientov s hmotnosťou ≥ 8 – 20 kg; 40 mg u pacientov
s hmotnosťou ≥ 20 – 45 kg; 80 mg u pacientov s hmotnosťou > 45 kg) a 60 pacientov bolo liečených placebom.
Profil nežiaducich reakcií pozorovaný v tejto pediatrickej štúdii bol vo všeobecnosti zhodný
s profilom u dospelých
(pozri tabuľku vyššie). Najčastejšími nežiaducimi reakciami, ktoré sa vyskytli
(s frekvenciou ≥ 1 %) u pacientov užívajúcich Revatio (v kombinovaných dávkach) a s frekvenciou
> 1 % u pacientov užívajúcich placebo, boli pyrexia, infekcia horných dýchacích ciest (každý 11,5 %), vracanie (10,9 %), zvýšená erekcia (zahŕňajúca spontánnu penilnú erekciu u mužských pacientov)
(9,0 %), nauzea, bronchitída (každý 4,6 %), faryngitída (4,0 %), rinorea (3,4 %), a pneumónia, rinitída
(každý 2,9 %).
Z 234 pediatrických pacientov liečených v krátkodobej, placebom kontrolovanej štúdii 220 pacientov vstúpilo do dlhodobej pokračovacej štúdie. Pacienti, ktorí boli na aktívnej liečbe sildenafilom, pokračovali v rovnakej dávkovacej schéme, kým pacienti, ktorým bolo v krátkodobej štúdii podané placebo, boli náhodne zaradení do liečby sildenafilom.
Najčastejšie nežiaduce reakcie hlásené počas celého trvania krátkodobej a dlhodobej štúdie boli vo všeobecnosti podobné ako tie, ktoré sa pozorovali v krátkodobej štúdii. Nežiaduce reakcie hlásené u > 10 % z 229 pacientov liečených sildenafilom (skupina s kombinovanými dávkami, vrátane
9 pacientov, ktorí nepokračovali v dlhodobej štúdii) boli infekcia horných dýchacích ciest (31 %), bolesť hlavy (26 %), vracanie (22 %), bronchitída (20 %), faryngitída (18 %), pyrexia (17 %), hnačka (15 %), chrípka a epistaxa (12 % v oboch prípadoch). Väčšina týchto nežiaducich reakcií bola považovaná za miernu až stredne závažnú.
Závažné nežiaduce udalosti boli hlásené u 94 (41 %) z 229 pacientov užívajúcich sildenafil.
Z 94 pacientov, ktorí hlásili závažné nežiaduce udalosti bolo 14/55 (25,5 %) pacientov v skupine
s nízkou dávkou, 35/74 (47,3 %) v skupine so strednou dávkou a 45/100 (45 %) v skupine s vysokou dávkou. Najčastejšie závažné nežiaduce udalosti, ktoré sa vyskytli s frekvenciou ≥ 1 % u pacientov užívajúcich sildenafil (kombinované dávky) boli pneumónia (7,4 %), zlyhanie srdca, pľúcna hypertenzia (5,2 % v oboch prípadoch), infekcia horných dýchacích ciest (3,1 %), zlyhanie pravej komory srdca, gastroenteritída (2,6 % v oboch prípadoch), synkopa, bronchitída, bronchpneumónia, pľúcna artériová hypertenzia (2,2 % vo všetkých prípadoch), bolesť na hrudi, zubný kaz (1,7 %
v oboch prípadoch) a kardiogénny šok, vírusová gastroenteritída, infekcia močových ciest (1,3 % vo všetkých prípadoch).
Nasledovné závažné nežiaduce udalosti boli považované za súvisiace s liečbou: enterokolitída, konvulzia, hypersenzitivita, stridor, hypoxia, neurosenzorická hluchota a ventrikulárna arytmia.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV štúdiách so zdravými dobrovoľníkmi boli po podaní jednorazových dávok do 800 mg nežiaduce reakcie podobné ako pri podaní nižších dávok, ale vyskytovali sa častejšie a boli závažnejšie. Jednorazové dávky 200 mg viedli k vyššiemu výskytu nežiaducich reakcií (bolesť hlavy, návaly, závrat, dyspepsia, nazálna kongescia a zmena videnia).
V prípade predávkovania sa majú podľa potreby zaviesť štandardné podporné opatrenia. Keďže sildenafil je pevne viazaný na bielkoviny plazmy a neeliminuje sa močom, nie je pravdepodobné, že by renálna dialýza mala urýchliť klírens sildenafilu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Urologiká, lieky používané pri poruchách erekcie, ATC kód: G04BE03
MechanizmusúčinkuSildenafil je účinný a selektívny inhibítor fosfodiesterázy typu 5 (PDE5), špecifickej pre cyklický guanozínmonofosfát (cGMP). Je to enzým, ktorý zodpovedá za degradáciu cGMP. Okrem prítomnosti
tohto enzýmu v kavernóznom telese penisu sa PDE5 nachádza aj v cievnom riečisku pľúc. Sildenafil
preto zvyšuje cGMP v bunkách hladkých svalov pľúcnych ciev, čo vedie k ich relaxácii. U pacientov
s pľúcnou artériovou hypertenziou to môže viesť k vazodilatácii v pľúcnom riečisku, a v menšej miere aj ku vazodilatácii v systémovom obehu.
Farmakodynamický účinokŠtúdie
in vitro preukázali, že sildenafil je selektívny pre PDE5. Jeho účinok je výraznejší na PDE5 ako na ostatné známe fosfodiesterázy. Sildenafil je 10-krát selektívnejší pre PDE5 ako pre PDE6, ktorá sa podieľa na fototransdukcii v retine. Má 80-krát vyššiu selektivitu pre PDE5 než pre PDE1 a viac ako
700-krát vyššiu selektivitu pre PDE5 než pre PDE2, 3, 4, 7, 8, 9, 10 a 11. Obzvlášť, sildenafil má
4 000-krát vyššiu selektivitu pre PDE5 ako pre PDE3, cAMP špecifickú izoformu fosfodiesterázy, ktorá sa podieľa na kontrole kontraktility srdcového svalu.
Sildenafil spôsobuje mierny a prechodný pokles tlaku krvi, vo väčšine prípadov bez klinického významu. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s artériovou hypertenziou poklesol systolický tlak oproti východiskovej hodnote v priemere o 9,4 mmHg a diastolický o 9,1 mmHg. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s pľúcnou artériovou hypertenziou sa pozorovali menej výrazné zmeny v poklese tlaku krvi (oba, systolický, ako aj diastolický tlak sa znížili
o 2 mmHg). Pri odporúčanej dávke 20 mg trikrát denne neboli pozorované žiadne poklesy systolického alebo diastolického tlaku.
Podávanie jednorazových dávok až do 100 mg u zdravých dobrovoľníkov neviedlo k žiadnemu klinicky relevantnému účinku na EKG. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s pľúcnou artériovou hypertenziou sa nepozorovali žiadne klinicky významné zmeny na EKG.
V štúdii zameranej na hemodynamické účinky jednorazovej perorálnej dávky 100 mg sildenafilu
u 14 pacientov s ťažkou koronárnou artériovou chorobou (coronary artery disease, CAD) (> 70 %
stenóza aspoň jednej koronárnej artérie) poklesol stredný pokojový systolický krvný tlak o 7 %
a diastolický krvný tlak o 6 % v porovnaní s východiskovými hodnotami. Stredný pľúcny systolický tlak krvi poklesol o 9 %. Sildenafil nemal vplyv na srdcový výdaj a neviedol ku zhoršeniu krvného
prietoku cez stenózne koronárne artérie.
U niektorých pacientov sa jednu hodinu po podaní dávky 100 mg sildenafilu pri použití Farnsworthovho-Munsellovho testu so 100 farebnými odtieňmi pozorovali mierne a prechodné rozdiely v rozlišovaní farieb (modrá/zelená). Dve hodiny po podaní sa už nezaznamenali žiadne účinky. Možný mechanizmus tejto zmeny v rozlišovaní farieb súvisí s inhibíciou PDE6, ktorá hrá úlohu vo fototransdukčnej kaskáde retiny. Sildenafil neovplyvňuje ani ostrosť, ani kontrast videnia. V placebom kontrolovanej štúdii s malým počtom pacientov s dokumentovaným včasným štádiom vekom podmienenej makulárnej degenerácie (n = 9) neboli vo vykonaných testoch videnia (ostrosť videnia, Amslerova mriežka, rozlíšenie farieb pri simulovanom dopravnom osvetlení, Humpreyho perimeter a fotostres) dokázané žiadne významné zmeny vplyvom sildenafilu (jednorazová dávka
100 mg).
Klinická účinnosť a bezpečnosť
Účinnosťudospelýchpacientovspľúcnouartériovouhypertenziou(PAH)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 278 pacientmi s primárnou pľúcnou hypertenziou, PAH spojenou s ochorením spojivového tkaniva a PAH po
chirurgickej liečbe vrodenej srdcovej chyby. Pacienti boli randomizovaní do jednej zo štyroch
liečených skupín: placebo, sildenafil 20 mg, sildenafil 40 mg alebo sildenafil 80 mg trikrát denne.
Z 278 randomizovaných pacientov dostalo 277 pacientov aspoň jednu dávku skúšaného lieku. Súbor pacientov tvorilo 68 mužov (25 %) a 209 žien (75 %) s priemerným vekom 49 rokov (v rozpätí
18 – 81 rokov) a východiskovými hodnotami 6-minútového testu chôdze od 100 do 450 metrov
vrátane (priemer: 344 metrov). 175 (63 %) zaradených pacientov malo diagnostikovanú primárnu pľúcnu hypertenziu, 84 (30 %) malo diagnostikovanú PAH spojenú s ochorením spojivového tkaniva
a 18 (7 %) pacientov malo diagnostikovanú PAH následne po chirurgickej korekcii vrodenej srdcovej
chyby. Väčšina pacientov mala funkčný stupeň II podľa WHO (107/277, 39 %) alebo
III (160/377, 58 %) s priemernými východiskovými hodnotami pri 6-minútovej chôdzi 378 metrov, resp. 326 metrov; menej pacientov malo stupeň I (1/277, 0,4 %) alebo IV (9/277, 3 %) ako
východiskovú hodnotu. Do štúdie neboli zaradení pacienti s ejekčnou frakciou ľavej komory < 45 %
alebo s frakčným skrátením ľavej komory < 0,2.
Sildenafil (alebo placebo) sa pridal k základnej liečbe pacientov, ktorá mohla zahŕňať kombináciu antikoagulancií, digoxínu, blokátorov kalciových kanálov, diuretík alebo kyslíka. Nesmeli sa používať prostacyklín, analógy prostacyklínu a antagonisty endotelínového receptora ako prídavná liečba, a ani
doplnková liečba arginínom. Pacienti, u ktorých predtým zlyhala liečba bosentanom, boli zo štúdie vyradení.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze (6MWD) v 12. týždni liečby. Vo všetkých troch skupinách so sildenafilom bolo v porovnaní so skupinami s placebom zaznamenané štatisticky významné predĺženie 6MWD. Pre jednotlivé dávky sildenafilu 20 mg, 40 mg a 80 mg TID predstavovalo zlepšenie 6MWD po korekcii na placebo 45 metrov (p < 0,0001), 46 metrov
(p < 0,0001) a 50 metrov (p < 0,0001). Medzi jednotlivými dávkami sildenafilu nebol v účinku signifikantný rozdiel. U pacientov s východiskovým 6MWD < 325 m bola pozorovaná lepšia účinnosť pri vyšších dávkach (zlepšenie po korekcii na placebo 58 metrov, 65 metrov a 87 metrov pre dávky
20 mg, 40 mg a 80 mg TID v uvedenom poradí).
Pri analýze podľa funkčného stupňa WHO sa pozorovalo v skupine s dávkou 20 mg štatisticky významné predĺženie 6MWD. Pre stupeň II a III predstavovalo zlepšenie po korekcii na placebo
49 metrov (p = 0,0007) a 45 metrov (p = 0,0031).
Zlepšenie 6MWD bolo zjavné po 4 týždňoch liečby a tento účinok pretrvával aj v 8. a 12. týždni. Výsledky boli všeobecne zhodné v podskupinách rozdelených podľa etiológie (primárnej PAH a PAH spojenej s ochorením spojivového tkaniva), funkčného stupňa podľa WHO, pohlavia, rasy, lokálnych údajov, stredného tlaku v pľúcnici (mPAP) a indexovanej pľúcnej vaskulárnej rezistencie (PVRI).
Pacienti pri všetkých dávkovaniach sildenafilu dosiahli v porovnaní s placebom štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP) a pľúcnej vaskulárnej rezistencie (PVR). Účinky liečby korigovanej na placebo s mPAP predstavovali -2,7 mmHg (p = 0,04), -3,0 mmHg (p = 0,01) a
-5,1 mmHg (p < 0,0001) pre sildenafil v dávke 20 mg, 40 mg a 80 mg TID v uvedenom poradí. Účinky liečby korigovanej na placebo s PVR predstavovali -178 dyn.sec/cm5 (p=0,0051), -195 dyn.sec/cm5 (p=0,0017) a -320 dyn.sec/cm5 (p<0,0001) pre sildenafil v dávke 20 mg, 40 mg a 80 mg TID v uvedenom poradí. Percentuálne zníženie PVR (11,2 %, 12,9 %, 23,3 %) v 12. týždni pre sildenafil 20 mg, 40 mg a 80 mg TID bolo proporcionálne výraznejšie ako zníženie systémovej vaskulárnej rezistencie (SVR) (7,2 %, 5,9 %, 14,4 %). Účinok sildenafilu na mortalitu nie je známy.
U väčšieho percenta pacientov sa preukázalo zlepšenie aspoň o jednu funkčnú triedu WHO v 12. týždni pri každej z dávok sildenafilu (t.j. 28 %, 36 % a 42 % osôb, ktoré užívali dávky 20 mg, 40 mg a 80 mg TID sildenafilu, v uvedenom poradí) v porovnaní s placebom (7 %). Zodpovedajúce pomery šancí boli 2,92 (p=0,0087), 4,32 (p=0,0004) a 5,75 (p<0,0001).
Dlhodobéúdajeoprežívanípopuláciebezinejliečby
Pacienti zaradení do pivotnej štúdie boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 87 % pacientov dávku 80 mg TID. V pivotnej štúdii bolo liečených
Revatiom celkovo 207 pacientov a ich dlhodobé prežívanie bolo hodnotené minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-
Meierových kriviek 96 %, 91 % a 82 %. Prežívanie pacientov s funkčným stupňom II podľa WHO pri vstupe do štúdie bolo po 1, 2 a 3 rokoch 99 %, 91 % a 84 % a u pacientov s funkčným stupňom III podľa WHO pri úvode do štúdie bolo 94 %, 90 % a 81 %.
ÚčinnosťudospelýchpacientovsPAH(pripoužitívkombináciisepoprostenolom)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 267 pacientmi s PAH, ktorí boli stabilizovaní intravenózne podávaným epoprostenolom. Pacientov s PAH tvorili pacienti s primárnou pľúcnou artériovou hypertenziou (212/267, 79 %) a PAH spojenou s ochorením spojivového tkaniva (55/267, 21 %).Väčšina pacientov mala funkčný stupeň II podľa WHO (68/267,
26 %) alebo III (175/267, 66 %); menej pacientov malo stupeň I (3/267, 1 %) alebo IV (16/267, 6 %)
ako východiskovú hodnotu; u niekoľkých pacientov funkčný stupeň podľa WHO nebol známy. Pacienti boli randomizovaní do skupiny s placebom alebo sildenafilom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg, a potom na 80 mg trikrát denne podľa znášanlivosti) pri použití v kombinácii s intravenózne podávaným epoprostenolom.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze v 16. týždni liečby. U sildenafilu bolo
v porovnaní s placebom zaznamenané štatisticky významné predĺženie vzdialenosti pri 6-minútovej
chôdzi. Priemerné zlepšenie testu chôdze po korekcii na placebo predstavovalo 26 metrov v prospech sildenafilu (95 % CI: 10,8, 41,2) (p = 0,0009). U pacientov s východiskovou hodnotou testu chôdze
³ 325 metrov bol účinok liečby 38,4 metra v prospech sildenafilu; u pacientov s východiskovou hodnotou testu chôdze < 325 metrov bol účinok liečby 2,3 metra v prospech placeba. U pacientov
s primárnou PAH bol účinok liečby 31,1 metra v porovnaní so 7,7 metra u pacientov s PAH spojenou
s ochorením spojivového tkaniva. Rozdiel vo výsledkoch medzi týmito randomizovanými podskupinami mohol vzniknúť náhodou vzhľadom na ich obmedzenú veľkosť reprezentovanej vzorky.
Pacienti liečení sildenafilom dosiahli v porovnaní s pacientmi užívajúcimi placebo štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP). Priemerný účinok liečby po korekcii na placebo predstavoval - 3,9 mmHg v prospech sildenafilu (95 % CI: - 5,7, - 2,1) (p = 0,00003). Sekundárnym hodnoteným parametrom bol čas do klinického zhoršenia stavu definovaný ako čas od randomizácie do vzniku prvej príhody klinického zhoršenia (smrť, transplantácia pľúc, začatie liečby bosentanom alebo klinické zhoršenie vyžadujúce úpravu liečby epoprostenolom). Liečba sildenafilom signifikantne oddialila čas do klinického zhoršenia PAH v porovnaní s placebom (p = 0,0074). Klinické zhoršenie nastalo u 23 pacientov v placebovej skupine (17,6 %) v porovnaní s 8 pacientmi
v skupine užívajúcej sildenafil (6,0 %).
Dlhodobéúdajeoprežívanívzákladnejepoprostenolovejštúdii
Pacienti zaradení do štúdie s prídavnou liečbu epoprostenolom boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 68 % pacientov dávku 80 mg TID. V úvodnej štúdii bolo liečených Revatiom celkovo 134 pacientov a ich dlhodobé prežívanie bolo hodnotené minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-Meierových kriviek 92 %, 81 % a 74 %.
Bezpečnosť a účinnosť u dospelých pacientov s PAH (pri použití v kombinácii s bosentanom) Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia so 103 klinicky stabilizovanými pacientmi s PAH (funkčný stupeň II a III podľa WHO), ktorí boli liečení bosentanom minimálne počas troch mesiacov. Pacienti s PAH zahŕňali pacientov s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. Pacienti boli randomizovaní do skupín s placebom alebo sildenafilom (20 mg trikrát denne), v kombinácii s bosentanom (62,5 – 125 mg dvakrát denne). Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vzdialenosti dosiahnutej pri 6-minútovej chôdzi (6MWD) v 12. týždni liečby. Výsledky ukazujú, že neexistuje významný rozdiel medzi priemernou zmenou voči východiskovej hodnote testu 6MWD pozorovanou pri podávaní sildenafilu (20 mg trikrát denne) 13,62 m (95 % CI: - 3,89 až 31,12)
a placeba 14,08 m (95 % CI: - 1,78 až 29,95).
Rozdiely vo vzdialenosti dosiahnutej pri 6-minútovej chôdzi boli pozorované medzi pacientmi
s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. U pacientov s primárnou PAH (67 pacientov) boli priemerné zmeny voči východiskovej hodnote 26,39 m (95 % CI: 10,70 až 42,08) v skupine so sildenafilom a 11,84 m (95 % CI: - 8,83 až 32,52) v skupine s placebom. U pacientov
s PAH spojenou s ochorením spojivového tkaniva (36 pacientov) boli však priemerné zmeny voči východiskovej hodnote 18,32 m (95 % CI: - 65,66 až 29,02) v skupine so sildenafilom a 17,50 m
(95 % CI: - 9,41 až 44,41) v skupine s placebom.
Z celkového hľadiska boli nežiaduce udalosti vo všeobecnosti podobné v oboch liečebných skupinách (sildenafil v kombinácii s bosentanom v porovnaní so samotným bosentanom) a v súlade so známym bezpečnostným profilom sildenafilu pri použití v monoterapii (pozri časti 4.4 a 4.5).
Pediatrickápopulácia
V randomizovanej, dvojito zaslepenej, multicentrickej, placebom kontrolovanej štúdii porovnávajúcej paralelné skupiny s rôznym rozsahom dávok bolo celkovo liečených celkovo 234 pacientov vo veku
hypertenziu (PPH) [33 %] alebo sekundárnu pľúcnu artériovú hypertenziu (PAH) k vrodenej srdcovej chybe [systémovo-pľúcny skrat 37 %, chirurgická korekcia 30 %].V tejto štúdii 63 z 234 (27 %) pacientov malo < 7 rokov (z nich nízku dávku sildenafilu užívali = 2; strednú dávku = 17; vysokú dávku = 28; placebo = 16 pacienti) a 171 z 234 (73 %) pacientov malo 7 rokov a viac (z nich nízku dávku sildenafilu užívali = 40; strednú dávku = 38; a vysokú dávku = 49, placebo = 44 pacienti). Najviac pacientov malo na začiatku liečby funkčný stupeň I podľa WHO (75/234, 32 %) alebo
II (120/234, 51 %); menej pacientov malo funkčný stupeň III (35/234, 15 %) alebo IV (1/234, 0,4 %);
a niekoľko pacientov (3/234, 1,3 %) malo funkčný stupeň podľa WHO neznámy.
Pacienti boli bez špecifickej PAH liečby a užívanie prostacyklínu, analógov prostacyklínu
a antagonistov endotelínového receptora a ani doplnková liečba arginínom, nitráty, alfablokátory a silné CYP450 3A4 inhibítory neboli povolené.
Primárnym cieľom štúdie bolo posúdiť účinnosť 16-týždňovej chronickej liečby perorálnym sildenafilom u detí a dospievajúcich zameranej na zlepšenie tolerancie fyzickej záťaže meranej spiroergometrickým testom (Cardiopulmonary Exercise Test, CPET) u pacientov, ktorí boli v rámci
ich vývoja schopný test vykonať (n = 115). Sekundárne ciele zahŕňali hemodynamické monitorovanie, hodnotenie symptómov, funkčný stupeň podľa WHO, zmenu základnej liečby a meranie kvality
života.
Pacienti boli zaradení do jednej z troch skupín liečených sildenafilom v dávkovacej schéme s nízkou (10 mg), strednou (10 – 40 mg) alebo vysokou dávkou (20 – 80 mg) Revatia podávanou trikrát denne alebo s placebom. Aktuálne dávky podávané v danej skupine boli závislé od telesnej hmotnosti (pozri časť 4.8). Podiel pacientov užívajúcich na začiatku štúdie podporné lieky (antikoagulanciá, digoxín, blokátory kalciového kanála, diuretiká a/alebo kyslík) bol podobný v kombinovanej skupine pacientov liečených sildenafilom (47,7 %) a v skupine pacientov liečených placebom (41,7%).
Primárnym cieľom v skupinách s kombinovanou dávkou bola percentuálna zmena spotreby kyslíka na vrchole záťaže (VO2) od začiatku liečby po 16. týždeň korigovaná voči placebu, hodnotená spiroergometrickým (CPET) testom (tabuľka 2). Celkovo 106 z 234 pacientov (45 %) bolo hodnotiteľných CPET testom, tento počet zahŕňal deti vo veku ≥ 7 rokov a deti vývojovo schopné vykonať test. Deti vo veku < 7 rokov (užívajúce kombinovanú dávku sildenafilu = 47; placebo = 16) boli hodnotiteľné len pre sekundárne ciele. Priemerné hodnoty spotreby kyslíka na vrchole záťaže (VO2) na začiatku štúdie boli porovnateľné vo všetkých skupinách pacientov liečených sildenafilom (17,37 až 18,03 ml/kg/min) a mierne zvýšené v skupine pacientov liečených placebom
(20,02 ml/kg/min). Výsledky hlavnej analýzy (skupiny s kombinovanou dávkou versus placebo) neboli štatisticky signifikantné (p = 0,056) (pozri tabuľku 2). Odhadovaný rozdiel medzi strednou dávkou sildenafilu a placebom bol 11,33 % (95 % Cl: 1,72 až 20,94) (pozri tabuľku 2).
Tabuľka 2: % zmena VO2 oproti vstupnému vyšetreniu, korigovaná voči placebu
Liečebná skupina Odhadovaný rozdiel 95 % interval spoľahlivosti
Nízka dávka (n = 24) Stredná dávka (n = 26) Vysoká dávka (n = 27)
3,81 -6,11, 13,73
11,33 1,72, 20,94
7,98 -1,64, 17,60
Skupiny s kombinovanou dávkou
(
n = 77)
7,71
(p = 0,056)
-0,19, 15,60
n = 29 pre skupinu s placebom
Odhady založené na ANCOVA s úpravami pre kovarianty vrcholovej VO
2
na začiatku štúdie, etiológie a telesnej hmotnosti.
Zlepšenie závislé od dávky bolo pozorované u indexovanej pľúcnej vaskulárnej rezistencie
(Pulmonary vascular resistance index, PVRI) a stredného tlaku v pľúcnici (Mean pulmonary arterial
pressure, mPAP). V oboch skupinách pacientov so strednou a vysokou dávkou sildenafilu sa pozorovalo zníženie PVRI v porovnaní s placebom o 18 % (95 % Cl: 2 % až 32 %) a o 27 % (95 % Cl:
14 % až 39 %), zatiaľ čo v skupine pacientov s nízkou dávkou nebol pozorovaný signifikantný rozdiel
oproti placebu (rozdiel 2 %). V skupinách pacientov s vysokou a strednou dávkou boli zobrazené zmeny mPAP od začiatku sledovania v porovnaní s placebom o -3,5 mmHg (95 % Cl: -8,9, 1,9)
a o -7,3 mmHg (95% Cl: -12,4, -2,1), zatiaľ čo v skupine pacientov s nízkou dávkou sa pozorovala malá zmena oproti placebu (zmena o 1,6 mmHg). Zlepšenie sa pozorovalo pri srdcovom indexe vo všetkých troch skupinách so sildenafilom v porovnaní s placebom, a to o 10 %, 4 % a 15 % v skupine
s nízkou, strednou a vysokou dávkou.
Signifikantné zlepšenie funkčného stupňa sa preukázalo len u pacientov v skupine s vysokou dávkou
v porovnaní s placebom. Pomery šancí v skupinách pacientov liečených sildenafilom v nízkej, strednej a vysokej dávke v porovnaní s placebom boli 0,6 (95 % CI: 0,18, 2,01), 2,25 (95 % CI: 0,75, 6,69)
a 4,52 (95 % CI: 1,56, 13,10).
Údaje z dlhodobej pokračovacej štúdie
Z 234 pediatrických pacientov liečených v krátkodobej, placebom kontrolovanej štúdii, vstúpilo 220
pacientov do dlhodobej pokračovacej štúdie. Pacienti, ktorým bolo v krátkodobej štúdii podávené placebo, boli náhodne zaradení do liečby sildenafilom. Pacienti s hmotnosťou ≤ 20 kg boli zaradení do
skupín so strednou alebo vysokou dávkou (1:1) a pacienti s hmotnosťou > 20 kg boli zaradení do
skupín s nízkou, strednou alebo vysokou dávkou (1:1:1). Z celkového počtu 229 pacientov, ktorí dostávali sildenafil, bolo v skupinách s nízkou, strednou a vysokou dávkou 55, 74 a 100 pacientov. Počas krátkodobej a dlhodobej štúdie sa celkové trvanie liečby od začiatku dvojitého zaslepenia
u jednotlivých pacientov pohybovalo v rozmedzí 3 až 3 129 dní. V liečebnej skupine so sildenafilom bolo stredné trvanie liečby 1 696 dní (s výnimkou 5 pacientov, ktorí v dvojito zaslepenej štúdii
dostávali placebo a neboli liečení v dlhodobej pokračovacej štúdii).
Odhady prežívania po 3 rokoch pomocou Kaplanových-Meierových kriviek u pacientov s hmotnosťou
> 20 kg na začiatku sledovania boli 94 %, 93 % a 85 % v skupinách s nízkou, strednou a vysokou dávkou; u pacientov s hmotnosťou ≤ 20 kg na začiatku sledovania boli odhady prežívania v skupinách
so strednou a vysokou dávkou 94 % a 93 % (pozri časti 4.4 a 4.8).
Počas štúdie bolo hlásených celkovo 42 úmrtí, či už v období počas liečby alebo hlásených v rámci ďalšieho sledovania prežívania. K 37 úmrtiam došlo pred rozhodnutím Výboru pre monitorovanie údajov postupne redukovať dávkovanie u pacientov na nižšie dávky na základe pozorovanej nerovnováhy úmrtnosti pri zvýšených dávkach sildenafilu. Z týchto 37 úmrtí bol počet úmrtí (%)
v skupinách s nízkou, strednou a vysokou dávkou 5/55 (9,1 %), 10/74 (13,5 %) a 22/100 (22 %).
Následne bolo hlásených ďalších 5 úmrtí. Príčiny úmrtí boli v súvislosti s PAH. U pediatrických pacientov s PAH sa nesmú používať vyššie než odporúčané dávky (pozri časti 4.2, a 4.4).
Spotreba kyslíka na vrchole záťaže (VO2) bola hodnotená 1 rok po začatí placebom kontrolovanej štúdie. Z pacientov liečených sildenafilom, ktorí boli v rámci svojho stupňa vývoja schopní vykonať spiroergometrické vyšetrenie CPET, 59/114 pacientov (52 %) nepreukázalo žiadne zhoršenie vrcholovej VO2 oproti začiatku liečby sildenafilom. Podobne 191 z 229 pacientov (83 %), ktorí užívali sildenafil, si podľa posúdenia po 1 roku buď uchovalo, alebo zlepšilo svoj funkčný stupeň podľa
WHO.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre Revatio
s ohľadom na novorodencov s pľúcnou artériovou hypertenziou (pozri časť 4.2 pre informáciu o pediatrickom použití)
5.2 Farmakokinetické vlastnosti
Absorpcia
Sildenafil sa rýchlo vstrebáva. Maximálne plazmatické koncentrácie sa dosahujú do 30 – 120 minút
(v priemere 60 minút) po perorálnom užití lieku nalačno. Priemerná absolútna perorálna biologická dostupnosť je 41 % (v rozmedzí 25 – 63 %). Pri perorálnom podávaní sildenafilu trikrát denne sa
zvyšuje hodnota AUC a Cmax úmerne dávke v rozmedzí 20 – 40 mg. Pri perorálnom podávaní 80 mg sildenafilu trikrát denne sa pozorovalo väčšie ako dávkovo úmerné zvýšenie jeho plazmatických hladín. Biologická dostupnosť sildenafilu po perorálnom podaní 80 mg trikrát denne bola u pacientov s pľúcnou artériovou hypertenziou v priemerne o 43 % (90 % IS: 27 % - 60 %) vyššia v porovnaní
s nižšími dávkami.
Ak sa sildenafil užíva súčasne s jedlom, rýchlosť absorpcie sa zníži, pričom priemerné oneskorenie tmax je 60 minút a priemerné zníženie Cmax je 29 %, avšak rozsah absorpcie nie je významne ovplyvnený (AUC je znížená o 11 %).
Distribúcia
Priemerný distribučný objem v rovnovážnom stave (Vss) sildenafilu je 105 l, čo naznačuje distribúciu do tkanív. Pri perorálnom podávaní 20 mg sildenafilu trikrát denne sú jeho priemerné maximálne celkové plazmatické koncentrácie v rovnovážnom stave približne 113 ng/ml. Sildenafil a jeho hlavný cirkulujúci N-desmetylmetabolit sa viaže v 96 % na plazmatické bielkoviny. Väzba na bielkoviny nie je závislá od celkových koncentrácií lieku.
Biotransformácia
Sildenafil je metabolizovaný predovšetkým hepatálnymi mikrozomálnymi izoenzýmami CYP3A4 (hlavná metabolická cesta) a CYP2C9 (vedľajšia metabolická cesta). Hlavný cirkulujúci metabolit
sildenafilu je výsledkom N-demetylácie sildenafilu. Tento metabolit má profil selektivity pre
fosfodiesterázu podobný sildenafilu a účinnosť in vitro na PDE5 približne 50 % v porovnaní
s materským liečivom. N-desmetylmetabolit je ďalej metabolizovaný a terminálny polčas je približne
4 h. U pacientov s pľúcnou artériovou hypertenziou plazmatické koncentrácie N-desmetylmetabolitu predstavujú približne 72 % koncentrácie sildenafilu pri podávaní 20 mg trikrát denne (čo sa premieta
do 36 % podielu na farmakologickom účinku sildenafilu). Následné ovplyvnenie účinnosti nie je
známe.
Eliminácia
Celkový telový klírens sildenafilu je 41 l/h a terminálny fázový polčas 3 – 5 h. Tak po perorálnom, ako aj po intravenóznom podaní sa sildenafil vylučuje vo forme metabolitov predovšetkým do stolice
(približne 80 % podanej perorálnej dávky) a v menšej miere do moču (približne 13 % podanej perorálnej dávky).
Farmakokinetika u osobitných skupín pacientov
Staršípacienti
U zdravých starších dobrovoľníkov (65-ročných a starších) bol znížený klírens sildenafilu, čo viedlo
k zvýšeniu plazmatických koncentrácií sildenafilu a aktívneho N-desmetylmetabolitu o približne 90 % v porovnaní s hodnotami u mladších zdravých dobrovoľníkov (18 – 45-ročných). Vzhľadom na rozdiely vo väzbe na plazmatické bielkoviny, ktoré sú podmienené vekom, bolo zodpovedajúce zvýšenie plazmatických koncentrácií voľného sildenafilu približne 40 %.
Renálnainsuficiencia
U dobrovoľníkov s ľahkým a stredne ťažkým poškodením funkcie obličiek (klírens kreatinínu =
30 – 80 ml/min) nebola zmenená farmakokinetika sildenafilu po podaní jednorazovej perorálnej dávky
50 mg. U dobrovoľníkov s ťažkým poškodením funkcie obličiek (klírens kreatinínu < 30 ml/min) bol klírens sildenafilu znížený a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia funkcie obličiek, sa AUC zvýšila o 100 % a Cmax o 88 %. Okrem toho hodnoty AUC a Cmax
N-desmetylmetabolitu sa signifikantne zvýšili o 200 % a o 79 % u jedincov s ťažkým poškodením
funkcie obličiek v porovnaní s jedincami s normálnou funkciou obličiek.
Hepatálnainsuficiencia
U dobrovoľníkov s ľahkou a stredne ťažkou cirhózou pečene (stupeň A a B podľa
Childovho-Pughovho skóre) sa klírens sildenafilu znížil a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia funkcie pečene, sa AUC zvýšila o 85 % a Cmax o 47 %. Navyše hodnoty AUC a Cmax pre N-desmetylmetabolit boli signifikantne vyššie o 154 %, resp. o 87 % u pacientov
s cirhózou pečene v porovnaní s jedincami s normálnymi pečeňovými funkciami. Farmakokinetika sildenafilu u pacientov s ťažkým poškodením funkcie pečene nebola študovaná.
Populačnáfarmakokinetika
U pacientov s pľúcnou artériovou hypertenziou boli priemerné rovnovážne koncentrácie pri sledovaných dávkach 20 – 80 mg trikrát denne o 20 – 50 % vyššie než v porovnávanej skupine
zdravých dobrovoľníkov. Cmax dosiahla dvojnásobnú hodnotu oproti zdravým dobrovoľníkom. Oba výsledky napovedajú, že pacienti s pľúcnou artériovou hypertenziou majú oproti zdravým dobrovoľníkom nižší klírens a/alebo vyššiu biologickú dostupnosť sildenafilu per os.
Pediatrickápopulácia
Z analýzy farmakokinetického profilu sildenafilu u pacientov zaradených do pediatrických klinických skúšaní sa telesná hmotnosť ukázala ako dobrý prediktor expozície lieku u detí. Odhadované hodnoty
polčasu koncentrácie sildenafilu v plazme sa pohybovali v rozmedzí 4,2 až 4,4 hodiny u pacientov
s hmotnosťou v rozmedzí od 10 do 70 kg a neukázali žiadne rozdiely, ktoré by sa javili ako klinicky relevantné. Odhadovaná Cmax po perorálnom podaní jednorazovej dávky sildenafilu 20 mg pre pacientov s hmotnosťou 70, 20 a 10 kg bola 49, 104 a 165 ng/ml. Odhadovaná Cmax po perorálnom podaní jednorazovej dávky sildenafilu 10 mg pre pacientov s hmotnosťou 70, 20 a 10 kg bola
24, 53 a 85 ng/ml. Odhadovaný Tmax bol približne 1 hodina a bol takmer nezávislý od telesnej hmotnosti.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
U mláďat potkanov, ktoré boli pre- a postnatálne liečené sildenafilom v dávke 60 mg/kg, sa pozoroval menší počet mláďat vo vrhu, nižšia hmotnosť mláďat v 1. deň a znížené prežívanie do 4. dňa pri expozíciách, ktoré boli približne 50-násobkom očakávanej expozície u človeka pri dávke 20 mg trikrát denne. Účinky v predklinických štúdiách sa pozorovali pri expozíciách považovaných za dostatočne vyššie, než je maximálna expozícia u ľudí, čo poukazuje na malý význam týchto zistení pre klinické použitie.
U zvierat neboli pri klinicky relevantných expozíciách pozorované žiadne nežiaduce reakcie
s možným významom pre klinické použitie, ktoré by neboli tiež pozorované v klinických štúdiách.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety: Mikrokryštalická celulóza Hydrogénfosforečnan vápenatý (bezvodý) Sodná soľ kroskarmelózy Magnéziumstearát
Filmová vrstva: Hypromelóza
Oxid titaničitý (E 171) Monohydrát laktózy
Triacetín
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
5 rokov
6.4 Špeciálne upozornenia na uchovávanieUchovávajte pri teplote neprevyšujúcej 30 °C. Uchovávajte v pôvodnom balení na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah baleniaPVC/hliníkové blistre s 90 tabletami
Veľkosť balenia 90 tabliet v papierovej škatuľke.
90 x 1 tableta v PVC/hliníkových perforovaných blistroch umožňujúcich oddelenie jednotlivej dávky.
PVC/hliníkové blistre s 300 tabletami.
Veľkosť balenia 300 tabliet v papierovej škatuľke.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomŽiadne zvláštne požiadavky na likvidáciu
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/05/318/001
EU/1/05/318/004
EU/1/05/318/005
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. október 2005
Dátum posledného predĺženia: 23. september 2010
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Revatio 0,8 mg/ml injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každý ml roztoku obsahuje 0,8 mg sildenafilu (vo forme citrátu). Každá 20 ml injekčná liekovka obsahuje 12,5 ml roztoku (10 mg sildenafilu, vo forme citrátu).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok
Číry, bezfarebný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Revatio injekčný roztok je určený na liečbu dospelých pacientov (≥ 18 rokov) s pľúcnou artériovou hypertenziou, ktorým bolo predpísané perorálne Revatio a ktorí sú dočasne neschopní užívať perorálne lieky, ale inak sú klinicky a hemodynamicky stabilizovaní.
Revatio (perorálne) je indikované na liečbu dospelých pacientov s pľúcnou artériovou hypertenziou, klasifikovanou ako funkčný stupeň II a III podľa WHO, za účelom zlepšenia tolerancie fyzickej záťaže. Účinnosť sa potvrdila pri primárnej pľúcnej hypertenzii a pľúcnej hypertenzii spojenej
s ochorením spojivového tkaniva.
4.2 Dávkovanie a spôsob podávania
Liečbu má začať a monitorovať len lekár, ktorý má skúsenosti s liečbou pľúcnej arteriálnej hypertenzie. V prípade zhoršenia klinického stavu napriek liečbe Revatiom, treba zvážiť alternatívne možnosti liečby.
Revatio injekčný roztok sa má podávať pacientom, ktorým už bolo predpísané perorálne Revatio, ako náhrada perorálneho podávania za podmienok, kedy nie sú dočasne schopní užívať Revatio perorálne.
Bezpečnosť a účinnosť dávok vyšších ako 12,5 ml (10 mg) TID neboli stanovené. Dávkovanie
Dospelí
Odporúčaná dávka je 10 mg (zodpovedajúca 12,5 ml) trikrát denne podávaná ako intravenózna bolusová injekcia (pozri časť 6.6).
Predpokladá sa, že dávka 10 mg Revatia injekčného roztoku zabezpečí expozíciu sildenafilu a jeho
N-desmetylmetabolitu a farmakologické účinky porovnateľné s účinkami 20 mg perorálnej dávky.
Pacientiužívajúciinélieky
Vo všeobecnosti sa má akákoľvek úprava dávky vykonávať až po dôkladnom zhodnotení prínosu a rizika. Úprava znížením dávky na 10 mg dvakrát denne sa má zvážiť, keď sa sildenafil podáva pacientom, ktorí už užívajú inhibítory CYP3A4, ako sú erytromycín alebo sachinavir. Úprava
znížením dávky na 10 mg jedenkrát denne sa odporúča v prípade súčasného podávania so silnejšími inhibítormi CYP3A4 klaritromycínom, telitromycínom a nefazodónom. Pre použitie sildenafilu s najsilnejšími inhibítormi CYP3A4, pozri časť 4.3. Úpravy dávky sildenafilu sa môžu vyžadovať, keď sa podáva s induktormi CYP3A4 (pozri časť 4.5).
Osobitné skupiny pacientov
Staršípacienti (≥65rokov)
Úprava dávky sa nevyžaduje u starších pacientov. Podľa nameraných vzdialeností pri 6-minútovej chôdzi by klinická účinnosť mohla byť menšia u starších pacientov.
Poškodeniefunkcieobličiek
Úprava začiatočnej dávky sa nevyžaduje u pacientov s poškodením funkcie obličiek, vrátane ťažkého
poškodenia funkcie obličiek (klírens kreatinínu < 30 ml/min). Úprava znížením dávky na 10 mg dvakrát denne sa má zvážiť po dôkladnom vyhodnotení prínosu a rizika, len ak liečba nie je dobre tolerovaná.
Poškodeniefunkciepečene
Úprava začiatočnej dávky sa nevyžaduje u pacientov s poškodením funkcie pečene (stupeň A a B podľa Childovho-Pughovho skóre). Úprava znížením dávky na 10 mg dvakrát denne sa má zvážiť po dôkladnom vyhodnotení prínosu a rizika, len ak liečba nie je dobre tolerovaná.
Revatio je kontraindikované u pacientov s ťažkým poškodením funkcie pečene (stupeň C podľa
Childovho-Pughovho skóre) (pozri časť 4.3).
Pediatrickápopulácia
Revatio injekčný roztok sa neodporúča používať u detí mladších ako 18 rokov kvôli nedostatočným údajom o bezpečnosti a účinnosti.
Prerušenie liečby
Obmedzené údaje naznačujú, že náhle prerušenie liečby perorálnym Revatiom nie je spojené
s následným zhoršením pľúcnej arteriálnej hypertenzie. Aby sa však predišlo možnému výskytu náhleho zhoršenia klinického stavu počas vynechania liečby, má sa zvážiť postupná redukcia dávok.
Počas prerušenia liečby sa odporúča intenzívne monitorovanie stavu.
Spôsob podávania
Revatio injekčný roztok je určený na intravenózne použitie ako bolusová injekcia. Pokyny na použitie, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na sildenafil alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Súčasné podávanie s donormi oxidu dusnatého (ako napríklad amylnitritom) alebo s nitrátmi v akejkoľvek forme v dôsledku hypotenzívneho účinku nitrátov (pozri časť 5.1).
Súbežné podanie PDE5 inhibítorov, vrátane sildenafilu, so stimulátormi guanylátcyklázy, akým je napr. riociguát, je kontraindikované, pretože môže viesť k symptomatickej hypotenzii (pozri časť 4.5).
Kombinácia s najsilnejšími z inhibítorov CYP3A4 (napr. ketokonazolom, itrakonazolom, ritonavirom) (pozri časť 4.5).
Pacienti, ktorí majú stratu videnia v jednom oku v dôsledku neartériovej prednej ischemickej neuropatie zrakového nervu (non-arteritic anterior ischaemic optic neuropathy, NAION) bez ohľadu na to, či táto príhoda súvisela alebo nesúvisela s predchádzajúcou expozíciou inhibítoru PDE5 (pozri časť 4.4).
Bezpečnosť sildenafilu sa nesledovala u nasledovných podskupín pacientov, a preto je jeho použitie u nich kontraindikované:
ťažké poškodenie funkcie pečene,
nedávno prekonaná cievna mozgová príhoda alebo infarkt myokardu, ťažká hypotenzia (tlak krvi < 90/50 mmHg) na začiatku liečby.
4.4 Osobitné upozornenia a opatrenia pri používaní
Nie sú dostupné žiadne klinické údaje o i.v. podaní sildenafilu klinicky alebo hemodynamicky nestabilným pacientom. Preto sa jeho použitie u týchto pacientov neodporúča.
Účinnosť Revatia u pacientov s ťažkou pľúcnou artériovou hypertenziou (funkčný stupeň IV) nebola potvrdená. Ak dôjde k zhoršeniu klinického stavu, má sa zvážiť použitie liečby odporúčanej pre ťažký stupeň ochorenia (napr. epoprostenol) (pozri časť 4.2).
Pomer prínosu/rizika liečby sildenafilom nebol stanovený u pacientov s funkčným stupňom I pľúcnej artériovej hypertenzie podľa WHO.
Štúdie so sildenafilom sa vykonali u foriem pľúcnej artériovej hypertenzie spojenej s primárnym (idiopatickým) ochorením spojivového tkaniva alebo s formami PAH spojenými s vrodenou chorobou srdca (pozri časť 5.1). Použitie sildenafilu u iných foriem PAH sa neodporúča.
Retinispigmentosa
Bezpečnosť sildenafilu sa neštudovala u pacientov s hereditárnymi degeneratívnymi ochoreniami retiny, ako sú retinitis pigmentosa (menšina z týchto pacientov má genetickú poruchu retinálnej fosfodiesterázy), a preto sa jeho použitie neodporúča.
Vazodilatačné pôsobenie
Pri predpisovaní sildenafilu má lekár dôkladne zvážiť, či pacient netrpí takým základným ochorením, ktorého priebeh by mohli mierne až stredne silné vazodilatačné účinky sildenafilu nepriaznivo
ovplyvniť, napríklad u pacientov s hypotenziou, pacientov s hypovolémiou, so závažnou obštrukciou výtokovej časti ľavej komory alebo autonómnou dysfunkciou (pozri časť 4.4).
Kardiovaskulárne rizikové faktory
Po uvedení sildenafilu určeného na erektilnú dysfunkciu u mužov na trh boli v časovej súvislosti
s užitím sildenafilu hlásené závažné kardiovaskulárne príhody vrátane infarktu myokardu, nestabilnej angíny pectoris, náhlej srdcovej smrti, komorovej arytmie, cerebrovaskulárnej hemorágie, tranzitórneho ischemického ataku, hypertenzie a hypotenzie. U väčšiny týchto pacientov, ale nie
u všetkých, boli prítomné už existujúce kardiovaskulárne rizikové faktory. Mnohé z týchto hlásených príhod vznikli počas sexuálneho styku alebo krátko po ňom a niekoľko z nich sa vyskytlo krátko po
užití sildenafilu, ale bez sexuálnej aktivity. Nie je možné určiť, či tieto príhody priamo súvisia s uvedenými alebo inými faktormi.
Priapizmus
Sildenafil sa má používať s opatrnosťou u pacientov s anatomickou deformáciou penisu (ako angulácia, kavernózna fibróza alebo Peyronieho choroba) alebo u pacientov s ochoreniami, ktoré
predisponujú k priapizmu (ako kosáčikovitá anémia, mnohonásobný myelóm alebo leukémia).
Po uvedení sildenafilu na trh boli hlásené predĺžené erekcie a priapizmus. V prípade, že erekcia trvá dlhšie ako 4 hodiny, má pacient ihneď vyhľadať lekársku pomoc. Ak priapizmus nie je liečený okamžite, môže dôjsť k poškodeniu tkaniva penisu a trvalej strate potencie (pozri časť 4.8).
Vazo-okluzívna kríza u pacientov s kosáčikovitou anémiou
Sildenafil sa nemá používať u pacientov so sekundárnou pľúcnou hypertenziou pri kosáčikovitej anémii. V klinickej štúdii bol počet vazookluzívnych kríz vyžadujúcich hospitalizáciu hlásený
častejšie u pacientov, ktorí užívali Revatio ako u tých, ktorí boli na placebe, čo viedlo k predčasnému
Udalosti súvisiace so zrakom
V súvislosti s užitím sildenafilu a ostatných inhibítorov PDE5 boli spontánne hlásené prípady porúch zraku. V súvislosti s užitím sildenafilu a ostatných inhibítorov PDE5 boli spontánne a v observačnej
štúdii hlásené prípady zriedkavého ochorenia, neartériovej prednej ischemickej neuropatie zrakového
nervu (pozri časť 4.8).
V prípade akéjkoľvek náhlej poruchy videnia sa má liečba ukončiť a zvážiť alternatívna liečba (pozri časť 4.3).
Alfa-blokátory
Opatrnosť sa odporúča, keď sa sildenafil podáva pacientom liečeným alfablokátorom, keďže súčasné podávanie môže viesť k symptomatickej hypotenzii u vnímavých jedincov (pozri časť 4.5). Pacienti
majú byť hemodynamicky stabilizovaní liečbou alfablokátormi ešte pred začatím liečby sildenafilom, aby sa minimalizovalo riziko vzniku posturálnej hypotenzie. Lekár má informovať pacientov, ako
postupovať pri objavení sa príznakov posturálnej hypotenzie.
Poruchy krvácania
Štúdie in vitro s humánnymi krvnými doštičkami naznačujú, že sildenafil potencuje antiagregačný účinok nitroprusidu sodného. Nie sú žiadne údaje o bezpečnosti podania sildenafilu pacientom
s poruchami krvácania alebo s aktívnym peptickým vredom. Preto sa má sildenafil u týchto pacientov podávať iba po dôslednom zvážení prínosu a rizika liečby.
Antagonisty vitamínu K
U pacientov s pľúcnou artériovou hypertenziou môže existovať potenciálne zvýšené riziko krvácania, keď sa liečba sildenafilom zavedie u pacientov, ktorí už užívajú antagonistu vitamínu K, zvlášť
u pacientov s pľúcnou artériovou hypertenziou ako sekundárnym ochorením pri ochorení spojivového
tkaniva.
Venookluzívne ochorenie
Nie sú dostupné údaje o použití sildenafilu u pacientov s pľúcnou artériovou hypertenziou spojenou s pľúcnym venookluzívnym ochorením. Avšak boli hlásené prípady život ohrozujúceho edému pľúc pri liečbe vazodilatátormi (hlavne prostacyklínom) u týchto pacientov. Z toho vyplýva, že ak sa
u pacientov s pľúcnou hypertenziou, ktorým bol podaný sildenafil, objavia znaky edému pľúc, má sa zvážiť možnosť súčasného výskytu venookluzívnej choroby.
Použitie sildenafilu s bosentanom
Účinnosť sildenafilu u pacientov liečených bosentanom nebola presvedčivo preukázaná (pozri časti
4.5 a 5.1).
Súčasné používanie s inými inhibítormi PDE5
Bezpečnosť a účinnosť sildenafilu pri súčasnom podaní s inými PDE5 inhibítormi, vrátane Viagry, u pacientov s PAH nebola študovaná a takéto súčasné používanie sa neodporúča (pozri časť 4.5).
4.5 Liekové a iné interakcie
Keďže nebolo inak vopred špecifikované, liekové interakčné štúdie s perorálnym sildenafilom sa vykonali u zdravých dospelých jedincov mužského pohlavia. Tieto výsledky sa vzťahujú aj na iné populácie a cesty podania.
Účinky iných liekov na intravenózny sildenafil
Predpoklady založené na farmakokinetickom modeli naznačujú, že liekové interakcie s inhibítormi
CYP3A4 by sa mali vyskytovať zriedkavejšie ako interakcie pozorované po podaní perorálneho sildenafilu. U intravenózneho sildenafilu sa v porovnaní s perorálnym sildenafilom očakáva znížená závažnosť interakcie, pretože interakcie v prípade perorálneho sildenafilu sú, aspoň čiastočne, podmienené jeho prvým prechodom pečeňou.
Účinky iných liekov na perorálny sildenafil
Štúdiein
vitro
Sildenafil je v rozhodujúcej miere metabolizovaný izoformami 3A4 (hlavná metabolická cesta) a 2C9 (vedľajšia metabolická cesta) cytochrómu P450 (CYP). Inhibítory týchto izoenzýmov môžu preto znížiť klírens sildenafilu, zatiaľ čo induktory týchto izoenzýmov môžu klírens sildenafilu zvýšiť. Tieto odporúčania, pozri časti 4.2 a 4.3.
Štúdiein vivo
Hodnotilo sa súčasné podávanie perorálneho sildenafilu a intravenózneho epoprostenolu (pozri časti
4.8 a 5.1).
V kontrolovaných klinických štúdiách nebola sledovaná účinnosť a bezpečnosť sildenafilu súčasne podávaného s inými terapeutickými modalitami na pľúcnu artériovú hypertenziu (napr. ambrisentanom, iloprostom). Preto sa v prípade súčasného podávania odporúča opatrnosť.
U pacientov s pľúcnou artériovou hypertenziou sa neskúmala bezpečnosť a účinnosť sildenafilu, ak sa podávalo súčasne s PDE5 inhibítormi (pozri časť 4.4)..
Populačná farmakokinetická analýza údajov z klinických štúdií týkajúcich sa pľúcnej artériovej hypertenzie ukázala pokles klírensu sildenafilu a/alebo zvýšenie jeho biologickej dostupnosti po perorálnom podaní, ak sa podával súčasne so substrátmi CYP3A4, resp. súčasne s kombináciou substrátov CYP3A4 a betablokátorov. Toto boli jediné faktory, ktoré štatisticky signifikantne ovplyvnili farmakokinetiku perorálneho sildenafilu u pacientov s pľúcnou artériovou hypertenziou. Expozícia sildenafilu u pacientov užívajúcich substráty CYP3A4 bola vyššia o 43 % a u pacientov užívajúcich kombináciu substrátov CYP3A4 a betablokátorov bola vyššia o 66 %, v porovnaní
s pacientmi, ktorí neužívali tieto skupiny liekov. Expozícia sildenafilu bola 5-krát vyššia pri perorálnej dávke 80 mg trikrát denne v porovnaní s expozíciou pri perorálnej dávke 20 mg trikrát denne. Toto
rozpätie koncentrácií zahŕňa zvýšenie expozície sildenafilu pozorované v špecificky zostavených
interakčných štúdiách s inhibítormi CYP3A4 (s výnimkou najsilnejších inhibítorov CYP3A4, napr. ketokonazolu, itrakonazolu, ritonaviru).
Zdá sa , že induktory CYP3A4 majú podstatný vplyv na farmakokinetiku perorálneho sildenafilu u pacientov s pľúcnou artériovou hypertenziou, čo bolo potvrdené aj in vivo štúdiou sledujúcou interakcie s induktorom CYP3A4 bosentanom. Súčasné podávanie bosentanu (stredne silný induktor CYP3A4, CYP2C9 a pravdepodobne CYP2C19) 125 mg dvakrát denne s perorálnym sildenafilom
80 mg trikrát denne (v rovnovážnom stave), ktoré sa súčasne podávali zdravým dobrovoľníkom počas
6 dní, viedlo k poklesu AUC sildenafilu o 63 %.
Populačná farmakokinetická analýza údajov týkajúcich sa sildenafilu z klinických štúdií s dospelými pacientmi s PAH vrátane 12-týždňovej štúdie na posúdenie účinnosti a bezpečnosti sildenafilu podávaného perorálne v dávke 20 mg trikrát denne po pridaní k stabilnej dávke bosentanu (62,5 mg –
125 mg dvakrát denne) ukázala pokles expozície sildenafilu pri súčasnom podávaní s bosentanom podobný tomu, ktorý sa pozoroval u zdravých dobrovoľníkov (pozri časti 4.4 a 5.1).
Účinnosť sildenafilu musí byť presne monitorovaná u pacientov, ktorí súčasne užívajú silné induktory
CYP3A4, ako sú karbamazepín, fenytoín, fenobarbital, ľubovník a rifampicín.
Súčasné podávanie inhibítora HIV proteáz, ritonaviru, ktorý je veľmi silný inhibítor cytochrómu P450, v rovnovážnom stave (500 mg dvakrát denne) a perorálneho sildenafilu (100 mg jednorazová dávka) viedlo k 300 % (4-násobnému) vzostupu Cmax sildenafilu a k 1 000 % (11-násobnému) vzostupu AUC sildenafilu v plazme. Po uplynutí 24 hodín boli plazmatické koncentrácie sildenafilu ešte stále
približne 200 ng/ml, v porovnaní s približne 5 ng/ml, ak bol sildenafil podaný samostatne. Tieto údaje sú v súlade s výraznými účinkami ritonaviru na široké spektrum substrátov P450. Vzhľadom na tieto farmakokinetické výsledky je súčasné podávanie sildenafilu a ritonaviru kontraindikované u pacientov s pľúcnou artériovou hypertenziou (pozri časť 4.3).
Súčasné podávanie inhibítora HIV proteáz, sachinaviru, inhibítora CYP3A4 v rovnovážnom stave (1 200 mg trikrát denne) a perorálneho sildenafilu (100 mg jednorazová dávka) viedlo k 140 % vzostupu Cmax sildenafilu a k 210 % vzostupu AUC sildenafilu. Sildenafil neovplyvňuje farmakokinetiku sachinaviru. Odporúčania pre dávkovanie, pozri časť 4.2.
Ak sa perorálny sildenafil podával jednorazovo v dávke 100 mg spolu s erytromycínom, stredne silným inhibítorom CYP3A4, v rovnovážnom stave (500 mg dvakrát denne 5 dní), zaznamenal sa
182 % vzostup systémovej expozície sildenafilu (AUC). Odporúčania pre dávkovanie, pozri časť 4.2.
U zdravých dobrovoľníkov mužského pohlavia sa nedokázal vplyv azitromycínu (500 mg denne počas
3 dní) na AUC, Cmax, tmax, eliminačnú rýchlostnú konštantu alebo následne na polčas perorálneho sildenafilu alebo jeho hlavného cirkulujúceho metabolitu. Nevyžaduje sa úprava dávky. Pri súčasnom podávaní perorálneho sildenafilu (50 mg) a cimetidínu (800 mg), ktorý je inhibítorom cytochrómu P450 a nešpecifickým inhibítorom CYP3A4, zdravým dobrovoľníkom sa zaznamenal 56 % vzostup plazmatickej koncentrácie sildenafilu. Nevyžaduje sa úprava dávky.
Očakáva sa, že najsilnejšie z inhibítorov CYP3A4, ako sú ketokonazol a itrakonazol, by mali mať podobné účinky ako ritonavir (pozri časť 4.3). V prípade inhibítorov CYP3A4, ako sú klaritromycín, telitromycín a nefazodón, sa očakáva účinok s intenzitou medzi ritonavirom a inhibítormi CYP3A4, ako sú sachinavir alebo erytromycín, predpokladá sa sedemnásobné zvýšenie expozície. Preto sa pri použití inhibítorov CYP3A4 odporúča úprava dávkovania (pozri časť 4.2).
Populačná farmakokinetická analýza u pacientov s pľúcnou artériovou hypertenziou užívajúcich perorálny sildenafil ukázala, že súčasné podávanie betablokátorov v kombinácii so substrátmi CYP3A4 môže vyústiť do dodatočného zvýšenia expozície sildenafilu v porovnaní s podávaním samotných substrátov CYP3A4.
Grapefruitová šťava je slabým inhibítorom metabolizmu prostredníctvom CYP3A4 v črevnej stene
a môže vyvolať mierny vzostup plazmatických hladín perorálneho sildenafilu. Nevyžaduje sa úprava dávky, ale súčasné užívanie sildenafilu s grapefruitovou šťavou sa neodporúča.
Biologická dostupnosť perorálneho sildenafilu nebola ovplyvnená podaním jednorazových dávok antacíd (hydroxidu horečnatého/hydroxidu hlinitého).
Súčasné podávanie s perorálnymi kontraceptívami (etinylestradiol 30 μg a levonorgestrel 150 μg)
neovplyvnilo farmakokinetiku perorálneho sildenafilu.
Nikorandil je hybrid aktivátora draslíkových kanálov a nitrátu. Vzhľadom na nitrátovú zložku má potenciál pre závažné interakcie so sildenafilom (pozri časť 4.3).
Účinky perorálneho sildenafilu na iné lieky
Štúdieinvitro
Sildenafil je slabým inhibítorom (IC50 > 150 μmol/l) izoforiem 1A2, 2C9, 2C19, 2D6, 2E1 a 3A4
cytochrómu P450.
Nie sú žiadne údaje o interakcii sildenafilu s nešpecifickými inhibítormi fosfodiesterázy, ako sú teofylín alebo dipyridamol.
Štúdieinvivo
Nezaznamenali sa žiadne signifikantné interakcie perorálneho sildenafilu (50 mg) s tolbutamidom
(250 mg) alebo s warfarínom (40 mg), liekmi, ktoré sú metabolizované prostredníctvom CYP2C9.
Perorálny sildenafil nemal významný vplyv na expozíciu atorvastatínu (AUC zvýšené o 11 %), čo naznačuje, že sildenafil nemá klinicky významný účinok na CYP3A4.
Medzi sildenafilom (v jednotlivej perorálnej dávke 100 mg) a acenokumarolom sa nepozorovali žiadne interakcie.
Perorálny sildenafil (50 mg) nepotencioval predĺženie času krvácania zapríčineného kyselinou acetylsalicylovou (150 mg).
Perorálny sildenafil (50 mg) nepotencioval hypotenzívny účinok alkoholu u zdravých dobrovoľníkov, ktorí mali priemernú maximálnu koncentráciu alkoholu v krvi 80 mg/dl.
V štúdii so zdravými dobrovoľníkmi vyvolal perorálny sildenafil v rovnovážnom stave (80 mg trikrát denne) zvýšenie AUC bosentanu o 50 % (125 mg dvakrát denne).
Populačná farmakokinetická analýza údajov zo štúdie s dospelými pacientmi s PAH na základnej liečbe bosentanom (62,5 mg – 125 mg dvakrát denne) ukázala zvýšenie (20 % (95 % CI: 9,8 – 30,8) AUC bosentanu pri súčasnom podávaní sildenafilu v rovnovážnom stave (20 mg trikrát denne)
v menšom rozsahu ako to, ktoré sa pozorovalo u zdravých dobrovoľníkov, ktorým bol podávaný bosentan súčasne so sildenafilom v dávke 80 mg trikrát denne (pozri časti 4.4 a 5.1)
V špecifickej interakčnej štúdii u pacientov s hypertenziou, ktorí súčasne užívali amplodipín
s perorálnym sildenafilom (100 mg), sa zaznamenalo ďalšie zníženie systolického tlaku krvi v ľahu o 8 mmHg. Zodpovedajúce ďalšie zníženie diastolického tlaku krvi v ľahu bolo o 7 mmHg. Toto
ďalšie zníženie tlaku krvi malo podobný rozsah, ako keď sa sildenafil podával zdravým dobrovoľníkom samostatne.
V troch špecifických liekových interakčných štúdiách sa pacientom s benígnou hyperpláziou prostaty
(BPH) stabilizovanou na liečbe doxazosínom súbežne podával alfablokátor doxazosín (4 mg, resp.
8 mg) a perorálny sildenafil (25 mg, 50 mg alebo 100 mg). V týchto štúdiách sa u sledovanej populácie pozoroval dodatočný priemerný pokles systolického a diastolického tlaku krvi v ľahu
o 7/7 mmHg, 9/5 mmHg, resp. 8/4 mmHg, ako aj dodatočný priemerný pokles tlaku krvi v stoji o 6/6
mmHg, 11/4 mmHg, resp. 4/5 mmHg. Keď sa sildenafil a doxazosín súbežne podávali pacientom stabilizovaným na liečbe doxazosínom, hlásenia o výskyte symptomatickej posturálnej hypotenzie
u pacientov boli ojedinelé. Tieto zahŕňali závraty a stratu rovnováhy, ale nie synkopu. Súčasné
podávanie sildenafilu pacientom užívajúcim alfablokátory môže viesť u vnímavých jedincov k symptomatickej hypotenzii (pozri časť 4.4).
Sildenafil (100 mg v jednorazovej perorálnej dávke) neovplyvnil farmakokinetiku inhibítora HIV
proteáz v rovnovážnom stave, sachinaviru, ktorý je substrátom/inhibítorom CYP3A4.
V súlade so známym účinkom sildenafilu na metabolickú cestu oxid dusnatý/cGMP (pozri časť 5.1) sa preukázalo, že sildenafil potencuje hypotenzívny účinok nitrátov, a preto je jeho súčasné podanie
s donormi oxidu dusnatého alebo nitrátmi v akejkoľvek forme kontraindikované (pozri časť 4.3).
Riociguát
Predklinické štúdie ukázali aditívny systémový účinok znižujúci krvný tlak, keď sa inhibítory PDE5 podávali súčasne s riociguátom. Klinické štúdie preukázali, že riociguát zosilňuje hypotenzívne účinky inhibítorov PDE5. V skúšanej populácii nebol nájdený žiadny dôkaz o priaznivom klinickom účinku spomínanej kombinácie. Súčasné užívanie riociguátu s PDE5 inhibítormi, vrátane sildenafilu, je kontraindikované (pozri časť 4.3).
Perorálny sildenafil nemal klinicky významný vplyv na plazmatické hladiny perorálnych kontraceptív
(etinylestradiol 30 μg a levonorgestrel 150 μg).
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku a antikoncepcia u mužov a žien
Vzhľadom na nedostatok údajov o účinnosti Revatia u gravidných žien sa Revatio neodporúča užívať u žien v reprodukčnom veku, pokiaľ nepoužívajú súčasne vhodnú antikoncepciu.
Gravidita
Nie sú k dispozícii žiadne údaje o použití sildenafilu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu a embryonálny/fetálny vývoj. Štúdie
na zvieratách preukázali toxicitu na postnatálny vývoj (pozri časť 5.3).
Vzhľadom na nedostatok údajov sa má Revatio používať u gravidných žien iba v nevyhnutných prípadoch.
Dojčenie
Nie sú k dispozícii žiadne adekvátne a dobre kontrolované štúdie u dojčiacich žien. Údaje od jednej dojčiacej ženy naznačujú, že sildenafil a jeho aktívny metabolit, N-demetylsildenafil, sa vo veľmi malých množstvách vylučujú do materského mlieka. Nie sú dostupné žiadne klinické údaje týkajúce sa nežiaducich účinkov u dojčených detí, ale nepredpokladá sa, že by požité množstvá spôsobovali
nejaké nežiaduce účinky. Predpisujúci lekári majú starostlivo zvážiť klinickú potrebu matky užívať sildenafil a akékoľvek potenciálne nežiaduce účinky u dojčeného dieťaťa.
Fertilita
Predklinické údaje získané na základe obvyklých farmakologických štúdií fertility neodhalili žiadne osobitné riziko pre ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Revatio má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Keďže závrat a zmenené videnie boli hlásené v klinických štúdiách so sildenafilom, pacienti predtým, ako budú viesť vozidlá a obsluhovať stroje, majú poznať, ako reagujú na podanie Revatia.
4.8 Nežiaduce účinky
Nežiaduce reakcie vyplývajúce z použitia intravenózneho Revatia sú podobné tým, ktoré sú spojené s používaním perorálneho Revatia. Pretože existujú obmedzené údaje týkajúce sa použitia intravenózneho Revatia a keďže farmakokinetické modely predpokladajú, že liekové formy 20 mg perorálne a 10 mg intravenózne budú mať za následok podobné plazmatické hladiny, informácia
o bezpečnosti intravenózneho Revatia sa opiera o skúsenosti s perorálnym Revatiom.
Intravenózne podanie
Predpokladá sa, že Revatio injekčný roztok v dávke 10 mg má za následok celkový účinok, ktorý zodpovedá spoločnému pôsobeniu voľného sildenafilu a jeho N-desmetylmetabolitu a ich kombinovaný farmakologický účinok je porovnateľný s účinkom 20 mg perorálnej dávky.
Štúdia A1481262 bola otvorená štúdia s jedným centrom s jednorazovou dávkou, ktorá posudzovala bezpečnosť, tolerabilitu a farmakokinetiku jednorazovej intravenóznej dávky sildenafilu (10 mg) podanej vo forme bolusovej injekcie pacientom s pľúcnou artériovou hypertenziou (PAH), ktorí už boli stabilizovaní a užívali 3-krát denne perorálne Revatio 20 mg.
Celkovo 10 pacientov s PAH bolo zaradených do štúdie a ukončilo ju. Priemerné zmeny systolického a diastolického tlaku krvi závislé od zmeny polohy a od času boli malé (< 10 mmHg) a vrátili sa na pôvodné hodnoty po 2 hodinách. S týmito zmenami neboli spojené žiadne symptómy hypotenzie. Priemerné zmeny vo frekvencii srdca boli klinicky nevýznamné. U 2 pacientov boli zaznamenané spolu 3 nežiaduce reakcie (začervenanie, flatulencia a návaly tepla). Jedna závažná nežiaduca reakcia

bola hlásená u pacienta so závažnou ischemickou kardiomyopatiou, u ktorého došlo k fibrilácii komôr a úmrtiu 6 dní po štúdii; Toto bolo posúdené ako nesúvisiace so skúmaným liekom.
Perorálne podávanieV pivotnej, placebom kontrolovanej štúdii s Revatiom pri pľúcnej artériovej hypertenzii bolo
207 pacientov randomizovaných a liečených dávkami 20 mg, 40 mg alebo 80 mg perorálneho Revatia TID a 70 pacientov bolo randomizovaných na placebo. Liečba trvala 12 týždňov. Celková frekvencia prerušenia liečby u pacientov liečených sildenafilom v dávkach 20 mg, 40 mg a 80 mg TID bola
2,9 %, 3,0 % a 8,5 % v uvedenom poradí, v porovnaní s 2,9 % u placeba. Z 277 pacientov liečených v pivotnej štúdii prešlo do dlhodobej pokračovacej štúdie 259 pacientov. V nej sa podávali dávky do
80 mg trikrát denne (4-krát vyššie ako odporúčaná dávka 20 mg trikrát denne) a po 3 rokoch 87 % zo
183 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V placebom kontrolovanej štúdii s Revatiom ako doplnkom k intravenóznemu epoprostenolu
u pľúcnej artériovej hypertenzie bolo celkovo 134 pacientov liečených perorálnym Revatiom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg a potom na 80 mg trikrát denne podľa
znášanlivosti) a epoprostenolom a 131 pacientov bolo liečených placebom a epoprostenolom. Dĺžka
liečby bola 16 týždňov. Celková frekvencia prerušenia liečby z dôvodu nežiaducich účinkov
u pacientov liečených sildenafilom/epoprostenolom bola 5,2 % v porovnaní s 10,7 % u pacientov liečených placebom/epoprostenolom. Nedávno hlásenými nežiaducimi liekovými reakciami, ktoré sa vyskytli oveľa častejšie v skupine so sildenafilom/epoprostenolom, bola okulárna hyperémia, rozmazané videnie, upchatý nos, nočné potenie, bolesť chrbta a sucho v ústach. Známe nežiaduce účinky bolesť hlavy, návaly tepla, bolesti v končatinách a edém boli zaznamenané s vyššou frekvenciou u pacientov liečených sildenafilom/epoprostenolom v porovnaní s pacientmi liečenými placebom/epoprostenolom. Z pacientov, ktorí ukončili úvodnú liečbu, prešlo do dlhodobej pokračovacej štúdie 242 pacientov. V nej sa podávali dávky do 80 mg TID a po 3 rokoch 68 % zo
133 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V dvoch placebom kontrolovaných štúdiách s perorálnym Revatiom boli nežiaduce účinky obyčajne mierne až stredne závažné. Najčastejšie hlásenými nežiaducimi účinkami, ktoré sa vyskytli (³ 10 %) u Revatia v porovnaní s placebom boli bolesti hlavy, návaly tepla, dyspepsia, hnačka a bolesť
v končatinách.
Zoznam nežiaducich reakcií usporiadaný do tabuľkyNežiaduce reakcie, ktoré sa vyskytli u > 1 % pacientov liečených Revatiom a boli častejšie (> 1 %
rozdiel) u Revatia v pivotnej štúdii alebo v spoločnom súbore údajov z oboch placebom kontrolovaných štúdií pri liečbe pľúcnej artériovej hypertenzie Revatiom v perorálnych dávkach
20, 40 alebo 80 mg TID, sú uvedené v tabuľke nižšie podľa triedy a skupín frekvencií (veľmi časté
(³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100) a neznáme (z dostupných údajov)). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Hlásenia po uvedení lieku na trh sú uvedené kurzívou.
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakciaInfekcie a nákazyČasté celulitída, chrípka, zápal priedušiek,
zápal prinosových dutín, rinitída, gastroenteritída
Poruchy krvi a lymfatického systémuČasté anémia
Poruchy metabolizmu a výživyČasté retencia tekutín
Psychické poruchy
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakcia
Časté nespavosť, úzkosť
Poruchy nervového systému
Veľmi časté bolesť hlavy
Časté migréna, tremor, parestézie, pocit pálenia, hypestézia
Poruchy oka
Časté krvácanie do sietnice, poškodenie zraku, rozmazané videnie, fotofóbia, chromatopsia, cyanopsia, podráždenie
očí, okulárna hyperémia
Neznáme nearteritická predná ischemická neuropatia zrakového nervu (NAION)
*, oklúzia ciev sietnice*, poruchy
v zornom poli*
Menej časté zníženie zrakovej ostrosti, diplopia, neprirodzené pocity v oku
Poruchy ucha a labyrintu
Časté závrat
Neznáme náhla strata sluchu
Poruchy ciev
Veľmi časté návaly tepla
Neznáme hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté epistaxa, kašeľ, upchatý nos
Poruchy gastrointestinálneho traktu
Veľmi časté hnačka, dyspepsia
Časté gastritída, gastroezofageálna refluxná choroba, hemoroidy, nafúknutie brucha, sucho v ústach
Poruchy kože a podkožného tkaniva
Časté
Neznáme
alopécia, erytém, nočné potenie
vyrážka
 Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Veľmi časté bolesť v končatinách
Časté myalgia, bolesť chrbta
Poruchy obličiek a močových ciestMenej časté hematúria
Poruchy reprodukčného systému a prsníkovMenej časté penilné krvácanie, hematospermia,
gynekomastia
Neznáme
priapizmus, zvýšená erekciaCelkové poruchy a reakcie v mieste podaniaČasté horúčka
* Tieto nežiaduce účinky/reakcie boli hlásené s neznámou frekvenciou u pacientov užívajúcich sildenafil na liečbu erektilnej dysfunkcie u mužov (MED).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV štúdiách so zdravými dobrovoľníkmi boli po podaní jednorazových perorálnych dávok do 800 mg nežiaduce reakcie podobné ako pri podaní nižších dávok, ale vyskytovali sa častejšie a boli závažnejšie. Jednorazové perorálne dávky 200 mg viedli k vyššiemu výskytu nežiaducich reakcií (bolesť hlavy, návaly, závrat, dyspepsia, nazálna kongescia a zmena videnia).
V prípade predávkovania sa majú podľa potreby zaviesť štandardné podporné opatrenia. Keďže sildenafil je pevne viazaný na bielkoviny plazmy a neeliminuje sa močom, nie je pravdepodobné, že by renálna dialýza mala urýchliť klírens sildenafilu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Urologiká, lieky používané pri poruchách erekcie, ATC kód: G04BE03
MechanizmusúčinkuSildenafil je účinný a selektívny inhibítor fosfodiesterázy typu 5 (PDE5), špecifickej pre cyklický guanozínmonofosfát (cGMP). Je to enzým, ktorý zodpovedá za degradáciu cGMP. Okrem prítomnosti tohto enzýmu v kavernóznom telese penisu sa PDE5 nachádza aj v cievnom riečisku pľúc. Sildenafil preto zvyšuje cGMP v bunkách hladkých svalov pľúcnych ciev, čo vedie k ich relaxácii. U pacientov
s pľúcnou artériovou hypertenziou to môže viesť k vazodilatácii v pľúcnom riečisku, a v menšej miere aj ku vazodilatácii v systémovom obehu.
Farmakodynamický účinokŠtúdie
in vitro preukázali, že sildenafil je selektívny pre PDE5. Jeho účinok je výraznejší na PDE5 ako na ostatné známe fosfodiesterázy. Sildenafil je 10-krát selektívnejší pre PDE5 ako pre PDE6, ktorá sa podieľa na fototransdukcii v retine. Má 80-krát vyššiu selektivitu pre PDE5 než pre PDE1 a viac ako
700-krát vyššiu selektivitu pre PDE5 než pre PDE2, 3, 4, 7, 8, 9, 10 a 11. Obzvlášť, sildenafil má
4 000-krát vyššiu selektivitu pre PDE5 ako pre PDE3, cAMP špecifickú izoformu fosfodiesterázy, ktorá sa podieľa na kontrole kontraktility srdcového svalu.
Sildenafil spôsobuje mierny a prechodný pokles tlaku krvi, vo väčšine prípadov bez klinického významu. Pri dlhodobom perorálnom podávaní 80 mg trikrát denne pacientom s artériovou hypertenziou poklesol systolický tlak oproti východiskovej hodnote v priemere o 9,4 mmHg
a diastolický o 9,1 mmHg. Pri dlhodobom perorálnom podávaní 80 mg trikrát denne pacientom
s pľúcnou artériovou hypertenziou sa pozorovali menej výrazné zmeny v poklese tlaku krvi (oba, systolický ako aj diastolický tlak, sa znížili o 2 mmHg). Pri odporúčanej perorálnej dávke 20 mg
trikrát denne neboli pozorované žiadne poklesy systolického alebo diastolického tlaku.
Podávanie jednorazových dávok až do 100 mg u zdravých dobrovoľníkov neviedlo k žiadnemu klinicky relevantnému účinku na EKG. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s pľúcnou artériovou hypertenziou sa nepozorovali žiadne klinicky významné zmeny na EKG.
V štúdii zameranej na hemodynamické účinky jednorazovej perorálnej dávky 100 mg sildenafilu
u 14 pacientov s ťažkou koronárnou artériovou chorobou (coronary artery disease, CAD) (> 70 %
stenóza aspoň jednej koronárnej artérie) poklesol stredný pokojový systolický krvný tlak o 7 %
a diastolický krvný tlak o 6 % v porovnaní s východiskovými hodnotami. Stredný pľúcny systolický tlak krvi poklesol o 9 %. Sildenafil nemal vplyv na srdcový výdaj a neviedol ku zhoršeniu krvného prietoku cez stenózne koronárne artérie.
U niektorých pacientov sa jednu hodinu po podaní dávky 100 mg sildenafilu pri použití Farnsworthovho-Munsellovho testu so 100 farebnými odtieňmi pozorovali mierne a prechodné rozdiely v rozlišovaní farieb (modrá/zelená). Dve hodiny po podaní sa už nezaznamenali žiadne účinky. Možný mechanizmus tejto zmeny v rozlišovaní farieb súvisí s inhibíciou PDE6, ktorá hrá úlohu vo fototransdukčnej kaskáde retiny. Sildenafil neovplyvňuje ani ostrosť, ani kontrast videnia. V placebom kontrolovanej štúdii s malým počtom pacientov s dokumentovaným včasným štádiom vekom podmienenej makulárnej degenerácie (n = 9) neboli vo vykonaných testoch videnia (ostrosť videnia, Amslerova mriežka, rozlíšenie farieb pri simulovanom dopravnom osvetlení, Humpreyho perimeter a fotostres) dokázané žiadne významné zmeny vplyvom sildenafilu (jednorazová dávka
100 mg).
Klinická účinnosť a bezpečnosť
Účinnosťintravenóznehosildenafiluudospelýchpacientovspľúcnouartériovouhypertenziou(PAH)
Predpokladá sa, že Revatio injekčný roztok v dávke 10 mg má za následok celkový účinok, ktorý zodpovedá spoločnému pôsobeniu voľného sildenafilu a jeho N-desmetylmetabolitu a ich kombinovaný farmakologický účinok je porovnateľný s účinkom 20 mg perorálnej dávky. Tento je založený len na farmakokinetických údajoch (pozri časť 5.2 Farmakokinetické vlastnosti). Dôsledky následnej nižšej expozície aktívnemu N-desmetylmetabolitu pozorované po opakovanom i.v. podaní Revatia neboli zdokumentované. Neboli vykonané žiadne klinické štúdie s cieľom preukázať, že tieto aplikačné formy majú porovnateľnú účinnosť.
Štúdia A1481262 bola otvorená štúdia s jedným centrom s jednorazovou dávkou, ktorá hodnotila bezpečnosť, tolerabilitu a farmakokinetiku jednorazovej intravenóznej dávky sildenafilu (10 mg) podanej vo forme bolusovej injekcie pacientom s PAH, ktorí už boli stabilizovaní a užívali 3-krát denne perorálne Revatio 20 mg.
Celkovo 10 pacientov s PAH bolo zaradených do štúdie a ukončilo ju. Osem pacientov užívalo bosentan a jeden pacient užíval okrem bosentanu a Revatia ešte treprostinil. Po podaní lieku sa zaznamenal v 30., 60. 120., 180. a 360. minúte tlak krvi v sede, v stoji a pulzová frekvencia. Priemerné zmeny tlaku krvi v sede oproti východiskovým hodnotám boli najväčšie po 1 hodine:
-9,1 mm Hg (SD ± 12,5) pre systolický tlak krvi a -3,0 (SD ± 4.9) mm Hg pre diastolický tlak krvi. Priemerné zmeny systolického a diastolického tlaku krvi, závislé na zmene polohy a čase, boli malé (< 10 mmHg) a po 2 hodinách sa vrátili na pôvodné hodnoty.
Účinnosťperorálnehosildenafiluudospelýchpacientovspľúcnouartériovouhypertenziou(PAH)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 278 pacientmi s primárnou pľúcnou hypertenziou, PAH spojenou s ochorením spojivového tkaniva a PAH po chirurgickej liečbe vrodenej srdcovej chyby. Pacienti boli randomizovaní do jednej zo štyroch liečených skupín: placebo, sildenafil 20 mg, sildenafil 40 mg alebo sildenafil 80 mg trikrát denne.
Z 278 randomizovaných pacientov dostalo 277 pacientov aspoň jednu dávku skúšaného lieku. Súbor pacientov tvorilo 68 mužov (25 %) a 209 žien (75 %) s priemerným vekom 49 rokov (v rozpätí
18 – 81 rokov) a východiskovými hodnotami 6-minútového testu chôdze od 100 do 450 metrov
vrátane (priemer: 344 metrov). 175 (63 %) zaradených pacientov malo diagnostikovanú primárnu pľúcnu hypertenziu, 84 (30 %) malo diagnostikovanú PAH spojenú s ochorením spojivového tkaniva a 18 (7 %) pacientov malo diagnostikovanú PAH následne po chirurgickej korekcii vrodenej srdcovej chyby. Väčšina pacientov mala funkčný stupeň II podľa WHO (107/277, 39 %) alebo
III (160/377, 58 %) s priemernými východiskovými hodnotami pri 6-minútovej chôdzi 378 metrov, resp. 326 metrov; menej pacientov malo stupeň I (1/277, 0,4 %) alebo IV (9/277, 3 %) ako
východiskovú hodnotu. Do štúdie neboli zaradení pacienti s ejekčnou frakciou ľavej komory < 45 %
alebo s frakčným skrátením ľavej komory < 0,2.
Sildenafil (alebo placebo) sa pridal k základnej liečbe pacientov, ktorá mohla zahŕňať kombináciu antikoagulancií, digoxínu, blokátorov kalciových kanálov, diuretík alebo kyslíka. Nesmeli sa používať prostacyklín, analógy prostacyklínu a antagonisty endotelínového receptora ako prídavná liečba, a ani doplnková liečba arginínom. Pacienti, u ktorých predtým zlyhala liečba bosentanom, boli zo štúdie vyradení.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze (6MWD) v 12. týždni liečby. Vo všetkých troch skupinách so sildenafilom bolo v porovnaní so skupinami s placebom zaznamenané štatisticky významné predĺženie 6MWD. Pre jednotlivé dávky sildenafilu 20 mg, 40 mg a 80 mg TID predstavovalo zlepšenie 6MWD po korekcii na placebo 45 metrov (p < 0,0001), 46 metrov
(p < 0,0001) a 50 metrov (p < 0,0001). Medzi jednotlivými dávkami sildenafilu nebol v účinku signifikantný rozdiel. U pacientov s východiskovým 6MWD < 325 m bola pozorovaná lepšia účinnosť
pri vyšších dávkach (zlepšenie po korekcii na placebo 58 metrov, 65 metrov a 87 metrov pre dávky
20 mg, 40 mg a 80 mg TID v uvedenom poradí).
Pri analýze podľa funkčného stupňa WHO sa pozorovalo v skupine s dávkou 20 mg štatisticky významné predĺženie 6MWD. Pre stupeň II a III predstavovalo zlepšenie po korekcii na placebo
49 metrov (p = 0,0007) a 45 metrov (p = 0,0031).
Zlepšenie 6MWD bolo zjavné po 4 týždňoch liečby a tento účinok pretrvával aj v 8. a 12. týždni. Výsledky boli všeobecne zhodné v podskupinách rozdelených podľa etiológie (primárnej PAH a PAH spojenej s ochorením spojivového tkaniva), funkčného stupňa podľa WHO, pohlavia, rasy, lokálnych údajov, stredného tlaku v pľúcnici (mPAP) a indexovanej pľúcnej vaskulárnej rezistencie (PVRI).
Pacienti pri všetkých dávkovaniach sildenafilu dosiahli v porovnaní s placebom štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP) a pľúcnej vaskulárnej rezistencie (PVR). Účinky liečby korigovanej na placebo s mPAP predstavovali -2,7 mmHg (p = 0,04), -3,0 mmHg (p = 0,01)
a -5,1 mmHg (p < 0,0001) pre sildenafil v dávke 20 mg, 40 mg a 80 mg TID v uvedenom poradí. Účinky liečby korigovanej na placebo s PVR predstavovali -178 dyn.sec/cm5 (p=0,0051),
-195 dyn.sec/cm5 (p=0,0017) a -320 dyn.sec/cm5 (p<0,0001) pre sildenafil v dávke 20 mg, 40 mg
a 80 mg TID v uvedenom poradí. Percentuálne zníženie PVR (11,2 %, 12,9 %, 23,3 %) v 12. týždni pre sildenafil 20 mg, 40 mg a 80 mg TID bolo proporcionálne výraznejšie ako zníženie systémovej
vaskulárnej rezistencie (SVR) (7,2 %, 5,9 %, 14,4 %). Účinok sildenafilu na mortalitu nie je známy.
U väčšieho percenta pacientov sa preukázalo zlepšenie aspoň o jednu funkčnú triedu WHO v 12. týždni pri každej z dávok sildenafilu (t.j. 28 %, 36 % a 42 % osôb, ktoré užívali dávky 20 mg, 40 mg a 80 mg TID sildenafilu, v uvedenom poradí) v porovnaní s placebom (7 %). Zodpovedajúce pomery šancí boli 2,92 (p=0,0087), 4,32 (p=0,0004) a 5,75 (p<0,0001).
Dlhodobéúdajeoprežívanípopuláciebezinejliečby
Pacienti zaradení do pivotnej štúdie s perorálnym podávaním boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 87 % pacientov dávku 80 mg TID. V pivotnej
štúdii bolo liečených Revatiom celkovo 207 pacientov a ich dlhodobé prežívanie bolo hodnotené
minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-Meierových kriviek 96 %, 91 % a 82 %. Prežívanie pacientov s funkčným
stupňom II podľa WHO pri vstupe do štúdie bolo po 1, 2 a 3 rokoch 99 %, 91 % a 84 % a u pacientov
s funkčným stupňom III podľa WHO pri úvode do štúdie bolo 94 %, 90 % a 81 %.
ÚčinnosťperorálnehosildenafiluudospelýchpacientovsPAH(pripoužitívkombináciis epoprostenolom)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 267 pacientmi
s PAH, ktorí boli stabilizovaní intravenózne podávaným epoprostenolom. Pacientov s PAH tvorili pacienti s primárnou pľúcnou artériovou hypertenziou (212/267, 79 %) a PAH spojenou s ochorením spojivového tkaniva (55/267, 21 %).Väčšina pacientov mala funkčný stupeň II podľa WHO (68/267,
26 %) alebo III (175/267, 66 %); menej pacientov malo stupeň I (3/267, 1 %) alebo IV (16/267, 6 %)
ako východiskovú hodnotu; u niekoľkých pacientov funkčný stupeň podľa WHO nebol známy.
Pacienti boli randomizovaní do skupiny s placebom alebo sildenafilom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg, a potom na 80 mg trikrát denne podľa znášanlivosti) pri použití v kombinácii s intravenózne podávaným epoprostenolom.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze v 16. týždni liečby. U sildenafilu bolo
v porovnaní s placebom zaznamenané štatisticky významné predĺženie vzdialenosti pri 6-minútovej chôdzi. Priemerné zlepšenie testu chôdze po korekcii na placebo predstavovalo 26 metrov v prospech
sildenafilu (95 % CI: 10,8, 41,2) (p = 0,0009). U pacientov s východiskovou hodnotou testu chôdze
³ 325 metrov bol účinok liečby 38,4 metra v prospech sildenafilu; u pacientov s východiskovou hodnotou testu chôdze < 325 metrov bol účinok liečby 2,3 metra v prospech placeba. U pacientov
s primárnou PAH bol účinok liečby 31,1 metra v porovnaní so 7,7 metra u pacientov s PAH spojenou s ochorením spojivového tkaniva. Rozdiel vo výsledkoch medzi týmito randomizovanými
podskupinami mohol vzniknúť náhodou vzhľadom na ich obmedzenú veľkosť reprezentovanej vzorky.
Pacienti liečení sildenafilom dosiahli v porovnaní s pacientmi užívajúcimi placebo štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP). Priemerný účinok liečby po korekcii na placebo predstavoval - 3,9 mmHg v prospech sildenafilu (95 % CI: - 5,7, - 2,1) (p = 0,00003). Sekundárnym hodnoteným parametrom bol čas do klinického zhoršenia stavu definovaný ako čas od randomizácie do vzniku prvej príhody klinického zhoršenia (smrť, transplantácia pľúc, začatie liečby bosentanom alebo klinické zhoršenie vyžadujúce úpravu liečby epoprostenolom). Liečba sildenafilom signifikantne oddialila čas do klinického zhoršenia PAH v porovnaní s placebom (p = 0,0074). Klinické zhoršenie nastalo u 23 pacientov v placebovej skupine (17,6 %) v porovnaní s 8 pacientmi
v skupine užívajúcej sildenafil (6,0 %).
Dlhodobéúdajeoprežívanívzákladnejepoprostenolovejštúdii
Pacienti zaradení do štúdie s prídavnou liečbu epoprostenolom boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 68 % pacientov dávku 80 mg TID. V úvodnej
štúdii bolo liečených Revatiom celkovo 134 pacientov a ich dlhodobé prežívanie bolo hodnotené
minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-Meierových kriviek 92 %, 81 % a 74 %.
Bezpečnosť a účinnosť u dospelých pacientov s PAH (pri použití v kombinácii s bosentanom) Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia so 103 klinicky stabilizovanými pacientmi s PAH (funkčný stupeň II a III podľa WHO), ktorí boli liečení bosentanom minimálne počas troch mesiacov. Pacienti s PAH zahŕňali pacientov s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. Pacienti boli randomizovaní do skupín s placebom alebo sildenafilom (20 mg trikrát denne), v kombinácii s bosentanom (62,5 – 125 mg dvakrát denne). Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vzdialenosti dosiahnutej pri 6-minútovej chôdzi (6MWD) v 12. týždni liečby. Výsledky ukazujú, že neexistuje významný rozdiel medzi priemernou zmenou voči východiskovej hodnote testu 6MWD pozorovanou pri podávaní sildenafilu (20 mg trikrát denne) 13,62 m (95 % CI: - 3,89 až 31,12)
a placeba 14,08 m (95 % CI: - 1,78 až 29,95).
Rozdiely vo vzdialenosti dosiahnutej pri 6-minútovej chôdzi boli pozorované medzi pacientmi
s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. U pacientov s primárnou PAH (67 pacientov) boli priemerné zmeny voči východiskovej hodnote 26,39 m (95 % CI: 10,70 až 42,08)
v skupine so sildenafilom a 11,84 m (95 % CI: - 8,83 až 32,52) v skupine s placebom. U pacientov
s PAH spojenou s ochorením spojivového tkaniva (36 pacientov) boli však priemerné zmeny voči východiskovej hodnote 18,32 m (95 % CI: 65,66 až 29,02) v skupine so sildenafilom a 17,50 m (95 %
CI: -9,41 až 44,41) v skupine s placebom.
Z celkového hľadiska boli nežiaduce udalosti vo všeobecnosti podobné v oboch liečebných skupinách (sildenafil v kombinácii s bosentanom v porovnaní so samotným bosentanom) a v súlade so známym bezpečnostným profilom sildenafilu pri použití v monoterapii (pozri časti 4.4 a 4.5).
5.2 Farmakokinetické vlastnosti
Absorpcia
Priemerná absolútna hodnota biologickej dostupnosti sildenafilu po perorálnom podaní je 41 % (v rozmedzí 25 – 63 %). V štúdii A1481262 bolo Cmax 248 ng/ml, Cl (klírens) 30,3 l/h a AUC(0-8)
330 ng h/ml. Pre N-desmetylmetabolit bola hodnota Cmax 30,8 ng/ml a AUC(0-8) 147 ng h/ml.
Distribúcia
Priemerný distribučný objem v rovnovážnom stave (Vss) sildenafilu je 105 l, čo naznačuje distribúciu do tkanív. Pri perorálnom podávaní 20 mg sildenafilu trikrát denne sú jeho priemerné maximálne celkové plazmatické koncentrácie v rovnovážnom stave približne 113 ng/ml. Sildenafil a jeho hlavný cirkulujúci N-desmetylmetabolit sa viaže v 96 % na plazmatické bielkoviny. Väzba na bielkoviny nie je závislá od celkových koncentrácií lieku.
Biostransformácia
Sildenafil je metabolizovaný predovšetkým hepatálnymi mikrozomálnymi izoenzýmami CYP3A4 (hlavná metabolická cesta) a CYP2C9 (vedľajšia metabolická cesta). Hlavný cirkulujúci metabolit sildenafilu je výsledkom N-demetylácie sildenafilu. Tento metabolit má profil selektivity pre fosfodiesterázu podobný sildenafilu a účinnosť in vitro na PDE5 približne 50 % v porovnaní
s materským liečivom. N-desmetylmetabolit je ďalej metabolizovaný a terminálny polčas je približne
4 h. U pacientov s pľúcnou artériovou hypertenziou plazmatické koncentrácie N-desmetylmetabolitu predstavujú približne 72 % koncentrácie sildenafilu pri perorálnom podávaní 20 mg trikrát denne (čo
sa premieta do 36 % podielu na farmakologickom účinku sildenafilu). Následné ovplyvnenie účinnosti
nie je známe. U zdravých dobrovoľníkov sú plazmatické hladiny N-desmetylmetabolitu po intravenóznom podaní sildenafilu signifikantne nižšie ako tie, ktoré sa pozorovali po jeho perorálnom podaní. V rovnovážnom stave predstavujú plazmatické koncentrácie N-desmetylmetabolitu po i.v. podaní približne 16 % oproti 61 % po perorálnom podaní sildenafilu.
Eliminácia
Celkový telový klírens sildenafilu je 41 l/h a terminálny fázový polčas 3 – 5 h. Tak po perorálnom, ako aj po intravenóznom podaní sa sildenafil vylučuje vo forme metabolitov predovšetkým do stolice
(približne 80 % podanej perorálnej dávky) a v menšej miere do moču (približne 13 % podanej
perorálnej dávky).
Farmakokinetika u osobitných skupín pacientov
Staršípacienti
U zdravých starších dobrovoľníkov (65-ročných a starších) bol znížený klírens sildenafilu, čo viedlo
k zvýšeniu plazmatických koncentrácií sildenafilu a aktívneho N-desmetylmetabolitu o približne 90 %
v porovnaní s hodnotami u mladších zdravých dobrovoľníkov (18 – 45-ročných). Vzhľadom na rozdiely vo väzbe na plazmatické bielkoviny, ktoré sú podmienené vekom, bolo zodpovedajúce
zvýšenie plazmatických koncentrácií voľného sildenafilu približne 40 %.
Renálnainsuficiencia
U dobrovoľníkov s ľahkým a stredne ťažkým poškodením funkcie obličiek (klírens kreatinínu =
30 – 80 ml/min) nebola zmenená farmakokinetika sildenafilu po podaní jednorazovej perorálnej dávky
50 mg. U dobrovoľníkov s ťažkým poškodením funkcie obličiek (klírens kreatinínu < 30 ml/min) bol klírens sildenafilu znížený a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia
funkcie obličiek, sa AUC zvýšila o 100 % a Cmax o 88 %. Okrem toho hodnoty AUC a Cmax
N-desmetylmetabolitu sa signifikantne zvýšili o 200 % a o 79 % u jedincov s ťažkým poškodením
funkcie obličiek v porovnaní s jedincami s normálnou funkciou obličiek.
Hepatálnainsuficiencia
U dobrovoľníkov s ľahkou a stredne ťažkou cirhózou pečene (stupeň A a B podľa
Childovho-Pughovho skóre) sa klírens sildenafilu znížil a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia funkcie pečene, sa AUC zvýšila o 85 % a Cmax o 47 %. Navyše hodnoty AUC a Cmax pre N-desmetylmetabolit boli signifikantne vyššie o 154 %, resp. o 87 % u pacientov
s cirhózou pečene v porovnaní s jedincami s normálnymi pečeňovými funkciami. Farmakokinetika sildenafilu u pacientov s ťažkým poškodením funkcie pečene nebola študovaná.
Populačnáfarmakokinetika
U pacientov s pľúcnou artériovou hypertenziou boli priemerné rovnovážne koncentrácie pri sledovaných perorálnych dávkach 20 – 80 mg trikrát denne o 20 – 50 % vyššie než v porovnávanej
skupine zdravých dobrovoľníkov. Cmax dosiahla dvojnásobnú hodnotu oproti zdravým dobrovoľníkom. Oba výsledky napovedajú, že pacienti s pľúcnou artériovou hypertenziou majú oproti zdravým dobrovoľníkom nižší klírens a/alebo vyššiu biologickú dostupnosť sildenafilu per os.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
U mláďat potkanov, ktoré boli pre- a postnatálne liečené sildenafilom v dávke 60 mg/kg, sa pozoroval menší počet mláďat vo vrhu, nižšia hmotnosť mláďat v 1. deň a znížené prežívanie do 4. dňa pri expozíciách, ktoré boli približne 50-násobkom očakávanej intravenóznej expozície u človeka pri
dávke 10 mg trikrát denne. Účinky v predklinických štúdiách sa pozorovali pri expozíciách považovaných za dostatočne vyššie, než je maximálna expozícia u ľudí, čo poukazuje na malý význam
týchto zistení pre klinické použitie.
U zvierat neboli pri klinicky relevantných expozíciách pozorované žiadne nežiaduce reakcie
s možným významom pre klinické použitie, ktoré by neboli tiež pozorované v klinických štúdiách.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
glukóza
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi alebo intravenóznymi roztokmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Každé balenie obsahuje jednu 20 ml injekčnú liekovku z priehľadného skla typu I s chlórbutylovou gumovou zátkou a hliníkovým pečatením.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Tento liek nevyžaduje riedenie alebo rekonštitúciu pred použitím.
Jedna 20 ml injekčná liekovka obsahuje 10 mg sildenafilu (vo forme citrátu). Odporúčaná dávka
10 mg vyžaduje objem 12,5 ml na podanie formou intravenóznej bolusovej injekcie. Chemická a fyzikálna kompatibilita bola dokázaná s nasledovnými roztokmi:
5 % roztok glukózy
izotonický roztok chloridu sodného 9 mg/ml (0,9 %) Ringerov roztok s laktátom
roztok 5 % glukózy/0,45 % chloridu sodného
roztok 5 % glukózy/Ringerovho roztoku s laktátom roztok 5 % glukózy/20 mmol chloridu draselného
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/05/318/002
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. október 2005
Dátum posledného predĺženia: 23. september 2010
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Revatio 10 mg/ml prášok na perorálnu suspenziu
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Po rekonštitúcii každý ml perorálnej suspenzie obsahuje 10 mg sildenafilu (vo forme citrátu). Jedna fľaška rekonštituovanej perorálnej suspenzie (112 ml) obsahuje 1,12 g sildenafilu (vo forme citrátu).
Pomocné látky so známym účinkom
Každý ml perorálnej suspenzie obsahuje 250 mg sorbitolu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Prášok na perorálnu suspenziu. Biely až sivobiely prášok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Dospelí
Liečba dospelých pacientov s pľúcnou artériovou hypertenziou, klasifikovanou ako funkčný stupeň II
a III podľa WHO, za účelom zlepšenia tolerancie fyzickej záťaže. Účinnosť sa potvrdila pri primárnej pľúcnej hypertenzii a pľúcnej hypertenzii spojenej s ochorením spojivového tkaniva.
Pediatrická populácia
Liečba pediatrických pacientov vo veku 1 až 17 rokov s pľúcnou artériovou hypertenziou. Účinnosť
v zmysle zlepšenia tolerancie fyzickej záťaže alebo pľúcnej hemodynamiky sa potvrdila pri primárnej pľúcnej hypertenzii a pľúcnej hypertenzii spojenej s vrodenou srdcovou chybou (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Liečbu má začať a monitorovať len lekár, ktorý má skúsenosti s liečbou pľúcnej arteriálnej hypertenzie. V prípade zhoršenia klinického stavu napriek liečbe Revatiom, treba zvážiť alternatívne možnosti liečby.
Dávkovanie
Dospelí
Odporúčaná dávka je 20 mg trikrát denne (TID). Lekári majú poradiť pacientom, ktorí zabudnú užiť
Revatio, aby užili dávku čo možno najskôr a potom pokračovali s normálnou dávkou. Pacienti nesmú užiť dvojnásobnú dávku, aby nahradili vynechanú dávku.
Pediatrickápopulácia(voveku1až17rokov)
U pediatrických pacientov vo veku 1 až 17 rokov je odporúčaná dávka u pacientov s hmotnosťou
≤ 20 kg je 10 mg (1 ml zriedenej suspenzie) trikrát denne a u pacientov s hmotnosťou > 20 kg je
20 mg (2 ml ziredenej suspenzie alebo 1 tableta) trikrát denne. U detí a dospievajúcich s pľúcnou artériovou hypertenziou (PAH) sa nesmú používať vyššie než odporúčané dávky (pozri tiež časti 4.4
a 5.1).
Pacienti
užívajúci
iné
lieky
Vo všeobecnosti sa má akákoľvek úprava dávky vykonávať až po dôkladnom zhodnotení prínosu a rizika. Úprava znížením dávky na 20 mg dvakrát denne sa má zvážiť, keď sa sildenafil podáva pacientom, ktorí už užívajú inhibítory CYP3A4, ako sú erytromycín alebo sachinavir. Úprava znížením dávky na 20 mg jedenkrát denne sa odporúča v prípade súčasného podávania so silnejšími inhibítormi CYP3A4 klaritromycínom, telitromycínom a nefazodónom. Pre použitie sildenafilu s najsilnejšími inhibítormi CYP3A4, pozri časť 4.3. Úpravy dávky sildenafilu sa môžu vyžadovať, keď sa podáva s induktormi CYP3A4 (pozri časť 4.5).
Osobitné skupiny pacientov
Staršípacienti(≥65rokov)
Úprava dávky sa nevyžaduje u starších pacientov. Podľa nameraných vzdialeností pri 6-minútovej chôdzi by klinická účinnosť mohla byť menšia u starších pacientov.
Poškodeniefunkcieobličiek
Úprava začiatočnej dávky sa nevyžaduje u pacientov s poškodením funkcie obličiek, vrátane ťažkého
poškodenia funkcie obličiek (klírens kreatinínu < 30 ml/min). Úprava znížením dávky na 20 mg dvakrát denne sa má zvážiť po dôkladnom vyhodnotení prínosu a rizika, len ak liečba nie je dobre tolerovaná.
Poškodeniefunkciepečene
Úprava začiatočnej dávky sa nevyžaduje u pacientov s poškodením funkcie pečene (stupeň A a B
podľa Childovho-Pughovho skóre). Úprava znížením dávky na 20 mg dvakrát denne sa má zvážiť po dôkladnom vyhodnotení prínosu a rizika, len ak liečba nie je dobre tolerovaná.
Revatio je kontraindikované u pacientov s ťažkým poškodením funkcie pečene (stupeň C podľa
Childovho-Pughovho skóre) (pozri časť 4.3).
Pediatrickápopulácia
Bezpečnosť a účinnosť Revatia u detí mladších ako 1 rok neboli stanovené. K dispozícii nie sú žiadne údaje.
Prerušenie liečby
Obmedzené údaje naznačujú, že náhle prerušenie liečby Revatiom nie je spojené s následným zhoršením pľúcnej arteriálnej hypertenzie. Aby sa však predišlo možnému výskytu náhleho zhoršenia
klinického stavu počas vynechania liečby, má sa zvážiť postupná redukcia dávok. Počas prerušenia
liečby sa odporúča intenzívne monitorovanie stavu.
Spôsob podávania
Revatio prášok na perorálnu suspenziu je určený len na perorálne použitie. Konštituovaná perorálna suspenzia (biela perorálna suspenzia hroznovej príchuti) sa má užívať s odstupom približne
6 až 8 hodín, s jedlom alebo bez jedla.
Pred odobratím požadovanej dávky, dôkladne pretrepávajte fľašku minimálne počas 10sekúnd. Pokyny na prípravu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Súčasné podávanie s donormi oxidu dusnatého (ako napríklad amylnitritom) alebo s nitrátmi
v akejkoľvek forme v dôsledku hypotenzívneho účinku nitrátov (pozri časť 5.1).
Súbežné podanie PDE5 inhibítorov, vrátane sildenafilu, so stimulátormi guanylátcyklázy, akým je
Kombinácia s najsilnejšími z inhibítorov CYP3A4 (napr. ketokonazolom, itrakonazolom, ritonavirom) (pozri časť 4.5).
Pacienti, ktorí majú stratu videnia v jednom oku v dôsledku neartériovej prednej ischemickej neuropatie zrakového nervu (non-arteritic anterior ischaemic optic neuropathy, NAION) bez ohľadu
na to, či táto príhoda súvisela alebo nesúvisela s predchádzajúcou expozíciou inhibítoru PDE5 (pozri časť 4.4).
Bezpečnosť sildenafilu sa nesledovala u nasledovných podskupín pacientov, a preto je jeho použitie u nich kontraindikované:
ťažké poškodenie funkcie pečene,
nedávno prekonaná cievna mozgová príhoda alebo infarkt myokardu, ťažká hypotenzia (tlak krvi < 90/50 mmHg) na začiatku liečby.
4.4 Osobitné upozornenia a opatrenia pri používaní
Účinnosť Revatia u pacientov s ťažkou pľúcnou artériovou hypertenziou (funkčný stupeň IV) nebola potvrdená. Ak dôjde k zhoršeniu klinického stavu, má sa zvážiť použitie liečby odporúčanej pre ťažký stupeň ochorenia (napr. epoprostenol) (pozri časť 4.2).
Pomer prínosu/rizika liečby sildenafilom nebol stanovený u pacientov s funkčným stupňom I pľúcnej artériovej hypertenzie podľa WHO.
Štúdie so sildenafilom sa vykonali u foriem pľúcnej artériovej hypertenzie spojenej s primárnym (idiopatickým) ochorením spojivového tkaniva alebo s formami PAH spojenými s vrodenou chorobou srdca (pozri časť 5.1). Použitie sildenafilu u iných foriem PAH sa neodporúča.
V dlhodobej pediatrickej pokračovacej štúdii sa pozoroval nárast úmrtí u pacientov, ktorým sa podávali dávky vyššie, než je odporúčaná dávka. Preto sa dávky vyššie než odporúčané dávky nesmú používať u detí a dospievajúcich s PAH (pozri tiež časti 4.2 a 5.1).
Retinitispigmentosa
Bezpečnosť sildenafilu sa neštudovala u pacientov s hereditárnymi degeneratívnymi ochoreniami retiny, ako sú retinitis pigmentosa (menšina z týchto pacientov má genetickú poruchu retinálnej fosfodiesterázy), a preto sa jeho použitie neodporúča.
Vazodilatačný účinok
Pri predpisovaní sildenafilu musí lekár dôkladne zvážiť, či pacient netrpí takým základným ochorením, ktorého priebeh by mohli mierne až stredne silné vazodilatačné účinky sildenafilu nepriaznivo ovplyvniť, napríklad u pacientov s hypotenziou, pacientov s hypovolémiou, so závažnou obštrukciou výtokovej časti ľavej komory alebo autonómnou dysfunkciou (pozri časť 4.4).
Kardiovaskulárne rizikové faktory
Po uvedení sildenafilu určeného na erektilnú dysfunkciu u mužov na trh boli v časovej súvislosti
s užitím sildenafilu hlásené závažné kardiovaskulárne príhody vrátane infarktu myokardu, nestabilnej angíny pectoris, náhlej srdcovej smrti, komorovej arytmie, cerebrovaskulárnej hemorágie,
tranzitórneho ischemického ataku, hypertenzie a hypotenzie. U väčšiny týchto pacientov, ale nie
u všetkých, boli prítomné už existujúce kardiovaskulárne rizikové faktory. Mnohé z týchto hlásených príhod vznikli počas sexuálneho styku alebo krátko po ňom a niekoľko z nich sa vyskytlo krátko po užití sildenafilu, ale bez sexuálnej aktivity. Nie je možné určiť, či tieto príhody priamo súvisia
s uvedenými alebo inými faktormi.
Priapizmus
Sildenafil sa má používať s opatrnosťou u pacientov s anatomickou deformáciou penisu (ako angulácia, kavernózna fibróza alebo Peyronieho choroba) alebo u pacientov s ochoreniami, ktoré
predisponujú k priapizmu (ako kosáčikovitá anémia, mnohonásobný myelóm alebo leukémia).
Po uvedení sildenafilu na trh boli hlásené predĺžené erekcie a priapizmus. V prípade, že erekcia trvá dlhšie ako 4 hodiny, má pacient ihneď vyhľadať lekársku pomoc. Ak priapizmus nie je liečený okamžite, môže dôjsť k poškodeniu tkaniva penisu a trvalej strate potencie (pozri časť 4.8).
Vazo-okluzívna kríza u pacientov s kosáčikovitou anémiou
Sildenafil sa nemá používať u pacientov so sekundárnou pľúcnou hypertenziou pri kosáčikovitej anémii. V klinickej štúdii bol počet vazookluzívnych kríz vyžadujúcich hospitalizáciu hlásený častejšie u pacientov, ktorí užívali Revatio ako u tých, ktorí boli na placebe, čo viedlo k predčasnému ukončeniu štúdie.
Udalosti súvisiace so zrakom
V súvislosti s užitím sildenafilu a ostatných inhibítorov PDE5 boli spontánne hlásené prípady porúch zraku. V súvislosti s užitím sildenafilu a ostatných inhibítorov PDE5 boli spontánne a v observačnej
štúdii hlásené prípady zriedkavého ochorenia, neartériovej prednej ischemickej neuropatie zrakového nervu (pozri časť 4.8).
V prípade akéjkoľvek náhlej poruchy videnia sa má liečba ukončiť a zvážiť alternatívna liečba (pozri časť 4.3).
Alfa-blokátory
Opatrnosť sa odporúča, keď sa sildenafil podáva pacientom liečeným alfablokátorom, keďže súčasné podávanie môže viesť k symptomatickej hypotenzii u vnímavých jedincov (pozri časť 4.5). Pacienti majú byť hemodynamicky stabilizovaní liečbou alfablokátormi ešte pred začatím liečby sildenafilom, aby sa minimalizovalo riziko vzniku posturálnej hypotenzie. Lekár má informovať pacientov, ako postupovať pri objavení sa príznakov posturálnej hypotenzie.
Poruchy krvácania
Štúdie in vitro s humánnymi krvnými doštičkami naznačujú, že sildenafil potencuje antiagregačný účinok nitroprusidu sodného. Nie sú žiadne údaje o bezpečnosti podania sildenafilu pacientom
s poruchami krvácania alebo s aktívnym peptickým vredom. Preto sa má sildenafil u týchto pacientov
podávať iba po dôslednom zvážení prínosu a rizika liečby.
Antagonisty vitamínu K
U pacientov s pľúcnou artériovou hypertenziou môže existovať potenciálne zvýšené riziko krvácania, keď sa liečba sildenafilom zavedie u pacientov, ktorí už užívajú antagonistu vitamínu K, zvlášť
u pacientov s pľúcnou artériovou hypertenziou ako sekundárnym ochorením pri ochorení spojivového tkaniva.
Venookluzívne ochorenie
Nie sú dostupné údaje o použití sildenafilu u pacientov s pľúcnou hypertenziou spojenou s pľúcnym venookluzívnym ochorením. Avšak boli hlásené prípady život ohrozujúceho edému pľúc pri liečbe
vazodilatátormi (hlavne prostacyklínom) u týchto pacientov. Z toho vyplýva, že ak sa u pacientov
s pľúcnou hypertenziou, ktorým bol podaný sildenafil, objavia znaky edému pľúc, má sa zvážiť možnosť súčasného výskytu venookluzívnej choroby.
Fruktózová intolerancia
Prášok obsahuje sorbitol. Pacienti so zriedkavými dedičnými problémami fruktózovej intolerancie nesmú užívať tento liek.
Použitie sildenafilu s bosentanom
Účinnosť sildenafilu u pacientov liečených bosentanom nebola presvedčivo preukázaná (pozri časti
4.5 a 5.1).
Súčasné používanie s inými inhibítormi PDE5
Bezpečnosť a účinnosť sildenafilu pri súčasnom podaní s inými PDE5 inhibítormi, vrátane Viagry, u pacientov s PAH nebola študovaná a takéto súčasné používanie sa neodporúča (pozri časť 4.5).
4.5 Liekové a iné interakcie
Účinky iných liekov na sildenafil
Štúdiein
vitro
Sildenafil je v rozhodujúcej miere metabolizovaný izoformami 3A4 (hlavná metabolická cesta) a 2C9 (vedľajšia metabolická cesta) cytochrómu P450 (CYP). Inhibítory týchto izoenzýmov môžu preto znížiť klírens sildenafilu, zatiaľ čo induktory týchto izoenzýmov môžu klírens sildenafilu zvýšiť. Tieto odporúčania, pozri časti 4.2 a 4.3.
Štúdieinvivo'
Hodnotilo sa súčasné podávanie perorálneho sildenafilu a intravenózneho epoprostenolu (pozri časti
4.8 a 5.1).
V kontrolovaných klinických štúdiách sa neskúmala účinnosť a bezpečnosť sildenafilu súčasne podávaného s inými terapeutickými modalitami na pľúcnu artériovú hypertenziu (napr. ambrisentanom, iloprostom). Preto sa v prípade súčasného podávania odporúča opatrnosť.
U pacientov s pľúcnou artériovou hypertenziou sa neskúmala bezpečnosť a účinnosť sildenafilu, ak sa podávalo súčasne s inhibítormi PDE5 (pozri časť 4.4).
Populačná farmakokinetická analýza údajov z klinických štúdií týkajúcich sa pľúcnej artériovej hypertenzie ukázala pokles klírensu sildenafilu a/alebo zvýšenie jeho biologickej dostupnosti po perorálnom podaní, ak sa podával súčasne so substrátmi CYP3A4, resp. súčasne s kombináciou substrátov CYP3A4 a betablokátorov. Toto boli jediné faktory, ktoré štatisticky signifikantne ovplyvnili farmakokinetiku sildenafilu u pacientov s pľúcnou artériovou hypertenziou. Expozícia sildenafilu u pacientov užívajúcich substráty CYP3A4 bola vyššia o 43 % a u pacientov užívajúcich kombináciu substrátov CYP3A4 a betablokátorov bola vyššia o 66 % v porovnaní s pacientmi, ktorí neužívali tieto skupiny liekov. Expozícia sildenafilu bola 5-krát vyššia pri dávke 80 mg trikrát denne v porovnaní s expozíciou pri dávke 20 mg trikrát denne. Toto rozpätie koncentrácií zahŕňa zvýšenie expozície sildenafilu pozorované v špecificky zostavených interakčných štúdiách s inhibítormi CYP3A4 (s výnimkou najsilnejších inhibítorov CYP3A4, napr. ketokonazolu, itrakonazolu, ritonaviru).
Zdá sa, že induktory CYP3A4 majú podstatný vplyv na farmakokinetiku sildenafilu u pacientov s pľúcnou artériovou hypertenziou, čo bolo potvrdené aj in vivo štúdiou sledujúcou interakcie
s induktorom CYP3A4 bosentanom.
Súčasné podávanie bosentanu (stredne silný induktor CYP3A4, CYP2C9 a pravdepodobne CYP2C19)
125 mg dvakrát denne so sildenafilom 80 mg trikrát denne (v rovnovážnom stave), ktoré sa súčasne podávali zdravým dobrovoľníkom počas 6 dní, viedlo k poklesu AUC sildenafilu o 63 %.
Populačná farmakokinetická analýza údajov týkajúcich sa sildenafilu z klinických štúdií s dospelými pacientmi s PAH vrátane 12-týždňovej štúdie na posúdenie účinnosti a bezpečnosti sildenafilu podávaného perorálne v dávke 20 mg trikrát denne po pridaní k stabilnej dávke bosentanu (62,5 mg –
125 mg dvakrát denne) ukázala pokles expozície sildenafilu pri súčasnom podávaní s bosentanom podobný tomu, ktorý sa pozoroval u zdravých dobrovoľníkov (pozri časti 4.4 a 5.1).
Účinnosť sildenafilu musí byť presne monitorovaná u pacientov, ktorí súčasne užívajú silné induktory
CYP3A4, ako sú karbamazepín, fenytoín, fenobarbital, ľubovník a rifampicín.
Súčasné podávanie inhibítora HIV proteáz, ritonaviru, ktorý je veľmi silný inhibítor cytochrómu P450, v rovnovážnom stave (500 mg dvakrát denne) a sildenafilu (100 mg jednorazová dávka) viedlo
k 300 % (4-násobnému) vzostupu Cmax sildenafilu a k 1 000 % (11-násobnému) vzostupu AUC sildenafilu v plazme. Po uplynutí 24 hodín boli plazmatické koncentrácie sildenafilu ešte stále približne 200 ng/ml, v porovnaní s približne 5 ng/ml, ak bol sildenafil podaný samostatne. Tieto údaje sú v súlade s výraznými účinkami ritonaviru na široké spektrum substrátov P450. Vzhľadom na tieto
farmakokinetické výsledky je súčasné podávanie sildenafilu a ritonaviru kontraindikované u pacientov s pľúcnou artériovou hypertenziou (pozri časť 4.3).
Súčasné podávanie inhibítora HIV proteáz, sachinaviru, inhibítora CYP3A4 v rovnovážnom stave (1 200 mg trikrát denne) a sildenafilu (100 mg jednorazová dávka) viedlo k 140 % vzostupu Cmax sildenafilu a k 210 % vzostupu AUC sildenafilu. Sildenafil neovplyvňuje farmakokinetiku sachinaviru. Odporúčania pre dávkovanie, pozri časť 4.2.
Ak sa sildenafil podával jednorazovo v dávke 100 mg spolu s erytromycínom, stredne silným inhibítorom CYP3A4, v rovnovážnom stave (500 mg dvakrát denne 5 dní), zaznamenal sa 182 % vzostup systémovej expozície sildenafilu (AUC). Odporúčania pre dávkovanie, pozri časť 4.2.
U zdravých dobrovoľníkov mužského pohlavia sa nedokázal vplyv azitromycínu (500 mg denne počas
3 dní) na AUC, Cmax, tmax, eliminačnú rýchlostnú konštantu alebo následne na polčas sildenafilu alebo jeho hlavného cirkulujúceho metabolitu. Nevyžaduje sa úprava dávky. Pri súčasnom podávaní sildenafilu (50 mg) a cimetidínu (800 mg), ktorý je inhibítorom cytochrómu P450 a nešpecifickým inhibítorom CYP3A4, zdravým dobrovoľníkom sa zaznamenal 56 % vzostup plazmatickej koncentrácie sildenafilu. Nevyžaduje sa úprava dávky.
Očakáva sa, že najsilnejšie z inhibítorov CYP3A4, ako sú ketokonazol a itrakonazol, by mali mať podobné účinky ako ritonavir (pozri časť 4.3). V prípade inhibítorov CYP3A4, ako sú klaritromycín, telitromycín a nefazodón, sa očakáva účinok s intenzitou medzi ritonavirom a inhibítormi CYP3A4, ako sú sachinavir alebo erytromycín, predpokladá sa sedemnásobné zvýšenie expozície. Preto sa pri použití inhibítorov CYP3A4 odporúča úprava dávkovania (pozri časť 4.2).
Populačná farmakokinetická analýza u pacientov s pľúcnou artériovou hypertenziou ukázala, že súčasné podávanie betablokátorov v kombinácii so substrátmi CYP3A4 môže vyústiť do dodatočného zvýšenia expozície sildenafilu v porovnaní s podávaním samotných substrátov CYP3A4.
Grapefruitová šťava je slabým inhibítorom metabolizmu prostredníctvom CYP3A4 v črevnej stene a môže vyvolať mierny vzostup plazmatických hladín sildenafilu. Nevyžaduje sa úprava dávky, ale súčasné užívanie sildenafilu s grapefruitovou šťavou sa neodporúča.
Biologická dostupnosť sildenafilu nebola ovplyvnená podaním jednorazových dávok antacíd
(hydroxidu horečnatého/hydroxidu hlinitého).
Súčasné podávanie s perorálnymi kontraceptívami (etinylestradiol 30 μg a levonorgestrel 150 μg)
neovplyvnilo farmakokinetiku sildenafilu.
Nikorandil je hybrid aktivátora draslíkových kanálov a nitrátu. Vzhľadom na nitrátovú zložku má potenciál pre závažné interakcie so sildenafilom (pozri časť 4.3).
Účinky sildenafilu na iné lieky
Štúdieinvitro
Sildenafil je slabým inhibítorom (IC50 > 150 μmol/l) izoforiem 1A2, 2C9, 2C19, 2D6, 2E1 a 3A4
cytochrómu P450.
Nie sú žiadne údaje o interakcii sildenafilu s nešpecifickými inhibítormi fosfodiesterázy, ako sú teofylín alebo dipyridamol.
Štúdieinvivo
Nezaznamenali sa žiadne signifikantné interakcie sildenafilu (50 mg) s tolbutamidom (250 mg) alebo s warfarínom (40 mg), liekmi, ktoré sú metabolizované prostredníctvom CYP2C9.
Sildenafil nemal významný vplyv na expozíciu atorvastatínu (AUC zvýšené o 11 %), čo naznačuje, že
sildenafil nemá klinicky významný účinok na CYP3A4.
Medzi sildenafilom (v jednotlivej dávke 100 mg) a acenokumarolom sa nepozorovali žiadne interakcie.
Sildenafil (50 mg) nepotencioval predĺženie času krvácania zapríčineného kyselinou acetylsalicylovou
(150 mg).
Sildenafil (50 mg) nepotencioval hypotenzívny účinok alkoholu u zdravých dobrovoľníkov, ktorí mali priemernú maximálnu koncentráciu alkoholu v krvi 80 mg/dl.
V štúdii so zdravými dobrovoľníkmi vyvolal sildenafil v rovnovážnom stave (80 mg trikrát denne)
zvýšenie AUC bosentanu o 50 % (125 mg dvakrát denne).
Populačná farmakokinetická analýza údajov zo štúdie s dospelými pacientmi s PAH na základnej liečbe bosentanom (62,5 mg – 125 mg dvakrát denne) ukázala zvýšenie (20 % (95 % CI: 9,8 – 30,8) AUC bosentanu pri súčasnom podávaní sildenafilu v rovnovážnom stave (20 mg trikrát denne)
v menšom rozsahu ako to, ktoré sa pozorovalo u zdravých dobrovoľníkov, ktorým bol podávaný bosentan súčasne so sildenafilom v dávke 80 mg trikrát denne (pozri časti 4.4 a 5.1).
V špecifickej interakčnej štúdii u pacientov s hypertenziou, ktorí súčasne užívali amplodipín so sildenafilom (100 mg), sa zaznamenalo ďalšie zníženie systolického tlaku krvi v ľahu o 8 mmHg. Zodpovedajúce ďalšie zníženie diastolického tlaku krvi v ľahu bolo o 7 mmHg. Toto ďalšie zníženie tlaku krvi malo podobný rozsah, ako keď sa sildenafil podával zdravým dobrovoľníkom samostatne.
V troch špecifických liekových interakčných štúdiách sa pacientom s benígnou hyperpláziou prostaty
(BPH) stabilizovanou na liečbe doxazosínom súbežne podával alfablokátor doxazosín (4 mg, resp.
8 mg) a sildenafil (25 mg, 50 mg alebo 100 mg). V týchto štúdiách sa u sledovanej populácie pozoroval dodatočný priemerný pokles systolického a diastolického tlaku krvi v ľahu o 7/7 mmHg,
9/5 mmHg, resp. 8/4 mmHg, ako aj dodatočný priemerný pokles tlaku krvi v stoji o 6/6 mmHg,
11/4 mmHg, resp. 4/5 mmHg. Keď sa sildenafil a doxazosín súbežne podávali pacientom stabilizovaným na liečbe doxazosínom, hlásenia o výskyte symptomatickej posturálnej hypotenzie
u pacientov boli ojedinelé. Tieto zahŕňali závraty a stratu rovnováhy, ale nie synkopu. Súčasné podávanie sildenafilu pacientom užívajúcim alfablokátory môže viesť u vnímavých jedincov
k symptomatickej hypotenzii (pozri časť 4.4).
Sildenafil (100 mg v jednorazovej dávke) neovplyvnil farmakokinetiku inhibítora HIV proteáz v rovnovážnom stave, sachinaviru, ktorý je substrátom/inhibítorom CYP3A4.
V súlade so známym účinkom sildenafilu na metabolickú cestu oxid dusnatý/cGMP (pozri časť 5.1) sa preukázalo, že sildenafil potencuje hypotenzívny účinok nitrátov, a preto je jeho súčasné podanie
s donormi oxidu dusnatého alebo nitrátmi v akejkoľvek forme kontraindikované (pozri časť 4.3).
Riociguát
Predklinické štúdie ukázali aditívny systémový účinok znižujúci krvný tlak, keď sa inhibítory PDE5 podávali súčasne s riociguátom. Klinické štúdie preukázali, že riociguát zosilňuje hypotenzívne účinky inhibítorov PDE5. V skúšanej populácii nebol nájdený žiadny dôkaz o priaznivom klinickom účinku spomínanej kombinácie. Súčasné užívanie riociguátu s PDE5 inhibítormi, vrátane sildenafilu, je kontraindikované (pozri časť 4.3).
Sildenafil nemal klinicky významný vplyv na plazmatické hladiny perorálnych kontraceptív
(etinylestradiol 30 μg a levonorgestrel 150 μg).
Pediatrická popolúcia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy v reprodukčnom veku a antikoncepcia u mužov a žien
Vzhľadom na nedostatok údajov o účinnosti Revatia u gravidných žien sa Revatio neodporúča užívať u žien v reprodukčnom veku, pokiaľ nepoužívajú súčasne vhodnú antikoncepciu.
Gravidita
Nie sú k dispozícii žiadne údaje o použití sildenafilu u gravidných žien. Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky na graviditu a embryonálny/fetálny vývoj. Štúdie
na zvieratách preukázali toxicitu na postnatálny vývoj (pozri časť 5.3).
Vzhľadom na nedostatok údajov sa má Revatio používať u gravidných žien iba v nevyhnutných prípadoch.
Dojčenie
Nie sú k dispozícii žiadne adekvátne a dobre kontrolované štúdie u dojčiacich žien. Údaje od jednej dojčiacej ženy naznačujú, že sildenafil a jeho aktívny metabolit, N-demetylsildenafil, sa vo veľmi malých množstvách vylučujú do materského mlieka. Nie sú dostupné žiadne klinické údaje týkajúce sa nežiaducich účinkov u dojčených detí, ale nepredpokladá sa, že by požité množstvá spôsobovali
nejaké nežiaduce účinky. Predpisujúci lekári majústarostlivo zvážiť klinickú potrebu matky užívať sildenafil a akékoľvek potenciálne nežiaduce účinky u dojčeného dieťaťa.
Fertilita
Predklinické údaje získané na základe obvyklých farmakologických štúdií fertility neodhalili žiadne osobitné riziko pre ľudí (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Revatio má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Keďže závrat a zmenené videnie boli hlásené v klinických štúdiách so sildenafilom, pacienti predtým, ako budú viesť vozidlá a obsluhovať stroje, majú poznať, ako reagujú na podanie Revatia.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
V pivotnej, placebom kontrolovanej štúdii s Revatiom pri pľúcnej artériovej hypertenzii bolo
207 pacientov randomizovaných a liečených dávkami 20 mg, 40 mg alebo 80 mg Revatia TID a
70 pacientov bolo randomizovaných na placebo. Liečba trvala 12 týždňov. Celková frekvencia prerušenia liečby u pacientov liečených sildenafilom v dávkach 20 mg, 40 mg a 80 mg TID bola
2,9 %, 3,0 % a 8,5 % v uvedenom poradí, v porovnaní s 2,9 % u placeba. Z 277 pacientov liečených v pivotnej štúdii prešlo do dlhodobej pokračovacej štúdie 259 pacientov. V nej sa podávali dávky do
80 mg trikrát denne (4-krát vyššie ako odporúčaná dávka 20 mg trikrát denne) a po 3 rokoch 87 % zo
183 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V placebom kontrolovanej štúdii s Revatiom ako doplnkom k intravenóznemu epoprostenolu u pľúcnej artériovej hypertenzie bolo celkovo 134 pacientov liečených Revatiom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg a potom na 80 mg trikrát denne podľa znášanlivosti) a epoprostenolom a 131 pacientov bolo liečených placebom a epoprostenolom. Dĺžka liečby bola 16 týždňov. Celková frekvencia prerušenia liečby z dôvodu nežiaducich účinkov
u pacientov liečených sildenafilom/epoprostenolom bola 5,2 % v porovnaní s 10,7 % u pacientov liečených placebom/epoprostenolom. Nedávno hlásenými nežiaducimi reakciami, ktoré sa vyskytli oveľa častejšie v skupine so sildenafilom/epoprostenolom, bola okulárna hyperémia, rozmazané videnie, upchatý nos, nočné potenie, bolesť chrbta a sucho v ústach. Známe nežiaduce reakcie bolesť hlavy, návaly tepla, bolesti v končatinách a edém boli zaznamenané s vyššou frekvenciou u pacientov liečených sildenafilom/epoprostenolom v porovnaní s pacientmi liečenými placebom/epoprostenolom. Z pacientov, ktorí ukončili úvodnú liečbu, prešlo do dlhodobej pokračovacej štúdie 242 pacientov.
V nej sa podávali dávky do 80 mg TID a po 3 rokoch 68 % zo 133 pacientov na skúmanej liečbe užívalo 80 mg Revatia TID.
V dvoch placebom kontrolovaných štúdiách boli nežiaduce účinky obyčajne mierne až stredne závažné. Najčastejšie hlásenými nežiaducimi účinkami, ktoré sa vyskytli (³ 10 %) u Revatia
v porovnaní s placebom boli bolesti hlavy, návaly tepla, dyspepsia, hnačka a bolesť v končatinách.
Zoznam nežiaducich reakcií usporiadaný do tabuľkyNežiaduce reakcie, ktoré sa vyskytli u > 1 % pacientov liečených Revatiom a boli častejšie (> 1 % rozdiel) u Revatia v pivotnej štúdii alebo v spoločnom súbore údajov z oboch placebom kontrolovaných štúdií pri liečbe pľúcnej artériovej hypertenzie Revatiom v dávkach
20, 40 alebo 80 mg TID, sú uvedené v tabuľke nižšie podľa triedy a skupín frekvencií (veľmi časté
(³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100) a neznáme (z dostupných údajov)). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Hlásenia po uvedení lieku na trh sú uvedené kurzívou.
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakciaInfekcie a nákazyČasté celulitída, chrípka, zápal priedušiek, zápal prinosových dutín, rinitída,
gastroenteritída
Poruchy krvi a lymfatického systémuČasté anémia
Poruchy metabolizmu a výživyČasté retencia tekutín
Psychické poruchyČasté nespavosť, úzkosť
Poruchy nervového systémuVeľmi časté bolesť hlavy
Časté migréna, tremor, parestézie, pocit pálenia, hypestézia
Poruchy okaČasté krvácanie do sietnice, poškodenie zraku, rozmazané videnie, fotofóbia, chromatopsia, cyanopsia, podráždenie
očí, okulárna hyperémia
Menej časté zníženie zrakovej ostrosti, diplopia, neprirodzené pocity v oku
Neznáme nearteritická predná ischemická neuropatia zrakového nervu (NAION)
*, oklúzia ciev sietnice
*, poruchy
v zornom poli
*Poruchy ucha a labyrintuČasté závrat
Neznáme
náhla strata sluchuPoruchy cievVeľmi časté návaly tepla
MedDRA trieda orgánových systémov (V.14.0) Nežiaduca reakcia
Neznáme hypotenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté epistaxa, kašeľ, upchatý nos
Poruchy gastrointestinálneho traktu
Veľmi časté hnačka, dyspepsia
Časté gastritída, gastroezofageálna refluxná choroba, hemoroidy, nafúknutie
brucha, sucho v ústach
Poruchy kože a podkožného tkaniva
Časté
Neznáme
alopécia, erytém, nočné potenie
vyrážka
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Veľmi časté bolesť v končatinách
Časté myalgia, bolesť chrbta
Poruchy obličiek a močových ciestMenej časté hematúria
Poruchy reprodukčného systému a prsníkovMenej časté penilné krvácanie, hematospermia, gynekomastia
Neznáme
priapizmus, zvýšená erekciaCelkové poruchy a reakcie v mieste podaniaČasté horúčka
* Tieto nežiaduce účinky/reakcie boli hlásené s neznámou frekvenciou u pacientov užívajúcich sildenafil na liečbu erektilnej dysfunkcie u mužov (MED).
Pediatrická populáciaV placebom kontrolovanej štúdii s Revatiom, do ktorej boli zaradení pacienti s pľúcnou artériovou hypertenziou vo veku 1 až 17 rokov, bolo liečených celkovo 174 pacientov trikrát denne v dávkovacej schéme s Revatiom buď s nízkou (10 mg u pacientov s hmotnosťou > 20 kg; žiadni pacienti
s hmotnosťou ≤ 20 kg nedostávali nízku dávku), strednou (10 mg u pacientov s hmotnosťou
≥ 8 – 20 kg; 20 mg u pacientov s hmotnosťou ≥ 20 – 45 kg; 40 mg u pacientov s hmotnosťou > 45 kg)
alebo vysokou dávkou (20 mg u pacientov s hmotnosťou ≥ 8 – 20 kg; 40 mg u pacientov
s hmotnosťou ≥ 20 – 45 kg; 80 mg u pacientov s hmotnosťou > 45 kg) a 60 pacientov bolo liečených placebom.
Profil nežiaducich reakcií pozorovaný v tejto pediatrickej štúdii bol vo všeobecnosti zhodný
s profilom u dospelých
(pozri tabuľku vyššie). Najčastejšími nežiaducimi reakciami, ktoré sa vyskytli
(s frekvenciou ≥ 1 %) u pacientov užívajúcich Revatio (v kombinovaných dávkach) a s frekvenciou
> 1 % u pacientov užívajúcich placebo, boli pyrexia, infekcia horných dýchacích ciest (každý 11,5 %), vracanie (10,9 %), zvýšená erekcia (zahŕňajúca spontánnu penilnú erekciu u mužských pacientov)
(9,0 %), nauzea, bronchitída (každý 4,6 %), faryngitída (4,0 %), rinorea (3,4 %), a pneumónia, rinitída
(každý 2,9 %).
Z 234 pediatrických pacientov liečených v krátkodobej, placebom kontrolovanej štúdii 220 pacientov vstúpilo do dlhodobej pokračovacej štúdie. Pacienti, ktorí boli na aktívnej liečbe sildenafilom, pokračovali v rovnakej dávkovacej schéme, kým pacienti, ktorým bolo v krátkodobej štúdii podané placebo, boli náhodne zaradení do liečby sildenafilom.
Najčastejšie nežiaduce reakcie hlásené počas celého trvania krátkodobej a dlhodobej štúdie boli vo všeobecnosti podobné ako tie, ktoré sa pozorovali v krátkodobej štúdii. Nežiaduce reakcie hlásené u > 10 % z 229 pacientov liečených sildenafilom (skupina s kombinovanými dávkami, vrátane
9 pacientov, ktorí nepokračovali v dlhodobej štúdii) boli infekcia horných dýchacích ciest (31 %), bolesť hlavy (26 %), vracanie (22 %), bronchitída (20 %), faryngitída (18 %), pyrexia (17 %), hnačka (15 %), chrípka a epistaxa (12 % v oboch prípadoch). Väčšina týchto nežiaducich reakcií bola považovaná za miernu až stredne závažnú.
Závažné nežiaduce udalosti boli hlásené u 94 (41 %) z 229 pacientov užívajúcich sildenafil.
Z 94 pacientov, ktorí hlásili závažné nežiaduce udalosti bolo 14/55 (25,5 %) pacientov v skupine
s nízkou dávkou, 35/74 (47,3 %) v skupine so strednou dávkou a 45/100 (45 %) v skupine s vysokou dávkou. Najčastejšie závažné nežiaduce udalosti, ktoré sa vyskytli s frekvenciou ≥ 1 % u pacientov užívajúcich sildenafil (kombinované dávky) boli pneumónia (7,4 %), zlyhanie srdca, pľúcna hypertenzia (5,2 % v oboch prípadoch), infekcia horných dýchacích ciest (3,1 %), zlyhanie pravej komory srdca, gastroenteritída (2,6 % v oboch prípadoch), synkopa, bronchitída, bronchpneumónia, pľúcna artériová hypertenzia (2,2 % vo všetkých prípadoch), bolesť na hrudi, zubný kaz (1,7 %
v oboch prípadoch) a kardiogénny šok, vírusová gastroenteritída, infekcia močových ciest (1,3 % vo všetkých prípadoch).
Nasledovné závažné nežiaduce udalosti boli považované za súvisiace s liečbou: enterokolitída, konvulzia, hypersenzitivita, stridor, hypoxia, neurosenzorická hluchota a ventrikulárna arytmia.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV štúdiách so zdravými dobrovoľníkmi boli po podaní jednorazových dávok do 800 mg nežiaduce reakcie podobné ako pri podaní nižších dávok, ale vyskytovali sa častejšie a boli závažnejšie. Jednorazové dávky 200 mg viedli k vyššiemu výskytu nežiaducich reakcií (bolesť hlavy, návaly, závrat, dyspepsia, nazálna kongescia a zmena videnia).
V prípade predávkovania sa majú podľa potreby zaviesť štandardné podporné opatrenia. Keďže sildenafil je pevne viazaný na bielkoviny plazmy a neeliminuje sa močom, nie je pravdepodobné, že by renálna dialýza mala urýchliť klírens sildenafilu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Urologiká, lieky používané pri poruchách erekcie, ATC kód: G04BE03
MechanizmusúčinkuSildenafil je účinný a selektívny inhibítor fosfodiesterázy typu 5 (PDE5), špecifickej pre cyklický guanozínmonofosfát (cGMP). Je to enzým, ktorý zodpovedá za degradáciu cGMP. Okrem prítomnosti
tohto enzýmu v kavernóznom telese penisu sa PDE5 nachádza aj v cievnom riečisku pľúc. Sildenafil
preto zvyšuje cGMP v bunkách hladkých svalov pľúcnych ciev, čo vedie k ich relaxácii. U pacientov
s pľúcnou artériovou hypertenziou to môže viesť k vazodilatácii v pľúcnom riečisku, a v menšej miere aj ku vazodilatácii v systémovom obehu.
Farmakodynamický účinokŠtúdie
in vitro preukázali, že sildenafil je selektívny pre PDE5. Jeho účinok je výraznejší na PDE5 ako na ostatné známe fosfodiesterázy. Sildenafil je 10-krát selektívnejší pre PDE5 ako pre PDE6, ktorá sa podieľa na fototransdukcii v retine. Má 80-krát vyššiu selektivitu pre PDE5 než pre PDE1 a viac ako
700-krát vyššiu selektivitu pre PDE5 než pre PDE2, 3, 4, 7, 8, 9, 10 a 11. Obzvlášť, sildenafil má
4 000-krát vyššiu selektivitu pre PDE5 ako pre PDE3, cAMP špecifickú izoformu fosfodiesterázy, ktorá sa podieľa na kontrole kontraktility srdcového svalu.
Sildenafil spôsobuje mierny a prechodný pokles tlaku krvi, vo väčšine prípadov bez klinického významu. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s artériovou hypertenziou poklesol systolický tlak oproti východiskovej hodnote v priemere o 9,4 mm Hg a diastolický o 9,1 mm Hg. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s pľúcnou artériovou hypertenziou sa pozorovali menej výrazné zmeny v poklese tlaku krvi (oba, systolický, ako aj diastolický tlak sa znížili o 2 mm Hg). Pri odporúčanej dávke 20 mg trikrát denne neboli pozorované žiadne poklesy systolického alebo diastolického tlaku.
Podávanie jednorazových dávok až do 100 mg u zdravých dobrovoľníkov neviedlo k žiadnemu klinicky relevantnému účinku na EKG. Pri dlhodobom podávaní 80 mg trikrát denne pacientom s pľúcnou artériovou hypertenziou sa nepozorovali žiadne klinicky významné zmeny na EKG.
V štúdii zameranej na hemodynamické účinky jednorazovej perorálnej dávky 100 mg sildenafilu
u 14 pacientov s ťažkou koronárnou artériovou chorobou (coronary artery disease, CAD) (> 70 %
stenóza aspoň jednej koronárnej artérie) poklesol stredný pokojový systolický krvný tlak o 7 %
a diastolický krvný tlak o 6 % v porovnaní s východiskovými hodnotami. Stredný pľúcny systolický tlak krvi poklesol o 9 %. Sildenafil nemal vplyv na srdcový výdaj a neviedol ku zhoršeniu krvného
prietoku cez stenózne koronárne artérie.
U niektorých pacientov sa jednu hodinu po podaní dávky 100 mg sildenafilu pri použití Farnsworthovho-Munsellovho testu so 100 farebnými odtieňmi pozorovali mierne a prechodné rozdiely v rozlišovaní farieb (modrá/zelená). Dve hodiny po podaní sa už nezaznamenali žiadne účinky. Možný mechanizmus tejto zmeny v rozlišovaní farieb súvisí s inhibíciou PDE6, ktorá hrá úlohu vo fototransdukčnej kaskáde retiny. Sildenafil neovplyvňuje ani ostrosť, ani kontrast videnia. V placebom kontrolovanej štúdii s malým počtom pacientov s dokumentovaným včasným štádiom vekom podmienenej makulárnej degenerácie (n = 9) neboli vo vykonaných testoch videnia (ostrosť videnia, Amslerova mriežka, rozlíšenie farieb pri simulovanom dopravnom osvetlení, Humpreyho perimeter a fotostres) dokázané žiadne významné zmeny vplyvom sildenafilu (jednorazová dávka
100 mg).
Klinická účinnosť a bezpečnosť
Účinnosťudospelýchpacientovspľúcnouartériovouhypertenziou(PAH)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 278 pacientmi s primárnou pľúcnou hypertenziou, PAH spojenou s ochorením spojivového tkaniva a PAH po
chirurgickej liečbe vrodenej srdcovej chyby. Pacienti boli randomizovaní do jednej zo štyroch
liečených skupín: placebo, sildenafil 20 mg, sildenafil 40 mg alebo sildenafil 80 mg trikrát denne.
Z 278 randomizovaných pacientov dostalo 277 pacientov aspoň jednu dávku skúšaného lieku. Súbor pacientov tvorilo 68 mužov (25 %) a 209 žien (75 %) s priemerným vekom 49 rokov (v rozpätí
18 – 81 rokov) a východiskovými hodnotami 6-minútového testu chôdze od 100 do 450 metrov
vrátane (priemer: 344 metrov). 175 (63 %) zaradených pacientov malo diagnostikovanú primárnu pľúcnu hypertenziu, 84 (30 %) malo diagnostikovanú PAH spojenú s ochorením spojivového tkaniva
a 18 (7 %) pacientov malo diagnostikovanú PAH následne po chirurgickej korekcii vrodenej srdcovej
chyby. Väčšina pacientov mala funkčný stupeň II podľa WHO (107/277, 39 %) alebo
III (160/377, 58 %) s priemernými východiskovými hodnotami pri 6-minútovej chôdzi 378 metrov, resp. 326 metrov; menej pacientov malo stupeň I (1/277, 0,4 %) alebo IV (9/277, 3 %) ako
východiskovú hodnotu. Do štúdie neboli zaradení pacienti s ejekčnou frakciou ľavej komory < 45 %
alebo s frakčným skrátením ľavej komory < 0,2.
Sildenafil (alebo placebo) sa pridal k základnej liečbe pacientov, ktorá mohla zahŕňať kombináciu antikoagulancií, digoxínu, blokátorov kalciových kanálov, diuretík alebo kyslíka. Nesmeli sa používať prostacyklín, analógy prostacyklínu a antagonisty endotelínového receptora ako prídavná liečba, a ani
doplnková liečba arginínom. Pacienti, u ktorých predtým zlyhala liečba bosentanom, boli zo štúdie vyradení.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze (6MWD) v 12. týždni liečby. Vo všetkých troch skupinách so sildenafilom bolo v porovnaní so skupinami s placebom zaznamenané štatisticky významné predĺženie 6MWD. Pre jednotlivé dávky sildenafilu 20 mg, 40 mg a 80 mg TID predstavovalo zlepšenie 6MWD po korekcii na placebo 45 metrov (p < 0,0001), 46 metrov
(p < 0,0001) a 50 metrov (p < 0,0001). Medzi jednotlivými dávkami sildenafilu nebol v účinku signifikantný rozdiel. U pacientov s východiskovým 6MWD < 325 m bola pozorovaná lepšia účinnosť pri vyšších dávkach (zlepšenie po korekcii na placebo 58 metrov, 65 metrov a 87 metrov pre dávky
20 mg, 40 mg a 80 mg TID v uvedenom poradí).
Pri analýze podľa funkčného stupňa WHO sa pozorovalo v skupine s dávkou 20 mg štatisticky významné predĺženie 6MWD. Pre stupeň II a III predstavovalo zlepšenie po korekcii na placebo
49 metrov (p = 0,0007) a 45 metrov (p = 0,0031).
Zlepšenie 6MWD bolo zjavné po 4 týždňoch liečby a tento účinok pretrvával aj v 8. a 12. týždni. Výsledky boli všeobecne zhodné v podskupinách rozdelených podľa etiológie (primárnej PAH a PAH spojenej s ochorením spojivového tkaniva), funkčného stupňa podľa WHO, pohlavia, rasy, lokálnych údajov, stredného tlaku v pľúcnici (mPAP) a indexovanej pľúcnej vaskulárnej rezistencie (PVRI).
Pacienti pri všetkých dávkovaniach sildenafilu dosiahli v porovnaní s placebom štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP) a pľúcnej vaskulárnej rezistencie (PVR). Účinky liečby korigovanej na placebo s mPAP predstavovali -2,7 mmHg (p = 0,04), -3,0 mmHg (p = 0,01) a
-5,1 mmHg (p < 0,0001) pre sildenafil v dávke 20 mg, 40 mg a 80 mg TID v uvedenom poradí. Účinky liečby korigovanej na placebo s PVR predstavovali -178 dyn.sec/cm5 (p=0,0051),
-195 dyn.sec/cm5 (p=0,0017) a -320 dyn.sec/cm5 (p<0,0001) pre sildenafil v dávke 20 mg, 40 mg
a 80 mg TID v uvedenom poradí. Percentuálne zníženie PVR (11,2 %, 12,9 %, 23,3 %) v 12. týždni pre sildenafil 20 mg, 40 mg a 80 mg TID bolo proporcionálne výraznejšie ako zníženie systémovej
vaskulárnej rezistencie (SVR) (7,2 %, 5,9 %, 14,4 %). Účinok sildenafilu na mortalitu nie je známy.
U väčšieho percenta pacientov sa preukázalo zlepšenie aspoň o jednu funkčnú triedu WHO
v 12. týždni pri každej z dávok sildenafilu (t.j. 28 %, 36 % a 42 % osôb, ktoré užívali dávky 20 mg,
40 mga 80 mg TID sildenafilu, v uvedenom poradí) v porovnaní s placebom (7 %). Zodpovedajúce pomery šancí boli 2,92 (p=0,0087), 4,32 (p=0,0004) a 5,75 (p<0,0001).
Dlhodobéúdajeoprežívanípopuláciebezinejliečby
Pacienti zaradení do pivotnej štúdie boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 87 % pacientov dávku 80 mg TID. V pivotnej štúdii bolo liečených
Revatiom celkovo 207 pacientov a ich dlhodobé prežívanie bolo hodnotené minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-
Meierových kriviek 96 %, 91 % a 82 %. Prežívanie pacientov s funkčným stupňom II podľa WHO pri vstupe do štúdie bolo po 1, 2 a 3 rokoch 99 %, 91 % a 84 % a u pacientov s funkčným stupňom III podľa WHO pri úvode do štúdie bolo 94 %, 90 % a 81 %.
ÚčinnosťudospelýchpacientovsPAH(pripoužitívkombináciisepoprostenolom)
Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia s 267 pacientmi s PAH, ktorí boli stabilizovaní intravenózne podávaným epoprostenolom. Pacientov s PAH tvorili pacienti s primárnou pľúcnou artériovou hypertenziou (212/267, 79 %) a PAH spojenou s ochorením spojivového tkaniva (55/267, 21 %).Väčšina pacientov mala funkčný stupeň II podľa WHO (68/267,
26 %) alebo III (175/267, 66 %); menej pacientov malo stupeň I (3/267, 1 %) alebo IV (16/267, 6 %)
ako východiskovú hodnotu; u niekoľkých pacientov funkčný stupeň podľa WHO nebol známy. Pacienti boli randomizovaní do skupiny s placebom alebo sildenafilom (s vopred stanoveným zvyšovaním dávky začínajúcim z 20 mg na 40 mg, a potom na 80 mg trikrát denne podľa znášanlivosti) pri použití v kombinácii s intravenózne podávaným epoprostenolom.
Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vo vzdialenosti dosiahnutej pri 6-minútovom teste chôdze v 16. týždni liečby. U sildenafilu bolo
v porovnaní s placebom zaznamenané štatisticky významné predĺženie vzdialenosti pri 6-minútovej
chôdzi. Priemerné zlepšenie testu chôdze po korekcii na placebo predstavovalo 26 metrov v prospech sildenafilu (95 % CI: 10,8, 41,2) (p = 0,0009). U pacientov s východiskovou hodnotou testu chôdze
³ 325 metrov bol účinok liečby 38,4 metra v prospech sildenafilu; u pacientov s východiskovou hodnotou testu chôdze < 325 metrov bol účinok liečby 2,3 metra v prospech placeba. U pacientov
s primárnou PAH bol účinok liečby 31,1 metra v porovnaní so 7,7 metra u pacientov s PAH spojenou
s ochorením spojivového tkaniva. Rozdiel vo výsledkoch medzi týmito randomizovanými podskupinami mohol vzniknúť náhodou vzhľadom na ich obmedzenú veľkosť reprezentovanej vzorky.
Pacienti liečení sildenafilom dosiahli v porovnaní s pacientmi užívajúcimi placebo štatisticky významné zníženie stredného tlaku v pľúcnici (mPAP). Priemerný účinok liečby po korekcii na placebo predstavoval - 3,9 mmHg v prospech sildenafilu (95 % CI: - 5,7, - 2,1) (p = 0,00003). Sekundárnym hodnoteným parametrom bol čas do klinického zhoršenia stavu definovaný ako čas od randomizácie do vzniku prvej príhody klinického zhoršenia (smrť, transplantácia pľúc, začatie liečby bosentanom alebo klinické zhoršenie vyžadujúce úpravu liečby epoprostenolom). Liečba sildenafilom signifikantne oddialila čas do klinického zhoršenia PAH v porovnaní s placebom (p = 0,0074). Klinické zhoršenie nastalo u 23 pacientov v placebovej skupine (17,6 %) v porovnaní s 8 pacientmi
v skupine užívajúcej sildenafil (6,0 %).
Dlhodobéúdajeoprežívanívzákladnejepoprostenolovejštúdii
Pacienti zaradení do štúdie s prídavnou liečbu epoprostenolom boli vhodní na zaradenie do dlhodobej otvorenej pokračovacej štúdie. Po 3 rokoch užívalo 68 % pacientov dávku 80 mg TID. V úvodnej štúdii bolo liečených Revatiom celkovo 134 pacientov a ich dlhodobé prežívanie bolo hodnotené minimálne počas 3 rokov. V tejto populácii boli 1-ročné, 2-ročné a 3-ročné odhady prežívania pomocou Kaplanových-Meierových kriviek 92 %, 81 % a 74 %.
Bezpečnosť a účinnosť u dospelých pacientov s PAH (pri použití v kombinácii s bosentanom) Bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia so 103 klinicky stabilizovanými pacientmi s PAH (funkčný stupeň II a III podľa WHO), ktorí boli liečení bosentanom minimálne počas troch mesiacov. Pacienti s PAH zahŕňali pacientov s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. Pacienti boli randomizovaní do skupín s placebom alebo sildenafilom (20 mg trikrát denne), v kombinácii s bosentanom (62,5 – 125 mg dvakrát denne). Z hľadiska účinnosti bola primárnym hodnoteným parametrom zmena voči východiskovej hodnote vzdialenosti dosiahnutej pri 6-minútovej chôdzi (6MWD) v 12. týždni liečby. Výsledky ukazujú, že neexistuje významný rozdiel medzi priemernou zmenou voči východiskovej hodnote testu 6MWD pozorovanou pri podávaní sildenafilu (20 mg trikrát denne) 13,62 m (95 % CI: - 3,89 až 31,12)
a placeba 14,08 m (95 % CI: - 1,78 až 29,95).
Rozdiely vo vzdialenosti dosiahnutej pri 6-minútovej chôdzi boli pozorované medzi pacientmi
s primárnou PAH a PAH spojenou s ochorením spojivového tkaniva. U pacientov s primárnou PAH (67 pacientov) boli priemerné zmeny voči východiskovej hodnote 26,39 m (95 % CI: 10,70 až 42,08) v skupine so sildenafilom a 11,84 m (95 % CI: - 8,83 až 32,52) v skupine s placebom. U pacientov
s PAH spojenou s ochorením spojivového tkaniva (36 pacientov) boli však priemerné zmeny voči východiskovej hodnote 18,32 m (95 % CI: - 65,66 až 29,02) v skupine so sildenafilom a 17,50 m
(95 % CI: - 9,41 až 44,41) v skupine s placebom.
Z celkového hľadiska boli nežiaduce udalosti vo všeobecnosti podobné v oboch liečebných skupinách (sildenafil v kombinácii s bosentanom v porovnaní so samotným bosentanom) a v súlade so známym bezpečnostným profilom sildenafilu pri použití v monoterapii (pozri časti 4.4 a 4.5).
Pediatrickápopulácia
V randomizovanej, dvojito zaslepenej, multicentrickej, placebom kontrolovanej štúdii porovnávajúcej paralelné skupiny s rôznym rozsahom dávok bolo celkovo liečených celkovo 234 pacientov vo veku
hypertenziu (PPH) [33 %] alebo sekundárnu pľúcnu artériovú hypertenziu (PAH) k vrodenej srdcovej chybe [systémovo-pľúcny skrat 37 %, chirurgická korekcia 30 %]. V tejto štúdii 63 z 234 (27 %) pacientov malo < 7 rokov (z nich nízku dávku sildenafilu užívali = 2; strednú dávku = 17; vysokú dávku = 28; placebo = 16 pacienti) a 171 z 234 (73 %) pacientov malo 7 rokov a viac (z nich nízku dávku sildenafilu užívali = 40; strednú dávku = 38; a vysokú dávku = 49, placebo = 44 pacienti). Najviac pacientov malo na začiatku liečby funkčný stupeň I podľa WHO (75/234, 32 %) alebo
II (120/234, 51 %); menej pacientov malo funkčný stupeň III (35/234, 15 %) alebo IV (1/234, 0,4 %);
a niekoľko pacientov (3/234, 1,3 %) malo funkčný stupeň podľa WHO neznámy.
Pacienti boli bez špecifickej PAH liečby a užívanie prostacyklínu, analógov prostacyklínu
a antagonistov endotelínového receptora a ani doplnková liečba arginínom, nitráty, alfablokátory a silné CYP450 3A4 inhibítory neboli povolené.
Primárnym cieľom štúdie bolo posúdiť účinnosť 16-týždňovej chronickej liečby perorálnym sildenafilom u detí a dospievajúcich zameranej na zlepšenie tolerancie fyzickej záťaže meranej spiroergometrickým testom (Cardiopulmonary Exercise Test, CPET) u pacientov, ktorí boli v rámci
ich vývoja schopný test vykonať (n = 115). Sekundárne ciele zahŕňali hemodynamické monitorovanie, hodnotenie symptómov, funkčný stupeň podľa WHO, zmenu základnej liečby a meranie kvality
života.
Pacienti boli zaradení do jednej z troch skupín liečených sildenafilom v dávkovacej schéme s nízkou (10 mg), strednou (10 – 40 mg) alebo vysokou dávkou (20 – 80 mg) Revatia podávanou trikrát denne alebo s placebom. Aktuálne dávky podávané v danej skupine boli závislé od telesnej hmotnosti (pozri časť 4.8). Podiel pacientov užívajúcich na začiatku štúdie podporné lieky (antikoagulanciá, digoxín, blokátory kalciového kanála, diuretiká a/alebo kyslík) bol podobný v kombinovanej skupine pacientov liečených sildenafilom (47,7 %) a v skupine pacientov liečených placebom (41,7%).
Primárnym cieľom v skupinách s kombinovanou dávkou bola percentuálna zmena spotreby kyslíka na vrchole záťaže (VO2) od začiatku liečby po 16. týždeň korigovaná voči placebu, hodnotená spiroergometrickým (CPET) testom (tabuľka 2). Celkovo 106 z 234 pacientov (45 %) bolo hodnotiteľných CPET testom, tento počet zahŕňal deti vo veku ≥ 7 rokov a deti vývojovo schopné vykonať test. Deti vo veku < 7 rokov (užívajúce kombinovanú dávku sildenafilu = 47; placebo = 16) boli hodnotiteľné len pre sekundárne ciele. Priemerné hodnoty spotreby kyslíka na vrchole záťaže (VO2) na začiatku štúdie boli porovnateľné vo všetkých skupinách pacientov liečených sildenafilom (17,37 až 18,03 ml/kg/min) a mierne zvýšené v skupine pacientov liečených placebom
(20,02 ml/kg/min). Výsledky hlavnej analýzy (skupiny s kombinovanou dávkou versus placebo) neboli štatisticky signifikantné (p = 0,056) (pozri tabuľku 2). Odhadovaný rozdiel medzi strednou dávkou sildenafilu a placebom bol 11,33 % (95 % Cl: 1,72 až 20,94) (pozri tabuľku 2).
Tabuľka 2: % zmena VO2 oproti vstupnému vyšetreniu, korigovaná voči placebu
Liečebná skupina Odhadovaný rozdiel 95 % interval spoľahlivosti
Nízka dávka (n = 24) Stredná dávka (n = 26) Vysoká dávka (n = 27)
3,81 -6,11, 13,73
11,33 1,72, 20,94
7,98 -1,64, 17,60
Skupiny s kombinovanou dávkou
(
n = 77)
7,71
(p = 0,056)
-0,19, 15,60
n = 29 pre skupinu s placebom
Odhady založené na ANCOVA s úpravami pre kovarianty vrcholovej VO
2
na začiatku štúdie, etiológie a telesnej hmotnosti.
Zlepšenie závislé od dávky bolo pozorované u indexovanej pľúcnej vaskulárnej rezistencie
(Pulmonary vascular resistance index, PVRI) a stredného tlaku v pľúcnici (Mean pulmonary arterial
pressure, mPAP). V oboch skupinách pacientov so strednou a vysokou dávkou sildenafilu sa pozorovalo zníženie PVRI v porovnaní s placebom o 18 % (95 % Cl: 2 % až 32 %) a o 27 % (95 % Cl:
14 % až 39 %), zatiaľ čo v skupine pacientov s nízkou dávkou nebol pozorovaný signifikantný rozdiel
oproti placebu (rozdiel 2 %). V skupinách pacientov s vysokou a strednou dávkou boli zobrazené zmeny mPAP od začiatku sledovania v porovnaní s placebom o -3,5 mmHg (95 % Cl: -8,9, 1,9)
a o -7,3 mmHg (95% Cl: -12,4, -2,1), zatiaľ čo v skupine pacientov s nízkou dávkou sa pozorovala malá zmena oproti placebu (zmena o 1,6 mmHg). Zlepšenie sa pozorovalo pri srdcovom indexe vo všetkých troch skupinách so sildenafilom v porovnaní s placebom, a to o 10 %, 4 % a 15 % v skupine
s nízkou, strednou a vysokou dávkou.
Signifikantné zlepšenie funkčného stupňa sa preukázalo len u pacientov v skupine s vysokou dávkou
v porovnaní s placebom. Pomery šancí v skupinách pacientov liečených sildenafilom v nízkej, strednej a vysokej dávke v porovnaní s placebom boli 0,6 (95 % CI: 0,18, 2,01), 2,25 (95 % CI: 0,75, 6,69)
a 4,52 (95 % CI: 1,56, 13,10).
Údaje z dlhodobej pokračovacej štúdie
Z 234 pediatrických pacientov liečených v krátkodobej, placebom kontrolovanej štúdii, vstúpilo 220
pacientov do dlhodobej pokračovacej štúdie. Pacienti, ktorým bolo v krátkodobej štúdii podávané placebo, boli náhodne zaradení do liečby sildenafilom. Pacienti s hmotnosťou ≤ 20 kg boli zaradení do
skupín so strednou alebo vysokou dávkou (1:1) a pacienti s hmotnosťou > 20 kg boli zaradení do
skupín s nízkou, strednou alebo vysokou dávkou (1:1:1). Z celkového počtu 229 pacientov, ktorí dostávali sildenafil, bolo v skupinách s nízkou, strednou a vysokou dávkou 55, 74 a 100 pacientov. Počas krátkodobej a dlhodobej štúdie sa celkové trvanie liečby od začiatku dvojitého zaslepenia
u jednotlivých pacientov pohybovalo v rozmedzí 3 až 3 129 dní. V liečebnej skupine so sildenafilom bolo stredné trvanie liečby 1 696 dní (s výnimkou 5 pacientov, ktorí v dvojito zaslepenej štúdii
dostávali placebo a neboli liečení v dlhodobej pokračovacej štúdii).
Odhady prežívania po 3 rokoch pomocou Kaplanových-Meierových kriviek u pacientov s hmotnosťou
> 20 kg na začiatku sledovania boli 94 %, 93 % a 85 % v skupinách s nízkou, strednou a vysokou dávkou; u pacientov s hmotnosťou ≤ 20 kg na začiatku sledovania boli odhady prežívania v skupinách
so strednou a vysokou dávkou 94 % a 93 % (pozri časti 4.4 a 4.8).
Počas štúdie bolo hlásených celkovo 42 úmrtí, či už v období počas liečby alebo hlásených v rámci ďalšieho sledovania prežívania. K 37 úmrtiam došlo pred rozhodnutím Výboru pre monitorovanie údajov postupne redukovať dávkovanie u pacientov na nižšie dávky na základe pozorovanej nerovnováhy úmrtnosti pri zvýšených dávkach sildenafilu. Z týchto 37 úmrtí bol počet úmrtí (%)
v skupinách s nízkou, strednou a vysokou dávkou 5/55 (9,1 %), 10/74 (13,5 %) a 22/100 (22 %).
Následne bolo hlásených ďalších 5 úmrtí. Príčiny úmrtí boli v súvislosti s PAH. U pediatrických pacientov s PAH sa nesmú používať vyššie než odporúčané dávky (pozri časti 4.2, a 4.4).
Spotreba kyslíka na vrchole záťaže (VO2) bola hodnotená 1 rok po začatí placebom kontrolovanej štúdie. Z pacientov liečených sildenafilom, ktorí boli v rámci svojho stupňa vývoja schopní vykonať spiroergometrické vyšetrenie CPET, 59/114 pacientov (52 %) nepreukázalo žiadne zhoršenie vrcholovej VO2 oproti začiatku liečby sildenafilom. Podobne 191 z 229 pacientov (83 %), ktorí užívali sildenafil, si podľa posúdenia po 1 roku buď uchovalo, alebo zlepšilo svoj funkčný stupeň podľa
WHO.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií pre Revatio
s ohľadom na novorodencov s pľúcnou artériovou hypertenziou (pozri časť 4.2 pre informáciu o pediatrickom použití)
5.2 Farmakokinetické vlastnosti
Absorpcia
Sildenafil sa rýchlo vstrebáva. Maximálne plazmatické koncentrácie sa dosahujú do 30 – 120 minút
(v priemere 60 minút) po perorálnom užití lieku nalačno. Priemerná absolútna perorálna biologická dostupnosť je 41 % (v rozmedzí 25 – 63 %). Pri perorálnom podávaní sildenafilu trikrát denne sa
zvyšuje hodnota AUC a Cmax úmerne dávke v rozmedzí 20 – 40 mg. Pri perorálnom podávaní 80 mg sildenafilu trikrát denne sa pozorovalo väčšie ako dávkovo úmerné zvýšenie jeho plazmatických hladín. Biologická dostupnosť sildenafilu po perorálnom podaní 80 mg trikrát denne bola u pacientov s pľúcnou artériovou hypertenziou v priemerne o 43 % (90 % IS: 27 % – 60 %) vyššia v porovnaní
s nižšími dávkami.
Ak sa sildenafil užíva súčasne s jedlom, rýchlosť absorpcie sa zníži, pričom priemerné oneskorenie tmax je 60 minút a priemerné zníženie Cmax je 29 %, avšak rozsah absorpcie nie je významne ovplyvnený (AUC je znížená o 11 %).
Distribúcia
Priemerný distribučný objem v rovnovážnom stave (Vss) sildenafilu je 105 l, čo naznačuje distribúciu do tkanív. Pri perorálnom podávaní 20 mg sildenafilu trikrát denne sú jeho priemerné maximálne celkové plazmatické koncentrácie v rovnovážnom stave približne 113 ng/ml. Sildenafil a jeho hlavný cirkulujúci N-desmetylmetabolit sa viaže v 96 % na plazmatické bielkoviny. Väzba na bielkoviny nie je závislá od celkových koncentrácií lieku.
Biotransformácia
Sildenafil je metabolizovaný predovšetkým hepatálnymi mikrozomálnymi izoenzýmami CYP3A4 (hlavná metabolická cesta) a CYP2C9 (vedľajšia metabolická cesta). Hlavný cirkulujúci metabolit
sildenafilu je výsledkom N-demetylácie sildenafilu. Tento metabolit má profil selektivity pre
fosfodiesterázu podobný sildenafilu a účinnosť in vitro na PDE5 približne 50 % v porovnaní
s materským liečivom. N-desmetylmetabolit je ďalej metabolizovaný a terminálny polčas je približne
4 h. U pacientov s pľúcnou artériovou hypertenziou plazmatické koncentrácie N-desmetylmetabolitu predstavujú približne 72 % koncentrácie sildenafilu pri podávaní 20 mg trikrát denne (čo sa premieta
do 36 % podielu na farmakologickom účinku sildenafilu). Následné ovplyvnenie účinnosti nie je
známe.
Eliminácia
Celkový telový klírens sildenafilu je 41 l/h a terminálny fázový polčas 3 – 5 h. Tak po perorálnom, ako aj po intravenóznom podaní sa sildenafil vylučuje vo forme metabolitov predovšetkým do stolice
(približne 80 % podanej perorálnej dávky) a v menšej miere do moču (približne 13 % podanej perorálnej dávky).
Farmakokinetika u osobitných skupín pacientov
Staršípacienti
U zdravých starších dobrovoľníkov (65-ročných a starších) bol znížený klírens sildenafilu, čo viedlo
k zvýšeniu plazmatických koncentrácií sildenafilu a aktívneho N-desmetylmetabolitu o približne 90 % v porovnaní s hodnotami u mladších zdravých dobrovoľníkov (18 – 45-ročných). Vzhľadom na rozdiely vo väzbe na plazmatické bielkoviny, ktoré sú podmienené vekom, bolo zodpovedajúce zvýšenie plazmatických koncentrácií voľného sildenafilu približne 40 %.
Renálnainsuficiencia
U dobrovoľníkov s ľahkým a stredne ťažkým poškodením funkcie obličiek (klírens kreatinínu =
30 – 80 ml/min) nebola zmenená farmakokinetika sildenafilu po podaní jednorazovej perorálnej dávky
50 mg. U dobrovoľníkov s ťažkým poškodením funkcie obličiek (klírens kreatinínu < 30 ml/min) bol klírens sildenafilu znížený a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia funkcie obličiek, sa AUC zvýšila o 100 % a Cmax o 88 %. Okrem toho hodnoty AUC a Cmax
N-desmetylmetabolitu sa signifikantne zvýšili o 200 % a o 79 % u jedincov s ťažkým poškodením
funkcie obličiek v porovnaní s jedincami s normálnou funkciou obličiek.
Hepatálnainsuficiencia
U dobrovoľníkov s ľahkou a stredne ťažkou cirhózou pečene (stupeň A a B podľa
Childovho-Pughovho skóre) sa klírens sildenafilu znížil a v porovnaní s dobrovoľníkmi rovnakého veku, ale bez poškodenia funkcie pečene, sa AUC zvýšila o 85 % a Cmax o 47 %. Navyše hodnoty AUC a Cmax pre N-desmetylmetabolit boli signifikantne vyššie o 154 %, resp. o 87 % u pacientov
s cirhózou pečene v porovnaní s jedincami s normálnymi pečeňovými funkciami. Farmakokinetika sildenafilu u pacientov s ťažkým poškodením funkcie pečene nebola študovaná.
Populačnáfarmakokinetika
U pacientov s pľúcnou artériovou hypertenziou boli priemerné rovnovážne koncentrácie pri sledovaných dávkach 20 – 80 mg trikrát denne o 20 – 50 % vyššie než v porovnávanej skupine
zdravých dobrovoľníkov. Cmax dosiahla dvojnásobnú hodnotu oproti zdravým dobrovoľníkom. Oba výsledky napovedajú, že pacienti s pľúcnou artériovou hypertenziou majú oproti zdravým dobrovoľníkom nižší klírens a/alebo vyššiu biologickú dostupnosť sildenafilu per os.
Pediatrickápopulácia
Z analýzy farmakokinetického profilu sildenafilu u pacientov zaradených do pediatrických klinických skúšaní sa telesná hmotnosť ukázala ako dobrý prediktor expozície lieku u detí. Odhadované hodnoty
polčasu koncentrácie sildenafilu v plazme sa pohybovali v rozmedzí 4,2 až 4,4 hodiny u pacientov
s hmotnosťou v rozmedzí od 10 do 70 kg a neukázali žiadne rozdiely, ktoré by sa javili ako klinicky relevantné. Odhadovaná Cmax po perorálnom podaní jednorazovej dávky sildenafilu 20 mg pre pacientov s hmotnosťou 70, 20 a 10 kg bola 49, 104 a 165 ng/ml. Odhadovaná Cmax po perorálnom podaní jednorazovej dávky sildenafilu 10 mg pre pacientov s hmotnosťou 70, 20 a 10 kg bola
24, 53 a 85 ng/ml. Odhadovaný Tmax bol približne 1 hodina a bol takmer nezávislý od telesnej hmotnosti.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
U mláďat potkanov, ktoré boli pre- a postnatálne liečené sildenafilom v dávke 60 mg/kg, sa pozoroval menší počet mláďat vo vrhu, nižšia hmotnosť mláďat v 1. deň a znížené prežívanie do 4. dňa pri expozíciách, ktoré boli približne 50-násobkom očakávanej expozície u človeka pri dávke 20 mg trikrát denne. Účinky v predklinických štúdiách sa pozorovali pri expozíciách považovaných za dostatočne vyššie, než je maximálna expozícia u ľudí, čo poukazuje na malý význam týchto zistení pre klinické použitie.
U zvierat neboli pri klinicky relevantných expozíciách pozorované žiadne nežiaduce reakcie
s možným významom pre klinické použitie, ktoré by neboli tiež pozorované v klinických štúdiách.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok na perorálnu suspenziu: Sorbitol
Bezvodá kyselina citrónová
Sukralóza Nátriumcitrát Xantánová guma
Oxid titaničitý (E171) Nátriumbenzoát (E211)
Bezvodý koloidný oxid kremičitý
Hroznová príchuť: Maltodextrín
Koncentrát hroznovej šťavy
Arabská guma
Koncentrát ananásovej šťavy
Bezvodá kyselina citrónová
Prírodná príchuť
6.2 Inkompatibility
Neaplikovateľné
6.3 Čas použiteľnosti
2 roky
Po rekonštitúcii je perorálna suspenzia stabilná 30 dní.
6.4 Špeciálne upozornenia na uchovávanie
Prášok
Uchovávajte pri teplote neprevyšujúcej 30 °C.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
Perorálna suspenzia
Uchovávajte pri teplote do 30 °C alebo v chladničke (2° až 8 °C). Neuchovávajte v mrazničke. Podmienky na uchovávanie zloženej perorálnej suspenzie, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Jedna 125 ml jantárová sklenená fľaška (s polypropylénovým skrutkovacím uzáverom) obsahuje
32,27 g prášku na perorálnu suspenziu.
Jedna rekonštituovaná fľaška obsahuje 112 ml perorálnej suspenzie, z čoho 90 ml je určených pre dávkovanie a podanie.
Veľkosť balenia: 1 fľaška
Každé balenie obsahuje aj polypropylénovú odmerku (s označením 30 ml), polypropylénovú perorálnu dávkovaciu striekačku (3 ml) s HDPE piestom a LDPE adaptérom zatlačeným vo fľaške.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Odporúča sa, aby lekárnik pacientovi pred vydaním lieku rekonštituoval Revatio perorálnu suspenziu. Pokyny na rekonštitúciu
Poznámka: Celkový objem 90 ml (3 x 30 ml) vody, bez ohľadu na dávku, ktorú užívate, sa má použiť na rekonštitúciu obsahu fľašky
1. Poklepte po fľaške, aby sa uvoľnil prášok.
2. Odstráňte kryt.
3. Odmerajte 30 ml vody naplnením odmerky (priloženej v škatuľke) po značku a potom nalejte vodu do fľašky. Pomocou odmerky odmerajte ďalších 30 ml vody a pridajte ju do fľašky. (obrázok 1).
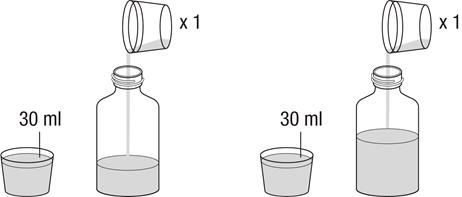
obrázok 1
4. Nasaďte kryt a dôkladne fľašku pretrepávajte minimálne počas 30 sekúnd. (obrázok 2).
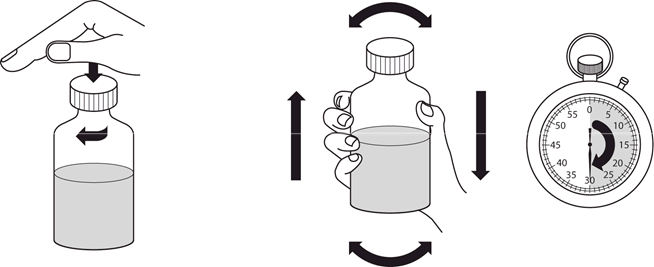
obrázok 2
5. Odstráňte kryt.
6. Pomocou odmerky odmerajte ďalších 30 ml vody a pridajte ju do fľašky. Vždy pridajte celkovo
90 ml (3 x 30 ml) vody bez ohľadu na dávku, ktorú užívate. (obrázok 3).
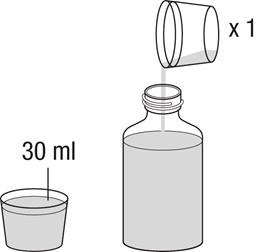
obrázok 3
7. Nasaďte kryt a dôkladne fľašku pretrepávajte minimálne počas 30 sekúnd. (obrázok 4).
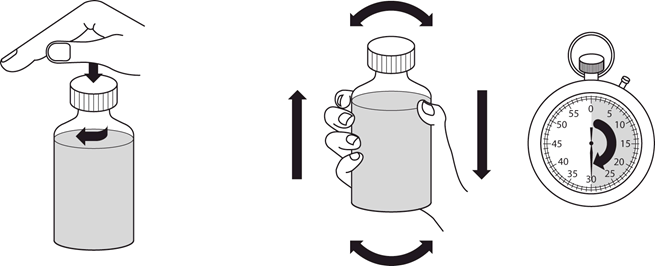
obrázok 4
8. Odstráňte kryt.
9. Zatlačte adaptér fľašky do hrdla fľašky (tak ako je to uvedené nižšie na obrázku 5). Adaptér je priložený, môžete teda naplniť perorálnu dávkovaciu striekačku s liekom z fľašky. Nasaďte kryt na fľašku.
obrázok 5
10. Zapíšte si dátum exspirácie pripravenej perorálnej suspenzie na štítok fľašky (dátum exspirácie pripravenej perorálnej suspenzie je 30 dní od dátumu prípravy). Po uplynutí tohto dátumu sa má akákoľvek nepoužitá perorálna suspenzia zlikvidovať alebo vrátiť do lekárne.
Pokyny pre použitie
1. Pred použitím dôkladne pretrepávajte zatvorenú fľašku pripravenej perorálnej suspenzie minimálne počas 10 sekúnd. Odstráňte kryt. (obrázok 6).
obrázok 6
2. S fľaškou v zvislej polohe, na rovnej ploche, vložte hrot perorálnej dávkovacej striekačky do adaptéra. (obrázok 7).
obrázok 7
3. Otočte fľašku hore dnom a pritom perorálnu dávkovaciu striekačku držte pripevnenú. Pomaly vyťahujte piest perorálnej dávkovacej striekačky po odmernú značku, ktorá označuje vašu dávku (natiahnutie 1 ml poskytuje dávku 10 mg, natiahnutie 2 ml poskytuje dávku 20 mg). Na
odmeranie presnej dávky má byť vrchná hrana piestu zarovnaná s príslušnou odmernou značkou
na perorálnej dávkovacej striekačke. (obrázok 8).
obrázok 8
4. Ak spozorujete veľké bubliny, pomaly zatlačte piest naspäť do striekačky. Tým vrátite liek naspäť do fľašky. Znovu zopakujte krok 3.
5. Otočte fľašku znovu hore dnom s pripevnenou perorálnou dávkovacou striekačkou. Vyberte perorálnu dávkovaciu striekačku z fľašky.
6. Vložte hrot perorálnej dávkovacej striekačky do úst. Nasmerujte hrot perorálnej dávkovacej
striekačky na vnútornú stranu líca. POMALY stláčajte piest perorálnej dávkovacej striekačky. Nestriekajte liek rýchlo. Ak sa liek bude podávať dieťaťu, pred podaním lieku sa uistite, že dieťa sedí alebo stojí vzpriamene. (obrázok 9).
obrázok 9
7. Nasaďte kryt na fľašku, pričom adaptér fľašky nechajte pripevnený. Perorálnu dávkovaciu striekačku umyte podľa nižšie uvedených pokynov.
Čistenie a uchovávanie striekačky:
1. Striekačka sa má umyť po každej dávke. Vytiahnite piest zo striekačky a umyte obe časti vodou.
2. Vysušte obe časti. Zatlačte piest naspäť do striekačky. Uchovávajte ju na čistom bezpečnom mieste spolu s liekom.
Po rekonštitúcii sa má perorálna suspenzia podávať len pomocou perorálnej dávkovacej striekačky priloženej v každom balení. Ďalšie podrobné pokyny pre použitie sú uvedené v písomnej informácii pre používateľa.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIPfizer Limited, Sandwich, Kent CT13 9NJ, Veľká Británia
8. REGISTRAČNÉ ČÍSLOEU/1/05/318/003
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. október 2005
Dátum posledného predĺženia: 23. september 2010
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.