
ených
injekčných striekačiek podaných za sebou do 30 minút.
Repatha 140 mg injekčný roztok naplnený v injekčnom pere
Dávka 420 mg raz mesačne alebo každé 2 týždne sa má aplikovať vo forme troch naplnených injekčných pier podaných za sebou do 30 minút.
Repatha 420 mg injekčný roztok v náplni
Dávka 420 mg raz mesačne alebo každé 2 týždne sa má aplikovať vo forme jednej náplne s automatizovaným minidávkovačom.
Repatha je určená na samopodávanie pacientom po príslušnom zaškolení. Podanie Repathy môže uskutočniť aj osoba, ktorá bola vyškolená na podanie lieku.
Iba na jednorazové použitie.
Pokyny na podávanie, pozri časť 6.6 a „Pokyny na používanie“ pribalené v škatuľke.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Porucha funkcieobličiek
Pacienti so závažnou poruchou funkcie obličiek (definovanou ako eGFR < 30 ml/min/1,73 m2) neboli
skúmaní (pozri časť 5.3). Repatha sa má používať opatrne u pacientov so závažnou poruchou funkcie obličiek.
Porucha funkciepečene
U pacientov so stredne závažnou poruchou funkcie pečene bolo pozorované zníženie celkovej
expozície evolokumabu, čo môže viesť k menšiemu účinku na zníženie LDL-C. Preto treba u týchto
pacientov zabezpečiť dôkladné monitorovanie.
Pacienti so závažnou poruchou funkcie pečene (trieda C podľa Childovej-Pughovej klasifikácie) neboli skúmaní (pozri časť 5.2). Repatha sa má používať opatrne u pacientov so závažnou poruchou funkcie pečene.
Suchý prírodný kaučuk
Repatha 140 mg injekčný roztok naplnený v injekčnej striekačke
Kryt ihly sklenej naplnenej injekčnej striekačky je zhotovený zo suchého prírodného kaučuku (derivát
latexu), ktorý môže vyvolať alergické reakcie.
Repatha 140 mg injekčný roztok naplnený v injekčnom pere
Kryt ihly naplneného injekčného pera je zhotovený zo suchého prírodného kaučuku (derivát latexu),
ktorý môže vyvolať alergické reakcie.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
S Repathou sa nevykonali žiadne formálne interakčné štúdie.
V klinických štúdiách s Repathou sa hodnotila farmakokinetická interakcia medzi statínmi a evolokumabom. Približne 20 % zvýšenie klírensu evolokumabu sa pozorovalo u pacientov, ktorým sa súbežne podávali statíny. Tento zvýšený klírens je sčasti sprostredkovaný statínmi zvyšujúcimi koncentráciu proproteínu konvertáza subtilizín/kexín 9 (PCSK9), ktorý neovplyvnil nepriaznivo farmakodynamický účinok evolokumabu na lipidy. Pri používaní spolu s Repathou úpravy dávky statínov nie sú potrebné.
Nevykonali sa štúdie o farmakokinetickej a farmakodynamickej interakcii medzi Repathou a liekmi
znižujúcimi lipidy inými ako sú statíny a ezetimib.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití Repathy u gravidných žien.
Štúdie na zvieratách nepreukázali priame ani nepriame účinky na reprodukčnú toxicitu (pozri
časť 5.3).
Repatha sa nemá používať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu
evolokumabom.
Dojčenie
Nie je známe, či sa evolokumab vylučuje do materského mlieka u ľudí.
Riziko pre novorodenca/dojča nemožno vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu Repathou, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre matku.
Fertilita
Nie sú dostupné žiadne údaje o vplyve evolokumabu na fertilitu u ľudí. Štúdie na zvieratách
nepreukázali účinky na koncové ukazovatele fertility vyjadrené ako plocha pod krivkou závislosti koncentrácie od času (AUC) pri hladinách expozícií oveľa vyšších ako u pacientov, ktorí dostávali
evolokumab v dávke 420 mg raz mesačne (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Repatha nemá známy vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
Najčastejšie hlásené nežiaduce reakcie pri odporúčaných dávkach počas pivotných štúdií s primárnou
hypercholesterolémiou a zmiešanou dyslipidémiou boli nazofaryngitída (4,8 %), infekcia horných dýchacích ciest (3,2 %), bolesť chrbta (3,1 %), artralgia (2,2 %), chrípka (2,3 %) a nauzea (2,1 %).
Profil bezpečnosti v populácii s homozygotnou familiárnou hypercholesterolémiou sa zhodoval
s profilom bezpečnosti preukázaným v populácii s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou.
Súhrn nežiaducich reakcií uvedený v tabuľke
Nežiaduce reakcie hlásené v pivotných kontrolovaných klinických štúdiách u pacientov s primárnou
hypercholesterolémiou a zmiešanou dyslipidémiou a homozygotnou familiárnou hypercholesterolémiou sú uvedené podľa triedy orgánových systémov a frekvencie v tabuľke 1 nižšie
na základe nasledujúcej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000).
T
abuľka 1. Nežiaduce reakcie pri liečbe Repathou
T
rieda orgánových systémov
(
T
O
S
) MedDRA
N
ežiaduce reakcie Trieda frekvencie
Infekcie a nákazy Chrípka Časté
Nazofaryngitída Časté
Infekcia horných dýchacích ciest
Časté
Poruchy imunitného systému Vyrážka Časté
Urtikária Menej časté
Poruchy gastrointestinálneho traktu Nauzea Časté
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva Celkové poruchy a reakcie v mieste
podania
Bolesť chrbta Časté Artralgia Časté Reakcie v mieste vpichu1 Časté
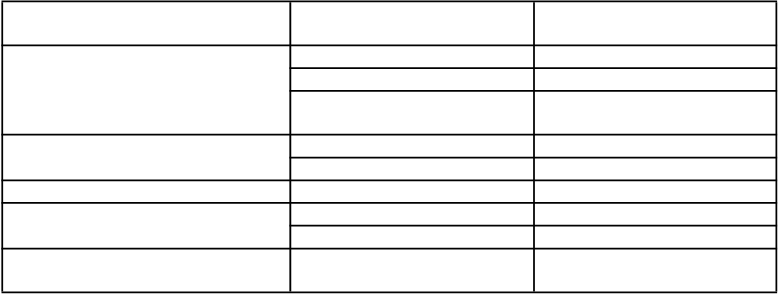
1Pozri časť Opis vybraných nežiaducich reakcií
Opis vybranýchnežiaducichreakciíReakcie v mieste vpichuNajčastejšie reakcie v mieste vpichu boli erytém, bolesť a modrina v mieste vpichu.
Pediatrická populáciaSkúsenosti s Repathou u pediatrických pacientov sú obmedzené. Do klinických štúdií bolo zaradených
štrnásť pacientov vo veku ≥ 12 až < 18 rokov s homozygotnou familiárnou hypercholesterolémiou. Medzi dospievajúcimi a dospelými pacientmi s homozygotnou familiárnou hypercholesterolémiou sa nepozoroval žiadny rozdiel v bezpečnosti.
Bezpečnosť a účinnosť Repathy u pediatrických pacientov s primárnou hypercholesterolémiou a
zmiešanou dyslipidémiou neboli stanovené.
Staršia populáciaHoci u pacientov vo veku 75 rokov a starších sa nepozorovali problémy z hľadiska bezpečnosti, údaje
v tejto vekovej podskupine sú obmedzené.
Z celkového počtu 6 026 pacientov v klinických štúdiách s Repathou bolo 1 779 (30 %) vo veku ≥ 65 rokov, zatiaľ čo 223 (4 %) bolo vo veku ≥ 75 rokov. Medzi týmito pacientmi a mladšími pacientmi sa nepozorovali celkové rozdiely v bezpečnosti alebo účinnosti.
ImunogenitaV klinických štúdiách 0,1 % pacientov (7 z 4 846 pacientov s primárnou hyperlipidémiou a zmiešanou
dyslipidémiou a 0 z 80 pacientov s homozygotnou familiárnou hypercholesterolémiou) liečených najmenej jednou dávkou Repathy malo pozitívny test na vývoj viažucich protilátok (4 z týchto pacientov mali krátkodobé protilátky). Pacienti, u ktorých sa zistili pozitívne viažuce protilátky v sére, boli ďalej hodnotení z hľadiska neutralizujúcich protilátok a u žiadneho pacienta sa nezistili pozitívne neutralizujúce protilátky. Prítomnosť viažucich protilátok proti evolokumabu neovplyvnila farmakokinetický profil, klinickú odpoveď ani bezpečnosť Repathy.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
V štúdiách na zvieratách pri expozíciách až 300-násobne vyšších ako expozície u pacientov liečených
Repathou v dávke 420 mg raz mesačne sa nepozorovali žiadne nežiaduce účinky.
Neexistuje špecifická liečba predávkovania Repathou. V prípade predávkovania sa má pacient liečiť
symptomaticky a podľa potreby sa majú vykonať podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné látky upravujúce lipidy. ATC kód: C10AX13
Mechanizmus účinku
Evolokumab sa selektívne viaže na PCSK9 a cirkulujúcemu proteínu PCSK9 bráni naviazať sa na
receptor lipoproteínu s nízkou hustotou (LDLR) na povrchu pečeňových buniek, čím zabraňuje
degradácii LDL receptorov sprostredkovanej proteínom PCSK9. Zvýšenie počtu LDL receptorov
v pečeni má za následok súvisiace zníženie LDL cholesterolu (LDL-C) v sére.
Farmakodynamické účinky
V klinických štúdiách Repatha znížila neviazaný PCSK9, LDL-C, TC, ApoB, non-HDL-C, TC/HDL-
C, ApoB/ApoA1, VLDL-C, TG a Lp(a) a zvýšila HDL-C a ApoA1 u pacientov s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou.
Jednorazové subkutánne podanie Repathy v dávke 140 mg alebo 420 mg viedlo k maximálnemu potlačeniu cirkulujúceho neviazaného PCSK9 do 4 hodín, za ktorým nasledovalo zníženie LDL-C dosahujúce priemernú najnižšiu hodnotu (nadir) odpovede do 14 a 21 dní, v uvedenom poradí. Zmeny v neviazanom PCSK9 a sérových lipoproteínov boli reverzibilné po prerušení liečby Repathou. Počas vymývania evolokumabu sa nepozoroval nárast neviazaného PCSK9 alebo LDL-C nad východiskové hodnoty, čo nasvedčuje, že kompenzačné mechanizmy na zvýšenie tvorby PCSK9 a LDL-C sa počas liečby nevyskytujú.
Subkutánne režimy dávok 140 mg každé 2 týždne a 420 mg raz mesačne boli pri priemernom znížení LDL-C (priemer 10. a 12. týždňa) ekvivalentné, čo viedlo k 72 % až 57 % zníženiu od začiatku liečby v porovnaní s placebom. Liečba Repathou používanou samostatne alebo v kombinácii s inou liečbou znižujúcou lipidy viedla k podobnému zníženiu LDL-C. Účinok zníženia LDL-C je dlhotrvajúci; najdlhšie zmerané trvanie bolo 112 týždňov.
Klinická účinnosťpriprimárnej hypercholesterolémii a zmiešanej dyslipidémii
Zníženie LDL-C približne o 55 % až 75 % sa s Repathou dosiahlo už 1. týždeň a počas dlhodobej
liečby sa udržalo. Maximálna odpoveď sa zvyčajne dosiahla do 1 až 2 týždňov po podávaní dávok
140 mg každé 2 týždne a 420 mg raz mesačne.
V 80 % – 85 % všetkých pacientov liečených jednou alebo druhou dávkou Repathy preukázala ≥ 50 % zníženie LDL-C v priemere 10. a 12. týždňa. Až 99 % pacientov liečených jednou alebo druhou dávkou Repathy dosiahlo LDL-C < 2,6 mmol/l a až 95 % dosiahlo LDL-C < 1,8 mmol/l v priemere
10. a 12. týždňa.
Repatha bola v oboch dávkach účinná oproti placebu a ezetimibu vo všetkých podskupinách, pričom medzi podskupinami sa nepozorovali žiadne významné rozdiely napríklad z hľadiska veku, rasy, pohlavia, regiónu, indexu telesnej hmotnosti, rizika podľa Národného cholesterolového edukačného
programu (NCEP, National Cholesterol Education Program risk), súčasného stavu fajčenia, východiskových rizikových faktorov koronárneho ochorenia srdca (KOS), rodinnej anamnézy predčasného KOS, stavu glukózovej tolerancie (t. j. diabetes mellitus 2. typu, metabolický syndróm alebo ani jedno z nich), hypertenzie, dávky a účinnosti statínu, neviazaného východiskového PCSK9, východiskových hladín LDL-C a východiskových hladín TG.
Repatha znížila LDL-C, non-HDL-C, Apo B, TC, Lp(a), VLDL-C, TG, TC/HDL-C a ApoB/ApoA1 a
zvýšila HDL-C u pacientov so zmiešanou dyslipidémiou.
Repatha bola pri znižovaní LDL-C, TC, ApoB, non-HDL-C, Lp(a), TC/HDL-C a ApoB/ApoA1
superiórna voči ezetimibu.
Kombinácia so statínom a statínom s inou liečbou znižujúcou lipidy
LAPLACE-2 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná 12-týždňová
štúdia s 1 896 pacientmi s primárnou hypercholesterolémiou alebo zmiešanou dyslipidémiou, ktorí boli randomizovaní na užívanie Repathy v kombinácii so statínmi (rosuvastatínom, simvastatínom alebo atorvastatínom). Repatha sa porovnávala s placebom v skupine s rosuvastatínom a simvastatínom a s placebom a ezetimibom v skupine s atorvastatínom.
Repatha významne znížila LDL-C od začiatku liečby po priemer 10. a 12. týždňa v porovnaní
s placebom v skupine s rosuvastatínom a simvastatínom a v porovnaní s placebom a ezetimibom
v skupine s atorvastatínom (p < 0,001). Repatha významne znížila TC, ApoB, non-HDL-C, TC/HDL- C, ApoB/ApoA1, VLDL-C, TG a Lp(a) a zvýšila HDL-C od začiatku liečby po priemer 10. a 12.
týždňa v porovnaní s placebom v skupine s rosuvastatínom a simvastatínom (p < 0,05) a významne
znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 a Lp(a) v porovnaní s placebom a ezetimibom v skupine s atorvastatínom (p < 0,001) (pozri tabuľky 2 a 3).
RUTHERFORD-2 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom kontrolovaná 12-týždňová štúdia s 329 pacientmi s heterozygotnou familiárnou hypercholesterolémiou, ktorí dostávali liečbu znižujúcu lipidy. Repatha významne znížila LDL-C od začiatku liečby po priemer 10. a 12. týždňa v porovnaní s placebom (p < 0,001). Repatha významne znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 VLDL-C, TG a Lp(a) a zvýšila HDL-C a ApoA1 od začiatku liečby po priemer 10. a 12. týždňa v porovnaní s placebom (p < 0,05) (pozri tabuľku 2).
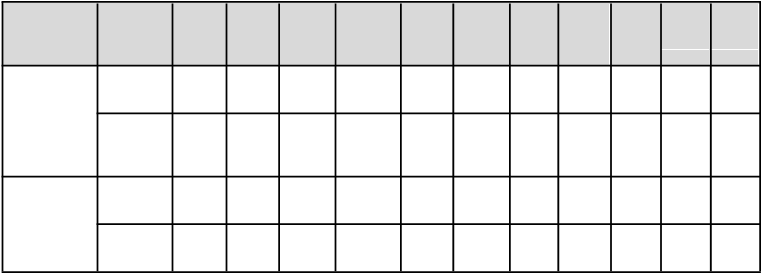
Tabuľka 2: Liečebné účinky Repathy v porovnaní s placebom u pacientov s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou – priemerná percentuálna zmena od začiatku liečby po priemer 10. a 12. týždňa (%, CI 95 %)
Štúd
i
a Dávkovací
režim
L
D
L
-C
(
%
)
N
o
n-
HDL-C
Ap
o B
(
%
)
TC
(
%
)
L
p(
a
)
(
%
)
V
L
D
L
-
C
HDL-
C
TG
(
%
)
Ap
o
A
1
(
%
)
Pomer
T
C
/
Pomer
Ap
oB
/
(
%
)
(
%
)
(
%
)
HDL-C ApoA1
LAPLACE-2
(HMD)
140 mg
Q2W
−72b
(−75,
−60b
(−63,
−56b
(−58,
−41b
(−43,
−30b
(−35,
−18b
(−23,
b −17b
6 (−22,
%
b −45b
3 (−47,
%
−56b
(−59,
(skupiny
s rosuvastatínom, simvastatínom a atorvastatínom
spolu)
RUTHERFORD-
2 (HeFH)
(N = 555)
420 mg
QM
(N = 562)
140 mg
Q2W
(N = 110)
420 mg
QM
(N = 110)
−69)
−69b
(−73,
−65)
−61b
(−67,
−55)
−66b
(−72,
−61)
−58)
−60b
(−63,
−57)
−56b
(−61,
−51)
−60b
(−65,
−55)
−53)
−56b
(−58,
−53)
−49b
(−54,
−44)
−55b
(−60,
−50)
−39)
−40b
(−42,
−37)
−42b
(−46,
−38)
−44b
(−48,
−40)
−25)
−27b
(−31,
−24)
−31b
(−38,
−24)
−31b
(−38,
−24)
−14)
−22b
(−28,
−17)
−23b
(−29,
−16)
−16b
(−23,
−8)
(4, 8)
8b
(6,
10)
8b
(4,
12)
9b
(5,
14)
−13)
−23b
(−28,
−17)
−22b
(−29,
−15)
−17b
(−24, −9)
(1, 5)
5b
(3, 7)
7a
(3, 12)
5a
(1, 9)
−42)
−46b
(−48,
−43)
−47b
(−51,
−42)
−49b
(−54,
−44)
−53)
−58b
(−60,
−55)
−53 (−58,
−48)
−56b
(−61,
−50)
Legenda: Q2W = jedenkrát každé 2 týždne, QM = raz mesačne, HMD = primárna hypercholesterolémia a zmiešaná dyslipidémia; HeFH = heterozygotná familiárna hypercholesterolémia; a hodnota p < 0,05 pri porovnaní s placebom. b hodnota p < 0,001 pri porovnaní s placebom.
Pacienti netolerujúci statíny
GAUSS-2 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, ezetimibom
kontrolovaná 12-týždňová štúdia s 307 pacientmi, ktorí netolerujú statíny alebo nedokážu tolerovať účinnú dávku statínu. Repatha významne znížila LDL-C v porovnaní s ezetimibom (p < 0,001). Repatha významne znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 a Lp(a) od začiatku liečby po priemer 10. a 12. týždňa v porovnaní s ezetimibom (p < 0,001) (pozri tabuľku 3).
Liečba bez statínov
MENDEL-2 bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom
a ezetimibom kontrolovaná 12- týždňová štúdia s Repathou so 614 pacientmi s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou. Repatha významne znížila LDL-C od začiatku
liečby po priemer 10. a 12. týždňa v porovnaní s placebom aj ezetimibom (p < 0,001). Repatha
významne znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 a Lp(a) od začiatku liečby po
priemer 10. a 12. týždňa v porovnaní s placebom aj ezetimibom (p < 0,001) (pozri tabuľku 3).
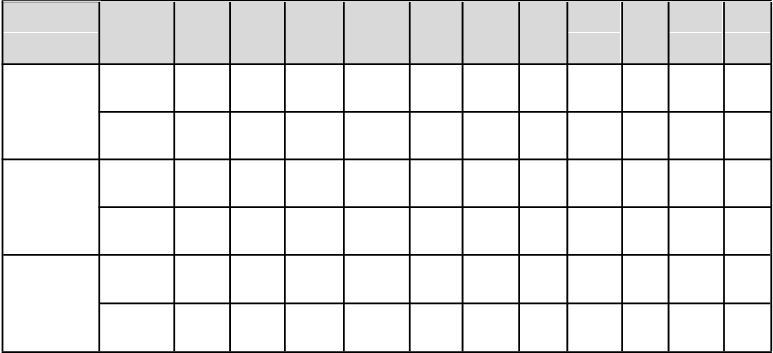
Tabuľka 3: Liečebné účinky Repathy v porovnaní s ezetimibom u pacientov s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou – priemerná percentuálna zmena od začiatku liečby po priemer 10. a 12. týždňa (%, 95 % CI)
Štúd
i
a Dávkovací
režim
L
D
L
-
C
(
%
)
N
o
n-
HDL-C (%)
Ap
o B
(
%
)
TC
(
%
)
L
p(
a
)
(
%
)
V
L
D
L
-
C (%)
HDL
-
C (%)
TG
(
%
)
Ap
o
A
1
(
%
)
Pomer
T
C
/ HDL-C
%
Pomer
Ap
oB
/ ApoA1
%
LAPLACE-2 (HMD)
140 mg
Q2W
−43c
(−50,
−34c
(−39,
−34c
(−38,
−23c
(−26,
−30c
(−35, −1
7c
(4, −2
c −27c
7 (−30,
−38c
(−42,
(skupiny
s atorvastatínom spolu)
GAUSS-2 (intolerancia statínov)
MENDEL-2 (liečba bez statínov)
(N = 219)
420 mg
QM
(N = 220)
140 mg
Q2W
(N = 103)
420 mg
QM
(N = 102)
140 mg
Q2W
(N = 153)
420 mg
QM
(N = 153)
−37)
−46c
(−51,
−40)
−38b
(−44, −33)
−39b
(-44, −35)
−40b
(−44,
−37)
−41b
(−44,
−37)
−30)
−39c
(−43,
−34)
−32b
(−36,
−27)
−35b
(−39,
−31)
−36b
(−39,
−32)
−35b
(−38,
−33)
−30)
−40c
(−44,
−36)
−32b
(−37,
−27)
−35b
(−40,
−30)
−34b
(−37,
−30)
−35b
(−38,
−31)
−19)
−25c
(−29,
−22)
−24b
(−28,
−20)
−26b
(−30,
−23)
−25b
(−28,
−22)
−25b
(−28,
−23)
−25)
−33c
(−41,
−26)
−24b
(−31,
−17)
−25b
(−34,
−17)
−22b
(−29,
−16)
−20b
(−27,
−13)
(−7, 5)
−7 (−20,
6)
−2 (−10,
7)
−4 (−13,
6)
−7 (−14,
1)
−10 (−19,
−1)
10)
8c
(5,
12)
5 (1,
10)
6 (1,
10)
6a
(3, 9)
4 (1, 7)
(−9, 5)
−8 (−21, 5)
−3 (−11, 6)
−6 (−17, 4)
−9 (−16, −1)
−9 (−18, 0)
(4, 9)
7c
(2, 11)
5a
(2, 9)
3 (−1, 7)
3 (0, 6)
4a
(1, 7)
−23)
−30c
(−34,
−26)
−27b
(−32,
−23)
−30b
(−35,
−25)
−29b
(−32,
−26)
−28b
(−31,
−24)
−34)
−42c
(−47,
−38)
−35b
(−40,
−30)
−36b
(−42,
−31)
−35b
(−39,
−31)
−37b
(−41,
−32)
Legenda: Q2W = jedenkrát každé 2 týždne, QM = raz mesačne, HMD = primárna hypercholesterolémia a zmiešaná dyslipidémia, a hodnota p < 0,05 pri porovnaní s ezetimibom, b hodnota p < 0,001 pri porovnaní s ezetimibom,
c nominálna hodnota p < 0,001 pri porovnaní s ezetimibom.
Dlhodobá účinnosť priprimárnej hypercholesterolémii a zmiešanej dyslipidémii
DESCARTES bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom
kontrolovaná 52-týždňová štúdia s 901 pacientmi s hyperlipidémiou, ktorí dostávali samotnú diétu, atorvastatín alebo kombináciu atorvastatínu s ezetimibom. Repatha v dávke 420 mg raz mesačne významne znížila LDL-C od začiatku liečby po 52. týždeň v porovnaní s placebom (p < 0,001). Liečebné účinky sa udržali celý 1 rok, ako dokazuje zníženie LDL-C od 12. do 52. týždňa. Zníženie LDL-C od začiatku liečby po 52. týždeň v porovnaní s placebom bolo konzistentné vo všetkých základných liečbach znižujúcich lipidy, optimalizovaných z hľadiska LDL-C a kardiovaskulárneho rizika.
Repatha významne znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG a Lp(a)
a zvýšila HDL-C a ApoA1 v 52. týždni v porovnaní s placebom (p < 0,001) (tabuľka 4).
T
abuľka 4: Liečebné účinky Repathy v porovnaní s placebom u pacientov s primárnou hypercholesterolémiou a zmiešanou dyslipidémiou – priemerná percentuálna zmena od začiatku liečby po 52. týždeň (%, 95 % CI)
Štúd
i
a Dávkovací
re
ž
im
DESCART 420 mg QM
LDL-C(%)−59b
Non-HDL-C (%)−50b
Apo B(%)−44b
TC (%)
(%)−33b
Lp(a)(%)−22b
VLDL-C (%)−29b
HDL-C (%)5b
TG(%)−12b
ApoA1(%)3a
PomerTC/ HDL-C%−37b
PomerApoB/ ApoA1%−46b
ES (N = 599)
(−64, −55) (−54, −46) (−48, −41)
(−36,
−31)
(−26,
−19)
(−40,
−18)
(3, 8)
(−17, −6)
(1, 5)
(−40,
−34)
(−50,
−43)
Legenda: QM = raz mesačne, a nominálna hodnota p < 0,001 pri porovnaní s placebom, b hodnota p < 0,001 pri porovnaní s placebom.
OSLER a OSLER-2 sú dva prebiehajúce, randomizované, kontrolované, otvorené predĺženia štúdií
hodnotiace dlhodobú bezpečnosť a účinnosť Repathy u pacientov, ktorí ukončili liečbu v „materskej“
štúdii. V každom predĺžení štúdie boli pacienti randomizovaní v pomere 2 : 1 na absolvovanie buď liečby Repathou a štandardnej liečby (skupina s evolokumabom), alebo iba štandardnej liečby (kontrolná skupina) prvý rok štúdie. Na konci prvého roka (52. týždeň v štúdii OSLER a 48. týždeň
v štúdii OSLER-2) pacienti spĺňali podmienky na vstup do celého obdobia liečby Repathou, v ktorom
všetci účastníci mohli dostávať otvorenú liečbu Repathou buď ďalšie 4 roky (OSLER), alebo 1 rok
(OSLER-2).
V štúdii OSLER bolo zaradených spolu 1 324 pacientov. Repatha v dávke 420 mg raz mesačne významne znížila LDL-C od začiatku liečby do 12. týždňa a 52. týždňa v porovnaní s kontrolnou skupinou (nominálna hodnota p < 0,001). Liečebné účinky sa udržali 124 týždňov, ako dokazuje zníženie LDL-C od 12. týždňa v materskej štúdii do 112. týždňa v otvorenom predĺžení štúdie.
V štúdii OSLER-2 bolo zaradených spolu 2 928 pacientov. Repatha významne znížila LDL-C od
začiatku liečby do 12. týždňa v porovnaní s kontrolnou skupinou (nominálna hodnota p < 0,001). Liečebné účinky sa udržali, ako dokazuje zníženie LDL-C od 12. týždňa do 24. týždňa v otvorenom predĺžení štúdie. Repatha významne znížila TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG a Lp(a) a zvýšila HDL-C a ApoA1 od začiatku liečby do 52. týždňa v štúdii OSLER a do 24. týždňa v štúdii OSLER-2 v porovnaní s kontrolnou skupinou (nominálna hodnota p < 0,001). LDL-C a ďalšie lipidové parametre sa vrátili k východiskovým hodnotám do 12 týždňov po prerušení liečby Repathou na začiatku štúdie OSLER alebo OSLER-2 bez dôkazu tzv. rebound fenoménu.
TAUSSIG je prebiehajúce multicentrické, otvorené, 5-ročné predĺženie štúdie, ktoré hodnotí dlhodobú bezpečnosť a účinnosť Repathy ako adjuvantnej liečby k inej liečbe znižujúcej lipidy u pacientov so závažnou familiárnou hypercholesterolémiou vrátane homozygotnej familiárnej hypercholesterolémie. Do štúdie TAUSSIG bolo zaradených spolu 102 pacientov so závažnou familiárnou hypercholesterolémiou a 96 pacientov s homozygotnou familiárnou hypercholesterolémiou. Všetci pacienti v štúdii boli primárne liečení Repathou v dávke 420 mg raz mesačne okrem pacientov, ktorí pri zaraďovaní dostávali aferézu a s Repathou začali v dávke 420 mg jedenkrát každé 2 týždne. Frekvencia dávkovania u pacientov bez aferézy sa mohla titrovať nahor až do 420 mg jedenkrát každé
2 týždne na základe odpovede LDL-C a hladín PCSK9. Dlhodobé používanie Repathy preukázalo pretrvávajúci liečebný účinok, o čom svedčí zníženie LDL-C u pacientov so závažnou familiárnou
hypercholesterolémiou (tabuľka 5).
Aj zmeny v ďalších lipidových parametroch (TC, ApoB, non-HDL-C, TC/HDL-C a ApoB/ApoA1) preukázali pretrvávajúci účinok dlhodobého podávania Repathy u pacientov so závažnou familiárnou hypercholesterolémiou.
T
abuľka 5: Účinok Repathy na LDL-C u pacientov so závažnou familiárnou
hypercholesterolémiou – medián percentuálnej zmeny od začiatku liečby do 36. týždňa OPŠ
P
opulácia pacientov
(
N
)
12. týždeň OPŠ
(
n = 16)
24. týždeň OPŠ
(
n = 8)
36. týždeň OPŠ
(
n = 5)
Závažná FH (N = 102) −47 −45 −48
Legenda: OPŠ = otvorené predĺženie štúdie, N (n) = počet hodnotiteľných pacientov (N) a pacientov
s hodnotami LDL pozorovanými na osobitnej naplánovanej návšteve (n) v skupine so závažnou familiárnou hypercholesterolémiou na predbežnú analýzu.
Klinická relevancia pretrvávajúcich veľmi nízkych hladín LDL-C (t. j. < 0,65 mmol/l [< 25 mg/dl]) vrátane dlhodobej bezpečnosti ešte nebola stanovená. Dostupné údaje preukazujú, že medzi profilmi bezpečnosti u pacientov s hladinami LDL-C < 0,65 mmol/l a profilmi bezpečnosti v prípade vyšších hodnôt LDL-C nie sú klinicky významné rozdiely, pozri časť 4.8.
Klinická účinnosťprihomozygotnej familiárnej hypercholesterolémii
TESLA bola medzinárodná, multicentrická, dvojito zaslepená, randomizovaná, placebom
kontrolovaná 12-týždňová štúdia so 49 pacientmi s homozygotnou familiárnou hypercholesterolémiou vo veku 12 až 65 rokov. Repatha v dávke 420 mg raz mesačne ako adjuvantná liečba k inej liečbe
znižujúcej lipidy (napr. statíny, sekvestranty žlčových kyselín) významne znížila LDL-C a ApoB po
12. týždni v porovnaní s placebom (p < 0,001) (tabuľka 6). Aj zmeny v ďalších lipidových
parametroch (TC, non-HDL-C, TC/HDL-C a ApoB/ApoA1) preukázali liečebný účinok podávania
Repathy pacientom s homozygotnou familiárnou hypercholesterolémiou.
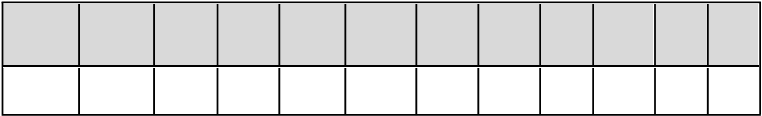
Tabuľka 6: Liečebné účinky Repathy v porovnaní s placebom u pacientov s homozygotnou familiárnou hypercholesterolémiou – priemerná percentuálna zmena od začiatku liečby do 12. týždňa (%, CI 95 %)
Štúd
i
a Dávkovací
režim
L
D
L
-
C
(
%
)
N
o
n-HDL-
C (%)
Ap
o B
(
%
)
TC
 (
%
)
(
%
)
'
L
p(
a
)
(
%
)
V
L
D
L
-
C (%)
HDL-
C (%)
TG
(
%
)
Pomer
T
C
/ HDL-C
%
Pomer
Ap
oB
/ ApoA1
%
TESLA
420 mg
−32b
−30a
−23b
−27a
−12
−44
−0,1
0,3
−26a
−28a
(HoFH)
QM
(N = 33)
(−45, −19) (−42, −18) (−35, −11)
(−38,
−16)
(−25, 2)
(−128,
40)
(−9, 9)
(−15, 16)
(−38,
−14)
(−39,
−17)

Legenda: HoFH = homozygotná familiárna hypercholesterolémia; QM = raz mesačne; a nominálna hodnota p
< 0,001 pri porovnaní s placebom; b hodnota p < 0,001 pri porovnaní s placebom.
Dlhodobá účinnosť prihomozygotnej familiárnej hypercholesterolémiiV štúdii TAUSSIG dlhodobé používanie Repathy preukázalo trvalý liečebný účinok, o čom svedčí
zníženie LDL-C približne o 20 % až 30 % u pacientov s homozygotnou familiárnou hypercholesterolémiou, ktorí nie sú liečení aferézou, a približne o 15 % až 25 % u pacientov
s homozygotnou familiárnou hypercholesterolémiou, ktorí sú liečení aferézou (tabuľka 7). Aj zmeny
v ďalších lipidových parametroch (TC, ApoB, non-HDL-C, TC/HDL-C a ApoB/ApoA1) preukázali pretrvávajúci účinok dlhodobého podávania Repathy u pacientov s homozygotnou familiárnou hypercholesterolémiou. Zníženie LDL-C a zmeny ďalších lipidových parametrov u 13 dospievajúcich pacientov (vo veku ≥ 12 až < 18 rokov) s homozygotnou familiárnou hypercholesterolémiou sú porovnateľné so znížením LDL-C a zmenami ďalších lipidových parametrov v celkovej populácii pacientov s homozygotnou familiárnou hypercholesterolémiou.
T
abuľka 7: Účinok Repathy na LDL-C u pacientov s homozygotnou familiárnou hypercholesterolémiou – priemerná percentuálna zmena od začiatku liečby do 36. týždňa OPŠ
Populácia pacientov
(
N)
12
. týždeň OPŠ 24. týždeň OPŠ 36. týždeň OPŠ
HoFH (N = 96)
Bez aferézy
(N = 65)
S aferézou
(N = 31)
−20
(n = 70)
−22
(n = 46)
−17
(n = 24)
−23
(n = 46)
−24
(n = 33)
−20
(n = 13)
−24
(n = 30)
−24
(n = 27)
−21 (n = 3)

Legenda: OPŠ = otvorené predĺženie štúdie; N (n) = počet hodnotiteľných pacientov (N) a pacientov
s hodnotami LDL pozorovanými na osobitnej naplánovanej návšteve (n) v skupine s HoFH na predbežnú
analýzu.
Účinok Repathy na kardiovaskulárnu morbiditu a mortalitu ešte nebol preukázaný.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Repathou vo
všetkých podskupinách pediatrickej populácie v liečbe zmiešanej dyslipidémie.
Európska agentúra pre lieky udelila odklad povinnosti predložiť výsledky štúdií s Repathou v jednej alebo vo viacerých podskupinách pediatrickej populácie v liečbe zvýšeného cholesterolu.
O používaní Repathy v pediatrickej populácii sú dostupné obmedzené údaje. Do klinických skúšaní bolo zaradených štrnásť dospievajúcich pacientov vo veku ≥ 12 až < 18 rokov s homozygotnou familiárnou hypercholesterolémiou. Medzi dospievajúcimi a dospelými pacientmi s homozygotnou familiárnou hypercholesterolémiou sa nepozorovali žiadne celkové rozdiely v bezpečnosti a účinnosti.
Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnostiAbsorpcia a distribúciaPo jednej subkutánnej dávke 140 mg alebo 420 mg Repathy podanej zdravým dospelým sa stredná
hodnota maximálnych sérových koncentrácií dosiahla za 3 až 4 dni. Podanie jednorazovej subkutánnej dávky 140 mg viedlo k priemernej hodnote Cmax (SD, štandardná odchýlka) 13,0 (10,4) μg/ml a priemernej hodnote AUClast (SD) 96,5 (78,7) deň•μg/ml. Podanie jednorazovej subkutánnej dávky
420 mg viedlo k priemernej hodnote Cmax (SD) 46,0 (17,2) μg/ml a priemernej hodnote AUClast (SD)
842 (333) deň•μg/ml. Tri subkutánne 140 mg dávky boli biologicky rovnocenné s jednorazovou subkutánnou 420 mg dávkou. Absolútna biologická dostupnosť po podaní s.c. dávok sa na základe farmakokinetických modelov stanovila na 72 %.
Po jednorazovej intravenóznej 420 mg dávke Repathy sa priemerný (SD) objem distribúcie
v rovnovážnom stave odhadol na 3,3 (0,5) l, čo nasvedčuje, že evolokumab má obmedzenú distribúciu v tkanivách.
BiotransformáciaRepathu tvoria výhradne aminokyseliny a sacharidy ako prirodzený imunoglobulín a nie je
pravdepodobné, že by bol eliminovaný prostredníctvom metabolických mechanizmov pečene. Predpokladá sa, že jeho metabolizmus a eliminácia sledujú dráhy klírensu imunoglobulínu, čo vedie
k degradácii na malé peptidy a jednotlivé aminokyseliny.
Eliminácia
Odhaduje sa, že evolokumab má účinný polčas 11 až 17 dní.
U pacientov s primárnou hypercholesterolémiou alebo zmiešanou dyslipidémiou, ktorí užívali vysoké dávky statínov, bola systémová expozícia evolokumabu mierne nižšia ako u pacientov užívajúcich statíny v nízkej až stredne veľkej dávke (pomer AUClast 0,74 [90 % CI 0,29; 1,9]). Približne 20 % zvýšenie klírensu je sčasti sprostredkované statínmi zvyšujúcimi koncentráciu PCSK9, čo neovplyvnilo nepriaznivo farmakodynamický účinok evolokumabu na lipidy. Populačná farmakokinetická analýza nepreukázala výrazné rozdiely v koncentráciách evolokumabu v sére
u hypercholesterolemických pacientov (non-familiárna hypercholesterolémia alebo familiárna hypercholesterolémia), ktorí súbežne dostávali statíny.
Linearita/nelinearita
Po jednorazovej 420 mg intravenóznej dávke bol priemerný (SD) systémový klírens odhadnutý na
12 (2) ml/h. V klinických štúdiách s opakovaným subkutánnym podávaním dávok počas 12 týždňov sa pozorovalo zvýšenie expozície úmerne s dávkou pri dávkovaní 140 mg a viac. Približne dvoj- až
trojnásobná kumulácia sa pozorovala v minimálnych sérových koncentráciách (Cmin (SD) 7,21 (6,6))
po 140 mg dávkach každé 2 týždne alebo po 420 mg dávkach podaných mesačne (Cmin (SD) 11,2 (10,8)) a minimálne sérové koncentrácie dosiahli rovnovážny stav po 12 týždňoch podávania dávok.
V sérových koncentráciách sa za obdobie 124 týždňov nepozorovali zmeny závislé od času.
Porucha funkcieobličiek
U pacientov s miernou až stredne závažnou poruchou funkcie obličiek nie je potrebná úprava dávky.
Populačná farmakokinetická analýza integrovaných údajov z klinických štúdií s Repathou neodhalila žiadny rozdiel vo farmakokinetike evolokumabu u pacientov s miernou až stredne závažnou poruchou funkcie obličiek oproti pacientom bez poruchy funkcie obličiek. Repatha nebola skúmaná u pacientov so závažnou poruchou funkcie obličiek (pozri časť 4.4).
Porucha funkciepečene
U pacientov s miernou poruchou funkcie pečene (triedy A podľa Childovej-Pughovej klasifikácie) nie
je potrebná úprava dávky. Jednorazové 140 mg subkutánne dávky Repathy sa skúmali u 8 pacientov
s miernou poruchou funkcie pečene, u 8 pacientov so stredne závažnou poruchou funkcie pečene a u 8
zdravých účastníkov. Zistilo sa, že expozícia evolokumabu je približne o 40 % – 50 % nižšia
v porovnaní so zdravými účastníkmi. Zistilo sa však, že východiskové hladiny PCSK9 a stupeň
a časový priebeh neutralizácie PCSK9 sú podobné medzi pacientmi s miernou alebo stredne závažnou poruchou funkcie pečene a zdravými dobrovoľníkmi. To malo za následok podobný časový priebeh
a podobnú mieru absolútneho zníženia LDL-C. Repatha sa neskúmala u pacientov so závažnou
poruchou funkcie pečene (triedy C podľa Childovej-Pughovej klasifikácie) (pozri časť 4.4).
Telesná hmotnosť
Telesná hmotnosť bola v populačnej PK analýze významným kovariátom ovplyvňujúcim minimálne
koncentrácie evolokumabu, vplyv na zníženie LDL-C sa však nepozoroval. Po opakovanom subkutánnom podaní dávky 140 mg každé 2 týždne boli minimálne koncentrácie v 12. týždni o 147 % vyššie u pacientov vážiacich 69 kg a o 70 % nižšie u pacientov vážiacich 93 kg ako u typického účastníka vážiaceho 81 kg. Menší vplyv telesnej hmotnosti sa pozoroval pri opakovanom subkutánnom podaní evolokumabu v dávke 420 mg mesačne.
I
né osobitné populácie
Populačné farmakokinetické analýzy nasvedčujú, že z hľadiska veku, rasy alebo pohlavia nie sú
potrebné úpravy dávky. Farmakokinetiku evolokumabu ovplyvnila telesná hmotnosť bez významného
účinku na zníženie LDL-C. Preto na základe telesnej hmotnosti nie sú potrebné úpravy dávky.
5.3 Predklinické údaje o bezpečnosti
Evolokumab nebol karcinogénny pri škrečkoch s expozíciami oveľa vyššími ako u pacientov, ktorí dostávajú evolokumab v dávke 420 mg raz mesačne. Mutagénny potenciál evolokumabu sa nehodnotil.
Pri škrečkoch a opiciach rodu cynomolgus s expozíciami oveľa vyššími ako u pacientov, ktorí dostávali evolokumab v dávke 420 mg raz mesačne, sa nepozoroval žiadny účinok na samčiu alebo samičiu fertilitu.
Pri opiciach rodu cynomolgus s expozíciami oveľa vyššími ako u pacientov, ktorí dostávali evolokumab v dávke 420 mg raz mesačne, sa nepozoroval žiadny účinok na embryonálno-fetálny alebo postnatálny vývoj (do veku 6 mesiacov).
Okrem zníženej protilátkovej odpovede závislej od T-buniek pri opiciach rodu cynomolgus imunizovaných hemocyanínom KLH (keyhole limpet hemocyanin) po 3 mesiacoch liečby evolokumabom sa nepozorovali žiadne nežiaduce účinky pri škrečkoch (až 3 mesiace) a opiciach rodu cynomolgus (až 6 mesiacov) s expozíciami oveľa vyššími ako u pacientov, ktorí dostávali evolokumab v dávke 420 mg raz mesačne. V týchto štúdiách sa pozoroval zamýšľaný farmakologický účinok zníženého sérového LDL-C a celkového cholesterolu a po ukončení liečby bol reverzibilný.
V kombinácii s rosuvastatínom za 3 mesiace sa nepozorovali žiadne nežiaduce účinky pri opiciach rodu cynomolgus s expozíciami oveľa vyššími ako u pacientov, ktorí dostávali evolokumab v dávke
420 mg raz mesačne. Zníženia sérového LDL-C a celkového cholesterolu boli výraznejšie ako
zníženia pozorované už v minulosti s evolokumabom samotným a po ukončení liečby boli
reverzibilné.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
prolín
ľadová kyselina octová polysorbát 80
hydroxid sodný (na úpravu PH)
voda na injekciu
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Repatha 140 mg injekčnýroztoknaplnenýv injekčnejstriekačke
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Repatha 140 mg injekčnýroztoknaplnenýv injekčnompere
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Repatha 420 mg injekčnýroztokv náplni
Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou.
Po vybraní z chladničky sa Repatha môže uchovávať pri izbovej teplote (do 25 °C) v pôvodnom obale a musí sa použiť v priebehu 1 mesiaca.
6.5 Druh obalu a obsah balenia
Repatha 140 mg injekčný roztok naplnený v injekčnejstriekačke
Jeden ml roztoku v jednorazovej naplnenej injekčnej striekačke zo skla typu I a s ihlou hrúbky 27 G
z nehrdzavejúcej ocele.
Kryt ihly naplnenej injekčnej striekačky je zo suchého prírodného kaučuku (derivát latexu, pozri časť
4.4).
Balenie po jednej naplnenej injekčnej striekačke.
Repatha 140 mg injekčný roztok naplnený v injekčnompere
Jeden ml roztoku v jednorazovom naplnenom injekčnom pere zo skla typu I a s ihlou hrúbky 27 G
z nehrdzavejúcej ocele.
Kryt ihly naplneného injekčného pera je zo suchého prírodného kaučuku (derivát latexu, pozri časť
4.4).
Balenia po jednom, dvoch, troch naplnených perách alebo multibalenie šiestich (3 x 2) naplnených
pier.
Repatha 420 mg injekčný roztok v náplni
3,5 ml roztoku v jednorazovej náplni z cykloolefínového polyméru s elastomérovou membránou a
piestom ako materiálmi, ktoré sa dostanú do kontaktu s liekom, a s viečkom zo živice. Súčasťou náplne je výsuvný skrutkový mechanizmus. K zostave náplne je pribalená pomôcka na podávanie. Materiál pre dráhu tekutiny v pomôcke na dávkovanie je z nehrdzavejúcej ocele a polyvinylchloridu bez DEHP, s ihlou hrúbky 29 G z nehrdzavejúcej ocele. Pomôcka na podávanie obsahuje batérie
s oxidom strieborným-zinkom, ako aj lepiacu náplasť zhotovenú z polyesterovej pásky s akrylátovým lepidlom. Pomôcka na podávanie je určená na použitie iba s dodanou zostavou náplne, ktorá je naplnená 3,5 ml roztoku.
Balenie po jednej náplni/automatizovanom minidávkovači alebo multibalenie troch (3 x 1)
náplní/automatizovaných minidávkovačov.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pred podaním sa má roztok skontrolovať. Roztok neaplikujte, ak obsahuje čiastočky, je zakalený alebo zmenil farbu. Pred aplikáciou injekcie nechajte liek dosiahnuť izbovú teplotu (do 25 °C), aby ste predišli nepríjemnému pocitu v mieste vpichu. Aplikujte celý obsah.
Nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAmgen Europe B.V. Minervum 7061
4817 ZK Breda
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)Repatha 140 mg injekčný roztok naplnený v injekčnejstriekačkeEU/1/15/1016/001 - 1 naplnená injekčná striekačka
Repatha 140 mg injekčný roztok naplnený v injekčnompereEU/1/15/1016/002 - 1 naplnené pero
EU/1/15/1016/003 - 2 naplnené perá
EU/1/15/1016/004 - 3 naplnené perá
EU/1/15/1016/005 - 6 (3 x 2) naplnených pier (multibalenie)
Repatha 420 mg injekčný roztok v náplniEU/1/15/1016/006 - 1 náplň s pribaleným automatizovaným minidávkovačom
EU/1/15/1016/007 – 3 (3 x 1) náplne s pribalenými automatizovanými minidávkovačmi (multibalenie)
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 17. júla 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1, 4817 ZK, Breda, Holandsko